synthetic aspects of porphyrin and metalloporphyrin...
TRANSCRIPT

7 Synthetic Aspects of Porphyrin and Metalloporphyrin Chemistry Tilak P. Wijesekera Sun Company, Inc .• Marcus /loo k.. Pennsylvania
David Dolphin Ulliversity 0/ British Columbia, Vancouver. British Columbia. Canada
I. INTRODUCTION
Nature has found a prominent place for porphyrins and related macrocycles such as chlori ll s, bacteriochlorins. a nd l:orrins (Figure I), particularly in their metallatcd forms. Iron protoporphyrin IX is the prosthetic group of heme proteins that serve diversified biological roles (Figure 2) including oxygen and elec tron transport and storage (hemoglobin and myoglobin) , elecIron transfer (cylochromes), 3m.l biocatalysis (catalase, peroxidase, lignin peroxidase, cytochrome P450). The nonmetallated ("rree base") porphyrins arc generally present in organisms as precursors of melalloporphyrins and are acculllulated and/or excreted in (;ertain physiological disorders sllch as porphyrias [1]. T he preferential localizaLion of certain porphyrins in tumor cells, coupled with the ability of porphyr ins to act as pholOsensitizcrs in the production of highly reactive singlet oxygen (which in turn exerts a cytotoxic action), has been used extensively in the novc!treatmenl of malignant tissues and pathogens, a technique referred 10 as photodynamic therapy (PDT) 12,31.
Catalyt ic act ivity of heme enzymes is known to be controlled by changes assoc iated with the macrocydc and by modifications of the protein matrix, which in addition to providing a suitable environment for substrate bindi ng may also provide axial ligands to the metal center. As such, synthet ic mctaJloporphyrins have been used extensively as biomimetic agents and thereby in
193

194
7 'I •
(A) Fiacher
PORPHYFUN NUCLEUS
Chlorin
WIJESEKERA AND DOLPHIN
(B) Porphyrin 1-24
ALTERNATIVE NUllBERING SCHEItES
H
Bacteriochlorin Corrin
Figure 1 Tetrapynolic macrocydes and their lIumbering systems .
H,C
H,C
HOze C02H
Heme ([ron Proloporphyrin IX)
Hemoglobin Myoglobin
Cytochromes
Catalase
Peroxidases (ligninose)
Cytochrome Oxidase
0 ... 0
0, I F e(n)P .. II' Fe(n)P
Fe(m)P ~ Fe(n)P --- 0 II
Fe(m)P~ Fe(rv)P·· + H,O
,H20 1
F.(nilP + 0,
o +-
II 'Hb Fe(IV)P+- Fe(nilP 2H+ 20
Cytochrome P-450 RH + 02 >- . ROH + H20
,.-Figure 2 Biological roles of selected heme proteins.

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 195
attempts to understand the complex biological processes involved in enzymatic reactions. Furthermore such studies help in the design and development of protein. free in vitro catalysts which cou ld be used to catalyze chemical reactions of industrial importance. In this context, cytochrome P450, the enzyme responsib le for ca talyzing the hydroxylation of nonpolar substrates (including drugs and pollutants) to yield parLially water-soluble products , is probably the enzyme that has received the most attention of both chemists and biochemists {4J . Because of its role ill dioxygcn activation. understanding and success fully mimicking the catalytic cycle of cytochrome P450 has becOllle extremely important in providing new and less expensive routes to hydroxylation of alkanes and epoxidation or olerins, both of which are industrially important reactions. Another enzyme that has received much allention, especia ll y recently, is lignin peroxidase [51. produced by the lignolytk fungus Phanerocliaele crysosporium, which is responsible for the degradation of the biopolymer lignin. This is primarily due to the requirement for an environmentally acceptable delignification process as a substitute for the chlorine-based blc3f..:h ing currently used in the pulp and paper indust ry.
With the increase in interest in porphyr ins and mctalloporphyrins from chemists of diverse backgrounds the necessity to access variously substituted porphyrins has been greater than in the past. Apart from the simplest symmetric porphyrins, most useful derivatives either arc not commercially avail able or arc priced beyond reach of an average researcher. In this chapter we wi ll review the currently available major synthetic routes to porphyrins and suggest approaches to the construction of the macrocycle based on the subslitucnts required al the periphery . It is appropriale that some of the salient features of porphyrins and metalloporphyrins be d iscussed first.
II . STRUCTURE AND PROPERTIES OF PORPHYRINS AND METALLOPORPHYRINS
The porphyrin nucleus (Figure I) is a cyclic tetrapyrro lic system consisting of a 20-carbon skeleton, the four pyrro!cs being linkcd by sillgle carbon a[Q1ll bridges. The nomcnclature commonly used in porphyrin chemistry (6) was developed by Hans Fischer and is based 011 the numbering system shown in Figure lA, where (he pyrrolic (13) positions are numbered from I to 8 and the bridging (meso) positions named LY , Ii, )', and o. Fischer nomenclature involves a large number of trivial names based on the substituents, which, however, do not convey any structural inrormation. In addition, it involves a "type-isomer" system to distinguish between the dirferent positional isomers. The inability of the Fischer system to name the large number of synthetic and newly isolated porphyrins led to the adopt ion of a systematic nomenclature based on the 1-24 numbering system (Figure 18), developed by a

196 WIJESEKERA AND DOLPHIN
joint commission on biochemical nomenclature, consisting of the International Union of Pure and Applied Chemistry (lUPAC) and the International Union of Biochemistry (lUll) [7[.
The porphyrin macrocycle contains a total of 22 1'r electrons with 1811" electrons in direct conjugation, conforming 10 Huckcl 's 4n + 2 rule for arom31iciLy. The two peripheral double bonds .:,\' and fi l7 of rings Band D. respectively (Figure 1 B) arc cross-conjugated and can therefore be reduced while retaining the aromaticity of the molecule. As sUl::h, chloTin (the parent macrocycie of a variety of chlorophylls) with one peripheral double bond reduced and bacteriochlorin with both double bonds reduced still retain the aromalicity of the macrocycle. or the 12 peripheral posi tions available for substitution, the reactivity of the eight pyrrolic positions (j3) differ rrom that or the rour bridging (meso) positions. It is also important to note that although porphyrins react primarily by substitution (clcctrophilic or radical) due to the aromatic nature of the macrocycle, the ~ 7 and .::l,11 double bonds. which are not part of the direct conjugation, are known to react differently under certain reaction conditions [8] .
Porphyrins exhibit characteristic absorption properties in the UV-visible region. The metal-free, their diprolonated form s, as well as metallatcd porphyrins have an intense absorption ncar 400 11m «( - lOS), referred to as the Sorel band, which is characteristic of the macrocyclic systelll. In addition, the metal-free systems exhibil four less intense bands designated IV, III, II, and I, between 450 and 700 11111. The intensity and the exact positions of these bands are dependent on the solvent as well as the c..:oncentratiol1. More importantly, correlations have been showll to exist between the relative intensities of the four bands and the nature and positions o f the porphyrin side chains [9).
Four basic types of spectra have been observed based on the relative intensi ties of the four visible bands (Figure 3). Goutcrman [9J proposed an interpretation of these spectral differences based 011 the perturbations of the 'IT-electron levels by the electronic nature of the peripheral substituents.
A. Elio Type
This type of spectrum is characterized by a I V > III > Il > 1 order of intensity. Porphyrins in which six or more 13 positions have alkyl substiluents with the other two being ullsubstituted will exhibit an etio-type spectrum irrespecti ve of the relative orientations of the substilucnts. In addition to etioporphyrin isomers (from which the lIame is derived), most naturally occurring porphyrins such as copro-, uro-, hcmato-, proto-, and deutcroporphyrins exhibit this type of spec..:truill.

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 197
RHODO
500 550 600 650 500 550 600 650
OXORHODO PHYllO
500 550 600 650 500 550 600 650
Figure 3 Typical absorption spectra or nonmctallated porphyrins.
B. Rhodo Type
Onc strongly electron-withdrawing group (e.g., form yl. acetyl, or carboxyl) conjugated with the porphyrin ring causes a change in the intensity pattern (Ill > IV > II > I) resulting in the rhoda-type spectrum (named after rhodoporphyrin). In addi tion, this produces a bathochromic shift o f a ll bands in the spectrum. The rhooofying effect o f one elect ron-withdrawing group is canceled by the presence of a similar group on a n adjacent pyrrolic group, rcsulLing in an clio-type spectrum . However , the effect on the red shift of absorption maxima is additive. We have shown that a rhodofying effect is al so produced when the macrocycle is d istorted by sho rt-st rap bridging, even without the int roduction of any elect ron-w ithdrawing groups [10].
C. Oxorhodo Type
This spectral pattern, in which the intensities o f the absorption maxima follow the order III > 11 > IV > I, is characteristic of porphyrins having

198 WIJESEKERA AND DOLPHIN
two electron-withdrawing groups on diagonally opposite pyrrole rings. This can be viewed as a further enhancement o f the rhotlofying effecl.
0, Phyllo Type
This speclral pallern (IV > II > III > I) na med afler phylloporphyrin is dist inguished from the clio type by less intense bands III and I. Two substitulion pa tterns on the periphery are known to produce the phylio-LyPC spectrum: (a) a single meso-alkyl substitution and (b) four or more unsubstituted (3 positions .
As shown in Figure 4a, porphin (the unsubstituted parent porphyrin) exhibits a phyllotype speelru", 1490, 520, 560, 568(sh), 614; Soret: 394 IlIn] while ,3-octaethylpo rphy rin (OEPH1) exhibits a typical ctio-type spectrum (Figure 4b; 498, 534, 566, 620; Soret: 400 nm). However, most of the synthetic porphyrins, particularly those used in ca talytic studies, exhibit spectral patterns that are different from a ny of the fo ur lypes given above. The un· substituted meso·tetraphenylporphyrin (TPPH2) shows a modified clio· type four-band speclrum (Figure 4c; 514, 550, 588, 644; Soret: 418nm) wit It an intense band IV . Tetraphenylporphyrins carrying ortho subslituenls on the phenyl groups show a significant reduction in the intensities o f bands III and I relat ive to the ~orrespollding para·subs tituted systems [11 ,121. This is part icularly so for the orfho·halogen (F. CI, llr)-substituted porphyr ins in which relat ive band intensi ties change to IV > 11 > III > I. This may be attributed to the restricted rotation of the phenyl groups resu lting frol11 steric int eraction of the orlho· halogcn substituent and the /1·pyrrole hydrogen. meso-Tetrakis(pentaOuorophenyl)porphyrin (TPFPPH2) with limited ro· tation exhibits a spect rum (506, 582; Sorel: 412 11m) in which bands III (536 sh) and I (636 wk) arc suppressed, while both meso-tetrakis(2.6-dichloro· pltenyl)porpltyrin (TDCPPH,; 512, 588; Soret: 41 8 nm) and lIleso-tetrakis(pentachlorophenyl)porphyrin (TPCPPH,; 514, 590; Soret: 420 nm), whose bulky orlilo-chioro substituenLs prevent free rotation of the phenyl groups, exhibit essentially two-band (corresponding to bands IV and II) spectra.
At low pH , the inner nitrogens o f porphyrins are protonated to produce dit.:ationic species whose visible spectra diller from those of the correspond ing free base. As shown in Figure 4a, protonation changes the phyllo-type four ~band spectrum of porphin to a single strong absorption at 544 11m with shoulders at 516 and 570 11m while the cLio-type four-band spectrum 0 1" octaethylporphyrin changes to a two-band spectrum (548 and 590 nm) with a sho ulder at 568 nm (Figure 4b) . In ac id, unsubstitutcd te traphenylporphyrin shows a single broad band at 654 nrn with a shoulder at 600 nm (Figure 4c) while the benzene ring- subst ituted tetraphcnylporphyrins show a red shih

§ N HN
• I a • u u c c 0 0
"' ~ -e 0 • • "' "' « «
t-- ~ I I =,
500 600 700
Wavelength (nm)
t;t' NH HN-
• • b
500 600 700
Wavelength (nm)
• u c 0 -e 0 • "' «
500
c
r' , , , , , , JA. \.
600 700
Wavelength (nm)
Figure 4 Absorption spectra of selected porphyrins: (a) porphine; (b) octaethylporphyrin; (c) tetra phenyl porphyrin. ~~_. free base; ----, prolonated dication.
~ Z .... ::t !!1 n :I> VI "D a VI o " "D o '" "D ::t <
'" z (') ::t m ;:
i!l '" <
~
'" '"

200 WIJESEKERA AND DOLPHIN
of the two-band spectra [e.g. , 576 and 624 nm for both meso-tctraki s(2,6-dichlorophenyJ)porphyrin and meso-tetrak is(penlaO uorophenyl)porph yri It J.
The porphyrin dianion, formed by (he removal o f the inner NH protons, acts as a tetradentate ligand capable of binding a wide variety of metal ions. Di va lent metals such as Z n l-t . Mg2t , C uh , Fez>, Co2 ' . a nd Nj!~ form electrically neutra l complexes which may either have a square-planar or octahedral geomet ry depending on the presence of axial ligation. Trivalent metals such as Fe3 t and MnJt carry anionic axial ligands to form elect rically neutral penl3t:oordinate squa re-pyramidal complexes or hexacoordina te complexes with a neut ral sixth ligand . Metallation leads lo a simplification of the visible
a
• 0 c 0
£ g ~ ~
300 400 500 600 700
Wovelength (nm)
b
• 0 c 0
£ ~ ~
,o0 400 500 600
Wovelength (nm)
C
· 0 c 0 -e ~ ~
300 400 500 600 700
Wo .... elenglh (om)
Figure 5 Absorption spect ra of l11eso ·tctraki s(penta nuorophcllyl)porphyrinato-iron( IJI ): (a) chloride; (b) hydroxide; (c) I'-OXO dimcr.

