denversom.wikispaces.com · web viewlearning objectives: unit ii blood and lymph. week. 5....
TRANSCRIPT

Learning Objectives: Unit II Blood and LymphWeek 5Transfusion Medicine:
1. Describe the basic process of donor qualification and blood collection relating specific steps to blood safety.
a. Volunteer blood donation (no significant monetary incentives) provides the first level of safety (for blood recipients). At time of donation the donor completes: questionnaire about current and past illnesses, surgery, travel, vaccination (donor and recipient safety) and high-risk behavior (recipient safety). Donor is encouraged to call back if they have any symptoms of a virus or other infection after blood donation to prevent the spread of the disease (recipient safety). Donors complete abbreviated physical exam to reaffirm their health: vitals, general appearance, skin and upper extremities, hematocrit screening, and platelet count (only for apheresis platelet donor) (donor and recipient safety). Skin preparation also reduces risk of bacterial contamination of blood products (donor and recipient safety).
2. Identify the basic components derived from blood donation, explain the biological characteristics of each component, and compare the optimal storage environment and storage time for each component.
3. Differentiate the specific indications for each of the basic blood components: packed red cells, fresh frozen plasma, and platelets.
Blood Component
Biological Characteristic Storage Temp
Storage Time Indications
Whole Blood
Red cells well maintained (but platelets and neutrophils degenerate by 24-48 hours). Loss of clotting factors more slowly. HCT: 36-40%. Volume: 500-575 mL
4-6° C 35 days Massive transfusions to replace O2 carrying capacity and blood volume. Must be cross-matched and infused through microaggregate filter over 2-4 hours or more rapidly with acute blood loss.
Packed RBCs (PRBC)
Whole blood without the plasma, special soln added to ↑ATP levels, HCT: 70% in 200-250 mL. Leukoreduced avoid adverse rxn. RBC’s glycerolized and frozen at -80° C for 10 years
4-6° C 35 days or longer (42 days)
Transfused for oxygen carrying capacity, for chronic anemia or acute blood loss, must be cross-matched and administered like whole blood, Not widely used, $$. For transfusion with rare RBC antibodies.
Fresh Frozen Plasma (FFP)
Acellular product with ˃80% of all pro- and anti-coagulant proteins + complement factors
-18° C 1 year Treats coagulopathy related to procoag deficient (DIC, liver failure, vitK def). ABO type specific. Administered over 1-3 hours.
Cryoprecipitate
Made from FFP frozen quickly at -80° C and allowed to sit for 18 hours at 4° C. Centrifuge
-18° C 1 year Low or absent fibrinogen. Replacement for Factor XIII deficiency. ABO type specific
1

and remove cryo-poor plasma. Contains 80-100 U Factor VIII/bag, equivalent vwF, 200-250 mg fibrinogen and ↑ levels of Factor XIII than in plasma.
of compatible. Infuse over 30-45 minutes.
Platelet concentrates
Contain platelets for TX of bleeding. Leukoreduced products avoids adverse rxn.1. random donor unit (RDU): 5x10^10 platelets in 50mL2. apheresis platelet (avoid multiple donors) 3x10^11 platelets in 200-300 mL
22-24° C 5-7 days in gas permeable bags with gentle agitation to prevent platelets from clumping
Bleeding associated with thrombocytopenia and/or platelet dysfunction, ABO type specific/compatible. Infuse over 30-45 minutes.
Granulocyte (WBC) concentrates
Collected by apheresis procedure, 2x10^10-2x10^11 neutrophils (depends if donor received dose of G-CSF), HCT: 3-5%, Volume: 200-300mL. Platelets contaminate.
Room temperature
NO STORAGE, transfused within 8-12 hours
Severe bacterial or fungal infections. ABO, Rh(D) type specific and crossmatch compatible. Infused ˂ 4hours.
4. Discuss the basic blood groups (ABO and Rh) and contrast the different compatibility requirements of basic blood components.
a. Alloantigens: blood groups on the surface of RBCs are antigens which are biochemically and immunologically distinct and inherited independently. Not functionally different. A, B, O and AB are alloantigens for the ABO blood group.
b. These are polysaccharides. Addition of fucose to the H antigen basic sugar chain= O, further addition of N-acetylgalactosamine to H antigen= A and addition of D-galactose= B. 44% of whites are A, 44% are O. By 12 months old, individuals develop naturally occurring agglutinins (IgM or IgG) to antigens they do not express (type A has anti-B)
c. Routine testing for ABO and Rh(D). For RBCs, additional antigens include Rh (C, E); Kell; Lewis (Lea,b); Kidd (Jka,b); Duffy (Fya,b); MNSs.
d. Rh antigens: complicated system consisting of 3 pairs of alleles inherited with low crossover between alleles. C/c D/- E/e
e. Two genes for 3 alleles: RHD, RHCE, (RHAG). Gene frequency: D (84%), C (70%), E (30%), c (80%), e (97%). Routine testing for D (Caucasian: 85% D positive, 15% D negative). Immunogenicity: D˃˃C, E, e, c ˃ others.
2
Black Box: fucose
Black Square: N-acetyl-galactosamin
White Diamond: D-galactose

5. Explain the basic rules of blood administration.a. Pre-transfusion testing for red cell transfusions
i. Donor cells and transfusion recipient are typed for ABO and Rh(D), serum is screened for antibodies other than ABO. Major cross-match completed: recipient’s serum and donor RBC’s are missed and evaluated for agglutination. This all takes about 45 minutes and reduces possibility of immediate hemolytic transfusion reactions. Urgent condition (ie: exsanguinating hemorrhage/trauma): give O, Rh(D) negative, for males and non-childbearing women: give O, Rh(D). Abbreviated testing (˂45 minutes): Do no complete cross-match, risk = 1/17,000.
6. Explain the infectious risks of blood transfusion and describe testing strategies to reduce risks of specific agents.
a. Standard tests completed on all donations include testing for antibodies against and/or antigen for infectious disease. Nucleic acid amplification test (NAT) look for Hepatitis C virus, HIV and West Nile Virus. Family, social, travel and exposure history are asked to screen for Creutzfeldt Jakob Disease (CJD) or variant CJD.
b. CMV transmission is LOW with seronegative or leukoreduced products.c. Chagas disease screening is done based on region of donation (closer to South America
gets checked, ie: TX, FL, CA)d. Screening test for the future: parvovirus, Epstein-Barr Virus, dengue and malaria.e. Donors are deferred for travel to malaria area or defined malaria disease based on
travel and personal medical history. f. Risks: ˂1/100,000 (syphilis), ˂1/1,000,000 (HepA), ˂1/200,000 (HepB), ˂1/2,000,000
(HepC), ˂1/2,000,000 (HIV), ˂1/2,500,000 (HTLV-I/II), ˂1/300,000 (West Nile).
7. Classify the non-infectious adverse events of transfusion, assess a constellation of symptoms and signs of the prototypic reactions, and describe the clinical management.
a. Febrile non-hemolytic transfusion reactioni. Usually caused by leukoagglutinins in recipient cytokines or other biologically1
active compounds.ii. Fever. May also involve chills.
iii. Supportive-antipyretics (Tylenol). Consider leukocyte-poor products for future. Risk per transfusion, 1:200.
b. Allergy/Anaphylactic reactioni. Most causes not identified. In IgA-deficient individuals, reaction occurs as a
result of antibodies to IgA.ii. Itching, hives, occasionally chills and fever. In severe reactions, may see signs of
anaphylaxis: dyspnea, pulmonary edema.iii. Mild to moderate reactions: diphenhydramine. More severe reactions:
epinephrine subcutaneously and steroids intravenously. Risk for mild to moderate allergic reactions, 1:400-1:200; severe anaphylactic reactions, 1:150,000.
c. Immediate hemolytic transfusion reactioni. Preformed alloantibodies (most commonly to ABO) and occasionally
autoantibodies cause rapid intravascular hemolysis of transfused cells with activation of clotting (DIC), activation of inflammatory mediators, and acute renal failure.
3

ii. Fever, chills, nausea, chest pain, back pain, pain at transfusion site, hypotension, dyspnea, oliguria, dark urine.
iii. The risk of this type of reaction overall is low (1:30,000), but the mortality rate is high (up to 40%). Stop the transfusion; maintain renal output with intravenous fluids and diuretics (furosemide or mannitol); treat DIC with heparin; and institute other appropriate supportive measures
d. Delayed hemolytic reactioni. Formation of alloantibodies after transfusion and resultant destruction of
transfused red cells, usually by extravascular hemolysis.ii. Fever, jaundice, anemia. A small percentage may develop chronic hemolysis.
iii. Detection, definition, and documentation (for future transfusions). Supportive care. Risk, 1:2,500.
e. Transfusion related lung injuryi. Acute lung injury occurring within 4 hrs after transfusion. Two sets of factors
interact to produce the syndrome. Patient factors: infection, surgery, cytokine therapy. Blood component factors: lipids, antibodies, cytokines. Two groups of factors interact during transfusion to result in lung injury indistinguishable from ARDS.2
ii. Tachypnea, dyspnea, hypoxia. Diffuse interstitial markings. Cardiac evaluation normal.
iii. May consider younger products: packed red blood cells ≤ 2 weeks, platelets ≤ 3 days, washing components to prevent syndrome. Management: supportive care. Risk, 1:2,000-1:5,000 per transfusion.
f. Dilutional Coagulopathyi. Massive blood loss and transfusion with replacement with fluids or blood
components and deficient clotting factors.ii. Bleeding.
iii. Replacement of clotting factors or platelets with appropriate blood components.
g. Bacterial Contaminationi. Contamination of units results in growth of bacteria or production of clinically
significant levels of endotoxin.ii. Chills, high fever, hypotension, other symptoms of sepsis or endotoxemia.
iii. Stop transfusion; make aggressive attempts to identify organism; provide vigorous supportive medical care including antibiotics. (Sepsis mortality <1/500,000)
h. Iron Overloadi. There is no physiologic mechanism to excrete excess iron. Target organs include
liver, heart, and endocrine organs. In patients receiving red cell transfusions over long periods of time, there is an increase in iron burden.
ii. Signs and symptoms of dysfunctional organs affected by the iron.iii. Chronic administration of iron chelator such as Deferoxamine or Exjade.
i. Graft-vs-Hosti. Lymphocytes from donor transfused in an immune-incompetent host.
ii. Syndrome can involve a variety of organs, usually skin, liver, gastrointestinal tract, and bone marrow.
iii. Preventive management: Irradiation (> 1500 cGy) of cellular blood components transfused to individuals with congenital or acquired immunodeficiency
4

syndromes, intrauterine transfusion, very premature infants, and when donors are relatives of the recipient
Disorders of Granulocyte/Monocyte Numbers1. Identify the basic morphologic features of neutrophils, eosinophils, basophils, monocytes, and
macrophages and explain their production, distribution, and turnover.Cell Type Image DescriptionNeutrophils Produced in marrow, storage pool for host
defense (10-14 days). Released into peripheral blood (6 hr.), then move to tissues. Turnover 1-2 days.Major component of innate immune system: non-specific defense against microbes. Important response to tissue injury.
Eosinophils Produced in bone marrow under influence IL-5. Morphologic features, mitotic and storage pool like neutrophils. Mature cell 12-14 micron dia. Large “eosinophilic” granules, bi-lobed nuclei. Released into peripheral blood, move to external surfaces (tracheobronchial tree, GI tract, etc.). Survive for weeks.Can function as phagocyte.Roles allergies, parasite infection, response to tumors: may be immuno-enhancing or immuno-suppressive.
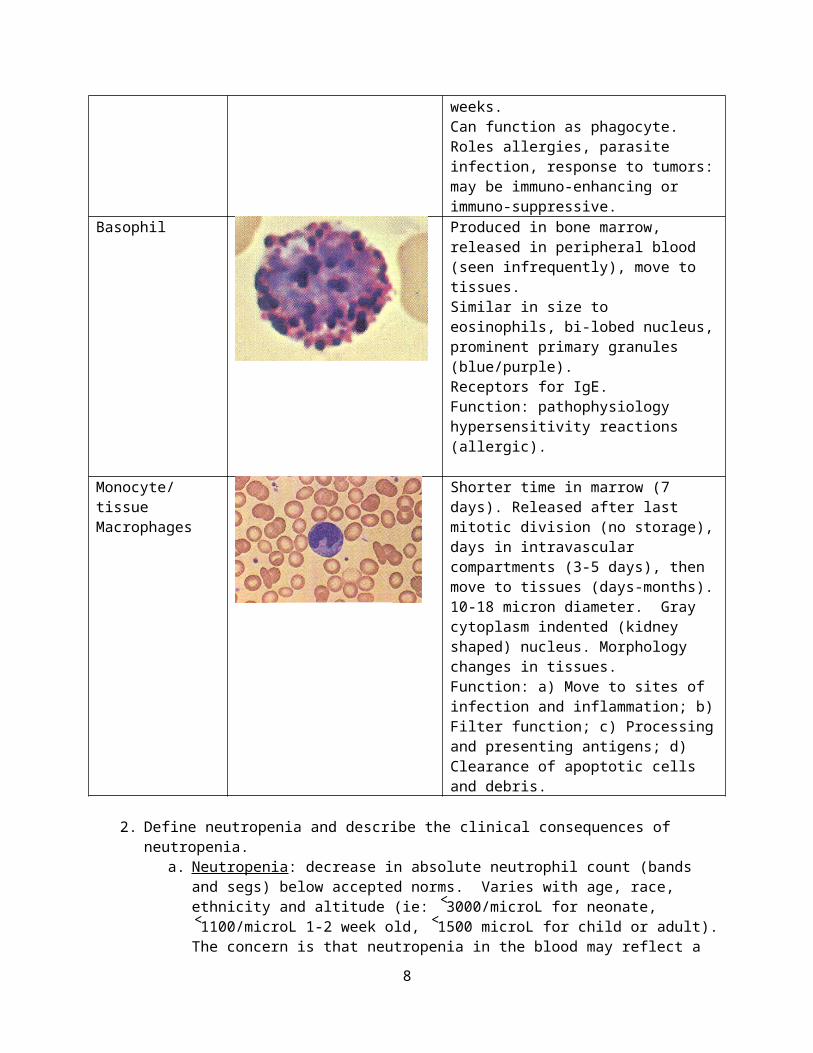
Basophil Produced in bone marrow, released in peripheral blood (seen infrequently), move to tissues.Similar in size to eosinophils, bi-lobed nucleus, prominent primary granules (blue/purple).Receptors for IgE.Function: pathophysiology hypersensitivity reactions (allergic).
Monocyte/tissue Macrophages
Shorter time in marrow (7 days). Released after last mitotic division (no storage), days in intravascular compartments (3-5 days), then move to tissues (days-months).10-18 micron diameter. Gray cytoplasm indented (kidney shaped) nucleus. Morphology changes in tissues.Function: a) Move to sites of infection and inflammation; b) Filter function; c) Processing and presenting antigens; d) Clearance of apoptotic cells and debris.
5

