vladislav navrátil, lukáš pawera -...
TRANSCRIPT

Masarykova univerzita Pedagogická fakulta
Fotokatalýza v praxi a ve škole Vladislav Navrátil, Lukáš Pawera
Brno 2014

Tato publikace byla vytvořena v rámci projektu Moduly jako prostředek inovace v integraci výuky moderní fyziky a chemie reg. č.: CZ.1.07/2.2.00/28.0182.
Recenzovali: RNDr. Renata Holubová, CSc., Prof. RNDr. Vratislav Kapička, DrSc.
© 2014 Vladislav Navrátil, Lukáš Pawera © 2014 Masarykova univerzita
ISBN 978-80-210-7157-5

3
Obsah
TEORETICKÁ ČÁST
1 Úvod ...................................................................................................................................... 4
2 Povrchové napětí kapalin ...................................................................................................... 4
2.1 Základní představy .......................................................................................................... 4
2.2 Kohezní (vnitřní) tlak kapalin . ........................................................................................ 7
2.3 Jevy na rozhraní kapalina – pevná látka – plyn .............................................................. 9
2.4 Využití povrchového napětí .......................................................................................... 13
2.5 Úhel smáčení ................................................................................................................. 16
2.6 Měření povrchové energie ............................................................................................ 19
3 Polovodiče. ........................................................................................................................... 21
3.1 Vlastní polovodič. .......................................................................................................... 21
3.2 Příměsové polovodiče . .................................................................................................. 24
3.3 Přechod PN, dioda ........................................................................................................ 24
3.4 Voltampérová charakteristika diody, speciální diody. ................................................... 28
3.5 Tranzistor ...................................................................................................................... 29
4 Katalýza – základní pojmy .................................................................................................. 31
4.1 Homogenní katalýza ..................................................................................................... 32
4.2. Fotochemické reakce .................................................................................................... 33
5 Fotokatalýza ........................................................................................................................ 35
5.1 Objev fotokatalýzy ........................................................................................................ 35
5.2 Mechanismus fotokatalýzy ........................................................................................... 35
5.3 Oxid titaničitý ................................................................................................................ 38
5.4 Zdroje záření ................................................................................................................. 39
5.5 Využití fotokatalýzy ...................................................................................................... 41
6 Závěr . ................................................................................................................................... 48
Literatura ................................................................................................................................ 48
PRAKTICKÉ ÚLOHY
Název modulu: Fotokatalýza .................................................................................................. 49
Demonstrační pokusy „Povrchové napětí kapalin“ ................................................................. 50
Základní měření povrchových jevů ........................................................................................ 52
Fotokatalytické vlastnosti TiO2 povrchů ................................................................................. 54
Shrnutí .................................................................................................................................... 55
Glosář pojmů .......................................................................................................................... 57
Rejstřík ................................................................................................................................... 59

4
Teoretická část
1 Úvod Slovo fotokatalýza je složeno ze dvou slov: foto = světlo a katalýza = proces, který je charakterizován tím, že látka, známá jako katalyzátor, usnadňuje rychlost reakce. Je třeba upřesnit, že fotokatalýza probíhá na povrchu polovodičů.
Teorie i praxe fotokatalýzy je typickou interdisciplinární oblastí, zahrnující oblast fyziky, chemie, biologie, environmentalistiky apod. Její teorie je založena na několika základních pilířích, z nichž na úvod této práce uvedeme dva, teorii a praxi povrchového napětí kapalin a povrchové energie pevných látek a stručný přehled katalýzy. Závěr potom bude pojednávat o fotokatalýze a možnostech jejího využití zejména v oblasti zlepšování životního prostředí.
2 Povrchové napětí kapalin
2.1 Základní představy Základními vlastnostmi kapalin je jejich tvarová nestálost (přizpůsobují se tvaru nádoby) a přitom hustota nepříliš odlišná od hustoty jejich pevné fáze. Z toho důvodu můžeme usoudit na to, že přitažlivé síly mezi molekulami v kapalinách (tzv. kohezní síly) jsou velké. Experiment ukazuje, že zejména v případě malých množství kapalin (drobné kapičky rtuti na skle, mlha, emulze oleje ve vodě, apod.), nebo ve stavu bez tíže, se kapaliny snaží zaujmout geometrický tvar koule, tj. objektu s minimálním povrchem při daném objemu. Kapaliny se tedy chovají tak, jakoby na jejich povrchu byla tenká blanka, která je stlačuje. Vznik blanky si můžeme názorně představit podle Obr. 2.1 (Laplace): mějme volný povrch kapaliny v nádobce a položme si otázku, jaké síly působí na molekulu částice, nacházející se uvnitř kapaliny. Síly mezi molekulami kapaliny klesají poměrně rychle se vzdáleností, takže na naši vybranou molekulu působí pouze molekuly, nacházející se v malé kouli, opsané kolem ní (tzv. sféra molekulárního působení). V důsledku symetrického rozložení sousedních molekul je výslednice všech sil rovna nule. Jinak je tomu u molekul, které se nachází na povrchu kapaliny, nebo blízko u něho ve vzdálenosti menší, nebo rovné poloměru sféry molekulárního působení. U nich přitažlivé síly mají nenulovou výslednici, působící směrem do kapaliny (v horní polokouli či vrchlíku koule o poloměru sféry molekulárního působení se nachází velmi málo molekul vzduchu, nebo par kapaliny). Proti této výslednici působí síly odpudivé, takže ve výsledku se jedná o statický stav a tedy nedochází k pohybu molekul povrchové vrstvy směrem do kapaliny.
Obr. 2.1 Vznik povrchového napětí kapalin [2]

5
Obr. 2.2 Obr. 2.3
Obr. 2.4 Obr 2.5
Obr. 2.2–2.5 Jevy povrchového napětí kapalin v přírodě [6,7]
Z této jednoduché představy se dá soudit na to, že povrchová vrstva kapaliny bude mít jiné fyzikální vlastnosti, než její objem. Takovou teoretickou představu lze snadno dokázat pomocí mnoha jednoduchých a přitom dostatečně průkazných jevů (Obr. 2.2–2.5). Na hladině kapaliny „plavou“ tělíska, která mají vyšší hustotu než kapalina (drobné mince, žiletka, jehla), po hladině pobíhají drobní vodní živočichové, apod.
Kvantitativně popisujeme povrchové napětí kapalin pomocí veličiny, zvané koeficient povrchového napětí , který definujeme jako sílu F, působící kolmo na délkový element l, nacházející se na povrchu kapaliny:
S
E
l
F
∆∆=
∆∆=σ
(2.1)
nebo jako povrchovou energii ∆Ε plošné jednotky ∆S povrchu kapaliny (pojem povrchová
energie upřesníme později).
Pokusme se kvalitativně odhadnout, na kterých parametrech prostředí bude napětí záviset. Zřejmě to bude poměr hustot obou prostředí (ρ2 / ρ1). Růst teploty T se projeví nejen v poklesu hustoty kapaliny, ale i vzrůstem hustoty par nad ní. Podobně tomu bude i se závislostí na barometrickém tlaku b vzduchu nad kapalinou a na veličině, kterou můžeme nazvat jako těkavost kapaliny (τ).

6
Lze tedy pro σ psát:
≈≈≈≈= ,......1
,1
,1
,1
2
τρρσ
bTf (2.2)
Vliv tlaku a těkavosti lze dokázat pomocí jednoduchého pokusu, se kterým se jistě už mnozí mimoděk setkali, v učebnicích však popsán není. Větší nádobu (demižon) pečlivě vymyjeme vodou. Poté zpozorujeme, že na jejím vnitřním povrchu zůstaly usazeny kapky vody. Protože jsou v klidu, platí rovnováha sil na ně působících, tj. síly tíhové směrem dolů, a síly povrchového napětí spolu s adhezí mezi kapičkami a sklem směrem nahoru (v místě styku kapičky s povrchem nádoby). Na kvalitě prostředí nad kapičkami závisí pouze síla povrchového napětí, takže kápneme-li do nádoby líh (funguje i se slivovicí – tak byl jev pozorován v praxi), zjistíme, že většina kapiček sklouzne ke dnu nádoby, neboť síla povrchového napětí se zmenší (zvýší se hustota plynu nad kapičkami). Dá se očekávat, že podobně se projeví i vzrůst atmosférického tlaku uvnitř nádoby. Zde je však třeba uvážit její mechanickou pevnost, neboť zvýšení tlaku musí být dostatečné. Podobného jevu využívají i skláři při vrtání děr do skla (pečlivě omyté sklo umístí do vody, čímž sníží jeho povrchové napětí a sklo se při vrtání nerozbije).
Pro ilustraci jsou v Tab. 2.1 uvedeny hodnoty koeficientu povrchového napětí pro několik vybraných kapalin [11].
Tab. 2.1 [11]
Kapalina .103 N.m-1 Kapalina .103 N.m-1
Aceton 23,3 Methylalkohol 22,6
Ethylalkohol 22,3 Olej olivový 33,0
Ethylether 16,96 Petrolej 27,0
Glycerin 65,7 Rtuť 472,0
Chlorbenzen 33,3 Sirouhlík 32,2
Chloroform 27,14 Toluen 28,4
Kyselina octová 27,4 Voda 72,75
Koeficient povrchového napětí σ je tedy veličinou, závislou na různých parametrech. Přesnější kvalitativní rozbor na základě modelu na Obr. 2.1 vede k závěru, že σ bude záviset na rozdílu hustoty kapaliny a plynu nad ní, na teplotě, barometrickém tlaku, čistotě kapaliny, atd.
Závislost na rozdílu hustot studoval již Laplace a dospěl ke vztahu
n
parkapC )( ρρσ −=
(2.3),
kde n = 2 (Van der Waals uvádí n = 3 a Bačinskij n = 4)

7
Teplotní závislostí σ se zabýval např. Eötvös:
)(3
2TT
v
Kk −=σ
(2.4),
kde v je měrný objem kapaliny, Tk je její kritická teplota a K je konstanta, blízká 2,1. Teplotní závislost povrchového napětí vody je uvedena v Tab. 2 Hodnoty σ v intervalu t < 100 0C byly změřeny při tlaku 105 Pa a hodnoty v intervalu 100 0C < t < 374,15 0C (kritická teplota vody) se vztahují k tlaku nasycených vodních par. Tab. 2.2 [11]
t 0C .103
N.m-1 t 0C 103
N.m-1 t 0C
103
N.m-1 t 0C
103
N.m-1
0 75,6 70 64,4 180 42,3 320 9,9
10 74,2 80 62,6 200 37,8 340 5,7
20 72,76 90 60,7 220 33,2 360 2,0
30 71,2 100 58,8 240 28,6 374,15 0,0
40 69,6 120 54,9 260 23,8
50 67,9 140 50,8 280 19,1
60 66,2 160 46,6 300 14,4
2.2 Kohezní (vnitřní) tlak kapalin Z načrtnutého kvalitativního modelu plyne, že kapalina je stlačena silou, jejíž podstatou je povrchové napětí kapalin a která se nazývá kohezní tlak kapalin. Velikost tohoto tlaku lze určit ze základních termodynamických představ: vnitřní energie určitého množství kapaliny je funkcí teploty T, objemu V a velikosti povrchu kapaliny A. Lze tedy psát:
dAA
UdV
V
UdT
T
UdU
VTATAV ,,,
∂∂+
∂∂+
∂∂= (2.5)
Člen dVV
U
AT ,
∂∂
představuje práci, potřebnou k překonání mezimolekulárních sil při změně
objemu kapaliny. Má rozměr energie, přičemž samotná derivace TV
U
∂∂
má rozměr tlaku.

8
Nazveme ji kohezní (vnitřní) tlak kapalin:
i
T
pV
U =
∂∂
(2.6)
Tento tlak je mírou mezimolekulárních sil, které rozhodují o chování uvažované kapaliny. Ze základních termodynamických úvah [10] lze odvodit, že platí:
pT
pTp
V
U
V
i
T
−
∂∂==
∂∂
(2.7)
kde p je vnější tlak.
Pomocí definic koeficientů tepelné roztažnosti pT
V
V
∂∂= 1α a stlačitelnosti
T
p
V
V
∂∂−= 1β lze poslední rovnici upravit na tvar
βα=
∂∂
∂∂
−=
∂∂
T
p
V
p
V
T
V
T
p
(2.8)
Spojením vztahů (2.7) a (2.8) dostaneme definiční vztah pro kohezní tlak:
pTpi −=βα
(2.9)
Poměr α / β je mnohem větší, než vnější tlak p [10] a proto lze tlak zanedbat. Vychází nám tedy pro kohezní tlak výraz
βα
Tpi = (2.10)

9
Hodnoty kohezního tlaku pro některé kapaliny jsou uvedeny v Tab. 2.3 [10].
Kapalina Kohezní tlak Pi.10
-8 Pa Kapalina
Kohezní tlak Pi.10
-8 Pa
aceton 5,146 chloroform 3,708
benzen 3,689 metanol 13,068
etanol 6,686 rtuť 31,400
éter 5,693 tetrachlormetan 3,354
n-heptan 2,543 voda 17,930
Z Tab. 2.3 plyne jednoduchý kvalitativní závěr: povrchové napětí kapalin je natolik velké, že kapaliny jsou jím stlačeny tak, že se nám jeví jako téměř nestlačitelné.
2.3 Jevy na rozhraní kapalina – pevná látka – plyn Vybraná částice kapaliny koná pohyb, připomínající na jedné straně chaotický pohyb částic v plynu a kmitavý pohyb částic v pevných látkách na straně druhé: jistý čas kmitá kolem rovnovážné polohy, poté se posune („prodere“) mezi ostatními částicemi na jiné místo a opět kmitá. S klesající teplotou klesá energie zejména chaotického pohybu, takže po dosažení bodu tuhnutí jí zůstává pouze pohyb kmitavý. Naskýtá se otázka: zmizí po ztuhnutí kapaliny její povrchové napětí? Vycházíme-li z modelu vzniku povrchového napětí, dojdeme k závěru, že i pevné látky mají povrchové napětí. Označme tedy plynné prostředí jako 1, kapalné jako 2 a pevné jako 3. Příslušná povrchová napětí mezi jednotlivými prostředími pak budou σ12, σ13 a σ23 a budou se při vzájemném skládání chovat jako vektory. Přehled základních situací je uveden na Obr. 2.6–2.9.
Praktickým důsledkem těchto jevů je elevace a deprese v kapilárách (Obr. 2.10) a tvar kapek jedné kapaliny na povrchu kapaliny druhé, bublinek v kapalinách, apod. (Obr. 2.8).
Na stejném principu jsou založeny i metody měření povrchového napětí, používané zejména ve školních laboratořích:
Obr. 2.6 Obr. 2.7

