translocation of the papillomavirus l2/vdna complex across...
TRANSCRIPT

Translocation of the papillomavirus L2/vDNA complexacross the limiting membrane requires the onset of mitosis
Item Type Article
Authors Calton, Christine M.; Bronnimann, Matthew P.; Manson, ArianaR.; Li, Shuaizhi; Chapman, Janice A.; Suarez-Berumen, Marcela;Williamson, Tatum R.; Molugu, Sudheer K.; Bernal, Ricardo A.;Campos, Samuel K.
Citation Translocation of the papillomavirus L2/vDNA complex acrossthe limiting membrane requires the onset of mitosis 2017, 13(5):e1006200 PLOS Pathogens
DOI 10.1371/journal.ppat.1006200
Publisher PUBLIC LIBRARY SCIENCE
Journal PLOS Pathogens
Rights © 2017 Calton et al. This is an open access article distributedunder the terms of the Creative Commons Attribution License.
Download date 20/06/2018 08:58:34
Link to Item http://hdl.handle.net/10150/624634

RESEARCH ARTICLE
Translocation of the papillomavirus L2/vDNA
complex across the limiting membrane
requires the onset of mitosis
Christine M. Calton1☯, Matthew P. Bronnimann2☯, Ariana R. Manson3, Shuaizhi Li4, Janice
A. Chapman1, Marcela Suarez-Berumen3, Tatum R. Williamson3, Sudheer K. Molugu5,
Ricardo A. Bernal5, Samuel K. Campos1,2,3,6*
1 BIO5 Institute, University of Arizona, Tucson, Arizona, United States of America, 2 Department of
Immunobiology, University of Arizona, Tucson, Arizona, United States of America, 3 Department of Molecular
& Cellular Biology, University of Arizona, Tucson, Arizona, United States of America, 4 Department of Cellular
& Molecular Medicine, University of Arizona, Tucson, Arizona, United States of America, 5 Department of
Chemistry, University of Texas at El Paso, El Paso, Texas, United States of America, 6 Cancer Biology
Graduate Interdisciplinary Program, University of Arizona, Tucson, Arizona, United States of America
☯ These authors contributed equally to this work.
Abstract
The human papillomavirus type 16 (HPV16) L2 protein acts as a chaperone to ensure that
the viral genome (vDNA) traffics from endosomes to the trans-Golgi network (TGN) and
eventually the nucleus, where HPV replication occurs. En route to the nucleus, the L2/vDNA
complex must translocate across limiting intracellular membranes. The details of this critical
process remain poorly characterized. We have developed a system based on subcellular
compartmentalization of the enzyme BirA and its cognate substrate to detect membrane
translocation of L2-BirA from incoming virions. We find that L2 translocation requires trans-
port to the TGN and is strictly dependent on entry into mitosis, coinciding with mitotic entry in
synchronized cells. Cell cycle arrest causes retention of L2/vDNA at the TGN; only release
and progression past G2/M enables translocation across the limiting membrane and subse-
quent infection. Microscopy of EdU-labeled vDNA reveals a rapid and dramatic shift in
vDNA localization during early mitosis. At late G2/early prophase vDNA egresses from the
TGN to a pericentriolar location, accumulating there through prometaphase where it begins
to associate with condensed chromosomes. By metaphase and throughout anaphase the
vDNA is seen bound to the mitotic chromosomes, ensuring distribution into both daughter
nuclei. Mutations in a newly defined chromatin binding region of L2 potently blocked translo-
cation, suggesting that translocation is dependent on chromatin binding during prometa-
phase. This represents the first time a virus has been shown to functionally couple the
penetration of limiting membranes to cellular mitosis, explaining in part the tropism of HPV
for mitotic basal keratinocytes.
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 1 / 29
a1111111111
a1111111111
a1111111111
a1111111111
a1111111111
OPENACCESS
Citation: Calton CM, Bronnimann MP, Manson AR,
Li S, Chapman JA, Suarez-Berumen M, et al.
(2017) Translocation of the papillomavirus L2/
vDNA complex across the limiting membrane
requires the onset of mitosis. PLoS Pathog 13(5):
e1006200. https://doi.org/10.1371/journal.
ppat.1006200
Editor: Craig Meyers, Penn State University School
of Medicine, UNITED STATES
Received: September 9, 2016
Accepted: January 25, 2017
Published: May 2, 2017
Copyright: © 2017 Calton et al. This is an open
access article distributed under the terms of the
Creative Commons Attribution License, which
permits unrestricted use, distribution, and
reproduction in any medium, provided the original
author and source are credited.
Data Availability Statement: All relevant data are
within the paper and its Supporting Information
files.
Funding: SKC is supported by grant
1R01AI108751-01 from the National Institute for
Allergy and Infectious Diseases. ARM and MSB
were funded in part by a grant to UA from HHMI
(52006942) that supports the Undergraduate
Biology Research Program, UBRP. RAB is
supported by grant NIH-NIGMS SC3GM113805

Author summary
Human papillomaviruses (HPVs) are the most prevalent sexually transmitted infections
and are responsible for about 5% of total human cancers worldwide. These small DNA
viruses must enter cells and deliver their genomes to the host cell nucleus to successfully
establish an infection. A major barrier that HPVs and other non-enveloped viruses must
overcome is the limiting membrane—the host cellular membrane through which the viral
genome must be transported en route to the nucleus. Here we have developed a novel
enzyme-dependent assay to study what is called penetration or translocation of the limit-
ing membrane by the HPV16 minor capsid protein L2. We find that translocation is
strictly dependent on host cell division, specifically progression into mitosis, and occurs
post trans-Golgi-network localization. After exiting the trans-Golgi network the viral
genomes are seen decorating the segregating mitotic chromosomes, to ensure the equal
partitioning into and infection of the daughter cells. The remarkable timing with, and
absolute dependence of, this event on mitosis likely dictates the natural tropism of these
viruses for the replicating basal keratinocytes of differentiating epithelial tissues. Further
work will be necessary to fully understand the molecular mechanisms of this extraordi-
nary mitosis-dependent phenomenon.
Introduction
Human papillomaviruses (HPVs) are small, double-stranded DNA viruses that infect the basal
keratinocytes of differentiating epidermal or mucosal epithelium. Most HPV infections are
asymptomatic or cause benign lesions. However, several HPV types, termed high-risk HPVs,
are associated with essentially all cases of cervical cancer, as well as a significant number of
anogenital and oropharyngeal cancers [1]. HPV16 is the most common high-risk type and
accounts for over 60% of cervical cancer cases. Combined, HPVs are responsible for approxi-
mately 5% of all human cancers [2].
The HPV16 capsid is a non-enveloped 55 nm icosahedral structure that consists of 72 pen-
tamers of the major capsid protein L1. Within the particle are approximately 20–40 copies of
the minor capsid protein L2, in complex with a circularized, 8 kb chromatinized genome [3,
4]. Despite its designation as a minor capsid protein, L2 plays important roles in capsid assem-
bly and genome encapsidation [5–7], and is essential for establishing infection within the cell
[8].
Initial attachment of HPV16 is mediated via electrostatic interactions with heparan sulfate
proteoglycans (HSPGs) on either the cell-surface or extracellular matrix (ECM) or through
transient binding to ECM resident laminin 332 [9–14]. Binding to HSPGs induces conforma-
tional changes in the capsid that are detectable by the exposure of masked epitopes in both L1
and L2 and facilitate cleavage of these capsid proteins by the host proteases kallikrein-8 and
furin respectively [10, 15, 16]. These conformational changes are also believed to mediate
transfer of the virion to an entry receptor complex that allows endocytosis in an actin-depen-
dent manner [17, 18].
Once inside the cell, HPV traffics through the endosomal pathway, where acidification pro-
motes further capsid disassembly and degradation [15, 19–21]. Host cyclophilins facilitate
release of the L2/vDNA complex from the remnants of the L1 capsid structure [22], thereby
enabling transport to the trans-Golgi network (TGN) and possibly other distal compartments
via retrograde trafficking pathways [23–26]. Retrograde transport to the TGN is dependent
upon the activity of furin and γ-secretase [23, 25], as well as interactions between L2 and
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 2 / 29
from the National Institute of General Medical
Sciences, grant NSF-MRI-0923437 from the
National Science Foundation, and grant AH-1649
from the Welch Foundation. The funders had no
role in study design, data collection and analysis,
decision to publish, or preparation of the
manuscript.
Competing interests: The authors have declared
that no competing interests exist.

cytosolic host trafficking factors including sorting nexin 17 (SNX17), sorting nexin 27
(SNX27), and the retromer complex [27–30]. Based on immunofluorescence microscopy and
colocalization studies, TGN/Golgi localization of L2/vDNA has been proposed to be necessary
for translocation to occur, but the details of this translocation event remain poorly defined [23,
31]. After exiting the TGN, the L2/vDNA complex traffics to the nucleus where it associates
with PML bodies and initiates early viral transcription and replication [8]. This event is poorly
defined but thought to require mitotic breakdown of the nuclear envelope, as cell cycle inhibi-
tors block nuclear localization of L2/vDNA and productive infection [31–33]. Prior work has
suggested that L2 can bind to mitotic chromatin as a means to ensure nuclear localization in
the infected daughter cells [31]. Following up on this idea, the accompanying manuscript by
the Schelhaas group defines a 147 residue (aa 188–334) chromatin binding region of HPV16
L2 that is necessary and sufficient for the binding of L2 to mitotic chromosomes [34]. Notably,
the association of L2 with mitotic chromatin requires progression into prometaphase, and is
likely indirect, involving an unknown host mitotic factor or factors.
How L2 and the viral genome make the leap from the TGN to the nucleus is unclear. A
recent report presents evidence that L2/vDNA remains in a vesicular compartment after egress
from the TGN during mitosis [35]. At some point on its journey to the nucleus, the L2/vDNA
complex must physically penetrate or translocate across a limiting lipid membrane within the
cell. A detailed understanding of this translocation has been hampered by a lack of suitable
techniques that are sensitive enough to detect the event. In this report, we have developed a
novel assay to monitor L2 translocation that depends on fusion of L2 to the bacterial biotin
ligase BirA [36]. The assay is based on the compartmentalization of a specific BirA enzyme-
substrate reaction. Virions encapsidating the L2-BirA fusion are used to infect host cells that
stably express a cytosolically localized BirA substrate. Only when L2 translocates across the
limiting membrane will the BirA encounter and biotinylate the substrate. Translocation is
therefore directly correlated to substrate biotinylation, which can easily be detected by western
blotting and neutravidin staining.
Using this simple but innovative assay, we show that inhibition of the cellular processes and
factors that are essential for L2/vDNA trafficking to the TGN, including furin, endosome acid-
ification, cyclophilins, and γ-secretase, potently block L2 translocation. Treatments that induce
cell cycle arrest also inhibit L2 translocation and cause vDNA to accumulate at the TGN. We
find that mitotic entry is sufficient for L2 translocation to occur and that translocation coin-
cides with mitosis in synchronized cells. Furthermore, during the onset of mitosis vDNA
moves rapidly from the TGN to a pericentriolar region before associating with mitotic host
chromosomes during prometaphase, ensuring infection of both daughter cells. Finally, here
and in the accompanying manuscript, we present evidence that translocation is blocked in
three different mutants of the newly defined chromatin binding region of L2 [34], suggesting a
functional link between the ability to bind chromatin and L2/vDNA translocation. Based on
these findings we propose a model whereby chromatin binding of L2 in prometaphase is a nec-
essary prerequisite for translocation of L2/vDNA across limiting membranes.
Results
Development of a BirA-based translocation system
Prior efforts to study L2/vDNA translocation across the limiting membrane have relied heavily
on confocal microscopy and colocalization of EdU-labeled vDNA with subcellular markers
[23, 31, 37]. While informative, these methods are unsuitable for drawing definitive conclu-
sions on the lumenal versus cytosolic state of the L2/vDNA complex. To detect egress of
virion-derived L2 out of vesicular compartments, we developed a membrane translocation
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 3 / 29

