systematic optimization and modification on human primary
TRANSCRIPT

Systematic Optimization and Modification on Human Primary T Cells
CitationShui, Yifang Ivana. 2019. Systematic Optimization and Modification on Human Primary T Cells. Master's thesis, Harvard Medical School.
Permanent linkhttp://nrs.harvard.edu/urn-3:HUL.InstRepos:42057392
Terms of UseThis article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http://nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of-use#LAA
Share Your StoryThe Harvard community has made this article openly available.Please share how this access benefits you. Submit a story .
Accessibility

Systematic Optimization and Modification on Human Primary T Cells
Yifang Shui
A Thesis Submitted to the Faculty of
The Harvard Medical School
in Partial Fulfillment of the Requirements
for the Degree of Master of Medical Sciences in Immunology
Harvard University
Boston, Massachusetts.
May, 2019

ii
Thesis Advisor: Dr. Mark Cobbold Yifang Shui
Systematic Optimization and Modification on Human Primary T Cells
Abstract
Adoptive T cell transfer (ACT) has become a promising and integral therapeutic option for patients with leukemia and lymphoma. However, challenges remain in the application of this mode of therapy to other cancer types.
One inherent obstacle is the limited replicative lifespan of T-cells with little progress in the development of durable engraftment of T-cells. Decades of work have been devoted to improvements in the genetic modification of prolonging T-cells lifespan yielding multiple technologies. Universal viral vectors (e.g., lentivirus) system are at a disadvantage for low capacity, lengthy manufacturing, and low cost-performance for both research and clinical use. Attractive new transposons system (e.g., piggyBac) provide better options, but integration efficiency was limited, and the therapeutic engineering for human primary T cells remains clouded.
In this study, we are aiming to identify genetic modification that enhances the function and survival of primary human T cells. To do this, we have established a systematic Leap-In/Neon engineering system. We were able to show that this system was able to efficiently and stably modify human T cells. We next generated a list of all recurrent mutations that have been associated with T-cell lymphoproliferative states. Finally, we introduced these genes into primary human T cells using the transposons and tested the function and characteristics of the integrated cells. Our system may thus be used to express multiple integrated genes and/or combinations that will enhance the function and survival of primary human T cells.
We showed the stable transduction of Jurkat and primary T-cells using the Leap-In/Neon engineering system with efficiencies of ~60% and ~20% respectively. We have identified 6 gene candidates that can confer proliferative and/or survival advantages to transfected cells in vitro. Moreover, we demonstrated that the single candidate gene STAT3 Y640F and the gene candidate combo of Fas-TM-41BB and Casp7-DN may exert a tremendous pro-proliferation advantage on primary CD8+ T cell modification with maintained original cell characteristics and functions.

iii
Systematic Optimization and Modification on Human Primary T Cells
Table of Contents
1. Chapter 1: Background 1.1. Background
1.1.1. Cancer Immunotherapy 1.1.2. T cells in cancer immunology and immunotherapy 1.1.3. Genetic alterations landscape in T cell neoplasm
1.1.3.1. LGL 1.1.3.2. ATL 1.1.3.3. AITL 1.1.3.4. T-PLL 1.1.3.5. CTCL
1.1.4. Oncogenic signaling pathways in the cancer genome atlas 1.1.5. Apoptosis pathways 1.1.6. Survival pathways 1.1.7. Cytokines 1.1.8. T cell signaling pathways 1.1.9. T cell co-stimulation pathways 1.1.10. Other oncogenic pathways
2. Chapter 2: Data and Methods 2.1. Questions addressed in this thesis 2.2. Materials and Methods 2.3. Results
2.3.1. Leap-In/Neon Engineering on Jurkat cell Line 2.3.2. The first attempt of Leap-In/Neon Engineering on human primary T cell
transfection 2.3.3. Optimizations for Leap-In/Neon Engineering on human primary T cell
transfection 2.3.4. Establish optimal Leap-In/Neon Engineering System for human primary T cell
modification 2.3.5. Target gene pathway and transfecting gene list for human primary T cells
modification 2.3.6. Effects of transfected gene STAT3 Y640F on human primary T cell 2.3.7. Transfecting gene combos list for human primary T cells modification 2.3.8. Effects of transfected gene combo Fas-TM-41BB + Casp7-DN on human primary
T cell 3. Chapter 3: Discussion and Perspectives
3.1. Brief Discussion 3.2. Limitations 3.3. Future Research
4. Bibliography 5. Appendices

iv
Figures
Fig.1 Leap-In Transposase & Leap-In/Neon Engineering System
Fig.2 Transfection feasibility & efficiency testing on Jurkat cell Line
Fig.3 Post-Transfection Expression Duration of Jurkat Cell Line
Fig.4 Expression Stability of Jurkat Cell Line
Fig.5 First Attempt on Human Primary T Cell Transfection
Fig.6 Transfection Optimization on Transposon/Transposase Ratio in Primary T Cell
Fig. 7 Transfection Optimization on Electroporation Parameter in Primary T Cell
Fig. 8 Transfection Optimization on Promoter in Primary T Cell
Fig.9 Optimal Strategies for human primary T cell modification
Fig.10 Optimal Leap-In/Neon Transfection Engineering System
Fig.11 Mutational Landscape of T Cell Neoplasm
Fig.12 Target Gene Pathway for Human Primary T Cell Modification
Fig.13 Single Gene List for Human Primary T Cell Modification
Fig.14 STAT3 Y640F Single Gene Performance on Transfected Human Primary T cell
Fig.15 Gene Combo List for Human Primary T Cell modification
Fig.16 Fas-TM-41BB + Casp7-DN Combo Gene Performance on Human Primary T Cell

v
Tables
Tab. 1 2016 WHO classification of mature lymphoid, histiocytic, and dendritic neoplasms

vi
Acknowledgments
Here I would like to express my immense gratitude to Dr. Mark Cobbold for providing such a excellent opportunity to take this T cell engineering project, and I’m more than grateful to have Dr. Feng Shi for guidance and encouragement in carrying out this project.
Thanks to my fellows in Cobbold Lab for the stimulating discussions, for helping move this project further and for all the fun we have had with our 3D printing machine. In particular, I am grateful to Dr. David G. Millar for helping with TEAC assay and Sean Sepulveda for the guidance on immunophenotyping.
Moreover, I need to thank Dr. Jeremy Minshull, CEO from ATUM, who kindly fund and collaborate on this project. Also, I need to thank my workfellow Meggie Lee from ATUM, who helped and took charge of the plasmid construction.
Last but not least, I would like to thank my family: my parents and to my dear for supporting me spiritually throughout writing this thesis and my life in general.
This work was conducted with support from Students in the Master of Medical Sciences in Immunology program of Harvard Medical School. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard University and its affiliated academic health care centers.

Chapter 1: Background
1.1. Background
1.1.1. Cancer immunology
Cancer is a genetic disease of the unrestricted growth of cells. The uncontrolled nature of proliferation is initiated by mutations that activate oncogenic drivers and is also accompanied by genetic or non-genetic activation or inactivation of genes that may promote or suppress the proliferation process of tumor cells (Chen and Mellman, 2017).
Accumulation of these oncogenic mutations affects tumor cell proliferation. The mutational burden of tumors, as an essential effect factor for oncogenesis, provides a selective advantage to the population of the tumor cells by increasing genetic diversity and accelerating evolutionary fitness especially in the setting of immune recognition. It endows the tumor cells with hallmarks of cancer such as sustained proliferation, resistance to cell death, replicative immortality, genomic instability, evasion of growth suppressors and immune escape (Hanahan and Weinberg, 2011). Eventually, it may determine a person’s immune response to cancer and, at least partly, the immunotherapy (Palucka and Coussens, 2016).
The immune response to cancer is dependent on T cells that are specific for cancer-associated antigens. Cancer is visible to the immune system, i.e., antigenic/immunogenic (Palucka and Coussens, 2016). Of the mutations mentioned above, some of the nonsynonymous point mutations generated peptides that bound to major histocompatibility complex (MHC) molecules with high affinity. A few of these peptides are highly immunogenic containing the mutations that can be exposed to the T cell receptor (TCR) or mutations that create new anchor residues that increase binding affinity for MHC class I molecules. Also, some of the most immunogenic peptides are associated with frameshift mutations, insertion or deletion which can even create new protein or peptide sequences that are recognized as foreign by the immune system, i.e., neoantigens. Neoantigen then released and captured by dendritic cells (DCs) for processing with immunogenic signals. DCs present captured antigens on MHC I and MHC II molecules to T cells, resulting in T cell priming and activation of effector T cell responses.
Furthermore, the activated effector T cells traffic to and infiltrate the tumor. The mutant peptides are recognized by antigen-specific tumor infiltrating lymphocytes (TILs) by its TCR. Eventually, these cancer cells get killed and release additional tumor-associated antigens to increase the breadth and depth of the response in subsequent revolutions of the cycle (Chen and Mellman, 2013).

2
Although most tumor variants are eliminated, a few subclones with the ability to avoid immune detection and/or possessing mechanisms to suppress or evade the immune system often exist and mediate escape mechanisms to allow the tumor to progress.
More interestingly, by supporting chronic inflammation, shaping tumor immunogenicity, and suppressing antitumor immunity, the immune system can also conversely promote tumor progression. This dual paradoxical role of the immune system in suppressing or promoting tumor growth is termed cancer immunoediting and consists of three phases: elimination, equilibrium, and escape (Schreiber, Old, and Smyth, 2011). Throughout the elimination phase, though, the constant pressure from the adaptive immune system together with the genetic instability of tumor cells can select for tumor subclones with reduced immunogenicity that can evade immune recognition and destruction (Teng et al., 2015). These immune-edited tumors can as thus enter the escape phase, in which their growth is unrestrained, and disease becomes clinically apparent.