2. Define neutropenia and describe the clinical consequences of neutropenia.a. Neutropenia : decrease in absolute neutrophil count (bands and segs) below accepted
norms. Varies with age, race, ethnicity and altitude (ie: ˂3000/microL for neonate, ˂1100/microL 1-2 week old, ˂1500 microL for child or adult). The concern is that neutropenia in the blood may reflect a decrease in the marrow myeloid pools to such an extent that delivery of neutrophils to infected tissues is decreased enough to shift the balance in favor of the microbes.
i. Values : ANC between normal and 500/microL= mild risk for infection, ˂500/microL = moderate to severe risk for infection and ˂250/microL = very severe risk.
3. Diagram the major causes and differentiate the major acquired or congenital/genetic disorders of neutropenia.
a. Decreased bone marrow productioni. PRIMARY
1. Kostmann Syndrome: severe peripheral neutropenia and decrease in myeloid production beyond promyelocytes leads to high risk for infection and death before age 2 unless patient receives aggressive management. Patients survive to develop myeloid leukemia or myelodysplastic syndrome (MDS). Severe neutropenia, monocytosis, eosinophilia, myeloid hypoplasia, recurrent purulent infections within first few month (S. aureus, E.coli, pseudomonas). Possible mechanism: elastase gene mutation (ELA-1) or HAX-1 gene mutations and early apoptosis of precursors. Inheritance is AR or AD or sporadic. 19p13.3/ELA-2: Heterozygous mutation opposite active site. Many single base mutations.
2. Shwachman Diamond syndrome: neutropenia, pancreatic insufficiency with fat malabsorption, bony abnormalities (metaphyseal chondrodysplasia and Erlenmeyer flask deformities of long bones) and growth delay. 50% have aplastic anemia of MDS/leukemia. Inheritance is AR. Abnormalities=FAS associated apoptosis in precursors and possible defect in nurse cells in marrow stroma providing support for developing myeloid cells. May have defect in SBDS gene (chromosome 7), 3 main mutations, 75% exon 2.
3. Cyclic neutropenia: severe peripheral neutropenia for 5-7 days with 15-25 day cycles. Recurrent fevers, pharyngitis, gingivitis, and mouth ulcers may accompany fevers. Other times during cycle, ANC is normal and there is no risk for infection. Liked to apoptosis of precursors in marrow and mutations in the elastase gene. Inheritance: AR, AD and sporadic. 19p13.3/ELA-2: Mutations cluster near active site. Most single base mutations.
ii. SECONDARY
6

1. Drug-induced : chemotherapy drugs used for malignant conditionsdirect effects on myeloid precursors and stem cells and leaves patient with reduced production and at high risk for infection (other cytopenias present), chloramphenicol also has direct effect on precursors. Drug can cause immune response (penicillin, other antibiotics, day-week onset, acute symptoms, recurrence with small dose), toxic response (phenothiazine, week to months onset, toxicity to cells), or hypersensitivity response (onset weeks to months, associated rash, fever, hepatitis, nephritis, aplastic anemia, direct toxicity to cells).
2. Viral infection: EBV, varicella, measles, CMV, hepatitis, HIV, suppress the bone marrow production or cause an increase in turnover peripherally (related to consumption in tissue or due to antibiotic effects).
3. Nutrtional deficiencies: folate, B12, copper and protein calorie malnutrition can cause ineffective myelopoiesis and neutropenia, other cytopenias present and megaloblastic changes in marrow.
iii. Other: 1. Chediak-Higashi syndrome (AR, 1q42.1-42.2: LYST (CHS1). Variety of
mutations in coding region), specific granule deficiency, glycogenosis Ib (AR, 11q23/G6PT1: point mutations), aminoaciduria, X-linked agammaglobulinemia, hyper-IgM syndrome, WHIM syndrome and idiopathic neutropenia.
b. Increased turnover of neutrophilsi. IMMUNE
1. Chronic benign neutropenia of childhood : production of antibodies that cross react with neutrophils, children (8-11 months) have very low ANC chronically but may increase their counts in association with infection. No increased risk for infection and the neutropenia resolves after 6-54 months, avg: 20 months).
2. Autoimmune neutropenia : antibodies to specific determinants on the neutrophil, in association with lupus, Evan’s Syndrome or Felty’s Syndrome. Antibodies to RBCs, platelets or coagulation proteins may be seen.
3. Alloimmune neutropenia : passive transfer of antibody from mother’s circulation attacking babies cells neutropenia. Transplacental passage of neonatal cells with contain antigens not expressed by maternal cells into maternal circulation sensitizes the mother to produce antibodies against the infant’s antigens. Accumulation of IgG antibodies by the fetus provides a pool of antibodies which bind the infant’s neutrophils and cause neutropenia. May last 2-4 weeks, occasionally 3-4 months.
ii. NON-IMMUNE 1. Infection : most common, usually acute and resolves in days to months.
Mechanism: Increased utilization, Complement mediated margination, Marrow suppression/failure, direct effect, Cytokine/chemokine induced margination, Antibody production. VIRAL CAUSES: Hepatitis, roseola, parvovirus, RSV, HIV, CMV, influenza. BACTERIAL: gram (-) sepsis, brucellosis, tuberculosis, typhoid, tularemia, paratyphoid. FUNGAL: histoplasmosis. PROTOZOAN: malaria, Leishmaniasis.
7
2. Splenomegaly and hypersplenism : related to excessive sequestration of neutrophils in the spleen and may be associated with sequestration of red cells and platelets.
3. Pseudoneutropenia : severe infection, bacterial pathogens and activation of complement (C5a) excessive demargination of neutrophils.
4. Discuss major treatment strategies including growth factors for treating neutropenia.Neutropenia treatment starts with attempting to diagnose the specific type of neutropenia that
is in question. Once this information is garnered, a plan can be formulated targeted at the specific cause of the neutropenia.
With chemotherapy induced neutropenia, severe congenital syndromes, or undiagnosed cases with and ANC<500 and evidence of infection, appropriate cultures should be obtained and prompt administration of broad spectrum antibiotics should be started. Attempts should be made to localize the infection and if the infective agent is identified, a switch to more specific antibiotics should be made.
- G-CSF at a dose of 3-5 μg/kg should be given to help normalize production, increase the neutrophil count in all compartments, and prevent infection. Chronic administration of G-CSF on a daily or every other day basis may be prescribed to prevent future infections.
-If antibody syndromes are implicated, one should consider intravenous gamma globulin (IVIG) as they sometimes yield a response in these cases.
5. Define leukocytosis and provide reasons for a high white blood cell count. Describe the term “left shift” and what it indicates.
Leukocytosis – an increase in the total white blood cell count beyond normal values. The main causes include infection, inflammation, non-specific physiologic stess, or malignancy such as leukemia.
A “left shift” is a term used to refer to a change in the WBC differential that results in an increase in an increase in the number of segs and bands and maybe even metamyelocytes or myelocytes (myeloid precursors) which are usually only found in the marrow.
6. Define eosinophilia, basophilia, and monocytosis, and point out major causes for each.
Eosiniphilia – Absolute count of Eosinophils >350/μl3 main causes: Allergies or allergic disorders (hay fever, asthma, hives)
Parasitic InfectionsDrug Reactions (usually allergic)
Rarer causes = pemphigus, tumors or malignancies, infections like chronic hepatitis
Basophilia – An increase in peripheral basophils- Usually seen in drug or food hypersensitivity- Also seen in urticarial (aka. hives)- Can be seen with infection or inflammation (rheumatoid arthritis,
influenza, varicella, smallpox, TB)- Also seen in myeloproliferative diseases (CML, myeloid metaplasia)
8

Disorders of Granulocytes/Monocyte Function1. Describe the normal functions of neutrophils, including adherence, chemotaxis, ingestion, and
degranulation/microbicidal activity.
a. Neutrophil function is critical to the first response of the host. They move in the laminar flow of blood but are initially pulled to areas of infection by interacting with endothelial cells in a rolling motionfirm adhesion mediated by adhesion proteins. Cells pass through the endothelial junctions (diapedesis) and move
towards offending organisms (chemotaxis). At infection site, microbe (opsonized by C3b or antibody) is enveloped by pseudopods and phagosome is formed encasing the ingested particle. Granules of each class fuse with phagolysosome and oxidase enzyme system is assembled in membrane initiating the respiratory burst and generating ROS (superoxide, hydrogen peroxide). The ROS and oxygen independent mechanisms (defensin, lysozyme, proteases) lead to the death and dissolution of microbe.
9

2. Explain how neutrophil function is affected in the following disorders: Disorder Clinical Functional Defect Molecular DefectLeukocyte Adhesion Deficiency I
ADHERENCE DISORDER
Recurrent soft tissue infections (skin, mucous membranes), gingivitis, periodontitis, cellulitis, abscesses. Delayed separation of umbilical cord because you need neutrophils to infiltrate the cord so it can fall off, but neutrophils can’t go outside capillaries). Poor wound healing
Neutrophilia (cells stuck, can’t get out of circulation to tissues). Decreased adherence to endothelial surface leading to defect in neutrophil movement to infected tissue sites.
Complete or partial deficiency of CD18 resulting in lack of expression of CD11b/CD18. AR.
Leukocyte Adhesion Deficiency II
ADHERENCE DISORDER
Recurrent infections, mental retardation, short stature, craniofacial abnormalities
Neutrophilia decreased rolling on endothelial surfaces as prelude to tight adherence. RBC also affected, abnormal ABH antigens
Abnormal transferase resulting in abnormal fucosylation of adhesion molecules (Sialyl LeX) and poor interaction with selectins. AR. Bombay phenotype.
Actin DysfunctionCHEMOTACTIC DISORDER
Recurrent severe infections, poor wound healing
↓chemotaxis, ↓ingestion
↓actin assembly. AR???
Specific Granule deficiency
GRANULE DEFECT DISORDER
Recurrent skin and deep tissue infections
↓chemotaxis, ↓microbicidal activity
Failure to produce specific granules or their contents, defect in transcription factor results in ↓ production of specific granule proteins. AR.
Myeloperoxidase Generally healthy. Increase Partial or complete Post-translational
10

deficiency
GRANULE DEFECT DISORDER
fungal infections with associated with diabetes
deficiency of myeloperoxidase. Mild defect in killing bacteria, significant defect in Candida killing
modification defect in processing protein. AR.
Chediak-Higashi syndrome
GRANULE DEFECT DISORDER
Oculocutaneous albinism, nystagmus photophobia. Recurrent skin/resp tract infections. Lymphoproliferative phase associated with EBV infection, fever, hepatosplenomegaly and hemophagocytic disorder. Neurodegenerative as adult.
Neutropenia. Giant granules in all leukocytes. Abnormal degranulation. Major defect in movement, also decreased degranulation and microbicidal activity
Alterations in membrane fusion with formation of giant, leaky granules. Metabolic abnormalities in microtubule assembly. CHS gene identified, AR.
Chronic granulomatous disease
BACTERICIDAL ACTIVITY/OXYGEN RADIAL PRODUCTION DISORDER
Recurrent purulent infections with catalase + bacteria (Staph aureus, Aspergillus) and fungi involving skin and mucous membranes. Leads to granulomas (MAC’s cannot eliminate organism) Deep infections of lung, liver, spleen, lymph nodes, bones.
Neutrophilia. Normal adherence, chemotaxis, ingestion and degranulation. Defect in oxidase enzyme system. No toxic oxygen metabolites produced (therefore, cannot kill catalase +). Neutrophils do not produce reactive oxygen radicals.
Defects in 1 of 4 oxidase components. Absent cytochrome b558 associated with gp91 phox (65% pts) sex-linked. P22phox (AR), or absent p47phox (AR), p67phox (AR).
Treatment: prophylactic antibiotics/antifungals, stem cell transplantation
3. Describe the NADPH oxidase enzyme system, techniques used to determine its activity, and the consequences of a defect in one of its components.
a. NADPH oxidase enzyme system is a membrane-bound enzyme complex. Under normal circumstances, the complex is latent in neutrophils and is activated to assemble in the membranes during respiratory burst. The system generates superoxide by
11

transferring electrons from NADPH inside the cell across the membrane and coupling these to molecular oxygen to produce superoxide (a reactive free radical). Superoxide can be make in phagosomes, which contain bacteria/fungi. In phagosome, superoxide can spontaneously form H2O2 that will undergo further reactions to generate ROS’s.
b. Diagnostic tests for NADPH oxidase defect: i. Nitro blue tetrazolium chloride (NBT dye reduction test): in CDG, neutrophil is
unable to make ROS or radicals required for bacterial killing, therefore UNABLE to reduce the dye (no blue), the higher the blue score the better the cell is at producing ROS.
ii. Dihydrorhodamine (DHR) test: whole blood is stained with DHR, incubated and stimulated to produce superoxide radicals (respiratory burst) which reduce DHR to rhodamine in cells with normal function.
c. Defective NADPH oxidase enzyme systems lead to Chronic Granulomatous Diseases (CGD). Low capacity to phagocytose which leads to recurrent infections.
4. Characterize the types of infections you might expect to see with defects of phagocyte function or complement.
a. Defects in Phagocyte Function: i. Very high rates of bacterial and fungal infections, especially with atypical or
unusual microorganisms (Asperigillis, disseminated candida and other gram negative organisms, Cepacia Burkholderi)
ii. Infections of catalase positive organisms in patients with CGD iii. Peridontal disease in children and infections of exceptional severityiv. Recurrent infections in areas of the body that are in contact with the microbial
world on the regular b. Defects in Complement:
i. Similar bacteria infections as might be seen with antibody deficiency (pyogenic organisms, H. Influenzae, S. pneumonia)
ii. Terminal complement deficiencies (C5-C9) see problems with Neisseria organisms
5. Discuss tests which would characterize a phagocyte or complement problem. Differentiate between screening or confirmatory tests.
PhagocytesScreening:
1. CBC, Differential2. Review of morphology3. Bactericidal Activity4. Chemotaxis Assay5. Expression of CD11b/CD186. NBT dye reduction or DHR
oxidation
Confirmatory/Detailed:1. Adherence to inert surface or endothelial cells.
Measure CD11b/CD18, L-selection, Sialyl LeX2. Response to chemoattractants: shape change,
change in direction or rate of movement. Evaluate Actin Assembly.
3. Ingestion of labeled particles or bacteria. Degranulation of specific and azurophilic components.
4. Bactericidal/candidicidal activity. Production of
12