assay that utilizes an L2 fusion to the Escherichia coli biotin ligase BirA [36, 38] (Fig 1A).
HaCaT keratinocytes were transfected with pCIP-NES-GFP-BAP to isolate a subclone that sta-
bly expresses cytosolically localized GFP fused to a 15 amino acid biotin acceptor peptide
(HaCaT GFP-BAP cells, Fig 1B). BAP is an engineered BirA-specific substrate that cannot be
biotinylated by holocarboxylase synthetase, the orthologous mammalian biotin ligase [39–41].
In this system, L2-BirA must traverse the limiting membrane to encounter cytosolic GFP-BAP.
BirA-dependent biotinylation of GFP-BAP is therefore a direct readout of L2-BirA membrane
translocation. Luciferase expressing HPV16 L2-BirA pseudovirions (PsV) were generated as
described in Materials and Methods. L2-BirA particles incorporate L2 at levels similar to wild
type (wt) particles (Fig 1C) and exhibit normal capsid morphology when examined by trans-
mission electron microscopy (Fig 1D). In vitro biotin ligase reactions were performed with
PsV containing wt L2 or the non-infectious R9,12K furin cleavage site mutant L2 [42]. Both
were capable of biotinylating BAP-tagged maltose binding protein (Fig 1E), demonstrating
that BirA retains activity in the context of an L2 fusion and that the purified PsV contain active
BirA enzyme. Infection of HaCaT GFP-BAP cells with L2-BirA results in biotinylation of
GFP-BAP and luciferase expression in a dose-dependent manner (Fig 1F). L2-BirA is less
infectious than PsV lacking the large C-terminal BirA fusion (Fig 1G), and we have observed
particle instability after prolonged storage at 4˚C. It is therefore recommended that aliquots be
stored at -80˚C and that the concentration of virus be verified before each use. All GFP-BAP
biotinylation and L2-BirA infection experiments herein were performed with fresh aliquots of
virus, at a non-saturating multiplicity of infection (MOI) according to the curve in Fig 1F.
To ensure that GFP-BAP biotinylation results only from encapsidated BirA protein and not
from expression of trace amounts of the 9.5 kb L2-BirA plasmid that may have been packaged
during PsV production, HaCaT GFP-BAP cells were infected with L2-BirA particles under
conditions where nascent protein synthesis was blocked, either with actinomycin D to inhibit
mRNA transcription or cycloheximide to block translation (S1A Fig). Inhibition of either tran-
scription or translation did not alter the levels of GFP-BAP biotinylation (S1B and S1C Fig),
but caused a drastic reduction in luciferase expression (S1C Fig). Thus, the translocation signal
is due to L2-BirA protein from incoming capsids rather than nascent L2-BirA synthesis.
L2 translocation precedes infection and requires endosome acidification
and cyclophilin activity
To examine the kinetics of L2-BirA translocation and infection, we monitored biotinylation
and luciferase expression in HaCaT GFP-BAP cells over time. GFP-BAP biotinylation was first
detectable at 8 hours post-infection (Fig 2A). Luciferase expression in L2-BirA-infected cells
was first observed at 10 hours post-infection and the infection profile was virtually indistin-
guishable from that of an L2-HA PsV (Fig 2B). Thus, translocation signal preceded reporter
expression from the luciferase-expressing vDNA as expected, and infection kinetics of the
virus are not affected by the large L2-BirA fusion. Prior work has shown that EdU-labeled
vDNA signal partitions from L1 capsid somewhere between 6-12h post infection [23] and L2
localizes to the TGN somewhere between 8-16h post infection by proximity ligation assay [25].
Thus, the faint detection of L2 translocation at 8h in our system is consistent with the timing
of L2/vDNA arrival at the TGN.
Uncoating is an important step in HPV entry that releases the L2/vDNA complex from the
L1 capsid to allow for subsequent trafficking to the TGN and the nucleus [43]. Breakdown of
the HPV capsid requires endosome acidification [15, 19, 20] and dissociation of L2/vDNA
from L1 pentamers requires the activity of host cyclophilins [22]. As a verification of the
L2-BirA assay, we examined how L2 translocation is affected by conditions that block
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 4 / 29
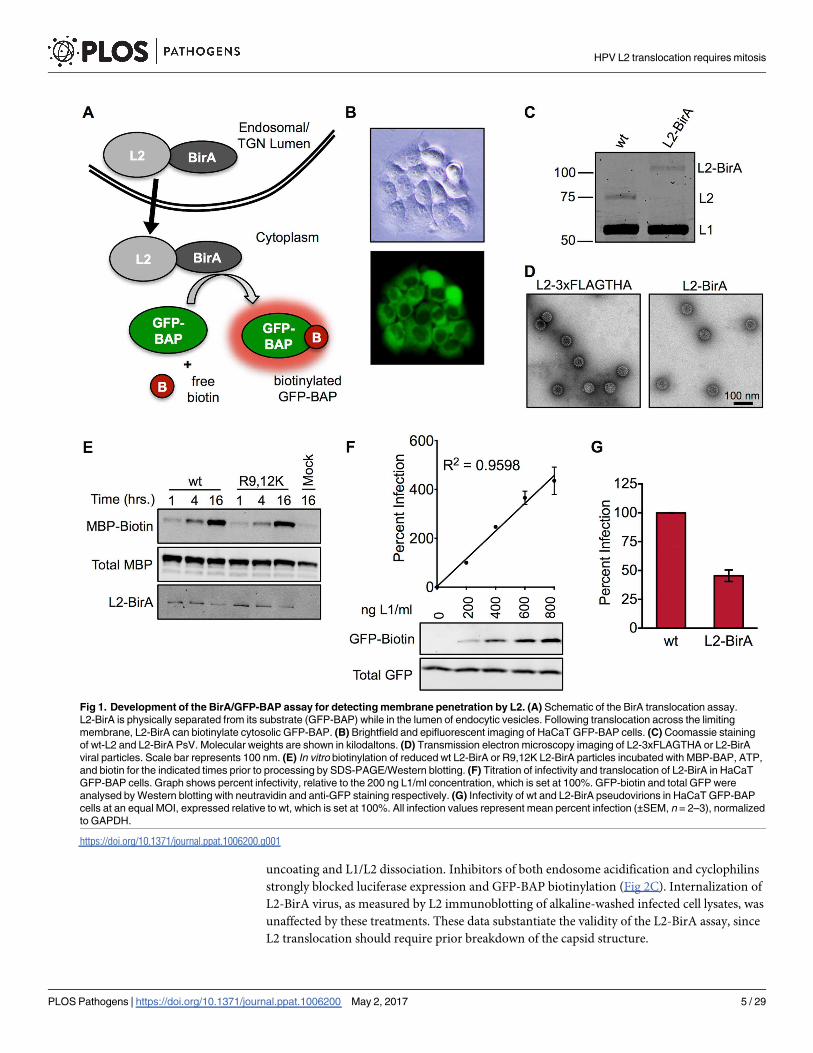
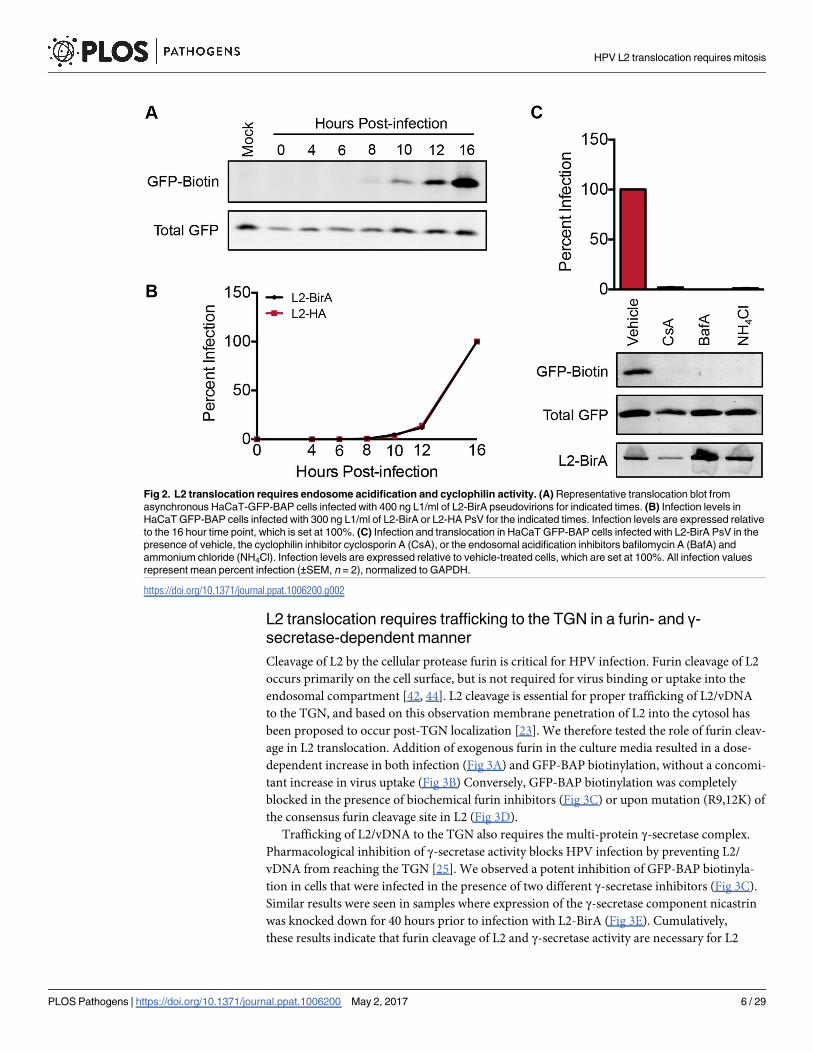
uncoating and L1/L2 dissociation. Inhibitors of both endosome acidification and cyclophilins
strongly blocked luciferase expression and GFP-BAP biotinylation (Fig 2C). Internalization of
L2-BirA virus, as measured by L2 immunoblotting of alkaline-washed infected cell lysates, was
unaffected by these treatments. These data substantiate the validity of the L2-BirA assay, since
L2 translocation should require prior breakdown of the capsid structure.
Fig 1. Development of the BirA/GFP-BAP assay for detecting membrane penetration by L2. (A) Schematic of the BirA translocation assay.
L2-BirA is physically separated from its substrate (GFP-BAP) while in the lumen of endocytic vesicles. Following translocation across the limiting
membrane, L2-BirA can biotinylate cytosolic GFP-BAP. (B) Brightfield and epifluorescent imaging of HaCaT GFP-BAP cells. (C) Coomassie staining
of wt-L2 and L2-BirA PsV. Molecular weights are shown in kilodaltons. (D) Transmission electron microscopy imaging of L2-3xFLAGTHA or L2-BirA
viral particles. Scale bar represents 100 nm. (E) In vitro biotinylation of reduced wt L2-BirA or R9,12K L2-BirA particles incubated with MBP-BAP, ATP,
and biotin for the indicated times prior to processing by SDS-PAGE/Western blotting. (F) Titration of infectivity and translocation of L2-BirA in HaCaT
GFP-BAP cells. Graph shows percent infectivity, relative to the 200 ng L1/ml concentration, which is set at 100%. GFP-biotin and total GFP were
analysed by Western blotting with neutravidin and anti-GFP staining respectively. (G) Infectivity of wt and L2-BirA pseudovirions in HaCaT GFP-BAP
cells at an equal MOI, expressed relative to wt, which is set at 100%. All infection values represent mean percent infection (±SEM, n = 2–3), normalized
to GAPDH.
https://doi.org/10.1371/journal.ppat.1006200.g001
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 5 / 29
L2 translocation requires trafficking to the TGN in a furin- and γ-secretase-dependent manner
Cleavage of L2 by the cellular protease furin is critical for HPV infection. Furin cleavage of L2
occurs primarily on the cell surface, but is not required for virus binding or uptake into the
endosomal compartment [42, 44]. L2 cleavage is essential for proper trafficking of L2/vDNA
to the TGN, and based on this observation membrane penetration of L2 into the cytosol has
been proposed to occur post-TGN localization [23]. We therefore tested the role of furin cleav-
age in L2 translocation. Addition of exogenous furin in the culture media resulted in a dose-
dependent increase in both infection (Fig 3A) and GFP-BAP biotinylation, without a concomi-
tant increase in virus uptake (Fig 3B) Conversely, GFP-BAP biotinylation was completely
blocked in the presence of biochemical furin inhibitors (Fig 3C) or upon mutation (R9,12K) of
the consensus furin cleavage site in L2 (Fig 3D).
Trafficking of L2/vDNA to the TGN also requires the multi-protein γ-secretase complex.
Pharmacological inhibition of γ-secretase activity blocks HPV infection by preventing L2/
vDNA from reaching the TGN [25]. We observed a potent inhibition of GFP-BAP biotinyla-
tion in cells that were infected in the presence of two different γ-secretase inhibitors (Fig 3C).
Similar results were seen in samples where expression of the γ-secretase component nicastrin
was knocked down for 40 hours prior to infection with L2-BirA (Fig 3E). Cumulatively,
these results indicate that furin cleavage of L2 and γ-secretase activity are necessary for L2
Fig 2. L2 translocation requires endosome acidification and cyclophilin activity. (A) Representative translocation blot from
asynchronous HaCaT-GFP-BAP cells infected with 400 ng L1/ml of L2-BirA pseudovirions for indicated times. (B) Infection levels in
HaCaT GFP-BAP cells infected with 300 ng L1/ml of L2-BirA or L2-HA PsV for the indicated times. Infection levels are expressed relative
to the 16 hour time point, which is set at 100%. (C) Infection and translocation in HaCaT GFP-BAP cells infected with L2-BirA PsV in the
presence of vehicle, the cyclophilin inhibitor cyclosporin A (CsA), or the endosomal acidification inhibitors bafilomycin A (BafA) and
ammonium chloride (NH4Cl). Infection levels are expressed relative to vehicle-treated cells, which are set at 100%. All infection values
represent mean percent infection (±SEM, n = 2), normalized to GAPDH.
https://doi.org/10.1371/journal.ppat.1006200.g002
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 6 / 29
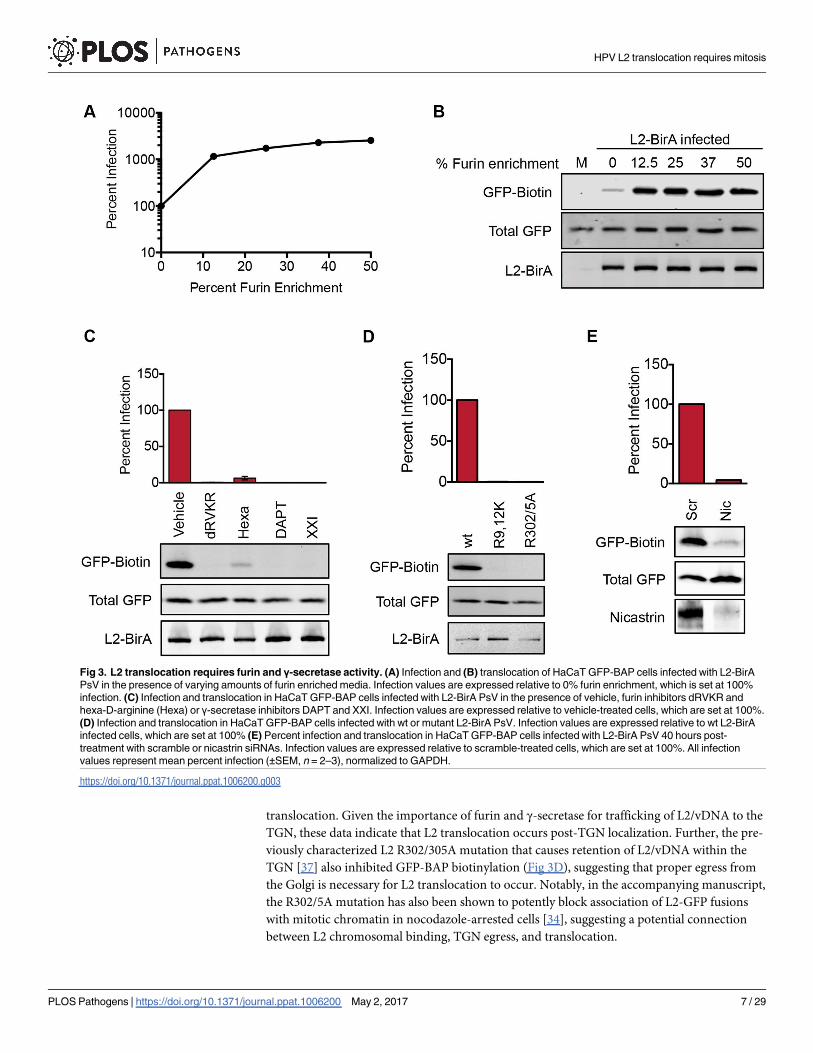
translocation. Given the importance of furin and γ-secretase for trafficking of L2/vDNA to the
TGN, these data indicate that L2 translocation occurs post-TGN localization. Further, the pre-
viously characterized L2 R302/305A mutation that causes retention of L2/vDNA within the
TGN [37] also inhibited GFP-BAP biotinylation (Fig 3D), suggesting that proper egress from
the Golgi is necessary for L2 translocation to occur. Notably, in the accompanying manuscript,
the R302/5A mutation has also been shown to potently block association of L2-GFP fusions
with mitotic chromatin in nocodazole-arrested cells [34], suggesting a potential connection
between L2 chromosomal binding, TGN egress, and translocation.
Fig 3. L2 translocation requires furin and γ-secretase activity. (A) Infection and (B) translocation of HaCaT GFP-BAP cells infected with L2-BirA
PsV in the presence of varying amounts of furin enriched media. Infection values are expressed relative to 0% furin enrichment, which is set at 100%
infection. (C) Infection and translocation in HaCaT GFP-BAP cells infected with L2-BirA PsV in the presence of vehicle, furin inhibitors dRVKR and
hexa-D-arginine (Hexa) or γ-secretase inhibitors DAPT and XXI. Infection values are expressed relative to vehicle-treated cells, which are set at 100%.
(D) Infection and translocation in HaCaT GFP-BAP cells infected with wt or mutant L2-BirA PsV. Infection values are expressed relative to wt L2-BirA
infected cells, which are set at 100% (E) Percent infection and translocation in HaCaT GFP-BAP cells infected with L2-BirA PsV 40 hours post-
treatment with scramble or nicastrin siRNAs. Infection values are expressed relative to scramble-treated cells, which are set at 100%. All infection
values represent mean percent infection (±SEM, n = 2–3), normalized to GAPDH.
https://doi.org/10.1371/journal.ppat.1006200.g003
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 7 / 29