10
Obr. 2.8
Obr. 2.9
Obr. 2.6–2.9 Některé jevy na rozhraní tří prostředí [2]
Obr. 2.10 Metoda kapilární elevace [2]
a) Metoda kapilární elevace
Z experimentálního pozorování víme, že hladina kapaliny v kapiláře, případně u stěny nádoby, nemá rovinný tvar, ale její hladina je buď vypouklá (konvexní), nebo vydutá (konkávní) – viz Obr. 2.6–2.10.
Obr. 2.11.1 Obr. 2.11.2 Obr. 2.11.3

11
Z jednoduché úvahy o podstatě povrchového napětí je jasné, že v případě situace, znázorněné na Obr. 2.11,2 působí povrchová vrstva na kapalinu přídavným tlakem, který se přičítá ke koheznímu tlaku, odpovídajícímu rovinnému povrchu (Obr. 2.11,1). V případě, znázorněném na Obr. 2.11,3 se naopak tento tlak odečítá. Tento přídavný tlak 2.11, 2 a 3 nazýváme tlakem kapilárním. Kapilární tlak je tím větší, čím menší je poloměr křivosti povrchu kapaliny R a v obecném případě je roven [1,2,3]:
+=
21
11
RRpk σ
(2.11)
Kde R1 a R2 jsou poloměry křivosti plochy kapaliny ve dvou na sebe kolmých směrech. Poloměr křivosti je kladný pro konvexní povrch a záporný pro povrch konkávní. Pro případ kulového vrchlíku má rovnice (2.11) tvar
Rpk
σ2±=
(2.12)
Pro případ vzestupu v kapiláře (Obr. 2.10) lze psát pro rovnováhu tlaků v kapiláře a mimo ni:
gh
Rpnpb kohkoh ..
2 ρσm±+=+ (2.13)
Odtud dostáváme
2
... gRh ρσ =
(2.14)
Místo poloměru R kulového menisku lze zavést poloměr r kapiláry (Obr. 2.10):
( )222 yRrR −+= , odkud
,2
22
y
yrR
+= nebo θcos
rR = (2.15)
Dosazením (2.15) do (2.14) dostaneme
( )y
yrghnebo
grh
4
..,
cos2
... 22 +== ρσθ
ρσ (2.16)

12
A v případě dokonalého smáčení vnitřní stěny kapiláry (cosθ =1) vztah
2
... rgh ρσ = . (2.17)
V případě nedokonalého smáčení musíme určit i úhel a výpočet provedeme podle vztahu (2.15).
b) Metoda odtrhávací
Tato metoda vychází přímo z definice povrchového napětí (Obr. 2.1, 2.12). Z kapaliny je pomalu vytahován drátěný rámeček a citlivými vahami se změří síla F, které je třeba k odtržení pomyslné blány od tenkého drátu délky l. Povrchové napětí pak vypočteme podle vztahu
l
F
2=σ
(2.18)
(jedná se o dvě plochy, obepínající drátek).
V případě kruhového prstence použijeme vztahu
r
F
.4πσ = (2.19)
Obr. 2.12 Metoda odtrhávací [2]

13
Obr 2.13 Metoda kapková [2] c) Metoda kapková
V tomto případě odkapává kapalina z tlustostěnné kapiláry (Obr. 2.13). Když tíha kapky m. g překročí sílu povrchového napětí, působící na zúženém profilu kapičky o poloměru r, kapička odpadne. Protože zmíněný poloměr r je obtížně měřitelný a po odtržení kapičky zůstane na kapiláře malé množství kapaliny, používá se tato metoda jako relativní. Předpokládejme, že jak poloměr r, tak i zmíněné zbytkové množství dvou kapalin jsou pro obě kapaliny stejné. Změříme potom hmotnost stejného počtu kapiček obou kapalin (m1 a m2). Známe-li povrchové napětí jedné z kapalin 2 (např. vody), můžeme určit σ1 z úměry
2
1
2
1
m
m=
σσ
. (2.20)
2.4 Využití povrchového napětí Kapitola, pojednávající o povrchovém napětí často ve fyzikálních učebnicích neprávem chybí. Vyhnuli se jí např. autoři světoznámého kurzu fyziky Halliday, Resnick a Walker, nezmiňuje se o ní R. Feynman a není ani v tzv. Berkleyském kurzu. Přitom na jevu povrchového napětí jsou založeny takové významné oblasti chemie, jako je koloidní chemie, katalýza, teorie adsorpce, apod. Bez znalosti povrchového napětí nemůžeme ani vysvětlit mnohé biologické jevy, jako je např. objasnění transportu kapalin v rostlinách a půdě, nebo četné jevy atmosférické. Jevy povrchového napětí jsou podstatou veškeré chemie výroby pracích prášků a mycích prostředků. Voda má totiž vysoké povrchové napětí a to je třeba snížit pomocí vhodné příměsi (mýdla či saponátu). I při využití tzv. herbicidů v zemědělství je třeba vhodnými přísadami upravit povrchové napětí roztoku herbicidu tak, aby sklouznul po kulturní rostlině a ulpěl na plevelech (a tím je zničil).
V technické praxi se povrchového napětí využívá v takových tradičních výrobních postupech, jako je broušení, vrtání a leštění. V těžebním průmyslu používaná metoda flotace není nic jiného, než vhodně upravené povrchové napětí vody tak, aby hlušina byla smáčena a klesla ke dnu a těžená ruda, která je v ní jemně rozptýlená, smáčena nebyla (zůstane na povrchu kapaliny jako pěna).
Povrchově aktivní látky
Povrchová energie se snaží dosáhnout své minimální hodnoty. K tomu může dojít buď tak, že se zmenší plocha povrchu, nebo se sníží povrchové napětí σ. Proto dodáme-li do kapaliny A s povrchovým napětím σΑ kapalinu B s povrchovým napětím σB < σΑ, bude se

14
kapalina B adsorbovat přednostně v povrchové vrstvě a povrchové napětí roztoku klesne. Takovou kapalinu B nazýváme povrchově aktivní (např. mýdlo a saponáty). Jiné látky naopak zvyšují povrchové napětí (např. roztok cukru, roztoky některých solí). Takové látky se koncentrují naopak mimo povrchovou vrstvu kapaliny A (tedy uvnitř). Takže dodáme-li např. sůl do mýdlového roztoku, dojde k intenzivnímu vytěsnění mýdla na povrch roztoku.
Poznámka k pojmu „povrchová energie“
Je známo, že přírodní systémy, složené z velkého počtu částic se řídí dvěma základními principy: principem minima energie a principem maxima entropie (neuspořádanosti). Příkladem může být rozložení vakancí v pevných látkách. Pokud by krystal měl minimální energii, nenacházely by se v něm žádné vakance. V tomto případě by ale byl maximálně uspořádán, měl by tedy minimální entropii. Naopak, kdyby měl krystal maximální entropii, byly by atomy, z nichž se skládá, rozloženy neuspořádaně v prostoru. Skutečností je stav (jakýsi kompromis), charakterizovaný tzv. rovnovážnou koncentrací vakancí. Odtud plyne zajímavý experimentální (i teoretický) výsledek – při teplotě vyšší, než 0 K nelze vyrobit materiál bez vakancí.
Podobně je tomu i v případě povrchového napětí. Neřídí se pouze minimem vnitřní energie, ale lze ho vyjádřit jako
STw .−=σ (2.21) kde w je povrchová vnitřní energie, T je teplota a S je povrchová entropie. σ je tedy veličina, známá z termodynamiky jako volná energie. Nadále budeme ale užívat tradičního názvu povrchová energie, který se již vžil. Budeme mít ale na paměti, že správný název je povrchová volná energie. Ta je v případě kapalin totožná s povrchovým napětím na rozdíl od pevných látek, kde je mezi oběma veličinami nepříliš velký rozdíl daný tím, že atomy v kapalinách jsou na rozdíl od pevných látek pohyblivé. Pokusme se na základě jednoduchého modelu odhadnout velikost povrchové energie pevné látky, krystalizující v prosté kubické mřížce (Obr 2.14). Vzdálenost mezi částicemi (atomy, molekuly) v mřížce označme a. Z tohoto modelu plyne jednoduchý výsledek, že všechny částice na povrchu krystalu mají nenasycené vazby na rozdíl od částic, nacházejících se uvnitř krystalu. Je-li celková vazebná energie částice uvnitř krystalu rovna ε, potom tatáž energie pro částici na povrchu bude pouze 5ε/6. Zbytek, tj. ε/6 odpovídá povrchové (vazebné) energii, připadající na jednu částici. Vazebná energie může být odhadnuta jako ε = h. m, kde h je měrné teplo vypařování
(sublimace) a 3.a
N
Mm
A
mol ρ== je hmotnost jedné částice (Mmol je hmotnost molu a NA je
Avogadrova konstanta). Podělíme-li molekulovou povrchovou energii ε / 6 plochou průřezu částice a2
, dojdeme k následujícímu odhadu pro hustotu povrchové energie:
ah
a
mh
a..
6
1.
6
16
1
22ρ
εσ ≈≈= (2.22)

15
Protože sublimační tepla pevných látek nebývají tabelována, budeme tento model aplikovat na odhad povrchového napětí vody. Pro vodu je ρ = 103 kg.m-3, měrné teplo vypařování h = 24.106 J.kg-1 a poloměr a = 1,76.10-10 m. Potom získáme výpočtem hodnotu velmi blízkou hodnotě tabelované (0,072 J.m-2).
Obr. 2.14 K určení povrchové energie látek
Vztah mezi povrchovým napětím (povrchovou energií) a základními termodynamickými funkcemi lze přímo odvodit ze základních termodynamických principů. Podle první věty termodynamické platí
WQdU δδ −= (2.23) kde dU je přírůstek vnitřní energie soustavy, δQ je dodané teplo a δW je vykonaná práce.
Pro vratné děje lze potom psát
dU = T.dS – p.dV - δWrev (2.24) kde dS je změna entropie soustavy, p je tlak a dV je změna objemu soustavy. δWrev je potom vratná práce, vykonaná soustavou. V případě kapalin platí vztah
-δWrev = σ.dA (2.25) (práce se projeví ve zvětšení povrchu A. Symbol A zde dále používáme výjimečně místo obvyklého S a to z důvodu, aby nedošlo k záměně s entropií S. Znaménko „–„ v posledním vztahu značí, že ke zvětšení povrchu je třeba systému práci dodat). Ze vztahů (2.23) a (2.24) dostaneme
dU – T.dS + p.dV = σ.dA (2.26)

16
Volná energie (Helmholtzova funkce) je definována vztahem F = U – T.S (2.27) takže dF = dU – T.dS – S.dT (2.28) Spojením rovnic (4) a (6) dostaneme
dF + S.dT + p.dV = dU – T.dS + p.dV = σ.dA (2.29) Odtud
VTA
F
,
∂∂=σ (2.30)
Podobně pomocí Gibbsovy volné entalpie, definované vztahem G = U + p.V – T.S (2.31) Dostáváme dG = dU + p.dV + V.dp – T.dS – S.Dt (2.32) a z (2.26) a (2.31) dostaneme
dG + S.dT – V.dp = dU – T.dS + p.dV = σ.dA (2.33) Odtud plyne druhý vztah pro σ:
pTA
G
,
∂∂=σ (2.34)
Oba vztahy (2.30) a (2.34) lze chápat jako termodynamické definice povrchového napětí (povrchové energie).
2.5 Úhel smáčení S minimem Helmholtzovy volné energie, nebo Gibbsovy volné entalpie úzce souvisí fyzikálně velmi zajímavá situace, která nastává v místě styku tří prostředí, pevného, kapalného a plynného. Tato situace je znázorněna na Obr. 2.6–2.10 a 2.15. Vyjdeme právě z Obr. 2.15 a z obecných termodynamických principů a odvodíme vztah mezi koeficienty povrchového napětí σ12, σ13 α σ23 , kde číslice 1, 2 a 3 přiřazujeme podle Obr. 2.6 prostředí plynnému (1), kapalnému (2) a pevnému (3).

