untersuchungen am system nmmo/h o/cellulose · weitaus häufigste organische makromolekül der...
TRANSCRIPT

Untersuchungen am System NMMO/H 2O/Cellulose
Tevfik Cibik

Untersuchungen am System NMMO/H2O/Cellulose
vorgelegt von Diplom-Physiker Tevfik Cibik
aus Tuzluca
Von der Fakultät III - Prozesswissenschaften - der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Naturwissenschaften - Dr. rer. nat. -
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr. H. Schubert Berichter: Prof. Dr. G. Hinrichsen Berichter: Dr. habil. H.-P. Fink Tag der wissenschaftlichen Aussprache: 14.11. 2003
Berlin 2003 D83

Abstract
Die vorliegende Arbeit befasst sich mit der Untersuchung des Zweistoffsystems N-
Methylmorpholin-N-oxid (NMMO)/H2O und des Dreistoffsystems NMMO/H2O/Cellulose
sowie mit der Herstellung und Charakterisierung von faserverstärkten Cellulosefolien.
Das binäre System wird mittels Dynamischer Differenzkalorimetrie und Röntgenweit-
winkel-Diffraktometrie untersucht und dadurch das Schmelzverhalten und die Phasen-
zusammensetzung dieses Systems im festen Zustand als Funktion des NMMO/H2O-
Verhältnisses bestimmt.
Den Schwerpunkt der Arbeit bildet die Untersuchung des ternären Stoffsystems
NMMO/H2O/Cellulose hinsichtlich der Löslichkeit der Cellulose im Lösemittelsystem
NMMO/H2O. Hierzu werden NMMO/H2O/Cellulose-Gemische mit unterschiedlichen
NMMO/H2O-Zusammensetzungen, aber einer konstanten Cellulosekonzentration von
8% präpariert und durch Löseversuche bei verschiedenen Temperaturen und Zeiten
untersucht. Der Quell- bzw. Lösezustand der in diesen Stoffgemischen enthaltenen
Cellulose wird mit Hilfe des DSC, der Polarisationsmikroskopie sowie der Röntgen-
weitwinkel-Diffraktometrie in Abhängigkeit vom NMMO/H2O-Gehalt, von der Tem-
peratur und der Einwirkzeit bestimmt. Zudem wird das Schmelzverhalten des Dreistoff-
systems als Funktion der NMMO/H2O-Zusammensetzung mittels DSC ermittelt und mit
dem des Zweistoffsystems verglichen, um Vorstellungen über die Wechselwirkung von
NMMO mit Wasser und Cellulose abzuleiten.
Die Meßergebnisse über die Löslichkeit der Cellulose im Lösemittelsystem NMMO/H2O
werden in einem Phasendiagramm für das ternäre System zusammengestellt. Beson-
ders wichtig ist dabei der schmale Phasenbereich im Diagramm, in dem die Cellulose
nur partiell gelöst wird.
An Hand des ternären Phasendiagramms, insbesondere unter Nutzung des schmalen
Phasengebiets des unvollständigen Lösens wird ein Laborverfahren vorgestellt, das die
Herstellung eines Verbundes (faserverstärkte Cellulosefolien) aus cellulosischen Fasern
und einer Cellulosematrix aus Aminoxidlösung ermöglicht. Ziel dieser Entwicklung ist
es, in eine Aminoxidlösung, die aus dem Lösemittelsystem NMMO, H2O sowie gelöster
Cellulose besteht, bei leicht veränderten Systemparametern Cellulosefasern hinzu-
zusetzen, die dann nicht mehr aufgelöst werden und nach dem Fällprozeß als Verstär-
kungsfasern erhalten bleiben.
Die mechanischen Eigenschaften (Zugfestigkeit, E-Modul) der hergestellten Cellulose-
folien werden in Zugversuchen bestimmt sowie deren morphologische Struktur mit
Hilfe der Elektronenmikroskopie charakterisiert.

Inhaltsverzeichnis
1 EINLEITUNG UND AUFGABENSTELLUNG .............................................................................. 1
2 GRUNDLAGEN ........................................................................................................................ 4
2.1 CELLULOSE.............................................................................................................................. 4
2.1.1 MOLEKULARE STRUKTUR DER CELLULOSE................................................................................ 5
2.1.2 KRISTALLSTRUKTUR DER CELLULOSE ........................................................................................ 7
2.1.3 ÜBERMOLEKULARE STRUKTUR DER NATIVEN CELLULOSE ........................................................ 8
2.2 CELLULOSELÖSEMITTEL....................................................................................................... 10
2.2.1 EINTEILUNG DER CELLULOSELÖSEMITTEL ............................................................................... 11
2.2.2 N-METHYLMORPHOLIN-N-OXID.............................................................................................. 13
2.2.3 MODELLVORSTELLUNGEN ZUM LÖSEMECHANISMUS DER CELLULOSE .................................. 18
2.3 CELLULOSE-REGENERATVERFAHREN.................................................................................. 21
2.3.1 DAS AMINOXIDVERFAHREN ................................................................................................... 21
2.3.2 DAS VISKOSEVERFAHREN ....................................................................................................... 22
2.3.3 DAS CARBAMATVERFAHREN.................................................................................................. 23
2.4 THERMODYNAMIK VON POLYMERSYSTEMEN ................................................................. 24
2.4.1 STOFFSYSTEME....................................................................................................................... 24
2.4.2 PHASEN .................................................................................................................................. 24
2.4.3 HOMOGENE UND HETEROGENE STOFFGEMISCHE ................................................................. 25
2.4.4 KRISTALLISATIONS- UND SCHMELZVERHALTEN VON POLYMEREN ......................................... 26
3 UNTERSUCHUNGSMETHODEN............................................................................................ 29
3.1 DYNAMISCHE DIFFERENZKALORIMETRIE (DSC) ................................................................ 29
3.1.1 MEßPRINZIP ............................................................................................................................ 29
3.1.2 MEßABLAUF UND EINFLUßFAKTOREN .................................................................................... 30
3.1.3 WÄRMESTROM UND ENTHALPIEÄNDERUNG.......................................................................... 30
3.2 ERGÄNZENDE MEßMETHODEN........................................................................................... 32
3.2.1 RÖNTGENBEUGUNG............................................................................................................... 32
3.2.2 LICHT- UND ELEKTRONENMIKROSKOPIE, REFRAKTOMETRIE UND MECHANISCHE PRÜFUNG . 33

4 UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ....................................... 34
4.1 DSC-MESSUNGEN AM SYSTEM NMMO/H2O...................................................................... 34
4.1.1 PROBENMATERIAL, PRÄPARATION UND VERSUCHSDURCHFÜHRUNG.................................... 35
4.1.2 AUSWERTUNG UND ERGEBNISSE ........................................................................................... 37
4.1.3 DISKUSSSION.......................................................................................................................... 41
4.2 ERGÄNZENDE EXPERIMENTE MITTELS RÖNTGENBEUGUNG ............................................ 43
4.2.1 EXPERIMENTELLES ZU DEN RÖNTGENWEITWINKELUNTERSUCHUNGEN ................................. 43
4.2.2 AUSWERTUNG UND ERGEBNISSE ........................................................................................... 43
4.3 DAS PHASENDIAGRAMM FÜR DAS BINÄRE SYSTEM NMMO/H2O .................................. 47
5 UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE................ 48
5.1 DSC-MESSUNGEN AM SYSTEM NMMO/H2O/CELLULOSE ................................................. 48
5.1.1 PROBENMATERIAL, PRÄPARATION UND PROBENBEHANDLUNG............................................. 49
5.1.2 AUSWERTUNG UND ERGEBNISSE ........................................................................................... 50
5.1.3 DISKUSSION............................................................................................................................ 57
5.2 ERGÄNZENDE MESSUNGEN AM TERNÄREN SYSTEM NMMO/H2O/CELLULOSE ............. 60
5.2.1 LICHTMIKROSKOPISCHE UNTERSUCHUNGEN AM SYSTEM NMMO/H2O/CELLULOSE .... 60
5.2.1.1 EXPERIMENTELLES ZU DEN LICHTMIKROSKOPISCHEN UNTERSUCHUNGEN ............................ 60
5.2.1.2 ERGEBNISSE............................................................................................................................ 63
5.2.2 RÖNTGENBEUGUNG AM STOFFSYSTEM NMMO/H2O/CELLULOSE .................................. 68
5.2.2.1 EXPERIMENTELLES ZU DEN RÖNTGENWEITWINKELUNTERSUCHUNGEN ................................. 68
5.2.2.2 ERGEBNISSE............................................................................................................................ 69
5.3 DAS PHASENDIAGRAMM FÜR DAS TERNÄRE SYSTEM NMMO/H2O/CELLULOSE........... 73
5.4 VERGLEICH DER SYSTEME NMMO/H2O - NMMO/H2O/CELLULOSE HINSICHTLICH DER THERMISCHEN EIGENSCHAFTEN......................................................................................... 75
6 HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN............... 80
6.1 EXPERIMENTELLES ............................................................................................................... 81
6.1.1 VERWENDETE MATERIALIEN ................................................................................................... 81
6.1.2 HERSTELLUNG DER CELLULOSELÖSUNG (MATRIXLÖSUNG) .................................................... 81
6.1.3 HERSTELLUNG DER VERBUNDLÖSUNG ................................................................................... 82
6.1.4 EXTRUSION DER VERBUNDLÖSUNGEN ZU CELLULOSEFOLIEN................................................. 84

6.2 ERGEBNISSE .......................................................................................................................... 86
6.2.1 MECHANISCHE EIGENSCHAFTEN DER FASERVERSTÄRKTEN BLASFOLIEN................................ 86
6.2.2 STRUKTURCHARAKTERISIERUNG ............................................................................................ 88
6.3 DISKUSSION.......................................................................................................................... 91
7 ZUSAMMENFASSUNG.......................................................................................................... 93
8 LITERATURVERZEICHNIS ...................................................................................................... 97

EINLEITUNG UND AUFGABENSTELLUNG ___________________________________________________________________________________________________
1
1 Einleitung und Aufgabenstellung
Cellulose ist der Hauptbestandteil der pflanzlichen Zellmembran und damit das
weitaus häufigste organische Makromolekül der Erde. Sie nimmt als mengenmäßig
wichtigstes Biopolymer eine besondere Stellung unter den nachwachsenden Rohstof-
fen ein. Wegen ihrer biologischen Abbaubarkeit, ihres mengenmäßigen Potentials und
aufgrund ihrer physikalisch-chemischen Eigenschaften findet die Cellulose nach wie
vor großes praktisches und wissenschaftliches Interesse. Industriell wird Cellulose als
Ausgangsmaterial für die Herstellung einer Reihe von Celluloseprodukten wie Cellulo-
seregenerate (Cellulose-Regeneratfasern, Folien, Membranen, Wursthüllen und andere
Celluloseformkörper) und Cellulosederivate (Celluloseether, Celluloseester) für unter-
schiedliche Einsatzgebiete genutzt.
Aufgrund ihrer fibrillären Struktur, die durch ausgeprägte zwischenmolekulare Wasser-
stoffbrückenbindungen gekennzeichnet ist, ist die Cellulose weder in Wasser noch in
üblichen organischen Lösemitteln löslich. Sie läßt sich auch nicht zersetzungsfrei
schmelzen. Daher ist zu ihrer technischen Verarbeitung entweder eine chemische Um-
setzung (Derivatisierung) oder die Auflösung in einem geeigneten Lösemittel und eine
anschließende Wiederausfällung (Regenerierung) erforderlich.
Im Laufe der letzten 25 Jahre wurde ein Verfahren zur Auflösung und Verformung von
Cellulose entwickelt, das für die industrielle Nutzung besonders aussichtsreich und
interessant ist. Es ist das Aminoxidverfahren, das auch unter dem Namen Lyocell- oder
NMMO-Verfahren bekannt ist. Dieses neuartige und flexible Verfahren weist gegen-
über dem herkömmlichen Viskoseverfahren in ökologischer und wirtschaftlicher Hin-
sicht Vorteile auf und basiert auf dem Lösemittelsystem N-Methylmorpholin-N-oxid
(NMMO).
Das Besondere an diesem Lösemittel ist, daß es Cellulose direkt, d. h. ohne Derivati-
sierung, ohne Komplexierung und ohne spezielle Aktivierung physikalisch lösen kann.
Nach seinem Einsatz im Verfahrensprozeß kann das Lösemittel NMMO vollständig
wiedergewonnen und zur Lösungsherstellung wieder verwendet werden. Das Amin-
oxidverfahren, das inzwischen technisch gereift ist, stellt die Grundlage für einen
Entwicklungssprung der Celluloseverarbeitung dar und bietet damit Chancen für die
Herstellung neuer cellulosischer Produkte, wie z. B. faserverstärkte Verbundwerkstoffe
aus cellulosischem Material.

EINLEITUNG UND AUFGABENSTELLUNG ___________________________________________________________________________________________________
2
Die mechanischen Eigenschaften der Celluloseprodukte sind für die Anwendung
substantiell und in erster Linie maßgeblich für die Aussichten dieser Produkte auf dem
Markt.
In der vorliegenden Arbeit war eine Methode zu erarbeiten, die eine Herstellung eines
Verbundes aus cellulosischen Fasern und einer Cellulosematrix aus Aminoxidlösung
ermöglicht. Ziel dieser Entwicklung war es, in eine Cellulosematrix, bestehend aus
Aminoxid, H2O und gelöster Cellulose, weitere unaufgelöste Cellulosefasern einzu-
arbeiten und hierdurch eine stabile und weiterverarbeitbare Verbundlösung zu präpa-
rieren. In einem zweiten Schritt sollte aus dieser Verbundlösung unter Anwendung des
Aminoxid- und Blasextrusionsverfahrens ein cellulosischer Verbundwerkstoff (faserver-
stärkte Celluloseblasfolien) hergestellt werden. Durch die Einarbeitung der hochfesten
cellulosischen Fasern in die Cellulosematrix sollte eine Verbesserung der mechanischen
Eigenschaften (Zugfestigkeit, E-Modul) der Cellulosefolien erreicht werden. Deshalb
mußte sichergestellt werden, daß die zusätzlich eingebrachten Cellulosefasern durch
das in der Matrixlösung befindliche Aminoxid nicht aufgelöst werden und eine Verstär-
kung der Matrix verursachen. Um aber keine zusätzlichen Probleme hinsichtlich der
Faser-Matrix-Haftung zu bekommen, sollten die Cellulosefasern in der Matrix jedoch
"angelöst", d. h. geringfügig gelöst werden. Hierdurch sollte beim Verarbeitungs-
prozeß (Koagulation) eine feste Einbettung der Cellulosefasern in die Matrix herbei-
geführt werden.
Ein wesentlicher Grund, für die Herstellung eines solchen Verbundes ausschließlich cel-
lulosisches Material zu verwenden, liegt in der Absicht, diesen Verbundwerkstoff unter
anderem als Verpackungsmaterial für Lebensmittel, z. B. als Wursthüllen, zu nutzen.
Für die Realisierung dieser Überlegungen waren grundlegende thermische Unter-
suchungen am Zweistoffsystem NMMO/H2O und am Dreistoffsystem NMMO/H2O/-
Cellulose notwendig. Die Untersuchungen hatten das Ziel, für die Aufstellung eines
Phasendiagramms für das Grundsystem NMMO/H2O/Cellulose die nötigen Erkennt-
nisse und Daten zu erhalten. Im Mittelpunkt stand dabei die Bestimmung des Einflus-
ses der Temperatur, der Zeit und des NMMO/H2O-Verhältnisses auf die Löslichkeit von
Cellulose im System NMMO/H2O. Das aufgestellte Phasendiagramm für das Dreistoff-
system sollte schließlich die quantitativen Bedingungen für eine Quellung, eine teil-
weise sowie eine vollständige Auflösung der Cellulose im Lösemittelsystem NMMO/-
H2O beinhalten.

EINLEITUNG UND AUFGABENSTELLUNG ___________________________________________________________________________________________________
3
Ein weiterer Schwerpunkt dieser Arbeit war die differentialkalorimetrische und rönt-
genographische Untersuchung des Zweistoffsystems NMMO/H2O. Der Zweck dieser
Untersuchungen bestand einmal darin, die kristallinen Hydrate, die NMMO mit Wasser
bildet, in Abhängigkeit vom NMMO/H2O-Verhältnis zu bestimmen und ein Phasendia-
gramm für das Zweistoffsystem NMMO/H2O zu konstruieren. Zum anderen sollten die
thermischen Eigenschaften des Zweistoffsystems NMMO/H2O mit denen des Dreistoff-
systems NMMO/H2O/Cellulose verglichen werden, um Aussagen über die im Löse-
prozeß erfolgte Wechselwirkung zwischen den Molekülen des Lösemittelsystems
NMMO/H2O und den Cellulosemolekülen zu treffen. Zudem sollte der Vergleich dazu
genutzt werden, den Löseprozeß bzw. Lösemechanismus der Cellulose im Lösemittel-
system NMMO/H2O zu interpretieren.
Eine zentrale Aufgabe der vorliegenden Arbeit war es, insbesondere den Bereich im
Phasendiagramm zu untersuchen, in dem die Cellulose zwar "angelöst", aber nicht
komplett gelöst wird. Die Bestimmung dieses kleinen Phasengebiets stellte den Aus-
gangspunkt für die Präparation der oben erwähnten Verbundlösung und damit den
Schlüssel für die Herstellung von faserverstärkten Blasfolien aus rein cellulosischem
Material dar.

GRUNDLAGEN ___________________________________________________________________________________________________
4
2 Grundlagen
2.1 Cellulose
Cellulose ist die häufigste, natürlich vorkommende makromolekulare Verbindung der
Erde. Sie ist Hauptbestandteil nahezu aller pflanzlichen Zellwände. Als Gerüstsubstanz
der Pflanzen stellt sie das mengenmäßig wichtigste Naturprodukt und den bedeu-
tendsten nachwachsenden Rohstoff dar. Sie besitzt einen breiten Anwendungsbereich.
So wird Cellulose als Ausgangsmaterial für zahlreiche industrielle Cellulose-Erzeugnisse
wie Textilfasern, Folien, Membranen, Papier und andere Formkörper eingesetzt. Diese
Produkte haben vor allem den Vorteil, daß sie umweltfreundlich und für ihre Einsatz-
zwecke hervorragend geeignet sind. Die Cellulose erlangt wegen ihrer biologischen
Abbaubarkeit und auf Grund ihrer physikalisch-chemischen Eigenschaften immer mehr
Interesse für bestimmte Spezialanwendungen (z. B. Cellulose-Derivate, faserverstärkte
Verbundwerkstoffe für die Automobilindustrie) [1, 2, 3a].
Die wichtigsten Rohstoffquellen der Cellulose sind Baumwolle, Bastfasern (Ramie,
Flachs, Hanf, Jute) und Holz. Der Gehalt an Cellulose beträgt in Baumwollfasern bis zu
95 %, in Bastpflanzen zwischen 60 - 80 % und in Holz in der Größenordnung von 40
bis 50 % [3b]. Neben Pflanzen produzieren aber auch niedere Organismen wie
Bakterien und Algen hochreine Cellulose.
Natürlich vorkommende Cellulose hat eine variierende Kettenlänge mit Durchschnitts-
Polymerisationsgraden (DP) zwischen 1000 und 15000. Der DP einer Celluloseprobe
hängt von ihrer Herkunft und ihrer Vorbehandlung (Aufschlußverfahren) ab. Baum-
woll-Cellulose weist einen DP von 1.000 - 3.000 [4], Regeneratcellulose einen DP
zwischen 250 - 500 auf [5].
Im Unterschied zu anderen Polysacchariden ist die Cellulose trotz ihres hydrophilen
Charakters sowohl in Wasser als auch in anderen üblichen organischen Lösemitteln
wie Ethanol, Dimethylsulfoxid und Tetrahydrofuran nicht löslich. Sie ist zudem nicht
zersetzungsfrei schmelzbar und kann deshalb auch nicht mit konventionellen thermo-
plastischen Verfahren verarbeitet werden. Daher ist zu ihrer technischen Verarbeitung
entweder eine chemische Umwandlung (Derivatisierung) oder ein geeignetes Löse-
mittel erforderlich [6].

GRUNDLAGEN ___________________________________________________________________________________________________
5
Ein großer Vorteil der Cellulose im Gegensatz zu anderen natürlichen Polymeren ist,
daß sie eine relativ gut untersuchte Substanz ist. Seit den ersten brauchbaren Studien
von Payen im Jahre 1838 über Cellulose [7] ist sie Gegenstand von zahlreichen wissen-
schaftlichen Untersuchungen. Aktuelle Arbeiten, die sich direkt oder indirekt mit der
Cellulose befassen, können als Indiz für das andauernde Interesse angesehen werden.
2.1.1 Molekulare Struktur der Cellulose
Cellulose hat die chemische Zusammensetzung (C6H10O5)n, d. h. der Kohlenstoffanteil
in der Cellulose beträgt etwa 44,4 %, der Wasserstoffanteil ca. 6,2 % und der Sauer-
stoffanteil annähernd 49,3 % (Monomereinheit, M = 162,14 g/mol). Sie ist aus D-
Glucopyranose-Einheiten aufgebaut, die über ß(1-4)-Bindungen miteinander verknüpft
sind [1].
Der chemische Aufbau (Konstitution) des Cellulosemolküls wurde 1928 von Haworth
[8] und Freudenberg [9] aufgeklärt. Die Polymernatur des Cellulosmoleküls dagegen
wurde durch die grundlegenden Arbeiten H. Staudingers [10] bekannt.
Cellulose gehört zu den unverzweigten Polysacchariden. Ihre kleinste Wiederholungs-
einheit innerhalb der Kette bildet das Disaccharid Cellobiose, die durch die Verbindung
zweier ß-Glucose-Einheiten über eine Sauerstoffbrücke entsteht (Abb. 2-1). Die Gluco-
pyranose-Ringe der Cellulosekette befinden sich, wie Infrarot-, Röntgenstruktur- und
Kernresonanzuntersuchungen zeigen, in der energetisch günstigen 4C1-Sesselkonfor-
mation. Jede Monomereinheit trägt an den C-2 und C-3 Atomen je eine sekundäre
und am C-6 Atom eine primäre Hydroxylgruppe. Die Hydroxylgruppen bzw. die
CH2OH-Gruppen sind parallel und die H-Atome senkrecht zur Ebene des Glucose-
Ringes angeordnet [11].
Abb. 2-1 Ausschnitt aus einer Cellulosekette

GRUNDLAGEN ___________________________________________________________________________________________________
6
Neben den ß(1-4)-Bindungen zwischen benachbarten Glucose-Einheiten existieren
intramolekulare Wasserstoffbrückenbindungen innerhalb des gleichen Moleküls. Diese
Art von H-Bindung kommt einmal zwischen der Hydroxylgruppe an C-2 und der
Hydroxylgruppe an C-6 des angrenzenden Glucopyranose-Ringes und zum anderen
zwischen der Hydroxylgruppe an C-3 und dem Ringsauerstoff eines benachbarten
Glucopyranose-Ringes vor (Abb. 2-2). Durch diese Bindungen wird eine Versteifung
der einzelnen Celluloseketten verursacht [12, 13].
Darüber hinaus sind intermolekulare H-Brücken zwischen benachbarten Cellulose-
ketten möglich. Derartige H-Bindungen, die den Zusammenhalt der Celluloseketten
sichern, können zwischen der Hydroxylgruppe an C-6 der einen Kette und der Hy-
droxylgruppe an C-3 des benachbarten Cellulosemoleküls ausgebildet werden [14].
Abb. 2-2 Wasserstoffbrücken-Bindungssystem der Cellulose Die Gesamtheit der H-Brückenbindungen bildet in der Cellulose ein dreidimensionales
Netz, das für die Ausbildung übermolekularer Strukturen und somit für den fibrillären
Aufbau der Cellulose verantwortlich ist. Die physikalischen Eigenschaften, wie z. B. die
Festigkeit der Cellulose, werden durch das System der Wasserstoffbrückenbindungen
entscheidend mitbeeinflußt.

GRUNDLAGEN ___________________________________________________________________________________________________
7
2.1.2 Kristallstruktur der Cellulose
Röntgenographische Untersuchungen an Celluloseproben ergeben, daß die Cellulose
im festen Zustand aus röntgenamorphen und hochgeordneten, kristallinen Anteilen
besteht. Sie ist demzufolge partiell kristallin und hat einen Kristallanteil von bis zu
60%. Die Gitterstruktur in den kristallinen Regionen der Cellulose kommt durch die
regelmäßige ß-(1-4)-glykosidische Verknüpfung der Glucose-Einheiten und die perio-
dische Ausbildung intra- und intermolekularer Wasserstoffbrücken entlang den Cellu-
loseketten zustande. Neben den kristallinen Bereichen existieren aber auch nicht-
kristalline, d. h. weniger geordnete, amorphe Bereiche (sowie Hohlräume) in der Cellu-
lose [15, 16].
Die Cellulose ist in der Lage, verschiedene Kristallgitter zu bilden. Man unterscheidet
hauptsächlich zwischen vier unterschiedlichen Kristallgitter-Modifikationen, die mit
Cellulose I, II, III und IV bezeichnet werden [17].
Aufgrund von Strukturuntersuchungen an nativer Cellulose, die bis Anfang 1980 von
einigen Arbeitsgruppen durchgeführt wurden, wird die Cellulose I mit dem monokli-
nen Kristallgitter der Raumgruppe P21 dargestellt [14, 15, 18]. Die Cellulosemoleküle
sind in dieser Cellulose-Modifikation parallel zueinander gerichtet [13].
In nativer Cellulose liegt - nach neueren Erkenntnissen - eine Mischung aus den beiden
Cellulose-Modifikationen Iα und Iβ vor [19]. Die Iα-Form besitzt ein triklines, die Iβ-Form
ein monoklines Gitter. Abhängig von der Art und Herkunft der Cellulose überwiegt
entweder der Iα-Anteil oder der Iβ-Anteil. Der Anteil an Iα-Modifikation überwiegt in
Cellulose, die von Algen und Bakterien produziert wird, der Iβ-Anteil in der von höhe-
ren Pflanzen (Baumwolle und Holzcellulose) gebildeten Cellulose.
Die für technische Prozesse und diese Arbeit wichtige Cellulose-Modifikation II erhält
man entweder durch Auflösung von Cellulose I in einem Lösemittel und anschließende
Ausfällung (Regenerierung) oder durch Aufquellung der Cellulose I in konzentrierter
Natriumhydroxidlösung und nachfolgende Entfernung des Quellmittels (Mercerisie-
rung) [14, 20]. Eine derartige Umwandlung von Cellulose I in Cellulose II hat eine Än-
derung im Wasserstoffbrückenbindungs-System zur Folge und kann sich auf das Löse-
verhalten der Cellulose in den verschiedenen Lösemitteln unterschiedlich auswirken
[21, 22]. Dieser Gittertyp unterscheidet sich vom Cellulose-I-Typ außerdem durch den

GRUNDLAGEN ___________________________________________________________________________________________________
8
Richtungssinn benachbarter Celluloseketten und stellt mit seinen antiparallelen Ketten
die thermodynamisch stabilere Modifikation dar [14].
Außer der Cellulose-I- und der Cellulose-II-Modifikation werden in der Literatur die
Kristallgitter-Modifikationen Cellulose III und Cellulose IV beschrieben. Cellulose III ent-
steht, wenn native Cellulose mit flüssigem Ammoniak oder primären aliphatischen
Aminen behandelt und anschließend das zur Umwandlung verwendete Mittel entfernt
wird. Diese Kristallgitterform steht in naher Verwandtschaft zur Cellulose-II-Modifika-
tion [18]. Die Cellulose-IV-Gitterstruktur bildet sich beim Erhitzen von Cellulose in
Glyzerin bei Temperaturen um 200 °C. Sie steht der Cellulose-I-Modifikation nahe [23,
24].
Für die Untersuchungen in dieser Arbeit sind lediglich die Modifikationen I und II von
Bedeutung.
2.1.3 Übermolekulare Struktur der nativen Cellulose
Unter der übermolekularen Struktur soll hier der Feinbau bzw. die innere Struktur der
Cellulose-Mikrofibrillen verstanden werden. Aus diversen elektronenmikroskopischen
Aufnahmen an nativer Cellulose ist bekannt, das die Cellulose in Fibrillen organisiert
vorliegt [25, 26]. Diese sogenannten Mikrofibrillen weisen je nach Celluloseart Längen
bis zu mehreren µm und Querschnittsabmessungen von 10 nm bis 100 nm auf und
werden allgemein als morphologische Grundbaueinheiten angesehen [16].
Über den inneren Aufbau der Cellulose-Mikrofibrillen gibt es unterschiedliche Vorstel-
lungen. Nach einer Auffassung, die sich auf hochauflösende elektronenmikroskopische
Untersuchungen stützt, setzen sich die Mikrofibrillen aus Untereinheiten, den Elemen-
tarfibrillen, zusammen. Diese Elementarfibrillen haben einen regelmäßigen kristallinen
Aufbau, besitzen einheitliche Querschnittsmaße von 3.5 nm x 3.5 nm und werden aus
etwa 30 Celluloseketten gebildet (Einphasenmodell) [26, 27].
Experimentelle Befunde aus EM- und Röntgenbeugungsuntersuchungen an unter-
schiedlichen nativen Cellulosen ergaben jedoch, daß sie (mit Ausnahme der Valonia-
Cellulose) einen merklichen Anteil an nichtkristallinen geordneten Kettensegmenten
aufweisen [28]. Diese Tatsache spricht mehr für ein Zweiphasenmodell der über-
molekularen Struktur der Cellulose. Ein solches Modell, das weitgehend Anerkennung
gefunden hat, ist das sogenannte Fransenfibrillarmodell. Nach dieser Auffassung

GRUNDLAGEN ___________________________________________________________________________________________________
9
bestehen die Mikrofibrillen aus abwechselnd kristallinen und amorphen Regionen, die
von Cellulosemolekülen durchlaufen werden (kristallin-amorphes Zweiphasenmodell).
Über gewisse Segmentlängen legen sich die kettenförmigen Makromoleküle in kristal-
linen Bereichen zusammen und bilden dadurch Elementarkristallite. Wegen ihrer Länge
durchlaufen die Celluloseketten mehrmals kristalline und amorphe Bereiche [29, 30,
31].
Abb. 2-3 Schematische Darstellung der Fransenfibrillarstruktur von Cellulose nach Hearle [32] Die Abb. 2-3 verdeutlicht die übermolekulare Struktur der Cellulose nach dem Fran-
senfibrillarmodell.