During the course of this work, we observed some interesting anomalies with siRNA trans-
fection in the BirA translocation system. Initial siRNA experiments utilized a transfection pro-
tocol that our lab had previously developed for use in HaCaT cells [45]. In this protocol, when
HaCaT GFP-BAP cells were infected with L2-BirA virus at 24 hours post-transfection, nicas-
trin siRNAs potently blocked infection with the L2-BirA virus (S2A Fig). However, no corre-
sponding decrease in GFP-BAP biotinylation was detected (S2B Fig). These seemingly
contradictive results led us to hypothesize that a temporary perturbation of membrane integ-
rity created by cationic lipid-based transfection reagents [46, 47] may cause aberrant transloca-
tion signal by leakage of GFP-BAP into vesicular compartments, which would for allow non-
physiological interactions between L2-BirA and GFP-BAP. Indeed, GFP-BAP biotinylation
still occurred in siRNA-transfected cells that were infected with L2-BirA in the presence of γ-
secretase inhibitor XXI (S2C Fig), which causes a potent block in L2 translocation in the
absence of siRNA transfection (Fig 3C). GFP-BAP biotinylation was also observed in siRNA-
transfected cells that were infected with L2-BirA virus containing the R302/305A Golgi-reten-
tion mutant, which is normally unable to translocate (S2D Fig). Combined, these findings
demonstrate that non-physiological L2 translocation signal can be induced during transfec-
tion. These issues were overcome by increasing the length of time between transfection and
infection from 24 to 40 hours and adding additional washes to ensure removal of the transfec-
tion reagent (compare S2A and S2B Fig with Fig 3E). These experiments illustrate that care
must be used when combining the L2-BirA assay with reagents that potentially perturb lipid
membrane integrity and suggest that targets with short-lived knock down effects are not suit-
able for use with the system. However, these findings also demonstrate the sensitivity of the
L2-BirA assay as a measure of L2-BirA and GFP-BAP compartmentalization.
Cell cycle arrest blocks L2 translocation and causes vDNA to
accumulate at the TGN
Prior research has demonstrated that inhibitors of the cell cycle abrogate PsV infection and
block nuclear localization of the L2/genome complex [31–33]. We tested different cell cycle
inhibitors in the L2-BirA assay, with the expectation that L2 translocation would still occur,
since movement into the nucleus is presumed to be a post-translocation event. Aphidicolin
and hydroxyurea were used to arrest cells in S phase; purvalanol A and kbNB 142–70 were uti-
lized to block cell cycle progression at late G2; and monastrol, was used to arrest cells in mito-
sis (Fig 4A). Cell cycle status was analyzed by flow cytometry to ensure that all of the inhibitors
Fig 4. Cell cycle arrest blocks L2 translocation. (A) Diagram depicting where the inhibitors used in this study block cell cycle progression:
aphidicolin (Aphi), hydroxyurea (HU), purvalanol A (PurA), kbNB 142–70 (kbNB), or monastrol (Mon). (B) Percent infection and (C) translocation in
HaCaT GFP-BAP cells infected with L2-BirA in the presence of various cell cycle inhibitors. Infection values represent mean percent infection
(±SEM, n = 3), normalized to GAPDH and expressed relative to vehicle-treated cells, which are set at 100%.
https://doi.org/10.1371/journal.ppat.1006200.g004
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 8 / 29

behaved as expected (S3 Fig). As previously reported [31, 32], all of the inhibitors except mon-
astrol blocked HPV infection (Fig 4B). Surprisingly, the S and late G2 inhibitors also potently
inhibited GFP-GAP biotinylation (Fig 4C). Only monastrol, which arrests cells in mitosis after
nuclear envelope breakdown, permitted biotinylation to occur at levels similar to those seen in
cells treated with the vehicle DMSO.
The translocation block observed above suggests that cell cycle inhibitors may impede or
alter normal intracellular trafficking of L2 and prevent it from gaining access to the cytosol.
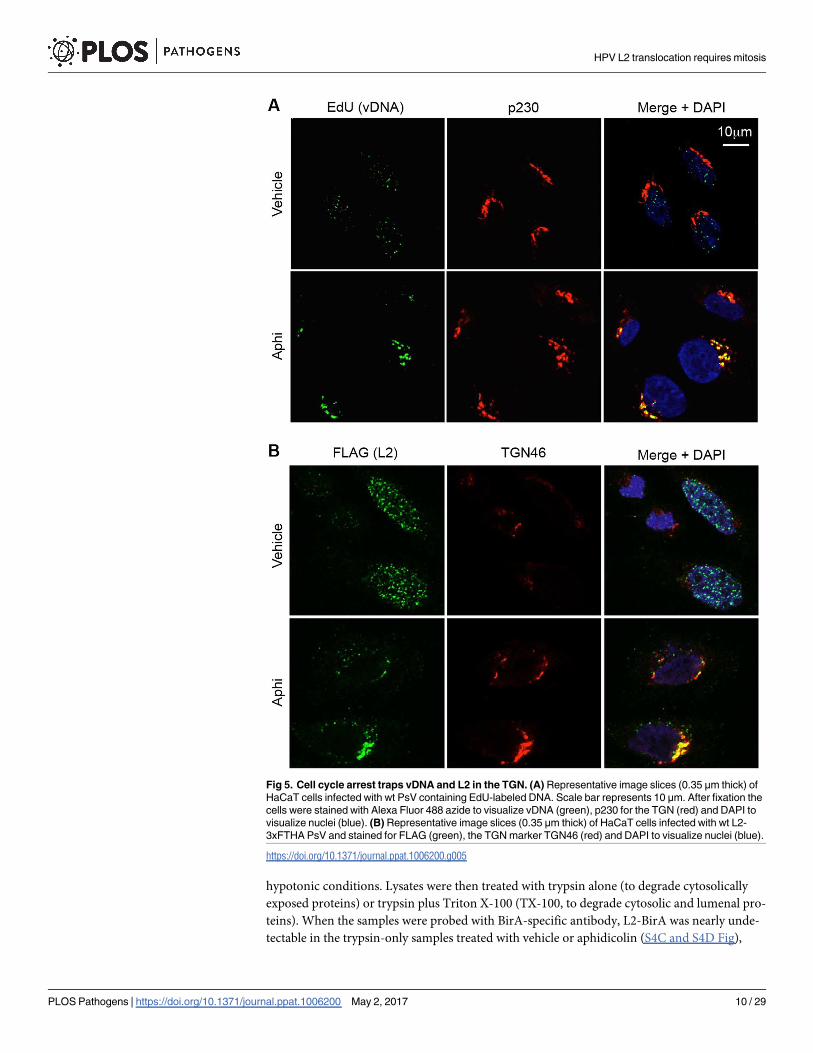
Since L2 traffics in complex with the viral genome, we examined where vDNA localized in cell
cycle arrested cells. HaCaT cells were infected with wt HPV16 containing EdU-labeled vDNA
to allow for direct detection of vDNA by confocal microscopy. In vehicle-treated cells vDNA
was readily detectable in both the TGN and nuclei of infected cells (Fig 5A). However, when
cells were infected in the presence of the S phase inhibitor aphidicolin, vDNA was almost
undetectable in the nucleus. Instead, consistent with prior observations [31], S phase arrest
caused the vDNA to accumulate with the TGN protein p230. A similar accumulation of L2
with the TGN protein TGN46 was observed in aphidicolin-treated cells infected with PsV con-
taining epitope-tagged L2-3xFLAG-thrombin-HA (L2-3xFTHA) (Fig 5B). TGN46 was used
for experiments with L2-3xFTHA virus due to species incompatibility between the FLAG and
p230 antibodies, which were both derived from mice. Collectively, the western blot and
microscopy data suggest that inhibiting cell cycle progression blocks translocation of L2/
vDNA, causing accumulation at the TGN.
L2-BirA adopts a membrane-spanning topology during infection
L2 contains several short peptide motifs that mediate interactions with various cytosolic sort-
ing factors to facilitate proper subcellular and retrograde trafficking of L2/vDNA from endo-
somes to the TGN [28, 29]. We have previously identified a critical transmembrane domain
(TMD) upstream of the sorting motifs near the N-terminus of L2 (S4A Fig) [48], suggesting
that L2 spans cellular membranes to expose these sorting motifs to their cytosolic interactions
partners. A recent report used L2 immunofluorescence microscopy with selective plasma
membrane permeabilization to confirm that L2 can indeed use this TMD during infection to
span vesicular membranes with a lumenal N-terminal domain (residues 1–45) and residues
immediately downstream of the TMD (residues 68–170) residing in the cytosol, which is con-
sistent with, but not proof of, L2 adopting a type-I transmembrane protein topology [49]. This
report also performed trypsin digestion assays with the L2 Golgi retention mutant of HPV18
(R295/298A, which is equivalent to the R302/305A mutant for HPV16 L2) to show that L2
spans across the TGN membrane, and that endosome acidification is required for L2 to span
host membranes [49]. However, the previous study did not thoroughly determine the topology
of the extreme C-terminus of L2, leaving open the possibility that C-terminal portions of L2
could be lumenal at the TGN. Indeed, S-phase block traps the L2/vDNA complex at the TGN
but also potently blocks GFP-BAP biotinylation (Figs 4 and 5), suggesting that the BirA por-
tion of the L2-BirA fusion is not exposed to the cytosol under these conditions. Moreover, if
the C-terminal BirA fusion were exposed to the cytosol during early trafficking, we would
expect to see translocation signal appear en route to the TGN, much earlier than 8 hours (Fig
2A).
One potential explanation is that the BirA fusion totally prevents L2 from spanning across
host membranes. To test this hypothesis, we performed a trypsin digestion assay on HaCaT
GFP-BAP cells infected with L2-BirA in the presence of DMSO, aphidicolin, or ammonium
chloride (experimental details outlined in S4B Fig). At 22 hours post-infection external virus
was washed off and the plasma membrane disrupted by passage through a needle under
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 9 / 29
hypotonic conditions. Lysates were then treated with trypsin alone (to degrade cytosolically
exposed proteins) or trypsin plus Triton X-100 (TX-100, to degrade cytosolic and lumenal pro-
teins). When the samples were probed with BirA-specific antibody, L2-BirA was nearly unde-
tectable in the trypsin-only samples treated with vehicle or aphidicolin (S4C and S4D Fig),
Fig 5. Cell cycle arrest traps vDNA and L2 in the TGN. (A) Representative image slices (0.35 μm thick) of
HaCaT cells infected with wt PsV containing EdU-labeled DNA. Scale bar represents 10 μm. After fixation the
cells were stained with Alexa Fluor 488 azide to visualize vDNA (green), p230 for the TGN (red) and DAPI to
visualize nuclei (blue). (B) Representative image slices (0.35 μm thick) of HaCaT cells infected with wt L2-
3xFTHA PsV and stained for FLAG (green), the TGN marker TGN46 (red) and DAPI to visualize nuclei (blue).
https://doi.org/10.1371/journal.ppat.1006200.g005
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 10 / 29