3
1.1.2. Cancer immunotherapy
Several decades of fundamental immunology advances along with cancer clinical trial data leading up to the point that many of the mechanisms and pathways restraining anti-tumor immunity have been uncovered. As such, although still early days, we could begin to unravel how to subvert cancer with a varies combination of immunotherapeutic approaches. Albeit with limitations and anticipated side effects, cancer immunotherapy has emerged as a rapidly progressing and transformative approach in the clinic to treat a wide variety of cancers. One important feature is it provides a degree of sustained clinical benefits, which is rarely observed with other treatments such as chemotherapy.
The general goal of cancer immunotherapy is to activate and enhance the immune response against cancer (Chen, 2018). Currently approved immunotherapies can be classified into two broad groups: active immunotherapy and passive immunotherapy based up a mechanism of action (Papaioannou, 2016).
Active immunotherapies engage the immune system by stimulating effector (or even memory) functions in patients with a responsive immune system upon challenge. Prominent examples include the development of cancer vaccines, including neoantigen/adjuvant vaccines, dendritic vaccines, and allogeneic whole cell vaccines, and the engagement of checkpoint inhibitors to the host immune system (Galluzzi, 2014). Also, recently adoptive cell transfer (ACT) using ex vivo activated and expanded tumor infiltrated lymphocytes (TILs) or the genetically engineered T cell (e.g., chimeric antigen receptor (CAR) - T cell and T cell receptor (TCR) - T cell) treatments.
Passive immunotherapy, in contrast, optimize the immune system to be more effective on patients with weak, unresponsive, or of low responsiveness immune systems. This category includes the infusion of checkpoint inhibitors and other tumor-specific antibodies, immune-checkpoint blockade, the systemic administration of recombinant immunostimulatory cytokines and other agents.
Preclinical studies have shown that only by targeting multiple non-redundant immune pathways in order to fully activate endogenous tumor immunity to enable iterative revolutions of the cancer immunity cycle can be efficient (Smyth et al., 2016). The type, location, and density of immune cells present in the tumor microenvironment (TME) are now recognized to be prognostic of a patient’s overall survival (OS) duration and predictive of their response to treatment (Hridesh Banerjee, Héctor Nieves-Rosado, 2018).

4
1.1.3. Genetic alterations landscape in T cell neoplasm
T-cells not only offer protection against the emergence of malignancies throughout the body but also they are capable of becoming transformed and changing into a malignancy by themselves.
T-cell neoplasms represent a rare but important and highly heterogeneous group of cancers (Watanabe et al., 2019). It contains precursor T-lymphoblastic lymphoma/leukemias which starts in thymus from thymocytes, for example, T-cell acute lymphoblastic leukemia/lymphoma (T-ALL); and peripheral T cell lymphomas (PTCL) that develop from more mature forms of T cells, such as, T-cell large granular lymphocyte leukemia (LGL), adult T-cell leukemia/lymphoma (ATL), T-cell prolymphocytic leukemia/lymphoma (T-PLL), angioimmunoblastic T-cell lymphoma (AITL). (Steven H. Swerdlow, 2016, Supplementary Data, Table1)
Moreover, like other types of tumor, T cell malignancies are well characterized by a feature of increased mutational burden. Recently, with the advance of molecular cytogenetics, large-scale expression analysis, Illumina Next-Generation Sequencing (NGS) and other comprehensive genomics services, investigations using integrated genomic sequencing (IGS) approach have helped to reclassify these diseases (see below). This knowledge of the mutational landscape of T cell neoplasm could provide more insights into T cell cancer evolution and heterogeneity. Moreover, more genotyping data obtained from healthy and diseased samples are uncovering new information on the genomic and transcriptomic profile along with diseased phenotypes and transcriptional sequences of copy number variations.
For the therapeutic approach, even though the landscape of oncogenic pathways is now primarily identified, clinical targeting remains unsatisfied. For the preliminary research, although genetically modified mice and primary-derived xenografts are available and can be used for preclinical studies in T-ALL, such in-vivo models are mainly missing in other more mature T cell malignancies inhibiting the process.

5
In this study, we summarized and listed some discoveries on mutational landscapes of several T cell neoplasms as follow:
▪ Large Granulocytic Leukemia (LGL)
LGL is believed to begin as an antigen-driven immune response followed by the constitutive activation of cytotoxic T lymphocytes or NK cells. Frequent somatic STAT3 mutations have previously been reported in large granular lymphocytic leukemia (LGL) (Olson et al., 2012). Other findings later validated this with another cohort reported up to 40% of LGL patients harbor mutations in the STAT3 gene, whereas 2% of them carry STAT5 mutations. Exome sequencing in a cohort of 113 LGL patients reveals that at least one mutation, for example, PTPRT directly affects STAT3 pathway or like BCL11B, SLIT2, and NRP1 indirectly alternate the T-cell activation (Andersson et al., 2013). Recently, these shreds of evidence are strengthened by another research of activating STAT3 in LGL, and they found that LGL patients without STAT3 SH2-domain mutations harbor novel activation mutations in the DNA-binding and coiled-coil domain of STAT3 (Andersson et al., 2015). Besides, previous studies (Fig.1) also highlight the dysregulations of other apoptotic pathways such as FAS/FASL and the activation of survival pathways like Janus Kinase/signal transducer and activator of transcription (JAK/STAT), IL-15, PDGF, phosphatidylinositol-3-kinase (PI3K)/AKT/mTOR, and RAS/Raf/MEK/ERK pathways (Sokol, 2007; Zhang and Jr, 2012)

6
Figure 1. The signaling network underlying LGL lymphoma pathogenesis (Zhang and Jr, 2012)
1.1.4. THE GENOMIC LANDSCAPE OF PEDIATRIC AND YOUNG
ADULT T-LINEAGE ACUTE LYMPHOBLASTIC LEUKEMIA
▪ Adult T-cell leukemia/lymphoma (ATL)
Even though HTLV-1 infection is a hallmark of ATL and prerequisite for its diagnosis, but HTLV-1 alone is still not enough for ATL development. Recent studies first demonstrated the clinical effects of new genetic abnormalities regarding the genetic profiling data. Besides ZEB1, recent integrated analysis suggest that aggressive (lymphoma) subtypes were associated with an increased burden of genetic and epigenetic alternations, higher frequencies of TP53 and IRF4 mutations, and many copy number alternations (CNAs), including PD-L1 amplification and CDKN2A deletions, compared with indolent chronic subtypes (Kataoka, Iwanaga, Yasunaga, Nagata, Kitanaka, Kameda and Yoshimitsu, 2018).
▪ Angioimmunoblastic T-cell lymphoma (AITL)
Up to now, the biology of AITL has become gradually understood as a result of the multistep and multilineage tumorigenesis concept: premalignant cells having epigenetic mutations evolve into the tumor and TILs through the clonal selection of the mutated cells (Fukumoto et al., 2018). The genetics of angioimmunoblastic T-cell lymphoma (AITL) is very poorly understood. Based on mutational landscape study across 219 genes in 85 cases revealed that AITL is characterized by high frequencies of overlapping mutations in epigenetic modifiers, such as TET2, IDH2, and DNMT3A. Three of four mutations in STAT3 also occurred at codons 614 and 661, which are known to confer gain-of-function in CTCL and LGL (Odejide et al., 2014).
▪ T-cell prolymphocytic leukemia/lymphoma (T-PLL)
T-PLL is an aggressive neoplasm of mature T lymphoma. Rearrangements between the TCL1A/B locus on chromosome 144, or its homolog MTCP1 on chromosome X and the T-cell receptor locus on chromosome 143, are characteristic of T-PLL (Dearden, 2009). Recent integrated genomic sequencing data reveals the mutational landscape of T-PLL and identified gain-of-function mutations involving IL2RG, JAK1/3, and STAT5B as well as deleterious mutations affecting EZH2, FBXW10, and CHEK2 in T-PLL.

7
Besides, evidence has been shown for pharmacologic targeting of primary T-PLL cells with the STAT5 inhibitor pimozide leads to apoptosis (Kiel et al., 2014).
▪ Cutaneous T-Cell Lymphoma (CTCL)
CTCL is a malignancy of skin-homing T cells. Loss of the normal TCR repertoire leads to immunosuppression and opportunistic infections, which are the most common disease-related causes of death. The previous investigation identified deletions of TP53 and CDKN2A and amplification of MYC (Laharanne et al., 2010). Recently exome and whole genome DNA sequencing and RNA sequencing have been performed on purified CTCL and matched normal cells (Choi et al., 2015). Results showed frequent deletions and damaging SSNVs in chromatin-modifying genes, such as ARIDA, CTCF, and DNMT3A (Couronné, 2012). Many mutated genes also contribute to other T cell neoplasm, for example, CD28 and RHOA were also reported in PTCL (Yoo, Sung, S. H. Lee, et al., 2014), STAT5B also reported in LGL (Rajala et al., 2013), ZEB1 also reported in ATL (Hill, Browne, and Tulchinsky, 2013). Also widely noticed, in CTCL, recurrent mutations in the PLCγ1 catalytic domain have been observed in 21% of cutaneous T-cell lymphomas and are associated with downstream signaling and NFAT activation (Vaqué et al., 2014).
1.1.5. Oncogenic Signaling Pathways in The Cancer Genome Atlas
▪ Apoptosis Pathway
Apoptosis is a mechanism by which cells are destined to death of either intrinsic signals or extrinsic signals with interactions of pro-apoptosis factors and anti-apoptosis factors. It plays a significant role in immune response, positive and negative T cell selection, and cytotoxic death of target cells. When apoptosis pathways are impaired or are not tightly regulated, for example, an imbalance in the anti-apoptotic and pro-apoptotic factors has been taken place, diseases such as cancer can ensue. Besides, current therapies target on the modulation of the apoptotic death pathways to regulate the immune response. In this study, we will focus on the process of T cell apoptosis in response to cancer progression.
Intrinsic pathway, also known as mitochondria pathway. Internal DNA damage signals through the pro-apoptotic molecule, Bax migrate to the mitochondrial outer membrane, act as an antagonist to the Bcl-2 family protein, which normally expressed on the membrane for an anti-apoptosis purpose, and the interaction leads to cytochrome c release by making pores. Cytochrome c then binds to apoptotic protease activating factor-1 (Apaf-1) and form apoptosome complex. Apoptosome complex then binds and activate

