zur existenz von tetramethylammoniumamalgam
TRANSCRIPT

Zur Existenz von Tetramethylammoniumamalgam
Barbara Alberta und Martin Jansenb*a Gieûen, Institut fuÈ r Anorganische und Analytische Chemieb Stuttgart, Max-Planck-Institut fuÈ r FestkoÈ rperforschung
Bei der Redaktion eingegangen am 17. April 2000.
InhaltsuÈ bersicht. Es konnte nachgewiesen werden, daûQuecksilber bei kathodischer Reduktion (substituiertes)Ammonium einlagert. Hierzu wurde Tetramethylammo-niumtetrafluoroborat in Acetonitril bei ±36 °C an einer Hg-Kathode elektrochemisch umgesetzt. Die Zusammensetzungdes Produktes wurde zu [N(CH3)4]+ ´ Hg5 ´ e± eingegrenzt.
Durch cyclovoltammetrische Messungen sowie massenspek-trometrische Untersuchungen der Zersetzungsproduktewurde der Einbau von [N(CH3)4] eindeutig belegt. Das Tet-ramethylammoniumamalgam ist roÈ ntgen- und neutronen-amorph, diamagnetisch und zersetzt sich oberhalb von±34 °C.
The Existence of Tetramethylammonium Amalgam
Abstract. It has been proven that mercury incorporates(substituted) ammonium at cathodic reduction. Tetra-methylammonium tetrafluoroborate was reacted electrochem-ically in acetonitrile at ±36 °C on a Hg-cathode. The com-position of the product has been estimated to about[N(CH3)4]+ ´ Hg5 ´ e±. The incorporation of [N(CH3)4] was
confirmed by cyclovoltammetry and mass spectroscopy ofthe decomposition products. The tetramethylammoniumamalgam is amorphous (X-ray and neutron diffraction), dia-magnetic, and decomposes at ±34 °C.
Keywords: Tetramethylammonium amalgam
Einleitung
Seebeck [1], Davy [2] und Berzelius [3] waren um 1808die ersten, die die Frage untersuchten, ob es ein denAlkalimetallen analoges freies Ammonium ¹NH4ª ge-ben koÈ nne. Die elektrochemische Reduktion von Am-moniumionen schien der geeignete Weg zu sein, zufreiem Ammonium zu gelangen, und vor allem diesersynthetische Zugang wurde in den folgenden Jahrenvon Ruff, Moissan und anderen vielfach erprobt undbeschrieben [4±15]. Bis heute ist die Diskussion dieserFrage aktuell, wie eine Glosse, die 1994 in Nature er-schien, beispielhaft zeigt, in der Jones anmerkt, daûder Jupiter moÈ glicherweise aus metallischem Ammo-nium oder Tetramethylammonium bestehe [16].
KoÈ nnte man die Frage nach der Existenz einerQuecksilberlegierung des Ammoniums, eines Ammo-niumamalgams, beantworten und auûerdem analysie-ren, wie die Konstitution und elektronische Situationeiner solchen Verbindung zu beschreiben ist, waÈredies ein erster, wichtiger Schritt auf dem Weg zum¹Ammoniummetallª.
Es ist denkbar, daû reduzierte Ammoniumspeziesstabiler sind, wenn die Wasserstoffatome in ihnendurch Alkyl- oder Arylreste substituiert sind, und be-
reits 1866 begannen Pfeil und Leffmann mit der Un-tersuchung von Amalgamen substituierten Ammoni-ums [17]. In der Folgezeit wurde eine Reihe weiterersolcher AnlaÈufe zur Synthese und Charakterisierungvon Alkylammoniumamalgamen unternommen[18±35].
