the long intergenic non-coding rna (lincrna) … · 26 long intergenic non-coding rnas (lincrnas)...
TRANSCRIPT

1
Short title: 1
Long non-coding RNA landscape of soybean genome 2
Corresponding authors: 3
Prem L. Bhalla 4
Email: [email protected] 5
Tel. +61 03 8344 9651 6
Full title: 7
The long intergenic non-coding RNA (lincRNA) landscape of the 8
soybean genome 9
Authors: 10
Agnieszka A. Golicz1 11
Mohan B. Singh1 12
Prem L. Bhalla1 13
1Plant Molecular Biology and Biotechnology Laboratory, Faculty of Veterinary and 14
Agricultural Sciences, University of Melbourne, Parkville, Melbourne, VIC, Australia. 15
One sentence summary: 16
The soybean genome encodes over 6,000 long intergenic non-coding RNAs implicated in 17
many biological processes including transcription, development and possibly influencing 18
agronomic traits. 19
Author contributions: 20
AAG: Designed the experiments, performed the analysis, wrote the manuscript; MBS: 21
Conceived research, wrote the manuscript; PLB: Conceived research, wrote the manuscript 22
Funding: Financial support for this work was obtained from the ARC Discovery Grant ARC 23
DP0988972 24
Plant Physiology Preview. Published on December 28, 2017, as DOI:10.1104/pp.17.01657
Copyright 2017 by the American Society of Plant Biologists
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

2
Abstract 25
Long intergenic non-coding RNAs (lincRNAs) are emerging as important regulators of 26
diverse biological processes. However, our understanding of lincRNA abundance and 27
function remains very limited especially for agriculturally important plants. Soybean is a 28
major legume crop plant providing over a half of global oilseed production. Moreover, 29
soybean can form symbiotic relationships with Rhizobium bacteria to fix atmospheric 30
nitrogen. Soybean has a complex paleopolyploid genome and exhibits many vegetative and 31
floral development complexities. Soybean cultivars have photoperiod requirements restricting 32
its use and productivity. Molecular regulators of these legume-specific developmental 33
processes remain enigmatic. Long non-coding RNAs may play important regulatory roles in 34
soybean growth and development. In this study over one billion RNASeq read pairs from 37 35
samples representing nine tissues were used to discover 6,018 lincRNA loci. The lincRNAs 36
were shorter than protein-coding transcripts, had lower expression levels and more sample 37
specific expression. Few of the loci were found to be conserved in two other legume species 38
(chickpea and Medicago), but almost two hundred homeologous lincRNA in the soybean 39
genome were detected. Protein-coding gene-lincRNA co-expression analysis suggested an 40
involvement of lincRNAs in stress response, signal transduction and developmental 41
processes. Positional analysis of lincRNA loci implicated involvement in transcriptional 42
regulation. lincRNA expression from centromeric regions was observed especially in actively 43
dividing tissues suggesting possible roles in cell division. Integration of publicly available 44
genome-wide association data with the lincRNA map of the soybean genome uncovered 23 45
lincRNAs potentially associated with agronomic traits. 46
Introduction 47
Recently, it has been elucidated that eukaryotic genomes, including plant genomes, encode a 48
multitude of non-coding RNAs (ncRNAs) (Chekanova et al., 2007; Kapranov et al., 2007). 49
One class of ncRNAs are long non-coding RNAs (lncRNAs), which are defined as transcripts 50
>200 bp in length and harbouring no discernible coding potential (Jin et al., 2013; Wang et 51
al., 2014; Chekanova, 2015). The relative location of lncRNAs to protein-coding genes 52
identifies a further subgroup known as long intergenic noncoding RNAs – lincRNAs, which 53
do not overlap protein-coding genes. LncRNAs were long considered little beyond 54
transcriptional noise; however current evidence points to important roles in diverse biological 55
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

3
processes across eukaryotes (Van Werven et al., 2012; Ulitsky and Bartel, 2013; Flynn and 56
Chang, 2014). In Arabidopsis and rice s have been shown to be involved in flowering time 57
regulation, reproduction and root organogenesis (Swiezewski et al., 2009; Cifuentes-Rojas et 58
al., 2011; Heo and Sung, 2011; Ariel et al., 2014; Bardou et al., 2014; Matzke et al., 2014; 59
Wang et al., 2014; Zhang et al., 2014; Berry and Dean, 2015; Khemka et al., 2016). LncRNA 60
are found both in the nucleus and cytoplasm, which suggests a diversity of modes of action, 61
including chromatin modification (Heo et al., 2013), acting as decoys preventing access of 62
regulatory proteins, including splicing machinery, and miRNAs to their true RNA and DNA 63
targets (Franco-Zorrilla et al., 2007; Wu et al., 2013; Bardou et al., 2014) and acting as 64
scaffolds for assembly of larger protein-RNA complexes (Lai et al., 2013; Pefanis et al., 65
2015). Recently, a large number of lncRNAs has been found to be associated with ribosomes 66
and co-expressed with ribosomal proteins, although not translated, which suggests possible 67
roles in translation regulation (Carlevaro-Fita et al., 2016). Despite increasing efforts (Jin et 68
al., 2013; Szcześniak et al., 2016) plant specific lncRNA databases are scarce, and lncRNA 69
genome wide discovery and especially functional annotation in agriculture important plant 70
species remain unavailable. 71
Legumes are a large family of plant species characterized by butterfly-like flowers and pod-72
shaped shaped fruits. They provide an invaluable contribution to ecosystems due to their 73
ability to form symbiotic relationships with Rhizobium bacteria. This symbiosis results in 74
dinitrogen capture from the air and its subsequent fixation, making legumes one of the major 75
sources of bioavailable nitrogen. Legume seeds are a second, after cereals, source of human 76
and animal food and include soybeans (Glycine max), peanuts (Arachis hypogaea), garden 77
peas (Pisum sativum), and broad beans (Vici faba). Additionally, soybean is responsible for 78
over a half of global oilseed production. Due to its economic importance as a source of food 79
and oils soybean has increasingly become a target of genomic and transcriptomic research 80
efforts. Sequencing of the soybean genome revealed its complex paleopolyploid structure 81
(Schmutz et al., 2010). Although comparison between soybean and the model plant species 82
Arabidopsis thaliana can be drawn, the two species are suggested to have diverged from a 83
common ancestor 92 million years ago (Zhu et al., 2003) and soybean has undergone at least 84
two genome duplication events resulting in homeologous relationships between 85
chromosomes and gene loci (Shoemaker et al., 2006). One of the most interesting questions 86
from the genomics point of view is ‘Which genomic features of soybean define its 87
characteristics and are responsible for its vegetative and floral complexities? Considering that 88
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

4
many of the key developmental control genes in soybean exist in multiple copies, a complex 89
interplay and additional control for ‘fine-tuning’ are expected. Our recent awareness of 90
prevalence and importance of long non-coding RNAs highlight that these may play important 91
regulatory roles in soybean growth and development. lncRNAs could provide the additional 92
level of control and signal integration, which is missing when only protein-coding genes are 93
considered. 94
This study presents first genome-wide discovery, characterization and functional annotation 95
of long intergenic non-coding RNAs in the soybean genome. Genome-wide lincRNA 96
discovery was performed using a combination of de novo, and reference guided assembly 97
approaches generating a most comprehensive lincRNA database. Comparative analysis 98
between soybean lincRNAs and other legume species was performed to identify lincRNAs 99
which could play universal roles in all legumes and the lincRNAs which are soybean-100
specific. Functional analysis was conducted to uncover biological processes that could be 101
influenced by lincRNA action. Finally, publicly available genome-wide association data was 102
used to further characterize the lincRNAs discovered and find potential links to agronomic 103
traits. 104
Results and discussion 105
Genome-wide discovery of 6,018 long non-coding intergenic loci 106
lincRNAs are a class of RNA molecules, which are >200 bp long and have no discernible 107
coding potential. High throughput technologies offer an opportunity for both coding and 108
noncoding transcript detection and quantification. In total 1,025,323,161 read pairs from 37 109
soybean samples were used in the analysis. The soybean sampled tissues included 28 samples 110
representing stem (germination and trefoil stage), flower (flower bud, unopened flower, 111
florescence and five days after flowering), leaf bud (germination, trefoil and differentiation 112
stage), leaf (trefoil, flower bud differentiation stage and senescent leaves), pod (three, four 113
and five weeks), seed (three, five, six, eight and 10 weeks) seed and pod (two, three and four 114
weeks), shoot meristem (flower bud differentiation stage), cotyledon (germination and trefoil 115
stage) and root (Shen et al., 2014). Additionally, nine samples (four from leaf tissue and five 116
from shoot apical meristem tissue (SAM)) representing time points during the floral transition 117
period following short day treatment (Wong et al., 2013) were used. Both de novo and 118
reference guided transcriptome assembly strategies were applied. StringTie reference guided 119
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

