thermal plasma processing -...
TRANSCRIPT

THERMAL PLASMA PROCESSING
OF FINE GRAINED
MATERIALS
by
MURALIDHARAN RAMACHANDRAN
RAMANA G. REDDY, COMMITTEE CHAIR
GARRY W. WARREN MARK L. WEAVER
YUEBIN GUO UDAY K. VAIDYA
A DISSERTATION
Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy
in the Department of Metallurgical and Materials Engineering in the Graduate School of
The University of Alabama
TUSCALOOSA, ALABAMA
2012

Copyright Muralidharan Ramachandran 2012 ALL RIGHTS RESERVED

ii
ABSTRACT
Synthesis of advanced ceramic materials has been systematically investigated using non-
transferred thermal plasma reactor. Low cost oxide feed materials has been used as the solid feed
to the reactor while methane (CH4) and argon (Ar) were used as reducing and carrier gases,
respectively.
A 2D computational fluid dynamics (CFD) based mathematical model was developed to
understand the flow and temperature profiles inside the reactor. The concept was extended to a
3D model and a comparison was made with the results from 2D model. The velocity increases
linearly with increase in the pressure in both the 2D and the 3D models while the maximum
velocity from 3D model is lower by about 35-40 m/s at any given pressure. A decrease in the
residence time was observed in the 3D model compared to the 2D model. An intermediate
plasma gas pressure of 45 psi was used in experiments to ensure high temperatures and residence
times in the reactor.
Thermochemical calculations have been carried out to determine the molar ratio of the
reducing gas to be used in TiO2-B2O3-CH4 and SiO2-CH4 system. The maximum theoretical
yield of TiB2 of about 82 mol% was obtained at a molar ratio of TiO2:B2O3:CH4 = 1:1:5.
Maximum theoretical yield of SiC of about 97 mol% was obtained at a molar ratio of
SiO2:CH4=1:3 at a temperature of 1520ºC.
Experiments were carried out using thermal plasma reactor to synthesize TiB2 and SiC.
The maximum yield of TiB2 of about 40 mol% was obtained with a feed molar ratio of

iii
TiO2:B2O3:CH4 = 1:1:5 and a power of 23.4 kW. Relatively higher solid feed rates increased the
yield of TiB2. The TiB2 spherical particles formed are in the range of 20-100 nm. A change in
crystal structure was observed in TiO2 from anatase to rutile.
Experiments using a molar ratio of SiO2:CH4 = 1:2 produced maximum yield of SiC of
about 65 mol% at a solid feed rate of 5 g/min. Mostly spherical morphology with some nanorods
have been observed. The presence of Si had been observed and was quantified using XRD, HR-
TEM, Raman and XPS.

iv
DEDICATION
This dissertation is dedicated to my mom, dad and brother.

v
LIST OF ABBREVIATIONS AND SYMBOLS
Density
r Radius
t Time
Velocity vector
Sm Source of any mass added from dispersed second phase
x
and r
Axial and radial velocities respectively
p Static pressure
τ Stress tensor
g Gravitational force
F External body forces
t Turbulent (or Eddy) viscosity
Gk Generation of turbulent kinetic energy (TKE) due to mean velocity gradients
Gb Generation of TKE due to buoyancy
YM Contribution of fluctuating dilatation in compressible turbulence to the overall dissipation rate
k Turbulent kinetic energy
Turbulence dissipation rate
keff Effective thermal conductivity.

vi
kt Turbulent thermal conductivity
Jj Diffusion flux of species j
Sh Heat of chemical reaction or any other volumetric heat source
h Sensible enthalpy
Yj Mass fraction of species j
hj Sensible enthalpy of species j
Specific dissipation rate
G Generation of
Yk and Y Dissipation of k and due to turbulence respectively
Sk and S User-defined sources terms for k and respectively
k and Turbulent Prandtl numbers for k and respectively
* Coefficient
Intermittency
S Strain rate magnitude
Flength Empirical correlation that controls the length of the transition length
Vorticity magnitude
G Total Gibbs energy of the system
Gi0 Standard molar Gibbs energy of species i at temperature T and pressure P
ni Number of moles of species i
Pi Partial pressure of species i
Xi Mole fraction of species i
i Activity coefficient of species i.
ITiB2 Intensity of TiB2

vii
ITiO2 Intensity of TiO2
IB2O3 Intensity of B2O3
IC Intensity of C
cTiB2 Volume fraction of TiB2
cTiO2 Volume fraction of TiO2
cB2O3 Volume fraction of B2O3
cC Volume fraction of C
RTiB2 Volume of inverse unit cell lattices of TiB2
RTiO2 Volume of inverse unit cell lattices of TiO2
RB2O3 Volume of inverse unit cell lattices of B2O3
RC Volume of inverse unit cell lattices of C

viii
ACKNOWLEDGMENTS
I would like to take this opportunity to express my sincere gratitude to my advisor and
chairman of this dissertation committee, Dr. Ramana G. Reddy, for his constant support and
guidance throughout my graduate study and research work at the University of Alabama.
I would like to thank Dr. Garry W. Warren, Dr. Mark L. Weaver, Dr. Yuebin Guo, and
Dr. Uday K. Vaidya for serving on my dissertation committee and providing valuable
suggestions and insights.
I’m thankful to the faculty, staff and students at the Metallurgical and Materials
Engineering department for their timely help. Special thanks to Dr. Divakar Mantha, research
engineer, for extensive technical discussions, comments and encouragement. I would like to
thank Dr. Muhammad Ali Rob Sharif for his help with the computational fluid dynamic
modeling of the plasma reactor. I thank the members of the central analytical facility (CAF) for
their constant help during characterization.
Thanks to ACIPCO and the University of Alabama for the financial support.
A special thanks to my parents, brother, and all my friends for their constant moral
support in all my endeavors.

ix
CONTENTS
ABSTRACT ................................................................................................ ii
DEDICATION ........................................................................................... iv
LIST OF ABBREVIATIONS AND SYMBOLS ........................................v
ACKNOWLEDGMENTS ....................................................................... viii
LIST OF TABLES ................................................................................... xiii
LIST OF FIGURES ...................................................................................xv
1. INTRODUCTION ...................................................................................1
2. LITERATURE REVIEW AND RESEARCH OBJECTIVES ................8
2.1 Plasma synthesis ..............................................................................8
2.2 Plasma sources for material synthesis..............................................9
2.3 Advanced material synthesis using thermal plasma ......................11
2.4 Properties and synthesis of titanium diboride ................................11
2.5 Properties and synthesis of silicon carbide ....................................12
2.6 Research objectives ........................................................................15
3. MATHEMATICAL MODEL ................................................................17
3.1 2D plasma reactor model ...............................................................17
3.2 Defining equations in 2D model ....................................................18
3.3 Grid refinement in 2D model .........................................................21
3.4 Results from 2D plasma reactor model ..........................................25

x
3.5 3D plasma reactor model ...............................................................31
3.6 Defining equations in 3D model ....................................................32
3.7 Turbulence and grid refinement in 3D model ................................34
3.8 Results from 3D plasma reactor model ..........................................44
4. EXPERIMENTAL SETUP ....................................................................49
4.1 Plasma power source ......................................................................49
4.2 Plasma reactor system ....................................................................54
4.2.1 Reaction zone ........................................................................54
4.2.2 Quench zone..........................................................................56
4.2.3 Filter zone .............................................................................57
4.3 Powder feeder ................................................................................58
4.4 Water source ..................................................................................59
4.5 Gas source ......................................................................................62
4.6 Raw materials.................................................................................63
4.7 Characterization of product powders .............................................66
4.7.1 X-Ray Diffraction (XRD) ....................................................66
4.7.2 Scanning Electron Microscope (SEM) ................................66
4.7.3 Transmission Electron Microscopy (TEM) .........................66
4.7.4 Differential Scanning Calorimetry (DSC) ...........................67
4.7.5 Raman Spectra .....................................................................67
4.7.6 X-Ray Photoelectron Spectroscopy (XPS) ..........................68
4.7.7 Thermo-Gravimetric and Differential Thermal Analyzer (TG-DTA) ............................................................................68
5. SYNTHESIS OF TITANIUM DIBORIDE ...........................................69

xi
5.1 Thermochemical calculations ........................................................69
5.2 Synthesis and characterization of titanium diboride ......................75
5.2.1 Phase and morphology of solid feed ....................................75
5.2.2 Phase, composition and morphology of product powders ...76
5.2.3 Phase transformation ............................................................83
5.2.4 Particle size reduction ..........................................................84
6. SYNTHESIS OF SILICON CARBIDE.................................................85
6.1 Thermochemical calculations ........................................................85
6.2 Synthesis and characterization of silicon carbide ..........................89
6.2.1 Phase and composition of product powders.........................89
6.2.2 Morphology of product powders .........................................98
6.2.3 Particle size reduction ........................................................103
6.2.4 Qualitative analysis of product powders ............................103
6.2.5 Post-processing of product powders ..................................106
7. CONCLUSIONS AND FUTURE WORK ..........................................108
7.1 CFD modeling of plasma reactor .................................................108
7.2 Synthesis of titanium diboride .....................................................109
7.3 Synthesis of silicon carbide .........................................................110
7.4 Future work ..................................................................................111
7.4.1 Modeling of plasma reactor ..........................................111
7.4.2 Synthesis of TiB2 ..........................................................112
7.4.3 Separation of Si, SiC and SiO2 .....................................112
REFERENCES ........................................................................................114

xii
APPENDIX ..............................................................................................121
A.1 INSTRUCTIONS FOR THE OPERATION OF PLASMA POWER SOURCE IN NON-TRANSFERRED MODE ............121
A.2 PROPERTIES OF GASES USED IN MODELING ..................125
A.3 CALCULATION OF VOLUME FRACTION USING DIRECT COMPARISON METHOD .........................................128

xiii
LIST OF TABLES
3.1 Initial and boundary condition values used during grid refinement studies .........................................................................23
3.2 Values of pressures used in determining the velocity and temperature profiles ............................................................................25
3.3 Average residence time in the plasma reactor as a function of plasma inlet pressure ...........................................................................30
3.4 Average residence time in the plasma reactor using 3D model as a function of plasma inlet pressure .....................................................48
4.1 Flow rate of cooling water for various parts of the plasma reactor system ..........................................................................61
4.2 Programmable gas controller set points in the thermal plasma power source for various types of torch ..................................63
5.1 Theoretical Yield of TiB2 and by-products in mol% as a function of molar ratio of methane in feed ........................................................72
5.2 Experimental design for the production of TiB2 using thermal plasma reactor ........................................................................75
5.3 Yield of product powders at different feed rates with a methane molar ratio of 5 in the feed ...................................................................78
6.1 Experimental Conditions for the production of SiC using thermal plasma .....................................................................................89
6.2 Weight fraction of product powders from experiment 1 .....................96
A.2.1 Viscosity (piecewise-linear) of argon ............................................125
A.2.2 Thermal conductivity (piecewise-linear) of argon .........................125
A.2.3 Heat capacity, CP (piecewise-linear) of methane ...........................126

xiv
A.2.4 Viscosity (piecewise-linear) of methane ........................................127
A.2.5 Thermal conductivity (piecewise-linear) of methane ....................127
A.3.1 The values of diffraction constants used in the calculation of volume fraction of product powders from TiB2 experiments ........129

xv
LIST OF FIGURES
1.1 Conceptual diagram of aerosol combustion process [3] ........................2
2.1 Schematic temperature distribution in a DC plasma arc (a) and RF discharge (b). ..................................................................................10
3.1 Schematic of the thermal plasma reactor. ............................................21
3.2 Grid of the plasma reactor with 4154 nodes (X=30; Y=133). .............21
3.3 Grid of the plasma reactor with 7257 nodes (X=40; Y=176). .............21
3.4 Grid of the plasma reactor with 11322 nodes (X=50; Y=221). ...........21
3.5 Velocity (a), stream function (b) and temperature (c) at a distance of X=1.8 cm from the plasma source for various
grid configurations. ..............................................................................22
3.6 Contour of temperature (a) and velocity (b) at a plasma inlet pressure of 45 psi. ................................................................................26
3.7 Effect of pressure on the inlet velocity of the plasma. .........................27
3.8 Profiles obtained from different areas of the reactor denoted by the line numbers. ........................................................................................28
3.9 Velocity profile along radial (a) and axial (b) directions at inlet pressure of 45 psi. ........................................................................29
3.10 Assume Flow Path of Solid Feed .......................................................30
3.11 Various grid sizes for grid refinement studies in 3D thermal plasma reactor: (a) 8013 nodes, (b) 11516 nodes and (c) 18936 nodes .........34
3.12 Velocity profile at the cross-section of the feed port: (a) standard k- model, (b) standard k- model and (c) Transition SST model ...35

xvi
3.13 Velocity profile at the symmetry of the reactor: (a) standard k- model, (b) standard k- model and (c) Transition SST model ..........36
3.14 Line profiles created in the reactor to compare the modeling results from various case studies ........................................................37
3.15 Comparison of velocity magnitudes along the feed entry point using the three turbulence models .....................................................38
3.16 Comparison of velocity magnitudes along the center of reactor using the three turbulence models .....................................................39
3.17 Velocity profile at the cross-section of the feed port: (a) 8013 nodes, (b) 11516 nodes and (c) 18936 nodes ....................................40
3.18 Velocity profile at the symmetry of the reactor: (a) 8013 nodes, (b) 11516 nodes and (c) 18936 nodes ...............................................41
3.19 Comparison of velocity magnitudes along the feed entry point using three different grid sizes ..........................................................42
3.20 Comparison of velocity magnitudes along the center of reactor using three different grid sizes ..........................................................43
3.21 Comparison of velocity magnitudes along the feed entry point using five different plasma inlet gas pressures ..................................45
3.22 Comparison of velocity magnitudes along the center of the reactor using five different plasma inlet gas pressures ..................................46
3.23 Effect of pressure on the inlet velocity of the plasma and comparison between the 2D model and the 3D model ......................47
3.24 Assume flow path for solid feed in 3D model ...................................48
4.1 Schematic of thermal plasma processing system .................................50
4.2 Schematic of non-transferred (left) and transferred (right) plasma arc torches ............................................................................................50
4.3 Photograph of plasma power source control panel ..............................51
4.4 Photograph of the new (left) and used (right) electrode assembly in a non-transferred plasma torch .............................................................52

xvii
4.5 Photograph of the electronic gas flow rate monitor on the plasma source ..................................................................................................53
4.6 Photograph of non-transferred thermal plasma reactor system ...........54
4.7 Photograph of the reaction zone ..........................................................55
4.8 Photograph of the two concentric copper quenching coils in the quench zone .........................................................................................56
4.9 Photograph of cloth filter in filter zone ................................................57
4.10 Photograph of the powder feeder (left) and the circuit of the control panel (right) ...........................................................................58
4.11 Photograph of cooling water system and roughing pump assembly ............................................................................................60
4.12 Flow meters controlling the flow rate of cooling water to various parts of the plasma reactor system ....................................................61
4.13 Photograph of gas supply system consisting of plasma, carrier and reducing gases .............................................................................62
4.14 Calibration of powder feeder for the mixture of TiO2 and B2O3 as the solid feed .................................................................................64
4.15 Calibration of powder feeder using SiO2 as the solid feed ................65
5.1 Formation of various product phases at a molar ratio of (a) TiO2:B2O3:CH4=1:1:4 and (b) TiO2:B2O3:CH4=1:1:5 ...................70
5.2 Formation of various product phases at a molar ratio of (a) TiO2:B2O3:CH4=1:1:6 and (b) TiO2:B2O3:CH4=1:1:7 ...................71
5.3 Theoretical yield of TiB2 and C at various molar ratio’s of methane to solid feed .......................................................................74
5.4 X-Ray diffraction pattern of 1:1 molar TiO2:B2O3 ..............................75
5.5 SEM images of solid feed powder at a molar ratio of TiO2:B2O3 = 1:1 ...................................................................................76
5.6 XRD patterns of product powders obtained from different experiments ..........................................................................................77

xviii
5.7 SEM images of product powders obtained from experiment 4 ...........79
5.8 SEM electron image (top) of the product powders obtained from experiment 4 and the corresponding EDS spectra (bottom) ................80
5.9 TEM image of the product powder obtained from experiment 4 ........81
5.10 HRTEM of product powders showing lattice fringes of hexagonal B2O3 in the (1 0 0) plane obtained from experiment 4 ......................82
5.11 STEM image (left) of product powders obtained from experiment 4 and the corresponding EDS spectra (right) .................83
6.1 Thermochemical calculations to determine stable phases at 1220⁰C as a function of molar ratio of methane to solid feed .........................85
6.2 Thermochemical calculations to determine stable phases at 1520⁰C as a function of molar ratio of methane to solid feed .........................86
6.3 Thermochemical calculations to determine stable phases at 2120⁰C as a function of molar ratio of methane to solid feed .........................87
6.4 Thermochemical calculations to determine stable phases at 3010⁰C as a function of molar ratio of methane to solid feed ..........................88
6.5 XRD pattern of product powders formed at various molar ratios of methane to solid feed and at a solid feed rate of 5g SiO2/min .............90
6.6 XRD patterns of product powders formed at various molar ratios of methane to solid feed and at a solid feed rate of 4g SiO2/min .............91
6.7 Experimental yield of product powders formed at various molar ratios of methane to solid feed and a solid feed rate of 5g/min ...........92
6.8 Experimental yield of product powders formed at various molar ratios of methane to solid feed and a solid feed rate of 4g/min ...........92
6.9 Effect of plasma power on the experimental yield at a feed rate of 5 g/min SiO2 and a molar ratio of SiO2:CH4 = 1:1 ..............................93
6.10 Effect of SiO2 feed rate on experimental yield at a power of 21.6 kW and a molar ratio of SiO2:CH4 = 1:2 ...................................94
6.11 Comparison of theoretical yield of SiC at various temperatures to experimental yield at a solid feed rate of 5g/min ................................94

xix
6.12 Heat capacity, sample heat flow and baseline heat flow for product powders from experiment 1 using DSC ...............................97
6.13 Comparison of heat capacity measured experimentally with those calculated using equation (29) ..........................................................98
6.14 SEM image showing the morphology of the product powders from experiment 1 ..............................................................................99
6.15 SEM image showing the morphology of the product powders from experiment 5 ..............................................................................99
6.16 SEM image showing the morphology of the product powders from experiment 7 ............................................................................100
6.17 TEM image showing the morphology of the product powders from experiment 1 ............................................................................101
6.18 (a) TEM image showing the morphology of the product powders from experiment 2; (b) and (c) HRTEM showing presence of Si and (d) SiO2 single crystal with insets of magnified image and electron diffraction pattern ...............................................................102
6.19 Raman spectra of product powders from experiments 1, 2 and 3 using a solid feed rate of 5g/min .....................................................104
6.20 XPS spectra of product powders from experiment 1 showing the Si 2p peak resolved into SiO2, SiC and Si peaks ............................105
6.21 Weight loss and heat flow curves from TG-DTA for the product powders from experiment 1 ...............................................106

1
CHAPTER 1
INTRODUCTION
Advanced ceramic materials are of enormous interest due to a wide variety of
applications in many key engineering fields such as electrical and electronics, optical,
mechanical, and chemical, to name a few. There are a number of commercial production
techniques that are in use depending on the size, shape and type of material. These ceramic
materials, in general, can be classified into three different categories namely, oxide ceramics,
non-oxide ceramics and ceramic based composites. Some of the most commonly used oxide
ceramics include but not limited to titanium dioxide, nickel oxide, cobalt oxide, molybdenum
oxide, yttria, zirconia, silica, etc. Other mixed oxides such as barium titanate, doped lanthanum
gallate, yttria stabilized zirconia, strontium doped LaMnO3 (LSM), yttrium barium copper oxide
(YBCO, commonly known as the 1-2-3 compound), are some of the examples of other oxide
based advanced ceramic materials.
One of the traditional ways of synthesizing mixed oxide is using high temperature ‘heat
and beat’ solid state synthesis route. The oxides are mixed together, heated to a temperature
followed by crushing / milling. This is done in cycles with increasing temperature in each cycle
till the final product is the desired single phase mixed oxide. This process is very time and
energy consuming and due to the extensive high temperature treatment of the material, the final
product size is in tens to hundreds of microns. Flux and hydrothermal synthesis routes [1] have
been reported to have relatively lower processing temperature between 200 and 600ºC.

