the structure of an asymmetric dimer relevant to the mode of action of the glycopeptide antibiotics
TRANSCRIPT
The structure of an asymmetric dimer relevant to themode of action of the glycopeptide antibiotics
Patrick Groves, Mark S Searle, Joel P Mackayt and Dudley H Williams*Cambridge Centre for Molecular Recognition, University Chemical Laboratories, Lensfield Road, Cambridge, CB2 1 EW, UK
Background: Glycopeptide antibiotics of the vancomycingroup are of crucial clinical importance in the treatment ofmethicillin resistant Staphylococcus aureus (MRSA) - theoften lethal 'super-bug' - characterized by its resistance toa wide range of antibiotics in common use. The antibioticsexert their physiological action by blocking cell wallsynthesis through recognition of nascent cell wall mucopep-tides terminating in the sequence -D-Ala-D-Ala. Evidencesuggests that the antibiotics are able to enhance their bio-logical activity by the formation ofhomodimers, and this issupported by the observation that dimerization and peptidebinding in vitro are cooperative phenomena. The basis ofthis enhancement is not understood at the molecular level.Results: The first detailed structure of a dimeric glycopeptide
antibiotic, that of eremomycin, is presented based uponsolution NMR data. The overall structure of the dimercomplex is asymmetric. The source of this asymmetry - aparallel alignment and mutual interaction of the disaccha-rides - appears to promote dimerization through specificsugar-sugar recognition.Conclusions: A molecular basis for the observed coopera-tivity of cell wall peptide binding by eremomycin is evidentfrom these studies of the dimer. The carboxylate anion ofthe cell wall component, which is crucial to binding, formsan amide-mediated ion-pair interaction to the alkylammo-nium ion of the ring 6 sugar in the other half of the dimermaking the structure and positioning of this sugar importantin mediating cooperativity.
Structure 15 August 1994, 2:747-754
Key words: asymmetric dimer, cooperativity, glycopeptide antibiotics, NMR structure
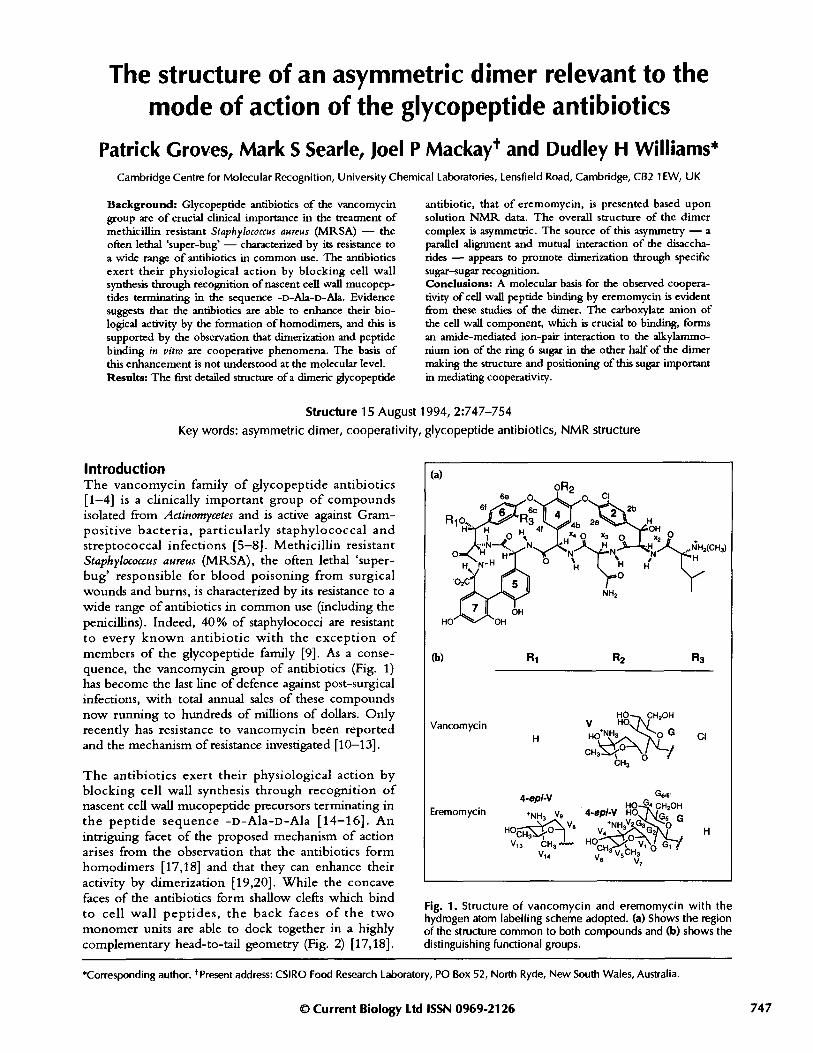
IntroductionThe vancomycin family of glycopeptide antibiotics[1-4] is a clinically important group of compoundsisolated from Actinomycetes and is active against Gram-positive bacteria, particularly staphylococcal andstreptococcal infections [5-8]. Methicillin resistantStaphylococcus aureus (MRSA), the often lethal 'super-bug' responsible for blood poisoning from surgicalwounds and burns, is characterized by its resistance to awide range of antibiotics in common use (including thepenicillins). Indeed, 40% of staphylococci are resistantto every known antibiotic with the exception ofmembers of the glycopeptide family [9]. As a conse-quence, the vancomycin group of antibiotics (Fig. 1)has become the last line of defence against post-surgicalinfections, with total annual sales of these compoundsnow running to hundreds of millions of dollars. Onlyrecently has resistance to vancomycin been reportedand the mechanism of resistance investigated [10-13].