suggesting that L2 has been degraded because it is in a membrane-spanning conformation
with the bulk of the protein exposed to the cytosol. In contrast, the levels of L2-BirA detected
in ammonium chloride treated cells were significantly higher than those seen with vehicle or
aphidicolin, indicating that ammonium chloride treatment largely prevented L2-BirA degra-
dation in the trypsin-only sample. Staining for the ER lumenal protein BiP showed significant
degradation only in samples containing TX-100 plus trypsin (S4C Fig), indicating that vesicu-
lar membrane integrity was largely maintained throughout the assay. In summary, these data
confirm the findings of DiGuiseppe et al. [49] and indicate that, similar to wt L2, L2-BirA
adopts a membrane-spanning conformation post-endosome acidification. The fact that
L2-BirA fails to give translocation signal pre-Golgi or in the presence of cell cycle inhibitors is
indirect evidence that both the N- and C-termini of L2-BirA (including the BirA moiety) are
indeed lumenal in this membrane-spanning conformation, a notion that will require further
experimentation to confirm.
L2/genome egress from the TGN requires progression into mitosis
Several pieces of evidence suggest that cell cycle arrest prevents L2/vDNA trafficking to the
nucleus by preventing the complex from trafficking past the TGN. We therefore designed an
experiment to test whether cell cycle progression is sufficient for L2/vDNA to exit the TGN,
penetrate the limiting membrane, and traffic to the nucleus (Fig 6A). Briefly, HaCaT GFP-BAP
cells were infected in the presence of aphidicolin to allow L2/vDNA to accumulate at the TGN.
At 24 hours post-infection, aphidicolin and external virus were washed off, and different drugs
were added. The infection was allowed to proceed for an additional 24 hours before samples
were collected to analyze translocation and infection. When cells were treated with drugs that
inhibit HPV infection at points prior to Golgi localization, biotinylation levels were similar to
those of vehicle-treated samples (Fig 6B), indicating that L2-BirA can now access GFP-BAP in
the cytosol. Infection levels were also restored under these conditions (Fig 6C), indicating that
the vDNA successfully egressed from the TGN to the nucleus. These results demonstrate that
once the L2/vDNA complex reaches the Golgi, it has trafficked beyond its need for early infec-
tion requirements such as endosome acidification, cyclophilins, furin, and γ-secretase. How-
ever, GFP-BAP biotinylation and infection were still inhibited in cells that received a second
aphidicolin treatment and thus remained in S phase arrest. GFP-BAP biotinylation and infec-
tion were also blocked by purvalanol A treatment, suggesting that progression from S to late
G2 was not sufficient for L2 translocation to occur. In contrast, monastrol treatment restored
GFP-BAP biotinylation and infection to levels seen in vehicle-treated samples. These findings
indicate that cell cycle progression through the early stages of mitosis is sufficient for L2/
vDNA to egress from the TGN.
L2 translocation coincides with mitosis
Since progression into mitosis appears sufficient for L2 to breach the limiting membrane, we
hypothesized that translocation would coincide with entry into mitosis. We therefore performed
cell cycle synchronizations to examine when L2 translocation occurred relative to mitosis. To do
this, we infected HaCaT-GFP-BAP cells with L2-BirA virus in the presence of aphidicolin to halt
cells in S phase. At 12 hours post-infection, the aphidicolin was washed out, and the cell cycle
was allowed to continue. Mitotic progression was monitored by phosphorylation of histone H3
at serine 10, a modification that is necessary for initiating chromatin condensation at the G2/M
transition [50, 51]. After a small spike in phospho-H3 levels at 6 hours, phospho-H3 levels are
significantly higher at 9 hours post-release, eventually tapering off around 12 hours post-release
as the majority of the cell population exits mitosis (Fig 7A and 7B). GFP-BAP biotinylation levels
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 11 / 29

correlate very well to phospho-H3 levels, gradually rising after release from aphidicolin, and are
significantly higher than baseline at 10 hours post-release. Cells that were synchronized with a
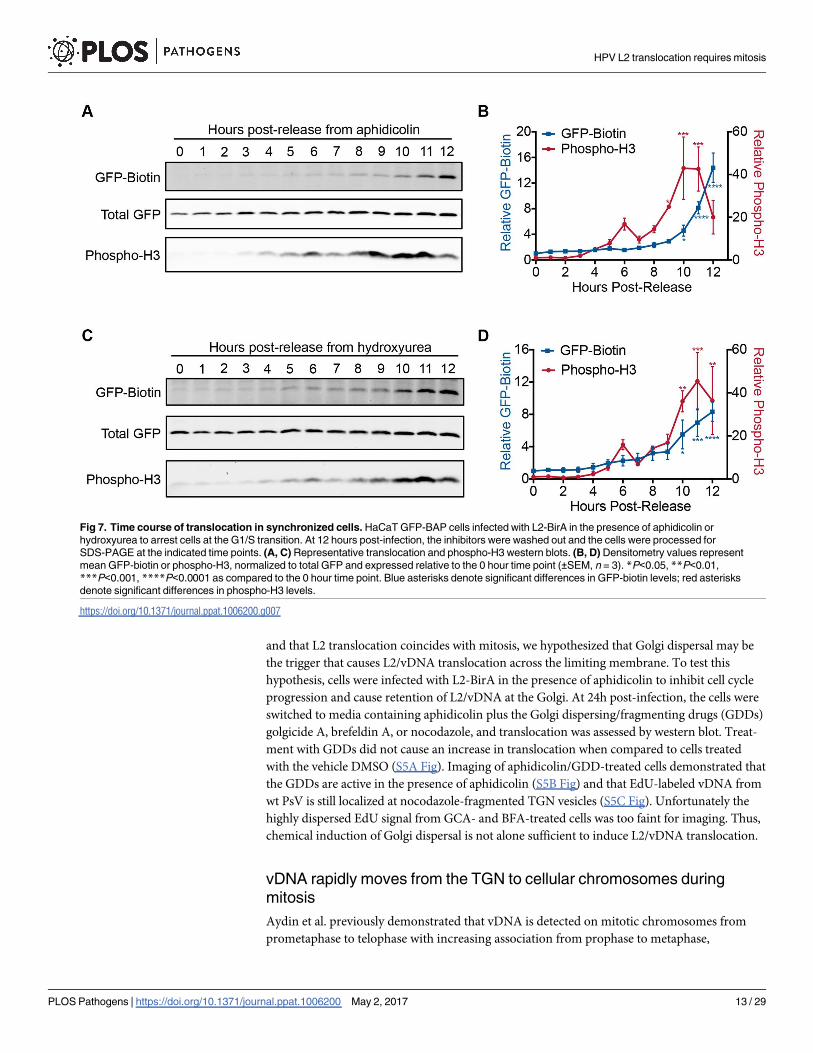
different S phase inhibitor, hydroxyurea, produced similar results (Fig 7C and 7D). Thus the
timing of L2-BirA cytosolic exposure coincides with or slightly lags behind the onset of mitosis.
Chemical dispersal of the Golgi is insufficient for L2 translocation
Fragmentation and dispersal of the Golgi apparatus is a necessary step in early mitosis [52, 53].
Given that cell cycle inhibition blocks L2 translocation and vDNA accumulation in the Golgi
Fig 6. Mitotic progression is sufficient for L2 translocation to occur. (A) Schematic of the aphidicolin washout assay. (B) Representative
translocation blot in the presence of various drugs post-aphidicolin washout. (C) Infection levels in the presence of various drugs post-aphidicolin
washout. Values represent mean percent infection (±SEM, n = 3), normalized to GAPDH and expressed relative to vehicle-treated cells, which are set
at 100%.
https://doi.org/10.1371/journal.ppat.1006200.g006
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 12 / 29
and that L2 translocation coincides with mitosis, we hypothesized that Golgi dispersal may be
the trigger that causes L2/vDNA translocation across the limiting membrane. To test this
hypothesis, cells were infected with L2-BirA in the presence of aphidicolin to inhibit cell cycle
progression and cause retention of L2/vDNA at the Golgi. At 24h post-infection, the cells were
switched to media containing aphidicolin plus the Golgi dispersing/fragmenting drugs (GDDs)
golgicide A, brefeldin A, or nocodazole, and translocation was assessed by western blot. Treat-
ment with GDDs did not cause an increase in translocation when compared to cells treated
with the vehicle DMSO (S5A Fig). Imaging of aphidicolin/GDD-treated cells demonstrated that
the GDDs are active in the presence of aphidicolin (S5B Fig) and that EdU-labeled vDNA from
wt PsV is still localized at nocodazole-fragmented TGN vesicles (S5C Fig). Unfortunately the
highly dispersed EdU signal from GCA- and BFA-treated cells was too faint for imaging. Thus,
chemical induction of Golgi dispersal is not alone sufficient to induce L2/vDNA translocation.
vDNA rapidly moves from the TGN to cellular chromosomes during
mitosis
Aydin et al. previously demonstrated that vDNA is detected on mitotic chromosomes from
prometaphase to telophase with increasing association from prophase to metaphase,
Fig 7. Time course of translocation in synchronized cells. HaCaT GFP-BAP cells infected with L2-BirA in the presence of aphidicolin or
hydroxyurea to arrest cells at the G1/S transition. At 12 hours post-infection, the inhibitors were washed out and the cells were processed for
SDS-PAGE at the indicated time points. (A, C) Representative translocation and phospho-H3 western blots. (B, D) Densitometry values represent
mean GFP-biotin or phospho-H3, normalized to total GFP and expressed relative to the 0 hour time point (±SEM, n = 3). *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001 as compared to the 0 hour time point. Blue asterisks denote significant differences in GFP-biotin levels; red asterisks
denote significant differences in phospho-H3 levels.
https://doi.org/10.1371/journal.ppat.1006200.g007
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 13 / 29

whereafter the association remained constant [31]. Thus, the localization of vDNA to host
chromosomes is likely triggered by mitosis. We therefore followed the subcellular localization
of EdU-labeled vDNA in cells released from aphidicolin synchronization. In interphase cells,
EdU signal overlaps with p230, indicating that the viral genome is still localized at the TGN
(Figs 8, S6 and S7). However, as the cells progress to prophase, there is a dramatic shift in EdU
localization. As cells reach late G2, around 9 hours post release, the centrioles have begun to
separate and EdU signal can start to be seen distinct from p230 positive structures. By pro-
phase most of the costaining with the TGN is lost and instead most of the vDNA is now local-
ized in the vicinity of the centrioles (Figs 8 and S6). After pericentriolar localization, the
association of vDNA with mitotic chromosomes begins in prometaphase, and by metaphase
the vast majority of vDNA is found decorating condensed mitotic chromosomes (Figs 8 and
S7). The vDNA remains associated with host chromosomes throughout the remaining stages
of mitosis. The microscopy data indicate that during mitosis the viral genome undergoes a dra-
matic change in subcellular localization, first moving from the TGN to a pericentriolar region,
followed by another shift onto host chromosomes. Thus, similar to L2 translocation, vDNA
movement from the TGN to host chromosomes coincides with mitosis.
Discussion
A major barrier that all nonenveloped viruses must overcome is transport of their nucleic acid
genomes across limiting membranes during infection of host cells. The papillomaviruses have
evolved the L2 minor capsid protein, which complexes with the vDNA to ensure proper sub-
cellular trafficking and subsequent penetration of limiting membranes. In this work we
describe the development of the first direct assay for membrane translocation of L2, based on
the bacterial biotin ligase BirA and its cognate BAP substrate. Our system involves the infec-
tion of GFP-BAP-expressing cells with virions encapsidating an L2-BirA fusion. Limiting
membranes separate the lumenal virion-associated BirA from the cytosolic GFP-BAP sub-
strate. Thus, biotinylation of GFP-BAP during infection is a direct consequence of L2 translo-
cation across the limiting membrane. The enzymatic signal-amplifying nature of the system
was intentional to favor the sensitive detection L2-BirA translocation. Similar BirA/BAP-based
systems could be useful for trafficking and penetration studies of other viruses containing cap-
sid proteins permissive to heterologous fusions, like adenovirus and the pIX capsid protein
[54]. Likewise, we anticipate and are working towards extending the system to enable tracking
of subcellular L2-BirA trafficking by engineering host cells to express BAP-tagged substrates in
specific subcellular compartments.
L2-BirA PsV recapitulated the trafficking and infection kinetics of unmodified wt PsV and
L2-BirA translocation was sensitive to pharmacological inhibitors that interfere with normal
subcellular transport of L2/vDNA to the TGN (Figs 2 and 3). These data strongly suggest that
localization to the TGN is a prerequisite to translocation, which has long been assumed, but
never directly tested. Cell cycle arrest experiments revealed a strict requirement for progres-
sion into mitosis for L2-BirA translocation to occur (Fig 4). In agreement with previously pub-
lished data [31], S phase block resulted in accumulation of vDNA at the TGN (Fig 5). Only
upon release from S phase and entry into mitosis did vDNA egress from the TGN (Fig 8). Like-
wise, release from S phase was necessary to restore L2-BirA translocation (Fig 6). Synchroniza-
tion timecourse experiments with two different S phase blockers, aphidicolin or hydroxyurea,
revealed that translocation signal coincided with or lagged slightly behind the appearance of
phosphorylated histone H3, a marker of progression into mitosis (Fig 7). Collectively these
data strongly suggest that translocation occurs post-TGN localization and requires the onset of
mitosis.
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 14 / 29
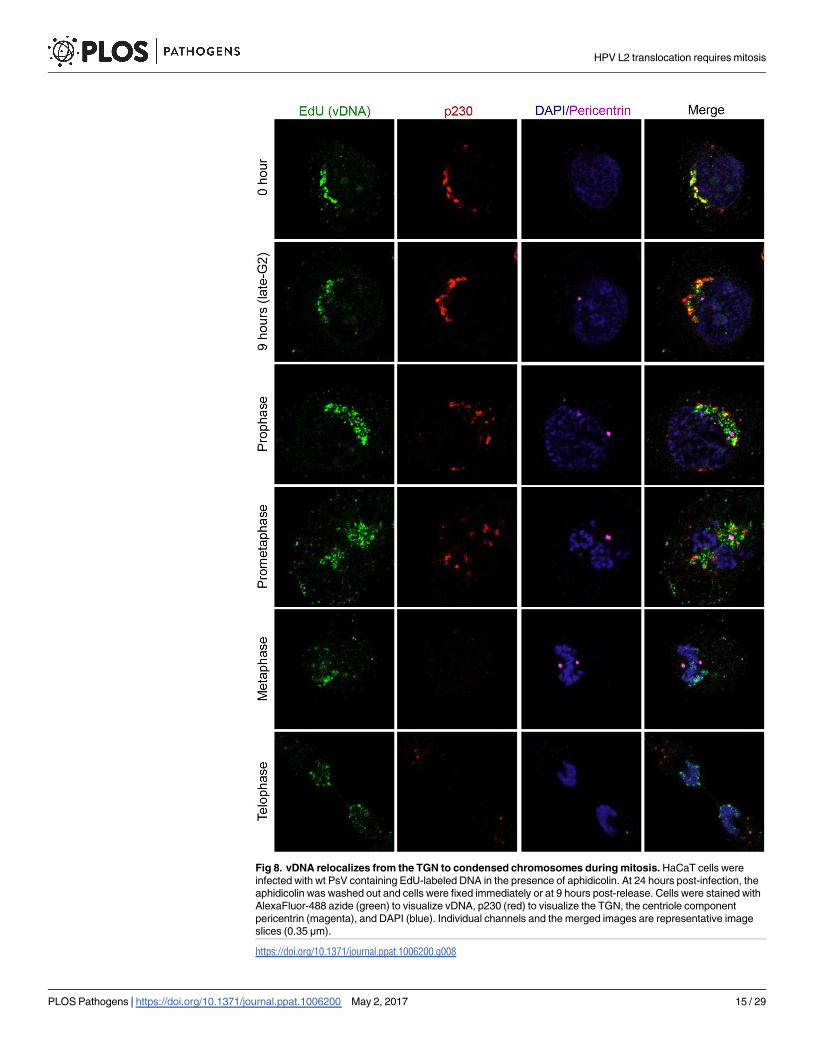
Fig 8. vDNA relocalizes from the TGN to condensed chromosomes during mitosis. HaCaT cells were
infected with wt PsV containing EdU-labeled DNA in the presence of aphidicolin. At 24 hours post-infection, the
aphidicolin was washed out and cells were fixed immediately or at 9 hours post-release. Cells were stained with
AlexaFluor-488 azide (green) to visualize vDNA, p230 (red) to visualize the TGN, the centriole component
pericentrin (magenta), and DAPI (blue). Individual channels and the merged images are representative image
slices (0.35 μm).
https://doi.org/10.1371/journal.ppat.1006200.g008
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 15 / 29

