alternative splicing and tissue-specific elastin · pdf filealternative splicing and...
TRANSCRIPT

1
Alternative Splicing and Tissue-specific Elastin Misassembly act as Biological Modifiers of Human Elastin Gene Frame Shift Mutations Associated with Dominant Cutis Laxa
Hideki Sugitani1, Eiichi Hirano1, Russell H. Knutsen1, Adrian Shifren1, Jessica E. Wagenseil2,
Christopher Ciliberto1, Beth A. Kozel3, Zsolt Urban4, Elaine C. Davis5, Thomas J. Broekelmann1, and Robert P. Mecham1
1Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, MO.
2Department of Biomedical Engineering, St. Louis University, St. Louis, MO. 3Department of Pediatrics, Washington University School of Medicine, St. Louis, MO. 4Department of Human Genetics, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA. 5Department of Anatomy and Cell Biology, McGill University, Montreal, Quebec, Canada
Running Title: Elastic fiber assembly and human disease
Corresponding Author: Robert Mecham, Washington University School of Medicine, Department of Cell Biology and Physiology, Campus Box 8228, 660 South Euclid Ave. St. Louis, MO 63110, Tel: 314-362-2254, Email: [email protected] Keywords: elastin, extracellular matrix, cutis laxa, alternative splicing Background: A humanized mouse was developed to study elastin assembly and the pathogenesis of cutis laxa. Results: Mutant transcripts incorporate into elastic fibers of skin and lung with adverse effects, but not aorta. Conclusions: Elastin frame-shift mutations alter elastin assembly domains. Significance: The mechanism of elastic fiber assembly may not be the same in all tissues. SUMMARY Elastin is the extracellular matrix protein in vertebrates that provides elastic recoil to blood vessels, the lung, and skin. Because the elastin gene has undergone significant changes in the primate lineage, modeling elastin diseases in non-human animals can be problematic. To investigate the pathophysiology underlying a class of elastin gene mutations leading to autosomal dominant cutis laxa, we engineered a cutis laxa mutation (single base deletion) into the human elastin gene contained in a bacterial artificial chromosome. When expressed as a transgene in mice, mutant elastin was incorporated into elastic fibers in the skin and lung with adverse effects on tissue function. In contrast, only low levels of mutant protein incorporated into aortic elastin, which explains
why the vasculature is relatively unaffected in this disease. RNA stability studies found that alternative exon splicing acts as a modifier of disease severity by influencing the spectrum of mutant transcripts that survive nonsense-mediated decay. Our results confirm the critical role of the C-terminal region of tropoelastin in elastic fiber assembly and suggest tissue-specific differences in the elastin assembly pathway.
The extracellular matrix protein elastin appeared in evolution concurrent with the closed circulatory system and allowed the heart to evolve into an efficient multi-chambered pump by enabling the arterial tree to support pulsatile flow. In a similar way, elastin enabled the vertebrate lung to become a more efficient gas exchange organ by allowing the respiratory apparatus to store potential energy created by contraction of the diaphragm during inhalation and use that energy to drive lung recoil during exhalation (1). The critical role of elastin in vertebrate tissue function is underscored by the fact the mice lacking elastin do not survive past the first few hours of life (2).
There is a single elastin gene in mammals (ELN) that encodes from 34-36 exons (depending on species) with a relatively simple promoter that is regulated mostly in an on-off fashion during development (3, 4). Like many other mammalian genes, ELN undergoes extensive alternative splicing,
http://www.jbc.org/cgi/doi/10.1074/jbc.M111.327940The latest version is at JBC Papers in Press. Published on May 9, 2012 as Manuscript M111.327940
Copyright 2012 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

2
which results in isoform diversity. Although the functional consequences of alternative splicing are unclear, the tight regulation of this process during development suggests an important role for variant transcripts in elastin function (5, 6).
Elastin is secreted as a monomer (tropoelastin) that is crosslinked in the extracellular space to form the functional polymer. The process of tropoelastin secretion and elastic fiber assembly is complicated and not all steps are understood (3). Proteins known to be important in the assembly process include members of the fibulin, fibrillin, and lysyl oxidase families. Functional mapping studies have identified domains in the C-terminal region of the tropoelastin protein, encoded by exons 30 and 36, that are critical for tropoelastin self-association and for interaction with cells and accessory assembly proteins (7). Loss of function mutations that delete this region of the gene transcript (e.g., premature termination, multi-exon deletion, etc.) lead to elastin haploinsufficiency and supravalvular aortic stenosis (SVAS, OMIM 185500), an autosomal dominant disease in humans that predominantly affects the large elastic vessels in the vascular system (8-12).