17
Systém v rovnováze má za stálého objemu a teploty minimální volnou energii, takže platí:
dT = 0, dV = 0, dF = 0 (2.35)
Systém se skládá ze tří fází a tak musí platit
dF = dF(12) + dF(13) + dF(23) = 0 (2.36)
kde indexy 1,2,3 se opět vztahují k plynné, kapalné a pevné fázi.
Z rovnic (2.35) a (2.36) dostaneme
dF = σ12.dA12 + σ13.dA13 + σ23.dA23 = 0 (2.37)
Přičemž veličiny (plochy) dA12, dA13 a dA23 jsou na sobě závislé (o co se zvětší styčná plocha mezi kapalinou a tuhou fází, o to se zmenší plocha mezi fází tuhou a plynnou):
dA23 = -dA13 (2.38)
Obr. 2.15 K určení úhlu smáčení [10]
Z Obr. 2.15 plyne, že
A23 = π.ρ2 = π[ r2 – (r – v)
2 ] = π.v(2r – v) (2.39)
A
A12 = 2π.r.v2 (2.40)
Vztahy (2.39) a (2.40) napíšeme nyní v diferenciálním tvaru
dA23 = 2π(r.dv + v.dr – v.dv) = 2π[ (r – v)dv + v.dr (2.41)
dA12 = 2π (r.dv + v.dr) (2.42)

18
Z podmínky stálého objemu kapky plyne
konstvrvV =−= ).3(.
3
1 2π
(2.43)
a
0)33..6(
322 =−+= dvvdrvdvvrdV
π
(2.44)
Odtud dostaneme:
v.dr = (v-2r)dv (2.45)
Z rovnic (2.41) a (2.42) dostaneme
dA23 = 2πr.dv (2.46) a
dA12 =2π( v – r) dv (2.47)
Protože
v – r = -a = -r.cosΘ (2.48) lze psát
dA12 = -2πr.cosΘ.dv (2.49) Vydělením rovnic (2.49) a (2.46) dostaneme
Θ= cos23
12
dA
dA (2.50)
Odtud
dA12 = dA23.cosΘ (2.51) Ze vztahů (3), (3´) a (15) dostáváme
(σ23 - σ 13 + σ12.cosΘ)dA23 = 0 (2.52) a nakonec dostáváme
12
2313cosσ
σσ −=Θ (2.53)
Z tohoto vztahu plyne, že je-li σ13 > σ23 má cosΘ kladnou hodnotu, úhel θ < 900 a kapalina smáčí tuhý povrch. Je-li σ23 > σ13, je cos Θ záporný, úhel Θ > 900 a kapalina povrch nesmáčí.

19
2.6 Měření povrchové energie: pomocí přístroje „Surface Energy Evaluation System“ Advex Instruments
Přístroj Surface Energy Evaluation System (SEE) byl vyvinut v Ústavu fyzikální elektroniky Přírodovědecké fakulty Masarykovy Univerzity v Brně [14] (Obr. 15). Jeho principem je přesné měření kontaktního úhlu na rozhraní pevná látka – kapalina – plyn.
Základní charakteristiky systému jsou následující:
• jedná se o velmi přesnou metodu měření kontaktního úhlu a tím i povrchové energie pevné látky (nebo povrchového napětí kapaliny).
• Použitá metoda eliminuje chyby, charakteristické pro metody, založené na manuálním měření kontaktního úhlu.
• Přístroj lze snadné připojit k PC.
• Měření využívá speciálního software, umožňujícího získat okamžité výsledky měření.
• Barevné obrázky rozhraní pevná látka – kapalina – plyn umožňují sledovat časový průběh profilu rozhraní. To umožňuje přesně fitovat profil rozhraní a stanovit jeho parametry (úhly).
• Pomocí software obdržíme přímo velikost kontaktního úhlu.
• Velikost kontaktního úhlu lze postupně uchovat v paměti počítače a tak určit jeho časový vývoj.
• Pomocí software lze určit povrchovou energii na základě nejčastěji používaných teoretických modelů [5] včetně odhadu chyb.
• Program využívá databáze cejchovacích (vztažných) kapalin, případně pevných povrchů. [5]
• Podrobnější popis měření uvádí přímo software, dodávané k přístroji.
Obr. 2.16 Surface Energy Evaluation System, celkový pohled [5]

20
Obr. 2.17 Přístroj SEE [5]
Obr. 2.18 Příklad měření úhlu smáčení [5]
Požadavky na referenční kapaliny, pomocí nichž lze určit kontaktní úhly jsou následující:
- Kapalina nesmí reagovat s povrchem pevné látky. - Kapalina musí mít dobře definované a stabilní povrchové napětí. - Kapalina nesmí být toxická. - Povrchové napětí referenční kapaliny musí být větší, než povrchová energie pevné
látky. - Kapalina musí mít vysokou čistotu

21
Literatura [1] ILKOVIČ, D.: Fyzika (1958)
[2] HLAVIČKA, A a kol.: Fyzika pro pedagogické fakulty (1971)
[3] HORÁK, Z., KRUPKA, F.: Fyzika (1976)
[4] HAJKO, V., DANIEL – SZABÓ, D.: Základy fyziky (1980)
[5] BURŠÍKOVÁ, V., SŤAHEL, P., NAVRÁTIL, Z., BURŠÍK, J., JANČA, J.: Surface Energy Evaluation of Plasma Treated Material by Contact Angle Measurement. Masaryk University, Brno 2004.
[6] MATVĚJEV, A. N.: Molekuljarnaja fizika (1987)
[7] GUBRECHT, H.: Mechanik, Akustik, Wärme (1990)
[8] MOORE, W. J.: Fyzikální chemie (1972)
[9] KIREEV, V. A.: Kurz fizičeskoj chimii (1975)
[10] HÁLA, E., REISER, A.: Fysikální chemie. NČSAV Praha 1960.
[11] BROŽ, J., ROSKOVEC, V., VALOUCH, M.: Fyzikální a matematické tabulky. SNTL Praha, 1980.
3 Polovodiče Jak uvidíme v dalších kapitolách, dochází k jevu fotokatalýzy pouze v polovodičích. Proto považujeme za vhodné uvést nyní krátkou a pokud možno názorně sestavenou představu o jevech, doprovázejících a podmiňujících vedení elektřiny v polovodičích.
Teorii vedení elektřiny v polovodičích lze podat v zásadě dvěma způsoby. První vychází z klasických představ vedení elektrického proudu jako záporně a kladně nabitých bodových nábojů (elektronů a děr). Tento způsob je jistě velmi názorný, vhodný k vysvětlení této látky na základní a střední škole, není však úplný. Druhý způsob vychází ze závěrů kvantové teorie pevných látek (konkrétně z pásové teorie pevných látek). Tímto druhým způsobem je vysvětlena teorie vedení elektrického proudu v polovodičích například v knize autorů Halliday, D, Resnick, R, Walker, J „Fyzika“ až po objasnění základů kvantové fyziky a můžeme jenom doporučit čtenáři, aby se s ní seznámil.
3.1 Vlastní polovodič Vytvoříme-li jednoduchý obvod, složený z baterie, měděných přívodů a žárovky, bude žárovka svítit, což je známkou toho, že vodičem protéká elektrický proud. Nahradíme-li část vodiče tyčinkou z křemíku, proud poteče až tehdy, ohřejeme-li ji např. plamenem plynového hořáku. Křemík patří k tomu druhu materiálu, který se co do své elektrické vodivosti nachází mezi vodiči a izolátory, tj. k polovodičům (kdybychom křemíkovou tyčinku nahradili tyčinkou keramickou, proud by neprotékal ani při vysoké teplotě).
Rozdíl mezi vodiči, polovodiči a izolátory lze vyjádřit i kvantitativně: řádová hodnota vodivosti kovů je cca 10-8–10-6 Ω.m, polovodičů cca 10-3 Ω.m a izolátorů 1012–1015 Ω.m.
V polovodičích a izolátorech se na rozdíl od kovů nemohou pohybovat valenční elektrony volně – jsou vázané tzv. kovalentní vazbou. Například u čtyřvazného křemíku je každý z valenčních elektronů jednoho atomu vázán touto vazbou s obdobným elektronem atomu sousedního (Obr. 3.1). Základními vlastnostmi kovalentní vazby je její směrovost, nasycenost a sdílení elektronů. Její podstata nemůže být dokonale objasněna na základě klasické fyziky a bude probrána podrobněji v posledním dílu učebnice, zabývající se kvantovou fyzikou.

22
Obr.3.1 Vlastní polovodič
Podle nastíněného obrázku krystalu polovodiče, vázaného kovalentní vazbou, by v něm neměla existovat možnost vedení elektrického proudu. Z praxe však víme, že polovodiče elektrický proud vedou. Vysvětlujeme si to tak, že dojde k uvolnění původně vázaného elektronu do oblasti mezi atomy. Může se tak stát např. působením světelného kvanta, (Obr. 3.2), nebo vlivem teploty. Takto uvolněný elektron se pohybuje polovodičem podobně, jako tomu bylo u vodičů, tj. proti směru intenzity elektrického pole – tzv. elektronová vodivost (Obr. 3.3). Jiný způsob vodivosti spočívá v tom, že na místo, které zůstalo po uvolněném elektronu (které je nyní kladné) se dostane elektron, odtržený např. od sousedního, nebo jiného blízkého atomu. Tento děj může pokračovat dále a pro jednoduchost si můžeme představit, že se „pohybuje“ ono kladné místo („díra“) a to ve směru intenzity elektrického pole. Takovou vodivost nazýváme děrovou.
Obr. 3.2 Mechanismus uvolnění elektronu světelným kvantem Elektronová nebo děrová vodivost čistých polovodičů je označována jako vlastní vodivost a zmíněné polovodiče jsou tzv. vlastní polovodiče. V tomto případě hovoříme o vzniku páru elektron – díra. Poněvadž podle II. termodynamické věty nelze vyrobit monokrystal bez cizích příměsí a vakancí (bodových poruch), je možný i vznik nosičů nábojů díky těmto nečistotám (minoritní nosiče nábojů).

23
Obr. 3.3 Elektronová vodivost v polovodičích Vlastní vodivost polovodičů není příliš využívána v praxi. Protože vlastní vodivost roste dosti značně s rostoucí teplotou, jsou vlastní polovodiče využívány jako čidla teploměrů, zejména v oblasti nízkých teplot. Z hlediska fyzikálně – chemického se prvky, které mají polovodivé vlastnosti, nacházejí ve zcela určité části Mendělejevovy periodické tabulky prvků (Tab. 3.1). Zde jsou uvedeny tzv. elementární polovodiče, vykazující vlastní vodivost.
V polovodičích dochází i k ději opačnému - k uvolňování elektronů z kovalentních vazeb, zvanému rekombinace (elektron + díra → neutrální atom). Celkový obraz vedení proudu v polovodičích je tedy takový, že v každém okamžiku sice existuje určitý počet volných elektronů a děr, ale volné elektrony se stále obměňují – nové vznikají a jiné rekombinují s dírami. Výsledkem je stav, zvaný dynamická rovnováha. Jako dobu života označujeme průměrnou dobu mezi vznikem páru elektron – díra a rekombinací na neutrální atom. Tato doba může být různá a může být vhodně regulována. V polovodičových součástkách bývá v rozmezí 10 µs – 0,1 µs.
G P
II III IV V VI VII VIII
II Be B C N O F
III Al Si P S Cl
IV Ca Ge As Se Br
V In Sn Sb Te J
VI Pb Bi Po At
Tab. 3.1 Elementární polovodiče v Mendělejevově tabulce prvků

24
3.2 Příměsové polovodiče Pomocí základních termodynamických principů lze, jak již bylo řečeno, dokázat, že nelze vyrobit krystal, ve kterém by nebyly přítomny žádné bodové poruchy (vakance, atomy nečistot). Pokud je cizí atom nežádoucí, hovoříme o nečistotě, pokud je přidán záměrně, hovoříme o příměsi. Vodivost typu N, donory. Vpravíme-li do krystalu polovodiče, složeného ze čtyřvazných atomů, pětivazné atomy (např. fosforu, velmi často pomocí difúze), naváží se tyto atomy kovalentní vazbou na své sousedy (Si, Ge, apod.). Pátý valenční elektron však bude slabě vázaný, snadno se uvolní a stane se z něho vodivostní elektron. Taková vodivost se nazývá vodivostí typu N (negativní) a příměsi, dodávající vodivostní elektrony do polovodiče, se nazývají donory (z latinského donor = dárce). (Obr. 3.4) Vodivost typu P, akceptory. Zavedeme-li do monokrystalu čistého křemíku nebo germania trojvazný prvek (např. kadmium), způsobí tento prvek tzv. děrovou vodivost (Obr. 3.5) a pohybující se částice budou kladné díry, pohybující se ve směru elektrického pole (fyzicky to však budou opět elektrony, jak jsme již dříve vysvětlili).
Hlubší zamyšlení nám objasní jeden z podstatných rozdílů mezi vodivostí vodičů a polovodičů: vodivost čistých vodičů je vysoká a malé procento příměsí ji nijak nezmění. Vodivost čistých polovodičů je velmi malá a již velmi malé procento příměsí ji značně zvýší. Jako silně dotovaný označujeme takový krystal, ve kterém na každých 102–104 atomů základního prvku připadá jeden atom příměsi. Ve slabě dotovaných krystalech připadá jeden dotující atom na 106–109 atomů základní matrice. Difúzní a vodivostní proud v polovodičích. Podobně jako v případě vodičů, pohybují se i v polovodičích nosiče proudu vlivem působení elektrického pole (což jsme již diskutovali). Druhou příčinou pohybu nosičů nábojů v polovodičích může být jev, zvaný difúze. Difúze je podmíněna teplotou systému (roste s teplotou, neboť nosiče nábojů jsou více „postrkovány“ kmity mříže) a koncentračním spádem nosičů nábojů (proudí z míst s vyšší koncentrací do míst s koncentrací nižší). Takový proud se nazývá proudem difúzním.
Obr. 3.4 Polovodič typu N Obr. 3.5 Polovodič typu P
3.3 Přechod PN, dioda Dotujeme-li část krystalu čistého polovodiče (Si,Ge) atomy pětivazného prvku (P,As,Sb), získáme polovodič typu N. Dotujeme-li zbylou část krystalu atomy trojvazného prvku (B,Al,Ga,In), dostaneme polovodič typu P. Část krystalu, rozdělující obě oblasti s různým typem vodivosti, nazýváme PN přechod. Mějme nyní krystal polovodiče, v jehož levé části jsme vytvořili pomocí nějakého technologického postupu (budou diskutovány

