the extracellular signal-regulated kinase pathway phosphorylates
TRANSCRIPT

MOLECULAR AND CELLULAR BIOLOGY, July 1996, p. 3967–3979 Vol. 16, No. 70270-7306/96/$04.0010Copyright q 1996, American Society for Microbiology
The Extracellular Signal-Regulated Kinase Pathway PhosphorylatesAML1, an Acute Myeloid Leukemia Gene Product, and
Potentially Regulates Its Transactivation AbilityTOMOYUKI TANAKA,1,2† MINEO KUROKAWA,1,2 KOHJIRO UEKI,1 KOZO TANAKA,1 YOICHI IMAI,1
KINUKO MITANI,1 KENJI OKAZAKI,3‡ NORIYUKI SAGATA,3§ YOSHIO YAZAKI,1
YOICHI SHIBATA,2 TAKASHI KADOWAKI,1 AND HISAMARU HIRAI1*
Third Department of Internal Medicine1 and Department of Transfusion Medicine and Immunohematology,2
Faculty of Medicine, University of Tokyo, Bunkyo-ku, Tokyo 113, and Division of Molecular Genetics,Institute of Life Science, Kurume University, Kurume, Fukuoka 830,3 Japan
Received 11 January 1996/Returned for modification 22 February 1996/Accepted 3 April 1996
AML1 (also called PEBP2aB, CBFA2, or CBFa2) is one of the most frequently disrupted genes in chromo-some abnormalities seen in human leukemias. It has been reported that AML1 plays several pivotal roles inmyeloid hematopoietic differentiation and other biological phenomena, probably through the transcriptionalregulation of various relevant genes. Here, we investigated the mechanism of regulation of AML1 functionsthrough signal transduction pathways. The results showed that AML1 is phosphorylated in vivo on two serineresidues within the proline-, serine-, and threonine-rich region, with dependence on the activation of extra-cellular signal-regulated kinase (ERK) and with interleukin-3 stimulation in a hematopoietic cell line. Thesein vivo phosphorylation sites of AML1 were phosphorylated directly in vitro by ERK. Although differencesbetween wild-type AML1 and phosphorylation site mutants in DNA-binding affinity were not observed, we haveshown that ERK-dependent phosphorylation potentiates the transactivation ability of AML1. Furthermore thephosphorylation site mutations reduced the transforming capacity of AML1 in fibroblast cells. These dataindicate that AML1 functions are potentially regulated by ERK, which is activated by cytokine and growthfactor stimuli. This study provides some important clues for clarifying unidentified facets of the regulatorymechanism of AML1 function.
The AML1 gene was first identified on chromosome 21 asthe gene that is disrupted in the (8;21)(q22;q22) translocation(56, 66), which is one of the most frequent chromosome ab-normalities associated with human acute myelogenous leu-kemia. In t(8;21)(q22;q22), the gene rearrangement results inthe production of an AML1/MTG8(ETO) fusion protein (19,20, 54). We and another group previously reported that theAML1 gene is also disrupted in t(3;21)(q26;q22), which isfound in the blastic crisis phase of chronic myelogenous leu-kemia and therapy-related acute myelogenous leukemia (53,59–62). Recently, it was reported that the AML1 gene is re-arranged in acute lymphoblastic leukemia carrying t(12;21)(p12;q22) (24, 77). On the other hand, PEBP2aB (also calledCBFA2 or CBFa2), a mouse homolog of AML1, was first iden-tified as the gene encoding a member of the polyomavirus en-hancer-binding protein (PEBP) 2a family or the core-bindingfactor (CBF) a (or A) family of the Moloney leukemia virusenhancer (6, 94). PEBP2 is a heterodimer composed of PEBP2aand PEBP2b (also called CBFB or CBFb) (63, 95). HumanPEBP2b is known to be disrupted in the inv(16)(p13q22) chro-
mosome abnormality associated with acute myelogenous leu-kemia (46, 47).These findings suggest that the structural alteration of
AML1 triggers the leukemic transformation and that intactAML1 may play important roles in hematopoietic cell differ-entiation and proliferation. We have shown that AML1 regu-lates myeloid cell differentiation and transcriptional activationantagonistically by two alternative spliced forms, suggestingthat a transactivation property of AML1 is necessary for my-eloid cell differentiation (90). It has also been reported thatAML1 regulates the transcription of various genes which areimportant in hematopoiesis, such as those for myeloperoxi-dase, leukocyte elastase (58), the receptor for macrophagecolony-stimulating factor (99, 100), granulocyte-macrophagecolony-stimulating factor (85), and T-cell receptors (TCRs)(23, 29, 30, 84, 97). It was recently shown that mice lackingAML1 die during midembryonic development, secondary tothe complete absence of liver-derived hematopoiesis (70). Fur-thermore, we and other groups have demonstrated that thefusion proteins produced in t(3;21) and t(8;21) leukemic cellsdominantly suppress the functions of intact AML1 and inhibitmyeloid cell differentiation (21, 51, 80, 87). In addition, it hasbeen suggested that AML1 also plays several important rolesin thymus development (81), muscle development (102), fibro-blast transformation (42), and polyomavirus replication (9, 79).From these data, it is anticipated that the AML1 functionscould be regulated by intracellular signal transduction path-ways responding to the extracellular environment, such as thepresence of cytokines and growth factors.Within the AML1 protein, there are two functional domains
that have been identified already. The runt homology domain,which is so designated because it is highly homologous to the
* Corresponding author. Mailing address: The Third Department ofInternal Medicine, Faculty of Medicine, University of Tokyo, 7-3-1Hongo, Bunkyo-ku, Tokyo 113, Japan. Phone: 81-3-3815-5411, ext.3102 or 3116. Fax: 81-3-5689-7286. Electronic mail address: [email protected].† Present address: Research Institute of Molecular Pathology, A-
1030 Vienna, Austria.‡ Present address: Department of Molecular Biology, Biomolecular
Engineering Research Institute, Suita, Osaka 565, Japan.§ Present address: Department of Biology, Faculty of Science, Ky-
ushu University, Higashi-ku, Fukuoka 812-81, Japan.
3967

corresponding region within the product of the Drosophilapair-rule gene runt (15), is responsible for DNA binding to aconsensus sequence (called a PEBP2/CBF site), as well as forheterodimer formation with PEBP2b (33, 50, 65), nuclear ac-cumulation of protein (48), and association with the Ets-1transcription factor (23). The PST region, which is so calledbecause it is rich in proline, serine, and threonine residues, isessential for the process of transactivation by AML1 (4, 90).However, the PST region is not a usual transactivation domainbecause it does not show transactivation through the Gal4-binding DNA sequence when fused to the DNA-binding do-main of Gal4 (64, 89). The role of the PST region in transac-tivation remains to be elucidated. However, no informationabout the mechanism of regulating transactivation by AML1has been available.Extracellular signal-regulated kinase (ERK), a member of
the mitogen-activated protein kinases (MAPKs), is a signalingmolecule common to pathways that regulate proliferation anddifferentiation in diverse cell types, including hematopoieticcells (16, 49). ERK is activated by a wide range of cytokine andgrowth factor stimuli. Several nuclear transcription factorshave been identified as in vivo substrates of ERK (31, 32).These transcription factors exhibit alterations of their func-tions, such as DNA-binding ability and transactivation capac-ity, depending on their phosphorylation by ERK. In thepresent study, we investigated the regulation of AML1 func-tions with dependence on ERK activation. It was shown thatAML1 is phosphorylated in vivo by ERK activation on twoserine residues within the PST region. It was also suggestedthat ERK-dependent phosphorylation potentiates the transac-tivation ability of AML1. This is the first report to identify aregulatory mechanism of AML1 via signal transduction path-ways. This study has provided important insight needed toclarify unidentified facets of AML1 functions in leukemogen-esis, hematopoiesis, and other biological phenomena.
MATERIALS AND METHODS
Plasmid construction. The pME-AML1a and pME-AML1 plasmids were con-structed by ligation of human AML1a and AML1b cDNAs, respectively, to thepME18S expression vector as described previously (90). The pME-AML1DEco47III and pME-AML1DPstI expression vectors of AML1b deletion mutantswere constructed as described elsewhere (42). For construction of pME-AML1DBstPI, we deleted the BstPI (in AML1b cDNA)-SpeI (in pME18S) frag-ment from pME-AML1, filled the resultant plasmid with a Klenow fragment, andreligated it. The Escherichia coli expression plasmid of a maltose-binding protein(MB)–wild-type AML1 chimeric protein was constructed, as reported previously(90). The AML1b cDNA was also inserted into the pSRaMSVtkneo vector (57)and used to produce an AML1-expressing retroviral vector as described previ-ously (42). The AML1 mutants S249A, S266A, S249A/S266A, and T273A/S276Awere obtained by replacing the serine (and threonine) residues with alanines bythe site-directed mutagenesis method for the AML1b cDNA (40). These mutantAML1 cDNAs were inserted into pME18S, pMAL-c2 (New England Biolabs),and pSRaMSVtkneo, replacing wild-type AML1b cDNA. For tagging AML1 orits mutants at the NH2 terminus, the influenza virus hemagglutinin (HA) epitope(YPYDVPDYA) was inserted after the first methionine codon by PCR (39).These HA-tagged cDNAs were inserted into the pME18S and pSRaMSVtkneovectors. pCMVMK, which is an expression vector of a rat ERK1-ERK2 chimericprotein (with the amino-terminal 20 residues derived only from ERK2), wasobtained by ligation of the chimeric cDNA into the pRc/CMV vector (Invitro-gen) as described previously (93). An ERK(KN) mutant was made by replacingthe conserved lysine 71 residue (of ERK1), which is involved in phosphatetransfer, with arginine by the site-directed mutagenesis method for the above-mentioned ERK cDNA. The ERK(KN) cDNA was inserted into the pRc/CMVvector and named pCMVMK(KN). The dominant-negative and constitutivelyactive mutants of MEK1 (MEK1DN and MEK1EE, respectively) were made bysite-directed mutagenesis and inserted into the pactEF expression vector, asdescribed previously (68). The construction of the Tww-tk-Luc and Tmm-tk-Lucreporter plasmids, which have the TCRb enhancer (regions 3 and 4, includingtwo PEBP2/CBF sites and mutated sites, respectively) inserted immediatelyupstream of the herpes simplex virus thymidine kinase promoter followed by thefirefly luciferase cDNA, has been described previously (65, 90).Metabolic labeling and immunoprecipitation. COS-7 cells were cultured and
transfected with an expression plasmid(s) by the DEAE-dextran method asdescribed previously (87). For metabolic labeling, COS-7 cells were cultured for30 to 35 h after transfection in Dulbecco’s modified Eagle’s medium (DMEM)containing 10% fetal calf serum (FCS) and then transferred to and incubated for12 to 15 h in DMEM containing 0.1% FCS. They were then transferred to andcultured for 3 to 4 h in methionine- or phosphate-free DMEM supplementedwith 0.1% FCS (dialyzed against 150 mM NaCl) plus 50 mCi of [35S]methionine(Tran-35S label; ICN) per ml or 400 mCi of [32P]phosphate (Phosphorus-32;Amersham) per ml, respectively. They were then either not treated or treated(for 5 min) with 10% FCS (dialyzed against 150 mM NaCl) plus 100 ng ofrecombinant human epidermal growth factor (EGF) (Wakunaga) per ml andharvested in 350 ml of lysis buffer C (50 mM HEPES [N-2-hydroxyethylpipera-zine-N9-2-ethanesulfonic acid] [pH 7.4], 100 mM NaCl, 1 mM EDTA, 20 mMNaF, 20 mM sodium PPi, 1% Triton, 10 nM okadaic acid, 10 U of aprotinin perml, and 2 mM phenylmethylsulfonyl fluoride) per 100-mm-diameter culture dish.Immunoprecipitation was carried out with an anti-PST serum (86) and proteinA-Sepharose (Pharmacia), and immunoprecipitates were analyzed by sodiumdodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and autora-diography, by standard methods (27). The bands which showed ERK activation-dependent shifts were excised from the gel and subjected to phosphoamino acidanalyses by methods described elsewhere (8).Retrovirus infection of BaF3 cells. A mouse pro-B cell line, BaF3, was main-
tained in RPMI 1640 medium supplemented with 10% FCS and 0.25 ng of mouseinterleukin 3 (IL-3) (a generous gift from Kirin Brewery) per ml (38, 82).Retroviral stocks were prepared from COS-7 cells, which were transfected withpSRaMSVtkneo or its derivative expression vectors of HA-tagged AML1 and itsmutant, together with a c2 packaging plasmid (57). BaF3 cells was incubatedwith the COS-7 cell condition media containing retroviruses in the presence of 8mg of Polybrene per ml for 10 h. The retrovirus-infected BaF cells were selectedwith G418 (1 mg/ml) for at least 10 days.Western blotting (immunoblotting). COS-7 cells were cultured for 30 to 35 h
after transfection in DMEM containing 10% FCS, transferred to and incubatedfor 15 to 18 h in DMEM supplemented with 0.1% FCS, treated or not with 10%FCS plus 100 ng of recombinant human EGF per ml, and harvested in lysis bufferC. BaF3 cells, which were infected with retrovirus and subsequently selected withG418, were incubated for 8 h in RPMI 1640 containing 0.5% FCS, treated or notwith 10 ng of mouse IL-3 per ml, and harvested in the lysis buffer describedpreviously (88). Cell lysates, including 70 mg of protein, were subjected to SDS-PAGE and Western blotting as described previously (88). Anti-MB/AML1 se-rum (1:500 dilution), HA.11 anti-HA serum (BabCo, 1:1,000 dilution), and aC92anti-ERK serum (1:1,000 dilution) were used for detection of the proteins (90,91).In vitro kinase assay. 3Y1 rat fibroblast cells were cultured in DMEM con-
taining 10% bovine serum. The 3Y1 cells or transfected COS-7 cells were incu-bated for 15 to 18 h in DMEM containing 0.1% BS or FCS, respectively, treatedor not with 100 ng of recombinant human EGF per ml (plus 10% FCS in the caseof COS-7 cells), and harvested in lysis buffer C. Cell lysates were incubated withaC92 anti-ERK serum and protein A-Sepharose. They were then immunopre-cipitated and subjected to an in vitro kinase reaction, as described previously(91), with myelin basic protein (Sigma) or the MB–wild-type AML1 (or mutant)protein as the substrate.Protein interaction assay. The MB–wild-type AML1 (or S249A/S266A) pro-
tein (2 mg) was produced in E. coli, immobilized on amylose resin (New EnglandBiolabs), as described previously (90) and was mixed with cell lysates (containing700 mg of protein) in lysis buffer C. After extensive washings with lysis buffer C,the bound proteins were eluted with SDS-PAGE sample buffer and subjected toSDS-PAGE and Western blotting (27).EMSA and luciferase assay. P19 mouse embryonal carcinoma cells were cul-
tured as described elsewhere (78). Nuclear extracts were prepared from COS-7and P19 cells, which were transfected with plasmids, and subjected to electro-phoretic mobility shift assay (EMSA) as described previously (22, 90). Theprocedures for preparing nuclear extracts have been described previously (87,90), except that mixtures of peptidase inhibitors (including diisopropylfluoro-phosphate, benzamidine, spermidine, antipain, leupeptin, pepstatin A, and soy-bean trypsin inhibitor in concentrations described elsewhere [58]) were added tothe lysates prior to the procedure. The double-stranded oligonucleotide probesM4 and M24 were used in this study (22, 90).P19 cells were transfected with reporter plasmids (containing the luciferase
gene) and expression vectors by the calcium phosphate precipitation method,cultured in medium containing 10% FCS for 30 to 36 h, and then harvested andsubjected to the luciferase assay as described previously (88). A plasmid express-ing b-galactosidase was cotransfected as an internal control of transfection effi-ciency, and the data were normalized to the b-galactosidase activity, as describedpreviously (88).Colony formation assay. The colony formation assay was performed according
to procedures described elsewhere (41, 42). Briefly, retroviral stocks were pre-pared from COS-7 cells, which were transfected with pSRaMSVtkneo or itsderivative expression vectors of AML1 and its mutants, together with a c2
packaging plasmid (57). AML1 and its mutants (S249A, S266A, S249A/S266A,and T273A/S276A) showed equivalent expression levels in COS-7 cells whentransfected into the cells by the pSRaMSVtkneo vector (data not shown). Con-dition media containing viruses were harvested, and the virus titers were nor-