A second class of elastin mutation is linked to an autosomal dominant form of cutis laxa (ADCL, OMIM 123700) and arises from nucleotide deletion, insertion, or exon-splicing errors. These mutations produce missense sequence, usually in the 3’ end of the transcript that encodes the sequences important for fiber assembly and elastin function (13-16). In contrast to loss of function associated with deletion of the 3’ region, evidence suggests a dominant-negative mechanism for ADCL where the mutant tropoelastin adversely affects assembly and function of protein from the normal allele (17, 18).
The clinical phenotype of ADCL is variable, ranging from loose and sagging skin—the defining phenotype in this disease—to severe pulmonary complication and mild internal organ involvement (15, 16, 19-21). An interesting question is why this type of elastin mutation greatly affects skin and lung, whereas the aorta, with a much higher elastin concentration, is relatively spared. This is opposite to what is seen in SVAS where elastin loss-of-function mutations severely affect the aorta and other elastic vessels but have little consequence in the skin.
To better understand the molecular pathology of ADCL and to learn more about how elastin gene mutations influence elastic fiber assembly, we used
recombineering to introduce a well-characterized ADCL mutation—a single base deletion in exon 30 at position 2012 (2012∆G) (16)—into the human elastin gene in a bacterial artificial chromosome (BAC). The mutant elastin BAC (hBACCL) and a BAC containing the wild-type human gene (hBACWT) were then express as transgenes in mice. It was necessary to use the human gene for this study because the elastin gene has undergone significant changes in the primate lineage (22). The human elastin gene has two fewer exons than mouse, attributable to the sequential loss of exons 34 and 35 during primate evolution (23). In addition, although still contained in the human gene, exon 22 is never included in the human elastin transcript but is found in the mouse transcript. Another difference is that the 3’ exons around the CL mutations are frequently spliced out in humans but not in mice (22), and the presence or absence of these spliced exons has a functional consequence in terms of transcript length and stability. These differences suggest that human mutations introduced into the mouse elastin gene will have a different biological effect and might not be relevant for functional analysis of the human disease. Importantly, the human BAC elastin transgene retains the human alternative splicing pattern when expressed in the mouse (22), which makes the human BAC mouse a relevant model to study how ELN mutations impact tissue function. Our findings show that mutant transcript is expressed appropriately in all elastin-rich tissues and that alternative splicing of down-stream exons generates multiple transcripts with different lengths of missense sequence and different stabilities. Analysis of tissues from animals expressing the hBACCL transgene found that the mutant protein is incorporated into elastic fibers in skin and lung with adverse effects, but low levels of incorporation were observed in the aorta, which explains why the vasculature is relatively unaffected in this disease as a consequence of this mutation. These results suggest that the process of elastin assembly may be different in the aorta compared to lung and skin, which challenges the current thinking of a common fiber assembly mechanism in all tissues. EXPERIMENTAL PROCEDURES Engineering a single base deletion synonymous to a known human cutis laxa mutation into the human BAC elastin gene
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

3
To introduce the cutis laxa mutation (deletion of nucleotide 2012) into the ELN-BAC gene (clone CTB-51J22, Research Genetics), we used a two-step RedE/RedT-recombination technique (24) in E. coli strain DY380 that expresses bacteriophage proteins for homologous recombination (kindly provided by Dr. Neal Copeland, National Cancer Institute, Frederick, MD). The overall strategy is illustrated in Supplemental Figure 1. The first step was recombination between a cloned genomic elastin fragment containing both a single base deletion at position 2012 sequence and a sacB-Kan fusion gene. This construct was prepared by cloning a 1.9 kb genomic DNA fragment containing elastin exons 28, 29, and 30 from hBACWT into a pUC vector. Guanine at BAC position 30494 (position 2012 in exon 30 of the elastin gene) was deleted using PCR. A sacB-Kan fusion gene, constructed by PCR using the pKOV-Kan vector as a template (kindly provided by Dr. John Heath, Birmingham, UK) (25) was inserted into an AccI site in the 1.9 kb elastin construct located in intron 29. The construct was then trimmed with BmgBi and PpuMi giving a 626 bp construct (nucleotides 30345-30970).