Alle diese Arbeiten zeigen, daû die (substituierten)Ammoniumamalgame ausnahmslos sehr empfindlichgegenuÈ ber Sauerstoff, Feuchtigkeit, Temperaturenoberhalb 0 °C und mechanischer Belastung sind. Un-klar bleibt, ob die Substanzen uÈ berhaupt in Abwesen-heit eines aÈuûeren elektrischen Potentials uÈ ber laÈnge-re Zeit stabil sind. Eine Isolierung von unzersetztenAmmoniumamalgamen scheint den publizierten Ana-lyseergebnissen zufolge bislang nicht gelungen zu sein.
Wir beschreiben nun unser Vorgehen bei derSynthese, Isolierung, Identifizierung und Charakteri-sierung von Tetramethylammoniumamalgam, sowieeinige unserer Versuche, die Substanz mittels Beu-gungsmethoden zu untersuchen [36].
Ergebnisse und Diskussion
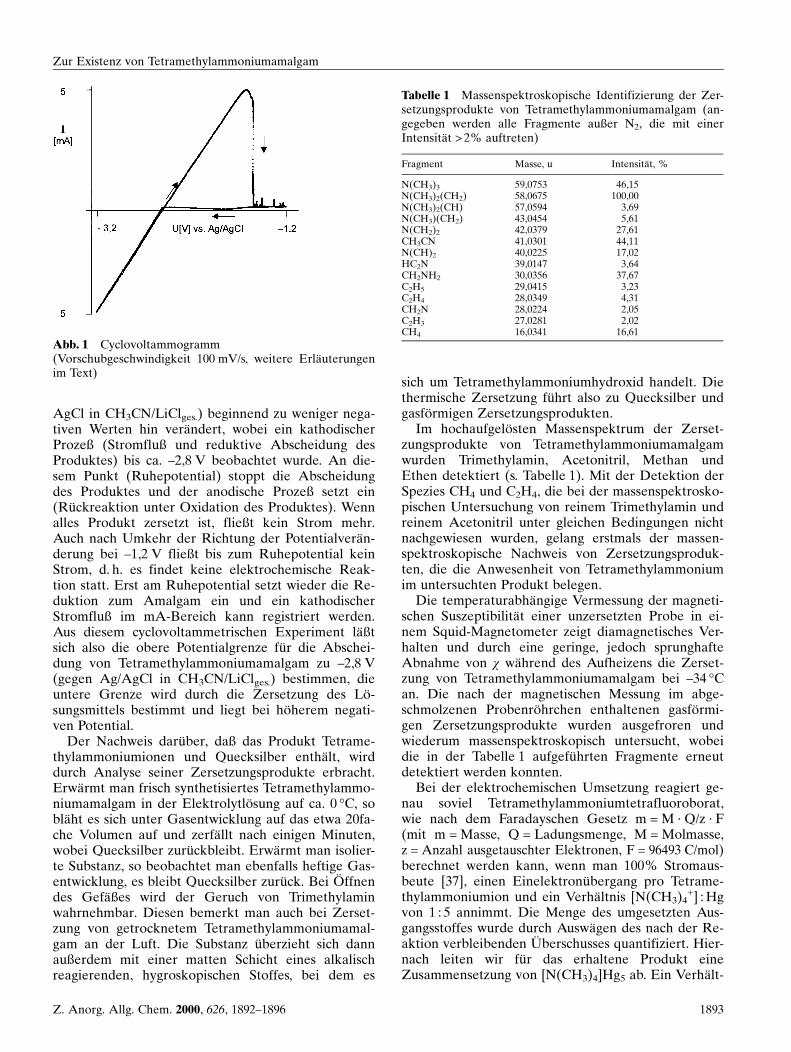
WaÈhrend der elektrochemischen Umsetzung von Tet-ramethylammoniumtetrafluoroborat in Acetonitril imH-Rohr wachsen bei einer Temperatur von ±36 °C aufdem fluÈ ssigen Quecksilber (Kathode) silbrigglaÈnzendeDendriten. Die Bildung des Produktes kann visuellund cyclovoltammetrisch (Abb. 1) verfolgt werden.Hierzu wurde das Potential von ±3,2 V (gegen Ag/
1892 Ó WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2000 0044±2313/00/6261892±1896 $ 17.50+.50/0 Z. Anorg. Allg. Chem. 2000, 626, 1892±1896
* Prof. Dr. M. Jansen,Max-Planck-Institut fuÈ r FestkoÈ rperforschung,Heisenbergstraûe 1,D-70569 Stuttgart,Fax 07 11-6 89 15 02e-mail: [email protected]
Zur Existenz von Tetramethylammoniumamalgam
AgCl in CH3CN/LiClges.) beginnend zu weniger nega-tiven Werten hin veraÈndert, wobei ein kathodischerProzeû (Stromfluû und reduktive Abscheidung desProduktes) bis ca. ±2,8 V beobachtet wurde. An die-sem Punkt (Ruhepotential) stoppt die Abscheidungdes Produktes und der anodische Prozeû setzt ein(RuÈ ckreaktion unter Oxidation des Produktes). Wennalles Produkt zersetzt ist, flieût kein Strom mehr.Auch nach Umkehr der Richtung der PotentialveraÈn-derung bei ±1,2 V flieût bis zum Ruhepotential keinStrom, d. h. es