zasady i metody klasycznej i molekularnej diagnostyki ... i metody.....pdf · klasycznej i...
TRANSCRIPT

Beata Nowakowska, 24.10.2016
Zasady i metody klasycznej i molekularnej
diagnostyki cytogenetycznej

Cytogenetyka
Klasyczne badanie cytogenetyczne - opiera
się na analizie chromosomów w stadium
metafazy
Dział genetyki zajmujący się badaniem chromosomów. Koncentruje się zarówno na kształcie jak i liczbie chromosomów oraz na
ich dziedziczeniu.
cytogenetyka molekularna - umożliwia badanie
materiału genetycznego w stadium interfazy

Rola i znaczenie badań cytogenetycznych
pre- i postnatalnego rozpoznania chorób i
zespołów uwarunkowanych aberracjami
chromosomowymi
identyfikacji rodzin ryzyka genetycznego
Badanie cytogenetyczne stanowi podstawę:
poznawania etiologii i patomechanizmu chorób
genetycznych

Od chromosomu do nukleotydu Cytogenetyka Sekwencjonowanie
CGH NGS
SNP + CNV (?)
CNV

Rozdzielczość metod cytogenetyki molekularnej
Sekwencjonowanie DNA- 1pz
FISH - 40-250 kpz
Klasyczne metody oceny kariotypu >4
Mpz
HR-CGH >3 Mpz Mikromacierze CGH – 1 Mpz – 20 pz
MLPA - 40-60 pz

Metody cytogenetyki klasycznej - hodowla limfocytów krwi obwodowej

Limfocyty – Tylko limfocyty ulegają pobudzeniu przez czynnik wzrostu
Limfocyty T
odpowiedzialne za odpowiedź odpornościową komórkową
U osoby zdrowej stanowią ~70% populacji wszystkich limfocytów
Limfocyty B
odpowiedzialne za rozpoznanie antygenu i wytwarzanie przeciwciał

Analiza prążkowa chromosomów
Każdy chromosom ma specyficzny dla siebie obraz (wzór) prążkowy, czyli układ poprzecznych prążków, różniących się wielkością i intensywnością zabarwienia.
W haploidalnym zestawie chromosomów metafazowych można wyróżnić około 400 prążków.
Chromosomy z wcześniejszych stadiów podziału chromosomowego, prometafazowe lub profazowe, wykazują, zależnie od stopnia kondensacji, 500-1250 prążków.
Wielkość najmniejszej aberracji która może być w nim rozpoznana odpowiada 7-10 Mpz, przy 400 prążkach i zwiększa się do 2-5 Mpz przy 850 prążkach.

6p21.3 region 2 prążek 1 subprążek 2
Każda para chromosomów musi być przeanalizowana •co najmniej dwukrotnie (każdy region prążkowy) •przy zalecanej rozdzielczości na podstawie wzorców z ISCN

46,XX

Kariotyp prawidłowy męski, rozdzielczość 450 prążków

Kariotyp prawidłowy męski, rozdzielczość 750 prążków

47,XY,+21

Trisomia 21 (zespół Downa), 47,XY,+21

47,XY,+21

47,XXY

47,XXX

47,XY,+mar

46,X,del(X)(q21)
del(X) X

46,X,del(X)(q21)

46,X,t(X;3)(p21.1;12.1)

3 der(3) der(X) X
t(X;3)(p21.1;12.1)

45,XY,der(13;14)(q10;q10)

46,XY,add(7)(q36)

Translokacja Robertsonowska zrównoważona, 46,XY,der(13;14)(p11,p11)

Translokacja Robertsonowska niezrównoważona, trisomia 13 (zespół Patau)
46,XY,der(13;14)(q10;q10),+13

Translokacja wzajemna zrównoważona, 46,XX,t(4;7)(p15.2;q11.2)

Delecja w obrębie długiego ramienia chromosomu 18 pary, 46,XY,del(18)(q21.3)

Metody cytogenetyki molekularnej

Technika FISH umożliwia identyfikację specyficznych
sekwencji DNA w chromosomach metafazowych, jądrach interfazowych
lub skrawkach tkanek znajdujących się w preparatach cytologicznych.