3968 TANAKA ET AL. MOL. CELL. BIOL.

malized prior to infection. NIH 3T3 cells were exposed to viral stocks mixed withPolybrene, selected by G418 resistance for 10 days, and seeded in DMEMcontaining 0.3% agarose and 20% FCS. Colonies were counted after 14 days ofculture in soft agar if they were larger than 0.25 mm in diameter.
RESULTS
AML1 is phosphorylated in vivo with dependence on theactivation of ERK. To analyze the role of the AML1 protein inthe signal transduction pathway, we used COS-7 cells trans-fected with AML1 because AML1 shows a very low level ofexpression of endogenous protein in various cells and, accord-ingly, has never been detected in any cell types that we have sofar investigated, by Western blotting or immunoprecipitation.The starting point of this study was the observation that AML1shows a faint size shift in Western blotting with an anti-AML1serum for detection of the protein when COS-7 cells trans-fected with AML1 are serum starved and treated with EGF(data not shown, but see Fig. 4A, lane 3). Growth factor-induced phosphorylation of several transcription factors is fre-quently detected as size-shifted bands of proteins in SDS-PAGE (12, 14, 67, 72, 73, 92). In order to reveal possibleposttranslational modification of AML1 protein in the signaltransduction pathway, which begins with the binding of growthfactors to their receptors, we investigated whether the phos-phorylation of AML1 is induced by the activation of ERK,which is known to mediate the signal from the receptors and tophosphorylate several transcription factors.A transient-transfection assay was employed to express ERK
in COS-7 cells. First, the kinase activity of ERK was evaluatedby an in vitro kinase assay with immunoprecipitates with anti-ERK antibody and myelin basic proteins as the substrate. Themost prominent ERK activity was detected when ERK-trans-fected COS-7 cells were stimulated by EGF plus FCS (Fig. 1A,lane 4). This shows that growth factors (EGF plus FCS) canpotentiate the kinase activity of the overexpressed ERK inCOS-7 cells. The endogenous ERK was also activated uponEGF-FCS stimulation (Fig. 1A, lane 3). Next, AML1 was tran-siently expressed with ERK in COS-7 cells and immunopre-cipitated by an anti-AML1 antibody (Fig. 1B). When COS-7cells were labeled with [35S]methionine, we observed equiva-lent amounts of AML1 expression, irrespective of ERK coex-pression or EGF-FCS stimulation (Fig. 1B, lanes 3 to 6). Inaddition to the main band of AML1, a few faint retarded bandsappeared when ERK was overexpressed and activated by EGF
plus FCS (Fig. 1B, lane 6). These shifted bands turned out tobe phosphorylated forms of AML1 when [32P]phosphate label-ing was carried out with COS-7 cells (Fig. 1B, lane 12). Suchphosphorylation was scarcely induced when a kinase-negativeERK mutant [ERK(KN)] (76) was used instead of wild-typeERK (labeling data not shown; see band shift data in Fig. 6B,row d, lanes 5 and 6). These data indicate that activation ofERK induces phosphorylation of AML1. A long exposure ofthe autoradiogram in the [32P]phosphate labeling experimentindicated that AML1 shows a faint size-shifted band uponEGF-FCS stimulation without ERK overexpression (data notshown, but see Fig. 4A, lane 3), possibly mirroring phosphor-ylation by an endogenous ERK. Furthermore, we have ob-served the constitutive phosphorylation of AML1, with or with-out ERK activation, as a band showing the same mobility as amain [35S]methionine-labeled band (Fig. 1B, lanes 9 to 12).S-249 and S-266 in AML1 are major and minor ERK-de-
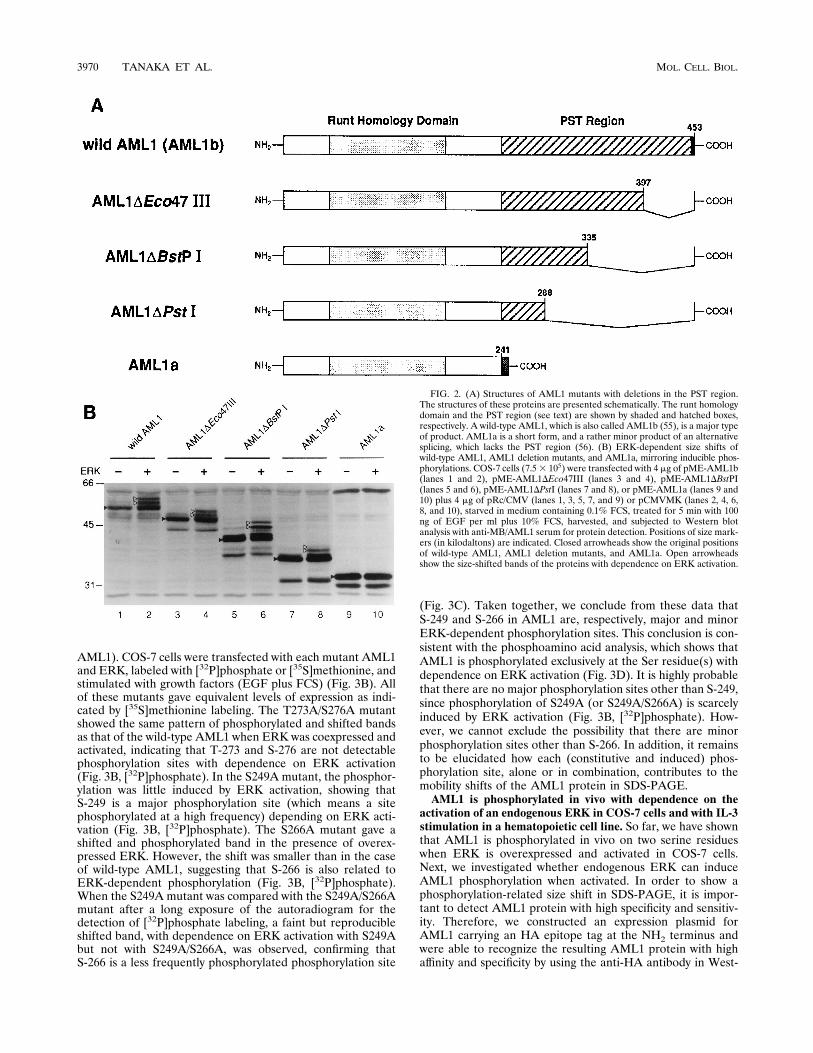
pendent phosphorylation sites, respectively.When AML1 wascoexpressed with activated ERK, a few retarded bands wereobserved in the Western blot analysis with whole lysates ofCOS-7 cells, mirroring the phosphorylated forms of AML1(Fig. 2B, lane 2). In order to make an approximate estimationof the phosphorylation sites in AML1, AML1 deletion mutantsand AML1a were expressed with ERK in COS-7 cells andsubsequently investigated for size shifts in SDS-PAGE (Fig.2A). When three types of AML1 mutants with deletions in thePST region were expressed, retarded bands were induced inalmost the same pattern as that in the case of whole AML1, inthe presence of the activated ERK (Fig. 2B, lanes 3 to 8).However, we did not detect such a size shift induction by ERKwhen AML1a was expressed (Fig. 2B, lanes 9 and 10). Thesedata suggest that a major ERK-dependent phosphorylationsite(s) exists within the region of amino acids 241 to 288 inAML1, although we cannot completely rule out the existenceof other phosphorylation sites outside this region.Ser/Thr-Pro is a minimal consensus sequence for phosphor-
ylation by ERK. Therefore, there are four potential phosphor-ylation sites within the sequence of amino acids 241 to 288, ifAML1 is directly phosphorylated by ERK (Fig. 3A). To deter-mine whether these candidate sites are phosphorylated de-pending on ERK activation, we replaced the Ser or Thr resi-due(s) with Ala at one or two sites by in vitro mutagenesis andmade the AML1 mutants S249A, S266A, S249A/S266A, andT273A/S276A (numbers designate the replaced residues in
FIG. 1. (A) ERK activities in COS-7 cells. COS-7 cells (7.5 3 105) were transfected with 4 mg of pRc/CMV (lanes 1 and 3) or pCMVMK (lanes 2 and 4), starvedin medium containing 0.1% FCS, treated for 5 min with 100 ng of EGF per ml plus 10% FCS (lanes 3 and 4) or not treated (lanes 1 and 2), harvested, and subjectedto an in vitro ERK assay with myelin basic protein (MBP) as the substrate followed by SDS-PAGE and autoradiography. (B) [35S]methionine (lanes 1 to 6) and[32P]phosphate (lanes 7 to 12) labeling of AML1 protein. COS-7 cells (7.5 3 105) were transfected with 4 mg of pME-AML1 (lanes 3 to 6 and 9 to 12) or pME18S(lanes 1, 2, 7, and 8) together with 4 mg of pRc/CMV (lanes 1, 3, 5, 7, 9, and 11) or pCMVMK (lanes 2, 4, 6, 8, 10, and 12), starved in medium containing 0.1% FCS,subjected to metabolic labeling, treated for 5 min with 100 ng of EGF per ml plus 10% FCS (lanes 1, 2, 5 to 8, 11, and 12) or not treated (lanes 3, 4, 9, and 10), harvested,immunoprecipitated with an anti-PST serum, and subjected to SDS-PAGE and autoradiography. The arrow on the left shows immunoprecipitated [35S]methionine-labeled AML1. The arrows on the right show [32P]phosphate-labeled forms of AML1. Positions of size markers are indicated.