O2-, H2O2, or other oxidants
5. Studies for specific molecular defects in oxidase or other cell constituents
ComplementScreening:
1. C3, CH502. Quantitative Igs3. Lymphocyte numbers
Confirmatory/Detailed:1. Measure specific complement components in
the alternative or classical pathway2. Perform a detailed evaluation of the adaptive
immune response
6. Discuss management strategies for patients with innate immune disorders.
For patients with innate immune disorders management consists of the following:1. Anticipate infections and make AGGRESSIVE attempts to define the causative agent of
the infection.2. Remember that surgical procedures to treat infections can be either diagnostic or
therapeutic or both.3. When an infection is anticipated, the prompt initiation of broad spectrum antibiotics is
critical. A switch to more specific antibiotics can be made once a microbial diagnosis is known.
4. If a sever quantitative disorder of neutrophils is present, G-CSF can be used at a dose of 3 μg/kg/day to resolve neutropenia.
5. Certain syndromes of neutrophil dysfunction (INFγ for CGD) may benefit from prophylactic antibiotics or cytokine therapy.
6. Transplantation with hematopoietic stem cells can reconstitute neutrophil numbers and function
7. Gene therapy is in the preliminary phase of study. Proof of concept has demonstrated reconstitution of neutrophil structure and function, but specific problems still need to be resolved.
Type II Immunopathology1. Describe the molecular and cellular details of the immunologic mechanism by which tissue
damage occurs in a Type II (“cytotoxic antibody”) reaction.a. Type II: due to actions of antibodies directed against a specific target tissue or cell, it is
one of the forms of autoimmunity. b. “Stimulatory hypersensitivity”: if autoantibody is directed against a cell-surface
receptor, it may behave as an agonist, mimicking whatever hormone or factor normally works at that receptor. Example: long-acting thyroid stimulator that mimics TSH and causes hyperthyroidism.
c. Complement-mediated damage: antibody to Ach receptor at neuromuscular endplate (in myasthenia gravia). This antibody may block transmission from nerve to muscle or increase receptor turnover, but ends up destroying the endplate by complement/phagocyte-mediated mechanisms.
d. Tissues can be damaged by lysis (AIHA), phagocytosis (ATP), or by release of phagocytes’ lysosomal enzymes and ROS’s (Goodpasture’s)
2. Give an example of a Type II mechanism disease of muscle, kidney, heart, red cells, platelets, lung, thyroid, pancreatic islets.
13

a. Myasthenia gravis (muscle): progressive muscle weakness because patients make antibody to myson and to ACh receptor (AChr). Antibody to myosin has no role in pathogenesis (only diagnostically). The antibody to the alpha-unit of the AChr does the damage--complement and neutrophil-mediated. The antibody to myosin may be the result of the release of sequestered myosin antigen (secondary to cell damage).
b. Rheumatic heart disease (heart): heart disease after infection with streptococcal pyrogenes due to a cross reaction between a Group A streptococcus M protein antigen and a structure on the heart’s endothelial lining, probably laminin on heart values, followed by neutrophil-mediated tissue destruction. Rheumatic fever is the same disease with widespread manifestations, including skin and CNS.
c. Dressler’s syndrome (heart): persistent cardiac pain, fever, malaise and pericardial ffusion seen after a heart attack or heart surgery. Patients make autoantibody which reacts with the heart pericardial or myocardial antigens immune response. Gets better as heart heals, TX with anti-inflammatory.
d. Goodpasture’s Disease (kidney): formation of autoantibodies to lung and kidney basement membrane (non-living connective tissues framework upon which the endothelial cells of capillaries sit). Type IV collagen is the antigen that is shared between the lung and kidney basement membranes (no other organs involved). Patient has persistent glomerulonephritis and pneumonitis with pulmonary hemorrhages. The antibody is directed against the basement membrane, not trapped as clumps, so the staining by IF is sharp and linear (not lumpy-bumpy as in Type III, immune complex conditions)
e. Autoimmune thrombocytopenia purpura (ATP) (platelets): bleeding abnormality due to destruction of platelets by autoantibody. Platelets are opsonized and their destruction (in the spleen) is rapid. Platelets are needed for blood clotting. TX: suppress immune system and/or remove the spleen. ATP is seen in young healthy people weeks after a viral infection or in older people in association with many other autoantibodies.
f. Autoimmune hemolytic anemia (AIHA) (RBCs): may follow a viral infection or be associated with autoimmune syndrome or cancer. Many drugs (PEN, methyldopa, chlorpromaxine, quinidine) can induce AIHA, usually temporarily. In paroxysmal cold hemoglobinuria (PCH) patient experiences hemolysis after exposure to cold.
g. Systemic lupus erythematosus (multifactorial): patients make antibody to nuclear and nucleolar proteins, DNA, RNA, erythrocytes, clotting factors, platelets, skin and T cells. Antibody to dsDNA is pathogenic and may explain the kidney disease and the facial butterfly rash with local immune complex formation near sun-damaged, DNA releasing skin cells. Incidence in USA: 1/3500 (higher in black, Hispanic and Asian populations).
h. Hashimoto’s thyroiditis (thyroid): inflammatory disease of thyroid where there is B and T cell immunity to various thyroid antigens, including thyroglobulin. The antibodies to thyroid antigens are destructive, not stimulatory. The thyroid is infiltrated with T cells. T cell damage allows antibodies access to normally sequestered antigens makes condition worse. Results in HYPOthyroidism.
i. Graves’ disease: hyperthyroidism causes by long-acting thyroid stimulator (LATS), a IgG antibody to the TSH receptor on thyroid cells. LATS binds to the receptors, it mimics TSH and cause cell to secrete thyroid hormones. Normal feedback cannot occur.
3. Describe the fluorescent antibody tests which would allow you to make the diagnosis of Goodpasture's Syndrome, given: patient's kidney biopsy, normal kidney biopsy, patient's serum, and fluoresceinated goat antisera to human IgG and complement.
14

a. Direct Test : take patient kidney (will already have antibody on its glomerular basement membrane)and add the fluorescently labeled goat antisera to human IgG. If patient has Goodpasture’s, you will observe binding alone the glomerulus because there was already antibody on the kidney that enable the goat anti-human antibody to bind.
b. Indirect Test : take a normal kidney biopsy and place on slide, add patient serum, add fluorescently labeled goal antisera to human IgG. If patient has Goodpasture’s the antibody in their serum with bind with basement membrane on normal biopsy and the fluorescent probe will light up the regions. In Goodpasture’s the antibody is directed against the basement membrane, not trapped in clumps, so the staining is sharp and linear, not “lumpy-bumpy” (as in Type III immune complex conditions)
LUMPY-BUMPY, TYPE III LINEAR, TYPE II
4. Distinguish between the “lumpy-bumpy” and linear immunofluorescent patterns in terms of the most probable immunopathologies they represent.
a. Linear immunofluorescent patterns indicate that the antibody is binding to a specific structure, like the basement membrane. The fluorescent antibody will show a clear structure that its coating. This is type II immunopathology, antibody against self.
b. Lumpy-bumpy patters indicate immune complex pathologies, whereby antigen and antibody clump together and precipitate. They do not line any particular structures, they just bind together in large groups. This is type III immunopathology.
5. Describe how you could tell, using fluorescent antibodies and biopsies of patient's kidney, if Type II or Type III immunopathology was involved. Name the antibodies you would use and the fluorescent patterns you would see.
a. Same as above?????
6. Given patient's serum, fluorescent antibody to human immunoglobulins, and slices of normal kidney, describe how you could tell if the patient's glomerulonephritis was due to Goodpasture's disease or SLE.
a. Add the fluorescent antibody to a preparation of normal kidney and patient serum. If the fluorescent antibodies fluoresced in a linear manner, one would think Type II and be leaning toward GoodPasture’s disease. If the fluorescent antibodies fluoresced in lumpy, bumpy fashion, one would be forced to think Type II and be leaning Systemic Lupus Erythematosus. This is essentially the application of the logic of learning objective #5 distinguishing between Type II and Type III fluorescence.
15

7. Describe how autoimmune disease could result from: The innocent bystander phenomenon, cross-reaction of a foreign antigen with self, coupling self antigen with a foreign antigenic “carrier,” emergence of a forbidden clone, exposure of a sequestered antigen, failure of regulatory T cells.
a. Innocent Bystander – Example is TB – The infectious bacteria are localized in the lungs and thus the lung tissue is damaged as the body tries to rid itself of the infection. The immune system spares nothing in its quest to exterminate foreign beasts, not even itself. Thus, infected tissue can often be damaged as the body seeks to expunge the infective agent, but in doing so does major damage to different parts of the body.
b. Cross-reaction of a foreign antigen with self : Example is Rheumatic Heart Disease. Antibody is produced against some devilish antigen and the antibody does a fine job of cleaning, destroying, and generally obliterating the antigen. However, the antibodies produced against the antigen also have some affinity for some self tissue (in the case of RHD, the heart valves). The antibody attacks the self-tissue, thinking it is “bad,” and all hell breaks loose as the body is being attacked by itself. It should be noted that by the time a patient develops clinical symptoms, the antigen that triggered everything may be long gone. The process is most likely being maintained by continued autoimmune responses to normally-sequestered antigens that are hemorrhaging from damaged cells.
c. Coupling Self Antigen with a Foreign Antigenic “Carrier” – Fundamental Idea is that B-Cells and T-Cells can be switched on by different antigens
i. An anti-self B-cell escapes detection and binds a self protein that happens to have a foreign antigen attached. Normally, the self-protein would not cause Tfh cells to give the B-cell the go ahead to activated, but because the self-protein has foreign protein attached a problem arises. If the B-cell, which bound self, presents the foreign antigen on its surface, T-cells will indeed send the appropriate signals to activate the B-cell thinking that the B-cell has found something foreign. However, the B-cell has actually been activated against the self-protein it originally bound. Again, the fundamental idea is that B-cells don’t necessarily see or get excited about the same antigens/epitopes as T-cells and this can create trouble.
d. Emergence of a Forbidden Clone – Example Myasthenia Gravis – Clones somehow escape the normal clonal abortion mechanisms and mature so that encounters with antigen are immunizing.
e. Exposure of a Sequestered Antigen – Example – Men who get mumps becoming sterilei. Sequestered antigens are usually hidden nice and cozy inside the bellies of their
sequestering beast cells. But, if they are somehow allowed to get out they can immunize cells and lead to an immune response. This usually happens when an immune response in initiated in the region where the antigen is sequestered. Damage to tissue where the cells that have sequestered antigens within reside can lead to the release of sequestered antigens and a further immune response.
f. Failure of a Regulatory T-Cell : It is currently being speculated that an appropriate balance of Th1, Th17, Tfh, Th2, and Treg activity is ESSENTIAL to an appropriate immune response. Some think that the balance can be perturbed so that self/non-self discrimination breaks down and the body starts attacking itself. Recent experiments that cause shifts in the T-cell balance show promise.
8. Identify “Rheumatoid Factor” and describe its molecular nature.
16

a. Rheumatoid Factor (RF) provided the initial evidence that rheumatoid arthritis was autoimmune. RF can be detected by adding a patient’s serum to microscopic beads coated with normal human IgG. In the presence of RF, the beads agglutinate. Thus, RF is IgM anti-IgG. Although it is a useful biomarker, it may not cause much actual joint damage.
9. Name the condition in which antibody stimulates rather than inhibits or harms its target cell. State the common name of the antibody.
a. Graves’ Disease aka. Hyperthyroidism. Long-Acting Thyroid Stimulator: (LATS) is an IgG antibody that mimics TSH and binds the TSH receptor causing the cell to secrete thyroid hormones. The normal feedback controls that work on TSH have no effect on LATS and thus the thyroid is over-stimulated and hyperthyroidism results.
10. Discuss how the Aire gene is involved in preventing autoimmune disease.a. The Aire gene is quite useful in that it causes thymic stromal genes to express a wide
variety of otherwise-inexplicable “out-of-lace” peptides so that reactive T cells may be removed from the repertoire. These out of place peptides are ones that are produced and present in other parts of the body and are made in the thymus for the singular purpose of helping to negatively select anti-self T-cells. Aire is so important that Aire-deficient people develop several auto-immune diseases
Hemostasis I1. Identify the elements that compose the hemostatic system. Understand the basic paradigm for
coagulation factor activation.a. Hemostasis : normal process whereby an injury to a blood vessel triggers a series of
enzymatic reactions resulting in the formation of platelet and fibrin plugs at the site of injury when then stems the loss of blood. Highly controlled process so that clot remains at the site of injury and is not propagated throughout the vascular system. After clot is formed, other enzymes will dissolve the clot gradually as wound healing advances.
b. Components of hemostatic system : coagulation factors (soluble + circulating), platelets (circulating, non-active), endothelium—vessel wall.
c. Coagulation Activation : vessel wall injury, vessel constricts to divert blood flow, exposed collagen triggers accumulation and activation of platelets, exposed tissue factor initiates thrombin generation, von Willebrand factor released from endothelium + bind to collage, thrombin (factor IIa) activates platelets and converts fibrinogen to fibrin, which forms clot. Coagulation factors circulate in the blood as zymogens. Exposure of the sub-endothelial layers initiates cascade in which coagulation factors are cleaved into active forms.
2. Describe the blood clotting pathway. List which components of the system are vitamin K dependent factors. Distinguish the extrinsic and intrinsic pathways and describe the screening tests to measure both (PT and APTT, respectively).
a. Role of Platelets : Collage exposed, platelets adhere (von Willebrand and platelet membrane protein are involved), platelets activated by generation of thrombin at site of injury, as platelets activated the receptors for fibrinogen are exposed. Activated platelets change shape and release ADP, vasoactive amines and form thromboxane A2. Thromboxane contracts smooth muscle and causes vasoconstriction of the vessel to
17

