updating the y-chromosomal phylogenetic tree for forensic applications based on whole genome snps
TRANSCRIPT

Forensic Science International Genetics xxx (2013) xxxndashxxx
G Model
FSIGEN-977 No of Pages 8
Updating the Y-chromosomal phylogenetic tree for forensicapplications based on whole genome SNPs
A Van Geystelen ab1 R Decorte ac MHD Larmuseau acd1a UZ Leuven Laboratory of Forensic Genetics and Molecular Archaeology Leuven Belgiumb KU Leuven Department of Biology Laboratory of Socioecology and Social Evolution Leuven Belgiumc KU Leuven Department of Imaging amp Pathology Forensic Medicine Leuven Belgiumd KU Leuven Department of Biology Laboratory of Biodiversity and Evolutionary Genomics Leuven Belgium
A R T I C L E I N F O
Article history
Keywords
Haploid markers
Y-chromosome
Phylogenetic tree
Bio-informatics
Whole-genome SNP calling
Y-SNPs
A B S T R A C T
The Y-chromosomal phylogenetic tree has a wide variety of important forensic applications and
therefore it needs to be state-of-the-art Nevertheless since the last lsquoofficialrsquo published tree many
publications reported additional Y-chromosomal lineages and other phylogenetic topologies Therefore
it is difficult for forensic scientists to interpret those reports and use an up-to-date tree and
corresponding nomenclature in their daily work Whole genome sequencing (WGS) data is useful to
verify and optimise the current phylogenetic tree for haploid markers The AMY-tree software is the first
open access program which analyses WGS data for Y-chromosomal phylogenetic applications Here all
published information is collected in a phylogenetic tree and the correctness of this tree is checked based
on the first large analysis of 747 WGS samples with AMY-tree The obtained result is one phylogenetic
tree with all peer-reviewed reported Y-SNPs without the observed recurrent and ambiguous mutations
Nevertheless the results showed that currently only the genomes of a limited set of Y-chromosomal
(sub-)haplogroups is available and that many newly reported Y-SNPs based on WGS projects are false
positives even with high sequencing coverage methods This study demonstrates the usefulness of AMY-
tree in the process of checking the quality of the present Y-chromosomal tree and it accentuates the
difficulties to enlarge this tree based on only WGS methods
2013 Elsevier Ireland Ltd All rights reserved
Contents lists available at SciVerse ScienceDirect
Forensic Science International Genetics
jou r nal h o mep ag e w ww e lsev ier co m loc ate fs ig
1 Introduction
A state-of-the-art phylogeny of the human Y-chromosomebased on bi-allelic polymorphisms is an essential tool for forensicgenetics Forensic scientists are taking advantage of the Y-chromosomal phylogenetic tree in their daily work eg bychecking the quality of datasets or by assigning geographicallandscapes to specific lineages [12] Y-chromosomal singlenucleotide polymorphisms (Y-SNPs) have a great capacity fordetecting geographical origins as many lineages defined by Y-SNPs show a strong continent-specific [34] and even intra-continent-specific distribution [5ndash7] Their usefulness is illus-trated by the fact that Y-SNP data are now also included in Y-chromosomal forensic databases such as in the YHRD database[8] Therefore an up-to-date extended Y-chromosomal phylogeny
Corresponding author at Katholieke Universiteit Leuven Forensic Medicine
Kapucijnenvoer 33 Bndash3000 Leuven Belgium Fax +32 0 16324575
E-mail address maartenlarmuseaubiokuleuvenbe (MHD Larmuseau)1 Both authors contributed equally to this study
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
1872-4973$ ndash see front matter 2013 Elsevier Ireland Ltd All rights reserved
httpdxdoiorg101016jfsigen201303010
based on these bi-allelic markers which are preferably unambig-uous and non-recurrent but which have a high discriminationpower is required for forensic applications
Since the publication of the latest lsquoofficialrsquo Y-chromosomalphylogenetic tree by Karafet et al [4] a continuous wave of newpeer-reviewed articles which report changes to this tree arepublished These changes include a new root and new basal clades[910] modifications of the global backbone [311] differentphylogenetic topologies within a haplogroup [12ndash14] newlydescribed sub-haplogroups [15ndash17] or other phylogenetic posi-tions for a certain mutation [18] As these publications are notcoordinated different names are given to the Y-chromosomallineages for which the phylogenetic position is given in differenttopologies Therefore the currently overall reported Y-chromo-somal tree is not clear and this makes it difficult for forensicresearchers to use a uniform phylogenetic tree Hence newinitiatives to ensure more continuity in the report of the mostrecent phylogenetic Y-chromosomal tree are needed
Large whole genome sequencing (WGS) projects such as the1000 Genomes Project [1920] bring an opportunity to introducethe required uniformity in the reporting of the haploid
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
TP FP TN FN
Measures
Expected stateancestralmutant
State in reference genome
Select Y-SNPs based on determined haplogroup
Expected situaon in samplecall ednot call ed
Actual situaon in samplecall ednot call ed
Fig 1 Workflow of the quality assessment algorithm of the AMY-tree version 11
First certain Y-SNPs of the Y-chromosomal phylogenetic tree are selected based on
the determined haplogroup of the first run in order to avoid too much false positive
Y-SNPs Next the expected state (ancestral or mutant) of the selected SNPs is
determined based on the haplogroup and the phylogenetic tree These expectations
of state are converted to expectations of called or not called based on the SNP state in
the reference genome These expectations are then compared to the actually called
SNPs of the sample such that the number of true positives (TP) false positives (FP)
true negatives (TN) and false negatives (FN) will be determined Finally several
different measures will be calculated
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx2
G Model
FSIGEN-977 No of Pages 8
Y-chromosomal tree The analysis of whole Y-chromosomes withinmale genomes allows verification and optimisation of thecurrently used phylogenetic tree WGS data has already provedto be useful in verifying and optimising the phylogeny of the otherhaploid markers in the human genome ie the mitochondrial DNA(mtDNA) Relevant ambiguous markers and back-mutations whichinfluence the interpretation of previous forensic and evolutionarygenetic studies were detected based on these data [21] Recently anew Y-chromosomal phylogenetic tree was built after a tabula rasa
of the present Y-chromosomal tree by using only Y-SNPs fromavailable WGS male samples [22] By comparing this newphylogenetic tree with the currently used one the backbone ofthe currently used phylogenetic tree was confirmed in this studyHowever this new tree is not useful for forensic research becausethere is no link between currently used and newly reportedlineages Furthermore the set of used genomes is not a goodrepresentation of all existing Y-chromosomal (sub-)haplogroupsand geographical regions There are also still too much falsepositive SNP calls in this WGS dataset Alternatively the AMY-treesoftware is the first open access program which academics andforensic professionals can use to verify and optimise the currentlyused Y-chromosomal tree by using WGS data [23] The first AMY-tree analysis was done based on 118 WGS samples and provedalready its usefulness to verify and to optimise the present Y-chromosomal phylogenetic tree [23]
The aim of this study is to perform the largest reportedscreening of male genomes for Y-chromosomal phylogeneticapplications based on the AMY-tree software Firstly we wantto merge all newly Y-SNPs from recent peer-reviewed publicationssince the latest lsquoofficialrsquo Y-chromosomal phylogeny [4] into onesingle tree which is useful for forensic applications Secondly thisupdated Y-chromosomal phylogenetic tree needs to be checked forrecurrent mutations ambiguous SNPs and other difficulties for the(forensic) application of the tree Thirdly this study also wants tofind out for which Y-chromosomal (sub-)haplogroups there isalready WGS data available Finally investigating the possibilitiesto enlarge the Y-chromosomal phylogenetic tree based on thecurrent Y-SNP detections in WGS data is the last aim of this study
2 Materials and methods
21 Updated phylogenetic tree
The latest updated phylogenetic tree of the Y-chromosome as itwas published by Van Geystelen et al [23] was manually updatedbased on recent descriptions of new Y-SNPs in academic researchpapers like Pamjav et al [16] and Scozzari et al [9] As the exactphylogenetic position of a few new Y-SNPs was not given theirposition needed to be determined based on the results of AMY-treeof all WGS samples Next also recurrent mutations ambiguousSNP-loci and wrongly defined mutation conversions within thenewly updated Y-chromosomal tree were ascertained based onthose AMY-tree results
22 WGS Y-SNPs dataset
In order to check the manually updated phylogenetic tree andto optimise the AMY-tree software a large dataset of wholegenome Y-SNP calls was assembled This dataset consists of 747samples which represent 660 males as several genomes wereanalysed in different projects Within this dataset the genomes ofeight males whose fatherrsquos genome was also sequenced arepresent The SNP calls were collected from four large WGS projectsand several individual genome projects (Supplementary MaterialsTable S1) These projects differ from each other based on the usednext-generation sequencing (NGS) platforms and sequence cover-
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
age First Complete Genomics made the SNP calls of 35 wholegenomes of males available (httpwwwcompletegenomicscompublic-data69-Genomes04 Jan 2013) those genomes weresequenced with a high sequencing coverage on the CompleteGenomics Analysis (CGA) Platform [24] Second the PersonalGenome Project (PGP) and Singapore Sequencing Malay Project(SSMP) also used this CGA platform PGP is a project started toobtain and openly share human genome sequences in combinationwith health information At the moment 40 male genomes wereavailable (wwwpersonalgenomesorg 04 Jan 2013) The SSMP onthe other hand wanted to characterise the polymorphic variants inthe population of Malays an Austronesian group present inSoutheast Asia and Oceania Recently the Y-SNP calls of 46 Malayswere made publically available [25] Next the 1000 GenomesProject aims to provide a comprehensive resource on humangenetic variation by sequencing more than 1000 human genomesIn 2010 SNP calls of 77 males were made available in the pilotphase [20] and two years later a set of 526 SNPs profiles werepublished as result of phase 1 of the Project [19] As the 1000Genomes project aims to sequence a large number of people thesequencing coverage was lower than in the other projects Finally23 additional samples were collected from several single genomeprojects [26ndash35 36 and unpublished genomes of Guy Froyen]
23 AMY-tree modifications
Several modifications to the AMY-tree software version 10 [23]were made for the assessment of the SNP calling quality in WGSdata This was necessary as the quality of the SNP calling influencesthe AMY-tree analysis of a sample and therefore also theinterpretation of the result of the analysis [23] The extra qualityassessment is based on the results of the first AMY-tree run of acertain sample This assessment assumes that the used phyloge-netic tree is correct and that the assigned haplogroup after the firstAMY-tree run is the actual haplogroup of that sample
The algorithm for the extra quality assessment is simple andcomprehensible as shown in Fig 1 First all Y-SNPs of thephylogenetic tree are selected except if the determined haplogroupof the first run is a paragroup (eg R1b1b2) In the case of aparagroup all Y-SNPs which are in sub-nodes of the main group ofthis paragroup are excluded from the selection This is done inorder to remove the influence of too much false positive SNP calls
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
R-M198 M198 M417 M512 M514 M515 Page7
R-M56R-M157 1
R-M87
R-P98
R-PK5
R-M43 4
R-Page68
R-Z28 0
R-Z93
R-M458
M56
M157 1
M87 M20 4
P98
PK5
M43 4
M45 8
Page68
Z28 0
Z93
R-M33 4M33 4
Fig 2 Overview of the position of the newly added sub-haplogroups R-Z280 and R-
Z93 (given in bold) within R-M198
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 3
G Model
FSIGEN-977 No of Pages 8
because when the haplogroup is not at the correct phylogeneticlevel too much false positives andor false negatives mutant Y-SNPs would be detected Next the expected state of all selected Y-SNPs is determined whereby all Y-SNPs in the path from theassigned sub-haplogroup till root are expected to be mutant andall others are expected to be ancestral The state of each Y-SNP isalso determined in the reference genome Thereafter theseexpectations of Y-SNP state are converted into expectations oflsquocalledrsquo or lsquonot calledrsquo in the next step based on the expected stateand the state in the reference genome Consider a Y-SNP in thepath from sub-haplogroup till root if the status of that SNP in thereference sequence is mutant the Y-SNP is expected to be lsquonotcalledrsquo else the expectation is lsquocalledrsquo Consider a Y-SNP not in thepath from sub-haplogroup till root if the status of that Y-SNP inthe reference sequence is mutant the Y-SNP is expected to belsquocalledrsquo else the expectation is lsquonot calledrsquo Then these expecta-tions are compared to the state in the sample such that thenumber of true positive false positive true negative and falsenegative SNP calls will be determined At last the quality of asample is expressed in several measures of quality Matthewscorrelation coefficient (MCC) accuracy sensitivity specificityprecision recall and F1-score (Supplementary method) When theMCC is larger or equal to 095 the SNP calling quality is calledexcellent otherwise it is called low The quality will be given in theoutput file of AMY-tree When the SNP calling quality is lowcaution has to be taken about the result of the AMY-tree analysisdue to the high occurrence of false negative and false positiveSNPs When the quality is excellent the results of AMY-tree areconsidered to be valuable for the control of the currently usedphylogenetic tree of the Y-chromosome and for the increase of itsresolution As such a better Y-SNP call quality assessment isimplemented in AMY-tree version 11 compared to the earlierversion [23]
Next when a sample was run in AMY-tree in the lsquosufficientrsquomode such that the reference genome was taken into account butthe determined haplogroup belongs to R-M269 and the MCC islower than 095 a second AMY-tree run needs to be executed but inthe lsquoinsufficientrsquo mode This is important as a MCC lower than 095indicates that this result of the first run is too much influenced bythe reference genome Finally another small modification to AMY-tree is based on the fact that Z381 L2 as well as L20 are mutant inthe reference genome and although AMY-tree version 10 alreadyhad a quite complex system to filter out the influence of thereference genome even on samples belonging to haplogroup R itwas not yet efficient enough Therefore when both Z381 and L2 orboth Z381 and L20 are mutant in the first AMY-tree run eg theancestral SNP was not called the sample will be handled asinsufficient in a second AMY-tree run such that the Y-SNPs of thereference genome are not used anymore when determining thehaplogroup By including these modifications even more certaintyis build in AMY-tree version 11 in comparison with the previousversion [23] for samples belonging to a R-M269 sub-haplogroupthe reference genome is only taken into account when the SNPcalling quality is excellent after the first run in the lsquosufficientrsquomode
The cut-offs to assess the Y-SNP calling quality is optimisedbased on all 747 genomes by performing several test runs andmanual analyses of the genomes and by checking it with the resultsin Van Geystelen et al [23] and with the publications of thegenomes whereby a Y-chromosomal analysis was already per-formed
24 Y-SNP detecting
The Y-SNPs which are present in the WGS samples but whichare not yet included in the updated phylogenetic tree were
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
detected by AMY-tree Only those Y-SNPs from WGS samples withan excellent Y-SNP calling quality were used But that was not theonly constraint for the potentially relevant Y-SNPs they must alsobe positioned in the unique regions of the Y-chromosome as readmapping and variant detection difficulties are expected due to thehigh frequency of repeated sequences on the Y-chromosome So Y-SNPs in the pseudoautosomal heterochromatic X-transposed andampliconic segments [37] of the male-specific part of the genomeas reported by Wei et al [22] were excluded
3 Results
In total 747 samples are analysed by the updated version of theAMY-tree software with the updated phylogenetic Y-chromosomaltree There are 131 samples of 126 individuals with an excellent Y-SNP calling quality ie MCC 095 which are mostly obtainedfrom Complete Genomics and the Personal Genome Project Theremaining 616 samples have a low calling quality and are mostlyobtained from the 1000 Genomes Project pilot and phase 1 (TableS1)
31 Updated tree for forensic applications
The state-of-the-art Y-chromosomal tree is manually updatedbased on all published Y-SNPs from academic studies After theAMY-tree runs of all 747 samples all Y-SNPs of which no exactphylogenetic position was given in the publications could now beincluded in the updated phylogenetic tree for example there weretwo new Y-SNPs reported in [16] within sub-haplogroup R-M198without clear phylogenetic positions (Fig 2)
The results with an excellent SNP calling quality also showedseveral recurrent Y-SNPs in the phylogenetic tree which cause thedetermination of multiple haplogroups for some samples Afterruling out that the recurrent SNPs are sample- or project-specificthese SNPs are removed from the phylogenetic tree an overview ofthe three observed recurrent SNPs of which enough evidence wasavailable is given in Table S2 These modifications led to the finalupdated tree version 11 which includes 359 Y-chromosomallineages and 721 Y-SNP markers The final tree and itscorresponding mutation conversions for all the Y-SNPs in the treecan be found in Tables S3 and S4
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
00 01 02 03 04 05 06 07 08 09 10
0
80
160
Number of samples
Mahewrsquos corr elaon coefficient
Fig 3 Distribution of the Matthews correlation coefficient of the 730 samples for which an unambiguous haplogroup could be determined
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx4
G Model
FSIGEN-977 No of Pages 8
32 Sub-haplogroup determining
The determined haplogroups of all 747 samples obtained by theAMY-tree analysis based on the final updated tree version 11 isgiven in Table S1 Only for 14 samples no haplogroup can bedetermined and for three other samples multiple haplogroups aredetermined by AMY-tree The distribution of the MCC of the 730samples for which an unambiguous haplogroup is determined isgiven in Fig 3 Only a minority of the samples has an excellent SNPcalling quality with a MCC 095 a MCC lower than 095 meansthat less than 975 of the negative and positive predictions arecorrect Overall 17 different haplogroups and 106 sub-hap-logroups are present in the dataset When considering only thesamples of excellent quality 10 different haplogroups and 47 sub-haplogroups remain Fig 4 and Table S5 give an overview of those(sub-)haplogroups and their frequencies
The samples of paternally related samples present in thedataset are of particular interest because they are considered torepresent the same Y-chromosome All the samples of one familywith eight members sequenced by Complete Genomics aredetermined to belong to R-P312 The paternal grandfather ofthat family is also analysed in the 1000 Genomes project and therehe is determined as P [P-92R7] The first attempt of AMY-tree todetermine the sub-haplogroup of that sample of 1000 Genomes ledto the sub-haplogroup R-L2 which has a higher phylogenetic levelthan the haplogroups of the Complete Genomics samples
A B C D E F G H I J
0
55
110
165
All geno mes
Good quali ty genomes
Numbe r of availab le WGS ge nomes
Hap lo
Fig 4 Frequency of whole genome sequencing (WGS) genomes per haplogroup in the dat
MCC 095 (grey)
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
However as the MCC value of that sample is smaller than 095the sub-haplogroup determination is done again but without theinfluence of the reference genome This led to the final sub-haplogroup P-92R7 which has a less accurate phylogenetic levelthan that of Complete Genomics Thus the influence of thereference genome can sometimes cause a too high or too lowphylogenetic level when the new modifications which were madeto AMY-tree version 11 would not have been applied
33 Detecting new Y-SNPs
The large amount of available samples leads to a huge amountof newly reported Y-SNPs ie Y-SNPs that are not yet present in theupdated phylogenetic tree version 11 In total 108681 new Y-SNPsare reported in all 660 male genomes when an individual isanalysed in more than one project only the sample with the highestMCC value is used The majority of the SNPs appears in only a fewsamples 62 appears in only one sample and 16 is present in twosamples as shown in Fig 5A In the 126 male genomes with anexcellent Y-SNP calling quality 50430 new Y-SNPs are reportedThese SNPs also come with a high frequency of low occurrences inthe excellent genomes 57 is unique and 11 appears in twosamples When only the regions within the Y-chromosome whichare identified as unique are taken into account a much lowernumber of new Y-SNPs is detected In total 35503 new Y-SNPs arereported in the 660 male genomes and 15208 new Y-SNPs in the
K L M N O P Q R S T
g roups
aset of all samples (black) and in the dataset of samples with an excellent quality ie
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
1 2 3 4 5 6 7 8 9 10 more
Numbe r of occ urr ences of Y-SNP
0
12000
24000
0
35000
70000
Number of Y-SNPsWhole Y-chromosome
Uniqu e regions Y-chromosome
A
B
All genomes
Excell ent qu ali ty genomes
Fig 5 Number of new Y-SNPs per number of occurrence in the full WGS dataset (A) SNPs in the whole Y-chromosome and (B) SNPs in only the identified unique regions of the
Y-chromosome The grey bars indicate the SNPs in all genomes and the black bars indicate the SNPs from samples with an excellent quality ie MCC 095
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 5
G Model
FSIGEN-977 No of Pages 8
genomes with excellent Y-SNP calling quality The same patterns ofoccurrence for these Y-SNPs are observed as with the non-filteredY-SNPs the majority of the new reported Y-SNPs appeared in onlyone or two samples of the WGS dataset as shown in Fig 5B
The genomes of eight males whose biological fatherrsquos genome isalso sequenced are present in the dataset one family of eightmales including the father the son and the six grandsons next toone fatherndashson pair In the family of eight paternal relatives 5155new SNPs are reported on the whole Y-chromosome and a largenumber of these SNPs is found in only one of the eight individualsas shown in Fig 6A The number of Y-SNPs decreases every time
0
500
1000
1500
2000
2500
0
1 2 3 4 5
Numbe r of o
0
20
40
60
80
100
120
Number of Y-SNPsA
B
8-member fam
All SNPs
Famil y-un iqu e SNP
Fig 6 Number of new Y-SNPs per occurrence in the eight paternally related samples and t
(B) SNPs in only the identified unique regions of the Y-chromosome The grey bars indica
uniquersquo they do not occur in any of the other samples with an excellent quality ie M
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
the number of samples in which the Y-SNP occurs increases exceptfor the occurrence in all-except-one and all samples of the familyWhen all Y-SNPs which also occur in any other sample withexcellent SNP calling quality are removed only less than 20 of theY-SNPs remains but the distribution of the number of Y-SNPs peroccurrence stays the same as the black bars in Fig 6A show Thesame comparison is made when only the SNPs in the unique part ofthe Y-chromosome are selected the same pattern as with all SNPsis visible in Fig 6B However for each number of occurrences thenumber of truly unique SNPs ie SNPs that do not occur in othergenomes outside the family is much higher Within the fatherndashson
6 7 8 1 2
cc urr ences of Y-SNP
Whole Y-chromosome
Uniqu e regions Y-chromosome
il y Father-son pair
s
he fatherndashson pair of Complete Genomics (A) SNPs in the whole Y-chromosome and
te the SNPs found in the family The black bars indicate the SNPs which are lsquofamily-
CC 095
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx6
G Model
FSIGEN-977 No of Pages 8
pair 2181 new SNPs are reported on the whole Y-chromosome forwhich the difference between occurrence in only one and bothgenomes is relatively small also for the proportion of SNPs whichdo not occur in the other genomes with an excellent Y-SNP quality(Fig 6A) Remarkably there are more new SNPs present in bothsamples than in one sample when the Y-SNPs which are notlocated in the unique region of the Y-chromosome are removed(Fig 6B) The effect of the increasing number of unique Y-SNPs thatdo not occur in other genomes as seen with the previous family isalso in the fatherndashson pair present
4 Discussion
The present study realises the first large screening of malegenomes for phylogenetic applications of the Y-chromosomeBased on this screening of 747 male samples an update has beenmade of the AMY-tree software and of a state-of-the-art Y-chromosomal phylogenetic tree was established for forensicscientists Furthermore also recommendations for future sequenc-ing projects dealing with a broader selection panel of Y-chromosomal haplogroup samples and for the validation of newlydetected Y-SNPs are made
First the large screening of the 747 male Y-chromosomesamples revealed that the SNP calling quality of a few samples wasoverestimated in AMY-tree version 10 As these samples belong tosub-haplogroup R-M269 they are very similar to the referencegenome which is used to estimate the SNP calling quality of asample [23] To remove this SNP calling quality overestimation theinfluence of the reference genome is excluded for all samplesbelonging R-M269 with a low SNP call quality That is why severalmodifications are made and implemented in AMY-tree version 11