The bacteria were prepared for the first recombination step by eletroporating the complete unmodified elastin BAC into DY380 cells using a Gene Pulser® II Electroporation System (Bio-rad) at 2.5 kV, 25 µF, 200 Ω in a 0.2 cm cuvette. Electroporated cells were grown in LB media containing chloramphenicol at 30ºC to prevent the expression of bacteriophage proteins. Recombination was initiated by activating the BAC-containing DY380 cells at 42ºC for 45 minutes then washing in cold water. The 626 bp genomic fragment containing the mutation and the sacB-Kan cassette were then introduced into the cells by electroporation (1.8 kV, 20 µF, 200 Ω in 0.1 cm cuvette). After incubating the electroporated cells at 30˚C for 1.5 hours to facilitate recombination, the bacteria were spread onto LB agar plates containing 20 µg/ml chloramphenicol and 20 µg/ml kanamycin. Surviving cells were harvested and used for the second recombination step, which involved electroporation into the cells of the mutant genomic fragment that lacked the sacB-Kan cassette. Selection of recombinants was done by growing the cells on LB agar plates containing 20 µg/ml chloramphenicol and 7% sucrose, which kills cells that still contain the sacB gene.
Appropriate recombination reactions were confirmed by digestion with Bgl I followed by ethidium bromide staining and Southern blot analysis. BAC DNA, obtained using a mini prep kit (Qiagen), was digested with Bgl I endonuclease and run on a 0.7% agarose gel. DNA was transferred from the gel onto a Hybond N+ nylon membrane and hybridized with a 32P-labeled probe specific for elastin intron 27. The probe corresponding to nucleotides 31538-32520 was made by PCR using forward primer 5’-CATTCCAGCCTGGGCGACAGAGCAAG-3’ and reverse primer 5’-GGCCTGGACGACAGAGCGAGACTGTA-3’. hBACCL DNA purification and generation of transgenic mice
hBACCL circular DNA was isolated using a Large-construct kit (Qiagen). After the last ethanol precipitation step, DNA was dissolved in injection buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 0.25 mM EDTA) and then dialyzed on a floating 0.1 µm Millipore membrane filter against the injection buffer. Isolated hBACCL DNA was injected at a concentration of 1 ng/ml into fertilized mouse hybrid C57BL/6—C3H/F1 oocytes, which were implanted into the uterus of pseudopregnant foster mothers. After birth, potential founders were screened for the presence of the transgene by PCR on mouse tail DNA using the human elastin specific primers forward 5’-GATCAGAATCTCTAGGACTGAATAGGCCAG-3’ and reverse 5’-GCTACTAGGGATACTAAGGGGGAATAATGG-3’. PCR reactions were performed in 25 µ l total volume containing 50 mM Tris-HCl (pH 9.2), 16 mM ammonium sulfate, 3.5 mM MgCl2, 0.1% Tween-20, 0.2 mM dNTP, 2.5 U KlentaqLA, and 1 µl mouse tail DNA. Mouse tail DNA was prepared by digesting 2–3 mm of mouse tail tissue overnight at 55°C in 200 µl tail digestion buffer [50 mM Tris-HCl (pH 8.0), 1 mM CaCl2, 1% Tween-20] plus 25 µl proteinase K (10 mg/ml). Animals positive for the transgene were mated to WT animals (C57BL/6) to stabilize the line. hBACCL mice were also bread into the hBACWT line to generate hBACCL- hBACWT double transgenics.
The generation and characterization of Eln+/- mice has been previously reported (2). The genotype at the mouse Eln locus was assessed by PCR
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4
analysis using specific primer sets: Primer A (DL1), 5’-GGTTGTTCAGACTACAATCTGACC-3’; primer B (DL2), 5’- CAACTTTGCCCAAATGACTCTCC-3’; primer C (DL5), 5’- GAGAGGTATAGAGGGAAGACTTGCT-3’. Used together, primers B and C are specific to the wild-type mouse Eln locus and yields a 477-bp fragment, whereas primer A is specific to the targeted mouse Eln locus and yields a 250-bp fragment when used with primer B (22). hBACCL- hBACWT double transgenic mice were genotyped by direct sequencing, using the primer 5’- ATCTTGAGCAAAGACGTGCCA-3’ after PCR amplification (see above).