GRUNDLAGEN ___________________________________________________________________________________________________
10
2.2 Celluloselösemittel
In der Cellulosekette sind die drei funktionellen Hydroxylgruppen in jeder Anhydro-
glucose-Einheit durch inter- und intramolekulare Wasserstoffbrückenbindungen blok-
kiert. Die Wasserstoffbrücken und andere sekundäre zwischenmolekulare Bindungs-
kräfte, vorwiegend van der Waals-Kräfte, sowie die lineare Struktur der Cellulose-
makromoleküle schaffen die Grundlage für die Ausbildung einer ausgeprägten über-
molekularen Ordnung in der Cellulose. Diese Faktoren (d. h. die Absättigung der freien
OH-Gruppen über H-Brücken, die sekundären, zwischenmolekularen Wechselwirkun-
gen zwischen und innerhalb der Cellulosemoleküle und die damit zusammenhängende
Entstehung geordneter übermolekularer- und morphologischer Strukturen) sind die
Ursache dafür, daß Cellulose in keinem der üblichen Lösemittel löslich ist und sich
auch nicht unzersetzt schmelzen läßt [33, 34]. Deshalb ist für ihre technische Verar-
beitung zu Celluloseverformungsprodukten entweder eine Derivatisierung oder der
Einsatz eines speziellen Lösemittels notwendig. Aber auch zur Durchführung der Cha-
rakterisierung der molekularen Eigenschaften ist ein Lösemittel erforderlich, in der die
Cellulose molekulardispers aufgelöst vorliegen sollte.
Es gibt hauptsächlich drei Möglichkeiten, den nachwachsenden Rohstoff Cellulose zu
Verformungsprodukten (wie z. B. Fasern, Folien) zu verarbeiten:
1) Direkte Überführung der Cellulose in Lösung - ohne Derivatisierung - mit Hilfe
eines Lösemittels, Formgebung und anschließende Trennung der Cellulose vom
Lösemittel (Regenerierung).
2) Überführung der Cellulose in Lösung nach vorheriger oder gleichzeitiger Bildung
eines instabilen Cellulosederivats mittels eines Lösemittelsystems, Formgebung und
Zersetzung zu unsubstituierter Cellulose (z. B. Viskoseverfahren).
3) Überführung der Cellulose in Lösung nach vorheriger oder gleichzeitiger Deriva-
tisierung der Cellulose zu einem stabilen Hauptvalenzderivat in einem geeigneten
Lösemittelsystem. Nach dem Formgebungsprozeß entsteht bei diesem Verfahren -
im Gegensatz zu den anderen beiden - ein Cellulosederivat-Formkörper, der die
chemischen und physikalischen Eigenschaften des Derivats hat (z. B. Cellulose-
acetatfasern).

GRUNDLAGEN ___________________________________________________________________________________________________
11
Abb. 2-4 Verfahren zur Herstellung von Formkörpern aus Cellulose [35] 2.2.1 Einteilung der Celluloselösemittel
Im Laufe der Zeit wurden in der Celluloseforschung verschiedene Lösemittel für die
Cellulose entwickelt, charakterisiert und angewendet [36]. Diese können nach be-
stimmten Kriterien klassifiziert werden. In der Literatur werden Celluloselösemittel
nach wäßrigen und nichtwäßrigen [37], nach der Art und Weise ihrer Wechselwirkung
mit der Cellulose [38], nach derivatisierenden und nichtderivatisierenden [39] sowie
nach umweltbelastenden und umweltfreundlichen [40] Lösemittelsystemen eingeteilt.
Eine Unterscheidung der Lösemittelsysteme vorzugsweise nach derivatisierend und
nichtderivatisierend wirkenden Systemen wird für die vorliegende Arbeit als sinnvoll
und hilfreich angesehen. Aus diesem Grunde sind in der Tab. 2-1 beispielhaft einige
Celluloselösemittel in zwei Klassen aufgeteilt [39, 41].
Unter derivatisierenden Lösemitteln werden solche verstanden, die die Cellulose unter
Bildung instabiler Hauptvalenzverbindungen auflösen und aus denen die Cellulose
durch geeignete Fällmittel wie z. B. Wasser relativ rasch unter Zerfall des Derivats rege-
neriert werden kann. D. h. Systeme, mit denen stabile Hauptvalenzderivate der Cellu-
lose entstehen, werden i. a. nicht zu den Celluloselösemitteln gerechnet [42].
Lösung in "inertem LM" Cellulosederivat
Formgebungsprozeß
Derivatspaltung
Cellulosederivat-Formkörper
Cellulose-Formkörper
Cellulose
Cellulosederivat
1 2 3
FormgebungsprozeßFormgebungsprozeß

GRUNDLAGEN ___________________________________________________________________________________________________
12
Tab. 2-1 Beispiele für Celluloselösemittel
DERIVATISIERENDE SYSTEME
WEITERE MERKMALE
N2O4/DMF (Distickstofftetroxid/Dimethylformamid)
nichtwäßrig/mehrkomponentig
NOHSO4/DMF (Nitrosylhydrogensulfat/Dimethylformamid
nichtwäßrig/mehrkomponentig
(CH3)3SiCl/DMF (Trimethylchlorsilan/Dimethylformamid)
nichtwäßrig/mehrkomponentig
(CH2O)x/DMSO (Paraformaldehyd/Dimethylsulfoxid)
nichtwäßrig/mehrkomponentig
NICHTDERIVATISIERENDE SYSTEME
WEITERE MERKMALE
Cuoxam (Kupferoxidammoniak=Kupfer(II)-Tetraamin-Hydroxid)
wäßrig/mehrkomp./metallbasiert
ZnCl2/H2O (wäßrige Zinkchloridlösung)
wäßrig/zweikomp./metallbasiert
NH3/NH4SCN (Ammoniumthiocyanat in flüssigem Ammoniak)
flüssig/zweikomponentig
DMSO/Methylamin (Dimethylsulfoxid/Methylamin)
nichtwäßrig/zweikomponentig
DMAc/LiCl (Dimethylacetamid/Lithiumchlorid)
nichtwäßrig/mehrkomponentig, organisches LM*
NMMO⋅1H2O (N-Methylmorpholin-N-oxid-Monohydrat
wäßrig/zweikomponentig organisches LM
*LM = Lösemittel
An ein Lösemittel, das industriell für Celluloseauflösung und -verarbeitung verwendet
werden soll, werden eine Reihe von Anforderungen gestellt. Diese sind im einzelnen
[43]:
- Das Lösemittel sollte aus einer oder höchstens zwei Komponenten bestehen, damit
die Rückgewinnung nicht kompliziert und kostenintensiv wird.
- Es muß eine hohe Lösekraft für Cellulose besitzen, um Lösungen mit hoher Cellulose-
konzentration (10 - 15 %) herstellen zu können.
- Eine direkte Auflösung der Cellulose, (d. h. ohne chemische Umsetzung) durch das
Lösemittel ist erwünscht.
- Die Rückgewinnung der Lösemittelbestandteile sollte einfach zu realisieren sein, und
die Rückgewinnungsverluste des Lösemittels sollten so gering wie möglich ausfallen.

GRUNDLAGEN ___________________________________________________________________________________________________
13
- Das Lösemittel sollte außerdem in dem zum Auflösen von Cellulose oder zum
Verarbeiten der Schmelzlösung notwendigen Temperaturbereich chemisch so stabil
sein, daß es für Perioden von mehreren hundert Rückgewinnungszyklen eingesetzt
werden kann.
- Weitere Anforderungen an das Lösemittel sind, daß damit brauchbare Endprodukt-
eigenschaften erreicht werden und im Verfahrensprozeß keine oder nur eine geringe
Umweltbelastung entsteht.
2.2.2 N-Methylmorpholin-N-oxid
Die ersten Arbeiten, die das Ziel hatten, Cellulose mit Hilfe von Aminoxiden zu lösen,
wurden von Graenacher und Sallmann durchgeführt [44]. In ihrer Patentschrift von
1939 schlugen sie verschiedene tertiäre Amine zur Herstellung von Aminoxiden als
Lösemittel für Cellulose vor.
Nach einer Zeitspanne von 30 Jahren meldete Johnson ein Patent an, in dem er eine
Liste von zyklischen, tertiären Aminen zur Herstellung von Aminoxiden als Lösemittel
für mehrere synthetische und natürliche Polymere, darunter auch Cellulose, bekannt-
gab [45]. 1970 wurde erstmalig das N-Methylmorpholin-N-oxid-System als Cellulose-
Lösemittel in einem US-Patent genannt [46].
Seit Anfang der 70er Jahre befaßten sich verschiedene Arbeitsgruppen, insbesondere
eine amerikanische und eine französische, mit Aminoxiden als mögliche Lösemittel für
Cellulose. Sie fanden unabhängig voneinander heraus, daß von den in Frage kommen-
den Aminoxiden das N-Methylmorpholin-N-oxid-System die Kriterien, die sie für ein
optimales Lösemittel aufgestellt hatten, am besten erfüllte [47, 48, 49, 50]. Deshalb
entschieden sich beide Gruppen für das N-Methylmorpholin-N-oxid (NMMO) als Löse-
mittel für Cellulose und stellten im Laufe ihrer Studien wichtige und grundlegende
Daten über Löseverhalten und chemisch-physikalische Eigenschaften von NMMO
bereit [51].
Die Erkenntnis, daß bei höheren Temperaturen und bei längeren Verweilzeiten der
Cellulose in dem Zweistoffgemisch NMMO/H2O eine Zersetzung des Lösemittels und
ein Abbau der Cellulose stattfindet, verzögerte das Bestreben, das NMMO-Verfahren
kommerziell zu nutzen. Schließlich wurden Anfang der 80er Jahre spezielle Verbindun-
gen als Stabilisatoren für Celluloselösungen in Aminoxiden entwickelt, die der Zerset-

GRUNDLAGEN ___________________________________________________________________________________________________
14
zung des NMMO und dem Abbau des Polymerisationsgrades der Cellulose bei hohen
Temperaturen erheblich entgegenwirkten [52]. Damit wurde ein Hinderungsgrund für
eine großtechnische Umsetzung des Aminoxid-Verfahrens behoben.
Die erste NMMO-Versuchsanlage wurde von der Firma Courtaulds in diesem Zeitraum
in Coventry (UK) errichtet. Nach erfolgreichen Entwicklungsarbeiten in der Versuchs-
anlage wurde mit dem Bau einer Aminoxid-Großanlage Anfang 1990 in Mobile
(Alabama, USA) begonnen und um die Mitte der 90er Jahre die Produktion von Cellu-
lose-Regeneratfasern (Tencel) aufgenommen. Auch in Europa wurde in jenem Zeitab-
schnitt (Lenzing AG, Österreich) mit einer großtechnischen Produktion von Regenerat-
cellulose-Fasern (Lyocell) mittels NMMO-Technologie begonnen [51].
In der Vielzahl der zwischen 1977 und 1996 publizierten Patentanmeldungen und
Veröffentlichungen zum Thema spiegelte sich das große Interesse an diesem Verfahren
wider [53]. Dies zeigt auch zugleich, daß sich das Aminoxid-Verfahren als eine alter-
native Technologie zum Viskoseverfahren weltweit allmählich durchsetzen könnte.

GRUNDLAGEN ___________________________________________________________________________________________________
15
Eigenschaften des NMMO
N-Methylmorpholin-N-oxid ist eine farblose, kristalline Substanz, die durch Oxidation
des tertiären Amins N-Methylmorpholin mit Wasserstoffperoxid großtechnisch herge-
stellt wird (z. B. von der BASF AG Ludwigshafen, Huntsman Corporation) [54].
N-Methylmorpholin N-Methylmorpholin-N-oxid Abb. 2-5 Herstellung von N-Methylmorpholin-N-oxid ausgehend von N-Methylmorpholin und
Wasserstoffperoxid
Es verfügt (wegen seines niedrigen Nitrosamingehaltes unterhalb 50 ppb) über
keinerlei toxikologische Eigenschaften [51]. Wasserfreies NMMO hat die chemische
Zusammensetzung C5H11NO2 und besitzt ein monoklines Gitter (Raumgruppe P21) mit
den Gitterkonstanten a = 0,989 nm, b = 0,662 nm, c = 0,511 nm, ß = 111,54 ° [55].
NMMO schmilzt bei 172 °C und zerfällt oberhalb seines Schmelzpunktes explosions-
artig. Durch die Anwesenheit von Schwermetallionen (z. B. Kupfer- und Eisenionen)
oder organischen Reduktionsmitteln, zu denen auch die Cellulose gehört, kann eine
spontane Zersetzung des NMMO auch bei niedrigeren Temperaturen (d. h. ab etwa
125 °C) erfolgen. Dabei fallen als Zerfallsprodukte hauptsächlich N-Methylmorpholin,
Morpholin und Kohlendioxid an, wobei als Nebenprodukte in geringem Ausmaß unter
anderem Formaldehyd, Ameisensäure und N-Formylmorpholin entstehen. Der Zerfall
des NMMO bei hohen Lösetemperaturen läßt sich aber durch geeignete Zusätze in
ausreichendem Maße reduzieren [56].
NMMO ist stark hygroskopisch und in Verbindung mit Wasser (als zweiter Lösemittel-
komponente) besser handhabbar als wasserfreies NMMO. Es bildet zwei stabile,
kristalline Hydrate, und zwar einmal das NMMO-Monohydrat (NMMO-MH), das eine
höhere Lösekraft für Cellulose und deshalb für die technische Nutzung mehr Relevanz
besitzt [50]. Das Molverhältnis von NMMO zu H2O beträgt im NMMO-MH 1:1;
entsprechend enthält es 13,3 Ma% H2O.

GRUNDLAGEN ___________________________________________________________________________________________________
16
Neben dem Monohydrat bildet das NMMO ein zweites kristallines Hydrat, das 2,5-
Hydrat (mit zwei NMMO-Molekülen auf fünf Wassermoleküle, d. h. ein Molverhältnis
von 2:5 bzw. 1:2,5; der H2O-Gehalt beträgt hier 28 Ma%) [57]. Die Wasserbindung
erfolgt exotherm. Die Hydrate weisen unterschiedliche Schmelzpunkte auf. Das
NMMO-Monoydrat hat einen Schmelzpunkt von etwa 72 °C, das 2,5-Hydrat von ca.
39 °C. Das heißt, mit Zunahme der Wasserbindung an das NMMO resultiert eine
Schmelzpunkterniedrigung, die wiederum die sinkende Bindungsstärke zwischen
NMMO und den Wassermolekülen anzeigt [56].
NMMO-MH hat im festen kristallinen Zustand einen monoklinen Gitteraufbau
(Raumgruppe P21) mit folgenden Abmessungen der Elementarzelle: a = 2,548 nm, b =
0,604 nm, c = 0,919 nm, ß = 99,88 ° [57]. Sowohl in der Struktur des reinen NMMO
als auch in der von NMMO-MH liegt der Morpholin-Ring in der Sesselkonformation vor
- wie auch die Cellulosemonomereinheit. Die N-O-Gruppe am Morpholin-Ring befindet
sich in axialer, die Methylgruppe des Morpholin-Ringes in äquatorialer Position [55].
Aufgrund dieser Konfiguration und der (geringen) Größe der Methylgruppe ist das
Sauerstoffatom der N-O-Gruppe für Baugruppen einer Substanz - wie z. B. für die Hy-
droxylgruppen der Cellulose - sterisch unbehindert zugänglich. Deshalb besitzt das
NMMO-Molekül gute Donatoreigenschaften [35].
Eine andere strukturelle Besonderheit des NMMO-Moleküls ist seine hochpolare
Beschaffenheit. Deswegen verfügt NMMO über eine hohe Basizität. Der starke Dipol-
Charakter des NMMO-Moleküls resultiert aus der N-O-Bindung, die kürzer als eine
übliche N-O-Einfachbindung ist. UV-spektroskopische Studien belegen, daß das Sauer-
stoffatom der N-Oxidgruppe die lösewirksame Komponente darstellt und N vielmehr
eine stabilisierende Funktion hat [58]. Das für NMMO und NMMO-MH rechnerisch
ermittelte Dipolmoment beträgt 4,4 D [59].
Eine weitere wichtige Struktureigenschaft des NMMO-Moleküls besteht darin, daß die
N-Oxide des Morpholin-Ringes ein bis zwei Wasserstoffbrückenbindungen ausbilden
können. Insbesondere beim NMMO-MH wurde ein ausgeprägtes H-Brückensystem
festgestellt, in dem die Wassermoleküle mit der N-Oxidgruppe gekoppelt sind.
Hierbei induziert die N-Oxidgruppe eine spezielle Wasserstruktur, in der Wassermole-
küle über H-Bindungen miteinander und mit den N-Oxiden verbunden sind (Abb. 2-6,
2-7) [55].

GRUNDLAGEN ___________________________________________________________________________________________________
17
NMMO ⋅ 1 H2O
Abb. 2-6 Strukturausschnitt des N-Methylmorpholin-N-oxid-Monohydrats [35]
NMMO ⋅ 2,5 H2O Abb. 2-7 Strukturausschnitt des N-Methylmorpholin-N-oxid-2,5-Hydrats [35]
Die oben aufgezählten Eigenschaften des NMMO-Moleküls (u. a. die Polarität,
Basizität, räumliche Struktur) hängen mit dessen Fähigkeit, Polymere wie z. B. Cellu-
lose zu lösen, eng zusammen.

GRUNDLAGEN ___________________________________________________________________________________________________
18
2.2.3 Modellvorstellungen zum Lösemechanismus der Cellulose
In der Literatur finden sich im wesentlichen zwei verschiedene Modellvorstellungen
zum Lösevorgang der Cellulose in Lösemittelsystemen, die im Laufe der Zeit teilweise
weiterentwickelt wurden [6]:
- das Elektronen-Donator-Akzeptor-(EDA)-Konzept, das von Nakao [60] vorgeschla-
gen und später von Philipp und Schleicher erweitert wurde [61], und
- das Säure-Base-Konzept, das auf Turbak et al. zurückgeht [62].
Nach dem EDA-Konzept sind an der Donator-Akzeptor-Wechselwirkung H- und O-
Atome der Hydroxylgruppen sowie Ring- und Brücken-O-Atome der Cellulose beteiligt,
wobei die H-Atome als Elektronenakzeptoren, die O-Atome als Elektronendonatoren
wirken. Das Lösemittel besitzt Donator- und Akzeptor-Zentren, die sowohl in einer
Molekülspezies als auch in verschiedenen Lösemittelkomponenten lokalisiert sein
können.
Die Grundüberlegung des Säure-Base-Konzepts ist die, daß Cellulosemoleküle teils
sauer, teils basisch sind und in Abhängigkeit von der chemischen Umgebung sowohl
als Säure wie auch als Base reagieren können [63].
Beide Modellvorstellungen werden in der Literatur für die Deutung der Wechsel-
wirkungen zwischen Cellulose und Lösemittel herangezogen. Einige Autoren bevorzu-
gen das erste, andere wiederum das zweite Konzept [42, 58].
In Systemen, in denen Cellulose unter Bildung instabiler Hauptvalenzderivate (derivati-
sierende Systeme) in Lösung überführt wird (z. B. NaOH/CS2/H2O), kann der Löse-
prozeß vereinfacht in folgender Weise skizziert werden [64]:
- "Andiffusion" des Lösemittels durch das Porensystem der Cellulose,
- Aufspaltung zwischenfibrillärer Bindungen,
- Modifizierung des Wasserstoffbrückensystems der Cellulose,
- Entfernen des Wasserstoffs von den OH-Gruppen der Cellulose,
- Ersetzen des Wasserstoffs durch verschiedene funktionelle Gruppen, die mehr oder
weniger polar sind,
- Wechselwirkung der Substituenten mit der Hauptkomponente des Lösemittel-
systems,
- Auflösung der Cellulose.

GRUNDLAGEN ___________________________________________________________________________________________________
19
Dabei können diese Teilvorgänge nacheinander, nebeneinander und nach verschiede-
nen Mechanismen ablaufen.
Im allgemeinen ist eine Überführung der Cellulose in Lösung nur möglich, wenn die
zwischenmolekularen Anziehungskräfte zwischen Cellulose und Lösemittel größer sind
als die der Einzelkomponenten.
Für Systeme, die Cellulose allein über zwischenmolekulare Wechselwirkungen (nicht-
derivatisierende Systeme) aufzulösen vermögen, wird zur Erklärung des Lösevorgangs
vorwiegend das EDA-Konzept angewendet [42]. Für Lösemittelsysteme wie z. B. das
NMMO und deren Wechselwirkung mit Cellulose wird ein Modell für den Löseprozeß
favorisiert, das sich insbesondere auf spektroskopische Untersuchungen stützt und
vom allgemeinen EDA-Konzept etwas abweicht [65]. Danach ist eine Auflösung der
Cellulose von der Partikeloberfläche her durch Abtragen von Molekülschichten - wie es
von niedermolekularen Substanzen bekannt ist - nicht möglich. Die Lösemittel-
Moleküle müssen zunächst in das Innere der Cellulosepartikel eindringen und mit den
Hydroxylgruppen der Cellulose wechselwirken, damit eine Auflösung überhaupt zu-
stande kommt. Das kann nur mit solchen Lösemittelsystemen realisiert werden, deren
Lösemittelbestandteile aufgrund der Molekülgröße und -geometrie dazu imstande
sind. Zudem findet die Wechselwirkung der aktiven Lösekomponente des Lösemittels
bevorzugt mit den primären Hydroxylgruppen (C-6-Atom der Anhydroglucoseeinheit)
der Cellulose statt [42, 66].
Diese Modellvorstellung soll nun im folgenden eigens für den Lösevorgang der
Cellulose in NMMO beschrieben werden [6]:
Nach Kontakt des wäßrigen NMMO-MH mit Cellulose diffundiert dieses in das
Porensystem der Cellulose.
Die zwischenfibrillären Bindungen der Cellulose werden durch die NMMO-Mole-
küle aufgebrochen.
Gleichzeitig findet eine starke Quellung der Cellulose statt, die eine Auflockerung
bzw. Aufweitung der Cellulosestruktur bewirkt. Dadurch können die NMMO-
Moleküle in das Cellulosegitter eindringen.
Die inter- und intramolekularen H-Brücken der Cellulose werden durch sukzessive
Wechselwirkung mit dem NMMO gesprengt. Das geschieht derart, daß die löse-
wirksame N-O-Gruppe des NMMO-Moleküls sowohl das Wasserstoffatom als auch

GRUNDLAGEN ___________________________________________________________________________________________________
20
das Sauerstoffatom der Cellulosehydroxylgruppe attackiert, wobei das Donator-
zentrum (O-) des NMMO-Moleküls am Wasserstoffatom und das Akzeptorzentrum
(N+) am Sauerstoffatom der Cellulosehydroxylgruppen angreift.
Danach blockieren die N-O-Gruppen die zugehörigen OH-Gruppen der Cellulose
und bilden einen Komplex, einen Celluloselösemittel-Komplex mit sogenannter
kryptoionischer Wasserstoffbrückenbindung zum Wasserstoffatom der Cellulose-
hydroxylgruppen, vorzugsweise zum Wasserstoffatom der C-6-Hydroxylgruppe.
Die derart gebildeten Celluloselösemittel-Komplexe werden dann solvatisiert. Als
Folge davon diffundieren die mit NMMO komplexierten, kinetisch frei beweglichen
Celluloseketten auseinander.
Auch wenn in der vorliegenden Arbeit die Lösemechanismen nicht eingehend unter-
sucht wurden, sind sie für die Interpretation der im Rahmen der Dissertation durch-
geführten DSC-Messungen am binären und ternären Stoffsystem NMMO/H2O bzw.
NMMO/H2O/Cellulose von großer Bedeutung.

GRUNDLAGEN ___________________________________________________________________________________________________
21
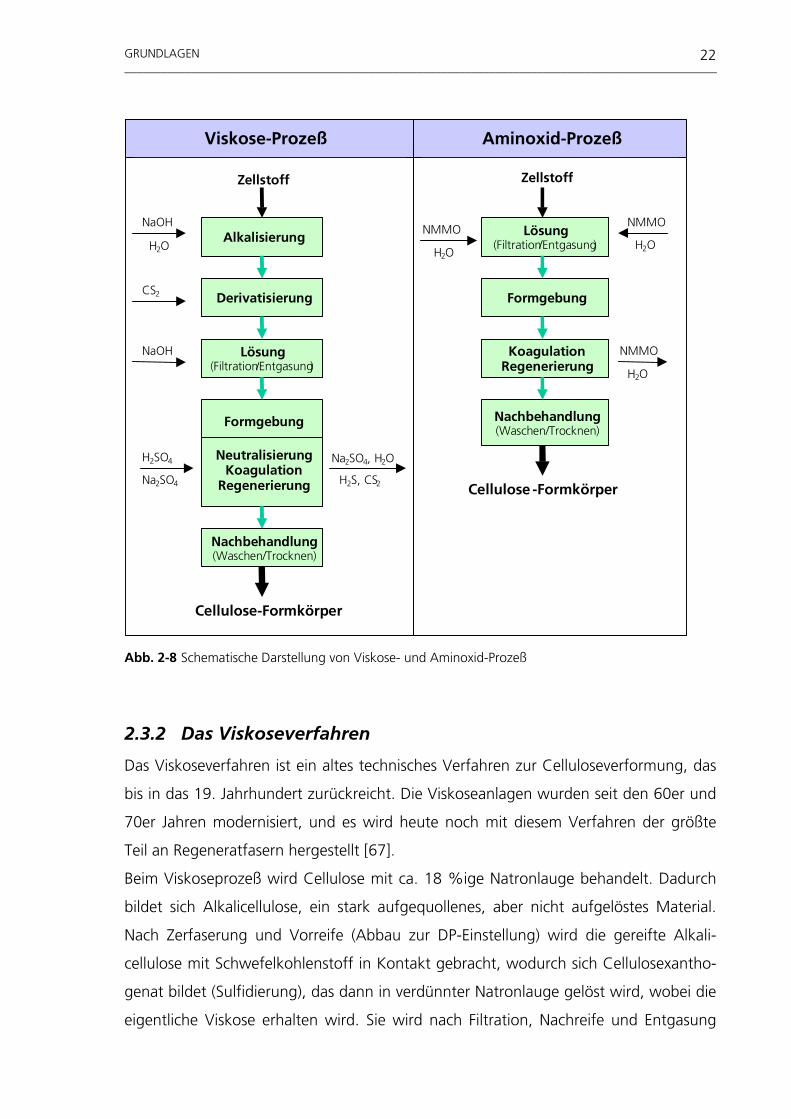
2.3 Cellulose-Regeneratverfahren
2.3.1 Das Aminoxidverfahren
Das Aminoxidverfahren, das in der Literatur auch als Lyocell- oder NMMO-Verfahren
bezeichnet wird, ist eine neuartige Methode, um Cellulose zu lösen und daraus Rege-
nerat-Formkörper herzustellen. Das Verfahren basiert auf der Verwendung von NMMO
als Lösemittel für Cellulose. Die Besonderheiten dieses Verfahrens sind:
• Die Auflösung, Formgebung und Regenerierung der Cellulose findet ohne eine
chemische Umsetzung statt.
• Das gesamte Verfahren besteht aus einigen wenigen Prozeßstufen.
• Es werden keine toxischen Chemikalien verwendet.
• Das Lösemittel N-Methylmorpholin-N-oxid wird nach Einsatz im Verfahrensprozeß
aufgearbeitet und wieder zur Lösungsherstellung eingesetzt.
Das Aminoxidverfahren kann grundsätzlich in folgende Prozeßstufen eingeteilt wer-
den:
− Lösen:
Zellstoff wird in einer wäßrigen Lösung von NMMO unter Erhitzen und Wasser-
entzug gelöst. Es entsteht dadurch eine homogene, viskose Lösung von Cellulose in
geschmolzenem Aminoxid-Monohydrat.
− Formgebung:
Nach Filtration und Entgasen wird die Schmelzlösung bei Temperaturen um 100 °C
verformt, in dem die hochviskose Lösung durch eine Loch- oder Schlitzdüse in einen
Luftspalt gepreßt wird (z. B. Spinnfaserherstellung, Folienextrusion, u. ä.).
− Koagulation:
Der Formkörper wird dann in ein Koagulationsbad (verdünnte Aminoxidlösung)
geführt, in dem die Cellulose regeneriert und das NMMO aus dem Cellulose-
Formkörper ausgetrieben wird. Die Aminoxidlösung des Koagulationsbades wird
nach Aufarbeitung wieder zur Lösungsherstellung eingesetzt.
− Nachbehandlung:
Zum Entfernen des restlichen NMMO aus dem Formkörper wird dieser in weiteren
Bädern mit Wasser gewaschen und anschließend getrocknet. Das NMMO wird aus
den Fäll- und Waschbädern durch Destillation wieder zurückgewonnen.
GRUNDLAGEN ___________________________________________________________________________________________________
22
Abb. 2-8 Schematische Darstellung von Viskose- und Aminoxid-Prozeß 2.3.2 Das Viskoseverfahren
Das Viskoseverfahren ist ein altes technisches Verfahren zur Celluloseverformung, das
bis in das 19. Jahrhundert zurückreicht. Die Viskoseanlagen wurden seit den 60er und
70er Jahren modernisiert, und es wird heute noch mit diesem Verfahren der größte
Teil an Regeneratfasern hergestellt [67].
Beim Viskoseprozeß wird Cellulose mit ca. 18 %ige Natronlauge behandelt. Dadurch
bildet sich Alkalicellulose, ein stark aufgequollenes, aber nicht aufgelöstes Material.
Nach Zerfaserung und Vorreife (Abbau zur DP-Einstellung) wird die gereifte Alkali-
cellulose mit Schwefelkohlenstoff in Kontakt gebracht, wodurch sich Cellulosexantho-
genat bildet (Sulfidierung), das dann in verdünnter Natronlauge gelöst wird, wobei die
eigentliche Viskose erhalten wird. Sie wird nach Filtration, Nachreife und Entgasung
Lösung(Filtration/Entgasung)
Formgebung
KoagulationRegenerierung
Nachbehandlung(Waschen/Trocknen)
Zellstoff
NMMO
H2O
NMMO
H2O
Cellulose -Formkörper
NMMO
H2O
Viskose-Prozeß
Alkalisierung
Derivatisierung
Lösung(Filtration/Entgasung)
Nachbehandlung(Waschen/Trocknen)
Zellstoff
NaOH
H2O
Cellulose-Formkörper
CS2
NaOH
Formgebung
NeutralisierungKoagulation
Regenerierung
Na2SO4, H2O
H2S, CS2
H2SO4
Na2SO4
Aminoxid-Prozeß

GRUNDLAGEN ___________________________________________________________________________________________________
23
durch eine oder mehrere Düsenöffnungen in der Gestalt des gewünschten Produktes
in eine wäßrige Lösung aus Salz und Säure (Schwefelsäure, Natriumsulfat, Zinksalze)
gepreßt. Das Salz fördert die Koagulation, die Säure neutralisiert das Alkali und zer-
setzt das Cellulosexanthogenat. Hierbei findet die Regenerierung der Cellulose zum
Viskose-Formkörper statt. Anschließend wird eine Reihe von Nachbehandlungen (Ent-
schwefelung, Bleiche, Wäsche) vorgenommen [68, 69].
Bei diesem Verfahren wird also die Cellulose in ein lösliches Derivat überführt, das sich
zu einem Formkörper verarbeiten läßt. Von Vorteil ist die große Vielseitigkeit des
Eigenschaftsprofils durch Variation der Verfahrensparameter. So können z. B. Cellu-
lose-Regeneratprodukte mit einem niedrigen oder hohen Elastizitätsmodul hergestellt
werden. Nachteilig wirken sich hierbei Umweltbelastungen durch CS2 und Schwer-
metalle aus. Nur durch enorme technische Aufwendungen können diese Umweltbe-
lastungen vermieden werden.
2.3.3 Das Carbamatverfahren
Beim Carbamatverfahren wird die Cellulose - wie beim Viskoseprozeß - zunächst mit
Natronlauge vorbehandelt, um eine Aktivierung und Vorreife der Cellulose zu erzielen.
Ein Teil des Alkalis wird aus der Alkalicellulose wieder entfernt, und die Alkalicellulose
wird mit Harnstoff gemischt. Diese Mischung kann dann in einem inerten Lösemittel
(z. B. Xylol) auf Temperaturen über 130 °C erwärmt werden, wobei sich Cellulose-
carbamat bildet. Das Lösemittel wird durch Destillation zurückgewonnen. Das Cellu-
losecarbamat wird gewaschen, dann in verdünnter Natronlauge gelöst, entgast und
gefiltert, wodurch eine viskose Carbamatlösung erhalten wird. Die Lösung wird über
eine formgebende Düsenöffnung in ein schwefelsaures, salzhaltiges (Natriumsulfat)
Bad geführt, in dem zunächst eine Koagulation des Cellulose-Carbamat-Formkörpers
erfolgt. In einer weiteren Verfahrensstufe findet die eigentliche Regenerierung der
Cellulose unter Zersetzung der Carbamatgruppen statt. Dies geschieht in einer stark
verdünnten Natronlauge unter Bildung von Ammoniak. Durch anschließende Nachbe-
handlungen wird der restliche Harnstoff aus dem Cellulose-Formkörper entfernt und
das Restcarbamat zersetzt [70, 71].