8
caspase-9 results in the cleave of downstream caspase-3 and caspase-7. The result of the caspase cascade is the degradation of chromosomal DNA and cell phagocytosis (Susan, 2007).
Extrinsic pathway, in contrast, is mediated by the death receptors and its specific ligand. An example of this is the Fas/FasL-dependent signaling pathway, which recruiting the Fas-associated death domain (FADD) and procaspase-8 to the Fas. Accumulation of several procaspase-8 molecules triggers the autocatalysis of caspase-8; this also results in the cleave of downstream caspase-3 and caspase-7, eventually leads to apoptosis of T cell. Moreover, chronic stimulation of the T cells could lead to terminal differentiation of T cell express programmed death 1 (PD-1) and T cell immunoglobulin mucin 3 (Tim3), referred to as exhausted T cells (Sakuishi et al., 2010), and leads to immunosuppression, thereby affecting the immune response.
These T cells fail to proliferate, and cannot function as effector cells; that is, they fail to elicit a cytotoxic response to antigen stimulation, which results in unhindered tumor growth.
▪ Survival Pathway
JAK/STAT signaling pathway is altered commonly in cancer includes T cell neoplasm such as T-LGL. As mentioned above, constitutive activation of STAT3 plays a fundamental pathogenic role in LGL leukemia. Besides, a high amount of IL-6 in LGL patients is able to activate the JAK-STAT3 pathway, and trigger clonal proliferation and expansion by DCs (Teramo et al., 2013).
The PI3K/ALT/mTOR signaling pathway is essential in regulating normal cell proliferation, metabolism, and survival. Pathogenic somatic activating mutations and gains/losses of function in the PI3K/AKT/mTOR pathway are the most commonly identified in many different solid and hematological tumors or cancers (Porta et al., 2014). The gain of function mutation of genes involved includes PIK3CA, PIK3R2, AKT1, AKT2, AKT3, and mTOR have been reported to cause a spectrum of overgrowth syndrome including lymphatic overgrowth (Keppler-Noreuil et
al., 2014). The components of this pathway exhibit significant cross-talk with many other pathways, including Ras family of small GTPase proteins/mitogen-activated protein kinases (Ras/MAPK) pathway (Keppler-Noreuil et al., 2016). Interestingly it has been demonstrated that in nelarabine-resistant T-ALL cell lines, there is hyperactivation of PI3K/AKT/mTOR, ERK1/2 and Bcl2 signaling in response to nelarabine treatment (Lonetti et al., 2016). PI3Ks are lipid kinases that phosphorylate the 3-hydroxyl group of phosphoinositides. PI3K converts phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-triphosphate (PIP3), which interacts with AKT at the cellular membrane where it is phosphorylated and activated. Moreover, key negative regulators of

9
the PI3K-Akt pathway includes tumor suppressor phosphatase and tensin homolog (PTEN) which dephosphorylates PIP3, germline loss-of-function mutations of these genes are established to cause PTEN Hamartoma Tumor Syndrome (PHTS) (Weigelt and Downward, 2012).
RAS/Raf/MEK/ERK/MAPK signaling pathway regulates multiple critical cellular functions including proliferation, growth, and senescence (McCubrey et al., 2007). This pathway has been reported in human cancer as a result of abnormal activation of receptor tyrosine kinases or gain-of-function mutations mainly in the RAS or RAF genes (Santarpia, Lippman and El-Naggar, 2012). Particularly as mentioned above, RHOA, a member of the RAS family, binds GTP in its active form. The G17V mutation, located in the GTP-binding pocket, prevents GTP binding and thus acts as a dominant negative. The functional loss of RHOA in T-cell progenitors in the thymus culminates in the development of T-cell lymphoblastic leukemia in mice, thus confirming a central role for RhoA in T-cell malignancies (Fukumoto et al., 2018).
▪ Cytokines
Cytokines are another dependency for the generation of a robust stimulation, in order to initially stimulate the proliferation of naïve T cells and the development of a fully competent T-cell response.
IL-2 is a pleiotropic cytokine that drives T cell growth, augments NK cytolytic activity, and mediates activation-induced cell death. IL-2 signals via the heterodimerization of the IL-2R β/γc cytoplasmic domains leads to the activation of at least 3 major signaling pathways: PI3K/AKT, Ras-MAPK, and JAK-STAT.
IL-15 is cytokine that utilizes a non-transducing, private α-chain and a common IL-2/IL-15 βγ-signal transducing complex intracellularly and exists in a transmembrane form to function as a growth and survival factor. IL-15 and IL-15R mediate the use of the JAK1/JAK3/STAT5 signaling pathway, which supports T cell and NK cell homeostasis and expansion (Mishra et al., 2014). In addition, IL-15 triggers other signaling pathways in T lymphocytes, including the PI3K/AKT, and the Ras/MAPK pathways which lead to mitogenic and antiapoptotic signals (Abadie and Jabri, 2014). Increased level of IL-15 showed in CTCL patients in a clinical stage-dependent manner, indicates a pathogenic role in CTCL. Besides, overexpression of IL-15 in the transgenic mouse model induces a spontaneous CTCL that mimics the human neoplasm (Mishra et al., 2016).
IL‐7 plays a vital role in the differentiation of pre‐T cells into mature thymocytes and controlling the survival and homeostatic proliferation of naive T cells in the periphery (Mazzucchelli, Riva and Durum, 2012). Two main pathways are activated by IL-7: the JAK/STAT5 pathway and the PI3K/Akt/mTOR mammalian target of rapamycin

10
pathway. Subsequently, studies have demonstrated that IL-7 stimulates a significant increase in DNA synthesis and prevents the caspase pathways that lead to spontaneous apoptosis. Data from recent studies demonstrated that around 10% of T-ALL patients display IL7R gain-of-function mutations (Ribeiro et al. 2013). The MAPK cascade could also be activated with IL-7 signaling in T-ALL (Tal et al., 2014).
Although still in early days, the understanding of pathogenesis of T-cell neoplasm has come to a theoretical model that it needs 3 signals, recurrent genetic modification in antigen (“signal 1”), costimulatory (“signal 2”) or cytokine receptors (“signal 3”), and the tyrosine kinases and other signaling proteins they active (Wilcox, 2016). In other words, many of these genetic modifications do not the only function autonomously by the cell itself, but also require interaction with the tumor environment. Especially in the concept of T cell neoplasm, the different fields of cancer genomics and immunology are converging.
As we mentioned above, genetic alternations in signaling pathways that control cell-cycle progression, apoptosis, and cell growth could contribute to typical hallmarks of cancer, but the mechanisms and co-occurrence of alternations in these pathways differ between each tumor types. Many studies, including The Cancer Genome Atlas (TCGA), profiled mutations, copy-number changes, mRNA expression, gene fusions, and DNA methylations, and analyzed the mechanisms and patterns of somatic mutations in several pathways.
Here in this study, we summarized some canonical pathways as follow:
▪ T-cell Receptor Signaling Pathway
The TCR is a multimeric complex composed of two ligand-binding glycoproteins containing variable regions (αβ- or γδ-TCR heterodimers), which are expressed on the cell surface in association with four CD3 molecules. TCR-αβ or TCR-γδ recognizes the antigen, whereas CD3 molecules control assembly and signal transduction. Each of the CD3γ, CD3δ, and CD3ɛ subunits contain one ITAM, and CD3ζ contains three ITAMs (Guy and Vignali, 2009). Upon receptor ligation, two tyrosine residues within each ITAM are rapidly phosphorylated by a member of the src-family PTK, transforming them into high-affinity ligands for Syk PTK such as ZAP-70. The coordinated actions of Src and Syk PTK initiate a cascade of signals that ultimately leads to cell proliferation, cytokine secretion, and effector functions (Palacios and Weiss, 2004). However, propagation and termination of TCR signaling need T-cell signaling molecules (for example, PKCθ, Lck and ZAP-70) to form a cluster at the site of cell-cell contact, generating an immunological synapse. The inducible T-cell kinase–SYK associates constitutively with lipid rafts in T cells and triggers antigen-independent phosphorylation

11
of TCR proximal proteins, leading to activation of downstream pathways and acute cellular outcomes that correspond to regular TCR ligation.
Notable examples like the chromosomal translocation t(5;9) (q33;q22) that generates the interleukin-2-inducible T-cell kinase (ITK)–spleen tyrosine kinase (SYK) fusion tyrosine kinase has been identified in a subgroup of PTCL (Feldman et al., 2008). Whereas, LCK, ZAP-70, LAT, and ITK are expressed in almost all PTCL, NOS, and AITL (Rodríguez-Pinilla et al., 2013).
The widespread expression of both TCR and its downstream kinases and adaptor proteins indicate that malignant T cells may exploit both the growth and survival signals they provide, including nuclear factor-κB (NF-κB), nuclear factor of activated T cells (NFAT), PI3K, RAS, and mitogen-activated protein kinase (MAPK) pathways.
▪ T-cell co-stimulation Pathway
Other engaging the TCR alone is insufficient to fully activate naïve T cells and initiate a fully competent immune response, it can only lead to a state of antigen unresponsiveness (i.e., anergy), in the absence of the second signal provided by costimulatory receptors. The multiple costimulatory receptors belong to either the immunoglobulin (Ig) superfamily or tumor necrosis factor receptor (TNFR) superfamily.
CD28 is the founding member of Ig superfamily expressing as a homodimer on most human T cells. By binding SH2- and SH3- domain-containing proteins, assembly and activation of these kinases and adaptor proteins on its cytoplasmic domain play an integral role in signal initiation and activation via PI3K/AKT, NF-κB, NFAT, and MAPK pathway. Continuously, this promotes cytokine production, transcriptionally upregulate pro-survival genes (e.g., Bcl-XL), regulate the actin cytoskeleton and formation of the immunologic synapse. More recently, recurrent mutations in both the CD28 extracellular (D124V, F51V) and intracytoplasmic (T195P) domains have been observed in a minority of PTCL (Yoo, Sung, et al., 2014). Mutations in the extracellular domain (particularly D124V) increase the binding affinity to CD28 ligands, including CD86, while mutations in the intracytoplasmic domain enhance binding to the adaptor proteins Grb2 and GADS/GRAP2.
ICOS, which is highly expressed on follicular helper T cells (Tfh), is induced following TCR and CD28 engagement, its signaling is initiated upon engagement by its ligand, CD275 (i.e., B7-H2) expressed by professional antigen-presenting cells and various nonhematopoietic cells (Wilcox et al., 2012). Particularly AITL, originate from clonally expanded Tfh cells and highly express ICOS (Bisig et al., 2013). Moreover, ICOS is inducibly expressed in an NPM-ALK and STAT3 dependent manner in ALK+