5
assembly resulted in 68,190 loci and 160,337 transcripts. Trinity assemblies rendered 120
448,338 transcripts using de novo and 337,955 transcripts using reference guided approach. 121
The PASA comprehensive transcript database built using StringTie and Trinity assemblies 122
comprised 147,825 loci and 293,537 transcripts. Both StringTie and PASA annotations were 123
subjected to lincRNA discovery pipeline and PASA derived lincRNAs, which did not appear 124
in StringTie annotation were used to supplement StringTie derived lincRNAs (Fig. S1). Loci 125
were considered to encode lincRNAs if they did not produce any protein-coding transcripts 126
(ORF size ≤ 100 amino acids and no similarity to protein-coding genes) and did not overlap 127
any protein-coding loci. The lincRNAs were filtered to remove loci producing transcripts 128
with similarity to tRNAs, rRNAs and snoRNAs (58 loci) found in Rfam database, transcripts 129
which were nested (entirety contained) within other lincRNAs (63 loci), transcripts which 130
overlapped protein-coding genes in Gmax_275_v2.0 genome annotation (126 loci). 131
lincRNAs are known to be expressed at low levels (Li et al., 2014; Zhang et al., 2014; Hao et 132
al., 2015). Choosing an expression cut-off requires balancing a trade-off between retaining 133
the largest possible set of lincRNAs and discarding the spurious transcription and mapping 134
artefacts. Two lincRNA sets were generated. The larger set (9,766 loci) with a permissive 135
cut-off >0.1 FPKM in a least one of the samples (Table S2) and a filtered set generated using 136
more stringent FPKM cut-off (≥1.0 FPKM in at least one of samples or ≥0.5 FPKM in at 137
least two samples or gene size of at least 1000 bp). The filtered lincRNA set consisted of 138
6,018 lincRNA loci (6,134 transcripts), including 3,435 StringTie derived and 2,583 PASA 139
derived loci (Table S3). The full set is provided for the benefit of the readers but only the 140
filtered lincRNA set was used in the analysis. 141
lincRNAs have distinct properties when compared to protein-coding genes 142
The lincRNA and protein-coding loci were examined for main gene characteristics. The 143
lincRNA transcripts were on average shorter than protein-coding transcripts (Fig. 1A). The 144
median length of lincRNA transcripts was 320 bp (mean: 467.3 bp), whereas the median 145
length of protein-coding transcripts was 3,657 bp (mean: 4,450 bp). The lincRNA transcripts 146
contained a lower number of exons than protein-coding transcripts (Fig. 1C). The majority of 147
lincRNA transcripts (90.3%) contained a single exon. The maximum number of exons found 148
in a lincRNA transcript was 4. The lincRNA genes had a lower number of isoforms compared 149
to protein-coding genes (Fig. 1B). A vast majority of lincRNA genes (98.5%) had a single 150
isoform. Finally, lincRNAs showed lower overall expression levels compared to coding 151
genes (Fig. 1D). The observations are consistent with lincRNA studies in other plant species. 152
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.
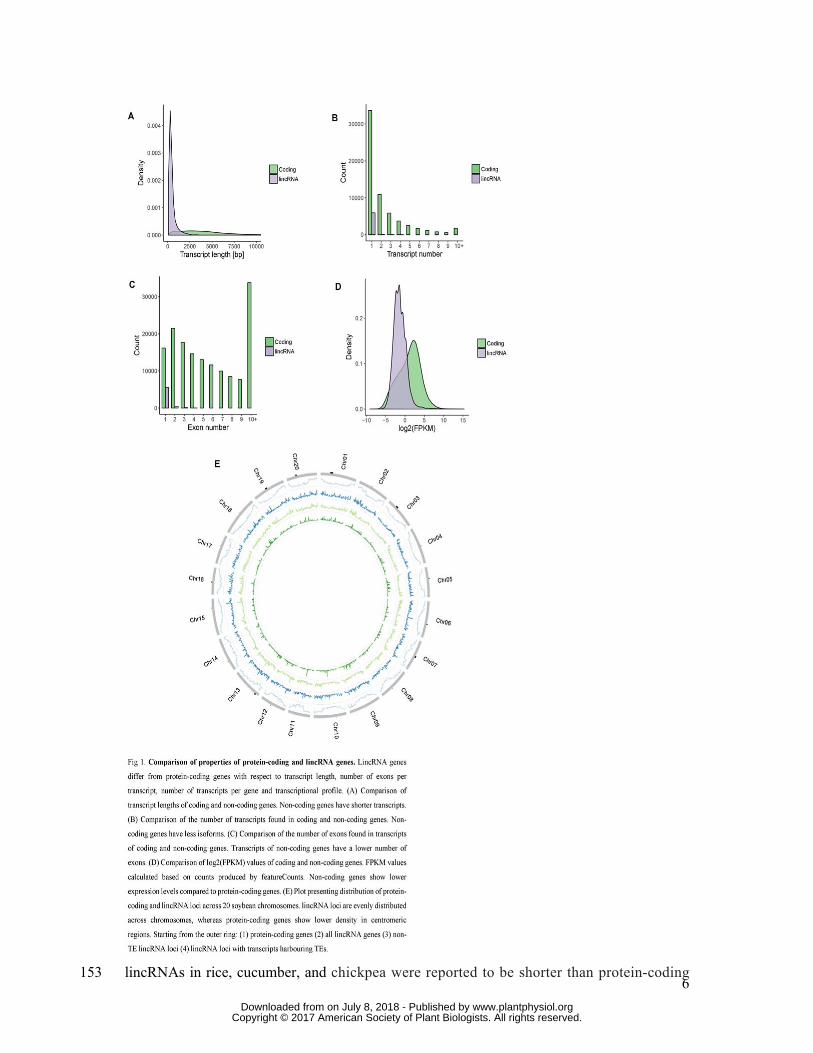
6
lincRNAs in rice, cucumber, and chickpea were reported to be shorter than protein-coding 153
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

7
genes (Zhang et al., 2014; Hao et al., 2015; Khemka et al., 2016). lincRNAs in cucumber, 154
maize, and chickpea were reported to have predominantly one exon only (Li et al., 2014; 155
Hao et al., 2015; Khemka et al., 2016). Also, low expression levels of lincRNAs were 156
observed in Arabidopsis, rice and maize (Liu et al., 2012; Li et al., 2014; Zhang et al., 157
2014; Hao et al., 2015). Although usually lacking sequence homology (Hao et al., 2015; 158
Mohammadin et al., 2015; Wang et al., 2015) lincRNAs appear to share similar 159
characteristics across different species which include short length, a low number of exons 160
and splice variants. 161
Centromeric regions of soybean chromosomes show lincRNA expression 162
The distribution of lincRNAs across chromosomes can provide clues regarding possible 163
functions and mechanisms of action. For example, lincRNAs located among protein-164
coding genes could modulate expression of their neighbours, while lincRNAs found close 165
to centromeres or in gene deserts may act distally or have additional roles. Centromeric 166
regions of soybean chromosomes are enriched in transposable elements (TEs) and depleted 167
in protein-coding loci (Schmutz et al., 2010). In contrast, lincRNA loci display an even 168
distribution across chromosomes (Fig. 1E), with active transcription from centromeric 169
regions. lincRNAs transcribed from centromeric regions have been implicated to play roles 170
in centromere maintenance and cellular division (Rošić and Erhardt, 2016). In total, 32 171
centromeric (as defined by transcription from regions delimited by GmCent-1 and 172
GmCent-2 repeats) lincRNAs on chromosomes 1,3,5,7,13,16,17,19 were identified. The 173
number of lincRNAs identified was weakly positively correlated with the identified 174
centromere size (rho=0.25). No centromeric lincRNAs were expressed in all the samples, 175
and the median number of samples showing centromeric lincRNA expression was seven. 176
The median expression value in samples which expressed centromeric lincRNA (FPKM > 177
0.1) was 0.31 FPKM. Centromeric lincRNAs showed higher transcriptional activity in 178
actively dividing tissues (flower bud, leaf bud and shoot apical meristem, Mann-Witney U 179
test, p-value < 0.01), (Fig. 2B). The most common transposable element type found within 180
centromeric lincRNAs was LTR Gypsy retrotransposon (Fig. 2C) which is consistent with 181
high prevalence of Gypsy transposable elements in the vicinity of centromeres (Schmutz et 182
al., 2010). 183
Although centromeric lincRNA expression was observed, similarly to rice and maize 184
(Wang et al., 2015), the majority of lincRNAs were found relatively close to neighbouring 185
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

8
protein-coding genes. The median distance from lincRNA to protein-coding gene was 186
1,064 bp (mean distance: 3,497 bp). LincRNAs found a short distance from protein-coding 187
genes could modulate their expression by actively recruiting activators, repressors, 188
epigenetic modifiers or simply by transcription from the lincRNA locus (Wang and Chang, 189
2011; Kornienko et al., 2013). 190
Nearly a fifth of lincRNA transcripts has sequence similarity to transposable elements 191
The relatively high abundance of lincRNAs proximal to centromeres sparked an 192
investigation of the contribution of transposable elements to lincRNA transcript 193
composition. In total, 18.3% of lincRNA transcripts were predicted to harbor TEs, and a 194
higher proportion of lincRNAs than coding transcripts (10.8%) contained TEs. For 195
transcripts, which harboured TEs, TEs contributed a larger amount of sequence to 196
lincRNAs (median lincRNA coverage by TEs was 100%, mean: 82.8%) than to protein-197
coding transcripts (median coding transcript coverage by TEs: 19%, mean: 36.5%). A 198
similar pattern was observed in the human genome, where 2/3 of mature noncoding 199
transcripts showed similarity to TEs and TEs were found to contribute signals essential for 200
biogenesis of many lncRNAs (Kapusta et al., 2013). The lincRNAs were found harbor 201
more retrotransposons than DNA transposons (Fig. 2A), which reflects the overall TE 202
landscape of the soybean genome (Du et al., 2010; Schmutz et al., 2010). 203
Soybean lincRNAs have low levels of sequence and positional conservation in chickpea 204
and Medicago 205
Information on about conservation of lincRNAs across species can provide further inputs 206
regarding their possible functions and the processes they are involved in. If a lincRNA is well 207
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

