structure and organization of the pel genes from erwinia
TRANSCRIPT

Vol. 170, No. 8
Structure and Organization of the pel Genes fromErwinia chrysanthemi EC16
S. J. TAMAKI, S. GOLD, M. ROBESON, S. MANULIS, AND N. T. KEEN*
Department of Plant Pathology, University of California, Riverside, California 92521
Received 2 February 1988/Accepted 21 April 1988
The peLA and pelC genes from Erwinia chrysanthemi EC16 were sequenced and overexpressed in Escherichiacoli cells. These genes and two others from the same strain that were characterized previously encodecatalytically related pectate lyase proteins that are involved with the maceration and soft-rotting of plant tissue.The pel genes of strain EC16 were organized as two loosely linked clusters, with two structurally homologousgenes in each. The peLlAE cluster also contained the remains of an additional pel gene, the 5' portion of whichhad been removed by a prior deletion event. Each of the four functional pel genes but not the deleted one
contained an efficient rho-independent transcriptional terminator after the translational stop. These and otherdata indicate that the pel genes are all independently regulated despite their structural homology and tandemclustered organization. Two of the genes, pelA and pelE, encoded proteins that differed greatly in theirisoelectric points and ability to macerate plant tissue. A recombinant gene constructed with the 5' portion ofpelE and the 3' portion of pelA yielded a chimeric protein with high pectate lyase activity but relatively lowmaceration activity. This result raised the possibility that the poor maceration ability of the pelA gene productmay involve other properties in addition to its low isoelectric point.
Substantial evidence (4, 5, 13) has established that theproduction of several pectate lyase (PL) proteins is causallyinvolved in the soft-rotting disease of plant tissue caused byErwinia chrysanthemi. Furthermore, high-level productionof the E. chrysanthemi PLe protein enabled Escherichia colicells to efficiently macerate potato tuber tissue (12, 19).Erwinia spp. secrete several additional enzymes that attackhigher-plant cell walls or membranes. These include xyla-nase, cellulase, protease, phospholipase, pectin lyase, andpectin esterase, but their role in pathogenicity has not yetbeen established (13).Most strains of E. chrysanthemi studied to date produce
five different PLs encoded by unique pel genes (14), butBarras et al. (2) demonstrated that strain EC16 producesonly four different PL proteins. The pel genes encoding theseproteins have been found to occur in two clusters on the E.chrysanthemi EC16 chromosome (1, 2, 11, 12). We initiallyisolated two different cosmid clones, pPEL3 and pPEL7,which encoded different PL proteins (11). It was subse-quently shown that cosmid clone pPEL3 encoded two dif-ferent PL proteins, but a 6.6-kilobase (kb) subclone(pPEL34) contained only one gene (2; Thurn and Chatterjee,personal communication; Lei and Wilcox, personal commu-nication). We previously sequenced one of these genes(peiB) as well as the 3' portion of a closely linked gene,assumed to be pelC (12). In this paper we report the fullsequence of the pelC gene.Plasmid pPEL74, a subclone of cosmid pPEL7 (11), was
also found to contain two different pel genes (2; Collmer,personal communication). We previously sequenced one ofthese genes, pelE, and overexpressed it in Escherichia coli(12). In this study we subcloned and sequenced the pelAgene, which is closely linked to pelE in pPEL74. Despiteconsiderable homology, the isoelectric points of the proteinproducts encoded by these genes are considerably different(pH 4.6 for PLa and pH 9.8 for PLe). The pelA protein is alsoca. 1,000 times less efficient in maceration of plant tissue than
* Corresponding author.
the pelE protein. In order to determine whether this differencewas due to the isoelectric point or to some other feature of theproteins, we constructed recombinant genes and tested theresultant proteins for maceration activity in plant tissue.
MATERIALS AND METHODS
Recombinant DNA methods. The E. coli strains, phages,and plasmids used are shown in Table 1. Plasmid constructswere generally made by the soft agarose cloning method ofCrouse et al. (6). DNA-modifying enzymes were purchasedfrom New England Biolabs, Bethesda Research Laborato-ries, or Boehringer Mannheim. Transformation of E. colicells and miniboil plasmid extractions were done as de-scribed previously (12). Large-scale plasmid isolations were
generally done by the alkaline lysis method (16).DNA sequencing. A series of exonuclease III (ExoIII)/S1
nuclease deletions were made on both orientations of DNAfragments cloned in pUC118 or pUC119 by the method ofHenikoff (8). Following plasmid religation, deletions were
transformed into strain DH5ot. Appropriate deletions were
selected and transformed into E. coli MV1193, which was
then transfected with lambda M13K07, and templates ofsingle-stranded plasmid DNA were isolated (27). Thesetemplates were sequenced by the dideoxy method as de-scribed previously (12). Data were analyzed by the BIONETsystem (supplied through Intelligenetics Corp., MountainView, Calif.) or by the programs of Pustell and Kafatos (20).
Cell culture and plant maceration assays. E. coli cellscarrying various plasmids were grown to the stationaryphase at 28°C in 15 ml of LB medium with the appropriateantibiotics and additives. Cells were recovered by centrifu-gation, and periplasmic fractions were prepared as describedpreviously (11). Plant tissue maceration assays were per-formed with cucumber fruit mesocarp tissue as describedpreviously (24).
Analytical techniques. Sodium dodecyl sulfate (SDS)-poly-acrylamide gels of whole E. coli cells were run as describedpreviously (12). Thin-layer electrofocusing of PLs from E.coli periplasmic fractions and polygalacturonate overlay as-
3468
JOURNAL OF BACTERIOLOGY, Aug. 1988, p. 3468-34780021-9193/88/083468-11$02.00/0Copyright © 1988, American Society for Microbiology

E. CHRYSANTHEMI EC16 pel GENES 3469
TABLE 1. Bacterial strains, phage, and plasmids
Strain, phage, or plasmid Description Source or reference
E. coliDH5a
MV1193
endAl hsdRJ7 (rK MK+) supE44 thi-J X- recAl gyrArelAl 480dlacZAM15, A(lacZYA-argF)U169
A(lac-proAB) thi rpsL endA sbcB15 hspR4A(srl-recA)306::TnJO (Tet') [F'::traD36 proABlacIqZAMJ5]
F- hsdS20 (hsdR hsdM) recAJ3 ara-14 proA2 lacYlgalK2 rpsL20 (Strr) xyl-5 mtl-l supE44 X-
HB101 (lacJq y+) [X xis (ASalI-XhoI)Kil c1857](A[lac pro] F' lacPZAM]5 pro')
HB101
D1210ABMH71-18
Phage lambda M13K07
Bethesda Research Laboratories
Messing, unpublished
16
723
27
PlasmidspBR322pUC18 and pUC19pUC118 andpUC119
pBluescript KSpJRD184pINK1pNH8apUR290
pPEL3pPEL401
pPEL402
pPEL403
pPEL405
pPEL406
pPEL407
pPEL410
pPEL413
pPEL74
pPEL7421
pPEL7422
pPEL712
pPEL743
pPEL748
pPEL760
Cloning plasmidCloning plasmidPlasmid for production of single-stranded DNA
Cloning plasmidCloning plasmidExpression vectorExpression vectorPlasmid containing lacZ with a polylinker site at the 3'end of the gene for construction of gene fusions