Second an update of the currently used Y-chromosomalphylogenetic tree was realised based on the large database of747 available samples At the moment this tree is the most state-of-the-art tree applicable for forensic geneticists all Y-SNPs whichare reported in academic publications till today are included andall ambiguous markers are excluded to avoid wrong Y-SNPinterpretations (Table S3) As often the case in the literature thephylogenetic relationship between new Y-SNPs and earlierreported Y-SNPs in the same (sub-)haplogroup are not given Byusing the AMY-tree results it is possible to find out the concretephylogenetic level of each Y-SNP relative to the other alreadyknown SNPs For example Pamjav et al [16] described andgenotyped two new Y-SNPs Z93 and Z280 within R-M198although the exact phylogenetic positions in relationship withthe other lineages within R-M198 were not given The presence ofboth Z93 and Z80 is checked in all samples belonging to the sub-haplogroup R-M198 Z93 occurred in three R-M198 samples andin none of the other samples Also Z280 does not occur in anysample except in one R-M198 sample Therefore both Z93 andZ280 are placed in the phylogenetic tree as sub-haplogroups of R-M198 as shown in Fig 2 The choice is made for a phylogenetic treein table format as described by Van Geystelen et al [23] instead of abranching diagram because the tree is very large and will becomelarger in the future Therefore the table format can be adaptedmore easily than the diagram and it is also more manageable
Not only newly reported Y-SNPs are included in the newupdated tree but also ambiguous Y-SNPs are excluded as they cancomplicate the Y-chromosomal applications for forensic studiesAs previously described [38] the most relevant ambiguous Y-SNPsare recurrent SNPs which have a paralogous distribution along thephylogenetic tree and which have thus mutant alleles in at leasttwo independent Y-chromosomal lineages Based on the screeningof the 747 analysed samples three Y-SNPs are recognised for thefirst time as recurrent (Table S2) There are no other indications forrecurrent mutations based on the present WGS dataset Therefore
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
we may assume in most cases that males which both have themutant allele for a Y-SNP used in the updated tree (Table S3) havereceived this mutant allele from one common ancestor and not byconvergent evolution Next also all Y-SNPs which could not beanalysed by WGS for one reason or another are excluded from theupdated tree although it may be possible to genotype these SNPscorrectly with Sanger sequencing methods For example allhundred E-M2 samples in the dataset did not reveal the mutantallele for Y-SNP V95 as expected by earlier publications [1439]The reason for this remarkable result may be that V95 is not wellvalidated but more likely it is the result of a bad SNP calling in theY-chromosomal region around V95 by the current WGS methodsFor some Y-SNPs also a mutation conversion is found which isdifferent from the reported one For example another ancestraland mutant allele are observed for Y-SNP M426 than reported byRootsi et al [40] Since the Y-chromosome has a very complexorigin it also has a lot of non-unique regions which complicate theanalysis of WGS data [37] The current reference genome GRCh37shows the evolution of the Y-chromosome and its numerousresulting non-unique regions So the reason for this wrong analysisof M426 may be the position of this SNP in one of the non-uniqueregions of the Y-chromosome as defined earlier [2237] To have anunambiguous phylogenetic tree we excluded all the Y-SNPs forwhich no reliable signal with the WGS methods could be found Inthe end an updated Y-chromosomal tree which includes 359 Y-chromosomal lineages and more than 721 Y-SNP markers (TableS3) is obtained and this tree is the basic tool to develop andoptimise Y-SNP-multiplexes for forensic applications [4142]
Third the distribution of the analysed haplogroups perancestral continent in the current dataset corresponds with theknown distributions [34] Nevertheless there is not yet arepresentative set of all phylogenetic sub-haplogroups availableIn total 17 haplogroups and 106 sub-haplogroups are reported inthe analysis of the whole dataset However the SNP calling qualityis low (MCC lt 095) for most of the analysed samples as most dataare obtained from the 1000 Genomes project and therefore thesub-haplogroup determination of these data is not completelyreliable When considering only the samples of excellent quality(MCC 095) 10 haplogroups and 47 sub-haplogroups remain andthis corresponds with only 13 of the total number lineagesdescribed so far Most of the Y-chromosomes are assigned tohaplogroups E O and R Therefore when the set of WGS Y-chromosomes will be enlarged the current phylogenetic tree aswell as the analysis of Wei et al [22] to calibrate the tree can beoptimised
Finally the screening of the dataset also revealed that new insilico and in vitro methods are required to verify new Y-SNPs basedon WGS methods As earlier mentioned [23] a huge number of newY-SNPs are false positives when the genomes were sequenced withlow coverage and consequently the called SNPs will have a lowquality These false positive SNP calls disturb the determination ofthe correct (sub-)haplogroup of the sample consequently theAMY-tree software has to correct for them by applying severaladditional methods in the analysis [23] The high number of falsepositive Y-SNPs ndash even detected in WGS samples with an excellentSNP calling quality ndash is observed by comparing genomes ofpaternal relatives Within the eight paternally related samplesbelonging to one family which are genotyped by CompleteGenomics 949 newly reported Y-SNPs in the full Y-chromosomeand 88 ones in the unique regions of the Y-chromosome are foundin at least one but not all family members (Fig 6) Despite the highcoverage of these genomes and the excellent SNP calling qualitythis is still a very high number of newly reported Y-SNPs which aremost likely to be false based on the mutation rate on the Y-chromosomes calculated based on a deep-rooting pedigree [43]and based on human-chimpanzee comparisons [44] A similar
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 7
G Model
FSIGEN-977 No of Pages 8
conclusion can be made based on the fatherndashson pair in the datasetwhich is also sequenced with a high coverage by CompleteGenomics (Fig 6)
The Y-SNP results show that adding new lineages to the Y-chromosomal phylogenetic tree only based on WGS is not evidentTherefore making tabula rasa and building a new tree based on allWGS Y-chromosomes as done by Wei et al [22] is not an optionwhen the tree is going to be used in forensics Each new Y-SNP andconsequently each Y-chromosomal lineage has to be validatedindependently from the WGS data before adding them to theupdated phylogenetic tree The validation status of many potentialpolymorphic Y-SNPs (gt1 in population) is often unclear but thiscan be resolved by the sharing of genomic data among geneticgenealogists which are interested in finding new Y-SNPs to resolvetheir particular paternal ancestry [45] Therefore this is an area inwhich closer collaborations between amateurs and forensicacademics could prove to be particularly useful [13] Neverthelessit is required that new in silico methods will be designed to selectgood and relevant candidates of Y-SNPs for the validation Forexample an interesting criterion is the position of the Y-SNPs it ismore interesting to validate only the SNPs located in the uniqueregions of the Y-chromosome as there are many non-uniqueregions due to the evolutionary history of this chromosome [37]The lower number of false positive SNP calls in these uniqueregions in comparison with those in the full Y-chromosome isclearly demonstrated in the fatherndashson pair The number of new Y-SNPs reported in only one of the two samples is higher than thenew ones reported in both samples based on the whole Y-chromosome (Fig 6A) in contrast with the situation based on theunique regions of the Y-chromosome (Fig 6B)
5 Conclusions
Based on the largest screening of male genomes with 747samples in total the most up-to-date Y-chromosomal phylogenet-ic tree for forensic applications is compiled Future publicationswhich will report new Y-SNPs have to situate their phylogeneticpositions in this tree to guarantee the continuity between old andnew publications At this moment forensic scientists as well asevolutionary biologists and genetic genealogists are lost in themany reports of newly described Y-chromosomal lineages [46]Therefore initiatives as AMY-tree which optimise the phylogenybased on peer-reviewed publications are required [23] This isalready the case for the mitochondrial genome with the Phylotreeinitiative of van Oven and Kayser [47] Nevertheless to optimisethe current updated phylogenetic tree for the human Y-chromo-some more high quality genomes of a broader set of (sub-)haplogroups than the frequent haplogroups E O and R arerequired Also a higher effort in the validation of reported Y-SNPsby new in silico and in vitro methods is required
Authorsrsquo contributions
Research design amp supervision MHDL programming AVGwriting MHDL amp AVG Commenting on manuscript AVG RD ampMHDL
Conflict of interest
The authors declare no conflict of interest
Acknowledgements
The authors want to thank Tom Wenseleers Manfred KayserJean-Jacques Cassiman Tom Havenith Hendrik Larmuseau and
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
Lucrece Lernout for useful discussions and comments Thanks alsoto Guy Froyen (VIB KU Leuven) Richard Rocca (independentresearcher) Cuiping Pan (Stanford University) and Andreas Keller(Saarland University) for providing us yet unpublished andpublished called SNPs of whole genome sequencing projectsMaarten HD Larmuseau is postdoctoral fellow of the FWO-Vlaanderen (Research Foundation Flanders) This study wasfunded by the KU Leuven BOF Centre of Excellence Financing onlsquoEco and socio evolutionary dynamicsrsquo (Project number PF201007) and on lsquoCentre for Archaeological Sciences 2 (CAS 2) ndash Newmethods for research in demography and interregional exchangersquo
Appendix A Supplementary data
Supplementary data associated with this article can be found in
the online version at httpdxdoiorg101016jfsigen201303010
References
[1] M Kayser Y-chromosomal markers in forensic genetics in RWD Rapley (Ed)Molecular Forensics John Wiley amp Sons Ltd Chichesters 2007 pp 141ndash161
[2] JM Butler Chapter 13 Y-Chromosomal DNA Testing in JM Butler (Ed) Ad-vanced Topics in Forensic DNA Typing Methodology Academic Press London2011 pp 371ndash403
[3] J Chiaroni PA Underhill LL Cavalli-Sforza Y chromosome diversity humanexpansion drift and cultural evolution Proc Natl Acad Sci USA 106 (2009)20174ndash20179
[4] TM Karafet FL Mendez MB Meilerman PA Underhill SL Zegura MFHammer New binary polymorphisms reshape and increase resolution of thehuman Y chromosomal haplogroup tree Genome Res 18 (2008) 830ndash838
[5] MHD Larmuseau N Vanderheyden M Jacobs M Coomans L Larno R DecorteMicro-geographic distribution of Y-chromosomal variation in the central-westernEuropean region Brabant Forensic Sci Int Genet 5 (2011) 95ndash99
[6] F Cruciani B Trombetta C Antonelli R Pascone G Valesini V Scalzi G Vona BMelegh B Zagradisnik G Assum et al Strong intra- and inter-continentaldifferentiation revealed by Y chromosome SNPs M269 U106 and U152 ForensicSci Int Genet 5 (2011) E49ndashE52
[7] MHD Larmuseau J Vanoverbeke G Gielis N Vanderheyden HFM LarmuseauR Decorte In the name of the migrant father ndash analysis of surname originidentifies historic admixture events undetectable from genealogical recordsHeredity 109 (2012) 90ndash95
[8] S Willuweit L Roewer Y chromosome haplotype reference database (YHRD)update Forensic Sci Int Genet 1 (2007) 83ndash87
[9] R Scozzari A Massaia E DrsquoAtanasio NM Myres UA Perego B Trombetta FCruciani Molecular dissection of the basal clades in the human Y chromosomephylogenetic tree Plos ONE 7 (2012) e49170
[10] F Cruciani B Trombetta A Massaia G Destro-Bisol D Sellitto R Scozzari Arevised root for the human Y chromosomal phylogenetic tree the origin ofpatrilineal diversity in Africa Am J Hum Genet 88 (2011) 814ndash818
[11] S Fornarino M Pala V Battaglia R Maranta A Achilli G Modiano A Torroni OSemino SA Santachiara-Benerecetti Mitochondrial and Y-chromosome diversi-ty of the Tharus (Nepal) a reservoir of genetic variation BMC Evol Biol 9 (2009)154
[12] FL Mendez TM Karafet T Krahn H Ostrer H Soodyall MF Hammer Increasedresolution of Y chromosome haplogroup T defines relationships among popula-tions of the Near East Europe and Africa Hum Biol 83 (2011) 39ndash53
[13] LM Sims D Garvey J Ballantyne Improved resolution haplogroup G phylogenyin the Y-chromosome revealed by a set of newly characterized SNPs Plos ONE 4(2009) e5792
[14] B Trombetta F Cruciani D Sellitto R Scozzari A new topology of the human Ychromosome haplogroup E1b1 (E-P2) revealed through the use of newly charac-terized binary polymorphisms PLoS ONE 6 (2011) e16073
[15] MS Jota DR Lacerda JR Sandoval PPR Vieira SS Santos-Lopes R Bisso-Machado VR Paixao-Cortes S Revollo C Paz-Y-Mino R Fujita et al A newsubhaplogroup of native American Y-chromosomes from the Andes Am J PhysAnthropol 146 (2011) 553ndash559
[16] H Pamjav T Feher E Nemeth Z Padar Brief communication new Y-chromo-some binary markers improve phylogenetic resolution within haplogroup R1a1Am J Phys Anthropol 149 (2012) 611ndash615
[17] NM Myres S Rootsi AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova A Pshenichnov B Yunusbayev et al A major Y-chromosomehaplogroup R1b Holocene era founder effect in Central and Western Europe Eur JHum Genet 19 (2011) 95ndash101
[18] S Yan CC Wang H Li SL Li L Jin G Consortium An updated tree of Y-chromosome Haplogroup O and revised phylogenetic positions of mutations P164and PK4 Eur J Hum Genet 19 (2011) 1013ndash1015
[19] T 1000 Genomes Project Consortium An integrated map of genetic variation from1092 human genomes Nature 491 (2012) 56ndash65
[20] DL Altshuler RM Durbin GR Abecasis DR Bentley A Chakravarti AG ClarkFS Collins FM De la Vega P Donnelly M Egholm et al A map of human
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx8
G Model
FSIGEN-977 No of Pages 8
genome variation from population-scale sequencing Nature 467 (2010)1061ndash1073
[21] AT Duggan M Stoneking A highly unstable recent mutation in human mtDNAAm J Hum Genet 9 (2013) 279ndash284
[22] W Wei Q Ayub Y Chen S McCarthy Y Hou I Carbone Y Xue C Tyler-Smith Acalibrated human Y-chromosomal phylogeny based on resequencing GenomeRes 23 (2013) 388ndash395
[23] A Van Geystelen R Decorte MHD Larmuseau AMY-tree an algorithm to usewhole genome SNP calling for Y chromosomal phylogenetic applications BMCGenomics 14 (2013) 101
[24] R Drmanac AB Sparks MJ Callow AL Halpern NL Burns BG Kermani PCarnevali I Nazarenko GB Nilsen G Yeung et al Human genome sequencingusing unchained base reads on self-assembling DNA nanoarrays Science 327(2010) 78ndash81
[25] L-P Wong RT-H Ong W-T Poh X Liu P Chen RQ Li KK-Y Lam NE PillaiK-S Sim H Xu et al Deep whole-genome sequencing of 100 Southeast AsianMalays Am J Hum Genet 92 (2013) 1ndash15
[26] SC Schuster W Miller A Ratan LP Tomsho B Giardine LR Kasson RS HarrisDC Petersen FQ Zhao J Qi et al Complete Khoisan and Bantu genomes fromsouthern Africa Nature 463 (2010) 943ndash947
[27] P Tong JGD Prendergast AJ Lohan SM Farrington S Cronin N Friel DGBradley O Hardiman A Evans JF Wilson et al Sequencing and analysis of anIrish human genome Genome Biol 11 (2010) R91
[28] A Keller A Graefen M Ball M Matzas V Boisguerin F Maixner P Leidinger CBackes R Khairat M Forster et al New insights into the Tyrolean Icemanrsquos originand phenotype as inferred by whole-genome sequencing Nature Commun 3(2012) 698
[29] R Chen GI Mias J Li-Pook-Than LH Jiang HYK Lam R Chen E Miriami KJKarczewski M Hariharan FE Dewey et al Personal omics profiling revealsdynamic molecular and medical phenotypes Cell 148 (2012) 1293ndash1307
[30] JM Rothberg W Hinz TM Rearick J Schultz W Mileski M Davey JH LeamonK Johnson MJ Milgrew M Edwards et al An integrated semiconductor deviceenabling non-optical genome sequencing Nature 475 (2011) 348ndash352
[31] D Pushkarev NF Neff SR Quake Single-molecule sequencing of an individualhuman genome Nat Biotechnol 27 (2009) 847ndash850
[32] M Rasmussen YR Li S Lindgreen JS Pedersen A Albrechtsen I Moltke MMetspalu E Metspalu T Kivisild R Gupta et al Ancient human genomesequence of an extinct Palaeo-Eskimo Nature 463 (2010) 757ndash762
[33] SM Ahn TH Kim S Lee D Kim H Ghang DS Kim BC Kim SY Kim WY KimC Kim et al The first Korean genome sequence and analysis full genomesequencing for a socio-ethnic group Genome Res 19 (2009) 1622ndash1629
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
[34] DA Wheeler M Srinivasan M Egholm Y Shen L Chen A McGuire W He YJChen V Makhijani GT Roth et al The complete genome of an individual bymassively parallel DNA sequencing Nature 452 (2008) U5ndashU872
[35] J Wang W Wang RQ Li YR Li G Tian L Goodman W Fan JQ Zhang J Li JBZhang et al The diploid genome sequence of an Asian individual Nature 456(2008) U1ndashU60
[36] BA Peters BG Kermani AB Sparks O Alferov P Hong A Alexeev Y Jiang FDahl YT Tang J Haas et al Accurate whole-genome sequencing and haplotyp-ing from 10 to 20 human cells Nature 487 (2012) 190ndash195
[37] H Skaletsky T Kuroda-Kawaguchi PJ Minx HS Cordum L Hillier LG Brown SRepping T Pyntikova J Ali T Bieri et al The male-specific region of the human Ychromosome is a mosaic of discrete sequence classes Nature 423 (2003) U2ndashU825
[38] SM Adams TE King E Bosch MA Jobling The case of the unreliable SNPrecurrent back-mutation of Y-chromosomal markers P25 through gene conver-sion Forensic Sci Int 159 (2006) 14ndash20
[39] B Trombetta F Cruciani PA Underhill D Sellitto R Scozzari Footprints of X-to-Y gene conversion in recent human evolution Mol Biol Evol 27 (2010) 714ndash725
[40] S Rootsi NM Myres AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova K Varendi H Sahakyan et al Distinguishing the co-ancestries ofhaplogroup G Y-chromosomes in the populations of Europe and the CaucasusEur J Hum Genet 20 (2012) 1275ndash1282
[41] S Caratti S Gino C Torre C Robino Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application Int J Legal Med 123(2009) 357ndash360
[42] C Bouakaze C Keyser S Amory E Crubezy B Ludes First successful assay of Y-SNP typing by SNaPshot minisequencing on ancient DNA Int J Legal Med 121(2007) 493ndash499
[43] YL Xue QJ Wang Q Long BL Ng H Swerdlow J Burton C Skuce R Taylor ZAbdellah YL Zhao et al Human Y chromosome base-substitution mutation ratemeasured by direct sequencing in a deep-rooting pedigree Curr Biol 19 (2009)1453ndash1457
[44] Y Kuroki A Toyoda H Noguchi TD Taylor T Itoh DS Kim DW Kim SH ChoiIC Kim HH Choi Comparative analysis of chimpanzee and human Y chromo-somes unveils complex evolutionary pathway Nat Genet 38 (2006) 158ndash167
[45] TE King MA Jobling Whatrsquos in a name Y chromosomes surnames and thegenetic genealogy revolution Trends Genet 25 (2009) 351ndash360
[46] MHD Larmuseau A Van Geystelen M van Oven R Decorte Genetic genealogycomes of age ndash perspectives on the use of deep-rooted pedigrees in humanpopulation genetics Am J Phys Anthropol 150 (2013) 505ndash511
[47] M van Oven M Kayser Updated comprehensive phylogenetic tree of globalhuman mitochondrial DNA variation Hum Mutat 30 (2008) E386ndashE394
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
- Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs
-
- Introduction
- Materials and methods
-
- Updated phylogenetic tree
- WGS Y-SNPs dataset
- AMY-tree modifications
- Y-SNP detecting
-
- Results
-
- Updated tree for forensic applications
- Sub-haplogroup determining
- Detecting new Y-SNPs
-
- Discussion
- Conclusions
- Authorsrsquo contributions
- Conflict of interest
- Acknowledgements
- Supplementary data
-
- References
-

TP FP TN FN
Measures
Expected stateancestralmutant
State in reference genome
Select Y-SNPs based on determined haplogroup
Expected situaon in samplecall ednot call ed
Actual situaon in samplecall ednot call ed
Fig 1 Workflow of the quality assessment algorithm of the AMY-tree version 11
First certain Y-SNPs of the Y-chromosomal phylogenetic tree are selected based on
the determined haplogroup of the first run in order to avoid too much false positive
Y-SNPs Next the expected state (ancestral or mutant) of the selected SNPs is
determined based on the haplogroup and the phylogenetic tree These expectations
of state are converted to expectations of called or not called based on the SNP state in
the reference genome These expectations are then compared to the actually called
SNPs of the sample such that the number of true positives (TP) false positives (FP)
true negatives (TN) and false negatives (FN) will be determined Finally several
different measures will be calculated
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx2
G Model
FSIGEN-977 No of Pages 8
Y-chromosomal tree The analysis of whole Y-chromosomes withinmale genomes allows verification and optimisation of thecurrently used phylogenetic tree WGS data has already provedto be useful in verifying and optimising the phylogeny of the otherhaploid markers in the human genome ie the mitochondrial DNA(mtDNA) Relevant ambiguous markers and back-mutations whichinfluence the interpretation of previous forensic and evolutionarygenetic studies were detected based on these data [21] Recently anew Y-chromosomal phylogenetic tree was built after a tabula rasa
of the present Y-chromosomal tree by using only Y-SNPs fromavailable WGS male samples [22] By comparing this newphylogenetic tree with the currently used one the backbone ofthe currently used phylogenetic tree was confirmed in this studyHowever this new tree is not useful for forensic research becausethere is no link between currently used and newly reportedlineages Furthermore the set of used genomes is not a goodrepresentation of all existing Y-chromosomal (sub-)haplogroupsand geographical regions There are also still too much falsepositive SNP calls in this WGS dataset Alternatively the AMY-treesoftware is the first open access program which academics andforensic professionals can use to verify and optimise the currentlyused Y-chromosomal tree by using WGS data [23] The first AMY-tree analysis was done based on 118 WGS samples and provedalready its usefulness to verify and to optimise the present Y-chromosomal phylogenetic tree [23]
The aim of this study is to perform the largest reportedscreening of male genomes for Y-chromosomal phylogeneticapplications based on the AMY-tree software Firstly we wantto merge all newly Y-SNPs from recent peer-reviewed publicationssince the latest lsquoofficialrsquo Y-chromosomal phylogeny [4] into onesingle tree which is useful for forensic applications Secondly thisupdated Y-chromosomal phylogenetic tree needs to be checked forrecurrent mutations ambiguous SNPs and other difficulties for the(forensic) application of the tree Thirdly this study also wants tofind out for which Y-chromosomal (sub-)haplogroups there isalready WGS data available Finally investigating the possibilitiesto enlarge the Y-chromosomal phylogenetic tree based on thecurrent Y-SNP detections in WGS data is the last aim of this study
2 Materials and methods
21 Updated phylogenetic tree
The latest updated phylogenetic tree of the Y-chromosome as itwas published by Van Geystelen et al [23] was manually updatedbased on recent descriptions of new Y-SNPs in academic researchpapers like Pamjav et al [16] and Scozzari et al [9] As the exactphylogenetic position of a few new Y-SNPs was not given theirposition needed to be determined based on the results of AMY-treeof all WGS samples Next also recurrent mutations ambiguousSNP-loci and wrongly defined mutation conversions within thenewly updated Y-chromosomal tree were ascertained based onthose AMY-tree results
22 WGS Y-SNPs dataset
In order to check the manually updated phylogenetic tree andto optimise the AMY-tree software a large dataset of wholegenome Y-SNP calls was assembled This dataset consists of 747samples which represent 660 males as several genomes wereanalysed in different projects Within this dataset the genomes ofeight males whose fatherrsquos genome was also sequenced arepresent The SNP calls were collected from four large WGS projectsand several individual genome projects (Supplementary MaterialsTable S1) These projects differ from each other based on the usednext-generation sequencing (NGS) platforms and sequence cover-
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
age First Complete Genomics made the SNP calls of 35 wholegenomes of males available (httpwwwcompletegenomicscompublic-data69-Genomes04 Jan 2013) those genomes weresequenced with a high sequencing coverage on the CompleteGenomics Analysis (CGA) Platform [24] Second the PersonalGenome Project (PGP) and Singapore Sequencing Malay Project(SSMP) also used this CGA platform PGP is a project started toobtain and openly share human genome sequences in combinationwith health information At the moment 40 male genomes wereavailable (wwwpersonalgenomesorg 04 Jan 2013) The SSMP onthe other hand wanted to characterise the polymorphic variants inthe population of Malays an Austronesian group present inSoutheast Asia and Oceania Recently the Y-SNP calls of 46 Malayswere made publically available [25] Next the 1000 GenomesProject aims to provide a comprehensive resource on humangenetic variation by sequencing more than 1000 human genomesIn 2010 SNP calls of 77 males were made available in the pilotphase [20] and two years later a set of 526 SNPs profiles werepublished as result of phase 1 of the Project [19] As the 1000Genomes project aims to sequence a large number of people thesequencing coverage was lower than in the other projects Finally23 additional samples were collected from several single genomeprojects [26ndash35 36 and unpublished genomes of Guy Froyen]
23 AMY-tree modifications
Several modifications to the AMY-tree software version 10 [23]were made for the assessment of the SNP calling quality in WGSdata This was necessary as the quality of the SNP calling influencesthe AMY-tree analysis of a sample and therefore also theinterpretation of the result of the analysis [23] The extra qualityassessment is based on the results of the first AMY-tree run of acertain sample This assessment assumes that the used phyloge-netic tree is correct and that the assigned haplogroup after the firstAMY-tree run is the actual haplogroup of that sample
The algorithm for the extra quality assessment is simple andcomprehensible as shown in Fig 1 First all Y-SNPs of thephylogenetic tree are selected except if the determined haplogroupof the first run is a paragroup (eg R1b1b2) In the case of aparagroup all Y-SNPs which are in sub-nodes of the main group ofthis paragroup are excluded from the selection This is done inorder to remove the influence of too much false positive SNP calls
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
R-M198 M198 M417 M512 M514 M515 Page7
R-M56R-M157 1
R-M87
R-P98
R-PK5
R-M43 4
R-Page68
R-Z28 0
R-Z93
R-M458
M56
M157 1
M87 M20 4
P98
PK5
M43 4
M45 8
Page68
Z28 0
Z93
R-M33 4M33 4
Fig 2 Overview of the position of the newly added sub-haplogroups R-Z280 and R-
Z93 (given in bold) within R-M198
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 3
G Model
FSIGEN-977 No of Pages 8