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 201
region of the absorption spectrum while retaining the Sorel absorption near 400 nm, primarily oue to the change in the conjugated ring symmetry. For octacthylporphyrin and other porphyrins exhibiting ctio-type spectra, coordination of a divalent metal results in two bands designated {j and a (between 500 and 600 11m), whose peak positions and relative intensities depend on the metal as well as the nature of the porphyrin ligand (e.g., OEPCu: 524, 560; Sorct: 398 nm). In the tetraphenylporphyrin series, the visible region consists essentially of a single strong band with two shoulders [TPPCu: 502(sh), 538, 572(sh); Sorel: 414 11m]. 111 the case of metalloporphyrins of trivalent metal ions, the UV-visible spectrum is significantly changed by the nature of the axial anionic ligand. meso-Tetrakis(pentafluorophenyl)porphyrinatoiron(III), (TPFPPFeX; Figure 5a) shows a characteristic split Soret (350 and 410 nm) with two weak bands at 502 and 628 nm when the axial ligand X ~ CI - . When X ~ OH - (Figure 5b), a strong Sorel band is observed at 406 11m with a weak band at 563 nm [131. When steric factors permit , as in the case of TPFPPFeX, Fe(III)porphyrins are capable of forming oxo-bridged dimers (Ji-oxo) whose UV-visible spectrum (e.g. , Figure 5c) differs from that of the hydroxo complex in that the So ret and the {J bands arc blue-shifted. C hromatographic purification of the hemin (following metal inser tion) generally leads to the exchange of the axial ligand but the hemin ca n be regenerated by treating the resulting hydroxide (or the p.-oxo dimer) with hydrochloric acid.
Changes ill the conjugation path affect the optical spectra of the porphyrin macrocycle significantly. Reduction of the one peripheral double bond, although not affecting the aromaticity of the molecule, produces a visible spectrum characterized by an intense long-wavelength absorption at ). ~ 650-680 nm (chloril1s) while reduction of both t!.' and t!." double bonds produces a strong absorption at A - 740 nm (bacteriochlorins).
III. SYNTHESIS OF PORPHYRINS
A. Construction of the Macrocycle
Since the landmark synthesis of proto hemin (iron protoporphyrin IX) by Hans Fischer in 1929 (which earned him the Nobel Prize), significant progress has been made in the fi eld of porphyrin synthesis [14,15] especially through the efforts of A. W. Johnson, G. W. Kenner, S. F. MacDonald , and R. B. Woodward. During the fir st half of the 20th century, synth.esis of porphyrins, like other areas of organic synthesis, was driven by the requirement for the establishment of structures of natural products. Each synthesis was based on the availability of st ructurally similar starting matcrials and thc possibility of the introduction and modification of substituents by known chemical reactions. Since the mid-1960s, a new and more systema tic approach to syn-

202 WIJESEKERA AND DOLPHIN
thesis has been adopted that focuses on the structural features of the target molecule rather than those of the starl ing material. Popularly known as retrosymhelic analysis, this approach transforms a synthetic target, in a stepwise manner, into progressively simpler sys tems that eventually lead to a readily available starlillg material [l6J. The necessity to synthesize variously substituted porphyrins via multistep syntheses has resulted in proper planning by rctrosynthelic analysis.
B. Symmetry and Substitution of the Target Molecule
The substitution pattern Oil the 12 peripheral positions of the target porphyrin molecule should form the bas is of the synthetic strategy employed. Several elegant synthetic methods are available for the cons truction of the porphyrin macrocycie and a retrosynthetic analysis of the target system will narrow down the choices available even though cer tain routes arc more convenient and/ or produce higher yields than others. It is important to realize at the very outset that while certain substitucnts can be introduced after macrocyci ization, others have to be already present in the pyrrolic precursor fragments.
The three most important factors to be taken into account in designing the synthesis are (a) the nature of the substituent; carbon- or heteroatombonded group, (b) the position of the substituent ; {1 or meso, and (c) the overall symmetry of the molecule. Although most heteroatom substituents such as halogens, nitro, and sulfonic acid groups can be readily introduced onto the preformed porphyrin macrocycle because of the electrophilic (or radical) nature of their activity. carbon-bonded substituents such as alkyl a nd aryl as well as their derivatives usually need to be incorporated before or during macrocyclization. Furthermore, if such a substituent is required on a {3 position , that group or an interconver tible functional group has to be incorporated onto the pyrrole subunit during its synthesis from acyclic precursors. TIle most important factor, however. is the overall symmetry of the Illacrocycle. Symmetrically placed peripheral substituents in the target molecule allow a synthetic chemist to use a simple one- or two-step method for the construction of the macrocycie while highly unsymmetric systems such as those found in nature require multistep manipulations that require the skill s of an experienced porphyrin chemist.
C. Retrosynthetic Analysis
Based on the currently available routes fo r the const ruction of the porphyrin macrocycie, retrosynthetic analysis of the target porphyrin would lead to several simple pyrrolic sys tems (Scheme 1). The choice of the route explored would depend on the nature and position of the substituent as well as how

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 203
R R
~ ~ , ~ NH N- , NH
~ , N-
R R R R R HN'
R N .'
R R
a a a R R (;('0 , dec Qq;)
R-C C-R RO R
R-C C-R
vco Np C-JJ , R R
[;c crQ C C
bJ) =
0 b c d
Scheme 1
that group would affect the reactivity of the pyrrolic precursor. This is particularly important in the case of electron-withdrawing p subslitucnts since such groups reduce the electron density of the pyrrolc, thereby affecting the clectrophilic reactivity at the (X position. The following skeletal disconnections lead to several simple synthetic precursors for porphyrins.
Type I (Scheme la): This approach disconnects the macrocyclc into four pyrrolc units and the meso carbons in a single step. This is possible only in highly symmetric porphyrins where the meso substituenls as well as the {J substilucnts are identical. In the synthetic direction, the most suitable functional group for the meso carbon is the aldehyde group with the substituent R attached to it. The pyrrolc should be unsubstituted at the a position while the {3 positions may either be unsubstituted or may carry substituents that do not affect its reactivity. Although it is possible to prepare porphyrins having either different R groups or two different {3 substituents on each pyrrole, such approaches lead to a mixture of porphyrin products andlor isomers lhat will require tedious separation procedures.
Type 2 (Scheme /b): This approach differs from type I in tha' the meso carbon is already attached to the 2 position of the pyrrole, which in turn carries the R group. In the synthetic direction this involves a self-condensation of a 2-substituted pyrrole via a pyrryJ carbo cation that undergoes a head-to-tail coupling of four units to give the porphyrin.
Type 3 (Scheme lc): This disconnection leads to two identical dipyrrolic units and two meso carbons carrying substituent (R) groups. The dipyrrolic system is a 5,5'-unsubstituted dipyrromethanc which can be coupled

204 WIJESEKERA AND DOLPHIN
with an appropriate aldehyde. This approach can be used where the target molecule carries different {3 SubSlilUcnts (symmetrically placed) and different meso groups in the opposite positions.
Type 4 (Scheme Jd) : This approach leads to dipyrrolic starling materials either directly by a simultaneous two-carbon disconnection or via linear tetrapyrroies by /wu one-carbon disconnections. This is undoubtedly the best method if the target molecule is unsymmetrically substituted. The linear telrapyrroJes can be buill in a stepwise manner sLarting from monopyrrolcs that carry the required {3 substilucnLs and cyci izcd in the final step to give the porphyrin.
D. Synthetic Precursors
The above-mentioned disconncctions lead LO three Iypes of pyrrolic precursors to porphyrins: monopyrroles, dipyrrolic systems, and linear tetrapyrroles. In order to transform these intermediates 10 porphyrins, they should be appropriately substituted at the u positions and the !1 substitucnts should be so positioned that the reactivity of the fragment is not affected. The COIll
mon precursors onc encounters during porphyrin synthesis are givcn in Figure 6.
I. Dipyrromelhanes
Dipyrrolllethanes contain two pyrrolic units linked by a methylene (CHz) bridge (Figure 6a). They resemble monopyrrolcs in their reactivity especially in that the a position is more reactive than the {3 position . They are prepared from monopyrroles by acid-catalyzed coupling methods but may undergo rearrangement to give isomeric mixtures under certain acidic conditions. In the coupling of dipyrromethanes to yield porphyrins, the meso carbons are introduced as formyl groups, either directly linked to it (Scheme Ib) or coupled as an aldehyde (Scheme Ic).
2. Dip yrromel helles
Dipyrromethenes contain two pyrrolic units linked by a methine (CH) bridge and hence are oxidized forms of dipyrromethanes. They are more stable in their protonated form. Figure 6b gives the four common dipyrromethcne pairs that are used for coupling where the meso carbons are provided by methyl or bromomethyl groups. A dipyrrole of lesser importance is dipyrroketone where two pyrroles are linked by a carbonyl group.
J. Linear Telrapy rroles
TIle nomenclature of linear tetrapyrroles is related to that of bile pigments hen~c the prefix "bila") and the numbering is in accordance with the lUPAC

~ }- NHHN -< H H
H H
~ }-NH HN-<
OHC CHO
~HH,.-(, " NH HN CHO H
a
~ ~ }- NH H~,,< }-NH H~,,<
Br CH2 Br BrH2 C CH2 Br I
Sr BrH2~C -1\1-1 H ~
" '" ~
~ y.. NH H~~ Br CH3
III
H3 C Sr
}",NHH~ ~
b
II Br Bf }",NH H~ ~
~ }-NH H~,,<
H3C CH3 IV
Sr Sr
}",NH H~ ~
! ) 7. (Z I' 17 II
11/1.. 5' n , loll .f\\L4ls1ln \. "N~N~N~N;>
H H H H
Bilane
n n ", F\ n " N;>...,..AN ~N"~N ;>
H H b H Bilene-b
n F\ n F\ "'N 'A....::?' N~N~f'{,)
H II. H C
BHadiene-a ,c
~ H II b c
Bilatriene -a ,b ,c
c Figure 6 Common pyrrolic precursors to porphyrins: (a) dipyrromethanes ; (b) dipyrromethenes; (c) li near tetrapyrroles.
~ Z .... :z: m .... o » (fJ ...,
a a " ..., a '" ..., :z: -<
'" z (") :z: m I: en .... '" -<
~ '"

206 WIJESEKERA AND DOLPHIN
recommendations (7). The stabi lity of the tetrapyrrole increases with the introduction of unsaluralion at the bridging carbons 5, 10, and 15 (a, b, c; Figure 6c) but the stability of saturated systems can be increased by lhe in· Iroduction of electron-withdrawing substit ucnts. Bilencs band biladicncs a,e arc the most common ly used linear tctrapyrroles in porphyrin synthesis.
IV. GENERAL SYNTHETIC METHODS
As slaled ear lier, the best approach to the synthesis of a desired porphyrin system is to analyze the target molecule retrosynthelically based on the symmetry of the system and the lype and relative position of the substitucnts. This will lead to the most suitable precursors which can be used according Lo the established synthetic procedures. The synthetic methods currently available are grouped according to the precursors used.
A. Cocondensation of a Pyrrole and an Aldehyde
One of the most commonly used methods for the synthesis of porphyrins has been the reaction of pyrrole (or a 3,4-disubstituted pyrrole) with an aliphatic or aromatic aldehyde. This method, which produces meso-tetrasubstituted porphyrins, has limited synthetic value due to the symmetr ic nature of the porphyrin produced and the restrictions on the type (and position) of the substituent that can be introduced. However, due to the ease of carrying out the reaction, Ihis method has been used extensively to prepare symmetric porphyrins, especially for catalytic sLudies.
meso-Tetrasubstituted porphyrins (4; A ::= H; Scheme 2) were first prepared in 1936 by Rothmund who heated pyrrole (I; A ~ H) and an a ldehyde (2) in pyridine in a sealed tube at ISO°C for 24 h (l71. This reaction, which was later extended to include more than 25 aliphatic and aromatic aldehydes {18,191, had earlier been used to prepare the unsubstituted system porphin (4; A = R = H) in 0.012% yield by reacting pyrrole and formaldehyde in methanol and pyridine [20}. The yields of porphyrins from this reaction were in general less than 30/0 and the product was contaminated with the corresponding chlodn, the 2,3-dihydroporphyrin. Ball ct al. i2l] obtained improved yields for TPPHz by adding zinc acetate to Ihe reaction mixture, a modificat ion that was later used by Badger et al. [III 10 prepare several orlho-substituted tetraphenylporphyrins. However, the major drawbacks in this synthetic procedure continued to be the low yields and the difficulty of introducing sensitive substitucnts (using the appropriate benzaldehydes) due to the severe reaction condi tions employed.
A major improvemenl in this area of cocondensation of pyrrole and aldehydes came from the work of Adler, Longo, and coworkers {22,23] when they showed that the yields of porphyrins call be greatly increased by carrying

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 207
A A f.)
N H
+ R H
' C / II o 2
..
3
Scheme 2
A R A
A A
R R
A
4
out the reaction in acidic media, open to air. In renuxing propionic acid, this condensation gave yields of greater than 20OJo, where the product crystallized out 011 cooling, thus simplifying the purification procedure. Although higher yields (30- 40%) were obtained by using acetic acid as the solvent , the prodm:l did not crystallize out due to the higher pK" of acetic acid, making the iso lation and purification more difficult. As in the Rothmund synthesis, the isolated porphyrin was contaminated by the corresponding chloTin, which is indicated by the presence of a strong band - 650 nm in the absorption spectrum. Although this can be separated from the porphyrin by chromatography, significant loss of material occurs due to adsorption. The recommended method of purification is to treal the porphyrin/ chlorin mixture with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) in order to oxidize the chlorin to the porphyrin [24J. The progress of the reaction can be monitored by the disappearance of the band ncar 650 nm and subsequent chromatography on alumina removes the oxidanllo yield a 95 + 0,10 recovery of the pure material. Several ortllO-. meta-, and para-phenyl- subs tituted porphyrins have been prepared using this method [1 2).
Another general synthetic method for meso-tetraphenylporphyrins was developed recently by Lindsey et al. (25J. This takes into account the fact that under equilibrium conditions, the acid catalyzed cOIllJensation proceeds via the porphyrinogen 3. The intermediacy of the porphyrinogen in the AdlerLongo synthesis had been shown by Dolphin [26], who was able to isolate