12
ALCL, and ligand engagement was associated with increased ALCL proliferation (Zhang et al., 2011).
1.1.6. Genetic modification of primary human cells
Most T cell engineering studies have been engaging viruses for a very long time, although very efficient, it still leads to several drawbacks relating their immunogenicity, the limitation of transferred DNA amount and some safety concerns. Meanwhile however, the use of non-viral gene modification method in lymphocytes, especially the use of plasmid vectors for the ex vivo genetic modification of non-transformed human tissue/cell types for therapeutic purposes has been hindered by the low efficiency of currently available plasmid transfection systems and by the toxicity that many plasmid transfection reagents have for primary cell types, in particular, lymphocytes (Van Tendeloo et al.) which are notoriously refractory to most kinds of non-viral DNA delivery system. Hence, there is a need for a better modification system to maximize gene editing efficiency in human primary T cells.
Physical method, such as electroporation, has been raised as alternative strategy performing on plasmid DNA encoding target genes for transiently introducing genes of interest into T lymphocytes. Bell et al. (Bell MP, 2001) demonstrated 57% transgene expression with 42% viability for electroporation of in vitro-activated DO11.10 murine T lymphocytes with a relatively high amount of plasmid DNA. Van Tendeloo et al. (Van Tendeloo VF et al. 2000) electroporated plasmid DNA into phytohemagglutinin (PHA)-stimulated human primary peripheral blood lymphocytes (PBLs) at optimized conditions and observed 16% gene expression with 70% mortality. Lai W. reported that 20–35% transgene expression with 75% viability was detected by transfecting resting and activated murine CD4 T cells using Nucleofection, a modified electroporation method with several limitations on the buffers (Lai W, 2001). Nevertheless, there were still some limitations: (1) in this approach the T cells require activation before gene transfer; (2) random integration after electroporation was of low efficiency; (3) the probability of co-electro-transfect and co-electro-integrate of gene pairs and combos of interest is rarely low.
The concept of using a transposon system as a genomic integration technique has also received increasing attention during the past decade. The development of different transposon system has broadened the applications of electroporation. However, systematic studies of the transposon system adapted electroporation for human primary T lymphocyte engineering have not been performed.
Transposon system such as sleeping beauty(SB) and piggyBac recently has been used as a promising tool for high-level, persistent transgene expression that can be

13
delivered in a nonviral plasmid form. SB is a synthetic DNA transposon of the Tc1/mariner superfamily that was “reawakened” by Hackett and colleagues through molecular reconstruction and mediates DNA transfer via a “cut-and-paste” mechanism.27 SB transposase mediates transposition by recognition of the short inverted/direct repeat (IR/DR) sequences of the transposon, resulting in the excision of the transposon and insertion into a TA-dinucleotide sequence in chromosomal DNA. The SB transposase has an N-terminal DNA-binding domain, a nuclear localization signal, and a C-terminal catalytic domain that is characterized by the DDE motif (containing 2 invariable aspartic acid residues and a glutamic acid residue) and is responsible for excision and subsequent integration of transposon DNA. The SB transposase can be introduced by expression of a transposase-encoding DNA molecule either on the same DNA molecule as the one containing the SB transposon (cis delivery) or on a separate DNA molecule (trans delivery). SB transposons have been shown to mediate transposition and long-term expression in a wide range of vertebrate cells and tissues, including cultured mammalian cells, mouse liver and lung tissues, mouse embryonic stem cells, and in mammalian germ-line transgenesis and insertional mutagenesis.
Leap In® transposases and synthetic transposons from ATUM is a novel transposon system which gives access to easily implemented cell line development workflow. ATUM has identified two orthologous transposases, designated Leap-In 1 (LPN1) and Leap-In 2 (LPN2), that stably integrate synthetic transposons into the host genome with close to 100% integration frequency and an absolute minimum of concatemers or other genetic instabilities. (Boldog et al.)
The Leap-In transposase mRNA is co-transfected with the synthetic transposon, which can encode up to 4 unique open reading frames (ORFs) flanked by inverted terminal repeats into the cell. By translated and acts integrating into the host genome, Leap-In transposases enable integration of single copies of the entire synthetic transposon that carries our target gene of interest into multiple transcriptionally active genomic loci. Herein the integration structure with single copy transposon has been made. Thus it also prevents instability resulting from concatemer recombination or repeat-induced silencing. Random non-homologous recombination always drives integration exhibit the rearrangements and contactmerization; however, do not exhibit on Leap-In contracts. By maintain the structural integrity of the sequence integrated into the host genome, this system ensures the regulatory elements remain associated with the appropriate open reading frames.
Neon® Transfection System has been widely implemented for electroporation in a variety of cell models and especially hard-to-transfect cells, such as primary cells and stem cells. The optimal transfection efficiency in human primary T cell was reported to be reached over 95% for RNA (EGFP mRNA) and 85% for DNA (pcDNAEF1a-emGFP) with high viability using certain Neon® programs followed by activation/expansion in

14
OpTmizer media with 2% human serum and CD3/CD28 Dynabeads. (Chang et al., 2017).
2. Chapter 2:
2.1. Short Introduction
T cell-intrinsic properties, for instance, durability, longevity, and functionality, play decisive roles in cancer immunology and the efficacy of cancer immunotherapy. T cell neoplasm, which represents a group of drastically disordered T cell with unrestricted proliferation, is an excellent example for T cells with the extraordinary ability of cell growth. Apoptosis and survival activity, which both regulates and is regulated by numerous different cellular signaling pathways and epigenetics, profoundly influences the qualities of T cell proliferation and other functions.
On the one hand, the recent studies shown multiple gene targets, which occurred in different T cell neoplasm mutational landscape, play prominent roles as driver or regulators of leukemia cell growth and metabolism. On the other hand, ways and means of genetic modification on human T cells gained extensive attention as CAR-T and ACT cancer therapy developed in these years.
Given this, this scholarly project sought to identify ways to equip human primary T cell ex vivo with improved durability and longevity by T cell neoplasm gene mutations regulating T cell apoptosis and survival, the short-term specific aims are:
a. To build a systematic modification & optimization system on human primary T cells.
b. To genetically modify primary T cell genome knock-in with mutated gene targets list.
c. To identify gene mutations of interest that facilitates T cell proliferation and survival.
d. To determine if these genetically modified T cells with great proliferation ability remain other T cell qualities and functions.

15
2.2. Materials and Methods
Antibodies
All antibodies used in the study for fluorescence-activated cell sorting and flow cytometry are listed below. CD8 CD19
TSF Cell line
T cell cancer cell line constructed in the lab by Feng Shi and can stably stimulate CD3/CD28secret IL-2, IL-7, and IL-15 simultaneously when culture with other cells in a proper density. Used in this study as T cell stimulation substitution for cytokines.
Human primary T cells Isolation and Stimulation
Primary human T Cells were isolated from healthy human donors from fresh whole blood. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood samples by Ficoll centrifugation using SepmMate tubes (STEMCELL, per manufacture’s instructions). T cells were isolated from PBMCs by negative magnetic selection using EasySep Human T Cell Isolation Kit (STEMCELL, per manufacturer’s instructions).
When frozen cells were used, previously isolated T cells that had been frozen were thawed, and then stimulated with TSF cells and handled as described for freshly isolated samples. Fresh blood was taken from healthy human donors under a protocol approved by the MGH. All patients and healthy donors provided informed consent.
Primary human T cell culture
Unless otherwise noted, bulk T cells were cultured in XVivo15 medium (STEMCELL) with 5% fetal bovine serum (FBS), 50 μM 2-mercaptoethanol, and 10 μM N-acetyl l-cystine. Immediately after isolation, T cells were stimulated for 3 days with TSF, along with a cytokine cocktail of IL-2 at 200 U ml−1 (), IL-7 at 5 ng ml−1 (), and

16
IL-15 at 5 ng ml−1 (). After electroporation, T cells were cultured in media with IL-2 at 500 U ml−1. Throughout the culture period, T cells were maintained at an approximate density of 1 million cells per ml of media. Every 2–3 days after electroporation, additional media was added, along with additional fresh IL-2 to bring the final concentration to 500 U ml−1, and cells were transferred to larger culture vessels as necessary to maintain a density of 1 million cells per ml.
Flow cytometry and cell sorting
Flow cytometric analysis was performed on a BD LSRFortessa X-20 Cytometer or an LSRII flow cytometer (BD). FACS was performed on the FACS Aria platform (BD). Surface staining for flow cytometry and cell sorting was performed by pelleting cells and resuspending in 25 μl of FACS buffer (2% FBS in PBS) with antibodies at the indicated concentrations (Supplementary Table 2) for 20 min at 4 °C in the dark. Cells were washed once in FACS buffer before resuspension. Data were analyzed with FlowJo v.10.
Immunophenotyping
Blood samples were collected from cancer patients into Streck tubes, which contained a tissue fixative. Each blood sample was then aliquoted and frozen down at -80°C. In preparation for flow analysis, each frozen blood aliquot was thawed in a 37°C water bath and then washed 2X with PBS containing 2% serum and 5mM in order to remove the red blood cells, which had been lysed by the freezing process (Ficoll density gradient centrifugation was not performed so as not to risk losing any more cells). The “purified” PBMCs were then divided among the round bottom tubes with fewer cells being put into the unstained control than in the Panel 1-3 tubes. The Panel 1-3 cocktails were prepared for 100µL/test (1:100 dilution) with a % error of +1 test per panel (e.g. if 5 samples were in the experiment, each cocktail would be prepared as if there were 6 samples). The cells were spun down at 1500 RPM for 7 minutes after which the media was aspirated off and each cell pellet was resuspended with the relevant cocktail (except for the unstained control). The cells were allowed to stain for 30 minutes in the dark at 4°C after which they were washed in 1mL of PBS containing 2% serum and 5mM EDTA and then resuspended in 200µL. The stained samples were then run on a Fortessa X-20 analyzer, and the data was processed in FlowJo v.10.
T cell-Engaging Antibody Circuits (TEAC) Assay
Cytolytic T cell function was assessed by the T cell-engaging antibody circuits (TEAC) assay. Cobbold Lab was able to create a T cell activation drug called TEAC with improved toxicity profile by splitting the T cell activation domain. Here, EpCAM-CD3 VH and EpCAM-CD3 VL were used for creating TEAC; each of them contains an scFv to a surface antigen on the tumor cell. By binding the surface antigen on the tumor cells and CD3 epsilon chain on T cells, the TEAC pair can build direct contact with tumor cells and help the signal cascade transduce through CD3 complex. Eventually, if the T cell is well functional, these signals will lead to T cell activation and function, in this case, cytolytic function assessed by IFN- gamma by ELISA. T cells transfected with Fas-

17
TM-41BB + Cas7-DN combo were tested, T cells from a normal donor were used for the negative control, cells are plated out in triplicate. EpCAM Bi-specific T cell antibodies (BiTE) labeled tumor cells are used for the positive controls, and untreated tumor cells are used for the negative controls for the TEAC. Stock TEAC was diluted to 10x concentrations of 7 dose-response range using Ab fragment. 20ul of EpCAM-CD3 VH and 20ul of EpCAM-CD3 VL (at same concentrations) were added to 15ml Falcon tube and the same done for each TEAC concentration (seven Falcon tubes in total). 40ul of PBS was added for the untreated control and 40ul of BiTE (1ul BiTE + 39ul PBS) for the positive control. Then, the cells were counted, wash, and resuspend to 1x106 per ml in fresh media and add 100ul per well. Cells incubated at room temperature for 30 mins. All cells were then washed to remove excess unbound TEAC. Next, T cells were counted, washed, and resuspended to 2.5 x105cells per ml and add 100ul to each well using a multi-channel pipette. Plates were incubated overnight at 37℃; the supernatant was then assessed for the presence of IFN gamma by ELISA.