2
Synthesis of Sr- and Li- doped lanthanum orthogallate is reported in literature [2] using
glycine- metal nitrate soft chemistry route. Stoichiometric metal nitrates were dissolved in water
and mixed with glycine, as fuel for combustion, and ammonium nitrate, as an initiator. The
initial synthesis temperature was very low, about 200ºC. The calcined powders were about 100-
200 nm in size and the crystallites in sintered pellets were about 0.5-1 m. In spite of low
temperature synthesis and finer grain sizes, the cost of starting materials, requirement of large
reactor volumes and batch processing make its commercial feasibility questionable.
Figure 1.1: Conceptual diagram of aerosol combustion process [3].
Aerosol combustion synthesis of MgO, ZrO2 and Yttria Stabilized Zirconia (YSZ) has
been reported in literature [3]. This concept has been patented [4-6] with the modification of the
actual apparatus. Similar to the metal nitrate-glycine soft chemistry route mentioned earlier, the
aerosol combustion synthesis route uses magnesium nitrate for MgO, zirconium nitrate for ZrO2,

3
and zirconyl chloride and yttrium nitrate for YSZ. These raw materials are mixed with sucrose
and the thus formed aerosol is sprayed into the reactor. It is a relatively high temperature process
and the temperature is in the range of 1000-1500 K. In spite of high temperature processing, due
to low residence time in the reactor, the particle size is small of the order of tens of nanometers.
The drawbacks of this processing technique are the low through-put and scalability to
commercial applications.
Yttria stabilized zirconia is one of the widely used solid electrolytes in solid oxide fuel
cells. It has good oxygen ion conductivity at about 1000ºC. Several researchers [7-12] have
studied the synthesis of YSZ to obtain small grain size, spherical morphology and sinterability at
relatively low temperatures. Thin film deposition of YSZ on -alumina substrate has also been
reported [13]. Electrochemical deposition of CeO2 thin films [14], microemulsion mediated
synthesis of mixed oxide powders [15], sol-gel synthesis of nano-TiO2 [16] and polyacrylamide
gel synthesis of -alumina [17], are some of the other techniques used in the synthesis of nano
scale oxide ceramic materials.
Carbides are an important class of non-oxide ceramics. Traditionally, carbides are made
by carbothermic reduction of corresponding metal oxide by carbon at relatively high
temperatures of about 1500-2000ºC. Transition metal carbide synthesis using alkalide reduction
[18] has been reported at low temperature with an annealing temperature varying between 950-
1200ºC for various carbides such as Fe3C, VC, TaC, and TiC nanocrystals. Low temperature
synthesis and shorter annealing time maintains the fine crystallite size in the range of 2-25 nm
with a relatively high surface area.
Carbon nanotubes have excellent properties. It is one of the most used materials in fiber-
reinforced polymer or metal composites. A synthesis procedure has been published [19] where

4
the carbon nanotube is reacted with volatile metal oxide or a volatile metal halide to form
nanorods of corresponding metal carbides. Synthesis of narrow rods with diameters ranging
between 2-30 nm and lengths up to 20 m has been reported. It is interesting to note that bulk
properties of the carbides (TiC – metal; NbC – superconductor; Fe3C – ferromagnetic; SiC –
semiconductor; BCx – insulator) are still maintained in the nanorod structures.
Precious metal catalysts are usually used as anode catalysts for hydrogen oxidation in
polymer electrolyte fuel cells. These were replaced by a nanoscale WC catalyst by Yang et. al.
[20]. They obtained high current densities of 0.9 A/cm2 at 80ºC and 3 atm at a relatively low WC
catalyst loading of about 0.5 mg/cm2. The nanoscale tungsten carbide was synthesized by
chemically reduced ball milling. A mixture of WO3, Mg and C powders were mixed in 1:6:1
ratio and ball milled for about 2 days. MgO formed as a byproduct is removed using HCl acid
solution.
Ceramic composites are a class of materials where the ceramic material is dispersed in a
matrix. The matrix can be a metal, alloy, polymer or another ceramic. The advantage of the
ceramic composites is that the dispersed ceramic material increases the strength and hardness of
the composite. The presence of matrix, on the other hand, can also influence the ceramic
material. It is shown that the photocatalytic activity of a TiO2/activated carbon composite is more
than that of pure TiO2 [21]. It is reported that the presence of activated carbon reduces the grain
growth of TiO2 particles during the sol-gel synthesis while a phase transformation from anatase
to rutile phase is observed.
Composites of Ag/YBCO superconductors have been reported in the literature [22]. It is
important to have a composite as the mechanical strength of pure superconductors is poor.
Synthesis of Y123 (YBa2Cu3O7) and Y123 + 40% Y211 (Y2BaCuO5) were carried out using

5
Y2O3, BaCO3 and CuO using solid state synthesis at a calcination temperature of 900ºC for 30h.
The product from the previous step was mixed with 0.5 wt% Pt and 3-30 wt% Ag2O. The
synthesis of Ag/YBCO composite was carried out using a melt growth method by partially
melting the sample and varying the maximum and the hold temperatures. Silver free regions
were observed in some of the samples. The effect of the starting material (Y123 or Y123 + 40
wt% Y211), maximum temperature, and holding temperature on the dispersion of silver in the
melt was made.
A multi-walled carbon nanotube (CNT) – alumina composite has been synthesized [23]
with the CNT’s highly ordered inside the alumina matrix. A three step synthesis method was
used. First, nano-channel alumina (NCA) template was developed on aluminum at 650ºC with
the diameter of the hexagonal close-packed pore controlled by the experimental anodization
conditions. Cobalt catalyst was deposited in the nano pores as a second step. This is followed in
the third step by the growth of parallel, highly-ordered carbon nanotubes by the pyrolysis of
acetylene. The removal of NCA template using a mixture of acids revealed that nanotubes were
parallel with a diameter distribution within 5% of mean diameter.
Iron-cementite nanocomposite was synthesized [24] using mechanosynthesis technique
from elemental iron and graphite powders. Finer grain sizes (about 7-8 nm) of iron and cementite
were obtained by ball-milling for 10 hours. The consolidated material also had relatively smaller
grain size of about 40 nm due to use of intermediate temperature and high pressure. While the
material had increased hardness, the magnetic properties were unaltered due to presence of finer
grains.
One of the widely used techniques for the production of nanoscale materials is the
conventional metal – catalytic vapor-liquid-solid (VLS) process [25]. This method is

6
implemented in different forms for the production of a variety of materials. Carbon nanotubes
using arc discharge [26], silicon nanowires [27] using a solution method, hot filament chemical
vapor deposition and laser ablation of a target [28,29] for large scale production, thermal
evaporation are some of the other processing techniques for the synthesis of nanoscale materials.
Oxide-assisted growth (OAG) [29-33] has been used for some materials (silicon nanowires) for
its large scale production. Some of the early synthesis methods include photolithography [34]
and scanning tunneling microscopy (STM) [35]. Some of the high temperature processes such as
solid state processing of B4C-Al composite is done at high temperature (1180⁰C) and high
pressure (20 MPa) [36], but yields coarse grained material.
There are two major factors in the synthesis of nanoscale materials. The thermodynamics
of a process determines the feasibility of a process. Among all the competing reactions in the
process, the one with the least Gibbs energy will be the one that will be the most favorable
reaction. The nucleation and growth are specific to the production technique and will depend on
a lot of process control variables.
The research objective of this project is to synthesize fine grained advanced ceramic
materials such as TiB2, and SiC in a thermal plasma reactor using low cost oxide feed materials
such as TiO2, B2O3 and SiO2 and methane as a reducing gas. Thermochemical calculations will
be performed on individual feed material-reducing gas system to evaluate the feasibility of
desired product formation and to obtain an estimate of optimum conditions and composition of
starting materials. Several experimental process parameters such as the molar ratio of the solid
feed to the reducing gas, the solid feed rate and the plasma power on the size, shape and amount
of the product powders will be systematically examined. A computational fluid dynamics (CFD)
based mathematical model will be developed to understand the flow inside the reactor. Several

7
characterization techniques such as XRD, SEM, TEM and EDS will be used in characterizing the
size, morphology, amount and orientation of the product powders. Thermal properties and
weight loss measurements will be done using TG-DTA and DSC to understand the constituents
of the product powders. Raman and XPS spectroscopic techniques will be used to determine the
presence of bonds and substantiation of the chemical binding energies, respectively.

8
CHAPTER 2
LITERATURE REVIEW AND RESEARCH OBJECTIVES
2.1 Plasma synthesis
Plasma synthesis is one of the methods of producing nanoscale materials. Plasma is
defined, in general, as a gas that is partially or fully ionized containing electrons, ions, neutral
atoms and/or molecules. There can be two possible states of plasma, either thermal or non-
thermal. Non-thermal plasmas are characterized by their low temperature while thermal plasmas
have relatively very high temperatures. Both the types of plasmas have been used successfully in
the synthesis of fine grained materials and thin films. In the case of thermal plasma, partial
thermal equilibrium is attained between the electrons and the heavy particles of the plasma
plume. In the mid to late twentieth century, thermal plasmas have been tested and used
extensively in many applications such as, extractive metallurgy, process metallurgy, plasma
spray coatings, plasma welding and cutting, synthesis of advanced materials, toxic and hazardous
waste treatment, etc. Plasma spray coating was an $800 million industry as reported in 1990 [37]
and rose up to a $1.35 billion in 1997 [38].
Synthesis of advanced materials has become one of the most important applications of
thermal plasma due to its high temperature, clean reaction environment, and use of inexpensive
feed materials. High purity products are obtained faster due to the enhanced reaction kinetics.
Rapid quenching from very high temperatures creates a steep temperature gradient aiding the
formation of fine sized particles. These fine particles can reach near-theoretical density on

9
sintering leading to improved mechanical properties. In spite of the numerous advantages of the
thermal plasma processing technique, there are some inherent drawbacks, such as engineering
and design difficulties, that can be overcome only with a detailed understanding of specific
reaction mechanisms. High installation and power consumption costs, recycling costs of the
process (off) gases from the plasma reactor are some of the other major concerns that need to be
addressed for an efficient operation of the process. The processes taking place inside the reactor
and reaction mechanisms are also difficult to understand and are specific to the reactor system.
2.2 Plasma sources for material synthesis
Material synthesis using thermal plasma can be done using various types of plasma
sources. The most used plasma sources in practice fall under the following categories: (1) high
intensity ac/dc arcs; (2) high frequency discharges; (3) Hybrid plasmas; (4) Reactive Submerged
Arc (RSA) plasma. In the high intensity DC arc plasma, both transferred and non-transferred
torches are used in material synthesis. In non-transferred arc plasma, the plasma arc is generated
between two electrodes and the chemical reaction occurs downstream. Transferred arc plasma is
originated using two electrodes, but the arc is transferred from one of the electrodes to the
material to be heated. There are two types of high frequency discharges used in the material
synthesis, namely, Radio Frequency (RF) inductively coupled plasma (ICP) and microwave
generated plasma (MWP). The ICP works in the range of kHz to MHz while the MWP works at
GHz and are generally called as electrodeless discharges. Hybrid plasmas are useful in various
applications. The ICP can be used in conjunction with either DC plasma or ICP plasma. DC
plasma has high temperature at the core of the plasma stream, while the ICP has an offset from
the center. A schematic of the temperature distribution in DC thermal plasma and RF plasma is
shown in Figure 2.1. The combination of these two plasmas will generate high temperatures

10
across the cross-section of the reactor. The combination of ICP-ICP plasmas is useful in
maintaining high temperatures in longer reactors. In reactive submerged arc (RSA) plasma, the
electrodes are immersed in a dielectric fluid. The arc formed between the electrodes vaporizes
the electrode and is quenched in liquid to form desired products.
Figure 2.1: Schematic temperature distribution in a DC plasma arc (a) and RF discharge (b).
(a)
(b)

11
2.3 Advanced material synthesis using thermal plasma
Literature is abundant on the types of plasma arcs and their uses and advantages in
materials production [39-41]. Production of AlN [42], ”-Al2O3 and diamond [43] have been
reported in the literature. Metallic and ceramic compounds such as magnesium, titanium carbide,
silicon carbide, boron carbide and zinc ferrite have been synthesized using this processing
technique [39-49]. Several composite materials such as Fe-TiN, Fe-TiC, Fe(Ti)-TiC, Al(Ti)-TiC,
and Al-SiC [50-52] have also been successfully synthesized. The use of thermal plasma for the
production of fine and ultrafine powders has been patented by Celik et al. in 2002 [53]. The
process variables such as plasma power, powder feed rate, and molar ratio of the reactants
influence the product phases and their yield.
2.4 Properties and synthesis of titanium diboride
The transition metal boride, TiB2, is of interest due to its exceptional
characteristics. The melting temperature of TiB2 is 3498 ±20 K [54-56]. The hardness of titanium
diboride is 25 GPa at a Vickers indentation load of 5N while the thermal and electrical
conductivity are 96 W/m.K and ~107 S/m, respectively, at 293 K [57]. Along with the high
melting point, high hardness, good electrical and thermal conductivity, titanium diboride also
exhibits good corrosion resistance and good oxidation resistance or high thermal resistance in
different media, making it an excellent candidate for parts requiring high wear resistance [58]. A
material with such excellent properties has been the focus of production using various techniques
such as electrodeposition from fused salt [59], induction plasma [60], plasma spray synthesis
[61] and many others [62-64]. The production of TiB2 using thermal plasma has been carried out
earlier by other researchers from ilmenite concentrates with a TiB2 yield of about 33% [65] and
from TiO2 and B2O3 with a yield of about 92% [66]. The high yield in the latter literature was

12
obtained by a series of post processing techniques used to remove the by-products. Titanium
diboride can also be synthesized using various high-temperature techniques such as direct
reaction between titanium and boron (Spark Plasma Sintering (SPS) of elemental powders) [67],
carbothermic reduction (thermite reaction using a pyrolant also known as Self-propagating High-
temperature Synthesis-SHS) between titanium dioxide and boron oxide [68], hydrogen reduction
of boron and titanium halides, etc. One of the classic solid state reactions to produce titanium
diboride is the borothermic reduction of titanium dioxide explained in reaction (1) below:
2TiO2 + B4C + 3C 2TiB2 + 4CO (1)
Even though the above mentioned synthesis method is well established, the titanium
diboride produced is larger in size and requires subsequent milling to reduce the particle size.
The sinterability of TiB2 [69] using sintering aids and properties of sintered material [70] have
also been reported in literature. Several synthesis procedures have been proposed by researchers
for the production of nano-crystalline TiB2 such as solution phase reaction of NaBH4 and TiCl4
at 1173-1373 K [71], Mechanical alloying of Ti and B powders [72], and solvothermal reaction
in benzene using amorphous boron powder, TiCl4 and Na at 673 K [73].
2.5 Properties and synthesis of silicon carbide
Silicon carbide is another material of interest due to is excellent properties. Some of the
properties include high strength, hardness and elastic modulus. This low density ceramic also
possesses high thermal conductivity, lower thermal expansion coefficient and excellent
resistance to thermal shock. It also has chemical inertness to numerous corrosive media. Such
excellent properties of silicon carbide make it a good candidate for application in numerous

13
fields. It is also a good semiconductor material with its conductivity dependant on the type and
amount of dopant used.
Silicon carbide can be synthesized using one of the oldest methods known as Acheson
method [74]. In this process silica, carbon, sawdust and common salt are mixed and heated in a
resistive heating furnace at 2700⁰C [75]. After allowing reaction to occur, the temperature is
slowly decreased. The final product of this process is mainly 6H-SiC. The product yield and
process reproducibility are not conducive for commercial production of SiC.
Lely method [76] followed later by improved Lely method [77-78] use carbon crucible
concentrically covered with a porous layer of SiC. The charge is loaded into the crucible and
heated in a furnace to about 2500⁰C. SiC platelets form on the inner side of the porous SiC layer.
The predominant phase of SiC is still 6H-SiC similar to Acheson method. The lack of control
over spontaneous nucleation and low yield are major drawbacks of the Lely and improved Lely
processes. Hence this process is also not commercially viable.
Seeded sublimation growth technique, also known as the modified Lely method [79],
uses a seed, and source material in a graphite crucible at temperatures between 1800 – 2600⁰C in
argon atmosphere at 10-4 to 760 Torr. In one configuration, the seed is at the bottom and source
material is on the top separated by a cylindrical graphite diaphragm [80]. Diffusion controls the
kinetics of species transport and the difference in temperature between the seed and the source
acts as a driving force, where the seed temperature is maintained slightly lower than the source
temperature. In the second configuration [81], seed is at the top and the source is at the bottom
and there is no graphite diaphragm used. Due to the high yield of this process [82], it is used
extensively today as a commercial production technique for SiC.

14
Sublimation sandwich method [83], Chemical Vapor Deposition (CVD) [84], and Liquid
Phase Epitaxy (LPE) [80,85] are some of the other techniques by which SiC can be synthesized.
As SiC is also used as a semiconductor material, doping is usually done during the synthesis of
the material. Aluminum for p-type and N2/Si3N4 for n-type are the most commonly used dopants.
Vanadium doping is used to make semi-insulating SiC. SiC exists in three major polymorphs,
3C-SiC (also known as -SiC), 4H-SiC and 6H-SiC (also known as -SiC). -SiC exists in
hexagonal crystal structure while -SiC is in cubic – zinc blende structure.
The current research work will concentrate on the production of fine grained materials
using thermal plasma processing technique. A 2D mathematical model will be developed to
determine the flow parameters inside the reactor. The model will be used to determine the effect
of plasma gas inlet pressure on the peak velocity of the flow inside the reactor. The concept will
be extended to obtain a 3D model to predict more accurate flow parameters. This will be
followed by thermochemical calculations to predict the optimum conditions for maximum yield
of product, synthesis experiments using thermal plasma reactor and characterization of the
product powders for phase, morphology and any properties, if possible.

15
2.6 Research objectives
The following are the specific research objectives of this research:
(i) Mathematical Model:
(a) To develop a two-dimensional (2D) mathematical model of the plasma reactor to
predict the flow and temperature profiles inside the reactor which is otherwise
difficult to determine experimentally.
(b) To refine the grid for specific reactor conditions and reaction parameters and to
optimize the grid size to obtain accurate results at a nominal time.
(c) To investigate the effect of the inlet pressure of the plasma gas on the velocity
profile inside the reactor.
(d) To obtain a three-dimensional (3D) mathematical model to determine, more
accurately, the flow profile inside the reactor.
(e) To investigate the effect of the inlet plasma gas pressure on the flow inside the
reactor.

16
(ii) Synthesis and Characterization of Materials:
(a) Thermochemical Calculations: To estimate the feasibility of synthesis of two
specific materials (TiB2 and SiC) from low cost oxide feed (B2O3, TiO2 and
SiO2) and methane as reducing gas using the concept of minimization of
Gibbs energy. These calculations enable the prediction of formation of
feasible products including the types and amounts of by-products.
(b) Thermal Plasma Synthesis: To run the thermal plasma experiments using the
low cost oxide feed and reducing gas to synthesize ceramic materials. To
investigate the effect of process parameters such as plasma input power (or the
processing temperature), the feed rate of the solid feed, and molar ratio of the
solid feed to the reducing gas, on the product yield. To optimize the process
parameters for higher product yield, and lower by-products.
(c) Characterization: To characterize the product powders using techniques such
as X-Ray Diffraction (XRD), Scanning Electron Microscopy (SEM), Energy
Dispersive Spectroscopy (EDS), and Transmission Electron Microscopy
(TEM). These techniques will characterize the products and by-products (if
present) for phase and morphology. Further characterization techniques such
as Thermo-Gravimetric and Differential Thermal Analysis (TG-DTA),
Differential Scanning Calorimetry (DSC), X-ray Photoelectron Spectroscopy
(XPS), etc, will be used to further substantiate the phase, morphology and post
processing of product powders.