stop leakage of blood. Platelet has receptor for coagulation proteins and forms a surface for coagulation cascade.
b. Clot Formation : sub-endothelial components exposed (collagen, TF, (-) charged surface), procoagulants circulate as zymogens, goal of coagulation is to make thrombin (Factor IIa) to convert fibrinogen to fibrin. Thrombin promotes platelet aggregation, activates Factor V and Factor VIII in cascade, and activates fibrinolytic factors to lyse the clot. The coagulation factors come together to form complexes on phospholipid surfaces (platelet membrane or vessel wall) at site of injury. In each complex there is a large molecular weight cofactor (tissue factor, Factor VIII, Factor V) the orients the enzymes and substrates. Next component is Vitamin K dependent factors and Calcium. With all components: efficiency of the production of activated factors is increased.
c. Vitamin K Dependent Factors (the serine proteases): Factors X, IX, VII, and II (prothrombin). Undergo vitamin K dependent gamma carboxylation. Reaction occurs in the liver.
d. Common Pathway : Xa with V, Ca++ and phospholipid convert prothrombin (II) to thrombin (IIa). Xa is generated by Factors IXa and VIIIa OR by tissue factor and VIIa. “Prothrombinase complex” occurs on the platelet and other membranes. Thrombin has many substrates both procoagulant and anticoagulant.
e. Intrinsic Pathway : INDEPENDENT of VII, contact activation of XII activates XI. XIa activates IX, IXa with VIII activates X.
f. Extrinsic Pathway : precipitating event is exposure of TISSUE FACTOR to blood. Tissue factor is a membrane protein which acts as a cofactor for VIIa. Phospholipid, tissue factor and VIIa, cleaves X or IX to Xa or IXa.
g. Protime (PT) : affected by VII, X, II and fibrinogen.h. Activated partial thromboplastin time (aPTT) : affected by XII, XI, IX, VIII and X.i. Thrombin time (TT) : affected by deficient or abnormal fibrinogen.
18

3. Explain how thrombin acts as the central regulation point for coagulation. Describe normal homeostasis in the coagulation system.
a. Thrombin converts fibrinogen to fibrin, promotes platelet aggregation, activates cofactors, activates fibrinolytic factors and protein C.
b. The activation of clotting cascade and of platelets is inhibited by an intact vascular endothelium and by continuous blood flow which washes away platelets and any activated factors. Also, natural anticoagulant proteins, anti-thrombin III (inactivates thrombin and Factors XII, XI, IX and X). Thrombin can activate protein C (regulatory protein) that cleaves Factors Va and VIIIa (cofactors of coagulation).
4. Explain the function of platelets in hemostasis. Describe the process of platelet aggregation.a. Platelet function:
i. Platelets adhere to the site of vessel injury where collagen is exposed. Plasma protein, von Willebrand factor and a platelet membrane protein are all involved with adherence.
ii. The platelets become activated by generation of thrombin at the site of injury. As activation occurs, receptors for fibrinogen are also exposed.
iii. The platelets change shape, release ADP, vasoactive amines, and form thromboxane A2. Thromboxane contracts smooth muscle and causes vasoconstriction of the vessel at the site of injury helping to stop the leakage of blood.
iv. Platelets have receptors for some of the coagulation proteins and also provide surfaces for the coagulation cascade to occur on end result being increased production of thrombin.
b. Process of platelet aggregation:i. Thrombin promotes platelet aggregation so that while fibrin is being hastily
formed, platelets are slamming into one another and starting to seal the injury in the vessel. Eventually fibrin and platelets form a pretty decent plug that seals the injury entirely. Von Willebrand factor and fibrinogen help the platelets to connect to one another.
5. Explain the contribution and influence of endothelial cells on coagulation.a. Endothelial cells are extremely important in coagulation as they get the entire process
started. If endothelium and endothelial cells are fully intact the clotting cascade is inhibited and continuous blood flow washes away platelets and any factors that were incorrectly activated. However, when an endothelium is damaged the sub-endothelium is exposed and sub-endothelial components such as collagen, tissue factor, or negatively charged surfaces) cause pro-coagulant proteins to initiate the clotting cascade.
6. Describe the regulatory mechanisms of coagulation, i.e., protein C pathway, antithrombin, fibrinolytic pathway.
a. Regulation of the cascade is extremely important as a person does not just want clots to randomly start forming all over the place (ie. thrombosis).
19

b. The initial layer of regulation comes from the endothelium and blood flow. When the endothelium is intact it inhibits the clotting cascade and blood flow draws any mistakenly activated factors or platelets along on their merry way.
c. Protein C Pathway : Activated by thrombin i. Cleaves Factors Va and VIIIa (the cofactors of coagulation) thus helping to stave
off a potential clotting cascadeii. protein S is a cofactor to protein C
d. Anti-thrombin III – a protein that binds with high affinity to heparin and thrombin inactivating them (mostly works inactivating thrombin)
i. Inactivates Factors XII, XI, IX, and X
Fibrinolytic pathway – Fibrin is degraded by the enzyme plasmin e. Plasminogen activated by tissue plasminogen
activator (tPA) to form plasminf. plasmin leads to the degradation of fibrin into
fibrin split products and D-dimerg. Fibrinolysis can be inhibited by plasminogen
activator inhibitors and alpha 2-antiplasmin
Type IV Immunopathology1. Define Type IV immunopathology.
a. T cell mediated immunity and delayed hypersensitivity (in contrast to immediate hypersensitivity), do not require antibody or B-cells, but usually disease involve both Type IV and antibody-mediated phenomena. Examples of Type IV: rejection of allografts, raft vs. host disease (the reverse of allograft rejection), a positive tuberculin skin test, resistance to Mycobacterium tuberculosis, resistance to fungal infections, contact dermatitis, etc.
b. Poison Ivy example: i. You are exposed to poison ivy and the oil (urushiol) penetrates your intact skin
and becomes associated with MHC Class II on dendritic cells (binds directly or binds peptides which then get presented). DC cell travels to draining lymph node and “presents” is MHC to precursors that develop into Th1 and Th17 cells. These cells divide in the usual way, but by the time increased numbers of them are in circulation, the antigen has been washed off or worn off and there is no reaction. This is the initiation phage when you become “immunized” or “sensitized” to poison ivy. You might not know it!
ii. Reencounter with poison ivy: the urushiol again associated with MHC on APCs, but this time there are memory T cells from the expanded clones throughout the body and they get activated in the area where the oil has been deposited. The memory T cells secrete IFNgamma, recruit macrophages and a firm red area of inflammation is visible in 6-12 hours, peaking 24-48 hours later, thus it is DELAYED.
iii. Key point about memory T cells: persisting cells in a clone that was expanded by antigen contact. There are more of them in a sensitized person than in a naïve
20

person. They also have a lower activation threshold so it takes less anatigen to elicit a reaction (compared to the first immunization).
2. Describe the cellular and molecular events following intradermal injection of tuberculin antigen into a person who have cell-mediated immunity to it. Justify calling the process ‘delayed hypersensitivity’. Characterize the cells that would be seen in a 48-hour biopsy of the site with regard to whether T cells or macrophages predominate.
a. Tuberculin skin testing (Mantoux): 0.1 mL of purified protein derivative (PPD) of M. tuberculosis antigens is injected intradermally.
b. TB antigen is taken up by local macrophages and dendritic cells (remember? APCs with Class II MHCs). The APCs “present” the antigen to Th1 cells and if the patient has an increased number of anti-tuberculosis Th1 cells, they will come and get stimulated by the APCs Th1 cells produce IFNgamma and attract more macrophages.
c. The PPD test is read after 48 hours. The diameter of the induration (firm raised part that represents cellular infiltrate—which is FULL of macrophages if a reaction occurred because one Th1 cell can attract 1000 macrophages) is measured and 15 mm is positive, 5-10 mm can even be positive under certain conditions (ie: immunocompromised patient).
d. Keep in mind a person can have a positive PPD if they have been immunized with the Bacille Calmette-Guerin (BCG) vaccine when they were born (done in lots of countries, but not the USA because our kids are not at a high risk of getting TB).
3. Explain why a person usually has no observed symptoms when first exposed to poison ivy.a. The initiation phase of an immune response follows the first exposure to the antigen
and the person is “immunized”. They may not KNOW they have been immunized because by the time the number of T cells are increased throughout the circulation, the antigen (poison ivy oil) has usually been washed off or worn off the skin, thus no red area of inflammation occurs. Their body has done everything correctly, but it just was too slow to elicit a response to the first exposure.
4. Discuss how a chemical or small peptide might not need to be processed through an antigen-presenting cell to be presented by that cell to T cells.
a. Processing of antigen is required to reduce the antigen to an epitope-sized particle capable of interacting with the MHC. If the antigen is already reduced to the size of the epitope, as with small chemicals or peptides, it may associate directly with the MHC without processing. Antigens can also bind to a peptide which is then presented on an MHC.
5. Discuss in principle how T cell immunity could be measured in vitro.a. Whole blood or WBCs can be incubated with antigen in cell culture and activation
observed, look at cell numbers for proliferation, look at cell size for activation “blast transformation”, look at DNA synthesis using radiolabeled precursors, measure cytokines released into the media
b. QuantiFERON-TB: purified M. tuberculosis-specific (human, NOT cow like the PPD) proteins are added to sample of whole blood and after incubation the IFNgamma levels are measured in the medium with ELISA. The test will be negative in people vaccinated
21

with BCG, allowing you to distinguish between infection and previous immunization. The test will be positive if you have sufficient T cells against human M. tuberculosis.
6. Explain why TB skin tests can be administered repeatedly to the same subject.a. The dose of PPD to elicit a positive reaction in an immune person is much lower than
would be required to immunize him/her. Therefore, the TB skin test does NOT immunize a person and they can be repeated with the subject becoming positive.
7. Differentiate between a first-set and second-set graft rejection.This is a similar idea to memory cells during infection. In a “first-set” graft rejection, a recipient
rejects a graft in 10-20 days because 5-10% of the recipient T cells recognize the graft MHCs as self MHC plus antigen, thus becoming activated. During the response anti-graft Th1 and CTLs are boosted and rejection occurs in full force. In a “second-set” graft rejection another skin graft from the same donor is placed upon the same recipient. This time the graft is rejected in 5-10 days. This quicker rejection is a result of memory T-cells recognizing the graft and being able to more quickly up-regulate a response. If another unrelated donor gave a graft, it would be rejected in 10-20 days suggesting that the response is specific.
8. Define hyperacute rejection.A hyperacute rejection is one in which a graft is rejected before it even has time to “heal in.”
This is sometimes called a “white graft” reaction because the graft stays white and bloodless. A hyperacute rejection results from the development of antibodies against histocompatibility antigens. Hyperacute rejections are common in xenografts (from another species) because humans have a pre-existing antibody against ubiquitous carbohydrate epitopes present in many animal species, but not humans.
9. Discuss how autoimmunity can result from environmental exposure to tissues that cross-react with human organs.
The example presented about this concerns the brain. The brain is antigenic, but not immunogenic to its owner because during T-cell development there is not brain protein presented causing the negative selection of anti-brain T-cells. Thus, anti-brain T-cells exist in everyone’s T-cell repertoire, they just are hardly ever stimulated because the brain is well defended and well protected. However, if the T-cells were to somehow get stimulated against the brain, they would attack it. This is believed to have happened in a meat packing plant in Minnesota where several workers charged with the task of blowing the brains out of pig heads developed severe neurological problems. It is believed that the workers inhaled pig brain pieces and their own cells presented them to T-cells as antigens. Because pig brain and human brain proteins are similar, the T-cells that were activated against the pig brain cross-reacted with the human brains of the workers and caused the neurological issues. Mouse studies have shown that this is possible and some researchers believe a mechanism similar to this may be the underlying cause of MS.
10. Discuss the requirements for graft-versus-host disease to occur.Three conditions must be met for graft versus host disease to occur:
1) The graft must contain immunocompetent T-cells (bone marrow does)
2) There must be at least one antigen in the host which the graft’s T-cells can recognize
22