because when the haplogroup is not at the correct phylogeneticlevel too much false positives andor false negatives mutant Y-SNPs would be detected Next the expected state of all selected Y-SNPs is determined whereby all Y-SNPs in the path from theassigned sub-haplogroup till root are expected to be mutant andall others are expected to be ancestral The state of each Y-SNP isalso determined in the reference genome Thereafter theseexpectations of Y-SNP state are converted into expectations oflsquocalledrsquo or lsquonot calledrsquo in the next step based on the expected stateand the state in the reference genome Consider a Y-SNP in thepath from sub-haplogroup till root if the status of that SNP in thereference sequence is mutant the Y-SNP is expected to be lsquonotcalledrsquo else the expectation is lsquocalledrsquo Consider a Y-SNP not in thepath from sub-haplogroup till root if the status of that Y-SNP inthe reference sequence is mutant the Y-SNP is expected to belsquocalledrsquo else the expectation is lsquonot calledrsquo Then these expecta-tions are compared to the state in the sample such that thenumber of true positive false positive true negative and falsenegative SNP calls will be determined At last the quality of asample is expressed in several measures of quality Matthewscorrelation coefficient (MCC) accuracy sensitivity specificityprecision recall and F1-score (Supplementary method) When theMCC is larger or equal to 095 the SNP calling quality is calledexcellent otherwise it is called low The quality will be given in theoutput file of AMY-tree When the SNP calling quality is lowcaution has to be taken about the result of the AMY-tree analysisdue to the high occurrence of false negative and false positiveSNPs When the quality is excellent the results of AMY-tree areconsidered to be valuable for the control of the currently usedphylogenetic tree of the Y-chromosome and for the increase of itsresolution As such a better Y-SNP call quality assessment isimplemented in AMY-tree version 11 compared to the earlierversion [23]
Next when a sample was run in AMY-tree in the lsquosufficientrsquomode such that the reference genome was taken into account butthe determined haplogroup belongs to R-M269 and the MCC islower than 095 a second AMY-tree run needs to be executed but inthe lsquoinsufficientrsquo mode This is important as a MCC lower than 095indicates that this result of the first run is too much influenced bythe reference genome Finally another small modification to AMY-tree is based on the fact that Z381 L2 as well as L20 are mutant inthe reference genome and although AMY-tree version 10 alreadyhad a quite complex system to filter out the influence of thereference genome even on samples belonging to haplogroup R itwas not yet efficient enough Therefore when both Z381 and L2 orboth Z381 and L20 are mutant in the first AMY-tree run eg theancestral SNP was not called the sample will be handled asinsufficient in a second AMY-tree run such that the Y-SNPs of thereference genome are not used anymore when determining thehaplogroup By including these modifications even more certaintyis build in AMY-tree version 11 in comparison with the previousversion [23] for samples belonging to a R-M269 sub-haplogroupthe reference genome is only taken into account when the SNPcalling quality is excellent after the first run in the lsquosufficientrsquomode
The cut-offs to assess the Y-SNP calling quality is optimisedbased on all 747 genomes by performing several test runs andmanual analyses of the genomes and by checking it with the resultsin Van Geystelen et al [23] and with the publications of thegenomes whereby a Y-chromosomal analysis was already per-formed
24 Y-SNP detecting
The Y-SNPs which are present in the WGS samples but whichare not yet included in the updated phylogenetic tree were
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
detected by AMY-tree Only those Y-SNPs from WGS samples withan excellent Y-SNP calling quality were used But that was not theonly constraint for the potentially relevant Y-SNPs they must alsobe positioned in the unique regions of the Y-chromosome as readmapping and variant detection difficulties are expected due to thehigh frequency of repeated sequences on the Y-chromosome So Y-SNPs in the pseudoautosomal heterochromatic X-transposed andampliconic segments [37] of the male-specific part of the genomeas reported by Wei et al [22] were excluded
3 Results
In total 747 samples are analysed by the updated version of theAMY-tree software with the updated phylogenetic Y-chromosomaltree There are 131 samples of 126 individuals with an excellent Y-SNP calling quality ie MCC 095 which are mostly obtainedfrom Complete Genomics and the Personal Genome Project Theremaining 616 samples have a low calling quality and are mostlyobtained from the 1000 Genomes Project pilot and phase 1 (TableS1)
31 Updated tree for forensic applications
The state-of-the-art Y-chromosomal tree is manually updatedbased on all published Y-SNPs from academic studies After theAMY-tree runs of all 747 samples all Y-SNPs of which no exactphylogenetic position was given in the publications could now beincluded in the updated phylogenetic tree for example there weretwo new Y-SNPs reported in [16] within sub-haplogroup R-M198without clear phylogenetic positions (Fig 2)
The results with an excellent SNP calling quality also showedseveral recurrent Y-SNPs in the phylogenetic tree which cause thedetermination of multiple haplogroups for some samples Afterruling out that the recurrent SNPs are sample- or project-specificthese SNPs are removed from the phylogenetic tree an overview ofthe three observed recurrent SNPs of which enough evidence wasavailable is given in Table S2 These modifications led to the finalupdated tree version 11 which includes 359 Y-chromosomallineages and 721 Y-SNP markers The final tree and itscorresponding mutation conversions for all the Y-SNPs in the treecan be found in Tables S3 and S4
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
00 01 02 03 04 05 06 07 08 09 10
0
80
160
Number of samples
Mahewrsquos corr elaon coefficient
Fig 3 Distribution of the Matthews correlation coefficient of the 730 samples for which an unambiguous haplogroup could be determined
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx4
G Model
FSIGEN-977 No of Pages 8
32 Sub-haplogroup determining
The determined haplogroups of all 747 samples obtained by theAMY-tree analysis based on the final updated tree version 11 isgiven in Table S1 Only for 14 samples no haplogroup can bedetermined and for three other samples multiple haplogroups aredetermined by AMY-tree The distribution of the MCC of the 730samples for which an unambiguous haplogroup is determined isgiven in Fig 3 Only a minority of the samples has an excellent SNPcalling quality with a MCC 095 a MCC lower than 095 meansthat less than 975 of the negative and positive predictions arecorrect Overall 17 different haplogroups and 106 sub-hap-logroups are present in the dataset When considering only thesamples of excellent quality 10 different haplogroups and 47 sub-haplogroups remain Fig 4 and Table S5 give an overview of those(sub-)haplogroups and their frequencies
The samples of paternally related samples present in thedataset are of particular interest because they are considered torepresent the same Y-chromosome All the samples of one familywith eight members sequenced by Complete Genomics aredetermined to belong to R-P312 The paternal grandfather ofthat family is also analysed in the 1000 Genomes project and therehe is determined as P [P-92R7] The first attempt of AMY-tree todetermine the sub-haplogroup of that sample of 1000 Genomes ledto the sub-haplogroup R-L2 which has a higher phylogenetic levelthan the haplogroups of the Complete Genomics samples
A B C D E F G H I J
0
55
110
165
All geno mes
Good quali ty genomes
Numbe r of availab le WGS ge nomes
Hap lo
Fig 4 Frequency of whole genome sequencing (WGS) genomes per haplogroup in the dat
MCC 095 (grey)
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
However as the MCC value of that sample is smaller than 095the sub-haplogroup determination is done again but without theinfluence of the reference genome This led to the final sub-haplogroup P-92R7 which has a less accurate phylogenetic levelthan that of Complete Genomics Thus the influence of thereference genome can sometimes cause a too high or too lowphylogenetic level when the new modifications which were madeto AMY-tree version 11 would not have been applied
33 Detecting new Y-SNPs
The large amount of available samples leads to a huge amountof newly reported Y-SNPs ie Y-SNPs that are not yet present in theupdated phylogenetic tree version 11 In total 108681 new Y-SNPsare reported in all 660 male genomes when an individual isanalysed in more than one project only the sample with the highestMCC value is used The majority of the SNPs appears in only a fewsamples 62 appears in only one sample and 16 is present in twosamples as shown in Fig 5A In the 126 male genomes with anexcellent Y-SNP calling quality 50430 new Y-SNPs are reportedThese SNPs also come with a high frequency of low occurrences inthe excellent genomes 57 is unique and 11 appears in twosamples When only the regions within the Y-chromosome whichare identified as unique are taken into account a much lowernumber of new Y-SNPs is detected In total 35503 new Y-SNPs arereported in the 660 male genomes and 15208 new Y-SNPs in the
K L M N O P Q R S T
g roups
aset of all samples (black) and in the dataset of samples with an excellent quality ie
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
1 2 3 4 5 6 7 8 9 10 more
Numbe r of occ urr ences of Y-SNP
0
12000
24000
0
35000
70000
Number of Y-SNPsWhole Y-chromosome
Uniqu e regions Y-chromosome
A
B
All genomes
Excell ent qu ali ty genomes
Fig 5 Number of new Y-SNPs per number of occurrence in the full WGS dataset (A) SNPs in the whole Y-chromosome and (B) SNPs in only the identified unique regions of the
Y-chromosome The grey bars indicate the SNPs in all genomes and the black bars indicate the SNPs from samples with an excellent quality ie MCC 095
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 5
G Model
FSIGEN-977 No of Pages 8
genomes with excellent Y-SNP calling quality The same patterns ofoccurrence for these Y-SNPs are observed as with the non-filteredY-SNPs the majority of the new reported Y-SNPs appeared in onlyone or two samples of the WGS dataset as shown in Fig 5B
The genomes of eight males whose biological fatherrsquos genome isalso sequenced are present in the dataset one family of eightmales including the father the son and the six grandsons next toone fatherndashson pair In the family of eight paternal relatives 5155new SNPs are reported on the whole Y-chromosome and a largenumber of these SNPs is found in only one of the eight individualsas shown in Fig 6A The number of Y-SNPs decreases every time
0
500
1000
1500
2000
2500
0
1 2 3 4 5
Numbe r of o
0
20
40
60
80
100
120
Number of Y-SNPsA
B
8-member fam
All SNPs
Famil y-un iqu e SNP
Fig 6 Number of new Y-SNPs per occurrence in the eight paternally related samples and t
(B) SNPs in only the identified unique regions of the Y-chromosome The grey bars indica
uniquersquo they do not occur in any of the other samples with an excellent quality ie M
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
the number of samples in which the Y-SNP occurs increases exceptfor the occurrence in all-except-one and all samples of the familyWhen all Y-SNPs which also occur in any other sample withexcellent SNP calling quality are removed only less than 20 of theY-SNPs remains but the distribution of the number of Y-SNPs peroccurrence stays the same as the black bars in Fig 6A show Thesame comparison is made when only the SNPs in the unique part ofthe Y-chromosome are selected the same pattern as with all SNPsis visible in Fig 6B However for each number of occurrences thenumber of truly unique SNPs ie SNPs that do not occur in othergenomes outside the family is much higher Within the fatherndashson
6 7 8 1 2
cc urr ences of Y-SNP
Whole Y-chromosome
Uniqu e regions Y-chromosome
il y Father-son pair
s
he fatherndashson pair of Complete Genomics (A) SNPs in the whole Y-chromosome and
te the SNPs found in the family The black bars indicate the SNPs which are lsquofamily-
CC 095
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx6
G Model
FSIGEN-977 No of Pages 8
pair 2181 new SNPs are reported on the whole Y-chromosome forwhich the difference between occurrence in only one and bothgenomes is relatively small also for the proportion of SNPs whichdo not occur in the other genomes with an excellent Y-SNP quality(Fig 6A) Remarkably there are more new SNPs present in bothsamples than in one sample when the Y-SNPs which are notlocated in the unique region of the Y-chromosome are removed(Fig 6B) The effect of the increasing number of unique Y-SNPs thatdo not occur in other genomes as seen with the previous family isalso in the fatherndashson pair present
4 Discussion
The present study realises the first large screening of malegenomes for phylogenetic applications of the Y-chromosomeBased on this screening of 747 male samples an update has beenmade of the AMY-tree software and of a state-of-the-art Y-chromosomal phylogenetic tree was established for forensicscientists Furthermore also recommendations for future sequenc-ing projects dealing with a broader selection panel of Y-chromosomal haplogroup samples and for the validation of newlydetected Y-SNPs are made
First the large screening of the 747 male Y-chromosomesamples revealed that the SNP calling quality of a few samples wasoverestimated in AMY-tree version 10 As these samples belong tosub-haplogroup R-M269 they are very similar to the referencegenome which is used to estimate the SNP calling quality of asample [23] To remove this SNP calling quality overestimation theinfluence of the reference genome is excluded for all samplesbelonging R-M269 with a low SNP call quality That is why severalmodifications are made and implemented in AMY-tree version 11
Second an update of the currently used Y-chromosomalphylogenetic tree was realised based on the large database of747 available samples At the moment this tree is the most state-of-the-art tree applicable for forensic geneticists all Y-SNPs whichare reported in academic publications till today are included andall ambiguous markers are excluded to avoid wrong Y-SNPinterpretations (Table S3) As often the case in the literature thephylogenetic relationship between new Y-SNPs and earlierreported Y-SNPs in the same (sub-)haplogroup are not given Byusing the AMY-tree results it is possible to find out the concretephylogenetic level of each Y-SNP relative to the other alreadyknown SNPs For example Pamjav et al [16] described andgenotyped two new Y-SNPs Z93 and Z280 within R-M198although the exact phylogenetic positions in relationship withthe other lineages within R-M198 were not given The presence ofboth Z93 and Z80 is checked in all samples belonging to the sub-haplogroup R-M198 Z93 occurred in three R-M198 samples andin none of the other samples Also Z280 does not occur in anysample except in one R-M198 sample Therefore both Z93 andZ280 are placed in the phylogenetic tree as sub-haplogroups of R-M198 as shown in Fig 2 The choice is made for a phylogenetic treein table format as described by Van Geystelen et al [23] instead of abranching diagram because the tree is very large and will becomelarger in the future Therefore the table format can be adaptedmore easily than the diagram and it is also more manageable
Not only newly reported Y-SNPs are included in the newupdated tree but also ambiguous Y-SNPs are excluded as they cancomplicate the Y-chromosomal applications for forensic studiesAs previously described [38] the most relevant ambiguous Y-SNPsare recurrent SNPs which have a paralogous distribution along thephylogenetic tree and which have thus mutant alleles in at leasttwo independent Y-chromosomal lineages Based on the screeningof the 747 analysed samples three Y-SNPs are recognised for thefirst time as recurrent (Table S2) There are no other indications forrecurrent mutations based on the present WGS dataset Therefore
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
we may assume in most cases that males which both have themutant allele for a Y-SNP used in the updated tree (Table S3) havereceived this mutant allele from one common ancestor and not byconvergent evolution Next also all Y-SNPs which could not beanalysed by WGS for one reason or another are excluded from theupdated tree although it may be possible to genotype these SNPscorrectly with Sanger sequencing methods For example allhundred E-M2 samples in the dataset did not reveal the mutantallele for Y-SNP V95 as expected by earlier publications [1439]The reason for this remarkable result may be that V95 is not wellvalidated but more likely it is the result of a bad SNP calling in theY-chromosomal region around V95 by the current WGS methodsFor some Y-SNPs also a mutation conversion is found which isdifferent from the reported one For example another ancestraland mutant allele are observed for Y-SNP M426 than reported byRootsi et al [40] Since the Y-chromosome has a very complexorigin it also has a lot of non-unique regions which complicate theanalysis of WGS data [37] The current reference genome GRCh37shows the evolution of the Y-chromosome and its numerousresulting non-unique regions So the reason for this wrong analysisof M426 may be the position of this SNP in one of the non-uniqueregions of the Y-chromosome as defined earlier [2237] To have anunambiguous phylogenetic tree we excluded all the Y-SNPs forwhich no reliable signal with the WGS methods could be found Inthe end an updated Y-chromosomal tree which includes 359 Y-chromosomal lineages and more than 721 Y-SNP markers (TableS3) is obtained and this tree is the basic tool to develop andoptimise Y-SNP-multiplexes for forensic applications [4142]
Third the distribution of the analysed haplogroups perancestral continent in the current dataset corresponds with theknown distributions [34] Nevertheless there is not yet arepresentative set of all phylogenetic sub-haplogroups availableIn total 17 haplogroups and 106 sub-haplogroups are reported inthe analysis of the whole dataset However the SNP calling qualityis low (MCC lt 095) for most of the analysed samples as most dataare obtained from the 1000 Genomes project and therefore thesub-haplogroup determination of these data is not completelyreliable When considering only the samples of excellent quality(MCC 095) 10 haplogroups and 47 sub-haplogroups remain andthis corresponds with only 13 of the total number lineagesdescribed so far Most of the Y-chromosomes are assigned tohaplogroups E O and R Therefore when the set of WGS Y-chromosomes will be enlarged the current phylogenetic tree aswell as the analysis of Wei et al [22] to calibrate the tree can beoptimised
Finally the screening of the dataset also revealed that new insilico and in vitro methods are required to verify new Y-SNPs basedon WGS methods As earlier mentioned [23] a huge number of newY-SNPs are false positives when the genomes were sequenced withlow coverage and consequently the called SNPs will have a lowquality These false positive SNP calls disturb the determination ofthe correct (sub-)haplogroup of the sample consequently theAMY-tree software has to correct for them by applying severaladditional methods in the analysis [23] The high number of falsepositive Y-SNPs ndash even detected in WGS samples with an excellentSNP calling quality ndash is observed by comparing genomes ofpaternal relatives Within the eight paternally related samplesbelonging to one family which are genotyped by CompleteGenomics 949 newly reported Y-SNPs in the full Y-chromosomeand 88 ones in the unique regions of the Y-chromosome are foundin at least one but not all family members (Fig 6) Despite the highcoverage of these genomes and the excellent SNP calling qualitythis is still a very high number of newly reported Y-SNPs which aremost likely to be false based on the mutation rate on the Y-chromosomes calculated based on a deep-rooting pedigree [43]and based on human-chimpanzee comparisons [44] A similar
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 7
G Model
FSIGEN-977 No of Pages 8
conclusion can be made based on the fatherndashson pair in the datasetwhich is also sequenced with a high coverage by CompleteGenomics (Fig 6)
The Y-SNP results show that adding new lineages to the Y-chromosomal phylogenetic tree only based on WGS is not evidentTherefore making tabula rasa and building a new tree based on allWGS Y-chromosomes as done by Wei et al [22] is not an optionwhen the tree is going to be used in forensics Each new Y-SNP andconsequently each Y-chromosomal lineage has to be validatedindependently from the WGS data before adding them to theupdated phylogenetic tree The validation status of many potentialpolymorphic Y-SNPs (gt1 in population) is often unclear but thiscan be resolved by the sharing of genomic data among geneticgenealogists which are interested in finding new Y-SNPs to resolvetheir particular paternal ancestry [45] Therefore this is an area inwhich closer collaborations between amateurs and forensicacademics could prove to be particularly useful [13] Neverthelessit is required that new in silico methods will be designed to selectgood and relevant candidates of Y-SNPs for the validation Forexample an interesting criterion is the position of the Y-SNPs it ismore interesting to validate only the SNPs located in the uniqueregions of the Y-chromosome as there are many non-uniqueregions due to the evolutionary history of this chromosome [37]The lower number of false positive SNP calls in these uniqueregions in comparison with those in the full Y-chromosome isclearly demonstrated in the fatherndashson pair The number of new Y-SNPs reported in only one of the two samples is higher than thenew ones reported in both samples based on the whole Y-chromosome (Fig 6A) in contrast with the situation based on theunique regions of the Y-chromosome (Fig 6B)
5 Conclusions
Based on the largest screening of male genomes with 747samples in total the most up-to-date Y-chromosomal phylogenet-ic tree for forensic applications is compiled Future publicationswhich will report new Y-SNPs have to situate their phylogeneticpositions in this tree to guarantee the continuity between old andnew publications At this moment forensic scientists as well asevolutionary biologists and genetic genealogists are lost in themany reports of newly described Y-chromosomal lineages [46]Therefore initiatives as AMY-tree which optimise the phylogenybased on peer-reviewed publications are required [23] This isalready the case for the mitochondrial genome with the Phylotreeinitiative of van Oven and Kayser [47] Nevertheless to optimisethe current updated phylogenetic tree for the human Y-chromo-some more high quality genomes of a broader set of (sub-)haplogroups than the frequent haplogroups E O and R arerequired Also a higher effort in the validation of reported Y-SNPsby new in silico and in vitro methods is required
Authorsrsquo contributions
Research design amp supervision MHDL programming AVGwriting MHDL amp AVG Commenting on manuscript AVG RD ampMHDL
Conflict of interest
The authors declare no conflict of interest
Acknowledgements
The authors want to thank Tom Wenseleers Manfred KayserJean-Jacques Cassiman Tom Havenith Hendrik Larmuseau and
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
Lucrece Lernout for useful discussions and comments Thanks alsoto Guy Froyen (VIB KU Leuven) Richard Rocca (independentresearcher) Cuiping Pan (Stanford University) and Andreas Keller(Saarland University) for providing us yet unpublished andpublished called SNPs of whole genome sequencing projectsMaarten HD Larmuseau is postdoctoral fellow of the FWO-Vlaanderen (Research Foundation Flanders) This study wasfunded by the KU Leuven BOF Centre of Excellence Financing onlsquoEco and socio evolutionary dynamicsrsquo (Project number PF201007) and on lsquoCentre for Archaeological Sciences 2 (CAS 2) ndash Newmethods for research in demography and interregional exchangersquo
Appendix A Supplementary data
Supplementary data associated with this article can be found in
the online version at httpdxdoiorg101016jfsigen201303010
References
[1] M Kayser Y-chromosomal markers in forensic genetics in RWD Rapley (Ed)Molecular Forensics John Wiley amp Sons Ltd Chichesters 2007 pp 141ndash161
[2] JM Butler Chapter 13 Y-Chromosomal DNA Testing in JM Butler (Ed) Ad-vanced Topics in Forensic DNA Typing Methodology Academic Press London2011 pp 371ndash403
[3] J Chiaroni PA Underhill LL Cavalli-Sforza Y chromosome diversity humanexpansion drift and cultural evolution Proc Natl Acad Sci USA 106 (2009)20174ndash20179
[4] TM Karafet FL Mendez MB Meilerman PA Underhill SL Zegura MFHammer New binary polymorphisms reshape and increase resolution of thehuman Y chromosomal haplogroup tree Genome Res 18 (2008) 830ndash838
[5] MHD Larmuseau N Vanderheyden M Jacobs M Coomans L Larno R DecorteMicro-geographic distribution of Y-chromosomal variation in the central-westernEuropean region Brabant Forensic Sci Int Genet 5 (2011) 95ndash99
[6] F Cruciani B Trombetta C Antonelli R Pascone G Valesini V Scalzi G Vona BMelegh B Zagradisnik G Assum et al Strong intra- and inter-continentaldifferentiation revealed by Y chromosome SNPs M269 U106 and U152 ForensicSci Int Genet 5 (2011) E49ndashE52
[7] MHD Larmuseau J Vanoverbeke G Gielis N Vanderheyden HFM LarmuseauR Decorte In the name of the migrant father ndash analysis of surname originidentifies historic admixture events undetectable from genealogical recordsHeredity 109 (2012) 90ndash95
[8] S Willuweit L Roewer Y chromosome haplotype reference database (YHRD)update Forensic Sci Int Genet 1 (2007) 83ndash87
[9] R Scozzari A Massaia E DrsquoAtanasio NM Myres UA Perego B Trombetta FCruciani Molecular dissection of the basal clades in the human Y chromosomephylogenetic tree Plos ONE 7 (2012) e49170
[10] F Cruciani B Trombetta A Massaia G Destro-Bisol D Sellitto R Scozzari Arevised root for the human Y chromosomal phylogenetic tree the origin ofpatrilineal diversity in Africa Am J Hum Genet 88 (2011) 814ndash818
[11] S Fornarino M Pala V Battaglia R Maranta A Achilli G Modiano A Torroni OSemino SA Santachiara-Benerecetti Mitochondrial and Y-chromosome diversi-ty of the Tharus (Nepal) a reservoir of genetic variation BMC Evol Biol 9 (2009)154
[12] FL Mendez TM Karafet T Krahn H Ostrer H Soodyall MF Hammer Increasedresolution of Y chromosome haplogroup T defines relationships among popula-tions of the Near East Europe and Africa Hum Biol 83 (2011) 39ndash53
[13] LM Sims D Garvey J Ballantyne Improved resolution haplogroup G phylogenyin the Y-chromosome revealed by a set of newly characterized SNPs Plos ONE 4(2009) e5792
[14] B Trombetta F Cruciani D Sellitto R Scozzari A new topology of the human Ychromosome haplogroup E1b1 (E-P2) revealed through the use of newly charac-terized binary polymorphisms PLoS ONE 6 (2011) e16073
