-nitrosoglutathione reductase s lymphocyte development requires
TRANSCRIPT

of March 28, 2018.This information is current as
-nitrosoglutathione ReductaseSLymphocyte Development Requires
Limin LiuWei Wei, Harry Ischiropoulos, Richard M. Locksley and Zhiyong Yang, Zhi-En Wang, Paschalis-Thomas Doulias,
ol.1000080http://www.jimmunol.org/content/early/2010/10/27/jimmun
published online 27 October 2010J Immunol
MaterialSupplementary
0.DC1http://www.jimmunol.org/content/suppl/2010/10/28/jimmunol.100008
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved.1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on March 28, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on March 28, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Lymphocyte Development Requires S-nitrosoglutathioneReductase
Zhiyong Yang,*,1 Zhi-En Wang,*,1 Paschalis-Thomas Doulias,†,‡ Wei Wei,*
Harry Ischiropoulos,†,‡ Richard M. Locksley,*,x,{ and Limin Liu*
NO is critical to immunity, but its role in the development of the immune system is unknown. In this study, we show that
S-nitrosoglutathione reductase (GSNOR), a protein key to the control of protein S-nitrosylation, is important for the development
of lymphocytes. Genetic deletion of GSNOR in mice results in significant decrease in both T and B lymphocytes in the periphery.
In thymus, GSNOR deficiency causes excessive protein S-nitrosylation, increases apoptosis, and reduces the number of CD4 single-
positive thymocytes. Lymphopenia and increase in S-nitrosylation and apoptosis in GSNOR-deficient mice are largely abolished
by genetic deletion of inducible NO synthase. Furthermore, the protection of lymphocyte development by GSNOR is apparently
intrinsic to hematopoietic cells. Thus, GSNOR, likely through regulation of S-nitrosylation and apoptosis, physiologically plays
a protective role in the development of the immune system. The Journal of Immunology, 2010, 185: 000–000.
Nitric oxide, best established as a major mediator of theimmune response, is increasingly recognized as an im-portant regulator of the immune system as well (1).
Inducible NO synthase (iNOS) has been detected in many cells ofthe immune system, although it is absent in most or all normallymphocytes (1). During immune responses, activated T cells, viaIFN-g and perhaps other factors, induce expression of iNOS inimmunosuppressive cells including macrophages (2, 3) and mes-enchymal stem cells (4, 5). NO or other reactive nitrogen speciesfrom iNOS in these cells during immune responses inhibit pro-liferation of T cells (2–5). NO bioactivity from iNOS has alsobeen shown to contribute to elimination of activated effectorT cells in the contraction phase of immune responses, probablythrough increasing T cell apoptosis (6, 7). Constitutive expressionof iNOS is detectable in thymus of immunologically naive mice(8, 9), although development of thymocytes and peripheral lym-phocytes appears to be normal in iNOS-deficient (iNOS2/2) mice(10, 11). Despite extensive study of NO in immunology, a role forNO important to the development of the immune system has notbeen shown.
NO bioactivity originating fromNO synthases involves a numberof reactive nitrogen species, including S-nitrosothiol (SNO) (12).S-nitrosylation, the covalent attachment of NO to cysteine sulfurthat forms SNO, has emerged as a major mechanism throughwhich NO modifies protein functions to exert control over a widerange of biological processes (13). S-nitrosoglutathione (GSNO),the main nonprotein SNO in cells, exists in equilibrium withprotein SNOs (14, 15). The ubiquitously expressed GSNO re-ductase (GSNOR, also known as alcohol dehydrogenase class III)degrades GSNO and, through the equilibrium between GSNOand S-nitrosylated proteins, regulates cellular levels of proteinS-nitrosylation (14–18). Genetic studies with targeted deletion ofGSNOR gene have established that NO bioactivity and functionare controlled not only at the level of synthesis by NOS butthrough enzymatic degradation by GSNOR (14–19). Studies ofGSNOR-deficient (GSNOR2/2) mice show that endogenousSNOs in systemic inflammation can cause apoptosis of lympho-cytes in thymus, spleen, and lymph nodes, indicating that en-dogenously formed SNOs may impair lymphocyte survival (15).During our study of GSNOR2/2 mice, we noticed that despite
being reared in a specific-pathogen–free facility, the animalssometimes suffered lung infection by opportunistic pathogens,thus appearing to be immunodeficient. In this study, we show thatGSNOR deficiency, likely through SNO originating from iNOS,impairs lymphocyte development and causes lymphopenia in na-ive GSNOR2/2 mice.
Materials and MethodsAnimals
GSNOR2/2 mice (15), after backcrossing 10 times to C57BL/6, were bredwith iNOS2/2 and eNOS2/2 mice (The Jackson Laboratory, Bar Harbor,ME) to obtain GSNOR2/2iNOS2/2 and GSNOR2/2eNOS2/2 mice, re-spectively. Both GSNOR2/2iNOS2/2 and GSNOR2/2eNOS2/2 mice arefertile, although litter sizes of GSNOR2/2eNOS2/2 mice are oftensmaller. B6.SJL-Ptprca (C57BL/6-Ly5.1) mice are obtained from the Na-tional Cancer Institute (Bethesda, MD), and TCRa-subunit–deficient(TCRa2/2) (20) mice are from The Jackson Laboratory. All mice weremaintained on normal mouse chow (5058 PicoLab Mouse Diet 20) ina specific-pathogen–free facility at the University of California, SanFrancisco (San Francisco, CA). Lung inflammation was analyzed bycounting inflammatory cells in airway lining fluid (17). Lung infection bythe opportunistic pathogenPasteurella pneumotropicawas detected throughPCR analysis by University of California (UC) Davis Comparative
*Department of Microbiology and Immunology, xHoward Hughes Medical Institute,and {Department of Medicine, University of California San Francisco, San Francisco,CA 94143; and †Department of Pediatrics and ‡Department of Pharmacology, Child-ren’s Hospital of Philadelphia Research Institute, University of Pennsylvania, Phil-adelphia, PA 19104
1Z.Y. and Z.-E.W. contributed equally to this work.
Received for publication January 11, 2010. Accepted for publication September 20,2010.
This work was supported by the Sandler Family Supporting Foundation (to L.L. andR.M.L.), National Institutes of Health Grants CA122359 (to L.L.) and HL54926 (toH.I.), and Howard Hughes Medical Institute (to R.M.L.).