17
CHAPTER 3
MATHEMATICAL MODEL
3.1 2D plasma reactor model:
The flow properties of plasma inside the reactor such as temperature and velocity at
various regions of the reactor are difficult to determine experimentally in a D. C. thermal plasma
jet. Some of the initial and the boundaries conditions, on the other hand, are well defined. These
conditions can be used in a mathematical model to determine the flow properties of plasma
inside the reactor. Estimation of flow properties of plasma is important as the particulate matter
injected into the reactor is surrounded by the plasma plume and hence the properties of particles
will be determined by the properties of plasma.
A systematic approach was used in determining the flow properties of plasma inside the
reactor. A two dimensional model of the reactor was developed using Fluent 6.3® [86]
considering the mass, momentum and energy conservation within the bounds of the reactor. A
2D grid was generated using Gambit [87]. A simple 2D model was generated using the plasma
gas Ar and reducing gas CH4 at the feed port. It is assumed that there is no solid feed at this point
(no species transport). A pressure based solver was used. The convergence was checked for the
conservation of mass, momentum, energy, turbulence and radiation. A simple 2-equation k-
model was used as an initial solver setting as it has been proven effective for thermal plasma
modeling [88-90]. A model using higher order turbulence equation such as the transition k- or
the transition shear stress transport (SST) model would give more accurate turbulence flow

18
profiles. This modification will be incorporated in the 3D model of the system. A P1 radiation
model was used to solve for the convergence of radiation.
The mathematical results obtained from modeling have to be validated using the results
obtained from experiments. These properties such as temperature and velocity, as mentioned
earlier, are difficult to measure and hence validation of the results becomes difficult. The
modeling results, while not helpful in the validation of results, can help in the design of
experiments and also in studying the effect of change of various well-established initial and
boundary conditions.
In this study, turbulent D. C. plasma jets will be considered during modeling, as it is the
type of plasma employed in the plasma synthesis of materials. The following assumptions are
made:
1. The plasma is in local thermodynamic equilibrium (LTE)
2. All the gases inside the reactor are considered to be ideal at all temperatures.
3. All the solid walls are immovable at all temperatures.
4. The solution obtained to the mathematical problem is at steady state in all the cases. No
time-dependant processes are considered.
5. The fluid flow formulation used in all the cases is compressible flow.
6. No species transport (or) secondary dispersed phase considered in the plasma flow.
3.2 Defining equations in 2D model:
The defining equations for the conservation of mass, or the continuity equation, are given
in the following equations. The general form of the equation is given in equation (2).
)2(. mSt

19
Sm – Source of any mass added from dispersed second phase. x
and r
are the axial and the
radial velocities.
The conservation of momentum in an inertial reference frame is given by
Where, p is the static pressure, τ is the stress tensor, g is the gravitational force and F is
the external body forces (also contains other sources). For the angular momentum, r or r2 =
constant and hence the conservation of angular momentum is given by
A standard k- turbulence model was used in the 2D model. More accurate higher order
turbulence models will be used in 3-D model of the plasma reactor. The transport equations for
the turbulent kinetic energy, k, and the turbulence dissipation rate, , are defined in the following
equations.
)4(mr
rx Srrx
)5(.. Fgpt
)6(2
rr
)7(kMbkjk
t
ji
i
SYGGx
k
xku
xk
t
)8(2
231
Sk
CGCGk
Cxx
uxt bk
j
t
ji
i
)3(0)(0.
t
oru

20
In the above equations, the turbulent (or Eddy) viscosity is defined as
Gk is generation of turbulent kinetic energy (TKE) due to mean velocity gradients; Gb is
generation of TKE due to buoyancy; YM is contribution of fluctuating dilatation in compressible
turbulence to the overall dissipation rate. C1, C2, C3, C are all constants. Standard values can
be used for the constants as a first approximation as follows. C1 =1.44, C2 = 1.92, C = 0.09, k
= 1.0, = 1.3. k and are the turbulent Prandtl numbers for k and , respectively.
The heat transfer (or the energy) equation for dissipation of energy through the reactor is
defined as
Where,
Effective conductivity, keff = k + kt,
kt is the turbulent thermal conductivity,
Jj is the diffusion flux of j,
Sh is the heat of chemical reaction or any other volumetric heat source,
h is the sensible enthalpy, Yj is the mass fraction of j and hj is the sensible enthalpy of j.
)9(2
kCt
)10(... hj
effjjeff SJhTkpEEt
Conduction Species
Diffusion
Viscous Dissipation
jjjhYh
phE
2
2

21
3.3 Grid refinement in 2D model:
Figure 3.1: Schematic of the thermal plasma reactor
Figure 3.2: Grid of the plasma reactor with 4154 nodes (X=30; Y=133)
Figure 3.3: Grid of the plasma reactor with 7257 nodes (X=40; Y=176)
Figure 3.4: Grid of the plasma reactor with 11322 nodes (X=50; Y=221)
1.8

22
Figure 3.5: Velocity (a), stream function (b) and temperature (c) at a distance of X=1.8 cm from
the plasma source for various grid configurations.
0100200300400500600700800
0.00 0.01 0.02 0.03 0.04 0.05 0.06 0.07 0.08 0.09 0.10
Vel
ocit
y M
agn
itu
de
(m/s
)
Distance in Y-Direction (m)
30 X 13340 X 17650 X 221
0
5
10
15
20
25
30
0.00 0.01 0.02 0.03 0.04 0.05 0.06 0.07 0.08 0.09 0.10
Str
eam
Fu
nct
ion
(k
g/s)
Distance in Y-Direction (m)
30 X 13340 X 17650 X 221
0
1000
2000
3000
4000
5000
6000
7000
0.00 0.01 0.02 0.03 0.04 0.05 0.06 0.07 0.08 0.09 0.10
Tem
per
atu
re (
ºC)
Distance in Y-Direction (m)
30 X 13340 X 17650 X 221
(a)
(b)
(c)

23
Table 3.1: Initial and boundary condition values used during grid refinement studies.
Position/Variable Value
Plasma inlet pressure 50 psi
Plasma gas Ar
Powder feeder gas pressure 14.6932 psi
Reducing gas CH4
Reactor outlet pressure Atmospheric
Solver Pressure based
Turbulence model Standard k-
Turbulence Specification Intensity and length scale*
- Plasma inlet 10% and 3.2 cm
- Powder feed inlet 10% and 1.6 cm
- Outlet 10% and 19.2 cm
Radiation model P1
Species Transport No
Gravity No
*length scale defined by hydraulic diameter, Dh = 4A/PW; A – Cross sectional area; PW – Wetted
perimeter
A schematic of the thermal plasma reactor is shown in Figure 3.1. The plasma plume
enters the reactor through the plasma port on the left side of the reactor. The port is located at the
center of the left side wall of the reactor. The feed enters the reactor through the feed port along

24
with carrying and reducing gases. All the products exit the reactor through the right side wall of
the reactor.
The geometry of 2D reactor and grid of various sizes were generated using Gambit [87].
Three different grid sizes were generated for grid refinement studies. The generated grids are
shown in Figures 3.2, 3.3 and 3.4 corresponding to 30x133 (4154 nodes), 40x176 (7257 nodes),
and 50x221 (11322 nodes), respectively. As can be seen from these figures, narrow grid spacing
was used at the plasma port and feed port to account for higher velocity at these places and a
smoother transition to lower velocity regions within the reactor. The initial and boundary
conditions used for grid refinement studies are listed in Table 3.1.
The solution to the CFD problem was obtained using Fluent [86]. Velocity, temperature
and stream function within the reactor was obtained for the three grid configurations used in the
study. An important cross-section in the reactor was chosen to analyze the results obtained from
the grid refinement studies. A distance of 1.8 cm from the plasma entry wall and across the
diameter of the reactor was chosen as it will account for the high velocity from the plasma
stream and any backflow from the feed stream. The results obtained from various grid sizes on
the velocity, stream function and temperature profiles at a distance of 1.8 cm are shown in Figure
3.5. As can be seen from the results, the size of the grid doesn’t affect the results while it
increases the number of iterations from 272, 375, and 641 with the increase in the number of
nodes from 4154, 7257, and 11322, respectively. Hence the processing time increases with the
increase in the number of nodes. Therefore, the grid with 7257 nodes was chosen to continue the
modeling to obtain as many data points (minimal intervals) at a nominal processing time.

25
3.4 Results from 2D plasma reactor model
Table 3.2: Values of pressures used in determining the velocity and temperature profiles.
Pressure (psi) Pressure (Pa) Temperature (K)
40 275790 6000
45 310264 7000
50 344737 8000
55 379211 9000
The change in pressure of the plasma gas, namely Ar, allows the increase in current
which in turn increases the power of the plasma plume. Increasing the current without a
corresponding increase in the pressure of gas will result in instability of the plasma plume. As a
first approximation, the pressure is increased in 5 psi segments as shown in Table 3.2 and
assumed that the corresponding increase in the temperature is 1000 K. Hence, at the plasma
entry, the pressures used were 40, 45, 50 and 55 psi and the corresponding inlet temperatures
used were 6000, 7000, 8000 and 9000 K. This is not accurate but will be a good starting point to
determine the initial flow and temperature profiles. Figure 3.6 shows the contours of temperature
and velocity inside the plasma reactor at inlet plasma pressure of 45 psi. The powder being fed
along with the carrying gas enters the reactor at ambient temperature. The images show that the
temperature at the outlet of reaction chamber is at least 4000 K. This is important information as
it determines the state of the feed powder before it enters the quenching chamber. The velocity
contour within the reactor at a plasma inlet pressure of 45 psi shows that velocity is a maximum
at the center of the reactor near plasma inlet. Due to the incoming flow from feed port, the
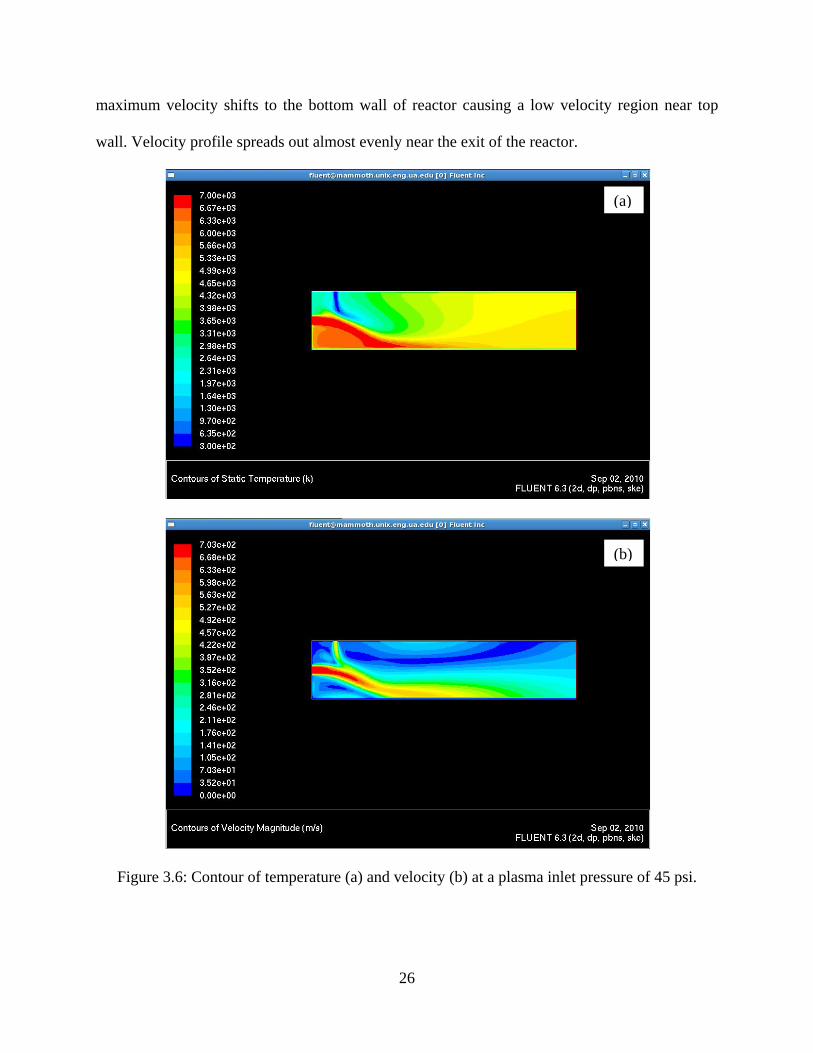
26
maximum velocity shifts to the bottom wall of reactor causing a low velocity region near top
wall. Velocity profile spreads out almost evenly near the exit of the reactor.
Figure 3.6: Contour of temperature (a) and velocity (b) at a plasma inlet pressure of 45 psi.
(a)
(b)

27
660
680
700
720
740
760
780
800
5000 6000 7000 8000 9000 10000
Vel
ocit
y (m
/s)
Temperature (K)
35 40 45 50 55 60Pressure of the plasma gas at inlet (psi)
Figure 3.7: Effect of pressure on the inlet velocity of the plasma.
The velocity profile inside the reactor is quite important as that of the temperature. This
determines the average residence time of the particles in the reactor. While the temperature
profile can be combined with thermodynamics of the reaction to determine its feasibility, the
velocity profile along with the physical properties of the powder such as its density, particle size,
etc, can be used in determining the residence time in the reactor. This when compared with the
vaporization time required for a given type of particle of a given size will determine the
feasibility and feed particle size range. Figure 3.7 shows the increase in the inlet velocity of the
plasma plume as a function of pressure of the plasma gas (Ar), plotted in the secondary x-axis.

28
As assumed earlier that the temperature is higher with increase in the plasma gas pressure, the
effect of temperature, plotted in the primary x-axis, is also shown on the inlet velocity.
Figure 3.8: Profiles obtained from different areas of the reactor denoted by line numbers.
To better understand the flow through the reactor chamber, sections were made at
different parts in the reactor as shown in the Figure 3.8. There are sections at a distance half way
to the solid feed point, at the solid feed entrance, half way of the entire reactor and at the end of
the reactor chamber, labeled as line 5, 6, 7 and 8 respectively. There is also a section made at the
center of the reactor along the flow of the plasma labeled as line 9. The flow profiles along these
labeled sections were obtained. The velocity along the radial lines 5, 6, 7 and 8 are plotted in
Figure 3.9 (a).
As can be seen, the velocity of plasma at the center is highest closer to the plasma entry.
At half way of the reactor length, the velocity is biased more towards the bottom of the reactor
which is an effect of the incoming carrying gas from the solid feed port. At the end of the
reactor, the velocity varies linearly along the radius of the reactor from about 100 m/s at the top
to about 230 m/s at the bottom. At the center of the reactor the velocity is very high closer to the
plasma torch and it starts to decrease exponentially right after the entry of the carrying gas from
Line 9
Line 5 Line 6
Line 7 Line 8

29
the solid feed inlet. It reaches a minimum and stabilizes at about a 140 m/s all the way to the end
of the reactor as can be seen from Figure 3.9 (b).
Figure 3.9: Velocity profile along radial (a) and axial (b) directions at inlet pressure of 45 psi.
0
0.012
0.024
0.036
0.048
0.06
0.072
0.084
0.096
0 100 200 300 400 500 600 700 800
Rad
ial D
ista
nce
(m
)
Velocity (m/s)
line 5line 6line 7line 8
0
100
200
300
400
500
600
700
800
0 0.044 0.088 0.132 0.176 0.22 0.264 0.308 0.352 0.396 0.44
Vel
ocit
y (m
/s)
Axial Distance (m)
line 9
(a)
(b)

30
Figure 3.10: Assume Flow Path of Solid Feed.
If the feed powder was fed through the feed port and as a first principle approximation if
the velocity of the particle is equal to velocity of the gasses and the plume in the reactor, the
residence time of the particles in the reactor can be calculated. The dark line inside the reactor
shown in Figure 3.10 is an approximate representation of the path of the solid feed. An estimate
of average velocity for flow from the powder feed port to the center of reactor and the flow
through the middle of reactor was estimated for various plasma gas pressures. Using these
average flow velocities, average residence times were calculated and are given in Table 3.3.
Table 3.3: Average residence time in the plasma reactor as a function of plasma inlet pressure.
Inlet Pressure (psi) Residence time (ms)
40 2.6974
45 2.5723
50 2.4201
55 2.3432

31
This residence times can be taken as the most conservative estimate as it does not take
into consideration the increase in density of the particulate matter compared to gasses and the
plasma plume. An estimate of the vaporization time of various feed materials of different sizes
and a comparison with residence time in the reactor will provide information on whether the feed
particles are completely vaporized.
3.5 3D plasma reactor model:
The flow properties of plasma plume in a three dimensional space is more accurate and is
a closer representation of actual plasma reactor. Hence a three-dimensional grid was generated
using Ansys 13.0® workbench [91] which is an integrated software that can generate the grid,
setup the computational fluid dynamics (CFD) problem, generate solution and compare results
from different case studies. A 3D axisymmetric geometry was generated using DesignModeler®.
Meshes of various sizes were created on the geometry using Meshing®. Fluent® 3d, pressure-
based, realizable k-epsilon version was used in the setup and solution of the CFD problem. The
results and post-processing of the obtained results such as generation of contours, and graphs,
etc., were done using CFD-Post®. DesignModeler®, Meshing®, Fluent® and CFD-Post® are all
parts of the Ansys 13.0 workbench®.
Assumptions similar to those made in the 2D model were used in the 3D model as a first
approximation. As the first step no species transport was considered in generating the flow field
of plasma plume inside reactor. This will help understand the velocity and temperature profiles
inside the reactor. The effect of particulate feed type and the solid feed rate into the reactor is a
separate case study.

32
3.6 Defining equations in 3D model:
Three different turbulent models were used in the 3D model, namely, k-, k- and
transition SST model. The transport equations for turbulent kinetic energy, k, and turbulence
dissipation rate, , for standard k- model are defined in equations (7) – (9) in section 3.2.
The transport equations for the standard k- model describing the turbulent kinetic
energy k and the specific dissipation rate are given below:
Where,
Gk - Generation of turbulence kinetic energy due to mean velocity gradients; G - Generation of
; Yk and Y – Dissipation of k and due to turbulence; Sk and S – User-defined sources terms
for k and .
Effective diffusivities for the k- model are defined in equations (13) and (14) while the
turbulent viscosity, t, using k and is defined in equation (15).
)11(kkkj
kj
ii
SYGx
k
xku
xk
t
)12( SYGxx
uxt jj
ii
)13(k
tk
)14(
t
)15(* k
t

33
)17(Rek
t
Where, k and are the turbulent Prandtl numbers for k and respectively.
At low Reynolds numbers, the coefficient, * defined in equation (16), helps in
damping the turbulent viscosity. At high Reynolds numbers, the coefficient is 1* * .
Model constants for the standard k- model are Rk = 6; *0 = i/3; i = 0.072; k = 2.0; = 2.0.
The following equations represent the transport equations for transition SST model for
intermittency, , given in equation (18). Defining equation for transition and destruction sources
are given in equations (19) and (20) respectively.
192 3
1
Consetlength FSFP
2011 PE
Where, S - Strain rate magnitude; Flength - Empirical correlation that controls the length of the
transition length.
212 12 turbFcP
22222 PcE
Where, is the vorticity magnitude. The constants for the intermittency equations are c1=0.03;
c2=50; c3=0.5; =1.0.
)18(2211
j
t
jj
j
xxEPEP
x
U
t
)16(/Re1
/Re*
*0*
kt
kt
R
R

34
3.7 Turbulence and grid refinement in 3D model:
Figure 3.11: Various grid sizes for grid refinement studies in 3D thermal plasma reactor: (a)
8013 nodes, (b) 11516 nodes and (c) 18936 nodes.
(a)
(b)
(c)

35
Figure 3.12: Velocity profile at the cross-section of the feed port: (a) standard k- model, (b)
standard k- model and (c) Transition SST model.
(a) (b)
(c)

36
Figure 3.13: Velocity profile at the symmetry of the reactor: (a) standard k- model, (b) standard
k- model and (c) Transition SST model.
(a)
(b)
(c)

37
A grid containing 5 turbulence layers and 8013 nodes was generated using Meshing
console of the Ansys Workbench as shown in Figure 3.11 (a). Three identical problems were
setup with the variation of the turbulence model and were solved using the Fluent console.
Figure 3.12 shows the velocity contours at the cross-section of feed port for three turbulence
models. Not much variation was observed in the velocity of plasma at the center of reactor in all
three cases. The major difference observed was in the velocity profile due to turbulence near the
wall. The standard k- model does not resolve the turbulence near the wall as well as the
standard k- model and is best resolved using the transition SST model.
Figure 3.14: Line profiles created in the reactor to compare the modeling results from various
case studies.
Contours of velocity magnitude were also obtained at the symmetry plane of the plasma
reactor. As can be seen from Figure 3.13 (a), the velocity profile is not well resolved in the case
of standard k- model, not only by the wall but also in the entire reactor. A comparison with the
standard k- and the transition SST models show that the resolution of turbulent velocity at the
38
wall, near the feed entry and through the reactor is better with the k- model and is best using
the transition SST model.
0 100 200 300 400 500 600 70010
8
6
4
2
0
Rad
ial D
ista
nce
(cm
)
Velocity (m/s)
k- Model k- Model SST Model
Figure 3.15: Comparison of velocity magnitudes along the feed entry point using the three
turbulence models.
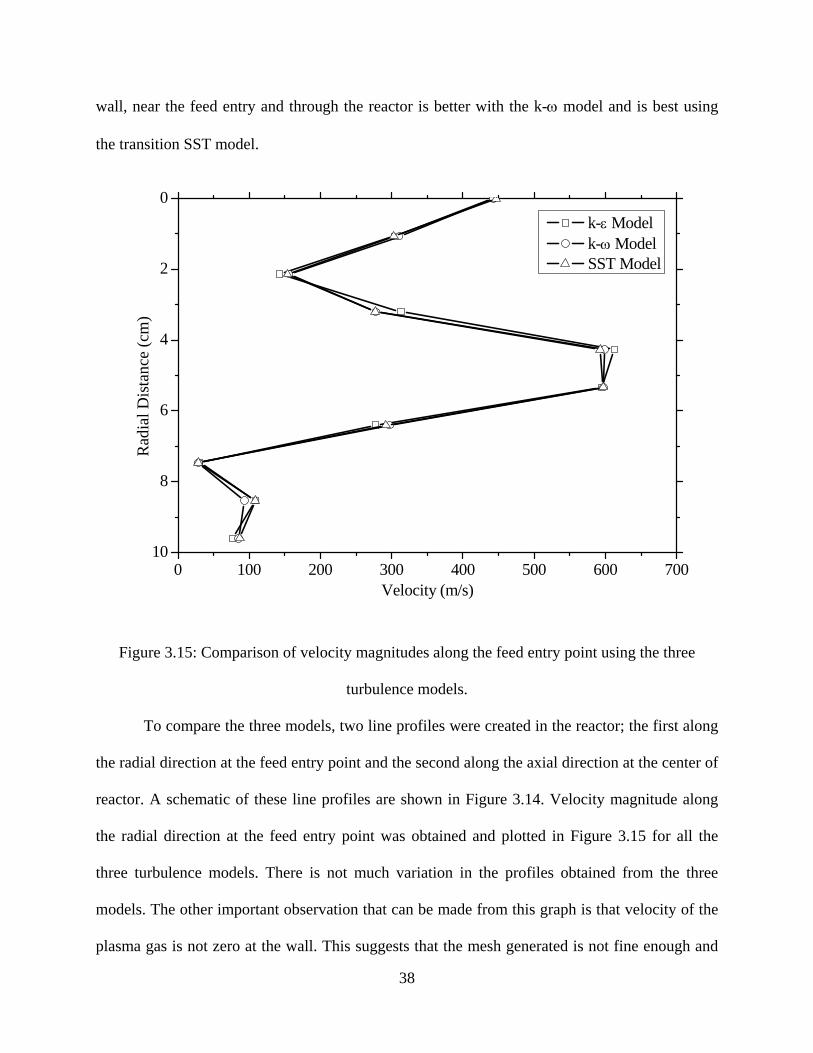
To compare the three models, two line profiles were created in the reactor; the first along
the radial direction at the feed entry point and the second along the axial direction at the center of
reactor. A schematic of these line profiles are shown in Figure 3.14. Velocity magnitude along
the radial direction at the feed entry point was obtained and plotted in Figure 3.15 for all the
three turbulence models. There is not much variation in the profiles obtained from the three
models. The other important observation that can be made from this graph is that velocity of the
plasma gas is not zero at the wall. This suggests that the mesh generated is not fine enough and

39
needs to be refined to obtain consistent and acceptable results. A plot of the velocity magnitude
at the center line of the reactor is shown in Figure 3.16. All the three models have approximately
similar velocity profile until the feed entry point near 4 cm. A variation, even though, small was
observed in the profiles after the powder feed point. The standard k- model shows highest
velocity along the center of the reactor while the standard k- model shows the lowest. Grid
refinement becomes necessary to obtain accurate and consistent results and the four equation
transition SST model will be used to refine the grids and to further solve the problem of effect of
plasma gas pressure on flow through the plasma reactor.
0 5 10 15 20 25 30 35 40 450
100
200
300
400
500
600
700
800
Vel
ocit
y (m
/s)
Axial Direction (cm)
k- Model k- Model SST Model
Figure 3.16: Comparison of velocity magnitudes along the center of reactor using the three
turbulence models.