The antibiotics exert their physiological action byblocking cell wall synthesis through recognition ofnascent cell wall mucopeptide precursors terminating inthe peptide sequence -D-Ala-D-Ala [14-16]. Anintriguing facet of the proposed mechanism of actionarises from the observation that the antibiotics formhomodimers [17,18] and that they can enhance theiractivity by dimerization [19,20]. While the concavefaces of the antibiotics form shallow clefts which bindto cell wall peptides, the back faces of the twomonomer units are able to dock together in a highlycomplementary head-to-tail geometry (Fig. 2) [17,18].
(a)
(b) R2 R3
VancomycinHO-. CH2OH
V HOES/
H HH 3 G
HH
4-epi-VEremomycin 'NH3 Vs
V13 CH3 -V14
G6/
HOG4
CH2OH4.epl _HO 0.. NHV
VH5 \ GH~7
V, V5 37
Cl
H
Fig. 1. Structure of vancomycin and eremomycin with thehydrogen atom labelling scheme adopted. (a) Shows the regionof the structure common to both compounds and (b) shows thedistinguishing functional groups.
© Current Biology Ltd ISSN 0969-2126
*Corresponding author. tPresent address: CSIRO Food Research Laboratory, PO Box 52, North Ryde, New South Wales, Australia.
747

748 Structure 1994, Vol 2 No 8
Fig. 2. Proposed hydrogen-bondingscheme in the eremomycin dimercomplex with the cell wall analogueN-acetyl-D-Ala-D-Ala. The backbonesof the two antiparallel monomers areshown in bold. Arrows representhydrogen bonds between the twohalves of the dimer, while dotted linesrepresent hydrogen bonds between thecell wall peptide and the antibiotic.
It is proposed that binding to a membrane-bound cellwall precursor at one site on the dimer serves to anchorthe dimer to the cell wall, making the second bindingevent effectively an intramolecular process, with anassociated chelate-like enhancement of binding affinityfor the cell wall components [20]. The observationsthat binding of cell wall analogues to the antibiotics invitro can promote dimerization (with the exception ofteicoplanin which does not dimerize [20]) and thatmembers of the group which dimerize strongly(Kdim10 5-10 6 M-1) show activity which is greater thanexpected on the basis of their binding affinity for thecell wall tripeptide di-N-acetyl-L-Lys-D-Ala-D-Ala invitro, provide strong support for the role of dimerizationin the mode of action [20]. Despite the fact that thechemical structures of a large number of glycopeptides,including vancomycin, have been known for 15 years,no relevant crystallographic data are available that beardirectly on the structure of the dimer. While earliermodels based upon solution NMR data [17,18], havepresented a description of interactions at the dimer
Fig. 3. Schematic views of the twopossible eremomycin monomericstructures (a), where the disaccharideunits are related by 180 ° rotationsabout the bond connecting theglucose sugar to residue 4 (see Fig. 1).The two possible symmetric dimerstructures are illustrated in (b) and (c),while the single asymmetric dimer isshown in (d); all are obtained fromdifferent combinations of themnn'mr .init cknu in in (a Tk. Aic_III IIUIIICI 1l11L 1 11V ... I, tI.. 11 l~ -
accharide consists of 4-epi-vancosamine (V) and glucose (G)sugar units. Thus, two sets of signals inthe NMR spectrum could be explainedeither in terms of a mixture of symmet-ncal almers n equal proponrons or acinli acvmmotrir rlimor
interface at the level of the glycopeptide backbone(Fig. 2), the role of the sugars [in particular the ring-4disaccharide of vancomycin and eremomycin (Fig. 1)]in promoting dimerization and cell wall recognition hasremained poorly defined.
ResultsNMR studies of eremomycinEremomycin has the largest dimerization constant of allthe glycopeptides so far studied (Kadm~10 6M- at 298K)[19]. The H NMR spectrum of the dimer consists oftwo sets of resonances of equal intensity due to species inslow exchange with each other on the chemical shifttimescale. Various combinations of the monomericcomplexes shown in Fig. 3a, differing in the orientationof the disaccharides, can lead to the dimer structuresshown in Figs 3b, 3c and 3d. The H NMR spectrumcan be rationalized in terms of either two symmetricalforms of the dimer present in equal proportions (Figs 3band 3c), or a single asymmetric dimer (Fig. 3d). The
:haride{a)
ligand /
(b)
1 disacc
-- _ -. 11 .......- "~'- _-_
Me
(C)
d

Dimer of glycopeptide antibiotic Groves et al. 749
largest differences in chemical shift between the twoforms occur for protons of the disaccharides and adjacentaromatic protons of residues 2, 4 and 6 suggesting that itis the orientation of the disaccharides that is fundamen-tally different in the two forms. The ambiguity inassigning two-dimensional (2D) NMR spectra of ere-momycin in complex with natural cell wall analogues haspreviously precluded a detailed description of the dimerstructure. This ambiguity has been resolved by bindingunnatural aromatic ligands which mimic the geometryand hydrogen-bonding pattern of the natural cell wallanalogues such as N-acetyl-D-Ala (Fig. 4a).Complexation-induced shifts from pyrrole-2-carboxylate(Fig. 4b) and indole-2-carboxylate (Fig. 4c) (binding
Fig. 4. The structures of (a) N-acetyl-D-Ala, (b) pyrrole-2-car-boxylate and (c) indole-2-carboxylate.
constants 340 ± 60M- 1 and 2200 ± +600M -1 , respectively,at 303 K) perturb the 1H NMR spectrum of the dimer tosuch an extent that two complete sets of antibiotic reso-nances have been assigned by 2D NMR methods. As aconsequence, intermolecular nuclear Overhauser effects(NOEs) have been identified at the dimer interface thatcan only be rationalized in terms of a parallel head-to-head alignment of the juxtaposed disaccharides,consistent with a single asymmetric dimer (Fig. 3d).