Address correspondence and reprint requests to Dr. Limin Liu, Department of Mi-crobiology and Immunology, University of California San Francisco, 513 ParnassusAvenue, HSE-201J, San Francisco, CA 94143. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this paper: Asc, ascorbate; BM, bone marrow; DP, double-positive; DTPA, diethylenetriamine pentaacetic acid; eNOS, endothelial NO syn-thase; GSNO, S-nitrosoglutathione; GSNOR, GSNO reductase; iNOS, inducibleNO synthase; KO, knockout; LC-MS/MS, liquid chromatography tandem mass spec-trometry; MRC, organomercury resin; MMTS, methyl methanethiosulfonate; SNO,S-nitrosothiol; SP, single-positive; UC, University of California; WT, wild type.
Copyright� 2010 by The American Association of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1000080
Published October 27, 2010, doi:10.4049/jimmunol.1000080 by guest on M
arch 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Pathology Laboratory (Davis, CA). Rederivation of GSNOR2/2 micethrough in vitro fertilization and embryo transfer was carried out byMurine Cryopreservation and Recovery Laboratory of UC Davis. Theexperimental protocol was approved by the Institutional Animal Careand Use Committee of University of California, San Francisco.
Flow cytometry
Complete blood count was performed by the UC Davis Comparative Pa-thologyLaboratory. Single-cell suspensionswere incubatedwith anti-CD16/CD32 blocking Ab (2.4G2) for 5 min at room temperature and then labeledwith the following Abs: allophycocyanin-Cy7–conjugated anti-CD4, FITC-conjugated anti-CD8a, PerCP-Cy5.5–conjugated anti-CD11b, allophyco-cyanin- or FITC-conjugated anti-CD19, allophycocyanin-conjugated anti-CD44, PE-conjugated anti-CD62L, allophycocyanin- or FITC-conjugatedanti-GR1, PE-conjugated anti-Ly5.1, and PerCP-Cy5.5–conjugated anti-Ly5.2. Flow cytometry was carried out using a BD Biosciences LSR-II(San Jose, CA) with live cells gated by DAPI exclusion, and lymphocytesgated on size and granularity on the basis of forward and side scatter.
Mixed bone marrow chimeras
Bone marrow (BM) from GSNOR2/2 mice (Ly5.2) was mixed at a 1:1ratio with BM from GSNOR-wild type (WT) C57BL/6-Ly5.1 mice, and2 3 106 cells were injected into lethally irradiated (2 3 600 rad)GSNOR2/2 or WT (Ly5.2) mice. Chimeras were provided with 2 g/Lneomycin sulfate and 100 mg/L polymyxin B in drinking water. Micewere analyzed at 16–20 wk after reconstitution.
Competitive reconstitution of CD4 cells
CD4 cells purified from spleen and mesenteric lymph node of GSNOR2/2
mice (Ly5.2) were mixed at 1:1 ratio with those purified from C57BL/6-Ly5.1 mice and injected intravenously into TCRa2/2 mice (5 3 106 cells/recipient). Mice were analyzed at 4 and 9 d after adoptive transfer.
TUNEL assay
Frozen tissue sections were prepared and assayed with In Situ Cell DeathDetection Kit (fluorescein) from Roche Applied Science (Mannheim, Ger-many), following the manufacturer’s protocol.
Selective enrichment and mass spectrometric identification ofS-nitrosylated proteins
Thymuses from three to four mice were combined and homogenized ina lysis solution (250 mM HEPES, pH 7.7, 1 mM diethylenetriaminepentaacetic acid [DTPA], 0.1 mM neocuproine, 1% Triton X-100, andprotease inhibitors) on ice. The homogenate was centrifuged at 13,0003 gfor 30 min at 4˚C, and the supernatant was collected. The amount ofprotein SNOs in the thymus extract was measured with the tri-iodidemethod, as described previously (21). An aliquot of the thymus extract,supplemented with 100 mM mannitol and 5 mM methyl methanethiosul-fonate (MMTS), was illuminated with a conventional UV transilluminatorfor 5 min on ice. Another aliquot of the extract was incubated with 1 mMascorbate (Asc)/0.1 mM CuSO4 for 30 min at 37˚C. UV photolysis and Cu/Asc treatment eliminate S-nitrosylation and were used to generate negativecontrols. Four milligrams of the UV-exposed, Cu/Asc-treated, or untreatedprotein extracts were precipitated with three volumes of acetone at 220˚Cfor 20 min, pelleted by centrifugation, and washed twice with 1 mL of220˚C acetone. The proteins were solubilized in blocking solution (250mM HEPES, pH 7.7, containing 1 mM DTPA, 0.1 mM neocuproine, 2.5%SDS, and 20 mM MMTS) and incubated at 50˚C for 30 min, with vor-texing every 4–5 min to block free thiols. After excess MMTS was re-moved by repeated (23) acetone precipitation, the blocked proteins wereresuspended in 250 mM MES, 1 mM DTPA, pH 6.0, and 1% SDS. Theproteins were loaded onto columns containing organomercury resin(MRC), which was generated by conjugation of r-amino-phenylmercuricacetate to N-hydroxysuccinimide–activated Affi-Gel 10 agarose beads andwas activated with 0.1 M NaHCO3 (pH 8.8). Whereas S-nitrosylatedproteins bound to MRC covalently through the SNO-mercury reaction(22), unbound proteins were removed by extensive washing with 50 mMTris-HCl, pH 7.4, 300 mM NaCl, 0.05% SDS, and 0.1% Triton X-100. Thebound proteins were eluted with 50 mM b-mercaptoethanol in TBS buffer(50 mM Tris-HCl, pH 7.4, 50 mM NaCl), separated by SDS-PAGE, andthen analyzed by liquid chromatography tandem mass spectrometry (21).Alternatively, to identify the specific site(s) of modification, the boundproteins were digested in column (1 mg trypsin in 0.1 M ammonium bi-carbonate, pH 7.4) in the dark at room temperature for 16 h. After ex-tensive wash with ammonium bicarbonate, mild performic acid treatment(3% in water at room temperature for 30 min) was used to release the
bound peptides and oxidize cysteine thiols to sulfonic acid (23). Under theworkflow used, 100% of cysteine-containing peptides were detected withthe sulfonic acid modification. The recovered peptides were analyzed byLC-MS/MS (21). Evaluation of SEQUEST peptide sequence assignmentsand protein identification were as described previously (21). Sequence-to-spectrum assignments from two biological replicates were combined inSCAFFOLD (21). The false identification rate (peptides identified in thenegative controls) was ,3%.
Statistical analysis
The data were analyzed with the two-tailed Student t test.