40
Grid refinement was done using three different grids as shown in Figure 3.11 using the
transition SST turbulence model. The first mesh with 8013 nodes was created with 5 layers by
the wall. The second mesh was created with 5 layers as well, but with reduced grid size, thus has
11516 nodes. The third mesh with 18936 nodes was created with same grid size as the second
mesh but with 10 layers near the wall.
Figure 3.17: Velocity profile at the cross-section of the feed port: (a) 8013 nodes, (b) 11516
nodes and (c) 18936 nodes.
(a) (b)
(c)

41
Figure 3.18: Velocity profile at the symmetry of the reactor: (a) 8013 nodes, (b) 11516 nodes and
(c) 18936 nodes.
(a)
(b)
(c)

42
Figure 3.17 shows the contours of velocity magnitude at the cross-section of the feed port
using various grid sizes. The contour using the mesh with 8013 nodes (grid 1) shows the shape of
the contour is a function of shape of the mesh. This is especially the case at the center of the
reactor where the velocity is relatively high. As the grid size is reduced, as in the case of 11516
nodes (grid 2) and 18936 nodes (grid 3), the shape of the contour is much smoother. The increase
in the number of turbulence layers from 5 in grid 2 to 10 in grid 3 creates a high velocity regime
in the top and the bottom of the reactor and restricts the flow of the mainstream fluid to the
center of the reactor.
0 100 200 300 400 500 600 70010
8
6
4
2
0
Rad
ial D
ista
nce
(cm
)
Velocity (m/s)
8013 Nodes 11516 Nodes 18936 Nodes
Figure 3.19: Comparison of velocity magnitudes along the feed entry point using three different
grid sizes.

43
The contours of velocity magnitude along the symmetry of the reactor are shown in
Figure 3.18 for the three grid sizes. The increase in the grid size from grid 1 to grid 2 improves
the shape of the contours obtained. On the other hand, increasing the number of turbulence layers
from 5 to 10 (grid 2 to grid 3) creates a high velocity regime at the top and the bottom of the
reactor along the feed entry port.
0 5 10 15 20 25 30 35 40 450
100
200
300
400
500
600
700
800
Vel
ocit
y (m
/s)
Axial Direction (cm)
8013 Nodes 11516 Nodes 18936 Nodes
Figure 3.20: Comparison of velocity magnitudes along the center of reactor using three different
grid sizes.
A comparison of the velocity profiles along the radial and the axial directions using the
three grid sizes is shown in Figures 3.19 and 3.20, respectively. It is seen from the radial velocity
profile that the increase in grid size from grid 1 to grid 2 updates the velocity at the wall to zero.
The velocity profile, otherwise, is similar to that obtained from grid 1. The increase in the

44
turbulence layers from 5 to 10 (from grid 2 to grid 3), creates a high velocity regime at the top
and the bottom of the reactor near the feed entry. This also reduces the mainstream velocity.
Similar observation is made using the velocity profile at the center line. The mainstream velocity
of grid 2 is higher than that of grid 1 and grid 3, the latter being lower due to increased
turbulence layers. Hence to obtain a conservative model, a grid with higher velocity will be
chosen for further analysis, which will lead to a smaller particle residence time in the reactor.
Hence grid 2 was chosen along with the transition SST turbulence model to determine the effect
of plasma inlet pressure on the velocity profile in the reactor.
3.8 Results from 3D plasma reactor model:
A grid with 11516 nodes and 5 turbulence layers and transition SST turbulence model
was used in the determination of effect of pressure of plasma gas. Five different plasma gas
pressures were considered, i.e. 35, 40, 45, 50 and 55 psi. Assumption similar to the 2D model
was made that a 5 psi increase in the plasma gas pressure can allow for an increase of plasma
temperature by 1000 K. Hence the temperatures used in the calculation were 5000 K, 6000 K,
7000 K, 8000 K, and 9000 K corresponding to the plasma gas pressures of 35, 40, 45, 50, and 55
psi, respectively.
The problem setup and solution was done using Fluent varying the inlet plasma gas
pressure and holding the rest of the parameters constant. Axial and radial line profiles were
created similar to that shown in Figure 3.14 for all the cases. The velocity profile for the radial
line along the feed entry is shown in Figure 3.21 for the five different gas pressures.

45
Figure 3.21: Comparison of velocity magnitudes along the feed entry point using five different
plasma inlet gas pressures.
The increase in the plasma gas pressure has an effect on the velocity in almost all places
in the reactor other than the wall. The increase in pressure from 35 psi to 55 psi, increases the
velocity at the feed entry point from about 440 m/s to about 493 m/s. The effect on the velocity
of mainstream is rather high and the velocity varies from about 493 m/s for a pressure of 35 psi
to about 624 m/s at 55 psi. Such increase in velocity will have enormous effect on the residence
time of particles in the reactor and will restrict the size of particle that can be used in
experiments.
0
2
4
6
8
100 100 200 300 400 500 600 700
Rad
ial D
irec
tion
(cm
)
Velocity (m/s)
SST model 35 psiSST model 40 psiSST model 45 psiSST model 50 psiSST model 55 psi

46
Figure 3.22: Comparison of velocity magnitudes along the center of the reactor using five
different plasma inlet gas pressures.
Similar observation was made when the velocity magnitude along the center of the
reactor was plotted as a function of the axial direction as shown in Figure 3.22. The velocity
increased from about 590 m/s at 35 psi to about 740 at 55 psi. The increase in the velocity at any
given point in the reactor seems to vary linearly as a function of the plasma inlet gas pressure.
The velocity at the exit of the reactor varies from 68 m/s to 86 m/s for a corresponding variation
in gas pressure from 35 psi to 55 psi.
0
100
200
300
400
500
600
700
800
0 5 10 15 20 25 30 35 40 45
Vel
ocit
y (m
/s)
Axial Direction (cm)
SST model 35 psiSST model 40 psiSST model 45 psiSST model 50 psiSST model 55 psi

47
580
600
620
640
660
680
700
720
740
760
780
800
5000 6000 7000 8000 9000
2D Model 3D Model
Temperature (K)
Vel
ocity
(m
/s)
35 40 45 50 55
Pressure (psi)
Figure 3.23: Effect of pressure on the inlet velocity of the plasma and comparison between the
2D model and the 3D model.
The maximum velocity at the inlet of the plasma torch is plotted as a function of the inlet
plasma gas pressure in Figure 3.23. Results from the 2D model were also plotted along with that
from the 3D model. Similar to the 2D model, the increase in velocity with the increase in the
inlet plasma gas pressure was observed to be almost linear. While the trend is similar to the 2D
model, the velocity at any given pressure was found to be lower by about 35 – 40 m/s in the 3D
model.

48
Figure 3.24: Assume flow path for solid feed in 3D model.
An assumed flow path of the solid feed similar to the 2D model was generated in the 3D
model as shown in Figure 3.24. Based on the average velocities from the powder feed port to the
center of the reactor and the flow through the center of the reactor, average residence times were
calculated for various plasma inlet gas pressures. The values of the residence times using the 3D
model is given in Table 3.4.
Table 3.4: Average residence time in the plasma reactor using 3D model as a function of plasma
inlet pressure.
Inlet Pressure (psi) Residence Time (ms)
35 2.1396
40 1.9919
45 1.8694
50 1.7639
55 1.6732
The residence time in the reactor is a strong function of the plasma inlet pressure. Hence
during experiments, a low enough plasma inlet gas pressure must be used to ensure a longer
residence time inside the reactor.

49
CHAPTER 4
EXPERIMENTAL SETUP
The non-transferred arc D. C. plasma reactor system was used in the synthesis of
materials. A detailed schematic of the reactor system is shown in Figure 4.1. The plasma reactor
system consists of the following parts:
(i) Plasma power source
(ii) Plasma reactor system
(iii) Powder feeder
(iv) Water source
(v) Gas source
4.1 Plasma power source
The plasma source, Model PT96 was manufactured by Plasma Energy Corporation. The
power source has a non-transferred plasma torch, Model PT-50C with a maximum power rating
of 45 kW and a transferred arc plasma torch that can be used with the transferred arc reactor
setup.
The two common types of plasma torches, namely, the transferred and the non-
transferred, vary in the positioning of the electrodes and hence, the way material is heated in the
reactor system. A schematic of the two different torches is shown in Figure 4.2.

50
Figure 4.1: Schematic of thermal plasma processing system.
Figure 4.2: Schematic of non-transferred (left) and transferred (right) plasma arc torches.

51
In the non-transferred type of plasma torch, anode and cathode are both parts of the torch
and the plasma arc is formed between them and is sustained till the current and voltage are
applied to the torch. The formed plasma plume and the hot gases travel through the plasma
reactor. The non-transferred mode is usually used in the synthesis of powders using a solid
powder feed.
Transferred plasma torch has a cathode similar to the non-transferred torch. The anode in
the transferred plasma torch is a collimator which helps in the formation of the plasma arc but
the plasma is eventually transferred to the material to be used and the work piece acts as an
anode during the operation of the reactor. The ionization and the principle of operation of the
plasma torch are otherwise similar to the non-transferred torch. Unlike the non-transferred torch,
a transferred plasma torch is used in melting and refining of materials. In case of solid powder
feed, the powder has to be made into a pellet before using the transferred torch.
Figure 4.3: Photograph of plasma power source control panel.

52
The plasma power source can be operated in the range of 15 kW – 90 kW, with a
maximum expandable power of up to 150 kW. Figure 4.3 shows the control panel of the plasma
power source. The controller can be used to operate both the transferred and the non-transferred
plasma torches. If the transfer mode is set to “DISABLE” as shown in the figure, the controller is
set to non-transferred mode of operation and “ENABLE” sets the controller to the transferred
mode of operation.
Figure 4.4: Photograph of the new (left) and used (right) electrode assembly in a non-transferred
plasma torch.
The non-transferred torch consists of a cathode and a consumable copper anode as shown
in Figure 4.4. The plasma arc is formed in between the anode and the cathode and moves through
the reactor. The size and the shape of the plasma formed and the temperature distribution inside
the plasma reactor is dependent on the shape of the anode. As the anode is consumable, it needs
to be replaced on a regular basis. The photograph on the right of Figure 4.4 shows an anode that
has been consumed as a result of regular use. Anode wear can also lead to instability of the
plasma plume during the operation of the plasma reactor.

53
Figure 4.5: Photograph of the electronic gas flow rate monitor on the plasma source.
The power source not only controls the current and voltage to the torch but also controls
the gas flow rate and the cooling water flow. The power source is also set to disable the
operation of the plasma torch without the minimum necessary conditions for the gas and the
water flow rates. An electronic display as shown in Figure 4.5 displays the present value of the
gas flow rate to the torch along with the set value and minimum value required for the operation
of the torch and the controller. Water flow meters are present in the back of the controller and the
cooling water flow rate to the torch is adjusted by the controller. Similar to the gas flow rates, the
water flow rates need to meet the minimum requirements for normal operation of the torch and
the controller.

54
4.2 Plasma reactor system
The non-transferred and the transferred thermal plasma reactor systems are both in-house
built systems. The photograph in Figure 4.6 shows the reactor setup for the non-transferred mode
of operation. It consists of the following zones: Reaction zone, Quench zone and Filter zone.
Figure 4.6: Photograph of non-transferred thermal plasma reactor system.
4.2.1 Reaction zone
The reaction zone is the most important part of the plasma reactor system. The solid
powder feed fed into the reactor evaporates in this part of the reactor. The reaction by which final
product powders are formed is in vapor phase. The reactor, hence, is designed and fabricated in-

55
house to ensure all the solid feed is vaporized. The outer jacket of the reaction zone is made of
316L stainless steel which is water cooled. The inner part of the reaction zone where the
interaction between the plasma plume and the solid feed occurs and vaporization takes place is
made of graphitic carbon. A heat balance, assuming the maximum temperature to be as high as
5000 – 6000 K, was made to determine the thickness and the outer diameters of the inner carbon
lining. Several layers of insulating alumina felt were placed in between the graphite tube and the
water-cooled stainless steel jacket.
Figure 4.7: Photograph of the reaction zone.
A photograph of the reaction zone is shown in Figure 4.7. The reactor system is set up
horizontally with the solid feed, carrying gas and the reducing gas entering the reactor through a
water-cooled feed port that is set up perpendicular to the circumference of the reactor. There are
numerous k-type thermocouples in the reaction zone that are set to read the temperature in the
alumina felt. During experiments, the temperatures at various spots in the reaction zone are

56
allowed to stabilize. This will ensure most of the heat from the plasma stream is utilized in
heating the powder feed and minimum heat loss occurs. One end of the reaction zone is
connected to the plasma torch while the other is attached to the quench zone.
4.2.2 Quench zone
Figure 4.8: Photograph of the two concentric copper quenching coils in the quench zone.
Second most important part of the plasma reactor system is the quench zone. The vapors
from the reaction chamber enter into the quenching chamber and the product powders nucleate
and grow on two water-cooled concentric copper coils as shown in Figure 4.8. The outer jacket is
made of 316L stainless steel and is not water-cooled. Two k-type thermocouples are inserted into
the outer jacket of the quench zone to obtain the temperature of hot gases. The water inlet and

57
the outlet are monitored constantly using thermocouples to ensure the temperature of the cooling
water doesn’t rise more than 2⁰C. Unlike the plasma power source the cooling water for the
reactor jacket and the quench tubes are obtained from the common laboratory water source and
the used cooling water is not recycled.
4.2.3 Filter zone
Figure 4.9: Photograph of cloth filter in filter zone.
The outer jacket of the filter zone is made of 316L stainless steel and contains two
thermocouples to measure the temperature of the outgoing gases. It also consists of a cloth filter
that helps in retaining any solid products that are not accumulated on the quenching coils. A
photograph of the cloth filter assembly in the filter zone is shown in Figure 4.9. While the
quenching coils help in the nucleation and growth of powders, it also serves to reduce the

58
temperature of outgoing flue gases. Hence, the flue gases do not affect the cloth filter and the
temperature of the flue gases is about 100⁰C.
4.3 Powder feeder
Figure 4.10: Photograph of the powder feeder (left) and the circuit of the control panel (right).
A computerized powder feeder, model 1270, was used to feed the reactor with the solid
powder feed. It was manufactured by Praxair Surface Technologies, Inc. The powder feeder is
loaded with the solid powder feed and gases are connected to it. The raw materials were sieved
through a 325 mesh (~45 m) and heated to 200ºC to get rid of any adsorbed moisture. A
photograph of the powder feeder and the circuit of the control panel are shown in Figure 4.10. It
has a wheel assembly that rotates during the operation of the powder feeder at particular preset
rotations per minute (RPM). This pushes the powder into a chamber where it is mixed with the

59
gases and the gases carry the solid particles out of the feeder and into the reactor. There are two
types of gases used, namely, the carrier gas and the reducing/reacting gas. The carrier gas used in
the experiments is argon. As the solid feed is oxide material in all of the experiments, methane is
used as a reducing gas.
The rate of solid feed depends on the type and of solid material used, the flow rate of the
carrying/reducing gas mixture and the RPM of the powder feeder wheel. The type and the size of
the starting material are fixed for experiments using similar raw materials. The flow rate of the
gas mixture (argon + methane) is set to 6 LPM. The only unfixed parameter is the RPM of the
powder feeder wheel. A calibration chart is made for each raw material type for the solid feed
rate as a function of RPM.
The powder feeder is also attached with a heating element that surrounds the container
with solid feed to remove any adsorbed moisture in the material with a maximum temperature
limit of 150⁰F. The maximum allowed pressure in the powder feeder chamber is 20 psi. If the
exit of the powder feeder is blocked due to agglomeration or other reasons, a built-in security
feature releases a safety valve reducing the pressure inside the chamber.
4.4 Water source
The cooling water to the plasma torch is regulated by the plasma power source. The water
supply to the plasma power source comes from a 500 gallon high density poly ethylene tank. A
photograph of the tank and a booster pump is shown in Figure 4.11, the latter being used in
pumping water to the plasma power source. The pressure of water is maintained no less than
200 psi on the controller while the controller controls the flow rate to the plasma torch and is
about 28 GPM. The requirement for the cooling water is to be less than 38⁰C above which a

60
safety circuit turns off the power to the plasma torch. The cooling water is returned to the tank
through the plasma power source and is re-circulated. Due to the large volume of the tank, the
water temperature never exceeds the upper limit of the instrument.
Figure 4.11: Photograph of cooling water system and roughing pump assembly.

61
Figure 4.12: Flow meters controlling the flow rate of cooling water to various parts of the plasma
reactor system.
Table 4.1: Flow rate of cooling water for various parts of the plasma reactor system
Part of reactor Flow rate (LPM)
Copper feed tube 1
Stainless steel reactor jacket 2
Copper quench coils 4.5
The cooling water to the reactor jacket, the feed tube and the quenching coils are supplied
by the common laboratory water supply. Due to geometric restrictions and cooling requirement

62
for each of these parts, various flow rates were designed based on heat transfer calculations.
Table 1 gives the values of cooling water set at various places in the plasma reactor system. A
photograph of the flow meters set to the required flow rates is shown in Figure 4.12.
4.5 Gas source
Figure 4.13: Photograph of gas supply system consisting of plasma, carrier and reducing gases.
The plasma gas used in the experiments is high purity argon. Other gases can be used as
the plasma gas such as nitrogen or helium. The volume of gas used for the operation of the
reactor per unit time is less for argon and hence was used for the experiments.
Figure 4.13 shows the gas supply system that supplies argon to the plasma power source
and the gas mixture of argon and methane to the powder feeder. Two cylinders of argon are
connected to the plasma source to ensure constant uninterrupted flow of gas to the torch. Digital
flow meters are connected to manifolds to obtain the flow rates of gases. One argon cylinder for
the carrying gas and one methane cylinder for the reducing gas are connected to the powder

