unidad 2 genoma
TRANSCRIPT

UNIVERSIDAD NACIONAL DE CHIMBORAZO
FACULTAD CIENCAS DE LA SALUDESCUELA DE MEDICINA
CÁTEDRA DE GENÉTICA MÉDICA
OCTAVO SEMESTRE “B”DRA. FERNANDA
ESCOBAR
EL GENOMA HUMANO: ESTRUCTURA Y FUNCIÓN DE
LOS GENES

MOSAICISMO

Es la presencia en UN individuo o un tejido
Debe dar al menos dos líneas celulares (genéticamente diferentes)
“PERO” proceden de un único cigoto
• Pueden originar clones de células genéticamente diferentes a partir a partir del cigoto original
• Debido a que una vez iniciada la mutación esta se da en toda la descendencia de la misma
MUTACIONES(PRE Y POSTNATAL)

Respecto a las mutaciones en genes únicos (células somática; células de la línea germinal):
• Manifestaciones cutáneas no son uniformes
• Aparecen de una manera parcheada
NEUROFIBROMATOSIS SEGMENTARIA
• En dos o mas hijos de progenitores infectados
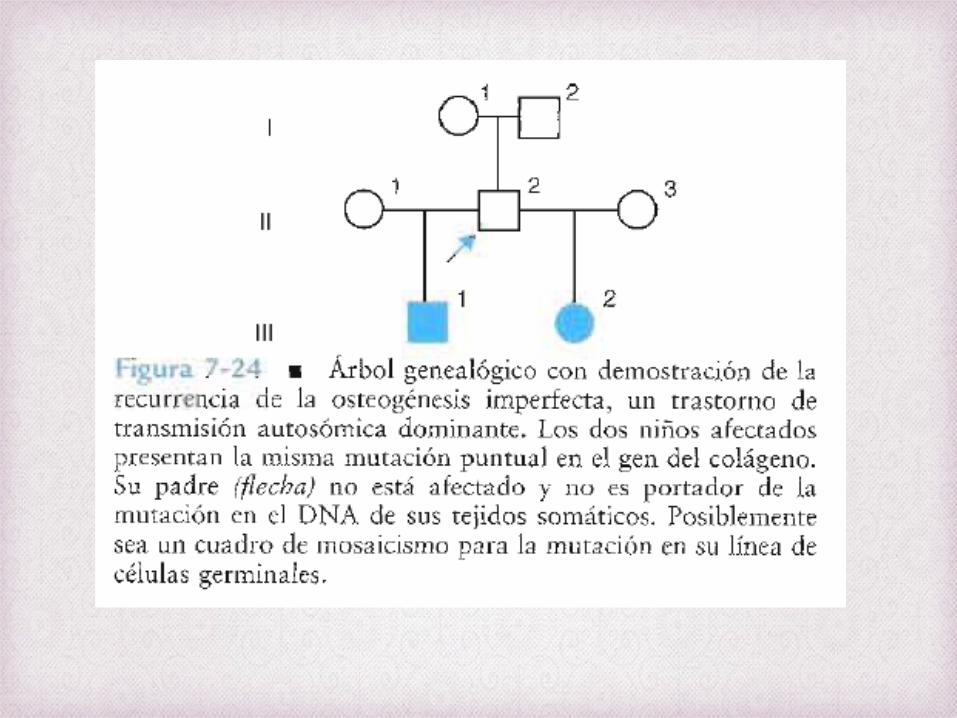
OSTEOGENESIS IMPERFECTA