3) The host must be relatively immunoincompetent or unable for some other reason (possibly genetic) to recognize the graft’s MHC antigens. If the host recognized the graft MHCs, the graft would be rejected too quickly for a graft vs. host reaction to occur.
11. Define the graft-versus-leukemia phenomenon.**Quick note: Patients with leukemia undergoing a bone marrow transplant often receive huge doses of radiation that would most likely in themselves be fatal resulting in complete destruction of the bone marrow (myeloablative – complete destruction of bone marrow).
Studies have shown that patients who receive T-depleted allogenic marrow or marrow from themselves during a bone marrow transplant have a higher rate of leukemia relapse than those who get allogenic bone marrow that still has T-cells. Thus, it is believed that graft-versus-leukemia reactions are somehow important for the success of a bone marrow transplant. The mechanisms underlying this phenomenon are not well understood.
12. Speculate on the role of HLA alleles in autoimmunity and chronic inflammatory diseases.It is believe that there is an association between HLA haplotype and autoimmunity. This has
been shown in Type 1 diabetes and celiac disease. The HLA alleles a person inherits determine the ways in which their antigen presenting cells display materials and their T-cells recognize presented materials. Thus, if a person’s HLA has certain amino acids arranged in ways that make them more likely to recognize self as foreign (β-cells in pancreas), that person will be more likely to develop autoimmune disease. Thus, there often is a genetic element in some autoimmune disorders.
Hemostasis Part 2: Defect1. List some of the major congenital or acquired disease states causing bleeding and/or clotting.
Disorder Factor Involved Phenotype Lab Results Notes
Hemophelia A*Most common cause of severe bleeding tendency
Hemophelia B
Factor VIII Deficiency
Factor IX Deficiency
1. Severe HemopheliaLess than 1% factor activity Spontaneous hemorrhage into joints,
muscles, soft tissues, retroperitoneal space, CNS
2. Moderate Hemophelia 2%-5% factor activity Trauma required to cause bleeding May get joint disease3. Mild Hemophelia >10% factor activity While only bleed after trauma, require
specific factor therapy to get resolution of the bleed & prior to surgery
Dx usually after surgery or bad trauma No chronic joint disease
Prolonged PTT Genetics: X-linked
Hemophelia A and B cause identical symptoms – need specific factor assay to distinguish
23

Factor XI Deficiency Factor XI Levels usually >5% so spontaneous bleeding rare
Classic presentation is post-operative hemorrhage
Prolonged PTT Genetics: Autosomal Recessive
Specific factor assay for XI to make Dx
Factor VII Deficiency Factor VII Causes severe bleeding disorder similar to hemophelia.
Classification similar to hemophelia.
Prolonged PT
Normal PTT
Genetics: Autosomal Recessive. Severe patients thought to be homozygous.
von Willebrand Disease
Type 1: vWF deficiency
Type 2: Abnormal vWF protein
Most common mild congenital bleeding disorder
vWF has 2 functions:o Adhere platelets to exposed
collageno Carry Factor VIII
Bleeding from mucosal membranes, nose bleeds, GI bleeds, menorrhagia.
No joint/muscle bleeding, but surgical complications w/o Tx with DDAVP or added Factor
Prolonged Bleeding Time
Decreased Factor VIII
Prolonged PTT if levels low enough to affect Factor VIII carrying capacity
For Dx, also test level of vWF antigen and test function of vWF (ristocetin cofactor)
Genetics: Autosomal Dominant
Disorder Factor Involved Phenotype Lab Results NotesAcquired Factor VIII Inhibitor
*There are also other factor inhibitors, but they are not discussed to any length in the notes
AB against Factor VIII
Soft tissue & muscle bleeding with marked hematomas of the skin or mucosal bleeding
Often these patients have other autoimmune illness, are postpartum, or of advanced age
Prolonged PTT Extremely Rare
Mixing testWhen normal plasma is mixed with the patient’s plasma, the PTT will not correct after two hours of incubation. This is because the antibody in the patient’s plasma will bind
24

and inhibit the normal Factor VIII and the PTT will remain prolonged.
Liver Disease: most factors made in liver, so abnl liver fxn can cause deficiencies of clotting factors
Factor V
Vitamin K-dependent factors: Factor II Factor VII Factor IX Factor X
Fibrinogen (very severe liver disease)
Not given *Prolonged PT*Normal to slightly prolonged PTT*Thrombin Time (if fibrinogen implicated)
Possible decr platelet count
Check to ensure that Vit K deficiencyexacerbating problem
Vit K Deficiency and Warfarin Administration
Vitamin K-dependent factors: Factor II Factor VII Factor IX Factor X
Seen in patients who have no oral intake of Vit K and have gut flora killed off by broad spectrum antibiotics
Prolonged PT
Normal to slightly prolonged PTT
Warfarin interferes with Vit K utilization
Disseminated Intravascular Coagulation (DIC)
Factor VIII
Factor V
Fibrinogen
Platelets
Coag cascade activated in the vascular system fibrin & platelet microthrombi form and plug capillaries
Tissue InfarctionAt same time, consumption of factors/platelets leads to coag factor deficiencies hemorrhage
Markedly fribrinogen level
Low platelet count
Fibrin Split Products (D-Dimers)
Prolonged PTT Prolonged Thrombin Time
If underlying cause corrected, fibrinogen will quickly return to normal and platelet count will rise
Disorder Factor Involved Phenotype Lab Results Notes
25

Anti-phospholipid Antibody Syndrome (APS)
Lupus Anticoagulant: IgG against phospholipids in platelet membrane or endothelial cell
No bleeding tendency
Thrombotic syndrome: DVT, PE, thrombotic stroke, recurrent miscarriage
Prolonged PTT that does not correct upon addition of normal plasma
Can also detect with dilute Russell’s Viper Venom Test
Common acquired abnormality. MUST be diagnosed to distinguish from other prolonged PTT syndromes as Tx is totally different
Antithrombin III Deficiency
Antithrombin III Recurrent venus thrombotic disease Antithrombin III activity
Genetics: Autosomal Dominant
Protein C Deficiency Protein C Recurrent venus thrombotic disease Protein C activity
Genetics: Autosomal Dominant Homozygo
us deficiencies fatal at birth
Protein S Deficiency Protein S Recurrent venus thrombotic disease Protein S free antigen
Genetics: Autosomal Dominant Homozygo
us deficiencies fatal at birth
Factor V Leiden Mutation in Factor V
Factor V not inactivated by protein C APC resistance assay
DNA analysis
More than one hypercoagulable condition required to cause patients to thrombose
2. Explain what the PT/INR, APTT, TT, bleeding time, and PFA are and what they are testing. Provide a differential diagnosis of an abnormal PT/INR, APTT, TT, bleeding time, and PFA. Describe some of the other tests used to evaluate patients with thrombotic or bleeding disorders.
a. Protime/International Normalized Ratio (PT/INR ): Measures extrinsic coagulation pathway. Calcium and thromboplastin (extract of tissue) added to citrated plasma and time to clot is measured in seconds. Thromboplastin varies by manufacturer and the time in seconds is normalized by adjusting for known potency. PT is 10 seconds
26

normally, INR= 1 is normal (INR=5=high chance of bleeding, INR=0.5= high chance of clotting). Prolonged with Factor II, VII, V, X, fibrinogen deficiencies, vitamin K deficiency, liver disease or patient on warfarin. If protime is more prolonged than PTT: liver disease, vit K deficiency, warfarin or rat poison
b. Activated Partial Thromboplastin Time (aPTT ): Measures intrinsic coagulation pathway. Surface activating agent and phospholipid are added to citrated plasma (citrate STOPS coagulation by binding up all the Ca++). Activated plasma is then recalcified and time to form thrombus (clot) is measured in seconds. Measures the entire pathway, EXCEPT Factor VII. Useful for VII, IX, XI and XII deficiencies. Sensitive to inhibition by heparin and fibrin split products. Normal value is ˂32 seconds. Prolonged PTT indicates: Hemophilia (deficiency in factor VIII, IX, or XI), acquired hemophilia, severe von Willebrand disease, heparin in sample (an anticoagulant), fibrin split products. Also can indicated Factor XII deficiency or lupus anticoagulant (both non-bleeding disorders). If PTT is relatively more prolonged than PT: Disseminated intravascular coagulation (DIC)
c. Thrombin Time (TT): excess thrombin is added to plasma, detects low or abnormal fibrinogen, fibrin split products or heparin, fibrinogen activity should also be measured. Normal is ˂18 seconds. Prolonged by: heparin, fibrin degradation products, factor XIII deficiency, and fibrinogen deficiency/abnormality.
d. Bleeding time : measures platelet function, vessel wall and skin integrity, a template device makes a cut on the forearm and time to clot is measured (2-9 minutes is normal). Prolonged time indicates: vascular disorders (thrombocytopenia, DIC, etc) and von Willebrand Disease--not by other coagulation factors such as haemophilia. Aspirin and other cyclooxygenase inhibitors can prolong bleeding time significantly.
e. Platelet Function Analyzer (PFA ): determines an in vitro bleeding time with agonistsf. Fibrinogeng. FSP h. Coagulation Factor Testing
i. Pooled normal plasma is 100% activity, normal are 60-150% activity, severe hemophilia= 0-1% spontaneous bleeds, mild hemophilia= ˃10% bleed with trauma and surgery, carriers=15-18% depends on levels
i. Mixing Test
3. Describe the clinical features and molecular basis for hemophilia A and B, factor VII deficiency, and von Willebrand disease.
i. Hemophilia: X-linked deficiency in Factor VIII (A) or IX (B), males primarily affected and carrier females often symptomatic, elevated PTT is only abnormal screening test. Many are new mutations. Diagnosis: screen males with
27

unexplained hematomas, bruises, bleeding. Complications: soft tissue hemotoma, joint bleeding, CNS bleeds (cause of death), retroperitoneal or psoas bleeds. Assay factor activity to distinguish A from B. Treatment: molecular diagnostics used, recombinant synthetic factor treatment is effective, genetic cure likely.
ii. Factor XI deficiency: PTT prolonged, post-operative bleeding, autosomal recessive, variable incidence in population.
iii. vWD: See above
4. Describe the role of liver disease in coagulopathy.a. Decreased synthesis of most factors, decreased vitamin K dependent carboxylation of II,
VII, IX and X, decreased fibrinogen production, consumption of plateletsb. Prolonged PT/INR (low II, VII, V, X), Prolonged PTT (low IX and X), Prolonged TT (low
fibrinogen and increased fibrinolysis). c. Treatment: replace factors with plasma infusion, augment vit K reactions, recombinant
factor VII (VII has the shortest half life), platelet transfusions
5. Describe disseminated intravascular coagulation (DIC) with its associated conditions. Understand diagnostic testing for DIC and associated conditions.
DIC occurs when the coagulation cascade is activated in the vascular system causing fibrin and platelet microthrombi to form plugs in capillaries which result in tissue infarction while at the same time, some factors and platelets are being consumed. Ultimately, this results in multiple coagulation factor deficiencies and hemorrhage is often the result. DIC can be caused by massive trauma, hemorrhagic or septic shock, amniotic fluid embolism, burns, acute leukemia, and drug reactions.
The most common laboratory findings are that the fibrinogen level has decreased and the platelet count is low. In thinking about the lab, think that the fibrinolytic (break down) system is activated to remove the fibrin-platelet microthrombi resulting in many fibrin split products. These prolong
the PTT assay as well as the thrombin time making these tests markedly prolonged. The key thing to note to distinguish DIC from liver disease is that in DIC the PTT is increased relative to the protime and that the protime is usually the least affected. When the underlying disorder is corrected, the fibrinogen will return to normal and the platelet count will rise. There are several potential clinical consequences of DIC, most of which are related to the production of platelet-fibrin thrombi and the consumption of coagulation factors.
28

6. Explain what a lupus anticoagulant is, how it affects coagulation, and ways to test for it.a. It should be noted first that a minority of people with lupus anticoagulant actually have
lupus. Lupus anticoagulant is a very common acquired abnormality which results in a hypercoaguable state. The “lupus anticoagulant” is an IgG antibody that binds phospholipid in the platelet or endothelial membrane. The presence of lupus anticoagulant results in syndrome called Antiphospholipid Antibody Syndrome (APS).
b. Lupus anticoagulant can be detected because its presence causes a prolonged PTT as a result of the lupus anticoagulant binding up all of the phospholipid used to initiate the PTT reaction. However, patients with lupus anticoagulant do not have a bleeding tendency. They instead have a thrombotic syndrome (though the notes give no details on what might cause this backwards logic). Lupus anticoagulant can cause deep vein thrombosis, pulmonary embolism, thrombotic stokes, and recurrent miscarriage (thrombotic disease of placental blood vessels – Remember the antibody is IgG). If lupus anticoagulant is mixed with a phospholipid source for binding (ie platelets), the antibody will bind to the phospholipid and be pulled out of the plasma. A re-run PTT will show a partial correction. Although the notes do not delve into why, the dilute Russell’s Viper Venom Test is also capable of identifying the lupus anticoagulant’s presence.
c. **Note: It is extremely important to distinguish patients with lupus anticoagulant because these patients have a thrombotic tendency and need to be treated in an entirely different (basically opposite) manner than other patients with prolonged PTT who have a bleeding tendency.
7. Explain how a 1:1 mixing study can distinguish a clotting factor deficiency from an inhibitor of coagulation.
a. *Note: The PTT for a clotting factor deficiency and an inhibitor of coagulation would be prolonged before a mixing test and the mixing test would be used to distinguish between the two.
b. If a clotting factor deficiency is present and the patient’s plasma is mixed 1:1 with normal plasma, the PTT will return to normal (no longer be prolonged) because the clotting factor in the normal plasma will make up for the missing factor in the patient’s blood. However, if an inhibitor of coagulation is present and the patient’s plasma is mixed 1:1 with normal plasma, the PTT will remain prolonged (unchanged) after 2 hours. Thus, a mixing test allows us to distinguish between a coagulation factor deficiency and an inhibitor of coagulation.
Rheumotology: Osteoarthritis 1. Describe the symptoms and signs, synovial fluid analysis, and x-ray features of osteoarthritis.
Symptoms of OA Signs of OA-Pain with use, improved with rest Localized Joint Tenderness-Stiffness – usually <30 mins worth Bony Enlargement – Heberden’s and
Bouchard’s nodes (DIPs and PIPs)- Squaring of 1st metacarpal joint
-Genu varus (bow legged)- Hallux valgus (big toe bunion)
-Cervical and lumbar spine sopndylosis- Relative preservation of function Crepitance (grating sensation or sound)
-Significant symptoms rare at < 40y/o Restricted Movement-Lack of systemic symptoms Variable swelling or instability
29