VOL. 16, 1996 ACTIVATED ERK PHOSPHORYLATES AML1 3969
AML1). COS-7 cells were transfected with each mutant AML1and ERK, labeled with [32P]phosphate or [35S]methionine, andstimulated with growth factors (EGF plus FCS) (Fig. 3B). Allof these mutants gave equivalent levels of expression as indi-cated by [35S]methionine labeling. The T273A/S276A mutantshowed the same pattern of phosphorylated and shifted bandsas that of the wild-type AML1 when ERK was coexpressed andactivated, indicating that T-273 and S-276 are not detectablephosphorylation sites with dependence on ERK activation(Fig. 3B, [32P]phosphate). In the S249A mutant, the phosphor-ylation was little induced by ERK activation, showing thatS-249 is a major phosphorylation site (which means a sitephosphorylated at a high frequency) depending on ERK acti-vation (Fig. 3B, [32P]phosphate). The S266A mutant gave ashifted and phosphorylated band in the presence of overex-pressed ERK. However, the shift was smaller than in the caseof wild-type AML1, suggesting that S-266 is also related toERK-dependent phosphorylation (Fig. 3B, [32P]phosphate).When the S249A mutant was compared with the S249A/S266Amutant after a long exposure of the autoradiogram for thedetection of [32P]phosphate labeling, a faint but reproducibleshifted band, with dependence on ERK activation with S249Abut not with S249A/S266A, was observed, confirming thatS-266 is a less frequently phosphorylated phosphorylation site
(Fig. 3C). Taken together, we conclude from these data thatS-249 and S-266 in AML1 are, respectively, major and minorERK-dependent phosphorylation sites. This conclusion is con-sistent with the phosphoamino acid analysis, which shows thatAML1 is phosphorylated exclusively at the Ser residue(s) withdependence on ERK activation (Fig. 3D). It is highly probablethat there are no major phosphorylation sites other than S-249,since phosphorylation of S249A (or S249A/S266A) is scarcelyinduced by ERK activation (Fig. 3B, [32P]phosphate). How-ever, we cannot exclude the possibility that there are minorphosphorylation sites other than S-266. In addition, it remainsto be elucidated how each (constitutive and induced) phos-phorylation site, alone or in combination, contributes to themobility shifts of the AML1 protein in SDS-PAGE.AML1 is phosphorylated in vivo with dependence on the
activation of an endogenous ERK in COS-7 cells and with IL-3stimulation in a hematopoietic cell line. So far, we have shownthat AML1 is phosphorylated in vivo on two serine residueswhen ERK is overexpressed and activated in COS-7 cells.Next, we investigated whether endogenous ERK can induceAML1 phosphorylation when activated. In order to show aphosphorylation-related size shift in SDS-PAGE, it is impor-tant to detect AML1 protein with high specificity and sensitiv-ity. Therefore, we constructed an expression plasmid forAML1 carrying an HA epitope tag at the NH2 terminus andwere able to recognize the resulting AML1 protein with highaffinity and specificity by using the anti-HA antibody in West-
FIG. 2. (A) Structures of AML1 mutants with deletions in the PST region.The structures of these proteins are presented schematically. The runt homologydomain and the PST region (see text) are shown by shaded and hatched boxes,respectively. A wild-type AML1, which is also called AML1b (55), is a major typeof product. AML1a is a short form, and a rather minor product of an alternativesplicing, which lacks the PST region (56). (B) ERK-dependent size shifts ofwild-type AML1, AML1 deletion mutants, and AML1a, mirroring inducible phos-phorylations. COS-7 cells (7.53 105) were transfected with 4 mg of pME-AML1b(lanes 1 and 2), pME-AML1DEco47III (lanes 3 and 4), pME-AML1DBstPI(lanes 5 and 6), pME-AML1DPstI (lanes 7 and 8), or pME-AML1a (lanes 9 and10) plus 4 mg of pRc/CMV (lanes 1, 3, 5, 7, and 9) or pCMVMK (lanes 2, 4, 6,8, and 10), starved in medium containing 0.1% FCS, treated for 5 min with 100ng of EGF per ml plus 10% FCS, harvested, and subjected to Western blotanalysis with anti-MB/AML1 serum for protein detection. Positions of size mark-ers (in kilodaltons) are indicated. Closed arrowheads show the original positionsof wild-type AML1, AML1 deletion mutants, and AML1a. Open arrowheadsshow the size-shifted bands of the proteins with dependence on ERK activation.
3970 TANAKA ET AL. MOL. CELL. BIOL.

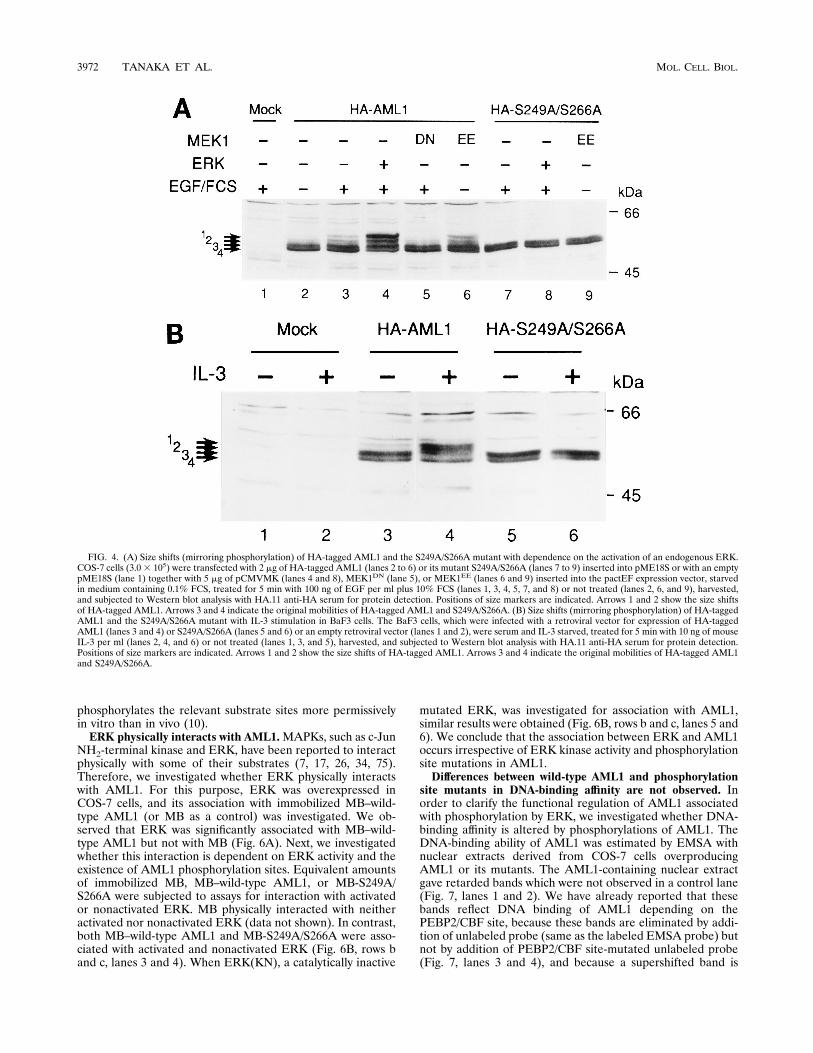
ern blotting. HA-tagged AML1 showed a subcellular localiza-tion and a transactivation ability similar to those of the originalAML1 (48, 89, 90). When COS-7 cells were transfected withHA-tagged AML1 plus ERK and stimulated with EGF plusFCS, we observed size shifts of AML1 mirroring its phosphor-ylation (Fig. 4A, lane 4). In the case of the HA-tagged S249A/S266A AML1 mutant, no size shifts were detected (Fig. 4A,lane 8). When AML1-expressing cells were treated with EGF-FCS without ERK transfection, a size shift, which is barelyseen without EGF-FCS treatment, was induced (Fig. 4A, lanes2 and 3, arrow 1). A similar size shift was detected withoutEGF-FCS treatment when a constitutively active form ofMEK1 (an activator of ERK) was coexpressed (Fig. 4A, lane 6,arrow 1) but not when cells were cotransfected with a domi-nant-negative form of MEK1 and treated with EGF-FCS (lane5). Such a size shift was not observed in the S249A/S266Amutant even when the cells were treated with EGF-FCS orcotransfected with a constitutively active form of MEK1 (Fig.4A, lanes 7 and 9). A smaller size shift, which was observed forAML1 without EGF-FCS treatment (Fig. 4A, lane 2, arrow 2)but not for the S249A/S266A mutant, probably reflects a weakphosphorylation of AML1 depending on endogenous ERK,since serum-starved COS-7 cells showed a remnant ERK ki-nase activity (Fig. 1, lane 1). Because MEK1 is a specificactivator for ERK (18, 52) and because growth factor stimuliinduce phosphorylation of AML1 at the same sites in the caseof ERK overexpression, we conclude that AML1 is phosphor-ylated in vivo with dependence on the activation of an endog-enous ERK in COS-7 cells.It has been reported that AML1 plays pivotal roles in he-
matopoietic cell differentiation and proliferation (70, 86, 90).Such findings prompted us to investigate whether AML1 isinducibly phosphorylated in hematopoietic cells. An IL-3-de-pendent mouse pro-B cell line, BaF3, was employed for thispurpose and was infected with a retroviral vector for expres-sion of HA-tagged AML1 or its phosphorylation site mutant(HA-tagged S249A/S266A). After selection for neo gene ex-pression of vectors, we established BaF3 cells which expressequivalent levels of HA-tagged AML1 and S249A/S266A (Fig.4B, lanes 3 and 5). It is already known that ERK can besubstantially activated with IL-3 stimulation in BaF3 cells (38,82). Therefore, established BaF3 cells were stimulated with
IL-3 after serum and IL-3 starvation. HA-tagged AML1showed the enhanced size shift bands, reflecting phosphoryla-tion (32P-labeling data not shown), when the cells were stim-ulated with IL-3 (Fig. 4B, lanes 3 and 4). In contrast, such achange was not seen in the case of HA-tagged S249A/S266A(Fig. 4B, lanes 5 and 6). These data indicate that AML1 isinducibly phosphorylated with IL-3 stimulation in a hemato-poietic cell line and suggest that AML1 is phosphorylated bythe ERK pathway in hematopoietic cells, because phosphory-lation is observed at the same sites as those of phosphorylationdepending on ERK activation in COS-7 cells.The S-249 and S-266 residues of AML1 are directly phos-