GRUNDLAGEN ___________________________________________________________________________________________________
24
2.4 Thermodynamik von Polymersystemen
Die Behandlung der Thermodynamik von binären und ternären Stoffsystemen ist für
die vorliegende Arbeit insofern wichtig, da der Phasenzustand des binären Systems
NMMO/H2O und das Phasen- und Löseverhalten der Cellulose im System NMMO/H2O
mittels der Kalorimetrie eingehend untersucht werden soll.
Bei der Beschreibung der thermodynamischen Zustände und Vorgänge, die in Polymer-
systemen auftreten, spielen grundlegende Begriffe wie Stoffsysteme, Stoffgemische,
Phasen, Kristallisations- und Schmelzverhalten sowie Enthalpieänderungen eine wichti-
ge Rolle. Zunächst wird auf diese Begriffe eingegangen. Für die Ausführungen in die-
sem Abschnitt wurden die folgenden Arbeiten verwendet: [1, 34, 72, 73, 74, 75, 76].
2.4.1 Stoffsysteme
Unter einem stofflichen System wird eine beliebige Menge Material verstanden, das
aus einem einzigen oder aus mehreren chemisch verschiedenen Bestandteilen (Stoff,
Komponente) besteht.
Um das betreffende System einfacher untersuchen und beschreiben zu können, wird
es in der Regel durch physikalische oder gedachte Wände von der Umgebung oder
anderen Systemen abgegrenzt. Dabei hängt es von der Art der Abgrenzung ab, ob es
sich um ein abgeschlossenes, geschlossenes oder offenes System handelt.
2.4.2 Phasen
Unter einer Phase werden - im thermodynamischen Sinne - all die homogenen Teile
eines Systems zusammengefaßt, die einheitliche chemische, strukturelle und physi-
kalische Eigenschaften besitzen.
In einem System sind die Phasen durch Grenzflächen voneinander getrennt. An den
Phasengrenzen ändern sich die Eigenschaften sprunghaft. Nach dem Aggregatzustand
werden feste, flüssige und gasförmige Phasen unterschieden. Nach der Zusammen-
setzung wird nach reinen Phasen, Phasengemischen und Mischphasen unterschieden.
Eine reine Phase besteht aus einer einzigen chemischen Substanz (Komponente),
welche die gleiche Stöchiometrie und die gleiche Beschaffenheit in allen ihren Orten

GRUNDLAGEN ___________________________________________________________________________________________________
25
aufweist. Reines Wasser oder reines Natriumchlorid sind Beispiele für reine Phasen-
systeme.
Ein Phasengemisch ist ein Stoffsystem, das aus mehreren unterschiedlichen, neben-
einander vorliegenden chemischen Substanzen (Phasen) zusammengesetzt ist. In den
jeweiligen Phasen sind die Mengenverhältnisse der Komponenten eindeutig festgelegt.
Unter einer Mischphase dagegen wird eine aus mehreren Komponenten zusammen-
gesetzte Phase verstanden, deren Komponenten sich in molekularer Form durch-
dringen (im festen Zustand spricht man von Mischkristallen). Das Wesentliche für eine
Mischphase ist, daß die Mengenverhältnisse der Komponenten in diesen Phasen große
Variationsbereiche aufweisen können. Denn auch eine reine Phase kann bei entspre-
chender Komponentenwahl aus mehreren Komponenten bestehen. Die Verhältnisse
der Komponenten sind hier hingegen durch die stöchiometrischen Koeffizienten fest-
gelegt.
Bestehen Stoffsysteme nur aus einer Phase, werden sie homogen genannt. Sind sie aus
mehreren Phasen zusammengesetzt, werden sie als heterogen bezeichnet.
2.4.3 Homogene und heterogene Stoffgemische
Ein aus mehreren Komponenten zusammengesetztes System oder Stoffgemisch hat
die besondere Eigenschaft, daß die Komponenten darin keine chemische Verbindung
eingehen und in der Regel durch physikalische Verfahren getrennt werden können.
Die Einzelkomponenten in Stoffgemischen können in allen Aggregatzuständen auf-
treten.
In einem homogenen Stoffgemisch, das makroskopisch auch aus einer Phase
besteht, bilden die darin enthaltenen Stoffe ein einheitliches Ganzes und sind nicht
durch Grenzflächen voneinander getrennt. Unter dem Mikroskop erscheint das homo-
gene System einheitlich und alle mechanisch abtrennbaren Teilchen haben gleiche
Stoffeigenschaften. Es unterscheidet sich von einer chemischen Verbindung dadurch,
daß es keinen einheitlichen Schmelz- und Siedepunkt aufweist. In einem solchen Stoff-
gemisch sind die einzelnen Komponenten nicht immer im gleichen Mischungs-
verhältnis vorhanden. Die physikalischen Eigenschaften eines homogenen Stoffge-
misches ändern sich daher mit dem Mischungsverhältnis.

GRUNDLAGEN ___________________________________________________________________________________________________
26
Ein derartiges, homogenes Stoffgemisch wird dann als Lösung bezeichnet, wenn eine
Komponente (Lösemittel) gegenüber den anderen (gelöste Stoffe) eine Sonderstellung
- z. B. durch einen großen Überschuß - einnimmt, und vor allem, wenn das Lösemittel
und die zu lösenden Stoffe sich im Aggregatzustand unterscheiden. Wenn jedoch in
einem homogenen Stoffgemisch die Komponenten in flüssigem Aggregatzustand
vorliegen, wird es als Mischung bezeichnet.
Beispiele für homogene Stoffgemische sind z. B. Luft für Gasgemische, das Methanol-
Aceton-Gemisch für Mischungen und die Kochsalzlösung für Lösungen.
In heterogenen Stoffgemischen, die aus mehreren Phasen bestehen, behalten die
Einzelkomponenten im Gegensatz zu den chemischen Verbindungen und den homo-
genen Stoffgemischen ihre charakteristischen Eigenschaften stets bei und lassen sich
zumindest mit Hilfe eines Mikroskops feststellen. Heterogene Systeme sind z. B. Ge-
menge (fest - fest), Suspensionen (flüssig - fest) oder Emulsionen (flüssig - flüssig).
Kolloidale Lösungen werden zu den mikroheterogenen Systemen gezählt.
2.4.4 Kristallisations- und Schmelzverhalten von Polymeren
Ob und in welchem Ausmaß Makromoleküle kristallisieren, d. h. sich zu einem drei-
dimensional-periodischen Gitter aneinanderlagern, hängt hauptsächlich von ihrer
chemisch-physikalischen Struktur sowie den vorliegenden äußeren thermodynami-
schen Bedingungen (T, p) ab. Unter dem Begriff Struktur sind hier die chemische Zu-
sammensetzung, die Symmetrie, die Taktizität sowie die Verzweigung an der Haupt-
kette des betreffenden Polymermoleküls zu verstehen. Zudem wirkt sich die Eigen-
schaft des Polymermoleküls, starke zwischenmolekulare Bindungen auszubilden, güns-
tig auf die Kristallisierbarkeit (Kristallisationsfähigkeit) aus.
Polymere mit unverzweigten Molekülketten und regelmäßig angeordneten Seiten-
gruppen, also lineare symmetrische und möglicherweise iso- oder syndiotaktische
Polymere können gut kristallisieren. Makromoleküle mit unregelmäßigen Grundbau-
steinen, sperrigen Seitengruppen oder mit einem hohen Verzweigungsgrad an der
Haupkette, erschweren in der Regel die dichte Packung, die für die Ausbildung einer
kristallinen Anordnung nötig ist. Derartige Kettentypen sind entweder schlecht oder
gar nicht kristallisierbar.

GRUNDLAGEN ___________________________________________________________________________________________________
27
Seitengruppen, die sich in einer regelmäßigen Anordnung entlang der Kette befinden
und nicht ins Gewicht fallen, stellen für die Kristallitbildung in der Regel kein Hindernis
dar. Molekülketten mit polaren Gruppen, die eine starke intermolekulare Anziehung
begünstigen, können zur besseren Kristallisation und deren Stabilität beitragen. Diese
Polymere können z. B. intermolekulare Wasserstoffbrückenbindungen ausbilden, die
die Stabilität des Kristallits wesentlich erhöhen.
Beispiele für gut kristallisierende Polymere sind lineares Polyethylen, Polytetrafluor-
ethylen und isotaktisches Polypropylen. Dagegen sind ataktisches Polyvinylchlorid,
ataktisches Polymethylmethacrylat und ataktisches Polystyrol Beispiele für nicht kristal-
lisierende Polymere.
Die Bildung einer dreidimensionalen geordneten Phase aus einem ungeordneten
Zustand (z. B. Schmelze, Lösung) ist ein Zweistufenprozeß. Im ersten Schritt der Kristal-
lisation entstehen unter entsprechenden Bedingungen (T, p, Verstreckung) Kristall-
keime. Die Bildung der Keime erfolgt entweder spontan durch Ansammlung von Mole-
külen bzw. Molekülsegmenten zu vereinzelten 'Clustern' infolge lokaler thermischer
Schwankungen (homogene Keimbildung) oder durch die Anwesenheit von "fremden
Grenzflächen" wie Staub, Gefäßwände oder Nukleierungsmittel (heterogene Keim-
bildung).
Für eine kristallisierende Polymerschmelze oder -lösung gilt, daß mit abnehmender
Temperatur die Wahrscheinlichkeit für die Entstehung von Kristallkeimen wächst.
Deshalb muß eine Lösung oder Schmelze zunächst unterkühlt werden, damit eine
genügend große Zahl an Keimen hervorgerufen wird. (Eine Kristallisation kann aber
auch durch Zusatz eines Keimbildners bei höheren Temperaturen induziert werden).
Die Keime werden aber erst dann stabil, wenn sie eine bestimmte kritische Dimension
überschritten haben. Erst dann wachsen sie kontinuierlich zu Kristalliten weiter. Ab
diesem Stadium setzt die zweite Stufe der Kristallisation ein, in der das Wachstum der
kristallinen Bereiche erfolgt. Eine theoretische Beschreibung des Kristallisationsver-
halten von Polymeren ist mit der Avrami-Gleichung möglich [77].
Je nach Polymertyp und Kristallisationsbedingungen (Konzentration, Temperatur)
wachsen die Keime in verschiedener Weise zu größeren Kristallstrukturen voran und
nehmen dabei verschiedene Formen an. Diese reichen von kugelförmigen bis zu faseri-
gen Überstrukturen. Überstrukturen wie z. B. Sphärolithe sind meist Mikrometer bis
Millimeter groß und besitzen einen mehr oder weniger geordneten inneren Aufbau.

GRUNDLAGEN ___________________________________________________________________________________________________
28
Selbst bei günstigen Kristallisationsbedingungen kristallisieren Polymere nie vollständig,
sondern immer nur partiell. Anzahl, Größe und Ordnungsgrad der kristallinen Bereiche
beeinflussen entscheidend das chemische, vor allem aber das physikalische und me-
chanische Verhalten teilkristalliner Polymere.
Im Gegensatz zu niedermolekularen Stoffen kristallisieren und schmelzen die
teilkristallinen makromolekularen Substanzen nicht bei bestimmten diskreten Tempera-
turen, sondern in breiten Temperaturbereichen. Dies liegt unter anderem daran, daß
Teile ein und desselben Kettenmoleküls sowohl in kristallinen als auch in amorphen
Bereichen liegen können. Das Schmelzverhalten hängt auch wesentlich von der Vor-
geschichte der Polymerprobe und deren Temperatur-Zeit-Behandlung während des
Schmelzvorgangs ab. Allgemein kann jedoch gesagt werden, daß je besser die mittlere
kristalline Ordnung im Polymer ist, desto enger das Schmelzintervall ist. Das heißt, daß
alle ordnungsverringernden Faktoren wie die Anwesenheit von Cokomponenten, Ver-
zweigungen und ein hoher Kurzkettenanteil das Schmelzintervall verbreitern. Schmel-
zen und Kristallisation sind bei Polymere keine thermodynamischen Gleichgewichts-
prozesse und müssen deshalb mit der Nichtgleichgewichts-Thermodynamik behandelt
werden.

UNTERSUCHUNGSMETHODEN ___________________________________________________________________________________________________
29
3 Untersuchungsmethoden
3.1 Dynamische Differenzkalorimetrie (DSC)
Die DSC (Differential Scanning Calorimetry) [78, 79, 80, 81, 82, 83] ist eine wichtige
physikalische Prüftechnik. Sie gehört zu den thermischen Analysemethoden und wird
eingesetzt, um thermische Materialeigenschaften zu ermitteln. Hauptanwendungen
liegen z. B. in der Bestimmung der spezifischen Wärmekapazität, der Reinheit, der
Polymorphie, der Glasumwandlungstemperatur und der Oxidationsstabilität. Zudem
können Aussagen über thermische Effekte, chemische Reaktionen und ihre Reaktions-
kinetik sowie über Schmelz- und Kristallisationsverhalten gemacht werden. Sie ist eine
relativ kostengünstige, schnelle und zuverlässige Meßtechnik. Ein großer Vorteil ist die
wenig aufwendige Probenvorbereitung. Mit einem derartigen Kalorimeter werden
Wärmeströme von und zu einer Probe in Differenz zu einer Referenzprobe gemessen.
3.1.1 Meßprinzip
Das Kalorimeter beim sogenannten Leistungskompensations-DSC besteht aus einer
Meßzelle aus zwei getrennten kleinen Öfen, wie in der schematischen Darstellung
Abb. 3-1 gezeigt.
Abb. 3-1 Prinzipieller Aufbau eines Dynamischen Leistungskompensations-Differenz-Kalorimeters; P Probe, R Vergleichsprobe, TP Temperatur der Probe, TR Temperatur der Vergleichsprobe, U Umgebung, PP der Probe zugeführte elektrische Heizleistung, PR der Vergleichsprobe zugeführte elektrische Heizleistung [78]

UNTERSUCHUNGSMETHODEN ___________________________________________________________________________________________________
30
Die Probenmeßstelle und die Vergleichs-(Referenz-) Meßstelle verfügen jeweils über
einen Temperaturfühler und eine Heizvorrichtung. Beide Seiten werden durch getrenn-
te Regler so mit Heizleistung versorgt, daß ihr Temperaturanstieg oder -abfall einem
gewünschten Programm folgt. Die thermisch induzierte Leistungsaufnahme, bzw.
abgabe der Probe wird dabei durch äquivalentes Vergrößern oder Vermindern der
Heizleistung eines Differenzheizkreises kompensiert [84]. Der dazu notwendige Heiz-
leistungsunterschied wird in eine proportionale elektrische Spannung umgewandelt,
die ihrerseits dem Wärmestrom zur Probe proportional ist [85].
3.1.2 Meßablauf und Einflußfaktoren
In dieser Arbeit wurden alle DSC-Messungen an einem Leistungskompensations-
Kalorimeter DSC 7 der Firma PerkinElmer Instruments durchgeführt. Die graphischen
Darstellungen und Auswertungen erfolgten mit der Software Pyris, Version 3.8. Die
Durchführung einer DSC-Messung geschah wie folgt:
- Probenpräparation,
- Einwiegen der Probe in einen Aluminiumtiegel,
- Verschließen des Tiegels mittels einer Pressvorrichtung,
- Einbringen des Probe- und Referenztiegels in die Meßzelle,
- Einstellen des Spülgasstroms (N2),
- Einstellen des gewünschten Meßprogramms.
Die geräte-und probenspezifischen Einflußfaktoren bei der Versuchsdurchführung
sind: Probenvorbehandlung, Einwaage, Heiz- bzw. Kühlrate, Spülgasart/-strom, Start-
/Endtemperatur, Tiegelart und die Referenzsubstanz.
3.1.3 Wärmestrom und Enthalpieänderung
Mit Hilfe der Kalorimetrie wird der Wärmestrom dQ/dt, d. h. die Änderung der Wär-
memenge Q pro Zeiteinheit dt gemessen. Dieser ist der spezifischen Wärmekapazität

UNTERSUCHUNGSMETHODEN ___________________________________________________________________________________________________
31
cp direkt proportional. Der Proportionalitätsfaktor ist die Heizrate v = dT/dt. Mit der
Probenmasse m ergibt sich:
(dQ/dt) ⋅ 1/m = v cp. (1) Für v = dT/dt in (1) eingesetzt, gilt:
(dQ/dt) ⋅ 1/m = (dT/dt) ⋅ cp . (2) Für praktische Anwendungen ist der Wärmeumsatz eines Stoffes zwischen zwei
Zuständen interessant. Diese Wärmemenge oder die Änderung der inneren Energie
eines Stoffes wird bei konstantem Druck als Enthalpie bezeichnet. Die innere Energie
ist bei allen Stoffen beim absoluten Nullpunkt der Temperatur gleich Null und nimmt
beim Erwärmen in charakteristischer Weise zu. Bei isobar geführten thermischen
Prozessen ist die Änderung der Wärmemenge dQ, die zwischen einem Stoff und seiner
Umgebung ausgetauscht wird, mit der zeitlichen Änderung der Enhalpie dH identisch:
(dH/dt)p = dQ/dt. (3) Setzt man (3) in (2) ein, so erhält man für die spezifische Wärmekapazität cp: cp = (dH/dT) ⋅ 1/m (4) oder nach dH umgeformt: dH = m ⋅ cpdT. (5) Durch Integration des Wärmestroms dH/dT oder der spez. Wärmekapazität cp über die
(zeitproportionale) Temperatur erhält man die Enthalpieänderung ∆H eines Stoffes:
∆H = (1/m) ⋅ ∫ cp⋅dT. (6) Um die Enthalpieänderung ∆H aus einer DSC-Kurve zu erhalten, muß die Fläche unter
der Kurve als Funktion der Zeit aufgetragen werden. Bei dynamischen Messungen ist
es aber üblich, auch die Temperaturinformation anzugeben. Deshalb wird in der Regel
eine zeitpropotionale Temperatur auf der Abszisse angegeben. Dabei handelt es sich
nicht um die Probentemperatur, sondern es ist je nach Hersteller die aus Starttem-
peratur und Heizgeschwindigkeit vom Programm errechnete Temperatur bzw. die
Temperatur der Referenzprobe.

UNTERSUCHUNGSMETHODEN ___________________________________________________________________________________________________
32
3.2 Ergänzende Meßmethoden
Neben der Dynamischen Differenzkalorimetrie kamen in dieser Arbeit folgende ergän-
zende Untersuchungsmethoden zur Anwendung: Röntgenweitwinkelbeugung,
Licht- und Elektronenmikroskopie, Refraktometrie und mechanische Prüfung.
3.2.1 Röntgenbeugung
Die Röntgenbeugung wurde zum einen angewendet, um die möglichen kristallinen
Phasen im binären System NMM/H2O als Funktion der Probenzusammensetzung zu
bestimmen. Zum anderen wurde damit der Quell- bzw. Lösezustand der im ternären
System NMMO/H2O/Cellulose enthaltenen Cellulose (Cellulose-I-II-Umwandlung) in Ab-
hängigkeit vom NMMO/H2O-Verhältnis, der Temperatur und der Einwirkzeit ermittelt.
Das Grundprinzip hierbei ist die Röntgenbeugung an polykristallinen Pulverproben:
Der aus einer Röntgenröhre austretende Primärstrahl wird durch Blenden zu einem
feinen Strahl ausgeblendet und trifft in senkrechter Transmission auf die Probe. Das
Beugungsbild bzw. die gestreute Intensitätsverteilung wird in Abhängigkeit vom dop-
pelten Streuwinkel 2ϑ auf einem ebenen Film, der senkrecht zum einfallenden Strahl
positioniert ist, aufgenommen.
Röntgenstrahlen, die das Raumgitter eines Kristalls treffen, werden von diesem
gebeugt, da die Atomabstände (Gitter- bzw. Netzebenenabstand d) im Kristallgitter in
der Größenordnung der Wellenlängen der Röntgenstrahlen liegen. Die Beugung ist
spezifisch für das Kristallgitter, d. h. jedem Gitter ist ein bestimmtes Beugungsbild zu-
geordnet.
Nach dem Huygensschen Prinzip wirkt jedes Atom (mit einer Elektronen-Dichte-
Verteilung), das von einfallenden Röntgenstrahlen getroffen wird, als Erregungszen-
trum einer neuen Elementarwelle. Diese Elementarwellen gelangen unter bestimmten
Winkeln zur konstruktiven Interferenz. Die Bedingung für eine konstruktive Interferenz
und damit die Beziehung zwischen dem Beugungswinkel ϑ, der Wellenlänge λ des
Röntgenstrahls und dem Netzebenenabstand d im Kristall gibt die Braggsche
Gleichung wieder:
n⋅λ = 2⋅dhkl⋅sinϑhkl n = 1, 2, 3 ... (7)

UNTERSUCHUNGSMETHODEN ___________________________________________________________________________________________________
33
Dabei sind
- n die Ordnungszahl des Reflexes,
- λ die Wellenlänge der Röntgenstrahlung (0.154 nm für CuKα)
- dhkl der Netzebenenabstand im Kristallgitter,
- hkl die Millerschen Indices,
- ϑhkl Glanzwinkel oder Braggsche Winkel.
Die verstärkten Wellen, die an den parallelen und äquidistanten Netzebenen im Kristall
gebeugt werden, rufen - bei zufälliger Orientierungsverteilung der Kristallite - Beu-
gungsringe hervor. Die Schärfe der Beugungsringe ist dabei umgekehrt proportional
zur Größe der Kristallite, das Verhältnis von Ringintensität und Untergrundstreuung
spiegelt den Kristallinitätsgrad der Probe wider.
3.2.2 Licht- und Elektronenmikroskopie, Refraktometrie und
mechanische Prüfung
Der Einfluß des NMMO/H2O-Verhältnisses, der Temperatur und der Zeit auf den Löse-
zustand der im Dreistoffsystem befindlichen Cellulose wurde außerdem mittels Licht-
mikroskopie untersucht.
Für die Bestimmung des NMMO-Gehalts im Zweistoff- und Dreistoffsystem kam die
Refraktometrie zum Einsatz. Hierbei wurden die NMMO-Gehalte der NMMO/H2O-
Gemische anhand der experimentell ermittelten Brechungsindices nNMMO aus einer
Tabelle, welche der Literatur entnommen wurde, abgelesen.
Mittels Raster- und Transmissionselektronenmikroskopie (TEM, REM) wurden die
Morphologie sowie die Faser-Matrix-Haftung von faserverstärkten Celluloseblasfolien,
die im Rahmen dieser Arbeit hergestellt wurden, untersucht. Die mechanischen Eigen-
schaften wie Festigkeit, Elastizitätsmodul und Dehnung der Blasfolien wurden in Zug-
versuchen durch mechanische Prüfung bestimmt.
Weitere experimentelle Details über die Messungen, die mit Hilfe der oben genannten
Meßmethoden durchgeführt wurden, werden in den einzelnen Abschnitten erläutert.

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
34
4 Untersuchungen am binären Stoffsystem NMMO/H2O
4.1 DSC-Messungen am System NMMO/H2O
Das Stoffsystem NMMO/H2O kann in Abhängigkeit von der Zusammensetzung sowohl
ein Löse- als auch ein Fällmittel für Cellulose und verschiedene andere Polymere sein.
Es ist interessant zu erfahren, bei welchen definierten Bedingungen das System Quell-,
bei welchen es Löse- und bei welchen es Fälleigenschaften besitzt. Um das feststellen
zu können, soll zunächst als Grundlage das binäre System NMMO/H2O thermodyna-
misch untersucht werden. Dabei soll das thermische Verhalten des Systems als Funk-
tion der Zusammensetzung geprüft werden. Zudem sollen anhand der DSC-Ergebnisse
Aussagen über die Wechselwirkung zwischen NMMO- und Wassermolekülen getrof-
fen werden.

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
35
4.1.1 Probenmaterial, Präparation und Versuchsdurchführung
Probenmaterial
Für die Untersuchungen des Systems NMMO/H2O wurde eine wäßrige NMMO-Lösung
von der Firma BASF verwendet, die NMMO für kommerzielle Zwecke in großen Men-
gen produziert. Außerdem wurde für die Experimente zum Vergleich eine zweite
NMMO-Lösung eingesetzt, die von der Forschungseinrichtung Thüringisches Institut
für Textil- und Kunststofforschung (TITK) e. V. bezogen wurde. Beide Lösungen
wurden mit einem NMMO-Gehalt von ca. 50 Ma% geliefert.
Probenpräparation
Die 50%igen NMMO-Lösungen wurden vor der Aufkonzentration mit einem Sieb und
einem Filtertuch (Kimwipes Lite 200) filtriert. Die quantitative Bestimmung des NMMO-
Gehalts im Zweistoffsystem NMMO/H2O erfolgte refraktometrisch mit einem Abbe-
Refraktometer (Krüss Optronic GmbH) durch Messung des Brechungsindex nNMMO bei
50 °C. In einem Laborreaktor (IKA, LR-2000 VP 7) wurde die Lösung durch Erhitzen
und durch Vakuumdestillation auf den jeweils gewünschten NMMO-Gehalt aufkon-
zentriert. Jede der so hergestellten Probe besaß schließlich eine genau definierte
NMMO/H2O-Zusammensetzung. Die Zusammensetzung der Proben wird in der Arbeit
über den NMMO-Gehalt in Ma% angegeben. Die Heiztemperatur der Lösung, bei der
eine Vakuumdestillation nötig war, variierte jeweils zwischen 65 und 115 °C. Bei Pro-
ben mit NMMO-Gehalten größer 85 Ma% konnte die Konzentrationsbestimmung
nicht mehr über den Brechungsindex nNMMO erfolgen, da die Lösung nach der Proben-
entnahme aus dem Reaktor abrupt auskristallisierte. Der NMMO-Gehalt dieser Proben
wurde mittels Elementaranalyse über den Stickstoffgehalt bestimmt.
Die NMMO/H2O-Proben wurden dann bei Raumtemperatur über 24 Stunden in luft-
dichten PE-Beuteln und Kunststofflaschen aufbewahrt. Damit sollte den Proben eine
ausreichende Zeit für Abkühlung und Kristallbildung gegeben werden. Anschließend
wurden sie im Kühlfach eines Kühlschranks bei ca. -18 °C gelagert. Dies war wegen
der hygroskopischen Beschaffenheit der NMMO-Lösungen erforderlich.