Intermolecular NOEs at the dimer interfaceA portion of the 50ms nuclear Overhauser effect spec-troscopy (NOESY) spectrum of the eremomycin dimercomplex with pyrrole-2-carboxylate (283K, pH7.0) isshown in Fig. 5. Both sets of 4-epi-vancosamine sugarresonances of the disaccharide are clearly resolved andindependently assigned by a combination of doublequantum filtered-correlated spectroscopy (DQF-COSY)and NOESY experiments. Strong intra-molecularNOEs, V1-- G2 and Vl*-4G2*, unambiguously identifythe glucose sugars with their respective disaccharides.(Each eremomycin monomer has the same atom labelswith asterisks distinguishing one eremomycin monomerfrom the other in the dimer complex: V, vancosamine;G, glucose.) Several intermolecular NOEs are observed
Fig. 5. Portion of the 50ms NOESY spectrum of the eremomycin dimer complex with pyrrole-2-carboxylate (283 K, pH 7.0). Eacheremomycin monomer has the same atom labels with asterisks distinguishing one eremomycin monomer from the other in the dimercomplex. The partial assignment of the epi-vancosamine sugars of the two disaccharides is highlighted, and several intermolecularNOEs identified in boxes (see text for details). NOEs are traced from V5 to both V6 and V7, and subsequently from V7 to V2e and Va(see Fig. 1). Dashed lines trace the correlations for one sugar (V), while solid lines trace the same pattern for its counterpart (V*.Weak chemical exchange cross-peaks between V6 and V6*, and V7 and V7* are labelled 'e'.
I (a) (b) (c) 1O H3C H
H O
G3 V2e*
g -------------t-------- - - -0!A , I V4 , IA 4~~~~~~~~~~~~~~~~~~~ I
1.5
ppm
'2.0
2.5
4.0 3.0 ppm 2.0
1-
I I

750 Structure 1994, Vol 2 No 8
Fig. 6. Conformation of the 4-epi-vancosamine sugar.Hydrogens are labelled V1to V7.
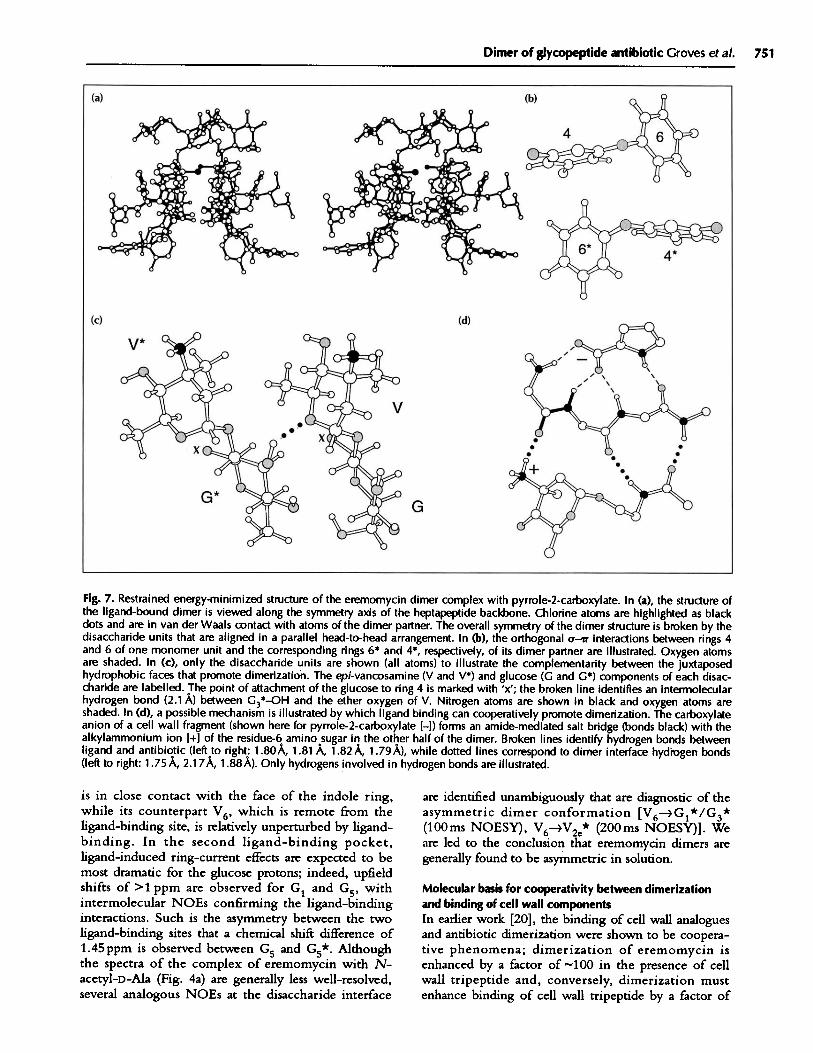
that are particularly noteworthy. The partial assignmentof the epi-vancosamine sugars of the two disaccharides ishighlighted in Fig. 5. For example, NOEs are tracedfrom V5 to both V6 and V7, and subsequently from V7to V2e and V2a (Fig. 6). The two sets of NOEs are well-resolved in 50ms data and permit an unambiguousassignment of two complete sets of resonances. It is nowevident that V6 gives an NOE cross-peak to the V2 e*resonance; i.e., the NOE V6--V 2e* is an intermolecularNOE (boxed cross-peak in Fig. 5) and arises from aninteraction at the dimer interface. By first examiningmodels of the two symmetric dimers (Figs 3b and 3c),we conclude that the NOE V6 -4V 2 * is incompatiblewith either symmetrical form since V6 and V2e* are, inboth cases, found on distal faces of the two disaccharideunits and are well separated. Additional intermolecularNOEs, V6-4V 7*, Gl* and G3* (Fig. 5), further supportthis conclusion. Only in the asymmetric conformationare different 'faces' of the two disaccharides broughtinto contact (Fig. 3d); thus, these NOEs (V6--V2e*,V7*, Gl* and G3*) define interactions at the dimerinterface and are uniquely characteristic of an asymmet-ric dimer. The hydrogens Gl* and G3 * are located onone face of the glucose sugar and, as is evident from thestructure of the complex shown in Fig. 7, the methylgroup V6 appears to nestle against the hydrophobic facepresented by the glucose sugar of its dimer partner.