ResultsSusceptibility to lung infection by opportunistic pathogens inGSNOR2/2 mice
When first generated and studied, GSNOR2/2 mice were freefrom lung infection and inflammation (17). However, lung in-flammation, indicative of infection, was recently detected in un-challenged GSNOR2/2 mice that were reared in a specific-pathogen–free facility (Supplemental Fig. 1). We also detectedlung infection by the opportunistic pathogen Pasteurella pneu-motropica in one of the three GSNOR2/2 mice studied. Lunginflammation was not detectable in GSNOR2/2 mice afterrederivation to get rid of opportunistic pathogens. Thus,GSNOR2/2 mice appear to be immunodeficient.
Lymphopenia in GSNOR2/2 mice
Although total counts of blood leukocytes, as with erythrocytesand platelets, are comparable between GSNOR2/2 and WT mice(Fig. 1A) (15), flow cytometric analyses showed significantlyfewer CD4 and CD8 T cells in the blood of GSNOR2/2 mice thanin WT control mice (Fig. 1B, 1C). Decrease in T cells, particularlyCD4 cells, was also observed in the spleen of GSNOR2/2 mice
FIGURE 1. Decrease in T lymphocytes in blood of GSNOR2/2 mice. A,
Numbers of WBCs, RBCs, and platelets in WT (white bars) and
GSNOR2/2 (knockout [KO]; black bars) mice. The data (mean 6 SE) are
from three WT and four KO age-matched mice. B, Percentage of CD4
(CD4+CD82CD192) T cells, CD8 (CD42CD8+CD192) T cells, CD19
(CD42CD82CD19+) B cells, and CD11b (CD11b+CD42CD82CD192)
cells in WBCs of WT and GSNOR2/2 mice. The data are mean (6SE)
from age-matched (5–12 wk) WT (n = 10–14) and KO (n = 13–17) mice.
ppp , 0.01, WT versus KO. C, Representative flow cytometric analysis of
total viable WBCs from WT and GSNOR2/2 mice.
2 LYMPHOCYTE DEVELOPMENT REQUIRES GSNOR
by guest on March 28, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
(Fig. 2C). Although B cell counts were slightly greater in the bloodof GSNOR2/2 mice than in WT control mice (Fig. 1), B cells weresignificantly fewer in the spleen of GSNOR2/2 mice (Fig. 2C),suggesting overall decrease of peripheral B cells in the knockoutmice. Ab production after immunization nevertheless appearedunimpaired in GSNOR2/2 mice (Supplemental Fig. 2). In contrastwith lymphocytes, myeloid cells including CD11b+ cells were notdecreased in either blood or spleen of GSNOR2/2mice (Figs. 1, 2).Thus, GSNOR deletion in mice results in lymphopenia.We used Abs to surface markers CD44 and CD62L to further
analyze CD4 T cells, the population decreased most prominentlyin GSNOR2/2 mice. Whereas the number of CD62L2CD44high
CD4 cells, the memory or memory-like cells, was comparablebetween GSNOR2/2 and WT mice, CD62L+ cells, in particular,the CD62L+CD44low naive cells, were significantly fewer in thespleen of GSNOR2/2 mice than in WT mice (Fig. 2D). Thus,GSNOR deficiency appears to cause decrease in the populationof naive CD4 T cells.
Deficiency of CD4 single-positive thymocytes in GSNOR2/2
mice
We investigated whether lymphopenia in GSNOR2/2 mice resultsfrom defective proliferation or abnormal death of the peripherallymphocytes. After adoptive transfer of equivalent numbers ofGSNOR2/2 and WT CD4 cells into TCRa2/2 mice, competitiverepopulation of the cells resulted in a 1:1 ratio of GSNOR2/2 toWTCD4 cells in recipient mice (Fig. 3 and data not shown), indicatingnormal growth of the GSNOR2/2 cells. In addition, data froma number of other analyses, including proliferation assaywith CFSEin vitro and TUNEL assay of spleen and lymph node, revealed nodefects in GSNOR2/2 T cells (data not shown and SupplementalFigs. 3, 4). Thus, lymphopenia in GSNOR2/2 mice is unlikely toresult from defects in peripheral lymphocytes.We analyzed thymocyte populations to determine whether
lymphopenia in GSNOR2/2 mice results from diminished pro-duction of lymphocytes from thymus. The total number of thy-mocytes in GSNOR2/2 mice was modestly decreased (Fig. 4A).
Whereas the proportions of double-positive (DP) and CD8 single-positive (SP) thymocytes in GSNOR2/2 mice were comparablewith those of WT mice, the proportion of CD4 SP thymocyteswas significantly lower in GSNOR2/2 than in WT mice (Fig. 4).Accordingly, DP and CD8 SP cell counts were affected modestlyin GSNOR2/2 mice, but the number of CD4 SP thymocytes wasmarkedly decreased (Supplemental Fig. 5). Thus, the develop-ment of CD4 SP thymocytes is significantly affected by GSNORdeficiency.
Lymphopenia in GSNOR2/2 mice is rescued by geneticdeletion of iNOS
To further establish the role of NO in disrupting lymphocyte de-velopment in GSNOR2/2 mice and to identify the source of NO,we analyzed lymphocytes in mice deficient in both GSNOR andiNOS or endothelial NOS (eNOS). Although lymphopenia was notrescued in GSNOR2/2eNOS2/2 mice (data not shown), deletionof iNOS in GSNOR2/2 mice led to significant recovery of pop-ulations of CD4 SP cells in thymus (Fig. 4), CD4 T cells in blood(Fig. 5A) and spleen (Fig. 5B), and B cells in spleen (Fig. 5B).Thus, lymphocyte development is apparently disrupted inGSNOR2/2 mice by SNO originating from iNOS.
FIGURE 2. Decrease in lymphocytes in the spleen of GSNOR2/2 mice.
Weight (A), total leukocyte counts (B), and CD4, CD8, CD19, and CD11b
cell counts (C) in spleens of WT and GSNOR2/2 (KO) mice. D, Flow
cytometric analysis of gated CD4 T cells. The data are mean (6SE) from
WT (n = 4–9) and knockout (KO; n = 4–11) mice. ppp , 0.01.