25
později) vodivost typu P a v pravé části vodivost typu N. Na Obr. 3.6 je taková situace schematicky zobrazena. V levé části krystalu jsou nosiči nábojů díry (+ v kroužku) a ionty akceptorů jsou tedy nabity záporně. Naopak v pravé části donory odevzdaly do krystalu elektrony a staly se z nich tedy kladně nabité ionty (barva je v obou oblastech různá, abychom tím vyjádřili rozdílné znaménko iontů akceptorů a donorů.
Volné díry i volné elektrony konají v krystalu chaotický pohyb, rekombinují, vznikají nové, atd. Protože v levé části krystalu je nadbytek děr, budou tyto difundovat do pravé části a naopak elektrony z pravé části budou difundovat do části levé (Obr. 3.7). Je třeba zdůraznit, že to není elektrická síla, která řídí přerozdělování nosičů nábojů, ale že je to difúze. Že tedy díry a elektrony se navzájem nepřitahují, jak by se na první pohled mohlo zdát – vždyť celý krystal je navenek neutrální (to bývá nejčastější chyba při interpretaci PN přechodu).
Difúzní proud po jisté době ustane, neboť po děrách zůstala záporně nabitá část krystalu, která je přitahuje zpět a po elektronech tomu je naopak. Dojde tedy opět k tzv. dynamické rovnováze, která má statistický charakter. Jiný způsob představy vzniku PN přechodu je následující: elektrony, které přešly z pravé části do části levé, se naváží na volné vazby u akceptorů (díry) a tím učiní oblast levé části poblíž PN přechodu zápornou a naopak po nich v pravé části poblíž PN přechodu zůstane část krystalu kladná (Obr. 3.8). Tak vzniká v části krystalu, těsně přiléhající k PN přechodu nevykompenzovaný náboj kladný a záporný – tzv. difúzní napětí. Oblast, ve které se nevykompenzovaný elektrický náboj nachází, se nazývá oblastí prostorového náboje. Je třeba zdůraznit, že tento prostorový náboj je tvořen ionty atomů příměsí, pevně zabudovanými v mřížce krystalu a tedy nepohyblivými a nelze proto jeho rozdíl potenciálů změřit například tak, že bychom se dotkli obou oblastí tenkými drátky a snažili se difúzní napětí změřit voltmetrem.
Obr. 3.6 PN přechod Všechny úvahy byly provedeny pro případ, kdy na krystal polovodiče nebylo přiloženo vnější napětí. Ke vzniku prostorového náboje dochází tedy v každém polovodičovém prvku ihned po vytvoření PN přechodu.

26
Obr. 3.7 Difúze na přechodu PN Obr. 3.8 Vznik oblasti prostorovéhonáboje PN přechod, připojený na vnější napětí. Připojíme-li nyní kladný pól baterie např. na oblast typu P a záporný na oblast typu N polovodiče, nebude při malých hodnotách vnějšího napětí procházet téměř žádný proud, neboť záporný prostorový náboj PN přechodu nemůže být elektrony překonán. Teprve po dosažení tzv. difúzního napětí (pro Si cca 0,8 V, pro Ge cca 0,4 V), začne proud prudce vzrůstat. Takové zapojení vnější baterie nazýváme zapojením v propustném směru (Obr. 3.9). Změníme-li polaritu vnější baterie na opačnou, získáme tzv. zapojení v závěrném
směru (Obr. 3.10). V tomto případě kladný pól baterie přitahuje elektrony z oblasti N a záporný pól díry z oblasti P, takže dojde k rozšíření oblasti prostorového náboje a přechodem PN nebude procházet téměř žádný proud. Záměrně jsme zdůraznili slovo téměř, neboť ve skutečnosti jistý velmi malý proud PN přechodem prochází a je třeba se zmínit o jeho podstatě, neboť byť by byl velmi malý, je důležitý. Vznik tohoto proudu lze vysvětlit následovně: již dříve jsme uvedli, že v celém prostoru krystalu existují rekombinační centra (atomy nečistot, vlastní vodivost polovodičů), která jsou zdrojem vzniku párů elektron – díra. Tato rekombinační centra dodávají do obvodu tzv. minoritní nosiče nábojů, které tak tvoří tzv. závěrný proud (Obr. 3.11). Pokud bychom zvyšovali napětí vnější baterie, může dojít ke vzniku lavin, vyvolaných minoritním nosiči proudu. Ty dosahují při vyšším napětí v závěrném směru tak vysoké rychlosti (a energie), že jsou schopné ionizovat nárazem neutrální atomy polovodiče. Tím vzroste počet nosičů nábojů, které se opět urychlí, a proud v závěrném směru roste velmi prudce. Tak velký proud způsobí průraz PN přechodu a zničí celý polovodičový prvek. Měření ukazují, že tzv. kritická hodnota intenzity elektrického pole, při níž dojde k průrazu, je 25 kV/mm. Odtud plyne, že pro reálné PN přechody, vyrobené v krystalech Si, nebo Ge, může být reálná hodnota závěrného napětí řádu několika desítek voltů.
Obr. 3.9 PN přechod, zapojený v propustném směru

27
Obr. 3.10 PN přechod, zapojený v závěrném směru
Obr. 3.11 K vysvětlení závěrného proudu v polovodičích
Poznámka. Zajímavým problémem je otázka, jaké faktory ovlivňují šířku oblasti prostorového náboje. V prvé řadě to bude jistě teplota, neboť teplota ovlivňuje difúzi, která je zodpovědná za vznik oblasti prostorového náboje. Podobně musí být druhým faktorem, určujícím šířku oblasti prostorového náboje koncentrace příměsí, která též silně ovlivňuje difúzi. Z jednoduchých úvah tedy plyne, že šířka oblasti prostorového náboje roste s teplotou a klesá s růstem koncentrace příměsí (k dosažení difúzního napětí stačí příměsi z užší oblasti poblíž přechodu PN).
Diody PππππN a PννννN. Z dosavadních úvah plyne, že nelze dosti dobře dosáhnout vysokého závěrného napětí (k tomu je potřebná velmi nízká koncentrace příměsí) a velkého proudu v propustném směru (k tomu je potřebná vysoká koncentrace příměsí). Aby odstranili tento nesoulad, navrhli Hall a Dunlop diodu nového typu, tzv. diodu PπN (nebo PνN). Taková dioda se skládá ze tří částí s různou vodivostí: silně dotované oblasti typu P, slabě dotované oblasti ν (donory) a nakonec opět silně dotované oblasti typu N (Obr. 3.12). Pokud takovou diodu zapojíme v propustném směru, nemění oblast ν nijak velikost proudu v propustném směru, neboť elektrony z oblastí N a ν (díry z P) ji snadno překonají. Naopak při zapojení v závěrném směru slabě dotovaná střední oblast ν způsobí značné rozšíření oblasti prostorového náboje, takže kritické intenzity elektrického pole (průrazného napětí) se dosahuje až při vysokých hodnotách závěrného napětí (cca 4000 V). Z právě uvedených důvodů se v současné době prakticky všechny polovodičové diody vyrábějí jako diody PπN, či PνN.

28
Obr. 3.12 Dunlopova dioda
3.4. Voltampérová charakteristika diody, speciální diody Z informací, týkajících se funkce PN přechodu a tedy i diod jako elektronických prvků, můžeme učinit kvalitativní závěr o tvaru tzv. voltampérové charakteristiky diody, což je závislost proudu, procházejícího diodou na napětí na ní. Typický tvar takové charakteristiky je uveden na Obr. 3.13. Je třeba poznamenat, že hodnoty, charakterizující kvalitativně křivku na tomto obrázku se mohou značně lišit pro různé konkrétní diody.
Obr. 3.13 Voltampérová charakteristika běžné a inverzní diody Zenerova dioda. V případě silně dotovaného přechodu PN, bude oblast prostorového náboje velmi úzká. V závěrném směru potom nedojde k průrazu, neboť nebude dosti prostoru pro vznik laviny. V praxi využíváme závěrné části voltampérové charakteristiky jako stabilizátoru napětí.
Inverzní dioda. Při ještě vyšším stupni dotace bude oblast prostorového náboje ještě užší, než u Zenerovy diody a průrazné napětí v závěrném směru bude nižší, než difúzní napětí ve směru propustném (Obr. 3.13). Inverzní diody se používají k usměrňování velmi nízkých střídavých napětí (dioda usměrňuje v závěrném směru – proto přívlastek inverzní).
Tunelová dioda. Ještě vyšší dotace příměsemi než tomu bylo v případě inverzní diody, je ovlivněna i voltampérová charakteristika v propustném směru a to tak, že na ní vznikne oblast se záporným odporem (Obr. 3.14). Mechanismus vedení proudu v této diodě lze objasnit pouze na základě poznatků z kvantové mechaniky pomocí tzv. tunelového jevu. Tunelové diody se používají při konstrukci vysokofrekvenčních generátorů jako zdroje elektromagnetických kmitů (jako oscilátory).

29
Obr. 3.14 Voltampérová charakteristika tunelové diody
Obr. 3.15 Princip činnosti tranzistoru [6]
3.5 Tranzistor V historickém kontextu lze polovodičové diody chápat jako mnohem úspornější a mnohem menší (co do objemu) náhrady diod vakuových. Vědci se analogicky snažili najít polovodičovou náhradu za vakuovou triodu, tj. elektronku se třemi elektrodami, která tím, že se stala základem zesilovačů a oscilátorů, předznamenala bouřlivý rozvoj radiotechniky v polovině minulého století.
Byli to američtí vědci, Bardeen, Brattain a Shockley, kteří tuto úlohu úspěšně vyřešili tím, že sestrojili tranzistor (a byli za tento vynález oceněni Nobelovou cenou). Tranzistor se skládá ze tří oblastí typu NPN, nebo PNP. Elektrické vývody těchto oblastí značíme jako báze, emitor a kolektor (Obr. 3.16). Tranzistor si můžeme představit jako dvě diody, zapojené proti sobě. V případě tranzistoru NPN, zapojeného v zapojení se společnou bází, je první dioda NP (editor – báze) zapojena v propustném směru a druhá dioda PN (báze – kolektor) je připojena na baterii s mnohem vyšším napětím ve směru závěrném. Elektrony z emitoru přicházejí do oblasti báze, která je konstruována jako úzká oblast mezi emitorem a kolektorem a proto jenom velmi malá část elektronů vytvoří tzv. proud báze Ib a převážná část jich přejde do oblasti kolektoru, kam je vtažena mnohem vyšším kolektorovým napětím a vytvoří tzv. kolektorový proud Ik. V zapojení se společnou bází (Obr. 3.16) je proudový
zesilovací činitel e
k
I
I=α poněkud menší, než 1, ovšem napěťové a výkonové zesílení je vždy
větší než 1. Snadno si můžeme podobnými úvahami odvodit, že při zapojení tranzistoru se společným emitorem, nebo se společným kolektorem je i proudový zesilovací činitel větší jak 1. Dokazuje to zapojení se společným emitorem na Obr. 3.17: malý proud báze řídí velký kolektorový proud.

30
Obr. 3.16 Zapojení transistoru se společnou bází
Obr. 3.17 Zapojení tranzistoru se společným emitorem
Literatura [1] Dioda, transistor a tyristor názorně. Programovaný kurs. SNTL Praha 1983.
[2] Halliday, D., Resnick, R., Walker, J.: Fyzika. VUTIUM Brno, PROMETHEUS Praha, 2000.
[3] Putilov, K.A.: Kurs fiziki II, Fizmatgiz 1959.
[4] Horák, Z., Krupka, F., Šindelář, V.: Technická fyzika, SNTL Praha 1960.
[5] Fuka, J, Havelka, B.: Elektřina a magnetismus. SPN Praha 1965.
[6] Encyklopedie Wikipedia.

31
4. Katalýza – základní pojmy Katalýza je definována jako jev změny rychlosti reakce, nebo jejího vzniku, ke kterým dochází vlivem některých látek, které nazýváme katalyzátory. Ty se účastní tohoto procesu, samy však zůstávají nezměněny.
Jak je známo, vliv katalyzátoru může být poměrně silný a jejich působením mohou být chemické reakce urychleny více než milionkrát. Vlivem katalyzátorů může docházet k reakcím, ke kterým by za daných podmínek bez nich nemohlo prakticky dojít. Katalýza je kladná, jestliže katalyzátor zvyšuje rychlost reakce a záporná, jestliže naopak katalyzátor tuto rychlost snižuje. Zpomalující působení záporných katalyzátorů je nezřídka podmíněno tím, že snižují aktivitu kladného katalyzátoru tak, že jej „otravují“. Dále se budeme věnovat přednostně kladné katalýze a budeme ji nazývat jednoduše katalýzou. Reakce, které jsou katalyticky urychlovány produkty, které vznikly v téže reakci, nazýváme autokatalytickými (sebe sama urychlujícími). Rychlost těchto reakcí roste s časem.
Pod pojmem katalýza chápeme jevy, lišící se značně co se týká mechanismu působení katalyzátorů.
Tak například v řetězových reakcích mohou kladné katalyzátory usnadňovat vznik řetězců (například působení par kovového sodíku na směs chlóru s vodíkem). Záporné katalyzátory mohou v řetězových reakcích působit tak, že naruší řetězce. Příkladem mohou být produkty rozkladu tetraethylu olova, nebo karbonylu železa, přidávané do benzínu kvůli odstranění možnosti předčasných zápalů ve válci motoru.
V některých jiných reakcích katalyzátorů se může vytvářet krátkodobá sloučenina katalyzátoru s jednou z reagujících látek – v tomto případě získáváme konečné produkty snadněni (s nižší aktivační energií). Předpokládejme například, že v obecné reakci
A + B = AB (4.1) Může katalyzátor K vzájemně reagovat s látkou A a vytvořit krátkodobou sloučeninu AK:
A + K = AK (4.2) Sloučenina AK vzájemně reaguje s B a může vytvořit AB tak, že vyloučí K ve volném stavu podle reakce
AK + B = AB + K (4.3) Jestliže je energie reakcí (b) a (c) podstatně menší, než energie reakce (a), potom vzájemné působení probíhá s vyšší rychlostí cestou přes vytváření přechodné sloučeniny typu AK a K je kladným katalyzátorem reakce (a). Avšak společné pro všechny katalytické procesy je to, že katalyzátory mohou vzbuzovat reakci, nebo měnit její rychlost, ale nikoliv
hranice jejího průběhu za daných podmínek, tj. nemění stav rovnováhy. Pouze usnadňují,
nebo zpomalují dosažení rovnováhy.
Při velké koncentraci katalyzátoru může dojít k jisté změně rovnovážné polohy, způsobené změnou složení prostředí (a nikoliv katalytickým působením). V důsledku nedostatečné unifikace terminologie se jindy řadí ke katalytickým vlivům i posun rovnováhy pod vlivem rozpouštědla, nebo změny velikosti povrchu dané fáze.
Katalyzátory vždy mění aktivační energii (snižují ji při kladné katalýze).