63
feeder. The flow rate to the powder feeder is small compared to that of the plasma gas flow rate
and hence one cylinder each will suffice the gas requirement of powder feeder. During
experimentation, the pressure of plasma argon gas at the cylinder outlet was maintained between
60 – 100 psi. The flow rate in LPM varies on the requirement at the plasma power source. There
are certain requirements on the starting, minimum and maximum pressure limits for the
programmable gas controller in the plasma power source to operate the torch and varies with the
type of torch. These values are given in the Table 2.
Table 4.2: Programmable gas controller set points in the thermal plasma power source for
various types of torch.
Type of torch Start Low High
Non-transferred 10 30 60
Transferred 20 30 60
4.6 Raw materials
Experiments for the synthesis of TiB2 were conducted using equi-molar quantities of
TiO2 (anatase, 99.9%, metals basis, Alfa-Aesar, MA, USA) and B2O3 (99.98%, metals basis,
Alfa-Aesar, MA, USA) that were mixed together and used as a powder feed to the plasma
reactor. Using argon as the carrying gas, methane as the reducing gas, 1:1 molar ratio TiO2:B2O3,
and total gas flow rate of 6 LPM, the powder feeder was calibrated for the solid feed rate in
g/min as a function of the powder feeder rotation speed in RPM as shown in Figure 4.14. A
linear fit for the graph was obtained and the two different solid feed rates, 2.15 g/min and 3.22
g/min were used for experiments. After the calibration of the powder feeder, the feed material is
fed to the reactor which is set to the required power level and the product powders are collected.
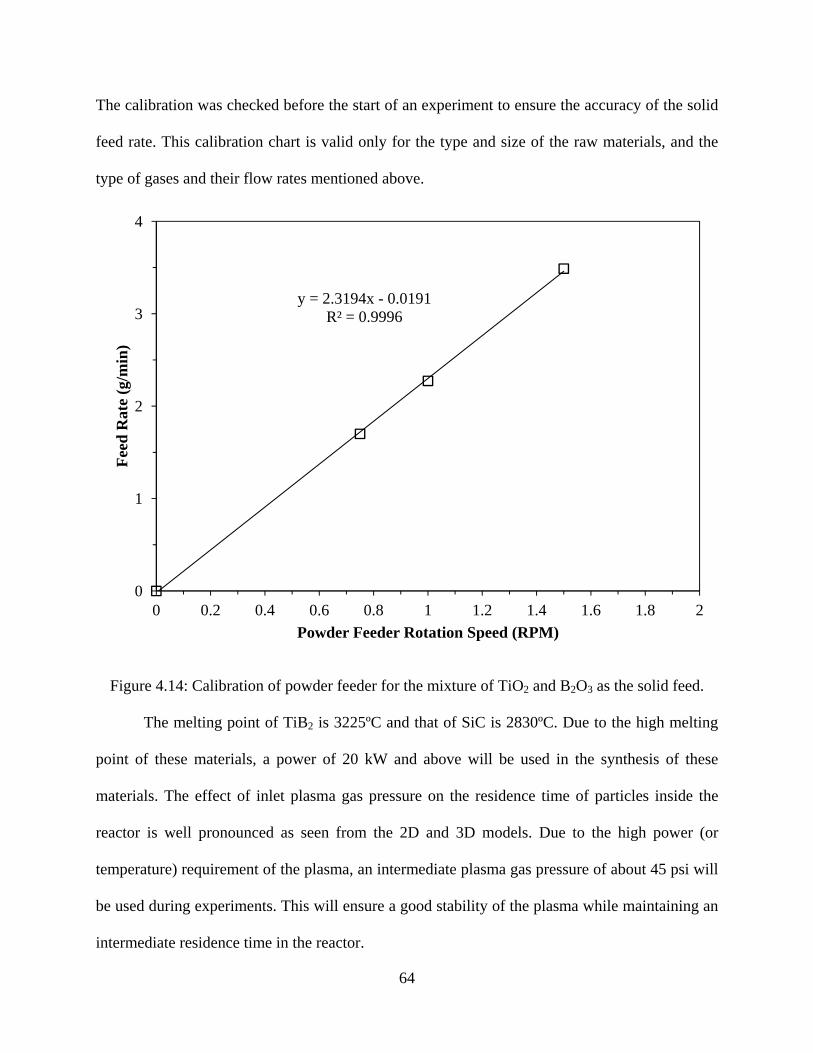
64
The calibration was checked before the start of an experiment to ensure the accuracy of the solid
feed rate. This calibration chart is valid only for the type and size of the raw materials, and the
type of gases and their flow rates mentioned above.
Figure 4.14: Calibration of powder feeder for the mixture of TiO2 and B2O3 as the solid feed.
The melting point of TiB2 is 3225ºC and that of SiC is 2830ºC. Due to the high melting
point of these materials, a power of 20 kW and above will be used in the synthesis of these
materials. The effect of inlet plasma gas pressure on the residence time of particles inside the
reactor is well pronounced as seen from the 2D and 3D models. Due to the high power (or
temperature) requirement of the plasma, an intermediate plasma gas pressure of about 45 psi will
be used during experiments. This will ensure a good stability of the plasma while maintaining an
intermediate residence time in the reactor.
y = 2.3194x - 0.0191R² = 0.9996
0
1
2
3
4
0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2
Fee
d R
ate
(g/m
in)
Powder Feeder Rotation Speed (RPM)

65
Experiments for the synthesis of SiC were conducted using SiO2 (99.9%, metals
basis, Alfa-Aesar, MA, USA) as a powder feed to the plasma reactor. The carrying gas used with
the powder feeder was ultra high purity Ar (AR UHP300 Gr 5.0, Airgas) and the reducing gas
was ultra high purity methane (ME UHP300 Gr 4.0, Airgas). The powder feeder was calibrated
to obtain the solid feed rate as a function of RPM as shown in Figure 4.15. The flow rate of gas
mixture, i.e. argon and methane, was maintained at 6 LPM. Two different solid feed rates, 4 and
5 g of SiO2/min were used in experiments. The calibration was checked before every experiment.
Figure 4.15: Calibration of powder feeder using SiO2 as the solid feed.
y = 2.8318xR² = 0.9989
0
2
4
6
8
10
12
14
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5
Fee
d R
ate
(g/m
in)
Powder Feeder Rotation Speed (RPM)

66
4.7 Characterization of product powders
4.7.1 X-Ray Diffraction (XRD)
The product powders obtained were characterized using X-Ray Diffraction (XRD),
model Phillips PW-1710 (TiB2 experiments) and model Phillips X’Pert MPD (SiC experiments),
for phase analysis and volume fraction calculation using direct comparison method. Both the
instruments use a monochromated Cu-K radiation with a wavelength of 1.54056 Å. The Phillips
PW-1710 was operated at 40kV and 35 mA while the Phillips X’Pert MPD was operated at 45
kV and 35 mA. SiO2 slide was used to hold the sample. Vacuum grease was applied to the slide
and the product powders were dispersed on it.
4.7.2 Scanning Electron Microscope (SEM)
Scanning Electron Microscope (SEM) fitted with Energy Dispersive Spectroscopy
(EDS), model JOEL 7000 FE SEM was used to determine the morphology and elemental
composition of the product powders. The product powders were dispersed in acetone and
ultrasonicated for 5 minutes. A flat top SEM sample stub was used with a double-sided Cu tape
attached to it for holding the sample. Two drops of the ultrasonicated liquid was added to the Cu
tape and allowed to air dry and then was analyzed using SEM and EDS for morphology and
elemental analysis.
4.7.3 Transmission Electron Microscopy (TEM)
Transmission Electron Microscopy (TEM), model Tecnai F-20 TEM was used to
determine the morphology of the product powders, while the Scanning Tunneling Electron
Microscopy (STEM) was used in elemental composition analysis. High Resolution TEM

67
(HRTEM) was used in observing the lattice fringes and subsequent calculation of d-spacing. The
product powders were dispersed in acetone and ultrasonicated for 5 minutes. One drop of the
liquid containing the sample was added to a 200-mesh holy carbon Cu TEM grid. The grid was
allowed to air dry and then analyzed for morphology, elemental analysis and lattice fringes.
4.7.4 Differential Scanning Calorimetry (DSC)
Differential Scanning Calorimetry (DSC), model Perkin Elmer Diamond, was used to
determine the heat capacity of the sample. The DSC instrument was calibrated using Indium as
standard before the measurement of heat capacity. The powder sample was homogenized and
about 5-10 mg was added to a aluminum DSC sample pan. It was covered with a aluminum lid
and crimped to maintain a air tight environment for measurements. The baseline heat flow was
measured using empty sample and reference pans. The sample side was loaded with the crimped
powder sample and reference side was loaded with an empty pan. An ‘Iso-Scan-Iso’ temperature
scan with an interval of 50ºC and a heating rate of 10ºC/min was used to determine the heat flow
curves of the sample and baseline. The calculation of heat capacity of sample is done by the
difference in the heat flow between the sample and the baseline.
4.7.5 Raman Spectra
Raman spectra of product powders were obtained with a Bruker Senterra Raman
microscope, using 785 nm wavelength, 1 mW laser source at a 50x magnification. The sample
powders were added to a glass slide and surface was flattened. Using a 50x lens, the sample was
focused. Using the above mentioned laser source settings, the spectra was obtained with three
repetitions per sample. The final spectrum of a sample is the average of three repetitions.

68
4.7.6 X-Ray Photoelectron Spectroscopy (XPS)
X-Ray Photoelectron Spectroscopy (XPS), model KRATOS AXIS 165 XPS/Auger, was
used to determine the chemical binding states of the product powders. Powder sample was
dispersed on a glass slide using vacuum grease. The sample was then focused with a CCD
camera and the XPS spectra were obtained. The sample was analyzed for C-1s, O-1s, Si-2s and
Si-2p binding energies.
4.7.7 Thermo-Gravimetric and Differential Thermal Analyzer (TG-DTA)
Perkin Elmer Diamond Thermo-Gravimetric and Differential Thermal Analyzer (TG-
DTA) was used to measure the weight change and the heat flow to or from the sample. Platinum
sample and reference pans were used during analysis. The instrument was calibrated using
indium, aluminum and tin for heat flow. The product powders were homogenized and about 15-
20 mg was added to the sample pan. The reference pan was added with approximately same
amount of alumina. The experiment was conducted in air atmosphere. A temperature ramp from
50ºC to 1100ºC was done at a heating rate of 10ºC/min. The weight gain/loss with corresponding
heat loss/gain was recorded as a function of temperature of the sample.

69
CHAPTER 5
SYNTHESIS OF TITANIUM DIBORIDE
5.1 Thermochemical calculations
Thermodynamic calculations were made on the TiO2-B2O3-CH4 system using HSC
Chemistry 5.1® [92]. In the TiO2-B2O3-CH4 system, the molar ratio of the solid feed TiO2 and
B2O3 was maintained a constant at 1:1. The reducing gas used was methane and the molar ratio
of reducing gas to that of the solid feed was varied between 4 and 7. The calculations are based
on the principle of minimization of Gibb’s energy [93] for a set of reactants at a given
temperature and pressure. The equation describing this principle is given in equation 23.
∑ ∑ ∑ (23)
G is the total Gibbs energy of the system, Gi0 is standard molar Gibbs energy of species i at
temperature T and pressure P, ni is number of moles of species i, Pi is the partial pressure of
species i, Xi is mole fraction of species i, and i is activity coefficient of species i.
Formation of various elements and compounds in different phases can be determined
using the thermochemical calculations as a function of temperature if pressure is maintained a
constant. Figure 5.1 and Figure 5.2 shows the stability of phases as a function of temperature
between 0 and 7000⁰C for molar ratio of solid feed to methane (TiO2:B2O3:CH4) varying from
1:1:4 to 1:1:7. It is observed in all the cases that the formation of TiB2 in various quantities
occurs between the temperatures of about 1000 – 3500⁰C.

70
Figure 5.1: Formation of various product phases at a molar ratio of (a) TiO2:B2O3:CH4=1:1:4 and
(b) TiO2:B2O3:CH4=1:1:5.
(a)
(b)

71
Figure 5.2: Formation of various product phases at a molar ratio of (a) TiO2:B2O3:CH4=1:1:6 and
(b) TiO2:B2O3:CH4=1:1:7.
(a)
(b)

72
If complete reduction of oxides takes place in the presence of methane to form elemental
titanium and boron in vapor phase with subsequent reaction of the elements to form titanium
diboride, the following stoichiometric reactions (equations 24-27) can be written for varying
molar ratio of methane.
4 4 8 (24)
5 5 10 (25)
6 5 12 (26)
7 5 2 14 (27)
Table 5.1: Theoretical yield of TiB2 and by-products in mol% as a function of molar ratio of
methane in feed.
Molar ratio
of Methane TiB2 TiO2 B2O3 B2O3(g) TiB B TiC TiO C
4 54.69 5.47 5.47 13.28 6.25 3.13 0.00 11.72 0.00
5 82.51 0.00 0.00 1.35 7.17 4.48 0.00 0.00 4.48
6 47.06 0.00 0.00 0.00 1.96 1.96 0.00 0.00 49.02
7 31.15 0.00 0.00 0.00 1.31 1.97 0.33 0.00 65.25
The results from thermochemical calculations are shown in Table 5.1 for the
TiO2:B2O3:CH4 system. At lower molar ratios of methane especially at 4, the conversion to TiB2
is not complete with the formation of some of the oxide phases such as TiO, TiO2 and B2O3
along with some boron and titanium boride (TiB) in the product. There is no carbon observed in
the systems with a methane molar ratio of 4 in the feed. At 4 moles of methane in the feed, the

73
formation of about 5 mol% of TiO2 and B2O3 each, about 13 mol% B2O3 (g), 6 mol% TiB, 3
mol% B and about 12 mol% TiO, was observed.
The formation of oxides, at 4 moles of methane in the feed, such as TiO2, TiO and B2O3
in the final products is attributed to the fact that the amount of carbon in the reactants is low and
is not sufficient to reduce the reactants in the reactor. With the increase in the molar ratio of
methane to 5, the oxide phases such as TiO2 and B2O3 disappear while TiB and B along with
some carbon are observed. It is also notable that the amount of TiB2 increased from about 55
mol% at a molar ratio of 4 to about 82 mol% at a molar ratio of 5. Subsequent increase in the
molar ratio of methane to 6 and 7 shows no oxide phases and the amount of TiB and B were
almost minimum in the final product. On the contrary, the only two major phases observed are
TiB2 and carbon, with substantial increase in the amount carbon to about 50 mol% at a molar
ratio of 6 and about 65 mol% at a molar ratio of 7. While 5 moles of methane has just sufficient
enough carbon to reduce the oxides, an increase in the moles of methane to 6 and 7 increases the
amount of carbon in the product powder. The formation of TiC in very small quantities is
observed at 7 moles of methane. The formation of TiB and B is seen in the products using higher
molar ratio of methane in the feed but are in significantly smaller quantities and can be
neglected.
Theoretical recovery of TiB2 and other phases as a function of varying methane mole
fraction shows that the yield of TiB2 is maximum (>80 mol %) when the molar ratio is 5 and at a
temperature of 1770⁰C (2043 K). The amount of TiB2 increases with the increase in molar ratio
of CH4 up to 5 moles as shown in Figure 5.3. Further increase in the molar ratio of CH4 increases
the amount of carbon formed in the final product. The yield of TiB2 decreases with increase in
methane content above 5 moles.
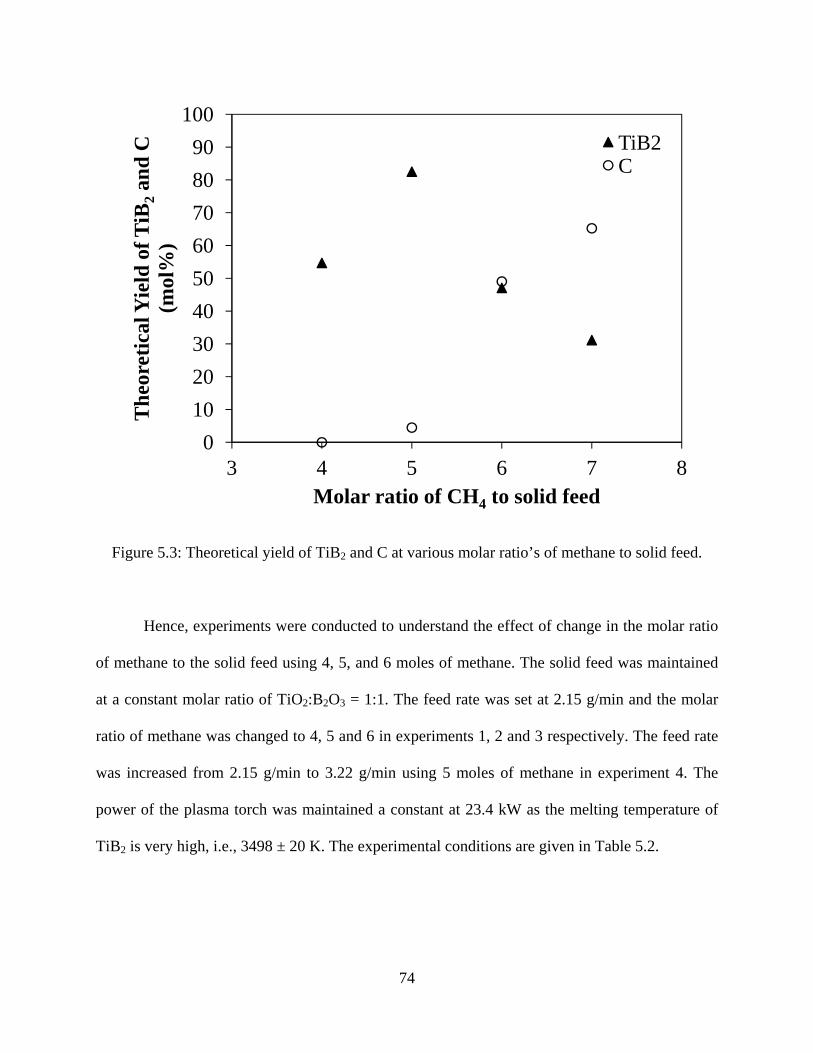
74
Figure 5.3: Theoretical yield of TiB2 and C at various molar ratio’s of methane to solid feed.
Hence, experiments were conducted to understand the effect of change in the molar ratio
of methane to the solid feed using 4, 5, and 6 moles of methane. The solid feed was maintained
at a constant molar ratio of TiO2:B2O3 = 1:1. The feed rate was set at 2.15 g/min and the molar
ratio of methane was changed to 4, 5 and 6 in experiments 1, 2 and 3 respectively. The feed rate
was increased from 2.15 g/min to 3.22 g/min using 5 moles of methane in experiment 4. The
power of the plasma torch was maintained a constant at 23.4 kW as the melting temperature of
TiB2 is very high, i.e., 3498 ± 20 K. The experimental conditions are given in Table 5.2.
0
10
20
30
40
50
60
70
80
90
100
3 4 5 6 7 8
Th
eore
tica
l Yie
ld o
f T
iB2
and
C
(mol
%)
Molar ratio of CH4 to solid feed
TiB2C

75
Table 5.2: Experimental design for the production of TiB2 using thermal plasma reactor.
Expt. # Raw Materials Molar Ratio Plasma Power (kW) Feed Rate (g/min)
1 TiO2:B2O3:CH4 1:1:4 23.4 2.15
2 TiO2:B2O3:CH4 1:1:5 23.4 2.15
3 TiO2:B2O3:CH4 1:1:6 23.4 2.15
4 TiO2:B2O3:CH4 1:1:5 23.4 3.22
5.2 Synthesis and characterization of titanium diboride
5.2.1 Phase and morphology of solid feed
The molar ratio of the solid feed TiO2:B2O3 was maintained at 1:1, consistent with the
thermochemical calculations. The mixture of the two oxides in the solid feed was analyzed using
XRD and the resulting pattern is shown in Figure 5.4.
Figure 5.4: X-Ray diffraction pattern of 1:1 molar TiO2:B2O3.

76
Figure 5.5: SEM images of solid feed powder at a molar ratio of TiO2:B2O3 = 1:1.
The morphology of the solid feed material in the molar ratio of TiO2:B2O3 = 1:1 was
analyzed using SEM. The images shown in Figure 5.5 show that the starting material was in the
size range of 20 – 50 m.
5.2.2 Phase, composition and morphology of product powders
Figure 5.6 shows the XRD patterns from the experiments conducted at different molar
ratio of methane and the feed rates. The amount of TiB2 in the final product in the case of
experiments 1 (4 moles of CH4 in feed) and 3 (6 moles of CH4 in feed), are relatively very less
compared to TiO2, B2O3 and C. Another important observation from the diffraction pattern is that
titanium dioxide in the product powder has changed from its original crystal structure of anatase
to its high temperature crystal structure, namely rutile. The anatase oxide phase was also present
in the product powders from experiments 1 and 3 but the amount was relatively small. This
suggests that the powders were vaporized and/or heated to very high temperatures and due to the
rapid quenching that takes place in the reactor TiO2 was condensed in its high temperature
structure. The experiments conducted with a methane molar ratio of 5 in feed yielded more TiB2

77
compared to those with molar ratios of 4 and 6 moles as can be seen from the XRD pattern
shown in Figure 5.6 (Expt-2).
20 30 40 50 60 70 80
*
0 Expt - 1
Expt - 2
Expt - 3
*
*
Inte
nsit
y (a
.u.)
Angle, 2 (degrees)
^
^
* *
*
x
0 0
0 - TiB2
* - TiO2 (a)
x - TiO2 (r)
^ - B2O
3
+ - C
^
x
0
0
+0
0 0 +
^
^
+ 0
*
*x* 0
*
*
^x
0
+ 0 +Expt - 4
Figure 5.6: XRD patterns of product powders obtained from different experiments.
Similar results were observed with increased solid feed rate of 3.22 g/min and a methane
molar ratio of 5 (Expt-4). The product phases such as TiC, TiB, and TiO that were obtained from
the thermodynamic simulation were not observed in the XRD patterns of product powders from
any of the experiments. Thus, we can conclude that all the phases that are thermodynamically
stable in a given system of reactions cannot be formed in the actual experiment using the thermal

78
plasma processing technique, but will serve as a good starting point for choosing experimental
conditions.
The volume fraction of the product powders were calculated using direct comparison
method from the XRD patterns. The volume fraction calculation was done on the product
powders in the condensed phase. Any products that escaped the reaction chamber in gaseous
phases are not accounted for. Similarly, amorphous phases that are present in the product
powders will not show peaks in the XRD pattern. They will rather add to the background and
hence, are not accounted for in these calculations. Some of the constants required to solve for the
volume fraction were obtained from Cullity [97]. The atomic positions of the elements in unit
cell were obtained from Pearson’s Handbook of Crystallographic Data [98]. The direct
comparison method was used in the calculation of the volume fractions of the product powders.
Equation (28) describes the equations for the product powders.
; ; ; 1 (28)
where, ITiB2, ITiO2, IB2O3 and IC represent the intensities of TiB2, TiO2, B2O3 and C respectively;
cTiB2, cTiO2, cB2O3 and cC represent the volume fractions of TiB2, TiO2, B2O3 and C respectively;
and RTiB2, RTiO2, RB2O3 and RC represent the volumes of inverse unit cell lattices of TiB2, TiO2,
B2O3 and C respectively.
Table 5.3: Yield of product powders at different feed rates with a methane molar ratio of 5 in the
feed.
Feed Rate (g/min) Yield TiB2 TiO2(r) B2O3 C
2.15 mol% 23.63 8.72 4.07 63.58
3.22 mol% 39.44 11.33 7.58 41.65

79
The values of the calculated volume fractions are reported in Table 5.3. With the increase
in the feed rate from 2.15 g/min to 3.22 g/min, the amount of TiB2 formed increased from about
24 mol% to about 40 mol%. It was also accompanied by the reduction in the amount of carbon in
the product powders from about 64 mol% to about 40 mol%.
Figure 5.7: SEM images of product powders obtained from experiment 4.