pHC79 cosmid clone containing pelB and pelC8.2-kb ClaI fragment from pPEL3 cloned into pBR322and containing the pelB and pelC genes
3.2-kb ClaI-AvaI fragment from pPEL401 cloned intothe same sites of pBR322 and containing only pelC
3.2-kb ClaI-XhoI fragment from pPEL401, containingthe pelC gene, cloned into the ClaI-XhoI sites ofpBluescript KS (anti orientation to the vector lacpromoter) (PL positive)
1.4-kb Sacl-Aval fragment of pPEL401 inserted (afterend-filling both termini with Klenow fragment) intothe HincIl site of pUC119 (anti orientation to thevector lac promoter) (PL positive)
1.4-kb HindIII-EcoRI insert of pPEL405 inserted intopUC119 in the opposite orientation (in the correctorientation behind the vector promoter) (PLpositive)
1.0-kb PstI fragment of pPEL405 cloned into pUC118(PL negative)
pelC expression plasmid containing a 1.2-kb fragmentobtained from pPEL406; an ExoIlI sequencingdeletion including 41 bp of DNA 5' to thetranslational start codon and extending to the filledAvaI terminus was removed from the pPEL406deletion clone with HindIII and EcoRlI and ligatedinto the same sites of pINK1 (PL positive)
pelC expression plasmid; a 1.4-kb BamHI-SphIfragment from pPEL406 was cloned into the samesites of pNH8a (in the correct orientation followingvector promoter inversion)
8.2-kb PstI fragment containing pelA and pelE, clonedin pBR329
1.2-kb EcoRI-SalI fragment carrying pelE only, clonedin the orientation downstream of the promoter inpUC8
Same construction as pPEL7421, except cloned in theopposite orientation to the promoter in pUC19
pPEL 7421 with deletion of a 360-bp internal EcoRVfragment (PL negative)
2.0-kb HindIII-SalI fragment from PEL74 cloned intopUC19; contains pelE and 5' DNA
Translational fusion containing the pelE gene frompPEL7421 fused to pINIII
1.2-kb EcoRI-SalI insert fragment from pPEL7421blunted with S1 nuclease and recloned into theSamI site of pUC119, in the orientation downstreamfrom the vector promoter (PL positive)
Continued on following page
162927
Stratagene912723
11This paper
This paper
This paper
This paper
This paper
This paper
This paper
This paper
11
12
This paper
12
12
12
This paper
VOL. 170, 1988
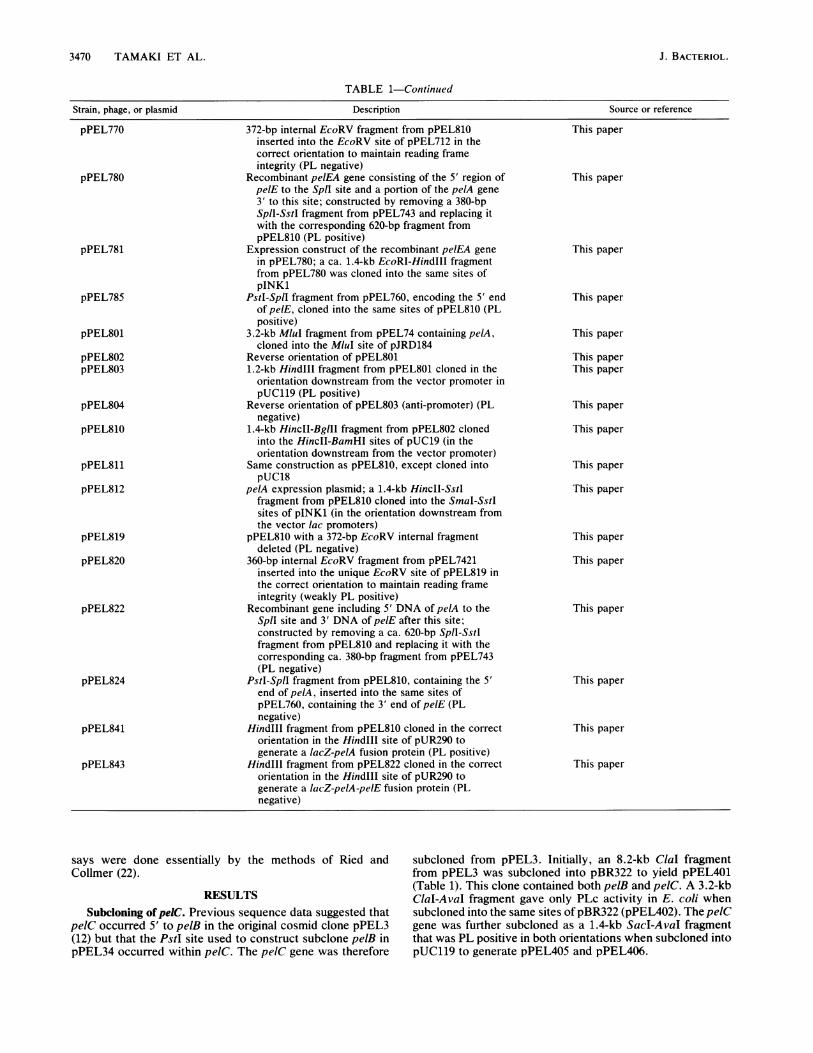
3470 TAMAKI ET AL.
TABLE 1-Continued
Strain, phage, or plasmid Description Source or reference
372-bp internal EcoRV fragment from pPEL810inserted into the EcoRV site of pPEL712 in thecorrect orientation to maintain reading frameintegrity (PL negative)
Recombinant pelEA gene consisting of the 5' region ofpelE to the SpIl site and a portion of the pelA gene3' to this site; constructed by removing a 380-bpSpll-SstI fragment from pPEL743 and replacing itwith the corresponding 620-bp fragment frompPEL810 (PL positive)
Expression construct of the recombinant pelEA genein pPEL780; a ca. 1.4-kb EcoRI-Hindlll fragmentfrom pPEL780 was cloned into the same sites ofpINK1
Pstl-SplI fragment from pPEL760, encoding the 5' endof pelE, cloned into the same sites of pPEL810 (PLpositive)
3.2-kb MluI fragment from pPEL74 containing pelA,cloned into the MluI site of pJRD184
Reverse orientation of pPEL8011.2-kb Hindlll fragment from pPEL801 cloned in the
orientation downstream from the vector promoter inpUC119 (PL positive)
Reverse orientation of pPEL803 (anti-promoter) (PLnegative)
1.4-kb HincII-BglIll fragment from pPEL802 clonedinto the HincII-BamHI sites of pUC19 (in theorientation downstream from the vector promoter)
Same construction as pPEL810, except cloned intopUC18
pelA expression plasmid; a 1.4-kb HincII-SstIfragment from pPEL810 cloned into the SmaI-SstIsites of pINK1 (in the orientation downstream fromthe vector lac promoters)
pPEL810 with a 372-bp EcoRV internal fragmentdeleted (PL negative)
360-bp internal EcoRV fragment from pPEL7421inserted into the unique EcoRV site of pPEL819 inthe correct orientation to maintain reading frameintegrity (weakly PL positive)
Recombinant gene including 5' DNA of pelA to theSplI site and 3' DNA of pelE after this site;constructed by removing a ca. 620-bp SplI-Sstlfragment from pPEL810 and replacing it with thecorresponding ca. 380-bp fragment from pPEL743(PL negative)
PstI-SpIl fragment from pPEL810, containing the 5'end of pelA, inserted into the same sites ofpPEL760, containing the 3' end of pelE (PLnegative)
HindIll fragment from pPEL810 cloned in the correctorientation in the HindIll site of pUR290 togenerate a lacZ-pelA fusion protein (PL positive)
HindIll fragment from pPEL822 cloned in the correctorientation in the HindlIl site of pUR290 togenerate a lacZ-pelA-pelE fusion protein (PLnegative)
This paper
This paper
This paper
This paper
This paper
This paperThis paper
This paper
This paper
This paper
This paper
This paper
This paper
This paper
This paper
This paper
This paper
says were done essentially byCollmer (22).
the methods of Ried and
RESULTSSubcloning of peiC. Previous sequence data suggested that
pelC occurred 5' to pelB in the original cosmid clone pPEL3(12) but that the PstI site used to construct subclone pelB inpPEL34 occurred within peIC. The pelC gene was therefore
subcloned from pPEL3. Initially, an 8.2-kb ClaI fragmentfrom pPEL3 was subcloned into pBR322 to yield pPEL401(Table 1). This clone contained both pelB and pe1C. A 3.2-kbClaI-AvaI fragment gave only PLc activity in E. coli whensubcloned into the same sites of pBR322 (pPEL402). The pelCgene was further subcloned as a 1.4-kb SacI-AvaI fragmentthat was PL positive in both orientations when subcloned intopUC119 to generate pPEL405 and pPEL406.
pPEL770
pPEL780
pPEL781
pPEL785
pPEL801
pPEL802pPEL803
pPEL804
pPEL810
pPEL811
pPEL812
pPEL819
pPEL820
pPEL822
pPEL824
pPEL841
pPEL843
J. BACTERIOL.

E. CHRYSANTHEMI EC16 pel GENES 3471
TABLE 2. PL produced in the periplasmic space of E. coli cellscarrying various pel gene constructs
PL activityaPlasmid Gene (U/g [fresh wt] of cells)
Uninduced Induced
pPEL401 pelC 50 136pPEL402 pelC 170 150pPEL403 pelC 1,200 1,040pPEL410 pelC 1,490 3,500pPEL413 pelC 20 2,400pPEL801 pelA 3 4pPEL803 pelA 13 22pPEL804 pelA <0.1 <0.1pPEL810 pelA 210 290pPEL812 pelA 1,470 2,925pPEL748 pelE 1,770 bpPEL781 pelEA 1,430 5,600pPEL822 pelAE <0.1 <0.1pPEL841 lacZ-pelA 2,150cpPEL843 lacZ-pelAE <O.1C
a DH5a cells were used in all experiments with the following exceptions:pPEL413, strain D1210X; pPEL748, HB101; and pPEL841 and pPEL843,strain BMH71-18. All cultures were grown at 28°C for ca. 20 h beforespheroplasting was done to obtain the periplasmic fraction (12). IPTG wasused for induction and added to 1 mM at culture initiation; IPTG was addedto all cultures containing pPEL413, and induction in this case was achieved byincubation at 42°C for 15 min when cultures had attained an A6M of ca. 0.7.Plating of cells from these cultures on pectate plates showed that >95% wereinduced to produce high levels of PL activity. PL activities were assayed inthe culture medium and cellular fractions as done previously (12). Since thesefractions each contained 10%o or less of the total PL activity in all cases, onlydata for the periplasmic fractions are reported.b-, Not determined.c Whole cells were lysed by sonication and used for the assay in these
cases.