Un individuo con MOSAICISMO
Puede estar presente en
algunos tejidos
Pero NO, en los gametos
(mosaicismo somático puro)
Se limita al linaje de células de los
gametos sin afectación
Puede estar presentes en las
células somáticas como en la de la línea germinativa
Según el momento en el que se de en el
proceso embriológico
TEORICAMENTE:
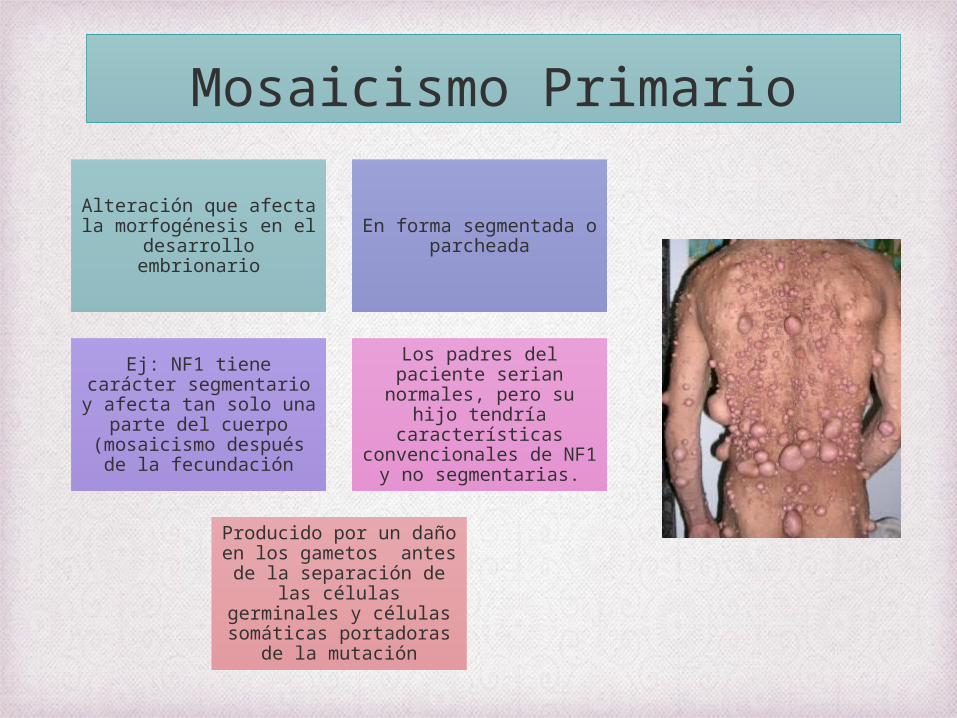
Mosaicismo Primario
Alteración que afecta la morfogénesis en el
desarrollo embrionario
En forma segmentada o parcheada
Ej: NF1 tiene carácter segmentario y afecta tan solo una parte del cuerpo (mosaicismo después de
la fecundación
Los padres del paciente serian normales, pero su
hijo tendría características
convencionales de NF1 y no segmentarias.
Producido por un daño en los gametos antes de la separación de las células
germinales y células somáticas portadoras de
la mutación

Mosaicismo en las Células Germinales
La probabilidad de que un trastorno
autosómico ligado al cromosoma X es baja
debido a que la aparición de
mutaciones nuevas es infrecuente
Sin embargo, hay casos de progenitores con fenotipos normales y
negativos a las pruebas moleculares para el
estado de portador con mas de un hijo con EAD
X.
Ej; Osteogenesis imperfecta con
mutaciones en los genes del colágeno tipo I ,
Hemofilia A y B, y DMD. Donde las madres no
presentan mutación en tejidos somáticos sino en sus células germinales.