Southern analysis, RNA isolation, and assessment of alternative splicing
To characterize the transgene by Southern analysis, 10 mg of genomic DNA was digested with Ssp I and subjected to electrophoresis in agarose and transferred to nitrocellulose membranes according to standard procedures. Radioactive probes were produced using a random priming kit obtained from Amersham, and the blots were hybridized with 32P-labeled 340-bp human elastin cDNA probe produced by PCR as described (22). Total RNA was isolated from aorta and lung using a TRIzol reagent kit (Invitrogen). The expression of human tropoelastin mRNA was confirmed by RT-PCR using primers specific for human elastin. Total RNA was reverse-transcribed using a 20-mer oligo (dT) primer and Superscript III reverse transcriptase (Invitrogen). PCR conditions were 95ºC for 5 min, at 95ºC for 30 sec, at 60ºC for 30 sec, at 72ºC for 1 min, 30 cycles.
Splice variants were assessed by semi-quantitative RT-PCR (21 cycles) with human- or mouse-specific primers located in exon 21, 25, 36. Amplification of mouse beta-actin provided an internal control (22). Human exon 21 forward primer 5’-TGCCGCCAAGGCTGCCAAGTA-3’ Human exon 25 reverse 5’-TGGTGGAGTTGCAGCTTGTGCAGCAAAAT-3’ Human exon 25 forward primer 5’-ATTTTGCTGCACAAGCTGCAACTCCACCA-3’ Human exon 36 reverse primer 5’-TTCCGGCCACAAGCTTTCCCCAG-3’
Analysis of exon 32 splicing using real-time quantitative PCR
Six-week old mice were euthanized with carbon dioxide, tissues harvested then immediately frozen for later processing. A square centimeter of skin was shaved then excised, and the lungs were removed by simple dissection. The aorta, from the heart to the diaphragm, was removed and loose connective tissue and fat was removed by careful dissection. RNA was isolated from the tissue using the Neucleospin RNA-XS kit (Macherey-Nagel Inc.). cDNA was reverse transcribed from 500ng mRNA using the Superscript II first–strand synthesis system (invitrogen) with oligo(dT)12-18 primers according to manufacturer’s instructions. Quantitative PC was performed on 10ng transcribed mRNA using an ABI Vii7 real-time PCR system and a common forward primer located in exon 28 of human elastin, a common reverse primer located in exon 36, and splice site specific TaqMan probes custom synthesized by Applied Biosystems Incorporated. The sequence of the primers and probes are: huELN 28F AGTAGGCATCCCAGGCGGTGT, huELN36R TCTCCACCAAGCAGTAGCACCAAC, huELN 31-32 6-Fam AAGCAGCTAAATA-CGGTGCTGCTGG MGB-NFQ, huELN 31-33 6Fam AGCAGCTAAATA-CGGAGTGGCAGCA MGB-NFQ. The dashes in the probes indicate the position of the splice junction. The primer-probe sets are human specific and show no amplification from wild-type mouse tissues (data not shown). TaqMan fast universal 2x PCR master mix (without UNG) from Applied Biosytems was used for the reactions with standard PCR conditions. Results of the amplification were normalized to cyclophilin-A, accession number: NM_008907.1 (ABI, PPIA-Fam Mm02342430_g1), and fold change between samples was calculated using the DDCT method (26) Immunohistochemistry and electron microscopic analyses
Frozen sections of ascending mouse aorta were stained with antibodies specific for mouse (antibody MRT6-17) or human (antibody HAE) elastin followed by detection with fluorescently labeled secondary antibodies. To reduce background autofluorescence from preexisting elastic fibers, sections were incubated with 0.5% pontamine sky blue in PBS for 5 mins followed by washing with
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

5
PBS prior to viewing (27). A Zeiss Axioscope microscope was used for fluorescence microscopy and the images were captured with an Axiocam digital camera using Axiovision software.