Overexpression of the pelC gene in E. coli. In order tooverexpress the pelC gene, various expression constructswere made. As noted in Table 2, E. coli cells carryingpPEL401 or pPEL402 produced only low levels of PLactivity, but a 3.2-kb subclone, pPEL403, produced higherlevels. This plasmid was constructed by cloning the insertDNA in the opposite orientation from the lac promoter ofpBluescript KS (Table 1). This result suggested that the pelCpromoter was active in E. coli because, as expected, cellscarrying pPEL403 did not exhibit increased PL production inthe presence of IPTG (isopropyl-,-D-thiogalactopyranoside)(Table 2). pPEL410, an expression construct in pINK1,contained only 41 base pairs (bp) of DNA 5' to pelC andresulted in high PLc production in the presence of IPTG.However, as previously observed with other constructs inthe pINKi expression plasmid (12), the uninduced level wasalso quite high (Table 2). Construct pPEL413 was con-structed with a reversible promoter vector, pNH8a. Expres-sion ofpelC by pPEL413 in the uninduced state was consid-erably lower than that by the pINK construct, pPL410(Table 2). The induced level of PLc in cells containingpPEL413 was nearly as high, however, as in those contain-ing pPEL410. Thus, pNH8a resulted in approximately 100-fold induction. PL activity was efficiently secreted to theperiplasmic space in all cases (Table 2).
Subcloming ofpelA. Subcloning disclosed that an additionalpel gene occurred 5' to pelE on pPL74. This gene, presumedto be pelA, was initially subcloned as a 3.2-kb MluI fragmentwhich expressed PL activity in both orientations whencloned into the MluI site of pJRD184 to yield pPEL801(Tables 1 and 2). Further subcloning showed that a 1.2-kbHindIll fragment from pPL801 gave low PL activity when
cloned downstream from the promoter in pUC19 (pPEL803)but yielded no detectable activity when cloned in the oppo-site orientation (pPEL804; Table 2). This was later found tobe due to the construction of a translational fusion inpPEL803, since the 5' HindIII site occurs inside the ATGstart codon (see Fig. 4).
High-level expression of the pelA gene in E. coli. A conve-nient HinclI site just 5' to the Shine-Dalgarno box of pelAwas located from sequencing data (see below) and used toconstruct pPEL810 in pUC19. This construct producedmoderate PL activity in E. coli (Table 2). The insert was alsotransferred to pINK1 to yield pPEL812. This construct ledto high PLa activity in the periplasmic space of E. coli cellswhen IPTG was supplied (Table 2). Significant expression(ca. 50% of the induced level) occurred in the absence ofIPTG.
Sequencing of peiC. The 1.4-kb insert in pPEL405 andpPEL406 was sequenced to identify the pelC gene (Fig. 1).As expected, a single open reading frame (ORF) was iden-tified that exhibited considerable homology with the pelBgene. As noted previously (12), pelC contains a sequenceafter the translational stop which would be expected tofunction as an efficient transcriptional terminator. The puta-tive signal peptide sequence of pelC also showed consider-able homology with that of pelB (Fig. 2), but the pelCcleavage site has not been definitely assigned by N-terminalamino acid sequencing of the mature protein. Significantoverall homology did not occur between the 5' noncodingDNA of pelC and that of pelB, but possible promoterelements were identified in both genes (Fig. 3). The pelCsequence, however, exhibited less similarity to the E. coliconsensus promoter. Unlike pelB (12), no identifiable catab-olite repressor-binding site was present in the 5' DNA ofpelC. The predicted molecular weight of the PLc preproteinwas 39,923 and that of the mature protein was 37,676. Thiscorresponds to values of 40,213 and 37,922, respectively, forPLb, determined previously (12). The proteins encoded bypelB and pelC had 84% amino acid identity, with twocompensatory single amino acid deletions occurring in whatwere otherwise colinear reading frames (Fig. 2).
Sequencing ofpeLU and detection of a deleted pel gene. The3.2-kb insert of pPEL801 contained a single long ORFcorresponding to pelA, located ca. 900 bp 5' to the start ofthe pelE gene (Fig. 4). No additional ORFs of significantlength were observed in either strand of the ca. 1 kb ofDNA5' to pelA. A Shine-Dalgarno box occurred just before theassumed start codon of the pelA gene at position 1099. The5' ends of the pelA and pelE coding regions were dissimilar,and the pelA gene product had a longer putative signalpeptide sequence than that of pelE (Fig. 5). However, thepredicted cleavage site of the PLa preprotein has not yetbeen confirmed by N-terminal amino acid sequencing of themature protein. The preprotein encoded by pelA had 393amino acids and a calculated molecular weight of 42,077.The putative mature protein contained 361 amino acids witha calculated weight of 38,756. The pelA gene product alsohad more amino acids at the amino-terminal end of themature protein than PLe (Fig. 5). Although several shortcompensatory deletions occurred in the remainder of thecoding regions of pelA and pelE, the mature proteins had62% amino acid identity and read colinearly (Fig. 4 and 5).Both genes terminated with TAA stop codons. There was arelatively large amount of intergenic DNA between theORFs constituting pelA and pelE (ca. 900 bp), and the 5'untranslated ends of both genes had unusually long stretchesof AT-rich DNA (e.g., base 830 to the translational start at
VOL. 170, 1988

3472 TAMAKI ET AL.
10 20 30 40 50 60* * a * * a
A GTC0C MA CTA TTA GM MT CTA TTC OAT CAA GAT AAT GMAA GrA ['T CAA [TA
70 80 90 100 110 120
T' AAT 10T TTG AlT TTT ACA CTA AGA C0 A G 10 GTT ACC OGA ATA GAT 100 CAA 0cc
130 140 150 160 170 180* I 1 I.
TTA OCA AM CAT ccC aGT CAA CAC OCA CTA OM G0G CAG QGA ATA ACT CAT C0C CAC AAC
190 200 210 220 230 240
ATA mT AM GTr TAG AGA AAA MA ATA CG OCT ATG AAA TCA CTC ATT ACC OCA ATT AOCMet Lys Sr Leu nle T1r Pro ne TDr
250 250 270 280 290 300
OCT COA C0 TTA CM1 000 CTC AOC CAA OCT TIG CT1 000 OCT AOC GAT ACC 0GT GOC TACAla Gly Lu Lae L Ala La Ser Gin PO Lae La Ala Ala Dr AsP Dr Gly Gly TYr
310 P I 320 330 340 350 3603 Pst *I *G00 OCT ACT OCA 000GooC MC 010 AaG a0 0 01GTC AOC AAG ACC OCA AcC lTC A1G CAAla Ala TDr Ala Gly Gly Asn Val Tr Gly Ala Val Ser Lys TDr Ala DTr Ser Met Gln
370 380 390 400 410 420* * * I I I
GAT ATC GTC AAT ATM AlT GAC G0C OCA CGT CTG GAT G0C MC GOC AAA AAA G1 AAA OOCAsp ne Val Asn fle ne Asp Ala Ala Arg Leu Asp Ala Asn Gly Lys Lys Val Lys Gly
430.
440 450 460 470 480* I * * I
000 000 TAT 000 CmG [GTA ATC AC TAT AGO 007 MC GMA OAC 11 ['G ATC AAC C0C GCTGly Ala Tyr Pro La Val ne Thr Tyr TDr Gly Aan Giu Asp Ser Lau ne Asn Ala Ala
490 500 510 520 530 540* I* I*I
G0C C AAT ATC 1C G0C CAG 0G AMC AAA GAC C00 GOT G0C GTA GMA ATC AAA GAG TICAla Ala Asn ne Cys Gly Gln Tr-p Ser Lys Asp Pro Arg Gly Val Glu ne Lys Glu Phe
550 560 570 580 990 600
ACC AAA G0C ATC ACC ATC ATT G0 0 MC00A TOT 110C0CC AAC TIC G0C ATC 1OG ATTDtr Lys Gly ne TDr ne ne Gly Ala Asn Gly Ser Ser Ala Asn Pte Gly ne Thp nle
610 620 630 640 650 660* I * * * I
AAG AAA TCC 10 GAT G01 G01 GTA CAG AAC A1 OUT AlC G0C TAC CM COG GacG0000 CTLys Lys Ser Ser Asp Val Val Val Gln Asn Met Arg ne Gly Tyr Leu Pro Gly Gly Ala
670 680 S 690 700 710 720
AAA GAT 00C GAC A1G A0 00C G0C GAC GAT TCG CG AAT GTC 10G GTT GAC CAT AAC GAALye Asp Gmy Asp met fle Ag Val Asp Asp Ser Pr Amn Val Trp Val Asp is Aan Glu
730 740 750 760 770 780* I I * * I
TI TTC 000 000 MC CAT ON] IC OAC GOC ACA COG CAC MC GAC ACC ACC mT GM T1CLeu Phe Ala Ala Asn Hs Glu Cys Asp Gly Dhr Pro Asp Asn Asp Dtr TDr Phe Glu Ser
790 800 810 820 830 840* * * * * I
G0C GTT GAT AlT AAG aGC G00 7C AAC ACC 0T0 AOC GTC0CC TAC AAC TAC AlT CAC OGTAla Val Asp fe Lys Gly Ala Ser Asn Ttr Val TDr Val Ser Tyr Asn Tyr ne His Gly
850 860 870 880 890 900
GTG AAA AAA G0 0UT cm GAT 0GT TCC AOC AGC AGT GAT AOC GOC 0OC AAT AT ACC TATVal Lys Lye Val Gly Lae Asp Gly Ser Ser Ser Ser Asp DTr Gly Arg Asn ne DTr Tyr
910 920 930 940 950 960* * * * * I
CAC CAT AAC TAC TAC AAC GAC GTT AAC C0C CGT cm COG TG CAA CGT 0GT 007 cm G0THis His Asn Tyr Tyr Asn Asp Val Asn Ala Arg Leu Pro Lau Gln Arg Gly Gly Leu Val
970 980 990 1000 1010 1020* * * * I
CAC OCT TAC AAC AAC cm TAC ACC AAC ATC ACC 007 TCC GOC cm AAC G1 0OC CAG AACHis Ala Tyr Asn Asn Leu Tyr TDr Asn ne Thr Gly Ser Gly Leu Asn Val Arg Gln Asn
1030 1040 1050 1060 1070 1080* * * * * £
G0C CAG OCA CTG ATC GAA AAC AAC 1G TICTG AAG 0G0 ATA AAC OCG GTA AOG TCC CGTGly Gln Ala Leu fle Glu Asn Asn Trp Pth Glu Lys Ala ne Asn Pro Val Ttr Ser Arg
1090 11°KpnI 1110 1120 1130 1140
TAT GAC GC AAA AAC TTC '¢FGT AC 1 G1 cm AAA 00C AAC AAC ATC AO AAA OG0Tyr Asp Gly Lys Asn Phe Gly Ttr Trp Val Leu Lys Giy Asn Asn ne Ttr Lys Pro Ala
1150 1160 1170 1180 1190 1200* * * * * I
GAC TTC TCT ACC TAC AGC AT ACC T10 ACG G0C GAC ACC AMG CCT TAT G1 AAT G00 GACAsp Phe Ser TDr Tyr Ser fle Tr Trp TDr Ala Asp Tr Lys Ro Tyr Val Asn Ala Asp
1210 1220 1230 1240 1250 1280* * * * * 1
AGC T10 ACC 110 AOC GOC AOC 7TC CCG ACC G01 GCC TAC AAC TAC AGC C00 G0C AGC OCASer Trp TDr Ser Dhr Giy Tr Phe Pry TDr Val Ala Tyr Asn Tyr Ser Pro Val Ser Ala
1270 1280 1290 1300 1310 1320* * I * * I
CAA mc Gm AM] GAC AAA CM CCT 0GC TAT G0C GOC G10 07T AAA AAT CM 0CC ACG CTGGln Cys Val Lys Asp Lys Leu Pro Giy Tyr Ala Giy Val Giy Lys Asn Leu Ala TDr Lau
1330 1340 1350 1360 1370 1380* * * * * I
ACC AOC ACA G0 &T AAA TAA TCT CAC OCA GAG GCA ¢¢C OCA 1TC 000 T&A CIC ACC 0G0Ttr Ser DTr Ala Cys Lys
1390 1400 1410 1420 1430TXhoTI 143 XhoIT[TrA11 CAC OCT AGOA00 GOC lOA AM0()OCTW CTr ['I TM CTAaCxI AlT CAC 11 W]A
FIG. 1. Sequence of the pelC gene and flanking DNA in pPEL405; the sequence 3' to the KpnI site was also determined previously (12).The Shine-Dalgamo sequence preceding pelC is underscored, and selected restriction sites are shown.