DISOMIA UNIPARENTAL
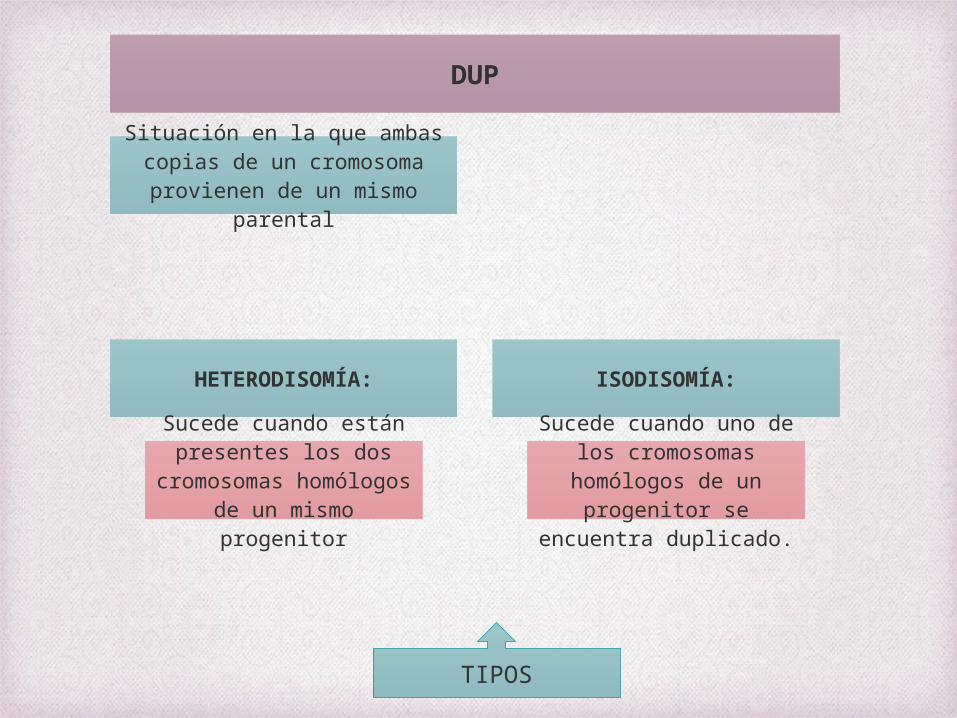
DUP
Situación en la que ambas copias de un cromosoma provienen de un mismo
parental
HETERODISOMÍA:
Sucede cuando están presentes los dos
cromosomas homólogos de un mismo progenitor
ISODISOMÍA:
Sucede cuando uno de los cromosomas homólogos de un
progenitor se encuentra duplicado.
TIPOS

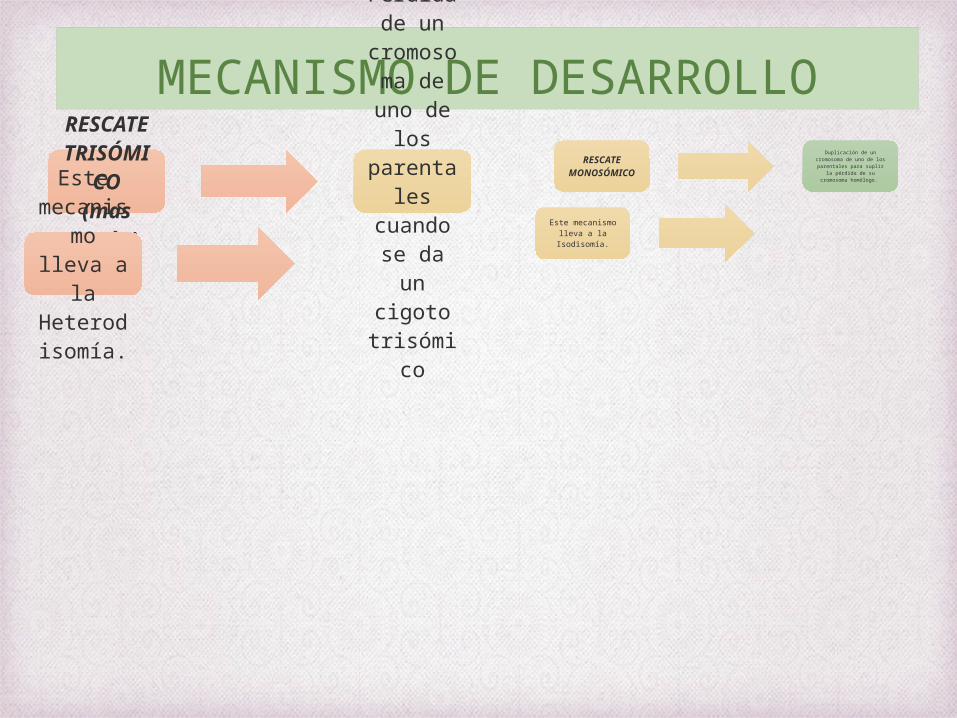
MECANISMO DE DESARROLLORESCAT
E TRISÓMICO(mas
común)
Pérdida de un
cromosoma de uno de
los parental
es cuando se da un cigoto
trisómico
Este mecanismo lleva
a la Heterodisomía.
RESCATE MONOSÓMI
CO
Duplicación de un cromosoma de uno de
los parentales para suplir la pérdida de su cromosoma homólogo.
Este mecanismo lleva a la
Isodisomía.