208 WIJESEKERA AND DOLPHIN
{J-octamcthyl-meso-letraphenylporphyrinogcn (3; A = CHJ ; R = CI'IHs) when 3,4-dimcthylpyrrolc was reacted with benzaldehyde under mild conditions (acetic acid, 50°C, 5 min). The porphyrin was obtained by treating it with an oxidizing agent (6 equivalents of iodine) since the oxidation of this Slcrically crowded porphyrinogcn in air was slow. The Lindsey modification (25J is a two-step one-pot procedure where pyrrole and the aldehyde are initially condensed with a trace of acid catalysis under anaerobic conditions to produce the porphyrinogen, which is oxidized to the porphyrin by subsequently added oxidant. Extensive reaction optimization studies have shown that maximum yields of the porphyrin (30-40%) arc obtained when equimolar concentrations (10 - 2 M) of pyrrole and aldehyde are condensed in the presence of boron trirluoride etherate (10- 3 M). The reaction is generally carried out in a chlorocarbon solvent at ambient temperature for I h before the oxidant (p-chloranil or DDQ) is added. In most instances; evaporation of the solvent followed by nash chromatography aHords the pure porphyrin.
Rothmund, Alder-Longo, and Lindsey methods (and modifications thereof) have all been used for the synthesis of meso-tetraphenylporphyrins with a wide variety of suhstituents on the phenyl groups. These include several biomimetic systems for heme oxygenation [27] as well as catalytic oxygen activation [28,29] .''lt is possible to prepare porphyrins carrying different groups al the meso positions by using mixed aldehydes in the cocondensalion with pyrrole . However, this results in the formation of isomeric mixtures which have to be separated in order to obtain the desired porphyrin. The mild reaction conditions of the Lindsey method allow porphyrins to be prepared ill good yields from sensitive aldehydes, but the optimum reaction concentration of 10- 2 M poses a problem for any large-scale synthesis. The Adler-Longo mcthod can therefore be considered complementary to the Lindsey method since it is amenable to scale-up for the synthesis of porphyrins in multigram quantities. However, the reaction of ortho-disubstituted benzaldehydes under the Adler- Longo conditions to produce the corresponding sterically hindered porphyrins has not mel with much success. onho· Uisubstitutcd phenylporphyrins have been used extensively as calalyst\ since the bulky orlho substiluents resist inactive woxo dimer formation and oxidative destruction during catalysis (Section VI). Although Longo et al. [30J reported the synthesis of both TPFPPHz (Adler-Longo conditions; 18°l(l spe'(;. trophotometric yield, I J % crude product yield) and TPCPPH2 (monochloro· acetic acid / benzene; 70/0 spectrophotometric yield), no yields have bcen rcported ror the pure product. Furthermore, strong absorption bands (659 and 663 nm, respecti vely) are reported for the two compounds indicating that the porphyrin is contaminated with the corresponding chlorin. Kim el aI. 1121 have reported the synthesis of TDCPPH, in 0.7% yield by rcnuxing

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 209
a mixture of 2.6·dichlorobenzaldchydc a nd pyrrole in benzene in the presence of acet ic acid. The yields have been improved using a modified Rothmund procedure (refluxing collidine instead of pyridine/scaled lube) whereby Lhe zinc complexes TPCPPZn and TDCPPZn have been synthesized in 4---100/ 0
yield [31,32J. The zi nc complexes are dcmclallaled with acid and treated wilh DDQ to obtain the pure porphyrin. However, under Lindsey conditions TDCPPH, and TPfPPH, have been synlhesized in 40-50'10 yield (33J. This method allows the synthesis of the free base porphyrin directly with no necess ity for removal of chlorin.
lIIeso-Telramcsilylporphyrin (TMPH2 , 4; A = H; R = (C,H,(CH,),J is a nother stcrically hindered porphyrin that has been used extensively for catalytic studies . Mcsitaldchydc (2,4,6*trimcthylbenzaldchydc) does not give the porphyrin product under either Adler- Longo or original Lindsey conditions, but has been converted to TMPHz in 4.5°"10 yield under modiried Rothmund conditions (34,35). By a modification of the original Lindsey procedure (using ethanol for cocatalysis) it has been shown (36,37] that TMPHz can be synthesized in up to 29% yield under mild reaclion conditions. Lindsey has extended this work to show that cocatalysis gives improved yields with 2-alkyl- , 2-alkoxy-, 2,6-dialkoxybcnzaldehydes (in addition lo 2,4,6-lrialkylbenzaldehyde) but ortlIO*halogen-substiluted benzaldehydes (e.g., 2,6-dichloro-, 2,6-difluoro-, and pcntafluorobenzaldehydes) show no increase in yields (33 J.
13-0ctasubsl ituted meso-tetraphenylporphyrins have a lso been sYlllhesized by cocolldcnsation of the 3,4-disubstitutcd pyrro!c and the appropriate benzaldehyde. Dolphin (26J prepared l3-octamcthyl-meso-tetraphenylporphyrin (4, A = CH,; R = c,.H,) in 43'10 yield by reacling 3,4-dimelhylpyrrole with benzaldehyde in refluxing acetic acid. whereas Evans and Smith (38J prepared the analogous octae thyl*meso*tet ra phenylporphyrin in 10070 yield starl ing from 3,4-diethyJpyrrole. Using 3,4-diphenylpyrrole and benzaklchyde, Mcdforth and Smith [39] prepared uodecaphenylporphyrin (4; A =
R = C"H j ) in 5.7070 yield under Lindsey conditions, whereas Takeda el al. (40J recemly obtained a 55% yield of this porphyrin under modified AdlerLongo conditions (refluxing in acet ic acid for 20 h followed by oxidation wilh DDQ).
A useful extension of the acid-catalyzed t.:ocondensations of pyrroles and aldehydes is the method developed by Cheng and LeGoff l41] to synthesize meso-ullsubstitUlcd ,:J-substi tu ted porphyrins. The reactions were carried out by refluxing ethanolic solutions of 3,4-disubstitutcd pyrrolcs with excess of formaldehyde in the presence of hydrochloric 01" hydrobromic acids. Aner several hours of reflux, the reaction mixtures were allowed to stand exposed to air to complete oxidation. Several meso-unsubslituted porphyrins carrying the same alkyl group o r different substiluellts (alkyl/ acetyl,

210 WIJESEKERA AND DOLPHIN
aJkyl / ester. anti phenyl / ester) in the two (3 positions of each pyrrole have been prepared in 64- 96% yield. When (he two (3 subsliLuenls are different , isomeric mixtures arc protiu<.:ed as expec ted . Changing the so lvent (to meth· anol or tetrahydrofuran) or lhe aldehyde (to a liphatic or aromat ic aldehydes) resulted in little or no porphyrin formation. Trcibs and Haberle [42) obtained 770/0 iJ-octamcthylporphyrin from the rcaUion of 3.4-dimethylpyrrole with formaldehyde in acetic acid and pyridine (Cheng a nd LeGorf report a 760/0 yield), while under similar reaclion conditions 3,4-diphcnylpyrro lc gave (:i-octaphenylporphyrin (OPPH, ; 4, A = C,H,; R = H) in 26% yield [43[. Recently, Takeda a nd coworkers [44) prepared OI'I' H, in 72% yield by condensing 3.4-diphenylpyrrole wi th formaldehyde in cthano l/ aq. HBr [d. 41) and oxidizing the ini tia ll y rormed porphyrinogcn wilh DDQ.
B. Self-Condensation of 2-Substituted Pyrroles
As shown in Scheme 3, this method uses the head-(O-lai l condensat ion o r a monopyrro!c 5 under acid co nditions. T he efficiency of this condensa tion reaction depends on the generation, stabi lity and reactivit y of the pyrryl-2-l:a rbo cation 6, produced by the removal or the leaving group (L) of the monopyrroJe 5. The 5 position of the pyrrole should be unsubstituled or should carry a carboxyl group (X = H or C02H), which would undergo facile elimination (decarboxylation) under the reaction condit ions . As is not the case in the cocondensat ion described earlier, the nature o r th is react ion allows the {3 substituents o f the pyrro lc to be different (A ~ B) and yet yield a single porphyrin product, although unuer certain acid concentrations the linear polypyrrolic intermediates a re known to undergo fragmentationl recombination at the bridging carbons lead ing to isomer rormation. Nature has chosen this method to cons truc t the common tetrapyrrolic macrocycie uroporphyrinogen using the monopyrroie porphobilinogen (5, A = CH2COzH; B = CH,CH,CO,H; R = H; X = H; L = NH,).
SiedcJ a nd Winkler [45) prepared several (l-substi tu ted porphyrins by heating 5-carboxy-3,4-dialkyl-2-hyoroxyn}ethylpyrroles ei ther dry or in 50 1-
B R A
~
¥tR- l ~Rl x N X H (l) H L A R B
5 6 7
Scheme 3

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 211
ution; Ihe 3.4-dilllethylpyrrole analog (5; A = B = CH,; R = H; X = CO,H; L = OH) produced a 46'70 yield or oetamethylparphyri n (OMP) on heating at 160- 170D C . However, if one or both {3 positions were unsubstituted, no porphyrin product was obtained. Octamcthyiporphyrin has also been prepared (as the copper complex) by reacting 2-aminomeLhyl-5-carboxy-3,4-dimethylpyrrole (5; A = B = CH,; R = H; X = CO,H; L = NH,) wil h coppcr(ll) acetate in refluxing methanol 146]. A template effect by the metal has been suggested because only traces of porphyrin were produced in the absence of metal. However, cx:taethyiporphyrill (DEP), one of the most widely used porphyrin model sys tems, ha<; also been made by a similar head-la-tail cundensation, without any added metal salts. The precu rsor pyrro!c used is 5-carbaxy-3,4-dielhyl-2-N,N-dilllethylaminomethylpyrrole [5, A = U = C,H, ; R = H ; X = CO,H; L = N(CH,hJ. This is reOuxed in near-neutral aqueous solution to give initially the porphyr inogen, which is oxidized to the porphyrin in air [47,4S}. The precursor pyrrolcs for these syntheses are prepared from 3,4-dialkyl-5-methylpyrrolc-2-carboxylates, which in lurn are obtained rrom acyclic precursors by standard Knorr reactions [49] . A recent report describes the synthesis of several 2-(substituted methyl) pyrroles and their transformations to porphyrins under Ilcar-neutral or basic condit ions [501·
Porphyrins carrying 6-0uoro substituents have also been synthesized by the cycJocondensalion of the appropriately substil uted pyrroles. 2,7,12, 17-Telrakis(tririuaromethyl)-3 ,8, 13,18-letraethylparphyrin (7, A = C, H,; B = CF1) has been prepared in 12010 yield (as its copper complex) by reOuxing 2-acetoxymethyl-5-carboxy-3-ethyl-4-trinuoromethylpyrroIe (5; A = C1H 1;
B = CF,; R = H; X = CO,H; L = OCOCH,) in acelie acid in the presence of copper acetate used as a template 151J. Once again, the starting pyrrolc has been prepared via a modified Knorr reaction 1521. The analogous 2-hydraxymethylpyrrolc (5, A = C, H, ; B = CF, ; R = H ; X = CO,H; L = OH), however, has been converted to the same porphyrin product in 30% yield by a room tempera ture reaetion in ethanol catalyzed by hydrobromic acid without added metal salts [53J. /l-Fluara-substitutcd 2-hydroxymethylpyrrale (5, A = C H,; B = F; R = H; X = CO,H; L = OH) has also been converted to the corresponding porphyrin, 2,7,12, 17-tetrafluoro-3 ,8,13 , 18-tetrarnethylporphyrin by refluxing in acetic acid in the presence of potassium fcrricyanide although in low 2% yield 1541. In the latter two cases, the starting pyrroles carrying the appropriate 11 subslituents were prepared by special reactions.
Another porphyrin synthesis that involves a self-condcllsation of a 2-hydroxymethylpyrrole uses as the starting material 3,4-disubstilulcd pyrrole-2-carboxylate esters. The estcr at the 2 position is reduced in situ wit h lithium aluminum hydride to give the corresponding 2-hydroxymethylpyrrole pre-

212 WIJESEKERA AND DOLPHIN
cursor (5, R = X = H; L = OH). which is cyc1ocondensed in dichloromethane (without isolation) using p-tolucncsulfonic acid as catalyst [55J. 11lis method has been used (0 synthes ize. in good yields, a series of porphyrins carrying a variety of (3 substituents including alkyl groups, substituted (2-, 3-,4- as well as 2,6 a nd 2,4,6-) phenyl groups and ester groups. It has been shown that lhe es ter group in the 0:' position of the starling pyrrole is select ively reduced by lithium aluminum hydride leaving any ester groups al the fl position unaffected [56J . The appropriately i3-substilutcd 5-unsubsti lulcu pyrrolc-2-carboxylate esters required for this synthesis are prepared by the reaction of nitroolelills or l3-acetoxynitro compounds with cr-isocyanoes ters in the presence of an organic base 157].
Self-condensa tio n of l1-unsubst ituted 2-hydroxymethylpyrro le (5, A = B = R = X = H; L = OH) has been used to prepare porphin it se lr. Krol [581 obtained yie lds of up to 5% by treating dilute solutions of the above pyrrole with potass ium pcrsulfate in glacial acetic add at 60°C . Longo et al. (591 reported yields as high as lSOJo for this synthesis by carrying o ut the cyciocondensation ill dilute ethylbenzene solutions at 100°C over a period of II days. Yalman [60] emphasized the imporlance of the metallemplate effect in his reported synthesis of copper porphin (1-20% yield) by adding a precooled solutio n of 2-hydroxymet hylpyrro le in cther/dimcthylformam idel acetic acid to a preheated (l45°C) mixture of excess copper acetate and dimeLhylformanlidc.
Recently, a new synthesis was been reported 1611 for the Illeso-tetrasubstituted porphyrins via the self-condensation of /J-unsubstitutcd 2-hydroxymcthylpyrrolcs 5 (A = B = H ; X = CO,H; l: = OH) where the R group is alkyl or aryl. The reactions have been carried out in relluxing propionic acid with yields ranging from 9% to 34070. Addition of zinc acetate to the reaction mixture improves the yield by 30-200%, which once agai n suggests a template effect of the metal. The R group was introduced in to the pyrrole precursor via a Vilsmeier-type acylation fo llowed by reduction with lilhium aluminum hydride.
C. Direct Coupling of Dipyrrolic Intermediates
This method, cOlllmonly referred to as the "2 + 2 synthesis," has been used ex tensively in porphyrin synthesis due ,to the variety of unsymmetric f3-subslituent patterns that can be accessed. It is important to note that only one of the two components needs 10 be symmetric to ensure a single porphyrin product. Dipyrromethanes and dipyrromethencs arc the widely used intermediates in the 2 + 2 syntheses and must carry suitable functional groups at the U ' positions in order to couple to each other. These can be prepared starLing from monopyrroles, which arc easily obtained by the classical Knorr reactions followed by functional group modifications 1491.