Given that the BirA translocation system is dependent on membrane compartmentaliza-
tion, the topology of L2 in the TGN membrane has important implications for the interpreta-
tion of the assay. We previously identified a transmembrane domain (TMD) near the N-
terminus of L2 that is essential for infection [48]. A recent report suggested L2 can span the
TGN membrane in a topology consistent with a type I transmembrane protein, with residues
immediately N-terminal of the TMD in the TGN lumen while residues immediately C-termi-
nal of the TMD are cytosolic [49]. However, the topology of the extreme C-terminus of L2 was
not directly assessed in this study or any others, leaving open the possibility that C-terminal
portions of L2 remain lumenal at the TGN. This possibility has important implications for the
L2-BirA system. If L2-BirA spans the TGN membrane in a type I topology as has been sug-
gested, BirA should biotinylate cytosolic GFP-BAP prior to complete translocation. However,
aphidicolin treatment, which traps L2/vDNA at the TGN (Fig 5), potently blocks GFP-BAP
biotinylation (Fig 4C), indicating that L2-BirA does not have access to GFP-BAP at the TGN.
Importantly, L2-BirA is still sensitive to trypsin digestion in the presence of aphidicolin (S4
Fig), demonstrating the BirA fusion does not prevent L2 from inserting into membranes.
Combined, these data suggest that if L2-BirA assumes a type I topology at the TGN then the
BirA portion may not be able to access its substrate, possibly due to steric hindrance from
proximal association to the membrane or binding of cellular sorting factors to the C-terminal
region of L2 [28, 29]. Alternatively, L2-BirA may span the TGN membrane twice, with both
the N- and C-termini remaining lumenal. Future work is necessary to elucidate the precise
topology, or topologies, that L2 adopts in cellular membranes. Regardless of its topology in the
membrane, our data support a model where BirA cannot access cytosolic GFP-BAP until
L2-BirA has passed the TGN and mitosis has commenced.
The Golgi apparatus undergoes dramatic and rapid changes during the transition from late
G2 to early M phase. The initial fragmentation is an important checkpoint for progression
through G2/M, and the eventual dispersal of Golgi into small cytosolic vesicles is believed to
enable equal partitioning of Golgi elements between daughter cells [52, 55–57]. We initially
hypothesized that these dynamic changes in Golgi morphology might be a trigger for L2 trans-
location. Contrary to our expectations, chemical fragmentation and dispersal of the Golgi was
alone insufficient to cause L2-BirA translocation (S5 Fig). However, it cannot be ruled out
that Golgi fragmentation in combination with processes unique to bona fide mitosis may be
required for translocation to occur. Indeed, many of the factors required for mitotic Golgi dis-
persal are only activated in late G2/M [57, 58].
Direct observation of EdU-labeled vDNA during wt PsV infection of synchronized HaCaT
cells corroborated our findings with the L2-BirA translocation system. These experiments
indicated that vDNA traffics to host chromosomes in a biphasic manner. vDNA was initially
colocalized with the TGN marker p230 during the aphidicolin block and at post-release times
prior to mitosis (Figs 8, S6 and S7). During the transition from late G2 to early prophase the
vDNA underwent a striking relocalization from the TGN to the pericentriolar region. These
changes were concomitant with Golgi fragmentation, chromosome condensation, and centri-
ole segregation (Fig 8). Given that centrosomes nucleate microtubule spindle formation dur-
ing mitosis, it is reasonable to hypothesize that L2/vDNA traffics along portions of these
microtubules. Indeed, some colocalization of vDNA with alpha tubulin-containing spindle-
like structures was recently observed in PsV-infected mitotic cells [35]. In contrast, the accom-
panying manuscript by Aydin et al. observed efficient localization of transiently expressed
L2-GFP fusion to mitotic chromosomes in the absence of microtubules [34]. Thus the ques-
tion of whether L2/vDNA localization to host chromosomes requires microtubule transport
remains unresolved, but the L2-chromosome interaction itself does not appear to require
microtubules.
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 16 / 29

The transient pericentriolar distribution of vDNA lasted through prophase and into prome-
taphase, at which time the vDNA had begun a second relocalization to mitotic chromosomes
(Figs 8 and S6). The vDNA was bound to chromosomes by metaphase, where it stayed
throughout the rest of mitosis, partitioning into the daughter cells during anaphase/telophase
(Figs 8 and S7). The distribution of vDNA on the condensed chromosomes was evocative of a
physical interaction or tethering, supporting the hypothesis of earlier work by Aydin et al.
[31]. Collectively the translocation and microscopy data support a model where L2/vDNA
accumulate at the TGN of interphase cells and undergo a rapid relocalization to the centriolar
region at the onset of mitosis in late G2/early prophase, followed by a shift to the condensed
chromosomes by metaphase. However, several important questions remain: Does L2/vDNA
traffic directly from the TGN to mitotic chromosomes, or does the complex pass through
other intracellular compartments on its way to the nucleus? Furthermore, does visual egress
from the TGN represent the translocation event? Or is the L2/vDNA complex still vesicle-
bound during its pericentriolar localization?
Given the short duration of time between the onset of mitosis and pericentriolar localiza-
tion of vDNA, it is reasonable to hypothesize that the viral genome takes a direct route from
TGN to the nucleus. However, the possibility that L2/vDNA traffic to other sites before reach-
ing the nucleus should not be ruled out. Day et al. observed slight overlap of HPV16 vDNA
with more distal cis/medial-Golgi markers giantin and GM130 [23]. Likewise Ishii et al.
reported partial colocalization of the related high risk HPV type 51 vDNA with the cis-Golgi
marker GM130 [26]. L2 has also been suggested to retrograde traffic beyond the Golgi, to the
ER based on proximity ligation assay with the lumenal ER residents BiP and protein disulfide
isomerase [25]. Whether these represent primary or alternative routes of infection, or even
unproductive dead ends is not clear.
During preparation of this manuscript, DiGiuseppe and colleagues published a paper
hypothesizing that vDNA remains in a vesicular compartment during mitosis after egress
from the TGN [35]. This hypothesis is based on nuclease protection assays and qPCR quantifi-
cation of vDNA levels during infection, and dual fluor-azide conjugation of EdU-labeled
vDNA in selectively and differentially permeabilized cells. They also present microscopy data
with the L2 R302/305A Golgi retention mutant showing visual egress of this mutant from
TGN markers during mitosis, but a failure to associate with mitotic chromosomes [35]. Their
prior work with this mutant showed enhanced vDNA/TGN colocalization in interphase cells
[37], suggesting this mutant is not really retained in the Golgi but may egress from the TGN
during mitosis in a vesicle-bound state only to become reabsorbed into the reforming TGN of
the daughter cells. Importantly, the companion paper by the Schelhaas group, using an assay
based on transient L2-GFP expression, defines a novel chromatin binding region (CBR) in L2
(residues 188–334), that is necessary and sufficient to promote L2 association with mitotic
chromosomes [34]. Mutagenesis experiments with this assay revealed that the R302/5A muta-
tion as well as IVAL286AAAA and RTR313EEE substitutions caused a striking defect in
mitotic chromosome binding. Like the R302/5A mutant, PsV containing the L2 RTR313EEE
and IVAL286AAAA mutants reach the TGN, but fail to bind mitotic chromosomes [34]. In
the accompanying manuscript these CBR mutations were tested for translocation in the
L2-BirA system. Strikingly, the chromatin binding activity and the translocation ability of
these CBR mutants are closely correlated; IVAL286AAAA, R302/5A, and RTR313EEE muta-
tions cause complete abrogation of both chromatin binding and translocation (Fig 3D and
[34]). These findings suggest that chromatin binding may be a necessary prerequisite for trans-
location of the L2/vDNA complex out of the TGN-derived vesicles. Additionally, the data pre-
sented here and in the companion paper demonstrate that the chromosomal association of
L2-GFP [34] and incoming L2/vDNA complexes (Figs 8, S6 and S7) both begin specifically
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 17 / 29
during prometaphase. Collectively, these data support the model of DiGiuseppe et al. that the
egress of vDNA from the TGN precedes true translocation of the L2/vDNA complex and fur-
thermore, that translocation is dependent on the L2 CBR binding an unknown mitotic chro-
mosome tethering factor that becomes available specifically during prometaphase.
In summary we have developed a novel system to directly study translocation of the L2/
vDNA complex during papillomavirus infection. We report, for the first time, that transloca-
tion requires TGN localization and entry into mitosis. Combined with microscopy of EdU-
labeled vDNA and the work of others [34, 35], our findings support a model where L2/vDNA
translocation occurs in mitosis after the visual egress of L2/vDNA from the TGN and is depen-
dent on the chromatin binding ability of L2 via a newly defined CBR. Mitosis is an enormously
dynamic and rapid process. Many drastic changes in subcellular structure and organization
occur within a short time: Golgi fragmentation and dispersal, chromosome condensation, cen-
triole segregation, spindle formation, nuclear envelope breakdown and remodeling of the ER
[59]. These dramatic processes occur in a highly coordinated and interconnected fashion. Elu-
cidating the specific mechanisms and host requirements for L2/vDNA translocation will be an
exciting challenge given the dynamic complexity of mitosis.
Materials and methods
Biochemical inhibitors
See Table 1 for a list of biochemical inhibitors used in this study.
Plasmid cloning
The pCIP-NES-GFP-BAP plasmid expresses cytosolic EGFP fused to the biotin acceptor pep-
tide (BAP), a specific substrate for the biotin protein ligase BirA [41]. Briefly, The EGFP cDNA
was PCR-amplified from pEGFP-N1 (Clontech) with primers encoding an N-terminally fused
PKI-type NES (NSNELALKLAGLDI) [60] and a C-terminally fused BAP (GLNDIFEAQ-
KIEWHE). The NES-GFP-BAP product was then NheI/BamHI cloned into the pCMV-IRES-
Table 1. Pharmacological inhibitors used in this study.
Compound Solvent Source (catalog #) Working Concentration
Actinomycin D DMSO Sigma Aldrich (A9415) 0.5 μg/mL
Aphidicolin DMSO Santa Cruz Biotechnology (201535) 6 μM
Bafilomycin A Methanol Millipore (196000) 100 nM
Brefeldin A DMSO Sigma (B6542) 150 nM
Cycloheximide DMSO Sigma (C104450) 10 μg/mL
Cyclosporin A DMSO Millipore (239835) 10 μM
DAPT DMSO Tocris (2634) 10 μM
dRVKR DMSO Millipore (344930) 25 μM
Golgicide A DMSO Santa Cruz Biotechnology (215103) 7.5 μM
Hexa-d-Arg Water Millipore (344931) 50 μM
Hydroxyurea PBS Sigma (H8627) 2 mM
Inhibitor XXI DMSO Millipore (565790) 200 nM
KbNB 142–70 DMSO Fisher (396210) 10μM
Monastrol DMSO Sigma (M8515) 50 μM
NH4Cl Water Santa Cruz Biotechnology (202936) 20 mM
Nocodazole DMSO Santa Cruz Biotechnology (3518) 5 μM
Purvalanol A DMSO Santa Cruz Biotechnology (224244) 12 μM
https://doi.org/10.1371/journal.ppat.1006200.t001
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 18 / 29

Puro plasmid backbone, a derivative of the lentiviral expression plasmid pCIG [61], modified
by insertion of a puromycin resistance gene downstream of the IRES sequence. pCMV-IRE-
S-Puro was a kind gift of Felicia Goodrum, University of Arizona. pXULL-L2-mycBirA is a
derivative of the HPV16 L1/L2 expression plasmid pXULL. Briefly, a codon optimized N-ter-
minally myc epitope tagged BirA gene was PCR-amplified from pcDNA3.1-mycBioID [62]
with primers introducing unique BspEI and SalI sites. This product was then cloned into
pXULL-L2-BKS, an intermediate construct containing in frame BspEI, KpnI, and SalI sites for
generating C-terminal fusions on L2. The R118G mutation of the mutant BirA was then
repaired back to R118 by Quickchange XL-II mutagenesis (Agilent #200522) to regenerate
wild type BirA. pcDNA3.1-mycBioID was a gift from Kyle Roux (Addgene plasmid # 35700).
p16shell.L2-3xFLAG-thrombin-HA (L2-3xFTHA) was a kind gift of Dan DiMaio and is
described in [25]. pXULL-L2-3xFTHA was constructed by PCR amplifying the 3xFTHA linker
with BspEI and SalI sites introduced to clone into pXULL-L2-BKS as a C-terminal fusion. An
additional KpnI site was engineered into the linker for fusion of inserts downstream of
3xFTHA. pXULL-L2-3xFTHA-BirA was generated by PCR amplifying BirA, flanked with
KpnI and SalI sites for ligation downstream of the 3xFTHA linker within pXULL-L2-3xFTHA.
pXULL-L2-HA was generated by introducing an HA tag fused to the C-terminus of L2 by
PCR, followed by subcloning the L2-HA back into pXULL with NotI and SalI.
Tissue culture
All cell lines were maintained at 37˚C with 5% CO2 and passaged every 2–3 days. 293TT (a
kind gift from Chris Buck, NCI, described in [63]) cells were cultured in Dulbecco’s modified
Eagle medium (DMEM) with high glucose and supplemented with 10% bovine growth serum
(BGS, HyClone SH30541.03) and antibiotic/antimycotic (Ab/Am, Sigma A5955). Furin-
secreting CHO furΔ1 cells and furin deficient FD11 cells (kind gifts of Patricia Day and Steven
Leppla respectively, both described in [64]) were maintained in high glucose DMEM with 10%
fetal bovine serum (FBS, HyClone SH30396.03), supplemented with 200 μM L-proline and
Ab/Am. Concentrated, active furin was isolated from CHOfurΔ1 conditioned media, diluted
into FD11 conditioned media as previously described [42]. HaCaT cells (a kind gift from Anne
Cress, University of Arizona, originally described in [65]) were grown in high glucose DMEM
supplemented with 10% FBS and Ab/Am.
Generation of NES-GFP-BAP HaCaT subclone
HaCaTs, plated to subconfluence in a 6-well plate were transfected with 2 μg pCIP-NES-GFP-
BAP using Lipofectamine 2000 (Life Technologies). After 48 hours of culturing and expansion
to a 10 cm dish, media was supplemented with 300–400 nM puromycin (HyClone) to begin
selection of clones stably expressing cytosolically localized GFP-BAP. Cells were further cul-
tured and expanded until most non-fluorescent clones died off. After one week of mainte-
nance, surviving cells were pooled and sparsely plated in 10cm dishes to isolate clonal
populations. Puromycin was gradually decreased from 400 nM to 200 nM during this selec-
tion. Individual clones were examined for GFP fluorescence and select clones were isolated
using cloning rings. An ideal clone and was subsequently expanded and banked as
HaCaT-GFP-BAP. These cells were maintained in DMEM supplemented with 10% FBS, 200
nM puromycin, and Ab/Am.
Pseudovirus production
Mutant viruses were generated by PCR-based methods or by site-directed mutagenesis using
the QuikChange XL-II kit. All mutations were verified by Sanger sequencing. PsV were
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 19 / 29