For electron microscopy, specimens were fixed in 3% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4), cut into small pieces, washed in buffer and postfixed sequentially en bloc in 1% osmium tetroxide, 2% tannic acid and 2% uranyl acetate (28). After dehydration through a graded series of methanol and infiltration with Epon, tissues were embedded in pure Epon and polymerized. Sixty-nm sections were counterstained with 7% methanolic uranyl acetate and lead citrate and viewed using a Tecnai 12 transmission electron microscope at 120 kV. Vessel, lung, and skin extensibility
For vessel extensibility measurements, isolated vessels from 3-month-old mice were cannulated and mounted on a pressure arteriograph (Danish Myotechnology, Copenhagen, Denmark). Vessels were transilluminated under an inverted microscope connected to a charged-coupled device camera and computerized system, allowing a continuous recording of changes in vessel diameter. Experimental details have been previously described in detail (29, 30). Assessment of respiratory system compliance was conducted as described (31). For mouse skin elasticity measurements, animals were anesthetized with 1% inhaled isoflurane. The abdomen of each mouse was shaved with electric hair clippers and Nair depilatory cream was applied to remove all remaining hair residue. The DermaLab suction cup apparatus (Cortex Technology) was then applied to the abdomen of each mouse using adhesive stickers. Pressure was applied in increasing increments to the small segment of skin under the suction cup causing the skin to be lifted into the apparatus. When the skin reached 2.5 mm, as detected by interference with a light beam emitted within the suction cup, the pressure was recorded and the skin released. This cycle is repeated four subsequent times. Tissue culture and protein synthesis inhibition
Cultured fibroblasts were obtained from skin explants of newborn mice. The cells were maintained in Dulbecco’s modified Eagle media supplemented with 100 units/ml penicillin, 100 µg/ml streptomycin, 2 mM glutamine, 0.1 mM
nonessential amino acid, and 10% fetal bovine serum in a 5% CO2 incubator at 37ºC. To assess RNA stability, cells were treated with the translation inhibitors puromycin (100 µg/ml) or anisomycin (100 µg/ml) for 4 hours prior to harvesting RNA. RNA levels were determined by semi-quantitative PCR as described above. Tissue extraction, western blot analysis, and desmosine analysis.
The aorta was dissected from 2-week-old mice and immediately homogenized in 1 ml RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS). The sample was centrifuged and total protein concentration of the supernatant determined using the BCA assay (Pierce). Three µg of total protein were loaded onto a 7.5% polyacrylamide gel. After electrophoresis, the gel was electroblotted to a nitrocellulose membrane followed by incubation with 5% nonfat milk in Tris-buffered saline containing 0.05% Tween 20 to block antibody nonspecific binding. The blot was incubated with HAE antibody or an antibody to mouse GAPDH (CEA-335, Stressgen) for 1 hour at room temperature and then, after washing, with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG. Reactive bands were detected with ECL detection reagents (GE Healthcare Life Sciences).