Several other NOEs provide further evidence for theproposed parallel alignment of the two disaccharideunits in the asymmetric dimer. The V*, 4-epi-van-cosamine sugar is positioned between the benzyl ringsof residues 2 and 6 of the same monomer unit such thatV6 * gives an intramolecular NOE to 2e*, while V7*gives an intramolecular NOE to 6c* (Fig. 1).Analogous NOEs are not observed from V6 and V7 to2e and 6c (as would be expected if the two disaccha-rides had symmetry-related orientations), but anintramolecular interaction is identified betweenV7 -2e, indicating that the disaccharide is rotated by~180 ° about the glucose ring-4 glycosidic bond(Fig. 1). An additional intermolecular NOE, between
V6--6 e*, places the disaccharides unambiguously in anasymmetric parallel alignment. The observation ofinterfacial NOEs V9a/V9e--2b* and V4, V9 e,V9a-)x3*, and V9 a*/V 9e*-- 2 b and V1 4 *, V9 e*,V9a*-x 3 place the residue-6 amino sugars in theappropriate orientation for forming a charged inter-molecular hydrogen bond (Fig. 2); other interactionsthat support the head-to-tail orientation of the peptidebackbone include the NOEs x4-6f* and x4*-46f.These valuable NOE data have been used subsequentlyas restraints for structure calculations.
Structure of the asymmetric dimerThe NMR structure of the asymmetric dimer incomplex with pyrrole-2-carboxylate is illustrated inFig. 7a. Figs 7b, 7c and 7d highlight features thatstabilize the complex and dimer. Electrostatic contribu-tions to dimerization arising from ru-ir interactions(where the C-H bond of one aromatic ring is insertedinto the 'r-electron cloud of a second aromatic ring)are clearly evident at the dimer interface (Fig. 7b). Theorthogonal rings 4 and 6 of one eremomycin monomerare optimally aligned with rings 6* and 4*, respectively,of their dimer partner, consistent with the largechemical shift changes to protons 6e and 6f observed ondimerization [2]. The relative orientations of the twodisaccharides are illustrated in Fig. 7c. The hydrophobicface of the epi-vancosamine sugar of one disaccharidenestles against the face of the disaccharide of itsopposite number. The NOEs V6-4V2 e*, V7*, G1* andG3* correspond to distances of less than 2.6A in thefinal minimized structure, including several van derWaals contacts. A bridging intermolecular hydrogenbond (2.1 A) between G3*-OH and the ether oxygenof V (Fig. 7c) is identified, indicative of a high comple-mentarity between the two disaccharides.
The parallel orientation of the disaccharides leads tosubtle differences in the nature of the interactions in thetwo ligand-binding pockets. While the methyl groupV6* 'caps' one ligand-binding site (Fig. 7a), and givesNOEs to ligand protons H3 and H4, its counterpart,V6, is involved in interactions at the dimer interface andgives a quite different pattern of NOEs to glucoseprotons of its dimer partner. In the second ligand-binding site, the glucose sugar provides equivalenthydrophobic capping interactions that promote ligandbinding (Fig. 7a), as evidenced by NOEs from ligandH3 to G1/G5 and H4 to G6'. Thus, complementaryhydrophobic interactions between ligand and disaccha-ride are evident in both ligand-binding pockets andinvolve burial of very similar non-polar surface areas;however, the origin of the interaction in each case israther different.
The most striking effect of the dimer asymmetry onligand-induced chemical shift changes is illustrated forthe binding of indole-2-carboxylate (Fig. 4c). Theindole ring produces a dramatic (>1 ppm upfield)ring-current effect on the chemical shift of V6*, which
V 2Va
HOHI
V 6V 7V 5
Ha
Dimer of glycopeptide antibiotic Groves et al. 751
Fig. 7. Restrained energy-minimized structure of the eremomycin dimer complex with pyrrole-2-carboxylate. In (a), the structure ofthe ligand-bound dimer is viewed along the symmetry axis of the heptapeptide backbone. Chlorine atoms are highlighted as blackdots and are in van der Waals contact with atoms of the dimer partner. The overall symmetry of the dimer structure is broken by thedisaccharide units that are aligned in a parallel head-to-head arrangement. In (b), the orthogonal o-r' interactions between rings 4and 6 of one monomer unit and the corresponding rings 6* and 4*, respectively, of its dimer partner are illustrated. Oxygen atomsare shaded. In (c), only the disaccharide units are shown (all atoms) to illustrate the complementarity between the juxtaposedhydrophobic faces that promote dimerization. The epi-vancosamine (V and V*) and glucose (G and G*) components of each disac-charide are labelled. The point of attachment of the glucose to ring 4 is marked with 'x'; the broken line identifies an intermolecularhydrogen bond (2.1 A) between G3*-OH and the ether oxygen of V. Nitrogen atoms are shown in black and oxygen atoms areshaded. In (d), a possible mechanism is illustrated by which ligand binding can cooperatively promote dimerization. The carboxylateanion of a cell wall fragment (shown here for pyrrole-2-carboxylate [-l) forms an amide-mediated salt bridge (bonds black) with thealkylammonium ion +] of the residue-6 amino sugar in the other half of the dimer. Broken lines identify hydrogen bonds betweenligand and antibiotic (left to right: 1.80A, 1.81 A, 1.82 A, 1.79 A), while dotted lines correspond to dimer interface hydrogen bonds(left to right: 1.75 A, 2.177A, 1.88A). Only hydrogens involved in hydrogen bonds are illustrated.