generated as previously described [66]. Briefly, 293TT cells were CaCO4 co-transfected with
the appropriate pXULL based plasmids and the luciferase reporter plasmid pGL3; virus was
then purified by CsCl gradient. The L1/L2 content of purified PsV was verified by SDS-PAGE
and Coomassie staining, as compared to bovine serum albumin protein standards. Pseudogen-
ome content was determined by SYBR green qPCR using primers specific for the luciferase
gene in pGL3. The capsid:genome ratios were all within the normal range for typical wild type
HPV16 preps. L2-BirA PsV were generated from pXULL constructs encoding either L2-myc-
BirA or L2-3xFTHA-BirA fusions. These L2-BirA containing PsV had comparable perfor-
mance in GFP-BAP translocation assays and were used interchangeably in this work.
pXULL-L2-BirA constructs are 9.5 kb in size, too large to be efficiently packaged within the
capsid using the 293TT PsV system [63, 67].
In vitro BirA biotinylation
Purified recombinant maltose binding protein substrate (MBP-BAP) was obtained from Avid-
ity (#BIS300). L2-BirA viruses were calculated to contain approximately 20 ng BirA per 500 ng
L1, assuming 24 molecules L2-BirA per virion. To disassemble the L1 capsid and make L2-BirA
accessible for reaction, 500 ng purified L2-BirA wt and R9,12K mutant PsV particles were
reduced in VSB + 16.7 mM DTT for 4 hours at room temperature. 50 μL reactions were then
set up in 50 mM bicine, 6.5 mM Tris, pH = 8.3, 10 mM ATP, 10 mM Mg-acetate, 50 μM D-bio-
tin, 5 μg MBP-BAP substrate (unbiotinylated or biotinylated positive control, Avidity #BIS300),
and 500 ng L1 (L2-BirA virus) of the DTT-reduced PsV. Reactions were stopped by the addi-
tion of SDS-PAGE loading buffer at 1, 4, and 16 hours. Samples were heated at 95˚C for 6 min-
utes prior to SDS-PAGE and western blot for biotinylated MBP, total MBP, and L2-BirA.
Infections
HaCaT GFP-BAP cells were plated at 50,000 cells per well in a 24-well plate. Cells were infected
the following day with L2-BirA virus at 2 x 108 viral genomes/well. At 24 hours post-infection
(p.i.), the cells were washed once with PBS and lysed in 100 μl reporter lysis buffer (Promega
E3971). Luciferase activity was measured on a DTX-800 multimode plate reader (Beckman
Coulter) using luciferase assay reagent (Promega E4550) according to the manufacturer’s
instructions. A fraction of these lysates were blotted for GAPDH to normalize luciferase activity.
Translocation assays
Unless otherwise stated, HaCaT GFP-BAP cells were plated at 50,000 cells per well in a 24-well
plate. The following day, the cells were infected with L2-BirA virus at 300 ng L1/ml. At 24
hours p.i., cells were lysed in 1X RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1%
NP40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with 1X reducing SDS-PAGE
loading buffer, 1X protease inhibitor cocktail (Sigma P1860), 1mM PMSF and 1X PhosSTOP
phosphatase inhibitor cocktail (Roche 04906845001). For experiments where intracellular lev-
els of L2 were assayed, cells were washed prior to lysing with alkaline PBS, pH 10.7 for 2.5 min-
utes to strip non-internalized virus off of the cell surface [18]. The samples were boiled at 95˚C
for 5 minutes and stored at -80˚C until further processing by western blot.
Western blotting
Samples were resolved by SDS-PAGE and transferred onto a 0.45 μm nitrocellulose membrane
for blotting. For GAPDH levels, blots were blocked in 5% non-fat powered milk dissolved in
Tris-buffered saline containing 0.1% Tween (TBST) and stained with anti-GAPDH (Cell
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 20 / 29

Signaling 2118) at 1:5000. To check for L2-BirA levels, blots were blocked in 5% milk/TBST
and stained with anti-L2 K4 at 1:5000 or anti-BirA (abcam 14002) 1:2000. For translocation,
blots were blocked in 100% Odyssey blocker buffer (Licor 927–40000) and stained sequentially
with neutravidin DyLight 800 (Pierce 22853) and anti-GFP (Clontech 6323770) at 1:5000 in
50% Odyssey blocker buffer/TBST. The lower portion of some translocation blots was cut off
and stained with anti-phopsho-H3 (Cell Signaling 3377) at 1:10,000 in 5% BSA/TBST. Nicas-
trin knockdown was verified by blotting with anti-nicastrin (Cell Signaling 5665) at 1:1000 in
5% bovine serum albumin (BSA)/TBST. MBP-BAP was stained using anti-MBP (New England
Biolabs E8030) at 1:10,000 in 1% BSA/TBST. Goat anti-rabbit DyLight 680 (Pierce 35568),
goat anti-mouse DyLight 680 (Pierce 35518), goat anti-mouse DyLight 800 (Pierce 35521), and
goat anti-chicken DyLight 680 Pierce (10074) were used as secondary antibodies at 1:10,000 in
5% milk/TBST. Blots were imaged on the Licor Odyssey Infrared Imaging System. Band inten-
sities were quantified by densitometry using ImageJ v1.48.
siRNA experiments
Pooled scramble (sc-37007) and nicastrin (sc-36036) siRNA duplexes were obtained from
Santa Cruz Biotechnologies. HaCaT GFP-BAP cells were plated in 24-well plates at 40,000 cells
per well in Ab/Am-free DMEM/10% FBS. The next day, cells were washed once with PBS and
the media replaced with OptiMEM (Life Technologies). Cells were transfected with 50 nM
siRNA using Lipofectamine RNAiMax (Life Technologies 13778150) according to the manu-
facturer’s instructions. At 18 hours post-transfection, siRNA/transfection reagent were washed
off by rinsing cells once with PBS and then adding Ab/Am-free DMEM/10% FBS to each well.
Cells were infected at 24 hours or 40 hours post-transfection in Ab/Am-free DMEM/10% FBS.
At 24 hours p.i., samples were collected for luciferase and western blotting as described above.
For infections occurring at 40 hours post-transfection, cells were washed at two additional
time points to further remove residual transfection reagent: 24 and 40 hours post-transfection.
Cell cycle analysis
Cell cycle status was analyzed by propidium iodide (PI) incorporation and flow cytometry.
Briefly, HaCaT GFP-BAP cells were treated with the appropriate inhibitors for 24 hours. Cells
were collected by trypsinization and pelleted at 1000 rpm for 10 minutes at 4˚C. The pellet was
resuspended in ice cold 70% ethanol to fix the cells and stored at -20˚C until ready for staining.
For PI staining, cells were pelleted at 2000 rpm for 15 minutes at 4˚C, resuspended in PBS, pH
7.4 containing 40 μg/mL PI and 500 μg/mL RNase A, and incubated at 37˚C for 30 minutes.
PI-stained cells were immediately analyzed using the BD Biosciences FACSCanto-II flow
cytometer and Diva 8.0 software.
EdU detection and immunofluorescence
Pseudovirions containing 5-ethynyl-2’-deoxyuridine (EdU) labeled pseudogenomes were
produced by CaPO4 transfection of 293TTs supplemented with 15 μM EdU as previously
described [45]. For infections, HaCaT cells were seeded on glass coverslips in 6-well plates at
100,000 cells per well. The following day, the cells were infected with 700ng of L1/ml of EdU-
labeled virus in the presence of various drugs. At 16 hours p.i., cells were washed once with
fresh media to remove unbound virus. Fresh media plus drugs were then added and the infec-
tion continued for an additional 14 hours. At 30 hour p.i., the cells were washed once in PBS,
pH 7.4 (PBS), then 1 x 2.5 min in PBS, pH 10.7 to remove surface-bound virus, then washed
twice in PBS, pH 7.4. In most cases the cells were fixed with 2% paraformaldehyde/PBS for 10
minutes at room temperature (RT) and permeabilized with 0.2% Triton X-100/PBS for 10
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 21 / 29

minutes at RT. In experiments where pericentrin was stained, the cells were fixed and permea-
bilized for 15 minutes in 100% methanol at -20˚C. The samples were blocked in 4% BSA/1%
goat serum/PBS overnight at 4˚C. EdU-labeled vDNA was then conjugated to Alexa Fluor
488-N3 using click chemistry according to manufacturer’s instructions (Life Technologies
C10337). For immunofluorescence, polyclonal rabbit anti-TGN46 (Sigma T7576), mouse anti-
p230 (BD Biosciences 611280), mouse anti-GM130 (BD Biosciences 610822), mouse anti-
FLAG (Sigma F3165) and rabbit anti-pericentrin (BioLegend 923701) were all used at 1:500.
Alexa Fluor-488, -555 and -647 labeled goat anti-mouse and goat anti-rabbit secondary anti-
bodies (Life Technologies A11029, A21424, A21429 and A21236) were used at 1:1,000.
Coverslips were mounted on glass slides with Prolong Antifade Diamond containing 40,6-dia-
midino-2-phenylindole (DAPI) (Life Technologies P36971).
Confocal and epifluorescence microscopy
Confocal microscopy was performed using a Zeiss LSM510 META system with a 405 nm laser
diode, a 488 nm argon laser and a 543 nm He/Ne1 laser or the Zeiss LSM880 system with 405
nm, 488 nm, 543 nm and 633 nm lasers. Samples were examined using a 63x objective, and Z-
stacks with a 0.35μm depth per plane were taken of each image. Representative single-plane
images were processed with the Zeiss META software or Zen Blue software and further pro-
cessed with Microsoft PowerPoint software. Epifluorescent micrographs were taken using an
Olympus IX71 inverted microscope with a 40x objective and xenon UV lamp.
Transmission electron microscopy
Each sample was applied to an ultra-thin carbon film over Lacey Carbon Support Film on
400-mesh copper grids (Ted Pella, Inc.) that were glow discharged for 1 minute. Excess solu-
tion was blotted with Whatman #1 filter paper and the grid was rapidly stained with 2% uranyl
acetate. The uranyl acetate was immediately blotted with Whatman #1 filter paper and rapidly
stained with 2% methylamine tungstate. The second stain was immediately blotted away with
Whatman #1 filter paper and allowed to air dry. Data was collected on a JEOL 3200FS micro-
scope operated at 300 kV. Images were acquired at a magnification of 46,000x and at 1.0 μm
underfocus using a Gatan UltraScan 4000 charge-coupled device (CCD) camera.
Aphidicolin release into pharmacological inhibitors
HaCaT GFP-BAP cells were plated at 50,000 cells per well in a 24-well tissue culture plate. The
following day, cells were infected on ice for 1 hour with wt L2-BirA virus at either 2 x 108 viral
genomes/well (for infection) or 150 ng L1/well (for translocation) in the presence of 6 μM
aphidicolin. After one hour, unbound virus was washed off, fresh media containing 6 μM aphi-
dicolin was added, and the cells were shifted to 37˚C. After 24 hours, the aphidicolin was
washed out by rinsing the cells one time with warm media, followed by one time with PBS, pH
10.7 for 2.5 minutes to remove non-internalized virus, and then one more time with warm
media. Fresh media containing different biochemical inhibitors was then added to the appro-
priate wells and the infection was allowed to proceed for an additional 24 hours. At this point
the samples were collected to measure infection by luciferase activity and translocation by
GFP-BAP biotinylation, as described above.
Infection with Golgi dispersing drugs
HaCaT GFP-BAP cells were plated at 50,000 cells per well. The following day, the cells were
infected with 150 ng L1/well of wt L2-BirA virus in the presence of 6 μM aphidicolin. At 24
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 22 / 29

hours p.i., fresh media was added containing aphidicolin plus one of the Golgi dispersing
drugs, or aphidicolin plus the vehicle DMSO. After 4 hours of GDD treatment, the samples
were lysed and translocation assessed by western blotting as described above. To ensure that
GDDs are still active in the presence of aphidicolin, a parallel set of cells on glass coverslips
were similarly treated, processed for immunofluorescence as described above, and imaged by
epipfluorescence microscopy. To test the effect of GDDs on vDNA trafficking, HaCaT cells
were plated on coverslips in 6-well plates at 100,000 cells per well. The next day, the cells were
infected with 700ng of L1/ml of EdU-labeled virus for a total of 30 hours in the presence of
aphidocolin. At 30 hours p.i., fresh media containing aphidocolin plus nocodozole or the vehi-
cle DMSO was added and the cells were cultured for an additional 4 hours. The samples were
then processed for EdU detection and immunofluorescence as described above and imaged by
confocal microscopy.
Trypsin digestion assay
HaCaT GFP-BAP cells were plated at 150,000 cells per well in a 6-well plate. Cells were infected
the following day with L2-BirA virus at 1 μg L1/well. At 22 hours p.i. the cells were washed
once with PBS pH 7.2. The cells were then washed with PBS pH 10.75 for 2.5 minutes to
remove surface bound virions. The cells were further washed 2x with PBS 7.2 to neutralize pH.
Next, 0.05% trypsin-EDTA was added and cells were incubated for 15 minutes at 37˚C to fur-
ther remove surface bound virions and lift cells off the plate. The trypsin was neutralized with
cDMEM and the cells were pelleted at 800 rpm for 5 minutes. Cells were then resuspended in
105 μl cold hypotonic lysis buffer (20 mM Tris-HCl, 10 mM EDTA, pH 8.5) and incubated on
ice for 15 minutes. The cells were then passed 15 times through a 25 gauge x 5/8” needle to lyse
the plasma membrane. Lysates were then split into four equal parts and exposed to final con-
centration of 0.04% trypsin-EDTA and 0.5% TX-100 or equivalent volumes of vehicle. The
lysates were incubated at 37˚C for 55 minutes. The lysates were then put on ice and a final con-
centration of 400 μg/ml trypsin inhibitor from soybean (Sigma T6522) was added to quench
the trypsin. Reducing SDS-PAGE loading buffer supplemented with a final concentration of 1
mM PMSF and 1X protease inhibitor cocktail (Sigma #P1860) was added to the samples. Sam-
ples were then boiled for 6 minutes at 95˚C and stored at -80˚C until further processing by
western blot.
Cell synchronization and translocation time course
HaCaT GFP-BAP cells were plated at 45,000 cells per well in 24-well plates. The next day, fresh
media containing 6 μM aphidicolin or 2 mM hydroxyurea was added to each well for 6 hours.
Cells were then infected with wt L2-BirA virus at 150 ng L1/well in the presence of 6 μM aphi-
dicolin or 2 mM hydroxyurea. At 12 hours p.i. (18 hours post-inhibitor addition), the synchro-
nized cells were released by washing each well twice with phosphate buffered saline, pH 7.4
(PBS) and adding fresh media to each well. Samples were collected and processed by western
blotting as described above.
Statistics
Statistical analyses were performed using Prism 6 (GraphPad Software). Significant differences
were determined by one-way ANOVA followed by Dunnett’s multiple comparisons test or
two-way ANOVA followed by Tukey’s multiple comparisons test.
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 23 / 29