base 1099 in pelA and position 2860 to the translational startof pelE at base 3198 [Fig. 4]; a particularly AT-rich stretchoccurred 5' to pelE between positions 2871 and 2931). Nosignificant homology was noted between the AT-rich regionspreceding pelA and pelE. Similar to pelB, pe1C, and pelE, aGC-rich palindromic sequence followed by a T repeat oc-curred after the translational stop ofpelA at positions 2295 to2320 (Fig. 4).The DNA sequence data (Fig. 4) disclosed the presence of
a short open reading frame beginning at ca. base 2325,immediately following the putative pelA transcriptional ter-minator. This ORF had considerable DNA and amino acidhomology with pelA and pelE (Fig. 5), but it encoded onlythe carboxy-terminal 132 amino acids of a putative PLprotein. The observed high homology and the similarity ofarrangement of this ORF and pelA and pelE with the pelADEclusters of other E. chrysanthemi strains (21) established
that it represents the remains of a pel gene, provisionallycalled pelD, in which about 2/3 of the 5' end has beendeleted, in strain EC16. Interestingly, a GC-rich palindromicsequence occurred at bases 2737 to 2766, following thetranslational stop ofpelD, but it was not followed by a stringof T or AT residues.
Construction of recombinant genes between peLA and pelE.Despite their considerable amino acid homology, the pro-teins encoded by pelA and pelE had widely different isoelec-tric points, 4.6 and 9.8, respectively. The pelA gene productwas also ca. 1,000-fold less efficient in maceration of potatotuber tissue than the pelE gene product (Table 3). In order toinvestigate whether this considerable difference in biologicactivity was due only to the isoelectric point disparity of thetwo proteins or to other reasons, recombinant pelAlE geneswere constructed. In the first constructs, a conserved inter-nal EcoRV fragment (Fig. 4 and 7) was interchanged, gener-
J. BACTERIOL.

E. CHRYSANTHEMI EC16 pel GENES 3473
1 MKSLITPIAAGLLLAFSQYSLAA-DTGGYTKTDGGDVSGAVKKTASSMQDIVNIIE
1 MKSLITPITAGLLLALSQPLLAATDTGGYAATAGGNVTGAVSKTATSMQDIVNIID
56 AAKVDANGKKVKGGAYPLVITYTGNEDSLINAAAANICGQWSKDARGVEIKDFTKG
57 AARLDANGKKVKGGAYPLVITYTGNEDSLINAAAANICGQWSKDPRGVEIKEFTKG
112 LTIIGANGSSANFGIWIVNSSDIVVRNMRIGYLPGGAQDGDMFRIDNSPNVWLDHN
113 ITIIGANGSSANFGIWIKKSSDVVVQNMRIGYLPGGAKDGDMIRVDDSPNVWVDHN
168 ELFAANHECDGTKDGDTTFESAIDIKKGATY-VTISYNYIHGVKKVGLSGFSSSDT
169 ELFAANHECDGTPDNDTTFESAVDIK-GASNTVTVSYNYIHGVKKVGLDGSSSSDT
223 AERNITYHHNIYSDVNARLPLQRGGNVHAYNNLYTGITSSGLNVRQNGKALIENNW
224 G-RNITYHHNYYNDVNARLPLQRGGLVHAYNNLYTNITGSGLNVRQNGQALIENNW
279 FENAVSPVTSRYDGSNFGTWVLKGNNITKPADFATYNITWTPDTKEYRNADTWTST
279 FEKAINPVTSRYDGKNFGTWVLKGNNITKPADPSTYSITWTADTKPYVNADSWTST
335 GTYPTVPYSYSPVSAQCVKDKLANYAGVGKNLATLASSACK
335 GTFPTVAYNYSPVSAQCVKDKLPGYAGVGKNLATLTSTACK
FIG. 2. Homology of the protein products of the pelB (top line)and pelC (bottom line) genes. Identical amino acids are connectedby two dots.
ating pPEL770 and pPEL820 (Table 1). Transformants car-rying the former construct, however, did not produce anydetectable PL activity in pectate plate tests, and cells withpPEL820 produced only very weak activity. These con-structs were therefore not investigated further. The pelA andpelE genes also contained conserved, unique SplI restrictionsites (Fig. 4 and 7), and constructions were therefore made inwhich the DNAs 3' to this site were interchanged. One of theresulting constructs, pPEL822, gave no detectable PL activ-ity in E. coli (Table 2), but the reciprocal construct(pPEL780) containing the 5' portion ofpelE and the 3' end ofpelA was active. E. coli cells containing pPEL780 gave highPL activity (data not shown), as did cells containing therecombinant gene cloned in the expression plasmid pINK1(pPEL781, Table 2).
In order to ensure that a inutation was not inadvertentlyintroduced during the construction of the PL-negativepPEL822, the reciprocal construct was made by uisingpPEL810 (containing pelA) and pPEL760 (containing pelE).The resulting chimeric genes in pPEL785 and pPEL824(Table 1) gave the same results in E. coli as the previousconstructs; pPEL785 led to high PL activity, while pPEL824gave no detectable activity (data not shown). Since theproteins encoded by pPEL822 and pPEL824 may not bestable in E. coli, the chimeric gene in pPEL822 was fused atthe HindIII site of the pelA portion at position 1109 (Fig. 4)to the 3' end of lacZ in pUR290 to generate pPEL843. E. colitransformants produced a strong band on SDS-polyacryl-amide gels at ca. 155 kilodaltons (kDa), the predicted weightof the lacZ fusion protein (data not shown). However, these
-35 -lopelB5' TATTGAA TGTATCCTTTTTGAGCTAAAAC GA 3'
.11 11111 111 l 11111 1 111 11lllpelC5' T TTTGAATGTTTGATTTTTACAC TAAGACJTGA 3'
TTGACA TATAATFIG. 3. Homology of possible promoter elements occurring 5' to
the coding regions ofpelB and pelC along with the consensus E. colipromoter sequence (below). Numbering begins with bases 511 (12)and 60 (Fig. 1) for pelB and peIC, respectively.