is in close contact with the face of the indole ring,while its counterpart V6 , which is remote from theligand-binding site, is relatively unperturbed by ligand-binding. In the second ligand-binding pocket,ligand-induced ring-current effects are expected to bemost dramatic for the glucose protons; indeed, upfieldshifts of >1 ppm are observed for G and G5 , withintermolecular NOEs confirming the ligand-bindinginteractions. Such is the asymmetry between the twoligand-binding sites that a chemical shift difference of1.45ppm is observed between G5 and G5*. Althoughthe spectra of the complex of eremomycin with N-acetyl-D-Ala (Fig. 4a) are generally less well-resolved,several analogous NOEs at the disaccharide interface
are identified unambiguously that are diagnostic of theasymmetric dimer conformation [V6 -- G l* /G 3*(100ms NOESY), V6-- V2e* (200ms NOESY)]. Weare led to the conclusion that eremomycin dimers aregenerally found to be asymmetric in solution.
Molecular basis for cooperativity between dimerizationand binding of cell wall componentsIn earlier work [20], the binding of cell wall analoguesand antibiotic dimerization were shown to be coopera-tive phenomena; dimerization of eremomycin isenhanced by a factor of -100 in the presence of cellwall tripeptide and, conversely, dimerization mustenhance binding of cell wall tripeptide by a factor of

752 Structure 1994, Vol 2 No 8
10 (100), providing a strong link between dimerizationand mode of action - binding to cell wall mucopep-tide. One manner in which this may be achieved isevident in Fig. 7d. The carboxylate anion of a cell wallfragment (shown here for the pyrrole-2-carboxylateanion) makes an ion-pair interaction with the alkylam-monium ion of the ring-6 amino sugar in the other halfof the dimer, with the interaction mediated through apolarizable amide group [17,19]. Thus, the structureand positioning of the residue-6 amino sugar appears tobe important. This example of cooperativity may berelevant to more general mechanisms involving dimer-ization in signalling [21-26], where a high specificity isattainable by a ligand that is able to simultaneously bindto the receptor and promote dimerization. Compoundsthat satisfy these stringent requirements are, of course,agonists. Thus, the cooperative effect of binding cellwall peptides on antibiotic dimerization may havefeatures in common with processes involving agonist-induced signal transduction.
Dimer asymmetry - the exception or the rule?Do glycopeptide antibiotics, in general, form asymmet-ric dimers relevant to their mode of action? NMRspectra of complexes of the antibiotic ristocetin Areveal several sets of signals that have been attributed totwo forms of an antibiotic dimer in equilibrium withthe monomer at 298K [27]; a dimerization constant of-500M - ' has been estimated for ristocetin A alone.The NMR data do reveal intermolecular NOEs thatare compatible with an asymmetric orientation of theantibiotic tetrasaccharides (unpublished data) and whichare consistent with the structure of eremomycinpresented here. We propose that glycopeptides thatdimerize are likely to do so in a manner analogous tothe stricture presented here, and that the asymmetricorientation of the ring-4 sugars appears to promotedimerization by sugar-sugar recognition. In the case oferemomycin, the manner in which dimerization ispromoted by structural epitopes which appear asadditions to the basic heptapeptide motif is striking.The introduction of sugars at two sites (rings 4 and 6)and a chlorine substituent (ring 2) enhances dimeriza-tion of eremomycin by a factor of -200 000 [19],leading to the conclusion that these epitopes have beenadded under selectional pressure to optimize activity bydimerization [28]. Further, members of the glycopep-tide group of antibiotics that dimerize strongly showactivity that is greater than expected on the basis oftheir binding affinities for cell wall peptides in vitro [20].In addition, their in vivo activity is difficult to antago-nize with exogenous cell wall peptide fragments (DABeauregard, DH Williams, DJC Knowles and MNAwynn, unpublished data), providing strong support forthe role of dimerization in the mode of action of theglycopeptide antibiotics.