[15] MS Jota DR Lacerda JR Sandoval PPR Vieira SS Santos-Lopes R Bisso-Machado VR Paixao-Cortes S Revollo C Paz-Y-Mino R Fujita et al A newsubhaplogroup of native American Y-chromosomes from the Andes Am J PhysAnthropol 146 (2011) 553ndash559
[16] H Pamjav T Feher E Nemeth Z Padar Brief communication new Y-chromo-some binary markers improve phylogenetic resolution within haplogroup R1a1Am J Phys Anthropol 149 (2012) 611ndash615
[17] NM Myres S Rootsi AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova A Pshenichnov B Yunusbayev et al A major Y-chromosomehaplogroup R1b Holocene era founder effect in Central and Western Europe Eur JHum Genet 19 (2011) 95ndash101
[18] S Yan CC Wang H Li SL Li L Jin G Consortium An updated tree of Y-chromosome Haplogroup O and revised phylogenetic positions of mutations P164and PK4 Eur J Hum Genet 19 (2011) 1013ndash1015
[19] T 1000 Genomes Project Consortium An integrated map of genetic variation from1092 human genomes Nature 491 (2012) 56ndash65
[20] DL Altshuler RM Durbin GR Abecasis DR Bentley A Chakravarti AG ClarkFS Collins FM De la Vega P Donnelly M Egholm et al A map of human
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx8
G Model
FSIGEN-977 No of Pages 8
genome variation from population-scale sequencing Nature 467 (2010)1061ndash1073
[21] AT Duggan M Stoneking A highly unstable recent mutation in human mtDNAAm J Hum Genet 9 (2013) 279ndash284
[22] W Wei Q Ayub Y Chen S McCarthy Y Hou I Carbone Y Xue C Tyler-Smith Acalibrated human Y-chromosomal phylogeny based on resequencing GenomeRes 23 (2013) 388ndash395
[23] A Van Geystelen R Decorte MHD Larmuseau AMY-tree an algorithm to usewhole genome SNP calling for Y chromosomal phylogenetic applications BMCGenomics 14 (2013) 101
[24] R Drmanac AB Sparks MJ Callow AL Halpern NL Burns BG Kermani PCarnevali I Nazarenko GB Nilsen G Yeung et al Human genome sequencingusing unchained base reads on self-assembling DNA nanoarrays Science 327(2010) 78ndash81
[25] L-P Wong RT-H Ong W-T Poh X Liu P Chen RQ Li KK-Y Lam NE PillaiK-S Sim H Xu et al Deep whole-genome sequencing of 100 Southeast AsianMalays Am J Hum Genet 92 (2013) 1ndash15
[26] SC Schuster W Miller A Ratan LP Tomsho B Giardine LR Kasson RS HarrisDC Petersen FQ Zhao J Qi et al Complete Khoisan and Bantu genomes fromsouthern Africa Nature 463 (2010) 943ndash947
[27] P Tong JGD Prendergast AJ Lohan SM Farrington S Cronin N Friel DGBradley O Hardiman A Evans JF Wilson et al Sequencing and analysis of anIrish human genome Genome Biol 11 (2010) R91
[28] A Keller A Graefen M Ball M Matzas V Boisguerin F Maixner P Leidinger CBackes R Khairat M Forster et al New insights into the Tyrolean Icemanrsquos originand phenotype as inferred by whole-genome sequencing Nature Commun 3(2012) 698
[29] R Chen GI Mias J Li-Pook-Than LH Jiang HYK Lam R Chen E Miriami KJKarczewski M Hariharan FE Dewey et al Personal omics profiling revealsdynamic molecular and medical phenotypes Cell 148 (2012) 1293ndash1307
[30] JM Rothberg W Hinz TM Rearick J Schultz W Mileski M Davey JH LeamonK Johnson MJ Milgrew M Edwards et al An integrated semiconductor deviceenabling non-optical genome sequencing Nature 475 (2011) 348ndash352
[31] D Pushkarev NF Neff SR Quake Single-molecule sequencing of an individualhuman genome Nat Biotechnol 27 (2009) 847ndash850
[32] M Rasmussen YR Li S Lindgreen JS Pedersen A Albrechtsen I Moltke MMetspalu E Metspalu T Kivisild R Gupta et al Ancient human genomesequence of an extinct Palaeo-Eskimo Nature 463 (2010) 757ndash762
[33] SM Ahn TH Kim S Lee D Kim H Ghang DS Kim BC Kim SY Kim WY KimC Kim et al The first Korean genome sequence and analysis full genomesequencing for a socio-ethnic group Genome Res 19 (2009) 1622ndash1629
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
[34] DA Wheeler M Srinivasan M Egholm Y Shen L Chen A McGuire W He YJChen V Makhijani GT Roth et al The complete genome of an individual bymassively parallel DNA sequencing Nature 452 (2008) U5ndashU872
[35] J Wang W Wang RQ Li YR Li G Tian L Goodman W Fan JQ Zhang J Li JBZhang et al The diploid genome sequence of an Asian individual Nature 456(2008) U1ndashU60
[36] BA Peters BG Kermani AB Sparks O Alferov P Hong A Alexeev Y Jiang FDahl YT Tang J Haas et al Accurate whole-genome sequencing and haplotyp-ing from 10 to 20 human cells Nature 487 (2012) 190ndash195
[37] H Skaletsky T Kuroda-Kawaguchi PJ Minx HS Cordum L Hillier LG Brown SRepping T Pyntikova J Ali T Bieri et al The male-specific region of the human Ychromosome is a mosaic of discrete sequence classes Nature 423 (2003) U2ndashU825
[38] SM Adams TE King E Bosch MA Jobling The case of the unreliable SNPrecurrent back-mutation of Y-chromosomal markers P25 through gene conver-sion Forensic Sci Int 159 (2006) 14ndash20
[39] B Trombetta F Cruciani PA Underhill D Sellitto R Scozzari Footprints of X-to-Y gene conversion in recent human evolution Mol Biol Evol 27 (2010) 714ndash725
[40] S Rootsi NM Myres AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova K Varendi H Sahakyan et al Distinguishing the co-ancestries ofhaplogroup G Y-chromosomes in the populations of Europe and the CaucasusEur J Hum Genet 20 (2012) 1275ndash1282
[41] S Caratti S Gino C Torre C Robino Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application Int J Legal Med 123(2009) 357ndash360
[42] C Bouakaze C Keyser S Amory E Crubezy B Ludes First successful assay of Y-SNP typing by SNaPshot minisequencing on ancient DNA Int J Legal Med 121(2007) 493ndash499
[43] YL Xue QJ Wang Q Long BL Ng H Swerdlow J Burton C Skuce R Taylor ZAbdellah YL Zhao et al Human Y chromosome base-substitution mutation ratemeasured by direct sequencing in a deep-rooting pedigree Curr Biol 19 (2009)1453ndash1457
[44] Y Kuroki A Toyoda H Noguchi TD Taylor T Itoh DS Kim DW Kim SH ChoiIC Kim HH Choi Comparative analysis of chimpanzee and human Y chromo-somes unveils complex evolutionary pathway Nat Genet 38 (2006) 158ndash167
[45] TE King MA Jobling Whatrsquos in a name Y chromosomes surnames and thegenetic genealogy revolution Trends Genet 25 (2009) 351ndash360
[46] MHD Larmuseau A Van Geystelen M van Oven R Decorte Genetic genealogycomes of age ndash perspectives on the use of deep-rooted pedigrees in humanpopulation genetics Am J Phys Anthropol 150 (2013) 505ndash511
[47] M van Oven M Kayser Updated comprehensive phylogenetic tree of globalhuman mitochondrial DNA variation Hum Mutat 30 (2008) E386ndashE394
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
- Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs
-
- Introduction
- Materials and methods
-
- Updated phylogenetic tree
- WGS Y-SNPs dataset
- AMY-tree modifications
- Y-SNP detecting
-
- Results
-
- Updated tree for forensic applications
- Sub-haplogroup determining
- Detecting new Y-SNPs
-
- Discussion
- Conclusions
- Authorsrsquo contributions
- Conflict of interest
- Acknowledgements
- Supplementary data
-
- References
-

R-M198 M198 M417 M512 M514 M515 Page7
R-M56R-M157 1
R-M87
R-P98
R-PK5
R-M43 4
R-Page68
R-Z28 0
R-Z93
R-M458
M56
M157 1
M87 M20 4
P98
PK5
M43 4
M45 8
Page68
Z28 0
Z93
R-M33 4M33 4
Fig 2 Overview of the position of the newly added sub-haplogroups R-Z280 and R-
Z93 (given in bold) within R-M198
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 3
G Model
FSIGEN-977 No of Pages 8
because when the haplogroup is not at the correct phylogeneticlevel too much false positives andor false negatives mutant Y-SNPs would be detected Next the expected state of all selected Y-SNPs is determined whereby all Y-SNPs in the path from theassigned sub-haplogroup till root are expected to be mutant andall others are expected to be ancestral The state of each Y-SNP isalso determined in the reference genome Thereafter theseexpectations of Y-SNP state are converted into expectations oflsquocalledrsquo or lsquonot calledrsquo in the next step based on the expected stateand the state in the reference genome Consider a Y-SNP in thepath from sub-haplogroup till root if the status of that SNP in thereference sequence is mutant the Y-SNP is expected to be lsquonotcalledrsquo else the expectation is lsquocalledrsquo Consider a Y-SNP not in thepath from sub-haplogroup till root if the status of that Y-SNP inthe reference sequence is mutant the Y-SNP is expected to belsquocalledrsquo else the expectation is lsquonot calledrsquo Then these expecta-tions are compared to the state in the sample such that thenumber of true positive false positive true negative and falsenegative SNP calls will be determined At last the quality of asample is expressed in several measures of quality Matthewscorrelation coefficient (MCC) accuracy sensitivity specificityprecision recall and F1-score (Supplementary method) When theMCC is larger or equal to 095 the SNP calling quality is calledexcellent otherwise it is called low The quality will be given in theoutput file of AMY-tree When the SNP calling quality is lowcaution has to be taken about the result of the AMY-tree analysisdue to the high occurrence of false negative and false positiveSNPs When the quality is excellent the results of AMY-tree areconsidered to be valuable for the control of the currently usedphylogenetic tree of the Y-chromosome and for the increase of itsresolution As such a better Y-SNP call quality assessment isimplemented in AMY-tree version 11 compared to the earlierversion [23]
Next when a sample was run in AMY-tree in the lsquosufficientrsquomode such that the reference genome was taken into account butthe determined haplogroup belongs to R-M269 and the MCC islower than 095 a second AMY-tree run needs to be executed but inthe lsquoinsufficientrsquo mode This is important as a MCC lower than 095indicates that this result of the first run is too much influenced bythe reference genome Finally another small modification to AMY-tree is based on the fact that Z381 L2 as well as L20 are mutant inthe reference genome and although AMY-tree version 10 alreadyhad a quite complex system to filter out the influence of thereference genome even on samples belonging to haplogroup R itwas not yet efficient enough Therefore when both Z381 and L2 orboth Z381 and L20 are mutant in the first AMY-tree run eg theancestral SNP was not called the sample will be handled asinsufficient in a second AMY-tree run such that the Y-SNPs of thereference genome are not used anymore when determining thehaplogroup By including these modifications even more certaintyis build in AMY-tree version 11 in comparison with the previousversion [23] for samples belonging to a R-M269 sub-haplogroupthe reference genome is only taken into account when the SNPcalling quality is excellent after the first run in the lsquosufficientrsquomode
The cut-offs to assess the Y-SNP calling quality is optimisedbased on all 747 genomes by performing several test runs andmanual analyses of the genomes and by checking it with the resultsin Van Geystelen et al [23] and with the publications of thegenomes whereby a Y-chromosomal analysis was already per-formed
24 Y-SNP detecting
The Y-SNPs which are present in the WGS samples but whichare not yet included in the updated phylogenetic tree were
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
detected by AMY-tree Only those Y-SNPs from WGS samples withan excellent Y-SNP calling quality were used But that was not theonly constraint for the potentially relevant Y-SNPs they must alsobe positioned in the unique regions of the Y-chromosome as readmapping and variant detection difficulties are expected due to thehigh frequency of repeated sequences on the Y-chromosome So Y-SNPs in the pseudoautosomal heterochromatic X-transposed andampliconic segments [37] of the male-specific part of the genomeas reported by Wei et al [22] were excluded
3 Results
In total 747 samples are analysed by the updated version of theAMY-tree software with the updated phylogenetic Y-chromosomaltree There are 131 samples of 126 individuals with an excellent Y-SNP calling quality ie MCC 095 which are mostly obtainedfrom Complete Genomics and the Personal Genome Project Theremaining 616 samples have a low calling quality and are mostlyobtained from the 1000 Genomes Project pilot and phase 1 (TableS1)
31 Updated tree for forensic applications
The state-of-the-art Y-chromosomal tree is manually updatedbased on all published Y-SNPs from academic studies After theAMY-tree runs of all 747 samples all Y-SNPs of which no exactphylogenetic position was given in the publications could now beincluded in the updated phylogenetic tree for example there weretwo new Y-SNPs reported in [16] within sub-haplogroup R-M198without clear phylogenetic positions (Fig 2)
The results with an excellent SNP calling quality also showedseveral recurrent Y-SNPs in the phylogenetic tree which cause thedetermination of multiple haplogroups for some samples Afterruling out that the recurrent SNPs are sample- or project-specificthese SNPs are removed from the phylogenetic tree an overview ofthe three observed recurrent SNPs of which enough evidence wasavailable is given in Table S2 These modifications led to the finalupdated tree version 11 which includes 359 Y-chromosomallineages and 721 Y-SNP markers The final tree and itscorresponding mutation conversions for all the Y-SNPs in the treecan be found in Tables S3 and S4
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
00 01 02 03 04 05 06 07 08 09 10
0
80
160
Number of samples
Mahewrsquos corr elaon coefficient
Fig 3 Distribution of the Matthews correlation coefficient of the 730 samples for which an unambiguous haplogroup could be determined
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx4
G Model
FSIGEN-977 No of Pages 8
32 Sub-haplogroup determining
The determined haplogroups of all 747 samples obtained by theAMY-tree analysis based on the final updated tree version 11 isgiven in Table S1 Only for 14 samples no haplogroup can bedetermined and for three other samples multiple haplogroups aredetermined by AMY-tree The distribution of the MCC of the 730samples for which an unambiguous haplogroup is determined isgiven in Fig 3 Only a minority of the samples has an excellent SNPcalling quality with a MCC 095 a MCC lower than 095 meansthat less than 975 of the negative and positive predictions arecorrect Overall 17 different haplogroups and 106 sub-hap-logroups are present in the dataset When considering only thesamples of excellent quality 10 different haplogroups and 47 sub-haplogroups remain Fig 4 and Table S5 give an overview of those(sub-)haplogroups and their frequencies
The samples of paternally related samples present in thedataset are of particular interest because they are considered torepresent the same Y-chromosome All the samples of one familywith eight members sequenced by Complete Genomics aredetermined to belong to R-P312 The paternal grandfather ofthat family is also analysed in the 1000 Genomes project and therehe is determined as P [P-92R7] The first attempt of AMY-tree todetermine the sub-haplogroup of that sample of 1000 Genomes ledto the sub-haplogroup R-L2 which has a higher phylogenetic levelthan the haplogroups of the Complete Genomics samples
A B C D E F G H I J
0
55
110
165
All geno mes
Good quali ty genomes
Numbe r of availab le WGS ge nomes
Hap lo
Fig 4 Frequency of whole genome sequencing (WGS) genomes per haplogroup in the dat
MCC 095 (grey)
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
However as the MCC value of that sample is smaller than 095the sub-haplogroup determination is done again but without theinfluence of the reference genome This led to the final sub-haplogroup P-92R7 which has a less accurate phylogenetic levelthan that of Complete Genomics Thus the influence of thereference genome can sometimes cause a too high or too lowphylogenetic level when the new modifications which were madeto AMY-tree version 11 would not have been applied
33 Detecting new Y-SNPs
The large amount of available samples leads to a huge amountof newly reported Y-SNPs ie Y-SNPs that are not yet present in theupdated phylogenetic tree version 11 In total 108681 new Y-SNPsare reported in all 660 male genomes when an individual isanalysed in more than one project only the sample with the highestMCC value is used The majority of the SNPs appears in only a fewsamples 62 appears in only one sample and 16 is present in twosamples as shown in Fig 5A In the 126 male genomes with anexcellent Y-SNP calling quality 50430 new Y-SNPs are reportedThese SNPs also come with a high frequency of low occurrences inthe excellent genomes 57 is unique and 11 appears in twosamples When only the regions within the Y-chromosome whichare identified as unique are taken into account a much lowernumber of new Y-SNPs is detected In total 35503 new Y-SNPs arereported in the 660 male genomes and 15208 new Y-SNPs in the
K L M N O P Q R S T
g roups
aset of all samples (black) and in the dataset of samples with an excellent quality ie
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
1 2 3 4 5 6 7 8 9 10 more
Numbe r of occ urr ences of Y-SNP
0
12000
24000
0
35000
70000
Number of Y-SNPsWhole Y-chromosome
Uniqu e regions Y-chromosome
A
B
All genomes
Excell ent qu ali ty genomes
Fig 5 Number of new Y-SNPs per number of occurrence in the full WGS dataset (A) SNPs in the whole Y-chromosome and (B) SNPs in only the identified unique regions of the
Y-chromosome The grey bars indicate the SNPs in all genomes and the black bars indicate the SNPs from samples with an excellent quality ie MCC 095
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 5
G Model
FSIGEN-977 No of Pages 8
genomes with excellent Y-SNP calling quality The same patterns ofoccurrence for these Y-SNPs are observed as with the non-filteredY-SNPs the majority of the new reported Y-SNPs appeared in onlyone or two samples of the WGS dataset as shown in Fig 5B
The genomes of eight males whose biological fatherrsquos genome isalso sequenced are present in the dataset one family of eightmales including the father the son and the six grandsons next toone fatherndashson pair In the family of eight paternal relatives 5155new SNPs are reported on the whole Y-chromosome and a largenumber of these SNPs is found in only one of the eight individualsas shown in Fig 6A The number of Y-SNPs decreases every time
0
500
1000
1500
2000
2500
0
1 2 3 4 5
Numbe r of o
0
20
40
60
80
100
120
Number of Y-SNPsA
B
8-member fam
All SNPs
Famil y-un iqu e SNP
Fig 6 Number of new Y-SNPs per occurrence in the eight paternally related samples and t
(B) SNPs in only the identified unique regions of the Y-chromosome The grey bars indica
uniquersquo they do not occur in any of the other samples with an excellent quality ie M
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
the number of samples in which the Y-SNP occurs increases exceptfor the occurrence in all-except-one and all samples of the familyWhen all Y-SNPs which also occur in any other sample withexcellent SNP calling quality are removed only less than 20 of theY-SNPs remains but the distribution of the number of Y-SNPs peroccurrence stays the same as the black bars in Fig 6A show Thesame comparison is made when only the SNPs in the unique part ofthe Y-chromosome are selected the same pattern as with all SNPsis visible in Fig 6B However for each number of occurrences thenumber of truly unique SNPs ie SNPs that do not occur in othergenomes outside the family is much higher Within the fatherndashson
6 7 8 1 2
cc urr ences of Y-SNP
Whole Y-chromosome
Uniqu e regions Y-chromosome
il y Father-son pair
s
he fatherndashson pair of Complete Genomics (A) SNPs in the whole Y-chromosome and
te the SNPs found in the family The black bars indicate the SNPs which are lsquofamily-
CC 095
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx6
G Model
FSIGEN-977 No of Pages 8
pair 2181 new SNPs are reported on the whole Y-chromosome forwhich the difference between occurrence in only one and bothgenomes is relatively small also for the proportion of SNPs whichdo not occur in the other genomes with an excellent Y-SNP quality(Fig 6A) Remarkably there are more new SNPs present in bothsamples than in one sample when the Y-SNPs which are notlocated in the unique region of the Y-chromosome are removed(Fig 6B) The effect of the increasing number of unique Y-SNPs thatdo not occur in other genomes as seen with the previous family isalso in the fatherndashson pair present
4 Discussion
The present study realises the first large screening of malegenomes for phylogenetic applications of the Y-chromosomeBased on this screening of 747 male samples an update has beenmade of the AMY-tree software and of a state-of-the-art Y-chromosomal phylogenetic tree was established for forensicscientists Furthermore also recommendations for future sequenc-ing projects dealing with a broader selection panel of Y-chromosomal haplogroup samples and for the validation of newlydetected Y-SNPs are made
First the large screening of the 747 male Y-chromosomesamples revealed that the SNP calling quality of a few samples wasoverestimated in AMY-tree version 10 As these samples belong tosub-haplogroup R-M269 they are very similar to the referencegenome which is used to estimate the SNP calling quality of asample [23] To remove this SNP calling quality overestimation theinfluence of the reference genome is excluded for all samplesbelonging R-M269 with a low SNP call quality That is why severalmodifications are made and implemented in AMY-tree version 11
Second an update of the currently used Y-chromosomalphylogenetic tree was realised based on the large database of747 available samples At the moment this tree is the most state-of-the-art tree applicable for forensic geneticists all Y-SNPs whichare reported in academic publications till today are included andall ambiguous markers are excluded to avoid wrong Y-SNPinterpretations (Table S3) As often the case in the literature thephylogenetic relationship between new Y-SNPs and earlierreported Y-SNPs in the same (sub-)haplogroup are not given Byusing the AMY-tree results it is possible to find out the concretephylogenetic level of each Y-SNP relative to the other alreadyknown SNPs For example Pamjav et al [16] described andgenotyped two new Y-SNPs Z93 and Z280 within R-M198although the exact phylogenetic positions in relationship withthe other lineages within R-M198 were not given The presence ofboth Z93 and Z80 is checked in all samples belonging to the sub-haplogroup R-M198 Z93 occurred in three R-M198 samples andin none of the other samples Also Z280 does not occur in anysample except in one R-M198 sample Therefore both Z93 andZ280 are placed in the phylogenetic tree as sub-haplogroups of R-M198 as shown in Fig 2 The choice is made for a phylogenetic treein table format as described by Van Geystelen et al [23] instead of abranching diagram because the tree is very large and will becomelarger in the future Therefore the table format can be adaptedmore easily than the diagram and it is also more manageable
Not only newly reported Y-SNPs are included in the newupdated tree but also ambiguous Y-SNPs are excluded as they cancomplicate the Y-chromosomal applications for forensic studiesAs previously described [38] the most relevant ambiguous Y-SNPsare recurrent SNPs which have a paralogous distribution along thephylogenetic tree and which have thus mutant alleles in at leasttwo independent Y-chromosomal lineages Based on the screeningof the 747 analysed samples three Y-SNPs are recognised for thefirst time as recurrent (Table S2) There are no other indications forrecurrent mutations based on the present WGS dataset Therefore
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
we may assume in most cases that males which both have themutant allele for a Y-SNP used in the updated tree (Table S3) havereceived this mutant allele from one common ancestor and not byconvergent evolution Next also all Y-SNPs which could not beanalysed by WGS for one reason or another are excluded from theupdated tree although it may be possible to genotype these SNPscorrectly with Sanger sequencing methods For example allhundred E-M2 samples in the dataset did not reveal the mutantallele for Y-SNP V95 as expected by earlier publications [1439]The reason for this remarkable result may be that V95 is not wellvalidated but more likely it is the result of a bad SNP calling in theY-chromosomal region around V95 by the current WGS methodsFor some Y-SNPs also a mutation conversion is found which isdifferent from the reported one For example another ancestraland mutant allele are observed for Y-SNP M426 than reported byRootsi et al [40] Since the Y-chromosome has a very complexorigin it also has a lot of non-unique regions which complicate theanalysis of WGS data [37] The current reference genome GRCh37shows the evolution of the Y-chromosome and its numerousresulting non-unique regions So the reason for this wrong analysisof M426 may be the position of this SNP in one of the non-uniqueregions of the Y-chromosome as defined earlier [2237] To have anunambiguous phylogenetic tree we excluded all the Y-SNPs forwhich no reliable signal with the WGS methods could be found Inthe end an updated Y-chromosomal tree which includes 359 Y-chromosomal lineages and more than 721 Y-SNP markers (TableS3) is obtained and this tree is the basic tool to develop andoptimise Y-SNP-multiplexes for forensic applications [4142]
Third the distribution of the analysed haplogroups perancestral continent in the current dataset corresponds with theknown distributions [34] Nevertheless there is not yet arepresentative set of all phylogenetic sub-haplogroups availableIn total 17 haplogroups and 106 sub-haplogroups are reported inthe analysis of the whole dataset However the SNP calling qualityis low (MCC lt 095) for most of the analysed samples as most dataare obtained from the 1000 Genomes project and therefore thesub-haplogroup determination of these data is not completelyreliable When considering only the samples of excellent quality(MCC 095) 10 haplogroups and 47 sub-haplogroups remain andthis corresponds with only 13 of the total number lineagesdescribed so far Most of the Y-chromosomes are assigned tohaplogroups E O and R Therefore when the set of WGS Y-chromosomes will be enlarged the current phylogenetic tree aswell as the analysis of Wei et al [22] to calibrate the tree can beoptimised
Finally the screening of the dataset also revealed that new insilico and in vitro methods are required to verify new Y-SNPs basedon WGS methods As earlier mentioned [23] a huge number of newY-SNPs are false positives when the genomes were sequenced withlow coverage and consequently the called SNPs will have a lowquality These false positive SNP calls disturb the determination ofthe correct (sub-)haplogroup of the sample consequently theAMY-tree software has to correct for them by applying severaladditional methods in the analysis [23] The high number of falsepositive Y-SNPs ndash even detected in WGS samples with an excellentSNP calling quality ndash is observed by comparing genomes ofpaternal relatives Within the eight paternally related samplesbelonging to one family which are genotyped by CompleteGenomics 949 newly reported Y-SNPs in the full Y-chromosomeand 88 ones in the unique regions of the Y-chromosome are foundin at least one but not all family members (Fig 6) Despite the highcoverage of these genomes and the excellent SNP calling qualitythis is still a very high number of newly reported Y-SNPs which aremost likely to be false based on the mutation rate on the Y-chromosomes calculated based on a deep-rooting pedigree [43]and based on human-chimpanzee comparisons [44] A similar