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
36
Vorversuche
An den NMMO/H2O-Proben wurden zunächst Vorversuche mit dem DSC realisiert, um
eine Übersicht über die zu erwartenden Schmelz- und Kristallisationstemperaturen zu
erhalten. Hierzu wurden die Proben mit unterschiedlichen Heiz- und Kühlraten im
Temperaturbereich zwischen -100 °C und 90 °C aufgewärmt oder abgekühlt. Bei einer
der zwei untersuchten NMMO/H2O-Probenserien konnte nach dem ersten Aufheiz-
vorgang keine Kristallisation während des Abkühlvorgangs beobachtet werden.
Obwohl die Kühlgeschwindigkeit von 40 °C/min auf 10 °C/min und 5 °C/min geändert
wurde und die Proben über 2 Stunden lang jeweils bei Temperaturen von -50, -80 und
-100 °C gehalten wurden, konnte keine Kristallisation festgestellt werden. Deshalb
wurde davon abgesehen, die Proben bei der DSC-Messung ein zweites Mal aufzu-
heizen. Für die Auswertung der Messungen wurde also generell die erste Aufheizkurve
verwendet. Da aber das Schmelzverhalten der zu untersuchenden Proben durch die
Vorgeschichte beeinflußt werden kann, wurden alle NMMO/H2O-Proben unter den
gleichen Bedingungen hergestellt, gelagert und unter den selben Voraussetzungen
sowie auf die gleiche Art und Weise mit dem DSC thermisch vermessen. Durch diese
Vorgehensweise sollte die Reproduzierbarkeit und die Interpretierbarkeit der DSC-
Ergebnisse gewährleistet sein.
Versuchsdurchführung
Für die DSC-Messungen wurde jedesmal eine Probenmasse zwischen 9 und 12 mg in
einen Al-Probentiegel eingewogen. Um einer unerwünschten Änderung der vorher
eingestellten Mengenverhältnisse von NMMO und H2O vorzubeugen, wurden die 50 µl
großen Tiegelpfännchen mit je einem Al-Deckel unter Zuhilfenahme einer speziellen
Pressvorrichtung hermetisch verschlossen. Die so präparierten Tiegel konnten dem In-
nendruck (schätzungsweise 2 bis 4 bar) standhalten. Die Messungen erfolgten unter
Stickstoff als Schutzgas mit einer Flußrate von 20 ml/min. Die Referenzmeßstelle des
Kalorimeters wurde mit einem leeren 50 µl Al-Tiegel bestückt.
Aufgrund der gewonnenen Erfahrungen aus den DSC-Vorversuchen mit NMMO/H2O-
Proben wurde mit folgendem Temperaturprogramm gearbeitet:
• Einsetzen der Probe bei Raumtemperatur,
• Abkühlung auf -50 °C mit einer Kühlrate von 5 °C/min,

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
37
• Halten bei -50 °C über 60 min,
• Aufheizung von -50 °C auf 90 °C mit einer Heizrate von 5 °C/min,
• Halten bei 90 °C über 1 min,
• Abkühlung von 90 °C auf 25 °C mit 40 °C/min Kühlrate.
4.1.2 Auswertung und Ergebnisse
An jeder NMMO/H2O-Probe mit definierter Zusammensetzung wurden jeweils drei
DSC-Messungen unter gleichen Versuchsbedingungen durchgeführt. In Abhängigkeit
von der Zusammensetzung wurden bei den DSC-Diagrammen zwei verschiedene
Typen von Kurvenverläufen beobachtet (vergleiche Abb. 4-1 und 4-2). Proben mit
NMMO-Gehalten zwischen 70 und 82 Ma% zeigen relativ flache und vergleichsweise
breite Schmelzpeaks. Die Schmelzpunkte und Schmelzenthalpien dieser Proben fallen
verhältnismäßig niedrig aus. Dagegen sind die Schmelzpeaks von Proben, die über ei-
nen NMMO-Gehalt zwischen 85 und 91 Ma% verfügen, relativ hoch und schmal. Die
Schmelzpunkt- und Schmelzenthalpiebeträge dieser Proben sind wesentlich größer als
die der Proben mit niedrigeren NMMO-Gehalten.
In Abb. 4-1 ist ein DSC-Diagramm einer Probe mit 78 Ma% dargestellt, das beispiel-
haft für die Proben mit NMMO-Gehalten kleiner 85 Ma% ist. In der Darstellung ist der
endotherme Peak während der Aufheizung zu sehen. Der Schmelzbereich dieser Probe
beginnt bereits bei einer niedrigen Temperatur (ca. 10 °C) und reicht bis ca. 70 °C.
Deshalb wurde die Schmelzwärme ∆H durch Integration der Peakfläche ab 12 °C er-
mittelt und beträgt 56 J/g. Das Maximum des Schmelzpeaks liegt bei 56 °C.
In der Abb. 4-2 ist eine zweite DSC-Messung einer Probe mit 91 Ma% NMMO-Gehalt
wiedergegeben. Das Thermogramm spiegelt den typischen Kurvenverlauf derjenigen
Proben wider, deren NMMO-Gehalt zwischen 85 und 91 Ma% beträgt. Diese Proben
mit höheren NMMO-Anteilen weisen relativ hohe und vergleichsweise schmale
Schmelzpeaks auf. Der Darstellung ist zu entnehmen, daß diese Probe im Temperatur-
bereich zwischen 70 und 80 °C schmilzt, die Peaktemperatur mit 76 °C und die
Schmelzenthalpie mit 132,5 J/g bestimmt wurden.

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
38
Abb. 4-1 Kurvenverlauf einer DSC-Messung an einer NMMO/H2O-Probe mit einem NMMO-Gehalt von 78 Ma% Tim Anfangstemperatur, Tfm Endtemperatur, Tpm Peaktemperatur, ∆H Schmelzenthalpie, Area Schmelzpeakfläche
-40 -20 0 20 40 60 80 100
20
25
30
35
40
45
50
55
60
NMMO/H2O-Gemisch
NMMO-Gehalt: 78 Ma%
Tim
= 12,24 °C
Tfm
= 70,02 °C
Tpm
= 56,36 °C
∆H = 55,70 J/gArea = 579,32 mJ
Hea
t Fl
ow E
ndo
Up
[mW
]
Temperature [°C]

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
39
Abb. 4-2 Kurvenverlauf einer DSC-Messung: Wärmestrom als Funktion der Temperatur beim Aufheizen von einer NMMO/H2O-Probe mit 91 Ma% NMMO-Gehalt
-40 -20 0 20 40 60 80 100
20
25
30
35
40
45
50
55
60
NMMO/H2O-Gemisch
NMMO-Gehalt: 91 Ma%
Tim
= 70,28 °C
Tfm
= 80,15 °C
Tpm
= 76,22 °C
∆H = 132,55 J/gArea = 1086,92 mJ
Hea
t Fl
ow E
ndo
Up
[mW
]
Temperature [°C]

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
40
In der Abb. 4-3 sind die aus den DSC-Messungen gewonnenen charakteristischen
Temperaturen als Schmelztemperaturen (Anfangstemperatur Tim und Peaktemperatur
Tpm) der NMMO/H2O-Proben als Funktion des NMMO-Gehalts aufgetragen. Die oben
beschriebenen zwei typischen DSC-Verläufe der NMMO/H2O-Proben sind auch in
dieser Darstellung gut zu sehen. Proben mit NMMO-Gehalten von 70 bis 82 Ma%
haben einen relativ breiten Schmelzpeak, andererseits weisen die Proben, deren
NMMO-Gehalt 85 Ma% oder größer ist, einen wesentlich schmaleren Schmelzpeak
auf.
Abb. 4-3 Temperaturen Tim und Tpm sowie die Schmelzenthalpie ∆H in Abhängigkeit vom NMMO-Gehalt
Der Abbildung ist außerdem zu entnehmen, daß mit Zunahme des NMMO-Gehalts der
NMMO/H2O-Proben auch deren Schmelztemperaturen ansteigen. Der Anstieg der
Schmelztemperaturen mit dem NMMO-Gehalt ist für die Proben mit NMMO-Gehalten
bis zu 85 Ma% beträchtlich. Die Schmelztemperaturen der Proben mit NMMO-Gehal-
ten von 85 Ma% und größer ändern sich dagegen nicht wesentlich.
70 75 80 85 90 95
0
20
40
60
80
100
120
140
160
180- das binäre System NMMO/H
2O -
Tim
Tpm
∆H
NMMO-Gehalt [Ma%]
Schm
elzt
empe
ratu
r T im
, Tpm
[°C
]
0
20
40
60
80
100
120
140
160
180
Schm
elze
ntha
lpie
∆H
[J/g
]

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
41
In der Abbildung sind ferner die aus den DSC-Messungen bestimmten Schmelzenthal-
pien der NMMO/H2O-Proben in Abhängigkeit vom NMMO-Gehalt aufgetragen. Diese
Darstellung zeigt im Prinzip einen ähnlichen Zusammenhang wie den für die Schmelz-
temperaturen. Die Schmelzenthalpien der Proben steigen mit wachsendem NMMO-
Gehalt ebenfalls an; der Zuwachs ist im Bereich zwischen 80 und 85 Ma% NMMO-
Gehalt relativ groß, und ab 85 Ma% ändern sich die Enthalpiewerte der Proben über
der Probenzusammensetzung nur wenig.
4.1.3 Diskusssion
Einige wichtige Beobachtungen und Ergebnisse, die mit der Dynamischen Differenz-
Kalorimetrie über das binäre Stoffsystem NMMO/H2O erzielt wurden, sollen an dieser
Stelle erörtert werden.
Zum besseren Verständnis ist es bedeutsam, einleitend zu erwähnen, daß die Proben
mit den hier untersuchten Zusammensetzungen, bis auf einige wenige Ausnahmen, in
festem kristallinen Zustand vorlagen.
Eine interessante Beobachtung ist das breite Schmelzintervall der NMMO/H2O-Proben
mit verhältnismäßig hohem Wasseranteil (NMMO-Gehalt: 72-82 Ma%) und der
schmale DSC-Peak der Proben mit relativ niedrigem Wassergehalt (NMMO-Gehalt: 85 -
91 Ma%). Diese verschiedenartigen Schmelzerscheinungen deuten auf unterschied-
liche Kristallitgrößenverteilungen in diesen Proben hin. Das breite Schmelzintervall ent-
spricht einer breiten Kristallitgrößenverteilung. Derartige Proben enthalten verschieden
große Kristallite, die bei verschiedenen Temperaturen aufschmelzen. Dagegen ist die
schmale DSC-Kurve einer engen Kristallitgrößenverteilung zuzuschreiben.
Eine andere Erklärung für dieses Phänomen wäre, daß die Proben mit breitem DSC-
Peak einen erheblichen Anteil von Kristallen enthalten, die in ihrem Aufbau unter-
schiedlich stark gestört sind. In solch einem Fall schmelzen die Kristalle je nach ihrer
kristallinen Ordnung bzw. Perfektion ebenfalls bei verschiedenen Temperaturen. Im
Vergleich zu diesen haben die Proben, die eine schmale DSC-Schmelzkurve aufweisen,
Kristalle mit besserer bzw. höherer kristalliner Ordnung.
Es ist aber auch denkbar, daß bei den Proben mit relativ hohem Wasseranteil sowohl
die breite Kristallitgrößenverteilung als auch der unterschiedlich gestörte Kristallaufbau
das breite Schmelzintervall gemeinsam verursachen. Entsprechend kann der schmale

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
42
Schmelzpeak bei den Proben mit wenig Wassergehalt durch eine enge Kristallitgrößen-
verteilung sowie einen besseren kristallinen Aufbau hervorgerufen werden.
Ein weiteres Ergebnis, das aus der Abb. 4-3 zu entnehmen ist, betrifft die Proben mit
NMMO-Gehalten zwischen 80 und 85 Ma%. Die verhältnismäßig steilen Anstiege der
Schmelztemperatur- und Enthalpiewerte dieser Proben lassen einen Übergangsbereich
erkennen. In diesem Bereich findet ebenso ein Wechsel von breiten zu schmalen DSC-
Kurven statt. Vermutlich sind diese Feststellungen ein Indiz für eine Änderung der
kristallinen Phase in den Proben. Es ist durchaus möglich, daß das NMMO/H2O-Verhält-
nis, das in diesem Übergangsbereich vorliegt, sich auf die Bildung einer neuen Kristall-
modifikation auswirkt.
Ein anderes erwähnenswertes Resultat hängt mit der Bindungsstärke zwischen den
NMMO- und den H2O-Molekülen zusammen. Die relativ niedrigen Schmelztempera-
turen und Schmelzenthalpien, die für die wasserreichen Proben bestimmt wurden und
die Zunahme dieser Meßgrößen mit Abnahme des Wasseranteils bzw. mit wachsen-
dem NMMO-Gehalt in den Proben weisen auf einen wichtigen Sachverhalt hin. Die
Bindung zwischen den NMMO- und den Wassermolekülen, die durch Wasserstoff-
brücken zustande kommt, ist in den wasserreichen Proben vergleichsweise schwach
und wird mit abnehmendem Wassergehalt bzw. mit steigendem NMMO-Gehalt in den
Proben eindeutig stärker.
In der Literatur wird hauptsächlich von zwei Hydraten des NMMO berichtet, dem was-
serreichen Hydrat NMMO⋅2,5 H2O und dem wasserarmen Monohydrat NMMO⋅1H2O.
Die NMMO/H2O-Zusammensetzung des 2,5-Hydrats beträgt 72/28 Ma%, die des Mo-
nohydrats 86,7/13,3 Ma%.
Unter Zuhilfenahme dieser Information läßt sich die hohe Bindungsstärke der wasser-
armen NMMO/H2O-Proben dadurch erklären, daß in diesen überwiegend die kristalline
Phase des Monohydrats vorliegt.

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
43
4.2 Ergänzende Experimente mittels Röntgenbeugung
Um herauszufinden, welche kristallinen Phasen bzw. NMMO-Hydrate in den präparier-
ten binären NMMO/H2O-Gemischen in Abhängigkeit von der NMMO/H2O-Zusammen-
setzung vorliegen, wurden an ihnen Röntgenweitwinkelbeugungsmessungen durch-
geführt.
4.2.1 Experimentelles zu den Röntgenweitwinkeluntersuchungen
Die Röntenbeugungsversuche an Proben des binären Stoffsystems wurden mit einer
universellen Planfilmkamera (Filmkammer-Meßplatz BRUKER AXS) durchgeführt, wobei
Röntgenweitwinkel-Diagramme unter den folgenden experimentellen Bedingungen
aufgenommen wurden: Cu-Kα-Strahlung, Ni-Filter, senkrechte Transmissionstechnik,
58 mm Abstand zwischen Probe und Film. Die Röntgenröhre wurde bei 40 kV und 40
mA betrieben, die Belichtungszeit betrug 60 Minuten.
Für die Untersuchung der NMMO/H2O-Proben mittels Röntgenbeugung wurde eine
spezielle temperierbare Meßzelle aus Edelstahl verwendet. Mit Hilfe eines Kryostaten
und eines Kältemittelkreislaufs wurde die Meßzelle auf eine Temperatur von -5 °C ge-
kühlt. Damit alle NMMO/H2O-Proben in festem kristallinen Zustand vermessen werden
konnten, war die Kühlung der Meßzelle mit eingeschlossener Probe notwendig. Denn
einige dieser Proben schmelzen bei höheren Temperaturen aufgrund ihrer niedrigen
Schmelztemperaturen auf.
Die Proben mußten in die Meßzelle zwischen zwei durchstrahlbaren Fensterfolien aus
Polyethylenterephthalat luftdicht eingeschlossen werden, weil die Proben hygrosko-
pisch sind.
4.2.2 Auswertung und Ergebnisse
Die einzelnen Lagen der gemessenen Beugungsreflexe der NMMO/H2O-Gemische
wurden durch Ausmessen der Abstände x zwischen Primärstrahl und Beugungsring auf
den Filmen bestimmt. Mittels der Gleichung tan 2θ = x/l und dem Wert l = 58 mm für
den Abstand zwischen Probe und Film wurden die Streuwinkel 2θ, die den gemesse-

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
44
nen Reflexlagen entsprachen, berechnet. Der maximale absolute Fehler für den Streu-
winkel 2θ beträgt ± 0,5°.
Abb. 4-4 zeigt Röntgenfilmaufnahmen von zwei NMMO/H2O-Gemischen, die kenn-
zeichnend für alle anderen NMMO/H2O-Proben sind.
a b
Abb. 4-4 Röntgen-Planfilmaufnahmen von NMMO/H2O-Proben, die im festen Zustand vermessen wurden; a) NMMO-Geh. 80 Ma%; b) NMMO-Geh. 91 Ma%
Für eine weitere Auswertung der ermittelten Röntgenbeugungsdaten der Gemische
wurde eine Software (PowderCell1.0) herangezogen. Mit diesem Programm lassen sich
die Kristallstruktur sowie die zu erwartenden Pulverdiffraktogramme von Substanzen
oder Gemischen, deren Kristalldaten bekannt sind, simulieren bzw. berechnen.
Die aus der Literatur bekannten Kristallstrukturdaten (relative Atomlage, Gitter-
konstanten, Raumgruppen-Nummer, Temperaturfaktor usw.) des reinen NMMO, des
NMMO-Monohydrats und des NMMO⋅2,5-Hydrats wurden in das genannte Programm
eingegeben und die Kristallstrukturen dieser Phasen generiert sowie die dazugehöri-
gen Beugungsdiagramme ermittelt. Durch Vergleich der so erhaltenen Beugungs-
diagramme (der drei NMMO-Modifikationen) mit den gemessenen Beugungsreflexen

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
45
von NMMO/H2O-Gemischen konnte eine qualitative Phasenanalyse dieser Proben
durchgeführt werden.
Abb. 4-5 Gemessene Beugungsreflexe (vertikale Striche) eines NMMO/H2O-Gemisches mit 80 Ma%
NMMO-Geh. und berechnetes Beugungsdiagramm des NMMO⋅2,5-Hydrats; die Abkürzungen stehen für die Intensitäten der Beugungsreflexe: st = stark, m = mittlel, sw = schwach
Es stellte sich heraus, daß in den NMMO/H2O-Gemischen, deren NMMO-Gehalt 72,
78, und 80 Ma% betrug, vorwiegend die kristalline Phase des NMMO⋅2,5-Hydrats
vorliegt. In den NMMO/H2O-Gemischen mit 85 und 91 Ma% NMMO-Konzentration
kommt dagegen hauptsächlich die Phase des NMMO-Monohydrats vor.
In der Abb. 4-5 sind das berechnete Pulverdiffraktogramm des NMMO⋅2,5-Hydrats
sowie die gemessenen Beugungsreflexe der NMMO/H2O-Probe mit 80 Ma% NMMO-
Gehalt dargestellt. Die Intensitäten der einzelnen Beugungsreflexe sind in drei Inten-
sitätsklassen eingeteilt: stark, mittel und schwach. Diese Darstellung ist zugleich bei-
spielhaft für die ermittelten Beugungsreflexe der Proben, deren NMMO-Gehalt 72 und
78 Ma% betrug.
Die Abb. 4-6 hingegen zeigt das berechnete Diffraktogramm des NMMO-Mono-
hydrats und die gemessenen Beugungsreflexe des NMMO/H2O-Gemisches, das einen
0 5 10 15 20 25 30 35 40 45
0
45
90
Inte
nsitä
t
2 theta [°]
berechnetes Beugungsdiagramm von NMMO·2,5H2O
gemessene Beugungsreflexe von NMMO·1,6H2O
(NMMO-Geh. = 80 Ma%)
msw
st st st
msw sw(100)
(002)
(-111) (013)/(202)
(014)
(-311)(-216)
(-222)

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
46
NMMO-Geh. von 91 Ma% besitzt. Das gleiche Resultat wurde auch für die Probe mit
85 Ma% beobachtet. Sowohl aus der obigen als auch aus dieser Auftragung ist
ersichtlich, daß die gemessenen Reflexe dieser Proben und die berechneten Beugungs-
reflexe der jeweiligen NMMO-Hydrate - innerhalb der Meßgenauigkeit - gut überein-
stimmen.
Abb. 4-6 Gemessene Beugungsreflexe eines NMMO/H2O-Gemisches mit 91 Ma% NMMO-Geh., darüber ist das Beugungsdiagramm des NMMO-Monohydrats zu sehen, das mit dem Programm PowderCell berechnet wurde
Der Vergleich der experimentell ermittelten Beugungsreflexe mit den berechneten
Beugungsdiagrammen ergab außerdem, daß in diesen Proben kein Phasengemisch
und auch keine Mischphase vorhanden ist, die sich möglicherweise aus den drei
bekannten kristallinen Phasen des NMMO zusammensetzen würden. Es wurden auch
keine Beugungsreflexe von den untersuchten Proben beobachtet, die mit dem
errechneten Beugungsdiagramm des reinen NMMO zusammenfallen.
0 5 10 15 20 25 30 35 40 45
0
50
100
150
Inte
nsitä
t
2 theta [°]
berechnetes Beugungsdiagramm von NMMO·1H2O
gemessene Beugungsreflexe von NMMO·0,65H2O
(NMMO-Geh.= 91 Ma%)
mst st
msw sw
(400)/(110)
(011)/(310) (-302)
(302)/(-502)
(020)(-712)

UNTERSUCHUNGEN AM BINÄREN STOFFSYSTEM NMMO/H2O ___________________________________________________________________________________________________
47
4.3 Das Phasendiagramm für das binäre System NMMO/H2O
Abb. 4-6 zeigt das Zustandsdiagramm des binären Stoffsystems NMMO/H2O für den
H2O-Konzentrationsbereich 10 - 30 Ma%. Im Diagramm sind die aus den DSC-Mes-
sungen ermittelten Schmelzpeaktemperaturen Tpm in Abhängigkeit vom NMMO/H2O-
Verhältnis dargestellt. Außerdem sind die Schmelzpeaktemperaturen des NMMO/H2O-
Systems als Kurve angegeben, die mit Hilfe der Gleichung [50]
1/ Tpm - 1/ T°pm = -(R/∆H) ⋅ ln x (8)
berechnet wurde. Dabei ist Tpm die Schmelzpeaktemperatur des betreffenden
NMMO/H2O-Gemsiches, T°pm die Schmelzpeaktemperatur des reinen NMMO-Hydrats
(NMMO⋅1H2O oder NMMO⋅2,5H2O), ∆H dessen Schmelzenthalpie, x dessen Molen-
bruch und R die allgemeine Gaskonstante. Der Abbildung kann entnommen werden,
daß die gemessenen Schmelzpunkte mit den berechneten Werten innerhalb der Meß-
genauigkeit gut übereinstimmen.
Abb. 4-6 Das Phasendiagramm des NMMO-H2O-Stoffsystems
0 5 10 15 20 25 30
40
60
80
100
120100 95 90 85 80 75 70
T pm [
°C]
H2O [Ma%]
experimentell ermittelte Schmelzpeaktemperaturen berechnete Schmelzpeaktemperaturen nach Gl. (8)
2,5H2O
28,0%
1H2O
13,3%
NMMO [Ma%]
fest
flüssigfest
flüssig

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE ___________________________________________________________________________________________________
48
5 Untersuchungen am ternären Stoffsystem NMMO/H2O/Cellulose
5.1 DSC-Messungen am System NMMO/H2O/Cellulose
Die Auflösung der Cellulose im Stoffsystem NMMO/H2O kann - theoretisch betrachtet -
von der Temperatur sowie dem Druck des Systems, der Behandlungsdauer der Cellu-
lose im NMMO/H2O-System, dem NMMO- bzw. dem Wassergehalt des NMMO/ H2O-
Gemisches, der Konzentration, der Molmasse sowie dem Kristallinitätsgrad der Cellulo-
se abhängen. In diesem Abschnitt soll experimentell der Zusammenhang zwischen der
Auflösung der Cellulose und den Einflußgrößen wie NMMO/H2O-Verhältnis, Tempera-
tur und Zeit geklärt werden. Ferner wird das thermische Verhalten des Dreistoff-
systems NMMO/H2O/Cellulose in Abhängigkeit von diesen Größen untersucht. Hierbei
wird nur ein Cellulosetyp eingesetzt und lediglich für eine Cellulosekonzentration das
vorliegende Stoffsystem betrachtet. Als Charakterisierungsmethoden kommen die
Dynamische Differenz-Kalorimetrie, die Lichtmikroskopie und die Röntgenweitwinkel-
beugung zum Einsatz. Ein wichtiges Ziel dabei ist, ein Phasendiagramm für das System
NMMO/H2O/Cellulose zu konstruieren, aus welchem die quantitativen Bedingungen
für die Quellung und Auflösung der Cellulose hervorgehen.

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE ___________________________________________________________________________________________________
49
5.1.1 Probenmaterial, Präparation und Probenbehandlung
Probenmaterial
Für die Prüfung des Stoffsystems NMMO/H2O/Cellulose wurde die gleiche wäßrige
NMMO-Lösung verwendet, die auch für das binäre System NMMO/H2O eingesetzt
wurde. Die Cellulose, die hier zur Anwendung kam und in Form von Zellstoffplatten
(Buckeye V60, Cuoxam-DP = 535) vorlag, wurde in einer Labor-Schneidemühle (Sieb-
größe 0.5 mm, Fa. Fritsch) zu feinen Fasern zermahlen und anschließend in einem
Trockenscharank 24 h bei 60 °C getrocknet.
Probenpräparation
Zunächst wurde die wäßrige NMMO-Lösung auf definierte NMMO/H2O-Zusammen-
setzungen aufkonzentriert. Die quantitative Bestimmung des NMMO-Gehalts der Lö-
sung erfolgte vor der Zugabe der Cellulosefasern mittels Refraktometrie oder Elemen-
taranalyse.
Um eine homogene Verteilung der Cellulosefasern in der NMMO-Lösung zu erhalten,
wurde die Cellulose mit der NMMO-Lösung in einem Labormixer ca. 2 Minuten lang
vermengt. Das Masseverhältnis zwischen der NMMO/H2O-Lösung und der Cellulose
wurde so gewählt, daß die Cellulosekonzentration - bezogen auf die Gesamtmenge
des NMMO/H2O/Cellulose-Gemisches (ca. 750 g) - für alle Proben 8 Ma% betrug. Die
so hergestellten Dreistoffgemische wurden in luftdichten PE-Beuteln bei Raumtem-
peratur aufbewahrt. Während des Abkühlens auf die Umgebungstemperatur kristalli-
sierte das System aus.
Probenpräparation durch Löseversuche
Die Löseversuche wurden in einem Meßkneter „Rheomix 600” in Verbindung mit
einem Meßantrieb (Drehmomentrheometer „Rheocord 9000”, Haake Meß-Technik
GmbH Karlsruhe) durchgeführt.
Der Meßkneter besaß eine dreigeteilte, einzeln temperierbare Kneterkammer von 120
cm3 Kammervolumen sowie zwei rotierende Nockenrotoren. Mit Hilfe eines Verschluß-
stempels konnte der Meßkneter luftdicht verschlossen werden. Der Kneterantrieb
erfolgte über eine Welle, die mit einem Drehmomentrheometer gekoppelt war. Rotor-
drehzahl, Massetemperatur und Versuchszeit wurden ständig erfaßt. Für die Messun-

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE ___________________________________________________________________________________________________
50
gen wurde der Meßkneter jedes Mal mit ca. 85 g NMMO/Wasser/Cellulose-Mischung
gefüllt. Die Mischungen hatten eine konstante Cellulosekonzentration von 8 Ma%,
jedoch unterschiedliche NMMO-Gehalte. Der Löseversuch wurde im einzelnen folgen-
dermaßen durchgeführt:
Die in festem kristallinem Zustand befindliche Probe mit definiertem NMMO-Gehalt
wurde mechanisch zerkleinert und in den Meßkneter eingefüllt. Am Meßkneter wurde
eine Temperatur zwischen 75 und 95 °C eingestellt und bei verschlossenem Kneter
und einer Rotordrehzahl von 10 U/min die Probe ca. 15 min lang gerührt. In dieser Zeit
wurde die Probe aufgeschmolzen und auf die eingestellte Massetempertur aufgeheizt.
Nach einer ersten Probenentnahme zu diesem Zeitpunkt wurde die Rotordrehzahl auf
30 U/min hochgesetzt und der eigentliche Versuch gestartet. Alle 20 min wurde der
Kneterantrieb unterbrochen, um eine kleine Menge an Probenmaterial zu entnehmen.
Der Versuch wurde nach 80 min Versuchsdauer beendet. Für eine fest eingestellte
Temperatur wurden dadurch fünf Proben mit unterschiedlichen Behandlungszeiten er-
halten. Diese Proben wurden in verschlossenen PE-Beuteln auf Raumtempertur abge-
kühlt. Nach 24-stündiger Lagerung bei Raumtemperatur wurden sie bis zu ihrer Cha-
rakterisierung bei -18 °C aufbewahrt.
Versuchsdurchführung mit DSC:
Die DSC-Messungen am Stoffsystem NMMO/H2O/Cellulose wurden auf die gleiche Art
und Weise durchgeführt wie die am binären Sytem (siehe voriger Abschnitt).
5.1.2 Auswertung und Ergebnisse
Bei den durchgeführten Löslichkeitsexperimenten wurde der Einfluß des NMMO- bzw.
des Wassergehalts, der Behandlungstemperatur TB und der Behandlungszeit tB unter-
sucht.
I. Einfluß des NMMO-Gehalts
Das thermische Verhalten der NMMO/H2O/Cellulose-Proben wurde mit dem DSC
geprüft. In Abhängigkeit von der Probenzusammensetzung (NMMO/H2O-Verhältnis)
wurden zwei verschiedene Typen von Schmelzkurven gemessen. Proben mit NMMO-