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 213
1. Porphyrins f rom Dipyrrometllelles
TIle 2 + 2 synt hesis using dipyrromethencs as intermediatcs formed the back· bone of Fischer's classical porphyrin syntheses [62J. The coupling reaction involved the fusion, in succinic acid, of one of four pairs of dipyrroillcthcnc intermediates shown in Figure 6b. The yields were genera lly low since the scnsiti ve {3 substilucnts did not survive the harsh reaction conditions. Selfcondensat ion o f dipyrromclhcncs has been used in the synthesis of porphyrins under milder reaction conditions [631 . The a ppropriately subst itu ted 5'-bromo-5-methyldipyrromethcnes (III) in rcnuxing rOrlnie acid (one equivalent of added bromine) produced etioporphyrin I a nd coproporphyri n I in up 10 60% yield, This synthesis can be generalized. to prepare porphyrins o f ~h symmetry using the more readily available 5'-carboxy-5-melhyldipyrromethene, which is brominatively det:a rboxyla ted under the reac tion condi tions. It has a lso been shown th.H the dipyrromelhcnc pair IV (Figure 6b) t:<ln bc used to synthesize porphyrins under similar react ion conditions [64) .
2. POIphyrills frum Dipyrromel/tanes
MacDonald [65) developed an allernative 2 .. · 2 synthesis that uses d ipyrromCll lanes, a procedure later modified by Kenncr I66J. This l11elllOd (Scheme 4) involves the condensation of 5,5'-diformyldipyrrometha nes (8) with 5,5'diullsubslitu!cd dipyrromethanes (9) under mild acid ca talysis, The intermedia te porphodimethcne (11) is rapidly oxidized to the porphyrin in air. Under the same condi tions, 5'-unsubstitutcd 5-formyldipyrrollicthanes (10) can undergo sel f-condensation to give porphyrins 12a or 12b of twofold ax ia l symmetry [67). This mcthod has been used successfully in the synthesis of highly deformed porph yrins [68), hindered capped porphyrins (69J, and fun ctionalized capped porphyrins (70J. Ogoshi and coworkers recent ly developed a mod ified 2 + 2 coupli ng of dipyrromethancs where instead of the 5,5'-di formyldipyrromethane (8) , a 5,5' -bis(hydroxybellzyl)dipyrro lllelhanc was coupled with a 5,5'-di unsubstituted d ipyrromethanc (9) to give unsymmetric 5, 15-diphcnylporphyrins [71],
A useful extension of this 2 ·t 2 coupling was reported by Ogoshi et al. [72J who cot:ondellsed (3-a lkyl-5,5'-ullsubslituted dipyrromclhanes (IJ) and aromat ic aldehydes (14) 10 prepare S,IS-diaryl -{3-oetaalkylporphyrills ( \6) at:cord ing to Scheme 5. By carrying out the reactions in reOuxing propionic aeid in the presence of zinc acetate, several porphyrins with substituted phenyl groups in the meso posit ions were obtained (as the zinc complex) in 15-25 11/0
yield, whi le the reaction in benzene wilh catalytic quantities of tr ifluoroacetic acid gave higher (30- 40%) yields of the des ired porphyrins a fte r ai r oxidation. As in the acid-catalyzed cocondensation o f pyrrole and bellzaldchydes, this reac tion was showl! by G unter and Mander (73J to procecd via a porphyrinogen in termedia te (15) , In the react ion o r l3- tctramcthyldi-

214 WIJESEKERA AND DOLPHIN
~: o 0
1-~H HW< \ ' or ii
C~C D D H H
9
H H
11 B
A/ A .
C C
D D 120
AMB~ 10 C D
~ NH HN I. H H
o
; ";I.; HMO H VNHHN~ o -" ..... A
C B 10
\ C
A D
D A C B
12b i) HI in CH,3C02H . then CH,3C0 2Na and 02
ii) TsOH in CH,30H/CH2CI2 . then CH, C02No and 02
B H H B
A ~ A ~~H ..;:.r
R- CHO OHC- R ~
....F~H ~""\- 14 A~A
B H H 8
13
Scheme 4
[ ;B H HB~ l _ B H H B
15
Scheme 5
B
B
16
B
B

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 215
pyrromethane (13, A = B = CH]) with orJho-ni lrobcnzaldehycte carried out in methanol wilh catalytic amounts of p-tolucncsulfonic acid, they isolated the porphyrinogcn in > 60070 yield and subsequently oxidized it quantitativel y to the porphyrin using a quinone oxidanl.
Several useful modifications of this synthesis have been reported, primarily to improve yields and increase the stability of the sensitive subslituents. Manka and Lawrence [74J reported the synthesis of a series of {1-unsubstilutcd 5, 15-diarylporphyrins (73- 92% yield) by (:a rrying out the cocondcnsation or 3,3',4,4'-unsubstitulcd dipyrromcthanc (13; A = Il = H) and substituted bCllzaldehydes in dichlormclhane at room tempera ture in the presence of catalytic amounts of trifluoroacetic acid, the in termediate being oxidized with p-chloranil. Maruyama and coworkers used milder conditions (trichloro· acetic acid/acetonitrile) to prepare a series of 5,15·diarylporphyrins (52-87% yield) carrying acid· labile substituents on the phenyl groups [75}. This system appears to be attractive for model building purposes, i.e., for sterica ll y hindered porphyrins [76], strapped porphyrins, and face-to· face d imers (77); for porphyrin oligomers for photosynthetic charge separation studies [78]; as well as for carotenoid-linked porphyrins used in intramolecular energy transrer studies [791.
D, Cyclization 01 Linear Tetrapyrroles
The need to synthesize porphyrins with totally unsymmetric 13 substitution led to the development of procedures involving the coupling of individual pyrroles in an unambiguous manner to give a linear tetrapyrrole, which is cyc1izcd in the final step to give the desired porphyrin. Of the four types of linear tetrapyrroles shown in Figure 6c, bilencs band biladienes ac are the most commonly used precursors to porphyrins [80).
I. Porphyrins from Bilelles b
Bilenes b (19) arc prepared [80,8 11 (Scheme 6) by Ihe ac id-catalyzed dccarboxylativc coupling of 5-carboxydipyrromethanc (17) with a 5-formyldipyrromethanc (18). The most commonly prepared bilenc is the I , 19-dimcthyl deri vat ive 19, which is readily cyciized to the copper complex of the porphyrin (20) using cupric saILs. The frce base porphyrin (21) is obtained by the removal of copper with st rong acids.
2. Porphyrins from Biladiel1es uc
Biladiencs ac are usually prepared and handled in their proLOnatcd rorm, commonly as the crystalline dihydrobromides. Their inherent stability makes them the most commonly used linear tetrapyrrole in porphyrin synthesis. Biladienes ac (24) were originally prepared by Johnson and Kay l82J by the
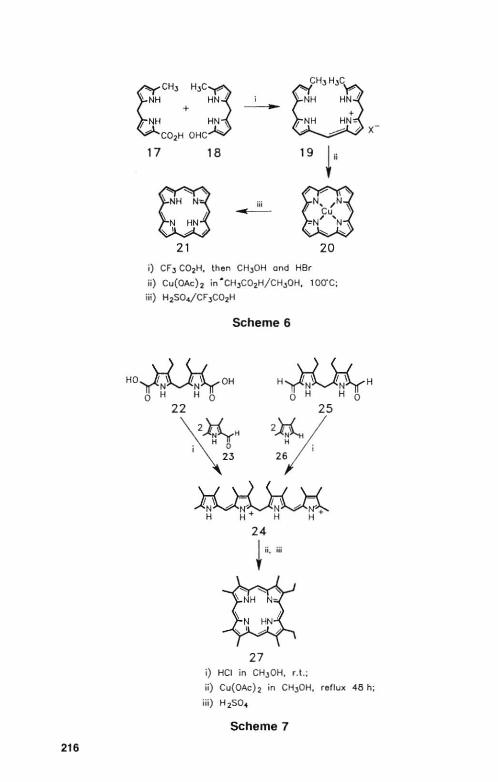
216
17 18
21
; -
iii -19 !'
20
i) CF J C02H, then CH,OH and Her
ji) Cu(OAch in · CH,)C02H/CHJOH. 100"C;
iii) H2S04/CF ,C02H
Scheme 6
x
HO 'rl \r{ OH YN~NY o H H 0 H~H
o H H 0
22 25
~¥rH ;\23 2~/ 2;1;
27 i) Hel in CH,OH, r.t.;
ii) Cu(OAcb in CH,)OH. reflux 48 h;
jii) H2S04
Scheme 7

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 217
acid-catalyzed (.;ondcnsalion of two equivalents of 2-formyJ-5-mcLhylpyrrolc (23) with a dipyrromethane-5.5'-dicarboxylic acid (22) or, alternatively, of 5-unsubslitutcd 2-lllethyipyrroie (26) with 5,5'-diformyidipyrromethane (25) (Scheme 7). Cyciizatioll of the biladienc was effected with cupric sails to give the porphyrin copper complex in 20-30070 yield.
The symmetry imposed on this sys tem, i.e. , the terminal rings having a symmetric arrangement of subslitucnts, has since been removed by a COI11 -pletely stepwise approach. Oifferenlially protected benzyl terl-butyldipyrromethallc-5.5'-dicarboxylatcs can be selectively dccstcrificd (by hydrogenolysis or acid cleavage) and coupled with a 2-fonnylpyrrolc to give initially a tripyrrin. Removal of the remaining ester allows the coupling of a different forlllyipyrroic to give the biladiene ac whose $-substituent arrangement can be totally unsymmelric [83-85[.
Johnson and coworkers (86] developed a regioselcctive synthesis of octaalkylporphyrins using l -bromo-19-methylbiladienes ac as the tetrapyrrolic precursor. As shown in Scheme 8, the biladicne is prepared from dipyrromethelles rather than dipyrromethanes. A 5'-bromo-5-bromomethyldipyrromethene 29 is reacted with a 5'-unsubstituted 5-methyldipyrromethcnc 28 in the presence of tin(lV) chlor ide to give the tin complex of the biladienc, which on treatment with mcthano lic hydrobromic acid produces the required biladienc 30 (as its dihydrobromide salt), in 80- 95070 yield. This is cyclized to the porphyrin either in refluxing o-dichlorobenzene (15 min) [86J or by allowing a solu tion in dimethylsulfoxide/pyridine to sland at room tempera-
8 C 8 C A~D ~ NH I-f\j -
D • A H,C H
28 i. ii
+ ~ 28r-
Sr CH2 Sf E
~ H
H ~ ~ ~ E G
30 F
G F t"i 29 8 C
A D
i) SnCI. /CH2CI 2• r.t. II) HBr/CHJOH H E
iii) O';OMSO/Pyridine. r. t. G F
Scheme 8

218 WIJESEKERA AND DOLPHIN
ture for several days in the dark 187]. The yields reported for the cyclizatiol1 step vary from 30% to 78%. Although thi s two-step procedure is by far the mildest for the synthesis of unsymmctr ic porphyrins in high yield, it was not explo ited to the fullest extent for some time due to the limited accessibility of the obligatory dipyrromethene com ponents 28 and 29. Significant improvements and modifications of some of the existing methodology for the synthes is of these dipyrromethellcs (hence the bi ladicllcs a nd porphyri ns) have been developed, the detai ls of which were reviewed rccenliy (88J. T his lwo·stcp procedure has been employed La synthesize several porphyrins in high yield; these include unsymmetr ic porphyrins, e.g., etioporphyrin III 189), singly and doubly linked porphyrin dimers 190,9 1) , /3-unsubstituled porphyr ins 192J as well as porphyrins with sensitive functional groups, e.g., mono- a nd di vinylporphyrins 192,93) and uroporphyrins I and III 194).
V. METALLATIONS AND DEMETALLATIONS OF PORPHYRINS
The tetradentatc anionic porphyrinato ligand gcnera ted by thc loss of the two inner NH protons is vcry versatile in it~ coordi nation chcmistry. and aJmost all metals and somc metaHoids have been combined wi th it. rive essen ~
tial stages can be idelllified in the synthes is o f a meta llopo rph yrin 195}:
A. Protonation-Deprotonation Equilibria
H~ p2+ .",. HlP · .",. HlP .",. HP - -== p2-
Deprotonalion of the free base porphyrin HzP uccurs so as to produce the dianion p2- which coordinatcs LO the metal ion. The presence of strong acids therefore retards metallation reactions and any reagent (or solvent) that absorbs the protons genera ted (decreases the acid ity or buffers the system) wi ll favor it.
B. Release of Metal Ion from the "Metal Carrier"
The metal carrier dissociates in the reaction medium produdllg a coord inatively unsaturated species which can combine with the dian ion p 2 - . The choice of the car rier depends on its activity. its availabilit y and its solubilit y in the solvent used to dissolve the porphyrin.
C. Formation of the MN, System
The init ial meLalla tion reaction involves the coordination of the four pyrrole nilrogen atoms to the metal. For divalent metal ions which prefer squareplanar arrangements (e .g., Ni, Cu), this produces a stable meta lloporphyrin complex.