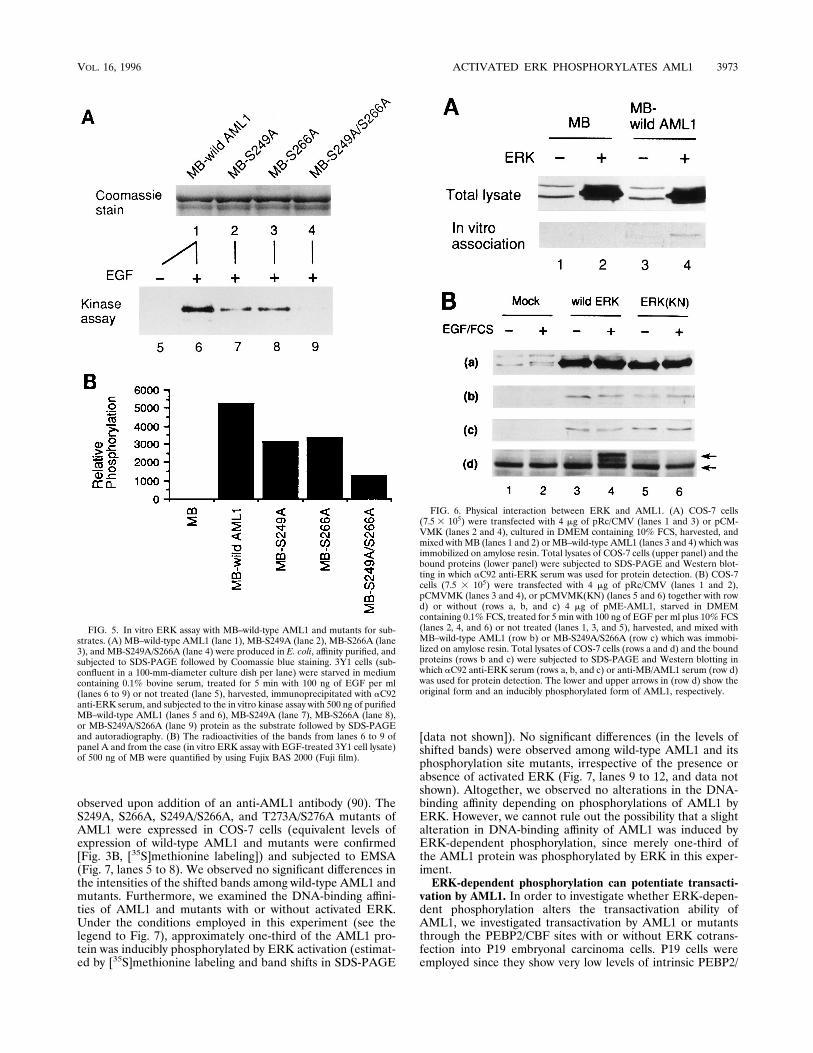
phorylated by ERK in vitro. It is quite likely that AML1 isdirectly phosphorylated by ERK at the S-249 and S-266 resi-dues, since these sites meet a minimal consensus sequence(Ser/Thr-Pro) for phosphorylation by ERK. In order to con-firm direct phosphorylation of these sites by ERK, the fusionprotein between MB and wild-type AML1 (or its mutants) wasproduced in E. coli, affinity purified (Fig. 5A, lanes 1 to 4), andused as a substrate for an in vitro ERK kinase assay. For thisassay, 3Y1 fibroblast cells were employed, because they show avery low level of ERK activity when starved for serum, whilethey exhibit markedly enhanced ERK activity when treatedwith EGF (28, 83). Prior to the assay, we observed that MB isnot phosphorylated by ERK in vitro (Fig. 5B). ERK was acti-vated by EGF treatment of 3Y1 cells (or not activated as acontrol), immunoprecipitated, and subjected to an in vitrokinase reaction in the presence of an equivalent amount ofMB–wild-type AML1 or mutants (MB-S249A, MB-S266A, andMB-S249A/S266A). It was observed that MB–wild-type AML1is phosphorylated in vitro by activated ERK but not withoutERK activation (Fig. 5A, lanes 5 and 6). Furthermore, thelevels of phosphorylation of MB-S249A and MB-S266A weredecreased compared with that of MB–wild-type AML1 (Fig.5A, lanes 6 to 8, and B). A further reduction of phosphoryla-tion was observed when MB-S249A/S266A was used as a sub-strate (Fig. 5A, lane 9, and B). These data indicate that theS-249 and S-266 residues of AML1 are directly phosphorylatedby ERK in vitro. A reduced but substantial level of phosphor-ylation of MB-S249A/S266A suggests that other sites in AML1are also phosphorylated in vitro in addition to S-249 and S-266.This finding is consistent with the fact that ERK sometimes
FIG. 3. (A) Potential phosphorylation sites in AML1 with dependence on ERK activation. Potential phosphorylation sites which meet a minimal consensusSer/Thr-Pro and reside within residues 241 to 288 are boxed. Numerals show the amino acid numbers in the AML1 protein. (B) Inducible phosphorylation of AML1mutants. COS-7 cells (7.5 3 105) were transfected with 4 mg of pME18S (Mock), pME-AML1 (wild-type AML1), or AML1 mutant cDNAs inserted into pME18S(S249A, S266A, S249A/S266A, or T273A/S276A) plus 4 mg of pRc/CMV (2) or pCMVMK (1), starved in the medium containing 0.1% FCS, subjected to metaboliclabeling with [32P]phosphate or [35S]methionine, treated for 5 min with 100 ng of EGF per ml plus 10% FCS, harvested, immunoprecipitated with anti-PST serum, andsubjected to SDS-PAGE followed by autoradiography. (C) Longer exposure of the lanes for S249A and S249A/S266A ([32P]phosphate labeling) in panel B. (D)Phosphoamino acid analysis of AML1 inducibly phosphorylated by ERK. The bands which were phosphorylated and shifted with dependence on ERK activation inFig. 1B, lane 12, were excised from the gel and subjected to phosphoamino acid analysis. The arrows show the directions of electrophoreses in buffers with the indicatedpHs. S, T, and Y, positions of phosphoserine, threonine, and tyrosine, respectively.
VOL. 16, 1996 ACTIVATED ERK PHOSPHORYLATES AML1 3971
phosphorylates the relevant substrate sites more permissivelyin vitro than in vivo (10).ERK physically interacts with AML1.MAPKs, such as c-Jun
NH2-terminal kinase and ERK, have been reported to interactphysically with some of their substrates (7, 17, 26, 34, 75).Therefore, we investigated whether ERK physically interactswith AML1. For this purpose, ERK was overexpressed inCOS-7 cells, and its association with immobilized MB–wild-type AML1 (or MB as a control) was investigated. We ob-served that ERK was significantly associated with MB–wild-type AML1 but not with MB (Fig. 6A). Next, we investigatedwhether this interaction is dependent on ERK activity and theexistence of AML1 phosphorylation sites. Equivalent amountsof immobilized MB, MB–wild-type AML1, or MB-S249A/S266A were subjected to assays for interaction with activatedor nonactivated ERK. MB physically interacted with neitheractivated nor nonactivated ERK (data not shown). In contrast,both MB–wild-type AML1 and MB-S249A/S266A were asso-ciated with activated and nonactivated ERK (Fig. 6B, rows band c, lanes 3 and 4). When ERK(KN), a catalytically inactive
mutated ERK, was investigated for association with AML1,similar results were obtained (Fig. 6B, rows b and c, lanes 5 and6). We conclude that the association between ERK and AML1occurs irrespective of ERK kinase activity and phosphorylationsite mutations in AML1.Differences between wild-type AML1 and phosphorylation
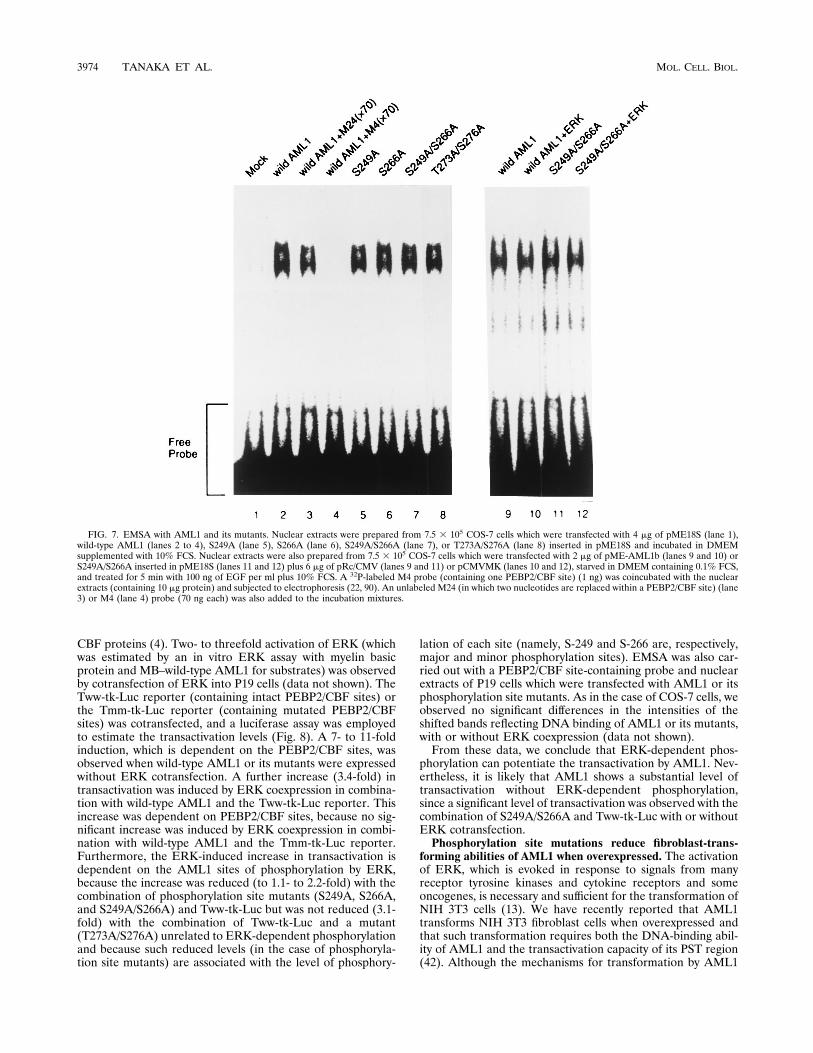
site mutants in DNA-binding affinity are not observed. Inorder to clarify the functional regulation of AML1 associatedwith phosphorylation by ERK, we investigated whether DNA-binding affinity is altered by phosphorylations of AML1. TheDNA-binding ability of AML1 was estimated by EMSA withnuclear extracts derived from COS-7 cells overproducingAML1 or its mutants. The AML1-containing nuclear extractgave retarded bands which were not observed in a control lane(Fig. 7, lanes 1 and 2). We have already reported that thesebands reflect DNA binding of AML1 depending on thePEBP2/CBF site, because these bands are eliminated by addi-tion of unlabeled probe (same as the labeled EMSA probe) butnot by addition of PEBP2/CBF site-mutated unlabeled probe(Fig. 7, lanes 3 and 4), and because a supershifted band is
FIG. 4. (A) Size shifts (mirroring phosphorylation) of HA-tagged AML1 and the S249A/S266A mutant with dependence on the activation of an endogenous ERK.COS-7 cells (3.03 105) were transfected with 2 mg of HA-tagged AML1 (lanes 2 to 6) or its mutant S249A/S266A (lanes 7 to 9) inserted into pME18S or with an emptypME18S (lane 1) together with 5 mg of pCMVMK (lanes 4 and 8), MEK1DN (lane 5), or MEK1EE (lanes 6 and 9) inserted into the pactEF expression vector, starvedin medium containing 0.1% FCS, treated for 5 min with 100 ng of EGF per ml plus 10% FCS (lanes 1, 3, 4, 5, 7, and 8) or not treated (lanes 2, 6, and 9), harvested,and subjected to Western blot analysis with HA.11 anti-HA serum for protein detection. Positions of size markers are indicated. Arrows 1 and 2 show the size shiftsof HA-tagged AML1. Arrows 3 and 4 indicate the original mobilities of HA-tagged AML1 and S249A/S266A. (B) Size shifts (mirroring phosphorylation) of HA-taggedAML1 and the S249A/S266A mutant with IL-3 stimulation in BaF3 cells. The BaF3 cells, which were infected with a retroviral vector for expression of HA-taggedAML1 (lanes 3 and 4) or S249A/S266A (lanes 5 and 6) or an empty retroviral vector (lanes 1 and 2), were serum and IL-3 starved, treated for 5 min with 10 ng of mouseIL-3 per ml (lanes 2, 4, and 6) or not treated (lanes 1, 3, and 5), harvested, and subjected to Western blot analysis with HA.11 anti-HA serum for protein detection.Positions of size markers are indicated. Arrows 1 and 2 show the size shifts of HA-tagged AML1. Arrows 3 and 4 indicate the original mobilities of HA-tagged AML1and S249A/S266A.