findet keine elektrochemische Reak-tion statt. Erst am Ruhepotential setzt wieder die Re-duktion zum Amalgam ein und ein kathodischerStromfluû im mA-Bereich kann registriert werden.Aus diesem cyclovoltammetrischen Experiment laÈûtsich also die obere Potentialgrenze fuÈ r die Abschei-dung von Tetramethylammoniumamalgam zu ±2,8 V(gegen Ag/AgCl in CH3CN/LiClges.) bestimmen, dieuntere Grenze wird durch die Zersetzung des LoÈ -sungsmittels bestimmt und liegt bei hoÈ herem negati-ven Potential.
Der Nachweis daruÈ ber, daû das Produkt Tetrame-thylammoniumionen und Quecksilber enthaÈ lt, wirddurch Analyse seiner Zersetzungsprodukte erbracht.ErwaÈrmt man frisch synthetisiertes Tetramethylammo-niumamalgam in der ElektrolytloÈ sung auf ca. 0 °C, soblaÈht es sich unter Gasentwicklung auf das etwa 20fa-che Volumen auf und zerfaÈ llt nach einigen Minuten,wobei Quecksilber zuruÈ ckbleibt. ErwaÈrmt man isolier-te Substanz, so beobachtet man ebenfalls heftige Gas-entwicklung, es bleibt Quecksilber zuruÈ ck. Bei Úffnendes GefaÈûes wird der Geruch von Trimethylaminwahrnehmbar. Diesen bemerkt man auch bei Zerset-zung von getrocknetem Tetramethylammoniumamal-gam an der Luft. Die Substanz uÈ berzieht sich dannauûerdem mit einer matten Schicht eines alkalischreagierenden, hygroskopischen Stoffes, bei dem es
sich um Tetramethylammoniumhydroxid handelt. Diethermische Zersetzung fuÈ hrt also zu Quecksilber undgasfoÈ rmigen Zersetzungsprodukten.
Im hochaufgeloÈ sten Massenspektrum der Zerset-zungsprodukte von Tetramethylammoniumamalgamwurden Trimethylamin, Acetonitril, Methan undEthen detektiert (s. Tabelle 1). Mit der Detektion derSpezies CH4 und C2H4, die bei der massenspektrosko-pischen Untersuchung von reinem Trimethylamin undreinem Acetonitril unter gleichen Bedingungen nichtnachgewiesen wurden, gelang erstmals der massen-spektroskopische Nachweis von Zersetzungsproduk-ten, die die Anwesenheit von Tetramethylammoniumim untersuchten Produkt belegen.