18
2.3. Results
2.3.1. Leap-In/Neon Engineering on Jurkat cell Line
Fig.1 Leap-In Transposase & Leap-In/Neon Engineering System
With the intent of setting up a platform for systematic modification on human primary T cells pursuing higher transfection efficiency with lower DNA toxicity in an easy consistent transgenesis manner, we build a new engineering system by combining the new Leap-In Transposon System with Neon Transfection System. The Leap-in/Neon Engineering System consists of a pair of Leap-in synthetic transposon vector and Leap-in transposases. We constructed 2 types of Leap-in transposon vector plasmids with respectively GFP and RFP selection marker to test 2 designated transposases, namely Leap-In 1 (LPN1) and Leap-In 2 (LPN2) (Fig. 1A, 1B). As demonstrated in (Fig. 1C), with the help of Neon electroporation, both transposon vector and transposase were transmitted into the cells. The Leap-In Transposases then translate, recognize transposon-specific inverted terminal repeats (ITRs). Next, under the direction of cut-and-paste mechanism, Leap-In transposases mediate integration of single copies of the entire synthetic transposon into multiple transcriptionally active genomic loci. The transposon
Leap-In Transposase & Leap-In/Neon Engineering System

19
can encode up to 4 unique open reading frames (ORFs) of gene targets of interest with another selection maker CD19 flanked by inverted terminal repeats. Since the CD19 selection marker is designed between two ITRs, it could be used as an indication of integration, while GFP/RFP expression level could represent the transfection efficiency because GFP/RFP reporter gene was designed inserting on the transposon backbone. Theoretically, this system could insert a gene expression cassette of interest from constitutive or inducible promoters with a variety of markers into different kinds of cells, in this study, human primary T cells, and makes transgenic primary T cell lines with only a single transfection.

20
Fig.2 Transfection feasibility & efficiency testing on Jurkat cell Line
To validate the feasibility of our Leap-In/Neon Engineering System model, we first tested it in a model cell line. Jurkat cell line was originally established in 1970 from the peripheral blood of a patient with acute T cell leukemia and later commonly used as model cells for T cells in cell biology research. According to manufactures’ protocol,

21
Jurkat cells can be efficiently transfected with Neon Transfection System (at a level of 94.2% transfection efficiency at the optimal parameter setting). Jurkat cells were transfected with each type of plasmid at a seeding density of 0.1 * 106 cell mL-1 as suggested. At our first attempt, two different transposases LPN1 and LPN2 were tested parallelly with empty GFP/RFP plasmid under 4 different plasmid/transposase ratio, 5:1, 10:1, 15:1, 20:1, which are previously reported ratios for Jurkat cells transfection (Troyanovsky et al., 2016). Preliminary results suggest CD19-GFP vector works better with LPN1 transposase, while CD19-RFP vector works better when paired with LPN2. The transfected cells were stimulated (day 0). CD19 and GFP/RFP expression were monitored, and flow cytometry tested on day1 and repeated every following week. The first attempt result shown that both types of transposon plasmids are successfully transfected. On day 4, the cells under a ratio of 15:1 on GFP plasmid and LPN1 transposase demonstrate a higher percentage of CD19 expression level than transfecting with plasmid only, it suggests transposase may help integrate transposon. The same results are shown by RFP plasmid with a ratio of 15:1 and 20:1, the transfection efficiency provided by our system is better than the ones previously determined for non-viral primary T cells transfection with I-PEI (Jérôme et al., 2018).

22
Fig.3 Post-Transfection Expression Duration of Jurkat Cell Line
To further verify if Leap-In/Neon could mediate integration of the designed transposon, we monitored the expression duration in Jurkat cells for 55 days. The result showed that no matter for the GFP transposon or RFP transposon group, among Jurkat cells only transfected with transposon plasmid but not transposase, the percentage of

23
CD19+ cells showed a drastic decrease of post-transfection, and the CD19 expression can be no longer detected 15 days after seeding (Fig.2A, 2B). It implies that without transposases, Neon electroporation can only mediate transient transfection, which will not support a long-lasting expression of the transfected vector. Compare to this, among Jurkat cells transfected with a pair of GFP transposon and LPN1 transposase in a different ratio, the percentage of CD19+ cells decreased post transfection, buts some of the cell still showed a persist expression after day15. The CD19 expression percentage in groups with a ratio of 10:1, 15:1, 20:1 even showed a mild increase after day 22. On day 55, CD19 expression from cells transfected with the GFP plasmid was detected in around 20% of the Jurkat cells with a 10:1 or 15:1 transposase ratio. Likewise, Jurkat cells transfected with a pair of RFP transposon and LPN2 transposase in different ratio performed in the same pattern with GFP transposon group. The percentage of CD19+ cells decreased drastically over time before stabling but demonstrated higher integration efficiency than the GFP vector at large. On day 55, around 40% of human is maintaining CD19+ expression with a 10:1 or 15:1 transposase ratio.
Fig.4 Expression Stability of Jurkat Cell Line
Moreover, to reinspect on the integration stability, we sorted the CD19+ Jurkat cells from day 55, keep culturing and monitoring them for more than 3 months. Results from day 104 demonstrated well integration stability in both settings, 94.4% of the transfected Jurkat cells in the RFP group are still CD19+, whereas 95.4% of Jurkat cells in the GFP

24
group keep maintaining a CD19 expression (Fig. 3). Taken together, the results showed that Leap-In/Neon Engineering System could transfect Jurkat cells, and the integration mediated by Leap-In transposase is stable. Besides, we can conclude that the transfection efficiency and integration level can be affected to an extent by the type of transposase and the ratio of transposon and transposase. LPN2 transposases performance is more sensitive to different ratios whereas LPN1 is more consistent in the long term.
Therefore, these results put forward for the first time that the Leap-In/Neon system is capable of mediate Jurkat cells transfection with a relatively high transfection efficiency and can maintain great stability for a long time.
2.3.2. The first attempt of Leap-In/Neon Engineering on human primary T
cell
Fig.5 First Attempt on Human Primary T Cell Transfection
One step forward, we next tested our Leap-In/Neon Engineering system for human primary T cells transfection. Much of the research in Primary T cells transfection had emphasized that primary T cell might not be efficiently transfected (Chang, Andronikou, and de Mollerat du Jeu, 2017). Similarly, our first attempt on human primary T cells gave corroborative evidence to this argument. Human primary CD8+ T cells were first sorted from a random normal donor (ND) 77 and ND78 PBMCs with a human CD8 positive selection. GFP vector plasmid was transfected parallelly in both ND77 and ND78 with a transposase ratio of 10:1. The same Neon electroporation setting with the Jurkat cell transfection was performed. Results showed that the integration efficiency of the vector with a GFP transposon was only 2.1% 14 days after reseeding, whereas the RFP transposon was present in only 0.3% of cells (Fig.4). This is probably a consequence from a low transfection efficiency, which can be an accumulated problem of several electroporation parameters. Inevitably, low integration level can also be a result from the

25
unsatisfying transposases and promoter performance. Further experiments on investigating the suitable transfection conditions for primary T cells are needed.
2.3.3. Optimizations for Leap-In/Neon Engineering on human primary T cell
There is a good possibility that with an optimized setting only designed for primary T cells could address a better performance of our Leap-In/Neon Engineering System. Therefore, we provide multiple solutions as follow for system optimization, including transposon/transposase ratio, electroporation settings, and alternative promoters. For these and subsequent experiments, we determine the optimal transfection conditions by evaluating transfection efficiency and cell viability represented by flow-cytometric analysis. The best transposase ratio and the promoter will be determined by monitoring the integration level for a duration.

26
Fig.6 Transfection Optimization on Transposon/Transposase Ratio in Primary T Cells
In terms of transposase ratio, we tested both GFP plasmid and RFP plasmid with 3 different ratios, 10:1, 20:1, 1:1. The transfection efficiency was determined by the percentage of GFP expression on day 1 post-transfection. Integration level indicated by the percentage of CD19 expression was monitored for over 18 days. Results showed that the ratio of 10:1 provided the best condition for both GFP and RFP plasmid (Fig. 6) However, there’s only a little difference between groups on both transfection efficiency and integration level, it implies that the transposase ratio may not be the most critical

27
factor on primary T cell transfection performance with Leap-In/Neon Engineering System. Therefore, we keep on the use the ratio of 10:1 for the following experiments.
Fig. 7 Transfection Optimization on Electroporation Parameter in Primary T Cells
As for electroporation parameters, we performed different conditions with altered voltages, periods, and plasmid amounts. The flow-cytometry test was conducted measuring transfection efficiency post transfection on day 1. The results indicated that higher voltage, more plasmid, longer duration lead to better transfection efficiency with the sacrifice of lower viability (Fig. 7). Of all the conditions we tested, a voltage of 1600v with a period of 10 ms electroporation resulted in relatively high GFP expression. Furthermore, under this condition, the DNA amount of 1 ug provided minimal toxicity. Hence, for the following experiments, we would keep performing electroporation under the condition of 1600v, 10ms, 3 pulses, 1 ug DNA plasmid.
28