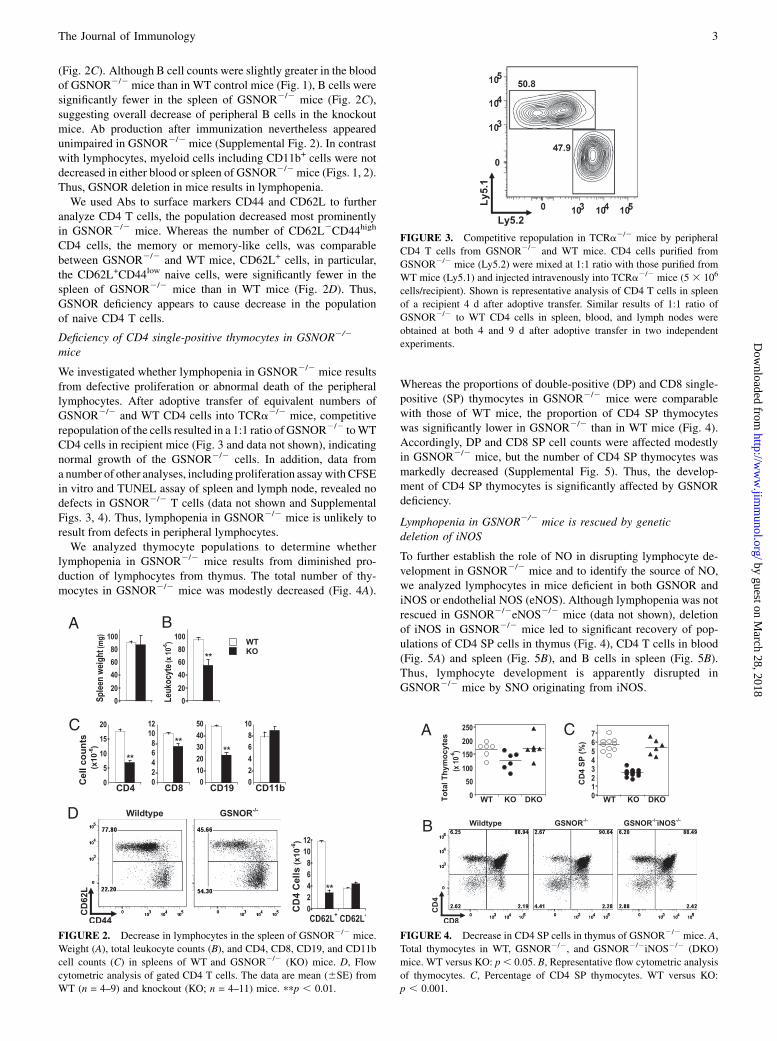
FIGURE 3. Competitive repopulation in TCRa2/2 mice by peripheral
CD4 T cells from GSNOR2/2 and WT mice. CD4 cells purified from
GSNOR2/2 mice (Ly5.2) were mixed at 1:1 ratio with those purified from
WT mice (Ly5.1) and injected intravenously into TCRa2/2 mice (5 3 106
cells/recipient). Shown is representative analysis of CD4 T cells in spleen
of a recipient 4 d after adoptive transfer. Similar results of 1:1 ratio of
GSNOR2/2 to WT CD4 cells in spleen, blood, and lymph nodes were
obtained at both 4 and 9 d after adoptive transfer in two independent
experiments.
FIGURE 4. Decrease in CD4 SP cells in thymus of GSNOR2/2 mice. A,
Total thymocytes in WT, GSNOR2/2, and GSNOR2/2iNOS2/2 (DKO)
mice. WT versus KO: p, 0.05. B, Representative flow cytometric analysis
of thymocytes. C, Percentage of CD4 SP thymocytes. WT versus KO:
p , 0.001.
The Journal of Immunology 3
by guest on March 28, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
Increase in apoptosis in thymus of GSNOR2/2 mice
We used the TUNEL assay to analyze apoptosis in thymuses ofGSNOR2/2 and control mice. The level of apoptosis inGSNOR2/2 thymus was significantly greater than that in WTcontrol mice (Fig. 6), indicating that the reduction of thymocytesmay result from elevated apoptosis in GSNOR2/2 mice. Fur-thermore, the increase in apoptosis in GSNOR2/2 thymus wascompletely abolished in GSNOR2/2iNOS2/2 mice. Thus, ele-vated apoptosis in GSNOR2/2 thymus is likely caused by iNOS-derived SNO, which is abolished by GSNOR in WT mice.
Protein S-nitrosylation is increased in thymus of GSNOR2/2
mice
Quantitative analysis indicated a significant increase in the level ofprotein SNOs in the thymus of GSNOR2/2 mice as compared withWT or GSNOR2/2iNOS2/2 mice (Supplemental Fig. 6A). Thelevel of protein SNOs in the GSNOR2/2iNOS2/2 thymus wasalmost 90% lower than that in GSNOR2/2 thymus, indicating thenecessity of iNOS-derived NO for the formation of protein SNOs.To identify endogenously S-nitrosylated proteins in thymus, weapplied a proteomic approach that uses the direct reaction ofSNOs with mercury to form a thiol-mercury bond (22). In thisassay, S-nitrosylated proteins are captured onto a column of MRCthrough the SNO-mercury reaction, released by b-mercaptoetha-nol, and analyzed by LC-MS/MS. Captured proteins were alsosubjected to in-column trypsin digestion to isolate the SNO pep-tides and then analyzed by LC-MS/MS to identify the site(s) ofS-nitrosylation. UV photolysis and copper-ascorbate treatment,which abolish S-nitrosylation, were included in control experi-ments to avoid false identification of SNOs. With this method, weidentified 96 sites of cysteine S-nitrosylation in 83 proteins in WTthymus and 160 sites in 132 proteins in GSNOR2/2 thymus(Supplemental Tables I, II). Thus, the number of S-nitrosylatedtargets increased under GSNOR deficiency. Importantly, only 11
sites of S-nitrosylation in 10 proteins were detected in GSNOR2/2
iNOS2/2 thymus (Supplemental Table III), demonstrating thatmost of the in vivo S-nitrosoproteome in the GSNOR2/2 thymuswas dependent on iNOS-derived NO. The S-nitrosylated proteinsin thymus included a few proteins important to apoptosis.Caspase-6, a mediator of apoptosis (24), was S-nitrosylated at twocysteine residues (cys146, cys271) in GSNOR2/2 thymus, but S-nitrosylated caspase-6 was not detected in WT or GSNOR2/2
iNOS2/2 thymus (Supplemental Tables I–III, SupplementalFig. 6B, 6C). Similarly to caspase-6, S-nitrosylation of voltage-dependent anion channel-1, a protein implicated in the control ofmitochondria-mediated apoptosis (25), was detected only inGSNOR2/2 thymus (Supplemental Tables I–III). In addition, S-nitrosylated GAPDH was detected in WT, GSNOR2/2, andGSNOR2/2iNOS2/2 thymus (Fig. 7, Supplemental Tables I–III).However, the intensity of the ion derived from S-nitrosylatedGAPDH was much greater in GSNOR2/2 than in WT andGSNOR2/2iNOS2/2 thymus (Fig. 7A). Taken together, our datasuggest that in thymus, GSNOR is important for preventing ex-cessive protein S-nitrosylation from iNOS activity.