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 7
G Model
FSIGEN-977 No of Pages 8
conclusion can be made based on the fatherndashson pair in the datasetwhich is also sequenced with a high coverage by CompleteGenomics (Fig 6)
The Y-SNP results show that adding new lineages to the Y-chromosomal phylogenetic tree only based on WGS is not evidentTherefore making tabula rasa and building a new tree based on allWGS Y-chromosomes as done by Wei et al [22] is not an optionwhen the tree is going to be used in forensics Each new Y-SNP andconsequently each Y-chromosomal lineage has to be validatedindependently from the WGS data before adding them to theupdated phylogenetic tree The validation status of many potentialpolymorphic Y-SNPs (gt1 in population) is often unclear but thiscan be resolved by the sharing of genomic data among geneticgenealogists which are interested in finding new Y-SNPs to resolvetheir particular paternal ancestry [45] Therefore this is an area inwhich closer collaborations between amateurs and forensicacademics could prove to be particularly useful [13] Neverthelessit is required that new in silico methods will be designed to selectgood and relevant candidates of Y-SNPs for the validation Forexample an interesting criterion is the position of the Y-SNPs it ismore interesting to validate only the SNPs located in the uniqueregions of the Y-chromosome as there are many non-uniqueregions due to the evolutionary history of this chromosome [37]The lower number of false positive SNP calls in these uniqueregions in comparison with those in the full Y-chromosome isclearly demonstrated in the fatherndashson pair The number of new Y-SNPs reported in only one of the two samples is higher than thenew ones reported in both samples based on the whole Y-chromosome (Fig 6A) in contrast with the situation based on theunique regions of the Y-chromosome (Fig 6B)
5 Conclusions
Based on the largest screening of male genomes with 747samples in total the most up-to-date Y-chromosomal phylogenet-ic tree for forensic applications is compiled Future publicationswhich will report new Y-SNPs have to situate their phylogeneticpositions in this tree to guarantee the continuity between old andnew publications At this moment forensic scientists as well asevolutionary biologists and genetic genealogists are lost in themany reports of newly described Y-chromosomal lineages [46]Therefore initiatives as AMY-tree which optimise the phylogenybased on peer-reviewed publications are required [23] This isalready the case for the mitochondrial genome with the Phylotreeinitiative of van Oven and Kayser [47] Nevertheless to optimisethe current updated phylogenetic tree for the human Y-chromo-some more high quality genomes of a broader set of (sub-)haplogroups than the frequent haplogroups E O and R arerequired Also a higher effort in the validation of reported Y-SNPsby new in silico and in vitro methods is required
Authorsrsquo contributions
Research design amp supervision MHDL programming AVGwriting MHDL amp AVG Commenting on manuscript AVG RD ampMHDL
Conflict of interest
The authors declare no conflict of interest
Acknowledgements
The authors want to thank Tom Wenseleers Manfred KayserJean-Jacques Cassiman Tom Havenith Hendrik Larmuseau and
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
Lucrece Lernout for useful discussions and comments Thanks alsoto Guy Froyen (VIB KU Leuven) Richard Rocca (independentresearcher) Cuiping Pan (Stanford University) and Andreas Keller(Saarland University) for providing us yet unpublished andpublished called SNPs of whole genome sequencing projectsMaarten HD Larmuseau is postdoctoral fellow of the FWO-Vlaanderen (Research Foundation Flanders) This study wasfunded by the KU Leuven BOF Centre of Excellence Financing onlsquoEco and socio evolutionary dynamicsrsquo (Project number PF201007) and on lsquoCentre for Archaeological Sciences 2 (CAS 2) ndash Newmethods for research in demography and interregional exchangersquo
Appendix A Supplementary data
Supplementary data associated with this article can be found in
the online version at httpdxdoiorg101016jfsigen201303010
References
[1] M Kayser Y-chromosomal markers in forensic genetics in RWD Rapley (Ed)Molecular Forensics John Wiley amp Sons Ltd Chichesters 2007 pp 141ndash161
[2] JM Butler Chapter 13 Y-Chromosomal DNA Testing in JM Butler (Ed) Ad-vanced Topics in Forensic DNA Typing Methodology Academic Press London2011 pp 371ndash403
[3] J Chiaroni PA Underhill LL Cavalli-Sforza Y chromosome diversity humanexpansion drift and cultural evolution Proc Natl Acad Sci USA 106 (2009)20174ndash20179
[4] TM Karafet FL Mendez MB Meilerman PA Underhill SL Zegura MFHammer New binary polymorphisms reshape and increase resolution of thehuman Y chromosomal haplogroup tree Genome Res 18 (2008) 830ndash838
[5] MHD Larmuseau N Vanderheyden M Jacobs M Coomans L Larno R DecorteMicro-geographic distribution of Y-chromosomal variation in the central-westernEuropean region Brabant Forensic Sci Int Genet 5 (2011) 95ndash99
[6] F Cruciani B Trombetta C Antonelli R Pascone G Valesini V Scalzi G Vona BMelegh B Zagradisnik G Assum et al Strong intra- and inter-continentaldifferentiation revealed by Y chromosome SNPs M269 U106 and U152 ForensicSci Int Genet 5 (2011) E49ndashE52
[7] MHD Larmuseau J Vanoverbeke G Gielis N Vanderheyden HFM LarmuseauR Decorte In the name of the migrant father ndash analysis of surname originidentifies historic admixture events undetectable from genealogical recordsHeredity 109 (2012) 90ndash95
[8] S Willuweit L Roewer Y chromosome haplotype reference database (YHRD)update Forensic Sci Int Genet 1 (2007) 83ndash87
[9] R Scozzari A Massaia E DrsquoAtanasio NM Myres UA Perego B Trombetta FCruciani Molecular dissection of the basal clades in the human Y chromosomephylogenetic tree Plos ONE 7 (2012) e49170
[10] F Cruciani B Trombetta A Massaia G Destro-Bisol D Sellitto R Scozzari Arevised root for the human Y chromosomal phylogenetic tree the origin ofpatrilineal diversity in Africa Am J Hum Genet 88 (2011) 814ndash818
[11] S Fornarino M Pala V Battaglia R Maranta A Achilli G Modiano A Torroni OSemino SA Santachiara-Benerecetti Mitochondrial and Y-chromosome diversi-ty of the Tharus (Nepal) a reservoir of genetic variation BMC Evol Biol 9 (2009)154
[12] FL Mendez TM Karafet T Krahn H Ostrer H Soodyall MF Hammer Increasedresolution of Y chromosome haplogroup T defines relationships among popula-tions of the Near East Europe and Africa Hum Biol 83 (2011) 39ndash53
[13] LM Sims D Garvey J Ballantyne Improved resolution haplogroup G phylogenyin the Y-chromosome revealed by a set of newly characterized SNPs Plos ONE 4(2009) e5792
[14] B Trombetta F Cruciani D Sellitto R Scozzari A new topology of the human Ychromosome haplogroup E1b1 (E-P2) revealed through the use of newly charac-terized binary polymorphisms PLoS ONE 6 (2011) e16073
[15] MS Jota DR Lacerda JR Sandoval PPR Vieira SS Santos-Lopes R Bisso-Machado VR Paixao-Cortes S Revollo C Paz-Y-Mino R Fujita et al A newsubhaplogroup of native American Y-chromosomes from the Andes Am J PhysAnthropol 146 (2011) 553ndash559
[16] H Pamjav T Feher E Nemeth Z Padar Brief communication new Y-chromo-some binary markers improve phylogenetic resolution within haplogroup R1a1Am J Phys Anthropol 149 (2012) 611ndash615
[17] NM Myres S Rootsi AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova A Pshenichnov B Yunusbayev et al A major Y-chromosomehaplogroup R1b Holocene era founder effect in Central and Western Europe Eur JHum Genet 19 (2011) 95ndash101
[18] S Yan CC Wang H Li SL Li L Jin G Consortium An updated tree of Y-chromosome Haplogroup O and revised phylogenetic positions of mutations P164and PK4 Eur J Hum Genet 19 (2011) 1013ndash1015
[19] T 1000 Genomes Project Consortium An integrated map of genetic variation from1092 human genomes Nature 491 (2012) 56ndash65
[20] DL Altshuler RM Durbin GR Abecasis DR Bentley A Chakravarti AG ClarkFS Collins FM De la Vega P Donnelly M Egholm et al A map of human
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx8
G Model
FSIGEN-977 No of Pages 8
genome variation from population-scale sequencing Nature 467 (2010)1061ndash1073
[21] AT Duggan M Stoneking A highly unstable recent mutation in human mtDNAAm J Hum Genet 9 (2013) 279ndash284
[22] W Wei Q Ayub Y Chen S McCarthy Y Hou I Carbone Y Xue C Tyler-Smith Acalibrated human Y-chromosomal phylogeny based on resequencing GenomeRes 23 (2013) 388ndash395
[23] A Van Geystelen R Decorte MHD Larmuseau AMY-tree an algorithm to usewhole genome SNP calling for Y chromosomal phylogenetic applications BMCGenomics 14 (2013) 101
[24] R Drmanac AB Sparks MJ Callow AL Halpern NL Burns BG Kermani PCarnevali I Nazarenko GB Nilsen G Yeung et al Human genome sequencingusing unchained base reads on self-assembling DNA nanoarrays Science 327(2010) 78ndash81
[25] L-P Wong RT-H Ong W-T Poh X Liu P Chen RQ Li KK-Y Lam NE PillaiK-S Sim H Xu et al Deep whole-genome sequencing of 100 Southeast AsianMalays Am J Hum Genet 92 (2013) 1ndash15
[26] SC Schuster W Miller A Ratan LP Tomsho B Giardine LR Kasson RS HarrisDC Petersen FQ Zhao J Qi et al Complete Khoisan and Bantu genomes fromsouthern Africa Nature 463 (2010) 943ndash947
[27] P Tong JGD Prendergast AJ Lohan SM Farrington S Cronin N Friel DGBradley O Hardiman A Evans JF Wilson et al Sequencing and analysis of anIrish human genome Genome Biol 11 (2010) R91
[28] A Keller A Graefen M Ball M Matzas V Boisguerin F Maixner P Leidinger CBackes R Khairat M Forster et al New insights into the Tyrolean Icemanrsquos originand phenotype as inferred by whole-genome sequencing Nature Commun 3(2012) 698
[29] R Chen GI Mias J Li-Pook-Than LH Jiang HYK Lam R Chen E Miriami KJKarczewski M Hariharan FE Dewey et al Personal omics profiling revealsdynamic molecular and medical phenotypes Cell 148 (2012) 1293ndash1307
[30] JM Rothberg W Hinz TM Rearick J Schultz W Mileski M Davey JH LeamonK Johnson MJ Milgrew M Edwards et al An integrated semiconductor deviceenabling non-optical genome sequencing Nature 475 (2011) 348ndash352
[31] D Pushkarev NF Neff SR Quake Single-molecule sequencing of an individualhuman genome Nat Biotechnol 27 (2009) 847ndash850
[32] M Rasmussen YR Li S Lindgreen JS Pedersen A Albrechtsen I Moltke MMetspalu E Metspalu T Kivisild R Gupta et al Ancient human genomesequence of an extinct Palaeo-Eskimo Nature 463 (2010) 757ndash762
[33] SM Ahn TH Kim S Lee D Kim H Ghang DS Kim BC Kim SY Kim WY KimC Kim et al The first Korean genome sequence and analysis full genomesequencing for a socio-ethnic group Genome Res 19 (2009) 1622ndash1629
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
[34] DA Wheeler M Srinivasan M Egholm Y Shen L Chen A McGuire W He YJChen V Makhijani GT Roth et al The complete genome of an individual bymassively parallel DNA sequencing Nature 452 (2008) U5ndashU872
[35] J Wang W Wang RQ Li YR Li G Tian L Goodman W Fan JQ Zhang J Li JBZhang et al The diploid genome sequence of an Asian individual Nature 456(2008) U1ndashU60
[36] BA Peters BG Kermani AB Sparks O Alferov P Hong A Alexeev Y Jiang FDahl YT Tang J Haas et al Accurate whole-genome sequencing and haplotyp-ing from 10 to 20 human cells Nature 487 (2012) 190ndash195
[37] H Skaletsky T Kuroda-Kawaguchi PJ Minx HS Cordum L Hillier LG Brown SRepping T Pyntikova J Ali T Bieri et al The male-specific region of the human Ychromosome is a mosaic of discrete sequence classes Nature 423 (2003) U2ndashU825
[38] SM Adams TE King E Bosch MA Jobling The case of the unreliable SNPrecurrent back-mutation of Y-chromosomal markers P25 through gene conver-sion Forensic Sci Int 159 (2006) 14ndash20
[39] B Trombetta F Cruciani PA Underhill D Sellitto R Scozzari Footprints of X-to-Y gene conversion in recent human evolution Mol Biol Evol 27 (2010) 714ndash725
[40] S Rootsi NM Myres AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova K Varendi H Sahakyan et al Distinguishing the co-ancestries ofhaplogroup G Y-chromosomes in the populations of Europe and the CaucasusEur J Hum Genet 20 (2012) 1275ndash1282
[41] S Caratti S Gino C Torre C Robino Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application Int J Legal Med 123(2009) 357ndash360
[42] C Bouakaze C Keyser S Amory E Crubezy B Ludes First successful assay of Y-SNP typing by SNaPshot minisequencing on ancient DNA Int J Legal Med 121(2007) 493ndash499
[43] YL Xue QJ Wang Q Long BL Ng H Swerdlow J Burton C Skuce R Taylor ZAbdellah YL Zhao et al Human Y chromosome base-substitution mutation ratemeasured by direct sequencing in a deep-rooting pedigree Curr Biol 19 (2009)1453ndash1457
[44] Y Kuroki A Toyoda H Noguchi TD Taylor T Itoh DS Kim DW Kim SH ChoiIC Kim HH Choi Comparative analysis of chimpanzee and human Y chromo-somes unveils complex evolutionary pathway Nat Genet 38 (2006) 158ndash167
[45] TE King MA Jobling Whatrsquos in a name Y chromosomes surnames and thegenetic genealogy revolution Trends Genet 25 (2009) 351ndash360
[46] MHD Larmuseau A Van Geystelen M van Oven R Decorte Genetic genealogycomes of age ndash perspectives on the use of deep-rooted pedigrees in humanpopulation genetics Am J Phys Anthropol 150 (2013) 505ndash511
[47] M van Oven M Kayser Updated comprehensive phylogenetic tree of globalhuman mitochondrial DNA variation Hum Mutat 30 (2008) E386ndashE394
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
- Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs
-
- Introduction
- Materials and methods
-
- Updated phylogenetic tree
- WGS Y-SNPs dataset
- AMY-tree modifications
- Y-SNP detecting
-
- Results
-
- Updated tree for forensic applications
- Sub-haplogroup determining
- Detecting new Y-SNPs
-
- Discussion
- Conclusions
- Authorsrsquo contributions
- Conflict of interest
- Acknowledgements
- Supplementary data
-
- References
-
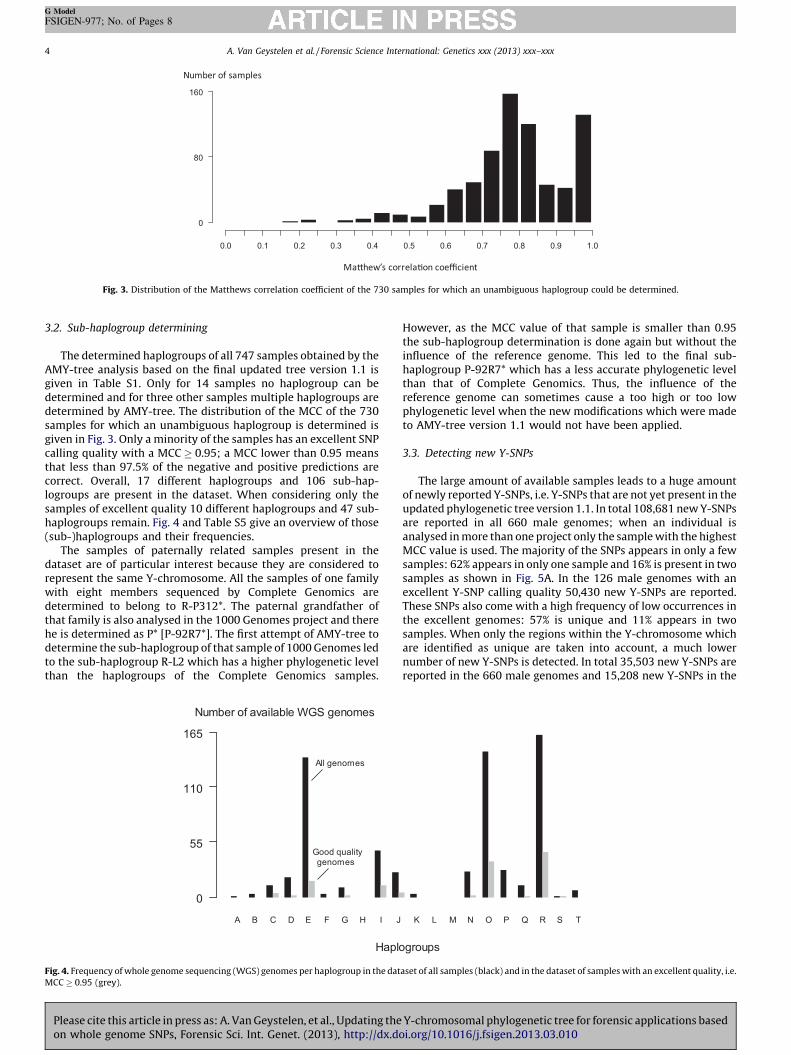
00 01 02 03 04 05 06 07 08 09 10
0
80
160
Number of samples
Mahewrsquos corr elaon coefficient
Fig 3 Distribution of the Matthews correlation coefficient of the 730 samples for which an unambiguous haplogroup could be determined
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx4
G Model
FSIGEN-977 No of Pages 8
32 Sub-haplogroup determining
The determined haplogroups of all 747 samples obtained by theAMY-tree analysis based on the final updated tree version 11 isgiven in Table S1 Only for 14 samples no haplogroup can bedetermined and for three other samples multiple haplogroups aredetermined by AMY-tree The distribution of the MCC of the 730samples for which an unambiguous haplogroup is determined isgiven in Fig 3 Only a minority of the samples has an excellent SNPcalling quality with a MCC 095 a MCC lower than 095 meansthat less than 975 of the negative and positive predictions arecorrect Overall 17 different haplogroups and 106 sub-hap-logroups are present in the dataset When considering only thesamples of excellent quality 10 different haplogroups and 47 sub-haplogroups remain Fig 4 and Table S5 give an overview of those(sub-)haplogroups and their frequencies
The samples of paternally related samples present in thedataset are of particular interest because they are considered torepresent the same Y-chromosome All the samples of one familywith eight members sequenced by Complete Genomics aredetermined to belong to R-P312 The paternal grandfather ofthat family is also analysed in the 1000 Genomes project and therehe is determined as P [P-92R7] The first attempt of AMY-tree todetermine the sub-haplogroup of that sample of 1000 Genomes ledto the sub-haplogroup R-L2 which has a higher phylogenetic levelthan the haplogroups of the Complete Genomics samples
A B C D E F G H I J
0
55
110
165
All geno mes
Good quali ty genomes
Numbe r of availab le WGS ge nomes
Hap lo
Fig 4 Frequency of whole genome sequencing (WGS) genomes per haplogroup in the dat
MCC 095 (grey)
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
However as the MCC value of that sample is smaller than 095the sub-haplogroup determination is done again but without theinfluence of the reference genome This led to the final sub-haplogroup P-92R7 which has a less accurate phylogenetic levelthan that of Complete Genomics Thus the influence of thereference genome can sometimes cause a too high or too lowphylogenetic level when the new modifications which were madeto AMY-tree version 11 would not have been applied
33 Detecting new Y-SNPs
The large amount of available samples leads to a huge amountof newly reported Y-SNPs ie Y-SNPs that are not yet present in theupdated phylogenetic tree version 11 In total 108681 new Y-SNPsare reported in all 660 male genomes when an individual isanalysed in more than one project only the sample with the highestMCC value is used The majority of the SNPs appears in only a fewsamples 62 appears in only one sample and 16 is present in twosamples as shown in Fig 5A In the 126 male genomes with anexcellent Y-SNP calling quality 50430 new Y-SNPs are reportedThese SNPs also come with a high frequency of low occurrences inthe excellent genomes 57 is unique and 11 appears in twosamples When only the regions within the Y-chromosome whichare identified as unique are taken into account a much lowernumber of new Y-SNPs is detected In total 35503 new Y-SNPs arereported in the 660 male genomes and 15208 new Y-SNPs in the
K L M N O P Q R S T
g roups
aset of all samples (black) and in the dataset of samples with an excellent quality ie
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
1 2 3 4 5 6 7 8 9 10 more
Numbe r of occ urr ences of Y-SNP
0
12000
24000
0
35000
70000
Number of Y-SNPsWhole Y-chromosome
Uniqu e regions Y-chromosome
A
B
All genomes
Excell ent qu ali ty genomes
Fig 5 Number of new Y-SNPs per number of occurrence in the full WGS dataset (A) SNPs in the whole Y-chromosome and (B) SNPs in only the identified unique regions of the
Y-chromosome The grey bars indicate the SNPs in all genomes and the black bars indicate the SNPs from samples with an excellent quality ie MCC 095
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 5
G Model
FSIGEN-977 No of Pages 8
genomes with excellent Y-SNP calling quality The same patterns ofoccurrence for these Y-SNPs are observed as with the non-filteredY-SNPs the majority of the new reported Y-SNPs appeared in onlyone or two samples of the WGS dataset as shown in Fig 5B
The genomes of eight males whose biological fatherrsquos genome isalso sequenced are present in the dataset one family of eightmales including the father the son and the six grandsons next toone fatherndashson pair In the family of eight paternal relatives 5155new SNPs are reported on the whole Y-chromosome and a largenumber of these SNPs is found in only one of the eight individualsas shown in Fig 6A The number of Y-SNPs decreases every time
0
500
1000
1500
2000
2500
0
1 2 3 4 5
Numbe r of o
0
20
40
60
80
100
120
Number of Y-SNPsA
B
8-member fam
All SNPs
Famil y-un iqu e SNP
Fig 6 Number of new Y-SNPs per occurrence in the eight paternally related samples and t
(B) SNPs in only the identified unique regions of the Y-chromosome The grey bars indica
uniquersquo they do not occur in any of the other samples with an excellent quality ie M
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
the number of samples in which the Y-SNP occurs increases exceptfor the occurrence in all-except-one and all samples of the familyWhen all Y-SNPs which also occur in any other sample withexcellent SNP calling quality are removed only less than 20 of theY-SNPs remains but the distribution of the number of Y-SNPs peroccurrence stays the same as the black bars in Fig 6A show Thesame comparison is made when only the SNPs in the unique part ofthe Y-chromosome are selected the same pattern as with all SNPsis visible in Fig 6B However for each number of occurrences thenumber of truly unique SNPs ie SNPs that do not occur in othergenomes outside the family is much higher Within the fatherndashson
6 7 8 1 2
cc urr ences of Y-SNP
Whole Y-chromosome
Uniqu e regions Y-chromosome
il y Father-son pair
s
he fatherndashson pair of Complete Genomics (A) SNPs in the whole Y-chromosome and
te the SNPs found in the family The black bars indicate the SNPs which are lsquofamily-
CC 095
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx6
G Model
FSIGEN-977 No of Pages 8
pair 2181 new SNPs are reported on the whole Y-chromosome forwhich the difference between occurrence in only one and bothgenomes is relatively small also for the proportion of SNPs whichdo not occur in the other genomes with an excellent Y-SNP quality(Fig 6A) Remarkably there are more new SNPs present in bothsamples than in one sample when the Y-SNPs which are notlocated in the unique region of the Y-chromosome are removed(Fig 6B) The effect of the increasing number of unique Y-SNPs thatdo not occur in other genomes as seen with the previous family isalso in the fatherndashson pair present
4 Discussion
The present study realises the first large screening of malegenomes for phylogenetic applications of the Y-chromosomeBased on this screening of 747 male samples an update has beenmade of the AMY-tree software and of a state-of-the-art Y-chromosomal phylogenetic tree was established for forensicscientists Furthermore also recommendations for future sequenc-ing projects dealing with a broader selection panel of Y-chromosomal haplogroup samples and for the validation of newlydetected Y-SNPs are made
First the large screening of the 747 male Y-chromosomesamples revealed that the SNP calling quality of a few samples wasoverestimated in AMY-tree version 10 As these samples belong tosub-haplogroup R-M269 they are very similar to the referencegenome which is used to estimate the SNP calling quality of asample [23] To remove this SNP calling quality overestimation theinfluence of the reference genome is excluded for all samplesbelonging R-M269 with a low SNP call quality That is why severalmodifications are made and implemented in AMY-tree version 11
Second an update of the currently used Y-chromosomalphylogenetic tree was realised based on the large database of747 available samples At the moment this tree is the most state-of-the-art tree applicable for forensic geneticists all Y-SNPs whichare reported in academic publications till today are included andall ambiguous markers are excluded to avoid wrong Y-SNPinterpretations (Table S3) As often the case in the literature thephylogenetic relationship between new Y-SNPs and earlierreported Y-SNPs in the same (sub-)haplogroup are not given Byusing the AMY-tree results it is possible to find out the concretephylogenetic level of each Y-SNP relative to the other alreadyknown SNPs For example Pamjav et al [16] described andgenotyped two new Y-SNPs Z93 and Z280 within R-M198although the exact phylogenetic positions in relationship withthe other lineages within R-M198 were not given The presence ofboth Z93 and Z80 is checked in all samples belonging to the sub-haplogroup R-M198 Z93 occurred in three R-M198 samples andin none of the other samples Also Z280 does not occur in anysample except in one R-M198 sample Therefore both Z93 andZ280 are placed in the phylogenetic tree as sub-haplogroups of R-M198 as shown in Fig 2 The choice is made for a phylogenetic treein table format as described by Van Geystelen et al [23] instead of abranching diagram because the tree is very large and will becomelarger in the future Therefore the table format can be adaptedmore easily than the diagram and it is also more manageable
Not only newly reported Y-SNPs are included in the newupdated tree but also ambiguous Y-SNPs are excluded as they cancomplicate the Y-chromosomal applications for forensic studiesAs previously described [38] the most relevant ambiguous Y-SNPsare recurrent SNPs which have a paralogous distribution along thephylogenetic tree and which have thus mutant alleles in at leasttwo independent Y-chromosomal lineages Based on the screeningof the 747 analysed samples three Y-SNPs are recognised for thefirst time as recurrent (Table S2) There are no other indications forrecurrent mutations based on the present WGS dataset Therefore
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
we may assume in most cases that males which both have themutant allele for a Y-SNP used in the updated tree (Table S3) havereceived this mutant allele from one common ancestor and not byconvergent evolution Next also all Y-SNPs which could not beanalysed by WGS for one reason or another are excluded from theupdated tree although it may be possible to genotype these SNPscorrectly with Sanger sequencing methods For example allhundred E-M2 samples in the dataset did not reveal the mutantallele for Y-SNP V95 as expected by earlier publications [1439]The reason for this remarkable result may be that V95 is not wellvalidated but more likely it is the result of a bad SNP calling in theY-chromosomal region around V95 by the current WGS methodsFor some Y-SNPs also a mutation conversion is found which isdifferent from the reported one For example another ancestraland mutant allele are observed for Y-SNP M426 than reported byRootsi et al [40] Since the Y-chromosome has a very complexorigin it also has a lot of non-unique regions which complicate theanalysis of WGS data [37] The current reference genome GRCh37shows the evolution of the Y-chromosome and its numerousresulting non-unique regions So the reason for this wrong analysisof M426 may be the position of this SNP in one of the non-uniqueregions of the Y-chromosome as defined earlier [2237] To have anunambiguous phylogenetic tree we excluded all the Y-SNPs forwhich no reliable signal with the WGS methods could be found Inthe end an updated Y-chromosomal tree which includes 359 Y-chromosomal lineages and more than 721 Y-SNP markers (TableS3) is obtained and this tree is the basic tool to develop andoptimise Y-SNP-multiplexes for forensic applications [4142]
Third the distribution of the analysed haplogroups perancestral continent in the current dataset corresponds with theknown distributions [34] Nevertheless there is not yet arepresentative set of all phylogenetic sub-haplogroups availableIn total 17 haplogroups and 106 sub-haplogroups are reported inthe analysis of the whole dataset However the SNP calling qualityis low (MCC lt 095) for most of the analysed samples as most dataare obtained from the 1000 Genomes project and therefore thesub-haplogroup determination of these data is not completelyreliable When considering only the samples of excellent quality(MCC 095) 10 haplogroups and 47 sub-haplogroups remain andthis corresponds with only 13 of the total number lineagesdescribed so far Most of the Y-chromosomes are assigned tohaplogroups E O and R Therefore when the set of WGS Y-chromosomes will be enlarged the current phylogenetic tree aswell as the analysis of Wei et al [22] to calibrate the tree can beoptimised