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 219
D. Charge Neutralization
For metal ions with a charge greater than 2' • the above step produces a charged metalloporphyrin. In order to attain electronculrality, ax ial coordinadination is built up with anionic ligands. For Fe3 + and CI - , the square-pyramidal PFeel that is fOfmed has the metal center coordinativcly saturated, thus producing a stable metalloporphyrin complex . It is important to note that chromatographic purification using added polar solvents (e.g., methanol) generally results in axiailigand exchange which can be distinguished by absorption spectroscopy (see Section II) . Treatment with dilute aqueous hydrochloric acid usually regenerales the chlor ide form.
E. Completion of the Coordination Sphere
For metal ions that prefer octahedral geometry, the coordination sphere is completed by further complexat ion wi th neutral donar ligands such as water, pyridine, etc .
As discussed in Section II, metal insertion leads to a change in the absorption spectrum, particularly in the 500- to 700-nm region. Absorption spectroscopy can therefore be used to monitor metal insertion reactions. Buchler [95) discussed in detai l the different melallating systems classified by solvent as well as the l11etal carrier. No single metal insertion procedure is applicable to a ll metals or porphyrins and special precaution should be taken with porphyrins carrying sensitive functional groups. Metallation is best carried out in solutions as concentrated as possible since dilute solutions tend to slow the bimolecular reaction significantly. This would result in longer reaction times, which for high-boiling solvent systems could lead to decomposition of starting material or product. For the catalytically useful l11etals such as iron and manganese, insert ion is carried out with the M(lI) sailS, but subsequent autoxidation under aerobic conditions produces the stab le higher oxidation states M(lJI). Some common mctallating methods arc brieOy discussed below 195.96J.
F. Chloroform/Methanol Method
A renuxing concentrated chloroform solution of the porphyrin is treated with a saturated solution of the metal(ll) acetate and the insertion is fol ~
lowed by absorption spectroscopy. Upon completion, the reaction mixture is concentrated and diluted with methanol to yield the metalloporphyrin in ncar-quantitative yield. For add-sensi ti ve systcms (e.g., zinc), it is recommended that some sodium acetate be added to buffer thc medium. This method has been lIsed to introduce divalent metal ions such as Zn(lJ), Co(Il) . Cu(II ). and Ni(II), the last of which requires longer reaction times.

220 WIJESEKERA AND DOLPHIN
Another organic solvent thai we have found useful. particularly for the insertion of iron to {J-alkyiporphyrins, is lelrahydrofuran (THF) because of (he good solubility properties of porphyrins in this solvent . Furthermore, both chloroform and THF are low-boiling solvents which reduce the thermal decomposition of sensitive substiluents.
G. Acetic Acidl Acetate Method [97 -99]
lllC metal salt, usually the acetate, is heated with the porphyrin in acetic acid at 100°C. The product is crystallized either directly by cooli ng or by the addition of wate r or methanol. This method is applicable to the synthesis of ITlctalloporphyrins of most divalent metal ions; Mn(Il) and Fe(lI) a re autoxidizcd to the more stable high oxidation state. Once aga in , the use of sodium acetate is recommended for the synt hesis of acid-sensi tive systems.
H. Dimethylformamide Method [100]
Adler et al. showed that several metals can be convenient ly introduced into porphyrins by refiuxing a metal salt (usually metal chlorides) in dimethylformamiue (OM F). DMF is a uniformly good solvent for both porphyrins and metal salts. The HCI produced in the reaction escapes at the renux temperature of the solvent. This is the method of choice for phenyl- and/or pyrrolc-substituted meso-tetraphcnylporphyrins primarily due to their insolubility in low-boiling solvents and also due to their higher thermal stability. The product is crystallized directly by dilution with water or with dilule He l (3 - 6 N) to dissolve the excess metal sa IL s in the reaction mixture .
A major disadvantage of this solvent is that at renux it decomposes into dimcthylamine which, though neutralizing the HCI produced in the reaction, can lead to complicating side reactions, if long reaction limes are required. This is particularly so in the case of highly electron-deficient halogenated porphyrin systems where dimethyJamine may act as a Ilucleophile leading either to halogen substitut ion (l01) or to porphyrin destruction. We have observed thaL (J-oclachloro-lnesO-letrak is( 4-5ul f OJlalophen yl)porphyrin undergoes rapid decomposition ir iron insertion is attempted in DMF 1102], This is probably due to the lack of steric protection at the meso positions (orlllo-phenyl positions being unsubstituled), allowing attack by a nudeophilc leading to ring opening.
I. Pyridine Method [103]
This is a useful method for mctalloporphyrins labile toward acetic acid since pyridine is a good solvent for porphyrins as well as metal salts. The metailoporphyrins are usually isolated as the pyridinates. Pyridine forms complexes with metals of high charge and consequently retards the melailation process.

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 221
J. Metal Carbonyl Method [l04,105J
111is method has been proven to be especially useful for the preparation o f mClalloporphyrills of groups VI-V III metals. The metal carbonyl or the carbonyl chloride is heated with the porphyrin in an inert so lvent such as benzene, toluene, or dccalin.
K. Acetylacetonate Method [106, 107J
Metal acctylacetonates make very useful "carriers" due to the ir ready avai labi lity, sol ubility in organic solvents, and the low pKJ of the acid li berated during the reaction. With meta l ions of high charge and small size , a weak ly acidic solvent (e.g., phenol) is required to liberate the active mctallating species. This method hal) been used with metals of groups ili a a nd IIl b.
The process of dcmetallation is favored by the presence of a(.;id since protonation o f the free base, once formed, drives the equilibrium in the forward direction. Metalloporphyrin stability is often defined in terms of the degree of resistan(.;e LO the displacement of the metal atom by acid. Sev· eral stability classes have been identified bascd on complete dcmetallation in (a) acet ic acid, (b) aqueous HCI·dichloromethane, and ((.;) sulfuric acid. While some divalent metals (e.g ., zinc) are readily removed by trinuoro· acetic acid in dichloromethane, others (e.g. , Cu) require 15- 25% sulfuric in trifluoroacetic acid or sometimes cven stronger (.;onditions.
In order to remove iron from porphyrins, the stable ferric sta te is best reduced LO the ferrous statc. Several reagents can be used for this purpose 196) and the choice is usually based on the sensit ivity of the subst itucnts. Reduct ion is commonly carried out by a saturated solution of ferrous sui· fate whereas demelallalion is effec ted by llydro(.;hloric acid or gaseous HCI [1081 ·
VI. BIOMIMETIC METALLOPORPHYRIN CATALYSTS
During the 19705 and early 1980s, the synthesis o r biomimetic rnetalloporphyrins was directed towards generating systems that could mimic the reversible binding of dioxygen to the Fe(l!) center of hemoglobi n and myoglobin l27J . The primary structural requirement therefore was the presence of sterk protection to prevent a second Fe(ll) syslem frolll approaching and reacting to produce all oxidized Ii-OXO Fe(I1I ) species. However, since the late 1970s when much of the attention was diverted Lo understanding the mechanism o f action o f hemoprotein catalysts , the structural requirements for the metalloporphyrin models a lso changed. Although cytochrome P450 continues to be the focus of attent ion in biomimetic studies, lignin peroxidase has a lso been studied extensively since its discovery in 1984. In order

222 WIJESEKERA AND DOLPHIN
to design dficienl model catalysts, it is necessary 10 understand as best as possible the mechanism by which the enzyme functions.
A. Catalytic Cycle of Lignin Peroxidase
Lignin peroxidase is an extracellular enzyme that catalyzes oxidations in lign in-related aromatic molecules. Much of the atlention that th is enzyme has received recently is not only due La it s oxidative depolymerizat ion of lignin but to its abi lity to degrade a variety of environmental pollu tants including polycyclic arom.lic hydrocarbons 11 09.1101. dibcnw·p·woxins (110], and chlorinated phenols (1 t 1]. The ca talytic cycle of lignin peroxidase (Figure 7) is initiated by a two-electron oxidation of the Fe(lll ) slale of the resting enzyme, by hydrogen peroxide. T he active high-valent iron-oxo species (compound I) so produced init iates lignin degradation by one-elec tron oxidations of methoxylated aromatic substructures of lignin, forming cationradical species . These undergo bond cleavage and further nonenzymatic reactions with water and/or oxygen leading to lignin degradation. The enzyme reverts to its resting Fe(lIl) slate via compound II , which is a one-electron oxidized form.
B. Catalytic Cycle of Cytochrome P450
Cytochrome P450 catalyzes the insertion of one oxygen atom from dioxygen into the substrate, the other being lost as water; hence its designation as a monooxygcnasc. Hydroxylation or alkanes and epoxidat ion of alkencs
o II
Fe(IV)Por+ •
COl/POUND I lignin
) H202 ~~.-
hgmnol< lignin 0
~ ./ II Fe(Ill)Por Fe(IV)Por
RESTING ENZYIIE COMPOUND II
Figure 7 Proposed catalytic cycle of lignin peroxidase.

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY
e-
Fc(w)Por _\......-""~-__ Fe(u)Por
R- OH't r A- O j 0,
R- H ~ ~A o 0 '"
o II •
F e(JV)Por + '~7--~-"""~-I
Fe(u)Por
H20 e; 2H+
Figure 8 Proposed catalytic cycle of cytochrome P450.
223
are the Iwo most imporlant reactions catalyzed by this enzyme. As shown in Figure 8, the resting enzyme Fe(lIl ) undergoes a one-electron reduction (0 the Fc(l l) state, which binds dioxygcll. Following another one-elcctron reduction , the rate-limiting enzymatic step. and subsequent prolonaLion of the terminal oxygen atom, a heterolytic cleavage of the 0-0 bond leads to the active oxygen complex, which is be lieved to be a high-valent iron- axo complex [41, the o ther oxygen atom being released as waler. Transfer of the oxygell alom to the substra te results in C-H bond hydroxylation or a lkene epoxidat ion regenerates the resting Fc(lll) slate of the enzyme.
C. Design of Biomimetic Metalloporphyrin Catalysts
The postulation or a high-valent iron-oxo system as the active species in the proposed catalytic cycles of both lignin peroxillase and cytochrome P450 have resu lt ed in similar structural requirements ror lhe synthetic meta lloporphyrin systems used in biomimetic studies. Understandi ng its naturc, stability, and reactivity is important in the design and synthesis of a suitable catalyst. The properties of compounds I of pcroxidases are consistent wi th an oxoiron( IV)porphyrin x-calion rad ical 1I1 2,1 13J while a similar structure has been postulatcll ror cytochrome £-'450 as well [4J. However, an oxoiron(lV) specics with a protein-centered radical (compound I or cytochrome c peroxidase) [1141 has not been ruled out.
11 has been shown I hal the oxidation of simple l11etalloporphyrins can give rise to similar highly oxidized species corresponding to the catalytic intermed ia te. Usi ng TMPFeCI and m-chloroperbenzoic acid, G roves c1 al.

224 WIJESEKERA AND DOLPHIN
IllS) prepared and character ized the Qxoiron(IV)porphyrin intermediate [FcIV = O(TMP») ! which has since been studied extensively. Morc recently. similar species have been generated and spectroscopically characterized for two other iron porphyrins, TlJCPPFcCI and meso-tetrakis(2,4,6-tr imethoxyphenyl)porphyrinatoiron(lIl) (116] . However. unlike in the enzyme syslem where the ac ti ve species is bound to the protein moiety and hel1<,;c protected, in homogeneous reaction media this highly reactive intermediate of simple Illclalloporphyrins is capable of self-destruct ion. In order to minimize such loss of ac tivity, catalyst design has been directed primarily to increasing the electron deficiency. while providing steric bulk around the metalloporphyrin sys tem. This has been shown to improve catalyst stability by res isting oxidat ive destruction.
It is important to note. however, that the generation of an active oxoiron(lV) species from the resting Fe(llI) state of the two enzymes differ, lignin peroxidase using hydrogen peroxide (a two-electron oxidant) directly. while cytochrome P450 uses molecular oxygen a nd two reducing equivalents provided by NADPI-I. Thus, mimicking the lignin peroxidase sys tem involves the direct use of a metalloporphyrin catalyst in the presence of hydrogen peroxide or a similar single-a tom oxygen transfer reagent. However. the catalytic cycle of cytochrome P450 is very difficult to mimic using simple metalloporphyrin systems with molecular oxygen and a reducing agent. Several model systems using Mn(lIl)porphyrins as catalysts have been reported [117], bUI with low rates and yields. Groves et al. [118J were the first to demonstrate thal a system consisting of TPPFeCI as the catalyst and iodosylbenzene as an oxygen atom donor was able La perform the reactions catalyzed by cytochrome P450, suggesting the generation o f the high-va lent intermediate. Jt is this shunting that has been studied extensively for epoxidation of alkenes, hydroxylation of alkanes, as well as the oxidation of othcr organk substra tes, although various combinations of metalloporphyrins and singleatom donor reagents (AO in Figure 8) have been used [11 9].
D. Synthesis of Electron-Deficient Porphyrins
Due to the ease of synthesis. lhe metalloporphyrin ca talysts used in biomimetic studies have been of the meso-tetraphenylpo rphyrin type. Since Groves et al. demonstrated that a high-valent oxoiron porphyrin sys tem could be observed wi th the TMPFe system [I15J . it was rea li zcd that bulky ortho substitue l1lS on the phenyl groups could be im portant in preventi ng oxidative self-destruction of the catalyst. By using TPFPPFe, C hang and Ebina 1120J showed thal elec tro n deficiency can enhance the stability of the ca talys t and also increase reactivity to the substrate by generating a more electrophilic oxoiron imermooiatc. Traylor et al. [31J showed that TDCPPFeCI is a very efficient catalyst since both electron deficiency and sterk bulk are