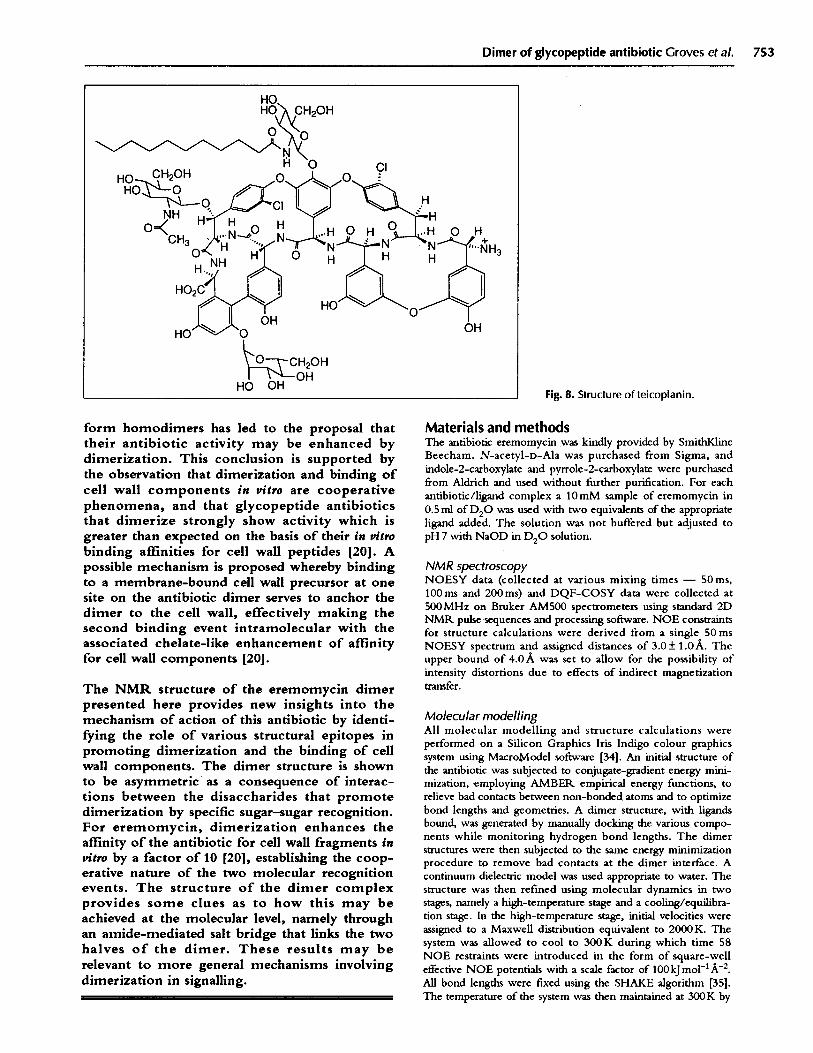
Teicoplanin (Fig. 8) is the only glycopeptide so farstudied that shows no measurable propensity todimerize, but it functions extremely effectively as an
antibiotic. Recently, it has been proposed [20] that partof the reason for its efficacy lies in the addition of astructural epitope - a C10 lipid side chain attached tothe residue-4 sugar substituent - that acts as amembrane anchor for the antibiotic. Thus, by attachingthe antibiotic to the cell membrane by a lipid anchor,the interaction of the antibiotic with cell wall peptidewould be effectively intramolecular and hence theaffinity is also enhanced by the chelate effect [20]. It isstriking that in precisely the case where dimerization isnot observed (i.e. for teicoplanin), a structural addition(the C 10 alkyl chain) is present promoting a functionallysimilar intramolecular event to enhance binding affinity.This hypothesis is, of course, consistent with the notionthat molecular elaborations requiring additionalenzymes and biosynthetic effort imply functional rolesfor these elaborations [28]. The above hypothesis is alsosupported by recent findings that teicoplanin is activeagainst bacterial strains possessing low-level vancomycinresistance (vanB phenotype) [29]. This resistance arisesfrom the substitution of a significant proportion of the-D-Ala-D-Ala terminating peptidoglycan precursors by-D-Ala-lactate sequences. The affinity of glycopeptidesfor such modified peptides is much diminished [30],which alone should significantly reduce their activity.However, in the case of glycopeptide dimers, a furtherfactor may act to decrease their activity; if a high pro-portion of the -D-Ala-D-Ala containing precursors aretransformed into -D-Ala-lactate, then the probability oftwo of the remaining -D-Ala-D-Ala peptides beingadjacent to each other is significantly reduced. Thus,the intramolecular advantage of dimerization presentedabove would be largely negated. However, teicoplaninin the analogous case does not require two adjacentprecursors to display a binding enhancement by anintramolecular binding process (as proposed above) andcould therefore retain antibiotic activity, as long as some-D-Ala-D-Ala-containing precursors were present.Further, semi-synthetic derivatives of vancomycin, inwhich the amino group of the residue-4 disaccharidehad been acylated or alkylated with C8 -C 10 lipids[31,32], were found to possess excellent activity againstvancomycin-resistant enterococci [33]. Interaction ofthe modified vancomycin with the cytoplasmicmembrane, in an analogous fashion to that proposedabove for teicoplanin, may play some part in theenhanced antibiotic activity.
Biological implicationsThe glycopeptide family of antibiotics hasassumed great clinical importance as the lastline of defence against often-lethal infectionsby the multiply drug-resistant 'super-bug'Staphylococcus aureus. The antibiotics exert theirphysiological action by blocking the cell wallsynthesis of Gram-positive bacteria by recogniz-ing and binding to nascent cell wall mucopeptideprecursors terminating in -D-Ala-D-Ala. Theobservation that many glycopeptide antibiotics
Dimer of glycopeptide antibiotic Groves et al. 753
Fig. 8. Structure of teicoplanin.
form homodimers has led to the proposal thattheir antibiotic activity may be enhanced bydimerization. This conclusion is supported bythe observation that dimerization and binding ofcell wall components in vitro are cooperativephenomena, and that glycopeptide antibioticsthat dimerize strongly show activity which isgreater than expected on the basis of their in vitrobinding affinities for cell wall peptides [20]. Apossible mechanism is proposed whereby bindingto a membrane-bound cell wall precursor at onesite on the antibiotic dimer serves to anchor thedimer to the cell wall, effectively making thesecond binding event intramolecular with theassociated chelate-like enhancement of affinityfor cell wall components [20].