9
conserved in a number of species, it can be assumed to play a generally important role. 208
Conversely, if a lincRNA is species specific, it may play a role unique to given organism or 209
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

10
provide a modulatory function which alters the otherwise conserved system. It has been noted 210
that the sequence conservation of lincRNAs is much lower than protein-coding genes (Hao et 211
al., 2015; Mohammadin et al., 2015), but higher levels of positional based conservation have 212
been postulated (Mohammadin et al., 2015; Wang et al., 2015). In total 6,018 soybean, 2,248 213
chickpea, 5,794 Medicago, and 6,480 Arabidopsis lincRNA were available for analysis. 214
Reciprocal best BLAST (RBB) comparison uncovered 143 soybean lincRNAs which have 215
sequence similarity to lincRNA in other species, with 4 lincRNAs showing similarity to 216
lincRNAs in both chickpea and Medicago. Because different tissue samples and discovery 217
pipelines were used, it is possible that some conserved lincRNA pairs were missed. To 218
address this soybean lincRNAs were compared against full genome assemblies which 219
resulted in the discovery of 787 additional loci with sequence similarity to genomes of other 220
species (Fig. 3A). Those could correspond to un-annotated non-coding transcripts. However, 221
in the absence of evidence of transcription, their function remains unknown, and those loci 222
were not be considered in further analysis. 223
Positional conservation between lncRNA loci has been suggested to extend across longer 224
evolutionary distances than sequence conservation (Mohammadin et al., 2015; Wang et al., 225
2015). A long non-coding RNA is often considered positionally conserved if found in the 226
same orientation (upstream or downstream) relative to orthologous protein-coding gene in at 227
least two species (Mohammadin et al., 2015; Wang et al., 2015). If the direction of 228
transcription of lincRNA is known, transcription from the same strand is also required. 229
However, it is conceivable that if a large number of lincRNAs is considered, a number of 230
those will show positional similarity across species (found in the same orientation relative to 231
protein-coding genes) by chance only, rather than as a result of evolutionary conservation. To 232
test this, the number of soybean lincRNAs which had positional similarity with chickpea, 233
Medicago, and A. thaliana lincRNAs was compared with control datasets constructed by 234
random redistribution of lincRNAs across genomes of all four species. Two properties of 235
lincRNA loci were considered while constructing the control datasets: (1) A proportion of 236
lincRNA loci is found in clusters of two or more loci (mirroring this property the in control 237
datasets will result in a more realistic distribution of lincRNA), (2) lincRNA loci are enriched 238
proximal to transcription factors (uneven distribution of lincRNAs relative to transcription 239
factors could affect the results if transcription factors are preferentially retained or lost from 240
syntenic regions). To accommodate those, four types of control datasets (5 simulations each, 241
20 datasets in total) were constructed: (1) Random re-distribution of lincRNA relative to 242
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

11
protein-coding loci, (2) Random re-distribution of lincRNA relative to protein-coding loci, 243
but maintaining the proportion of lincRNA found adjacent to transcription factors (3) 244
Random re-distribution of existing lincRNA clusters relative to protein-coding loci, (4) 245
Random re-distribution of existing lincRNA clusters relative to protein-coding loci, but 246
maintaining the proportion of lincRNA found adjacent to transcription factors. The true 247
biological lincRNA dataset and the simulated control datasets were analysed using the same 248
positional conservation discovery pipeline. The number of positionally similar lincRNAs in 249
the biological dataset (1,201) and the simulation datasets 1 and 2 were not significantly 250
different (Fisher test, p-value > 0.01 for majority comparisons, Fig. 3B). However, more 251
positionally similar lincRNAs were found in the biological dataset when compared to 252
simulation datasets 3 and 4 (Fisher test, p-value < 0.01 for all comparisons). Although, 253
comparison with simulation datasets 3 and 4 suggest that positional similarity observed is 254
somewhat higher than by chance alone the difference is not large (Fig. 3B). Results of 255
analyses of positional conservation ought to be interpreted with caution, especially across 256
larger evolutionary distances, and considered in conjunction with sequence similarity and 257
analysis of expression patterns. 258
Strong support for positional conservation of lincRNAs rather than chance positional 259
similarity would be any sequence similarity between transcripts. Comparison of positionally 260
similar transcript pairs uncovered 48 soybean lincRNAs which show positional similarity and 261
sequence similarity with other species. Sequence comparison of the positionally similar 262
lincRNA pairs in simulated datasets (100 simulated datasets using random re-distribution of 263
lincRNA relative to protein-coding loci) showed them to have no sequence similarity (median 264
number of pairs with sequence similarity per dataset: 0), suggesting that the sequence 265
similarity observed was not due to chance alone (permutation test, p-value < 0.01). 266
Subsequently, the 48 loci were analysed in more detail. Protein-coding genes and short RNA 267
primary transcripts are known to have higher conservation levels than long non-coding RNAs 268
(Hezroni et al., 2015). Some lncRNAs are known to be sRNA precursors. The 48 putative 269
conserved lincRNAs were inspected to check whether they (1) show similarity to 270
transposable elements (TEs), (2) could encode small conserved peptides which would be 271
missed by the lincRNA discovery pipeline (peptides < 100 aa and with no similarity to 272
proteins as evaluated by blastx) or (3) could be sRNA precursors. They were also compared 273
against NCBI RefSeq-RNA database to check for similarity with any other known ncRNAs. 274
Only one of the 48 lincRNAs had similarity to TEs (Table S5). Three of the 48 lincRNAs had 275
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

12
significant similarity to tens of sequences annotated as ncRNAs in RefSeq database. 276
However, a detailed analysis of the homologous region revealed it to contain a short 25 277
amino acid open reading frame (ORF) encoding a peptide RPL41 (ribosomal protein L41), 278
which was embedded within a much longer transcript. Because of the short length of the 279
peptide, transcripts carrying RPL41 were annotated as lncRNAs by the pipeline used in this 280
study as well as NCBI annotation pipeline. Following this discovery, the entire lincRNA 281
dataset was re-analysed to check for presence of other RPL41 ORFs. However, only 5 282
lincRNA loci (including the three conserved ones) were carrying RPL41 ORF. This finding 283
does suggest that some of the transcripts classified as lncRNAs based on the discovery 284
algorithm parameters used could, in fact, encode small peptides (Niazi and Valadkhan, 2012; 285
Ruiz-Orera et al., 2014; Nelson et al., 2016). Short of extremely well-conserved examples 286
like RPL41, in the absence of proteomic data these are impossible to discern. Six of the 287
lincRNAs showed 100% percentage identity to microRNA, suggesting that they could be 288
precursors of short RNAs. Re-analysis of the whole lincRNA dataset suggested that 56 of 289
lincRNAs could be microRNA precursors and the microRNA precursors were over-290
represented in the positionally and sequence conserved lincRNAs (Fisher test, p-value < 291
0.01). Finally, 19 lincRNAs showed similarity to other lncRNA transcripts in RefSeq and 292
those represented other species. 293
Almost two hundred of homeologous lincRNA loci can be traced to a soybean-lineage 294
specific whole genome duplication which occurred ~13 MYA 295
Soybean genome has a paleopolyploid structure resulting in extensive homeology across 296
chromosomes (Shoemaker et al., 2006). It has undergone two rounds of whole genome 297
duplications, a more ancient event which occurred ~59 million years ago (MYA) and 298
soybean-lineage-specific paleotetraploidization which took place ~13 MYA. As a result, 299
soybean genome is composed of large blocks of homeologous regions (Schmutz et al., 2010). 300
It is possible that akin to protein-coding loci, homeologous lincRNA loci in soybean genome 301
exist. Following a similar procedure for lincRNA positional similarity analysis performed 302
between species, analysis of positional similarity of lincRNA loci within soybean genome 303
was performed. Again, control datasets 1, 2, 3 and 4 were used to compare the number of 304
positionally similar lincRNA loci found to the number which would be expected by chance 305
alone. The number of positionally similar lincRNA loci in the true biological dataset was 306
significantly larger than the number found in any of the control datasets (Fisher test, p-value 307
< 0.01 for all comparisons, Fig. 3C). The difference between biological and control datasets 308
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