29
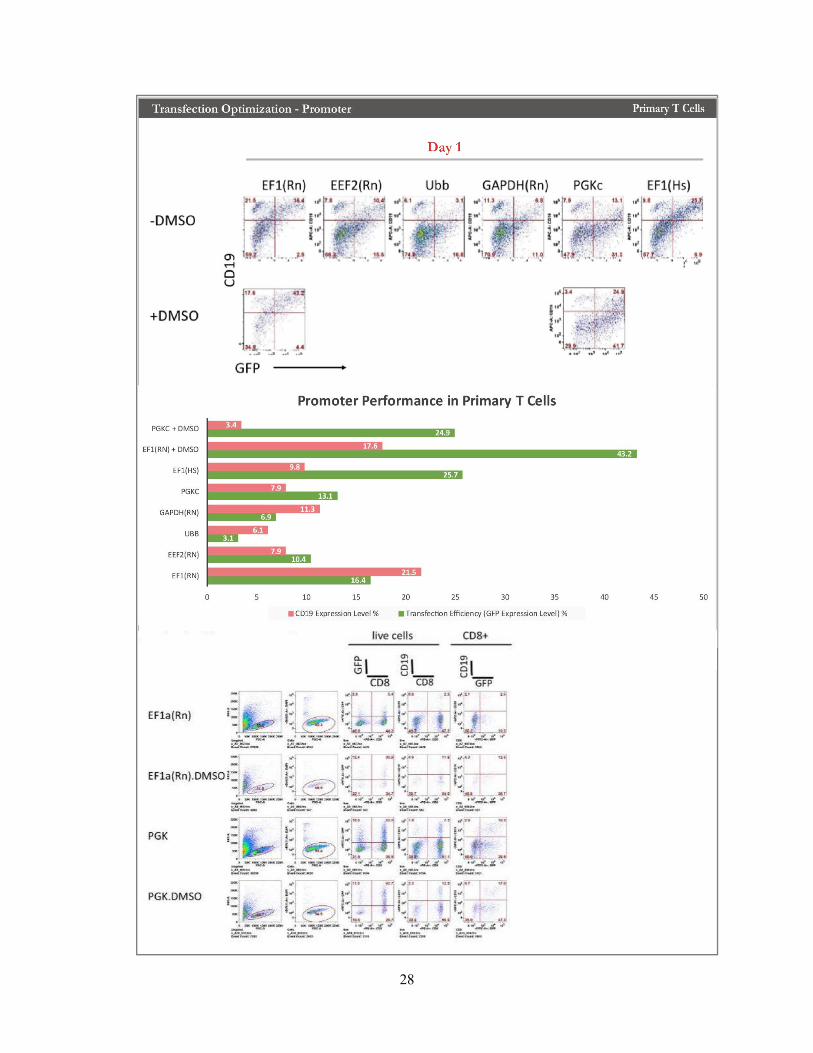
Fig. 8 Transfection Optimization on Promoter in Primary T Cell
The impact of promoter performances was also considered. We tried several different promoters according to references; the results confirmed that promoter performances had an impact on both transfection efficiency and the percentage of CD19 expression on human primary T cells. Among all the promoter that we tested, EFIa is the most efficient promoter under the current setting providing the highest transfection efficiency. Besides, treating with Dimethyl sulfoxide (DMSO) showed potential in increasing CD19 integrations. However, in the following monitoring experiments, promoter Phosphoglycerate Kinase (PGK) showed better CD19 integration level in CD8 T cells. Therefore, PGK will be used in the following experiment with a supplement of DMSO if necessary.
2.3.4. Establish optimal Leap-In/Neon Engineering System for human
primary T cell modification
Fig.9 Optimal Strategy for Human Primary T cell modification with Leap-In/Neon
Engineering

30
Taken together, we established an optimized strategy to perform human primary T cell modification with Leap-In/Neon Engineering System. First, consider that some of the previous transfections were conducted on normal donor PBMC, and a population of transfected B cell can be found on the flow results, we think it might be more reasonable to perform a CD8+ T cell purification/sorting before transfection to better serve the purpose of this project. Second, we primed the purified CD8 T cells with feeder cells TSF cell line, which was a lab constructed cell line by Feng Shi, and it could provide sufficient CD3/CD28 stimulation and secret cytokines IL-2, IL-7 and IL-15 for boosting initial CD8+ T proliferation post transfection. After 3 days, the primed CD8+ T cells were transfected with a pair of GFP-tagged transposon plasmid with LPN1 transposase, non-transposon GFP RNA will also be transfected as control. Theoretically, the post-transfection population of primary CD8 T cells consist of 3 kinds of cells, namely transfected, transfected but unintegrated, also transfected and integrated. We perform measurements with flow cytometry on day 1 to assess the transfection efficiency and cell viability. We selected the successfully transfected cells and cultured them with IL-2 in a standard T cell medium. Usually T cells with a limited lifespan will die out after a period without stimulations, non-transposon GFP RNA control will decrease to a background level owing to non-integration and degradation over this time. However, some primary CD8+ T cell transfected with specific genes prevented itself from apoptosis and gradually started proliferating when the transfected genes are integrated into the genome and stably expressed. Finally, we will focus on these transfected T cells with a favored proliferation

31
characteristic and keep monitoring the percentage of integration level along with their effects on the transfected T cells
Fig.10 Optimal Leap-In/Neon Transfection Engineering System
Moreover, to pursue a higher efficiency and a better quality for the following
modification with Leap-In/Neon Engineering System, several improvements were made to the system.
First, we deleted the selection marker outside transposon, and only preserve the inner selection marker and exchange it from CD19 to GFP in the transposon between 2 ITRs. Since we have already used a double marker to verify the system working as we hypothesized, previous kinetics results showed the outer marker would fall off quickly post-transfection because it can’t be integrated into the genome. Besides, the inner marker can also indicate transfection efficiency on day 1 post-transfection by itself, and GFP can provide an easier readout. Second, we replaced transposase plasmid with single transposase RNA which can easily be transmitted into the cells with higher efficiency and less damage; this will ensure more success bond of translated transposase protein with

32
ITRs and therefore increase the possibilities of more transposase-mediated integration of transposons.

33
2.3.5. Target gene pathway and transfecting gene list for human primary T
cell modification25

34
Fig.11 Mutational Landscape of T Cell Neoplasm
As we mentioned in the background, several gene mutations and epigenetic modifications in upstream regulators and downstream pathways are responsible for the T cell neoplasm cancer genesis. To select pro-proliferation gene targets for genetic modification on human primary T cells, here we reviewed and summed up the most unique mutational landscape in 4 types of T cell neoplasms, namely LGL, AITL, T-ALL, CTCL from 25 related papers. We listed mutations with a well-reported gain-of-function here with the signaling pathway they most often involved for a combatively more suitable serving for our Leap-In/Neon Engineering System, some mutations were reported repeatedly in different previous studies (Fig. 11). These neoplasms recapitulate a multistep oncogenic process associating apoptosis, survival, epigenetic deregulation, and second hit mutations affecting the TCR signaling and co-stimulation pathway. Especially in LGL, a disease related to prominent dysregulation on JAK-STAT pathway, here we listed not only the most significant mutation site STAT3 Y640F but also other STAT3 mutations such as D661V, D661H, N647I, K658N, and other JAK-STAT pathway related mutations PTPRT V995M and STAT5B. It’s worth mentioning that STAT3 K658N from JAK-STAT pathway along with another two JAK2 mutations JAK2 V617F, JAK2 G571S is also reported in AITL. BCL11b H126R engaging in T cell activation, FLT3 from PI3K/ATK/mTOR pathway and CD40LG are also valued. In AITL, we noticed TET2 R1261H, IDH2 R1/2, DNMT3A R38C from PI3K/ATK/mTOR regulating T cell activation are also mentioned in several other studies. Intriguingly, some of these relevant mutations are also reported in apparently healthy blood cells as well as these tumor cell samples (Santarpia, Lippman and El-Naggar, 2012), it may suggest differences and septicity of blood samples should also be considered, in the following studies we have designed experiments with samples from different donors to investigate.
To better understand how these noted mutations are involved in the signaling pathways and eventually lead to an ‘immortalized’ tumor characteristic, we summarize here the apoptosis, survival and T cell activation signaling pathway with the noted genes tagged (Fig. 12). Moreover, we don’t want to narrow our project down to only focus on what people have found already. With this engineering tool, we think it might be useful to try out other targets that didn’t appear above and investigate what would happen if we mutate some other genes upstream or downstream in the same pathway as well, since they work with the noted genes in the same network that everyone may have a mutual effect on others. Based on this concept, we designed the gene lists that consists of the desired gene candidates and the form of their mutations that we would like to modify on human primary T cells with our Leap-In/Neon Engineering System (Fig. 13).

35
Fig.12 Target Gene Pathway for Human Primary T Cell Modification
Fig.13 Single Gene List for Human Primary T Cells Transfection

36
2.3.6. Effects of transfected gene STAT3 Y640F on human primary T cell
Fig.14 STAT3 Y640F Single Gene Effects on Transfected Human Primary T cell
To determine if gene mutation candidates among our list have any effects on human primary T cell proliferation and survival, we first performed parallel modifications with plasmid constructs each containing one single gene in the list with the optimal strategy mentioned above. Then we conduct flow-cytometry 24h post-transfection to determine based on transfection efficiency if the constructs have been successfully transfected into cells. Furthermore, for the successfully transfected cells, we performed stimulation rapidly with TSF cells to rescue the population post DNA toxicity and electroporation.

37
Eventually, we monitored the status of the cell and performed flow cytometry testing every 1-2 week. Effects of gene candidates on these transfected cells were evaluated by transfection efficiency, cell viability, accumulating gene expression indicated by GFP percentage.
Of all the single gene candidates, the gene mutation candidate STAT3 Y640F start to show a significant increase in the percentage of GFP expression level after 28 days (Fig, 14). Interestingly, we noticed the increase, which is different from the rest, persists for over 2 months, as the percentage of GFP expression continuously accumulating the cells keep proliferating. On day 80, 96.7% of the STAT3 Y640F transfected human primary CD8 T cells are GFP+, which means 96.7% of the transfected cells are integrated with STAT3 Y640F mutations carried by the Leap-In transposon. And this integration appeared with a significant advantage on the cell status and a favorable proliferation characteristic which would not be anticipated in standard T cell medium. To the contrast, the control group, which is transfected with GFP RNA only, the cells population shrunk post transfection as soon as the GFP RNA degraded no more than 2 weeks, the cells then stop proliferating and start to die out.
Moreover, on Day 21, based on the newest flow cytometry results we got, we summarized their percentage of GFP expression, grouped the ones that greater than 8%, between 3% to 8%, and less than 3% (>2%). Compared to other gene candidates, We identified 6 gene candidates which showed an unusually high percentage of GFP expression indicating a considerable percentage of the cell are integrated with the target gene, namely PI3KCA(L1001P), STAT3 Y640F, CTNNB1(T41A), Mcl-1, Survivin BIRC5, and CD28 (D124E; T195P). (Fig. 14) With an average initial transfection efficiency and integration level (data not are shown), this could result from these genes can confer a proliferative advantage when transfected into human primary T cells. Therefore, we designed the next step proliferation assay to confirm in the future experiments.
More importantly, to make the better use of the system and the results we got so far, we wonder if the phenomenon would be recaptured or even better improved by transfecting gene mutations combos of STAT3 Y640F under the same setting. Moreover, we wonder what will happen to the cells if other possible gene combos are transfected. For this reason, we consider their current percentage as well as the role the played in the whole network, then designed several gene combos, transfected and tested them with the same method mentioned above (Fig.14).