RODZAJE SOND MOLEKULARNYCH
1. Sondy
malujące
2. Sondy specyficzne
do unikalnych
sekwencji DNA
3. Sondy specyficzne do
powtarzalnych sekwencji DNA
subtelomer
centromer
subtelomer
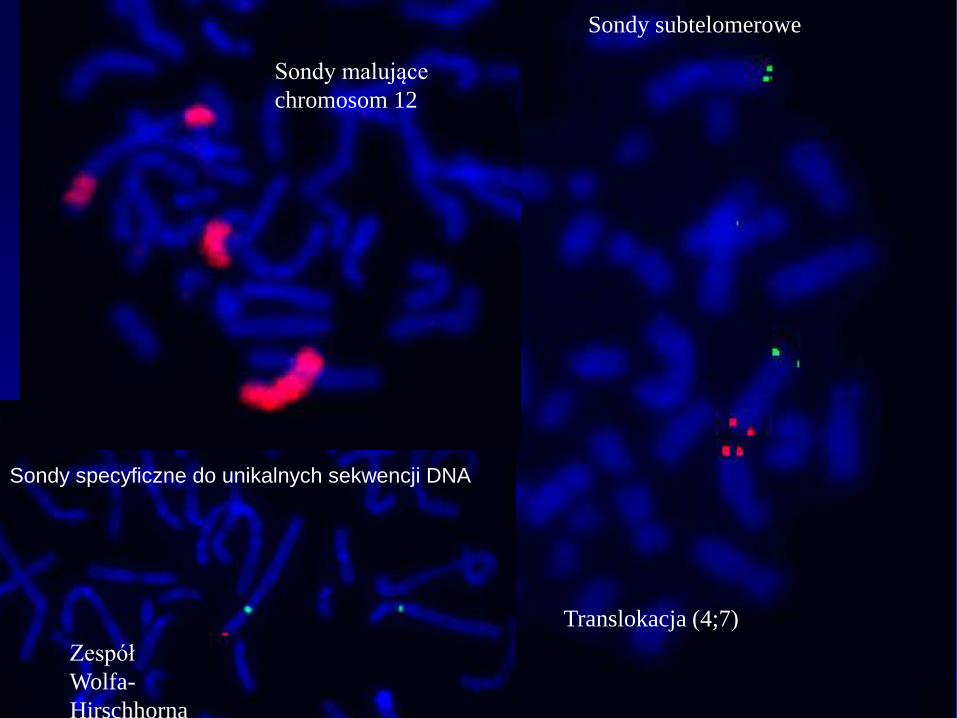
Zespół
Wolfa-
Hirschhorna
Sondy specyficzne do unikalnych sekwencji DNA
Translokacja (4;7)
Sondy subtelomerowe
Sondy malujące
chromosom 12

p-arm probes fluoresce green
q-arm probes fluoresce red
1 2 3 4 5 6 7 8
9 10 11 12 13 14 15 16
18 19 20 21 22 X/Y 17
Subtelomere Region-specific Probes

Sondy
subtelomerowe
Chromosomy 3 pary -
prawidłowe
Chromosomy 6 pary -
prawidłowe
Translokacja (4;7)

Multicolor - FISH

Rapid FISH
Prawidłowa liczba sygnałów
13 - sygnały zielone
21 - sygnały czerwone
Nieprawidłowa liczba sygnałów
3 sygnały zielone
– trisomia 13 pary
36

Metoda MLPA
Zalety
• Możliwość analizy ~45 loci w jednym badaniu
• Możliwość badania do 96 próbek równocześnie
• Wynik badania po 24 godz.
• Prostota, mały koszt
Zastosowanie
Diagnostyka:
• zespołów mikrodelecji/mikroduplikacji
• aberracji subtelomerowych
• aneuploidii w badaniach prenatalnych
MRC Holland http://www.mrc-holland.com

Starter Y do reakcji PCR
Sekwencja komplementarna do badanego regionu DNA
Starter X do reakcji PCR
Unikalnej długości sekwencje DNA
Sekwencja komplementarna do badanego regionu DNA
X 5’
5’ 3’ Badany region DNA A
Y
Badany region DNA B
X
5’
5’ 3’
Y
5’ 3’ 5’ 3’
X Y
5’ 3’
X Y
5’ 3’
Ligacja i amplifikacja
Ilościowa analiza powstałych tak produktów PCR umożliwia odzwierciedlenie liczby kopii analizowanych loci. (d) Rozróżnienie poszczególnych sond odbywa się na drodze rozdziału elektroforetycznego w sekwenatorze, dzięki temu, że każda z sond jest innej długości
Pojedyncza sonda zawiera dwa różne oligonukleotydy (20-43 nukleotydy), których miejsca wiązania w genomie znajdują się koło siebie. Sondy swoiste dla przylegających do siebie fragmentów DNA analizowanego regionu połączone są z krótkimi, unikalnymi sekwencjami DNA o różnej długości (19-370 nukleotydów), oflankowanymi sekwencjami komplementernymi do uniwersalnych starterów.
Schouten, J.P. et al. Nucl. Acid Res. 30, e57. More info on www.mrc-holland.com
Hybrydyzacja
38