13
suggested that at least 200 to 300 lincRNA loci with homeologs in the soybean genome were 309
to be expected. Sequences of the lincRNA pairs with positional similarity within soybean 310
genome were compared, which allowed identification of 103 pairs of homeologous loci 311
(Table S6). Sequence comparison of the positionally similar lincRNA pairs in simulated 312
datasets (100 simulated datasets using random re-distribution of lincRNA relative to protein-313
coding loci) showed them to have no sequence similarity (median number of pairs with 314
sequence similarity per dataset: 0), again suggesting that that the sequence similarity 315
observed was not due to chance alone (permutation test, p-value < 0.01). The number also 316
roughly corresponds to the predictions based on comparison of positional similarity in 317
biological and control datasets. 318
The age of homeologous blocks can be established using pairwise synonymous distance 319
(Ks values) of paralogues (Schlueter et al., 2004; Pfeil et al., 2005; Schmutz et al., 2010). In 320
case of soybean the Ks values of 0.06–0.39 correspond to 13-Myr genome duplication and 321
the Ks values of values of 0.40–0.80 to the 59-Myr genome duplication (Schmutz et al., 322
2010). The vast majority of Ks values of protein-coding gene pairs flanking homeologous 323
lincRNA loci fall within the 0.06–0.39 range (Fig. 3D), suggesting a more recent origin 324
resulting from the soybean-lineage-specific paleotetraploidization. It is also possible that that 325
some homeologous loci representing the ~59 MYA duplication do exist, but sequence 326
divergence prevents their identification. Taken together, results of inter- and intra-species 327
comparisons suggest that while a life-span of soybean lincRNA can exceed 15 MY it is 328
unlikely to extend over 60 MY. 329
Functional enrichment of proteins flanking homeologous loci revealed over-representation of 330
genes involved in response to abiotic stimuli including cellular response to phosphate 331
starvation and response to absence of light (Table S7). Finally, the co-expression of 332
homeologous lincRNA loci was significantly higher (Fig 3E, Mann-Whitney U test, p-value 333
< 0.01) when compared to a randomly selected lincRNA loci pairs, suggesting at least partial 334
conservation of expression patterns. 335
The lincRNAs show highly tissue specific expression 336
Expression of lincRNAs across all tissues was investigated using a combination of straight-337
forward counting method and Tau specificity index, which were recently shown to be most 338
successful methods of expression characterization (Kryuchkova-Mostacci and Robinson-339
Rechavi, 2017). lincRNAs displayed more tissue specific expression than protein-coding 340
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

14
genes (Fig. 4A). Any given lincRNAs was on average expressed in 8 samples (median: 6.0), 341
whereas any given protein-coding gene was on average expressed in 23 samples (median: 342
30). Only 27 lincRNAs were expressed in all the samples. The tissue with the highest number 343
of lincRNAs expressed (FPKM > 0.1) was floral tissue, followed by shoot apical meristem 344
and leaf, suggesting an active role of lincRNAs in flowering and developmental processes. 345
The sample with the highest number of lincRNAs expressed in total and uniquely was flower 346
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

15
bud (flower1; 1891 lincRNAs expressed in total, 51 expressed uniquely) (Fig. 4B, C, Table 347
S3). A large number of lincRNAs expressed in SAM are consistent with previous 348
observations in chickpea and other plants (Khemka et al., 2016). Overall, samples from the 349
same tissue show similar expression patterns (Fig 4D). Samples representing shoot apical 350
meristem (SAM), leaf, flower, and seed are grouped together. Nine of the samples from two 351
tissues (leaf and SAM) represent floral transition period following short day treatment. In 352
total 366 lincRNAs were uniquely expressed in the floral transition samples and of these 363 353
(99% of all lincRNAs) were expressed following short day treatment, with 89, 128 and 149 354
lincRNAs expressed in leaf only, SAM only and leaf and SAM respectively. These lincRNAs 355
represent an interesting target for the study of the mechanism of soybean floral transition. 356
The specificity of lincRNA expression can be better contextualized when compared with 357
different groups of protein-coding genes. The lincRNA tissue expression patterns were 358
compared with expression patterns of protein-coding genes representing different specificity 359
groups (transcription factors – high specificity, protein phosphorylation – medium specificity, 360
translation – low specificity). lincRNAs have higher tissue specificity than any of the protein-361
coding gene groups, but the expression pattern is closest to the transcription factors (Fig. 4E). 362
Transcription factors are known master regulators of gene expression and the parallels 363
observed can suggest similar roles of lincRNAs. The high tissue-specific lincRNA expression 364
supports the idea of their highly specialized, possible regulatory functions. It also allows for 365
the possibility of using lincRNAs as tissue type and state markers. 366
The lincRNA-protein-coding gene co-expression network and position of lincRNAs relative 367
to protein-coding neighbours allows functional annotation of non-coding RNAs 368
Functional annotation of long noncoding RNAs poses a considerable challenge. In case of 369
protein-coding genes often extensive information about the function of a gene in a model 370
organism is available, and sequence homology can be used to transfer existing annotation to 371
newly discovered loci. In the case of lincRNAs, very few functional assignments exist, and 372
lack of sequence homology hampers inter-species comparisons (Rinn and Chang, 2012; 373
Smith and Mattick, 2017). The primary form of annotation involves a construction of co-374
expression network and using a method of so-called ‘guilt-by-association’. Correlation of 375
expression between lincRNAs and protein-coding genes can imply involvement in common 376
biological processes. Spearman correlation between expression of lincRNA and protein-377
coding loci was calculated. Only significant correlations were used in the analysis (p-value 378
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

16
<0.05, p-value adjusted for multiple comparisons using method ‘holm’). The resulting 379
distribution of correlation coefficients is presented in Fig. S2A. The minimum absolute value 380
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

17
of correlation coefficient used in the analysis was 0.84. A higher number of positive than 381
negative correlations was observed and a large number of perfect correlations (rho=1) were 382
observed. A similar observation was made in a human lncRNA annotation project, noting a 383
higher number of positive correlations (Derrien et al., 2012). The high number of perfect 384
correlations being due to high tissue specificity of lincRNA expression. lincRNAs were 385
annotated using hub-based approach (Liao et al., 2011). GO enrichment analysis of protein-386
coding 1st-degree neighbours resulted in functional annotation of 1,574 lincRNAs (Table S8). 387
The summary of the GO annotation mapped to GOslim terms is presented in Fig. S2B. 388
Overall, lincRNAs are annotated with a range of functions including stress response, signal 389
transduction and DNA methylation. Genes which are specifically or highly expressed in a 390
given tissue, are considered likely to contribute to relevant biological processes (Boyle et al., 391
2017). Clustering of lincRNA based on their expression across tissues showed that genes 392
which have peak expression in a given tissue are likely to have overall similar expression 393
profiles (Fig. S3), implying involvement in common biological process. The lincRNAs have 394
been divided based on the tissue with peak expression (each set contained lincRNAs with 395
peak expression in a given tissue) and GO enrichment for each of the lincRNA sets (peak 396
expression in: cotyledon, shoot apical meristem, flower, leaf, leaf bud, pod, pod seed, seed, 397
stem, root, Fig. 5) was calculated. The enrichment of highly or specifically expressed 398
lincRNA functions correlated well with the tissue-associated biological processes. For 399
example, functionally annotated lincRNAs expressed in shoot apical meristem, floral tissue 400
and root were highly enriched with processes associated with regulation of photoperiodism, 401
sexual reproduction and phloem transport respectively. The results suggest possible 402
involvement of lincRNAs in tissue specific biological processes. 403
Finally, lincRNAs often exert their function on neighbouring protein-coding genes therefore 404
analysis of overrepresentation of classes of protein-coding genes flanking lincRNA loci 405
provides additional source of functional annotation. The genes flanking lincRNAs were 406
enriched in functions associated with transcription and development, suggesting possible 407
lincRNA involvement in these processes (Table S9). 408
Several lincRNAs are potentially related to agronomic traits 409
Genome-wide association studies (GWAS) have been successful in uncovering the genetic 410
basis of trait variation and linking casual loci to phenotypic traits. However, only a portion of 411
variants identified by GWA studies can be assigned to protein-coding genes (Sonah et al., 412
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