80
Figure 5.8: SEM electron image (top) of the product powders obtained from experiment 4 and
the corresponding EDS spectra (bottom).
The SEM micrograph of the product powders from experiment 4 is shown in Figure 5.7.
The particle sizes of these powders are in the range of 50-100 nm. The morphology of the
particles is about spherical and is uniform though out the sample. The Energy Dispersive Spectra
(EDS) and the corresponding electron image of the material are shown in Figure 5.8. The EDS
shows the presence of only titanium in the sample. The absence of oxygen and carbon in the

81
EDS proves that the titanium is not present as titanium oxide, titanium dioxide or titanium
carbide. Also, the absence of elemental titanium or boron from the XRD pattern of the sample
shows that the titanium is present either in the form of titanium boride (TiB) or titanium diboride
(TiB2) in the sample, as boron is not detectable using EDS. The absence of TiB phase from XRD
confirms the presence of only TiB2 in the sample.
Figure 5.9: TEM image of the product powder obtained from experiment 4.
The morphology of the material produced from experiment 4 was analyzed using TEM.
Figure 5.9 shows that the particles formed are in the range of 20-100 nm and are mostly spherical
as observed from the SEM images. A high resolution TEM image of the powder is shown in
Figure 5.10. The lattice fringes from the image were used in determining the d-spacing of the
crystal structure. From the calculations, it was found that the d-spacing was 3.75 Å, which
corresponds to the d-spacing of hexagonal boron oxide in the (1 0 0) plane.

82
Figure 5.10: HRTEM of product powders showing lattice fringes of hexagonal B2O3 in the (1 0
0) plane obtained from experiment 4.
STEM image of product powders from experiment 4 is shown in Figure 5.11. As the
major phases in the product powder (other than C) is TiB2 and TiO2, which have closer atomic
weight, phase contrast due to different phases is not as pronounced in the image. A spot EDS
was obtained from one of the brightest spots in the image which would correspond to a high
atomic number phase. It is marked in the STEM image with a red circle. The EDS corresponding
to the spot is shown on the right in Figure 5.11. Cu and Si are observed in the EDS which are
from the grid used to disperse the powders. Titanium is detected from the elemental analysis
similar to the EDS obtained from SEM-EDS. On the basis of a similar argument made earlier in
the SEM-EDS section, it can be concluded that the particle is titanium diboride.

83
Figure 5.11: STEM image (left) of product powders obtained from experiment 4 and the
corresponding EDS spectra (right).
5.2.3 Phase transformation
The product powders were relatively finer in size of the order of 20-100 nm as seen from
the morphology of the product powders shown in Figures 5.7 and 5.9. The phase transformation
of anatase to rutile, either through the transformation to brookite or a direct transformation, is
reported in literature [94] for particles less than 100 nm in size. The temperature of
transformation is relatively less and is about 1000 K. Similar observation was found in the
literature [95] for the transformation of anatase to rutile in the temperature range of 973 to 1073
K. The transformation of anatase to rutile has been reported [96] at a temperature as low as 923
K after heat treatment for 4 hours. They also reported that the increase in the temperature
decreases the heat treatment time. Since the temperatures achieved in the plasma reactor are very
high, of the order of thousands of degrees, the phase transformation from anatase to rutile occurs
instantaneously.

84
5.2.4 Particle size reduction
Another important observation can be made from the morphology of the product
powders. The initial particle size of the solid feed as shown in Figure 5.5 is about 45 m or less.
A high resolution TEM (HRTEM) image of unreacted boron oxide is shown in Figure 5.10. It is
seen that particle size of the chemically unreacted boron oxide has reduced to about tens of
nanometers from submicron scale in the raw material. This observation can be used to conclude
that thermal plasma can be used in reduction of particle size of materials. The actual mechanism
in which the size reduction takes place is unknown at this time. As mentioned earlier the
temperature inside the reactor is of the order of thousands of degrees. One of mechanism that is
proposed here, by which the size reduction takes place, might be complete vaporization of the
feed and re-condensation of the unreacted boron oxide. The second reaction proposed is that the
surface of the particle melts and vaporizes at high temperatures, but complete vaporization
doesn’t take place leading to the reduction in the particle size.

85
CHAPTER 6
SYNTHESIS OF SILICON CARBIDE
6.1 Thermochemical calculations
Thermodynamic calculations were made on the SiO2-CH4 system using HSC Chemistry
5.1® [92]. In the SiO2-CH4 system, the molar ratio was varied between 0.8 and 5. The
calculations are based on the principle of minimization of Gibb’s energy [93] for a set of
reactants at a given temperature and pressure as explained in equation (23) in chapter 5.
Figure 6.1: Thermochemical calculations to determine stable phases at 1220⁰C as a function of
molar ratio of methane to solid feed.
0.00.10.20.30.40.50.60.70.80.91.01.11.21.31.41.5
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5 6
Th
eoretical Yield
(mol%
)T
heo
reti
cal Y
ield
(m
ol%
)
Molar Ratio of Methane to Solid Feed
SiO2
C
SiC
Si
T = 1220ºC

86
The amount of theoretical product formation as a function of molar ratio of methane in
the feed at various temperatures was calculated and is shown in Figure 6.1. At 1220⁰C, the
temperature is relatively low and the amount of SiO2 reduced is relatively small as well. The
amount of carbon at 1220⁰C increases with increase in the amount of methane consistent with
the small amount of SiO2 reduced.
Figure 6.2: Thermochemical calculations to determine stable phases at 1520⁰C as a function of
molar ratio of methane to solid feed.
The equilibrium composition at 1520⁰C (Figure 6.2) shows an increase in the amount of
SiC with increase in the molar ratio of methane in the feed from 0.8 and reaches a maximum at
3. The SiC formation decreases with subsequent increase of molar ratio of methane from 3 to 5
with a corresponding increase in the amount of carbon and almost no SiO2 present.
0
10
20
30
40
50
60
70
80
90
100
0.5 1.5 2.5 3.5 4.5 5.5
Th
eore
tica
l Yie
ld (
mol
%)
Molar Ratio of Methane to Solid Feed
SiO2
C
SiC
SiO(g)
Si
T = 1520ºC

87
Figure 6.3: Thermochemical calculations to determine stable phases at 2120⁰C as a function of
molar ratio of methane to solid feed.
When the temperature is increased to 2120⁰C, SiO2 is converted to SiO in gaseous phase
(Figure 6.3). Silicon forms when molar ratio of methane was increased above 1 and is a
maximum at 2. SiC forms only when the molar ratio of methane is 3. The amount of C increases
and SiO decreases with the increase in the amount of methane. At extremely high temperature,
for example at 3010⁰C, there is no formation of SiC, with Si forming after a methane molar ratio
of 1 and has a maximum at 2 (Figure 6.4). The amount of SiO decreases and the amount of C
increases with the increase in the methane concentration in feed.
It is concluded from thermochemical calculations that formation of SiC is favorable at
lower molar ratios of methane and is a maximum at 3. Favorable temperatures for the formation
of SiC are from about 1500⁰C to 2100⁰C. Formation of Si is favored at lower molar ratios of
0
20
40
60
80
100
120
0.0 1.0 2.0 3.0 4.0 5.0 6.0
Th
eore
tica
l Yie
ld (
mol
%)
Molar Ratio of Methane to Solid Feed
SiO2SiCSiCSiO(g)
T = 2120ºC

88
methane as well but is a maximum at a molar ratio of 2. Formation temperatures favorable for Si
are from about 2100⁰C to 3000⁰C. Formation of carbon increases steadily with the increase of
methane in the feed. Relatively very low amount of SiC is formed at very low (<1500⁰C) and at
very high (>2100⁰C) temperatures. These observations led to the design of experiments with the
variation in the moles of methane between 0.8 and 3, two different plasma powers 18.9 and 21.6
kW and two different solid feed rates, 4 and 5 g of SiO2/min. The experimental conditions are
given in Table 6.1.
Figure 6.4: Thermochemical calculations to determine stable phases at 3010⁰C as a function of
molar ratio of methane to solid feed.
0
20
40
60
80
100
120
0.0 1.0 2.0 3.0 4.0 5.0 6.0
Th
eore
tica
l Yie
ld (
mol
%)
Molar Ratio of Methane to Solid Feed
Si
C
SiO(g)
T = 3010ºC

89
Table 6.1: Experimental Conditions for the production of SiC using thermal plasma
Expt. # Raw Materials Molar Ratio Plasma Power (kW) Feed Rate (g/min)
1 SiO2:CH4 1:1 21.6 5
2 SiO2:CH4 1:0.8 21.6 5
3 SiO2:CH4 1:1.5 21.6 5
4 SiO2:CH4 1:1 18.9 5
5 SiO2:CH4 1:2 21.6 5
6 SiO2:CH4 1:2.5 21.6 5
7 SiO2:CH4 1:1 21.6 4
8 SiO2:CH4 1:2 21.6 4
9 SiO2:CH4 1:3 21.6 4
6.2 Synthesis and characterization of silicon carbide
6.2.1 Phase and composition of product powders
X-ray diffraction was done on the product powders from the experiment to determine the
product phases. The XRD patterns from experiments using a constant plasma power of 21.6 kW
and a constant feed rate of 5 g/min are shown in Figure 6.5. SiC is the major phase in all the
experiments with the variation of molar ratio of methane, while the amount of Si, C and SiO2
varies. Figure 6.6 shows the XRD patterns of the product powders from experiments with a
constant feed rate of 4 g/min and a constant power of 21.6 kW. The amount of SiC is more in the
case of 1:1 molar ratio of SiO2:CH4 but decreases with increase in the amount of methane with a
corresponding increase in the amount of SiO2 and carbon in the product powders.

90
20 40 60 80 100 120
0
2000
4000
6000
8000
10000
12000
(1 0
1)#
(0 0
2)^
SiO2:CH
4 = 1:2.5
SiO2:CH
4 = 1:2
SiO2:CH
4 = 1:1.5
SiO2:CH
4 = 1:1
Inte
nsity
(a.
u.)
Angle, 2 (degrees)
SiO2:CH
4 = 1:0.8
Feed Rate = 5g SiO2/min
Power = 21.6 kW(1 1
1)*
(2 0
0)*
(2 2
0)*
(3 1
1)*
(2 2
2)*
(3 3
1)*
(4 2
0)*
(1 1
1)0
(2 2
0)0
* SiC0 Si ̂C
# SiO2
(3 1
1)0
(1 0
2)^
(1 0
0)^
(1 0
0)#
Figure 6.5: XRD pattern of product powders formed at various molar ratios of methane to solid
feed and at a solid feed rate of 5g SiO2/min.
The volume fractions were calculated using the direct comparison method as described
earlier and was converted to corresponding mole percent and is presented in Figure 6.7 for the
experimental yield of products at a feed rate of 5 g/min and a power of 21.6 kW. As can be seen
SiC is the major phase formed with a minimum of about 40 mol% and a maximum of about 65
mol% at corresponding methane molar ratios of 0.8 and 2 in the feed, respectively. The amount
of Si is a maximum at a methane molar ratio of 1 and is determined to be about 40 mol%. SiO2 in
the product powder varies between a minimum of about 5 mol% and a maximum of about 35
mol%. The amount of C is relatively low in the experiments using a feed rate of 5 g of SiO2/min
and is a maximum of about 20 mol% at a methane molar ratio of 2.5. It can be concluded that

91
relatively lower molar ratios of methane favors formation of Si while relatively higher molar
ratios of methane favors the formation of SiC at a feed rate of 5g SiO2/min.
20 40 60 80 100 120
0
1000
2000
3000
4000
5000* SiC0 Si ̂C
# SiO2
Feed Rate = 4g SiO2/min
Power = 21.6 kW
SiO2:CH
4 = 1:3
SiO2:CH
4 = 1:2
SiO2:CH
4 = 1:1
Inte
nsi
ty (
a.u
.)
Angle, 2 (degrees)
(1 0
0)#
(0 0
2)^
(1 0
1)#
(1 1
1)0
(1 1
1)*
(2 0
0)*
(2 2
0)*
(3 1
1)*
(2 2
2)*
(3 3
1)*
(4 2
0)*
(1 0
0)^
(1 0
2)^
(3 1
1)0
Figure 6.6: XRD patterns of product powders formed at various molar ratios of methane to solid
feed and at a solid feed rate of 4g SiO2/min.
Similar calculations were done on the product powders from the experiments conducted
at a solid feed rate of 4 g of SiO2/min. It can be seen from Figure 6.8 that the amount of SiC and
Si are both a maximum at a methane molar ratio of 1. The experimental yield of SiC and Si were
found to be about 55 mol% and 35 mol% respectively. The amount of SiO2 in the product
powders seemed to be almost a constant with change in the molar ratio of methane and is about
10 mol%.

92
Figure 6.7: Experimental yield of product powders formed at various molar ratios of methane to
solid feed and a solid feed rate of 5g/min.
Figure 6.8: Experimental yield of product powders formed at various molar ratios of methane to
solid feed and a solid feed rate of 4g/min.
0
10
20
30
40
50
60
70
0.5 1 1.5 2 2.5 3
Yie
ld (
mol
%)
Molar Ratio of Methane to Solid Feed
SiC ProductionFeed Rate 5 g/min
Power 21.6 kW
SiO2SiSiCC
0
10
20
30
40
50
60
70
0 1 2 3 4
Yie
ld (
mol
%)
Molar Ratio of Methane to Solid Feed
SiC ProductionFeed Rate 4 g/min
Power 21.6 kW
SiO2SiSiCC
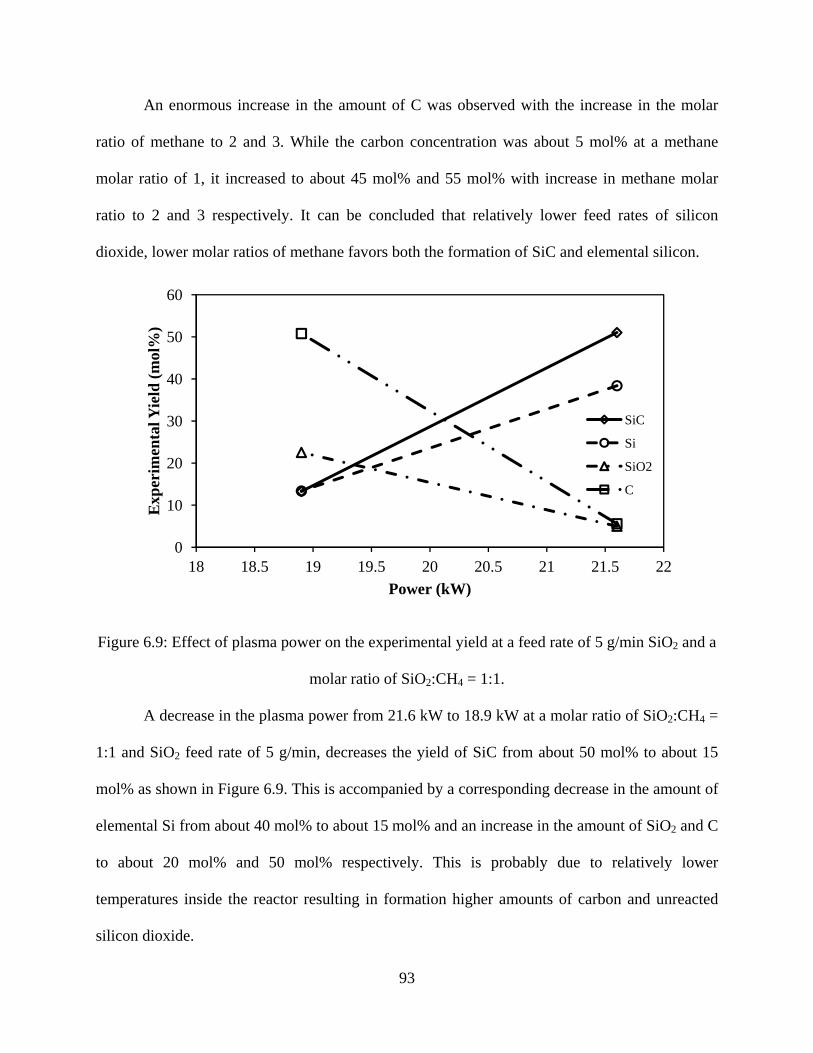
93
An enormous increase in the amount of C was observed with the increase in the molar
ratio of methane to 2 and 3. While the carbon concentration was about 5 mol% at a methane
molar ratio of 1, it increased to about 45 mol% and 55 mol% with increase in methane molar
ratio to 2 and 3 respectively. It can be concluded that relatively lower feed rates of silicon
dioxide, lower molar ratios of methane favors both the formation of SiC and elemental silicon.
Figure 6.9: Effect of plasma power on the experimental yield at a feed rate of 5 g/min SiO2 and a
molar ratio of SiO2:CH4 = 1:1.
A decrease in the plasma power from 21.6 kW to 18.9 kW at a molar ratio of SiO2:CH4 =
1:1 and SiO2 feed rate of 5 g/min, decreases the yield of SiC from about 50 mol% to about 15
mol% as shown in Figure 6.9. This is accompanied by a corresponding decrease in the amount of
elemental Si from about 40 mol% to about 15 mol% and an increase in the amount of SiO2 and C
to about 20 mol% and 50 mol% respectively. This is probably due to relatively lower
temperatures inside the reactor resulting in formation higher amounts of carbon and unreacted
silicon dioxide.
0
10
20
30
40
50
60
18 18.5 19 19.5 20 20.5 21 21.5 22
Exp
erim
enta
l Yie
ld (
mol
%)
Power (kW)
SiC
Si
SiO2
C

94
Figure 6.10: Effect of SiO2 feed rate on the yield of SiC at a power of 21.6 kW and a molar ratio
of SiO2:CH4 = 1:2.
Figure 6.11: Comparison of theoretical yield of SiC at various temperatures to experimental yield
at a solid feed rate of 5g/min.
0
10
20
30
40
50
60
70
3 4 5 6
Exp
erim
enta
l Yie
ld (
mol
%)
Feed Rate of SiO2 (g/min)
SiC
Si
SiO2
C
-10
0
10
20
30
40
50
60
70
80
90
100
0.5 1 1.5 2 2.5 3 3.5 4 4.5 5 5.5
Yie
ld (
mol
%)
Molar Ratio of CH4 to Solid Feed
1220 C - Theoretical1520 C - Theoretical2120 C - Theoretical3010 C - TheoreticalExperimental

95
The effect of SiO2 solid feed rate on the formation of product phases from experiments at
a molar ratio of SiO2:CH4 = 1:2 and a power of 21.6 kW is shown in Figure 6.10. Decrease in the
feed rate results in decreased amount of SiC and is almost half the amount formed at 5 g/min
feed rate. The amount of carbon, on the other hand, increased with the decrease in feed rate and
is almost twice as much compared to 5 g/min feed rate. The amount of elemental silicon in the
product powders remain almost the same while the amount of unreacted silicon dioxide has
doubled in quantity. A comparison with the yield at a molar ratio of SiO2:CH4 = 1:1 from
Figures 6.7 and 6.8 shows that the yield of product phases are almost similar. Hence, it can be
concluded that relatively higher molar ratios of methane and lower feed rate favors the formation
of carbon and unreacted SiO2, thus decreasing the yield of SiC.
A comparison of theoretical yield at various temperatures to the experimental yield of
product powders formed at various amounts of methane in feed and a solid feed rate of 5 g/min
was made. It is seen from Figure 6.11 that the experimental yield of SiC closely follows the
theoretical yield at 1520⁰C and is in between 1520⁰C to 2100⁰C. This observation suggests that
the temperature of the plasma reactor is about 1500⁰C where the formation of SiC takes place.
Theoretical calculation suggests the formation of elemental silicon at relatively higher
temperatures between 2100ºC and 3000ºC and at molar ratio of methane more than 1. But
experiments show the formation of Si at methane molar ratio of 1 or less. The formation of
SiO(g) is favorable at lower molar ratios as can be seen from Figures 6.3 and 6.4. Hence it is
possible that SiO2 vaporizes inside the reactor and reduces to SiO (g) at higher temperatures
which is subsequently reduced to Si (g). The gaseous Si condenses and nucleates on the quench
coils which are seen in the XRD patterns shown in Figure 6.5 and 6.6.

96
Heat capacity measurement can be used as a method to verify the composition of a
mixture. The composition of the product powders were obtained from XRD spectra using direct
comparison method. The heat capacity of the powder can be measured using DSC and compared
with the theoretically calculated heat capacity using a mixture rule. The heat capacity of a
mixture is the weight-averaged sum of the heat capacity of the constituents assuming no
interaction takes place between the constituents. It can be expressed in the form of an equation
given below.
∑ , (29)
Where CP(mixture) is the heat capacity of the mixture, wi is the weight fraction of the ith
component and CP,i is the heat capacity of the ith component. The heat capacities of pure
components were calculated using HSC chemistry between the temperatures of 50 and 350⁰C
[92]. The weight fractions of product powders from experiment 1 were calculated from
corresponding volume fraction obtained using direct comparison method and the density of pure
components and is given in Table 6.2.
Table 6.2: Weight fraction of product powders from experiment 1
Component Wt%
SiO2 9.44
Si 24.21
SiC 64.24
C 2.11
Total 100.00

97
Heat capacity measurement of product powders from experiment 1 was carried out using
a Perkin-Elmer Diamond DSC. The sample and the baseline heat flow, along with the heat
capacity of the material are shown in Figure 6.12. An ‘Iso-Scan-Iso’ temperature scan with an
interval of 50ºC and a heating rate of 10ºC/min was used to determine the heat flow curves of the
sample and baseline. The calculation of heat capacity of sample is done by the difference in the
heat flow between the sample and the baseline.
50 100 150 200 250 300 350
0
1
2
3
Hea
t Cap
acity
(J/
g*o C
)
Temperature (oC)
Specific Heat (J/g*°C)
-10
0
10
20
30
40
50
60
70
80
Sample Heat Flow Endo Up (mW) Baseline Heat Flow Endo Up (mW)
Hea
t Flo
w E
ndo
Up
(mW
)
Figure 6.12: Heat capacity, sample heat flow and baseline heat flow for product powders from
experiment 1 using DSC.