Delecja 22q11.2 39

Duplikacja 4p 40

Metoda porównawczej hybrydyzacji genomowej
Pozycja na chromosomie
Genomowe DNA
pacjenta
Genomowe DNA
referencyjne
Cot-1 DNA
addycja delecja


Chromosom
markerowy
Sondy centromerowe 8/9
Sondy centromerowe 13/21
Chromosom
mar

Sonda malX

Porównawcza hybrydyzacja genomowa (CGH) do mikromacierzy:
Genomowe DNA
pacjenta
Genomowe DNA
referencyjne Cot-1 DNA
Pozycja na chromosomie
1
Wzglę
dna
fluore
scencja
addycja delecja Profil intensywności
fluorescencji
Wartości progowe

Cy5 Cy3 Test Sample Reference Sample
Hybridization for 16 hrs
Porównawcza hybrydyzacja genomowa (CGH) do mikromacierzy:

Schemat analizy DNA genomowego techniką mikromacierzy CGH:
DNA referencyjne DNA testowane
Mieszanina sond
Hybrydyzacja do mikromacierzy
Fragment sekwencji DNA umieszczony na mikromacierzy
Wizualizacja wyniku
Duplikacja fragmentu
DNA
Względna fluorescencja
Wstępne przetwarzanie
danych
Fragmenty DNA umieszczone na
szkiełku podstawowym
wg. Chari i wsp. 2006


Mikromacierze DNA:
CGH: SNP:
identyfikacja zmiany liczby kopii fragmentów DNA (CNV)
identyfikacja zmiany liczby kopii fragmentów DNA (CNV)
identyfikacja disomii jednorodzicielskiej (UPD)
identyfikacja utraty heterozygotyczności (LOH)
identyfikacja mutacji punktowych

Rodzaje i zastosowanie CGH do mikromacierzy Mikromacierz skonstruowana dla określonego
regionu chromosomu -region 1p36 - Yu W i wsp.,2003, -region 15q –Locke i wsp.,2004, -region 18q – Veltman JA i wsp., 2004, -region 17p – Shaw CJ i wsp.,2004
Mikromacierz kliniczna, zawiera sondy specyficzne dla regionów krytycznych zespołów genetycznych oraz regiony subtelomerowe (Cheung SW i wsp.,2005, Shaffer L i wsp., 1999)
Mikromacierz sporządzona dla całego genomu (Li Ji wsp.,2003, Cai WW i wsp.,2002 )

Charakterystyka mikromacierzy stosowanych w badaniach
diagnostycznych

Mikromacierze selektywne vs. całogenomowe:
1. Targeted BAC Array 2. Whole Genome BAC Array 1MB 3. Whole Genome BAC Tiling Array 4. Oligo Targeted Array 5. Oligo Whole Genome Tiling Array

CNV- Copy Number Variations
Zmiany liczby kopii
fragmentów DNA łagodne CNVs oraz
patogenne CNVs
indel (insertion or deletion)

CNV (copy number variants) – zmiana liczby kopii sekwencji DNA
Large-scale copy number polymorphism in the human genome. Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Månér S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Science. 2004 Jul 23;305(5683):525-8. Detection of large-scale variation in the human genome. Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Nat Genet. 2004 Sep;36(9):949-51.

CNV: 290 fenotypowo prawidłowych osób pochodzących z
różnych grup etnicznych
1668 CNVs (średnio 11 CNVs na osobę)
średnia wielkość 340 - 465 kpz (100 kpz- 10 Mpz)
łącznie CNVs stanowiły 360 Mpz
co stanowi 12% ludzkiego genomu (obecnie w internetowej bazie zmiany łagodne pokrywają 29% genomu)
Uważa się, że CNVs są odpowiedzialne za
- ewoluję
- zmienność osobniczą ( np. HIV - Gonzalez et al. 2005 , choroby autoimmunologiczne - Yang et al. 2007)
- odpowiedź na leki (np. onkologia - Ouahchi et al. 2006)
- podatność na powstawanie chorób

Oddziaływanie CNVs na fenotyp:
Zmiana ekspresji genu
Modyfikację dawki produktu danego genu
Zaburzenie funkcji elementów regulatorowych
„Odsłonięcie” alleli recesywnych