32
V Tab. 4.1 jsou uvedeny příklady snížení aktivační energie pomocí katalyzátoru v některých reakcích.
Tab. 4.1 Snížení aktivační energie E* (v J/mol) při katalýze) [1]
Reakce Katalyzátor E* bez
katalyzátoru E*
s katalyzátorem
Rozklad amoniaku W 2,96.105 1,63.105
Rozklad oxidu dusíku Pt 2,45.105 1,36.105
Rozklad sacharózy v roztoku kyseliny solné
Invertáza (ferment)
1,07.105 3,9.104
Různé katalytické reakce se obvykle rozdělují na reakce homogenní katalýzy a reakce heterogenní katalýzy. K prvním z nich se vztahují takové reakce, ve kterých se katalyzátor nachází v homogenní plynné, nebo kapalné směsi s reagujícími látkami. V heterogenní katalýze se katalyzátor nachází ve formě samostatné fáze a vzájemné působení probíhá na jeho povrchu. V této hlavě probereme pouze homogenní katalytické reakce a nejdříve se budeme věnovat reakcím v plynech.
4.1 Homogenní katalýza Jako příklad homogenních katalytických reakcí v plynech může sloužit okysličování SO2 (nebo přesněji siřičité kyseliny) pomocí oxidů dusíku jako katalyzátoru, používaných v komorovém a věžovém způsobu výroby kyseliny sírové. V tomto procesu se okysličování SO2 uskutečňuje pomocí NO2 tak, že se okysličí vzdušným kyslíkem. Některé detaily tohoto procesu lze interpretovat různě, ale je jasné, že katalýza vytváří meziprodukty, ze kterých se poté regeneruje.
Podobným způsobem fungují katalyzátory i v řadě jiných reakcí. Například rozklad acetylaldehydu na metan a oxid uhličitý:
CH3CHO = CH4 + CO (4.4) Tento proces je katalyzován parami jódu. Při 518 0C je aktivační energie této reakce bez přítomnosti katalyzátoru rovna 1,90.105 J. V přítomnosti par jódu se sníží na 1,36.105 J a rychlostní konstanta reakce vzroste asi 10000 krát. K tomu dojde proto, že v přítomnosti jódových par má reakce dvě stádia:
CH3CHO + I2 = CH3I + HI + CO (4.5) CH3I + HI = CH4 + I2 (4.6) A pro tyto reakce je aktivační energie nižší. Jak je vidět, i v tomto případě působí katalyzátor při vytváření meziproduktů, ze kterých se poté úplně regeneruje. V mnoha případech se daří určit meziprodukty za použití citlivých metod zkoumání – např. metod spektrálních, apod.
Homogenní katalýza je v roztocích nejčastěji způsobována působením vodíkových, nebo hydroxylových iontů. Katalytické působení kyselin bylo objeveno K. Kirchhoffem (1811). Inverze cukru, máselnatění složitých esterů, hydrolytický rozklad amidů, acetylů

33
a mnoho jiných reakcí v roztocích je urychlováno vodíkovými ionty, přičemž zvýšení jejich koncentrace způsobuje přibližně přímoúměrné zvýšení rychlosti. V homogenních katalytických reakcích v roztocích je v mnoha případech také bezpečně stanoveno vytváření meziproduktů s účastí katalyzátorů. Tak např. N. I. Kobarev ukázal, že ionty Cr2O7
2, WO42,
MoO42, katalyzující rozklad peroxydu vodíku, vytváří s ním meziprodukty, které se rozpadají
s vyloučením kyslíku. Posledním procesem je určena kinetika celé reakce.
Spolu s vytvářením takových poměrně stabilních meziproduktů, jejichž přítomnost může být dokázána, hraje v homogenní katalýze roli i časově mnohem kratší vázání iontů a molekul, způsobené ion – dipólovým vzájemným působením, nebo vytvářením vodíkové vazby. Při takových vzájemných působeních může docházet k polarizaci těchto částic a změně jejich reakční schopnosti.
V nedávné době byla objevena nová katalytická metoda, založená na použití laseru. Zvýšení teploty způsobuje zintenzívnění kmitů všech atomů a skupin atomů a při dostatečném zahřátí vede k roztržení nejdříve těch nejslabších vazeb. Na rozdíl od toho působením paprsků laseru, díky velmi přesně nastavené frekvenci kmitů může dojít k zesílení kmitů určitého typu a nakonec rozrušení dané vazby, nikoliv nutně té nejslabší. Vysoká ostrost naladění laseru dovoluje v určitých případech selektivně vybrat narušení daného typu vazeb v molekulách určitého izomeru, nebo dokonce v molekulách, obsahujících určitý izotop daného prvku, což může být použito pro jejich rozdělení (selekci).
Tento typ katalýzy lze dát do souvislosti s fotokatalýzou.
4.2 Fotochemické reakce V tomto případě se jedná o reakce, které probíhají při působení světla. Přesně řečeno fotochemické jsou všechny reakce, ve kterých je energie, potřebná pro jejich průběh,
nebo vznik energie elektromagnetických kmitů – viditelného světla, ultrafialových paprsků,
nebo méně často paprsků infračervených. K takovým reakcím může docházet jak v plynech, tak i v kapalinách a pevných látkách
Fotochemické reakce mohou být různé. Patří k nim nejen reakce fotosyntézy uhlovodíků, ke kterým dochází v rostlinách za přítomnosti slunečního záření, ale i reakce, které jsou základem fotografického procesu, jevy luminiscence, apod.
Co do chemické podstaty jsou fotochemické reakce velmi různorodé. Vlivem světla může docházet k reakcím syntézy (fosgen, chlorovodík), rozklad (H2O2), okysličování a další.
F. Ch. Grottus (1817) stanovil, že chemicky aktivní je pouze to světlo, které je pohlcováno reakčním prostředím a K. A. Timirjazev (1875) ukázal, že množství produktů, získaných při dané fotochemické reakci je úměrné množství pohlcené světelné energie. Fotochemické působení světla spočívá v tom, že atomy nebo molekuly reagující látky pohlcujíc světlená kvanta se nabuzují, tj. zvyšuje se jejich vnitřní energie a zejména ta její forma, která má vliv na průběh dané reakce (v některých případech způsobuje i disociaci molekul).
Podle toho lze rozdělit fotochemické reakce na dvě skupiny:
1. reakce, které i bez působení světla mohou termodynamicky za daných podmínek probíhat samovolně (například reakce H2 + Cl2 = 2HCl.). V tomto případě hraje světlo roli pouze takovou, že nabudí reakci a pomáhají překonat (nebo snížit) její vysokou aktivační energii. Přitom množství látky v procesu nemusí být úměrné množství pohlcené světelné energie. Takové reakce nazýváme fotokatalytickými.
2. Reakce, které za daných podmínek nemohou termodynamicky probíhat samovolně, a pro jejich uskutečnění je třeba dodat energii zvnějšku. Tuto práci dodáváme

34
ve formě elektromagnetického záření. V případě těchto reakcí množství reagujících látek jsou úměrná pohlcené energii v souladu se zákonem fotochemické ekvivalence.
Nejdůležitější z fotochemických reakcí takového typu je fotosyntéza, ke které dochází v rostlinách. Tyto reakce jsou silně endotermické a jsou doprovázeny růstem izobaricko-izotermického potenciálu (∆G > 0). Bez světla nemohou samovolně probíhat. K této skupině reakcí patří také reakce fotochemického rozkladu látek (reakce fotolýzy). Tak např. pod vlivem ultrafialových paprsků v oblasti vlnových délek 160–220 nm dochází k částečnému rozkladu amoniaku, zejména při vysoké teplotě. Některé aldehydy a ketony jsou také podrobeny fotolýze za podobných podmínek. Vztah mezi množstvím pohlcené energie a množstvím reagujících látek je dán zákonem fotochemické ekvivalence, který odvodil na základě termodynamických úvah A. Einstein (1912). Ten je ve své podstatě vyjádřením zákona zachování energie, aplikovaným na dané procesy. Podle tohoto zákona každá molekula, reagující pod vlivem
světla, pohlcuje jedno kvantum záření, způsobující reakci. Tedy, množství energie
λν chN
hNE A
.. 0==
(4.7)
kde NA je Avogadrovo číslo, ν je frekvence kmitů, c je rychlost světla a λ vlnová délka. Tedy množství energie, pohlcené molekulou závisí na vlnové délce elektromagnetic-
kých kmitů a je jí nepřímo úměrné. Vyšší energii a vyšší chemickou aktivitu mají kmity s kratší vlnovou délkou. Ve viditelném světle jsou nejaktivnější fialové paprsky (λ kolem 400 nm). Pro ně je E = 2,97.105 J/mol. Nejméně aktivní je červená část spektra (λ ∼ 750 nm), pro ni je E ~ 1,6.105 J/mol. Proto lze málo citlivé (přesněji nesenzibilizované) fotomateriály – fotopapíry a jiné – vyvolávat při červeném světle.
Při experimentálním prověření zákonů fotochemické ekvivalence se často objevují zdánlivé rozpory s výsledky pokusu. Kritériem použitelnosti bývá často kvantový výtěžek reakce. Kvantovým výtěžkem φ nazýváme poměr počtu molekul, u kterých došlo k dané
přeměně, k počtu pohlcených kvant elektromagnetického záření:
Φ = (Počet zreagovaných molekul dané látky) / (Počet pohlcených světelných kvant)
Podle zákona fotochemické ekvivalence se musí kvantový výtěžek rovnat jedné. A skutečně, většina reakcí odpovídá této podmínce. Avšak spolu s tím se nezřídka setkáváme i s takovými reakcemi, u nichž je kvantový výtěžek různý od jedné, nahoru nebo dolů, přičemž v případě prvním se jedná často o značné odchylky.
Odklon na stranu vyšších hodnot pozorujeme v těch případech, kdy je reakce za daných podmínek řetězová, tj. když přeměna, způsobená pohlcením jednoho kvanta světla molekulou vede ke vzniku aktivních částic, volných radikálů, valenčně nenasycených atomů, nebo nabuzených molekul, které vzájemně interagujíc s jinými molekulami, mohou v nich způsobit podobné změny. V tomto případě, třebaže počáteční vzájemná působení odpovídají zákonu ekvivalence, celkový kvantový výtěžek produktu bude větší, než jedna.
Při nevelké délce řetězců nemůže být kvantový výtěžek o moc větší, než jedna, ale při velké délce může být celkem velký a například při fotochemickém uskutečnění reakce
H2 + Cl2 = 2 HCl se může rovnat 106. Protože délka řetězců závisí na podmínkách uskutečnění reakce, potom i kvantový výtěžek v takových případech na nich závisí.