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 225
incorporated into the molecule with orlllo-chiaro groups. In order to adapt this catalyst for use in aqueous media as a model for lignin peroxidase, Dolphin ct al. [121] prepared a water-soluble form by introducing sulfonic acid groups to the phenyl moieties, which also helped ill increasing the electron deficiency of the catalyst. The synthetic methods available for TMPFe, TPFPPFc, and TDCPPFc catalysts have already been discussed in Section IV.A. Sulfonalion of TDCPPH, has been carried Oul using fuming sulfuric acid at 165 °C for 7 h. Due to the directing influence of the ortho-chloro groups, sulronation takes place at the meta (3-) positions of the phenyl groups.
The development of the next generation of catalysts saw the introduction of more electron-withdrawing substituents into the catalyst molecule. Considering the electrophilic and radical nature of reactivity of Ihe porphyrin macrocycie, iJ-perhalogenation was the method of choice for creating greater electron deficiency in the system. Chlorination of TDCPP has been achieved by reacting its iron complex (31, M = FeCI, X = H), with chlorine gas at 140°C in the presence of anhydrous ferric chloride (I22}. The 24-IlJll red shift of the Soret band indicates lhe completion of the reactioll (20-30 min) and the purified /3-octachloroporphyrinatoiron (32, M = FeCI) has been isolated in > 85070 yield. The zinc complex (31; M = Zo, X = 1-1) has also
31
a lei SO~H
32 33
Scheme 9

226 WIJESEKERA AND DOLPHIN
F
F F
F 34 F
F F
F
Scheme 10
been chlorinated with N-chlorosuccinimidc 1123) in rcfluxing chlorocarbon solvents to give the analogous zinc complex (32; M = Zn) in 78% yield, but had required extended reaction limes (22 h). (:J Pcrchlorinatioll of TPFPP (34) has been achieved 1123, 124J via bOlh methods while Ihe truly perchloroporphyrin 37 has been prepared by the chlorination of TPCPPFe (36, M = FeCi) using the CI,/FeCi , procedure 11221. Chlorination of TMPZn (38, M = Zn, X = H) by N-chlorosuccinimidc. in rcnuxing methanol, produced 39 (M = Zo, X = Y = el) in which one meta position of each phenyl ring is chlorinated in addition to the e ight pyrrolic posit ions 1125] . Demctallalion with trinuoroacetic acid produced the free base porphyrin in 45 - 60% overall yield. 13 Chlorination of unsubstituted TPPFe (40, M = FeCi, X = H)

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 227
CI
CI CI
CI
CI
Scheme 11
could not be achieved usi ng the e ll/FeCI) sys tem due to destruction o f the macrocycle , while attempted chlorination of TPPFe, TPPZn, or TPPCu with N-chlo rosuccinimide produced the des ired ,B-octachlor inated porphyrin (41 , M ~ FeCi, Zn, or Cu) in low yield. However, T PPNi readed wilh Nchlorosuccinimide at 140°C to give 41 (M = Ni) in 750'/0 yield which was readil y demcta1lated and converted to the ferric complex in > 75% overall yield [1 22J. This confi rms the observation tha I the cent ra lly coordina ted metal exerts a d irective innuencc on peripheral substitut ion [1 26J. Heat ing the Ni COI11-
plex of 41 (M = Ni) in concent rated sulfuric add for 5 h followed by stirr ing a t rOO I11 tempera tu re fo r a fur ther 12 h produced , a fte r iro n insertion, the sulfonated hemin 42 (M = FcCI) , while the su lfonatio ll o f 32 to give 33

228
C
C
CH,
Scheme 12
WIJESEKERA AND DOLPHIN
required lhe use of fum ing sul furic acid 1121 , 122] . These compounds have been used as models for lignin peroxidase.
Perbromination of the porphyrin periphery has also been achieved with eit her bromine or N-bromosuccinimide. Octabromination of TDCP PZn (31, M = Zn. X = H) was first reported in reOuxing carbon tetrachloride (l g/ l00 rnl) using to equivalents of N-bromosuccinimide to give the desired prod uct 31 (M = Zn. X = llr) in 71 % yield II 27J. However. low solubility of the starting material in this solvent leads to ex tended reaction limes resulting in partial demetallation and incomplete bromination. The use of chloroform or tetrachloroct hene as the solvent has been recommended ill order to improve the yields [123]. TMP, as its zinc complex (38, M = Zn ,

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 229
40
41 42 Scheme 13
x = H), has also been oCLabrominated using 10 equivalents of N-bromosuccinimide in refluxing methanol solution (125). The product 38 (M = Zn, X = Bf) has been isolated in 50-65% yield. TPFPP, as its zinccompicx (34, M == ZI1), has been brominatcd wiLh N-bromosuccinimide in refluxing chloroCorm/ tetrachloroethane (l: 1) over a period of 48 h while triOuoroacetic acid wa>; slowly added [128). The oClabrolllinaloo product (35, M = Zn) has been isolated in 70% yield after zinc reinsert ion and purification . The chloroiroll complex 34 (M = FeCI), however. has been brominated with a 6 M solution of bromine (1 29], in dry degassed carbon tetrachloride by refluxing for 18 h, to give a 74 D/o yield of 3S (M = FeCI). Unsubstituted TPP. as its copper complex (40, M = C u, X = H), has been oClabromi nated (130) , by stirring a solu tion in chloroform/ carbon tetrachloride (1:1) with liquid bromine at room temperature (4 h) followed by the addition o f pyridine and further stirring (12 h). The produet40 (M = Cu, X = Br), isolated in 75"1, yield , was demetallaled with pcrchloric acid.

230 WIJESEKERA AND DOLPHIN
The effect of halogen substitution of the porphyrin ligand on the metal cenler has been demonstrated by cyclic voltammetry of the chioroiron(lll) complexes [131]. In general, electron-withdrawing chiaro subsLituenlS produce anodic shifts of rcdw.: tion potentials , but the crrecI was shown to be more pronounced for pyrrolic substitution. This rcOeds the case of reduelion of the metal center of Ii-halogenated porphyrins. While the positive shift of E ~'r for Felt I Fcl+ reduction of 31 (M = Fe, X = 1-1) compared to 40 (M = Fe, X = H) was marginal, the correspond ing shins for 32 (M = Fe) compared wil h 31 (M ~ Fe, X ~ H) as well as Ihal for 41 (M ~ Fe) compared (040 (M ::::; Fe, X = H) were approximately 0.4 V. Pcrchlorinalion of TPPFcCi (40, X ~ H) 10 TPCPPFeCI (37) produced a shift of 0.64 V.
The X-ray crys tallographic structures of !3-octabromo-TMPNi (38, M ~ Ni, X ~ I3r), TPFPPNi (35, M ~ Ni, X ~ I3r), as well as oclachlo roTPFPPCu (35, M = Cu, X = Cl) exhibit sadd le-shapc conformations. crcated by the buckling of the manocycie [128,132]. This distortion rrom planarity minimizes the nonbondcd interaction between f)- halogen atoms anu the orrho-phenyl subst itucnts.
E. Synthetic Metalloporphyrins for Commercial Applications
It is not the objective of this chapter to review the catalytk studies carricd out with the different metalloporphyrin model systems prepared to date, si nce that aspect will be discussed in several chapters of this volume (for a recent review, see Ref. 119). However, it should be noted thal some or the metalloporphyrin systems developed ror biomimetic studies have shown considerable promise in com mercial applications.
The water-soluble (sulfonated) polychlorinated Illetalloporphyrins prepared by Dolphin and coworkers as models for lignin peroxidase have also been examined for their ability to bleach kraft pulp [I22,lJ3]. This possible commercial applicatioll is particularly important due to the necessity to limit the currcnt use or chlorine in pulp bleaching. meso-Tetrakis(2,6-dichloro-3-sulfonalophenyl)-ti-uclachloroporphyrinaloiron(lll) [33, M ~ FeCll, in the presence of terr-butylhydroperoxide, has been shown to effect greater than 45% delignification or pulp when incubated for 18 h al60°C in pH = 4.8 burfer. A second possible appl ica tion that has been investigated is the decolorization of highly colored effluent rrom bleach plants, which is an environmental problem. Once again, the above catalyst / oxidant combination was capable of tlecolorizing (greater than 50%) undiluted effluent in less than 13 mill under appropriate comlilions. Further studies arc underway to determine the optimal reac tion conditions as well as the use or cheaper oxidants.

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 231
The hydroxylation of light alkanes to alcohols is an important reaction in the cOlllmercial production of fuel additives and other oxygenated chemicals . However. the major problem with biomimetic systems is that the use of single-alom oxygen transfer reagents to generate the active intermediate would not IlcI.:cssarily be economically viable. The work of Ellis and Lyons [132,134] is therefore unique in that oxidations of light alkanes to alcohols are carried out using activated metalJoporphyrins and molecular oxygen rather than single-atom donor reagents. Wilh J3-octachloro- and (J-oclabromo-mesO-LeL fa kis(pcntalluorophcnyl)porphyrinaloi Ton(ll I) as ca l alysts. it has been possible to oxidize isobutane with air even at room temperature to give lerl· but yl alcohol in > 90% selectivity with> 13,000 turnovers. The fact that catalysis is effected without the use of an added coreductant sug· gests that the mechanism is difrercnt frolll that proposed for cytochrome P450 itself. According to a postulated mechanism, the electron delkicncy in the porphyrin liga nd facilitates the reduction of the metal center to the Fe(ll) state , which binds dioxygen and subsequently produces the cata lyti cally active oxoiron(IV) species by homolytic cleavage of the It-PCroxo dimer. The Fe(ll) state and the oxoiron(lV) state are thought to be in equilibrium with a It-OXO dimer species, the position of this equilibrium being dependent on steric and electronic factors. Although no crystallographic data are yet available for the catalytically active hemins, it is likely that the enhanced catalytic activity observed in i3-halogenaled systems is related to the saddle shape of the ligand (observed for Ni and eu complexes (1321), which restricts its ability to form stable fI.-oxo dimer species. According 1O this mechanism, the catalyst essentially serves as a dioxygcnase rather than a monooxygenase (cytochrome P450) since both a to ms of molecula r oxygen arc transferred to the substrate via the oxoiron(IV) intermediate.
ACKNOWLEDGMENTS
This work was supported by the U.S. Department of Energy (MorganlOwll Energy Technology Center) , the Gas Research Ins titute and Sun Company (TPW), and by the Canadia n Natural Sciem.:es and Engineering Research Council (DD) .
REFERENCES
J. T . K. With, A short hi story or porphyrins and the porphyrias, Inl. 1. /Jive/utili . • II : 189 (1980).
2. D. Dolphin and E. Stcrubcrg, Medical applicati ons or dyes: a review of photodynamic therapy, in Infrared Absorbing Dyes (M . Matsuoka, cd .), Plenum Press, New York, 1990, p . l~J .

232 WIJESEKERA AND DOLPHIN
3. B. W . Henderson and T.J. Dougherty, How does photodynamic therapy work7 Pho/Dche"" PhUIOb;o:. , 55: 145 (1992).
4. P. R. Ortiz de Monlcllano, Oxygen activation and transfer, Cytochrome P-450: Structure, Mechanism alld Biodlemislry (p.R. Orti z de Montellano, ed.) , Plenum Press. New York. 1986.
5. M. Tien and T.K. Kirk. Lignin-degrading enzyme from Phallerochaefe cluysosporium: purification, characterization and catalytic properties of a unique H20 2-rcquiring oxygenase, Proc. Natl. Acad. Sci. USA, 81: 2280 (1984).
6. R. Donnett, Nomenclature. The Porphyrins. Vol. I (D. Dolphin, cd.), Academic Press, New York, 1978. p . I.
7. Nomenclature of tclrapyrrolcs; Reco mmendations of IUPAC-JUO Joint Commission on Biochemical Nomenclature, Eur. J. Biochem., 178: 277 (1988).
8. M.J . Crossley, P.L. Burn, 5.5. Chew , F.B. Cultance, and l.A. Newsom, Rcgiospecific imroducti on of rour subs titucnts LO porphyrin systems at antipodal pyrrolcnic positions. J. Cllem. Soc. Cirelli. Commufl., 1564 (1991).
9. M. Gou tcrman, Effects of substilUtion on the absorption spectra of porphin, 1. Cllem. Pllys .• 30: 1139 (1959).
10. T.P. Wijcsckera, J.B. Paine III , D. Dolphin, F.W.Il. Einstein, and T. Jones, Enforced deformation of porphyrins by short-strap br idging, J. Am. Chell/. Soc., 105: 6747 (1983).
II . G.M. Badger, R.A. Jones. and R.L. Laslett, The synthesis uf porphyrins by the Rothmund reaction, Allst. J. Chem. , 17: 1028 ( 1964).
12. J .D. Kim , J .J . Leonard, and F.R. Longo. A mechanistic stutl y of the synthesis and spectra l properties of meso-tc traarylporphyrins, J. Am. Chen/. Soc., 94: 3986 ( 1972).
13. K. Jcyarajah, A. Gold, G.E. Toney, and J.H. Helms, Preparation and characteri zation of the }(-oxo uimer and hydroxo complexes of (tetrakis(pentanuorophenyl)porphinatoJiron(III), IfI()rg. Chem., 25: 3516 (1986) .
14. D. Dolphin (cd.), The Porphyrins, Vols. I anti 2, Academic Press, New York, 1978.
15. A.H. Jackson and K.M . Smilh, The total synthesis of pyrro lc pigment s: 1973-1980, in Toral Sy"lhesi~i of Natural Products, Vol. 6 (J. ApSi mon. cd.), Wileyintersd ence. New York. 1984, p. 237.
16. E.J. Corey and H. -M. Che ll g, TIle Logic of Chemical S),flfllelis, Wiley, New York, 1989.
17. P . ROlhmund, A new porph yrin synthesis. The synthesis of porphin. J. Am. Chelll . Soc .. 58: 625 (19.16).
18. P. Rothmund, Porphyrin studies. III. The structure of the porphine ring system, J. Alii. Chelll. Soc., 6/: 2912 (1939).
19. P. Rothmuno and A.R . Mcnotti, Porphyrin studies. IV. The synthesis of tr.IJ. I ', o-tctraphenylporphine, J. Am. Chelll. Suc., 63: 267 (1941).
20. P. Rothrnund. f o rmation of porphyrins from pyrrole and aldehydes, J. Am. Cllem. Suc .. 57: 20 10 ( 1935).
21. R.I-I . Ilall, G. O. Dorough, and M. Cah' in, A fUTlher stud y of the porphine-like products of tlie reaction of benzaldehyde and Dyrro)e, J. Am. Clrem. Soc., 68: 2278 (1946) .