38
2.3.7. Transfecting gene combos list for human primary T cells modification
Fig.14 Gene Combo List for Human Primary T Cells Transfection

39
2.3.8. Effects of transfected gene combo Fas-TM-41BB + Casp7-DN on
human primary T cell
Fig.15 Fas-TM-41BB + Casp7-DN Combo Gene Performance on Human Primary T Cell
Of all the combo gene edits we made, the gene mutation combo of Fas-41BB and
Cas-7-DN (Fas4Cas7) led to a better result of 97.5% GFP expression in 54 days (Fig, 15.A). Fas-41BB is a synthetic molecule which expressed a Fas immunomodulatory

40
fusion protein (IFP) and engineered to express a 4-1BB with an ectodomain that dimerizes, replace Fas signaling and provide co-stimulation. In this way, this molecule could convert the inhibitory CD200R signal to a costimulatory CD28 signal. Previous studies have shown that the Fas-4-1BB IFP could exhibit enhanced accumulation and function in T cells in vitro (Oda et al., 2018). Cas7-DN is a dominant negative mutant of caspase 7 which abolish the function of caspase 7 and inhibit the function of the simultaneously expressed wild-type protein (dominant negative mutations). This approach has wide application to the study of several different kinds of proteins, including hormone receptors, oncogenes, and growth factor receptors (Sheppard, 1994). The result of 97.5% GFP expression is probably a consequence of the Fas-41BB death-to-live convert effect on co-stimulation pathway activation along with the enhancement provided by Cas-7-DN inhibiting the apoptosis pathway. It’s hard to rule out the accidental impact of random normal donor samples, we then performed the same transfection in two different random donors, ND 48 and ND51with ND55 as a control parallelly. So far, the result on day 24 of ND48 exert the same pattern of GFP expression level with ND55, whereas ND51even showed a GFP expression of 51% on day 24, more following results are needed to make a conclusion, but it indicates that the effects of Fas4Cas7 on ND55 is not an accidental event (Fig, 15B).
In addition, in order to determine if the Fas4Cas7 transfected cell is functional or not, we performed a cytotoxic functional test. The result of INF-ɤ level implied that Fas4Cas7 transfected cells could release INF-ɤ in response to CD3 stimulation at a level that similar to the normal donor T cell line, which means it remains the normal CD8+ T cell integrity and functions (Fig, 15C).
Furthermore, for the purpose of better under the impact of Fas4Cas7 gene mutation combo on human primary T cells, we performed immunophenotyping assay and T cell cytotoxicity assay. Results from immunophenotyping data showed ND55 Fas4Cas7 transfected sample exerted a phenotype of increased CD3+, prominent CD38+, abTCR+, and gdTCR+ population compared to ND55 PBMC control samples (Fig, 15D).
2.4. Brief Discussion
In this study, we have developed a Leap-In/Neon Engineering System for human primary T cell modifications. With this platform, we successfully modified human primary T cells with a list gene of interest and established several genes with a prominent pro-proliferation advantage. Result demonstrated that designed Leap-In vectors with genes could efficiently transpose transgenes from linear DNA fragments to human primary T cells.
Previous studies have shown that both the size of the entire plasmid and the size of the transposon had an impact on the transposon system like piggyBac, (Troyanovsky et
al., 2016), similar results were found in Leap-In/Neon modifications, and the larger plasmids or multiple plasmids had a lower initial transfection efficiency than the smaller

41
ones likely due to their decreased ability to penetrate the cell plasma membrane. However, surprisingly, with lower initial transfection efficiency, gene combos of Fas-TM-41BB exert unexpected pro-proliferation potentials with a maintained cell characteristic.
(Kataoka, et al., 2018) It’s also interesting that STAT3 Y640F stands out of a list of single genes on primary
T cell modifications. STAT3 Y640F is a hotspot gain-of-function mutation that was frequently observed across different disease and resulted in an enhanced STAT3 transcriptional activity (Andersson et al., 2015), especially LGL and AITL in the context of T cell neoplasm. (Teramo et al., 2013; Odejide et al., 2014), mutations involving S614 and G618 were also observed frequently in ATL case, all recurrently mutated residues are present around the phosphotyrosine-binding pocket. (Kataoka, et al., 2018). It was reported 38 patients out of 101 analyzed (37.6%) carrying STAT3 mutations in T-LGL patient, 24 cases presented Y640F (63.2%), 9 cases D661Y (23.7%), one case D661V (2.6%), one case N647I (2.6%) (Teramo et al., 2017). JAK–STAT3 signaling is crucial for cancer development in both tumor cells and the tumor microenvironment (Yu et al., 2014). Activated STAT3 persistently by transfect the gene mutation may trigger tumor progression through direct regulation of oncogenic gene expression. Apart from its oncogenic role in regulating gene expression in T cells, activated STAT3 in immune cells was believed to have results in inhibition of immune mediators and promotion of immunosuppressive factors (Wang et al., 2018).

42
3. Chapter 3:
3.1. Limitations
It should be noted that this study has examined only flow cytometry results on the percentage of gene expression level in the transfected CD8+ primary cells so far to determine whether there is integration. We are aware that it’s better for us to perform quantitative polymerase chain reaction (PCR) separately on host cell chromosomal DNA and episomal plasmid DNA to confirm this signal was due to plasmid backbone integration into the host cell genome but not prolonged stabilization of the episomal plasmid. Moreover, it may be useful to perform single-cell sequencing on these transfected CD8+ primary cells to prove the gene mutation fragment(s) has already been inserted into the cell genome, especially for the gene combos. Admittedly, it’s hard to confirm with our current data if and to what percentage of the transfected cells are double-gene inserted for the gene combos. Similarly, our current can’t make a conclusion on which kind of integration on these cells, single or double, helps to exert the effects on primary T cells. We can design two different fluorescent tags to manage to prove and maybe improve the result, or we can design constructs with two genes express together.
Besides, we must point out that we do not have data yet to prove these transfected cells or the maintenance of gene integration level, because the cells are still under proliferation. We will keep a monitor on these cells to see if the GFP expression level would decrease after a period. Moreover, this population of transfected proliferating cells stimulated with any specific CARs or another type of antigens to determine their functions as CD8+ T cells. However, we have plans to explore that in future experiments. Furthermore, on the purpose of confirmation and validation, parallel study with a better control group design will also be considered in every single experiment in the further.
Notwithstanding these limitations, this study does suggest for the first time that gene mutations and mutation combos could be transfected into human primary T cells with Leap-In/Neon transfection system, and some of the gene mutations could facilitate primary T cells proliferation and survival.
3.2. Future Directions
Despite its preliminary character, this study can indicate that some gene mutations such as STAT3 and gene mutation combos such as Fas-41BB and Cas7-DN could facilitate human primary T cell proliferation and survival.
However, this study can move a step forward if some questions could be better answered. In the future, we would focus on solving these questions as follow:
a. Improved gene combo transfection with constructs expressing two genes simultaneously to prove gene(s) integration editing to the cells
b. Augmented functional test with antigen-specific cytotoxicity assays to better understand the function and status of the cells

43
c. Express TCR in these transfected primary T cells d. In vivo engraftment of transfected primary T cells engineering
Supplementary data

44

45

46
References Abadie, V. and Jabri, B. (2014) ‘IL-15: a central regulator of celiac disease immunopathology’, Immunological Reviews, 260(1), pp. 221–234. doi: 10.1111/imr.12191.
Andersson, E. et al. (2015) ‘Activating somatic mutations outside the SH2-domain of STAT3 in LGL leukemia’, Leukemia. Macmillan Publishers Limited, 30(5), p. 1204. doi: 10.1038/leu.2015.263.
Andersson, E. I. et al. (2013) ‘Novel somatic mutations in large granular lymphocytic leukemia affecting the STAT-pathway and T-cell activation’, Blood Cancer Journal. Nature Publishing Group, 3(12), pp. e168-9. doi: 10.1038/bcj.2013.65.
Bisig, B. et al. (2013) ‘CD30-positive peripheral T-cell lymphomas share molecular and phenotypic features’, Haematologica, 98(8), pp. 1250–1258. doi: 10.3324/haematol.2012.081935.
Chang, S., Andronikou, N. and de Mollerat du Jeu, X. (2017) ‘Optimization of Gene Editing in Human Primary T Cells with Neon® Transfection System’, The Journal of Immunology, 198(1 Supplement).
Chen, D. S. and Mellman, I. (2013) ‘Oncology meets immunology: The cancer-immunity cycle’, Immunity, 39(1), pp. 1–10. doi: 10.1016/j.immuni.2013.07.012.
Chen, D. S. and Mellman, I. (2017) ‘Elements of cancer immunity and the cancer-immune set point’, Nature, 541(7637), pp. 321–330. doi: 10.1038/nature21349.
Choi, J. et al. (2015) ‘Genomic landscape of cutaneous T cell lymphoma’, 47(9), pp. 1011–1019.
Couronné, L., Bastard, C. and Bernard, O. A. (2012) ‘TET2 and DNMT3A Mutations in Human T-Cell Lymphoma’, New England Journal of Medicine, 366(1), pp. 95–96. doi: 10.1056/NEJMc1111708.
Dearden, C. E. (2009) ‘T-Cell Prolymphocytic Leukemia’, Clinical Lymphoma and Myeloma, 9, pp. S239–S243. doi: 10.3816/CLM.2009.s.018.
Feldman, A. L. et al. (2008) ‘Overexpression of Syk tyrosine kinase in peripheral T-cell lymphomas’, Leukemia, 22(6), pp. 1139–1143. doi: 10.1038/leu.2008.77.
Fukumoto, K. et al. (2018) ‘Review of the biologic and clinical significance of genetic mutations in angioimmunoblastic T-cell lymphoma’, Cancer Science, 109(3), pp. 490–496. doi: 10.1111/cas.13393.
Guy, C. S. and Vignali, D. A. A. (2009) ‘Organization of proximal signal initiation at the TCR:CD3 complex’, Immunological Reviews, 232(1), pp. 7–21. doi: 10.1111/j.1600-065X.2009.00843.x.
Hanahan, D. and Weinberg, R. A. (2011) ‘Hallmarks of Cancer: The Next Generation’, Cell, 144(5), pp. 646–674. doi: 10.1016/j.cell.2011.02.013.
Hill, L., Browne, G. and Tulchinsky, E. (2013) ‘ZEB/miR-200 feedback loop: at the crossroads of signal transduction in cancer.’, International journal of cancer, 132(4), pp. 745–54. doi: 10.1002/ijc.27708.
Hridesh Banerjee, Héctor Nieves-Rosado, L. P. K. (2018) ‘33rd Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2018) : Washington, D.C., USA. 7-11 November 2018.’, Journal for immunotherapy of cancer. BioMed Central, 6(Suppl 1), p. 115. doi: 10.1186/s40425-018-0423-x.
Jérôme, V. et al. (2018) ‘Non-Viral Transfection of Human T Lymphocytes’, Processes, 6(10), p. 188. doi: 10.3390/pr6100188.