98
The experimentally measured heat capacity was compared with that calculated using
equation (29) as shown in Figure 6.13. The measured heat capacities match quite well with the
theoretically calculated heat capacity. This further substantiates the accuracy of the composition
of the phases present in the sample calculated using the direct comparison method.
Figure 6.13: Comparison of heat capacity measured experimentally with those calculated using
equation (29).
6.2.2 Morphology of product powders
Scanning electron microscopy was used to determine the morphology of the product
powders from the experiments. Figure 6.14 shows the morphology of the products from
experiment 1 with methane molar ratio of 1, solid feed rate of 5 g/min and a power of 21.6 kW.
0.0
0.5
1.0
1.5
2.0
2.5
3.0
50 100 150 200 250 300 350
Hea
t C
apac
ity,
CP
(J/g
*⁰C
)
Temperature (⁰C)
Cp (mix)Experimental

99
Figure 6.14: SEM image showing the morphology of the product powders from experiment 1.
Figure 6.15: SEM image showing the morphology of the product powders from experiment 5.

100
Figure 6.16: SEM image showing the morphology of the product powders from experiment 7.
The particles are almost spherical with an average diameter varying between 200 and 500
nm. It can also be seen from the micrographs that formation of nanorods are observed in the
morphology. Figure 6.15, on the other hand, shows the morphology of powders from experiment
5 with a methane molar ratio of 2 and 5 g/min solid feed. Mostly spherical particulates are
observed in this case with an average particle size distribution between 200 and 500 nm.
It was also observed that with the increase in the amount of methane in the feed above 1,
the amount of nanorods observed were less. Correlating with the phases formed from the XRD
patterns, it is speculated that the formation of nanorods is due to the presence of Si in the product
powders. This speculation is further justified by the morphology of the product powders from
experiment 7 with 1 mole of methane and 4 g/min solid feed rate in Figure 6.16. There is about
35 mol% Si present in the product powder which might have favored the formation of the

101
nanorods. Particulate matter of spherical morphology was also observed with an average particle
size between 200 nm and 500 nm.
Figure 6.17: TEM image showing the morphology of the product powders from experiment 1.
Product powders from experiment 1 at a molar ratio of SiO2: CH4 = 1:1 and a feed rate of
5g/min were further analyzed using TEM for its morphological features. Nanorods of about 20 -
30 nm in diameter are observed as can be seen from Figure 6.17. Similar observation was made
from the powders obtained from experiment 2 at a molar ratio of SiO2: CH4 = 1:0.8. Figure 6.18
(a) shows nanorods with an average diameter of 20-30 nm. High resolution TEM images from
the same experiment show the presence of Si at the tip of a nanorod as seen in Figure 6.18 (b).
Further evidence for the presence of Si is shown in Figure 6.18 (c) with a small inclusion of SiO2
of about 5nm in size. There is substantial amount (about 25 mol%) of unreacted SiO2 present in
the product powders from experiment 2 with a methane molar ratio of 0.8 as seen from the XRD

102
analysis. A small single crystal SiO2 particle was observed in the TEM analysis shown in Figure
6.18 (d) with insets of a magnified image and the corresponding diffraction pattern.
Figure 6.18: (a) TEM image showing the morphology of the product powders from experiment
2; (b) and (c) HRTEM showing presence of Si and (d) SiO2 single crystal with insets of
magnified image and electron diffraction pattern.
(a) (b)
(c) (d)

103
6.2.3 Particle size reduction
An observation similar to that in the synthesis of titanium diboride can be made here. The
particle size of the initial solid feed was 45 m or less. Figure 6.18 (d) shows a high resolution
image of silicon dioxide particle from experiment 2. As can be seen from the image, the size of
the unreacted silicon dioxide particle is in tens of nanometers. While the actual mechanism is
unknown at this time, the two mechanisms proposed earlier, namely, vaporization and re-
condensation of unreacted solid feed and surface vaporization, are assumed to be a possible way
in which the particle size reduction takes place.
6.2.4 Qualitative analysis of product powders
Inelastic or Raman scattering using a laser source was used to qualitatively substantiate
the presence of various components in the product powders. Raman spectra of product powders
from experiments 1, 2 and 3 (methane molar ratios varying from 0.8 to 1.5 at 5 g/min feed rate)
were obtained as shown in Figure 6.19. Presence of carbon in all the samples is substantiated by
the peaks at wavenumbers 1302.17, 1573.89 and 2593.51 cm-1. The peaks at wavenumbers 789
and 925.63 cm-1 correspond to SiC. It is reported in the literature that these wavenumbers
correspond to SiC in nanorod form [99]. The peaks corresponding to SiC are observed only in
experiments 1 and 2 at methane molar ratios of 0.8 and 1 and not in experiment 3 which
corresponds to a methane molar ratio of 1.5.

104
500 1000 1500 2000 2500 3000 35000
50
100
150
200
250
300
350
400
450
500
550
600
650
700
Experiment 3
Experiment 2
Experiment 1
512.
49, S
i
Ram
an I
nten
sity
Wavenumber (cm-1)
Experiment 1 (1:1) Experiment 2 (1:0.8) Experiment 3 (1:1.5)
300.
32
925.
63, S
iC N
R
1302
.17,
car
bon
1573
.89,
car
bon
2593
.51,
car
bon
789,
SiC
NR
Feed Rate 5g/min
Figure 6.19: Raman spectra of product powders from experiments 1, 2 and 3 using a solid feed
rate of 5g/min.
Presence of Si in the samples from experiment 1 and 2 are substantiated by the presence
of peak at a wavenumber of 512.49 cm-1. The composition calculation using XRD pattern shows
relatively high amounts of Si in experiments with methane molar ratio of 1 or less. This not only
corroborates the volume fraction calculation from XRD using direct comparison method, but
also accounts for presence of higher amounts of nanorods at lower molar ratios of methane as
seen from SEM and TEM.

105
92 94 96 98 100 102 104 106 108 110 112 114500
1000
1500
2000
2500
3000
3500
4000
4500
5000
XPS Raw Data XPS Raw Data - Gaussian Fit SiO
2 Peak - Gaussian Fit
SiC Peak - Gaussian Fit Si Peak - Gaussian Fit
Binding Energy (eV)
Inte
nsity
(C
PS
)
Figure 6.20: XPS spectra of product powders from experiment 1 showing the Si 2p peak resolved
into SiO2, SiC and Si peaks.
XPS spectra measures the elemental composition of a material based on its binding
energy. The difference in the binding energies can be used to substantiate the presence of
specific compounds. Si 2p XPS spectra of product powders from experiment 1 were obtained as
shown in Figure 6.20. A peak at binding energy of about 101.6 eV corresponds to elemental Si
bonding, while the peak at about 104.8 eV corresponds to the SiC bonding. The peak at about
108 eV corresponds to SiO2 in the product powders. The peaks positions for SiO2 and SiC have
also been reported in the literature [100,101]. A small change in the peak positions of about 2-4

106
eV was observed which might be due to the fluorescence effect of the powders used to determine
the binding energy. This provides further substantiation for the presence of Si, SiC and SiO2 in
the product powders.
6.2.5 Post-processing of product powders
100 200 300 400 500 600 700 800 900 1000 1100-800
-700
-600
-500
-400
-300
-200
-100
0
100
200
300
400
500100 200 300 400 500 600 700 800 900 1000 1100
Onset = 536.870C
W = 0.052 mg
W = 2.005 mg
Temperature (0C)
Hea
t Flo
w E
ndo
Dow
n (m
W)
Temperature (0C)
Heat Flow Endo Down (mW)
W = 0.548 mg
Onset = 1005.130C
15
16
17
18
19
20
Wei
ght (
mg)
Weight (mg)
Figure 6.21: Weight loss and heat flow curves from TG-DTA for the product powders from
experiment 1.

107
The product powders from thermal plasma synthesis experiments contain many phases
such as SiC, Si, SiO2 and C. Post processing or phase separation of these powders become
important in obtaining high quality powders useful in various applications. One of the easiest
ways of removing carbon is by calcining the material in oxygen or air atmosphere where carbon
reacts with oxygen to form carbon dioxide gas and leaves the system. To determine the
feasibility of this process the powders from experiment 1 were heated from room temperature to
1100⁰C in atmospheric air in a TG-DTA experiment (Figure 6.18). A weight loss (3.09 wt%)
was observed at a temperature of 536.87⁰C. It is accompanied by an exothermic heat. It is
speculated that the weight loss is due to the reaction of carbon and oxygen to form carbon
dioxide.
An 11.3 wt% weight gain was observed between the temperatures of 536.87⁰C and
1100⁰C. It is also accompanied by a steady drop in the heat flow. This could be due to the
oxidation of Si that is present in the sample due to the presence of atmospheric air. A small
weight gain (about 0.3 wt%) was observed at a temperature of 1005.13⁰C with an accompanying
exothermic heat. This is probably due to a phase transformation reaction that might be taking
place and is speculated to be the transformation of high quartz to tridymite. Hence it is possible
to remove carbon from product powders by calcining it at a temperature of about 550ºC in air. It
should also be noted that any increase in temperature might lead to the oxidation of elemental Si
to SiO2.

108
CHAPTER 7
CONCLUSIONS AND FUTURE WORK
7.1 CFD modeling of plasma reactor
A two dimensional (2D) model depicting the flow profile inside the plasma reactor was
developed using Fluent. Grid refinement studies were performed and an optimum grid size was
chosen for analysis. The velocity and temperature profiles inside the reactor were obtained. The
effect of inlet plasma pressure on the inlet velocity of the plasma gas was studied. It was
observed that velocity increases almost linearly with the increase in pressure. An assumed flow
path was generated inside the reactor. A conservative estimate of the residence time inside the
reactor was calculated as a function of plasma inlet gas pressure.
A three dimensional (3D) model was developed extending the 2D model. Transition SST
turbulence model was used with various grid sizes and turbulence layers to perform grid
refinement study. An optimum grid size was chosen and the effect of plasma inlet pressure on
the inlet velocity of the plasma gas was estimated. It was observed that the variation in velocity
was almost linear with change in the inlet pressure similar to the 2D model. The velocity at any
given pressure was found to be lower by about 35-40 m/s in the 3D model compared to that of
the 2D model. While the maximum velocity is lower than the 2D model, the residence time in
the 3D model is lower as well. Based on the requirements for experimental synthesis an
intermediate plasma gas pressure of 45 psi was used to ensure high temperature and a moderate
residence time in the reactor.

109
7.2 Synthesis of titanium diboride
The production of TiB2 using thermal plasma was carried out using TiO2 and B2O3 as
solid feed and CH4 as the reducing gas holding the molar ratio of the solid feed at 1:1.
Thermodynamic simulation based on the minimization of Gibbs energy shows that maximum
yield of TiB2 was obtained at a methane molar ratio of 5 with solid feed molar ratio of TiO2:B2O3
= 1:1. The experiments using 4, 5 and 6 moles of CH4 at a power of 23.4 kW and a solid feed
rate of 2.15 g/min showed that the yield of TiB2 was more using 5 moles of CH4 and was about
24 mol%. The increase in the solid feed rate from 2.15 g/min to 3.22 g/min at a feed composition
of TiO2:B2O3:CH4 = 1:1:5 increased the yield of TiB2 to about 40 mol%. The product yield
obtained in thermodynamic simulations in a reaction system may not be obtained in a thermal
plasma reactor. Hence, it is concluded that a relatively higher solid feed rate and a molar ratio of
TiO2:B2O3:CH4 = 1:1:5 yielded the maximum yield of TiB2. The as-formed product yield of TiB2
in a thermal plasma reactor is higher than that obtained in an earlier research. A change in crystal
structure was observed in TiO2 from anatase to rutile. While increase in feed rate from 2.15
g/min to 3.22 g/min increased the TiB2 yield from about 24 mol% to about 40 mol%, it also
reduced the amount of carbon from ~64 mol% to ~40 mol%. The TiB2 spherical particles formed
are in the range of 20-100 nm. Particle size reduction of boron oxide was observed in final
product powder from submicron sizes in solid feed to tens of nanometers in final product.

110
7.3 Synthesis of silicon carbide
SiC was successfully produced using thermal plasma using SiO2 as the solid feed and
methane as the reducing gas. Thermochemical calculations suggest that the yield of SiC is a
maximum at a temperature of 1520⁰C and a molar ratio of SiO2:CH4 = 1:3. Formation of Si and
SiO(g) was also observed from the calculations. Experiments using a molar ratio of SiO2:CH4 =
1:2 produced maximum yield of SiC with a solid feed rate of 5 g/min and a power of 21.6 kW.
Average heat capacity of the product powders was about 0.75 – 1.00 J/g*⁰C in the temperature
range of 50-350⁰C as determined by the DSC experiments, which is consistent with the
theoretical heat capacity calculated using mixture rule on the heat capacities of pure components.
This confirms the mole fractions of the product phases calculated using XRD. The size and shape
of the final products changed with the change in the process parameters. Spherical particulate
matter of no greater than 500 nm and nanorods of about 20-30 nm in diameter were observed. It
was speculated from HRTEM that Si acts as a nucleating agent for the nanorods to grow.
Reduction in particle size of unreacted silicon dioxide was observed in the final product
powders. The size of silicon dioxide in the final product powder is in tens of nanometers which is
very less compared to the submicron size of the starting powder. Raman spectra confirm the
presence of elemental Si in the samples and the presence of SiC nanorods. XPS confirms the
presence of Si, SiO2 and SiC in the samples. TG-DTA experiments in air show that the carbon
present in the sample can be removed when calcined at a temperature less than 600⁰C, thus,
preventing the oxidation of Si.

111
7.4 Future work
7.4.1 Modeling of plasma reactor
The flow properties of the plasma gas inside the reactor were obtained using both a 2D
model and a 3D model. It is seen that the 3D model more accurately predicts the flow inside the
reactor. An analysis of effect of various turbulence models on the flow properties in the reactor
showed that the transition SST model was the best for use in the thermal plasma reactors. Grid
refinement study was done on the meshes generated considering the total number of nodes and
the number of turbulence layers. An optimum grid size was chosen and the flow properties
determined inside the plasma reactor. Based on the model generated, a secondary solid phase can
be added to the system and the following information can be generated.
(i) Effect of solid feed on the flow properties in the reactor.
(ii) Effect of particle size on the residence time in the reactor.
(iii) Effect of solid feed rate on the residence time in the reactor.
This information can be generated for various systems, such as, TiO2, B2O3, SiO2, MgO,
etc. As the physical and the chemical properties of these compounds and their mixtures differ
from each other, the modeling results will give a good starting point for experiments with
different starting materials. After the experiments, the validation of the model can be performed
by comparing the theoretical weight of products and that obtained from the experiments.

112
7.4.2 Synthesis of TiB2
In the current research project, the synthesis of titanium diboride was carried out using a
constant molar ratio of TiO2:B2O3 = 1:1 in the solid feed. Due to the low melting and the
vaporization temperatures of boron oxide, some of it might have left the reactor in gaseous
phase. Standard data shows that the melting point of boron oxide is 450ºC. This might have been
a reason for the low (about 40 mol%) yield of TiB2 compared to about 82 mol% yield from the
theoretical calculations. This could be overcome by using higher molar ratios of boron oxide in
the feed materials. Experiments can be setup to use similar conditions such as a plasma power of
23.4 kW, molar ratio of methane to solid feed at 5 and a solid feed rate of 3.22 g/min, but the
molar ratio of TiO2:B2O3 can be increased to 1:1.5 and 1:2. This will increase the amount of
boron in the system and thus an increase in the yield of titanium diboride.
7.4.3 Separation of Si, SiC and SiO2
The synthesis of SiC using thermal plasma has been carried out successfully. The final
powders from the experiments contain four major phases, namely, silicon, silicon carbide, silicon
dioxide and carbon. The carbon present in the material can be removed by calcining the powders
in air atmosphere at about 600⁰C. The rest of the phases can be separated using the concepts of
electrophoresis and gravitational settling. Zeta-potential is the defined as the potential difference
between the surface of the particle and the bulk liquid. Electrophoresis uses the concept the zeta
potential to move solid suspension in a liquid. The zeta potential and the electro-osmotic velocity
of silica as a function of pH were determined as early as 1991 [102]. Isoelectric point (i.e.p) is
defined as the point at which zeta potential is zero. The particles dispersed in the solution
become unstable and start to agglomerate near the i.e.p. It is shown in the literature that the zeta

113
potential of silica varies with pH and the type of solvent but never reaches zero. Zeta potential
also has medical applications [103]. It varies with the change in the surface ion group. Silicon
and silicon carbide, unlike silica has i.e.p. and is very close to each other at low pH values of
around 3. Hence a higher pH was used to separate silicon from silicon carbide from waste slurry
obtained from sawing silicon wafers using silicon carbide wheels [104-106]. The isoelectric
points (IEP) for commercial SiC of various grit sizes, G800, G1000 and G1200 are at pH values
of 4.8, 4.2 and 3.3 respectively [107]. Thus using the difference in the particle shape and size,
difference in the zeta potential and varying pH, the three phases, namely, Si, SiC, and SiO2 can
be separated using a setup combining electrophoresis and gravitational settling.

114
REFERENCES
[1] A. Stein, S. W. Keller, T. E. Mallouk, Science, 259 (1993) 1558-1564.
[2] A-M Azad, M. Ramachandran, N. Schweitzer, Solid State Ionics, 178 (2007) 1476–1486.
[3] Helble, J. J., Journal of Aerosol Science, 29 [5/6] (1998) 721-736,
[4] Helble, J. J., Moniz, G. A. and Morency, J. R. (1994) United States Patent number 5,358,695.
[5] Helble, J. J., Moniz, G. A. and Morse, T.F. (1995) United States Patent number 5,447,708.
[6] Helble, J. J., Moniz, G. A. and Morse, T.F. (1997) United States Patent number 5,599,511.
[7] P. Duran, J. Tartaj, J. F. Fernandez, M. Villegas, and C. Moure, Ceramics International, 25 (1999) 125-135.
[8] P. Duran, M. Villegas, F. Capel, P. Recio, and C. Moure, Journal of European Ceramic Society, 16 (1996) 945-952.
[9] M. Mazaheri, A. Simchi, M. Dourandish, F. Golestani-Fard, Ceramics International, 35 (2009) 547–554.
[10] G. S. A. M. Theunissen, A. J. A. Winnubst, and A. J. Burggraaf, Journal of the European Ceramic Society 11 (1993) 315-324.
[11] O. Vasylkiv, and Y. Sakka, Journal of American Ceramic Society, 84 [11] (2001) 2489-2494.
[12] D. S. Patil, K. Prabhakaran, C. Durgaprasad, N. M. Gokhale, A. B. Samui, and S. C. Sharma, Ceramics International, 35 (2009) 515–519.
[13] L. G. J. de Haart, Y. S. Lin, K. J. de Vries, and A. J. Burggraaf, Journal of the European Ceramic Society 8 (1991) 59-70.
[14] Y. Zhou, J. A. Switzer, Journal of Alloys and Compounds, 237 (1996) 1-5.

115
[15] H. Herrig and R. Hempelmann, Nano Structured Materials, 9 (1997) 241-244.
[16] B. Li, X. Wang, M. Yan, and L. Li, Materials Chemistry and Physics, 78 (2002) 184–188.
[17] H. Wang, L. Gao, W. Li, and Q. Li, Nano Structured Materials, 11 [8] (1999) 1263–1267.
[18] J. A. Nelson and M. J. Wagner, Chemistry of Materials, 14 (2002) 4460-4463.
[19] Dai H., Wong E. W., Lu Y. Z., Fan S., and Lieber C. M., Nature, 375 (1995) 769-772.
[20] X. G. Yang and C. Y. Wang, Applied Physics Letters, 86 (2005) 224104.
[21] Y. Li, S. Zhang, Q. Yu, and W. Yin, Applied Surface Science, 253 (2007) 9254–9258.
[22] Y. Nakamura, K. Tachibana, and H. Fujimoto, Physica C, 306 (1998) 259–270.
[23] J. Li, C. Papadopoulos, J. M. Xu, and M. Moskovits, Applied Physics Letters, 75 [3] (1999) 367-369.
[24] T. J. Goodwin, S. H. Yoo, P. Matteazzi, and J. R. Groza, Nano Structured Materials, 8 [5] (1991) 559-566.
[25] R. S. Wagner, W. C. Ellis, Applied Physics Letters, 4 (1964) 89.
[26] S. Iijima, Nature, 354 (1991) 56.
[27] J. D. Holmes, K. P. Johnston, R. C. Doty, B. A. Korgel, Science, 287 (2000) 1471.
[28] A. M. Morales, C. M. Lieber, Science, 279 (1998) 208.
[29] Y. F. Zhang, Y. H. Tang, N. Wang, D. P. Yu, C. S. Lee, I. Bello, S. T. Lee, Applied Physics Letters, 72 (1998) 1835.
[30] N. Wang, Y. F. Zhang, Y. H. Tang, C. S. Lee, S. T. Lee, Physical Review B, 58 (1998) R16024.
[31] S. T. Lee, Y. F. Zhang, N. Wang, Y. H. Tang, I. Bello, C. S. Lee, Y. W. Chung, Journal of Materials Research, 14 (1999) 4503.
[32] S. T. Lee, N. Wang, C. S. Lee, Material Science and Engineering A, 286 (2000) 16.
[33] S. T. Lee, N. Wang, Y. F. Zhang, Y. H. Tang, MRS Bulletin, 24 (1999) 36.