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE ___________________________________________________________________________________________________
51
Gehalten zwischen 70 und 75 Ma% haben nur einen Schmelzpeak, Proben mit
NMMO-Gehalten ≥ 78 Ma% besitzen hingegen zwei getrennte Schmelzpeaks. Diese
zwei Typen von DSC-Kurven sind in Abb. 5-2 exemplarisch anhand zweier DSC-
Diagramme dargestellt.
In der Abb. 5-1 sind die Schmelztemperaturen (Anfangstemperatur Tim und Peaktem-
peratur Tpm) dieser Proben als Funktion des NMMO-Gehalts aufgetragen. Die Anfangs-
temperaturen der ersten Schmelzpeaks ändern sich nicht wesentlich bei der Variation
des NMMO-Gehalts. Im Gegensatz dazu nehmen die Peaktemperaturen dieser Kurven
bei Erhöhung des NMMO-Gehalts in den Proben leicht ab. Eine damit zusammenhän-
gende Beobachtung ist die, daß die Breite des ersten Schmelzpeaks mit Zunahme des
NMMO-Anteils in den Proben kleiner wird.
Abb. 5-1 Schmelztemperaturen Tim und Tpm als Funktion des NMMO-Gehalts bei 8% Cellulose Die Schmelzpeaktemperaturen des zweiten Schmelzpeaks steigen mit Zunahme des
NMMO-Gehalts von etwa 55 auf 75 °C an, die Anfangstemperaturen hingegen zeigen
keine starke Änderung in Abhängigkeit von der NMMO/H2O-Zusammensetzung. Au-
ßerdem fallen die Schmelzintervalle der zweiten Schmelzpeaks breiter aus als die der
ersten Kurven.
70 75 80 85 900
20
40
60
80
100
120
Schm
elzt
empe
ratu
r T im
, Tpm
[°C
]
NMMO-Gehalt [Ma%]
1.Tim
1.Tpm
2.Tim
2.Tpm
- das ternäre System NMMO/H2O/Cellulose -

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE __________________________________________________________________________________________________________________________________________________________
52
Abb. 5-2 DSC-Schmelzkurven von zwei verschiedenen NMMO/H2O/Cellulose-Proben; beide Proben wurden bei 90 °C 60 min lang im Kneter behandelt
-40 -20 0 20 40 60 80 100
20
25
30
35
40
45
50
55
60
Hea
t Fl
ow E
ndo
Up
[mW
]
Tem perature [°C ]
N M M O /H2O /C e ll.-P roben
N M M O -G eh. 75 M a% T
pm = 41,3 °C
∆H = 134,7 J/g
N M M O -G eh. 85 M a% 1 .T
pm = 35,8 °C
1 .∆H = 14,4 J/g 2 .T
pm = 70,1 °C
2 .∆H = 105,3 J/g
1
2
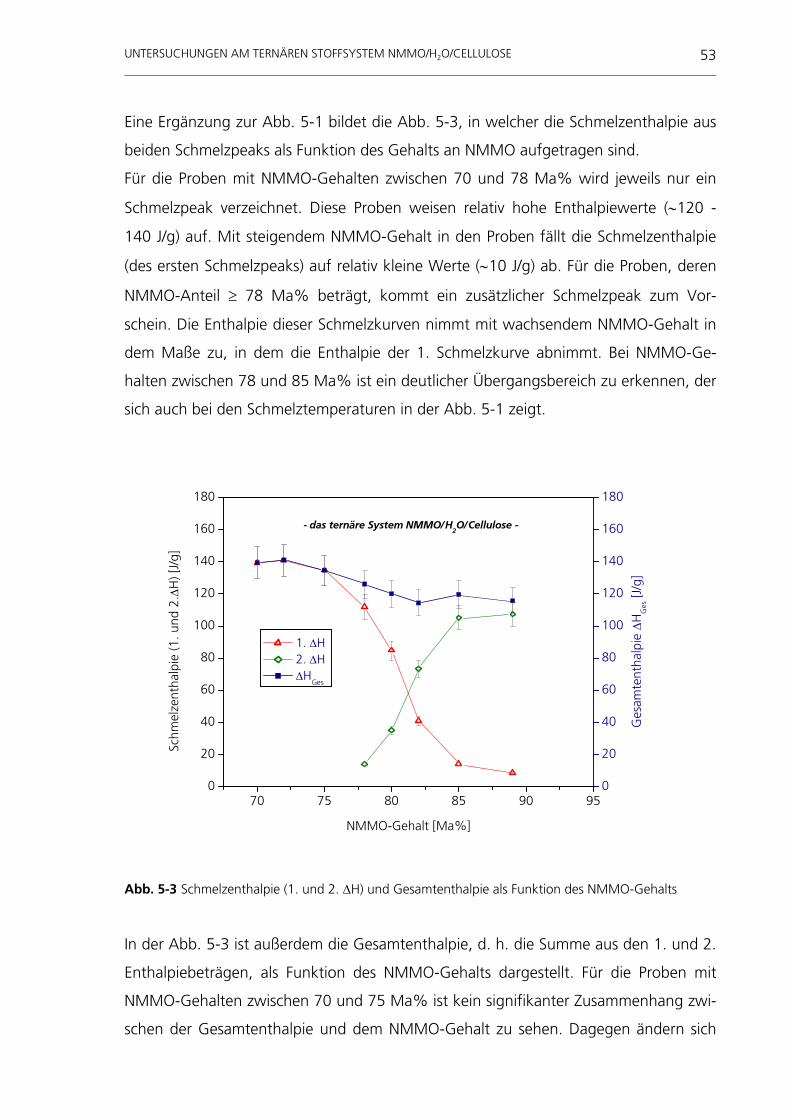
UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
53
Eine Ergänzung zur Abb. 5-1 bildet die Abb. 5-3, in welcher die Schmelzenthalpie aus
beiden Schmelzpeaks als Funktion des Gehalts an NMMO aufgetragen sind.
Für die Proben mit NMMO-Gehalten zwischen 70 und 78 Ma% wird jeweils nur ein
Schmelzpeak verzeichnet. Diese Proben weisen relativ hohe Enthalpiewerte (∼120 -
140 J/g) auf. Mit steigendem NMMO-Gehalt in den Proben fällt die Schmelzenthalpie
(des ersten Schmelzpeaks) auf relativ kleine Werte (∼10 J/g) ab. Für die Proben, deren
NMMO-Anteil ≥ 78 Ma% beträgt, kommt ein zusätzlicher Schmelzpeak zum Vor-
schein. Die Enthalpie dieser Schmelzkurven nimmt mit wachsendem NMMO-Gehalt in
dem Maße zu, in dem die Enthalpie der 1. Schmelzkurve abnimmt. Bei NMMO-Ge-
halten zwischen 78 und 85 Ma% ist ein deutlicher Übergangsbereich zu erkennen, der
sich auch bei den Schmelztemperaturen in der Abb. 5-1 zeigt.
Abb. 5-3 Schmelzenthalpie (1. und 2. ∆H) und Gesamtenthalpie als Funktion des NMMO-Gehalts In der Abb. 5-3 ist außerdem die Gesamtenthalpie, d. h. die Summe aus den 1. und 2.
Enthalpiebeträgen, als Funktion des NMMO-Gehalts dargestellt. Für die Proben mit
NMMO-Gehalten zwischen 70 und 75 Ma% ist kein signifikanter Zusammenhang zwi-
schen der Gesamtenthalpie und dem NMMO-Gehalt zu sehen. Dagegen ändern sich
70 75 80 85 90 950
20
40
60
80
100
120
140
160
180
1. ∆H 2. ∆H ∆H
Ges
NMMO-Gehalt [Ma%]
Schm
elze
ntha
lpie
(1. u
nd 2
.∆H
) [J/
g]
- das ternäre System NMMO/H2O/Cellulose -
0
20
40
60
80
100
120
140
160
180
Ges
amte
ntha
lpie
∆H
Ges [J
/g]

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
54
die Gesamtenthalpiebeträge der anderen Proben mit steigendem Gehalt an NMMO,
und zwar von 140 auf ca. 120 J/g.
II. Einfluß der Behandlungstemperatur TB
Zur Bewertung des Einflusses der Behandlungstempertur TB auf den Lösevorgang der
Cellulose im Lösemittelsystem NMMO/H2O wurde bei den Löslichkeitsversuchen die
Temperatur des NMMO/H2O/Cellulose-Gemisches (Massetemperatur am Meßkneter)
zwischen 75 und 95 °C in 5 °C-Abständen variiert. Diese Proben wurden mit dem DSC
vermessen. Die daraus bestimmten Schmelztemperaturen und -enthalpien wurden in
Abhängigkeit von der Behandlungstemperatur graphisch dargestellt. Es stellte sich für
alle hergestellten Proben heraus, daß weder die Schmelztemperaturen noch die
Schmelzenthalpien von der Behandlungstemperatur TB abhängen. Diese Feststellung
wird exemplarisch durch die folgenden Abbildungen 5-4 und 5-5, in denen nur für ei-
ne ausgewählte Probenserie die Schmelztemperaturen und -enthalpien dargestellt
sind, veranschaulicht.
Abb. 5-4 Schmelztemperaturen (Tim und Tpm) als Funktion der Behandlungstemperatur TB; NMMO/H2O/Cellulose-Proben, die einen NMMO-Gehalt von 80 Ma% besitzen und 60 min lang im Kneter behandelt wurden; 8% Celluloseanteil
75 80 85 90 95 10010
20
30
40
50
60
70
80
90
Schm
elzt
empe
ratu
r T im
, pm [°
C]
Behandlungstemperatur TB [°C]
1.Tim
1.Tpm
2.Tim
2.Tpm
- das ternäre System NMMO/H2O/ Cellulose -
/ NMMO-Geh. 80 Ma%, Beh.zeit 60 min /
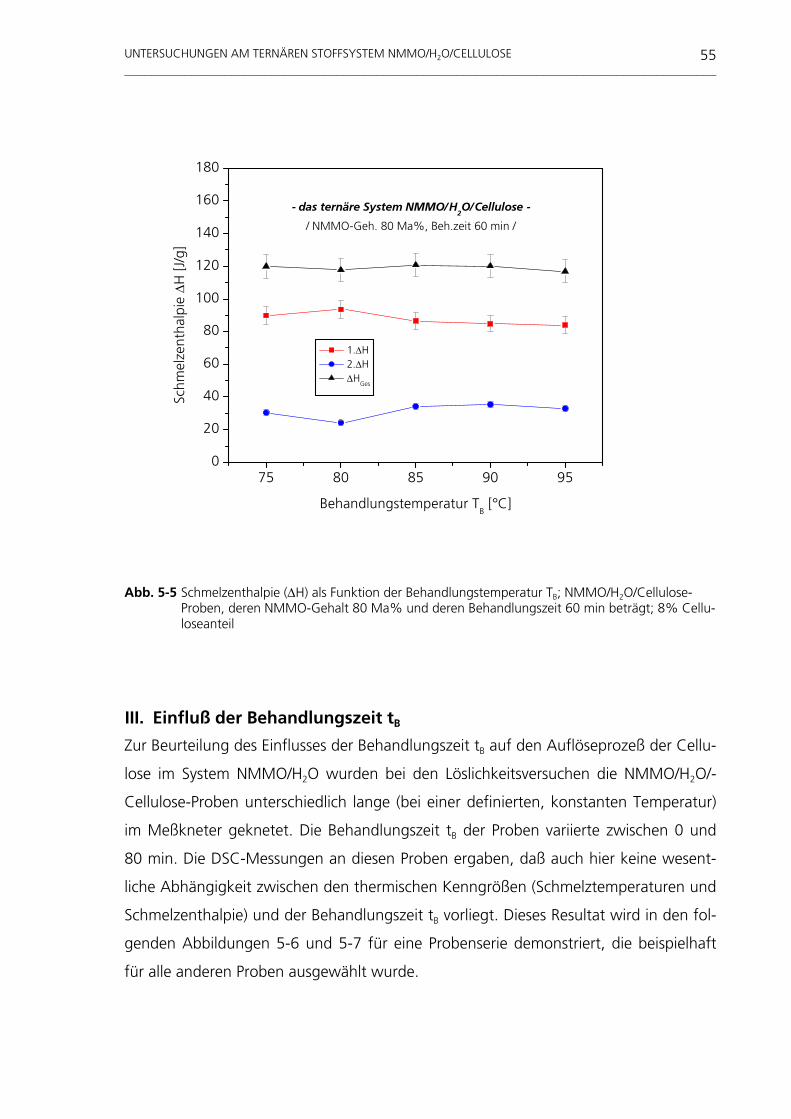
UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
55
Abb. 5-5 Schmelzenthalpie (∆H) als Funktion der Behandlungstemperatur TB; NMMO/H2O/Cellulose-
Proben, deren NMMO-Gehalt 80 Ma% und deren Behandlungszeit 60 min beträgt; 8% Cellu-loseanteil
III. Einfluß der Behandlungszeit tB
Zur Beurteilung des Einflusses der Behandlungszeit tB auf den Auflöseprozeß der Cellu-
lose im System NMMO/H2O wurden bei den Löslichkeitsversuchen die NMMO/H2O/-
Cellulose-Proben unterschiedlich lange (bei einer definierten, konstanten Temperatur)
im Meßkneter geknetet. Die Behandlungszeit tB der Proben variierte zwischen 0 und
80 min. Die DSC-Messungen an diesen Proben ergaben, daß auch hier keine wesent-
liche Abhängigkeit zwischen den thermischen Kenngrößen (Schmelztemperaturen und
Schmelzenthalpie) und der Behandlungszeit tB vorliegt. Dieses Resultat wird in den fol-
genden Abbildungen 5-6 und 5-7 für eine Probenserie demonstriert, die beispielhaft
für alle anderen Proben ausgewählt wurde.
75 80 85 90 950
20
40
60
80
100
120
140
160
180
Schm
elze
ntha
lpie
∆H
[J/g
]
Behandlungstemperatur TB [°C]
1.∆H 2.∆H ∆H
Ges
- das ternäre System NMMO/H2O/Cellulose -
/ NMMO-Geh. 80 Ma%, Beh.zeit 60 min /

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
56
Abb. 5-6 Schmelztemperaturen (Tim und Tpm) als Funktion der Behandlungszeit tB; NMMO/H2O/-Cellulose-Proben, die einen NMMO-Gehalt von 78 Ma% besitzen und bei 90 °C unterschiedlich lange im Kneter behandelt wurden; Celluloseanteil 8%
Abb. 5-7 Schmelzenthalpien (∆H) als Funktion der Behandlungszeit tB; NMMO/H2O/Cellul.-Proben, deren Behandlungtemperatur 90 °C beträgt; 78 Ma% NMMO, 8% Celluloseanteil
0 20 40 60 80
20
30
40
50
60
70
80
Schm
elzt
empe
ratu
ren
T im, T
pm [°
C]
Behandlungszeit tB [min]
1.Tim
1.Tpm
2.Tim
2.Tpm
- das ternäre System NMMO/H2O/Cellulose -
/ NMMO-Geh. 78 Ma%, Beh.temp. 90 °C /
0 20 40 60 800
20
40
60
80
100
120
140
160
180
200
Schm
elze
ntha
lpie
∆H
[J/g
]
Behandlungszeit tB [min]
1.∆H 2.∆H ∆H
Ges
- das ternäre System NMMO/H2O/Cellulose -
/ NMMO-Geh. 78 Ma%, Beh.temp. 90 °C /

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
57
5.1.3 Diskussion
Ein zentraler Punkt, der hier diskutiert werden soll, betrifft die Kristallisationsfähigkeit
des ternären Systems NMMO/H2O/Cellulose. Im Unterschied zum Zweistoffsystem
NMMO/H2O erstarrten alle NMMO/H2O/Cellulose-Proben nach der Probenherstellung
während des Abkühlens auf Raumtemperatur zu Kristallen. Eine mögliche Erklärung
hierfür ist, daß die Cellulose im vorliegenden System unter den hier eingestellten Be-
dingungen als Keimbildner wirkt und somit die Kristallisation initiiert.
Ein anderer Grund für diese Erscheinung könnte darin liegen, daß die Cellulose und
das NMMO um das in dem System vorhandene Wasser konkurrieren. Wenn die Cellu-
lose mit Wasser in Berührung kommt, nimmt sie solange Wasser auf und quillt da-
durch (Volumenvergrößerung), bis ein Gleichgewichtswert bzw. ein Quellwert erreicht
ist. (Dabei dringen die Wasser-Moleküle in die nicht-kristallinen Bereiche der Cellulose
und verursachen eine Spreizung der Celluloseketten.) Durch die Wasseraufnahme der
Cellulose könnte im Dreistoffsystem ein NMMO/H2O-Verhältnis entstehen, in welchem
der für die Wechselwirkung mit NMMO zur Verfügung stehende Wasseranteil bzw.
der Anteil an freien Wassermolekülen deutlich verringert wird. Für die NMMO-Mole-
küle würde hierdurch nur ein begrenzter Anteil der im Gesamtsystem vorhandenen
Wassermoleküle für eine Wechselwirkung (Ausbildung von Wasserstoffbrückenbin-
dungen) zur Verfügung stehen. Das würde sich wiederum günstig für die Kristall-
bildung des NMMO/H2O/Cellulose-Gemisches auswirken, da aus den Experimenten mit
dem binären System NMMO/H2O bekannt ist, daß dieses um so besser kristallisiert, je
geringer der Wasseranteil darin ist.
Für die folgenden Erörterungen dienen die Abbildungen 5.1 und 5.3 als Grundlage. Es
werden zunächst die Proben betrachtet, deren NMMO-Gehalt zwischen 70 und 75
Ma% liegt. Diese Proben haben die Gemeinsamkeit, daß für sie jeweils nur ein DSC-
Schmelzpeak verzeichnet wurde. Das ist ein Hinweis dafür, daß in diesen Proben ledig-
lich eine einzige kristalline Phase vorliegt, die bei ca. 44 °C aufschmilzt. Bei dieser Pha-
se handelt es sich um das Kristallgitter des NMMO⋅2,5-Hydrats.
Eine signifikante Änderung der Schmelztemperaturen und -enthalpie in Abhängigkeit
vom NMMO-Gehalt wird für diese Proben nicht beobachtet. Bemerkenswert ist aber,
daß die Schmelzpeaks dieser Proben relativ schmal und hoch ausfallen. Außerdem
wurde für jene Proben vergleichsweise hohe Enthalpiebeträge bestimmt, obwohl sie

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
58
kleine Schmelztemperaturwerte besitzen. Die Ursache dafür kann darin liegen, daß in
diesen Proben die Größenverteilung der Kristallite des 2,5-NMMO-Hydrats relativ eng
und zudem ihre kristalline Ordnung verhältnismäßig gut ausgeprägt ist, wodurch hohe
Enthalpiewerte zustande kommen.
Die Feststellungen bezüglich der Proben, deren NMMO-Gehalt ≥ 78 Ma% betragen,
und deren DSC-Thermogramme zwei Schmelzpeaks beinhalten, können folgenderma-
ßen gedeutet werden: Die zwei erhaltenen DSC-Peaks weisen auf zwei unterschied-
liche, nebeneinander vorliegende kristalline Phasen in den Proben hin. Es sind die
Kristallmodifikationen des 2,5-Hydrats und des Monohydrats des NMMO, deren
Schmelztemperaturen laut Literaturangabe entsprechend im Temperaturbereich 31 -
45 °C und 70 - 85 °C liegen.
Der Übergangsbereich, der sich bei NMMO-Gehalten zwischen 78 und 85 Ma%
offenbart, und die Enthalpiewerte des 1. und 2. Schmelzpeaks in diesem Bereich zei-
gen, daß mit Zunahme der NMMO-Konzentration in den Proben der kristalline Anteil
der Monohydratphase kontinuierlich ansteigt und im gleichen Maße der des 2,5-Hyd-
rats abnimmt.
Das Auftreten eines zweiten Schmelzpeaks in den DSC-Thermogrammen kann auch
ein Hinweis dafür sein, daß ab diesen NMMO-Gehalten die Auflösung der Cellulose im
NMMO/H2O-Gemisch beginnt. Dies würde für die Kristallmodifikation der Cellulose
eine Umwandlung von Cellulose I zu II bedeuten.
Eine weitere nennenswerte Beobachtung hängt mit den Proben zusammen, deren
NMMO-Gehalt ≥ 75 Ma% beträgt. Die Gesamtenthalpie dieser Proben geht mit Zu-
nahme der NMMO-Konzentration von 140 auf 120 J/g zurück. Dieses Resultat kann so
gedeutet werden, daß ein Teil der in der Probe enthaltenen NMMO-Moleküle an den
Hydroxylgruppen der Cellulose gebunden und folglich der Auflöseprozeß in Gang
gebracht wird. Dies heißt aber, daß für die Kristallbildung des NMMO/H2O-Gemisches
dieser Anteil an NMMO-Molekülen nicht mehr zur Verfügung steht und hierdurch ein
weiteres Kristallwachstum unterbunden wird. Deshalb fällt aller Wahrscheinlichkeit
nach die Gesamtschmelzenthalpie für diese Proben kleiner aus als für die Proben, bei
denen keine Celluloseauflösung stattfindet.
Mit dem DSC wurde hinsichtlich der Löslichkeit der Cellulose in den NMMO/H2O-
Gemischen lediglich die oben diskutierte Abhängigkeit vom Gehalt des NMMO gemes-
sen. Eine signifikante Abhängigkeit der mittels DSC bestimmten Größen (Schmelz-

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
59
temperaturen und -enthalpie) von der Behandlungszeit tB und der Behandlungs-
temperatur TB der Proben wurde jedoch nicht beobachtet. Diese Gegebenheit, daß
also bezüglich der Löslichkeit der Cellulose in den unterschiedlichen NMMO/H2O-
Gemischen mit Hilfe des DSC keine weiteren nennenswerten Abhängigkeiten gemes-
sen wurden, läßt den möglichen Schluß zu, daß das DSC als Meßapparat für diesen
Untersuchungszweck nicht die nötige Empfindlichkeit besitzt und deshalb auf den
Auflöseprozeß der Cellulose bzw. auf die Wechselwirkung zwischen dem Lösemittel-
system und der Cellulose nicht angemessen reagiert.
Im nächsten Abschnitt werden ergänzende Messungen mittels Röntgenweitwinkel-
streuung an ausgewählten NMMO/H2O/Cellulose-Gemischen durchgeführt, um die
eventuelle Auflösung der Cellulose in den genannten Gemischen nachzuweisen und
die für die Auflösung nötigen experimentellen Bedingungen genau anzugeben.

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
60
5.2 Ergänzende Messungen am ternären Stoffsystem NMMO/H2O/Cellulose
5.2.1 Lichtmikroskopische Untersuchungen am System NMMO/H2O/Cellulose
In diesem Abschnitt wird der Quell- bzw. Lösezustand der im Dreistoffgemisch
NMMO/H2O/Cellulose enthaltenen Cellulosefasern mit Hilfe der Lichtmikroskopie un-
tersucht. Der Zweck dieser Untersuchung besteht darin, den gelösten Anteil an einge-
setzten Cellulosefasern im Stoffsystem NMMO/H2O/Cellulose in Abhängigkeit von der
NMMO-Konzentration, der Behandlungstemperatur TB, sowie von der Behandlungszeit
tB zu bestimmen.
5.2.1.1 Experimentelles zu den lichtmikroskopischen Untersuchungen
Der Lösezustand der Cellulosefasern im Stoffgemisch NMMO/H2O/Cellulose wurde
mittels eines Lichtmikroskops Leica DMLM charakterisiert. Das Mikroskop war mit
einem regelbaren Heiztisch, einer Videokamera und einem rechnergestützten Bild-
analysesystem ausgestattet. Bei der Untersuchung der Proben wurde das Mikroskop im
Durchlichtverfahren im linear polarisierten Licht mit gekreuzten Polarisatoren betrie-
ben. Dabei wurden zwei Phasenkontrastobjektive (N Plan, 10x/0.25; N Plan, 20x/0.4)
mit zehnfacher bzw. zwanzigfacher Vergrößerung verwendet.
Aus den NMMO/H2O/Cellulose-Gemischen, die bei -20 °C in verschlossenen PE-Beuteln
aufbewahrt waren und in erstarrter bzw. kristalliner Form vorlagen, wurde jeweils eine
kleine Probe entnommen und auf einen Objektträger gelegt. Der Objektträger mit
dem Probenmaterial wurde dann auf dem Heiztisch auf eine konstante Temperatur
von 72 °C erwärmt. Nach dem Aufschmelzen wurde die Probe mit einem Deckglas
versehen und unter dem Mikroskop beobachtet. Mit einer Videokamera wurden für
jede Probe zwei bis drei Bilder von verschiedenen Stellen des Probenmaterials aufge-
nommen und im Rechner gespeichert.
Für die Auswertung der lichtmikroskopischen Aufnahmen von NMMO/H2O/Cellulose-
Gemischen wurden mikroskopische Referenzaufnahmen einer Reihe von NMMO/H2O/-
Cellulose-Proben verwendet, bei denen der gelöste Anteil an eingesetzten Cellulose-
fasern nach dem Erscheinungsbild visuell abgeschätzt wurde (Abb. 5-7). Als Bewer-

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
61
tungskriterien für den Lösezustand der Cellulosefasern wurden hierbei Faserkennwerte
wie Faserhäufigkeit, Faserlänge sowie Oberflächenzustand der Fasern (z. B. charak-
teristische Kugelquellungen an den Fasern) herangezogen. Die Lösezustände der Cel-
lulosefasern wurden dann entsprechend Tab. 5-1 festgelegt.
minimale Auflösung / 10% geringfügige Auflösung / 30% mittlere Auflösung / 50% starke Auflösung / 70%
sehr starke Auflösung / 90% vollständige Auflösung / 100%
Abb. 5-7 Lichtmikroskopische Referenzaufnahmen einer Reihe von NMMO/H2O/Cellulose-Proben, bei denen der gelöste Anteil an eingebrachten Cellulosefasern unterschiedlich groß ist
Die Auswertung des gelösten Celluloseanteils in den Proben erfolgte durch Vergleich
der aufgenommenen mikroskopischen Bilder mit den erwähnten Referenzbildern. Da-
zu wurden die Bilder mit dem Software-Programm Corel PHOTO-PAINT auf dem
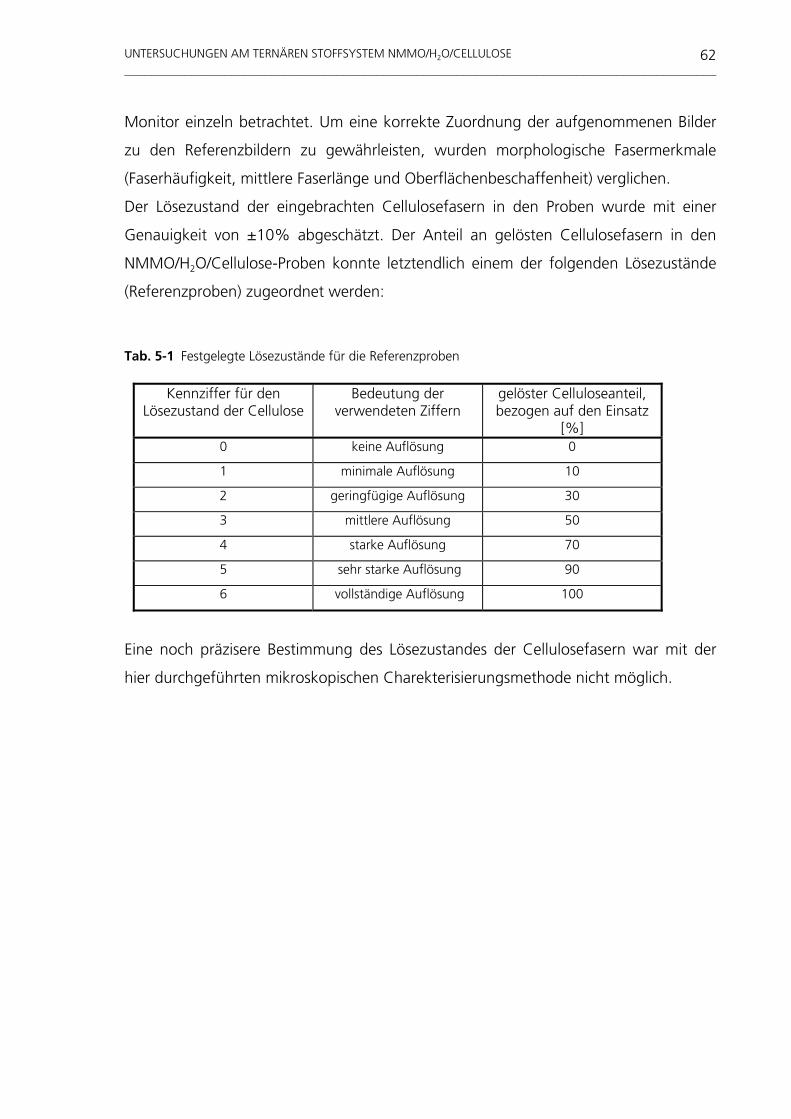
UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
62
Monitor einzeln betrachtet. Um eine korrekte Zuordnung der aufgenommenen Bilder
zu den Referenzbildern zu gewährleisten, wurden morphologische Fasermerkmale
(Faserhäufigkeit, mittlere Faserlänge und Oberflächenbeschaffenheit) verglichen.
Der Lösezustand der eingebrachten Cellulosefasern in den Proben wurde mit einer
Genauigkeit von ±10% abgeschätzt. Der Anteil an gelösten Cellulosefasern in den
NMMO/H2O/Cellulose-Proben konnte letztendlich einem der folgenden Lösezustände
(Referenzproben) zugeordnet werden:
Tab. 5-1 Festgelegte Lösezustände für die Referenzproben
Kennziffer für den Lösezustand der Cellulose
Bedeutung der verwendeten Ziffern
gelöster Celluloseanteil, bezogen auf den Einsatz
[%] 0 keine Auflösung 0
1 minimale Auflösung 10
2 geringfügige Auflösung 30
3 mittlere Auflösung 50
4 starke Auflösung 70
5 sehr starke Auflösung 90
6 vollständige Auflösung 100
Eine noch präzisere Bestimmung des Lösezustandes der Cellulosefasern war mit der
hier durchgeführten mikroskopischen Charekterisierungsmethode nicht möglich.