CONSECUENCIAS DE LA DUP
La Disomía Uniparental afecta a los individuos debido a los distintos patrones de metilación de los cromosomas paternos y maternos.
Algunos genes se encuentran silenciados en los cromosomas maternos y otros en los cromosomas paternos.
El poseer un gen silenciado en ambos alelos generados por la Disomía Uniparental causa problemas de salud y desarrollo.

SINDROMES

IMPRONTA GENÓMICA• Puede definirse como la expresión selectiva
de un gen según el origen parental del alelo (paterno o materno).
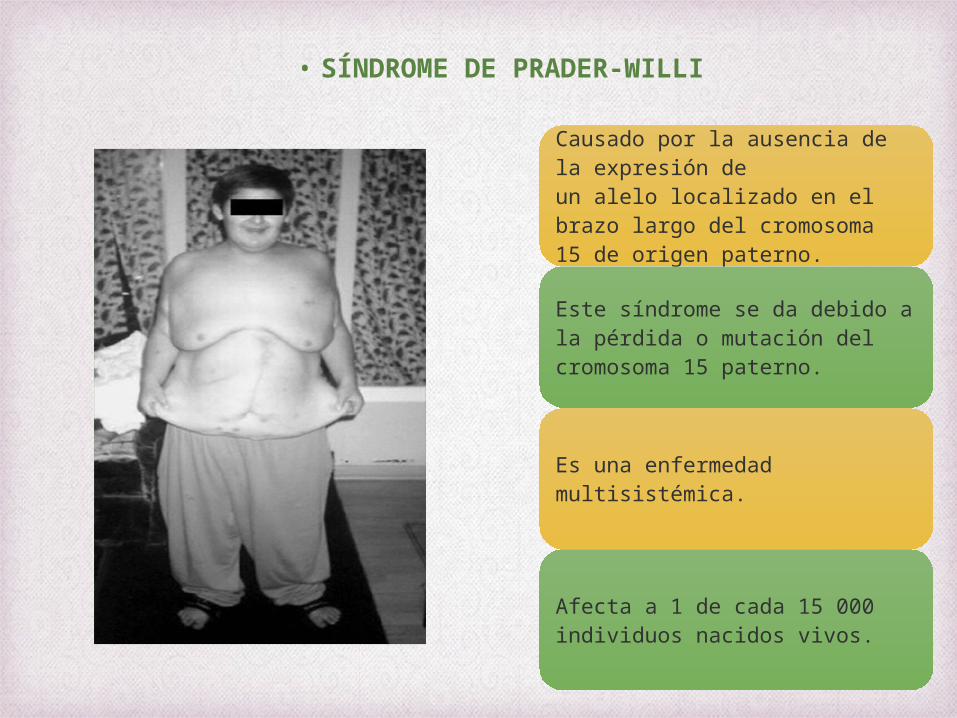