Synovial fluid analysis: Type I fluid (200-2000 WBCs, 25% Polymorphonuclear leukocytes, normal viscosity)Normal crystal exam and normal glucoseX-Ray Changes:
- Loss of cartilage space, Medial compartment disease of the knee
- Bony sclerosis (hardening, or increased bone deposition) and eburnation (increased bone deposition in areas of damaged cartilage), “Gull-wing” changes in interphalangeal joints, Hallux valgus (big toe bunion) w/o other metatarsal disease- Cystic changes of subchondral bone-Osteophyte formation-Altered shape of bone, horizontal osteophytes of the
spine-Joint effusion – non-inflammatory, decreased joint space superiorly with relative medial preservation in the hip
2. Discuss the risk factors for getting OA.a. ****Advanced Age is one of the strongest risk factors for OA*** b. -75% > 70 y/o have OAc. The overall frequency of OA is equal between males and females, however males are
more likely to get it younger than 45 and females are more likely to get it older than 45. Women also are more likely to get severe disease and more likely to develop Heberden’s or Bouchard’s nodes.
d. Occupational risk factors = conflicting datai. OA of hips, knees, shoulders frequency in miners
ii. OA of hands = frequency in weaversiii. However, some studies have found no increased frequency in other professions
elsewheree. Sports = no increased risk generally (exercise may, in fact, be protective)f. Trauma/ Previous Injury = Increased Risk of OA
30

g. !!!OBESITY!!!h. Genetics –some forms have a genetic predispositioni. Metabolic abnormalities of cartilage – hemochromatosis, Wilson’s disease, ochronosis
3. Explain the various theories on the pathogenesis of OAa. In the most basic understanding of osteoarthritis, the destructive factors overcome
constructive factors resulting in loss of cartilage. The cartilage and the chondrocyte are the main of focus of pathology in osteoarthritis. Cartilage is constantly remodeling by way of destructive factors such as metalloproteinases (collagenase-1, stromelysin-1, and gelatinase) and constructive production of Type II collagen and proteoglycans such as agrecan.
b. The chondrocyte has the ability to synthesize constructive materials such as collagen and proteoglycans. Chondrocytes can also synthesize products that degrade collagen and proteoglycans such as matrix metalloproteinases. Some as yet unknown injury to the chondrocyte occurs, resulting in increased activity of proteases and collagenases in the cartilage matrix. At the same time reparative mechanisms of collagen production and proteoglycan production fail. This results in decreased proteoglycan content, and decreased proteoglycan aggregation. Loss of hydrophilicity leads to decreased elasticity of the cartilage, disruption of collagen fibers and eventual loss of collagen with further damage to the cartilage. As a compensatory mechanism chondrocyte numbers increase initially resulting in hypercellular cartilage with decreased proteoglycan matrix between cells. As the disease progresses the chondrocytes die.
c. There are a number of cytokines and inflammatory mediators that can promote the progression of cartilage damage. These products are released from chondrocytes and synovial cells. Examples are interleukin -1 (IL-1) and tumor necrosis factor α (TNF α) that stimulates matrix metalloproteinases (MMP) and other mediators such as nitric oxide and prostaglandins. Nitric oxide also increases MMP production and causes chondrocyte apoptosis (cell death). Prostaglandins can also increase MMP production.
d. Initially in OA, the water content of cartilage increases. The collagen-proteoglycan network then begins to weaken and loosen its “weave.” Advanced OA may have a proteoglycan content of <50% of normal. In response chondrocytes initially increase in number, but then begin to die off.
e. Trauma or physical forces can cause damage to cartilage and chondrocytes causing them to release degradative enzymes that result in collagen fibrillation and cartilage breakdown.
f. There are hereditary forms of osteoarthritis. Recent data has shown, in some families with particularly early and aggressive forms of osteoarthritis, an abnormal type II collagen. Other abnormal collagens, such as type IX collagen, have also been found. The first report of a mutation in type II collagen was a substitution of one amino acid, cysteine (not found normally in human type II collagen), for another, arginine at position 519. Since then, over 37 mutations of type II collagen have been described.
4. Compare and contrast the aged joint with the OA joint.a. Cartilage is the main focus of pathology is OA. The changes observed in OA represent
the final commonpathway of a number of abnormalities that can occur in the collagen, proteoglycans, matric-proteins including the metalloproteinases and the chondrocytes. We think of OA as a degenerative process with some secondary inflammation, in reality
31

the inflammatory mediators plat a significant role in OA. Cartilage remodels over time because of destructive factors that degrade the components of the ECM and destroy cartilage like metalloproteinase (collagenease-1, stromelysin-1 and gelatinase) AND the constructive production of collagen (mainly Type II collagen) and proteoglycans (aggrecan). Chondrocytes are responsible for destruction and construction. In OA, the destruction ˃ construction.
b. Factors linked to destruction in articular cartilage:i. Focal mechanical stress : trauma, physical force, instability of joint, metabolic
abnormalities, crystal diseases can injure chondrocyte causing it to release degradative enzymes that leads to collagen fibrillation and matrix break down.
ii. Interleukin -1 (IL-1): promotes ECM degreadtion and decreases new matrix formation, specifically promotes degradation of type II collagen and aggrecanby stimulating chondrocytes to make metalloproteases (MMP). Also, stimulates other mediators like prostaglandins (PGE2), nitric oxide (NO) and interleukin-6. IL-1 sustains inflammation and cartilage degradation.
iii. Tumor necrosis factor alpha : similar to IL-1, stimulates production of matrix degrading proteinases and causes cartilage damage.
iv. Other pro-inflammatory involved with cartilage destruction: IL-6, IL-17 (Th17 T cells), IL-18 (macrophages, induces IL-1 and TNFalpha production).
v. Nitric Oxide (NO ): produced by endothelial cells and chondrocytes, it increases MMP production and inhibits proteoglycan synthesis. NO seems to induce chondrocyte apoptosis inhibiting the repair process.
vi. Inhibitory cytokines : IL-4, IL-10, IL-13, IL1 receptor antagonist (IL-1Ra) decrease production and activities of catabolic and proinflammatory cytokines in chondrocytes in vitro and suppresses cartilage destruction in vivo. TGF-beta and insulin-like growth factor maintain cartilage.
5. Discuss the treatment of OA as it relates to the pathophysiology.a. Diagnosis is often made very late in the disease because OA is defined by radiographic
change and symptoms. Are you treating for pain? Functional limitation? Look at age, comorbitity, clinical severity, individual preferences, cost.
b. To prevent cartilage loss: weight loss (if obese), modify activies/occupation, diminish weight bearing with crutches or cane, assistive devices, physical therapy, exercise.
c. Medications: topical agents, non-opioid analgesics (acetaminophen), anti-inflammatory agents, opioid analgesics, nutraceuticals and alternative therapies, intra-articular agents, muscle relaxants.
d. Intra-articular agents: corticosteroids, hyaluronic acide. Nutraceuticals: glucosamine and chondroitin sulfate. f. Surgery: arthroscopic, reparative, reconstruction or total joint replacementg. Alternative therapy: 80% of patients use! leech therapy, S-denosylmethionine (SAMe),
ginger extract, avocado/soybeanunsaponifiables, acupuncture, electromagnetic fields, magnets
Chronic Frustrated Immune Response and Regulation1. Describe the factors that regulate the differentiation of Th0 cells in the Peyer’s Patches to Th1,
Th2, or Th17 versus into Treg cells.a. TGFβ favors Treg : Submucosal Peyer’s Patches have abundant TGFβ which favors the
differentiation of Th0 cells into Treg cells. The dendritic cells in the Peyer’s Patch make
32

IL-10, which lso favors Treg development. Thus, these sites are rite in Treg’s which makes sense since they have bacteria, and immunogens coming through the M cells of the gut epithelium. Also common are Tfh that drive B cells to make IgA so that mucus layer closest to the gut is almost sterile.
b. TGFβ + IL-6 favors Th1 and Th17: TGFβ and IL-6 has been shown to down regulate Treg and up regulate Th1 and Th17. IL-6 is produced by epithelial and other cells in response to stress or damage. Therefore, we have adapted by setting a “cut-off”, above a certain level of stress/damage (IL-6), we switch from Treg to defensive mode with Th1 and Th17 and Th2.
c. TO THINK ABOUT: if you have insufficient Treg cells (the cells that suppress the action of other Th cells and produce potent suppressors: IL-10 and TGFβ) in your gut, lung or skin, you would be constantly responding to harmless things with Th1, Th17 and Th2. This would go on and on and on and on and on because it is IMPOSSIBLE to remove all of the “normal” bugs/organisms in your body. This would be SUPER BAD.
2. Discuss the relative influence of environment and genetics on the risk for inflammatory bowel disease.
a. Inflammatory bowel disease (IBD) includes Crohn’s Disease (affects the large and small intestine, especially terminal ileum, patchy) and ulcerative colitis (superficial in large intestine). Both diseases are thought to involve dysregulated immune responses, perhaps due to commensal bacteria, activated Th1, Th17 and Th2 against normal commensal organisms.
b. Genetically, there are 71 loci associated with risk of Crohn’s, 47 loci associated with risk of ulcerative colitis (identified through Genome Wide Association Studies). 28 of the loci are common between the two condition (NOD2 is one of the loci shared). The loci predict 23% of risk in Crohn’s and 16% in ulcerative colitis. Strong genetic component, but environment and bad luck also play a role. Concordance in monozygotic twin studies is 30-35% for Crohn’s and 10-15% for ulcerative colitis.
3. Discuss the pathogenesis of celiac disease, and the relative role played by antibody and T cells.a. Celiac disease, gluten-sensitive enteropathy, affects 1% of world population, is when the
body decides gluten is dangerous and must be destroyed. Infants present with malabsorption, diarrhea and failure to thrive. In adults, non-specific (anemia, rash, and osteoporosis) symptoms secondary to malabsorption as the villi in the gut atrophy. Diagnostic hallmark: antibody, tissue transglutaminase 2, to gut endomysium (lining that supports smooth muscle layer). This enzyme makes protein crosslinks through glutamines and in some people it couples to and can’t release digestion-resistant glutamine-rich gliadin (wheat) peptides and turns itself into a B cell auto-antigen by the illicit help mechanism. It is T-cell immunity to gliadin peptides that causes chronic inflammation. 90% of patients with celiac disease have HLA-DQ2 and the rest are HLA-DQ8 (these are the acidic and basic configuration of amino acids that allow gluten to be presented). But not everyone with these HLA’s gets celiac disease. Mix of genetics and environment. Treatable—just don’t eat gluten! Gut will revert back to normal!
4. Outline the Hygiene or Old Friends Hypothesis.a. The Hygiene or Old Friends Hypothesis was proposed in an attempt to explain the non-
uniformities in the world-wide increase in allergies and asthma (less of an increase in poor vs rich countries, rural vs urban, equatorial vs. northern). It has been suggested
33

that somehow living outside in the dirt and being exposed to predominate outdoor infections helps the immune system mature normally.
b. The more recent “Old Friends Hypothesis” posits a more complicated and evidence based theory saying that certain harmless microorgansims (namely non-TB mycobacteria, lactobacilli, and helminth worms) have been in humans for so long that they help to instruct our immune systems not to overreact to commensals or low-grade pathogens (ie. they help in the development of the correct number of Tregs). However, if during some critical stage in development (0-2 y/o?) a person does no interact with these harmless microorganisms, they may not develop the correct number of Tregs and be too ready activate a strong Th1, Th2 or Th17 response to an organism that poses a mere paltry threat or no threat at all.
5. Discuss the idea that switching Th1 to Th2 responses may be a way to treat certain autoimmune diseases.
a. Studies have shown that lowering the affinity of the tripartite MHC-peptide-TCR reaction shifts T cell responses from Th1 to Th2. That is, somehow altering the affinity of MHC for antigen can change the type of T-cell that respond. In mice a study showed that if mice with the arthritis associated allele HLA-DR-1 were immunized with the peptide collagen II, a Th1 response was stimulated and they would develop symptoms similar to rheumatoid arthritis. However, if mice were immunized with an altered peptide ligand (APL) that had 2 altered MHC-binding anchor peptides from collagen II (lowering affinity 100 fold), a Th2 response was obtained. Immunizing the mice with the analog and collagen II together actually prevented arthritis. Thus, somehow changing the binding affinity of ligands for MHC can alter the type of T-cell that responds and may ultimately be able to prevent or treat autoimmune diseases.
6. Define altered peptide ligands, and comment on their possible uses in the future.a. Altered peptide ligands (APL) – immunogenic peptides in which the T-cell receptor
contact sites have been manipulated. Another definition may be immunogenic peptide analogs in which MHC-binding anchor peptides are altered, thereby changing the affinity for MHC.
b. A very appealing aspect of the potential of APLs is the focus on a specific antigenic response rather than global immune suppression. APLs show huge potential for treatment of autoimmune diseases if they can be shown to not only prevent disease, but also work once a disease is established. This will require looking at if a T-cell response has already begin, can the type of T-cell responding still be altered (Th1 to Th2, where does Treg act?).
Pharmacology of Anticoagulation Therapy1. Describe the mechanism of action and pharmacokinetics of heparin and low molecular weight
heparins, and differences in management of patients on these therapies.**Heparin is naturally found in mast cell granules, extracted from pig intestine and cow lung for therapeutic usage.
Unfractionated Heparin Low Molecular Weight Heparins (LMWH)—lovenox, enoxaparin, dalteparin, nadroparin
Fondaparinux (Aristra)
34