3972 TANAKA ET AL. MOL. CELL. BIOL.
observed upon addition of an anti-AML1 antibody (90). TheS249A, S266A, S249A/S266A, and T273A/S276A mutants ofAML1 were expressed in COS-7 cells (equivalent levels ofexpression of wild-type AML1 and mutants were confirmed[Fig. 3B, [35S]methionine labeling]) and subjected to EMSA(Fig. 7, lanes 5 to 8). We observed no significant differences inthe intensities of the shifted bands among wild-type AML1 andmutants. Furthermore, we examined the DNA-binding affini-ties of AML1 and mutants with or without activated ERK.Under the conditions employed in this experiment (see thelegend to Fig. 7), approximately one-third of the AML1 pro-tein was inducibly phosphorylated by ERK activation (estimat-ed by [35S]methionine labeling and band shifts in SDS-PAGE
[data not shown]). No significant differences (in the levels ofshifted bands) were observed among wild-type AML1 and itsphosphorylation site mutants, irrespective of the presence orabsence of activated ERK (Fig. 7, lanes 9 to 12, and data notshown). Altogether, we observed no alterations in the DNA-binding affinity depending on phosphorylations of AML1 byERK. However, we cannot rule out the possibility that a slightalteration in DNA-binding affinity of AML1 was induced byERK-dependent phosphorylation, since merely one-third ofthe AML1 protein was phosphorylated by ERK in this exper-iment.ERK-dependent phosphorylation can potentiate transacti-
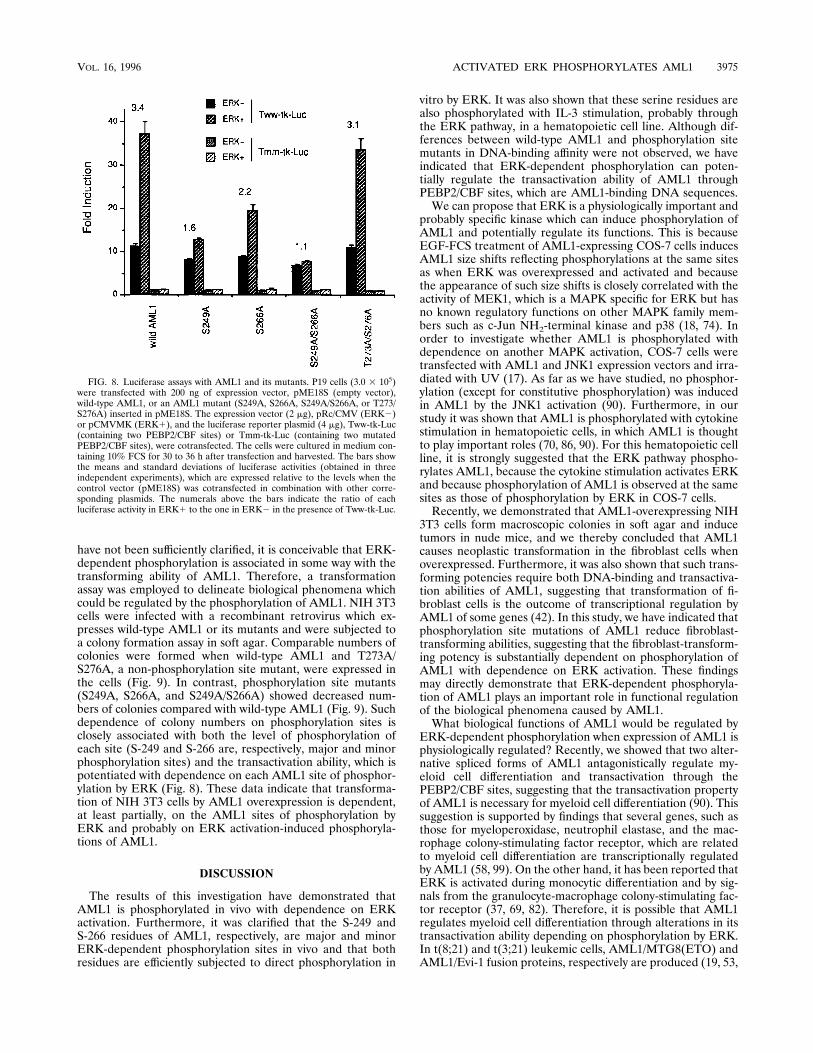
vation by AML1. In order to investigate whether ERK-depen-dent phosphorylation alters the transactivation ability ofAML1, we investigated transactivation by AML1 or mutantsthrough the PEBP2/CBF sites with or without ERK cotrans-fection into P19 embryonal carcinoma cells. P19 cells wereemployed since they show very low levels of intrinsic PEBP2/
FIG. 5. In vitro ERK assay with MB–wild-type AML1 and mutants for sub-strates. (A) MB–wild-type AML1 (lane 1), MB-S249A (lane 2), MB-S266A (lane3), and MB-S249A/S266A (lane 4) were produced in E. coli, affinity purified, andsubjected to SDS-PAGE followed by Coomassie blue staining. 3Y1 cells (sub-confluent in a 100-mm-diameter culture dish per lane) were starved in mediumcontaining 0.1% bovine serum, treated for 5 min with 100 ng of EGF per ml(lanes 6 to 9) or not treated (lane 5), harvested, immunoprecipitated with aC92anti-ERK serum, and subjected to the in vitro kinase assay with 500 ng of purifiedMB–wild-type AML1 (lanes 5 and 6), MB-S249A (lane 7), MB-S266A (lane 8),or MB-S249A/S266A (lane 9) protein as the substrate followed by SDS-PAGEand autoradiography. (B) The radioactivities of the bands from lanes 6 to 9 ofpanel A and from the case (in vitro ERK assay with EGF-treated 3Y1 cell lysate)of 500 ng of MB were quantified by using Fujix BAS 2000 (Fuji film).
FIG. 6. Physical interaction between ERK and AML1. (A) COS-7 cells(7.5 3 105) were transfected with 4 mg of pRc/CMV (lanes 1 and 3) or pCM-VMK (lanes 2 and 4), cultured in DMEM containing 10% FCS, harvested, andmixed with MB (lanes 1 and 2) or MB–wild-type AML1 (lanes 3 and 4) which wasimmobilized on amylose resin. Total lysates of COS-7 cells (upper panel) and thebound proteins (lower panel) were subjected to SDS-PAGE and Western blot-ting in which aC92 anti-ERK serum was used for protein detection. (B) COS-7cells (7.5 3 105) were transfected with 4 mg of pRc/CMV (lanes 1 and 2),pCMVMK (lanes 3 and 4), or pCMVMK(KN) (lanes 5 and 6) together with rowd) or without (rows a, b, and c) 4 mg of pME-AML1, starved in DMEMcontaining 0.1% FCS, treated for 5 min with 100 ng of EGF per ml plus 10% FCS(lanes 2, 4, and 6) or not treated (lanes 1, 3, and 5), harvested, and mixed withMB–wild-type AML1 (row b) or MB-S249A/S266A (row c) which was immobi-lized on amylose resin. Total lysates of COS-7 cells (rows a and d) and the boundproteins (rows b and c) were subjected to SDS-PAGE and Western blotting inwhich aC92 anti-ERK serum (rows a, b, and c) or anti-MB/AML1 serum (row d)was used for protein detection. The lower and upper arrows in (row d) show theoriginal form and an inducibly phosphorylated form of AML1, respectively.
VOL. 16, 1996 ACTIVATED ERK PHOSPHORYLATES AML1 3973
CBF proteins (4). Two- to threefold activation of ERK (whichwas estimated by an in vitro ERK assay with myelin basicprotein and MB–wild-type AML1 for substrates) was observedby cotransfection of ERK into P19 cells (data not shown). TheTww-tk-Luc reporter (containing intact PEBP2/CBF sites) orthe Tmm-tk-Luc reporter (containing mutated PEBP2/CBFsites) was cotransfected, and a luciferase assay was employedto estimate the transactivation levels (Fig. 8). A 7- to 11-foldinduction, which is dependent on the PEBP2/CBF sites, wasobserved when wild-type AML1 or its mutants were expressedwithout ERK cotransfection. A further increase (3.4-fold) intransactivation was induced by ERK coexpression in combina-tion with wild-type AML1 and the Tww-tk-Luc reporter. Thisincrease was dependent on PEBP2/CBF sites, because no sig-nificant increase was induced by ERK coexpression in combi-nation with wild-type AML1 and the Tmm-tk-Luc reporter.Furthermore, the ERK-induced increase in transactivation isdependent on the AML1 sites of phosphorylation by ERK,because the increase was reduced (to 1.1- to 2.2-fold) with thecombination of phosphorylation site mutants (S249A, S266A,and S249A/S266A) and Tww-tk-Luc but was not reduced (3.1-fold) with the combination of Tww-tk-Luc and a mutant(T273A/S276A) unrelated to ERK-dependent phosphorylationand because such reduced levels (in the case of phosphoryla-tion site mutants) are associated with the level of phosphory-
lation of each site (namely, S-249 and S-266 are, respectively,major and minor phosphorylation sites). EMSA was also car-ried out with a PEBP2/CBF site-containing probe and nuclearextracts of P19 cells which were transfected with AML1 or itsphosphorylation site mutants. As in the case of COS-7 cells, weobserved no significant differences in the intensities of theshifted bands reflecting DNA binding of AML1 or its mutants,with or without ERK coexpression (data not shown).From these data, we conclude that ERK-dependent phos-
phorylation can potentiate the transactivation by AML1. Nev-ertheless, it is likely that AML1 shows a substantial level oftransactivation without ERK-dependent phosphorylation,since a significant level of transactivation was observed with thecombination of S249A/S266A and Tww-tk-Luc with or withoutERK cotransfection.Phosphorylation site mutations reduce fibroblast-trans-
forming abilities of AML1 when overexpressed. The activationof ERK, which is evoked in response to signals from manyreceptor tyrosine kinases and cytokine receptors and someoncogenes, is necessary and sufficient for the transformation ofNIH 3T3 cells (13). We have recently reported that AML1transforms NIH 3T3 fibroblast cells when overexpressed andthat such transformation requires both the DNA-binding abil-ity of AML1 and the transactivation capacity of its PST region(42). Although the mechanisms for transformation by AML1
FIG. 7. EMSA with AML1 and its mutants. Nuclear extracts were prepared from 7.5 3 105 COS-7 cells which were transfected with 4 mg of pME18S (lane 1),wild-type AML1 (lanes 2 to 4), S249A (lane 5), S266A (lane 6), S249A/S266A (lane 7), or T273A/S276A (lane 8) inserted in pME18S and incubated in DMEMsupplemented with 10% FCS. Nuclear extracts were also prepared from 7.5 3 105 COS-7 cells which were transfected with 2 mg of pME-AML1b (lanes 9 and 10) orS249A/S266A inserted in pME18S (lanes 11 and 12) plus 6 mg of pRc/CMV (lanes 9 and 11) or pCMVMK (lanes 10 and 12), starved in DMEM containing 0.1% FCS,and treated for 5 min with 100 ng of EGF per ml plus 10% FCS. A 32P-labeled M4 probe (containing one PEBP2/CBF site) (1 ng) was coincubated with the nuclearextracts (containing 10 mg protein) and subjected to electrophoresis (22, 90). An unlabeled M24 (in which two nucleotides are replaced within a PEBP2/CBF site) (lane3) or M4 (lane 4) probe (70 ng each) was also added to the incubation mixtures.
3974 TANAKA ET AL. MOL. CELL. BIOL.
have not been sufficiently clarified, it is conceivable that ERK-dependent phosphorylation is associated in some way with thetransforming ability of AML1. Therefore, a transformationassay was employed to delineate biological phenomena whichcould be regulated by the phosphorylation of AML1. NIH 3T3cells were infected with a recombinant retrovirus which ex-presses wild-type AML1 or its mutants and were subjected toa colony formation assay in soft agar. Comparable numbers ofcolonies were formed when wild-type AML1 and T273A/S276A, a non-phosphorylation site mutant, were expressed inthe cells (Fig. 9). In contrast, phosphorylation site mutants(S249A, S266A, and S249A/S266A) showed decreased num-bers of colonies compared with wild-type AML1 (Fig. 9). Suchdependence of colony numbers on phosphorylation sites isclosely associated with both the level of phosphorylation ofeach site (S-249 and S-266 are, respectively, major and minorphosphorylation sites) and the transactivation ability, which ispotentiated with dependence on each AML1 site of phosphor-ylation by ERK (Fig. 8). These data indicate that transforma-tion of NIH 3T3 cells by AML1 overexpression is dependent,at least partially, on the AML1 sites of phosphorylation byERK and probably on ERK activation-induced phosphoryla-tions of AML1.