Die temperaturabhaÈngige Vermessung der magneti-schen SuszeptibilitaÈt einer unzersetzten Probe in ei-nem Squid-Magnetometer zeigt diamagnetisches Ver-halten und durch eine geringe, jedoch sprunghafteAbnahme von v waÈhrend des Aufheizens die Zerset-zung von Tetramethylammoniumamalgam bei ±34 °Can. Die nach der magnetischen Messung im abge-schmolzenen ProbenroÈ hrchen enthaltenen gasfoÈ rmi-gen Zersetzungsprodukte wurden ausgefroren undwiederum massenspektroskopisch untersucht, wobeidie in der Tabelle 1 aufgefuÈ hrten Fragmente erneutdetektiert werden konnten.
Bei der elektrochemischen Umsetzung reagiert ge-nau soviel Tetramethylammoniumtetrafluoroborat,wie nach dem Faradayschen Gesetz m = M ´ Q/z ´ F(mit m = Masse, Q = Ladungsmenge, M = Molmasse,z = Anzahl ausgetauschter Elektronen, F = 96493 C/mol)berechnet werden kann, wenn man 100% Stromaus-beute [37], einen EinelektronuÈ bergang pro Tetrame-thylammoniumion und ein VerhaÈ ltnis [N(CH3)4
+] : Hgvon 1 : 5 annimmt. Die Menge des umgesetzten Aus-gangsstoffes wurde durch AuswaÈgen des nach der Re-aktion verbleibenden Ûberschusses quantifiziert. Hier-nach leiten wir fuÈ r das erhaltene Produkt eineZusammensetzung von [N(CH3)4]Hg5 ab. Ein VerhaÈ lt-
Z. Anorg. Allg. Chem. 2000, 626, 1892±1896 1893
Abb. 1 Cyclovoltammogramm(Vorschubgeschwindigkeit 100 mV/s, weitere ErlaÈuterungenim Text)
Tabelle 1 Massenspektroskopische Identifizierung der Zer-setzungsprodukte von Tetramethylammoniumamalgam (an-gegeben werden alle Fragmente auûer N2, die mit einerIntensitaÈ t > 2% auftreten)
Fragment Masse, u IntensitaÈ t, %
N(CH3)3 59,0753 46,15N(CH3)2(CH2) 58,0675 100,00N(CH3)2(CH) 57,0594 3,69N(CH3)(CH2) 43,0454 5,61N(CH2)2 42,0379 27,61CH3CN 41,0301 44,11N(CH)2 40,0225 17,02HC2N 39,0147 3,64CH2NH2 30,0356 37,67C2H5 29,0415 3,23C2H4 28,0349 4,31CH2N 28,0224 2,05C2H3 27,0281 2,02CH4 16,0341 16,61

nis von Kation zu Metall von 1 : 5 wurde kuÈ rzlich vonSvetlicic et al. auch fuÈ r [DMP]Sn5 und [DMP]Pb5 for-muliert [38, 39].
Die Reduktionskraft des Produktes, also die Exi-stenz des auf das Produkt von der Kathode uÈ bertrage-nen Elektrons auch nach Unterbrechen des Stromesund insbesondere noch mehrere Stunden nach der imexperimentellen Teil beschriebenen Isolierung, belegtdie Reaktion mit dem Redoxindikator 9-Fluorenon.Dieser faÈrbt sich in Gegenwart reduzierender Speziesrot.