Finally the screening of the dataset also revealed that new insilico and in vitro methods are required to verify new Y-SNPs basedon WGS methods As earlier mentioned [23] a huge number of newY-SNPs are false positives when the genomes were sequenced withlow coverage and consequently the called SNPs will have a lowquality These false positive SNP calls disturb the determination ofthe correct (sub-)haplogroup of the sample consequently theAMY-tree software has to correct for them by applying severaladditional methods in the analysis [23] The high number of falsepositive Y-SNPs ndash even detected in WGS samples with an excellentSNP calling quality ndash is observed by comparing genomes ofpaternal relatives Within the eight paternally related samplesbelonging to one family which are genotyped by CompleteGenomics 949 newly reported Y-SNPs in the full Y-chromosomeand 88 ones in the unique regions of the Y-chromosome are foundin at least one but not all family members (Fig 6) Despite the highcoverage of these genomes and the excellent SNP calling qualitythis is still a very high number of newly reported Y-SNPs which aremost likely to be false based on the mutation rate on the Y-chromosomes calculated based on a deep-rooting pedigree [43]and based on human-chimpanzee comparisons [44] A similar
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 7
G Model
FSIGEN-977 No of Pages 8
conclusion can be made based on the fatherndashson pair in the datasetwhich is also sequenced with a high coverage by CompleteGenomics (Fig 6)
The Y-SNP results show that adding new lineages to the Y-chromosomal phylogenetic tree only based on WGS is not evidentTherefore making tabula rasa and building a new tree based on allWGS Y-chromosomes as done by Wei et al [22] is not an optionwhen the tree is going to be used in forensics Each new Y-SNP andconsequently each Y-chromosomal lineage has to be validatedindependently from the WGS data before adding them to theupdated phylogenetic tree The validation status of many potentialpolymorphic Y-SNPs (gt1 in population) is often unclear but thiscan be resolved by the sharing of genomic data among geneticgenealogists which are interested in finding new Y-SNPs to resolvetheir particular paternal ancestry [45] Therefore this is an area inwhich closer collaborations between amateurs and forensicacademics could prove to be particularly useful [13] Neverthelessit is required that new in silico methods will be designed to selectgood and relevant candidates of Y-SNPs for the validation Forexample an interesting criterion is the position of the Y-SNPs it ismore interesting to validate only the SNPs located in the uniqueregions of the Y-chromosome as there are many non-uniqueregions due to the evolutionary history of this chromosome [37]The lower number of false positive SNP calls in these uniqueregions in comparison with those in the full Y-chromosome isclearly demonstrated in the fatherndashson pair The number of new Y-SNPs reported in only one of the two samples is higher than thenew ones reported in both samples based on the whole Y-chromosome (Fig 6A) in contrast with the situation based on theunique regions of the Y-chromosome (Fig 6B)
5 Conclusions
Based on the largest screening of male genomes with 747samples in total the most up-to-date Y-chromosomal phylogenet-ic tree for forensic applications is compiled Future publicationswhich will report new Y-SNPs have to situate their phylogeneticpositions in this tree to guarantee the continuity between old andnew publications At this moment forensic scientists as well asevolutionary biologists and genetic genealogists are lost in themany reports of newly described Y-chromosomal lineages [46]Therefore initiatives as AMY-tree which optimise the phylogenybased on peer-reviewed publications are required [23] This isalready the case for the mitochondrial genome with the Phylotreeinitiative of van Oven and Kayser [47] Nevertheless to optimisethe current updated phylogenetic tree for the human Y-chromo-some more high quality genomes of a broader set of (sub-)haplogroups than the frequent haplogroups E O and R arerequired Also a higher effort in the validation of reported Y-SNPsby new in silico and in vitro methods is required
Authorsrsquo contributions
Research design amp supervision MHDL programming AVGwriting MHDL amp AVG Commenting on manuscript AVG RD ampMHDL
Conflict of interest
The authors declare no conflict of interest
Acknowledgements
The authors want to thank Tom Wenseleers Manfred KayserJean-Jacques Cassiman Tom Havenith Hendrik Larmuseau and
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
Lucrece Lernout for useful discussions and comments Thanks alsoto Guy Froyen (VIB KU Leuven) Richard Rocca (independentresearcher) Cuiping Pan (Stanford University) and Andreas Keller(Saarland University) for providing us yet unpublished andpublished called SNPs of whole genome sequencing projectsMaarten HD Larmuseau is postdoctoral fellow of the FWO-Vlaanderen (Research Foundation Flanders) This study wasfunded by the KU Leuven BOF Centre of Excellence Financing onlsquoEco and socio evolutionary dynamicsrsquo (Project number PF201007) and on lsquoCentre for Archaeological Sciences 2 (CAS 2) ndash Newmethods for research in demography and interregional exchangersquo
Appendix A Supplementary data
Supplementary data associated with this article can be found in
the online version at httpdxdoiorg101016jfsigen201303010
References
[1] M Kayser Y-chromosomal markers in forensic genetics in RWD Rapley (Ed)Molecular Forensics John Wiley amp Sons Ltd Chichesters 2007 pp 141ndash161
[2] JM Butler Chapter 13 Y-Chromosomal DNA Testing in JM Butler (Ed) Ad-vanced Topics in Forensic DNA Typing Methodology Academic Press London2011 pp 371ndash403
[3] J Chiaroni PA Underhill LL Cavalli-Sforza Y chromosome diversity humanexpansion drift and cultural evolution Proc Natl Acad Sci USA 106 (2009)20174ndash20179
[4] TM Karafet FL Mendez MB Meilerman PA Underhill SL Zegura MFHammer New binary polymorphisms reshape and increase resolution of thehuman Y chromosomal haplogroup tree Genome Res 18 (2008) 830ndash838
[5] MHD Larmuseau N Vanderheyden M Jacobs M Coomans L Larno R DecorteMicro-geographic distribution of Y-chromosomal variation in the central-westernEuropean region Brabant Forensic Sci Int Genet 5 (2011) 95ndash99
[6] F Cruciani B Trombetta C Antonelli R Pascone G Valesini V Scalzi G Vona BMelegh B Zagradisnik G Assum et al Strong intra- and inter-continentaldifferentiation revealed by Y chromosome SNPs M269 U106 and U152 ForensicSci Int Genet 5 (2011) E49ndashE52
[7] MHD Larmuseau J Vanoverbeke G Gielis N Vanderheyden HFM LarmuseauR Decorte In the name of the migrant father ndash analysis of surname originidentifies historic admixture events undetectable from genealogical recordsHeredity 109 (2012) 90ndash95
[8] S Willuweit L Roewer Y chromosome haplotype reference database (YHRD)update Forensic Sci Int Genet 1 (2007) 83ndash87
[9] R Scozzari A Massaia E DrsquoAtanasio NM Myres UA Perego B Trombetta FCruciani Molecular dissection of the basal clades in the human Y chromosomephylogenetic tree Plos ONE 7 (2012) e49170
[10] F Cruciani B Trombetta A Massaia G Destro-Bisol D Sellitto R Scozzari Arevised root for the human Y chromosomal phylogenetic tree the origin ofpatrilineal diversity in Africa Am J Hum Genet 88 (2011) 814ndash818
[11] S Fornarino M Pala V Battaglia R Maranta A Achilli G Modiano A Torroni OSemino SA Santachiara-Benerecetti Mitochondrial and Y-chromosome diversi-ty of the Tharus (Nepal) a reservoir of genetic variation BMC Evol Biol 9 (2009)154
[12] FL Mendez TM Karafet T Krahn H Ostrer H Soodyall MF Hammer Increasedresolution of Y chromosome haplogroup T defines relationships among popula-tions of the Near East Europe and Africa Hum Biol 83 (2011) 39ndash53
[13] LM Sims D Garvey J Ballantyne Improved resolution haplogroup G phylogenyin the Y-chromosome revealed by a set of newly characterized SNPs Plos ONE 4(2009) e5792
[14] B Trombetta F Cruciani D Sellitto R Scozzari A new topology of the human Ychromosome haplogroup E1b1 (E-P2) revealed through the use of newly charac-terized binary polymorphisms PLoS ONE 6 (2011) e16073
[15] MS Jota DR Lacerda JR Sandoval PPR Vieira SS Santos-Lopes R Bisso-Machado VR Paixao-Cortes S Revollo C Paz-Y-Mino R Fujita et al A newsubhaplogroup of native American Y-chromosomes from the Andes Am J PhysAnthropol 146 (2011) 553ndash559
[16] H Pamjav T Feher E Nemeth Z Padar Brief communication new Y-chromo-some binary markers improve phylogenetic resolution within haplogroup R1a1Am J Phys Anthropol 149 (2012) 611ndash615
[17] NM Myres S Rootsi AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova A Pshenichnov B Yunusbayev et al A major Y-chromosomehaplogroup R1b Holocene era founder effect in Central and Western Europe Eur JHum Genet 19 (2011) 95ndash101
[18] S Yan CC Wang H Li SL Li L Jin G Consortium An updated tree of Y-chromosome Haplogroup O and revised phylogenetic positions of mutations P164and PK4 Eur J Hum Genet 19 (2011) 1013ndash1015
[19] T 1000 Genomes Project Consortium An integrated map of genetic variation from1092 human genomes Nature 491 (2012) 56ndash65
[20] DL Altshuler RM Durbin GR Abecasis DR Bentley A Chakravarti AG ClarkFS Collins FM De la Vega P Donnelly M Egholm et al A map of human
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx8
G Model
FSIGEN-977 No of Pages 8
genome variation from population-scale sequencing Nature 467 (2010)1061ndash1073
[21] AT Duggan M Stoneking A highly unstable recent mutation in human mtDNAAm J Hum Genet 9 (2013) 279ndash284
[22] W Wei Q Ayub Y Chen S McCarthy Y Hou I Carbone Y Xue C Tyler-Smith Acalibrated human Y-chromosomal phylogeny based on resequencing GenomeRes 23 (2013) 388ndash395
[23] A Van Geystelen R Decorte MHD Larmuseau AMY-tree an algorithm to usewhole genome SNP calling for Y chromosomal phylogenetic applications BMCGenomics 14 (2013) 101
[24] R Drmanac AB Sparks MJ Callow AL Halpern NL Burns BG Kermani PCarnevali I Nazarenko GB Nilsen G Yeung et al Human genome sequencingusing unchained base reads on self-assembling DNA nanoarrays Science 327(2010) 78ndash81
[25] L-P Wong RT-H Ong W-T Poh X Liu P Chen RQ Li KK-Y Lam NE PillaiK-S Sim H Xu et al Deep whole-genome sequencing of 100 Southeast AsianMalays Am J Hum Genet 92 (2013) 1ndash15
[26] SC Schuster W Miller A Ratan LP Tomsho B Giardine LR Kasson RS HarrisDC Petersen FQ Zhao J Qi et al Complete Khoisan and Bantu genomes fromsouthern Africa Nature 463 (2010) 943ndash947
[27] P Tong JGD Prendergast AJ Lohan SM Farrington S Cronin N Friel DGBradley O Hardiman A Evans JF Wilson et al Sequencing and analysis of anIrish human genome Genome Biol 11 (2010) R91
[28] A Keller A Graefen M Ball M Matzas V Boisguerin F Maixner P Leidinger CBackes R Khairat M Forster et al New insights into the Tyrolean Icemanrsquos originand phenotype as inferred by whole-genome sequencing Nature Commun 3(2012) 698
[29] R Chen GI Mias J Li-Pook-Than LH Jiang HYK Lam R Chen E Miriami KJKarczewski M Hariharan FE Dewey et al Personal omics profiling revealsdynamic molecular and medical phenotypes Cell 148 (2012) 1293ndash1307
[30] JM Rothberg W Hinz TM Rearick J Schultz W Mileski M Davey JH LeamonK Johnson MJ Milgrew M Edwards et al An integrated semiconductor deviceenabling non-optical genome sequencing Nature 475 (2011) 348ndash352
[31] D Pushkarev NF Neff SR Quake Single-molecule sequencing of an individualhuman genome Nat Biotechnol 27 (2009) 847ndash850
[32] M Rasmussen YR Li S Lindgreen JS Pedersen A Albrechtsen I Moltke MMetspalu E Metspalu T Kivisild R Gupta et al Ancient human genomesequence of an extinct Palaeo-Eskimo Nature 463 (2010) 757ndash762
[33] SM Ahn TH Kim S Lee D Kim H Ghang DS Kim BC Kim SY Kim WY KimC Kim et al The first Korean genome sequence and analysis full genomesequencing for a socio-ethnic group Genome Res 19 (2009) 1622ndash1629
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
[34] DA Wheeler M Srinivasan M Egholm Y Shen L Chen A McGuire W He YJChen V Makhijani GT Roth et al The complete genome of an individual bymassively parallel DNA sequencing Nature 452 (2008) U5ndashU872
[35] J Wang W Wang RQ Li YR Li G Tian L Goodman W Fan JQ Zhang J Li JBZhang et al The diploid genome sequence of an Asian individual Nature 456(2008) U1ndashU60
[36] BA Peters BG Kermani AB Sparks O Alferov P Hong A Alexeev Y Jiang FDahl YT Tang J Haas et al Accurate whole-genome sequencing and haplotyp-ing from 10 to 20 human cells Nature 487 (2012) 190ndash195
[37] H Skaletsky T Kuroda-Kawaguchi PJ Minx HS Cordum L Hillier LG Brown SRepping T Pyntikova J Ali T Bieri et al The male-specific region of the human Ychromosome is a mosaic of discrete sequence classes Nature 423 (2003) U2ndashU825
[38] SM Adams TE King E Bosch MA Jobling The case of the unreliable SNPrecurrent back-mutation of Y-chromosomal markers P25 through gene conver-sion Forensic Sci Int 159 (2006) 14ndash20
[39] B Trombetta F Cruciani PA Underhill D Sellitto R Scozzari Footprints of X-to-Y gene conversion in recent human evolution Mol Biol Evol 27 (2010) 714ndash725
[40] S Rootsi NM Myres AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova K Varendi H Sahakyan et al Distinguishing the co-ancestries ofhaplogroup G Y-chromosomes in the populations of Europe and the CaucasusEur J Hum Genet 20 (2012) 1275ndash1282
[41] S Caratti S Gino C Torre C Robino Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application Int J Legal Med 123(2009) 357ndash360
[42] C Bouakaze C Keyser S Amory E Crubezy B Ludes First successful assay of Y-SNP typing by SNaPshot minisequencing on ancient DNA Int J Legal Med 121(2007) 493ndash499
[43] YL Xue QJ Wang Q Long BL Ng H Swerdlow J Burton C Skuce R Taylor ZAbdellah YL Zhao et al Human Y chromosome base-substitution mutation ratemeasured by direct sequencing in a deep-rooting pedigree Curr Biol 19 (2009)1453ndash1457
[44] Y Kuroki A Toyoda H Noguchi TD Taylor T Itoh DS Kim DW Kim SH ChoiIC Kim HH Choi Comparative analysis of chimpanzee and human Y chromo-somes unveils complex evolutionary pathway Nat Genet 38 (2006) 158ndash167
[45] TE King MA Jobling Whatrsquos in a name Y chromosomes surnames and thegenetic genealogy revolution Trends Genet 25 (2009) 351ndash360
[46] MHD Larmuseau A Van Geystelen M van Oven R Decorte Genetic genealogycomes of age ndash perspectives on the use of deep-rooted pedigrees in humanpopulation genetics Am J Phys Anthropol 150 (2013) 505ndash511
[47] M van Oven M Kayser Updated comprehensive phylogenetic tree of globalhuman mitochondrial DNA variation Hum Mutat 30 (2008) E386ndashE394
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
- Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs
-
- Introduction
- Materials and methods
-
- Updated phylogenetic tree
- WGS Y-SNPs dataset
- AMY-tree modifications
- Y-SNP detecting
-
- Results
-
- Updated tree for forensic applications
- Sub-haplogroup determining
- Detecting new Y-SNPs
-
- Discussion
- Conclusions
- Authorsrsquo contributions
- Conflict of interest
- Acknowledgements
- Supplementary data
-
- References
-

1 2 3 4 5 6 7 8 9 10 more
Numbe r of occ urr ences of Y-SNP
0
12000
24000
0
35000
70000
Number of Y-SNPsWhole Y-chromosome
Uniqu e regions Y-chromosome
A
B
All genomes
Excell ent qu ali ty genomes
Fig 5 Number of new Y-SNPs per number of occurrence in the full WGS dataset (A) SNPs in the whole Y-chromosome and (B) SNPs in only the identified unique regions of the
Y-chromosome The grey bars indicate the SNPs in all genomes and the black bars indicate the SNPs from samples with an excellent quality ie MCC 095
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 5
G Model
FSIGEN-977 No of Pages 8
genomes with excellent Y-SNP calling quality The same patterns ofoccurrence for these Y-SNPs are observed as with the non-filteredY-SNPs the majority of the new reported Y-SNPs appeared in onlyone or two samples of the WGS dataset as shown in Fig 5B
The genomes of eight males whose biological fatherrsquos genome isalso sequenced are present in the dataset one family of eightmales including the father the son and the six grandsons next toone fatherndashson pair In the family of eight paternal relatives 5155new SNPs are reported on the whole Y-chromosome and a largenumber of these SNPs is found in only one of the eight individualsas shown in Fig 6A The number of Y-SNPs decreases every time
0
500
1000
1500
2000
2500
0
1 2 3 4 5
Numbe r of o
0
20
40
60
80
100
120
Number of Y-SNPsA
B
8-member fam
All SNPs
Famil y-un iqu e SNP
Fig 6 Number of new Y-SNPs per occurrence in the eight paternally related samples and t
(B) SNPs in only the identified unique regions of the Y-chromosome The grey bars indica
uniquersquo they do not occur in any of the other samples with an excellent quality ie M
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
the number of samples in which the Y-SNP occurs increases exceptfor the occurrence in all-except-one and all samples of the familyWhen all Y-SNPs which also occur in any other sample withexcellent SNP calling quality are removed only less than 20 of theY-SNPs remains but the distribution of the number of Y-SNPs peroccurrence stays the same as the black bars in Fig 6A show Thesame comparison is made when only the SNPs in the unique part ofthe Y-chromosome are selected the same pattern as with all SNPsis visible in Fig 6B However for each number of occurrences thenumber of truly unique SNPs ie SNPs that do not occur in othergenomes outside the family is much higher Within the fatherndashson
6 7 8 1 2
cc urr ences of Y-SNP
Whole Y-chromosome
Uniqu e regions Y-chromosome
il y Father-son pair
s
he fatherndashson pair of Complete Genomics (A) SNPs in the whole Y-chromosome and
te the SNPs found in the family The black bars indicate the SNPs which are lsquofamily-
CC 095
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx6
G Model
FSIGEN-977 No of Pages 8
pair 2181 new SNPs are reported on the whole Y-chromosome forwhich the difference between occurrence in only one and bothgenomes is relatively small also for the proportion of SNPs whichdo not occur in the other genomes with an excellent Y-SNP quality(Fig 6A) Remarkably there are more new SNPs present in bothsamples than in one sample when the Y-SNPs which are notlocated in the unique region of the Y-chromosome are removed(Fig 6B) The effect of the increasing number of unique Y-SNPs thatdo not occur in other genomes as seen with the previous family isalso in the fatherndashson pair present
4 Discussion
The present study realises the first large screening of malegenomes for phylogenetic applications of the Y-chromosomeBased on this screening of 747 male samples an update has beenmade of the AMY-tree software and of a state-of-the-art Y-chromosomal phylogenetic tree was established for forensicscientists Furthermore also recommendations for future sequenc-ing projects dealing with a broader selection panel of Y-chromosomal haplogroup samples and for the validation of newlydetected Y-SNPs are made
First the large screening of the 747 male Y-chromosomesamples revealed that the SNP calling quality of a few samples wasoverestimated in AMY-tree version 10 As these samples belong tosub-haplogroup R-M269 they are very similar to the referencegenome which is used to estimate the SNP calling quality of asample [23] To remove this SNP calling quality overestimation theinfluence of the reference genome is excluded for all samplesbelonging R-M269 with a low SNP call quality That is why severalmodifications are made and implemented in AMY-tree version 11
Second an update of the currently used Y-chromosomalphylogenetic tree was realised based on the large database of747 available samples At the moment this tree is the most state-of-the-art tree applicable for forensic geneticists all Y-SNPs whichare reported in academic publications till today are included andall ambiguous markers are excluded to avoid wrong Y-SNPinterpretations (Table S3) As often the case in the literature thephylogenetic relationship between new Y-SNPs and earlierreported Y-SNPs in the same (sub-)haplogroup are not given Byusing the AMY-tree results it is possible to find out the concretephylogenetic level of each Y-SNP relative to the other alreadyknown SNPs For example Pamjav et al [16] described andgenotyped two new Y-SNPs Z93 and Z280 within R-M198although the exact phylogenetic positions in relationship withthe other lineages within R-M198 were not given The presence ofboth Z93 and Z80 is checked in all samples belonging to the sub-haplogroup R-M198 Z93 occurred in three R-M198 samples andin none of the other samples Also Z280 does not occur in anysample except in one R-M198 sample Therefore both Z93 andZ280 are placed in the phylogenetic tree as sub-haplogroups of R-M198 as shown in Fig 2 The choice is made for a phylogenetic treein table format as described by Van Geystelen et al [23] instead of abranching diagram because the tree is very large and will becomelarger in the future Therefore the table format can be adaptedmore easily than the diagram and it is also more manageable
Not only newly reported Y-SNPs are included in the newupdated tree but also ambiguous Y-SNPs are excluded as they cancomplicate the Y-chromosomal applications for forensic studiesAs previously described [38] the most relevant ambiguous Y-SNPsare recurrent SNPs which have a paralogous distribution along thephylogenetic tree and which have thus mutant alleles in at leasttwo independent Y-chromosomal lineages Based on the screeningof the 747 analysed samples three Y-SNPs are recognised for thefirst time as recurrent (Table S2) There are no other indications forrecurrent mutations based on the present WGS dataset Therefore
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
we may assume in most cases that males which both have themutant allele for a Y-SNP used in the updated tree (Table S3) havereceived this mutant allele from one common ancestor and not byconvergent evolution Next also all Y-SNPs which could not beanalysed by WGS for one reason or another are excluded from theupdated tree although it may be possible to genotype these SNPscorrectly with Sanger sequencing methods For example allhundred E-M2 samples in the dataset did not reveal the mutantallele for Y-SNP V95 as expected by earlier publications [1439]The reason for this remarkable result may be that V95 is not wellvalidated but more likely it is the result of a bad SNP calling in theY-chromosomal region around V95 by the current WGS methodsFor some Y-SNPs also a mutation conversion is found which isdifferent from the reported one For example another ancestraland mutant allele are observed for Y-SNP M426 than reported byRootsi et al [40] Since the Y-chromosome has a very complexorigin it also has a lot of non-unique regions which complicate theanalysis of WGS data [37] The current reference genome GRCh37shows the evolution of the Y-chromosome and its numerousresulting non-unique regions So the reason for this wrong analysisof M426 may be the position of this SNP in one of the non-uniqueregions of the Y-chromosome as defined earlier [2237] To have anunambiguous phylogenetic tree we excluded all the Y-SNPs forwhich no reliable signal with the WGS methods could be found Inthe end an updated Y-chromosomal tree which includes 359 Y-chromosomal lineages and more than 721 Y-SNP markers (TableS3) is obtained and this tree is the basic tool to develop andoptimise Y-SNP-multiplexes for forensic applications [4142]
Third the distribution of the analysed haplogroups perancestral continent in the current dataset corresponds with theknown distributions [34] Nevertheless there is not yet arepresentative set of all phylogenetic sub-haplogroups availableIn total 17 haplogroups and 106 sub-haplogroups are reported inthe analysis of the whole dataset However the SNP calling qualityis low (MCC lt 095) for most of the analysed samples as most dataare obtained from the 1000 Genomes project and therefore thesub-haplogroup determination of these data is not completelyreliable When considering only the samples of excellent quality(MCC 095) 10 haplogroups and 47 sub-haplogroups remain andthis corresponds with only 13 of the total number lineagesdescribed so far Most of the Y-chromosomes are assigned tohaplogroups E O and R Therefore when the set of WGS Y-chromosomes will be enlarged the current phylogenetic tree aswell as the analysis of Wei et al [22] to calibrate the tree can beoptimised
Finally the screening of the dataset also revealed that new insilico and in vitro methods are required to verify new Y-SNPs basedon WGS methods As earlier mentioned [23] a huge number of newY-SNPs are false positives when the genomes were sequenced withlow coverage and consequently the called SNPs will have a lowquality These false positive SNP calls disturb the determination ofthe correct (sub-)haplogroup of the sample consequently theAMY-tree software has to correct for them by applying severaladditional methods in the analysis [23] The high number of falsepositive Y-SNPs ndash even detected in WGS samples with an excellentSNP calling quality ndash is observed by comparing genomes ofpaternal relatives Within the eight paternally related samplesbelonging to one family which are genotyped by CompleteGenomics 949 newly reported Y-SNPs in the full Y-chromosomeand 88 ones in the unique regions of the Y-chromosome are foundin at least one but not all family members (Fig 6) Despite the highcoverage of these genomes and the excellent SNP calling qualitythis is still a very high number of newly reported Y-SNPs which aremost likely to be false based on the mutation rate on the Y-chromosomes calculated based on a deep-rooting pedigree [43]and based on human-chimpanzee comparisons [44] A similar
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 7
G Model
FSIGEN-977 No of Pages 8
conclusion can be made based on the fatherndashson pair in the datasetwhich is also sequenced with a high coverage by CompleteGenomics (Fig 6)