TABLE 3. Maceration of cucumber mesocarp tissue byperiplasmic fractions of E. coli cells containing overexpressed pel
genes or a recombinant pelEA gene
Minimal PL concnPlasmid Gene (U/ml) required
for macerationa
pPEL812 pelA 45.1pPEL781 pelEA 0.3pPEL748 pelE 0.007pPEL344 pelB 0.04pPEL410 pelC 0.05pPEL841 lacZ-pelA 38.4
a Maceration was determined by incubating serial dilutions of periplasmicfractions with cucumber mesocarp slices for 60 min at 37°C as describedpreviously (24). All assays used a total volume of 1 ml, and all solutions werebrought to 0.02 M Tris hydrochloride, pH 8.0. Five cucumber slices were usedper assay tube. Dilution series of the various enzymes were prepared, and theminimal amounts of PL which caused detectable cell separation during theexperiment are shown.
bacteria contained no detectable PL activity (Table 2). Thecorrespondingi HindIII fusion between lacZ of pUR290 andthe wild-type pelA gene (pPEL841) exhibited high PL activ-ity (Table 2). It therefore appeared that the chimeric pelA-pelE protein lacked PL activity but the recombinant pelE-pelA (pelEA) product (PLea) was fully active.
Characterization of the gene products of pelA, pe11, peiC,pelE, and peE:A. Periplasmic fractions were collected fromE. coli cells containing high-expression plasmid constructsof the four pel genes as well as the recombinant pelEA gene.Electrofocusing of these preparations on thin-layer gelsdisclosed that all of the constructs produced PL proteinswith the expected isoelectric points, as determined by Coo-massie blue staining, and these also gave PL activity follow-ing substrate overlays (data not shown). pelA, contained inpPEL812, produced the PLa protein with a pl of 4.6; pelB,carried by pPEL344, produced the PLb protein with a pl of8.8; pe1C, carried by pPEL410, produced the PLc proteinwith a pI of 9.0; and pelE, contained in pPEL748 (12),produced the PLe protein with an isoelectric point of ca. 9.8;the recombinant PLea protein encoded by pPEL781 gave apI of 7.05 on the electrofocusing gel.SDS gel electrophoresis of E. coli cells carrying various
expression plasmids showed that the pelC gene product ranas a 39-kDa proteins very close to the value found by DNAsequencing. This value was also close to that of the pelBprotein, as determined previously (12). The pelA gene prod-uct as well as the recombinant PLea encoded by pPEL781migrated more slowly in SDS gels (estimated weight, 45 kDa;data not shown) than the ca. 39 kDa determined for themature proteins from DNA sequence data. Thus, like PLe(12), PLa and PLea migrate anomalously slowly on SDSgels. This behavior with PLe was previously suspected to bedue to its relatively high pl (12), but pI can clearly notaccount for the behavior of PLa and PLea.
Maceration of plant tissue. Periplasmic fractions wereprepared from E. coli cells containing expression plasmidsfor various PL proteins and assayed for PL activity andmaceration activity on cucumber tissue slices. Several ex-periments with purified and dialyzed PLa and PLe showedthat ca. 1,000 times more units of PLa were required toproduce the same maceration as PLe (data not shown). Intests with periplasmic fractions from E. coli cells carryingexpression plasmids, it was observed that PLb and PLc gavesimilar maceration activity (Table 3), which was less thanthat of PLe, as noted previously (12). The recombinant PLea
VOL. 170, 1988

3474 TAMAKI ET AL. J. BACTERIOL.
10 20 30 40 50 60 1030 1040 1050 1060 1070 1080MIutI a0
A0oaA cm GTA w TFTi GA arCA A AaT mA M mc TM7 TMc A 0CC AMC rA ATA MT AA MT MC CrA TA ATA TAT AM GA ATT ATT mT AMC ACr TA GAT AM
70 80 90 100 110 120 Hincl 1090 1100pelA 1110HindN1120 113P 1140
MT T'r mc mC AT w7 mC CAT CT WCC&A lt TAA lT WCA Ar TTC AT Ir 0 AGT CAA CTA AM AA AAT AM AM AC MAAAA TCb OGA wr m rT C (C l ANotMb A laSrMyArgS Ph irkAgSir
130 140 150 160 170 180 1150 1160 1170 1180 1190 1200* * a * * a a * a * * *
mAACA 0CATram TACMC AACT CT TT mATrMAAAUTCACCcA WG MA TATCI CIU M MTI AICCa Gr MTAIDAoVT0T( TMVOC OGOC0 GML.ys Tyr Lw Lt Ala Ttr Lm no Ala GMy Met Va Ala 9' Mly Val Sir Ala Ala GMu
190 200 210 220 230 240 1210 1220 1230 12110 129D 1260* * * * * * * * * * * a
0CC (11 MrACA TMG lT AMA o 1ITmG ACr MC GOT GAT AGAWC7lf CAD M TTT TM10(117OC OAT MAA GM TM GMA Ivr 0oC(DC AC M0 900 7CC CM MT 0(1La Val Sur Asp Lys Ala Lw Glu Sir Ala Pr Dhr Val My lp Ala S9r 014 JAn My
250 260 270 20 290 300 1270 120 1290 1300 1310 1320* * * a * * * * * * * *
ITA CDC AM WA 1TrA CT ACA MC TAC ACA MD CIT CAm (C 70 WA TIT AT1 TMCACA CC 00 OCT WA C AOC GAC MT AMC TM AI ( AC MT AI MCPhe DTr Dhr Gy Gly Ala Ala Ala nr Sir App Aa no Tyr no Val Dr Am nIe 9-
310 320 330 340 350 360 Eco RI 1330 1340 1350 1360 1370 1380
CS OCr GA TITT TI mCA TTC 7C ACC MC AMT TT TIC AM 0C G ATT CmTr C GMA TID MOC AGT =C CTT AC aoc OCA GM GMAAM AIJC XT7 CA ATATiMA 0( MGlu Re Dr Si Ala Lwu Sir Ala Gly Ala Mlu Ala Ljp no no Gn no Lys Gy Dr
370 380 390 400 410 12040 1410 14 1430 140EcoR ' .AC MC(MT CMC AAC AMC ADAGGI TT ACMG aoC CACTTC TAT CA0BACOAAM GATAMCAOCGC AM CCTTMCACC ATTIC GM0GT C AAAOCC CCCA
no Asp no si Gy Gy TDr Pr Tyr Dr Asp Rph Ala Ap Gin Lys Ala Arg Sir Gin
430 440 g .jI40 1460 470 480 1450 1460 1170 1480 1190 1900* . BSql . . . . . . . .*
TIC CAC CATMTCMGACMAAAC t MTAMAT MTCAG 0TAMTMCC MTT AACATT0CA0 CC rAM MTCrI amD0 M3C AOCCGM AM TIT AI=Cne AmIle Pro Ala As Dr Ttr Val no Gly Lw Gy Dr Asp Ala Lys Rh no Am
490 500 510 520 530 ,540 1510 1520 1530 1540 15 1560* * * * * * * * * * a a
CMA CGT GaA G0 CM TID MT C AlT lAT 1 GmC 0G IMOOCrTTATCC MT TTA IM 0C ICT CM ATT a7 Ga 0r AIM GAC AC MT AAC GIC AD mcAIDT AMC 07C TATGly swr Lu nle ne Asp Gly Dr Amp GMy Dtr Ann Am Val no no kg Am Val Tyr
550 560 570 580 590 600 157 1580 150 160 1610 162* .BstERH . * a.0C 0CC MTAwTTm MAT 71m aAG MT 0C MA TD ATTM MT GTT 71 GTC ATC CM MC0 ATT GM (TA GM OM CM TC aM MA MrW MC0 M 0AC GM
ne Gin Dir ro ne AP Val Gmu fR H Tyr u ty GMy Asp Gy, lIp Am Ala GMu
610 620 630 640 650 660 1630 1640 1650 1660 1670 1680* * * * * * * * * * * a
CC ATA AM MlA TAT AOC CA CAAAATGACITIAT7TADAACAGGC OMrA TOG GACM CA AATMCAADMT=WACCACCCaTM MD2CA GAT CAT =ACM AClrP Asp Ala Mat Am noe Tr Amn y Ala HIB Ms Vol Tp no Amp Mis Vol Dr neo
670 680 690 700 710 720 1690 1700 1710 1720 1730 1740* * * * * * * * * * * §
OCT T TA TIT a ACC AAT AA TMC OUT CA =C TTA ImT OCA CO CDC ( GAT GAA AMT GM 0M AA TID MC GC GM AM TA MC MC MA GM 0(1 GM MC T A(1M CMSer Asp (y Am Ru TDr Asp Asp Mat Tyr Dr TDr Ly Asp Wy GMu DrT} r Vol Gn
730 740 750 7|60 T70 780 175E Ri'? 1770 1780 1790 1800* a a 0 0 . ECO a* * *tlOT TID AC TAT TT TAr AM TM MA MT Am TMAlT MA rA AMA T 0C CAT GM CO cmGOT AI A C wGM T 7 C AD mIAMc cm
His As Gy Aa Lw AsP no LpsAM Gy Swr AMp Tyr Vol Dr n Sir Ann Sr Lwu
790 800 810 820 830 840 1810 1820 1830 1840 1850 1860