Supporting information
S1 Fig. GFP-BAP is biotinylated by L2-BirA from incoming capsids. (A) Schematic of the
experimental setup. To minimize toxicity, an abbreviated 14 hour infection was performed,
with addition of actinomycin D (ActD) or cycloheximide (CHX) at 8.5 hours after the start of
infection, with the cells only exposed to drugs for 5.5 hours. (B) Representative translocation
blots for HaCaT GFP-BAP cells infected with L2-BirA PsV in the presence of vehicle (DMSO),
ActD, or CHX. (C) Infection and translocation levels in the presence of vehicle or inhibitors.
Infection values represent mean percent infection (±SEM, n = 2), normalized to GAPDH. Per-
cent translocation levels were quantified by densitometry of GFP-biotin bands, normalized to
total GFP band intensity. Percent infection and translocation are expressed relative to DMSO-
treated cells infected with L2-BirA, which are set at 100%.
(TIFF)
S2 Fig. Transfection reagent causes aberrant translocation signal. (A) Infection and (B)
translocation in HaCaT GFP-BAP cells that were transfected with scramble (scr) or nicastrin
(nic) specific siRNA for 24 hours and then infected with wt L2-BirA for an additional 24
hours. Infection values represent mean percent infection (±SEM, n = 2), normalized to
GAPDH and expressed relative to scramble-treated cells, which are set at 100%. (C) Transloca-
tion in HaCaT GFP-BAP cells treated with media, the transfection reagent RNAiMax alone, or
RNAiMax-conjugated siRNAs in the presence of the vehicle DMSO or γ-secretase inhibitor
XXI. (D) Infection and translocation in HaCaT GFP-BAP treated with media or scramble
siRNA for 24 hours and then infected with wt L2-BirA or R302/5-BirA for an additional 24
hours. Infection values represent mean percent infection (±SEM, n = 2), normalized to
GAPDH and expressed relative to the wt sample for each condition, which are set at 100%.
(TIFF)
S3 Fig. Verification of the cell cycle inhibitors used in this study. Flow cytometry of HaCaT
GFP-BAP cells treated with various cell cycle inhibitors or vehicle control for 24 hours, fixed
and analyzed for DNA content by propidium iodide. G1, S, and G2/M peaks are indicated on
the vehicle (DMSO) profile.
(TIFF)
S4 Fig. L2-BirA adopts a transmembrane topology post-endosome acidification. (A) Dia-
gram of L2-BirA fusion protein showing furin cleavage site and transmembrane domain
(TMD). (B) Diagram of the trypsin digestion assay experimental setup. Briefly, HaCaT
GFP-BAP cells were infected with L2-BirA PsV for 22 hours in the presence of DMSO, Aphi,
or NH4Cl. Cells were then washed with alkaline PBS and trypsinized to remove extracellular
virus and lift the cells from the dish. Cells were gently pelleted and lysed by shearing. Crude
lysate was aliquoted equally among four tubes for treatment ± trypsin and TX-100, and then
incubated for 55 minutes at 37˚C prior to processing for SDS-PAGE and western blot. (C)
Anti-BirA and anti-BiP stains of infected cell lysates, treated as indicated. (D) Densitometry
values represent mean L2-BirA levels, normalized to total BiP and expressed relative to
the -trypsin condition for vehicle, Aphi and NH4Cl (±SEM, n = 3).
(TIFF)
S5 Fig. Chemical disruption of the Golgi is insufficient to induce translocation. (A) Repre-
sentative translocation blot of HaCaT GFP-BAP cells infected in the presence of aphidicolin
for 24 hours and then treated with aphidicolin plus GDDs for 4 additional hours. (B) Repre-
sentative epifluorescent images of HaCaT cells treated with aphidicolin for 24 hours and then
treated with aphidicolin plus GDDs for an additional 4 hours. Cells were stained with anti-
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 24 / 29

GM130 (green, cis-Golgi marker) and TGN46 (red, trans-Golgi marker). (C) Representative
slices (0.35 μm) of HaCaT cells infected with wt PsV containing EdU-labeled DNA in the pres-
ence of aphidicolin for 30 hours, then exposed to aphidicolin plus nocodozole for 4 hours.
After fixation the cells were stained with Alexa Fluor 488 azide to visualize vDNA (green),
anti-p230 for the TGN (red), and DAPI to visualize nuclei (blue).
(TIFF)
S6 Fig. vDNA relocalizes from the TGN to a pericentriole region early in mitosis. Maxi-
mum intensity projections of HaCaT cells infected with wt PsV containing EdU-labeled DNA
in the presence of aphidicolin and processed as described in Fig 8. Representative images of
interphase cells (0 hours post-release) and late G2 and prophase (9 hours post-release) are
shown. White arrows indicate location of pericentrin.
(TIFF)
S7 Fig. vDNA relocalizes from centrioles to host chromosomes in late mitosis. Maximum
intensity projections of HaCaT cells infected with wt PsV containing EdU-labeled DNA in the
presence of aphidicolin and processed as described in Fig 8. Representative images of prometa-
phase and meta/telophase cells (9 hours post-release) are shown. White arrows indicate loca-
tion of pericentrin.
(TIF)
Acknowledgments
We gratefully acknowledge Dr. Martin Muller for the K4L220-38 monoclonal antibody, Dr.
Patricia Day for the furΔ1 furin-secreting CHO cells, and Dr. Steven Leppla for the furin defi-
cient FD11 CHO cells. We thank Chris Buck for the 293TT cells. We thank Anne Cress for the
HaCaT cells. We thank Felicia Goodrum, Kyle Roux, and Dan DiMaio for plasmid constructs.
We thank Mike Kuhns for pericentrin antibody. We thank Dena Yoder of the UA BIO5 Media
Facility, Patty Jansma of the UA ORD Imaging Core-Marley, and Paula Campbell and John
Fitch of the UACC/ARL Cytometry Core Facility which is funded by a UA Cancer Center Sup-
port Grant (CCSG—CA 023074). We thank Greg Rogers, David Elliott, Jean Wilson, and the
“UA trafficking group” for critical feedback on various aspects of this work. We thank the
Schelhaas group for critical reading of the manuscript.
Author Contributions
Conceived and designed the experiments: CMC MPB SKC.
Performed the experiments: CMC MPB JAC ARM SL MSB TRW SKM SKC.
Analyzed the data: CMC MPB SKC.
Contributed reagents/materials/analysis tools: SKM RAB.
Wrote the paper: CMC MPB SKC.
References
1. Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, et al. The biology and life-cycle of human
papillomaviruses. Vaccine. 2012; 30 Suppl 5:F55–70. Epub 2012/12/05.
2. Forman D, de Martel C, Lacey CJ, Soerjomataram I, Lortet-Tieulent J, Bruni L, et al. Global burden of
human papillomavirus and related diseases. Vaccine. 2012; 30 Suppl 5:F12–23. Epub 2012/12/05.
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 25 / 29

3. Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, et al. Arrangement of L2 within
the papillomavirus capsid. Journal of virology. 2008; 82(11):5190–7. Epub 2008/03/28. https://doi.org/
10.1128/JVI.02726-07 PMID: 18367526
4. Favre M, Breitburd F, Croissant O, Orth G. Chromatin-like structures obtained after alkaline disruption
of bovine and human papillomaviruses. Journal of virology. 1977; 21(3):1205–9. Epub 1977/03/01.
PMID: 191643
5. Day PM, Roden RB, Lowy DR, Schiller JT. The papillomavirus minor capsid protein, L2, induces locali-
zation of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML onco-
genic domains. Journal of virology. 1998; 72(1):142–50. Epub 1998/01/07. PMID: 9420209
6. Holmgren SC, Patterson NA, Ozbun MA, Lambert PF. The minor capsid protein L2 contributes to two
steps in the human papillomavirus type 31 life cycle. Journal of virology. 2005; 79(7):3938–48. Epub
2005/03/16. https://doi.org/10.1128/JVI.79.7.3938-3948.2005 PMID: 15767396
7. Ishii Y, Ozaki S, Tanaka K, Kanda T. Human papillomavirus 16 minor capsid protein L2 helps capsome-
res assemble independently of intercapsomeric disulfide bonding. Virus genes. 2005; 31(3):321–8.
Epub 2005/09/22. https://doi.org/10.1007/s11262-005-3250-3 PMID: 16175337
8. Day PM, Baker CC, Lowy DR, Schiller JT. Establishment of papillomavirus infection is enhanced by pro-
myelocytic leukemia protein (PML) expression. Proceedings of the National Academy of Sciences of
the United States of America. 2004; 101(39):14252–7. Epub 2004/09/24. https://doi.org/10.1073/pnas.
0404229101 PMID: 15383670
9. Broutian TR, Brendle SA, Christensen ND. Differential binding patterns to host cells associated with
particles of several human alphapapillomavirus types. The Journal of general virology. 2010; 91(Pt
2):531–40. Epub 2009/10/23. https://doi.org/10.1099/vir.0.012732-0 PMID: 19846678
10. Cerqueira C, Liu Y, Kuhling L, Chai W, Hafezi W, van Kuppevelt TH, et al. Heparin increases the infec-
tivity of Human Papillomavirus type 16 independent of cell surface proteoglycans and induces L1 epi-
tope exposure. Cellular microbiology. 2013; 15(11):1818–36. Epub 2013/04/23. https://doi.org/10.1111/
cmi.12150 PMID: 23601855
11. Culp TD, Cladel NM, Balogh KK, Budgeon LR, Mejia AF, Christensen ND. Papillomavirus particles
assembled in 293TT cells are infectious in vivo. Journal of virology. 2006; 80(22):11381–4. Epub 2006/
09/01. https://doi.org/10.1128/JVI.01328-06 PMID: 16943284
12. Johnson KM, Kines RC, Roberts JN, Lowy DR, Schiller JT, Day PM. Role of heparan sulfate in attach-
ment to and infection of the murine female genital tract by human papillomavirus. Journal of virology.
2009; 83(5):2067–74. Epub 2008/12/17. https://doi.org/10.1128/JVI.02190-08 PMID: 19073722
13. Kines RC, Thompson CD, Lowy DR, Schiller JT, Day PM. The initial steps leading to papillomavirus
infection occur on the basement membrane prior to cell surface binding. Proceedings of the National
Academy of Sciences of the United States of America. 2009; 106(48):20458–63. Epub 2009/11/19.
https://doi.org/10.1073/pnas.0908502106 PMID: 19920181
14. Richards KF, Bienkowska-Haba M, Dasgupta J, Chen XS, Sapp M. Multiple heparan sulfate
binding site engagements are required for the infectious entry of human papillomavirus type 16. Journal
of virology. 2013; 87(21):11426–37. Epub 2013/08/24. https://doi.org/10.1128/JVI.01721-13 PMID:
23966387
15. Cerqueira C, Samperio Ventayol P, Vogeley C, Schelhaas M. Kallikrein-8 Proteolytically Processes
Human Papillomaviruses in the Extracellular Space To Facilitate Entry into Host Cells. Journal of
virology. 2015; 89(14):7038–52. Epub 2015/05/01. https://doi.org/10.1128/JVI.00234-15 PMID:
25926655
16. Day PM, Gambhira R, Roden RB, Lowy DR, Schiller JT. Mechanisms of human papillomavirus type 16
neutralization by l2 cross-neutralizing and l1 type-specific antibodies. Journal of virology. 2008; 82
(9):4638–46. Epub 2008/02/29. https://doi.org/10.1128/JVI.00143-08 PMID: 18305047
17. Day PM, Schelhaas M. Concepts of papillomavirus entry into host cells. Current opinion in virology.
2014; 4:24–31. Epub 2014/02/15. https://doi.org/10.1016/j.coviro.2013.11.002 PMID: 24525291
18. Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, et al. Entry of human papillomavirus
type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS pathogens. 2012; 8
(4):e1002657. Epub 2012/04/27. https://doi.org/10.1371/journal.ppat.1002657 PMID: 22536154
19. Muller KH, Spoden GA, Scheffer KD, Brunnhofer R, De Brabander JK, Maier ME, et al. Inhibition by cel-
lular vacuolar ATPase impairs human papillomavirus uncoating and infection. Antimicrobial agents and
chemotherapy. 2014; 58(5):2905–11. Epub 2014/03/13. https://doi.org/10.1128/AAC.02284-13 PMID:
24614368
20. Smith JL, Campos SK, Wandinger-Ness A, Ozbun MA. Caveolin-1-dependent infectious entry of
human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for pH-
dependent uncoating. Journal of virology. 2008; 82(19):9505–12. Epub 2008/08/01. https://doi.org/10.
1128/JVI.01014-08 PMID: 18667513
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 26 / 29