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
63
5.2.1.2 Ergebnisse
I. Einfluß der NMMO-Konzentration auf das Löseverhalten der Cellulose
Die mit Hilfe der Lichtmikroskopie bestimmten Anteile an gelöster Cellulose im Stoff-
gemisch NMMO/H2O/Cellulose sind in Abhängigkeit vom NMMO-Gehalt für eine feste
Behandlungstemperatur und -zeit in der Abb. 5-8 graphisch dargestellt.
Abb. 5-8 Relativer Anteil an gelöster Cellulose im Gemisch NMMO/H2O/Cellulose in Abhängigkeit vom NMMO-Gehalt; Celluloseanteil im Gemisch 8 %
Aus der Auftragung geht hervor, daß die Cellulose in den NMMO/H2O/Cellulose-
Proben mit NMMO-Gehalten 70 und 72 Ma% keine Auflösung erfährt. Im NMMO-
Konzentrationsbereich zwischen 75 und 80 Ma% findet dagegen eine partielle Auf-
lösung der Cellulose in den Proben statt, wobei der gelöste Celluloseanteil in der Probe
mit 80 Ma% NMMO-Gehalt schon relativ hoch ist. Eine vollständige Auflösung der
Cellulose zeigen die Proben, die einen NMMO-Gehalt ≥ 82 Ma% aufweisen. Die Abb.
5-8 zeigt außerdem, daß die Proben im Konzentrationsbereich zwischen 78 - 82 Ma%
zur Klärung des Löseverhaltens der Cellulose im NMMO/H2O-Gemisch von besonde-
rem Interesse sind. Deshalb wurde an diesen Proben der Einfluß einer Temperatur- und
Zeitvariation untersucht, der an dieser Stelle aufgezeigt werden soll.
70 75 80 85 90
0
20
40
60
80
100
120
Beh.temp. 90 °CBeh.zeit 60 min
gelö
ster
Cel
lulo
sean
teil
[%]
NMMO-Gehalt [Ma%]

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
64
II. Einfluß von Lösetemperatur und -zeit
Für die Untersuchungen zum Einfluß von Temperatur und Zeit auf den Lösezustand
der Cellulose in den NMMO/H2O/Cellulose-Gemischen wurden in den Löseversuchen
die Temperaturstufen 75, 80, 85, 90 und 95 °C gewählt und Lösezeiten von 0 bis 80
min eingestellt.
• Proben mit 78 Ma% NMMO-Gehalt
Die Abb. 5-9 zeigt den Anteil an gelöster Cellulose in NMMO/H2O/Cellulose-Proben in
Abhängigkeit von der Behandlungstemperatur TB. Diese Proben enthalten 8% Cellu-
lose, 92% NMMO/H2O-Gemisch und wurden über 60 min im Meßkneter unter mecha-
nischer Vermengung behandelt.
Abb. 5-9 Relativer Anteil an gelöster Cellulose im Gemisch NMMO/H2O/Cellulose in Abhängigkeit von
der Behandlungstemperatur TB; Celluloseanteil im Gemisch 8 % Der Abbildung 5-9 ist zu entnehmen, daß nach 60 min Behandlungszeit der gelöste
Anteil an zugesetzter Cellulose in den bei 75, 80 und 85 °C behandelten Proben nur
minimal ist. Mit zunehmender Temperatur nimmt auch der Anteil an gelöster Cellulose
zu und erreicht schließlich einen Wert von 50% bei 95 °C.
75 80 85 90 95
0
20
40
60
80
100
NMMO-Geh. 78 Ma%Beh.zeit 60 min
gelö
ster
Cel
lulo
sean
teil
[%]
Behandlungstemperatur TB [°C]

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
65
Eine Variation der Lösezeit ergibt für diese Proben ein ähnliches Resultat hinsichtlich
des Löseszustandes der Cellulose. Es stellt sich heraus, daß die Cellulose in den
NMMO/H2O/Cellulose-Proben mit 78 Ma% NMMO-Gehalt lediglich partiell aufgelöst
wird (Abb. 5-10). Der gelöste Celluloseanteil ist nach 40 min Lösezeit mit ca. 10% un-
beträchtlich und steigt mit zunehmender Lösezeit auf maximal 50% an.
Abb. 5-10 Relativer Anteil an gelöster Cellulose im Gemisch NMMO/H2O/Cellulose in Abhängigkeit von der Behandlungszeit tB; Celluloseanteil im Gemisch 8 %
• Proben mit 80 Ma% NMMO-Gehalt
Die Abb. 5-11 zeigt für diese Proben die mittels Lichtmikroskopie ermittelten Löse-
zustände der Cellulose als Funktion der Behandlungstemperatur TB bei konstanter
Behandlungszeit von 60 min. Die Löslichkeit der Cellulose weist hier einen annähernd
linearen Verlauf über der Temperatur auf. Nach 60 min Behandlungsdauer ist bereits
mehr als die Hälfte der eingesetzten Cellulose im Temperaturbereich zwischen 75 und
85 °C aufgelöst. Für die eingestellten Temperaturen 90 und 95 °C ist der Anteil an
gelöster Cellulose mit 80 bzw. 90% relativ groß, aber eine hundertprozentige Auf-
lösung tritt unter den hier eingestellten Versuchsbedingungen noch nicht ein.
0 20 40 60 80
0
20
40
60
80
100
NMMO-Geh. 78 Ma%Beh.temp. 90 °C
gelö
ster
Cel
lul.a
ntei
l [%
]
Behandlungszeit tB [min]
0 20 40 60 80
0
20
40
60
80
100
NMMO-Geh. 78 Ma%Beh.temp. 90 °C
gelö
ster
Cel
lulo
sean
teil
[%]
Behandlungszeit tB [min]

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
66
Abb. 5-11 Relativer Anteil an gelöster Cellulose im Gemisch NMMO/H2O/Cellulose in Abhängigkeit von der Behandlungstemperatur TB; Behandlungszeit 60 min, Celluloseanteil im Gemisch 8 %
Abb. 5-12 Relativer Anteil an gelöster Cellulose im Gemisch NMMO/H2O/Cellulose in Abhängigkeit von
der Behandlungszeit tB; Celluloseanteil im Gemisch 8 %
75 80 85 90 95
0
20
40
60
80
100
NMMO-Geh. 80 Ma%Beh.zeit 60 minge
löst
er C
ellu
lose
ante
il [%
]
Behandlungstemperatur TB [°C]
0 20 40 60 80
0
20
40
60
80
100
NMMO-Geh. 80 Ma%Beh.temp. 90 °Cge
löst
er C
ellu
lose
ante
il [%
]
Behandlungszeit tB [min]

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
67
In Abb. 5-12 ist der Lösezustand der Cellulose in den NMMO/H2O/Cellulose-Proben,
die ebenfalls einen NMMO-Gehalt von 80 Ma% besitzen, als Funktion der Behand-
lungszeit tB aufgetragen. Die eingestellte Probentemperatur für diese Zeitvariation be-
trägt 90 °C.
Bemerkenswert ist hierbei die Feststellung, daß die Hälfte der eingesetzen Cellulose
bereits nach 20 min Behandlungszeit aufgelöst wird. Die Auflösung des restlichen Cel-
luloseanteils dauert hingegen 60 min. Eine vollständige Auflösung der Cellulose wird
auch hier innerhalb der Behandlungszeit von 80 min nicht beobachtet.
• Proben mit 82 Ma% NMMO-Gehalt
Tab. 5-2 Lösezustand von Cellulose im System NMMO/H2O in Abhängigkeit von der Behandlungs-temperatur TB und der Behandlungszeit tB; Cellulosekonzentration im Gemisch 8 %; NMMO-Gehalt 82 Ma%
Beh.temperatur TB
[°C]
Beh.zeit tB
[min]
gelöster Celluloseanteil, bezogen auf den Einsatz
[%]
75 60 90
80 60 100
80 0 0
80 20 90
80 60 100
Tab. 5-2 gibt den Lösezustand der Cellulose in den NMMO/H2O/Cellulose-Proben (mit
82 Ma% NMMO-Gehalt) für zwei verschiedene Behandlungstemperaturen und unter-
schiedliche Behandlungszeiten wider.
Bei dem System mit 82 Ma% NMMO zeigt sich, daß der gelöste Anteil an eingebrach-
ter Cellulose bereits bei 75 °C verhältnismäßig hoch ist. Für diese Temperatur beträgt
der gelöste Cellulosanteil in diesen Proben nach 60 min Behandlungszeit 90%. Eine
vollständige Auflösung der Cellulose tritt jedoch erst bei 80 °C für dieselbe Behand-
lungsdauer von 60 min ein.
Bei einer Variation der Behandlungszeit für die bei 80 °C behandelten Proben stellt
sich heraus, daß nach 20 min 90% der Cellulosefasern und nach 60 min die Gesamt-
heit der eingebrachten Cellulosefasern im gelösten Zustand vorliegt.

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
68
5.2.2 Röntgenbeugung am Stoffsystem NMMO/H2O/Cellulose
An einigen ausgewählten NMMO/H2O/Cellulose-Gemischen wurden Röntgenweitwin-
kelexperimente vorgenommen. Das Ziel dabei war, die Auflösung der Cellulose im
Lösemittelsystem NMMO/H2O als Funktion der Probenzusammensetzung (d. h. des
NMMO/H2O-Verhältnisses), der Temperatur und der Zeit aufzuzeigen. Ausgangspunkt
ist die Überlegung, daß infolge des Auflöseprozesses der Cellulose im Lösemittelge-
misch NMMO/H2O und anschließender Koagulation der Celluloselösung im Fällbad
eine Gitterumwandlung von Cellulose I in Cellulose II stattfindet, die mit der Röntgen-
diffraktometrie leicht nachweisbar ist.
5.2.2.1 Experimentelles zu den Röntgenweitwinkeluntersuchungen
Die Röntenbeugungsversuche an Proben des ternären Stoffsystems wurden mittels
Filmkamera unter den bei 4.2.1 angegebenen experimentellen Bedingungen aufge-
nommen, wobei hier der Abstand zwischen Probe und Film 60 mm und die Belich-
tungszeit 30 Minuten betrug.
Die Proben des ternären Stoffsystems NMMO/H2O/Cellulose wurden vor den Röntgen-
streuexperimenten mit destilliertem Wasser ausgewaschen, so daß nur noch Cellulose
zurückblieb. Diese wurde dann im Trockenschrank bei 40 °C über 24 h getrocknet.
Um die Cellulose-I-II-Umwandlung im Dreistoffsystem zu untersuchen, wurden vier
Probenreihen mittels RWWS vermessen. In zwei Reihen wurde die Umsetzung von
Cellulose I in Cellulose II in Abhängigkeit von der Behandlungstemperatur TB, in den
zwei anderen als Funktion der Behandlungszeit tB bestimmt.

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
69
5.2.2.2 Ergebnisse
Zum Zwecke der Auswertung der aufgenommenen Röntgenfilmaufnahmen wurden
Referenz-Streufilmaufnahmen einer definierten Mischungsreihe von Proben der beiden
Gittermodifikationen herangezogen [86]. Durch Vergleich der eigenen Planfilmauf-
nahmen mit den Referenz-Streuaufnahmen wurde geprüft, ob und in welchem Maße
eine Cellulose-I-II-Umwandlung in den Proben stattgefunden hat. Im Falle einer
Cellulose-Umsetzung wurde das Mengenverhältnis Cellulose I : Cellulose II anhand der
Planfilmaufnahmen mit einer Genauigkeit von ±10% bestimmt.
I. Cellulose-I-II-Umwandlung als Funktion der Behandlungstemperatur TB
Die kristalline Ordnung der nativen Cellulose I wird unter geeigneten Bedingungen
durch die Aufnahme von NMMO/H2O im Dreistoffgemisch NMMO/H2O/Cellulose zer-
stört, so daß die Cellulose in Lösung geht. Infolge von NMMO-Entzug (Waschen) ent-
steht Wassercellulose und nach Wasserentzug (Trocknen) Cellulose II, welche die
stabilste Cellulosemodifikation darstellt.
Bei den mit Röntgenbeugung untersuchten Proben handelt es sich um solche, bei
denen die Temperatur TB in Löslichkeitsexperimenten zwischen 75 und 95 °C in 5 °C-
Schritten variiert wurde. Ihr NMMO-Gehalt betrug 78 bzw. 80 Ma% und die Behand-
lungsdauer im Meßkneter 60 min.
• Proben mit 78 Ma% NMMO-Gehalt, Behandlungszeit 60 min
Die Planfilmaufnahmen der Proben, die bei 75, 80 und 85 °C behandelt wurden, zei-
gen keine erkennbare Änderung der Gittermodifikation. Die Gitterumwandlung der
Cellulose I setzt erst bei 90 °C ein. Der Cellulose-II-Anteil in dieser Probe wurde mit
30% bestimmt. Die bei 95 °C behandelte Probe hat einen höheren Anteil an umge-
setzter Cellulose. Der Anteil beträgt hierbei 50%.
• Proben mit 80 Ma% NMMO-Gehalt, Behandlungszeit 60 min
Die Proben der zweiten Probenserie, bei der ebenfalls eine Temperaturvariation vor-
genommen wurde, hatten einen NMMO-Gehalt von 80 Ma% und waren über eine

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
70
Zeitdauer von 60 min im Meßkneter bei den oben genannten Temperaturen
behandelt.
Abb. 5-13 zeigt die Röntgenfilmaufnahmen von Celluloseproben einer Reihe von Pro-
ben mit einem NMMO-Gehalt von 80 Ma% und einer Behandlungsdauer von 60 min.
Diese Celluloseproben waren Bestandteile von NMMO/H2O/Cellulose-Gemischen und
wurden später für die Röntgenuntersuchungen durch Waschen vom Lösemittelsystem
NMMO/H2O getrennt und anschließend getrocknet.
unbehandelt TB = 75 °C TB = 80 °C Cellulose I
TB = 85 °C TB = 90 °C TB = 95 °C Cellulose II
Abb. 5-13 Röntgen-Planfilmaufnahmen von V60-Zellstoffproben, die im Lösemittelgemisch NMMO/-
H2O (80 Ma% NMMO, 20 Ma% H2O) 60 min lang bei unterschiedlichen Temperaturen behandelt, neutralisiert und anschließend getrocknet wurden
Abb. 5-14 zeigt für diese Proben die aus den Röntgenaufnahme ermittelte Cellulose-II-
Anteile über der Behandlungstemperatur TB.

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
71
Die Auswertung der Röntgenstreudiagramme dieser Proben ergab, daß eine Cellulose-
I-II-Umwandlung bereits bei 75 °C eintritt. Der Cellulose-II-Anteil dieser Probe beträgt
30%. Die bei 80 °C behandelte Probe weist einen Cellulose-II-Anteil von 50% auf und
für die Probe, die bei 85 °C behandelt war, wird ein recht hoher Cellulose-II-Anteil von
70% festgestellt. Der Cellulose-II-Anteil steigt in der bei 90 °C behandelten Probe auf
80% an, und für die bei 95 °C behandelten Probe wird eine vollständige Umwandlung
der Cellulose I zu Cellulose II registriert.
Abb. 5-14 Aus den WAXS-Aufnahmen bestimmter Cellulose-II-Anteil der Proben in Abhängigkeit von der Behandlungstemperatur TB, NMMO-Geh. 80 Ma%, Behandlungszeit 60 min
II. Cellulose-I-II-Umwandlung als Funktion der Behandlungszeit tB
• Proben mit 78 Ma% NMMO-Gehalt, Behandlungstemperatur 90 °C
Die Auswertung der Röntgenaufnahmen dieser Proben ergab, daß eine Variation der
Behandlungszeit tB (0 - 80 min) keine wesentliche Umwandlung von Cellulose I zu II
zur Folge hatte. Eine Cellulose-I-II-Umwandlung weist nur die Probe mit 80 min Be-
handlungsdauer auf, die einen Cellulose-II-Anteil von 20% hat.
75 80 85 90 950
20
40
60
80
100
Cel
lulo
se-II
-Ant
eil [
%]
Behandlungstemperatur TB [°C]
NMMO-Geh. 80 Ma%Beh.zeit 60 min

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
72
• Proben mit 80 Ma% NMMO-Gehalt, Behandlungstemperatur 90 °C
Der Einfluß der Behandlungsdauer auf die Gitterumwandlung der Cellulose I tritt bei
diesen Proben stärker in Erscheinung als für die oben beschriebenen. Der aus den
Röntgenaufnahmen bestimmte Cellulose-II-Anteil der Proben ist als Funktion der Be-
handlungszeit tB in Abb. 5-15 dargestellt. Eine Gitterumwandlung der Cellulose I setzt
innerhalb von 20 min Behandlungszeit ein. Der Anteil an Cellulose II ist nach 20 min
mit 65% relativ hoch. Ab hier steigt der Cellulose-II-Anteil mit zunehmender Zeit linear
an. Nach 80 min Behandlungsdauer ist eine fast vollständige Umwandlung der Cellu-
lose I in das Gitter der Cellulose II erfolgt. Eine komplette Cellulose-I-II-Umwandlung
wird für diese Proben innerhalb von 80 min jedoch nicht beobachtet.
Abb. 5-15 Aus den WAXS-Aufnahmen bestimmter Cellulose-II-Anteil der Proben in Abhängigkeit von der Behandlungszeit tB, NMMO-Geh. 80 Ma%, Behandlungstemperatur 90 °C
0 20 40 60 80 100
0
20
40
60
80
100
Cel
lulo
se-II
-Ant
eil [
%]
Behandlungszeit tB [min]
NMMO-Geh. 80 Ma%Beh.temperatur 90 °C

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE _________________________________________________________________________________________
73
5.3 Das Phasendiagramm für das ternäre System NMMO/H2O/Cellulose
Die in diesem Abschnitt ermittelten Meßdaten über die Löslichkeit der Cellulose im Lö-
semittelgemisch NMMO/H2O sind in einem Zustandsdiagramm für das ternäre System
auf der nächsten Seite zusammengetragen. Auf der unteren Abszisse ist der H2O-
Gehalt, auf der oberen Abszisse der NMMO-Gehalt und auf der Ordinate die Tempera-
tur eingetragen. Der Celluloseanteil im System NMMO/H2O/Cellulose beträgt 8%. Die
Angaben im Phasendiagramm hinsichtlich der Löslichkeit der Cellulose im Lösemittel-
gemisch NMMO/H2O gelten für eine Einwirkzeit von 60 min.
Die Kurve 1 gibt die berechneten Schmelzpeaktemperaturen wieder, die für das binäre
System NMMO/H2O (nach Gleichung (8), Abschnitt 4.3) bestimmt wurden. Die um
diese Liquidus-Kurve verteilten Punkte sind die experimentell bestimmten Schmelz-
punkte für das binäre System. Die Kurve 2 stellt die Liquidus-Kurve für das reine Mo-
nohydrat dar, die der Arbeit von Chanzy et al. [50] entnommen wurde. Die gestrichel-
te Linie (Nr. 5) gibt die maximale Temperatur an, die bei den Löslichkeitsmessungen
eingestellt wurde.
Bei den Kurven 3 und 4 handelt es sich um die Kurven, welche die drei unterschied-
lichen Phasenbereiche für die Cellulose kennzeichnen. (Diese Kurven beruhen auf
Meßdaten aus den Löslichkeitsuntersuchungen aus 5.2.1 und 5.2.2.) Im Phasenbereich
A (H2O-Gehalt > 22 Ma%) kommt es bei Temperaturen < 90 °C lediglich zu einer
Quellung der Cellulose im Lösemittelsystem NMMO/H2O. Im Bereich B (H2O-Gehalt: 19
- 22 Ma%) löst sich die Cellulose im Temperaturintervall 70 - 95 °C teilweise auf, und
im Bereich C (H2O-Gehalt < 19 Ma%) wird die Cellulose bei Temperaturen > 75 °C
vollständig im NMMO/H2O-Gemisch aufgelöst.

UNTERSUCHUNGEN AM TERNÄREN STOFFSYSTEM NMMO/H2O/CELLULOSE __________________________________________________________________________________________________________________________________________
74
Abb. 5-16 Phasendiagramm des ternären Stoffsystems NMMO/H2O/Cellulose
0 5 10 15 20 25 3040
50
60
70
80
90
100
110
120
Tem
pera
tur
[°C
]
H2O [%]
Celluloseanteil im System 8% Einwirkzeit 60 min
A: reversible Quellung
B: teilweise Auflösung
C: vollständige Auflösung
ABC
NMMO [%]
100 95 90 85 80 75 70
1
2
3
4
5

VERGLEICH DER SYSTEME HINSICHTLICH DER THERMISCHEN EIGENSCHAFTEN _________________________________________________________________________________________
75
5.4 Vergleich der Systeme NMMO/H2O und NMMO/H2O/Cellulose hinsichtlich der thermischen Eigenschaften
In diesem Abschnitt der Arbeit werden das binäre und das ternäre Stoffsystem hin-
sichtlich ihres thermischen Verhaltens miteinander verglichen. Der Vergleich soll dazu
dienen, Aussagen über mögliche Wechselwirkungen zwischen dem Lösemittelsytem
NMMO/H2O und der Cellulose zu treffen.
Die Ergebnisse, die über das binäre und das ternäre System als Funktion der NMMO-
Konzentration erhalten und auf den vorangegangenen Seiten beschrieben wurden,
sollen in diesem Abschnitt gegenübergestellt und zusammenfassend diskutiert wer-
den; Grundlage dieser Diskussion sind die Abbildungen 5-17 bis 5-19. Für die Erörte-
rung der Ergebnisse wird es als vorteilhaft angesehen, die folgenden Abbildungen in
drei NMMO-Konzentrationsbereiche aufzuteilen, und zwar I: 70 - 78 Ma%, II: 78 - 85
Ma% und III: 85 - 91 Ma%.
In den Abbildungen 5-17 und 5-18 sind die Schmelzpeaktemperaturen und die
Schmelzenthalpien der beiden Systeme als Funktion der NMMO-Konzentration gra-
phisch dargestellt.
Abb. 5-17 Schmelzpeaktemperaturen (Tpm) des binären und des ternären Stoffsystems als Funktion des NMMO-Gehalts
70 75 80 85 90 950
20
40
60
80
100
120
Schm
elzp
eakt
empe
ratu
r T pm
[°C
]
NMMO-Gehalt [Ma%]
Tpm
/ binär
1.Tpm
/ ternär
2.Tpm
/ ternär
- Vergleich der Systeme: NMMO/H2O - NMMO/H
2O/Cellulose -

VERGLEICH DER SYSTEME HINSICHTLICH DER THERMISCHEN EIGENSCHAFTEN _________________________________________________________________________________________
76
Ein Vergleich der Schmelzpeaktemperaturen der beiden Stoffsysteme (siehe Abb. 5-17)
zeigt, daß durch die Anwesenheit der Cellulose im Lösemittelsystem NMMO/H2O die
Peaktemperaturen (2.Tpm) um ca. 5 - 10 °C abgesenkt wurden.
Die Schmelzenthalpien des binären Systems (Abb. 5-18) fallen im Konzentrations-
bereich I und II wesentlich kleiner aus als die des ternären Systems. Dieser Sachverhalt
ist auf das Kristallisationsverhalten der beiden Systeme zurückzuführen. Das binäre
System kristallisiert im Konzentrationsgebiet I und II bei weitem nicht so gut wie das
ternäre Stoffsystem.
Da das ternäre System ab 78 Ma% NMMO-Gehalt zwei unterschiedliche Schmelz-
peaks besitzt, sind auch in den Darstellungen zwei verschiedene Peaktemperaturen
(1.Tpm; 2.Tpm) bzw. zwei Schmelzenthalpien (1.∆H; 2.∆H) für dieses System angege-
ben.
Abb. 5-18 Schmelzenthalpien (∆H) des binären und des ternären Stoffsystems als Funktion des NMMO-Gehalts
Ein Resultat betrifft den Verlauf der Enthalpiewerte der 2. Schmelzpeaks des ternären
Systems. Der Abb. 5-18 ist zu entnehmen, daß die Enthalpiewerte, die den 2. Schmelz-
70 75 80 85 90 95
0
20
40
60
80
100
120
140
160
180
200
Schm
elze
ntha
lpie
∆H
[J/g
]
∆H/ binär 1.∆H/ ternär 2.∆H/ ternär
- Vergleich der Systeme: NMMO/H2O - NMMO/H
2O/Cellulose -
NMMO-Gehalt [Ma%]
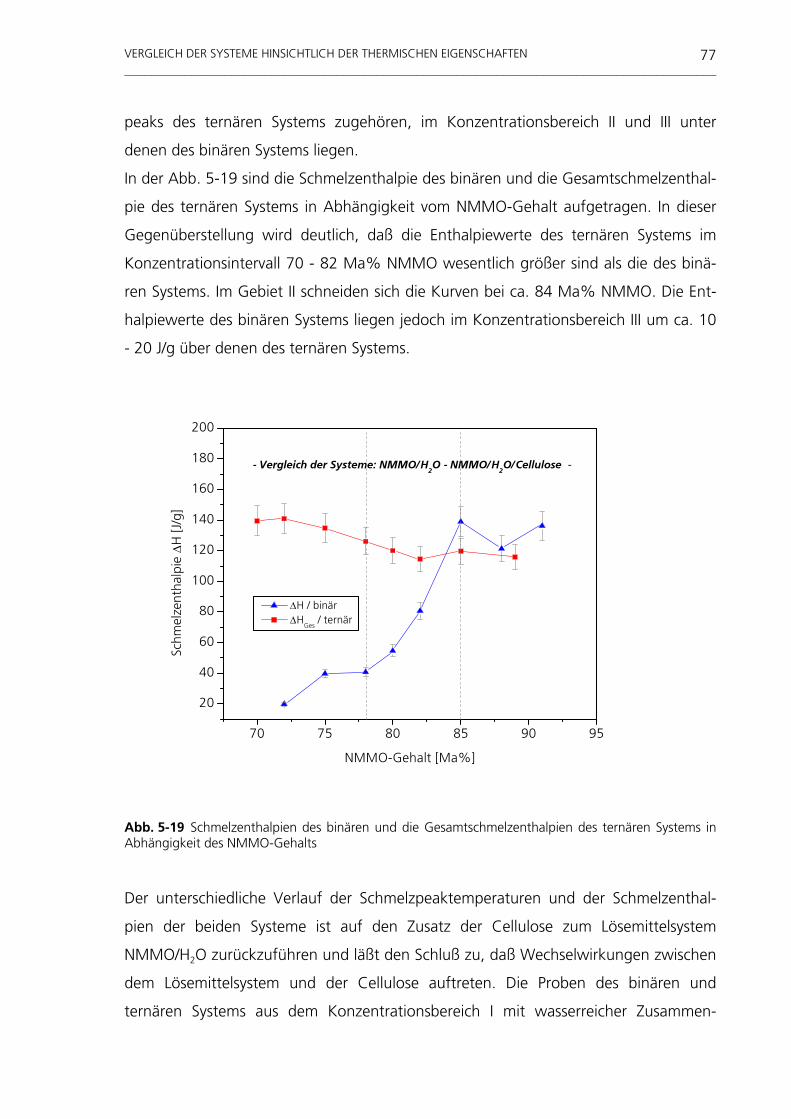
VERGLEICH DER SYSTEME HINSICHTLICH DER THERMISCHEN EIGENSCHAFTEN _________________________________________________________________________________________
77
peaks des ternären Systems zugehören, im Konzentrationsbereich II und III unter
denen des binären Systems liegen.
In der Abb. 5-19 sind die Schmelzenthalpie des binären und die Gesamtschmelzenthal-
pie des ternären Systems in Abhängigkeit vom NMMO-Gehalt aufgetragen. In dieser
Gegenüberstellung wird deutlich, daß die Enthalpiewerte des ternären Systems im
Konzentrationsintervall 70 - 82 Ma% NMMO wesentlich größer sind als die des binä-
ren Systems. Im Gebiet II schneiden sich die Kurven bei ca. 84 Ma% NMMO. Die Ent-
halpiewerte des binären Systems liegen jedoch im Konzentrationsbereich III um ca. 10
- 20 J/g über denen des ternären Systems.
Abb. 5-19 Schmelzenthalpien des binären und die Gesamtschmelzenthalpien des ternären Systems in Abhängigkeit des NMMO-Gehalts Der unterschiedliche Verlauf der Schmelzpeaktemperaturen und der Schmelzenthal-
pien der beiden Systeme ist auf den Zusatz der Cellulose zum Lösemittelsystem
NMMO/H2O zurückzuführen und läßt den Schluß zu, daß Wechselwirkungen zwischen
dem Lösemittelsystem und der Cellulose auftreten. Die Proben des binären und
ternären Systems aus dem Konzentrationsbereich I mit wasserreicher Zusammen-
70 75 80 85 90 95
20
40
60
80
100
120
140
160
180
200
Schm
elze
ntha
lpie
∆H
[J/g
]
NMMO-Gehalt [Ma%]
∆H / binär ∆H
Ges / ternär
- Vergleich der Systeme: NMMO/H2O - NMMO/H
2O/Cellulose -