AC DCAA TCCCTTC C C ACCAAA TT TATAITCAATATMA AC GAC CGJC G AAA AICMA C= CAC AGACA A GCCGGne Amp Gin His Asp Lys Dr Mat Lu noe MDy Hi* An Asp Dr Ann Si Ala Gin Asp
850 860 870 880 890 900 1870 1880 1890 1900 1910 19D
TMAGAAATAUA AA AAMIACATTAAMAAA OMOUT TICATTATT GCTAT CAT MA Am OC AAG cM CAT AMCMc TIC AAC AC TATIC AT UACCGU =C0Lys GMy Ls Lwu HB Val Dr Lwu Am Asn Vol Rh Am kg Val Ttr Gu Arg Ala
910 920 930 910 950 9S0 SpI 11930 1940 1950 1960 1970 1980
MA CAAT MA TMA MT GTA TIC ATT MC MAC OCT TAT 00 GAT ACC MC GA AAA IT CT QGC(10 TMC 0C AMC AMC 7A MAC MC (1UC AA WrMA 01 OAT 00 MAPm kg Val A Tyr Gy si no Hi ser Phe Am An Val Rh Lye Gy Asp Ala Iy
970 980 990 1000 1010 1020 1990 m20 2010 KpnM° 20 20
TA TTA Tm ITA AM A AT OAT TM TAT AI A£ AM mC 0DC TM CAT C T1M AM AT O (1TA CTM r TM CM TMCAmC 1 AI C MxC= 0C M (m =mAsP Pro Val yr AMg Tgr GMn Tyr sir PR Gly no Gy Dr ser Gly S Vol Lw Sir

E. CHRYSANTHEMI EC16 pel GENES 3475
2050 2060 2070 2080 2090 2100* 0 * * * 0
GAA 000 MC OC TTC AG ATT OCI MC CTG hAC 0cc AC MA 0CA TC AM MT GM AMGlu Gly An SerhRhe r ne Ala Amn Lu Ser Ala Ser Lys Ala Cys Lys Val Val Lys
2110 2120 2130 2140 2150 2160* * * 0 0 *
AAA TC AAC 001TCCATC TTC TCT GAC AAC MGT 1CC G1C CCl AAC G0C AOC GCC GTC GATLys PRe Aen Gly Serln Phe Ser Asp Asn Gly Ser Val Leu Asn Gly Se' Ala Val Asp
2170 2180 2190 2200 2210 2220* 0 0 * 0 0
CI T GOTMrMC (GT TICAOC GCC TAC ACC aOC AAG hIC OCT TAC ATE TAC GAT GTr CAGLeu Ser Gly Cys Gly Re Ser Ala Tyr DTr Set Lya le Pro [yr fle Tyr Asp Val Gin
2230 2240 2250 2280 2270 2280
COG ATG ACG ACC GMA CI CGM CAG 1TC ATT ACG GAC MAC 0CC 00TCT GOC AMA CMG TAAPr Met Itr Tr Glu Leu Ala Gin Set ne Thr Asp Asn Ala Gly Ser Gly Lys Leu
2290 2300 2310 2320 pel D2330 2340
TT1TMG (T ATC CMA CM0 Cm CCC X00 CAG 0am m TEA TTC OCrT TEAMC 00 TTC OACAla Leu AsnAm g Re Ap
2350 2360 2370 2380 2390 2400* 0 0 * * 0
GOT GT ACT C( 0CC TTA 0C TT aGOT AC GT CAT CAC AC AM CM T TAC ACC ¢CAM AM Dr P kg LeAkg Pe Gly ham Val EIa Ala Tyr han An Val Tyr Dr Gly
240Hi nd[2420 2430 2440 2450 2MO0OAT GTC MT CIT Am iAc 0Cc TAT CrG TIC AOC TTaO cAmCCahC AoCAGC CIO CIGAP Va hanAm lyM Ala Tyr Ag Tyr Gn Tyr Se PRe Gly Dr Se Gly Ser Leu Leu
2470 2480 2490 2500 2510 2520* 0 0 0 * 0
TCT GAG AAC AAT 0A TIT ACC ATE GAT AAC CIC AG AAG ATC AAC a0C 0CC OAT AAG GAASet Giu An Asn Ala Pe TDr fle Asp han Lou Lya Ly ne An (fly Ak hap MuGiu
2530 2540 2550BgIIff2%0 2570 2580
TGT AOC 01C ¢C MAA 00 mI MC 0 A C lTM AOAMA 00ATCAlT AT) MCCy Se Val Val Lye Ala Rhe an Gly Ls nle Re Sw hap y Gly Set fe ne An
2650 Ssp I 260 2670 26 2690z7*0CCT A TAT CI OCr CIAC AOCATC AOCMC wCAACwCALC AiC AwC AoC ANT
Pro Tyr Ly Tyr Ser Ala Gin Dr ne Tr Dr Asn Lau Ala Asn Ser ne Sew Ser Asn
Z710 Z720 2730 Z40 2750 2760* * * * * §
Ga Ga TAc aoC AAA cm TAA rr GA TTC ACT 1G GAA MGC 0CC OG AAA CG CG OCTAla Gly Tyr Gly Lys Leu -
Z770 Z780 z790 2800 2810 2820* 0 * * * 0
TO CIC CIC ACC AAG TCA IrA MA ItT CA ATT T7 TMA CAA= TAT CAG CO0 OGA AAA
2830 2840 2850 2860 2870 2880* * * 0 0 0
AOC AGT GTC TAT CAC MC AMA TAG 0GG CIT COCGCA TTA ATA AG0 CAC TTA MT TMA TAA
2890 2900 2910 2920 2930 2940* 0 0 * 0 0
AAT TAT ATT TAA TM MMA TG MT TAT TGT TTA ACG GT TTA TIT ATA TCA GOC M AAT
2950 2960 2970 2980 2990 3000* 0 * 0 0 0
TCC GGA TCA TAT GAC 1GA ATT TAA AG AAA ATT CMATA MCAAC CAT TAG CAA MG TTA
3010 302D 3030 3040 3050 3060* 0 * 0 0 0
COG GTC 1G ATC ACM GTT TAG ATA AAA TEA ACA ACA 0CC ATA AAA AAA A GAT GOAT
3070 3080 3090 3100 3110 3120* * * 0 * 0
cCAAA ACA TCT CCC OC AM TGC TTA AAA ATC CAC CCTT GCC G0G CAA MT CDC ATE
3130 3140EcoR3150 3160 3170 3180
1TCm IT CAC MA 0C0 CIT mT GM TTC TEA ACA ATG CAT TM GAT TAG CXC CTA CAG
2590 2600 2610 260 2630 2640 3190 peIE 320 MIu 1 3210
GOc0CC TAC AAC CG AAMCGGTTOC00CTTmGC TTO MCACC TAT 10GCCAAGAAT)C GA A1G AAA 00G TT AG AAA MCAAG GOTMy Ala Set Tyr han Lu han (ly Cy Gly Re Gy Re hsn Dr t'r S Ala J.ya neFIG. 4. Sequence of the pelA gene and associated 5' DNA, the 3' end of a deleted gene presumed to be pelD, and intergenic DNA between
the 3' end of pelD and the previously sequenced pelE gene (12). Sequence data shown are for the 3.2-kb insert of pPEL801. Selectedrestriction sites are shown. Palindromic sequences are indicated by arrows, and Shine-Dalgarno boxes are underscored. The start of thetruncated pelD gene at base 2325 was determined by homology with corresponding regions of pelA and pelE.
protein encoded by pPEL781 (pelEA) macerated tissue muchless efficiently than PLb, PLc, or PLe but was more activethan PLa (Table 3). The lacZ fusion protein with PLa frompPEL841 produced maceration similar to that of the wild-type PLa. However, it is not clear whether degradation ofthe fusion protein might have occurred in the cucumbertissue during the maceration assay. Thus, it cannot beconcluded with assurance that the much larger fusion proteinin fact has maceration properties similar to those of PLa.
DISCUSSION
In conjunction with our past work (12), the sequence datapresented here for the pelA and pelC genes from E. chry-santhemi EC16 reveal the structure and organization of allfour known genes encoding endo-PLs in this strain. Inaddition, the sequence data detected a deleted pel geneoccurring between pelA and pelE. The pel genes are orga-nized in two chromosomal clusters. The pelB and pelC genesare linked in tandem with ca. 500 bp of intergenic DNA (Fig.6). This is similar to the organization of the pelB and pelCgenes in two other E. chrysanthemi strains (21, 25), althoughthese genes have not been sequenced. Furthermore, we have
identified sequences with considerable homology in the 5'noncoding regions of the pelB and pelC genes which mayfunction as promoter elements (Fig. 3). Significantly, how-ever, overall homology between the 5' DNAs of the pelB andpelC genes is low. The high homology of the coding regions(84% amino acid identity, including the signal peptide se-quences) (Fig. 2) and the similarity of the protein products(pl values of 8.8 and 9.0) indicate that pelB arose via anevolutionary duplication of pe1C. It is unclear why such aduplication was tolerated in E. chrysanthemi, but it shouldbe noted that duplications of genes having high homologywith pelB and pelC have been shown to occur in the relatedbacterium Erwinia carotovora (15; Lei, Hin, Wang, andWilcox, submitted for publication).An evolutionary duplication event presumably also gave
rise to the clustered pelA and pelE genes of strain EC16, aswell as to the subsequently deleted pelD gene (Fig. 5 and 7).The pelAlE genes have diverged more than the pelBIC genes,so that the protein products have very different pI values (4.6for PLa and 9.8 for PLe). However, the mature PLa and PLeproteins have 62% amino acid identity, and the proteinproducts exhibit similar catalytic properties in vitro (2),although not in plant tissue (Table 3).