Desmosine evaluation in hydrolyzates of aortic tissue was kindly provided by Dr. Barry Starcher, University of Texas Health Science Center, Tyler, TX. Values were normalized to total protein determined by amino acid analysis as described (32). RESULTS Nomenclature of mouse lines
In this report, mWT refers to a non-transgenic mouse with the wild-type murine elastin gene (Eln+/+). mHet is a mouse with one functional elastin allele (Eln+/-). hBACWT refers to the wild-type human elastin gene (ELN) expressed from a BAC (22). hBACCL refers to the human elastin BAC containing the 2012∆G cutis laxa mutation. Compound genotypes are indicated by the presence or absence of each allele. For example, hBACWT-mWT represents the hBACWT transgene in the mWT background (ELN+; Eln+/+), hBACWT-mHet indicates the human wild-type transgene in the mouse heterozygous background (ELN+; Eln+/-),
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

6
and hBACWT-mNull is the human wild-type transgene in the null background (ELN+; Eln-/-). A similar nomenclature is used for the hBACCL background. Mutation of BAC human elastin gene
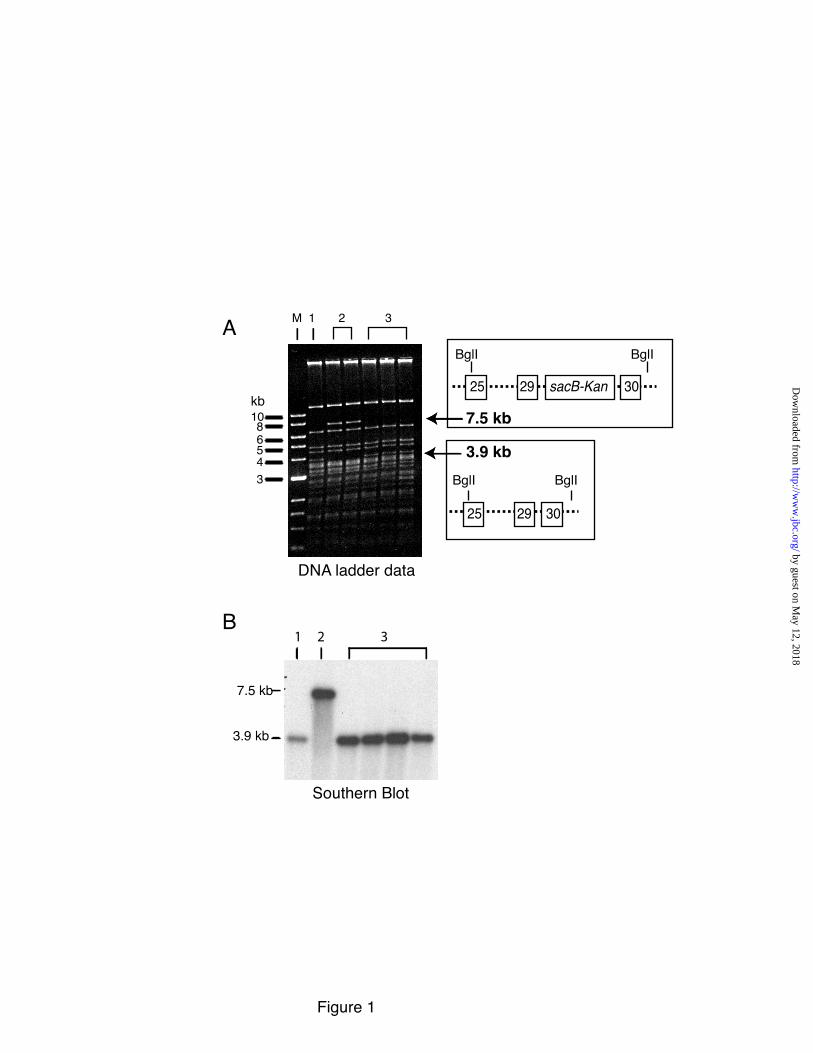
The two-step recombineering procedure used for BAC targeting provided for the introduction of a common ADCL mutation [a single base deletion at position 2012 (2012∆G) of exon 30 (16)] into the human elastin gene without leaving behind a drug-selectable marker at the targeted locus (33, 34) (Supplemental Figure 1). Targeting this region of the gene using homologous recombination was extremely difficult, most likely due to the enrichment of Alu sequences and repetitive elements in the surround introns (35). Correct insertion was confirmed by restriction digestion of hBAC DNA with Bgl1 followed by gel electrophoresis and ethidium bromide staining. Figure 1A shows a ladder pattern of fragments in the Bgl1 digest as well as a 3.9 kb fragment that corresponds to elastin base pairs 30115-33981 (exons 28-30, GeneBank-AC005056). This band shifted in size to 7.5 kb in intermediate clones after kanamycin selection, which is the expected size of the fragment containing the sacB-Kan selection cassette. After the second recombination step, the size of the band returned to 3.9 kb, confirming removal of the sacB-Kan cassette. Southern blot analysis of the Bgl1digests with probes located outside (Figure 1B) and inside (data not shown) of homologous recombination arms confirmed the correct size of the constructs. Moreover, PCR amplification of the final product followed by direct sequencing showed that nucleotide 2012G was deleted in the hBACCL but not hBACWT clone (Supplemental Figure 2). Together, these data confirm that a human elastin gene was generated that contains the 2012∆G cutis laxa mutation. Generation of the hBACCL transgenic mouse
Mouse hybrid C57BL/6—C3H/F1 oocytes injected with the hBACCL were screened for incorporation of the transgene by Southern blot analysis and three founder lines were identified (Figure 2A). The presence of the CL-human elastin gene was detected in all three lines, with lines 1 and 3 having the highest copy number. No Southern-
positive band was detected in nontransgenic WT mice. Line 1 was used in all subsequent studies.