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 233
22. A.D. Adler, F.R. Longo, and W. Shergalis, Mechanistic investigat ions of porphyrin synthesis. Preliminary studies on mS-lctraphcnylporphin, 1. Alii. Chem. Soc .. 86, 3145 (1964).
23. A.D. Adler, F.R. Longo, J. Finarclli, 1. Goldmachcr, J. Assour, and L. KOf ' sakoff. A simplified synthesis for mcso-tct raphcnylporphin, 1. Org. Chem., 32,476 (1967).
24. K. Rousseau and D. Dolphin, A purification of meso-Ictraphenylporphyrin, Terruliedron Lell., 4251 (1974).
25. 1.S. Lindsey. I.e. Schrciman, H.C. H SlI, P.e. Kearney. and A. M. Marguerettaz, Rothmund and Adler-Longo reactions rcvisislcd: synthesis of Lelraphenylporphyrins under equilibrium conditions, J. Org . Chem., 52: 827 (1987).
26. D. Dolphin, Porphyrinogens anti porphotlimetbenes. Intermediates in the synthesi s of meso-tetraphenylporphyrins from pyrroles anti benzaldehyde, 1. Heterocyclic Chelll., 7: 275 (1970).
27. U. Morgan and D. Dolphin, Synthesis and strut:ture of biomimetic porphyrins, in Structure and Bonding, Springer-Verlag, New York, 1987, p. 116.
28. D. Mansuy, P. Baltioni, and J.-P. Baltioni, Chcmi{";al model systems for drugmetabolizing cytochrome PA50 dependent lIlonooxygcnases, Eur. J. Bioehelll., 184,267 (1989).
29. D. Mansuy. Diomimetic catalysts for selective oxidation in organic chemistry, Pure Appl. Chem .• 62: 741 (1990).
30. F.R. Longo, M.G. Finarelli, and J.B. Kim, The Synthesis and Some Physical Properties of ms-Tetra(pentachlorophenyl)porphin, 1. Heterocyclic Chell/., 6: 927 (1969).
31. P.S. Traylor, D. Dolphin, and T.G. Traylor, Slerically protected hem ins with electronegative substituents: efficient catalysts for hydroxylation ami epoxidation, J. Chem. Soc. Chem. Commufl., 279 (1984).
32. M.M. Williamson, C.M. Prosser-McCartha, S. Mukundan, Jr., and c.L. Hill, Isolation and characterization of the principal kinetic products in the Rothmund synthesis of sterically hindered tetraarylporphyrins . Crystal and molecular st ructures of [tetrakis(2,6-dichlorophenyl)porphinatoJzinc(lI) and bis[(meso-2, 6-d ich I oro phen y 1)-5 -( 0,0' -d ich lorobenz y I)d i pyrromet hene J zi nc (II) com p lexes, II/org. Chem., 27: 1061 (1988).
33. J .S. Lindsey and R. W. Wagner, Investigation of the synthesis of ortho-substituted tctraphenylporphyrins, 1. Org. Chem., 54: 828 (1989).
34. S.S. Eaton and G.R. Eaton, Rotation of phenyl rings in metal complcxes of substituted tetraphenylporphyrins, J. Am. Chem. Suc., 97: 3660 (1975).
35 . J.T. Groves and T.E. Ncmo, Aliphatk hydroxylation catalyzed by iron porphyrin complexes, 1. Am. Chem. Soc., 105: 6243 (1983).
36. R.W. Wagner, D.S. Lawrence, and 1.S. Lindsey, An improvcd synthesis of tetramesitylporphyrin, Tetruhedron Lell., 28: 3069 (1987).
37. M. Kihn-Botulinski and B. Meunier, Synthesis oftetramesitylporphyrin, II/org. Cltem., 27: 209 (1988).
38. B. Evans and K.M. Smith, Sterically crowded porphyrins: meso-tetraphenyl octacthylporphyrin, T{'{rahedrvn Lett ., 18: 443 (1977).
39. c.J. Medforlh and K.M. Smith, The synthesis and solution conformation of dodecaphenylporphyrin, Tefruhedrol/ Lell., 31: 5583 (1990).

234 WIJESEKERA AND DOLPHIN
40. J. Takeda, T. Ohya. and M. Sato. A fcrrochclatasc transition-state model. Rapid incorporation of copper(lI) illlo nonpianar dodccaphenylporphyrill , IlIor~. Chem ., 31 : 2877 (1992).
41. D.O. Cheng and E. LeGorf, SYllIhcsis of substituted porphyrins, Telral/{x lrUI/ Le/I .• 17: 1469 ( 1977) .
42. A. Treibs and N. Haberle, Syn thesis and electronic spectra of ms-subslillilcd porphines. JustllS Liebigs AI/II. Chem ., 718: JS) (1968).
43. M. Friedman. OClaarylporphyrins, 1. Org. Chem .• 30: 859 ( 1965). 44. J. Takeda, T. Ohya. and M. Sato, A new synthesis of oclaaryiporphyrills: nalur
ally occuring porphyrin mimics, Chelll. PIlarm. Bull., 38: 264 (1990), 45. W. Siedci and F. Winkler, Oxidation of Dyrrole uC!rivatiycs with Pb letraace·
tatc. A new kind or porphyrin synthesis, JUSfUS Liebigs Anfl. Chelll., 554: 162 (1943).
46. G.M . Badger and A .V. Ward. The synthesis of porphyrins rrom 2-aminomcthyl pyrrolcs.llust. J. Cirelli .. 17; 649 ( 1964) .
47. J.B. Paine III , W.B. Kirshner , D.W. Moskowitz, and D. Dolphin, An improved synthesis or octaelhylporphyrin, J. Org. Clrem., 41 : 3857 (1976).
48. c. -u. Wallg and C. K. Chang, A convenient synthesis or pyrrole precursors for oClaalkylporphyrins. Synfhesis, 548 (1979) .
49. J.B. Paine III, Synthesis of pyrroles anu of porphyrins via single-step coupli ng o f dipyrrolic intermediate, in The Porphy rins (D. Dolphin, ed.l, Academic Press . New Yurko 1978, p. 101.
50. H. Kinoshita. S. Tanaka, N. Nishimori , H. Dejima, and K. Inomata, Synthesis or 2-(substitutcd mcthyl)-3,4-disubstitulcd pyrrolcs and their cOllversion into the corresponding porphyrins, 8ull. C"em. Soc. Jpn., 65: 2660 (1992).
51. M. HOl1lllla , K. Auyagi, Y. AoyaTlta, and H. Ogoshi , Electron deficient porphyrins I. Tetrakis(trinuoromethyll porphyri n and its metal complexes, Tefrohedrol/ Leu., 14: 4343 (1983) .
52. H. Ogoshi, M. l-Ioml1la, K. Yakota, 1-1. To i, and Y. Aoyama, Synthesis o f 13-trinuoromethylpyrroles, Tetrahedron Lell ., 24: 929 (1983).
53 . K. Aoyagi, H. Toi, Y. Aoyama, and H. Ogoshi, Facile syntheses of pcrnuoroalk ylporphyrins. Electron deficient porphyrins 11, Chemislry Leu., 1891 ( 1988).
54. H. Ollda, H. Toi, Y. Aoyama. and H. Ogoshi, Fluoropyrroles and tctranuuroporphyrins, Tetrahedron Leff., 26: 4221 (1985).
55. N. Ono, H. Kawamura, M. Bougauchi, and K. Maruyama, Porphyrin synthesis from nilrocompounds, Tefrahedron. 46: 7483 (1990).
56. N. Ono, K. Sugi, and T. Ogawa , Preparation of porphyrins having ester or nilTo functions, Chew. Express, 6: 869 (1991).
57. O.H.R. Barton, J. Kervagoret, and S.Z. Zard, A useful synthesis of pyrrolcs from nitroolcfins, Tetrahedron, 46: 7587 ( 1990).
58. S. Krol. A new synthesis of porphin, J. Org. Chem., 14: 2065 (1959) . 59. F.R. Longo, E.J. Thorne, A.D. Adler, and S. DYIll, Notes on the synthesis of
porphin, J. I/elerocyclic Cllelll., 12: 1305 (1975). 60. R.G. Yaiman, Prcparation or porphin, substitu tcd porphin and metal ,-=hclates
thereo f. U.S. Palent 3,579,533 (1971). 61. Y. Kuroda, H. Murase, Y. Suzuki, and H. Ogoshi, A new roule for Meso-sub
stituted porphyrin, Tetrahedron L ell., 30; 2411 (1989).

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 235
62. H. Fischer and l-1. O rth, Die Chemie des Py rruls. Vol. 2, Part 1, Johnson Reprint, New York . 1968.
63. K. Smith, Po rphyrins and bile pigments from brominalcu pyrromcthcncs, 1. Chem . Suc. Perkin Trails., I : 147 1 (1972) .
64 . 1. B. Paine III , C. K. Chang, and D. Dolphin , The synthesis o f porphyrins via dipyrromclhcncs, lIeferu(,),t'ies. 7: 831 (1977).
65. G. P. Arsenault. E. BullOC k, and S.F. Mac Uo nald , Pyrromcthancs and po rph yrins therefro m, 0111. J . Chem .. 82: 4384 (1 960) .
66. J.A.S. Cavcleiro . A.M.O'A Rocha Gonsa lves , G. W. Kenner, and K.M. Smith , Pyrrolcs and relat ed compounds. 33. Total synthesis of ueutcralcJ derivati ves of protoporphyrin IX for nuclear magnetic resonance studies of hemoprolci ns , 1. Chelll. Soc. Perkin Trails., I: 1771 (1974) .
67. A. Markovac and S.F. Macdonald , Syntheses with 5-dibromomethyl- and 5-fo rmylpyrromethenes , Can. J. Chem ., 43: 3364 (1 965).
68. T .P. Wijesekera . J .U. Pai ne III, and D. Do lph in, Improvcd synthcsis o f covalent ly strapped porphyrins. Applicatio n to highl y deformed porphyrin synthesis. J. Org. Chelll ., j3: 1345 (1988).
69. T.P. Wijcsckera, S. Oavid , J.U. Paine III , B. R. Jamcs, am.l D. Dolphin , Durenecapped po rphyrins: synthesis and cha rac.:tcrizatio ll , 0.111. J. Chelll ., 66: 2063 (1988).
70. II. Tang, T .P . Wijesekera , and 0. Ool phill. Synthcs i ~ and chara!.: tcrilatio ll o f deri vati zed capped porphyrins, Can. J. Chem ., 70: 1366 (1992).
71. T . Ema, Y. Kuroda, and H . Ogoshi , Se1cc.: ti vc synthescs of ullsymmctrkallllcsoar yJpo rphyrins, Tetrahedron Lell ., 32: 4529 (1 99 1).
72. 1-1. Ogos hi. H . Sugimoto , T . Nishiguchi , T . Watanabe, Y. Matsuda , and Z. -!. Yoshida , Synthesis o f 5-aryl- and 5. 15-diaryl-2 ,3,7 ,8 ,1 2, 13, 17 , 18-octaet hylporphyrins, Chemistry Lell., p. 29 (1978).
73. M .J. G unter and L. N. Mander, Synthesis and atropisomcr sepa ration o f po rphyrins contai ning fUllctionali zati oll a t the 5, 15-meso positions: applit:ation to the synthesis o f binuclear li gand systems, J. Org. Chem .. 46: 4792 (198 1).
74. J .S. Manka and D.S. Lawrence, High yie ld synthesis o f 5.1 5-di aryl porphyrins, Tetrahedron Leu .. 30: 6989 (1989) .
75. A. Osuka, T . Nagata, F. Kobayashi, and K. Maruyama. An improvcd synthesis o f 5. 15-di ary loctaalkylporphyrins , J. lIeteroqclic Chem., 27: 1657 (1990).
76. R. Young and C. K. Chang, Synthesis am) eharacterizalion of blocked and ligandappen'ded hemes derived from alropisollle ri c meso-d iphenylpo rphyrins, J. A m. Chem. Sm.: .• 107: 898 (198S).
77. A. Osuka, F. Ko bayashi, T. Nagata, and K. Maru yama, One-pot synthesis o f strapped porphyrins and face-to-face d imcric po rphyrins. Chelll . LeU., 287 (1 990).
78. T . Nagata, A. Osuka . and K. Maru yama , Synthesis and o pt ical properties of conformationally constrained Irimede and pentameric porphyrin arrays, J. Am. Chem. Soc, 112: 3054 (1990).
79. A. Osuka , U. Yamada, and K. Maru yama, Synthesis of !.:ollfo rmationally restriclcd caro tenoid-linked po rphyrins, Chelll . Lett ., p. 1905 (1990).
80. A.W. Johnso n, Synthesis o f porphyrins from 1, 19-dideoxy bi ladiencs-ac and 1,1 9-dideoxybilenes-b , in The Porphyrills, Vol. I (D. Dol phin. cd .) , Academic Press. New York , 1978, p . 235.