Structure
Proteoglycan containing covalently linked sulfated polysaccharide chains of various lengths. Mean MW=12000 daltons. It has highest (-) charge density of any macromolecule making it “sticky” with poor pharmacokinetics and poor bioavailability.
Produced by chemical or enzymatic depolymerization of heparin to 1/3 the size (4500 daltons), smaller so has better pharmacokinetics.
Synthetic pentasaccharide corresponding to minimal sequence in heparin for binding anti-thrombin.
Mechanism of Action
Binds to antithrombin III (a natural protease inhibitor in plasma that inactivates coagulation factors). **Only heparin with a least 18 saccharide units (˃5400 daltons) can bind antithrombin/thrombin complex**Heparin bound to antithrombin increases rate of thrombin INactivation by 1000X. Heparin accelerates rate of decay of IXa, Xa, XIIa by antithrombin III. This all prevents conversion of fibrinogen to fibrin. Once thrombin is bound to antithrombin/heparin complex, heparin is released to be recycled.
Selectively binds to and inactivates factor Xa by antithrombin. CANNOT inhibit thrombin by antithrombin.
Pharmacokineti
NOT ABSORBED FROM GI. Must be given IV (immediate) or SQ (delayed). Poor
Given SQ and effects are prolonged, longer t1/2. Better bioavailability. More predictable dose
35

cs pharmacokinetics, poor bioavailability. Short t1/2
(1-5 hr). Unpredictable dose response. This is all due to HIGH (-) charge. Requires in-patient care + monitoring. Drug of choice during pregnancy (does NOT cross placenta)
response. Outpatient (requires less monitoring).
Usage Venous thrombosis, pulmonary embolism (rapid onset). Used with oral anticoagulants (warfarin) and fibrinolytics. Fondaparinux approved for this use. Management of unstable angina or acute MI. During/after coronary angioplasty or stent placement. During surgery requiring cardiopulm bypass. Kidney dialysis.
2. Describe the complications associated with heparin therapy, including excessive bleeding and heparin-induced thrombocytopenia with associated thrombosis.
a. Bleedingi. Usually the anticoagulant effect of heparin wears off within hours of
discontinuing the drug. If life-threatening hemorrhage occurs, the effects of heparin and LMWH, can be reversed with PROTAMINE SULFATE, a positively charged compound that neutralizes heparin (binds up all the (-) charge).
b. Heparin-induced thrombocytopenia syndrome (HIT)i. In 3-5% of patients, 5-10 days after heparin use, their platelet count decreases
by ˃50%. Caused by antibodies to platelet factor 4/heparin complexes. Antibodies bind to and activate platelets resulting in prothrombotic state and clots develop: venous thromboembolism, arterial thrombosis, MI and stroke. Thrombocytopenia is less common with LMWH.
c. Allergic eventi. Due to contaminant oversulfated chondroitin sulfate that is rarely, if ever, found
in nature (what happened in China when large recall occurred)ii. Activation of the contact system (production of bradykinin and complement
activation)iii. Development of improved screening methods prevents this.
3. Describe the alternative anticoagulant therapies used for patients with heparin-induced thrombocytopenia.
a. Direct thrombin inhibitors: i. Argatroban (Novastan), a small molecule inhibitor
ii. Lepirudin (Refludan), recombinant form of hirubdin—anticoagulant from leeches.
4. Describe the mechanism of action, pharmacokinetic and uses of oral anticoagulant warfarin.a. Warfarin (Coumadin) is the most commonly used oral anti-coagulant. It is a derivative
of dicumarol (discovered as component in spoiled sweet clover that hemorrhage in cows). Initially made for rodent control. Nomenclature: Wisconsin Alumni Research Foundation. It is a vitamin K analogue.
b. Mechanism of Action: Inhibits enzymes that use vitamin K as a cofactor. The coagulation proteins that undergo vitamin K-dependent gamma carboxylation of N-terminal glutamates are: II, VII, IX and X. Warfarin inhibits that recycling of vitamin K to the reduced form (by reductases), thus vitamin K is depleted and the coagulation proteins that need vitamin K for gamma carboxylation cannot bind Ca++ and are non-functional.
36

c. Pharmacokinetics : Rapidly absorbed (90 minutes), good bioavailability, long half-life (36-48 hours). Disadvantage: Full antithrombotic effect of warfarin isn’t achieved until existing coagulation factors in circulation are removed (requires 2-3 days) because only NEWLY synthesized coagulation factors will be effected (SLOW ONSET).
d. Usage : prevent venous thromboembolism (in combo with heparin—which acts rapidly), systemic embolism in patients with prosthetic heart valves or atrial fibrillation, stroke, recurrent infarction or death in patients with acute MI.
5. Describe the adverse effects and potential complications associated with use of warfarin.a. Warfarin is an anti-coagulant. Thus, one of the major potential adverse affects is
hemorrhage. If hemorrhage occurs, warfarin administration is stopped, Vitamin K is administered, and sometimes plasma is transfused to replace coagulation factors. Without the replacement of coagulation factors it can take 24-48 hours for coagulation factors to be synthesized after warfarin administration is stopped.
b. Warfarin CANNOT be used in pregnancy as the drug crosses the placenta and is teratogenic (causes malformation of the embryo).
c. Warfarin also has quite a few drug-drug or drug-food interactions. They include:Drugs that Increase Warfarin action Drugs that Decrease Warfarin Action
Inhibitors of platelet function (aspirin) Increase metabolism by inducing metabolic liver enzymes (barbituates, rifampin)Decrease vitamin K synthesis by intestinal
microbes (Antibiotics)Displace warfarin from plasma proteins
(increase [Free Warfarin] in blood) –clofibrate and phenytoin
Decrease Warfarin absorption across GI tract (cholestyramine)
Reduce metabolism and elimination of warfarin by liver (cimetidine, amiodorone,
phenylbutazone)
37

6. Describe the relationship between mechanisms of action and speed of onset of action of heparin and oral anticoagulants
a. Heparin had a rapid onset of action because it works by binding antithrombin III, an inactivator of thrombin. Thrombin is an essential component of the coagulation cascade. Therefore, once it is inactivated by heparin bound antithrombin III, no coagulation can occur. Heparin is rapid because it causes the inactivation of a direct component of the coagulation cascade.
b. The oral anticoagulant warfarin has a slower onset of action because it acts on an enzyme that is part of the external machinery of the coagulation cascade (the enzyme that causes vitamin K reduction). Warfarin prevents Vitamin K from being reduced thereby preventing new coagulation factors, which require vitamin K mediated gamma carboxylation, from being manufactured. However, because it does not act on a direct component of the coagulation cascade, warfarin has a slower onset of action than other drugs. This is because it takes a bit of time for the coagulation cofactors that were present in circulation before warfarin administration to be degraded.
c. The new oral anticoagulants dabigatran etexilate and rivaroxaban have rapid rates of onset because they act directly to inhibit components of the coagulation cascade.
7. Describe the mechanisms of action and uses of fibrinolytic agents.a. Fibrinolytic agents work by converting plasminogen to plasmin, a protease that
degrades fibrin and fibrin clots.
-t-PA – binds to fibrin and increases cleavage of plasminogen to plasmin
-Urokinase – converts plasminogen to plasmin, but does not bind fibrin
-Streptokinase – forms a complex with plasminogen, which converts plasmin to plasminogen
Fibrinolytic agents are used to treat acute myocardial infarction (AMI) in combination with aspirin (Must be given within 6 hrs ideally and a max of 12 hrs after eent). They also are used to treat ischemic stroke (given within 3 hrs. of event), deep vein thrombosis (combo w/ warfarin/heparin), and pulmonary embolism.
8. Describe the mechanisms of action and uses of antiplatelet agents.a. Aspirin: inhibits formation of platelet products
i. Works by irreversibly inactivating cyclooxygenase preventing thromboxane A2 formation by platelets. Thromboxane A2 normally stimulates platelet activation
38

and aggregation. Aspirin’s effect is permanent and lasts the life of a platelet or around 7-10 days.
ii. Aspirin is used in combo with thrombolytic therapy after AMI and thrombotic stroke. It has been shown to prevent secondary events.
b. ADP receptor antagonists: bind ADP receptor on platelets and block platelet activation by ADP, thereby inhibiting the secretion of alpha granules and blocking the expression of adhesion proteins like GPIIb/IIIa
i. slow onset of action due to being prodrugs that must be metabolized in the liver to active intermediate
ii. Used in combination with aspirin to prevent cardiac events in patients with atherosclerosis and unstable angina
iii. Used in patients with aspirin intolerancec. Glycoprotein IIb/IIIa inhibitors: Glycoprotein IIb/IIIa are integrin (adhesion protein ) on
the surface of platelets that acts as a receptor for fibrinogen. Inhibitors of the integrin prevent platelet aggregation by preventing fibrinogen from binding.
i. use is decliningii. used for treatment during angioplasty and for unstable angina. Also used in
combo with aspirin and heparin to prevent recurrent MI or in combination with throbolitic drugs for “AMI.”
iii. Adverse effects like bleeding and thrombocytopenia can be reversed by platelet infusions.
Immunopathology Type I1. Define:
a. Atopic : prone to develop any of the range of allergic syndromes, an atopic individual may begin life with eczema, go on to have allergies to milk/fish/eggs, develop asthma or hay feer.
b. Immediate hypersensitivity : Hypersensitivity refers to excessive, undesirable (damaging, discomfort-producing and sometimes fatal) reactions produced by the normal immune system. Hypersensitivity reactions require a pre-sensitized (immune) state of the host.
i. Also called Type I hypersensitivity, immediate hypersensitivity is mediated by IgE
ii. The primary cellular component in this hypersensitivity is the mast cell or basophil. The reaction is amplified and/or modified by platelets, neutrophils and eosinophils.
1. A biopsy of the reaction site demonstrates mainly mast cells and eosinophils
iii. The mechanism of reaction involves preferential production of IgE, in response to certain antigens (often called allergens).
iv. The precise mechanism as to why some individuals are more prone to type-I hypersensitivity is not clear. However, it has been shown that such individuals preferentially produce more of TH2 cells that secrete IL-4, IL-5 and IL-13 which in turn favor IgE class switch. IgE has very high affinity for its receptor (Fcε; CD23) on mast cells and basophils.
c. Allergy : “other response”, similar to atopic, atypical immune response to environmental antigens, eventually becoming characterized by increased reactivity or hypersensitivity of the end-organs to inflammatory mediators and irritants.
39

d. Allergen : something that triggers an allergy, the “antigen”, plants/flowers, dust, animal dander, tobacco smoke
e. Anaphylaxis : a severe, life-threatening Type I hypersensitivity reactionf. Asthma : reversible bronchoconstrictive disease with progressive inflammation leading
to fibrosisg. Hives : redness, inflammation and edema of the skin associated with a Type I
hypersensitivity reaction (urticaria is long term hives)h. Wheal-and-flare reaction : development of a hive following intradermal injection of an
allergen, diameter can be used for allergy testing
2. State the approximate incidence of atopic diseases in the general population, and in individuals with allergic parents.
a. Allergic diseases are among the most common conditions. Estimates as high as 20% of all people have allergies, but 15% seem more likely to experience allergic symptoms at some time in their lives. Allergic seasonal rhinitis is the most common, food allergy, eczema in children and asthma. Incidence of asthma nearly doubled from 1980 to 1995 (and the increase is REAL, not just better diagnostics).
b. Strong multifactorial genetic component. 1 allergic parent = 35% risk of developing allergy. 2 allergic parents= 65%.
3. Describe the mechanism of IgE-mediated hypersensitivity in terms of: IgE attachment to basophils or mast cells; reaction with allergens; mediator release; effects of mediators on target tissues and cells.
a. WORMS and IgEi. A person with a worm infection will make IgG and IgE against the worm. IgG
binds the worm or its ova, activates complement (worms are impervious), C3a and C5a attract neutrophils, neutrophils seize the opsonized work and NOTHING HAPPENS. Neutrophils lack a helminthocidal mechanism.
ii. Worm shed antigen that diffused to nearby mast cells (which are LOADED with anti-helminth IgE---as soon as IgE is made it almost immediately binds to mast cells). IgE is cross-linked and the mast cells degranulate. Histamine smooth muscle contract in gu and violent peristalsis can expel worm. Late-phase response: prostaglandins and leukotrienes work as ECF-A (eosinophil chemotactic factor-anaphylaxis) and attract eosinophils in large numbers. Eosinophils (like other phagocytes) have Fc receptors of IgG (which are coating the worm). Eosinophil engages the opsonized worm and releases contents of granules, including Major Basic Protein (why eosinophils stain red), which is highly toxic to helminths.
iii. Second part of response: Th2 attracts eosinophil and macrophages but it makes IL-4, IL-5 and IL-13 which turns macrophages into alternatively activated macrophages, M2. These M2 cells are the ones that heal damage and wall off M1-resistant invaders. Therefore, even if the worm hatches, it will be in “jail”, totally walled off.
b. Type I Immunopathology and IgEi. Two phases: Immediate and Late
ii. IMMEDIATE: Type I immunopathology involves IgE and mast cells, roughly equivalent to allergy. In response to an allergen, Th2-like Tfh helps B cells switch to IgE production. IgE binds strongly to FcεR1 (receptors) on mast cell
40

surface (association constant of 10^-10, CRAZY STRONG—that’s why IgE plasma levels are so low, all secreted IgE is immediately bound). When 2 adjacent IgE molecules bound are cross-linked by allergen (must be at least divalent), the mast cell is signaled to release the contents of its granules, including histamine, heparin, enzymes and TNF. ***NOTE: normal people may have IgE on mast cells, but this IgE is the product of many weakly-activated clones, the chances of having 2 adjavent IgEs both specific for 2 epitopes on the same allergenic protein are small. Allergic person will have a few clones responding strongly, and be able to put 2 IgEs from the clones side by side.*** This causes local OR systemic vasodilation, increase permeability, gut and bronchial smooth muscle contraction (rapid onset: 15 minutes). This phase can be blocked by anti-histamines (receptor antagonists)
iii. LATE: activated mast cell does a series of enzymatic steps, phospholipase PLA2 cleaves arachidonic acid from membrane phospholipids, and arachidonic acid can be converted by cyclooxygenase pathway to prostaglandins, and by lipoxygenase to leukotrienes. These initiate inflammation, constrict bronchioles and are called the ECF-A (eosinophil chemotactic factor of anaphylaxis). Late phase is 4-10 hours after immediate and is not affected by anti-histamines, as it depends on prostaglandins, leukotrienes and cytokines.
4. Discuss the features that the various atopic diseases have in common which justify lumping them together.
a. Atopic diseases are characterized by an abnormal IgE response to an environmental antigen. Allergies, unlike Type IV immunopathologies, are mediated by Th2 cells. There is also a lot of cross reaction, it: foods that cause OAS contain proteins that are similar to and cross react with proteins in pollen, person with T-cell mediated contact dermatitis to latex has symptoms of IgE-mediated oral allergy to avocados, bananas (which have cross reactive antigen).
i. Asthma : bronchoconstrictive and inflammatory. ii. Eczema : dry, easily irritated skin, itch and rash that is self-worsening
iii. Oral allergy syndrome OAS : antigen passes through mucous membranes in mouth and gain access to mast cells, tingling lips/tongue, itching, swelling of lips. Symptoms are oral because the antigen is destroyed in the acidic stomach.
iv. Allergic rhinitis (seasonal ): runny nose, itchy eyes that we get Aug-Sept (ragweed season), due to pollen, cat or dogs.
5. Discuss the reasons for using glucocorticoids in asthma treatment.a. Asthma: reversible bronchoconstriction disease with progressive inflammation leading
to fibrosis. Tested for with spirometry (measures air flow, the FEV1, volume of air forcibly exhaled from full lungs in 1 second). Measure at baseline, and after bronchodilator, a significant improvement after bronchodilation bronchoconstrictive disease.
b. Glucocorticoids (inhaled), inhibit the production of arachidonic acid from phospholipids and block both PG and LI synthesis. are used to treat asthma early to prevent the late-phase reactants and Th2 cells present in the lung that are pro-inflammatory.
41