DISCUSSION
The results of this investigation have demonstrated thatAML1 is phosphorylated in vivo with dependence on ERKactivation. Furthermore, it was clarified that the S-249 andS-266 residues of AML1, respectively, are major and minorERK-dependent phosphorylation sites in vivo and that bothresidues are efficiently subjected to direct phosphorylation in
vitro by ERK. It was also shown that these serine residues arealso phosphorylated with IL-3 stimulation, probably throughthe ERK pathway, in a hematopoietic cell line. Although dif-ferences between wild-type AML1 and phosphorylation sitemutants in DNA-binding affinity were not observed, we haveindicated that ERK-dependent phosphorylation can poten-tially regulate the transactivation ability of AML1 throughPEBP2/CBF sites, which are AML1-binding DNA sequences.We can propose that ERK is a physiologically important and
probably specific kinase which can induce phosphorylation ofAML1 and potentially regulate its functions. This is becauseEGF-FCS treatment of AML1-expressing COS-7 cells inducesAML1 size shifts reflecting phosphorylations at the same sitesas when ERK was overexpressed and activated and becausethe appearance of such size shifts is closely correlated with theactivity of MEK1, which is a MAPK specific for ERK but hasno known regulatory functions on other MAPK family mem-bers such as c-Jun NH2-terminal kinase and p38 (18, 74). Inorder to investigate whether AML1 is phosphorylated withdependence on another MAPK activation, COS-7 cells weretransfected with AML1 and JNK1 expression vectors and irra-diated with UV (17). As far as we have studied, no phosphor-ylation (except for constitutive phosphorylation) was inducedin AML1 by the JNK1 activation (90). Furthermore, in ourstudy it was shown that AML1 is phosphorylated with cytokinestimulation in hematopoietic cells, in which AML1 is thoughtto play important roles (70, 86, 90). For this hematopoietic cellline, it is strongly suggested that the ERK pathway phospho-rylates AML1, because the cytokine stimulation activates ERKand because phosphorylation of AML1 is observed at the samesites as those of phosphorylation by ERK in COS-7 cells.Recently, we demonstrated that AML1-overexpressing NIH
3T3 cells form macroscopic colonies in soft agar and inducetumors in nude mice, and we thereby concluded that AML1causes neoplastic transformation in the fibroblast cells whenoverexpressed. Furthermore, it was also shown that such trans-forming potencies require both DNA-binding and transactiva-tion abilities of AML1, suggesting that transformation of fi-broblast cells is the outcome of transcriptional regulation byAML1 of some genes (42). In this study, we have indicated thatphosphorylation site mutations of AML1 reduce fibroblast-transforming abilities, suggesting that the fibroblast-transform-ing potency is substantially dependent on phosphorylation ofAML1 with dependence on ERK activation. These findingsmay directly demonstrate that ERK-dependent phosphoryla-tion of AML1 plays an important role in functional regulationof the biological phenomena caused by AML1.What biological functions of AML1 would be regulated by
ERK-dependent phosphorylation when expression of AML1 isphysiologically regulated? Recently, we showed that two alter-native spliced forms of AML1 antagonistically regulate my-eloid cell differentiation and transactivation through thePEBP2/CBF sites, suggesting that the transactivation propertyof AML1 is necessary for myeloid cell differentiation (90). Thissuggestion is supported by findings that several genes, such asthose for myeloperoxidase, neutrophil elastase, and the mac-rophage colony-stimulating factor receptor, which are relatedto myeloid cell differentiation are transcriptionally regulatedby AML1 (58, 99). On the other hand, it has been reported thatERK is activated during monocytic differentiation and by sig-nals from the granulocyte-macrophage colony-stimulating fac-tor receptor (37, 69, 82). Therefore, it is possible that AML1regulates myeloid cell differentiation through alterations in itstransactivation ability depending on phosphorylation by ERK.In t(8;21) and t(3;21) leukemic cells, AML1/MTG8(ETO) andAML1/Evi-1 fusion proteins, respectively are produced (19, 53,
FIG. 8. Luciferase assays with AML1 and its mutants. P19 cells (3.0 3 105)were transfected with 200 ng of expression vector, pME18S (empty vector),wild-type AML1, or an AML1 mutant (S249A, S266A, S249A/S266A, or T273/S276A) inserted in pME18S. The expression vector (2 mg), pRc/CMV (ERK2)or pCMVMK (ERK1), and the luciferase reporter plasmid (4 mg), Tww-tk-Luc(containing two PEBP2/CBF sites) or Tmm-tk-Luc (containing two mutatedPEBP2/CBF sites), were cotransfected. The cells were cultured in medium con-taining 10% FCS for 30 to 36 h after transfection and harvested. The bars showthe means and standard deviations of luciferase activities (obtained in threeindependent experiments), which are expressed relative to the levels when thecontrol vector (pME18S) was cotransfected in combination with other corre-sponding plasmids. The numerals above the bars indicate the ratio of eachluciferase activity in ERK1 to the one in ERK2 in the presence of Tww-tk-Luc.
VOL. 16, 1996 ACTIVATED ERK PHOSPHORYLATES AML1 3975

54, 60, 62). We noted that AML1/MTG8(ETO) and AML1/Evi-1 lose the ERK-dependent phosphorylation site-contain-ing region of AML1 by chromosomal rearrangement, and thissuggests that their transactivation abilities would not be poten-tiated by ERK. In fact, with or without ERK overexpression inP19 cells, the AML1/Evi-1 chimeric protein did not show anytransactivation through PEBP2/CBF-binding sites of theTCRb enhancer, while this fusion protein still demonstratedthe dominant-negative effects on transactivation by AML1 (87,89). Such loss in functional regulation of these chimeric pro-teins may play some role in leukemogenesis in leukemias witht(8;21) and t(3;21) chromosome abnormalities.In addition, it has been indicated that AML1 regulates tran-
scription of the granulocyte-macrophage colony-stimulatingfactor gene in T lymphocytes (85), depending on the 12-O-tetradecanoylphorbol 13-acetate stimulus, which potentiatesERK activity (1, 3). In this pathway, ERK-dependent phos-phorylation may be important for the activation of AML1. Inaddition, AML1 is known to be highly expressed in T cellsduring thymic development (81) and to play a pivotal role intranscriptional regulation of the TCR genes (23, 29, 30, 84, 97).It has also been reported that ERK activation is required insome steps of thymocyte differentiation (2). ERK-dependentfunctional regulation of AML1 may be important in thymocytedifferentiation.Furthermore, it has been reported that AML1 is expressed
in muscle cells (myoblasts, myotubes, and adult muscles) andthat its expression is regulated by electrical activity, which isessential for muscle development (102). It has also been sug-gested that ERK activity is important for muscle developmentand activation (36, 43, 101). In such phenomena seen in musclecells, ERK-dependent phosphorylation of AML1 may play apivotal role. Finally, it has been pointed out that AML1 stim-ulates polyomavirus DNA replication through binding to thePEBP2/CBF sites of the polyomavirus enhancer (9, 79). It isnot known whether AML1 is able to enhance DNA replication
in eukaryotic cells. However, stimulation of the growth of fi-broblast cells and their transformation by AML1 when over-expressed are compatible with the notion that AML1 enhancesDNA replication (42). In this system, we have shown that thepresence of ERK-dependent phosphorylation sites is impor-tant for AML1 to transform fibroblast cells, as mentionedabove. Such a finding is consistent with the facts that ERKpromotes mitogenesis in fibroblast cells (71) and that activa-tion of ERK is necessary for the transformation of NIH 3T3cells (13).In mammalian cells, two genes, AML2 (also called PEBP2aC,
CBFA3, or CBFa3) and AML3 (also called PEBP2aA, CBFA1,or CBFa1), show extensive sequence similarities to AML1.AML1, AML2, and AML3 show different expression patternsin various tissues and cell lines, suggesting that they havedivergent biological functions (5, 45, 54, 65, 96). We can pro-pose a potential mechanism for this functional difference. Ithas been found that AML2 has an internal deletion corre-sponding to exon 7B of AML1 (45, 55). This exon of AML1contains ERK-dependent phosphorylation sites which we haveidentified in the present study. Therefore, it is likely thatAML2 is not phosphorylated and thus that its transactivationactivity is not potentiated by ERK. In fact, we have observedthat AML2 does not show size shifts in SDS-PAGE, which areseen in the case of AML1 with dependence on phosphorylationin vivo, when ERK is activated (89). In contrast, the phosphor-ylation sites of AML1 are conserved also in AML3 (45, 65),suggesting that AML3 is regulated by ERK by a mechanismsimilar to that of AML1. The functional regulation of AML2and AML3 by ERK should be investigated further.What is the significance of ERK-AML1 physical interaction
irrespective of ERK activity and AML1 phosphorylation sites?It has been reported that ERK physically associates with sev-eral transcription factors which are phosphorylated by ERK,such as Elk-1, c-Myc, and c-Jun (7, 26, 75). At least the ERK–c-Myc interaction is independent of ERK activity and is not
FIG. 9. Colony formation assays with NIH 3T3 cells expressing AML1 and its mutants. NIH 3T3 cells were infected with the retroviral expression vector of AML1or its mutants (S249A, S266A, S249A/S266A, or T273A/S276A), selected by G418 resistance, seeded in soft agar, and cultured for 14 days. As a control, cells wereinfected with the empty retroviral vector and treated in the same procedure (Mock). (A) Photographs of representative culture dishes. (B) Colony count in eachcondition. Colonies were counted if they were larger than 0.25 mm in diameter. The bars show the means and standard deviations of colony counts (normalized to onesderived from 105 NIH 3T3 cells seeded in soft agar) which were obtained in three independent experiments.
3976 TANAKA ET AL. MOL. CELL. BIOL.

achieved fully by the c-Myc phosphorylation site alone (26).This interaction resembles the ERK-AML1 association iden-tified in this study. Recently, an attractive model about theassociation between MAPKs and their substrate transcriptionfactors, originally based on analyses of the JNK2–c-Jun inter-action (34, 35), has been proposed. According to this model,MAPK family members have docking elements, which partic-ipate in the binding to certain regions (probably outside phos-phoacceptor sites) of substrates, outside their catalytic units.This docking reaction facilitates the phosphorylation of sub-strate phosphoacceptors by the MAPK catalytic unit, resultingin enhanced specificity for phosphorylation of substrate tran-scription factors by a relevant MAPK when it is activated andenters the nuclei of cells (11, 25, 44). It has been reported thatthe AML1 protein is located in the nuclei within cells (48). Themode of ERK-AML1 interaction, clarified in the present study,is in good agreement with this model. Further investigationsshould be carried out to identify the ERK “docking site” andthe binding site of AML1. Alternatively, it is also possible thatERK indirectly associates with AML1 and that an unidentifiedfactor mediates such association.What mechanisms convert ERK-dependent phosphorylation
of AML1 into enhanced transactivation? It is possible thatsuch phosphorylation enhances AML1’s DNA-binding affinityor heterodimer formation with PEBP2b (CBFB or CBFb).However, the DNA-binding affinity is not remarkably alteredby the phosphorylation of AML1. In addition, we can predictthat heterodimer formation with PEBP2b (CBFB or CBFb) isnot significantly changed, since the heterodimer formation po-tentiates the DNA-binding affinity of AML1 (65, 98). From theresults of several recent studies focusing on enhancers of TCRgenes, it has been suggested that AML1 can manifest its fulltransactivator functions only when it coordinates the assemblyof a high-order enhancer complex (23, 29, 30, 84, 97). Forinstance, cooperative binding and transactivation by Ets-1 andAML1 are observed in the TCRb enhancer 3 and 4 regions(64, 84, 97), which we also used for the AML1 transactivationassay in this study. It is feasible that the phosphorylation ofAML1 by ERK regulates coordination with other transcriptionfactors.Our studies have identified, for the first time, a potential
regulatory mechanism for AML1 functions via the signal trans-duction pathways. Further analyses of AML1 functions andtheir regulatory mechanisms will provide essential insight intomany biological phenomena that are related to AML1 butwhich have not yet been adequately elucidated.
ACKNOWLEDGMENTS
We thank M. Ohki (National Cancer Center Research Institute,Tokyo, Japan) for providing us with AML1a cDNA, Y. Ito (KyotoUniversity, Kyoto, Japan) for his kind donation of the TCRb enhancergene, O. N. Witte (University of California, Los Angeles) for his gift ofthe pSRaMSVtkneo vector, R. Davis (Howard Hughes Medical Insti-tute, Boston, Mass.) for the JNK1 expression plasmid, and Y. Groner(The Weizmann Institute of Science, Rehovot, Israel) for AML2cDNA. We are also grateful to J. Nishida (Teikyo University, Tokyo,Japan), R. Sakai (Jichi Medical School, Tochigi, Japan), and K. Tobe(University of Tokyo, Tokyo, Japan) for helpful discussions, to N.Machiyama (University of Tokyo) for preparing the photographs, andto M. Shibazaki (University of Tokyo) for technical support.This work was supported in part by grants-in-aid from the Ministry
of Education, Science and Culture and from the Ministry of Healthand Welfare of Japan.
REFERENCES
1. Adams, P. D., and P. J. Parker. 1992. Activation of mitogen-activatedprotein (MAP) kinase by a MAP kinase-kinase. J. Biol. Chem. 267:13135–13137.
2. Alberola, I. J., K. A. Forbush, R. Seger, E. G. Krebs, and R. M. Perlmutter.1995. Selective requirement for MAP kinase activation in thymocyte dif-ferentiation. Nature (London) 373:620–623.
3. Alessandrini, A., C. M. Crews, and R. L. Erikson. 1992. Phorbol esterstimulates a protein-tyrosine/threonine kinase that phosphorylates and ac-tivates the Erk-1 gene product. Proc. Natl. Acad. Sci. USA 89:8200–8204.
4. Bae, S. C., E. Ogawa, M. Maruyama, H. Oka, M. Satake, K. Shigesada,N. A. Jenkins, D. J. Gilbert, N. G. Copeland, and Y. Ito. 1994. PEBP2aB/mouse AML1 consists of multiple isoforms that possess differential trans-activation potentials. Mol. Cell. Biol. 14:3242–3452.
5. Bae, S. C., E. Takahashi, Y. W. Zhang, E. Ogawa, K. Shigesada, Y. Namba,M. Sataka, and Y. Ito. 1995. Cloning, mapping and expression ofPEBP2aC, a third gene encoding the mammalian Runt domain. Gene159:245–248.
6. Bae, S. C., I. Y. Yamaguchi, E. Ogawa, M. Maruyama, M. Inuzuka, H.Kagoshima, K. Shigesada, M. Satake, and Y. Ito. 1993. Isolation ofPEBP2aB cDNA representing the mouse homolog of human acute myeloidleukemia gene, AML1. Oncogene 8:809–814.
7. Bernstein, L. R., D. K. Ferris, N. H. Colburn, and M. E. Sobel. 1994. Afamily of mitogen-activated protein kinase-related proteins interacts in vivowith activator protein-1 transcription factor. J. Biol. Chem. 269:9401–9404.