236 WIJESEKERA AND DOLPHIN
81. P .S. Clczy and L. van Thuc. The chemistry of pyrrolic compounds, The oxidative cyciizalion of deri vatives of 1.19-dideoxyb ilenes-b, Aust. J. Chem .. 37: 2085 (1984).
82. A.W. Johnson and I.T. Kay, The formal ion o f porphyrins by the cycl ization of bilcnes, J, Chem. Soc., 2418 (1961).
83. l.A.P.D. de Almeida, G.W . Kenner, J . Rimmer, and K.M. Smith, Pyrrolc and related compounds. 35. A stepwise general synthesis of unsymmet rically subst ituted porphyrins, Tetrahedron, 32: 1793 «(976).
84. J. Engel and A. Gossauer, Synthesis of new bi ladicne-ac deri vati ves alld their cyciizatio ll to uroporphyrin Ill, 12-decarboxyuro porphyrin III and ~hy riapor
phyrin III methyl esters, Uebigs Anf/ . Chem., 1637 (1976) . 85. K. M. Smith and O.W. Craig, Porphyrin synthesis through tripyrrins: An alter
native approach , J. Org. Chem., 48: 4302 (1983). 86. R.L.N. Harris . A.W. Johnson. and I.T. Kay. A stepwise synthesis orunsym
metrical porphyrins, J. Chem. Soc. C, 22 (1966). 87. P. Bamfield, R. L.N. Harris, A. W. Johnson, and I.T. Kay, Synthesis of rhodo
porphyrin XV diethyl ester and relaled porphines. J. CheNI. Soc. C, 1436 (1966). 88. T.P . Wijcsekera and D. Dolphin, l-Bromo-19-mcthylbiladicncs-ac; userul pre
cursors 10 porphyrins. Synletl, 235 (1990). 89. J.B. Paine Ill , J. Hiom, and D. Dolphin , Bromination ordipyrromethenes for
porphyrin synthesis, J. Org. Chem., 53: 2196 (1988) . 90. J.B. Paine III and D. Dolphin, Synlhesisorcovaicntly linked dimcric porphy
rins, Call. J. Chem., 56: 1710 ( 1978). 91. J. Hiom, J.B. Paine Ill, U. Zap r, and D. Dolphin. The synthesis of cofacial
porphyrin dimers, Call. J. Clrem. , 61: 2220 (J983). 91. P. Yon-Hin, T.P. Wijesckera, and D. Dolphin. Transformation or a mono
viny lporphyrin to benloporphyrins via Diels- Adler adducts, Tetrahedron Lea., 30: 6135 (1989) .
93. P . Yon-Hill, T.P . Wijesekera, and D. Dolphin. An efficient route to viny lporphyrins, Call. J. Chem., 68: 1867 (1990).
94. T.P. Wijesekcra and D. Dolphin, unpublished resu hs. 95. J. W. Buchler. Synthesis and properties of metalloporphyrins, in Tile Porplry
rins, Vol. 1 (D. Dolphin, cd.), Academic Press , New York. 1978, p. 389. 96. J.-H. Fuhrhop and K.M. Smith. Laboratory methods, in Porplryrills alld Mel
allQPurphyrills (K.M. Smith, cd.), Elsev ier, Amsterdam. 1975, p. 757. 97. J.O. Alben, W.H. Fuchsman, C.A. Beaudreau, and W.S. Caughey. Substitu led
dcuteroporphyrins. III. Iron( lI) derivatives. React ions wit h oxygen and preparations from chloro and rnethoxo hemins. Biochemistry, 7: 624 (1968) .
98. P.J. Crook, A.H. Jackson. and O.W. Kenner, Pyrroles and related compounds. 28. Synthesis of alpha+oxymesoporphyrin -IX, Justus Liebigs Ann. ChenJ., 748: 26 (1971).
99. J .W. Huchler and L. Puppe, Metal complexes with tetrapyrro le ligands. II. Metal chelates of alpha, gamma-dimeth yl, gamma-dihydrooclaethylporphinc via reductive methylation of (octacth)'lporphinato)zinc, Justus Liebigs Anll. Chell/., 740: 142 (1970).
100. A.D. Adler, F.R. Longo, F. Kalllpas. and J. Kim, 011 the preparat ion or metalloporphyrins. 1. Illorg. Nud. Clrem., 32: 2443 (1970).

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 237
101. K.M . Kadish, C. Araullo-McAdams, B.C. Han, and M.M. Franzen, Syntheses and spectroscopic characterization o f (T(P-Mc2N)F4PPJ H2 and IT(p-Mc2N)F4-prIM where T(p-Me2N)F"pP is the dianion of mcso-tetrakis{o,o,m,m-lclraIluoro-p-(dimelhylamino)phcnyilporphyrin and M = Co(I I) , C u(ll), or Ni( II), Strw..: tures of (T(p-MczN)F4PP)Co and [mcso-tetrakis(penlanuorophcnyl)porphinato lcoball(II), (TF, PP)Co. J . Am. Chem. Soc., 112: 8364 (1990) .
102. T.P. Wijcsckcra and D. Dolphin, unpublished observations. 103. J. -H . Fuhrho p and D. Mauzerall, Onc-elcctron oxidation of mctalloporphy
rins. J . Am. Cltem. Soc., 9J: 4174 (1969) . 104 . M . Tsulsui. M. lchakawa, F. Vohwi nkel, and K. Suzuki, Unusual metallo
porphyrins. 1. Preparation of chromium mesoporphyrin IX dimethyl ester, J . Am. Chem. Soc., 88: 854 (1966) .
105. M. Tsutsui, c. P. Hrung, D. Ostfeld, T.S. Srivastava, D.L. Cu ll cn, and E.F. Meyer Jr. , Unusual metalloporphyrins. 23. Unusual meta lloporphyrin complexes of rhenium and technicium, J. Am. Chenl. Soc., 75: 3952 (1975).
106. J.W. Buchler, M. Foltz, H. Habets, J . van Kaam, and K. Rohb<x:k, Metal complexes of tet rapyrrole ligands. 17. Octacoordinate zirconium and ha fnium po rphincs 4.:0nlaining axial 1,3-dikcto nal e liga nds, Chel1l. Her., JQ9: 1477 ( 1976).
107. J.W. Buchler , L. Puppe, K. Rohbock , and H. Schneehage , Metal complexes of oclaelhylporphine. Preparation, axial ligand substitution and red uct ion, Arm. N. Y. Acad. Sci. , 206: 116 (1973) .
108. D.O. Morell, J. Darrell, and P. S. Clczy , Prosthetic group of cytochrome oxidase. I. Purification as porphyrin a and conve rsion into hemin a , Biocl/em. J., 78: 79J (1961) .
109. J.A. Bumpas , M. Tien , D. Wright. and S. D. Aust, Oxidation o f persistent cnviro nmental pollutants by a wh ite rot fungus , Science. 228: 1434 (1985).
110. K.E. Hammel, O. Kalyanaraman, and T .K. Kirk, Oxidation o f polycyclic aromaLic hydroca rbons and dibenzo lpJdioxins by Phanerochaefe chrysosporiwlI ligninase. J . Bioi. Chem., 261: 16948 (1986) .
Ill . V.-B. Hu ynh, H.-M. Chang, T.W. Joyce, ami T.K. Kirk, Dechlorinatio n of chloroorganics by a white -rot fu ngus, Tapp; J .. 68: 98 (1985).
112. D. Dolphin , A . Forman, D.C. Borg, J. Fajer, and R. H. Felton, Compounds I of catalase and ho rse radish peroxidase: 1r ·cation rad icals, Proc. Nail. Acad. Sci. USA. 68: 614 (1971).
113. J .E. Roberts. B.M. H offman, R. Ruiter , and L.P. Hager. Elect ron-nuclear double resonance o f horse radish perox idase compound I. Dcte<.:tion o f the po rphyrin w--calio n radical. J. Bioi. Chem., 256: 2 11 8 (1981).
114. B.M. Hoffman, J .E. Roberts, C. H . Kang, and E. Margoliash, Electron paramagnetic and electron nuclear double resonance o f the hydrogcn peroxide compound o f cytoch rome c peroxidase. J. Bioi. Chem., 256: 6556 (1981).
11 5. J .T. Groves, R.C. Haushaher, M. Nakamura, T.E. Nemo, and U.J . Evans, Ifigh-valcllt iroll - porphyrin complexes related to peroxidase and cytochrome P-450, J . Am. Chern. $Oc., 103: 2884 (1981).
11 6. D. Mandon, R. Weiss , K. Jayarajah, A . Gold, J . Terner, E. Bill, and A.X. Trautwein, Models for peroxidase compound I: ge nerat io n and spectroscopic

238 WIJESEKERA AND DOLPHIN
characterization of new oxoferry l porphyrin .. -catio n radical species, Inurg. Chem., 3/: 4404 (1992) .
117. I. Tabushi. Reducti ve dioxygcn acti vatio n by use of artificia l P-450 systems, Coord. Chem. Rev" 86: I (1988).
118. J.T. Groves. T .E. Nemo, and R.S. Me yers , H ydroxylatio n and epoxidalion catalyzed by iron- porphine complexes. Oxygen transfer from iodosylbenzcnc, J. Am. Chem. Soc., 101: 1032 (1979).
119. B. Meunier, Melalloporphyrins as versatile cata lysts for oxidation reactions and oxidative DNA cleavage, ehell/. Rev .. 92: 1411 (1992).
120. C.K. Chang and F. Ebina, NIH shiH in haemin -iodosy lbcnzcnc mediated hydroxylalions, J . Chem. Soc. Chem. COli/mull .• 778 (1981).
121. D. Dolphin, T . Nakano, T.E. Maione, T .K. Kirk, and R.K. Farrell, Synthelie mOtielligllinascs, in Lig"in Enzymic and Microbia' Degradatiull (E. Odier, ed .), INRA, Paris, 1987, p. 157.
122. D. Dolphin, T . Nakano, T.K. Kirk , T .P . Wijesekera, R.L. Farrell , and T.E. Maio ne, Porphyrins and uses thereoL U.S. Patent 4.892,941 ( IWO) .
123 . A.M . d'A, Rocha Gonsalves. R.A .W . Jo hnstone, M.M. Pereira, J. Shaw, and A .J.F. do N. Sobral, Metal -assisted react ions. 22. Synthesis o f perhaJogenated porphyrins and their use as oxidation cata lysts, Tetrahedron Lell. , 32: 1355 (1991).
124. J .F. Bartoli, O. Brigaud, P. Baltioni, and D. Mansuy, Hydroxylation of linear alkanes catalyzed by iron porphyrins: partkular efficacy and regiosclecli vit y o f perhalogenated porphyrins, J. Chem . Suc. Cltem. Commull., 440 (1991).
125. P. Hoffm ann , A. Robert, and B. Meunier , Preparation and catalytic adivili es of Ihe manganese and iron derivat ives of BrBTMP and CI'2TMP, two robust porphyr in ligands obtained by halogenatio n o f lelramesityiporphyrill, Bull. Chem . Soc. Fr., 119: 85 (1992) .
126. M.M. Catalano, M.J . Crossley, /\:t.M . Harding, and L.G. King, Control o f reacti vit y at the porphyr in periphery by metal - io n coordinalion: a general method for specific nitration at the bcta-pyrrolic position of 5,IO, 15,20-tetra aryporphyrins, J. Chem. Soc. Chelll. Commun., 1535 (19S4).
127. T.G. Traylor and S. Tsuchiya, Perhalogcnatcd lelraphenylhemins: stable cata· Iys ts of high turnover catalytic hydroxylations, /nurg. Chem., 26: 1338 ( 1987).
128. D. Mandon, P. Ochsenbein, J. Fischer, R. Weiss , K. Jayarajah, R.N. Austin, A. Gold, P .S. While, O. Brigaud, P. Ballioni, and D. Mansuy, {3-Halogcnalcdpyrrole porphyrins. Molecular structures of 2,3,7,8 ,12,13,17,IS-octabromo-5, 10, 15,20-tctramesilylporph yrin, ni ckel(ll) 2,3,7 ,S, 12, 13, 17 ,IS-octabromo-5, IO, 15,20·tet ramesi tylporph yrin and l1ickcl( ll) 2,3,7,8,12, 13 , 17, 18-oc tabro mo-5.IO, 15,20-letrakis{penlaJ1uo rophenyl)porphyrin, /norg . Chem .. 31: 2044 (1992).
129. J .E. Lyons and P.E. Ellis, Jr. , Selec ti ve low temperature hydroxylat ion o f isobutane by molecular oxygen catalyzed by an iron pcrhaloporphyrin COIllplcx. Lalalysis Lell., 8: 45 (1991) .

SYNTHETIC ASPECTS OF PORPHYRIN CHEMISTRY 239
130. P. Bhyrappa and V. Krishnan, OctabromOlclraphcnyporphyrin and its metal derivatives: elect ron ic structure anti electrochemical properties, Illorg. CIU.!/II., 30: 239 (1991).
131. T. Wijcsckera, A . Matsumo to , D. Dolphin, and D. Lcxa. Perchlorinated and highly chlorinated Illcso-Icl raphcnylporphyrins. Aflgew. Chem. Inl. t:d. El1gl., 29: 1028 (1990).
132. J ,E. Lyons, P .E. Ellis Jr., R.W . Wagner, P.R. Thompson, H . B. Gray, M.E. Hughes, and J .A. Hodge, Air-oxidation o f light alkanes by first -row transilio n lIletal.~ in macrocyclic ligand environments. in Proceedings of American Chell/kat Society. Petroleum Chemistry Division , San Francisco, 1992, p. 307.
133. P.S. Skcrkcr. R.L. Farrell. D . Dolphin, F. Cui, and T . Wijesckera , Iliomimcl ic bleaching or kraft pu lps, in Applicatiolls of Biutechllology ill Pulp and Paper Mllllufacture (T.K. Kirk anJ H .-M. Chang , cds. ), Butterworth -Heinemann, Boston , 1990, p . 203.
134. P .E. Ellis Jr . and J.E . Lyons, Selec tive air ox ida tio n o r light alkanes cata lyzed by act ivated mClall oporphyrins- the search ror a suprabiotic system, Coord. C'''em. Rev., 105 : 181 (1 990).