In der Literatur findet sich mehrfach der Hinweisdarauf, daû Tetramethylammoniumamalgam eineStruktur wie Quecksilber ± leicht verzerrt ± aufweist.In der Tat zeigen die wenigen beschriebenen Diffrak-togramme von [N(CH3)4]Hgx Reflexe, die mit derStruktur von elementarem Quecksilber in Ûberein-stimmung zu bringen sind [24±26, 34]. Da in elementa-rem Quecksilber die kuÈ rzesten AbstaÈnde zwischenden Quecksilberatomen 347 und 299 pm betragen,scheint es ausgeschlossen, daû ein Tetramethylammo-niumion (mit einem Durchmesser von 550 bis 570 pm)auf einem Zwischengitterplatz oder substituierend indie Struktur von Quecksilber eingebaut werden kann,ohne daû in dieser die Anordnung der Atome ent-scheidend veraÈndert wird. Das zeigt, daû fruÈ here An-gaben zur Kristallstruktur von Tetramethylammo-niumamalgam vermutlich auf durch elementaresQuecksilber hervorgerufenen Beugungsereignissen be-ruhen.
In den jetzt durchgefuÈ hrten Experimenten hat sichTetramethylammoniumamalgam ausnahmslos als roÈ nt-genamorph erwiesen. Bei diesen Experimenten han-delte es sich um RoÈ ntgenbeugung an Substanz(±40 °C), die in Glaskapillaren ¹hineingewachsenªwar, und um RoÈ ntgenbeugung an einzelnen Bruch-stuÈ cken der Dendrite (Abb. 2), die in Glaskapillarenverankert und bei ±150 °C auf dem Einkristalldiffrak-tometer vermessen wurden (die Partikel wurden nachdem Beugungsexperiment darauf gepruÈ ft, ob es sichnoch um unzersetzte Substanz handelte, indem sie mit9-Fluorenon in Kontakt gebracht wurden) undschlieûlich um in situ-Neutronenbeugungsunter-suchungen mittels einer Elektrolysezelle aus Quarzmit eingeschmolzenen Elektroden, die den Dimensio-nen des D1B Neutronendiffraktometers (Grenoble)angepaût war. In ihr gelang es, bei ±36 °C Tetra-methylammoniumamalgam auf dem Diffraktometerzu synthetisieren und das dendritisch in den Neutro-nenstrahl gewachsene Produkt zu untersuchen. Keinesdieser Experimente lieferte Hinweise auf detektier-bare Beugungsereignisse.
Somit kann nach unseren Ergebnissen zwar die Exi-stenz und Zersetzungstemperatur von Tetramethylam-moniumamalgam als gesichert angesehen und dessenZusammensetzung eingegrenzt werden, seine Konsti-tution bleibt jedoch unklar.
Experimenteller Teil
Darstellung. In Anlehnung an die Arbeiten vonLittlehailes und Woodhall [25, 26] haben wir Tetra-methylammoniumamalgam auf elektrochemischenWege dargestellt, entsprechend der Reaktionsglei-chung:
x Hg + [N(CH3)4+] + e± = [N(CH3)4
+]Hgxe±
Die Elektrolysezelle (Abb. 3) wurde wie folgt geschal-tet:
7 (Pt)Hg/CH3CN + [N(CH3)4][BF4]/CH3CN +[N(CH3)4][BF4]/Pt +
Tetramethylammoniumtetrafluoroborat wurde alsAusgangssubstanz gewaÈhlt, weil Vorversuche mit Tet-ramethylammoniumchlorid ergeben hatten, daû dieChlorentwicklung an der Gegenelektrode die Pro-duktbildung bei laÈngeren Reaktionszeiten empfindlichstoÈ rt.
In ein vorbereitetes (ausgeheiztes) H-Rohr-GefaÈû wird imArgonstrom Quecksilber gegeben (35 g; Fa. Degussa, Frank-furt, SonderqualitaÈ t III, 99,9995%; die Destillation desQuecksilbers vor der Umsetzung beeinfluût das Reaktions-ergebnis nicht). Das GefaÈû wird noch einmal im Feinvaku-um mit der entleuchteten Bunsenbrennerflamme ausgeheizt,bevor im Argonstrom zwei Quickfit/Schliff-ÛbergangsstuÈ ckevon oben in das H-Rohr eingesetzt und durch die Verschrau-bungen (Teflondichtungen) glasummantelte Platinelektrodenin die durch eine G4-Glasfritte getrennten Schenkel desReaktionsgefaÈûes eingefuÈ hrt werden. Eine der Elektrodentaucht in das Quecksilber, so daû der Platindraht gaÈnzlichvon Quecksilber bedeckt ist (Arbeitselektrode). Als Gegen-elektrode ragte eine Platinwendel in den Anodenraum.