21. Grassel L, Fast LA, Scheffer KD, Boukhallouk F, Spoden GA, Tenzer S, et al. The CD63-Syntenin-1
Complex Controls Post-Endocytic Trafficking of Oncogenic Human Papillomaviruses. Scientific reports.
2016; 6:32337. Epub 2016/09/01. https://doi.org/10.1038/srep32337 PMID: 27578500
22. Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. Cyclophilins facilitate dissociation of the
human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. Journal of
virology. 2012; 86(18):9875–87. Epub 2012/07/05. https://doi.org/10.1128/JVI.00980-12 PMID: 22761365
23. Day PM, Thompson CD, Schowalter RM, Lowy DR, Schiller JT. Identification of a role for the trans-
Golgi network in human papillomavirus 16 pseudovirus infection. Journal of virology. 2013; 87(7):3862–
70. Epub 2013/01/25. https://doi.org/10.1128/JVI.03222-12 PMID: 23345514
24. Lipovsky A, Popa A, Pimienta G, Wyler M, Bhan A, Kuruvilla L, et al. Genome-wide siRNA screen identi-
fies the retromer as a cellular entry factor for human papillomavirus. Proceedings of the National Acad-
emy of Sciences of the United States of America. 2013; 110(18):7452–7. Epub 2013/04/10. https://doi.
org/10.1073/pnas.1302164110 PMID: 23569269
25. Zhang W, Kazakov T, Popa A, DiMaio D. Vesicular trafficking of incoming human papillomavirus 16 to
the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. mBio. 2014; 5(5):
e01777–14. Epub 2014/09/18. https://doi.org/10.1128/mBio.01777-14 PMID: 25227470
26. Ishii Y, Nakahara T, Kataoka M, Kusumoto-Matsuo R, Mori S, Takeuchi T, et al. Identification of
TRAPPC8 as a host factor required for human papillomavirus cell entry. PloS one. 2013; 8(11):e80297.
Epub 2013/11/19. https://doi.org/10.1371/journal.pone.0080297 PMID: 24244674
27. Bergant M, Banks L. SNX17 facilitates infection with diverse papillomavirus types. Journal of virology.
2013; 87(2):1270–3. Epub 2012/11/02. https://doi.org/10.1128/JVI.01991-12 PMID: 23115288
28. Bergant Marusic M, Ozbun MA, Campos SK, Myers MP, Banks L. Human papillomavirus L2 facilitates
viral escape from late endosomes via sorting nexin 17. Traffic. 2012; 13(3):455–67. Epub 2011/12/14.
https://doi.org/10.1111/j.1600-0854.2011.01320.x PMID: 22151726
29. Popa A, Zhang W, Harrison MS, Goodner K, Kazakov T, Goodwin EC, et al. Direct binding of retromer
to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection.
PLoS pathogens. 2015; 11(2):e1004699. Epub 2015/02/19. https://doi.org/10.1371/journal.ppat.
1004699 PMID: 25693203
30. Pim D, Broniarczyk J, Bergant M, Playford MP, Banks L. A Novel PDZ Domain Interaction Mediates the
Binding between Human Papillomavirus 16 L2 and Sorting Nexin 27 and Modulates Virion Trafficking.
Journal of virology. 2015; 89(20):10145–55. Epub 2015/07/24. https://doi.org/10.1128/JVI.01499-15
PMID: 26202251
31. Aydin I, Weber S, Snijder B, Samperio Ventayol P, Kuhbacher A, Becker M, et al. Large Scale RNAi
Reveals the Requirement of Nuclear Envelope Breakdown for Nuclear Import of Human Papillomavi-
ruses. PLoS pathogens. 2014; 10(5):e1004162. Epub 2014/05/31. https://doi.org/10.1371/journal.ppat.
1004162 PMID: 24874089
32. Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. Establishment of human papillomavirus infec-
tion requires cell cycle progression. PLoS pathogens. 2009; 5(2):e1000318. Epub 2009/02/28. https://
doi.org/10.1371/journal.ppat.1000318 PMID: 19247434
33. Broniarczyk J, Massimi P, Bergant M, Banks L. Human Papillomavirus Infectious Entry and Trafficking
Is a Rapid Process. Journal of virology. 2015; 89(17):8727–32. Epub 2015/06/13. https://doi.org/10.
1128/JVI.00722-15 PMID: 26063434
34. Aydin I, Villalonga-Planells R, Greune L, Bronnimann MP, Calton CM, Becker M, et al. A central region
in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane pene-
tration for mitotic nuclear entry. PLoS pathogens. 2017; 13(5):e1006308.
35. DiGiuseppe S, Luszczek W, Keiffer TR, Bienkowska-Haba M, Guion LG, Sapp MJ. Incoming human
papillomavirus type 16 genome resides in a vesicular compartment throughout mitosis. Proceedings of
the National Academy of Sciences of the United States of America. 2016; 113(22):6289–94. Epub
2016/05/18. https://doi.org/10.1073/pnas.1600638113 PMID: 27190090
36. Barker DF, Campbell AM. The birA gene of Escherichia coli encodes a biotin holoenzyme synthetase.
Journal of molecular biology. 1981; 146(4):451–67. Epub 1981/03/15. PMID: 7024555
37. DiGiuseppe S, Bienkowska-Haba M, Hilbig L, Sapp M. The nuclear retention signal of HPV16 L2 protein
is essential for incoming viral genome to transverse the trans-Golgi network. Virology. 2014; 458–
459:93–105. Epub 2014/06/15. https://doi.org/10.1016/j.virol.2014.04.024 PMID: 24928042
38. Howard PK, Shaw J, Otsuka AJ. Nucleotide sequence of the birA gene encoding the biotin operon
repressor and biotin holoenzyme synthetase functions of Escherichia coli. Gene. 1985; 35(3):321–31.
Epub 1985/01/01. PMID: 3899863
39. Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-cata-
lyzed biotinylation. Protein science: a publication of the Protein Society. 1999; 8(4):921–9. Epub 1999/
04/22.
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 27 / 29

40. Parrott MB, Adams KE, Mercier GT, Mok H, Campos SK, Barry MA. Metabolically biotinylated adenovi-
rus for cell targeting, ligand screening, and vector purification. Molecular therapy: the journal of the
American Society of Gene Therapy. 2003; 8(4):688–700. Epub 2003/10/08.
41. Schatz PJ. Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13
residue consensus peptide specifies biotinylation in Escherichia coli. Bio/technology. 1993; 11
(10):1138–43. Epub 1993/10/01. PMID: 7764094
42. Bronnimann MP, Calton CM, Chiquette SF, Li S, Lu M, Chapman JA, et al. Furin Cleavage of L2 During
Papillomavirus Infection: Minimal Dependence on Cyclophilins. Journal of virology. 2016. Epub 2016/
04/29.
43. Cerqueira C, Schelhaas M. Principles of polyoma- and papillomavirus uncoating. Medical microbiology
and immunology. 2012; 201(4):427–36. Epub 2012/09/25. https://doi.org/10.1007/s00430-012-0262-1
PMID: 23001401
44. Richards RM, Lowy DR, Schiller JT, Day PM. Cleavage of the papillomavirus minor capsid protein, L2,
at a furin consensus site is necessary for infection. Proceedings of the National Academy of Sciences of
the United States of America. 2006; 103(5):1522–7. Epub 2006/01/25. https://doi.org/10.1073/pnas.
0508815103 PMID: 16432208
45. Campos SK, Chapman JA, Deymier MJ, Bronnimann MP, Ozbun MA. Opposing effects of bacitracin on
human papillomavirus type 16 infection: enhancement of binding and entry and inhibition of endosomal
penetration. Journal of virology. 2012; 86(8):4169–81. Epub 2012/02/22. https://doi.org/10.1128/JVI.
05493-11 PMID: 22345461
46. Stebelska K, Dubielecka PM, Sikorski AF. The effect of PS content on the ability of natural membranes
to fuse with positively charged liposomes and lipoplexes. The Journal of membrane biology. 2005; 206
(3):203–14. Epub 2006/02/04. https://doi.org/10.1007/s00232-005-0793-0 PMID: 16456715
47. Wattiaux R, Jadot M, Warnier-Pirotte MT, Wattiaux-De Coninck S. Cationic lipids destabilize lysosomal
membrane in vitro. FEBS letters. 1997; 417(2):199–202. Epub 1997/12/12. PMID: 9395295
48. Bronnimann MP, Chapman JA, Park CK, Campos SK. A transmembrane domain and GxxxG motifs
within L2 are essential for papillomavirus infection. Journal of virology. 2013; 87(1):464–73. Epub 2012/
10/26. https://doi.org/10.1128/JVI.01539-12 PMID: 23097431
49. DiGiuseppe S, Keiffer TR, Bienkowska-Haba M, Luszczek W, Guion LG, Muller M, et al. Topography of
the Human Papillomavirus Minor Capsid Protein L2 during Vesicular Trafficking of Infectious Entry.
Journal of virology. 2015; 89(20):10442–52. Epub 2015/08/08. https://doi.org/10.1128/JVI.01588-15
PMID: 26246568
50. Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, et al. Mitosis-specific phosphor-
ylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in
an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997; 106
(6):348–60. Epub 1998/01/10. PMID: 9362543
51. Van Hooser A, Goodrich DW, Allis CD, Brinkley BR, Mancini MA. Histone H3 phosphorylation is
required for the initiation, but not maintenance, of mammalian chromosome condensation. Journal of
cell science. 1998; 111 (Pt 23):3497–506. Epub 1998/11/13.
52. Colanzi A, Corda D. Mitosis controls the Golgi and the Golgi controls mitosis. Current opinion in cell biol-
ogy. 2007; 19(4):386–93. Epub 2007/08/11. https://doi.org/10.1016/j.ceb.2007.06.002 PMID:
17689238
53. Colanzi A, Hidalgo Carcedo C, Persico A, Cericola C, Turacchio G, Bonazzi M, et al. The Golgi mitotic
checkpoint is controlled by BARS-dependent fission of the Golgi ribbon into separate stacks in G2. The
EMBO journal. 2007; 26(10):2465–76. Epub 2007/04/14. https://doi.org/10.1038/sj.emboj.7601686
PMID: 17431394
54. Marsh MP, Campos SK, Baker ML, Chen CY, Chiu W, Barry MA. Cryoelectron microscopy of protein
IX-modified adenoviruses suggests a new position for the C terminus of protein IX. Journal of virol-
ogy. 2006; 80(23):11881–6. Epub 2006/09/22. https://doi.org/10.1128/JVI.01471-06 PMID:
16987967
55. Cervigni RI, Bonavita R, Barretta ML, Spano D, Ayala I, Nakamura N, et al. JNK2 controls fragmenta-
tion of the Golgi complex and the G2/M transition through phosphorylation of GRASP65. Journal of
cell science. 2015; 128(12):2249–60. Epub 2015/05/08. https://doi.org/10.1242/jcs.164871 PMID:
25948586
56. Sutterlin C, Hsu P, Mallabiabarrena A, Malhotra V. Fragmentation and dispersal of the pericentriolar
Golgi complex is required for entry into mitosis in mammalian cells. Cell. 2002; 109(3):359–69. Epub
2002/05/23. PMID: 12015985
57. Corda D, Barretta ML, Cervigni RI, Colanzi A. Golgi complex fragmentation in G2/M transition: An
organelle-based cell-cycle checkpoint. IUBMB life. 2012; 64(8):661–70. Epub 2012/06/26. https://doi.
org/10.1002/iub.1054 PMID: 22730233
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 28 / 29

58. Valente C, Colanzi A. Mechanisms and Regulation of the Mitotic Inheritance of the Golgi Complex.
Frontiers in cell and developmental biology. 2015; 3:79. Epub 2016/01/07. https://doi.org/10.3389/fcell.
2015.00079 PMID: 26734607
59. Champion L, Linder MI, Kutay U. Cellular Reorganization during Mitotic Entry. Trends in cell biology.
2016. Epub 2016/08/17.
60. Guttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, et al. NES consensus redefined by
structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nature structural & molecu-
lar biology. 2010; 17(11):1367–76. Epub 2010/10/26.
61. Jabbour M, Campbell EM, Fares H, Lybarger L. Discrete domains of MARCH1 mediate its localization,
functional interactions, and posttranscriptional control of expression. Journal of immunology. 2009; 183
(10):6500–12. Epub 2009/11/03.
62. Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and
interacting proteins in mammalian cells. The Journal of cell biology. 2012; 196(6):801–10. Epub 2012/
03/14. https://doi.org/10.1083/jcb.201112098 PMID: 22412018
63. Buck CB, Pastrana DV, Lowy DR, Schiller JT. Efficient intracellular assembly of papillomaviral vectors.
Journal of virology. 2004; 78(2):751–7. Epub 2003/12/25. https://doi.org/10.1128/JVI.78.2.751-757.
2004 PMID: 14694107
64. Gordon VM, Klimpel KR, Arora N, Henderson MA, Leppla SH. Proteolytic activation of bacterial toxins
by eukaryotic cells is performed by furin and by additional cellular proteases. Infection and immunity.
1995; 63(1):82–7. Epub 1995/01/01. PMID: 7806387
65. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratiniza-
tion in a spontaneously immortalized aneuploid human keratinocyte cell line. The Journal of cell biology.
1988; 106(3):761–71. Epub 1988/03/01. PMID: 2450098
66. Campos SK, Ozbun MA. Two highly conserved cysteine residues in HPV16 L2 form an intramolecular
disulfide bond and are critical for infectivity in human keratinocytes. PloS one. 2009; 4(2):e4463. Epub
2009/02/14. https://doi.org/10.1371/journal.pone.0004463 PMID: 19214230
67. Buck CB, Thompson CD, Pang YY, Lowy DR, Schiller JT. Maturation of papillomavirus capsids. Journal
of virology. 2005; 79(5):2839–46. Epub 2005/02/15. https://doi.org/10.1128/JVI.79.5.2839-2846.2005
PMID: 15709003
HPV L2 translocation requires mitosis
PLOS Pathogens | https://doi.org/10.1371/journal.ppat.1006200 May 2, 2017 29 / 29