VOL. 170, 1988

3476 TAMAKI ET AL.
1 MICKASGRSFTRSSKYLLATLIAGIOASGVSAAELVSDKALESAPTVGWASQNGFT1 MKNTRVRSIGTKSLLAAVVTAALMATSAYAAVETDAATTGWATQNGGT
57 TGGA-AATSDNIYIVTNISEFTSAL-SAGAEAKIIQIKGTIDISGGTPYTDFADQK49 TGGAKAA---KAVEVKNISDFKKALNGTDSSAKIIKVTGPIDISGGKAYTSFDDQK
111 ARSQINIPANTTVIGLGTDAKFINGSLIIDGTDGTNNVIIRNVYIQTPIDVEPHYE::::: :: :::::1: ::::::0:G: ::L: :: :: :: :: ::::
102 ARSQISIPSNTTIIGVGSNGKFTNGSLVIKGVK---NVILRNLYIETPVDVAPHYE
167
155
KGDGWNAEWDAkNITNGAHHVWIDHVTISDGNFTDDNYTTKDGETYVQHDGALDIKSGDGWNAEWDAAVID-NSTNVWVDHVTISDGSFTDDKY TTDGEKYVQHDGALDIK
223 RGSDYVTISNSLIDQHDKTNLIGHNDTNSAQDKGKLHVTLFNNVFNRVTERAPRVR210 KGSDYVTISYSRFELHDKTILIGHSDSNGSQDSGKLRVTFHNNVFDRVTERAPRVR
1 ALNRFDRRTPRLR
279 YGSIHSFNNVFKGDAKDPVYRYQYSFGIGTSGSVLSEGNSFTIANLSASKACK---
266 FGSIHAYNNVYLGDVKHSVYPYLYSFGLGTSGSILSESNSFTLSNLKSIDGKNPEC14 FGNVHAYNNVYTGDVNHKAYRYQYSFG--TSGSLISENNAFTIDNLKKINGRDKEC
332 -VVKKFNGSIFSDNGSVLNG-SAVDLSGCGFSAYTS--KIPYIYDVQPMTTELAQS322 SIVKQFNSKVFSDKGSLVNGSTTTKLDTCGLTAY-KP-TLPYKYSAQTMTSSLATS68 SVVKAFNGKIFSDKGSIINGASY-NLNGCGFGFNTYSAKIPYKYSAQTITTNLANS
384 ITDNAGSGKL
376 INNNAGYGKL
123 ISSNAGYGKL
FIG. 5. Homology of the protein products of the pelA (top line)and pelE (second line) genes ofE. chrysanthemi EC16 and the 3' endof an ORF assumed to be the remains of the deleted pelD gene (thirdline).
The organization of the pelA and pelE genes in strain EC16differs from that in two other strains ofE. chrysanthemi (21).Whereas pelA and pelE are transcribed in the same directionof EC16 (Fig. 7), they appear to be transcribed divergently inthe other two strains, as determined by the polarity ofexpression of lacZ insertions (21). These strains also pro-duce a fifth PL protein, encoded by an additional gene, pelD,a functional copy of which does not occur in strain EC16 (2,22). This gene encodes a PL with a pl of about 9.3 and hasconsiderable homology with pelE (Kotoujansky, personalcommunication). The sequence data presented here estab-lish why this gene is not functional in strain EC16. The shortORF located immediately 3' to pelA (Fig. 4 and 5) has high
L 11 i ( 1i1 1 11
pel A 3' End1179 bp pel D
-394 bp
C1 11c 1,%14IV?a I I i( r a-
40 C,
pei C pel 81125 bp 1125 bp
I I I I I I I0
Kb2
coI I I
3
FIG. 6. Gene organization of the E. chrysanthemi Et116 pelB andpelC genes based on DNA sequence data presented here andelsewhere (12). A, Site of a deletion used in subcloning; for otherabbreviations, see the legend to Fig. 7.
homology to the corresponding regions of PLa and PLe andalmost certainly represents a remnant of the missing pelDgene. The deletion event presumed to have given rise to thetruncated pel gene must have occurred relatively recently,because no detectable genetic drift was observed in the ORF(Fig. 5). Thus, while the ORF does not appear to retain afunctional rho-independent transcriptional stop like theother pel genes, no mutations have yet occurred that de-stroyed ORF integrity (Fig. 4). The deletions and insertionsobserved in the truncated pelD gene relative to pelA andpelE were most likely introduced during its evolution as afunctional gene because the pelBIC and pelAlE pairs exhibitsimilar features (Fig. 2, 4, and 5).We previously observed that pelB and pelE have little
homology, despite the fact that their gene products bothcatalyze random eliminative cleavages of sodium polypec-tate (2). Two short regions of conserved amino acids werenoted in the two proteins, however. These conserved re-gions also occur in pelA and peiC, and one of them alsooccurs in the truncated pelD gene. The additional data haveshown that certain conserved amino acids also precederegion II (Fig. 8), and other amino acids between regions Iand II are evolutionarily conserved substitutions. Structuralor mutational analyses have not yet been undertaken todetermine whether these conserved regions are essential forenzymatic activity.The pelB, pe1C, and pelE genes of strain EC16 were
previously shown to possess sequences which would beexpected to function as rho-independent transcriptional ter-minators after the translational stops (12). This suggests thatthese genes are independently expressed, consistent withexpression and mutation studies (13, 21). We have nowobserved that a similar sequence follows the pelA gene (Fig.
rwt qNPI I10
psl E1155bp ORF X
0 2 3Kb
4 5
FIG. 7. Gene organization of the EC16 pelA and pelE genes and the deleted gene assumed to be pelD as determined from DNA sequencedata given here and elsewhere (12). ORF X has no known function. A pem gene encoding pectin esterase occurs ca. 2.0 kb to the right of thetranslational stop ofpelE (see map in reference 11). Arrows denote open reading frames; circles show translational initiation sites. Restrictionsite abbreviations: B, Bgll; Bs, BstEII; BII, BglII; D, DraI; Hc, HincII; Hd, HindIII; K, KpnI; M, MIuI; N, NotI; P, PstI; RI, EcoRI; RV,EcoRV; Sa, SalI; Sp, Spll; Ss, SstI; Sc, ScaI; X, XhoI.
J. BACTERIOL.

E. CHRYSANTHEMI EC16 pel GENES 3477VOL. 170,1988
chrysanthemi
Erwinia
pel B
pel C
pel A
pel E
3'pel D
pel A
pel B
189AIDIKKGATY-VTISYNYI190AVDIK-GASNTVTVSYNYI218ALDIKRGSDY-VTISNSLI205ALDIKKGSDY-VTISYSRF
30 VNARLPLQRGGNVHAYNNLYTG
29 VNARLPLQRGGLVHAYNNLYTN
34 VTERAPRVRYGSIBSFNNVFKG
34 VTERAPRVRFGSIKAYNNVYLG
RTPRLRFGNVHAYNNVYTG
189AVDIKKGSTN-VTVSYNYI189AVDIKKGSTN-VTVSYNYI
29 VNSRLPLQRGGQVHAYTNLYDG
29 VNSRLPLQRGGLVNAYTNLYDGcarotovoraFIG. 8. Amino acid homologies occurring in the four EC16 PL proteins as well as the carboxy terminus of PLd and the E. carotovora PLa
and PLb proteins, which have high overall homology with the EC16 PLb and PLc proteins (15; Lei et al., submitted). Boldface letters denoteconserved amino acids; numbers between regions show number of intervening amino acids.
4), which would be predicted to function as a rho-indepen-dent transcriptional terminator (10). This is of interest be-cause pelA is poorly expressed relative to the other pel geneswhen E. chrysanthemi is grown under laboratory cultureconditions (21, 22). A palindromic sequence also occurredafter the translational stop of the deleted pelD gene (Fig. 4),but this was not followed by a poly(T) sequence. Thus,unlike the other EC16 pel genes, the deleted pelD genewould not appear to contain a rho-independent transcrip-tional terminator (10). This may be relevant to preliminarysuggestions that the residual pelD ORF influences expres-sion of the downstream pelE gene (1; Manulis and Keen,unpublished).We previously identified an ORF (ORF X, Fig. 7) for
which the Shine-Dalgarno box was located only 20 bpdownstream from the transcriptional terminator of the EC16pelE gene (12). This would be assumed to seriously limittranscription of the second ORF unless it is coupled totranslation of pelE in the manner reported by Wright andHayward (28) for the rho-indpendent transcriptional termi-nator of the E. coli gal operon. The ORF following pelE didnot have significant homology with pelE but possessed asignal peptide sequence (12). We subcloned the region fromthe NotI site (Fig. 7) which occurs in one stem element of thepelE transcriptional terminator and 58 bp ahead of the ORFX start codon (12) to sites up to 3 kb downstream (data notshown). These constructs were assumed to destroy termina-tion function and allow expression of the ORF from thepUC19 lac promoter. All constructs, however, failed to yieldPL or detectable xylanase, protease, cellulase, or pectinlyase activities in E. coli cells when IPTG was added (datanot shown). Thus, although the identity of the ORF is notknown, it does not appear to encode a random chain-splittingPL.We also observed that a strain EC16 gene encoding a
pectin esterase occurs on a 2.2-kb SmaI-SstI fragmentlocated ca. 2.0 kb from the 3' end of the pelE gene on clonepPEL74 (11; unpublished observations). Significantly, otherE. chrysanthemi strains also contain a pectin esterase genethat maps at the same position (Kotoujansky, personalcommunication).