18
2015; Zhang et al., 2015; Zhou et al., 2015; Zhou et al., 2015). Some of the remaining 413
intergenic trait-associated variants can potentially be assigned to lincRNAs and serve as an 414
additional source of functional annotation. In total 316 SNPs identified as associated with 415
agronomic traits were used in the analysis. A lincRNA was identified as potentially related to 416
a trait if the SNP was found either within the lincRNA locus or the locus was closer to the 417
SNP than any other protein-coding gene. In total 23 lincRNA candidates have been identified 418
(Fig S4). Six of the lincRNAs overlapped trait-associated SNPs, the reminder was found in 419
close proximity (median distance 981 bp). The putative trait-related lincRNA are enriched in 420
multi-exon loci (Fisher test, p-value < 0.01). The SNPs proximal to candidate lincRNA loci 421
were related to traits such as number of days to flowering, number of days from flowering to 422
maturity and number of seeds per pod. 423
Several loci are typically found in the vicinity of a trait associated SNP and it is usually not 424
immediately obvious which may contribute to the trait. Accordingly, although the 23 425
lincRNAs were found closer to the SNP than any other protein-coding gene, it possible that a 426
more distal coding gene contributes to the trait instead of the lincRNA (an interaction 427
between the lincRNA and neighbouring protein-coding gene is also possible). To add more 428
confidence to the functional predictions, the genomic-position-only based analysis was 429
supplemented with investigation of expression patterns of neighbouring genes. For each of 430
the 23 putative, trait related lincRNAs the samples with peak expression for the lincRNA as 431
well as 5 upstream and downstream protein-coding genes were investigated. The lincRNAs 432
were considered more likely to influence the trait if they were showed peak expression in a 433
relevant tissue (for example, lincRNA associated with days to flowering being highly 434
expressed in shoot apical meristem upon short day treatment, Table S10, Fig. S4). As a result, 435
top six lincRNAs which were found in the vicinity of trait associated SNPs and showed 436
consistent expression patterns were analysed in more detail (Fig 6A). Interestingly, 4 out of 6 437
had a positionally similar lincRNAs in other species. Two of them showed expression in 438
similar tissue types across species (NC_GMAXST00018683, NC_GMAXPA00061260), for 439
the remaining two expression data in relevant tissue in chickpea and Medicago were 440
unavailable. One of the lincRNAs (NC_GMAXST00018683) which overlapped SNP 441
associated with the number of days to flowering and had peak expression in shoot apical 442
meristem upon short day treatment had positional similarity with lincRNA in chickpea (Fig. 443
6B). Comparison of expression patterns across samples in soybean and chickpea showed the 444
lincRNAs to be expressed in flower buds and shoot apical meristem (SAM) in both species 445
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

19
(Fig. 6B). The other lincRNA (NC_GMAXPA00061260) was found 223 bp from a SNP 446
associated with number of seeds per pod and again had a positionally similar lincRNA in 447
chickpea. Both lincRNAs showed peak expression in mature flowers (Fig 6C). The proximity 448
to trait associated SNPs, expression in relevant tissues and conservation of expression 449
patterns across species makes them likely candidate for trait related lincRNAs. Combination 450
of proximity to trait associated SNPs and expression profile, as well as intra-species 451
conservation has been successfully used for functional annotation of lncRNAs in other 452
species including human, zebrafish, rice and maize (Ulitsky et al., 2011; Gong et al., 2015; 453
Wang et al., 2015; Hon et al., 2017). In human, the study incorporating expression and 454
genetic data found that lncRNAs which harboured trait associated SNPs were also 455
specifically expressed in tissues relevant to the trait, leading the authors to conclude the 456
lncRNAs are likely functional and play important roles in disease (Hon et al., 2017). Further, 457
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

20
the putative functional lncRNAs also exhibited higher levels of conservation (Hon et al., 458
2017). Similarly, in maize, SNPs associated with leaf morphological traits were significantly 459
enriched in genomic loci encoding maize lincRNAs, leading the authors to suggest roles of 460
lincRNAs in control of agronomic traits (Wang et al., 2015). Even without the support of 461
GWAS data lncRNA conservation itself was also found to be indicative of functionality. In 462
zebrafish, lincRNAs selected based on their tissue-specific expression and synteny with 463
mammalian lincRNAs were shown to be important for developmental processes (Ulitsky et 464
al., 2011). Taken together, the availability of evidence from several sources and earlier 465
studies suggesting that the GWAS, expression profile and conservation evidence is highly 466
indicative of lncRNA functionality gives additional confidence in the functional predictions. 467
Conclusions 468
The soybean genome encodes several thousand of long intergenic non-coding RNAs, and 469
several lincRNAs may be related to agronomic traits. Further investigations on detailed 470
function and regulation including identification of interacting partners and regulators of the 471
lincRNAs will elucidate their mechanism of action. The study also provides evidence that the 472
network controlling and implementing biological processes in soybean involves complex 473
interactions between proteins and long and short non-coding RNAs. Further, this study 474
presents a first, comprehensive atlas of lincRNAs in the soybean genome and paves the way 475
for the future research. 476
Materials and Methods 477
Data 478
RNASeq sequence data corresponding to Sequence Read Archive (SRA) projects SRP020868 479
and PRJNA238493 were downloaded (full list of accessions can be found in Table S1). 480
Glycine max genome assembly (Gmax_275_v2.0) and corresponding annotation 481
(Gmax_275_Wm82.a2.v1) were downloaded from Phytozome v10. 482
lincRNA annotation 483
Reads were mapped to the reference genome using HISAT2 (Kim et al., 2015) v2.0.5 (--min-484
intronlen 20 --max-intronlen 2000). For each accession transcripts were assembled and 485
subsequently merged using StringTie (Pertea et al., 2015) v1.3.0 (--merge -F 0.5 -T 0.5 -G 486
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

21
Gmax_275_Wm82.a2.v1.gene_exons.gff3). Reads were also assembled using Trinity 487
(Grabherr et al., 2011) v2.3.2. Both de novo (--seqType fq --max_memory 50G --verbose --488
normalize_reads --trimmomatic --CPU 16) and reference guided (--genome_guided_bam --489
genome_guided_max_intron 10000 --max_memory 50G --verbose --CPU 16, reads trimmed 490
and normalized during de novo Trinity run were used) assemblies were performed. The 491
resulting StringTie and Trinity assemblies were supplied to PASA (Haas et al., 2003) in order 492
to build comprehensive transcriptome database using procedure as described in PASA user 493
guide (http://pasapipeline.github.io/). The aligner used was BLAT and 494
MAX_INTRON_LENGTH was set to 2000. StringTie only and PASA transcripts were 495
processed in parallel to identify potential lncRNAs (Fig. S1). Transcripts >200 bp in length 496
were subjected to ORF discovery using OrfPredictor (Min et al., 2005) v3.0. Transcripts with 497
ORF >300 bp (100 aa) were considered coding. Remaining transcripts were extracted and 498
subjected to DIAMOND (Buchfink et al., 2015) v0.8.25 blastx search (--more-sensitive --499
evalue 0.01) against NCBI nr database (obtained on the 23.10.2016). Transcript which had a 500
significant blastx hit were considered coding. The remaining transcripts fulfilling the three 501
criteria (1) Length >200 bp (2) ORF size <=300 bp (3) No significant blastx hit were 502
considered putative lncRNAs. A gene was considered coding if at least one transcript was 503
coding. A gene was considered non-coding if none of the transcripts were coding. The 504
positions of non-coding genes from StringTie and PASA annotations were compared against 505
positions of coding genes in both annotations. If the putative lncRNA gene did not overlap 506
any coding loci it was considered lincRNA gene. lincRNA loci from both annotations were 507
merged. If lincRNA loci from both annotations had positional overlap, StringTie annotation 508
was kept. Finally, reads mapping to gene were counted using Subread v1.5.1 featureCounts 509
(Liao et al., 2014) (-p -B -P -d 0 -D 1000) and FPKM values were calculated for each gene 510
(109*fragments mapped to exons/assigned fragments*total length of exons). lincRNAs which 511
did not have FPKM value larger than 0.1 in one of the samples were discarded. 512
lincRNA functional annotation 513
LincRNA functional annotation was performed by building lincRNA-protein-coding gene co-514
expression network. Co-expression was measured between identified lincRNA loci and 515
protein-coding loci from Gmax_275_Wm82.a2.v1 annotation updated by StringTie. FPKM 516
values were used for Spearman correlation calculation. Correlation coefficients and 517
corresponding p-values were calculated using corr.test function of R package Psych. 518
Adjustment for multiple comparisons was performed using method ‘holm’. Only lincRNA-519
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

22
protein-coding gene pairs with p-value < 0.05 were retained. All the protein partners were 520
functionally annotated using Blast2GO (Conesa et al., 2005) (nr subset corresponding to 521
"Arabidopsis"[porgn] OR "Oryza"[porgn] OR "Sorghum"[porgn] OR "Glycine"[porgn] OR 522
"Medicago"[porgn] OR "Brachypodium"[porgn]). For each of the lincRNAs all the proteins 523
which were significantly correlated were gathered and GO enrichment of biological processes 524
(BP) category was calculated using topGO (Alexa et al., 2006) v2.22.0. All proteins in 525
correlation with lincRNAs were used as background. Adjustment for multiple comparisons 526
was performed using method ‘weight’. GO terms which were significantly enriched were 527
assigned to the corresponding lincRNA as functional annotation (p-value cut-off 0.05). The 528
GO terms were mapped to the plant GOslim terms using Map2Slim option of owltools. 529
Sequence based similarity of lincRNAs 530
Sequence based similarity of lincRNAs was measured using reciprocal best BLAST, 531
BLAST+ v2.5.0 (-task blastn –evalue 1e-3). Best hits were identified by lowest e-value. 532
Coordinates of chickpea lincRNAs were obtained from Khemka et al. (Khemka et al., 2016), 533
Medicago lincRNAs from Wang et al. (Wang et al., 2015) and A. thaliana lincRNA from 534
http://chualab.rockefeller.edu/gbrowse2/homepage.html. The lincRNA sequences were 535
extracted from genome assemblies (chickpea Cicer_arietinum_GA_v1.0, Medicago Mt4.0v1 536
and A. thaliana TAIR9). Comparisons against genome sequence were performed using 537
BLAST+ v2.5.0 (-task dc-megablast –evalue 1e-3). In order to remove spurious hits due to 538
presence of transposable elements or repetitive sequences, lincRNAs which had more than 539
three matches in the genome were excluded. Additionally, the most significant HSP between 540
lincRNA and the genome was required to cover at least 10% of the lincRNA. 541
Transposable element (TE) composition of lincRNAs 542
The soybean TE database was obtained from SoyBase (SoyBase_TE_Fasta.txt). The 543
lincRNA transcripts were compared against the TE database using BLAST+ v2.2.30 (blastn -544
task megablast –evalue 1e-5). The 50,000 random non-overlapping intervals which did not 545
overlap lincRNAs were identified in the soybean genome using regioneR (Gel et al., 2016). 546
The corresponding sequences were extracted and compared against the TE database with the 547
same BLAST parameters as for lincRNAs, 548
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