Lymphopenia in GSNOR2/2 mice is rescued by WT BM
When BM of irradiated GSNOR2/2 mouse was reconstituted withmixed BM cells from WT and GSNOR2/2 mice, T cell deficiencyin thymus and peripheral tissues was rescued (Fig. 8). Thus, thefunction of GSNOR in hemopoietic cells is apparently sufficientfor normal development of T cells, and GSNOR in non-hemopoietic cells is not required.After competitive reconstitution in GSNOR2/2 mice with equal
numbers of GSNOR2/2 and WT BM cells, equal numbers of Gr1+
granulocytes were derived from the two donor sources (Fig. 8B).Thus, BM in recipient mice appeared to be reconstituted by WTand GSNOR2/2 hemopoietic stem cells equally well, and de-velopment of Gr1+ granulocytes from GSNOR2/2 stem cells wascomparable with that from WT stem cells. In contrast, both CD4SP cells in thymus (Fig. 8C) and CD4 T cells in peripheral tissues(Fig. 8B and data not shown) of the reconstituted mice were de-rived virtually completely from WT mice, indicating that
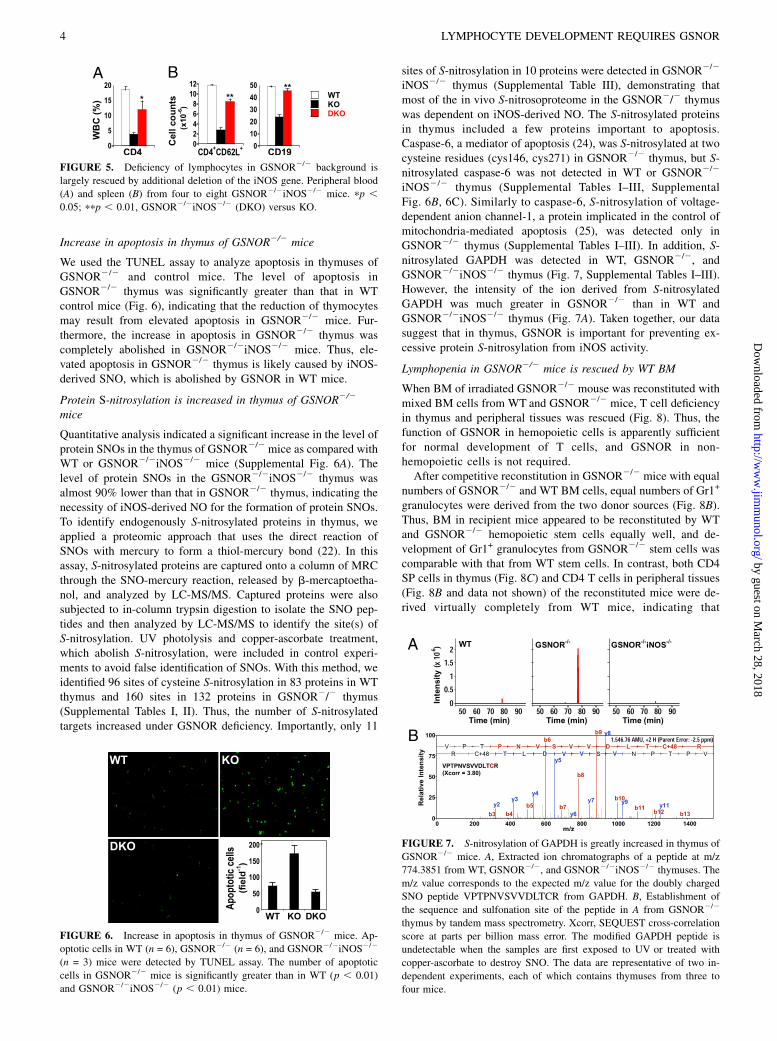
FIGURE 5. Deficiency of lymphocytes in GSNOR2/2 background is
largely rescued by additional deletion of the iNOS gene. Peripheral blood
(A) and spleen (B) from four to eight GSNOR2/2iNOS2/2 mice. pp ,0.05; ppp , 0.01, GSNOR2/2iNOS2/2 (DKO) versus KO.
FIGURE 6. Increase in apoptosis in thymus of GSNOR2/2 mice. Ap-
optotic cells in WT (n = 6), GSNOR2/2 (n = 6), and GSNOR2/2iNOS2/2
(n = 3) mice were detected by TUNEL assay. The number of apoptotic
cells in GSNOR2/2 mice is significantly greater than in WT (p , 0.01)
and GSNOR2/2iNOS2/2 (p , 0.01) mice.
FIGURE 7. S-nitrosylation of GAPDH is greatly increased in thymus of
GSNOR2/2 mice. A, Extracted ion chromatographs of a peptide at m/z
774.3851 from WT, GSNOR2/2, and GSNOR2/2iNOS2/2 thymuses. The
m/z value corresponds to the expected m/z value for the doubly charged
SNO peptide VPTPNVSVVDLTCR from GAPDH. B, Establishment of
the sequence and sulfonation site of the peptide in A from GSNOR2/2
thymus by tandem mass spectrometry. Xcorr, SEQUEST cross-correlation
score at parts per billion mass error. The modified GAPDH peptide is
undetectable when the samples are first exposed to UV or treated with
copper-ascorbate to destroy SNO. The data are representative of two in-
dependent experiments, each of which contains thymuses from three to
four mice.
4 LYMPHOCYTE DEVELOPMENT REQUIRES GSNOR
by guest on March 28, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
development of CD4 T cells is severely compromised by GSNORdeficiency. Surprisingly, DP and CD8 SP thymocytes and CD19+
B cells in the reconstituted mice were derived mostly from WTmice (Fig. 8 and data not shown). In addition, selective de-velopment of T and B cells from WT BM was also observed whencompetitive reconstitution experiments were performed in WTrecipient mice (data not shown). Thus, GSNOR deficiency inhemopoietic cells, at least in the competitive condition, signifi-cantly impaired development of B and T lymphocytes.