Tetramethylammoniumtetrafluoroborat [N(CH3)4][BF4](Fa. Merck-Schuchardt, Hohenbrunn, > 98%) wird 16 Stun-den bei 120 °C im Feinvakuum getrocknet und dann unterArgon in einen zuvor ausgeheizten Kolben gefuÈ llt (1 g).Frisch destilliertes Acetonitril (s. u.) wird mit einer Pipette(80 ml) aus einem mit Argon gefuÈ llten Kolben auf das Tet-ramethylammoniumtetrafluoroborat gegeben. Der Kolben
B. Albert, M. Jansen
1894 Z. Anorg. Allg. Chem. 2000, 626, 1892±1896
Abb. 2 Lichtmikroskopaufnahmen von Tetramethylammo-niumamalgam (65fache VergroÈ ûerung)

Zur Existenz von Tetramethylammoniumamalgam
wird verschlossen, der LoÈ sungsvorgang mit Ultraschall be-schleunigt und die LoÈ sung sofort im Argonstrom in dasH-Rohr-GefaÈû pipettiert. Nach Schlieûen der beiden dieElektrolysezelle mit der Schutzgasanlage verbindenden HaÈh-ne wird die Elektrolysezelle in ein EthanolkuÈ hlbad, welchesmit einem TauchkuÈ hlgeraÈ t auf ±36 °C gehalten wird, ge-taucht. Anwesenheit von Wasser in Acetonitril bedroht nichtnur die StabilitaÈ t der Zielverbindung, sondern beeinfluûtauûerdem den Potentialbereich, in welchem mit Acetonitrilunter VernachlaÈssigung der Zersetzung des LoÈ sungsmittelsgearbeitet werden kann. Zwischen Arbeits- und Gegen-elektrode kann eine Potentialdifferenz von 8 V (Beginn derElektrolyse) bis 12 V (Ende der Elektrolyse) abgegriffenwerden (Voltmeter, Modell Multimeter BBC M200K,Fa. Metrawatt, NuÈ rnberg). Die elektrochemische Zersetzungvon Acetonitril ist erkennbar an charakteristischen FarbaÈn-derungen (® gelb, rot, braun). Acetonitril wird uÈ ber Phos-phorpentoxid (Fa. Baker, Deventer) getrocknet (48 Stundenrefluxiert, anschlieûend uÈ ber eine 1 m lange Vigreuxkolonnein argongefuÈ llte Kolben destilliert, das Destillat erneut mitPhosphorpentoxid versetzt, wiederum 16 Stunden refluxiertund erneut abdestilliert).
Als Spannungsquelle diente ein Potentiostat (Modell 363,Fa. Eg & G Princeton Applied Research Corp., Princeton),der galvanostatisch betrieben wird. Eine StromstaÈrke von500 lA und eine Reaktionsdauer von 24 Stunden erwies sichals optimal. Eine laÈngere Elektrolysedauer ist wegen fort-schreitender Zersetzung des Elektrolyten nicht zu empfeh-len. Die Diffusion der farbigen Zersetzungsprodukte vonAcetonitril durch die Glasfritte in den Kathodenraum be-wirkt dann, daû das Produkt in sich zusammenfaÈ llt undschwarz wird.