47
Kataoka, K., Iwanaga, M., Yasunaga, J., Nagata, Y., Kitanaka, A., Kameda, T. and Yoshimitsu, M. (2018) ‘Prognostic relevance of integrated genetic pro fi ling in adult T-cell leukemia / lymphoma’, 131(2), pp. 215–226. doi: 10.1182/blood-2017-01-761874.
Kataoka, K., Iwanaga, M., Yasunaga, J., Nagata, Y., Kitanaka, A., Kameda, T., Yoshimitsu, M., et al. (2018a) ‘Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma’, Blood, 131(2), pp. 215–225. doi: 10.1182/blood-2017-01-761874.
Kataoka, K., Iwanaga, M., Yasunaga, J., Nagata, Y., Kitanaka, A., Kameda, T., Yoshimitsu, M., et al. (2018b) ‘Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma’, Blood, 131(2), pp. 215–225. doi: 10.1182/blood-2017-01-761874.
Keppler-Noreuil, K. M. et al. (2014) ‘Clinical delineation and natural history of the PIK3CA -related overgrowth spectrum’, American Journal of Medical Genetics Part A, 164(7), pp. 1713–1733. doi: 10.1002/ajmg.a.36552.
Keppler-Noreuil, K. M. et al. (2016) ‘Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies.’, American journal of medical genetics. Part C, Seminars in medical
genetics. NIH Public Access, 172(4), pp. 402–421. doi: 10.1002/ajmg.c.31531.
Kiel, M. J. et al. (2014) ‘Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia.’, Blood. The American Society of Hematology, 124(9), pp. 1460–72. doi: 10.1182/blood-2014-03-559542.
Laharanne, E. et al. (2010) ‘Genome-wide analysis of cutaneous T-cell lymphomas identifies three clinically relevant classes.’, The Journal of investigative dermatology, 130(6), pp. 1707–18. doi: 10.1038/jid.2010.8.
Lonetti, A. et al. (2016) ‘Improving nelarabine efficacy in T cell acute lymphoblastic leukemia by targeting aberrant PI3K/AKT/mTOR signaling pathway’, Journal of Hematology & Oncology, 9(1), p. 114. doi: 10.1186/s13045-016-0344-4.
Mazzucchelli, R. I., Riva, A. and Durum, S. K. (2012) ‘The human IL-7 receptor gene: Deletions, polymorphisms and mutations’, Seminars in Immunology, 24(3), pp. 225–230. doi: 10.1016/j.smim.2012.02.007.
McCubrey, J. A. et al. (2007) ‘Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance’, Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1773(8), pp. 1263–1284. doi: 10.1016/j.bbamcr.2006.10.001.
Mishra, A. et al. (2016) ‘Mechanism, Consequences, and Therapeutic Targeting of Abnormal IL15 Signaling in Cutaneous T-cell Lymphoma.’, Cancer discovery. NIH Public Access, 6(9), pp. 986–1005. doi: 10.1158/2159-8290.CD-15-1297.
Mishra, A., Sullivan, L. and Caligiuri, M. A. (2014) ‘Molecular Pathways: Interleukin-15 Signaling in Health and in Cancer’, Clinical Cancer Research, 20(8), pp. 2044–2050. doi: 10.1158/1078-0432.CCR-12-3603.
Oda, S. K. et al. (2018) ‘Cheating death: a Fas-41BB immunomodulatory fusion protein obviates a death signal to enhance T cell function and adoptive therapy targeting leukemia and solid tumors’, The Journal
of Immunology, 200(1 Supplement).
Odejide, O. et al. (2014) ‘A targeted mutational landscape of angioimmunoblastic T-cell lymphoma’,

48
Blood, 123(9), pp. 1293–1296. doi: 10.1182/blood-2013.
Olson, T. et al. (2012) ‘Mutations in Large Granular Lymphocytic Leukemia’, pp. 1905–1913.
Palacios, E. H. and Weiss, A. (2004) ‘Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation’, Oncogene, 23(48), pp. 7990–8000. doi: 10.1038/sj.onc.1208074.
Palucka, A. K. and Coussens, L. M. (2016) ‘The Basis of Oncoimmunology’, Cell. Elsevier Inc., 164(6), pp. 1233–1247. doi: 10.1016/j.cell.2016.01.049.
Porta, C., Paglino, C. and Mosca, A. (2014) ‘Targeting PI3K/Akt/mTOR Signaling in Cancer’, Frontiers
in Oncology, 4, p. 64. doi: 10.3389/fonc.2014.00064.
Rajala, H. L. M. et al. (2013) ‘Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia.’, Blood, 121(22), pp. 4541–50. doi: 10.1182/blood-2012-12-474577.
Ribeiro, D., Melão, A. and Barata, J. T. (2013) ‘IL-7R-mediated signaling in T-cell acute lymphoblastic leukemia’, Advances in Biological Regulation, 53(2), pp. 211–222. doi: 10.1016/j.jbior.2012.10.005.
Rodríguez-Pinilla, S. M. et al. (2013) ‘Loss of TCR-beta F1 and/or EZRIN expression is associated with unfavorable prognosis in nodal peripheral T-cell lymphomas.’, Blood cancer journal. Nature Publishing Group, 3(4), p. e111. doi: 10.1038/bcj.2013.10.
Sakuishi, K. et al. (2010) ‘Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity’, The Journal of Experimental Medicine, 207(10), pp. 2187–2194. doi: 10.1084/jem.20100643.
Santarpia, L., Lippman, S. M. and El-Naggar, A. K. (2012) ‘Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy.’, Expert opinion on therapeutic targets. NIH Public Access, 16(1), pp. 103–19. doi: 10.1517/14728222.2011.645805.
Schreiber, R. D., Old, L. J. and Smyth, M. J. (2011) ‘Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion’, Science, 331(6024), pp. 1565–1570. doi: 10.1126/science.1203486.
Sheppard, D. (1994) ‘Dominant negative mutants: tools for the study of protein function in vitro and in vivo.’, American Journal of Respiratory Cell and Molecular Biology, 11(1), pp. 1–6. doi: 10.1165/ajrcmb.11.1.8018332.
Smyth, M. J. et al. (2016) ‘Combination cancer immunotherapies tailored to the tumour microenvironment’, Nature Reviews Clinical Oncology, 13(3), pp. 143–158. doi: 10.1038/nrclinonc.2015.209.
Sokol, L., Loughran, T. P. and Leukemia, T. L. G. L. (2007) ‘Large Granular Lymphocyte Leukemia’, pp. 1–5.
Susan, E. (2007) ‘Apoptosis: A Reveiw of Programmed Cell Death’, Toxicologic Pathology, 35(4), pp. 496–516. doi: 10.1080/01926230701320337.
Tal, N. et al. (2014) ‘Interleukin 7 and thymic stromal lymphopoietin: from immunity to leukemia’, Cellular and Molecular Life Sciences, 71(3), pp. 365–378. doi: 10.1007/s00018-013-1337-x.
Teng, M. W. L. et al. (2015) ‘From mice to humans: developments in cancer immunoediting.’, The
Journal of clinical investigation. American Society for Clinical Investigation, 125(9), pp. 3338–46. doi:

49
10.1172/JCI80004.
Teramo, A. et al. (2013) ‘Intrinsic and extrinsic mechanisms contribute to maintain the JAK/STAT pathway aberrantly activated in T-type large granular lymphocyte leukemia’, Blood, 121(19), pp. 3843–3854. doi: 10.1182/blood-2012-07-441378.
Teramo, A. et al. (2017) ‘STAT3 mutation impacts biological and clinical features of T-LGL leukemia.’, Oncotarget. Impact Journals, LLC, 8(37), pp. 61876–61889. doi: 10.18632/oncotarget.18711.
Troyanovsky, B. et al. (2016) ‘The Functionality of Minimal PiggyBac Transposons in Mammalian Cells’, Molecular Therapy - Nucleic Acids. Cell Press, 5, p. e369. doi: 10.1038/MTNA.2016.76.
Vaqué, J. P. et al. (2014) ‘PLCG1 mutations in cutaneous T-cell lymphomas’, Blood, 123(13), pp. 2034–2043. doi: 10.1182/blood-2013-05-504308.
Wang, Y. et al. (2018) ‘The role of STAT3 in leading the crosstalk between human cancers and the immune system’, Cancer Letters, 415, pp. 117–128. doi: 10.1016/j.canlet.2017.12.003.
Watanabe, T. et al. (2019) ‘Editorial Introduction to the review series on T-cell malignancies’, 129(9), pp. 2019–2021. doi: 10.1182/blood-2017-01-741389.Lamy.
Weigelt, B. and Downward, J. (2012) ‘Genomic Determinants of PI3K Pathway Inhibitor Response in Cancer’, Frontiers in Oncology, 2, p. 109. doi: 10.3389/fonc.2012.00109.
Wilcox, R. A. et al. (2012) ‘The B7 Homologues and their Receptors in Hematologic Malignancies’, European Journal of Haematology, 88(6), pp. 465–475. doi: 10.1111/j.1600-0609.2012.01766.x.
Wilcox, R. A. (2016) ‘A three-signal model of T-cell lymphoma pathogenesis.’, American journal of
hematology. NIH Public Access, 91(1), pp. 113–22. doi: 10.1002/ajh.24203.
Yoo, H. Y., Sung, M. K., Lee, S. H., et al. (2014) ‘A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma.’, Nature genetics, 46(4), pp. 371–5. doi: 10.1038/ng.2916.
Yoo, H. Y., Sung, M. K., Lee, S. S. H., et al. (2014) ‘A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma.’, 46(4). doi: 10.1038/ng.2916.
Yu, H. et al. (2014) ‘Revisiting STAT3 signalling in cancer: new and unexpected biological functions’, Nature Reviews Cancer. Nature Publishing Group, 14(11), pp. 736–746. doi: 10.1038/nrc3818.
Zhang, D. and Jr, T. P. L. (2012) ‘Large granular lymphocytic leukemia : molecular pathogenesis , clinical manifestations , and treatment’, pp. 652–659.
Zhang, Q. et al. (2011) ‘Oncogenic tyrosine kinase NPM-ALK induces expression of the growth-promoting receptor ICOS’, Blood, 118(11), pp. 3062–3071. doi: 10.1182/blood-2011-01-332916.