The Y-SNP results show that adding new lineages to the Y-chromosomal phylogenetic tree only based on WGS is not evidentTherefore making tabula rasa and building a new tree based on allWGS Y-chromosomes as done by Wei et al [22] is not an optionwhen the tree is going to be used in forensics Each new Y-SNP andconsequently each Y-chromosomal lineage has to be validatedindependently from the WGS data before adding them to theupdated phylogenetic tree The validation status of many potentialpolymorphic Y-SNPs (gt1 in population) is often unclear but thiscan be resolved by the sharing of genomic data among geneticgenealogists which are interested in finding new Y-SNPs to resolvetheir particular paternal ancestry [45] Therefore this is an area inwhich closer collaborations between amateurs and forensicacademics could prove to be particularly useful [13] Neverthelessit is required that new in silico methods will be designed to selectgood and relevant candidates of Y-SNPs for the validation Forexample an interesting criterion is the position of the Y-SNPs it ismore interesting to validate only the SNPs located in the uniqueregions of the Y-chromosome as there are many non-uniqueregions due to the evolutionary history of this chromosome [37]The lower number of false positive SNP calls in these uniqueregions in comparison with those in the full Y-chromosome isclearly demonstrated in the fatherndashson pair The number of new Y-SNPs reported in only one of the two samples is higher than thenew ones reported in both samples based on the whole Y-chromosome (Fig 6A) in contrast with the situation based on theunique regions of the Y-chromosome (Fig 6B)
5 Conclusions
Based on the largest screening of male genomes with 747samples in total the most up-to-date Y-chromosomal phylogenet-ic tree for forensic applications is compiled Future publicationswhich will report new Y-SNPs have to situate their phylogeneticpositions in this tree to guarantee the continuity between old andnew publications At this moment forensic scientists as well asevolutionary biologists and genetic genealogists are lost in themany reports of newly described Y-chromosomal lineages [46]Therefore initiatives as AMY-tree which optimise the phylogenybased on peer-reviewed publications are required [23] This isalready the case for the mitochondrial genome with the Phylotreeinitiative of van Oven and Kayser [47] Nevertheless to optimisethe current updated phylogenetic tree for the human Y-chromo-some more high quality genomes of a broader set of (sub-)haplogroups than the frequent haplogroups E O and R arerequired Also a higher effort in the validation of reported Y-SNPsby new in silico and in vitro methods is required
Authorsrsquo contributions
Research design amp supervision MHDL programming AVGwriting MHDL amp AVG Commenting on manuscript AVG RD ampMHDL
Conflict of interest
The authors declare no conflict of interest
Acknowledgements
The authors want to thank Tom Wenseleers Manfred KayserJean-Jacques Cassiman Tom Havenith Hendrik Larmuseau and
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
Lucrece Lernout for useful discussions and comments Thanks alsoto Guy Froyen (VIB KU Leuven) Richard Rocca (independentresearcher) Cuiping Pan (Stanford University) and Andreas Keller(Saarland University) for providing us yet unpublished andpublished called SNPs of whole genome sequencing projectsMaarten HD Larmuseau is postdoctoral fellow of the FWO-Vlaanderen (Research Foundation Flanders) This study wasfunded by the KU Leuven BOF Centre of Excellence Financing onlsquoEco and socio evolutionary dynamicsrsquo (Project number PF201007) and on lsquoCentre for Archaeological Sciences 2 (CAS 2) ndash Newmethods for research in demography and interregional exchangersquo
Appendix A Supplementary data
Supplementary data associated with this article can be found in
the online version at httpdxdoiorg101016jfsigen201303010
References
[1] M Kayser Y-chromosomal markers in forensic genetics in RWD Rapley (Ed)Molecular Forensics John Wiley amp Sons Ltd Chichesters 2007 pp 141ndash161
[2] JM Butler Chapter 13 Y-Chromosomal DNA Testing in JM Butler (Ed) Ad-vanced Topics in Forensic DNA Typing Methodology Academic Press London2011 pp 371ndash403
[3] J Chiaroni PA Underhill LL Cavalli-Sforza Y chromosome diversity humanexpansion drift and cultural evolution Proc Natl Acad Sci USA 106 (2009)20174ndash20179
[4] TM Karafet FL Mendez MB Meilerman PA Underhill SL Zegura MFHammer New binary polymorphisms reshape and increase resolution of thehuman Y chromosomal haplogroup tree Genome Res 18 (2008) 830ndash838
[5] MHD Larmuseau N Vanderheyden M Jacobs M Coomans L Larno R DecorteMicro-geographic distribution of Y-chromosomal variation in the central-westernEuropean region Brabant Forensic Sci Int Genet 5 (2011) 95ndash99
[6] F Cruciani B Trombetta C Antonelli R Pascone G Valesini V Scalzi G Vona BMelegh B Zagradisnik G Assum et al Strong intra- and inter-continentaldifferentiation revealed by Y chromosome SNPs M269 U106 and U152 ForensicSci Int Genet 5 (2011) E49ndashE52
[7] MHD Larmuseau J Vanoverbeke G Gielis N Vanderheyden HFM LarmuseauR Decorte In the name of the migrant father ndash analysis of surname originidentifies historic admixture events undetectable from genealogical recordsHeredity 109 (2012) 90ndash95
[8] S Willuweit L Roewer Y chromosome haplotype reference database (YHRD)update Forensic Sci Int Genet 1 (2007) 83ndash87
[9] R Scozzari A Massaia E DrsquoAtanasio NM Myres UA Perego B Trombetta FCruciani Molecular dissection of the basal clades in the human Y chromosomephylogenetic tree Plos ONE 7 (2012) e49170
[10] F Cruciani B Trombetta A Massaia G Destro-Bisol D Sellitto R Scozzari Arevised root for the human Y chromosomal phylogenetic tree the origin ofpatrilineal diversity in Africa Am J Hum Genet 88 (2011) 814ndash818
[11] S Fornarino M Pala V Battaglia R Maranta A Achilli G Modiano A Torroni OSemino SA Santachiara-Benerecetti Mitochondrial and Y-chromosome diversi-ty of the Tharus (Nepal) a reservoir of genetic variation BMC Evol Biol 9 (2009)154
[12] FL Mendez TM Karafet T Krahn H Ostrer H Soodyall MF Hammer Increasedresolution of Y chromosome haplogroup T defines relationships among popula-tions of the Near East Europe and Africa Hum Biol 83 (2011) 39ndash53
[13] LM Sims D Garvey J Ballantyne Improved resolution haplogroup G phylogenyin the Y-chromosome revealed by a set of newly characterized SNPs Plos ONE 4(2009) e5792
[14] B Trombetta F Cruciani D Sellitto R Scozzari A new topology of the human Ychromosome haplogroup E1b1 (E-P2) revealed through the use of newly charac-terized binary polymorphisms PLoS ONE 6 (2011) e16073
[15] MS Jota DR Lacerda JR Sandoval PPR Vieira SS Santos-Lopes R Bisso-Machado VR Paixao-Cortes S Revollo C Paz-Y-Mino R Fujita et al A newsubhaplogroup of native American Y-chromosomes from the Andes Am J PhysAnthropol 146 (2011) 553ndash559
[16] H Pamjav T Feher E Nemeth Z Padar Brief communication new Y-chromo-some binary markers improve phylogenetic resolution within haplogroup R1a1Am J Phys Anthropol 149 (2012) 611ndash615
[17] NM Myres S Rootsi AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova A Pshenichnov B Yunusbayev et al A major Y-chromosomehaplogroup R1b Holocene era founder effect in Central and Western Europe Eur JHum Genet 19 (2011) 95ndash101
[18] S Yan CC Wang H Li SL Li L Jin G Consortium An updated tree of Y-chromosome Haplogroup O and revised phylogenetic positions of mutations P164and PK4 Eur J Hum Genet 19 (2011) 1013ndash1015
[19] T 1000 Genomes Project Consortium An integrated map of genetic variation from1092 human genomes Nature 491 (2012) 56ndash65
[20] DL Altshuler RM Durbin GR Abecasis DR Bentley A Chakravarti AG ClarkFS Collins FM De la Vega P Donnelly M Egholm et al A map of human
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx8
G Model
FSIGEN-977 No of Pages 8
genome variation from population-scale sequencing Nature 467 (2010)1061ndash1073
[21] AT Duggan M Stoneking A highly unstable recent mutation in human mtDNAAm J Hum Genet 9 (2013) 279ndash284
[22] W Wei Q Ayub Y Chen S McCarthy Y Hou I Carbone Y Xue C Tyler-Smith Acalibrated human Y-chromosomal phylogeny based on resequencing GenomeRes 23 (2013) 388ndash395
[23] A Van Geystelen R Decorte MHD Larmuseau AMY-tree an algorithm to usewhole genome SNP calling for Y chromosomal phylogenetic applications BMCGenomics 14 (2013) 101
[24] R Drmanac AB Sparks MJ Callow AL Halpern NL Burns BG Kermani PCarnevali I Nazarenko GB Nilsen G Yeung et al Human genome sequencingusing unchained base reads on self-assembling DNA nanoarrays Science 327(2010) 78ndash81
[25] L-P Wong RT-H Ong W-T Poh X Liu P Chen RQ Li KK-Y Lam NE PillaiK-S Sim H Xu et al Deep whole-genome sequencing of 100 Southeast AsianMalays Am J Hum Genet 92 (2013) 1ndash15
[26] SC Schuster W Miller A Ratan LP Tomsho B Giardine LR Kasson RS HarrisDC Petersen FQ Zhao J Qi et al Complete Khoisan and Bantu genomes fromsouthern Africa Nature 463 (2010) 943ndash947
[27] P Tong JGD Prendergast AJ Lohan SM Farrington S Cronin N Friel DGBradley O Hardiman A Evans JF Wilson et al Sequencing and analysis of anIrish human genome Genome Biol 11 (2010) R91
[28] A Keller A Graefen M Ball M Matzas V Boisguerin F Maixner P Leidinger CBackes R Khairat M Forster et al New insights into the Tyrolean Icemanrsquos originand phenotype as inferred by whole-genome sequencing Nature Commun 3(2012) 698
[29] R Chen GI Mias J Li-Pook-Than LH Jiang HYK Lam R Chen E Miriami KJKarczewski M Hariharan FE Dewey et al Personal omics profiling revealsdynamic molecular and medical phenotypes Cell 148 (2012) 1293ndash1307
[30] JM Rothberg W Hinz TM Rearick J Schultz W Mileski M Davey JH LeamonK Johnson MJ Milgrew M Edwards et al An integrated semiconductor deviceenabling non-optical genome sequencing Nature 475 (2011) 348ndash352
[31] D Pushkarev NF Neff SR Quake Single-molecule sequencing of an individualhuman genome Nat Biotechnol 27 (2009) 847ndash850
[32] M Rasmussen YR Li S Lindgreen JS Pedersen A Albrechtsen I Moltke MMetspalu E Metspalu T Kivisild R Gupta et al Ancient human genomesequence of an extinct Palaeo-Eskimo Nature 463 (2010) 757ndash762
[33] SM Ahn TH Kim S Lee D Kim H Ghang DS Kim BC Kim SY Kim WY KimC Kim et al The first Korean genome sequence and analysis full genomesequencing for a socio-ethnic group Genome Res 19 (2009) 1622ndash1629
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
[34] DA Wheeler M Srinivasan M Egholm Y Shen L Chen A McGuire W He YJChen V Makhijani GT Roth et al The complete genome of an individual bymassively parallel DNA sequencing Nature 452 (2008) U5ndashU872
[35] J Wang W Wang RQ Li YR Li G Tian L Goodman W Fan JQ Zhang J Li JBZhang et al The diploid genome sequence of an Asian individual Nature 456(2008) U1ndashU60
[36] BA Peters BG Kermani AB Sparks O Alferov P Hong A Alexeev Y Jiang FDahl YT Tang J Haas et al Accurate whole-genome sequencing and haplotyp-ing from 10 to 20 human cells Nature 487 (2012) 190ndash195
[37] H Skaletsky T Kuroda-Kawaguchi PJ Minx HS Cordum L Hillier LG Brown SRepping T Pyntikova J Ali T Bieri et al The male-specific region of the human Ychromosome is a mosaic of discrete sequence classes Nature 423 (2003) U2ndashU825
[38] SM Adams TE King E Bosch MA Jobling The case of the unreliable SNPrecurrent back-mutation of Y-chromosomal markers P25 through gene conver-sion Forensic Sci Int 159 (2006) 14ndash20
[39] B Trombetta F Cruciani PA Underhill D Sellitto R Scozzari Footprints of X-to-Y gene conversion in recent human evolution Mol Biol Evol 27 (2010) 714ndash725
[40] S Rootsi NM Myres AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova K Varendi H Sahakyan et al Distinguishing the co-ancestries ofhaplogroup G Y-chromosomes in the populations of Europe and the CaucasusEur J Hum Genet 20 (2012) 1275ndash1282
[41] S Caratti S Gino C Torre C Robino Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application Int J Legal Med 123(2009) 357ndash360
[42] C Bouakaze C Keyser S Amory E Crubezy B Ludes First successful assay of Y-SNP typing by SNaPshot minisequencing on ancient DNA Int J Legal Med 121(2007) 493ndash499
[43] YL Xue QJ Wang Q Long BL Ng H Swerdlow J Burton C Skuce R Taylor ZAbdellah YL Zhao et al Human Y chromosome base-substitution mutation ratemeasured by direct sequencing in a deep-rooting pedigree Curr Biol 19 (2009)1453ndash1457
[44] Y Kuroki A Toyoda H Noguchi TD Taylor T Itoh DS Kim DW Kim SH ChoiIC Kim HH Choi Comparative analysis of chimpanzee and human Y chromo-somes unveils complex evolutionary pathway Nat Genet 38 (2006) 158ndash167
[45] TE King MA Jobling Whatrsquos in a name Y chromosomes surnames and thegenetic genealogy revolution Trends Genet 25 (2009) 351ndash360
[46] MHD Larmuseau A Van Geystelen M van Oven R Decorte Genetic genealogycomes of age ndash perspectives on the use of deep-rooted pedigrees in humanpopulation genetics Am J Phys Anthropol 150 (2013) 505ndash511
[47] M van Oven M Kayser Updated comprehensive phylogenetic tree of globalhuman mitochondrial DNA variation Hum Mutat 30 (2008) E386ndashE394
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
- Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs
-
- Introduction
- Materials and methods
-
- Updated phylogenetic tree
- WGS Y-SNPs dataset
- AMY-tree modifications
- Y-SNP detecting
-
- Results
-
- Updated tree for forensic applications
- Sub-haplogroup determining
- Detecting new Y-SNPs
-
- Discussion
- Conclusions
- Authorsrsquo contributions
- Conflict of interest
- Acknowledgements
- Supplementary data
-
- References
-

A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx6
G Model
FSIGEN-977 No of Pages 8
pair 2181 new SNPs are reported on the whole Y-chromosome forwhich the difference between occurrence in only one and bothgenomes is relatively small also for the proportion of SNPs whichdo not occur in the other genomes with an excellent Y-SNP quality(Fig 6A) Remarkably there are more new SNPs present in bothsamples than in one sample when the Y-SNPs which are notlocated in the unique region of the Y-chromosome are removed(Fig 6B) The effect of the increasing number of unique Y-SNPs thatdo not occur in other genomes as seen with the previous family isalso in the fatherndashson pair present
4 Discussion
The present study realises the first large screening of malegenomes for phylogenetic applications of the Y-chromosomeBased on this screening of 747 male samples an update has beenmade of the AMY-tree software and of a state-of-the-art Y-chromosomal phylogenetic tree was established for forensicscientists Furthermore also recommendations for future sequenc-ing projects dealing with a broader selection panel of Y-chromosomal haplogroup samples and for the validation of newlydetected Y-SNPs are made
First the large screening of the 747 male Y-chromosomesamples revealed that the SNP calling quality of a few samples wasoverestimated in AMY-tree version 10 As these samples belong tosub-haplogroup R-M269 they are very similar to the referencegenome which is used to estimate the SNP calling quality of asample [23] To remove this SNP calling quality overestimation theinfluence of the reference genome is excluded for all samplesbelonging R-M269 with a low SNP call quality That is why severalmodifications are made and implemented in AMY-tree version 11
Second an update of the currently used Y-chromosomalphylogenetic tree was realised based on the large database of747 available samples At the moment this tree is the most state-of-the-art tree applicable for forensic geneticists all Y-SNPs whichare reported in academic publications till today are included andall ambiguous markers are excluded to avoid wrong Y-SNPinterpretations (Table S3) As often the case in the literature thephylogenetic relationship between new Y-SNPs and earlierreported Y-SNPs in the same (sub-)haplogroup are not given Byusing the AMY-tree results it is possible to find out the concretephylogenetic level of each Y-SNP relative to the other alreadyknown SNPs For example Pamjav et al [16] described andgenotyped two new Y-SNPs Z93 and Z280 within R-M198although the exact phylogenetic positions in relationship withthe other lineages within R-M198 were not given The presence ofboth Z93 and Z80 is checked in all samples belonging to the sub-haplogroup R-M198 Z93 occurred in three R-M198 samples andin none of the other samples Also Z280 does not occur in anysample except in one R-M198 sample Therefore both Z93 andZ280 are placed in the phylogenetic tree as sub-haplogroups of R-M198 as shown in Fig 2 The choice is made for a phylogenetic treein table format as described by Van Geystelen et al [23] instead of abranching diagram because the tree is very large and will becomelarger in the future Therefore the table format can be adaptedmore easily than the diagram and it is also more manageable
Not only newly reported Y-SNPs are included in the newupdated tree but also ambiguous Y-SNPs are excluded as they cancomplicate the Y-chromosomal applications for forensic studiesAs previously described [38] the most relevant ambiguous Y-SNPsare recurrent SNPs which have a paralogous distribution along thephylogenetic tree and which have thus mutant alleles in at leasttwo independent Y-chromosomal lineages Based on the screeningof the 747 analysed samples three Y-SNPs are recognised for thefirst time as recurrent (Table S2) There are no other indications forrecurrent mutations based on the present WGS dataset Therefore
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
we may assume in most cases that males which both have themutant allele for a Y-SNP used in the updated tree (Table S3) havereceived this mutant allele from one common ancestor and not byconvergent evolution Next also all Y-SNPs which could not beanalysed by WGS for one reason or another are excluded from theupdated tree although it may be possible to genotype these SNPscorrectly with Sanger sequencing methods For example allhundred E-M2 samples in the dataset did not reveal the mutantallele for Y-SNP V95 as expected by earlier publications [1439]The reason for this remarkable result may be that V95 is not wellvalidated but more likely it is the result of a bad SNP calling in theY-chromosomal region around V95 by the current WGS methodsFor some Y-SNPs also a mutation conversion is found which isdifferent from the reported one For example another ancestraland mutant allele are observed for Y-SNP M426 than reported byRootsi et al [40] Since the Y-chromosome has a very complexorigin it also has a lot of non-unique regions which complicate theanalysis of WGS data [37] The current reference genome GRCh37shows the evolution of the Y-chromosome and its numerousresulting non-unique regions So the reason for this wrong analysisof M426 may be the position of this SNP in one of the non-uniqueregions of the Y-chromosome as defined earlier [2237] To have anunambiguous phylogenetic tree we excluded all the Y-SNPs forwhich no reliable signal with the WGS methods could be found Inthe end an updated Y-chromosomal tree which includes 359 Y-chromosomal lineages and more than 721 Y-SNP markers (TableS3) is obtained and this tree is the basic tool to develop andoptimise Y-SNP-multiplexes for forensic applications [4142]
Third the distribution of the analysed haplogroups perancestral continent in the current dataset corresponds with theknown distributions [34] Nevertheless there is not yet arepresentative set of all phylogenetic sub-haplogroups availableIn total 17 haplogroups and 106 sub-haplogroups are reported inthe analysis of the whole dataset However the SNP calling qualityis low (MCC lt 095) for most of the analysed samples as most dataare obtained from the 1000 Genomes project and therefore thesub-haplogroup determination of these data is not completelyreliable When considering only the samples of excellent quality(MCC 095) 10 haplogroups and 47 sub-haplogroups remain andthis corresponds with only 13 of the total number lineagesdescribed so far Most of the Y-chromosomes are assigned tohaplogroups E O and R Therefore when the set of WGS Y-chromosomes will be enlarged the current phylogenetic tree aswell as the analysis of Wei et al [22] to calibrate the tree can beoptimised
Finally the screening of the dataset also revealed that new insilico and in vitro methods are required to verify new Y-SNPs basedon WGS methods As earlier mentioned [23] a huge number of newY-SNPs are false positives when the genomes were sequenced withlow coverage and consequently the called SNPs will have a lowquality These false positive SNP calls disturb the determination ofthe correct (sub-)haplogroup of the sample consequently theAMY-tree software has to correct for them by applying severaladditional methods in the analysis [23] The high number of falsepositive Y-SNPs ndash even detected in WGS samples with an excellentSNP calling quality ndash is observed by comparing genomes ofpaternal relatives Within the eight paternally related samplesbelonging to one family which are genotyped by CompleteGenomics 949 newly reported Y-SNPs in the full Y-chromosomeand 88 ones in the unique regions of the Y-chromosome are foundin at least one but not all family members (Fig 6) Despite the highcoverage of these genomes and the excellent SNP calling qualitythis is still a very high number of newly reported Y-SNPs which aremost likely to be false based on the mutation rate on the Y-chromosomes calculated based on a deep-rooting pedigree [43]and based on human-chimpanzee comparisons [44] A similar
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 7
G Model
FSIGEN-977 No of Pages 8
conclusion can be made based on the fatherndashson pair in the datasetwhich is also sequenced with a high coverage by CompleteGenomics (Fig 6)
The Y-SNP results show that adding new lineages to the Y-chromosomal phylogenetic tree only based on WGS is not evidentTherefore making tabula rasa and building a new tree based on allWGS Y-chromosomes as done by Wei et al [22] is not an optionwhen the tree is going to be used in forensics Each new Y-SNP andconsequently each Y-chromosomal lineage has to be validatedindependently from the WGS data before adding them to theupdated phylogenetic tree The validation status of many potentialpolymorphic Y-SNPs (gt1 in population) is often unclear but thiscan be resolved by the sharing of genomic data among geneticgenealogists which are interested in finding new Y-SNPs to resolvetheir particular paternal ancestry [45] Therefore this is an area inwhich closer collaborations between amateurs and forensicacademics could prove to be particularly useful [13] Neverthelessit is required that new in silico methods will be designed to selectgood and relevant candidates of Y-SNPs for the validation Forexample an interesting criterion is the position of the Y-SNPs it ismore interesting to validate only the SNPs located in the uniqueregions of the Y-chromosome as there are many non-uniqueregions due to the evolutionary history of this chromosome [37]The lower number of false positive SNP calls in these uniqueregions in comparison with those in the full Y-chromosome isclearly demonstrated in the fatherndashson pair The number of new Y-SNPs reported in only one of the two samples is higher than thenew ones reported in both samples based on the whole Y-chromosome (Fig 6A) in contrast with the situation based on theunique regions of the Y-chromosome (Fig 6B)
5 Conclusions
Based on the largest screening of male genomes with 747samples in total the most up-to-date Y-chromosomal phylogenet-ic tree for forensic applications is compiled Future publicationswhich will report new Y-SNPs have to situate their phylogeneticpositions in this tree to guarantee the continuity between old andnew publications At this moment forensic scientists as well asevolutionary biologists and genetic genealogists are lost in themany reports of newly described Y-chromosomal lineages [46]Therefore initiatives as AMY-tree which optimise the phylogenybased on peer-reviewed publications are required [23] This isalready the case for the mitochondrial genome with the Phylotreeinitiative of van Oven and Kayser [47] Nevertheless to optimisethe current updated phylogenetic tree for the human Y-chromo-some more high quality genomes of a broader set of (sub-)haplogroups than the frequent haplogroups E O and R arerequired Also a higher effort in the validation of reported Y-SNPsby new in silico and in vitro methods is required
Authorsrsquo contributions
Research design amp supervision MHDL programming AVGwriting MHDL amp AVG Commenting on manuscript AVG RD ampMHDL
Conflict of interest
The authors declare no conflict of interest
Acknowledgements
The authors want to thank Tom Wenseleers Manfred KayserJean-Jacques Cassiman Tom Havenith Hendrik Larmuseau and
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
Lucrece Lernout for useful discussions and comments Thanks alsoto Guy Froyen (VIB KU Leuven) Richard Rocca (independentresearcher) Cuiping Pan (Stanford University) and Andreas Keller(Saarland University) for providing us yet unpublished andpublished called SNPs of whole genome sequencing projectsMaarten HD Larmuseau is postdoctoral fellow of the FWO-Vlaanderen (Research Foundation Flanders) This study wasfunded by the KU Leuven BOF Centre of Excellence Financing onlsquoEco and socio evolutionary dynamicsrsquo (Project number PF201007) and on lsquoCentre for Archaeological Sciences 2 (CAS 2) ndash Newmethods for research in demography and interregional exchangersquo
Appendix A Supplementary data
Supplementary data associated with this article can be found in
the online version at httpdxdoiorg101016jfsigen201303010
References
[1] M Kayser Y-chromosomal markers in forensic genetics in RWD Rapley (Ed)Molecular Forensics John Wiley amp Sons Ltd Chichesters 2007 pp 141ndash161
[2] JM Butler Chapter 13 Y-Chromosomal DNA Testing in JM Butler (Ed) Ad-vanced Topics in Forensic DNA Typing Methodology Academic Press London2011 pp 371ndash403
[3] J Chiaroni PA Underhill LL Cavalli-Sforza Y chromosome diversity humanexpansion drift and cultural evolution Proc Natl Acad Sci USA 106 (2009)20174ndash20179
[4] TM Karafet FL Mendez MB Meilerman PA Underhill SL Zegura MFHammer New binary polymorphisms reshape and increase resolution of thehuman Y chromosomal haplogroup tree Genome Res 18 (2008) 830ndash838
[5] MHD Larmuseau N Vanderheyden M Jacobs M Coomans L Larno R DecorteMicro-geographic distribution of Y-chromosomal variation in the central-westernEuropean region Brabant Forensic Sci Int Genet 5 (2011) 95ndash99
[6] F Cruciani B Trombetta C Antonelli R Pascone G Valesini V Scalzi G Vona BMelegh B Zagradisnik G Assum et al Strong intra- and inter-continentaldifferentiation revealed by Y chromosome SNPs M269 U106 and U152 ForensicSci Int Genet 5 (2011) E49ndashE52
[7] MHD Larmuseau J Vanoverbeke G Gielis N Vanderheyden HFM LarmuseauR Decorte In the name of the migrant father ndash analysis of surname originidentifies historic admixture events undetectable from genealogical recordsHeredity 109 (2012) 90ndash95
[8] S Willuweit L Roewer Y chromosome haplotype reference database (YHRD)update Forensic Sci Int Genet 1 (2007) 83ndash87
[9] R Scozzari A Massaia E DrsquoAtanasio NM Myres UA Perego B Trombetta FCruciani Molecular dissection of the basal clades in the human Y chromosomephylogenetic tree Plos ONE 7 (2012) e49170
[10] F Cruciani B Trombetta A Massaia G Destro-Bisol D Sellitto R Scozzari Arevised root for the human Y chromosomal phylogenetic tree the origin ofpatrilineal diversity in Africa Am J Hum Genet 88 (2011) 814ndash818
[11] S Fornarino M Pala V Battaglia R Maranta A Achilli G Modiano A Torroni OSemino SA Santachiara-Benerecetti Mitochondrial and Y-chromosome diversi-ty of the Tharus (Nepal) a reservoir of genetic variation BMC Evol Biol 9 (2009)154