RT-PCR/Southern blot analysis of RNA from 4 week-old animals documented expression of the transgene in aorta, lung, heart and skin, with highest expression levels in the aorta. Semiquantitative RT-PCR showed that mRNA levels from the hBACCL transgene were approximately 50% lower than mRNA levels from the human WT transgene in hBACWT mice. This ratio was similar in aorta and lung of the two animals (Figure 2B). hBACCL expression decreased with increasing age in aorta and lung (data not shown) and was similar to the pattern of expression of the hBACWT transgene and the endogenous mouse gene (22). Cutis laxa-BAC transgene retains human-type alternative splicing
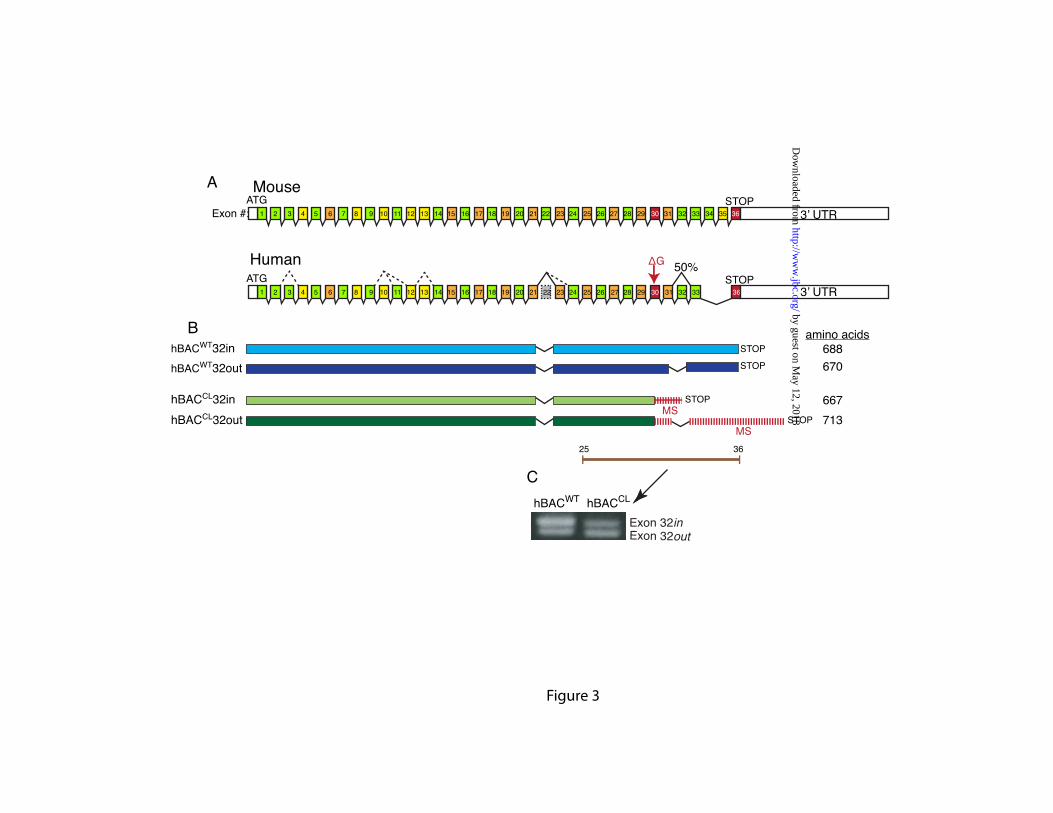
The cutis laxa single base deletion in exon 30 creates a frame shift and missense sequence that continues until encountering a premature stop codon in exon 32. If exon 32 is spliced out, the missense sequence continues into the 3’UTR (Figure 3A and 3B). To investigate the extent of alternative splicing in the human hBACCL transgene and to determine whether the CL mutation influences the splice pattern in the 3’ region of the gene, RT-PCR was performed on total RNA isolated from hBACCL and hBACWT mouse lung tissue using human-specific primers that span exons 25-36. Figure 3C depicts both full-length tropoelastin (hBACWT-ex32in) and tropoelastin lacking exon 32 (hBACWT-ex32out) detected in RNA from the hBACWT, with the higher molecular weight full-length transcript slightly more abundant than the lower molecular weight RNA with exon 32 spliced out. Bands of similar size and density were detected in RT-PCR products from the hBACCL mice, although the ratio of the two bands differed from what was observed for the WT gene. In hBACCL animals, the CL product that contained exon 32 was about half as abundant as the RNA with exon 32 spliced out, and was at much lower levels than the full-length transcript from the WT human hBAC (hBACWT-ex32in). No other bands were detected following RT-PCR with exon 21-36 primers, confirming that only exon 32 is spliced in this region of the gene and that the CL mutation does not alter the overall splicing pattern or induce splicing of exon 30 (Supplemental Figure 3). Similar to our earlier results (22), no splicing was
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

7
detected between exons 21-36 of the endogenous mouse elastin gene in either hBACWT or hBACCL transgenics. CL-mutant transcript undergoes partial nonsense-mediated decay when exon 32 is not spliced out
Reduced levels of full-length mutant (hBACCL-ex32in) transcript compared to the full-length transcript from the WT human gene (hBACWT-ex32in) or from the CL transcript with exon 32 spliced out (hBACCL-ex32out) were also found in skin fibroblasts from hBACWT and hBACCL animals using semiquantitative RT-PCR (Figure 4A). It is unlikely that this disparity results from differing levels of gene transcription, since the hBACWT and hBACCL transgenes are identical except for the single base deletion in the hBACCL. Although insertional effects or variation in copy number could be a reason for the differences, analysis of splice variant