• SÍNDROME DE PRADER-WILLI
Causado por la ausencia de la expresión de un alelo localizado en el brazo largo del cromosoma 15 de origen paterno.
Este síndrome se da debido a la pérdida o mutación del cromosoma 15 paterno.
Es una enfermedad multisistémica.
Afecta a 1 de cada 15 000 individuos nacidos vivos.
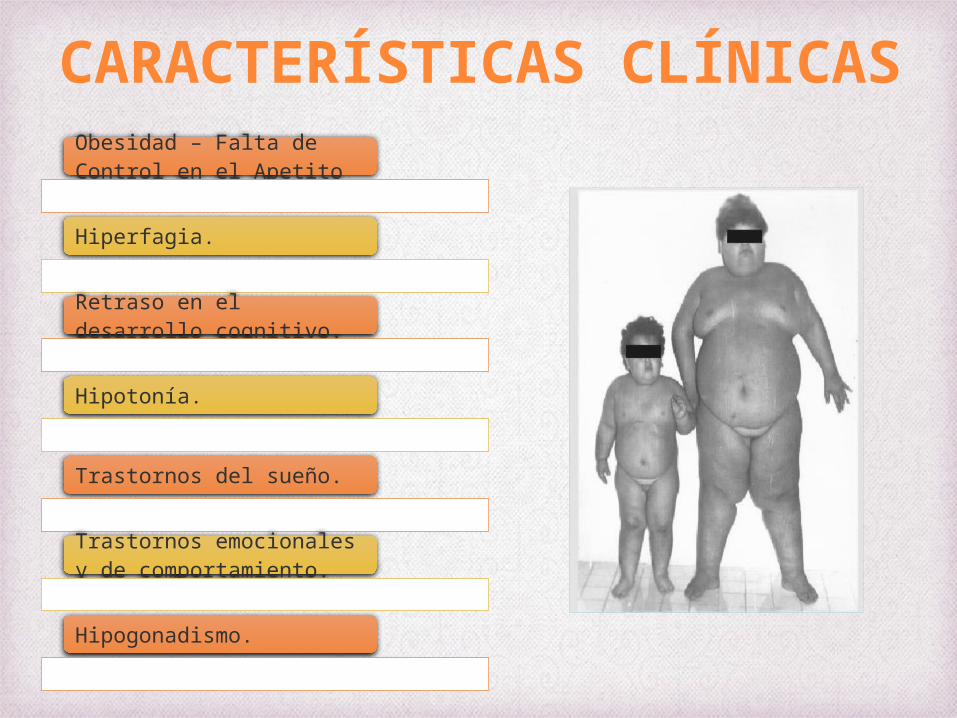
Obesidad – Falta de Control en el Apetito
Hiperfagia.
Retraso en el desarrollo cognitivo.
Hipotonía.
Trastornos del sueño.
Trastornos emocionales y de comportamiento.
Hipogonadismo.
CARACTERÍSTICAS CLÍNICAS