23
Centromeric lincRNA identification 549
Centromeres were identified by presence of two soybean centromere specific repeats 550
CentGm-1 and CentGm-2. CentGm-1 and CentGm-2 were compared against soybean 551
genome (Gmax_275_v2.0) using BLAST+ v2.2.30 (blastn -task megablast). The coordinates 552
of centromere for a given chromosome corresponded to 1st and 3rd quartile of CentGm-1 and 553
CentGm-2 match coordinates. LincRNAs which fell within centromeres were identified as 554
centromeric lincRNAs. 555
Position based similarity of lincRNAs 556
Syntenic blocks between genomes of soybean (Gmax_275_v2.0), chickpea 557
(Cicer_arietinum_GA_v1.0), Medicago (Mt4.0v1) and A. thaliana (TAIR10) were identified 558
using MCScanX (Wang et al., 2012). The syntenic blocks were used to identify positional 559
similarity between soybean lincRNAs and lincRNAs from other species. For each lincRNA 560
five protein-coding neighbours upstream and downstream were extracted. The neighbours 561
were then compared with collinear blocks identified by MCScanX. The lincRNA was said to 562
belong to a collinear block if at least three out of ten protein-coding neighbours were fund in 563
the block. lincRNAs from two species were said to be positionally similar if they belonged to 564
the same collinear block, at least one of the two pairs of flanking protein-coding genes were 565
identified as orthologous and the lincRNAs shared the same relative position (upstream or 566
downstream) with respect to the orthologous gene/genes. The lincRNA loci which shared 567
positional similarity were compared using BLAST+ v2.5.0 (-task blastn –evalue 1e-3). 568
Comparison against RefSeq RNA database (downloaded on: 27.06.2017) was also performed 569
with BLAST+ v2.5.0 (-task blastn –evalue 1e-3). 570
Generation of control datasets 571
The control datasets were generated by assigning existing lincRNA to new protein-coding 572
neighbours, taken from the pool of all protein-coding genes found in the genome. For datasets 573
1 and 2 coordinate sorted full list of protein-coding genes was shuffled using Linux shuf 574
function, which generates random permutations, and first n genes corresponding to the 575
number of lincRNAs in a given dataset were assigned to existing lincRNAs. The assigned 576
protein-coding gene became new downstream protein-coding neighbour and the new 577
lincRNA position was immediately upstream of the protein-coding gene assigned. For 578
datasets 3 and 4 the procedure was similar, but the existing lincRNA clusters were kept 579
together. 580
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

24
Calculation of synonymous substitution rate 581
The synonymous substitution rates were computed between pairs of genes identified as 582
homeologous by MCScanX. Proteins were aligned by Clustal Omega v1.2.0 (Sievers et al., 583
2011). The protein alignments were converted to nucleotide alignments using PAL2NAL v14 584
(Suyama et al., 2006). The Ks values were calculated using PAML (yn00) v4.7 (Yang, 2007). 585
Selections of protein groups for comparison of tissue specific expression with lincRNAs 586
The protein-coding genes were divided into three categories. Genes expressed in no more 587
than 15 samples (high specificity expression pattern), genes expressed in 16 to 35 samples 588
(medium specificity expression pattern) and genes expressed in more than 35 samples (low 589
specificity expression pattern). For each group enrichment of biological processes category 590
was performed using topGO (Alexa et al., 2006), using all protein-coding genes as 591
background. Adjustment for multiple comparisons was performed using method ‘weight’. For 592
each category a representative process was chosen (process with the highest number of 593
significant genes among top ten enriched GO terms). All the genes from a given category 594
annotated with representative process were gathered and Tau specificity indices were 595
calculated (Yanai et al., 2005). 596
Identification of lincRNAs potentially related to agronomic traits 597
The positions of single nucleotide polymorphisms (SNPs) associated with agronomic traits 598
identified by Zhou et al (Zhou et al., 2015)., Zhang et al. (Zhang et al., 2015), Zhou et al. 599
(Zhou et al., 2015), Sonah et al. (Sonah et al., 2015) and Fang et al. (Fang et al., 2017) were 600
obtained. Some of the SNPs were originally discovered against an older version of soybean 601
genome (NCBI accession GCA_000004515.1) therefore their coordinates were transferred to 602
the Gmax_275_v2.0 genome assembly using NCBI remap tool 603
(https://www.ncbi.nlm.nih.gov/genome/tools/remap). The lincRNA was consider potentially 604
related to agronomic trait if it either harboured a SNP identified in association studies or it 605
was closer to a SNP than any protein-coding gene and no further than 10 kb. 606
Code availability: The code used for generation of all the figures can be found 607
on:.https://github.com/agolicz/lncRNAs-Plots 608
Data availability: The dataset described in the manuscript can be downloaded from: 609
https://osf.io/d7qz2/ 610
Acknowledgements 611
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

25
Financial support for this work was obtained from the ARC Discovery Grant ARC 612 DP0988972 is gratefully acknowledged. This research was supported by Melbourne 613 Bioinformatics at the University of Melbourne, project UOM0033. 614
Figures 615
Fig 1. Comparison of properties of protein-coding and lincRNA genes. LincRNA genes 616
differ from protein-coding genes with respect to transcript length, number of exons per 617
transcript, number of transcripts per gene and transcriptional profile. (A) Comparison of 618
transcript lengths of coding and non-coding genes. Non-coding genes have shorter 619
transcripts. (B) Comparison of the number of transcripts found in coding and non-coding 620
genes. Non-coding genes have less isoforms. (C) Comparison of the number of exons found 621
in transcripts of coding and non-coding genes. Transcripts of non-coding genes have a lower 622
number of exons. (D) Comparison of log2(FPKM) values of coding and non-coding genes. 623
FPKM values calculated based on counts produced by featureCounts. Non-coding genes 624
show lower expression levels compared to protein-coding genes. (E) Plot presenting 625
distribution of protein-coding and lincRNA loci across 20 soybean chromosomes. lincRNA 626
loci are evenly distributed across chromosomes, whereas protein-coding genes show lower 627
density in centromeric regions. Starting from the outer ring: (1) protein-coding genes (2) all 628
lincRNA genes (3) non-TE lincRNA loci (4) lincRNA loci with transcripts harbouring TEs. 629
Fig 2. Transposable element composition of lincRNA. (A) Types of TEs found within 630
lincRNA transcripts and in 50,000 randomly selected regions of soybean genome. TE 631
composition of lincRNAs follows that of soybean genome. RLG = LTR Gypsy. RLC = LTR 632
Copia, RIu = LINE, RIL = LINE L1, DTT = Tc1-Mariner, DTO = PONG, DTM = Mutator, 633
DTH = PIF-Harbinger, DTC = CACTA, DHH = Helitron, * - p-value < 0.01, Fisher test. (B) 634
Expression patterns of lincRNAs located in centromeric regions (n=32). (C) TE composition 635
of centromeric lincRNAs. 636
Fig 3. Conservation of lincRNA loci in chickpea, Medicago and A. thaliana. (A) Number 637
of soybean lincRNA loci showing sequence similarity with lincRNAs or genomes of other 638
species. (B) Number of soybean lincRNA loci in biological and control datasets showing 639
positional similarity with lincRNAs in other species. (C) Number of soybean lincRNA loci in 640
biological and control datasets showing positional similarity with other lincRNAs in soybean 641
genome. (D) Ks values calculated for protein-coding gene pairs flanking homeologous 642
lincRNA loci and a random selection of homeologous protein-coding gene pairs (n=444). The 643
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