DiscussionThis study shows the unexpected result that normal developmentof lymphocytes, and hence of the adaptive immune system, de-pends on the function of GSNOR in the regulation of endogenousSNOs. We find that genetic deletion of GSNOR causes an increasein protein S-nitrosylation and cell apoptosis in thymus and wide-spread lymphopenia. The defects in the immune system arelargely prevented by further deletion of iNOS, indicating iNOS asthe origin of immunosuppressive SNOs. Furthermore, our resultssuggest that lymphocyte development may depend on functionalGSNOR in hemopoietic cells.Our study suggests that GSNOR deficiency causes apoptosis
through excessive protein S-nitrosylation from iNOS in thymus ofimmunologically unchallenged mice. Expression of iNOS hasbeen described in dendritic cells and other stromal cells in thymusof immunologically naive mice (8, 9, 26). Thymic expression of
iNOS may be increased by stimulation of TCR signaling withanti-CD3 Ab (8) or superantigen Staphylococcal enterotoxin B(9). Increased iNOS activity can cause apoptosis of thymocytes (8,27). We showed previously that thymocyte apoptosis caused byincreased iNOS activity in immune response was greatly increasedby GSNOR deficiency (15). We find in this study that GSNORdeficiency increases apoptosis from iNOS in thymus of immu-nologically naive mice. In these animals, GSNOR deficiency alsoresults in increase in thymic content of total protein SNOs, theabundance of S-nitrosylated proteins, and the level of S-nitro-sylation of proteins. When excessive protein S-nitrosylation isabolished by iNOS deletion, increase of thymic apoptosis is pre-vented, further supporting a causative role of dysregulated S-nitrosylation in apoptosis. Proteins with increased levels of S-nitrosylation in GSNOR2/2 thymus include a few involved in thecontrol of apoptosis. Caspase-6 is an important execution caspasein apoptosis (24), and voltage-dependent anion channel-1 is con-sidered important for the control of mitochondria-mediatedapoptosis (25). S-nitrosylation, which can modulate enzymaticactivity, channel conductivity, and protein-protein interaction (13),might modulate the function of caspase-6 or voltage-dependentanion channel-1 in apoptosis. S-nitrosylation of GAPDH has beenshown to promote nuclear accumulation of GAPDH and to causeapoptosis in macrophages and neurons (28). It is thus possible thatthe marked increase of GAPDH S-nitrosylation in GSNOR2/2
thymus may promote apoptosis of thymocytes. Because apoptosisis a key mechanism for the control of the development and numberof lymphocytes in thymus (29), increased apoptosis in thymus ofGSNOR2/2 mice may cause reduced thymic output of T cells andconsequently lymphopenia in periphery.T cell lymphopenia in GSNOR2/2 mice may result largely from
diminished thymic output. Reduction of CD4 and CD8 SP thy-mocytes in GSNOR2/2 mice is associated with reduction of CD4and CD8 T cells in periphery. In fact, the decreases of CD4 andCD8 SP cells in thymus are quantitatively comparable with thoseof CD4 and CD8 T cells, respectively, in secondary lymphoidorgans. Diminished thymic output typically has little effect on thenumber of memory T cells but often significantly decreases thenumber of naive T cells (30, 31), in part, because proliferation ofnaive T cells induced by lymphopenia converts them to memory-like T cells (32). Reduction of the naive T cell population with nochange in the number of memory phenotype cells in GSNOR2/2
mice thus provides further evidence suggesting reduced thymicoutput as the major cause of T cell lymphopenia. Alternatively,T cell lymphopenia in GSNOR2/2 mice might result from in-crease in lymphocyte death in periphery. However, apoptosis inperipheral lymphoid tissues appears unchanged from GSNORdeficiency. T cell lymphopenia also is not likely to result fromimpaired proliferation of T cells in periphery, because lympho-penia-induced proliferation of T cells is unlikely to produce naivephenotype T cells (32), and because proliferative responses ofT cells from GSNOR2/2 mice are similar to those of WT control.GSNOR appears to be particularly important to the development
of CD4 SP thymocytes. Both CD4 and CD8 SP thymocytes developfrom DP cells after positive selection. Because GSNOR deficiencycauses no change in the proportions of DP and CD8 SP cells inthymocytes, but a significant reduction in the proportion of CD4SP cells, GSNOR deficiency does not appear to affect the de-velopment of CD8 SP cells from DP cells or CD4-versus-CD8lineage choice. Instead, GSNOR deficiency may impair the gen-eration of CD4 SP cells from DP cells or the survival of CD4 SPlineage cells. A number of proteins important to CD4 SP lineagehave been identified in thymocytes, although most of the proteinsalso play important roles in the development of DP or CD8 SP cells
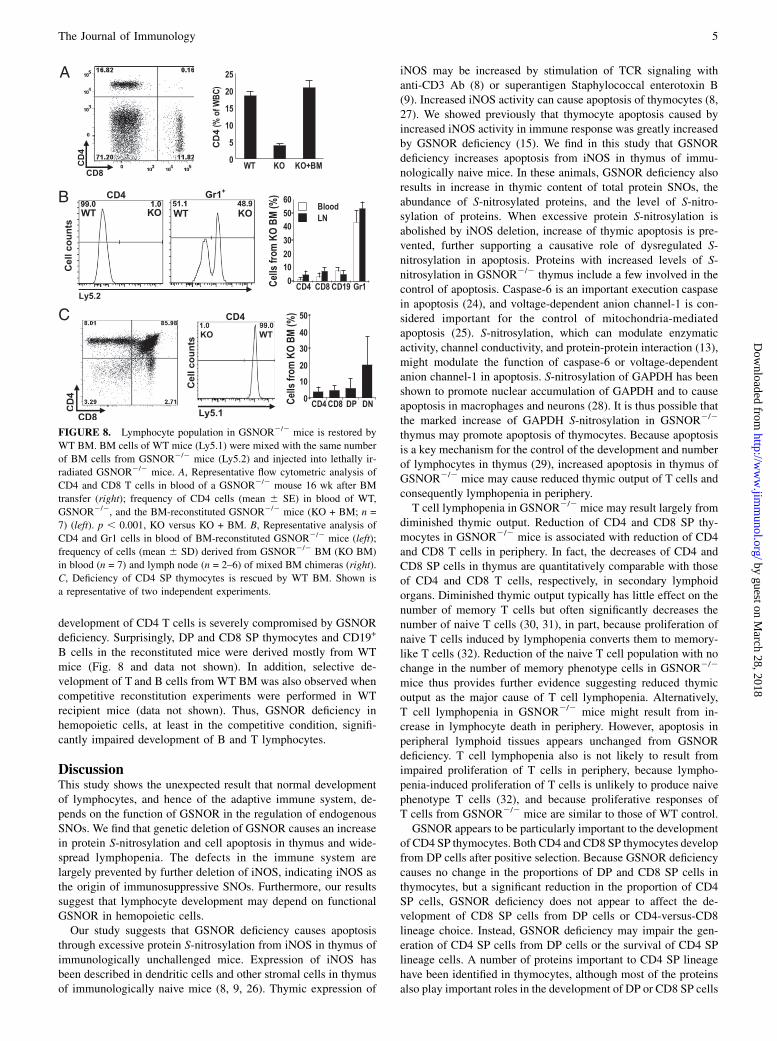
FIGURE 8. Lymphocyte population in GSNOR2/2 mice is restored by
WT BM. BM cells of WT mice (Ly5.1) were mixed with the same number
of BM cells from GSNOR2/2 mice (Ly5.2) and injected into lethally ir-
radiated GSNOR2/2 mice. A, Representative flow cytometric analysis of
CD4 and CD8 T cells in blood of a GSNOR2/2 mouse 16 wk after BM
transfer (right); frequency of CD4 cells (mean 6 SE) in blood of WT,
GSNOR2/2, and the BM-reconstituted GSNOR2/2 mice (KO + BM; n =
7) (left). p , 0.001, KO versus KO + BM. B, Representative analysis of
CD4 and Gr1 cells in blood of BM-reconstituted GSNOR2/2 mice (left);
frequency of cells (mean 6 SD) derived from GSNOR2/2 BM (KO BM)
in blood (n = 7) and lymph node (n = 2–6) of mixed BM chimeras (right).