Ustalenie rodzinnego pochodzenia zmiany
Bazy danych wariantów polimorficznych:
- Toronto Database of Genomic Variants http://projects.tcag.can/variation
Bazy danych osób z nieprawidłowym fenotypem:
- DECIPHER
http://www.sanger.ac.uk/PostGenomics/decipher/
Region bogaty w geny
Wielkość zmiany
Delecja czy duplikacja
Informacje od lekarza kierującego pacjenta na badania
Interpretacja wyników badań metodą aCGH

Interpretacja wyników badań metodą aCGH
Rodzicielskie pochodzenie CNV Mniejsze prawdopodobieństwo
patogenności (>99% dziedziczonych CNV to CNV łagodne) ale w interpretacji wyniku trzeba
uwzględnić: - Ocenę fenotypu rodzica, nosiciela CNV (zróżnicowanie
ekspresji, niepełna penetracja, mozaikowość – odziedziczone CNV np.1q21.1; 1q41q42; 3q29; 15q11.2; 15q13.2q13.3; 16p11.2; 16p13.11; 22q11.2 mogą być patogenne
- Czynniki epigenetyczne (CNV zawiera piętnowany locus, np. del UBE3A od matki skutkuje chorobą u dziecka)
- Możliwość ujawnienia się mutacji recesywnej gdy dotyczy drugiego allelu genu znajdującego się w obrębie CNV odziedziczonego od rodzica

Niepełna penetracja

McCarroll S.A. Human Molecular Genetics, 2008, Vol. 17, Review Issue 2
Niepełna penetracja

Możliwość ujawnienia się mutacji recesywnej

Interpretacja wyników badań metodą aCGH Ustalenie rodzinnego pochodzenia zmiany
Bazy danych wariantów polimorficznych:
- Toronto Database of Genomic Variants http://projects.tcag.can/variation
Bazy danych osób z nieprawidłowym fenotypem:
- DECIPHER
http://www.sanger.ac.uk/PostGenomics/decipher/
Region bogaty w geny
Wielkość zmiany
Delecja czy duplikacja
Informacje od lekarza kierującego pacjenta na badanie




A JHG 82, 181-187 2008

Interpretacja wyników badań metodą aCGH Ustalenie rodzinnego pochodzenia zmiany
Bazy danych wariantów polimorficznych:
- Toronto Database of Genomic Variants http://projects.tcag.can/variation
Bazy danych osób z nieprawidłowym fenotypem:
- DECIPHER
http://www.sanger.ac.uk/PostGenomics/decipher/
Region bogaty w geny
Wielkość zmiany
Delecja czy duplikacja
Informacje od lekarza kierującego pacjenta na badanie

10,7Mb delecja interstycjalna bez
efektu fenotypowego

5q14.3q15
MEF2C
d
a
c
b
Patient 2
Patient 1
PH critical region 5.8 Mb
TMEM161B FLJ11292 LYSMD3
GPR98
CETN3
AL050132
86,200,000-95,500,000
86 96
Position MB
91 87 88 89 90 93 92 94 95
RASA1
CCNH
LOC153364
POLR3G
ARRDC3 FLJ42709
NR2F1
FAM172A
POU5F2
AK130941
KIAA0825
C5orf36
CR590796
ANKRD32
MCTP1
CR614705
SPATA9
RHOBTB3
KIAA0878
FAM81B
TTC37
UNQ630
ARSK
GPR150
RFESD
GLRX
ELL2
Patient 3
Deletion: Cardoso et al.(2009) Patient 1
Deletion: Engels et al.(2009) Patient 1
Deletion: Engels et al.(2009) Patient 2
Deletion: Le Meur et al. (2009) Patient 2
Deletion: Le Meur et al.(2009) Patient 1
Deletion: Le Meur et al. (2009) Patient 3
Deletion: Le Meur et al. (2009) Patient 4
Deletion: Le Meur et al. (2009) Patient 5
Duplication: Le Meur et al. (2009) Patient 6
Deletion: Cardoso et al.(2009) Patient 3
Deletion: Cardoso et al.(2009) Patient 2
Deletion: Engels et al.(2009) Patient 3
Nowakowska i wsp., Am J Med. Genet Part B, 2010