Isolierung. Nach Beenden der Reaktion durch Unterbrechendes Stromflusses wird der Elektrolyt sofort im Argonstromabpipettiert und durch frisches, gekuÈ hltes Acetonitril ersetzt,
um nichtumgesetztes TetramethylammoniumtetrafluoroboratherauszuloÈ sen. Ein ummanteltes Glasrohr (Mantel evaku-iert), das mit drei SchliffansaÈ tzen versehen ist, wird mitdurchstroÈ mendem trockenen Stickstoff, der durch eine mitfluÈ ssigem Stickstoff gekuÈ hlte Kupferspirale geleitet wordenist, von innen auf unter ±40 °C gekuÈ hlt. Festes Produkt wirdim Argonstrom aus der Elektrolysezelle von der Quecksil-beroberflaÈche zusammen mit daruÈ berstehendem Acetonitrilentnommen und auf kuÈ rzestem Wege in den gekuÈ hltenStickstoffstrom im Glasrohr uÈ berfuÈ hrt.
Es kann durch ein Mikroskop betrachtet und mit Glas-staÈben vorsichtig durch einen der SchliffansaÈ tze in mit fluÈ ssi-gem Stickstoff gekuÈ hlte AufbewahrungsgefaÈûe geschobenwerden.
Charakterisierung. FuÈ r die cyclovoltammetrische Verfolgungder Produktbildung wurde die H-Rohr-Elektrolysezelle zurSynthese von [N(CH3)4] ´ Hg (Konzentration der LoÈ sung wieunter Synthesebedingungen) verwendet und zusaÈ tzlich uÈ bereine Luggin-Kapillare eine Bezugselektrode (Ag/AgCl inCH3CN/LiClges., mit tabelliertem Potential: ±0,19 V gegendie gesaÈttigte Kalomel-Elektrode) eingefuÈ hrt. Die Spannungwurde mit verschiedenen Vorschubgeschwindigkeiten zwi-schen ±1,2 V und ±3,2 V variiert.
Die gasfoÈ rmigen Zersetzungsprodukte, die beim ErwaÈr-men des Produktes auftraten, wurden massenspektrosko-pisch untersucht. Dazu wurden groûe SubstanzstuÈ cke ausder Isolierungsapparatur uÈ ber ein T-StuÈ ck in ein auf ±196 °CgekuÈ hltes einseitig zugeschmolzenes Glasrohr uÈ berfuÈ hrt.Dieses wurde dann im Argonstrom von der Apparatur abge-nommen, mit einem evakuierten, mit Young-Ventil ver-schlossenen Winkelrohr verbunden und uÈ ber den zweitenAbgriff des T-StuÈ ckes kurzzeitig evakuiert (±196 °C). Dannwurde das Young-Ventil geoÈ ffnet, das Winkelrohr gekuÈ hltund die die Substanz enthaltende Ampulle auf Raumtem-peratur erwaÈrmt, wobei die Substanz sich zersetzte, anschlie-ûend das Winkelrohr wieder verschlossen und an den Gas-einlaû des Massenspektrometers angesetzt. Es gelang,hochaufgeloÈ ste Spektren der Gasphase aufzunehmen. ZumVergleich wurden auch Massenspektren von Trimethylamin(Fa. Fluka) und Acetonitril (PCK Schwedt) aufgenommen.
Wir danken dem Fonds der Chemischen Industrie fuÈ r finan-zielle Hilfe und Dr. Alan Hewat vom ILL (Grenoble) fuÈ r sei-ne sehr kompetente und engagierte UnterstuÈ tzung bei derDurchfuÈ hrung des Neutronenbeugsexperiments, sowie HerrnDr. G. Eckhardt (Bonn) fuÈ r die massenspektroskopischenUntersuchungen.
Literatur
[1] T. J. Seebeck, Ann. Chim. (Paris) 1808, 66, 191.[2] H. Davy, Ann. Chim. (Paris) 1809, 70, 225 (Phil. Trans.