8. Boyle, W. J., P. van der Geer, and T. Hunter. 1991. Phosphopeptide map-ping and phosphoamino acid analysis by two-dimensional separation onthin-layer cellulose plates. Methods Enzymol. 201:110–149.
9. Chen, L. F., K. Ito, Y. Murakami, and Y. Ito. 1995. Stimulation of poly-omavirus DNA replication by a transcription factor PEBP2, abstr. 441, p.161. In Abstracts of the 54th Annual Meeting of the Japanese CancerAssociation 1995. Japanese Cancer Association, Tokyo.
10. Chen, R. H., C. Abate, and J. Blenis. 1993. Phosphorylation of the c-Fostransrepression domain by mitogen-activated protein kinase and 90-kDaribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 90:10952–10956.
11. Chen, R. H., C. Sarnecki, and J. Blenis. 1992. Nuclear localization andregulation of erk- and rsk-encoded protein kinases. Mol. Cell. Biol. 12:915–927.
12. Cheng, J. T., M. H. Cobb, and R. Baer. 1993. Phosphorylation of the TAL1oncoprotein by the extracellular-signal-regulated protein kinase ERK1.Mol. Cell. Biol. 13:801–808.
13. Cowley, S., H. Paterson, P. Kemp, and C. J. Marshall. 1994. Activation ofMAP kinase kinase is necessary and sufficient for PC12 differentiation andfor transformation of NIH3T3 cells. Cell 77:841–852.
14. Crossley, M., and S. H. Orkin. 1994. Phosphorylation of the erythroidtranscription factor GATA-1. J. Biol. Chem. 269:16589–16596.
15. Daga, A., J. E. Tighe, and F. Calabi. 1992. Leukaemia/Drosophila homol-ogy. Nature (London) 356:484.
16. Davis, R. J. 1993. The mitogen-activated protein kinase signal transductionpathway. J. Biol. Chem. 268:14553–14556.
17. Derijard, B., M. Hibi, I. H. Wu, T. Barrett, B. Su, T. Deng, M. Karin, andR. J. Davis. 1994. JNK1: a protein kinase stimulated by UV light andHa-Ras that binds and phosphorylates the c-Jun activation domain. Cell76:1025–1037.
18. Derijard, B., J. Raingeaud, T. Barrett, I. H. Wu, J. Han, R. J. Ulevitch, andR. J. Davis. 1995. Independent human MAP kinase signal transductionpathways defined by MEK and MKK isoforms. Science 267:682–685.
19. Erickson, P., J. Gao, K. S. Chang, T. Look, E. Whisenant, S. Raimondi, R.Lasher, J. Trujillo, J. Rowley, and H. Drabkin. 1992. Identification ofbreakpoints in t(8;21) acute myelogenous leukemia and isolation of a fusiontranscript, AML1/ETO, with similarity to Drosophila segmentation gene,runt. Blood 80:1825–1831.
20. Erickson, P., M. Robinson, G. Owens, and H. A. Drabkin. 1994. The ETOportion of acute myeloid leukemia t(8;21) fusion transcript encodes a highlyevolutionarily conserved, putative transcription factor. Cancer Res. 54:1782–1786.
21. Frank, R., J. Zhang, H. Uchida, S. Meyers, S. W. Hiebert, and S. D. Nimer.1995. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML1B. Oncogene 11:2667–274.
22. Furukawa, K., Y. Yamaguchi, E. Ogawa, K. Shigesada, M. Satake, and Y.Ito. 1990. A ubiquitous repressor interacting with an F9 cell-specific silencerand its functional suppression by differentiated cell-specific positive factors.Cell Growth Differ. 1:135–147.
23. Giese, K., C. Kingsley, J. R. Kirshner, and R. Grosschedl. 1995. Assemblyand function of a TCRa enhancer complex is dependent on LEF-1-inducedDNA bending and multiple protein-protein interactions. Genes Dev. 9:995–1008.
24. Golub, T. R., G. F. Barker, S. K. Bohlander, S. W. Hiebert, D. C. Ward,W. P. Bray, E. Morgan, S. C. Raimondi, J. D. Rowley, and D. G. Gilliland.1995. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 inacute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 92:4917–4921.
25. Gonzalez, F. A., A. Seth, D. L. Raden, D. S. Bowman, F. S. Fay, and R. J.
VOL. 16, 1996 ACTIVATED ERK PHOSPHORYLATES AML1 3977

Davis. 1993. Serum-induced translocation of mitogen-activated protein ki-nase to the cell surface ruffling membrane and the nucleus. J. Cell. Biol.122:1089–1101.
26. Gupta, S., and R. J. Davis. 1994. MAP kinase binds to the NH2-terminalactivation domain of c-Myc. FEBS Lett. 353:281–285.
27. Harlow, E., and D. Lane. 1988. Antibodies: a laboratory manual, p. 421–510. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
28. Hattori, S., M. Fukuda, T. Yamashita, S. Nakamura, Y. Gotoh, and E.Nishida. 1992. Activation of mitogen-activated protein kinase and its acti-vator by ras in intact cells and in a cell-free system. J. Biol. Chem. 267:20346–20351.
29. Hernandez, M. C., and M. S. Krangel. 1994. Regulation of the T-cellreceptor d enhancer by functional cooperation between c-Myb and core-binding factors. Mol. Cell. Biol. 14:473–483.
30. Hernandez, M. C., and M. S. Krangel. 1995. c-Myb and core-binding factor/PEBP2 display functional synergy but bind independently to adjacent sitesin the T-cell receptor d enhancer. Mol. Cell. Biol. 15:3090–3099.
31. Hill, C. S., and R. Treisman. 1995. Transcriptional regulation by extracel-lular signals: mechanisms and specificity. Cell 80:199–211.
32. Hunter, T., and M. Karin. 1992. The regulation of transcription by phos-phorylation. Cell 70:375–387.
33. Kagoshima, H., K. Shigesada, M. Satake, Y. Ito, H. Miyoshi, M. Ohki, M.Pepling, and P. Gergen. 1993. The Runt domain identifies a new family ofheteromeric transcriptional regulators. Trends Genet. 9:338–341.
34. Kallunki, T., B. Su, I. Tsigelny, H. K. Sluss, B. Derijard, G. Moore, R.Davis, and M. Karin. 1994. JNK2 contains a specificity-determining regionresponsible for efficient c-Jun binding and phosphorylation. Genes Dev.8:2996–3007.
35. Karin, M. 1995. The regulation of AP-1 activity by mitogen-activated pro-tein kinases. J. Biol. Chem. 270:16483–16486.
36. Khalil, R. A., C. B. Menice, C. L. Wang, and K. G. Morgan. 1995. Phos-photyrosine-dependent targeting of mitogen-activated protein kinase indifferentiated contractile vascular cells. Circ. Res. 76:1101–1108.
37. Kharbanda, S., A. Saleem, Y. Emoto, R. Stone, U. Rapp, and D. Kufe. 1994.Activation of Raf-1 and mitogen-activated protein kinase during monocyticdifferentiation of human myeloid leukemia cells. J. Biol. Chem. 269:872–878.
38. Kinoshita, T., T. Yokota, K. Arai, and A. Miyajima. 1995. Suppression ofapoptotic death in hematopoietic cells by signalling through the IL-3/GM-CSF receptors. EMBO J. 14:266–275.
39. Kolodziej, P. A., and R. A. Young. 1991. Epitope tagging and proteinsurveillance. Methods Enzymol. 194:508–519.
40. Kunkel, T. A., J. D. Roberts, and R. A. Zakour. 1987. Rapid and efficientsite-specific mutagenesis without phenotypic selection. Methods Enzymol.154:367–382.
41. Kurokawa, M., S. Ogawa, T. Tanaka, K. Mitani, Y. Yazaki, O. N. Witte, andH. Hirai. 1995. The AML1/Evi-1 fusion protein in the t(3;21) translocationexhibits transforming activity on Rat1 fibroblasts with dependence on Evi-1sequence. Oncogene 11:833–840.
42. Kurokawa, M., T. Tanaka, K. Tanaka, S. Ogawa, K. Mitani, Y. Yazaki, andH. Hirai. 1996. Overexpression of the AML1 proto-oncoprotein in NIH3T3cells leads to neoplastic transformation depending on the DNA-binding andtransactivational potencies. Oncogene 12:883–892.
43. LaBonne, C., B. Burke, and M. Whitman. 1995. Role of MAP kinase inmesoderm induction and axial patterning during Xenopus development.Development 121:1475–1486.
44. Lenormand, P., C. Sardet, G. Pages, G. L’Allemain, A. Brunet, and J.Pouyssegur. 1993. Growth factors induce nuclear translocation of MAPkinases (p42mapk and p44mapk) but not of their activator MAP kinasekinase (p45mapkk) in fibroblasts. J. Cell Biol. 122:1079–1088.
45. Levanon, D., V. Negreanu, Y. Bernstein, A. I. Bar, L. Avivi, and Y. Groner.1994. AML1, AML2, and AML3, the human members of the runt domaingene-family: cDNA structure, expression, and chromosomal localization.Genomics 23:425–432.
46. Liu, P., S. A. Tarle, A. Hajra, D. F. Claxton, P. Marlton, M. Freedman,M. J. Siciliano, and F. S. Collins. 1993. Fusion between transcription factorCBFb/PEBP2b and a myosin heavy chain in acute myeloid leukemia. Sci-ence 261:1041–1044.
47. Liu, P. P., A. Hajra, C. Wijmenga, and F. S. Collins. 1995. Molecularpathogenesis of the chromosome 16 inversion in the M4Eo. Blood 85:2289–2302.
48. Lu, J., M. Maruyama, M. Satake, S. C. Bae, E. Ogawa, H. Kagoshima, K.Shigesada, and Y. Ito. 1995. Subcellular localization of the a and b subunitsof the acute myeloid leukemia-linked transcription factor PEBP2/CBF.Mol. Cell. Biol. 15:1651–1661.
49. Marshall, C. J. 1995. Specificity of receptor tyrosine kinase signaling: tran-sient versus sustained extracellular signal-regulated kinase activation. Cell80:179–185.
50. Meyers, S., J. R. Downing, and S. W. Hiebert. 1993. Identification ofAML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: the runt homology domain is required for
DNA binding and protein-protein interactions. Mol. Cell. Biol. 13:6336–6345.
51. Meyers, S., N. Lenny, and S. W. Hiebert. 1995. The t(8;21) fusion proteininterferes with AML-1B-dependent transcriptional activation. Mol. Cell.Biol. 15:1974–1982.
52. Minden, A., A. Lin, M. McMahon, C. Lange-Carter, B. Derijard, R. J.Davis, G. L. Johnson, and M. Karin. 1994. Differential activation of ERKand JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science266:1719–1723.
53. Mitani, K., S. Ogawa, T. Tanaka, H. Miyoshi, M. Kurokawa, H. Mano, Y.Yazaki, M. Ohki, and H. Hirai. 1994. Generation of the AML1-EVI-1fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelo-cytic leukemia. EMBO J. 13:504–510.
54. Miyoshi, H., T. Kozu, K. Shimizu, K. Enomoto, N. Maseki, Y. Kaneko, N.Kamada, and M. Ohki. 1993. The t(8;21) translocation in acute myeloidleukemia results in production of an AML1-MTG8 fusion transcript.EMBO J. 12:2715–2721.
55. Miyoshi, H., M. Ohira, K. Shimizu, K. Mitani, H. Hirai, T. Imai, K.Yokoyama, E. Soeda, and M. Ohki. 1995. Alternative splicing and genomicstructure of the AML1 gene involved in acute myeloid leukemia. NucleicAcids Res. 23:2762–2769.
56. Miyoshi, H., K. Shimizu, T. Kozu, N. Maseki, Y. Kaneko, and M. Ohki.1991. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia areclustered within a limited region of a single gene, AML1. Proc. Natl. Acad.Sci. USA 88:10431–10434.
57. Muller, A. J., J. C. Young, A. M. Pendergast, M. Pondel, N. R. Landau,D. R. Littman, and O. N. Witte. 1991. BCR first exon sequences specificallyactivate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromo-some-positive human leukemias. Mol. Cell. Biol. 11:1785–1792.
58. Nuchprayoon, I., S. Meyers, L. M. Scott, J. Suzow, S. Hiebert, and A. D.Friedman. 1994. PEBP2/CBF, the murine homolog of the human myeloidAML1 and PEBP2b/CBFb proto-oncoproteins, regulates the murine my-eloperoxidase and neutrophil elastase genes in immature myeloid cells.Mol. Cell. Biol. 14:5558–5568.