6. Discuss intradermal skin tests with reference to procedure, safety and specificity.a. An intradermal test is useful because it is easy. A drop of allergen extract is placed on
the forearm and a needle is used to prick the epidermis through the drop. The results are observed at 15 to 20 minutes and the wheal/diameter of the central raised area is measured (5/15 mm).
b. For safety, testing with buffer is necessary to control for skin hyperreactivity. Patients should be observed for 20-30 minutes after the test to make sure no problems result.
c. A positive skin test does not necessarily mean symptoms are due to the allergen. Levels of sensitivity may be sub-clinical even with a positive test or symptoms might come from something that cross-reacts with the test extract.
7. Discuss specific immunotherapy of allergic disease, considering duration of effect, risk of anaphylaxis, and percent of patients obtaining significant relief.
Avoidance: The cornerstone of treatment – if you aren’t exposed you won’t get allergies. But, patient compliance is an issue.
Antihistamines: Acute treatment effective for early, histamine-dependent phase
Epinephrine: (Epi Pen) Powerful sympathomimetic catecholamine that constricts blood vessels and relaxes bronchial smooth muscle – first line treatment in emergencies. Carried by people with significant allergic symptoms (ie airway constriction).
Theophylline: inhaler, phosphodiesterase inhibitor that raises intracellular levels of cyclic AMP in bronchial smooth muscle causing relaxation. No longer 1st line drug.
Glucocorticoids: excellent when they can be kept local (pulmonary inhaler or ointment), but risky when systemic. Inhibit arachadonic acid production from phospholipids and block PG and LT synthesis – inhibit apoptosis of eosinophils.
Leukotriene inhibitors: Inhibit LT synthesis or block their binding to receptors. Great in asthma.
LABAs (Long-acting beta 2 agonists): reduce bronchoconstriction rapidly – often given with inhalable steroid. Fluticasone/salmeterol (Advair) is dominant drug $8 billion annually. Symbicort is fast catching up.
IgE blocker: treatment of moderate to severe asthma in people >12 y/o who do not respond to inhaled steroids. It uses a monoclonal antibody to mop up any IgE in the blood. The drug that does the work is called omalizumab (Xolair).
Immunotherapy: “allergy shots” – dilute solutions of allergen extracts give subcutaneously 2x a week with increasing concentrations as tolerated. 75% of people with seasonal rhinitis say it helps. The mechanism of actions is unclear although some believe that the route of administration favors IgG formation which clears the antigen before it can reach IgE coated mast cells. Others think desensitization by repeated exposure is the key. Although, the keen Treg may be the real savior.
42

8. Describe the immediate allergic reaction and the late-phase reaction in terms of: time course of the reaction mediators involved
Immediate phase: Within 15 minutes of the intradermal injection of an allergen, a positive wheal-and-flare response (hives) is very obvious. Histamine, the most important granule released, has a half life in tissue of only a minute so it is very short lived.
Almost all of the IgE in the body (except in extremely allergic people ) is bound to mast cells via the receptor FcεRI.
Mast cells loaded with IgE are triggered to release the contents of their granules when two adjacent IgE molecules are cross-inked by allergen (must be divalent). The granules contain histamine, heparin, enzymes and TNF. These are all preformed so action is very rapid.
Late Phase Reaction:Begins 4 to 10 hours after the immediate phase and is not affected by histamines.In addition to releasing granules, the activated mast cell initiates a series of enzymatic steps causing phospholipase PLA2 to cleave arachidonic acid from membrane phospholipids. Arachidonic acid can then be cleaved to prostaglandins by cyclooxygenase and to leukotrienes by lipoxygenase. These compounds initiate inflammation and constrict bronchioles. The are together called the “eosinophil chemotactic factor of anaphylaxis” (ECF-A) b/c good at attracting eosinophils.
Late phase is not affected by histamines and instead depends on prostaglandins, leukotrienes and cytokines. Since histamines are not a big cause of the bronchoconstriction in asthma, anti-histamines do not play a major role in asthma. Eczema also seems to be more of a chronic late phase Type I immunopathology needing anti-inflammatory treatment rather than anti-histamines.
9. Discuss the roles of IgG, IgE, M2 macrophages, and eosinophils in helminth immunity.A person with a helminth (worm) infection will produce boatloads of IgG and IgE. The IgG binds
the worm and activates complement (no effect on worm) causing neutrophils to be attracted; however neutrophils have no effect on worms. But, DON’T WORRY IgE will save the day!
IgE stuck to mast cells picks up antigens shed by the worms. The antigens cross link the IgE activating the mast cells to degranulate. The histamine that is released causes smooth muscle contractions that can drive the worm out by violent peristalsis. However, the fun comes in when the late phase reaction is activated and all parts of the ECF-A (prostaglandins and leukotrienes) send an invitation for eosinophils to come join the party. Eosinophils see the IgG opsonizing the surface of the worm and bind the IgG via their Fc receptors. In doing so they release Major Basic Protein which is highly toxic to helminthes. Meanwhile Tfh is stimulating the B cells in the lymph nodes to switch to IgE production and Th2 goes out in the body to attract eosinophils and macrophages. The suite of
lymphokines a Th2 makes (IL-4, IL-5, IL-13) turn macrophages into alternatively activated M2 macrophages which heal damage and wall off M1-resistant invaders.
Overview of Platelet Function and Approach to Patient with Bleeding
43

1. Review events occurring during hemostasis. Compare primary and secondary hemostasis.a. Primary hemostasis: adhesion, activation and aggregation of platelets to form platelet
plugb. Secondary hemostasis: platelet plug is stabilized by formation of fibrin network
generated through coagulation cascade
2. Diagram the structure of a mature platelet and show the location of: dense granules, alpha granules, glycoprotein Ib, glycoprotein IIb/IIIa, and phospholipids.
a. Small, anuclear discoid cell with mitochondria and granules. 2-3 microns in diameter. Comes from megakaryocytes. Maturation time 4-5 days, circulating time 9-10 days. 80% of platelets circulate and 0% are in spleen (assuming normal spleen, in hypersplenism 90% of platelets can be in spleen). Newly formed platelets are larger and called megathrombocytes. Extensive system of internal membrane tunnels called the surface-connected canalicular system (granules extruded through these during platelet aggregation and secretion).
b. Dense granules contain ATP, ADP, serotonin and calcium.c. Alpha-granules contain a number of protein essential for platelet function, including
procoagulant proteins (fibrinogen, factor V, von Willebrand factor, etc.), platelet specific factor for platelet activation, and growth factors such as platelet-derived growth factor.
d. Lysosomal granules contain acid hydrolases.
3. List three functions of platelets.a. Adhesion to vascular subendothelium at sites of injury to begin the hemostatic processb. Activation of intracellular signaling pathways leading to cytoskeletal changes and release
of intracellular granules to enhance platelet plug formationc. Aggregation to form the platelet plusd. Support of thrombin generation by providing a phospholipid surface for coagulation
cascade to take place
4. Construct a simple diagram that depicts the process of platelet adhesion. Include in the drawing subendothelial collagen, von Willebrand factor, and glycoprotein Ib. Explain why platelet adhesion to blood vessels does not occur under normal circumstances.
a. NORMALLY: endothelial cells of intact vessel prevent blood coagulation by secretion of a heparin-like molecule and through expression of thrombomodulin, which when bound
44

to thrombin activtes protein C and S. Intact endothelial cells prevent platelet aggregation by the secretion of nitric oxide and prostacyclin, inhibitors of platelet activation.
b. Adhesion : vessel injury subendothelium exposed circulating vWF adheres to damaged area. Under conditions of high shear flow, circulating platelets contact the subendothelium in a rolling fashion and adhere by interaction between GP1b on platelet surface and vWF. With exposure to agonists, thrombin, ADP, epinephrine, thromboxane A2 or to adhesive proteins such as collagen and vWF, the platelet GPIIb/IIIa increases affinity for vWF (tighter binding). GPVI directly interacts with collagen in subendothelium. Other ligands (collagen, laminin, fibronectin) interact with integrins on the platelet surface. All of this leads to firm adherence of platelet to subendothelial surface.
c.
5. Similarly, construct a simple diagram that shows the process of platelet aggregation; include the release reaction (ADP), thromboxane synthesis, ADP and thromboxane receptors, glycoprotein IIb/IIIa, and fibrinogen.
a. Activation : New platelets stick to activated platelets and are themselves activated through release of compounds (products of oxidation of arachidonic acid by cyclooxygenase pathway that includes thromboxane A2 and ADP released from dense granules) that further amplify platelet activation. Platelets change from disc shaped to spheres with extended pseudopods to increase surface area. Contents of platelet granules are released. Soluble agonists (thrombin, thromboxane A2, epinephrine, ADP) interact with their respective G protein couple platelet membrane receptors intracellular signaling and Ca++ mobilization. Ca++ activates phospholipase A1, which releases arachidonic acid from phospholipids. Cyclooxygenase (COX-1) then converts arachidonic acid to prostaglandin H2, which is converted to thromboxane A2 by thromboxane synthestase. Thromboxane A2 is released and amplifies platelet activation. With platelet activation, the phospholipid phosphatidylserine switches from inner to outer membrane leaflet (available to interact with clotting factors thrombin generation)
45

a. List and describe three mechanisms that could lead to thrombocytopenia.a. Thrombocytopenia (low platelet count) can be due to decreased platelet production,
increased platelet destruction, or consumption/sequestration of platelets in the spleen. b. Platelet production: can be caused by primary bone marrow disorders, myelodysplasia,
and leukemia. Invasion of bone marrow by metastatic cancer, myeofibrosis, or TB can also cause it. Toxins like chemotherapy and severe nutritional disorders like vitamin B12 or folate deficiency can also cause decreased platelet production. If bone marrow is affected, thrombocytopenia is a possibility.
c. Increased platelet destruction: most common cause is immune thrombocytopenic purpura Autoantibodies are made that are directed at platelet antigens leading to their removal by macrophages. There are two types; acute and chronic. Acute = more common in children and presents with nosebleeds or viral infection, usually resolves within 2 to 6 weeks sin tratamiento or with steroids. Chronic = more common in adults with autoimmune disorders, most requiring treatment. Treatment methods include corticosteroids, Intravenous immunoglobulin (IVIG), and splenectomy.
d. Consumption/sequestration of platelets in spleen: the notes describe this category a bit broader than just in the spleen. Thrombotic thrombocytopenic purpura (TTP) is the example given. It occurs when endothelial damage leads to an abnormally large release of abnormally large vWF from storage sites. The vWF is abnormally large because metalloprotease ADAMTS13, whose normal function is to digest large vWF multimers into small multimers, is absent These vWF molecules mediate platelet adhesion and aggregation forming diffuse arteriole plugs. The disease is treated by plasmapheresis to remove the large vWF multimers and replace the ADAMTS13.
7. Identify three methods of treating ITP and the mechanism by which they increase the platelet count.ITP = immune thrombocytopenic purpura3 treatments: Corticosteroids – slow the proliferation of the B-cell clone making the auto antibodies. Work within 7-10 days
46

Intravenous immunoglobulin (IVIG): acts by blocking splenic Fc receptors to prevent their binding to antibody-coated platelets – effect seen 1 to 2 days.Splenectomy: removes the site where the majority of autoantibody induced platelet removal is occurring. Lasting effects in 60 to 70% of patients.
8. Describe the molecular defect, typical clinical course, and general approach to treatment for a patient with Von Willebrand Disease.
vWD is the most common congenital bleeding disorder. It can be caused by autoantibodies against the von Willebrand Factor (vWF), an inadequate amount of vWf, or mutations in the vWF gene. Lack of or alteration in the vWF can lead to an abnormal platelet/endothelial interaction leading to a bleeding disorder. Since vWF is also a carrier for Factor VIII, severe vWD can lead to factor VIII deficiency and a disorder with secondary hemostasis.
Lab tests to identify vWD include bleeding time (PFA-100 prolonged), a factor VIII level, a von Willenbrand antigen test to measure the amount of von Wellenbrand protein, or a vWF activity test.
A common treatment of vWD is DDAVP (arginine vasopressin), which enhances release of vWF from endothelial stores. However, it only works for partial quantitative defects (Type 1 vWD). Type 2 (qualitative defects) and type 3 (near-complete absence of vWF) can be treated with factor replacement. Patients should avoid aspirin and other platelet inhibiting agents.
9. List important questions to ask when obtaining a bleeding history in a patient with excessive bleeding.***Questions concerning the type, frequency, and amount of bleeding are essential.
Does the patient displace excessive, prolonged, recurrent, or delayed bleeding?Has the patient ever had the opportunity to bleed excessively? (Trauma, surgery, skin lacerations)Is there a family history of significant bleeding?
She also provided the following chart:
Multiple bleeding sites? – suggests more severe generalized hemolytic disorder
47

10. List important laboratory studies to obtain when evaluating a patient with excessive bleeding.a. Platelet Countb. Blood Smearc. APTT – intrinsic coagulation pathwayd. PT/INR – extrinsic coagulation pathwaye. Thrombin clotting time (TCT) – fibrinogen defects, heparin effects, fibrin split productsf. Fibrinogen level
48