C, Deficiency of CD4 SP thymocytes is rescued by WT BM. Shown is
a representative of two independent experiments.
The Journal of Immunology 5
by guest on March 28, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

(33, 34). Conditional deletion of GATA-3 or c-Myb in DP thy-mocytes through CD4-Cre causes minimal changes in the per-centage of DP and CD8 SP cells but significantly reduces thepercentage of CD4 SP cells (35, 36). In addition, CD83 deficiencyin thymic epithelial cells has little effect on the development ofDP or CD8 SP thymocytes but significantly reduces the number ofCD4 SP cells (37). It remains to be determined whether deficiencyof GSNOR and GATA-3, c-Myb, or CD83 inhibits a common stepor pathway of CD4 SP development.Our results, particularly those from BM chimeras, suggest that
GSNOR may be important to the development of both T andB cells. GSNOR2/2 BM stem cells in the competitive re-constitution experiment gave rise to minimal DP, CD4 SP, andCD8 SP thymocytes, suggesting that GSNOR is important forT cell development not only at the stage of CD4 SP cells but at anearlier stage. This is consistent with reduction of total thymocytesin GSNOR2/2 mice. In addition, the marked deficiency of mostT and B cells, but no deficiency of granulocytes, from GSNOR2/2
BM stem cells may suggest defective development of commonlymphoid progenitors (38) of GSNOR2/2 mice. The immuno-suppressive effect could be a side effect of NO, or it might rep-resent dysregulated NO signaling in lymphocyte progenitors.Alternatively, the inhibitory mechanisms in T and B lineagecells might be unrelated. We reported earlier that GSNOR,through its control of S-nitrosylation, is essential for the survivalof lymphocytes in the inflammatory response (15). Our currentfindings demonstrate that GSNOR is also important for the de-velopment of lymphocytes, establishing a broad function forGSNOR in the immune system.
AcknowledgmentsWe thank Drs. Yan Zhang, Yanan Zhu, and Cliff Mcarthur for technical as-
sistance and Drs. Anthony DeFranco and Christopher Allen for helpful
comments.
DisclosuresThe authors have no financial conflicts of interest.
References1. Bogdan, C. 2001. Nitric oxide and the immune response. Nat. Immunol. 2: 907–
916.2. Albina, J. E., J. A. Abate, and W. L. Henry, Jr. 1991. Nitric oxide production is
required for murine resident peritoneal macrophages to suppress mitogen-stimulated T cell proliferation. Role of IFN-gamma in the induction of thenitric oxide-synthesizing pathway. J. Immunol. 147: 144–148.
3. Mills, C. D. 1991. Molecular basis of “suppressor” macrophages. Argininemetabolism via the nitric oxide synthetase pathway. J. Immunol. 146: 2719–2723.
4. Sato, K., K. Ozaki, I. Oh, A. Meguro, K. Hatanaka, T. Nagai, K. Muroi, andK. Ozawa. 2007. Nitric oxide plays a critical role in suppression of T-cell pro-liferation by mesenchymal stem cells. Blood 109: 228–234.
5. Ren, G., L. Zhang, X. Zhao, G. Xu, Y. Zhang, A. I. Roberts, R. C. Zhao, andY. Shi. 2008. Mesenchymal stem cell-mediated immunosuppression occurs viaconcerted action of chemokines and nitric oxide. Cell Stem Cell 2: 141–150.
6. Dalton, D. K., L. Haynes, C. Q. Chu, S. L. Swain, and S. Wittmer. 2000. In-terferon gamma eliminates responding CD4 T cells during mycobacterial in-fection by inducing apoptosis of activated CD4 T cells. J. Exp. Med. 192: 117–122.
7. Vig, M., S. Srivastava, U. Kandpal, H. Sade, V. Lewis, A. Sarin, A. George,V. Bal, J. M. Durdik, and S. Rath. 2004. Inducible nitric oxide synthase in T cellsregulates T cell death and immune memory. J. Clin. Invest. 113: 1734–1742.
8. Tai, X. G., K. Toyo-oka, N. Yamamoto, Y. Yashiro, J. Mu, T. Hamaoka, andH. Fujiwara. 1997. Expression of an inducible type of nitric oxide (NO) synthasein the thymus and involvement of NO in deletion of TCR-stimulated double-positive thymocytes. J. Immunol. 158: 4696–4703.
9. Virag, L., G. Hasko, A. L. Salzman, and C. Szabo. 1998. NADPH diaphorasehistochemistry detects inducible nitric oxide synthetase activity in the thymus ofnaive and staphylococcal enterotoxin B-stimulated mice. J. Histochem. Cyto-chem. 46: 787–791.
10. MacMicking, J. D., C. Nathan, G. Hom, N. Chartrain, D. S. Fletcher,M. Trumbauer, K. Stevens, Q. W. Xie, K. Sokol, N. Hutchinson, et al. 1995.
Altered responses to bacterial infection and endotoxic shock in mice lackinginducible nitric oxide synthase. Cell 81: 641–650.
11. Wei, X. Q., I. G. Charles, A. Smith, J. Ure, G. J. Feng, F. P. Huang, D. Xu,W. Muller, S. Moncada, and F. Y. Liew. 1995. Altered immune responses in micelacking inducible nitric oxide synthase. Nature 375: 408–411.