SÍNDROME DE ANGELMAN
Causado por una falla en la expresión de los genes localizados en 15q11-q13 (Mismos que causan el síndrome de Prader-Willi).
Este síndrome se da debido a la pérdida o mutación del cromosoma 15 materno.
Se presenta en 1 de cada 15 000 – 300 000 individuos.
Solo el 4% de los casos se deben a una Disomía Uniparental, siendo las grandes
deleciones las responsables del 70%-75% de los casos.

CARACTERÍSTICAS CLÍNICASRetraso en el desarrollo cognitivo. Déficit de atención.
Problemas lingüísticos o ausencia del habla.
Baja receptividad comunicativa.
Problemas en la coordinación motriz. Hipermotricidad. Ataxia
Aparente estado de alegría permanente.
Lengua - Mandíbula prominente. Babeo frecuente,

CLONACIÓN
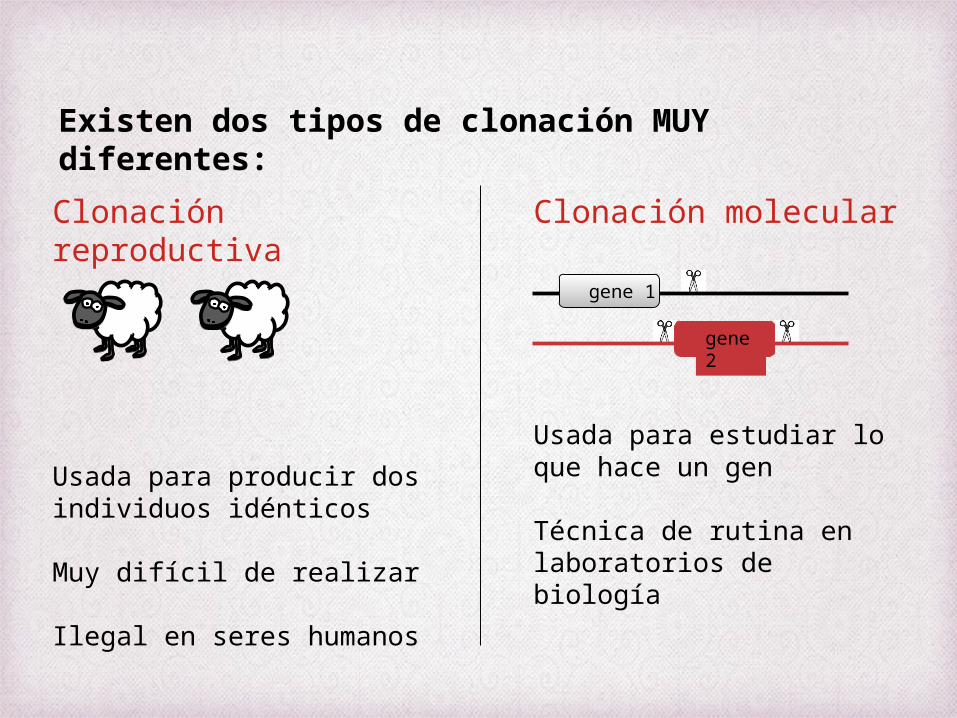
Existen dos tipos de clonación MUY diferentes:
Clonación reproductiva
Usada para producir dos individuos idénticos
Muy difícil de realizar
Ilegal en seres humanos
Clonación molecular
Usada para estudiar lo que hace un gen
Técnica de rutina en laboratorios de biología
gene 1
gene 2

Clonación reproductiva
sacar el núcleo y coger el resto de la célula
óvulo
tomar el núcleo(contiene el ADN)
célula del cuerpo
ClonarIdéntico al individuo que donó el núcleo
La oveja Dolly