116
[34] H. I. Liu, N. I. Maluf, R. F. W. Pease, Journal of Vacuum Science and Technology B, 10 (1992) 2846.
[35] T. Ono, H. Saitoh, M. Esashi, Applied Physics Letters, 70 (1997) 1852.
[36] D. C. Halverson, A. J. Pyzik, I. A. Aksay, and W. E. Snowden, Journal of American Ceramic Society, 72 [5] (1989) 775-80.
[37] D. Apelian, (1992). Materials Synthesis: A New Horizon for Plasma Processing. International Symposium on Thermal Plasma Applications in Materials and Metallurgical Processing, San Diego, California, USA, TMS, pp. 3-12.
[38] Thermal Spray Application for the Next Millennium – A Business Development Perspective: KASSABJI F., JACQ G., EDF, Direction des Etudes et Recherches, Dept. Systèmes Energétiques; MORET SUR LOING, FRANCE; DURAND, J., P., Magetex, France: Thermal Spray – Meeting the challenges of 21st century, Proceedings of the 15th International Thermal Spray Conference, Nice, France, 25-29 May 1998, pp. 1677-1680.
[39] P. R. Taylor, and S. A. Pirzada, Advanced Performance Materials, 1 (1994) 35.
[40] M. I. Boulos, IEEE Transactions on Plasma Science, 19[6] (1991) 1078.
[41] E. Pfender, Plasma Chemistry and Plasma Processing, 19[1] (1999) 1.
[42] S-M. Oh, and D-W. Park, Thin Solid Films, 316 (1998) 189.
[43] O. Fukumasa and S. Sakiyama, Surface and Coatings Technology, 131 (2000) 493.
[44] S. W. White and R. G. Reddy, Waste Processing of MgO Bag House Dust Using Plasma Arc Technology, in: EPD Congress, TMS, 1999, pp. 687-697.
[45] R. G. Reddy and L. V. M. Antony, JOM, 55 [3] (2003) 19.
[46] R. G. Reddy and L. V. M. Antony, Processing of SiC Nano Powders using Thermal Plasma Technique, in: Proceedings of the 2003 international conference on Nanotechnology: Scientific challenges and commercial opportunities, Rhode Island, 2003, pp. 47-54.
[47] I. Mohai, J. Szépvölgyi, I. Bertóti, M. Mohai, J. Gubicza, and T. Ungár, Solid State Ionics, 141-142 (2001) 163.
[48] L. Tong and R. G. Reddy, Scripta Materialia, 52 (2005) 1253.
[49] N. R. Thakkar, and R. G. Reddy, (2003): Thermal Plasma Processing of Boron Carbide Fine Powders. In Powder Materials: Current Research and Industrial Practices III. Ed. Marquis, F.D.S., TMS, Warrendale, PA, pp. 47 - 60.

117
[50] S. Niyomwas, B. Wu, and R. G. Reddy, Synthesis and Modeling of Fe-TiN Composites by Thermal Plasma Processing, in: C.Suryanarayana, N. N. Thadhani, and T. C. Lowe (Eds.), Ultrafine Grained Materials, TMS, 2000, pp. 89-98.
[51] L. V. M. Antony, Thermal Plasma Processing of Al-SiC Ultrafine Composites: Master’s Thesis, The University of Alabama, 2004.
[52] L. Tong and R. G. Reddy, Metallurgical and Materials Transactions B, 37B (2006) 531.
[53] U. S. Patent No. 6,379,419 B1, Date of Patent: Apr. 30, 2002.
[54] E. Rudy, Ternary Phase Equilibria in Transition Metal-Boron-Carbon-Silicon Systems, in: Part V, Compendium of Phase Diagram Data, Technical Report AFML-TR-65-2, Wright-Patterson Air Force Base, Ohio (1969) p. 198.
[55] A. E. McHale, (Ed.), Phase Equilibria Diagrams, American Ceramic Society, 10 (1994) 140.
[56] H. Duschanek, P. Rogl, and H. L. Lukas, Journal of Phase Equilibria, 16 (1995) 46.
[57] Munro R. G., Journal of Research of NIST, 105 (2000) 709.
[58] U.S. Patent No. 4,673,550, Date of Patent: Jun. 16, 1987.
[59] M. Makyta, V. Danek, G. M. Haarberg, J. Thonstad, Journal of Applied Electrochemistry, 26 (1996) 319.
[60] M. Shigeta, T. Watanabe, Thin Solid Films, 515 (2007) 4217.
[61] S. Dallaire, and B. Champagne, Thin Solid Films, 118 (1984) 477.
[62] T. Kodama, Y. Ikeda, and H. Tamura, Journal of Thermal Spray Technology, 8(4) (1999) 537.
[63] I. Song, L. Wang, M. Wixom, and L. T. Thompson, Journal of Materials Science, 35 (2000) 2611.
[64] A. M. Locci, R. Orru, G. Cao and Z. A. Munir, Journal of American Ceramic Society, 89(3) (2006) 848.
[65] P. R. Taylor, M. Manrique and S. A. Pirzada, Proceedings of the EPD Congress, TMS Annual Meeting, Las Vegas, Nevada, 1995, pp. 49-59.
[66] G. L. Watt, P. R. Taylor and S. A. Pirzada, Proceedings of the EPD Congress, TMS Annual Meeting, Las Vegas, Nevada, 1995, pp. 61-73.

118
[67] J. Schmidt et al., Science and Technology Advanced Materials, 8 (2007) 376.
[68] A. K. Khanra et al., Materials Letters, 58 (2004) 733.
[69] U. S. Patent No. 6,420,294 B1, Date of Patent: Jul. 16, 2002.
[70] B. Basu et al., International Materials Reviews, 51 (2006) 352.
[71] S. E. Bates et al., Journal of Materials Research, 10 (1995) 2599.
[72] A. Y. Hwang and J. K. Lee, Materials Letters, 54 (2002) 1.
[73] Y. Gu et al., Journal of Alloys and Compounds, 352 (2003) 325.
[74] Acheson A. G., British Patent 17:911, (1892).
[75] Knippenberg W. F., “Growth Phenomena in Silicon Carbide: Preparative Procedures.” Philips Research Reports, 18 (1966) 170–179.
[76] Lely J. A., Darstellung von Einkristallen von Siliziumcarbid und Beherrschung von Art und Menge der eingbauten Verunreinigungen. Berichte Deutche Keramik Geselshaft, 32 (1955) 229–231.
[77] Hamilton D. R., “The Growth of Silicon Carbide by Sublimation.” In: Connor JR, Smilestens J (eds.) Silicon Carbide, A High Temperature Semiconductor. Pergamon, Oxford, (1960) pp 45–51.
[78] Novikov V. P. and Ionov V. I., Growth of Crystals, 6b (1968) 9–21.
[79] Tairov Y. M. and Tsvetkov V. F., Journal of Crystal Growth, 43 (1978) 209–212.
[80] Ziegler G., Lanig P., Theis D. and Weyrich C., IEEE Transactions on Electron Device ED-30 (1983) 277–281.
[81] Glass R. C., Henshall D., Tsvetkov V. F. and Carter Jr C. H., MRS Bulletin 22 [3] (1997) 30–35.
[82] Tairov Yu. M., Materials Science and Engineering B, 29 (1995) 83–89.
[83] Karpov S. Yu., Makarov Yu. N., Mokhov E. N., Ramm M. G., Ramm M. S., Roenkov A. D., Talalaev R. A. and Vodakov Yu. A., Journal Crystal Growth 173 (1997) 408–416.
[84] Matsunami H. and Kimoto T., Materials Science and Engineering R, 20 (1997) 125–166.
[85] Epelbaum B. M., Hofmann D., Muller M. and Winnacker A., Materials Science Forum 338–342 (2000) 107–110.

119
[86] Fluent, version 6.3, Fluid Dynamics International, Fluid Dynamics Analysis Package. Fluent Inc. two-dimensional double precision (2ddp), pressure-based solver with standard k- model for turbulence.
[87] Gambit, Fluid Dynamics International, Fluid Dynamics Analysis Package, Evanston, IL.
[88] Li H-P. and Chen X., Journal of Physics D Applied Physics 34 (2001) L99–L102.
[89] Li H-P., Chen X., and Pfender E., Journal of Physics D Applied Physics 36 (2003) 1084–1096.
[90] Park J. M., Kim K. S., Hwang T. H., and Hong S. H., IEEE Transactions on Plasma Science, 32 (2004) 479–487.
[91] Ansys Workbench, version 13.0.
[92] HSC Chemistry Ver. 5.1, Copyright © Outokumpu Research Oy, Pori, Finland, A.Roine
[93] N. A. Gokcen, and R. G. Reddy, Thermodynamics, second ed., Plenum Publications, New York, 1996, pp. 203-243.
[94] H. Zhang and J. F. Banfield, Journal of Physical Chemistry B, 104 [15] (2000) 3481.
[95] N. Wetchakun, and S. Phanichphant, Current Applied Physics, 8 (2008) 343.
[96] W. Ma, Z. Lu, and M. Zhang, Applied Physics A, 66 (1998) 621.
[97] B. D. Cullity, Elements of X-Ray Diffraction, second ed., Addison-Wesley Publishing Co., 1978.
[98] P. Villars, Pearson's Handbook of Crystallographic Data for Intermetallic Phases, second ed., ASM International, Ohio, 1991.
[99] S. L. Zhang, B. F. Zhub, F. Huanga, Y. Yana, E. Y. Shangb, S. Fanc, and W. Hanc, Solid State Communications, 111 (1999) 647–651.
[100] Y. Ryu, Y. Tak, and K. Yong, Nanotechnology, 16 (2005) S370–S374.
[101] A. A. Galuska, J. C. Uht, and N. Marquez, Journal of Vacuum Science and Technology A, 6 [1] (1988) 110-122.
[102] C. Schwer and E. Kenndler, Analytical Chemistry, 63 (1991) 1801-1807.
[103] Y. Nitta, K. Okamoto, T. Nakatani, H. Hoshi, A. Homma, E. Tatsumi, and Y. Taenaka, Diamond & Related Materials, 17 (2008) 1972-1976.

120
[104] Y-F. Wu, and Y-M. Chen, Separation and Purification Technology, 68 (2009) 70-74.
[105] T-H. Tsai, Journal of Hazardous Materials, 189 (2011) 526-530.
[106] T-H. Tsai, Separation and Purification Technology, 78 (2011) 16-20.
[107] R. R. Rao, H. N. Roopa, and T. S. Kannan, Ceramics International, 25 (1999) 223-230.

121
APPENDIX
A.1 INSTRUCTIONS FOR THE OPERATION OF PLASMA POWER SOURCE IN NON-
TRANSFERRED MODE
START-UP
Check the power source and reactor connections:
Ensure water, gas, and power are connected properly from the power source to the torch
and well insulated.
Ensure the reactor is put together without any leaks and the exhaust is connected from the
filter chamber.
Ensure gas and water lines to the torch and the reactor are leak free.
Cooling water system:
Check the water level in the tank (approximately 200 gallons)
Ensure the water inlet and outlet valves are open on the back of the power supply.
Turn on the power to the pump.
Check for water leaks at the pump, other connections and at the torch.
Ensure the water pressure on the back of the power supply is at least 200 psi.
Turn on the water supply to the reactor from the wall.
Make sure there is no leak in the feed tube, jacket or the quench coils or any of the
connections.
Adjust the cooling water to the following values.

122
Table 3.1: Flow rate of cooling water for various parts of the plasma reactor system
Part of reactor Flow rate (LPM)
Copper feed tube 1
Stainless steel reactor jacket 2
Copper quench coils 4.5
Gas Supply:
Open the plasma (argon) gas cylinder and check for a minimum of 500 psig tank pressure
on the regulator gauge.
Always connect a second argon cylinder to ensure uninterrupted gas supply to the torch.
Set the outlet pressure at the regulator to a minimum of 60 psig.
Open the gas inlet valve on the power supply’s gas manifold and check for approximately
3 scfm flow rate.
The above mentioned flow rate will correspond to a minimum outlet pressure of 10 psig
at the torch. The following table gives the minimum gas pressures for the operation of the
power supply and the plasma torch.
Table 3.2: Programmable gas controller set points in the thermal plasma power source for
various types of torch.
Type of torch Start Low High
Non-transferred 10 30 60
Transferred 20 30 60

123
Set the flow rate of methane and argon to the powder feeder based on the molar
requirements of the experiment.
As the total flow rate is relatively low, 6 lpm, no minimum requirement of gas pressure is
required. Ensure adequate gas supply to maintain the molar ratio of the reducing gas to
the solid feed and the total solid feed rate.
Make sure the calibration of the powder feeder is still valid.
Power Supply:
Turn on the power to the plasma from the wall.
Turn the red/black circuit breaker at the power supply unit.
Set the transfer mode switch to “Disable” (=non-transferred, “Enable”=transferred).
Twist and pull the red emergency safety button.
Turn safety key switch to “Close” position. This will enable the corresponding non-
transferred or transferred circuit if the gas flow (10 psig), water flow (min 200 psig) meet
the minimum requirements.
Push black “reset” button to reset the alarms.
Push the green “start” button and the DC rectifiers will turn on with a voltage of 650V.
Set the starting potentiostat level to 5 which will correspond to startup current of about
100A.
Push the red “ignition” button to start the plasma. The minimum gas flow to start the
plasma torch is 30 psig.
Increasing the pressure (max 60 psig) will help stabilize the plasma and increase the
voltage with corresponding increase in the current (max 200A).

124
SHUT DOWN PROCEDURE
Turn off the powder feeder and its gas supplies.
Decrease the current to the startup level of 5 (approximately 100A).
Push the red “emergency stop” button. This will cease the plasma.
Turn the “Safety” key to “Open” position. This will turn off the circuits.
Decrease the gas flow rate to the startup level (about 10 psig).
Remove the plasma torch from the reactor.
Seal the reactor with the lid and the clamps.
Turn off the gas supply to the plasma torch.
Turn off the water supply pump after the torch gets to room temperature.
Turn off red/black circuit breaker at the power supply and the main power supply from
the wall.
Turn off the water supply to the reactor, quenching coils and the powder feed tube after a
cool down time of about 5 minutes.

125
A.2 PROPERTIES OF GASES USED IN MODELING
ARGON
Heat capacity, CP = 0.520 J/g/K = 20.786 J/mol/K. (constant)
Molecular Weight = 39.948 g/mol.
Density = 1.633 g/L (ideal gas)
Table A.2.1: Viscosity (piecewise-linear) of argon
Temperature (K) Viscosity * 106 (kg/m-s)
100 8.1
200 15.9
300 22.7
400 28.6
500 33.9
600 38.8
Table A.2.2: Thermal conductivity (piecewise-linear) of argon
Temperature (K) Thermal Conductivity (W/m/K)
100 6.3
200 12.4
300 17.7
400 22.4
500 26.5
600 30.3
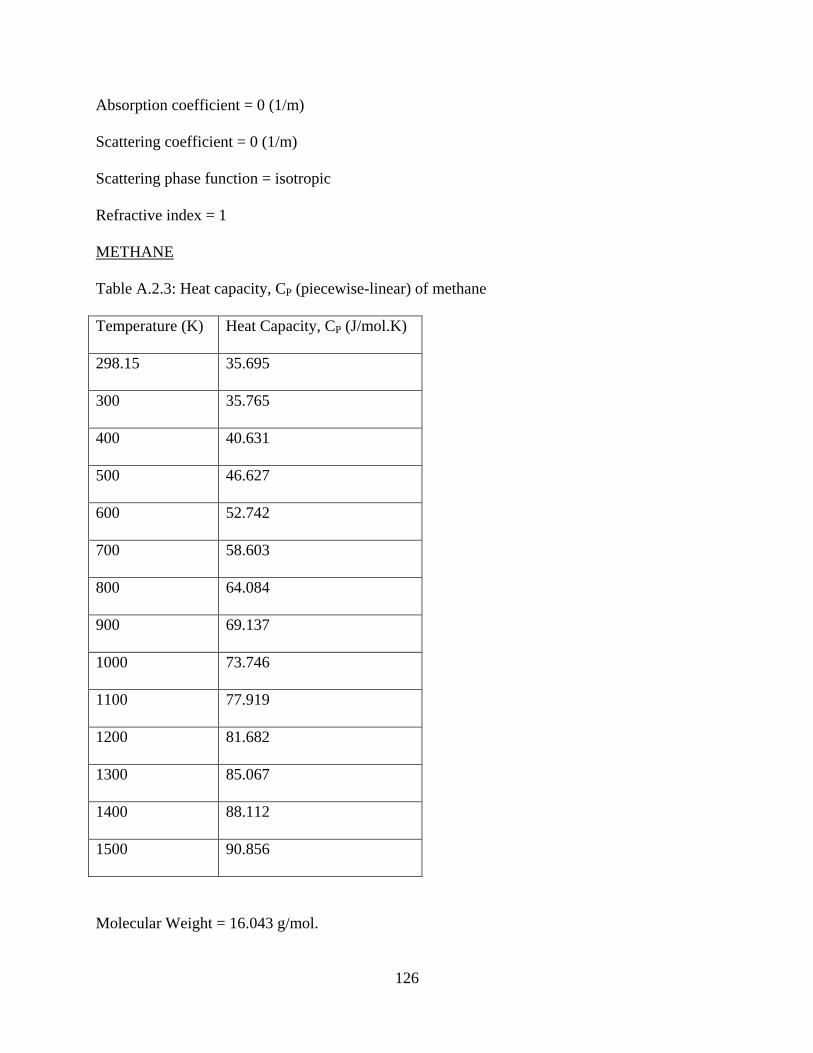
126
Absorption coefficient = 0 (1/m)
Scattering coefficient = 0 (1/m)
Scattering phase function = isotropic
Refractive index = 1
METHANE
Table A.2.3: Heat capacity, CP (piecewise-linear) of methane
Temperature (K) Heat Capacity, CP (J/mol.K)
298.15 35.695
300 35.765
400 40.631
500 46.627
600 52.742
700 58.603
800 64.084
900 69.137
1000 73.746
1100 77.919
1200 81.682
1300 85.067
1400 88.112
1500 90.856
Molecular Weight = 16.043 g/mol.

127
Density = 1.633 g/L (ideal gas)
Table A.2.4: Viscosity (piecewise-linear) of methane
Temperature (K) Viscosity * 106 (kg/m-s)
100 3.9
200 7.7
300 11.1
400 14.2
500 17.0
600 19.5
Table A.2.5: Thermal conductivity (piecewise-linear) of methane
Temperature (K) Thermal Conductivity (W/m/K)
100 10.4
200 21.8
300 34.4
400 50.0
500 68.4
600 88.6
Absorption coefficient = 0 (1/m)
Scattering coefficient = 0 (1/m)
Scattering phase function = isotropic
Refractive index = 1

128
A.3 CALCULATION OF VOLUME FRACTION USING DIRECT COMPARISON
METHOD
Phase analysis of the material was done using XRD. The volume fraction calculation was
done using direct comparison method [97]. The following equation is used in the determination
of the volume fractions of individual phases.
)30(1;; CBABB
CC
B
C
BB
AA
B
A ccccR
cR
I
I
cR
cR
I
I
IA, IB and IC – Intensities of A, B and C respectively
cA, cB and cC – Volume fractions of A, B and C respectively
RA, RB and RC – Volumes of inverse unit cell lattices of A, B and C respectively.
R depends on q, hkl and the kind of substance.
V is the volume of unit cell.
F is the Structure factor calculated using the atomic scattering factor, f, the hkl and the fractional
coordinates of the atoms uvw.
P is the Multiplicity factor for powder method.
e-2M is the temperature factor which is taken as 1 as the XRD was done at room temperature.
The values of F, f, fractional coordinates, L-P factor, and P were obtained from standard
literature [97].
)31(cossin
2cos11 22
22
2MePF
VR
)32(.cossin
2cos12
2
FactoronPolarizatiLorentz

129
Table A.3.1: The values of diffraction constants used in the calculation of volume fraction of
product powders from TiB2 experiments.
Material 2 hkl R
B2O3 26.006 011 3.280
TiO2(r) 27.911 110 4.485
C 41.859 100 1.192
TiB2 44.463 101 2.187
Table 5.3: Yield of product powders at different feed rates with 5 moles of methane in the feed.
Feed Rate (g/min) Yield TiB2 TiO2(r) B2O3 C
2.15 mol% 23.63 8.72 4.07 63.58
3.22 mol% 39.44 11.33 7.58 41.65
The volume fraction calculated using direct comparison method was converted to
corresponding mole fractions using the density and molecular weights of individual pure phases.
The converted result in mol% is shown in Table 5.3 for the synthesis of TiB2.