The NMR structure of the eremomycin dimerpresented here provides new insights into themechanism of action of this antibiotic by identi-fying the role of various structural epitopes inpromoting dimerization and the binding of cellwall components. The dimer structure is shownto be asymmetric as a consequence of interac-tions between the disaccharides that promotedimerization by specific sugar-sugar recognition.For eremomycin, dimerization enhances theaffinity of the antibiotic for cell wall fragments invitro by a factor of 10 [20], establishing the coop-erative nature of the two molecular recognitionevents. The structure of the dimer complexprovides some clues as to how this may beachieved at the molecular level, namely throughan amide-mediated salt bridge that links the twohalves of the dimer. These results may berelevant to more general mechanisms involvingdimerization in signalling.
Materials and methodsThe antibiotic eremomycin was kindly provided by SmithKlineBeecham. N-acetyl-D-Ala was purchased from Sigma, andindole-2-carboxylate and pyrrole-2-carboxylate were purchasedfrom Aldrich and used without further purification. For eachantibiotic/ligand complex a 10mM sample of eremomycin in0.5 ml of D2 0 was used with two equivalents of the appropriateligand added. The solution was not buffered but adjusted topH 7 with NaOD in D20 solution.
NMR spectroscopyNOESY data (collected at various mixing times - 50ms,100ms and 200ms) and DQF-COSY data were collected at500MHz on Bruker AM500 spectrometers using standard 2DNMR pulse sequences and processing software. NOE constraintsfor structure calculations were derived from a single 50 msNOESY spectrum and assigned distances of 3.0+ 1.0 A. Theupper bound of 4.OA was set to allow for the possibility ofintensity distortions due to effects of indirect magnetizationtransfer.
Molecular modellingAll molecular modelling and structure calculations wereperformed on a Silicon Graphics Iris Indigo colour graphicssystem using MacroModel software [34]. An initial structure ofthe antibiotic was subjected to conjugate-gradient energy mini-mization, employing AMBER empirical energy functions, torelieve bad contacts between non-bonded atoms and to optimizebond lengths and geometries. A dimer structure, with ligandsbound, was generated by manually docking the various compo-nents while monitoring hydrogen bond lengths. The dimerstructures were then subjected to the same energy minimizationprocedure to remove bad contacts at the dimer interface. Acontinuum dielectric model was used appropriate to water. Thestructure was then refined using molecular dynamics in twostages, namely a high-temperature stage and a cooling/equilibra-tion stage. In the high-temperature stage, initial velocities wereassigned to a Maxwell distribution equivalent to 2000K. Thesystem was allowed to cool to 300K during which time 58NOE restraints were introduced in the form of square-welleffective NOE potentials with a scale factor of 100kJmol-'A- 2.All bond lengths were fixed using the SHAKE algorithm [35].The temperature of the system was then maintained at 300K by

754 Structure 1994, Vol 2 No 8
resealing velocities every 0 .2 ps with the time step of the integra-tor set to 1.5fs. The total dynamics simulation time was 100ps.Several low-energy dynamics structures were further refinedusing restrained energy minimization with the final structuresshowing no violations of the distance bounds. In fact, themajority of restrained distances in the final minimized structuresfell well within the upper distance bound of 4.0A, consistentwith the observation of the corresponding NOEs in relativelyshort mixing time NOESY experiments.
Coordinates of the eremomycin dimer complex are availablefrom the authors on request.
Acknowledgements: We thank SERC, GLAXO, Xenova (PG),British Council and Merck OPM) for financial support and theBiomedical NMR Centre, Mill Hill, for access to NMRequipment.
References1. Kalman, J.R. & Williams, D.H. (1977). Structural and mode of
action studies on the antibiotic vancomycin. Evidence from270-MHz proton magnetic resonance. . Am. Chem. Soc. 99,2768-2774.
2. Spiri-Nakagawa, P., Tanaka, Y., Oiwa, R., Tanaka, H. & Omura, S.(1979). Studies on bacterial cell wall inhibitors VIII. Mode ofaction of a new antibiotic, azureomycin B in Bacillus cereus T.I. Antibiot. (Tokyo) 32, 995-1001.
3. Hunt, A.H. & Vernon, P.D. (1981). Complexation of acetyl-D-alanyl-D-alanine by antibiotic A35512B. . Antibiot. (Tokyo) 34,469-471.
4. Bardone, M.R., Paternoster, M. & Coronelli, L. (1978).Teicomycins, new antibiotics from Actinoplanes teicomyceticusspecies nova II. Extraction and chemical characterisation.I. Antibiot (Tokyo) 31, 170-177.
5. McHenry, M.C. & Gavan, T.L. (1983). Vancomycin. Pediatr. Clin.North Am. 30, 31-47.
6. Geraci, J.E. & Herman, P.E. (1983). Vancomycin. Mayo Clin. Proc.58, 88-91.
7. Wise, R. & Reeves, D.S., Eds. (1984). Vancomycin Therapy.J. Antimicrob. Chemother., 14 (suppl. D).
8. Foldes, M., Munro, R., Sorrell, T.C., Shankar, S. & Toohey, M.J.(1990). In vitro effects of vancomycin, rifampicin and fusidic acid,alone and in combination, against methicillin resistantStaphylococcus aureus. . Antibiot. Agents Chemother. 11, 21-26.
9. Brant, M., Wingert, P. & Hager, P. (1994). The end of antibiotics?Newsweek 123, 38-43.
10. Courvalin, P. (1990). The resistance of enterococci to glycopep-tides. Antimicrob. Agents Chemother. 34, 2291-2296.
11. Wade, J., Baillie, L., Rolando, N. & Casewell, M. (1992).Pristinamycin for Enterococcus faecium resistant to vancomycinand gentamicin. Lancet 339, 312-313.
12. French, G., Abdulla, Y., Heathcock, R., Poston, S. & Cameron, J.(1992). Vancomycin resistance in south London. Lancet 339,818-819.