35
Odklon na stranu nižších hodnot φ pozorujeme v těch případech, kdy kvanta světla, pohlcená molekulami dané látky, se částečně spotřebují na nějaké vedlejší procesy (kdy se například energie molekuly předá při srážce molekule jiné látky), nebo když jsou jako celek pohlcovány molekulami jiných látek, přítomných v soustavě. Tyto zdroje odchylek hrají důležitou roli při průběhu fotochemických reakcí v roztocích.
Kromě toho kvantový výtěžek se může zmenšit v důsledku samovolného průběhu opačného procesu. Tak například při reakcích fotochemické disociace molekul Br2 na atomy, tyto se mohou částečně znovu spojovat na molekuly (rekombinace atomů bromu).
Všechny tyto odchylky jsou způsobovány tedy různými sekundárními procesy. Primární přeměny, způsobené pohlcením světla, jsou dobře popsány zákonem fotochemické ekvivalence
Při pohlcení elektromagnetických kmitů látka získává zvnějšku energii a její izotermický potenciál se zvyšuje. Molekuly, které pohltily světlo, se stanou schopnými přeměny, kterou by bez toho nebyly schopny uskutečnit.
Význam fotochemických reakcí není vyčerpán typy reakcí, které jsme uvedli. Řada fotochemických reakcí je používána v chemickém průmyslu. Důležitým problémem do budoucna je možnost technického využití fotochemických reakcí v energetice s cílem využít energii slunečního záření.
Literatura [1] KIREEV, V. A.: Kurz fizičeskoj chimii (1975)
[2] HÁLA, E., REISER, A.: Fysikální chemie. NČSAV Praha 1960.
5 Fotokatalýza
5.1 Objev fotokatalýzy Rozvoj fotokatalýzy je spojen zejména se jménem japonského profesora Fujishimy, který v roce 1967 ještě jako postgraduální student objevil následující jev: do vodného roztoku umístil titanovou a platinovou elektrodu a celou soustavu osvětlil silným světlem [2]. Pozoroval, že na obou elektrodách se objevují bublinky plynu (později bylo zjištěno, že na titanové elektrodě se vyvíjí kyslík a na platině vodík). Tento jev, označený jako Honda-Fujishimův jev (H – F jev) byl zprvu přijat s nedůvěrou, kterou částečně podporovala i malá účinnost energetické konverze (0,3%). Jako zdroj energie tento jev bude zřejmě zatím nepoužitelný, ovšem velmi brzy po objevu byly nalezeny oblasti, v nichž lze H-F jev s úspěchem využít. Zasloužili se o to kromě prof. Fujishimy zejména Dr. Hashimoto a Dr. Watanabe (rozklad nečistot, antibakteriální účinky, atd. – viz další části článku). Přehled všech možností je uveden v [3,4].
5.2 Mechanismus fotokatalýzy Již jsme uvedli, že fotokatalýza probíhá na povrchu polovodiče. Polovodič se svým složením a fyzikálními vlastnostmi nejvíce blíží izolátorům. Obvykle jsou jako polovodiče označovány látky s pásem zakázaných energií nižším, než 3,2 eV. Je-li uvedený pás širší, hovoříme o izolátoru [5]. Z různých elementárních polovodičů a jiných látek s polovodivými vlastnostmi se pro fotokatalýzu nejlépe hodí oxid titaničitý TiO2. Ozáříme-li polovodič světlem, jehož kvanta mají energii vyšší, než je šířka zakázaného pásu energií, dojde v něm ke vzniku páru díra – elektron (elektron ve vodivostním, díra ve valenčním pásu). Pokud nějaký

36
vhodný akceptor nezachytí tyto defekty, dojde během několika nanosekund k jejich rekombinaci. V opačném případě se ukazuje, že díry (h+) ve valenčním pásu působí jako oxidanty a elektrony (e-) ve vodivostním pásu jako resultanty. Na povrchu polovodiče tedy současně probíhají dvě reakce, jichž se účastní látky zde adsorbované [2].
Obr. 5.1 Fotokatalytické zařízení na solární konverzi energie [4]
Obr. 5.2 Společné rysy fotokatalýzy a fotosyntézy [10]

37
Obr. 5.3 Fyzikální princip fotokatalýzy [10]
Obr. 5.4 Princip fotokatalýzy [4]

38
Obr. 5.5 Princip fotokatalýzy [9]
5.3 Oxid titaničitý Již bylo uvedeno, že oxid titaničitý se zatím jeví jako nejvhodnější polovodič pro fotokatalýzu. V Tab. 1. jsou uvedeny některé další oxidy s polovodivými vlastnostmi, které by též mohly sloužit k fotokatalytickým účelům (mají vhodnou šířku pásu zakázaných energií ∆E). Všechny však buď snadno podléhají korozi, nebo jsou chemicky nestabilní [7]. Dále podrobněji uvedeme základní vlastnosti TiO2, jako zatím nejslibnějšího oxidu, vhodného pro fotokatalýzu. K pozitivním vlastnostem oxidu titaničitého patří zejména jeho fotostabilita, korozivzdornost, netoxicita, vysoká fotokatalytická aktivita a cenová dostupnost. Podobně výjimečné mechanické vlastnosti má ostatně i čistý titan v kovové formě (korozivzdornost, nízká hustota, vysoká pevnost a snášenlivost s lidskou tkání). Přestože je po hliníku a železe nejrozšířenějším kovem v Zemské kůře, je v ní bohužel značně rozptýlen a téměř netvoří ložiska. Proto je čistý kovový titan zatím poměrně drahý.
Oxid titaničitý se v přírodě nachází ve třech modifikacích jako anatas, rutil a brookit. Anatas má strukturu tetragonální za nízkých teplot, rutil má tutéž strukturu za vysokých teplot. Brookit má strukturu ortorombickou.
Nejvhodnější fotokatalytické vlastnosti má TiO2 ve struktuře anatasu. Je tomu tak proto, že šířka pásu zakázaných energií je pro anatas 3,23 eV, což odpovídá UV záření o vlnové délce 388 nm, zatímco pro rutil je šířka zakázaného pásu energií 3,02 eV a tomu odpovídající vlnová délka UV záření je 413 nm. Elektrony, vzniklé v anatasu mají tedy větší redukční schopnost než elektrony v rutilu (mají vyšší energii).
Co se týká struktury, užívá se pro fotokatalytické účely oxid titaničitý ve dvou formách – ve formě suspenze bílého prášku a ve formě tenké vrstvy, nanesené na substrátu (např. na skle). Efektivita fotokatalýzy na tenké vrstvě je sice nižší, ale má lepší praktické využití, jak později zdůvodníme. Příprava fotoaktivních vrstev je v podstatě dvojí:
• chemický způsob (tzv. metoda sol – gel), [7]
• fyzikální způsob (např. v nízkoteplotním plazmatu). [8]

39
Tab. 5.1
Polovodič ∆∆∆∆E (eV)
ZnO 3,2
ZnS 3,6
α-Fe2O3 3,1
WO3 2,8
SrTiO3 3,2
5.4 Zdroje záření Otázkou podstaty světla se již od 17. století zabývali tak významní vědci, jako byl G. Galilei, I. Newton, Ch. Huyghens, T. Young a další. Velký pokrok v objasnění této otázky učinil až J. C. Maxwell, který dokázal, že světlo je postupná vlna, tvořená elektrickým a magnetickým polem – tzv. elektromagnetická vlna. Tím se optika značně přiblížila k elektromagnetismu (což bylo experimentálně potvrzeno pokusy H. Hertze).
Na Obr. 5.6 je uvedeno tzv. elektromagnetické spektrum, se kterým se setkáváme v běžném životě. Jsme pronikáni rádiovými a televizními signály, mikrovlnami radarů a mobilů. Interagujeme s viditelným světlem (očima), světlem infračerveným (pocit tepla na kůži), světlem ultrafialovým (při neopatrném slunění). Rentgenové záření námi proniká, aniž je nějak registrujeme, stejně jako záření radioaktivní či kosmické paprsky.
Viditelná část spektra byla a je námi snad nejvíce využívána a zahrnuje oblast vlnových délek od 430 nm po 690 nm.
Protože fotokatalýza je stimulována UV zářením, všimneme si dále podrobněji právě této oblasti elektromagnetického záření. Zdroji tohoto záření mohou být kromě Slunce (Obr. 5.7) rtuťové výbojky nízkotlaké a vysokotlaké, xenonové vysokotlaké výbojky, sodíkové výbojky a některé další speciální zdroje [5].
Ultrafialové záření
Ultrafialové záření se v elektromagnetickém spektru nachází v oblasti přiléhající k fialové části viditelného spektra a ze strany kratších vlnových délek je ohraničeno zářením rentgenovým. Za objevitele UV záření je pokládán německý fyzik J. W. Ritter (1801).
Podle biologických účinků se UV záření dělí zpravidla na tři základní oblasti:
UVA 400–320 nm dlouhovlnné
UVB 320–280 nm středovlnné
UVC pod 280 nm krátkovlnné
UV záření je z větší části pohlcováno ozónovou vrstvou, která se nachází ve stratosféře.
UVA záření má nejdelší vlnovou délku a tvoří většinu veškerého UV záření, které projde ozónovou vrstvou a dopadne na povrch Země (ozón a kyslík propustí na povrch Země asi 1/3 UVA záření). Někdy je vhodné dělit tuto oblast na dvě části, UVA I (340–400 nm) a UVA II (320–340 nm).

40
UVB záření je téměř veškeré absorbováno ozónovou vrstvou v atmosféře. Jeho účinky na živé organismy jsou biologicky významné a často mohou být zhoubné: rozkládá, nebo narušuje bílkoviny a jiné organické sloučeniny, což má vážné následky pro metabolismus jedince, narušení DNA a vznik rakoviny. Například zvýšení intenzity UVB záření o 2 % zvyšuje výskyt rakoviny kůže o 3–6%. Může dojít i k poškození očí (čípků a tyčinek) – tzv. sněžná slepota a šedý zákal. Negativně ovlivňuje růst zelených rostlin, účinnost procesu fotosyntézy apod. U dvou třetin hospodářských plodin snižuje jejich produkci.
UVC záření je nejtvrdší. Vytváří ozón tak, že z 3 molekul kyslíku O2 vytvoří dvě molekuly ozónu O3 a touto reakcí je absorbováno. Toto záření je prokazatelně karcinogenní a má charakter záření ionizujícího. Je také nazýváno germicidním (dezinfekčním) – užívá se k dezinfekci sálů ve zdravotnictví.
Obr. 5.6 Elektromagnetické spektrum
Obr. 5.6 Složení a energie Slunečního záření [3]

41
UV záření má však i pozitivní účinky. Je hybnou silou evoluce (vznik mutací), vytváří vitamín D v kůži a využívá se k léčení některých kožních nemocí (lupénka, vitiligo).
5.5 Využití fotokatalýzy Poněvadž účinnost procesu fotokatalýzy vody je po ozáření viditelným světlem pouze asi 0,3 %, je využití tohoto procesu pro energetické účely zatím neefektivní. Poměrně rychle však bylo nalezeno několik oblastí využití tohoto jevu a další možnosti nejsou ještě vyčerpány. Uveďme alespoň některé z nich (viz Obr. 5.7, 5.9).
Obr. 5.7 Stručný přehled využití fotokatalýzy v technické a environmentální praxi [4]
Obr. 5.8 Využití fotokatalýzy na TiO2 [10]

42
Obr. 5.9 Využití fotokatalýzy na TiO2 [10]
5.5.1 Ničení mikrobů a virů
Již jsme uvedli, že na povrchu TiO2 může po ozáření UV světlem dojít k rozkladu organických látek. Těmito látkami mohou být nejen ropné produkty, tabákový dehet, ale i živé organické látky, jako jsou bakterie, viry, houby a plísně. Tak například je známo, že v nemocnicích se rozmnožují patogenní bakterie, odolné proti většině běžných antibiotik (např. Staphylococus aureus). Tyto bakterie napadají zejména méně odolné a přestárlé pacienty. Proti mikrobům a virům byly vyvinuty oxidem titaničitým pokryté antimikrobiální dlaždice [4].
Praxe výroby těchto dlaždic je následující: na glazovanou dlaždici je nastříkána suspenze, obsahující jemný oxid titaničitý a poté vypálena při teplotě kolem 800 0C, takže dojde ke vzniku tenké vrstvičky TiO2 o tloušťce řádu mikrometrů. Trvanlivost takové vrstvičky je cca 10 let. Protože dlaždice, nacházející se v přítmí by neplnily svoji funkci, zdokonalili vědci technologii výroby dlaždic přídavkem kladně nabitých iontů stříbra či mědi. Roztok soli jednoho z těchto kovů se nastříká na povrch dlaždice s TiO2 a po ozáření UV světlem se velmi malé částice kovu pevně uchytí na povrchu dlaždic. Fotokatalytická metoda uchycení atomů Ag či Cu na povrchu má výhodu oproti klasické metodě, ve které jsou kovy přimíchány do glazury a poté vypáleny v tom, že hustota atomů kovů na povrchu je mnohem vyšší (atomy uvnitř glazury se chemických reakcí pochopitelně neúčastní – Obr. 5). Zajímavým experimentálním faktem je, že fotokatalytické antibakteriální dlaždice mají velmi slibnou trvanlivost.
Při praktickém testování účinnosti této metody ve srovnání s klasickou chemickou dezinfekcí bylo zjištěno, že množství bakterií na fotokatalytických dlaždicích kleslo na nulu, zatímco po chemické dezinfekci vždy nějaké bakterie přežívají [3].
Fotokatalytické antibakteriální dlaždice jsou ideálním obkladem do sprch a na toalety. Bylo například zjištěno, že tvorba nepříjemně páchnoucího amoniaku na veřejných záchodcích poklesla trvale na 5x nižší hodnotu.