Similar to the previous results with pelB and pelE, thepelA and pelC genes from strain EC16 could be satisfactorilyoverexpressed in E. coli cells with pINK1, an expressionplasmid derived from pINIII, constructed by Inouye andassociates (18). Also similar to the previous results, theuninduced level ofPL with these constructs was high despitethe presence of the lacI gene on the vector. Recently, Hasan
and Szybalski (7) constructed an invertible promoter vector,pNH8a. We found that this vector resulted in low uninducedlevels of PLc (Table 2) but led to induced levels that werecomparable to those of the pelC gene cloned in pINK1.
Barras et al. (2) reported that PLa is a much less efficientmacerating factor for plant tissue than PLe, but both pro-teins have similar catalytic properties in vitro. We confirmedthe poor maceration ability of the pelA protein (ca. 1,000times less effective than the pelE protein against cucumbermesocarp tissue [Table 3]). However, Kotoujansky (13)showed that the pelA gene of E. chrysanthemi 3937 isessential for full pathogenicity on African violet plants.Furthermore, the pelA gene appears to occur in all strains ofE. chrysanthemi tested to date (22), raising the possibilitythat it provides a selective advantage. These considerationsraise the question of the function of PLa in plant pathogen-esis. PLs with little plant-macerating ability and low plvalues, similar to pelA, have also been described for thenon-plant-pathogenic bacteria Yersinia pseudotuberculosisand Klebsiella pneumoniae (3, 17). The fact that thesebacteria infrequently encounter plant tissue raises the pos-sibility that the acidic PLs may have alternative but as yetunknown physiologic functions.
Since the pelA and pelE proteins from strain EC16 givesimilar specific activity in in vitro PL assays, the mostobvious explanation for the low macerating activity of thepelA protein is its low pl value, as suggested by Tanabe andKobayashi (26). This might impede the physical penetrationof the protein into the negatively charged plant cell wallmatrix. In order to test this possibility, we constructedseveral recombinant pelAlpelE genes. Three of these con-structs resulted in little or no PL activity in E. coli cells, butpPEL780 and pPEL785 led to PL activity at levels similar tothose of the parental pelA and pelE genes. The macerationactivity of the recombinant PLea protein was greater thanthat of PLa but considerably less than that of PLe (Table 3).The recombinant protein was also less efficient at tissuemaceration than PLb or PLc, which have pIs of 8.8 and 9.0,respectively. Furthermore, many PLs from erwinias andother organisms which efficiently macerate plant tissue pos-sess isoelectric points in the range of 7.0 to 8.5 (13). Sincethe pl of the recombinant PLea protein (pH 7.05) wasconsiderably higher than the pH of most plant intercellularfluids (pH 5 to 6), our data suggest that other factors inaddition to the low pl value ofPLa may contribute to its poormacerating efficiency. Additional work with recombinantgene constructions should further illuminate domains of the

3478 TAMAKI ET AL.
proteins required for PL activity, plant tissue maceration,and pathogenicity.
ACKNOWLEDGMENTS
This research was supported by USDA grant 86-CRCR-1-2233.Some sequence data were analyzed with the BIONET NationalComputer Resource of Molecular Biology, whose funding is pro-
vided by the Division of Research Resources, National Institutes ofHealth, grant U41 RR-01685-02.
LITERATURE CITED1. Barras, F., and A. K. Chatterjee. 1987. Genetic analysis of the
pelA-pelE cluster encoding the acidic and basic pectate lyases inErwinia chrysanthemi EC16. Mol. Gen. Genet. 209:615-617.
2. Barras, F., K. K. Thurn, and A. K. Chatterjee. 1987. Resolutionof four pectate lyase structural genes of Erwinia chrysanthemi(EC16) and characterization of the enzymes produced in Esch-erichia coli. Mol. Gen. Genet. 209:319-325.
3. Chatterjee, A. K., G. E. Buchanan, M. K. Behrens, and M. P.Starr. 1979. Synthesis and excretion of polygalacturonic acidtrans-eliminase in Erwinia, Yersinia and Klebsiella species.Can. J. Microbiol. 25:94-102.
4. Chatterjee, A. K., and A. K. Vidaver. 1986. Genetics of patho-genicity factors: application to phytopathogenic bacteria. Adv.Plant Pathol. 4:1-213.
5. Colimer, A., and N. T. Keen. 1986. The role of pectic enzymesin plant pathogenesis. Annu. Rev. Phytopathol. 24:383-409.
6. Crouse, G. F., A. Frischauf, and H. Lehrach. 1983. An inte-grated and simplified approach to cloning into plasmids andsingle-stranded phages. Methods Enzymol. 101:78-89.
7. Hasan, N., and W. Szybalski. 1987. Control of cloned geneexpression by promoter inversion in vivo: construction ofimproved vectors with a multiple cloning site and the Ptacpromoter. Gene 56:145-151.
8. Henikoff, S. 1984. Unidirectional digestion with exonuclease IIIcreates targeted breakpoints for DNA sequencing. Gene 28:351-359.
9. Heusterspreute, M., V. H. Thi, S. 0. Emery, S. Tournis-Gamble,N. Kennedy, and J. Davison. 1985. Vectors with restriction sitebanks. IV. pJRD184, a 3793-bp plasmid vector having 43 uniquecloning sites. Gene 39:299-304.
10. Holmes, W. M., T. Platt, and M. Rosenberg. 1983. Terminationof transcription in E. coli. Cell 32:1029-1032.
11. Keen, N. T., D. Dahlbeck, B. Staskawicz, and W. Belser. 1984.Molecular cloning of pectate lyase genes from Erwinia chrysan-themi and their expression in Escherichia coli. J. Bacteriol. 159:825-831.
12. Keen, N. T., and S. Tamaki. 1986. Structure of two pectate lyasegenes from Erwinia chrysanthemi EC16 and their high-levelexpression in Escherichia coli. J. Bacteriol. 168:595-606.
13. Kotoujansky, A. 1987. Molecular genetics of pathogenesis bysoft-rot Erwinias. Annu. Rev. Phytopathol. 25:405-430.
14. Kotoujansky, A., A. Diolez, M. Boccara, Y. Bertheau, T. Andro,and A. Coleno. 1985. Molecular cloning of Erwinia chrysanthemipectinase and cellulase structural genes. EMBO J. 4:781-785.
15. Lei, S.-P., H.-C. Lin, S.-S. Wang, J. Callaway, and G. Wilcox.1987. Characterization of the Erwinia carotovora pelB gene andits product, pectate lyase. J. Bacteriol. 169:4379-4383.
16. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
17. Manulis, S., D. Y. Kobayashi, and N. T. Keen. 1988. Molecularcloning and sequencing of a pectate lyase gene from Yersiniapseudotuberculosis. J. Bacteriol. 170:1825-1830.
18. Masui, Y., J. Coleman, and M. Inouye. 1983. Multipurposeexpression cloning vehicles in Escherichia coli, p. 15-32. In M.Inouye (ed.), Experimental manipulation of gene expression.Academic Press, Inc., New York.
19. Payne, J. H., C. Schoedel, N. T. Keen, and A. Collmer. 1987.Multiplication and virulence in plant tissues of Escherichia coliclones producing pectate lyase isozymes PLb and PLe at highlevels and of an Erwinia chrysanthemi mutant deficient in PLe.Appl. Environ. Microbiol. 53:2315-2320.
20. Pustell, J., and F. C. Kafatos. 1984. A convenient and adaptablepackage of computer programs for DNA and protein sequencemanagement, analysis and homology determination. NucleicAcids Res. 12:643-655.
21. Reverchon, S., F. Van Glsegem, M. Rouve, A. Kotoujansky, andJ. Robert-Baudouy. 1986. Organization of a pectate lyase genefamily in Erwinia chrysanthemi. Gene 49:215-224.
22. Ried, J. L., and A. CoUimer. 1986. Comparison of pecticenzymes produced by Erwinia chrysanthemi, Erwinia caroto-vora subsp. carotovora, and Erwinia carotovora subsp. atro-septica. Appl. Environ. Microbiol. 52:305-310.
23. Ruther, U., and B. Muller-Hill. 1983. Easy identification ofcDNA clones. EMBO J. 2:1791-1794.
24. Schlemmer, A. F., C. F. Ware, and N. T. Keen. 1987. Purifica-tion and characterization of a pectin lyase produced by Pseu-domonasfluorescens W51. J. Bacteriol. 169:4493-4498.
25. Schoedel, C., and A. Colimer. 1986. Evidence of homologybetween the pectate lyase-encoding pelB and pelC genes inErwinia chrysanthemi. J. Bacteriol. 167:117-123.
26. Tanabe, H., and Y. Kobayashi. 1987. Pectin lyase originatingfrom Erwinia chrysanthemi with a low maceration potential.Agric. Biol. Chem. 51:779-783.
27. Vieira, J., and J. Messing. 1987. Production of single-strandedplasmid DNA. Methods Enzymol. 153:3-11.
28. Wright, J. J., and R. S. Hayward. 1987. Transcriptional termi-nation at a fully rho-independent site in Escherichia coli isprevented by uninterrupted translation of the nascent RNA.EMBO J. 6:1115-1119.
29. Yanisch-Perron, C., J. Veira, and J. Messing. 1985. ImprovedM13 phage cloning vectors and host strains: nucleotide se-quences of the M13mpl8 and pUC19 vectors. Gene 33:103-119.
J. BACTERIOL.