12. Stamler, J. S., D. J. Singel, and J. Loscalzo. 1992. Biochemistry of nitric oxideand its redox-activated forms. Science 258: 1898–1902.
13. Hess, D. T., A. Matsumoto, S. O. Kim, H. E. Marshall, and J. S. Stamler. 2005.Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6:150–166.
14. Liu, L., A. Hausladen, M. Zeng, L. Que, J. Heitman, and J. S. Stamler. 2001. Ametabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature410: 490–494.
15. Liu, L., Y. Yan, M. Zeng, J. Zhang, M. A. Hanes, G. Ahearn, T. J. McMahon,T. Dickfeld, H. E. Marshall, L. G. Que, and J. S. Stamler. 2004. Essential roles ofS-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116: 617–628.
16. Feechan, A., E. Kwon, B. W. Yun, Y. Wang, J. A. Pallas, and G. J. Loake. 2005.A central role for S-nitrosothiols in plant disease resistance. Proc. Natl. Acad.Sci. USA 102: 8054–8059.
17. Que, L. G., L. Liu, Y. Yan, G. S. Whitehead, S. H. Gavett, D. A. Schwartz, andJ. S. Stamler. 2005. Protection from experimental asthma by an endogenousbronchodilator. Science 308: 1618–1621.
18. Wei, W., B. Li, M. A. Hanes, S. Kakar, X. Chen, and L. Liu. 2010. S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hep-atocarcinogenesis. Sci Transl Med 2: 19ra13.
19. Lima, B., G. K. Lam, L. Xie, D. L. Diesen, N. Villamizar, J. Nienaber,E. Messina, D. Bowles, C. D. Kontos, J. M. Hare, et al. 2009. EndogenousS-nitrosothiols protect against myocardial injury. Proc. Natl. Acad. Sci. USA106: 6297–6302.
20. Mombaerts, P., A. R. Clarke, M. A. Rudnicki, J. Iacomini, S. Itohara,J. J. Lafaille, L. Wang, Y. Ichikawa, R. Jaenisch, M. L. Hooper, et al. 1992.Mutations in T-cell antigen receptor genes alpha and beta block thymocyte de-velopment at different stages. Nature 360: 225–231.
21. Greco, T. M., R. Hodara, I. Parastatidis, H. F. Heijnen, M. K. Dennehy,D. C. Liebler, and H. Ischiropoulos. 2006. Identification of S-nitrosylation motifsby site-specific mapping of the S-nitrosocysteine proteome in human vascularsmooth muscle cells. Proc. Natl. Acad. Sci. USA 103: 7420–7425.
22. Saville, B. 1958. A scheme for the colorimetric determination of microgramamounts of thiols. Analyst (Lond.) 83: 670–672.
23. Pesavento, J. J., B. A. Garcia, J. A. Streeky, N. L. Kelleher, and C. A. Mizzen.2007. Mild performic acid oxidation enhances chromatographic and top downmass spectrometric analyses of histones. Mol. Cell. Proteomics 6: 1510–1526.
24. Degterev, A., M. Boyce, and J. Yuan. 2003. A decade of caspases. Oncogene 22:8543–8567.
25. Shoshan-Barmatz, V., V. De Pinto, M. Zweckstetter, Z. Raviv, N. Keinan, andN. Arbel. 2010. VDAC, a multi-functional mitochondrial protein regulating celllife and death. Mol. Aspects Med. 31: 227–285.
26. Aiello, S., M. Noris, G. Piccinini, S. Tomasoni, F. Casiraghi, S. Bonazzola,M. Mister, M. H. Sayegh, and G. Remuzzi. 2000. Thymic dendritic cells expressinducible nitric oxide synthase and generate nitric oxide in response to self- andalloantigens. J. Immunol. 164: 4649–4658.
27. Fehsel, K., K. D. Kroncke, K. L. Meyer, H. Huber, V. Wahn, and V. Kolb-Bachofen. 1995. Nitric oxide induces apoptosis in mouse thymocytes. J.Immunol. 155: 2858–2865.
28. Hara, M. R., N. Agrawal, S. F. Kim, M. B. Cascio, M. Fujimuro, Y. Ozeki,M. Takahashi, J. H. Cheah, S. K. Tankou, L. D. Hester, et al. 2005. S-nitrosylatedGAPDH initiates apoptotic cell death by nuclear translocation following Siah1binding. Nat. Cell Biol. 7: 665–674.
29. Marsden, V. S., and A. Strasser. 2003. Control of apoptosis in the immunesystem: Bcl-2, BH3-only proteins and more. Annu. Rev. Immunol. 21: 71–105.
30. Bourgeois, C., Z. Hao, K. Rajewsky, A. J. Potocnik, and B. Stockinger. 2008.Ablation of thymic export causes accelerated decay of naive CD4 T cells in theperiphery because of activation by environmental antigen. Proc. Natl. Acad. Sci.USA 105: 8691–8696.
31. Voehringer, D., H. E. Liang, and R. M. Locksley. 2008. Homeostasis and effectorfunction of lymphopenia-induced “memory-like” T cells in constitutively T cell-depleted mice. J. Immunol. 180: 4742–4753.
32. Jameson, S. C. 2002. Maintaining the norm: T-cell homeostasis. Nat. Rev.Immunol. 2: 547–556.
33. Ho, I. C., T. S. Tai, and S. Y. Pai. 2009. GATA3 and the T-cell lineage: essentialfunctions before and after T-helper-2-cell differentiation. Nat. Rev. Immunol. 9:125–135.
34. Rothenberg, E. V., and T. Taghon. 2005. Molecular genetics of T cell de-velopment. Annu. Rev. Immunol. 23: 601–649.
35. Pai, S. Y., M. L. Truitt, C. N. Ting, J. M. Leiden, L. H. Glimcher, and I. C. Ho.2003. Critical roles for transcription factor GATA-3 in thymocyte development.Immunity 19: 863–875.
36. Maurice, D., J. Hooper, G. Lang, and K. Weston. 2007. c-Myb regulates lineagechoice in developing thymocytes via its target gene Gata3. EMBO J. 26: 3629–3640.
37. Fujimoto, Y., L. Tu, A. S. Miller, C. Bock, M. Fujimoto, C. Doyle, D. A. Steeber,and T. F. Tedder. 2002. CD83 expression influences CD4+ T cell development inthe thymus. Cell 108: 755–767.
38. Laiosa, C. V., M. Stadtfeld, and T. Graf. 2006. Determinants of lymphoid-myeloid lineage diversification. Annu. Rev. Immunol. 24: 705–738.
6 LYMPHOCYTE DEVELOPMENT REQUIRES GSNOR
by guest on March 28, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from