43
Obr. 5.10 Fotoelektrické antibakteriální dlaždice [4]
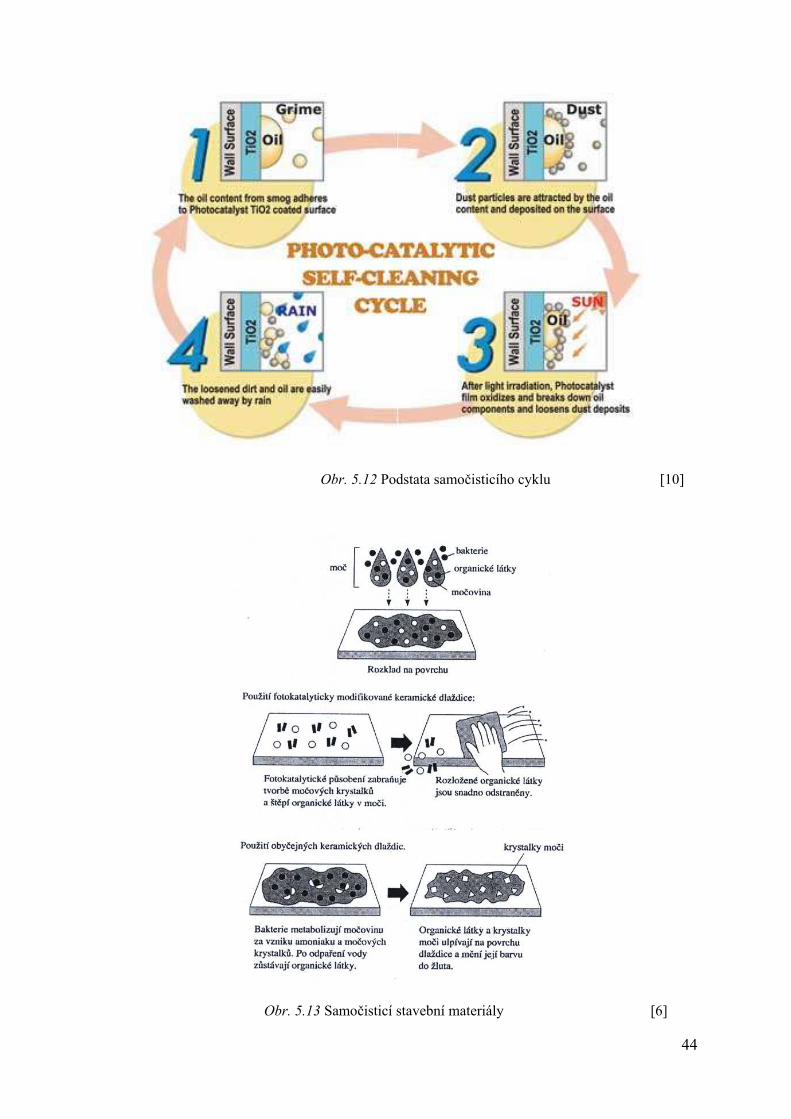
5.5.2 Samočisticí stavební materiály
Stěny budov, obložené kachlemi po čase ztrácejí svůj lesk a je potřeba je nákladně čistit. Usazeniny na stěnách jsou převážně organického původu a ukazuje se, že při použití fotokatalytických dlaždic je nutnost čištění nesrovnatelně nižší. Podobně jako na toaletách lze využít tyto dlaždice i v kuchyni, neboť kuchyňská odpadní voda obsahuje zejména organický odpad (Obr. 5.11–5.13).
Fotokatalytický účinek tenké vrstvy TiO2 pozorujeme i na křemenném skle, kde je deponován. Na běžném sodnovápenatém skle je fotokatalytický účinek TiO2 velmi slabý. Zprvu byla jistým technickým problémem průhlednost vrstvičky TiO2, ale i ten se podařilo vyřešit. [4] Samočisticí se sklo se již využívá například u lamp, osvětlujících tunely, kde je čištění obzvláště obtížné. Oxidem titaničitým lze pokrýt i některé tvrdé organické látky (např. tarpaulin) a jeho funkce je stejná, jako v předcházejících případech (možnost využití např. v automobilovém průmyslu).
Obr. 5.11 Samočisticí povrchy TiO2 [9]
44
Obr. 5.12 Podstata samočisticího cyklu [10]
Obr. 5.13 Samočisticí stavební materiály [6]

45
Obr. 5.14 K objasnění hydrofilnosti materiálů [4]
5.5.3 Superhydrofilnost
Hydrofobní materiály se vyznačují tím, že kapičky vody na nich mají velký kontaktní úhel (sklo ∼ 200, plasty ∼800). Naopak hydrofilní materiály vykazují tento úhel velmi malý. Tenká vrstva, tvořená TiO2 vykazuje velikost tohoto úhlu několik desítek stupňů. Osvětlíme-li jej však UV světlem, úhel se zmenší až téměř na nulu (kapičky se rozplynou). V tomto případě se jedná o tzv. superhydrofilní stav, který takovým zůstává až dva dny, i když není vystaven UV paprskům. Potom se kontaktní úhel začíná zvětšovat a povrch se stane opět hydrofobním. Další ozáření UV paprsky opět nastolí superhydrofilní stav. (Obr. 5.15) Využití tohoto jevu se přímo nabízí tam, kde nám vodní zamlžení nějakého objektu (zpětných zrcátek a čelních skel automobilů, zrcadel v koupelnách, apod.) ztěžuje nebo znemožňuje vidění.
Obr. 5.15 K možnosti fotokatalytického čistění vzduchu [4]

46
5.5.4 Fotokatalytické čistění vzduchu.
Zvláštností fotokatalytické reakce na povrchu TiO2 je skutečnost, že energie, která je k dispozici pro chemické děje, odpovídá vnitřní energii při teplotě 30 000 0C (ovšem ve velmi malém prostoru řádu rozměrů atomů až molekul, takže povrch TiO2 se na tuto teplotu neohřeje). Při takové teplotě dochází k okamžitému rozkladu organických látek na CO2 a H2O. V knize [4] je uvedena velmi názorná analogie, která tento děj přirovnává k odhození hořící zápalky do plaveckého bazénu. Oheň se uhasí okamžitě, ale teplota vody v bazénu se nezvýší. Pokud je ale v místě kontaktu např. komár, okamžitě shoří (komár = organická molekula). Protože fotokatalýza je proces kontinuální, je třeba tuto analogii doplnit představou, že zápalka stále hoří a pohybuje se po povrchu vody (fosforové zápalky tuto vlastnost mají), dokud na komára nenarazí. Spálí ho, ale nezhasne a pohybuje se dále.
Fotokatalytické čistění vzduchu je technicky nejvíce použitelné tam, kde se jedná o nízké koncentrace nepříjemně zapáchajících látek ve vzduchu (acetaldehyd, merkaptan, apod.).
Vzduch ve velkoměstech bývá zatížen velkým množstvím škodlivých plynů, jako je např. SO2, NO2, NO, apod., pocházejících zejména z automobilových motorů, elektráren a tepláren. Tyto plyny mohou být rozloženy pomocí TiO2, umístěného např. na stěnách budov ve městech. Při takovém rozkladu však vznikají některé škodlivé produkty, jako je slabá kyselina sírová a dusičná. V tomto případě je řešení jednoduché – fotokatalyticky působící plochy jsou umístěny venku na stěnách a střechách budov a vzniklé produkty jsou spláchnuty deštěm (Obr. 5.15).
Obr. 5.16 Fotokatalytické čištění vody [4]

47
5.5.5 Fotokatalytické čištění vody
Fotokatalytické reakce na povrchu práškového TiO2 lze využít i v případě čištění vody. Problémem však zůstává závěrečné odstranění takového prášku z již vyčištěné vody (filtrace je obtížná). Ve světe se zkouší několik možných systémů, například skleněné kuličky, potažené TiO2 (Obr. 9).

48
6 Závěr
Předložená práce si klade za cíl podnítit a motivovat studenty středních a vysokých škol ke studiu přírodních věd v čele s fyzikou. Jsou zde uvedeny základní principy fotokatalýzy, která se jeví jako velmi slibná metoda s významnými technickými a environmentálními aplikacemi. K pochopení základních principů fotokatalýzy jsou potřebné alespoň středoškolské znalosti fyziky a chemie. Zájemce o projektovou výuku z této oblasti si může vybrat i některé dílčí problémy, související s fotokatalýzou (chemická katalýza, povrchové napětí, povrchová energie, fyzika polovodičů, apod.). Široký výběr má zájemce i v oblasti vlastní fotokatalýzy – využití v průmyslu, environmentalistice, léčení rakoviny, podobnost fotokatalýzy s fotosyntézou, atd.
Literatura [1] NAVRÁTIL, V., SOLDÁN, M.: Univerzita v Roskilde – experiment ve vzdělávání.
Chemický občasník, PAIDO Brno, PdFMU, 6, (1992).
[2] FUJISHIMA, A, HONDA, K.: Electrochemical photolysis of water at a semiconductor electrode. Nature 238 (1972) 37–38.
[3] FUJISHIMA, A., RAO, T. N., TRYK, D. A.: Titanium dioxide photocatalysis. Journ. Of Photochemistry and Photobiology C: Photochemistry Reviews 1 (2000) 1–21.
[4] FUJISHIMA, A., HASHIMOTO, K., WATANABE, T.: TiO2 Fotokatalýza, základy a aplikace. Silikátový svaz, Praha 2002.
[5] HALLIDAY, D., RESNICK, R., WALKER, J.: Fyzika. Vutium Brno, Prometheus, 2000.
[6] HASHIMOTO, K., IRIE, H., FUJISHIMA, A.: TiO2 Photocatalysis: A Historical Overwiew and Future Prospects. Japanese Journal of Applied Physics. Vol. 44, No 12, (2005), 8269–8285.
[7] VESELÁ, M.: Fotokatalytická aktivita tištěných vrstev oxidu titaničitého. Diplomová práce, VUT Brno, Fakulta chemická, 2009.
[8] HOŠEK, T.: Využití bariérových výbojů při přípravě fotokatalytických TiO2 vrstev. Bakalářská práce, MU Brno 2008.

49
Praktické úlohy
Název modulu: Fotokatalýza Vstupní podmínky: základní kurz fyziky, základy chemie
Typ modulu: laboratorní měření, experimenty
Stručná anotace vymezující cíle modulu
Studenti budou seznámeni s dnešním stále běžnějším využitím fotokatalýzy jako jsou samočisticí a samosterilizující povrchy a seznámí se s podstatou jevu a testováním těchto povrchových vrstev. Protože důležitou vědní oblastí, podmiňující pochopení jevu fotokatalýzy je jev povrchového napětí a povrchové energie, je polovina modulu věnována těmto jevům. Jde o jednu z aktuálních oblastí zkoumání na rozhraní fyziky a chemie.
Předpokládané výsledky modulu
Modul sestává ze 4 praktických úloh, jejichž řešením se studenti seznámí s problematikou, související s jevem fotokatalýzy. Sem patří zejména jevy povrchového napětí kapalin a povrchové energie pevných látek, které úzce souvisejí s katalýzou a fotokatalýzou. Praktické zkušenosti studentů s danými jevy jim pomohou k lepšímu porozumění významu těchto jevů z hlediska fungování různých procesů ve fyzice, chemii, biologii a v technické praxi. Související problematika je vždy vysvětlena v teoretické části pracovních listů.
Obsah modulu/hodinová osnova
1. Teplotní závislost povrchového napětí kapalin (3 h)
2. Demonstrační pokusy „Povrchové napětí kapalin“ (4 h)
3. Povrchová energie pevných látek (4 h)
4. Základní vlastnosti fotokatalytického jevu (4 h)

50
Demonstrační pokusy „Povrchové napětí kapalin“
Úkol č. 1
Úkolem je ukázat kvalitativní závislost povrchového napětí kapalin na některých parametrech:
a) závislost na druhu kapaliny
Umístěte žiletku, jehlu, malou hliníkovou minci, kousek alobalu atd. opatrně na povrch různých kapalin (voda, líh, benzín, glycerín …; viz Tab. 1). Ne vždy se vám to podaří. Výsledek porovnejte s Tab. 1 a zdůvodněte.
Výsledek: při jisté dávce opatrnosti se daří umístit všechny výše uvedené předměty na klidný povrch vodní hladiny v Petriho misce. Je to proto, že voda má velké povrchové napětí. Umístění stejných předmětů na hladinu lihu, nebo benzínu se nedaří, neboť obě kapaliny mají mnohem nižší povrchové napětí, než voda.
b) Závislost na čistotě kapaliny
Na povrch vody umístěte tenké dřevěné tyčinky (zápalky) podle Obr. 1.1. Doprostřed obrazce kápněte kapku roztoku saponátu, nebo mýdla. Poté vyměňte vodu a zopakujte pokus tak, že kápnete doprostřed kapku roztoku cukru. Získané výsledky popište a vysvětlete. (Ze stejného důvodu se pohybuje po povrchu vodní hladiny lodička, vložíme-li do ní kousek mýdla).
Obr. 1.1 K závislosti povrchového napětí na čistotě kapaliny
Závěr: kápneme-li doprostřed obrazce, vytvořeného ze zápalek saponát, zápalky se od sebe oddálí ve směru od středu. Je tomu tak proto, že saponát vytvoří na povrchu tenkou vrstvu s nižším povrchovým napětím než voda a povrchová vrstva na obvodu obrazce, tvořená čistou vodou s vysokým povrchovým napětím táhne zápalky v radiálním směru.
Opačný pohyb zápalek (směrem ke středu) nastane tehdy, kápneme-li doprostřed kapku roztoku cukru. To je způsobeno tím, že roztok cukru zvyšuje povrchové napětí vody.
c) Plateauova tělesa
Do roztoku saponátu (osvědčil se např. tento poměr: 12 šálků vlažné vody, 1 šálek prostředku na mytí nádobí, 1 šálek kukuřičného škrobu, 2 polévkové lžíce prášku do pečiva)

51
umístěte opatrně (aby se na povrchu neutvořily bublinky) drátěný rámeček ve tvaru krychle (Obr. 1.2) Pokus proveďte několikrát a zdůvodněte, proč má blanka typický geometrický tvar. Výsledek se pokuste podpořit matematicky (minimální plocha).
Obr. 1.2 Minimální povrch, vytvořený blankou z roztoku saponátu
Výsledek měření: pokud byl povrch saponátového roztoku bez bublinek, skládal se vzniklý obrazec ze dvou komolých čtyřbokých jehlanů se společnou menší vodorovnou čtvercovou základnou, umístěnou uprostřed krychle. Označíme-li stranu této základny x a hranu krychle a, dostáváme pro celkovou plochu obou komolých čtyřbokých jehlanů výraz
222 24 ++−= xxxS (zde jsme zvolili ze délkovou jednotku délku hrany krychle), S je povrch celého obrazce.
Průběh této funkce (modrá) a její derivace (červená) je uveden na grafu na Obr. 1.3. Vidíme, že její minimum je rovno 0,408.a, kde a je délka hrany krychle. V mezích přesnosti měření souhlasí tento výsledek s naměřenými hodnotami (je třeba podotknout, že měření nebyla příliš přesná, neboť není možné se dotknout blanky a délku je potřeba odhadovat).
Obr. 1.3 Graf funkce 222 24 ++−= xxxS (Plateauova úloha). Funkce je vyznačena modře, její derivace červeně.