59. Nucifora, G., C. R. Begy, P. Erickson, H. A. Drabkin, and J. D. Rowley.1993. The 3;21 translocation in myelodysplasia results in a fusion transcriptbetween the AML1 gene and the gene for EAP, a highly conserved proteinassociated with the Epstein-Barr virus small RNA EBER 1. Proc. Natl.Acad. Sci. USA 90:7784–7788.
60. Nucifora, G., C. R. Begy, H. Kobayashi, D. Roulston, D. Claxton, B. J. Ped-ersen, E. Parganas, J. N. Ihle, and J. D. Rowley. 1994. Consistent intergenicsplicing and production of multiple transcripts between AML1 at 21q22 andunrelated genes at 3q26 in (3;21)(q26;q22) translocations. Proc. Natl. Acad.Sci. USA 91:4004–4008.
61. Nucifora, G., D. J. Birn, R. Espinosa III, P. Erickson, M. M. LeBeau, D.Roulston, T. W. McKeithan, H. Drabkin, and J. D. Rowley. 1993. Involve-ment of the AML1 gene in the t(3;21) in therapy-related leukemia and inchronic myeloid leukemia in blast crisis. Blood 81:2728–2734.
62. Nucifora, G., and J. D. Rowley. 1995. AML1 and the 8;21 and 3;21 trans-locations in acute and chronic myeloid leukemia. Blood 86:1–14.
63. Ogawa, E., M. Inuzuka, M. Maruyama, M. Satake, F. M. Naito, Y. Ito, andK. Shigesada. 1993. Molecular cloning and characterization of PEBP2b,the heterodimeric partner of a novel Drosophila runt-related DNA bindingprotein PEBP2a. Virology 194:314–331.
64. Ogawa, E., M. Maruyama, L. F. Chen, Y. Murakami, and Y. Ito. 1995.Analysis of the mechanism of transcription activation by PEBP2, abstr. 713,p. 230. In Abstracts of the 54th Annual Meeting of the Japanese CancerAssociation 1995. Japanese Cancer Association, Tokyo.
65. Ogawa, E., M. Maruyama, H. Kagoshima, M. Inuzuka, J. Lu, M. Satake, K.Shigesada, and Y. Ito. 1993. PEBP2/PEA2 represents a family of transcrip-tion factors homologous to the products of the Drosophila runt gene and thehuman AML1 gene. Proc. Natl. Acad. Sci. USA 90:6859–6863.
66. Ohki, M. 1993. Molecular basis of the t(8;21) translocation in acute myeloidleukaemia. Semin. Cancer Biol. 4:369–375.
67. Okazaki, K., and N. Sagata. 1995. The Mos/MAP kinase pathway stabilizesc-Fos by phosphorylation and augments its transforming activity in NIH3T3cells. EMBO J. 14:5048–5059.
68. Okazaki, K., and N. Sagata. 1995. MAP kinase activation is essential foroncogenic transformation of NIH3T3 cells by Mos. Oncogene 10:1149–1157.
69. Okuda, K., J. S. Sanghera, S. L. Pelech, Y. Kanakura, M. Hallek, J. D.Griffin, and B. J. Druker. 1992. Granulocyte-macrophage colony-stimulat-ing factor, interleukin-3, and steel factor induce rapid tyrosine phosphory-lation of p42 and p44 MAP kinase. Blood 79:2880–2887.
70. Okuda, T., J. Deursen, S. W. Hiebert, G. Grosbeld, and J. R. Downing.1996. AML1, the target of multiple chromosomal translocations in humanleukemia, is essential for normal fetal liver hematopoiesis. Cell 84:321–330.
71. Pages, G., P. Lenormand, G. L’Allemain, J. C. Chambard, S. Meloche, andJ. Pouyssegur. 1993. Mitogen-activated protein kinases p42mapk andp44mapk are required for fibroblast proliferation. Proc. Natl. Acad. Sci.USA 90:8319–8323.
72. Pulverer, B. J., J. M. Kyriakis, J. Avruch, E. Nikolakaki, and J. R.
3978 TANAKA ET AL. MOL. CELL. BIOL.

Woodgett. 1991. Phosphorylation of c-jun mediated by MAP kinases. Na-ture (London) 353:670–674.
73. Rabault, B., and J. Ghysdael. 1994. Calcium-induced phosphorylation ofETS1 inhibits its specific DNA binding activity. J. Biol. Chem. 269:28143–18151.
74. Raingeaud, J., A. J. Whitmarsh, T. Barrett, B. Derijard, and R. J. Davis.1996. MKK3- and MKK6-regulated gene expression is mediated by the p38mitogen-activated protein kinase signal transduction pathway. Mol. Cell.Biol. 16:1247–1255.
75. Rao, V. N., and E. S. Reddy. 1994. Elk-1 proteins interact with MAPkinases. Oncogene 9:1855–1860.
76. Robbins, D. J., E. Zhen, H. Owaki, C. A. Vanderbilt, D. Ebert, T. D.Geppert, and M. H. Cobb. 1993. Regulation and properties of extracellularsignal-regulated protein kinases 1 and 2 in vitro. J. Biol. Chem. 268:5097–5106.
77. Romana, S. P., M. Mauchauffe, C. M. Le, I. Chumakov, P. D. Le, R. Berger,and O. A. Bernard. 1995. The t(12;21) of acute lymphoblastic leukemiaresults in a tel-AML1 gene fusion. Blood 85:3662–3670.
78. Rudnicki, M. A., and M. W. McBurney. 1987. Cell culture methods andinduction of differentiation of embryonal carcinoma cell lines, p. 19–49. InE. J. Robertson (ed.), Teratocarcinomas and embryonic stem cells. IRLPress, Oxford.
79. Rutberg, S. E., L. R. Dolan, and Z. Ronai. 1994. PEBP2—a modulator ofpolyoma DNA replication. DNA Cell Biol. 13:865–874.
80. Sakakura, C., I. Y. Yamaguchi, M. Satake, S. C. Bae, A. Takahashi, E.Ogawa, A. Hagiwara, T. Takahashi, A. Murakami, K. Makino, T. Naka-gawa, N. Kamada and Y. Ito. 1994. Growth inhibition and induction ofdifferentiation of t(8;21) acute myeloid leukemia cells by the DNA-bindingdomain of PEBP2 and the AML1/MTG8(ETO)-specific antisense oligonu-cleotide. Proc. Natl. Acad. Sci. USA 91:11723–11727.
81. Satake, M., S. Nomura, I. Y. Yamaguchi, Y. Takahama, Y. Hashimoto, M.Niki, Y. Kitamura, and Y. Ito. 1995. Expression of the Runt domain-encoding PEBP2a genes in T cells during thymic development. Mol. Cell.Biol. 15:1662–1670.
82. Sato, N., K. Sakamaki, N. Terada, K. Arai, and A. Miyajima. 1993. Signaltransduction by the high-affinity GM-CSF receptor: two distinct cytoplasmicregions of the common b subunit responsible for different signaling. EMBOJ. 12:4181–4189.
83. Shirakabe, K., Y. Gotoh, and E. Nishida. 1992. A mitogen-activated protein(MAP) kinase activating factor in mammalian mitogen-stimulated cells ishomologous to Xenopus M phase MAP kinase activator. J. Biol. Chem.267:16685–16690.
84. Sun, W., B. J. Graves, and N. A. Speck. 1995. Transactivation of theMoloney murine leukemia virus and T-cell receptor b-chain enhancers byCbf and Ets requires intact binding sites for both proteins. J. Virol. 69:4941–4949.
85. Takahashi, A., M. Satake, I. Y. Yamaguchi, S. C. Bae, J. Lu, M. Maruyama,Y. W. Zhang, H. Oka, N. Arai, K. Arai, and Y. Ito. 1995. Positive andnegative regulation of granulocyte-macrophage colony-stimulating factorpromoter activity by AML1-related transcription factor, PEBP2. Blood86:607–616.
86. Tanaka, K., T. Tanaka, S. Ogawa, M. Kurokawa, K. Mitani, Y. Yazaki, andH. Hirai. 1995. Increased expression of AML1 during retinoic-acid-induceddifferentiation of U937 cells. Biochem. Biophys. Res. Commun. 211:1023–1030.
87. Tanaka, T., K. Mitani, M. Kurokawa, S. Ogawa, K. Tanaka, J. Nishida, Y.Yazaki, Y. Shibata, and H. Hirai. 1995. Dual functions of the AML1/Evi-1chimeric protein in the mechanism of leukemogenesis in t(3;21) leukemias.
Mol. Cell. Biol. 15:2383–2392.88. Tanaka, T., J. Nishida, K. Mitani, S. Ogawa, Y. Yazaki, and H. Hirai. 1994.
Evi-1 raises AP-1 activity and stimulates c-fos promoter transactivation withdependence on the second zinc finger domain. J. Biol. Chem. 269:24020–24026.
89. Tanaka, T., K. Tanaka, M. Kurokawa, and H. Hirai. Unpublished data.90. Tanaka, T., K. Tanaka, S. Ogawa, M. Kurokawa, K. Mitani, J. Nishida, Y.
Shibata, Y. Yazaki, and H. Hirai. 1995. An acute myeloid leukemia gene,AML1, regulates hematopoietic myeloid cell differentiation and transcrip-tional activation antagonistically by two alternative spliced forms. EMBO J.14:341–350.
91. Tobe, K., T. Kadowaki, H. Tamemoto, K. Ueki, K. Hara, O. Koshio, K.Momomura, Y. Gotoh, E. Nishida, Y. Akanuma, Y. Yazaki, and M. Kasuga.1991. Insulin and 12-O-tetradecanoylphorbol-13-acetate activation of twoimmunologically distinct myelin basic protein/microtubule-associated pro-tein 2 (MBP/MAP2) kinases via de novo phosphorylation of threonine andtyrosine residues. J. Biol. Chem. 266:24793–24803.
92. Towatari, M., G. E. May, R. Marais, G. R. Perkins, C. J. Marshall, S.Cowley, and T. Enver. 1995. Regulation of GATA-2 phosphorylation bymitogen-activated protein kinase and interleukin-3. J. Biol. Chem. 270:4101–4107.
93. Ueki, K., S. Matsuda, K. Tobe, Y. Gotoh, H. Tamemoto, M. Yachi, Y.Akanuma, Y. Yazaki, E. Nishida, and T. Kadowaki. 1994. Feedback regu-lation of mitogen-activated protein kinase kinase kinase activity of c-Raf-1by insulin and phorbol ester stimulation. J. Biol. Chem. 269:15756–15761.
94. Wang, S., Q. Wang, B. E. Crute, I. N. Melnikova, S. R. Keller, and N. A.Speck. 1993. Cloning and characterization of subunits of the T-cell receptorand murine leukemia virus enhancer core-binding factor. Mol. Cell. Biol.13:3324–3339.
95. Wang, S. W., and N. A. Speck. 1992. Purification of core-binding factor, aprotein that binds the conserved core site in murine leukemia virus enhanc-ers. Mol. Cell. Biol. 12:89–102.
96. Wijmenga, C., N. A. Speck, N. C. Dracopoli, M. H. Hofker, P. Liu, and F. S.Collins. 1995. Identification of a new murine runt domain-containing gene,Cbfa3. Genomics 26:611–614.
97. Wotton, D., J. Ghysdael, S. Wang, N. A. Speck, and M. J. Owen. 1994.Cooperative binding of Ets-1 and core binding factor to DNA. Mol. Cell.Biol. 14:840–850.
98. Zaiman, A. L., A. F. Lewis, B. E. Crute, N. A. Speck, and J. Lenz. 1995.Transcriptional activity of core binding factor a (AML1) and b subunits onmurine leukemia virus enhancer cores. J. Virol. 69:2898–2906.
99. Zhang, D. E., K. Fujioka, C. J. Hetherington, L. H. Shapiro, H. M. Chen,A. T. Look, and D. G. Tenen. 1994. Identification of a region which directsthe monocytic activity of the colony-stimulating factor 1 (macrophage col-ony-stimulating factor) receptor promoter and binds PEBP2/CBF (AML1).Mol. Cell. Biol. 14:8085–8095.
100. Zhang, D. E., C. J. Hetherington, S. Meyers, K. L. Rhoades, C. J. Larson,H. M. Chen, S. W. Hiebert, and D. G. Tenen. 1996. CCAAT enhancer-binding protein (C/EBP) and AML1 (CBFa2) synergistically activate themacrophage colony-stimulating factor receptor promoter. Mol. Cell. Biol.16:1231–1240.
101. Zhang, Y., S. Moreland, and R. S. Moreland. 1994. Regulation of vascularsmooth muscle contraction: myosin light chain phosphorylation dependentand independent pathways. Can. J. Physiol. Pharmacol. 72:1386–1391.
102. Zhu, X., J. E. Yeadon, and S, J. Burden. 1994. AML1 is expressed inskeletal muscle and is regulated by innervation. Mol. Cell. Biol. 14:8051–8057.
VOL. 16, 1996 ACTIVATED ERK PHOSPHORYLATES AML1 3979