VERGLEICH DER SYSTEME HINSICHTLICH DER THERMISCHEN EIGENSCHAFTEN _________________________________________________________________________________________
78
setzung bestehen hauptsächlich aus dem 2,5-Hydrat des NMMO und vermutlich zu
einem geringfügigen Teil aus einer Mischphase aus NMMO und H2O. In den NMMO/-
H2O/Cellulose-Proben können die NMMO-Moleküle entweder mit den Wasser-
molekülen oder mit den Cellulosemolekülen über Wasserstoffbrückenbindungen
wechselwirken. Die Messungen zeigen, daß die NMMO-Moleküle eine Wechsel-
wirkung mit den Wassermolekülen bevorzugen. Das heißt, im Dreistoffsystem
NMMO/H2O/Cellulose tritt zunächst eine Wechselwirkung zwischen den NMMO- und
den Wassermolekülen ein; erst dann kommt es zu einer Wechselwirkung zwischen
dem gebildeten NMMO-Hydrat und der Cellulose. Das ist auch der Grund dafür, daß
die Cellulose in NMMO-Hydraten mit NMMO-H2O-Zusammensetzungen wie im Kon-
zentrationsbereich I nicht in Lösung geht. Dort ist die Konzentration an frei verfüg-
baren Wassermolekülen so hoch, daß die NMMO-Moleküle mit diesen vorzugsweise
wechselwirken und dadurch das NMMO-Hydrat mit n = 2,5 ausbilden. Im Konzen-
trationsbereich I findet demzufolge lediglich eine begrenzte Quellung der Cellulose
statt. Die kristalline Struktur der Cellulose bleibt dabei erhalten. Die Quellung der Cel-
lulosefasern ist jedoch ein Indiz dafür, daß eine Wechselwirkung, wenn auch eine
schwache, zwischen den Molekülen des NMMO-Hydrats und den Cellulosemolekülen
stattfindet.
Der Konzentrationsbereich II stellt einen Übergangsbereich bezüglich der Hydrat-
phasenbildung des NMMO und dem kristallinen Zustand der Cellulose dar. Die Proben
aus diesem Bereich setzen sich - je nach ihrer Zusammensetzung - aus einem Anteil
aus 2,5-Hydrat, einem Anteil aus Monohydrat und vermutlich einem kleinen Bruchteil
aus einer Mischhydratphase mit variabler Hydratationszahl n zusammen. Aufgrund der
verhältnismäßig großen Änderungen der Schmelztemperatur- und Enthalpiewerte in
diesem Konzentrationsgebiet wird ein konkurrierender Bildungsprozeß für die beiden
Hydrate des NMMO angenommen. Zunächst überwiegt der Anteil des 2,5-Hydrats
(NMMO-Geh. 78 - 80 Ma%) - wie die Enthalpiebeträge dies zeigen -, aber mit steigen-
der NMMO-Konzenration nimmt dieser Anteil ab, wobei der Anteil der Monohydrat-
phase immer größer wird. Ab einem NMMO-Gehalt von 82 Ma% ist die Monohydrat-
phase dominierend.
Die Wechselwirkung des Lösemittelsystems mit der Cellulose nimmt in diesem Bereich
an Stärke zu, da ab diesem Mengenverhältnis von NMMO und H2O (NMMO-Gehalt ≥
78 Ma%) die Bildung der Monohydratphase und eventuell einer Mischphase aus

VERGLEICH DER SYSTEME HINSICHTLICH DER THERMISCHEN EIGENSCHAFTEN _________________________________________________________________________________________
79
NMMO und H2O in den Proben möglich ist. Diese Mischphase besitzt wahrscheinlich
eine variable Hydratationszahl n, die kleiner ist als die des 2,5-Hydrats. Besonders im
Konzentrationsbereich 78 - 82 Ma% an NMMO findet - bei den hier eingestellten
experimentellen Bedingungen - eine signifikante Veränderung hinsichtlich der Wech-
selwirkung zwischen den Lösemittel - und den Cellulosemolekülen statt. Diese Wech-
selwirkung macht sich dadurch bemerkbar, daß die zwischenmolekularen Anziehungs-
kräfte zwischen den Molekülen des Lösemittelsystems und der Cellulose größer wer-
den als die der Einzelkomponenten, d. h. der intramolekularen Nebenvalenzkräfte zwi-
schen den hydratisierten NMMO-Molekülen einerseits und denen der Cellulosemo-
leküle andererseits. Bei abnehmender H2O-Konzentration werden wegen zuneh-
mendem Mangel an freien H2O-Molekülen somit zunehmend Hydroxylgruppen der
Cellulose in die Wechselwirkung mit den NMMO-Molekülen einbezogen. Dies hat
dann zur Folge, daß die übermolekulare kristalline Ordnung der Cellulose nach und
nach abnimmt und eine Auflösung der Cellulose im Lösemittelsystem eintritt.
In den Proben aus dem Konzentrationsbereich III, die eine wasserarme Zusammen-
setzung besitzen, liegt fast ausschließlich die Phase des Monohydrats vor. Für eine
zusätzliche Wechselwirkung der NMMO-Moleküle mit H2O-Molekülen sind praktisch
keine weiteren Wassermoleküle vorhanden. Da aber die NO-Gruppe des NMMO-
Moleküls bis zu zwei stabile Wasserstoffbrückenbindungen mit hydroxylgruppenhalti-
gen Verbindungen eingehen kann, erfolgt in dem vorliegenden Fall die zweite Verbin-
dung mit den Hydroxylgruppen der Cellulosemoleküle. Weiterhin ist die räumliche
Struktur des Monohydrats hinsichtlich seiner Größe für den sterischen Zugang zu den
Hydroxylgruppen der Cellulose günstiger als die des NMMO-2,5Hydrats. Diese beiden
Eigenschaften des NMMO-Monohydrats können mögliche Erklärungen dafür sein, daß
die Wechselwirkung zwischen dem Lösemittelsystem NMMO/H2O und der Cellulose in
diesem Bereich am stärksten ist. Für die Cellulose heißt das, daß sie relativ rasch in Lö-
sung geht, und daß sie in diesen Proben in amorpher Form vorliegt, gleichgültig, ob
die Proben im festen (d. h. in erstarrter Schmelzelösung) oder im flüssigen Zustand
sind. (Der gesamte Löseprozeß erfolgt nach Zugabe der Cellulose zur NMMO/H2O-
Lösung bei einer Temperatur von ca. 90 °C innerhalb weniger Minuten.) Wie bereits
früher mittels Röntgen-RDF-Analyse gezeigt wurde [87], wird die nichtkristalline
Nahordnung des NMMO/H2O-Systems durch die gelöste Cellulose nur unwesentlich
gestört, jedenfalls bildet sich infolge der Cellulose keine qualitativ andere Struktur.

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
80
6 Herstellung und Prüfung von faserverstärkten Cellulosefolien
Gegenstand dieses Abschnitts ist der Einsatz des Aminoxid- und Blasextrusionsver-
fahrens, um einen Verbundwerkstoff aus Cellulose und cellulosischen Fasern herzu-
stellen. Es sollte zunächst geprüft werden, ob die Herstellung einer Verbundlösung
(aus NMMO, H2O, gelöster Cellulose sowie zusätzlich eingebrachten ungelösten
Cellulosefasern) und deren Verarbeitung zu Cellulosefolien überhaupt möglich ist. Bei
positivem Ausgang dieses Experiments sollten dann die hergestellten Cellulosefolien
auf ihre mechanischen und morphologischen Eigenschaften hin untersucht werden.
Das eigentliche Ziel dieser anwendungsbezogenen Untersuchung ist, die mechani-
schen Eigenschaften wie Festigkeit und E-Modul der Cellulosefolien durch die zusätz-
lich in die Cellulosematrix eingebrachten, hochorientierten Cellulosefasern zu verbes-
sern. Damit soll das leichte Weiterreißen der Folien an vorhandenen Rissen verhindert
und ein isotropes Verhalten der Folien bezüglich der Festigkeitseigenschaften erreicht
werden.
Unter Hinzunahme der in den letzten Abschnitten dargelegten Resultate (über das
Stoffsystem NMMO/H2O/Cellulose) sollte schließlich versucht werden, die Versuchser-
gebnisse dieses Abschnitts zu deuten.
Es ist bekannt, daß bei Verbunden, bei denen Fasern in eine Polymermatrix einge-
bracht werden, häufig Probleme bezüglich der Faser-Matrix-Haftung entstehen. Diese
Schwierigkeiten werden meistens durch einen aufwendigen Einsatz von Haftvermitt-
lern überwunden. Dadurch, daß hier aber ausschließlich Cellulosefasern in eine Cellu-
losematrix eingebracht werden, sollten die Probleme der Faser-Matrix-Haftung von
vornherein unterbunden werden. Es wird nämlich angenommen, daß die Oberflächen
der zugesetzten Fasern in der Celluloselösung gequollen oder geringfügig gelöst wer-
den. Hierdurch sollten dann die Fasern beim späteren Koagulationsprozeß fest in die
Cellulosematrix eingebettet werden.
Eine weitere Überlegung hierbei ist, daß durch das Einbringen von Cellulosefasern in
eine Cellulosematrix ein Verbundmaterial entsteht, das umweltfreundlich entsorgt
werden kann, da sowohl die Matrix als auch die Faser biologisch abbaubar sind.
Ein anderer Grund, bei der Herstellung eines solchen Verbundmaterials ausschließlich
Cellulose und cellulosisches Fasermaterial zu verwenden, liegt in der Absicht, diesen

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
81
Verbundwerkstoff unter anderem als Verpackungsmaterial für Lebensmittel (z. B. als
Wursthüllen) zu nutzen.
6.1 Experimentelles
6.1.1 Verwendete Materialien
Für die hier vorgenommenen Untersuchungen wurde der Zellstoff Buckeye V60, ein
Vorhydrolyse-Kiefernsulfatzellstoff, mit einem Cuoxam-DP von 535 und einem α-Cellu-
losegehalt von 95,5% eingesetzt.
Als Verstärkungswerkstoffe wurden die in Tab. 6-1 aufgeführten cellulosischen Fasern
verwendet. Bei den Mikrofasern, mittellange Fasern 1 und 2 sowie den Langfasern
handelt es sich um Holzfasern, beim Lyocell um eine Regeneratfaser.
Tab. 6-1 Eingesetzte Cellulosefasern
6.1.2 Herstellung der Celluloselösung (Matrixlösung)
Es wurde zunächst eine 8,5 %ige Celluloselösung, die im weiteren als Matrixlösung
bezeichnet wird, in einem Laborkneter hergestellt. Es war möglich, mit dem Labor-
kneter in einem Prozeßlauf 3,5 kg Lösung zu präparieren. Die Lösungsherstellung
basierte auf der Verwendung des Lösemittelsystems NMMO/H2O und wird im folgen-
den kurz beschrieben:
Der gemahlene Zellstoff V60 wurde in 50 %iger wäßriger NMMO-Lösung im Labor-
kneter getränkt. Zur Verhinderung der NMMO-Zersetzung und eines DP-Abbaus der
Cellulose wurde dem Gemisch 2 % (bezogen auf den Zellstoff) Propylgallat zugege-
ben. Unter einem Vakuum von 50 - 80 mbar und mechanischer Vermengung des
Fasertyp Kurzbezeichn. Für die Faser verw. Abkürz.
mittl. Länge [µm]
∅ [µm]
Mikrofaser BE 600-30 Mikro 30 18 Mittellange Faser1 B 700 R Fas.1 90 20 Mittellange Faser2 BC 200 Fas.2 300 20 Langfaser FIF 400 Fas.3 2.000 35 Bakteriencellul. / fzmb BC BC Lyocellfasern / Lenzing Lyocell Lyocell 6.000 12 (1.7 dtex) (Regenaratfaser)

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
82
Gemisches im Kneter wurde bei ansteigender Temperatur (bis maximal 95 °C) soviel
H2O abdestilliert, bis sich die Cellulose im NMMO/H2O-Gemisch auflöste. Infolge
dessen stieg die NMMO-Konzentration in der Lösung auf 87 - 88%, so daß im End-
effekt eine stabile Matrixlösung erhalten wurde. Die NMMO-Konzentration wurde
refraktometrisch über den Brechungsindex n bestimmt und die Qualität der Lösung
bzw. die vollständige Auflösung der Cellulose mit einem Polarisationsmikroskop über-
prüft.
6.1.3 Herstellung der Verbundlösung
In den vorangegangenen Kapiteln wurde die Grundlage für die experimentellen
Arbeiten dieses Abschnitts gelegt. Das thermische Verhalten des Stoffsystems NMMO/-
H2O/Cellulose sowie das Löseverhalten der im System enthaltenen Cellulose wurde
unter Variation der Temperatur, Zeit und Zusammensetzung ausführlich erkundet.
Durch die Zusammenstellung dieser Meßergebnisse war es möglich, ein Phasendia-
gramm für das betreffende System zu erstellen. Mit Hilfe dieses Phasendiagramms
konnte eine experimentelle Methode abgeleitet werden, die eine Möglichkeit zur
Herstellung einer Verbundlösung aus einer Cellulosematrix und einem cellulosischen
Fasermaterial bietet. Hier ging es in erster Linie zunächst darum, cellulosische Fasern in
eine Matrixlösung (aus Cellulose, NMMO und H2O) einzubringen und in Folge eine
weiterverarbeitbare Verbundlösung zu präparieren.
Bei der Präparation einer Verbundlösung aus einer Matrixlösung und einem cellulosi-
schen Fasermaterial müssen einige Punkte beachtet werden, damit die Herstellung von
Cellulosefolien mit den gewünschten Eigenschaften möglich wird. Dazu gehört die
Forderung, daß die Cellulosefasern, welche der Matrixlösung zugesetzt werden, in der
Matrix nicht aufgelöst werden und dadurch zur Verstärkung der Matrix beitragen kön-
nen. Damit sich aber eine gute Haftung zwischen Faser und Matrix einstellt, müssen
die Faseroberflächen in der Matrixlösung partiell gelöst werden.
Ein weiterer Punkt bezieht sich auf die Stabilität und die Weiterverarbeitbarkeit der
Verbundlösung zu faserverstärkten Folien. Die Faserzugabe zur Matrix muß auf eine
Art und Weise durchgeführt werden, daß es zu keiner Ausfällung der in der Matrix-
lösung gelösten Cellulose kommt.

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
83
Außerdem muß dafür gesorgt werden, daß die Fasern in der Matrix gleichmäßig ver-
teilt werden, damit eine nachfolgende Extrusion dieser Lösung zu dünnen Blasfolien
ohne große Komplikationen realisiert werden kann.
Um diesen Anforderungen gerecht zu werden, mußte auf das erstellte Phasen-
diagramm zurückgegriffen werden. Besonders wichtig war im Phasendiagramm (Abb.
5-16) der Bereich B, in dem die Cellulose nur minimal oder geringfügig gelöst wird.
Anhand der gewonnenen Kenntnisse über den erwähnten engen Lösebereich für die
Cellulose konnte ein Laborverfahren zur Präparation von brauchbaren Verbundlö-
sungen entwickelt werden. Das hier angewendete Verfahren zur Einbringung von
Cellulosefasern in eine Cellulosematrixlösung läßt sich auf folgende Weise skizzieren:
Die NMMO-Konzentration der Matrixlösung wurde durch Verdünnen mit H2O auf Werte zwischen 82 - 84% verringert.
Gleichzeitig wurde die Temperatur der Lösung auf 70 - 75 °C herabgesetzt.
Nachträglich wurden die Fasern entweder im trockenen oder im vorgequollenen Zustand der Matrixlösung zugegeben.
Dabei war es wichtig, die Fasern zur Matrix in geringen und konstanten Mengen sowie
kontinuierlich unter permanenter mechanischer Vermengung beizumischen. Schließ-
lich mußte eine bestimmte Zeit zwischen der Fertigstellung der Verbundlösung und
deren Verarbeitung zu Celluloseblasfolien genau eingehalten werden, damit es in der
Lösung nicht zu unerwünschten Reaktionen kommt.
Das Einbringen der Fasern im trockenen Zustand in die verdünnte Matrixlösung stellte
sich als das günstige Verfahren heraus. Im Falle der Bakteriencellulose-Fasern (BC-
Fasern) war dies aber nicht möglich. Die Bakteriencellulose in der Form, in der sie zur
Verfügung stand, mußte zunächst in einer wäßrigen Lösung mechanisch in kleine
Faserstücke dispergiert werden. Anschließend wurde der Dispersion durch Anlegen
eines Vakuums absichtlich nur begrenzt Wasser entzogen. Denn bei weiterem Was-
serentzug bildeten sich Agglomerationen von BC, deren homogene Verteilung in der
Matrixlösung sich später als sehr problematisch erwies. Deshalb wurde die BC im
nassen Zustand in Form eines Slurrys zur Matrix zugesetzt. Das Slurry bestand aus einer
Suspension aus BC-Teilchen und wäßriger NMMO-Lösung, deren Menge und Zusam-
mensetzung so eingestellt wurde, daß die gewünschte NMMO-, H2O- und Faserkon-
zentration in der Verbundlösung erreicht wurde.

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
84
Nach der Faserzugabe zur Matrixlösung konnte in einigen Fällen (für BC bzw. Lyocell)
eine starke bis vollständige Faserauflösung nicht verhindert werden.
6.1.4 Extrusion der Verbundlösungen zu Cellulosefolien
Die Verbundlösung wurde nach dem Folienblasverfahren zu Cellulosefolien [88] extru-
diert. Das Prinzip des Folienextrudierens wird im folgenden kurz dargestellt:
Die Lösung wird im Extruder auf die Verarbeitungstemperatur temperiert, durch eine
Filmblasdüse in einen Luftspalt gepreßt und dadurch zu einem Folienschlauch geformt,
wobei die Prozeßführung hier vertikal abwärts erfolgt. In einem Koagulationsbad, in
dem das Innenvolumen des Schlauchs ebenfalls mit dem Fällmittel gefüllt wird, wird er
zwischen zwei Abquetschwalzen zusammengelegt und in der Folge über mehrere Rol-
len abgezogen und in nachfolgende Waschbäder geführt. In Waschbädern wird das
restliche NMMO aus der Schlauchfolie entfernt und diese anschließend getrocknet.
Im Luftspalt zwischen Düse und Fällbad wird der Folienschlauch durch Aufbringen
eines Überdrucks in das Innenvolumen aufgeblasen. Dadurch kann eine Querreckung
eingestellt werden. Durch eine Variation der Abzugsgeschwindigkeit ist eine Längsrek-
kung der Folie möglich. Als Fällmittel wird in der Regel H2O oder wäßrige NMMO-Lö-
sung eingesetzt.
Die Herstellung der faserverstärkten Celluloseblasfolien konnte im Rahmen dieser Ar-
beit unter den folgenden Prozeßbedingungen realisiert werden:
- Temperatur der Blasdüse 85 °C
- Durchmesser der Düsenöffnung 22 mm
- Spaltenbreite der Düsenöffnung 1,5 mm
- Förderleistung der Spinnpumpe 1,2 cm3/u
- Umdrehungsgeschwindigkeit der Spinnpumpe 40 U/min
- Verwendete Lösungsmenge in einem Experiment 3 kg
- Abzugsverhältnis 1,3
- Durchmesser des Schlauches beim Trocknen 32 mm
- Dicke der Folien (trocken) 13 - 44 µm
Die Parameter der hergestellten faserverstärkten Blasfolien sind in der Tab. 6-2 zusam-
mengestellt. In der Tabelle sind ergänzend einige Parameter angegeben, welche die

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
85
einzelnen Blasfolien voneinander unterscheiden: die NMMO-Konzentration der Matrix-
und Verbundlösungen, die zur Verstärkung der Cellulosematrix eingebrachten cellulo-
sischen Fasertypen, der Fasergehalt in den Folien, die Methode der Faserzugabe sowie
der Lösezustand der eingebrachten Fasern in den Blasfolien.
Tab. 6-2 Parameter der extrudierten Celluloseblasfolien Cellul.konz.
der Matrixlsg.
[%]
NMMO-Konz. der
Matrixlsg. [%]
NMMO-Konz. der
Verbundlsg. [%]
Zugesetzte Fasern
Benutzte Abkürzung
für die Fasern
Faser- gehalt
[%]
Faser-zugabe
Lösezustandder
zugesetzten Fasern
8.5 87.5 83.0 Mikrofaser Mikro 5 naß 0 8.5 87.5 83.0 Mikrofaser Mikro 10 naß 0 8.5 87.0 83.0 Mikrofaser Mikro 25 naß 0 8.5 86.5 83.5 Mikrofaser Mikro 40 naß 0 8.5 87.0 83.0 mittellange
Faser 1 Fas.1 25 naß 0
8.5 86.5 83.0 mittellange Faser 2
Fas.2 25 naß 0
8.5 88.8 83.0 Langfaser Fas.3 25 naß 0 8.5 88.5 83.0 Bakterien-
cellulose BC 25 naß 2
8.5 88.8 84.0 Bakterien-cellulose
BC 22.5 naß 1
8.5 87.0→83.0 84.0 Lyocellfaser Lyocell 25 trocken 3 8.5 87.0→82.0 83.0 Lyocellfaser Lyocell 25 trocken 1 8.5 87.0→82.0 82.0 Referenzfolie ohne Fasern
1Die Kennziffern, die für den Lösezustand der zusätzlich eingebrachten Cellulosefasern
verwendet wurden, haben jeweils die folgende Bedeutung:
0 = keine, 1 = minimale, 2 = partielle, 3 = mittlere, 4 = starke, 5 = vollständige Auflösung.
Die Abschätzung des gelösten Anteils an beigemischten Cellulosefasern in den Blas-
folien wurde unmittelbar nach ihrer Extrusion durchgeführt. Dazu wurden die Blas-
folien, bevor sie getrocknet wurden, einzeln unter einem Polarisationsmikroskop be-
trachtet. Der Lösezustand der eingebrachten und in der Cellulosematrix eingebetteten
Cellulosefasern wurde dabei nach morphologischen Gesichtspunkten visuell beurteilt
und in der Folge einem der oben festgelegten Lösezustände zugeordnet.

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
86
6.2 Ergebnisse
6.2.1 Mechanische Eigenschaften der faserverstärkten Blasfolien
Die mechanischen Eigenschaften der trockenen und wiederbefeuchteten Blasfolien
wurden in Zugversuchen in Anlehnung an die Norm ISO 527-3 bestimmt. Die Festig-
keiten, die Bruchdehnungen sowie die E-Moduln der Folien wurden längs zur Maschi-
nenlaufrichtung mit Hilfe einer Universalprüfmaschine (Zwick Z 020) ermittelt. Für die
Messungen in Längsrichtung wurden die Folien in 10 mm breite Streifen geschnitten,
wobei die Einspannlänge 100 mm betrug.
Einfluß von Fasertyp und -gehalt auf die Eigenschaften der Verbundfolien
Abb. 6-1 Einfluß der eingesetzten Cellulosefasern auf Zugmodul und Zugfestigkeit (trocken, längs) von Celluloseblasfolien; (Fasergehalt: 25 Ma%)
Die Abb. 6-1 gibt die Auftragung des im Zugversuch ermittelten E-Moduls und der
Zugfestigkeit der faserverstärkten Blasfolien als Funktion der zugesetzten cellulosischen
Fasern wieder. Aus dieser Abbildung ist zu ersehen, daß nur zwei der verwendeten
Cellulosefasern eine Verstärkungswirkung für die Cellulosematrix aufweisen: Mikro-
pur
Mikr
oFa
s.1Fa
s.2Fa
s.3 BC
Lyoc
ell0
2000
4000
6000
8000
10000
E-Modul Zugfestigkeit
E-M
odul
[MPa
]
0
50
100
150
200
Zugf
estig
keit
[MPa
]

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
87
und Lyocellfasern. Ob der Verstärkungseffekt im Falle der Lyocellfasern durch die
eingebrachten Lyocellfasern verursacht wird, die in der Matrix nicht aufgelöst wurden,
oder aber durch deren teilweise Auflösung in der Matrixlösung, ist nicht eindeutig zu
beurteilen. Denn für die mit Lyocellfasern verstärkte Blasfolie ist der gelöste Anteil an
eingebrachten Lyocellfasern nach deren Extrusion mit 50% bewertet. Das bedeutet,
daß deshalb der Gesamtanteil an gelöster Cellulose in der Verbundlösung erhöht und
damit das Verhältnis von gelöster Cellulose zu ungelösten Cellulosefasern in dieser Fo-
lie verändert wurde. Dadurch könnte die Zunahme des E-Modul- und Festigkeitswertes
der Folie mitverursacht worden sein.
Dagegen scheinen die Mikrofasern eine Verstärkung für die Cellulosematrix hervorzu-
rufen. Denn die in die Cellulsoematrix eingebrachten Mikrofasern hatten im Unter-
schied zu den Lyocellfasern keinerlei Auflösung erfahren (siehe Tab. 6-2).
Abb. 6-2 Einfluß des Fasergehalts auf Zugmodul und Zugfestigkeit (trocken, längs) von faserverstärkten Celluloseblasfolien; (Eingesetzter Fasertyp: Mikrofasern)
In Abb. 6-2 ist die Abhängigkeit der Zugfestigkeit und des Zugmoduls der (mit
Mikrofasern verstärkten) Blasfolien vom Fasergehalt dargestellt. Für die mit 5% Mikro-
fasern hergestellte Folie wird eher ein Rückgang der mechanischen Werte beobachtet.
pur
5% 10%
25%
40%
0
2000
4000
6000
8000
10000
E-Modul Zugfestigkeit
E-M
odul
[MPa
]
0
50
100
150
200
Zugf
estig
keit
[MPa
]

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
88
Ein Fasergehalt von 10% wirkt sich hingegen positiv auf die Eigenschaften der Folien
aus. Sowohl der E-Modul als auch die Zugfestigkeit der Verbundfolien nehmen hier zu.
Eine Erhöhung des Fasergehalts auf 25% verbessert zwar die Steifheit der Blasfolien,
vermindert aber gleichzeitig die Zugfestigkeit. Bei weiterer Erhöhung des Gehalts an
cellulosischen Mikrofasern wird eine Verschlechterung des E-Moduls und der Zug-
festigkeit der Folie festgestellt.
6.2.2 Strukturcharakterisierung
Elektronenmikroskopische Untersuchungen an den faserverstärkten Folien wurden mit
dem Philips-Transmissionselektronenmikroskop CM 200 bei 120 kV und dem Raster-
elektronenmikroskop Jeol-JSM 6300 F bei 5 kV durchgeführt. Für die Charakterisie-
rung der Querschnittsmorphologie der faserverstärkten Blasfolien wurden TEM-Auf-
nahmen von Ultradünnschnitten von nassen Folienproben erstellt. Um auch Informa-
tionen über die Faser-Matrix-Haftung in den Verbundfolien zu erhalten, wurden von
Bruchflächen (Bruch in flüssigem N2) REM-Bilder ausgewählter Folien angefertigt.
Aus den TEM-Aufnahmen der Verbundfolien ist ersichtlich, daß einige der Blasfolien
einen symmetrischen, andere wiederum einen asymmetrischen Aufbau der fibrillären
Struktur über den Folienquerschnitt aufweisen. In dieser Matrixstruktur sind die Fasern
unregelmäßig verteilt. In Abhängigkeit vom Faseranteil sind Querschnitte von Fasern
zu erkennen, die in die Struktur der Matrix eingebettet sind (Abb. 6-3). In einigen
Folien sind die eingelagerten Fasern nicht eindeutig von der sie umgebenden fibrillären
Struktur der Matrix zu unterscheiden. Diese Fasern erscheinen angelöst. In der Mehr-
zahl der Folien jedoch sind die zugesetzten Fasern von der sie umgebenden Matrix-
struktur leicht zu unterscheiden. Charakteristisch für die mit Holzfasern (Mikrofasern,
Fas.1, Fas.2 und Fas.3) verstärkten Folien ist, daß von den eingelagerten Fasern jeweils
Querschnitte und keine Schräg- oder/und Längsschnitte erhalten wurden. Diese Fasern
sind in die Matrix so eingebettet, daß sie in Maschinenlaufrichtung orientiert sind.