52
Základní měření povrchových jevů 1 Pomůcky stalagmometrická trubice (tlustostěnná kapilára), laboratorní váhy, váženky, teploměr 2 Pracovní úkoly • Pomocí kapkové metody určete teplotní závislost povrchového napětí pro destilovanou
vodu. • Srovnáním s vodou určete při pokojové teplotě (20°C) povrchové napětí lihu stejnou
metodou. 3 Experimentální upořádání a metody Při odtrhávání kapky z konce svislé kapiláry platí, že kapka se odtrhne, jakmile tíhová síla překoná sílu vyvolanou povrchovým napětím. Je-li vnější poloměr R, hmotnost kapky m dostaneme
σπRmg 2=
Při odtrhávání se však neodtrhne kapka celá. Na kapiláře vždy zůstává poměrem stejná část kapky a to i pro různé kapaliny. Pokud tedy změříme hmotnosti M1 a M2 kapek dvou kapalin, platí
2
1
2
1
m
m
M
M=
kde, m1 a m2 jsou hmotnosti kapek dvou kapalin při úplném odtržení. Z předchozích vztahů lze odvodit, že
1
212
M
Mσσ =
kde σ2 resp. σ1 jsou příslušná povrchová napětí příslušející hmotnostem M1 a M2.

53
4 Postup • Tlustostěnnou kapiláru upevníme svisle do stojanu, spodním koncem asi 5 cm od povrchu
podložky. • Pomocí váženek a laboratorních vah zjišťujeme hmotnost odtržené kapky a to tak, že
necháme odkapat do váženek minimálně 10 kapek měřené kapaliny. • Nejprve změříme hmotnost kapky destilované vody Mv při teplotě 20°C a použijeme
tabulkovou hodnotu pro povrchové napětí vody σv = 72,75 ⋅ 10-3 Nm
-1. Při následných výpočtech budeme pracovat s touto referenční hodnotou hmotnosti.
• Teplotní závislost povrchového napětí vody zjišťujeme pro interval teplot od 0°C do 10°C
s krokem 10°C a následně vyneseme závislost do grafu. • Změříme povrchové napětí lihu pro teplotu 20°C 5 Výsledky 1. Zjišťování teplotní závislost povrchového napětí Teplotní závislost povrchového napětí vody σv
t [°C] m [mg] σv [N⋅m-1] ⋅ 10-3
0 98,86 ± 0,32 79,3 ± 0,6 10 93,99 ± 0,36 75,4 ± 0,6 20 90,67 ±±±± 0,69 72,75
30 81,05 ± 1,19 65,0 ± 1,0
Teplotní závislost povrchového napěí vody
60,00
65,00
70,00
75,00
80,00
85,00
0 5 10 15 20 25 30 35
teplota [°C]
σσ σσ [N
.m]1
0-3

54
3. Měření povrchového napětí alkoholu kapkovou metodou M2 LIHU = 35,25 ±±±± 0,10 mg σσσσLIHU = 28,3 ± 0,2 ⋅± 0,2 ⋅± 0,2 ⋅± 0,2 ⋅ 10-3 [N⋅⋅⋅⋅m-1]
6 Diskuze 7 Závěr
Úkol č. 3 Fotokatalytické vlastnosti TiO2 povrchů
V odstavci 4.5.3 je uvedeno, že hydrofobní materiály se vyznačují tím, že kapičky vody na nich mají velký kontaktní úhel (sklo ∼ 200, plasty ∼800). Naopak hydrofilní materiály vykazují tento úhel velmi malý.
Tenká vrstva, tvořená TiO2 vykazuje velikost tohoto úhlu několik desítek stupňů. Osvětlíme-li jej však UV světlem, úhel se zmenší až téměř na nulu (kapičky se rozplynou). V tomto případě se jedná o tzv. superhydrofilní stav, který takovým zůstává až dva dny, i když není vystaven UV paprskům. Potom se kontaktní úhel začíná zvětšovat a povrch se stane opět hydrofobním. Další ozáření UV paprsky opět nastolí superhydrofilní stav (Obr. 7)
Využití tohoto jevu se přímo nabízí tam, kde nám vodní zamlžení nějakého objektu (zpětných zrcátek a čelních skel automobilů, zrcadel v koupelnách, apod.) ztěžuje nebo znemožňuje vidění.
Prakticky se o tom přesvědčíme tak, že na sklo, pokryté tenkou TiO2 vrstvičkou kápneme kapku vody. Pomocí přístroje SEE změříme několikrát kontaktní úhel.Θ1. Pomocí zdroje UV záření (nehtové studio) ozáříme kapičku vody a znovu změříme kontaktní úhel Θ2, který bude blízký 00. Po určité době ∆τ (dny) budeme celý pokus opakovat se stejnou vrstvičkou - kontaktní úhel Θ3 bude opět nenulový. Pokud nám to umožní experimentální podmínky, můžeme studovat závislost Θ3 = φ (∆τ).

55
Shrnutí Již od šedesátých let minulého století je projektová výuka známa jako jedna
z nejefektivnějších vyučovacích metod a to na všech stupních škol. Zkušenosti vyučujících vedou k závěru, že jako nejplodnější se ukazují být témata, zahrnující v sobě několik vědních oblastí a mající praktický výstup. Téma „Fotokatalýza“ je právě takovým tématem: zahrnuje v sobě oblasti fyzikální (mechanika kapalin a pevných látek – povrchové napětí a povrchová energie a fyziku polovodičů), oblast chemickou (katalýza) a samozřejmě i potřebnou matematiku.
Závěrečná část práce je věnována možnostem praktického využití fotokatalýzy. Autoři doufají, že po přečtení monografie si žáci a studenti na všech stupních škol od základní až po vysokou, budou schopni vybrat některou její část a rozpracovat ji v rámci svého projektu. Zejména máme na mysli kapitolu, týkající se povrchového napětí kapalin a povrchové energie pevných látek. Právě tato vědní oblast, nacházející se na rozhraní fyziky a chemie má mnoho výhod:
1. Její teorie je poměrně jednoduchá a názorná, pochopitelná i pro žáky základní školy.
2. Využití povrchového napětí v domácnosti, technice a v zemědělství je velmi rozsáhlé.
3. Některé oblasti chemie, biologie a průmyslové technologie jsou založeny právě na jevu povrchového napětí.
4. Existuje nepřeberný počet přírodních jevů, jak z oblasti živé, tak i z oblasti neživé přírody, jejichž základ tvoří jevy povrchového napětí.
5. Pokusy, dokumentující povrchové napětí a povrchovou energii jsou velmi jednoduché a zároveň velmi názorné.
Jedním ze základních cílů předložené monografie je právě upozornění zejména na didaktické výhody téma povrchového napětí kapalin.
Klíčová slova: fotokatalýza, mezioborový experiment, laboratorní měření

56
Summary Since the sixties of the last century classroom project is known as one of the most
effective teaching methods at all levels of education. Experience of teachers leads to the conclusion that as prove to be fruitful topics involving themselves in several fields of science and of practical output. The theme of "Photocatalysis" is just such a theme: it implies the physical (mechanics of fluids and solids - surface tension and surface energy and semiconductor physics), the field of chemical (catalysis) and of course the necessary mathematics.
The final part is devoted to the practical use of photocatalysis. The authors hope that after reading pupils and students at all levels of schools from elementary to college, will be able to select any part of it and incorporate it as part of their projects. In particular, we refer to chapter on the surface tension of the liquids and the surface energy of solids. It is this area of science, on the interface of physics and chemistry has many advantages:
The first- theory is relatively simple and illustrative, of course for primary school pupils.
2nd- application of surface tension in the household, technology, and agriculture is very extensive.
3rd- some areas of chemistry, biology, and industrial technologies are based just on the phenomenon of surface tension.
4th-there are many natural phenomena, from both the living area and from the field of inanimate nature, whose basis is the phenomenon of surface tension.
5th- documenting surface tension and surface energy are very simple and very clear.
One of the fundamental objectives presented monograph is just a warning especially for educational benefits topic of surface tension of liquids.
Keywords: photocatalysis, interdisciplinary experiment, laboratory measurements

57
Glosář pojmů
1. Povrchové napětí je efekt, při kterém se povrch kapalin chová jako elastická fólie a snaží se dosáhnout co možná nejhladšího stavu s minimální plochou. To znamená, že se povrch tekutiny snaží dosáhnout stavu s nejmenší energií. Čím větší je povrchové napětí, tím „kulatější“ je kapička této kapaliny.
2. Kohezní tlak je tlak, vyvolaný silami povrchového napětí. Tento tlak je značný, takže se nám kapaliny jeví jako prakticky nestlačitelné. V důsledku existence povrchového napětí je v povrchové vrstvě nahromaděna i jistá energie, tzv. povrchová energie.
3. Helmholtzova volná energie se určí ze vztahu
,
kde je vnitřní energie, je termodynamická teplota a je entropie.
Volná energie je považována za tu část vnitřní energie systému, která je volná a lze ji přeměnit na práci, zatímco člen označuje část vnitřní energie, která je při dané teplotě vázána a nelze ji přeměnit na práci.
Totální diferenciál má tvar
Izotermicko-izochorický děj bude probíhat samovolně, pokud platí podmínka
,
přičemž pro vratné děje platí a pro nevratné děje .
4. Volná entalpie, jinak řečeno Gibbsova volná energie či jen Gibbsova energie nebo Gibbsova funkce, je jedním z termodynamických potenciálů, tedy extenzivní stavová
termodynamická veličina s rozměrem energie. Gibbsova energie je stavová funkce, která je zpravidla značená písmenem G, která popisuje chemické děje za podmínek konstantního tlaku, konstantní teploty a konstantního objemu, kdy entropie jako kritérium samovolnosti děje nevyhovuje. Přirozenými proměnnými jsou pro Gibbsovu energii termodynamická teplota, tlak a látkové množství.
Vzhledem k tomu, že je vhodná pro posuzování termodynamické rovnováhy soustav při konstantním tlaku a teplotě, je často využívaná pro charakteristiku přirozeného směru chemických reakcí - které zpravidla probíhají při atmosférickém tlaku a teplotě prostředí.
Ve všech vztazích je termodynamická teplota, je entropie, je tlak, je objem, je chemický potenciál a látkové množství i-té složky, je entalpie, je vnitřní
energie.
Gibbsovu energii lze vyjádřit z entalpie vztahem:
Z vnitřní energie lze Gibbsovu energii vyjádřit vztahem:

58
Diferenciál Gibbsovy energii lze v přirozených proměnných vyjádřit vztahem:
jehož výsledkem je, že úbytek Gibbsovy energie ∆G systému za konstantního tlaku a teploty, je roven maximální práci, kterou může systém odevzdat do okolí.
5. Katalyzátor (z řeckého καταλύτης katalýtés) je látka, vstupující do chemické reakce, urychluje ji (nebo zpomaluje), a přitom z ní vystupuje nezměněná. Slovo katalyzátor se někdy používá i v přeneseném smyslu pro jakoukoli událost, osobu apod., které přispějí k uskutečnění něčeho (např. „příjemná hudba byla katalyzátorem úspěšného jednání“. Nebo naopak „přítomnost ekologického aktivisty byla inhibitorem (neg. katalyzatoru) toho, že se strany nedohodly“). V živých organismech jako katalyzátory fungují enzymy nebo ribozymy.
6. Fotokatalýza je proces chemického rozkladu látek za přítomnosti fotokatalyzátoru a světelného záření. Principiálně vychází z fotolýzy, přirozeného rozkladu některých látek působením světla, urychlené přítomností fotokatalyzátoru. Je-li materiál s fotokatalytickými vlastnostmi vystaven světelnému záření vhodné vlnové délky, aktivuje se jeho povrch a spustí se charakteristická reakce. Primárně vzniklý volný pár elektron-díra a hydroxylové radikály sekundárně vznikající kontaktem excitované molekuly fotokatalyzátoru a vodní páry rozkládají přítomné organické a anorganické substance.
7. Hydrofilnost (také označovaná jako „smáčivost“) se stanoví podle styčného úhlu kapky vody s povrchem. Všechny povrchy běžně dostupné na trhu jsou hydrofobní (tj. vodu odpuzující).
Zdroje http://cs.wikipedia.org/wiki/Helmholtzova_voln%C3%A1_energie
http://cs.wikipedia.org/wiki/Povrchov%C3%A9_nap%C4%9Bt%C3%AD#Povrchov.C3.A1_
KIREEV, V. A.: Kurz fizičeskoj chimii (1975).
HÁLA, E., REISER, A.: Fysikální chemie. NČSAV Praha 1960.
FUJISHIMA, A., HASHIMOTO, K., WATANABE, T.: TiO2 Fotokatalýza, základy
a aplikace. Silikátový svaz, Praha 2002.

59
Rejstřík Avogadrova konstanta, 13 Avogadrovo číslo, 22 deprese, 8 elevace, 8, 9 entalpie, 14, 15, 43 entropie, 12 Fotochemické reakce, 22 Fotokatalýza, 2, 3, 24, 36, 44
hustota kapaliny, 5 kapilára, 10, 11, 39 Katalýza, 2, 19 Povrchové napětí, 2, 3, 10, 19, 36, 42 projektovou výuka, 35 smáčení, 10, 15, 18 sublimace, 13

Fotokatalýza v praxi a ve škole
prof. RNDr. Vladislav Navrátil, CSc., Mgr. Lukáš Pawera
Vydala Masarykova univerzita 2014
1. vydání, 2014
Náklad 200 výtisků
Tisk: Tiskárna Didot, spol. s r.o., Trnkova 119, 628 00 Brno-Líšeň
ISBN 978-80-210-7157-5