1808, 333/369, 354).[3] J. J. Berzelius, M. M. Pontin, Gilbert's Ann. 1810, 36,
261.[4] G. Michaud, Am. Chem. J. 1894, 16, 488.[5] A. Coehn, Z. Anorg. Chem. 1900, 25, 430.[6] A. Coehn, K. Dannenberg, Z. Phys. Chem. 1901, 38,
609.[7] O. Ruff, Chem. Ber. 1901, 34, 2604.[8] H. Moissan, C. R. Acad. Sci. 1901, 133, 713.[9] H. Moissan, C. R. Acad. Sci. 1901, 133, 715.
Z. Anorg. Allg. Chem. 2000, 626, 1892±1896 1895
Abb. 3 Elektrolysezelle zur Synthese von Tetramethylam-moniumamalgam

[10] H. Moissan, Bull. Soc. Chim. Fr. 1902, 27, 714.[11] G. McPhail Smith, Chem. Ber. 1907, 40, 2941.[12] H. Moissan, C. R. Acad. Sci. 1907, 144, 790.[13] A. J. Deyrup, J. Am. Chem. Soc. 1934, 56, 2594.[14] J. F. Chittum, H. Hunt, J. Phys. Chem. 1936, 40, 581.[15] R. J. Johnston, A. R. Ubbelohde, J. Chem. Soc. 1951,
1731.[16] D. Jones, Nature 1994, 368, 404.[17] J. S. Pfeil, H. Leffmann, Z. Chem. 1866, 542.[18] M. Le Blanc, Z. Phys. Chem. 1890, 5, 467.[19] Crotogino, Z. Elektrochem. 1901, 7, 648.[20] H. N. McCoy, W. C. Moore, Science 1909, 3, 315.[21] H. N. McCoy, W. C. Moore, J. Am. Chem. Soc. 1911, 33,
273.[22] G. B. Porter, J. Chem. Soc. 1954, 760.[23] H. Kulke, Diplomarbeit, Univ. Giessen 1954.[24] G. Brauer, G. DuÈ sing, Z. Anorg. Allg. Chem. 1964, 328,
154.[25] J. D. Littlehailes, B. J. Woodhall, J. Chem. Soc., Chem.
Commun. 1967, 665.[26] J. D. Littlehailes, B. J. Woodhall, Discuss. Faraday Soc.
1968, 45, 187.[27] J. Myatt, P. F. Todd, J. Chem. Soc., Chem. Commun.
1967, 1033.
[28] V. A. Smirnov, N. F. Sidorenko, D. P. Semchenko, SU-Pat. 257463, 1969 [C.A. 72:128150c].
[29] E. Kariv-Miller, C. Nanjundiah, J. Electroanal. Chem.1983, 147, 319.
[30] E. Kariv-Miller, C. Nanjundiah, J. Eaton, K. E. Swen-son, J. Electroanal. Chem. 1984, 167, 141.
[31] E. Kariv-Miller, R. Andruzzi, J. Electroanal. Chem.1985, 187, 175.
[32] E. Kariv-Miller, P. B. Lawin, Z. Vajtner, J. Electroanal.Chem. 1985, 195, 435.
[33] E. Kariv-Miller, V. Svetlicic, J. Electroanal. Chem. 1986,205, 319.
[34] E. Kariv-Miller, P. D. Christian, Dissertation P. D. Chri-stian, Minnesota 1990.
[35] E. Garcia, A. H. Cowley, A. J. Bard, J. Am. Chem. Soc.1986, 108, 6082.
[36] B. Albert, M. Jansen, Dissertation, B. Albert, Univ.Bonn 1995.
[37] H. Schmidt, J. Noack, Z. Anorg. Allg. Chem. 1958, 296,262.
[38] E. Kariv-Miller, P. D. Christian, V. Svetlicic, Langmuir1994, 10, 3338.
[39] E. Kariv-Miller, P. D. Christian, V. Svetlicic, Langmuir1995, 11, 1817.
B. Albert, M. Jansen
1896 Z. Anorg. Allg. Chem. 2000, 626, 1892±1896