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
89
In der Abb. 6-3 ist eine TEM-Aufnahme von einer faserverstärkten Cellulosefolie zu
sehen. Im rechten Bild ist die Aufnahme vergrößert dargestellt. Auf den Bildern ist der
Querschnitt einer Holzfaser zu erkennen (Nr. 1), die von der Matrix umgeben wird. Die
Holzfaser ist - wie bei Hohlfasern - durch ein Lumen in der Fasermitte (Nr.2) und durch
eine konzentrische Lamellierung (Nr. 3, schichtartig aneinandergereihte Hohlräume)
der Faser gekennzeichnet (typische Querschnittsmorphologie von Naturfasern).
Im rechten Bild ist der Übergang Faser-Matrix höher vergrößert abgebildet. Dort ist
beim Übergang von der Faser zur Matrix (Nr. 4) kein Spalt oder Hohlraum festzustel-
len, so daß man hier von einer guten Einbettung der Faser in der Matrix sprechen
kann.
Abb. 6-3 TEM-Aufnahme eines Ultradünnschnittes von einer faserverstärkten Celluloseblasfolie;
(Eingesetzter Fasertyp: Fas.2/ mittellange Fasern)
1
3
4
3
2
4
2

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
90
An Hand der REM-Aufnahmen (Abb. 6-4), die quer zur Probenlängsrichtung aufge-
nommen wurden, können folgende Aussagen über die Faser-Matrix-Haftung und über
die Faserorientierung in der Matrix getroffen werden: Es ist zu erkennen, daß zwischen
Faser und Matrix neben Hohlräumen viele Haftungspunkte bestehen, die auf eine be-
friedegende Haftung zwischen Faser und Matrix hinweisen. Die Aufnahmen beweisen
zugleich, daß die Fasern überwiegend im Randbereich der Folien (Abb. 6-4, Bild 110)
anzutreffen sind. Außerdem geht aus den REM-Bildern hervor, daß die Fasern in der
Matrix in Folienlängsrichtung orientiert sind.
Abb. 6-4 REM-Bilder von Bruchflächen der faserverstärkten Celluloseblasfolien (Bilder wurden quer zur
Probenlängsrichtung aufgenommen (eingesetzter Fasertyp: Mikrofasern)

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
91
6.3 Diskussion
Die Tatsache, daß unter den verwendeten Fasertypen eigentlich nur die Mikrofasern
eine Verstärkungswirkung für die Cellulosematrix hervorrufen, wirft die folgende Frage
auf: Woran liegt es, daß der Einsatz der anderen Fasertypen zu keiner merklichen Ver-
besserung der mechanischen Eigenschaften der Verbundfolien führt?
Die Ursache dafür liegt wahrscheinlich in der Ausdehnung bzw. Geometrie der einge-
setzten Fasern und der extrudierten Verbundfolien. Die Größen der hier verwendeten
Faserdimensionen sind mit denen der Foliendicken vergleichbar. Die mittlere Dicke der
extrudierten Verbundfolien beträgt z. B. 30 µm. Der mittlere Durchmesser der zur Ver-
stärkung eingesetzten Fasern liegt zwischen 18 und 35 µm, die mittlere Länge dieser
Fasern reicht von 30 bis 6000 µm. Die Herstellung der Verbundfolien mit den angege-
benen Faser- und Foliengeometrien führt wahrscheinlich dazu, daß durch die relativ
langen und breiten Fasern in der vergleichsweise dünnen Cellulosematrix Störstellen
eingebaut werden. Hierdurch wird möglicherweise jeder Punkt an der Folienober-
fläche, an dem ein Faserende oder -teil herausragt, auch zu einer Defektstelle in der
Verbundfolie. Jede dieser Störstellen trägt dementsprechend zur Minderung der ma-
kroskopischen mechanischen Eigenschaften der Folien bei.
Der Einsatz von Bakteriencellulose als Verstärkungssubstanz verringert den E-Modul
und die Festigkeit der betreffenden Verbundfolien erheblich. Eine Erklärung für diese
Verringerung liegt womöglich in der hohen wasserbindenden Kapazität der Bakterien-
cellulose. Aufgrund dieser Besonderheit der BC ist es denkbar, daß im Herstellungs-
prozeß (im Fäll- oder Waschbad) die BC-Fasern in der Cellulosematrix infolge der ho-
hen Wasseraufnahme stark gequollen werden. Diese Quellung könnte eine mecha-
nische Schädigung der Grenzfläche zwischen BC-Fasern und der Matrix bewirken. Die
mechanischen Schädigungen könnten zur Bildung von freien Oberflächen und inneren
Hohlräumen zwischen Faser und Matrix führen, die gegebenenfalls eine Herabsetzung
des Festigkeits- und Modulwertes verursachen.
Aus den bisherigen Ergebnissen zur Herstellung von faserverstärkten Celluloseblas-
folien lassen sich einige Schlußfolgerungen für weitere ausstehende Experimente ablei-
ten. Die Verbundlösungen, die in dieser Arbeit aus Cellulosefasern und Cellulosematrix
präpariert und zu Blasfolien extrudiert wurden, können ebenso zur Herstellung von
Gießfolien eingesetzt werden. Ein großer Vorteil wäre hierbei, daß die einzuhaltenden

HERSTELLUNG UND PRÜFUNG VON FASERVERSTÄRKTEN CELLULOSEFOLIEN _________________________________________________________________________________________
92
Anforderungen beim Verarbeitungsprozeß nicht so hoch sind wie beim Extrudieren
von Blasfolien. Jedoch muß auch hier darauf geachtet werden, daß die Abmessungen
(Dicke) der Gießfolien weitaus größer sind als die der Blasfolien.
Ein anderer Ansatzpunkt für eine Nutzung der in dieser Arbeit durchgeführten Mes-
sungen besteht darin, statt Cellulosefasern andere nicht-cellulosische Mikrofasern wie
z. B. Polyacrylnitrilfasern oder Polyamidfasern als Verstärkungsmaterial in eine NMMO/-
H2O/Cellulose-Matrix einzuarbeiten und daraus Blasfolien oder andere Celluloserege-
nerat-Formkörper herzustellen. In diesem Fall würde man die Schwierigkeiten, die
hinsichtlich der Auflösung von Cellulosefasern in NMMO auftreten, umgehen. An
diesen Folien müßte aber durch entsprechende Experimente die Faser-Matrix-Haftung
eingehend untersucht werden. Ein Nachteil wäre hierbei, daß diese Folien nicht biolo-
gisch abbaubar sind.
Eine weitere Möglichkeit, ein Verbundmaterial aus Fasern und cellulosischer Matrix
herzustellen, könnte darin bestehen, ein Faservlies (z. B. Flachsfaservlies) mit NMMO/
H2O/Cellulose-Lösung einseitig oder doppelseitig zu durchtränken bzw. zu beschich-
ten, dieses dann durch ein Fällbad zu führen und dabei die cellulosische Matrix auszu-
fällen. In einem weiteren Prozeßschritt könnte dieses Verbundmaterial mit Hilfe einer
Presse in die gewünschte Form gepresst werden. Anstelle des Faservlieses könnte mög-
licherweise auch Papier verwendet werden. Dadurch würde man z. B. eine schützende
Matrixschicht aus Cellulosefolie auf das Papiermaterial aufbringen. Die so erhaltenen
Verbunde würden im Unterschied zu den Blas- oder Gießfolien einen hohen Faser-
anteil und einen geringen Matrixanteil aufweisen.

ZUSAMMENFASSUNG _________________________________________________________________________________________
93
7 Zusammenfassung
Ausgehend von den Erfahrungen bei der Herstellung von Cellulosefolien aus Amin-
oxidlösungen sollte ein Verfahren zur Einarbeitung von cellulosischen Fasern in eine
Aminoxidlösung entwickelt werden. Die Aminoxidlösung, die aus dem Lösemittel-
system N-Methylmorpholin-N-oxid (NMMO), Wasser sowie gelöster Cellulose bestand,
stellte für die einzuarbeitenden Fasern eine Matrixlösung (Matrix) dar.
Die besondere Aufgabe bestand dabei darin, zum einen Cellulose (Zellstoff) in
NMMO/H2O in Lösung zu bringen, und zum anderen, bei leicht veränderten Systempa-
rametern Cellulosefasern hinzuzusetzen, die dann nicht mehr aufgelöst werden und
nach dem Fällprozeß als Verstärkungsfasern erhalten bleiben. Um entsprechende Ver-
arbeitungsbedingungen abzuleiten, waren grundsätzliche Daten über das thermische
Verhalten des ternären Stoffsystems NMMO/H2O/Cellulose erforderlich.
Im Mittelpunkt der vorliegenden Arbeit stand die differentialkalorimetrische Unter-
suchung des ternären Stoffsystems NMMO/H2O/Cellulose. Um jedoch ein einheitliches
und besseres Verständnis vom thermischen Verhalten des ternären Stoffsystems zu
erhalten, wurde zunächst das binäre System mittels Dynamischer Differenzkalorimetrie
und Röntgenweitwinkel-Diffraktometrie untersucht. Dadurch konnten das Schmelz-
verhalten und die Phasenzusammensetzung des Zweistoffsystems im festen Zustand in
Abhängigkeit vom NMMO/H2O-Verhältnis charakterisiert werden.
Das binäre System weist im NMMO-Konzentrationsbereich 72 - 82 Ma% relativ breite
und flache Schmelzintervalle auf. Diesem Sachverhalt entspricht eine breite Kristallit-
größenverteilung und/oder ein stark gestörter kristalliner Aufbau des Stoffsystems
NMMO/H2O. Die relativ niedrigen Schmelzenthalpien (10 - 80 J/g) des Systems in
diesem Konzentrationsbereich deuten auf eine vergleichsweise niedrige Bindungs-
stärke zwischen den NMMO- und H2O-Molekülen hin. Röntgenweitwinkelmessungen
belegen, daß das System in diesem Konzentrationsbereich vorwiegend aus der kristalli-
nen Phase des 2,5-Hydrats aufgebaut ist.
Im Konzentrationsbereich 85 - 91 Ma% an NMMO fallen dagegen die gemessenen
Schmelzpeaks des Systems verhältnismäßig schmal und hoch aus. Diesem Schmelz-
verhalten ist eine enge Kristallitgrößenverteilung und/oder eine hohe kristalline Ord-
nung des Stoffsystems zuzuschreiben. Für das System wurden in diesem Konzen-

ZUSAMMENFASSUNG _________________________________________________________________________________________
94
trationsbereich relativ hohe Schmelzenthalpien (ca. 130 J/g) bestimmt, die auf eine
vergleichsweise starke Bindung zwischen den NMMO- und H2O-Molekülen hinweisen.
Anhand von WAXS-Aufnahmen konnte gezeigt werden, daß das System in diesem
Bereich vorzugsweise die kristalline Phase des Monohydrats bildet.
Aus den ermittelten Daten wurde für das binäre Stoffsystem NMMO/H2O ein Phasen-
diagramm im NMMO-Konzentrationsbereich 70 bis 90 Ma% (H2O-Konzentrations-
bereich 10 bis 30 Ma%) aufgestellt.
Den Schwerpunkt der Arbeit bildete die Untersuchung des ternären Stoffsytems
NMMO/H2O/Cellulose hinsichtlich der Löslichkeit der Cellulose im Lösemittelsystem
NMMO/H2O. Hierzu wurde eine Reihe von NMMO/H2O/Cellulose-Gemischen mit un-
terschiedlichen NMMO/H2O-Zusammensetzungen, jedoch einer konstanten Cellulose-
konzentration von 8% präpariert. Für die Untersuchungen wurde als cellulosisches
Material durchgängig der Zellstoff Buckeye V60 mit einem mittleren Polymerisations-
grad von 535 (Cuoxam-DP) eingesetzt.
Bei den differentialkalorimetrischen Untersuchungen stellte sich heraus, daß das ther-
mische Verhalten des ternären Stoffystems hauptsächlich vom NMMO/H2O-Verhältnis
bestimmt wird.
Ein wesentliches Resultat der DSC-Untersuchungen über das ternäre Stoffsystem im
festen Zustand ist, daß für die Proben mit NMMO-Konzentrationen 72 - 75 Ma% nur
ein Schmelzpeak, für die Proben mit NMMO-Konzentrationen ≥ 78 Ma% hingegen
zwei Schmelzpeaks beobachtet werden. Die Bestimmung eines einzigen Schmelzpeaks
für die Proben ≤ 75 Ma% wurde dadurch begründet, daß in diesen Proben lediglich
eine Hydratphase, nämlich die des 2,5-Hydrats, vorkommt. Dagegen wurde die Auf-
nahme von zwei unterschiedlichen Schmelzkurven für die Proben mit NMMO-Kon-
zentrationen ≥ 78 Ma% als Indiz für die Anwesenheit von zwei unterschiedlichen, ne-
beneinander vorkommenden kristallinen Hydraten (2,5-Hydrat und das Monohydrat
des NMMO) angesehen.
Um den möglichen Einfluß der Temperatur und Zeit auf das Löseverhalten von Cellu-
lose im Lösemittelsystem NMMO/H2O zu untersuchen, wurden die Gemische mit un-
terschiedlichen NMMO/H2O-Zusammensetzungen in Löseversuchen bei verschiedenen
Temperaturen und Zeiten behandelt. Der Quell- bzw. Lösezustand der in diesen
Stoffgemischen enthaltenen Cellulose wurde mit Hilfe des DSC, der Polarisations-

ZUSAMMENFASSUNG _________________________________________________________________________________________
95
mikroskopie sowie der Röntgenweitwinkel-Diffraktometrie in Abhängigkeit vom
NMMO/H2O-Gehalt, von der Temperatur und der Einwirkzeit bestimmt.
Die Meßergebnisse wurden in einem Phasendiagramm für das ternäre System (8%
Cellulose) zusammengestellt. Bei der Erstellung des Phasendiagramms wurde das Au-
genmerk vor allem auf einen schmalen Bereich im Diagramm gerichtet, in dem die Cel-
lulose angelöst bzw. teilweise gelöst, aber nicht komplett aufgelöst wird.
Die Cellulose erfährt im Lösemittelsystem NMMO/H2O bei Konzentrationen < 78 Ma%
an NMMO ( > 22 Ma% an H2O) bei Temperaturen < 90 °C und einer Einwirkzeit von
60 min lediglich eine Quellung. Im Konzentrationsbereich 78 - 81 Ma% an NMMO, im
Temperaturintervall 70 - 95 °C und einer Einwirkzeit von 60 min wird die Cellulose im
System NMMO/H2O teilweise gelöst. Hingegen wird die Cellulose bei Konzentrationen
> 81 Ma% an NMMO, bei Temperaturen > 75 °C und bei einer Zeit von 60 min im
NMMO/H2O-Gemisch vollständig aufgelöst.
Vorstellungen über die Wechselwirkung von NMMO mit Wasser und Cellulose wurden
vor allem aus einem Vergleich des Verhaltens des binären Systems NMMO/H2O und
des ternären NMMO/H2O/Cellulose-Systems abgeleitet. Offensichtlich konkurrieren
NMMO- und Cellulosemoleküle um das vorhandene Wasser. Bei hinreichender Verrin-
gerung des Wasserangebotes im Bereich der NMMO-Monohydratbildung werden Hy-
droxylgruppen der Cellulose in die Wechselwirkung mit dem NMMO einbezogen, was
zum Eindringen von NMMO in die geordneten Bereiche der Cellulose und somit zur
Auflösung kristalliner Bereiche führt.
An Hand des ternären Phasendiagrammes, dabei insbesondere unter Nutzung des
schmalen Phasengebiets des unvollständigen Lösens konnte ein Laborverfahren abge-
leitet werden, das die Herstellung von faserverstärkten Verbundwerkstoffen ermög-
licht.
Das Verfahren läßt sich in drei Prozeßschritte unterteilen. Im ersten Schritt wird die
NMMO-Konzentration der Matrixlösung durch Verdünnen auf Werte zwischen 82 - 78
Ma% abgesenkt. Gleichzeitig wird die Temperatur der Lösung auf 70 - 75 °C herab-
gesetzt. Im zweiten Schritt wird eine Verbundlösung aus Matrixlösung und Fasern
präpariert. Dies erfolgt dadurch, daß der Matrixlösung - unter permanenter mechani-
scher Vermengung - kontinuierlich Cellulosefasern zugegeben werden. Im letzten
Schritt des Verfahrens wird die so hergestellte Verbundlösung unter Nutzung einer

ZUSAMMENFASSUNG _________________________________________________________________________________________
96
formgebenden Verarbeitungsmethode (z. B. Extrudieren, Gießen, Pressen) zu einem
faserverstärkten Verbundmaterial verformt.
Durch Anwendung dieser Methode konnten schließlich Verbundlösungen aus Cellulo-
sefasern und einer cellulosischen Matrixlösung präpariert und zu faserverstärkten Cel-
luloseblasfolien verarbeitet werden. Selbstverständlich läßt sich dieses Verfahren auch
zur Herstellung von anderen faserverstärkten Verbundwerkstoffen auf Cellulosebasis
einsetzen.
Die mechanischen Eigenschaften (Zugfestigkeit, E-Modul) der hergestellten Cellulose-
folien wurden in Zugversuchen bestimmt. Aus den Meßergebnissen ging hervor, daß
von den zur Verstärkung der Matrix eingesetzten cellulosischen Fasern die Mikrofasern
die höchste Verstärkungswirkung aufweisen. Der E-Modul der trockenen, mikrofaser-
verstärkten Blasfolien in Folienlängsrichtung wurde zu 9700 MPa, der von puren
Blasfolien ohne Fasern zu 7900 MPa ermittelt. Dies bedeutet eine Erhöhung des E-
Moduls um 20%.
Eine vorgenommene Variation des Mikrofasergehalts in den Folien ergab, daß sich ein
Fasergehalt von 10 und 25% am besten auf die Festigkeits- und E-Modulwerte der
Blasfolien auswirkt. Hierbei konnte der E-Modul um 20% und die Zugfestigkeit um
15% verbessert werden.
Mit Hilfe von TEM- und REM-Aufnahmen konnte gezeigt werden, daß zwischen den
eingebrachten Fasern und der Matrix viele Haftungspunkte bestehen, die auf eine
befriedigende Faser-Matrix-Haftung in den Folien hinweisen. Außerdem wurde heraus-
gefunden, daß die in die Struktur der Folien eingebetteten Cellulosefasern überwie-
gend in Folienlängsrichtung orientiert sind.
Ansatzpunkte für eine weitergehende Nutzung der Ergebnisse zur Herstellung von
faserverstärkten Formkörpern aus Cellulose wurden aufgezeigt.

LITERATURVERZEICHNIS _________________________________________________________________________________________
97
8 Literaturverzeichnis
[1] Römpp Lexikon Chemie - Version 2.0 (Georg Thieme Verlag, Stuttgart/New York, 1999)
[2] Atkins, P.W.:"Moleküle - Die chemischen Bausteine der Natur", Spektrum d. Wissensch. Verl.-Gesellschaft, Heidelberg, S. 113 - 115 (1988)
[3a] Niederstadt, G./ Herrmann, A.S./ Hanselka, H.: Digest: Moderne Werkstoffe, Spektrum der Wissensch., Digest; 3, 94 (1996)
[3b] Franz, G. in: Polysaccharide, W. Burchard (Hrsg.), Springer-Verlag Berlin, S. 1-14 (1985)
[4] Jayme, G./ Roffael, E./ Hasvold, K.K.: Das Papier 23, 597 (1969)
[5] Klemm, D./ Philipp, B./ Heinze, T./ Heinze, U./ Wagenknecht, W.: "Comprehensive Cellulose Chemistry", Vol. 1, Wiley-VCH, Weinheim 1998
[6] Berger, W./ Keck, M./ Philipp, B./ Schleicher, H.: Lenzinger Ber. 59, 88 (1985)
[7] Payen, A.: Troisième mémoire sur le développement végétaux. Extrait des mémoires de l`Académie Royale des Sciences. Tòme III des Savants Etrangères, Paris, Imprimerie Royale 1842
[8] Haworth, W.N.: Helv. Chim. Acta 11, 547 (1928)
[9] Freudenberg, K./ Braun, E.: Ann. 460, 288 (1928)
[10] Staudinger, H.:"Die hochmolekularen Verbindungen - Kautschuk und Cellulose", Springer, Berlin (1932)
[11] Ellefsen, O./ Tonnesen, B.A., in: "Cellulose and cellulose derivatives", Bikales, N.M./ Segal, L. (Hrsg.), Vol. V, Part IV, Wiley-Interscience, New York, S. 151-180 (1971)
[12] Liang, C.Y./ Marchessault, R.H.: J. Polym. Sci. 37, 385 (1959)
[13] Gardner, K.H./ Blackwell, J.: Biopolymers 13, 1975 (1974)
[14] Kolpak, F.J./ Blackwell, J.: Macromolecules 9, 273 (1976)
[15] Meyer, K.H./ Misch, L.: Helv. Chim. Acta 20, 232 (1937)
[16] Fink, H.-P./ Hofmann, D./ Purz, H.J.: Acta Polym. 41, 131 (1990)
[17] Zugenmaier, P.: Prog. Polym. Sci. 26, 1341 (2001)
[18] Marchessault, R.H./ Sarko, A., in: Advan. Carbohydr. Chem. 22, 421 (1967)
[19] VanderHart, D.L./ Atalla, R.H.:Macromolecules 17, 1465 (1984)
[20] Philipp, B/ Lehmann, R./ Ruscher, C.: Faserforsch. u. Textiltechn. 10, 22 (1959)
[21] Schleicher, H.: Habilitationsschrift, A. d. W. der DDR, Teltow-Seehof (1983)
[22] Kroon-Batenburg, L.M.J./ Kroon, J./ Northolt, M.G.: Das Papier 12, 640 (1990)
[23] Hess, K./ Kiessig, H.: Z. Phys. Chem. B 49, 235 (1941)
[24] Gardiner, E.S./ Sarko, A.: Can. J. Chem. 63, 173 (1985)

LITERATURVERZEICHNIS _________________________________________________________________________________________
98
[25] Frey-Wyssling, A./ Mühlethaler, K. : Makromol. Chem. 62, 25 (1963)
[26] Blackwell, J./ Kolpak, F.J.: Macromolecules 8, 322 (1975)
[27] Mühlethaler, K. : J. Polym. Sci., Part C 28, 305 (1969)
[28] Fengel, D.: Holzforschung 32, 37 (1978)
[29] Hearle, J.W.S.: J. Polym. Sci. 28, 432 (1958)
[30] Schurz, J.: Lenzinger Ber. 49, 15 (1980)
[31] Fink, H.-P./ Philipp, B.: J. Appl. Polym. Sci. 30, 3779 (1985)
[32] Hearle, J.W.S., in: Fiber Structure (Hrsg.: Hearle, J.W.S./ Peters, R.H.), Butterworth, London (1963)
[33] Balser, K. in: Polysaccharide, W. Burchard (Hrsg.), Springer-Verlag Berlin, S. 84-110 (1985)
[34] Philipp, B./ Reinisch, G.: Grundlagen der makromolekularen Chemie (Akademie- Verlag, Berlin, 1976)
[35] Herlinger, H./ Grynaeus, P./ Hirt, P., et al.: Lenzinger Ber. 69, 65 (1990)
[36] Gruber, E./ Gruber, R.: Das Papier 35, 133 (1981)
[37] Hudson, S.M./ Cuculo, J.A.: J. Macromol. Sci. - Rev. Macromol. Chem. C18, 1 (1980)
[38] Turbak, A.F./ Hammer, R.B./ Portnoy, N.A./ Davies, R.E., in: "Solvent Spun Rayon Modified Cellulose Fibres and Derivatives", Turbak, A.F.(Hrsg.), American Chemical Society, Washington DC 1977, S. 12
[39] Philipp, B.: J. Macromol. Sci.-Pure Appl. Chem. A30, 703 (1993)
[40] Schleicher, H./ Fink, H.-P.: Lenzinger Ber.74, 5 (1994)
[41] Heinze, Th./ Liebert, T.: Cellulose Chem. Technol. 32, 3 (1998]
[42] Schleicher, H./ Philipp, B./ Kabrelian, V.: Das Papier 42, 653 (1988)
[43] Hergert, H.L.: Das Papier 33, 562 (1979)
[44] Graenacher, Ch./ Sallmann, R.: U.S. Patent 2,179,181 (1939)
[45] Johnson, D.L.: U.S. Patent 3,447,939 (1969)
[46] Johnson, D.L.: U.S. Patent 3,508,941 (1970)
[47] Franks, N.E./ Varga, J.K.: U.S. Patent 4,145,532 (1979)
[48] McCorsley, C.C.: U.S. Patent 4,246,221 (1981)
[49] Chanzy, H./ Dubé, M./ Marchessault, R.H.: J. Polym. Letters Ed. 17, 219 (1979)
[50] Chanzy, H./ Nawrot, S/ Peguy, A/ Smith, P./ Chevalier, J.:J. Polym. Sci., Polym. Phys. Ed. 20, 1909 (1982)
[51] Walker, M./ Zimmerman, R.L./ Whitcombe, G.P./ Humbert, H.H.:Lenzinger Ber. 76, 76 (1997)
[52] Brandner, A./ Zengel, H.:DE 30 34 685 (1980)
[53] Wachsmann, U./ Diamantoglou, M.:Das Papier 12, 660 (1997)

LITERATURVERZEICHNIS _________________________________________________________________________________________
99
[54] Zimmerman, R.L.: U.S. Patent 5,216,154 (1993), assigned to Texaco Chemical Company
[55] Maia, E./ Peguy, A./ Pérez, S.: Acta Cryst. B37, 1858 (1981)
[56] Taeger, E./ Michels, Ch./ Nechwatal, A.: Das Papier 12, 784 (1991)
[57] Maia, E./ Pérez, S.: Acta Cryst. B38, 849 (1982)
[58] Berger, W./ Keck, M.: Lenzinger Ber. 57, 43 (1984)
[59] Kast, K.M./ Reiling, S./ Brickmann, J.: J. Molec. Struct. (Theochem) 453, 169 (1998)
[60] Nakao Sani, O.: To Kogyo 4, 128 (1971)
[61] Philipp, B./ Schleicher, H./Wagenknecht, W.: Cellulose Chem. Technol. 12, 529 (1978)
[62] Turbak et al.: Cellulose and Cellulose Derivatives, Interscience 1981
[63] Berger, W./ Keck, M./ Philipp, B.: Cellulose Chem. Technol. 22, 387 (1988)
[64] Seger, B.: Dissertation, Universität Freiburg i.Br. (1996)
[65] Berger, W./ Keck, M./ Do Van Sang /Philipp, B.: Z. phys. Chemie, Leipzig 266, 436 (1985)
[66] Berger, W./ Kabrelian, V./ Keck, M./ Kressler, J./ Herzog, K./ Scheller, D./ Mun Sang U./ Philipp, B.: Acta Polym. 41, 25 (1990)
[67] Vollbracht, L. Chemiefasern/Textilindustrie 91, 935 (1989)
[68] Götze, K.: Chemiefaser nach dem Viscoseverfahren (2 Bände), 3. Aufl., Springer, Berlin 1967
[69] Elias, H.-G.: Makromoleküle, (Industrielle Polymere und Synthesen), Bd.3, 6. Aufl., Wiley-VCH, Weinheim 2001
[70] Brück, M./ Gensrich, J.: Vortrag zum 3. Internationalen Symposium „Alternative Cellulose - Herstellen, Verformen, Eigenschaften” (Sept. 1998)
[71] Fink, H.-P./ Rihm, R./ Gensrich, J.: Vortrag zum 5. Internationalen Symposium „Alternative Cellulose - Herstellen, Verformen, Eigenschaften” (Sept. 2002)
[72] Cemic, L.: Thermodynamik in der Mineralogie. Eine Einführung, Springer-Verlag, Berlin/Heidelberg 1988
[73] Sommer, K.: Wissensspeicher Chemie, Volk und Wissen Verlag, Berlin 1996
[74] Cowie, J.M.G.: Chemie und Physik der Polymeren. Eine Einführung, Verlag Chemie, Weinheim/New York 1976
[75] Carlowitz, B.(Hrsg.): Die Kunststoffe. Chemie, Physik, Technologie (Kunststoff-Handbuch Bd.1), Carl Hanser Verlag, München/Wien 1990
[76] Elias, H.-G.: Makromoleküle, (Physikalische Strukturen und Eigenschaften), Bd.2, 6. Aufl., Wiley-VCH, Weinheim 2001
[77] Tieke, B.: Makromolekulare Chemie. Eine Einführung, VCH, Weinheim 1997

LITERATURVERZEICHNIS _________________________________________________________________________________________
100
[78] Hemminger, W./ Cammenga, H.: Methoden der Thermischen Analyse, Springer-Verlag Berlin/Heidelberg 1989
[79] Hemminger, W./ Höhne, G.: Grundlagen der Kalorimetrie, Verlag Chemie, Weinheim 1979
[80] Hoffmann, M./ Krömer, H./ Kuhn, R.: Polymeranalytik II: makromolekulare Strukturen, physikalische Methoden, Anwendungskriterien, Georg Thieme Verlag Stuttgart 1977
[81] Ehrenstein, G.W./ Riedel, G./ Trawiel, P: Praxis der thermischen Analyse von Kunststoffen, Carl Hanser Verlag, München/Wien 1998
[82] Ehrenstein, G.W: Kunststoff-Schadensanalyse: Methoden und Verfahren, Carl Hanser Verlag, München/Wien 1992
[83] Möhler, H./ Knappe, S.: Thermische Analyse für Polymerwerkstoffe, NETZSCH-Applikationsband 1998
[84] O`Neill, M.J.: Analytical Chemistry, 47, 630 (1975)
[85] Widmann, G./ Riesen, R.: Thermoanalyse: Anwendungen, Begriffe, Methoden, 3. Aufl., Hüthig Buch Verlag, Heidelberg 1990
[86] Fink, H.-P.: Dissertation B, A. d. W. zu Berlin, Teltow-Seehof (1990)
[87] Fink, H.-P./ Weigel, P./ Purz, H.J./ Ganster, J.: Prog. Polym. Sci. 26, 1473 (2001)
[88] Weigel, P./ Fink, H.-P.: Lenzinger Ber. 76, 115 (1997)

Danksagung
Diese Arbeit wurde im Fraunhofer-Institut für Angewandte Polymerforschung (IAP) in
der Abteilung Processing des Forschungsbereiches Native Polymere angefertigt.
Meinem Abteilungsleiter Herrn Dr. P. Weigel danke ich für die interessante Themen-
stellung und die fachliche Betreuung. Sein immerwährendes Interesse an meiner Arbeit
waren mir eine große Hilfe.
Herrn Prof. Dr. G. Hinrichsen von der Technischen Universität Berlin möchte ich auf-
richtig für die Übernahme des Referates sowie für sein Interesse am Fortgang dieser
Arbeit und seine wegweisenden Ratschläge danken.
Ganz besonders bedanke ich mich bei Herrn Dr. habil. H.-P. Fink für seine Unterstüt-
zung bei der Themenfindung sowie für die nützlichen Diskussionen und Hinweise.
Herrn Dr. J. Ganster danke ich für seine ständige Diskussions- und Hilfsbereitschaft
sowie für die kritische Durchsicht der Dissertation.
Bei Herrn Dr. W. Wagenknecht möchte ich mich für die hilfreichen Diskussionen über
die Cellulosechemie bedanken.
Den Mitarbeitern der Abteilungen Processing danke ich ganz herzlich für die stetige
Hilfsbereitschaft und das freundliche Arbeitsklima. Danken möchte ich auch den
Mitarbeitern der Abteilung Strukturcharakterisierung, die mich bei Messungen der
Röntgendiffraktometrie und der Elektronenmikroskopie unterstützt haben.
Ganz besonderer Dank gilt meiner Ehefrau Nurhayat, die in besonderer Weise die
Höhen und Tiefen bei der Bewerkstelligung dieser Arbeit miterleben durfte.
Meinen Eltern möchte ich herzlich danken, die mir jederzeit beistanden.

Lebenslauf
Tevfik Cibik Persönliche Daten:
Geburtsdatum: 1. Januar 1965 Geburtsort: Tuzluca, Türkei Staatsangehörigkeit: deutsch Familienstand: verheiratet, zwei Kinder, 12 und 15 Jahre alt Wohnort: Juliusstrasse 19 12051 Berlin
Schulbildung:
1971 - 1973 Besuch der türkischen Grundschule in Igdir, Türkei 1973 - 1978 Besuch des Deutschkursus und der Emil-Fischer-Grundschule
in Berlin 1978 - 1982 Besuch der Kurt-Löwenstein-Oberschule, der Helmholtz-
Oberschule und der Otto-Hahn-Oberschule in Berlin 1982 - 1984 Besuch der gymnasialen Oberstufe in Berlin 1984 Erwerb der allgemeinen Hochschulreife
Studium:
1985 - 1987 Studium des Studienfaches Elektrotechnik an der Techni-schen Universität Berlin
1987 - 1997 Studium des Studienganges Physik an der Technischen Uni-versität Berlin
1992 Zwölfwöchiges Industriepraktikum in der Werkstatt der Wil-helm-Foerster-Sternwarte in Berlin
1994 - 1996 Studien- und Diplomarbeit im Hahn-Meitner-Institut in Berlin April 1997 Studienabschluß als Diplom-Physiker
Aktuell:
1998 - 2003 Arbeit an der Dissertation im Fraunhofer-Institut für Ange-wandte Polymerforschung in Golm