Principios de Clonación Molecular
1) Sacar el ADN del núcleo
célula 1 célula 2
gen 1 gen 2
Sacar el ADN de la célula.
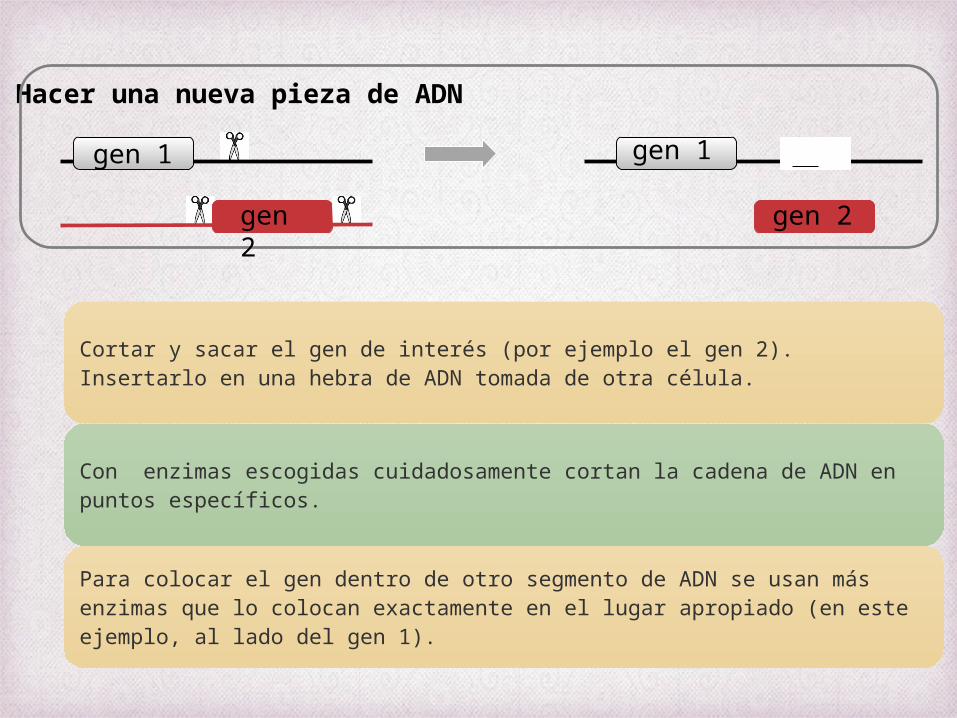
Cortar y sacar el gen de interés (por ejemplo el gen 2). Insertarlo en una hebra de ADN tomada de otra célula.
Con enzimas escogidas cuidadosamente cortan la cadena de ADN en puntos específicos.
Para colocar el gen dentro de otro segmento de ADN se usan más enzimas que lo colocan exactamente en el lugar apropiado (en este ejemplo, al lado del gen 1).
gen 1
gen 2
2) Hacer una nueva pieza de ADN
gen 1
gen 2
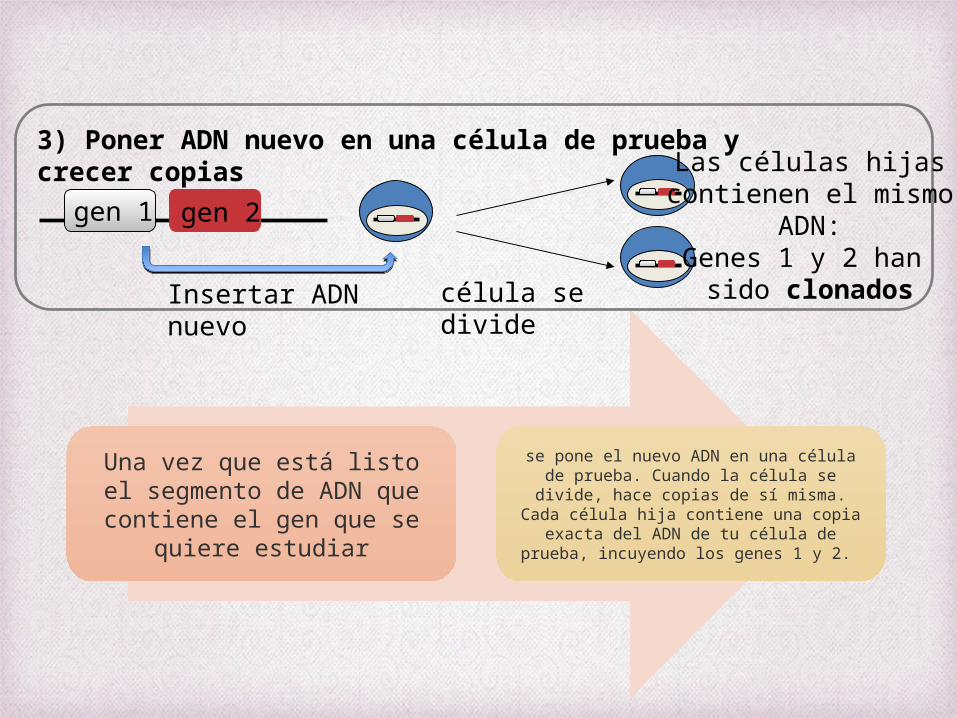
Una vez que está listo el segmento de ADN que
contiene el gen que se quiere estudiar
se pone el nuevo ADN en una célula de prueba. Cuando la célula se divide, hace copias de sí misma. Cada célula
hija contiene una copia exacta del ADN de tu célula de prueba, incuyendo los genes 1 y 2.
3) Poner ADN nuevo en una célula de prueba y crecer copias
gen 1
célula se divide
Las células hijascontienen el mismo
ADN:Genes 1 y 2 han
sido clonados
gen 2
Insertar ADN nuevo