26
distribution of Ks values representing random selection has two peaks corresponding two 644
duplication events. The protein pairs flanking homeologous loci mostly represent single, 645
more recent duplication. (E) Correlation of expression between homeologous lincRNA and a 646
random selection of lincRNA pairs (n=3,000). Homeologous loci have higher levels of co-647
expression. 648
Fig 4. Sample specific lincRNA expression. (A) Comparison of the number of samples 649
showing expression of coding and non-coding genes. Expression of non-coding genes is more 650
sample specific. (B) Number of total and unique lincRNA genes expressed in each tissue 651
(samples from different time points for the same tissue were combined). Tissues with the 652
highest total number of lincRNAs expressed and the highest number of uniquely expressed 653
lincRNAs are floral tissue and shoot apical meristem. (C) Number of lincRNAs expressed in 654
each sample. The sample with most lincRNAs expressed is flower1. (D) Heatmap showing 655
relationships between samples. Samples from the same tissue have similar lincRNA 656
expression profiles and cluster together. The colour corresponds to distance calculated as 1-657
cor(log1p(FPKM)). Clustering was performed using hclust, method=complete. (E) Tau 658
expression specificity index calculated for lincRNA loci and three groups of proteins 659
representing different biological processes. Higher values of Tau, correspond to more sample 660
specific expression. Cotyledon 1 – germination stage; cotyledon 2 – trefoil stage; flower 1 – 661
flower bud differentiation stage; flower 2 – flowering stage, bud before flowering; flower 3 – 662
flowering stage, florescence; flower 4 – flowering stage, 5 days after flowering; flower 5 – 663
flowering stage, florescence, different stage; leaf 1 – trefoil stage; leaf 2 - flower bud 664
differentiation stage; leaf 3 – senescent leaves; leafbud 1 – germination stage; leafbud 2 – 665
trefoil stage; leafbud 3 – flower bud differentiation stage; pod seed 1 – two weeks; pod seed 2 666
– three weeks; pod seed 3 – four weeks; pod 1 – three weeks; pod 2 – four weeks; pod 3 – 667
five weeks; seed 1 – three weeks; seed 2 – five weeks; seed 3 – six weeks; seed 4 – eight 668
weeks; seed 5 – ten weeks; shoot meristem – flower bud differentiation stage; stem 1- 669
germination; stem 2 – trefoil stage; root – germination stage; sam sd0 – shoot apical meristem 670
– before short day treatment; sam sd1-4 – shoot apical meristem – short days 1-4; leaf sd0 – 671
leaf – before short day treatment; leaf sd1-3 – leaf – short days 1-3. 672
Fig 5. Significantly enriched biological processes among lincRNAs which show peak 673
expression in a given tissue. Enrichment calculated using topGO, adjustment for multiple 674
comparisons using method ‘weight’, p-value < 0.01. 675
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

27
Fig 6. Analysis of potential trait-related lincRNAs. (A) For each of the putative trait related 676
lincRNAs the plot presents eleven genes found in close vicinity of trait associated SNP (1 677
putative trait related lincRNA + 5 downstream protein-coding genes + 5 upstream protein-678
coding genes). Each dot-point represents a gene (red – lincRNA, blue – protein-coding) and 679
is labelled with sample with peak expression (y axis represents actual expression value). (B, 680
C) Expression of lincRNA in soybean samples and its positional orthologue in chickpea. The 681
x axis corresponds to samples., the y axis corresponds to the FPKM values. 682
683
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

Parsed CitationsAlexa A, Rahnenführer J, Lengauer T (2006) Improved scoring of functional groups from gene expression data by decorrelating GOgraph structure. Bioinformatics 22: 1600-1607
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Ariel F, Jegu T, Latrasse D, Romero-Barrios N, Christ A, Benhamed M, Crespi M (2014) Noncoding transcription by alternative rnapolymerases dynamically regulates an auxin-driven chromatin loop. Molecular Cell 55: 383-396
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Bardou F, Ariel F, Simpson CG, Romero-Barrios N, Laporte P, Balzergue S, Brown JWS, Crespi M (2014) Long Noncoding RNAModulates Alternative Splicing Regulators in Arabidopsis. Developmental Cell 30: 166-176
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Berry S, Dean C (2015) Environmental perception and epigenetic memory: Mechanistic insight through FLC. Plant Journal 83: 133-148Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Boyle EA, Li YI, Pritchard JK (2017) An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169: 1177-1186Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Meth 12: 59-60Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Carlevaro-Fita J, Rahim A, Guigó R, Vardy LA, Johnson R (2016) Cytoplasmic long noncoding RNAs are frequently bound to anddegraded at ribosomes in human cells. RNA 22: 867-882
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Chekanova JA (2015) Long non-coding RNAs and their functions in plants. Current Opinion in Plant Biology 27: 207-216Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Chekanova JA, Gregory BD, Reverdatto SV, Chen H, Kumar R, Hooker T, Yazaki J, Li P, Skiba N, Peng Q, Alonso J, Brukhin V,Grossniklaus U, Ecker JR, Belostotsky DA (2007) Genome-Wide High-Resolution Mapping of Exosome Substrates Reveals HiddenFeatures in the Arabidopsis Transcriptome. Cell 131: 1340-1353
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Cifuentes-Rojas C, Kannan K, Tseng L, Shippen DE (2011) Two RNA subunits and POT1a are components of Arabidopsis telomerase.Proceedings of the National Academy of Sciences of the United States of America 108: 73-78
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization andanalysis in functional genomics research. Bioinformatics 21: 3674-3676
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L,Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR,Hubbard TJ, Notredame C, Harrow J, Guigó R (2012) The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their genestructure, evolution, and expression. Genome Research 22: 1775-1789
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Du J, Grant D, Tian Z, Nelson RT, Zhu L, Shoemaker RC, Ma J (2010) SoyTEdb: a comprehensive database of transposable elements in www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from
Copyright © 2017 American Society of Plant Biologists. All rights reserved.

the soybean genome. BMC Genomics 11: 113Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Fang C, Ma Y, Wu S, Liu Z, Wang Z, Yang R, Hu G, Zhou Z, Yu H, Zhang M, Pan Y, Zhou G, Ren H, Du W, Yan H, Wang Y, Han D, Shen Y,Liu S, Liu T, Zhang J, Qin H, Yuan J, Yuan X, Kong F, Liu B, Li J, Zhang Z, Wang G, Zhu B, Tian Z (2017) Genome-wide associationstudies dissect the genetic networks underlying agronomical traits in soybean. Genome Biology 18: 161
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Flynn RA, Chang HY (2014) Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 14: 752-761Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, García JA, Paz-Ares J (2007) Targetmimicry provides a new mechanism for regulation of microRNA activity. Nature Genetics 39: 1033-1037
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Gel B, Díez-Villanueva A, Serra E, Buschbeck M, Peinado MA, Malinverni R (2016) regioneR: an R/Bioconductor package for theassociation analysis of genomic regions based on permutation tests. Bioinformatics 32: 289-291
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Gong J, Liu W, Zhang J, Miao X, Guo A-Y (2015) lncRNASNP: a database of SNPs in lncRNAs and their potential functions in human andmouse. Nucleic Acids Research 43: D181-D186
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E,Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Trinity: reconstructinga full-length transcriptome without a genome from RNA-Seq data. Nature biotechnology 29: 644-652
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, Hannick LI, Maiti R, Ronning CM, Rusch DB, Town CD, Salzberg SL, White O(2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31: 5654-5666
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Hao Z, Fan C, Cheng T, Su Y, Wei Q, Li G (2015) Genome-wide identification, characterization and evolutionary analysis of longintergenic noncoding rnas in cucumber. PLoS ONE 10
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Heo JB, Lee YS, Sung S (2013) Epigenetic regulation by long noncoding RNAs in plants. Chromosome Research 21: 685-693Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Heo JB, Sung S (2011) Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science (New York, N.Y.) 331:76-79
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I (2015) Principles of long noncoding RNA evolution derived fromdirect comparison of transcriptomes in 17 species. Cell reports 11: 1110-1122
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Hon C-C, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJL, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J, Lizio M,Kawaji H, Kasukawa T, Itoh M, Burroughs AM, Noma S, Djebali S, Alam T, Medvedeva YA, Testa AC, Lipovich L, Yip C-W, Abugessaisa I,Mendez M, Hasegawa A, Tang D, Lassmann T, Heutink P, Babina M, Wells CA, Kojima S, Nakamura Y, Suzuki H, Daub CO, de Hoon
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

MJL, Arner E, Hayashizaki Y, Carninci P, Forrest ARR (2017) An atlas of human long non-coding RNAs with accurate 5′ ends. Nature543: 199
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Jin J, Liu J, Wang H, Wong L, Chua NH (2013) PLncDB: Plant long non-coding RNA database. Bioinformatics 29: 1068-1071Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermüller J, Hofacker IL, Bell I, Cheung E,Drenkow J, Dumais E, Patel S, Helt G, Ganesh M, Ghosh S, Piccolboni A, Sementchenko V, Tammana H, Gingeras TR (2007) RNA mapsreveal new RNA classes and a possible function for pervasive transcription. Science 316: 1484-1488
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, Yandell M, Feschotte C (2013) Transposable Elements Are MajorContributors to the Origin, Diversification, and Regulation of Vertebrate Long Noncoding RNAs. PLoS Genetics 9: e1003470
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Khemka N, Singh VK, Garg R, Jain M (2016) Genome-wide analysis of long intergenic non-coding RNAs in chickpea and their potentialrole in flower development. Scientific Reports 6
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Meth 12: 357-360Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Kornienko AE, Guenzl PM, Barlow DP, Pauler FM (2013) Gene regulation by the act of long non-coding RNA transcription. BMC Biology11
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Kryuchkova-Mostacci N, Robinson-Rechavi M (2017) A benchmark of gene expression tissue-specificity metrics. Briefings inBioinformatics 18: 205-214
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, Shiekhattar R (2013) Activating RNAs associate with Mediator toenhance chromatin architecture and transcription. Nature 494: 497-501
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Li L, Eichten SR, Shimizu R, Petsch K, Yeh CT, Wu W, Chettoor AM, Givan SA, Cole RA, Fowler JE, Evans MMS, Scanlon MJ, Yu J,Schnable PS, Timmermans MCP, Springer NM, Muehlbauer GJ (2014) Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biology 15
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Liao Q, Liu C, Yuan X, Kang S, Miao R, Xiao H, Zhao G, Luo H, Bu D, Zhao H, Skogerbø G, Wu Z, Zhao Y (2011) Large-scale prediction oflong non-coding RNA functions in a coding–non-coding gene co-expression network. Nucleic Acids Research 39: 3864-3878
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomicfeatures. Bioinformatics 30: 923-930
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Liu J, Jung C, Xu J, Wang H, Deng S, Bernad L, Arenas-Huertero C, Chua N-H (2012) Genome-Wide Analysis Uncovers Regulation ofLong Intergenic Noncoding RNAs in Arabidopsis. The Plant Cell 24: 4333-4345
Pubmed: Author and Title www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

CrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Matzke MA, Kanno T, Matzke AJ (2014) RNA-Directed DNA Methylation: The Evolution of a Complex Epigenetic Pathway in FloweringPlants. Annu. Rev. Plant Biol
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Min XJ, Butler G, Storms R, Tsang A (2005) OrfPredictor: Predicting protein-coding regions in EST-derived sequences. Nucleic AcidsResearch 33: W677-W680
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Mohammadin S, Edger PP, Pires JC, Schranz ME (2015) Positionally-conserved but sequence-diverged: identification of long non-coding RNAs in the Brassicaceae and Cleomaceae. BMC Plant Biology 15: 217
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, Cannon SC,Houser SR, Bassel-Duby R, Olson EN (2016) A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCAactivity in muscle. Science 351: 271
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Niazi F, Valadkhan S (2012) Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity andparallels with 3′ UTRs. RNA 18: 825-843
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, Federation A, Chao J, Elliott O, Liu ZP, Economides AN, Bradner JE, RabadanR, Basu U (2015) RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell 161: 774-789
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL (2015) StringTie enables improved reconstruction of atranscriptome from RNA-seq reads. Nat Biotech 33: 290-295
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Pfeil BE, Schlueter JA, Shoemaker RC, Doyle JJ, Page R (2005) Placing Paleopolyploidy in Relation to Taxon Divergence: APhylogenetic Analysis in Legumes Using 39 Gene Families. Systematic Biology 54: 441-454
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. In Annual Review of Biochemistry, Vol 81, pp 145-166Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Rošić S, Erhardt S (2016) No longer a nuisance: long non-coding RNAs join CENP-A in epigenetic centromere regulation. Cellular andMolecular Life Sciences 73: 1387-1398
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Ruiz-Orera J, Messeguer X, Subirana JA, Alba MM (2014) Long non-coding RNAs as a source of new peptides. eLife 3: e03523Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Schlueter JA, Dixon P, Granger C, Grant D, Clark L, Doyle JJ, Shoemaker RC (2004) Mining EST databases to resolve evolutionaryevents in major crop species. Genome 47: 868-876
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y,Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell- www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from
Copyright © 2017 American Society of Plant Biologists. All rights reserved.

Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang X-C, Shinozaki K, Nguyen HT, Wing RA,Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of the palaeopolyploidsoybean. Nature 463: 178-183
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Shen Y, Zhou Z, Wang Z, Li W, Fang C, Wu M, Ma Y, Liu T, Kong L-A, Peng D-L, Tian Z (2014) Global Dissection of Alternative Splicing inPaleopolyploid Soybean. The Plant Cell Online
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Shoemaker RC, Schlueter J, Doyle JJ (2006) Paleopolyploidy and gene duplication in soybean and other legumes. Current Opinion inPlant Biology 9: 104-109
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG(2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology7: 539-539
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Smith MA, Mattick JS (2017) Structural and Functional Annotation of Long Noncoding RNAs. In JM Keith, ed, Bioinformatics: Volume II:Structure, Function, and Applications. Springer New York, New York, NY, pp 65-85
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Sonah H, O'Donoughue L, Cober E, Rajcan I, Belzile F (2015) Identification of loci governing eight agronomic traits using a GBS-GWASapproach and validation by QTL mapping in soya bean. Plant Biotechnology Journal 13: 211-221
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Suyama M, Torrents D, Bork P (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codonalignments. Nucleic Acids Research 34: W609-W612
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Swiezewski S, Liu F, Magusin A, Dean C (2009) Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycombtarget. Nature 462: 799-802
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Szcześniak MW, Rosikiewicz W, Makałowska I (2016) CANTATAdb: A collection of plant long non-coding RNAs. Plant and CellPhysiology 57: e8
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Ulitsky I, Bartel DP (2013) LincRNAs: Genomics, evolution, and mechanisms. Cell 154: 26-46Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP (2011) Conserved Function of lincRNAs in Vertebrate Embryonic DevelopmentDespite Rapid Sequence Evolution. Cell 147: 1537-1550
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Van Werven FJ, Neuert G, Hendrick N, Lardenois A, Buratowski S, Van Oudenaarden A, Primig M, Amon A (2012) Transcription of twolong noncoding RNAs mediates mating-type control of gametogenesis in budding yeast. Cell 150: 1170-1181
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wang H, Chung PJ, Liu J, Jang IC, Kean MJ, Xu J, Chua NH (2014) Genome-wide identification of long noncoding natural antisensetranscripts and their responses to light in Arabidopsis. Genome Research 24: 444-453 www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from
Copyright © 2017 American Society of Plant Biologists. All rights reserved.

Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wang H, Niu QW, Wu HW, Liu J, Ye J, Yu N, Chua NH (2015) Analysis of non-coding transcriptome in rice and maize uncovers roles ofconserved lncRNAs associated with agriculture traits. Plant Journal 84: 404-416
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wang KC, Chang HY (2011) Molecular mechanisms of long noncoding RNAs. Molecular cell 43: 904-914Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wang T-Z, Liu M, Zhao M-G, Chen R, Zhang W-H (2015) Identification and characterization of long non-coding RNAs involved in osmoticand salt stress in Medicago truncatula using genome-wide high-throughput sequencing. BMC Plant Biology 15: 131
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wang Y, Fan X, Lin F, He G, Terzaghi W, Zhu D, Deng XW (2014) Arabidopsis noncoding RNA mediates control of photomorphogenesisby red light. Proceedings of the National Academy of Sciences of the United States of America 111: 10359-10364
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wang Y, Tang H, DeBarry JD, Tan X, Li J, Wang X, Lee T-h, Jin H, Marler B, Guo H, Kissinger JC, Paterson AH (2012) MCScanX: atoolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Research 40: e49-e49
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wong CE, Singh MB, Bhalla PL (2013) The Dynamics of Soybean Leaf and Shoot Apical Meristem Transcriptome Undergoing FloralInitiation Process. PLoS ONE 8
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Wu HJ, Wang ZM, Wang M, Wang XJ (2013) Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants.Plant Physiology 161: 1875-1884
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, Lancet D,Shmueli O (2005) Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification.Bioinformatics 21: 650-659
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Yang Z (2007) PAML 4: Phylogenetic Analysis by Maximum Likelihood. Molecular Biology and Evolution 24: 1586-1591Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Zhang J, Song Q, Cregan PB, Nelson RL, Wang X, Wu J, Jiang G-L (2015) Genome-wide association study for flowering time, maturitydates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genomics 16: 217
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Zhang YC, Liao JY, Li ZY, Yu Y, Zhang JP, Li QF, Qu LH, Shu WS, Chen YQ (2014) Genome-wide screening and functional analysisidentify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome biology 15: 512
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Zhou L, Wang S-B, Jian J, Geng Q-C, Wen J, Song Q, Wu Z, Li G-J, Liu Y-Q, Dunwell JM, Zhang J, Feng J-Y, Niu Y, Zhang L, Ren W-L,Zhang Y-M (2015) Identification of domestication-related loci associated with flowering time and seed size in soybean with the RAD-seqgenotyping method. Scientific Reports 5: 9350
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.

Zhou Z, Jiang Y, Wang Z, Gou Z, Lyu J, Li W, Yu Y, Shu L, Zhao Y, Ma Y, Fang C, Shen Y, Liu T, Li C, Li Q, Wu M, Wang M, Wu Y, Dong Y,Wan W, Wang X, Ding Z, Gao Y, Xiang H, Zhu B, Lee S-H, Wang W, Tian Z (2015) Resequencing 302 wild and cultivated accessionsidentifies genes related to domestication and improvement in soybean. Nat Biotech 33: 408-414
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
Zhu YL, Song QJ, Hyten DL, Van Tassell CP, Matukumalli LK, Grimm DR, Hyatt SM, Fickus EW, Young ND, Cregan PB (2003) Single-nucleotide polymorphisms in soybean. Genetics 163: 1123-1134
Pubmed: Author and TitleCrossRef: Author and TitleGoogle Scholar: Author Only Title Only Author and Title
www.plantphysiol.orgon July 8, 2018 - Published by Downloaded from Copyright © 2017 American Society of Plant Biologists. All rights reserved.