13. Bugg, T.D.H., Dutka-Malen, S., Arthur, M., Courvalin, P. & Walsh,C.T. (1991). Identification of vancomycin resistance protein Van Aas a -D-Ala-D-Ala ligase of altered substrate specificity.Biochemistry 30, 2017-2021.
14. Barna, J.C.J. & Williams, D.H. (1984). The structure and mode ofaction of the glycopeptide antibiotics of the vancomycin group.Annu. Rev. Microbiol. 38, 339-357.
15. Nieto, A. & Perkins, H.R. (1971). Modifications of the acyl-D-alanyl-D-alanine terminus affecting complex formation withvancomycin. Biochem. J. 123, 789-803.
16. Kannan, R., Harris, C.M., Harris, T.M., Waltho, J.P., Skelton, N.J.& Williams, D.H. (1988). Function of the amino sugar and the N-terminal amino acid of the antibiotic vancomycin in itscomplexation with cell wall peptides. J. Am. Chem. Soc. 110,2946-2953.
17. Waltho, J.P. & Williams, D.H. (1989). Aspects of molecular recog-nition: solvent exclusion and dimerization of the antibioticristocetin when bound to a model bacterial cell wall precursor. J.Am. Chem. Soc. 111, 2475-2480.
18. Gerhard, U., Mackay, J.P., Maplestone, R.A. & Williams, D.H.(1993). The role of the sugar and chlorine substituents in thedimerization of vancomycin antibiotics. . Am. Chem. Soc. 115,232-237.
19. Mackay, J.P., Gerhard, U., Beauregard, D.A., Maplestone, R.A. &Williams, D.H. (1994). Dissection of the contributions towardsdimerization of glycopeptide antibiotics. J. Am. Chem. Soc. 116,4573-4580.
20. Mackay, J.P., Gerhard, U., Beauregard, D.A., Westwell, M.S.,Searle, M.S. & Williams, D.H. (1994). Glycopeptide antibioticactivity and the possible role of dimerization: a model for biologi-cal signalling. . Am. Chem. Soc, 116, 4581-4590.
21. Ullrich, A. & Schlessinger, J. (1990). Signal transduction byreceptors with tyrosine kinase activity. Cell 61, 203-212.
22. Milburn, M.V., et a/., & Kim, S.-H. (1991). 3-Dimensional struc-tures of the ligand binding domain of the bacterial aspartatereceptor with and without ligand. Science 254, 1342-1347.
23. Canals, F. (1992). Signal transmission by epidermal growth factorreceptor: coincidence of activation and dimerization.Biochemistry 31, 4493-4501.
24. Spencer, D.M., Wandless, T.J., Schreiber, S.L. & Crabtree, G.R.(1993). Controlling signal transduction with synthetic ligands.Science 262, 1019-1024.
25. Zhou, M., et al., & Schlessinger, J. (1993). Real-time measure-ments of kinetics of EGF binding to soluble EGF receptormonomers and dimers support the dimerization model for receptoractivation. Biochemistry 32, 8193-8198.
26. Ohashi, H., Maruyama, K., Liu, Y.-C. & Yoshimuru, A. (1994).Ligand-induced activation of chimeric receptors between the ery-thropoietin receptor and tyrosine kinases. Proc. Natl. Acad. Sci.USA 91, 158-162.
27. Groves, P., Searle, M.S., Chicarelli-Robinson, I. & Williams, D.H.(1994). Recognition of the cell wall binding site of the van-comycin-group antibiotics by unnatural structural motifs. 1H NMRstudies of the effects of ligand binding on antibiotic dimerization.J. Chem. Soc. Perkin Trans. 1659-665.
28. Williams, D.H., Stone, M.J., Hauck, P.R. & Rahman, S.K. (1989).Why are secondary metabolites (natural products) biosynthesised?J. Nat. Prods. LLoydia 52, 1189-1208.
29. Arthur, M. & Courvalin, P. (1993). Genetics and mechanisms ofglycopeptide resistance in Enterococci. Antimicrob. AgentsChemother. 37, 1563-1571.
30. Bugg, T.D.H., Wright, G.D., Dutka-Malen, S., Arthur, M.,Courvalin, P. & Walsh, C.T. (1991). Molecular basis for van-comycin resistance in Enterococcus faecium BM4147 -biosynthesis of a depsipeptide peptidoglycan precursor by van-comycin resistance proteins Van H and Van A. Biochemistry 30,10408-10415.
31. Nagarajan, R., Schabel, A.A., Occolowitz, ).L., Counter, F.T. &Ott, J.L. (1988). Synthesis and antibacterial activity of N-acyl van-comycins. i. Antibiot. (Tokyo) 41, 1430-1438.
32. Nagarajan, R., Schabel, A.A., Occolowitz, .L., Counter, F.T.,Ott, J.L. & Felty-Duckworth, A.M. (1989). Synthesis and anti-bacterial evaluation of N-alkyl vancomycin. J. Antibiot. (Tokyo)42, 63-72.
33. Nagarajan, R. (1993). Structural activity relationships of van-comycin-type glycopeptide antibiotics. . Antibiot. (Tokyo) 46,1181-1195.
34. Mohamadi, F., et al., & Still, W. C. (1990). MacroModel - anintegrated software system for modelling organic and bioorganicmolecules using molecular mechanics. . Comput. Chem. 11,440-467.
35. Van Gunsteren, W. & Berendsen, H.J.C. (1990). Computer simu-lation of molecular dynamics: methodology, applications andperspectives in chemistry. Angew. Chem. 29, 992-1023.
Received: 6 Jun 1994; revisions requested: 4 ul 1994;revisions received: 7 Jul 1994. Accepted: 8 Jul 1994.