Aplicaciones de la clonación molecular:
embrión de ratón normal
sin el gen A
Quitar un gen para ver si algo Funciona distinto
Pérdida de función
Gen involucrado en coloración del ojo
ojo
Pérdida de función (conocida como “bloqueo de genes”):
una técnica común que ha sido muy útil para que los
científicos entiendan cómo
genes particulares están involucrados en el desarrollo de
enfermedades.
Se elimina o bloquea un gen para que deje de funcionar y los científicos observan lo que ocurre.

Gen reporteroAgregar un gen que
muestra cuando otro gen está trabajando
El gen está activo en Las partes azules
Gen reportero o trazador:
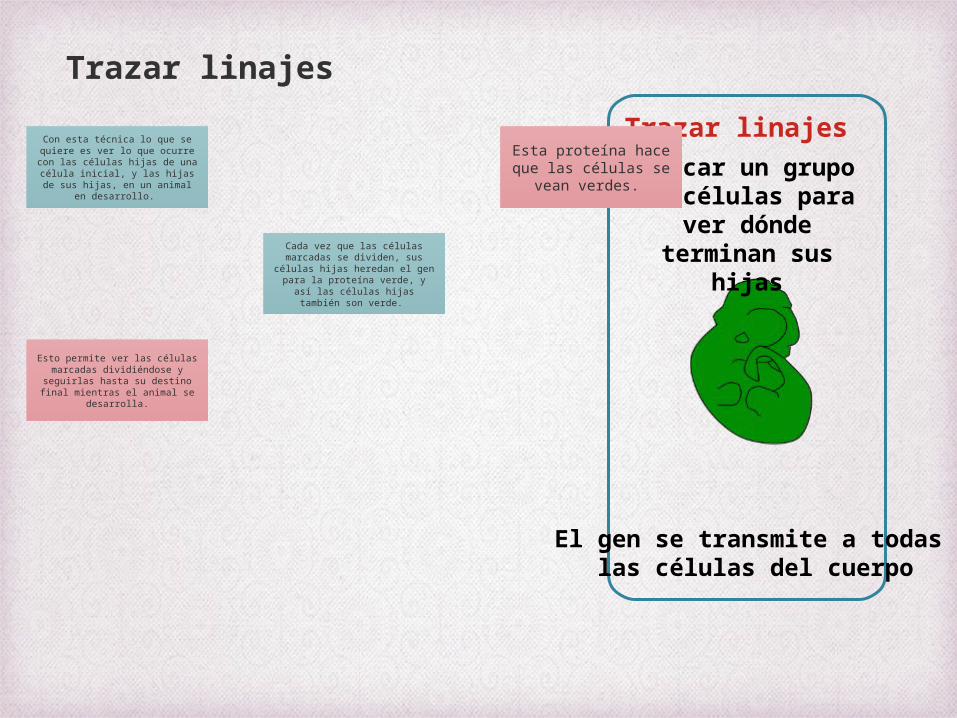
Trazar linajesmarcar un grupo de
células para ver dónde terminan sus hijas
El gen se transmite a todas las células del cuerpo
Trazar linajes
Con esta técnica lo que se quiere es ver lo que ocurre con las células hijas de una célula
inicial, y las hijas de sus hijas, en un animal en desarrollo.
Esta proteína hace que las células se
vean verdes.
Cada vez que las células marcadas se dividen, sus células
hijas heredan el gen para la proteína verde, y así las células
hijas también son verde.
Esto permite ver las células marcadas dividiéndose y
seguirlas hasta su destino final mientras el animal se desarrolla.