Średnia wielkość CNV
Wg. Buysse i wsp., Eur J Med. Genet., 2009

• Platformy stosowane do badań diagnostycznych w przypadkach
NI/ASD/WWR o nieznanej etiologii powinny być całogenomowe i
mieć rozdzielczość ≥ 400 kpz
Rekomendacje ISCA – The International Standard Cytogenomic Array
Consortium Opracowany na podstawie 35 badań w grupie 21 478 pacjentów z NI
/opóźnieniem rozwoju psychoruchowego/ chorobą ze spektrum zaburzeń
autystycznych (ASD) / wrodzonymi wadami rozwojowymi o nieznanej etiologii
Rekomenduje
Podstawa: Stosując rozdzielczość ~400 kpz wykrywamy wszystkie
znane i powtarzające się zmiany warunkujące choroby genomowe i
większość unikalnych, patogennych niezrównoważeń genomu oraz
niewielką liczbę łagodnych CNV
• Kariotyp konwencjonalny powinien być wykonywany w
przypadkach wywiadu rodzinnego obciążonego zrównoważoną
rearanżacją chromosomową lub licznymi poronieniami oraz w
przypadkach znanych zespołów aneuploidii (tris.21, 13, zespół
Turnera i Klinfeltera)

Noworodki z wielowadziem i/lub cechami dysmorfii ~ 14 - 28%
Niepełnosprawność intelektualna / opóźnienie rozwoju 9 – 13%
psychoruchowego (3 – 5% met. GTG)
CNV stwierdzane metodą aCGH w różnych grupach klinicznych
Szacuje się, że aż 15% chorób genetycznych uwarunkowana jest
submikroskopowym niezrównoważeniem genomu
Mikromacierze są ok. 3 krotnie skuteczniejsze w diagnostyce
niezrównoważenia genomu niż badania metodami
konwencjonalnymi

Wapner et al. June, 2014
Przydatność diagnostyczna metody aCGH w badaniach prenatalnych:

Internetowe bazy danych: Wapner et al. June, 2014 74

Lepsze poznanie i zrozumienie fenotypowej zmienności w znanych zespołach genetycznych
Wyjaśnianie genetycznej etiologii znanych zespołów (poprzez identyfikację genów odpowiedzialnych za patologię np. CHD7 w zespole CHARGE)
Diagnostykę rzadkich zespołów genetycznych
Odkrywanie nowych zespołów mikrodelecji/mikroduplikacji
Poznawanie znaczenia klinicznego mozaikowości (w 8 – 10% mozaikowość nie wykrywana badaniami konwencjonalnymi)
Wzbogacanie wiedzy o polimorfizmie genetycznym (częstość i rozmieszczenie w genomie CNV, 4000 loci), jego aspektach klinicznych np. znaczenia dla predyspozycji do chorób wieloczynnikowych (autyzm, Alzheimer, nowotwory)
Przydatność kliniczna badań na mikromacierzach
Badania na mikromacierzach umożliwiają:

Interpretation of array comparative genome hybridization data: a major challenge. Gijsbers AC, Schoumans J, Ruivenkamp CA. Cytogenet Genome Res. 2011;135(3-4):222-7

Porównawcza hybrydyzacja genomowa do mikromacierzy
(aCGH)
Zalety Ograniczenia
• Duża rozdzielczość • Nie wykrywa
zrównoważonych
rearanżacji i poliploidii
(~0.3% osób z NI ma
aberrację zrównoważoną)
• Precyzyjne mapowanie
miejsc złamań
• Wykrywanie duplikacji • Duża liczba wykrywanych
polimorfizmów
• Wykrywanie mozaikowatości
(10% BAC, 30% oligonukleotydowe)
• Problemy z interpretacją
wyników badań

Metoda
Rozdzielczość
Zalety
Ograniczenia
Problem Skutki
GTG 4 – 10 Mpz • Identyfikacja aberracji zrównoważonych
• Mała rozdzielczość • Subiektywizm oceny
•nie identyfikowane mikroaberracje • wysokie ryzyko błędu
FISH
40 - 250 kpz •Wysoka rozdzielczość •Łatwość wykonania •Cena
•ograniczona liczba analizowanych loci •konieczność znajomości regionu patologii
•Wieloetapowa identyfikacja aberracji
MLPA 40 - 60 pz
HR-CGH >3 Mpz •Analiza całego genomu •Identyfikacja dodatkowego materiału
•Mała rozdzielczość •Nie identyfikowane aberracje zrównoważone
•nie identyfikowane mikroaberracje •Nie identyfikowane translokacje zrównoważone
aCGH 1 Mpz do 10 - 20 kpz
•Duża rozdzielczość •Wykrywanie mozaikowości
•Nie identyfikowane aberracje zrównoważone •Wykrywanie polimorfizmów
•Nie identyfikowane translokacje zrównoważone •Trudności w interpretacji

1997 r.
79