[12] FL Mendez TM Karafet T Krahn H Ostrer H Soodyall MF Hammer Increasedresolution of Y chromosome haplogroup T defines relationships among popula-tions of the Near East Europe and Africa Hum Biol 83 (2011) 39ndash53
[13] LM Sims D Garvey J Ballantyne Improved resolution haplogroup G phylogenyin the Y-chromosome revealed by a set of newly characterized SNPs Plos ONE 4(2009) e5792
[14] B Trombetta F Cruciani D Sellitto R Scozzari A new topology of the human Ychromosome haplogroup E1b1 (E-P2) revealed through the use of newly charac-terized binary polymorphisms PLoS ONE 6 (2011) e16073
[15] MS Jota DR Lacerda JR Sandoval PPR Vieira SS Santos-Lopes R Bisso-Machado VR Paixao-Cortes S Revollo C Paz-Y-Mino R Fujita et al A newsubhaplogroup of native American Y-chromosomes from the Andes Am J PhysAnthropol 146 (2011) 553ndash559
[16] H Pamjav T Feher E Nemeth Z Padar Brief communication new Y-chromo-some binary markers improve phylogenetic resolution within haplogroup R1a1Am J Phys Anthropol 149 (2012) 611ndash615
[17] NM Myres S Rootsi AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova A Pshenichnov B Yunusbayev et al A major Y-chromosomehaplogroup R1b Holocene era founder effect in Central and Western Europe Eur JHum Genet 19 (2011) 95ndash101
[18] S Yan CC Wang H Li SL Li L Jin G Consortium An updated tree of Y-chromosome Haplogroup O and revised phylogenetic positions of mutations P164and PK4 Eur J Hum Genet 19 (2011) 1013ndash1015
[19] T 1000 Genomes Project Consortium An integrated map of genetic variation from1092 human genomes Nature 491 (2012) 56ndash65
[20] DL Altshuler RM Durbin GR Abecasis DR Bentley A Chakravarti AG ClarkFS Collins FM De la Vega P Donnelly M Egholm et al A map of human
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx8
G Model
FSIGEN-977 No of Pages 8
genome variation from population-scale sequencing Nature 467 (2010)1061ndash1073
[21] AT Duggan M Stoneking A highly unstable recent mutation in human mtDNAAm J Hum Genet 9 (2013) 279ndash284
[22] W Wei Q Ayub Y Chen S McCarthy Y Hou I Carbone Y Xue C Tyler-Smith Acalibrated human Y-chromosomal phylogeny based on resequencing GenomeRes 23 (2013) 388ndash395
[23] A Van Geystelen R Decorte MHD Larmuseau AMY-tree an algorithm to usewhole genome SNP calling for Y chromosomal phylogenetic applications BMCGenomics 14 (2013) 101
[24] R Drmanac AB Sparks MJ Callow AL Halpern NL Burns BG Kermani PCarnevali I Nazarenko GB Nilsen G Yeung et al Human genome sequencingusing unchained base reads on self-assembling DNA nanoarrays Science 327(2010) 78ndash81
[25] L-P Wong RT-H Ong W-T Poh X Liu P Chen RQ Li KK-Y Lam NE PillaiK-S Sim H Xu et al Deep whole-genome sequencing of 100 Southeast AsianMalays Am J Hum Genet 92 (2013) 1ndash15
[26] SC Schuster W Miller A Ratan LP Tomsho B Giardine LR Kasson RS HarrisDC Petersen FQ Zhao J Qi et al Complete Khoisan and Bantu genomes fromsouthern Africa Nature 463 (2010) 943ndash947
[27] P Tong JGD Prendergast AJ Lohan SM Farrington S Cronin N Friel DGBradley O Hardiman A Evans JF Wilson et al Sequencing and analysis of anIrish human genome Genome Biol 11 (2010) R91
[28] A Keller A Graefen M Ball M Matzas V Boisguerin F Maixner P Leidinger CBackes R Khairat M Forster et al New insights into the Tyrolean Icemanrsquos originand phenotype as inferred by whole-genome sequencing Nature Commun 3(2012) 698
[29] R Chen GI Mias J Li-Pook-Than LH Jiang HYK Lam R Chen E Miriami KJKarczewski M Hariharan FE Dewey et al Personal omics profiling revealsdynamic molecular and medical phenotypes Cell 148 (2012) 1293ndash1307
[30] JM Rothberg W Hinz TM Rearick J Schultz W Mileski M Davey JH LeamonK Johnson MJ Milgrew M Edwards et al An integrated semiconductor deviceenabling non-optical genome sequencing Nature 475 (2011) 348ndash352
[31] D Pushkarev NF Neff SR Quake Single-molecule sequencing of an individualhuman genome Nat Biotechnol 27 (2009) 847ndash850
[32] M Rasmussen YR Li S Lindgreen JS Pedersen A Albrechtsen I Moltke MMetspalu E Metspalu T Kivisild R Gupta et al Ancient human genomesequence of an extinct Palaeo-Eskimo Nature 463 (2010) 757ndash762
[33] SM Ahn TH Kim S Lee D Kim H Ghang DS Kim BC Kim SY Kim WY KimC Kim et al The first Korean genome sequence and analysis full genomesequencing for a socio-ethnic group Genome Res 19 (2009) 1622ndash1629
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
[34] DA Wheeler M Srinivasan M Egholm Y Shen L Chen A McGuire W He YJChen V Makhijani GT Roth et al The complete genome of an individual bymassively parallel DNA sequencing Nature 452 (2008) U5ndashU872
[35] J Wang W Wang RQ Li YR Li G Tian L Goodman W Fan JQ Zhang J Li JBZhang et al The diploid genome sequence of an Asian individual Nature 456(2008) U1ndashU60
[36] BA Peters BG Kermani AB Sparks O Alferov P Hong A Alexeev Y Jiang FDahl YT Tang J Haas et al Accurate whole-genome sequencing and haplotyp-ing from 10 to 20 human cells Nature 487 (2012) 190ndash195
[37] H Skaletsky T Kuroda-Kawaguchi PJ Minx HS Cordum L Hillier LG Brown SRepping T Pyntikova J Ali T Bieri et al The male-specific region of the human Ychromosome is a mosaic of discrete sequence classes Nature 423 (2003) U2ndashU825
[38] SM Adams TE King E Bosch MA Jobling The case of the unreliable SNPrecurrent back-mutation of Y-chromosomal markers P25 through gene conver-sion Forensic Sci Int 159 (2006) 14ndash20
[39] B Trombetta F Cruciani PA Underhill D Sellitto R Scozzari Footprints of X-to-Y gene conversion in recent human evolution Mol Biol Evol 27 (2010) 714ndash725
[40] S Rootsi NM Myres AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova K Varendi H Sahakyan et al Distinguishing the co-ancestries ofhaplogroup G Y-chromosomes in the populations of Europe and the CaucasusEur J Hum Genet 20 (2012) 1275ndash1282
[41] S Caratti S Gino C Torre C Robino Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application Int J Legal Med 123(2009) 357ndash360
[42] C Bouakaze C Keyser S Amory E Crubezy B Ludes First successful assay of Y-SNP typing by SNaPshot minisequencing on ancient DNA Int J Legal Med 121(2007) 493ndash499
[43] YL Xue QJ Wang Q Long BL Ng H Swerdlow J Burton C Skuce R Taylor ZAbdellah YL Zhao et al Human Y chromosome base-substitution mutation ratemeasured by direct sequencing in a deep-rooting pedigree Curr Biol 19 (2009)1453ndash1457
[44] Y Kuroki A Toyoda H Noguchi TD Taylor T Itoh DS Kim DW Kim SH ChoiIC Kim HH Choi Comparative analysis of chimpanzee and human Y chromo-somes unveils complex evolutionary pathway Nat Genet 38 (2006) 158ndash167
[45] TE King MA Jobling Whatrsquos in a name Y chromosomes surnames and thegenetic genealogy revolution Trends Genet 25 (2009) 351ndash360
[46] MHD Larmuseau A Van Geystelen M van Oven R Decorte Genetic genealogycomes of age ndash perspectives on the use of deep-rooted pedigrees in humanpopulation genetics Am J Phys Anthropol 150 (2013) 505ndash511
[47] M van Oven M Kayser Updated comprehensive phylogenetic tree of globalhuman mitochondrial DNA variation Hum Mutat 30 (2008) E386ndashE394
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
- Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs
-
- Introduction
- Materials and methods
-
- Updated phylogenetic tree
- WGS Y-SNPs dataset
- AMY-tree modifications
- Y-SNP detecting
-
- Results
-
- Updated tree for forensic applications
- Sub-haplogroup determining
- Detecting new Y-SNPs
-
- Discussion
- Conclusions
- Authorsrsquo contributions
- Conflict of interest
- Acknowledgements
- Supplementary data
-
- References
-

A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx 7
G Model
FSIGEN-977 No of Pages 8
conclusion can be made based on the fatherndashson pair in the datasetwhich is also sequenced with a high coverage by CompleteGenomics (Fig 6)
The Y-SNP results show that adding new lineages to the Y-chromosomal phylogenetic tree only based on WGS is not evidentTherefore making tabula rasa and building a new tree based on allWGS Y-chromosomes as done by Wei et al [22] is not an optionwhen the tree is going to be used in forensics Each new Y-SNP andconsequently each Y-chromosomal lineage has to be validatedindependently from the WGS data before adding them to theupdated phylogenetic tree The validation status of many potentialpolymorphic Y-SNPs (gt1 in population) is often unclear but thiscan be resolved by the sharing of genomic data among geneticgenealogists which are interested in finding new Y-SNPs to resolvetheir particular paternal ancestry [45] Therefore this is an area inwhich closer collaborations between amateurs and forensicacademics could prove to be particularly useful [13] Neverthelessit is required that new in silico methods will be designed to selectgood and relevant candidates of Y-SNPs for the validation Forexample an interesting criterion is the position of the Y-SNPs it ismore interesting to validate only the SNPs located in the uniqueregions of the Y-chromosome as there are many non-uniqueregions due to the evolutionary history of this chromosome [37]The lower number of false positive SNP calls in these uniqueregions in comparison with those in the full Y-chromosome isclearly demonstrated in the fatherndashson pair The number of new Y-SNPs reported in only one of the two samples is higher than thenew ones reported in both samples based on the whole Y-chromosome (Fig 6A) in contrast with the situation based on theunique regions of the Y-chromosome (Fig 6B)
5 Conclusions
Based on the largest screening of male genomes with 747samples in total the most up-to-date Y-chromosomal phylogenet-ic tree for forensic applications is compiled Future publicationswhich will report new Y-SNPs have to situate their phylogeneticpositions in this tree to guarantee the continuity between old andnew publications At this moment forensic scientists as well asevolutionary biologists and genetic genealogists are lost in themany reports of newly described Y-chromosomal lineages [46]Therefore initiatives as AMY-tree which optimise the phylogenybased on peer-reviewed publications are required [23] This isalready the case for the mitochondrial genome with the Phylotreeinitiative of van Oven and Kayser [47] Nevertheless to optimisethe current updated phylogenetic tree for the human Y-chromo-some more high quality genomes of a broader set of (sub-)haplogroups than the frequent haplogroups E O and R arerequired Also a higher effort in the validation of reported Y-SNPsby new in silico and in vitro methods is required
Authorsrsquo contributions
Research design amp supervision MHDL programming AVGwriting MHDL amp AVG Commenting on manuscript AVG RD ampMHDL
Conflict of interest
The authors declare no conflict of interest
Acknowledgements
The authors want to thank Tom Wenseleers Manfred KayserJean-Jacques Cassiman Tom Havenith Hendrik Larmuseau and
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
Lucrece Lernout for useful discussions and comments Thanks alsoto Guy Froyen (VIB KU Leuven) Richard Rocca (independentresearcher) Cuiping Pan (Stanford University) and Andreas Keller(Saarland University) for providing us yet unpublished andpublished called SNPs of whole genome sequencing projectsMaarten HD Larmuseau is postdoctoral fellow of the FWO-Vlaanderen (Research Foundation Flanders) This study wasfunded by the KU Leuven BOF Centre of Excellence Financing onlsquoEco and socio evolutionary dynamicsrsquo (Project number PF201007) and on lsquoCentre for Archaeological Sciences 2 (CAS 2) ndash Newmethods for research in demography and interregional exchangersquo
Appendix A Supplementary data
Supplementary data associated with this article can be found in
the online version at httpdxdoiorg101016jfsigen201303010
References
[1] M Kayser Y-chromosomal markers in forensic genetics in RWD Rapley (Ed)Molecular Forensics John Wiley amp Sons Ltd Chichesters 2007 pp 141ndash161
[2] JM Butler Chapter 13 Y-Chromosomal DNA Testing in JM Butler (Ed) Ad-vanced Topics in Forensic DNA Typing Methodology Academic Press London2011 pp 371ndash403
[3] J Chiaroni PA Underhill LL Cavalli-Sforza Y chromosome diversity humanexpansion drift and cultural evolution Proc Natl Acad Sci USA 106 (2009)20174ndash20179
[4] TM Karafet FL Mendez MB Meilerman PA Underhill SL Zegura MFHammer New binary polymorphisms reshape and increase resolution of thehuman Y chromosomal haplogroup tree Genome Res 18 (2008) 830ndash838
[5] MHD Larmuseau N Vanderheyden M Jacobs M Coomans L Larno R DecorteMicro-geographic distribution of Y-chromosomal variation in the central-westernEuropean region Brabant Forensic Sci Int Genet 5 (2011) 95ndash99
[6] F Cruciani B Trombetta C Antonelli R Pascone G Valesini V Scalzi G Vona BMelegh B Zagradisnik G Assum et al Strong intra- and inter-continentaldifferentiation revealed by Y chromosome SNPs M269 U106 and U152 ForensicSci Int Genet 5 (2011) E49ndashE52
[7] MHD Larmuseau J Vanoverbeke G Gielis N Vanderheyden HFM LarmuseauR Decorte In the name of the migrant father ndash analysis of surname originidentifies historic admixture events undetectable from genealogical recordsHeredity 109 (2012) 90ndash95
[8] S Willuweit L Roewer Y chromosome haplotype reference database (YHRD)update Forensic Sci Int Genet 1 (2007) 83ndash87
[9] R Scozzari A Massaia E DrsquoAtanasio NM Myres UA Perego B Trombetta FCruciani Molecular dissection of the basal clades in the human Y chromosomephylogenetic tree Plos ONE 7 (2012) e49170
[10] F Cruciani B Trombetta A Massaia G Destro-Bisol D Sellitto R Scozzari Arevised root for the human Y chromosomal phylogenetic tree the origin ofpatrilineal diversity in Africa Am J Hum Genet 88 (2011) 814ndash818
[11] S Fornarino M Pala V Battaglia R Maranta A Achilli G Modiano A Torroni OSemino SA Santachiara-Benerecetti Mitochondrial and Y-chromosome diversi-ty of the Tharus (Nepal) a reservoir of genetic variation BMC Evol Biol 9 (2009)154
[12] FL Mendez TM Karafet T Krahn H Ostrer H Soodyall MF Hammer Increasedresolution of Y chromosome haplogroup T defines relationships among popula-tions of the Near East Europe and Africa Hum Biol 83 (2011) 39ndash53
[13] LM Sims D Garvey J Ballantyne Improved resolution haplogroup G phylogenyin the Y-chromosome revealed by a set of newly characterized SNPs Plos ONE 4(2009) e5792
[14] B Trombetta F Cruciani D Sellitto R Scozzari A new topology of the human Ychromosome haplogroup E1b1 (E-P2) revealed through the use of newly charac-terized binary polymorphisms PLoS ONE 6 (2011) e16073
[15] MS Jota DR Lacerda JR Sandoval PPR Vieira SS Santos-Lopes R Bisso-Machado VR Paixao-Cortes S Revollo C Paz-Y-Mino R Fujita et al A newsubhaplogroup of native American Y-chromosomes from the Andes Am J PhysAnthropol 146 (2011) 553ndash559
[16] H Pamjav T Feher E Nemeth Z Padar Brief communication new Y-chromo-some binary markers improve phylogenetic resolution within haplogroup R1a1Am J Phys Anthropol 149 (2012) 611ndash615
[17] NM Myres S Rootsi AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova A Pshenichnov B Yunusbayev et al A major Y-chromosomehaplogroup R1b Holocene era founder effect in Central and Western Europe Eur JHum Genet 19 (2011) 95ndash101
[18] S Yan CC Wang H Li SL Li L Jin G Consortium An updated tree of Y-chromosome Haplogroup O and revised phylogenetic positions of mutations P164and PK4 Eur J Hum Genet 19 (2011) 1013ndash1015
[19] T 1000 Genomes Project Consortium An integrated map of genetic variation from1092 human genomes Nature 491 (2012) 56ndash65
[20] DL Altshuler RM Durbin GR Abecasis DR Bentley A Chakravarti AG ClarkFS Collins FM De la Vega P Donnelly M Egholm et al A map of human
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx8
G Model
FSIGEN-977 No of Pages 8
genome variation from population-scale sequencing Nature 467 (2010)1061ndash1073
[21] AT Duggan M Stoneking A highly unstable recent mutation in human mtDNAAm J Hum Genet 9 (2013) 279ndash284
[22] W Wei Q Ayub Y Chen S McCarthy Y Hou I Carbone Y Xue C Tyler-Smith Acalibrated human Y-chromosomal phylogeny based on resequencing GenomeRes 23 (2013) 388ndash395
[23] A Van Geystelen R Decorte MHD Larmuseau AMY-tree an algorithm to usewhole genome SNP calling for Y chromosomal phylogenetic applications BMCGenomics 14 (2013) 101
[24] R Drmanac AB Sparks MJ Callow AL Halpern NL Burns BG Kermani PCarnevali I Nazarenko GB Nilsen G Yeung et al Human genome sequencingusing unchained base reads on self-assembling DNA nanoarrays Science 327(2010) 78ndash81
[25] L-P Wong RT-H Ong W-T Poh X Liu P Chen RQ Li KK-Y Lam NE PillaiK-S Sim H Xu et al Deep whole-genome sequencing of 100 Southeast AsianMalays Am J Hum Genet 92 (2013) 1ndash15
[26] SC Schuster W Miller A Ratan LP Tomsho B Giardine LR Kasson RS HarrisDC Petersen FQ Zhao J Qi et al Complete Khoisan and Bantu genomes fromsouthern Africa Nature 463 (2010) 943ndash947
[27] P Tong JGD Prendergast AJ Lohan SM Farrington S Cronin N Friel DGBradley O Hardiman A Evans JF Wilson et al Sequencing and analysis of anIrish human genome Genome Biol 11 (2010) R91
[28] A Keller A Graefen M Ball M Matzas V Boisguerin F Maixner P Leidinger CBackes R Khairat M Forster et al New insights into the Tyrolean Icemanrsquos originand phenotype as inferred by whole-genome sequencing Nature Commun 3(2012) 698
[29] R Chen GI Mias J Li-Pook-Than LH Jiang HYK Lam R Chen E Miriami KJKarczewski M Hariharan FE Dewey et al Personal omics profiling revealsdynamic molecular and medical phenotypes Cell 148 (2012) 1293ndash1307
[30] JM Rothberg W Hinz TM Rearick J Schultz W Mileski M Davey JH LeamonK Johnson MJ Milgrew M Edwards et al An integrated semiconductor deviceenabling non-optical genome sequencing Nature 475 (2011) 348ndash352
[31] D Pushkarev NF Neff SR Quake Single-molecule sequencing of an individualhuman genome Nat Biotechnol 27 (2009) 847ndash850
[32] M Rasmussen YR Li S Lindgreen JS Pedersen A Albrechtsen I Moltke MMetspalu E Metspalu T Kivisild R Gupta et al Ancient human genomesequence of an extinct Palaeo-Eskimo Nature 463 (2010) 757ndash762
[33] SM Ahn TH Kim S Lee D Kim H Ghang DS Kim BC Kim SY Kim WY KimC Kim et al The first Korean genome sequence and analysis full genomesequencing for a socio-ethnic group Genome Res 19 (2009) 1622ndash1629
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
[34] DA Wheeler M Srinivasan M Egholm Y Shen L Chen A McGuire W He YJChen V Makhijani GT Roth et al The complete genome of an individual bymassively parallel DNA sequencing Nature 452 (2008) U5ndashU872
[35] J Wang W Wang RQ Li YR Li G Tian L Goodman W Fan JQ Zhang J Li JBZhang et al The diploid genome sequence of an Asian individual Nature 456(2008) U1ndashU60
[36] BA Peters BG Kermani AB Sparks O Alferov P Hong A Alexeev Y Jiang FDahl YT Tang J Haas et al Accurate whole-genome sequencing and haplotyp-ing from 10 to 20 human cells Nature 487 (2012) 190ndash195
[37] H Skaletsky T Kuroda-Kawaguchi PJ Minx HS Cordum L Hillier LG Brown SRepping T Pyntikova J Ali T Bieri et al The male-specific region of the human Ychromosome is a mosaic of discrete sequence classes Nature 423 (2003) U2ndashU825
[38] SM Adams TE King E Bosch MA Jobling The case of the unreliable SNPrecurrent back-mutation of Y-chromosomal markers P25 through gene conver-sion Forensic Sci Int 159 (2006) 14ndash20
[39] B Trombetta F Cruciani PA Underhill D Sellitto R Scozzari Footprints of X-to-Y gene conversion in recent human evolution Mol Biol Evol 27 (2010) 714ndash725
[40] S Rootsi NM Myres AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova K Varendi H Sahakyan et al Distinguishing the co-ancestries ofhaplogroup G Y-chromosomes in the populations of Europe and the CaucasusEur J Hum Genet 20 (2012) 1275ndash1282
[41] S Caratti S Gino C Torre C Robino Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application Int J Legal Med 123(2009) 357ndash360
[42] C Bouakaze C Keyser S Amory E Crubezy B Ludes First successful assay of Y-SNP typing by SNaPshot minisequencing on ancient DNA Int J Legal Med 121(2007) 493ndash499
[43] YL Xue QJ Wang Q Long BL Ng H Swerdlow J Burton C Skuce R Taylor ZAbdellah YL Zhao et al Human Y chromosome base-substitution mutation ratemeasured by direct sequencing in a deep-rooting pedigree Curr Biol 19 (2009)1453ndash1457
[44] Y Kuroki A Toyoda H Noguchi TD Taylor T Itoh DS Kim DW Kim SH ChoiIC Kim HH Choi Comparative analysis of chimpanzee and human Y chromo-somes unveils complex evolutionary pathway Nat Genet 38 (2006) 158ndash167
[45] TE King MA Jobling Whatrsquos in a name Y chromosomes surnames and thegenetic genealogy revolution Trends Genet 25 (2009) 351ndash360
[46] MHD Larmuseau A Van Geystelen M van Oven R Decorte Genetic genealogycomes of age ndash perspectives on the use of deep-rooted pedigrees in humanpopulation genetics Am J Phys Anthropol 150 (2013) 505ndash511
[47] M van Oven M Kayser Updated comprehensive phylogenetic tree of globalhuman mitochondrial DNA variation Hum Mutat 30 (2008) E386ndashE394
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
- Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs
-
- Introduction
- Materials and methods
-
- Updated phylogenetic tree
- WGS Y-SNPs dataset
- AMY-tree modifications
- Y-SNP detecting
-
- Results
-
- Updated tree for forensic applications
- Sub-haplogroup determining
- Detecting new Y-SNPs
-
- Discussion
- Conclusions
- Authorsrsquo contributions
- Conflict of interest
- Acknowledgements
- Supplementary data
-
- References
-

A Van Geystelen et al Forensic Science International Genetics xxx (2013) xxxndashxxx8
G Model
FSIGEN-977 No of Pages 8
genome variation from population-scale sequencing Nature 467 (2010)1061ndash1073
[21] AT Duggan M Stoneking A highly unstable recent mutation in human mtDNAAm J Hum Genet 9 (2013) 279ndash284
[22] W Wei Q Ayub Y Chen S McCarthy Y Hou I Carbone Y Xue C Tyler-Smith Acalibrated human Y-chromosomal phylogeny based on resequencing GenomeRes 23 (2013) 388ndash395
[23] A Van Geystelen R Decorte MHD Larmuseau AMY-tree an algorithm to usewhole genome SNP calling for Y chromosomal phylogenetic applications BMCGenomics 14 (2013) 101
[24] R Drmanac AB Sparks MJ Callow AL Halpern NL Burns BG Kermani PCarnevali I Nazarenko GB Nilsen G Yeung et al Human genome sequencingusing unchained base reads on self-assembling DNA nanoarrays Science 327(2010) 78ndash81
[25] L-P Wong RT-H Ong W-T Poh X Liu P Chen RQ Li KK-Y Lam NE PillaiK-S Sim H Xu et al Deep whole-genome sequencing of 100 Southeast AsianMalays Am J Hum Genet 92 (2013) 1ndash15
[26] SC Schuster W Miller A Ratan LP Tomsho B Giardine LR Kasson RS HarrisDC Petersen FQ Zhao J Qi et al Complete Khoisan and Bantu genomes fromsouthern Africa Nature 463 (2010) 943ndash947
[27] P Tong JGD Prendergast AJ Lohan SM Farrington S Cronin N Friel DGBradley O Hardiman A Evans JF Wilson et al Sequencing and analysis of anIrish human genome Genome Biol 11 (2010) R91
[28] A Keller A Graefen M Ball M Matzas V Boisguerin F Maixner P Leidinger CBackes R Khairat M Forster et al New insights into the Tyrolean Icemanrsquos originand phenotype as inferred by whole-genome sequencing Nature Commun 3(2012) 698
[29] R Chen GI Mias J Li-Pook-Than LH Jiang HYK Lam R Chen E Miriami KJKarczewski M Hariharan FE Dewey et al Personal omics profiling revealsdynamic molecular and medical phenotypes Cell 148 (2012) 1293ndash1307
[30] JM Rothberg W Hinz TM Rearick J Schultz W Mileski M Davey JH LeamonK Johnson MJ Milgrew M Edwards et al An integrated semiconductor deviceenabling non-optical genome sequencing Nature 475 (2011) 348ndash352
[31] D Pushkarev NF Neff SR Quake Single-molecule sequencing of an individualhuman genome Nat Biotechnol 27 (2009) 847ndash850
[32] M Rasmussen YR Li S Lindgreen JS Pedersen A Albrechtsen I Moltke MMetspalu E Metspalu T Kivisild R Gupta et al Ancient human genomesequence of an extinct Palaeo-Eskimo Nature 463 (2010) 757ndash762
[33] SM Ahn TH Kim S Lee D Kim H Ghang DS Kim BC Kim SY Kim WY KimC Kim et al The first Korean genome sequence and analysis full genomesequencing for a socio-ethnic group Genome Res 19 (2009) 1622ndash1629
Please cite this article in press as A Van Geystelen et al Updating theon whole genome SNPs Forensic Sci Int Genet (2013) httpdxdo
[34] DA Wheeler M Srinivasan M Egholm Y Shen L Chen A McGuire W He YJChen V Makhijani GT Roth et al The complete genome of an individual bymassively parallel DNA sequencing Nature 452 (2008) U5ndashU872
[35] J Wang W Wang RQ Li YR Li G Tian L Goodman W Fan JQ Zhang J Li JBZhang et al The diploid genome sequence of an Asian individual Nature 456(2008) U1ndashU60
[36] BA Peters BG Kermani AB Sparks O Alferov P Hong A Alexeev Y Jiang FDahl YT Tang J Haas et al Accurate whole-genome sequencing and haplotyp-ing from 10 to 20 human cells Nature 487 (2012) 190ndash195
[37] H Skaletsky T Kuroda-Kawaguchi PJ Minx HS Cordum L Hillier LG Brown SRepping T Pyntikova J Ali T Bieri et al The male-specific region of the human Ychromosome is a mosaic of discrete sequence classes Nature 423 (2003) U2ndashU825
[38] SM Adams TE King E Bosch MA Jobling The case of the unreliable SNPrecurrent back-mutation of Y-chromosomal markers P25 through gene conver-sion Forensic Sci Int 159 (2006) 14ndash20
[39] B Trombetta F Cruciani PA Underhill D Sellitto R Scozzari Footprints of X-to-Y gene conversion in recent human evolution Mol Biol Evol 27 (2010) 714ndash725
[40] S Rootsi NM Myres AA Lin M Jarve RJ King I Kutuev VM Cabrera EKKhusnutdinova K Varendi H Sahakyan et al Distinguishing the co-ancestries ofhaplogroup G Y-chromosomes in the populations of Europe and the CaucasusEur J Hum Genet 20 (2012) 1275ndash1282
[41] S Caratti S Gino C Torre C Robino Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application Int J Legal Med 123(2009) 357ndash360
[42] C Bouakaze C Keyser S Amory E Crubezy B Ludes First successful assay of Y-SNP typing by SNaPshot minisequencing on ancient DNA Int J Legal Med 121(2007) 493ndash499
[43] YL Xue QJ Wang Q Long BL Ng H Swerdlow J Burton C Skuce R Taylor ZAbdellah YL Zhao et al Human Y chromosome base-substitution mutation ratemeasured by direct sequencing in a deep-rooting pedigree Curr Biol 19 (2009)1453ndash1457
[44] Y Kuroki A Toyoda H Noguchi TD Taylor T Itoh DS Kim DW Kim SH ChoiIC Kim HH Choi Comparative analysis of chimpanzee and human Y chromo-somes unveils complex evolutionary pathway Nat Genet 38 (2006) 158ndash167
[45] TE King MA Jobling Whatrsquos in a name Y chromosomes surnames and thegenetic genealogy revolution Trends Genet 25 (2009) 351ndash360
[46] MHD Larmuseau A Van Geystelen M van Oven R Decorte Genetic genealogycomes of age ndash perspectives on the use of deep-rooted pedigrees in humanpopulation genetics Am J Phys Anthropol 150 (2013) 505ndash511
[47] M van Oven M Kayser Updated comprehensive phylogenetic tree of globalhuman mitochondrial DNA variation Hum Mutat 30 (2008) E386ndashE394
Y-chromosomal phylogenetic tree for forensic applications basediorg101016jfsigen201303010
- Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs
-
- Introduction
- Materials and methods
-
- Updated phylogenetic tree
- WGS Y-SNPs dataset
- AMY-tree modifications
- Y-SNP detecting
-
- Results
-
- Updated tree for forensic applications
- Sub-haplogroup determining
- Detecting new Y-SNPs
-
- Discussion
- Conclusions
- Authorsrsquo contributions
- Conflict of interest
- Acknowledgements
- Supplementary data
-
- References
-