abundance offers another explanation. Figure 4 shows that transcript levels with exon32out from both hBACWT and hBACCL is similar for both alleles, confirming approximately equal expression of both transgenes. However, the amount of transcript with exon32in from the hBACCL (a premature stop in the new reading frame) is considerably less (~80%) than exon32in from the hBACWT. The lower transcript levels suggest that mRNA from the mutant allele in both lung and skin is less stable when exon 32 is present. The existence of a premature termination codon in exon 32 of the mutant transcript makes it a candidate for degradation through nonsense-mediated decay (NMD), a RNA surveillance pathway that degrades transcripts with premature termination codons (36). To determine whether NMD was a factor in reducing hBACCL-ex32in mRNA stability, skin fibroblasts from the hBACWT and hBACCL animals were treated with the translation inhibitors puromycin or anisomycin. Since NMD is a translation-dependent process, these drugs can be used to increase the abundance of transcripts that otherwise would be degraded (36). Following treatment with either reagent, there were increased levels of the mutant full-length transcript (hBACCL-ex32in) with little change in the full-length transcript from the hBACWT (Figure 4B) or in levels of the mutant transcript with exon 32 spliced out (hBACCL-ex32out). This finding confirms that the hBACCL-ex32in transcript with a premature stop codon in
exon 32, but not the hBACCL-ex32out transcript with the longer missense sequence, undergoes substantial, but not complete, degradation by NMD.
Tissue-specific splicing in hBACWT and hBACCL
aorta, lung, and skin. A comparison of exon 32 splicing in aorta, lung,
and skin tissue from adult mice is shown in Figure 5. qPCR using primers in exons 28 and 36, and TaqMan probes specific for the exon 32in and 32out splice variants, confirmed less exon 32in than exon 32out transcript in all three tissues in both hBACCL
and hBACWT animals. The extent of exon 32 splicing was similar in the three hBACCL tissues but differed slightly in the hBACWT mouse where skin showed more splicing than aorta.
hBACWT, but not hBACCL, rescues the lethality associated with Eln-/- phenotype
In a previous study, we showed that human elastin from the hBACWT transgene can rescue the perinatal lethality of the mouse null (Eln-/-) phenotype (22). A small number of the hBACWT-mNull animals die shortly after birth, but most live past three months of age. In contrast, no viable animals were found when the hBACCL transgene was bred into the mouse null background (hBACCL-mNull) (Figure 6). Including the hBACCL transgene in the hBACWT-mNull background also had a detrimental effect on the ability of the hBACWT transgene to rescue the null phenotype, with only ~40% of the hBACWT-hBACCL-mNull animals surviving to two months of age (Figure 6). The inability of hBACCL to rescue the elastin-null phenotype suggests that the mutant protein is either incapable of forming functional elastic fibers or is rapidly degraded. Furthermore, the deleterious effect of the mutant protein when combined with WT elastin from the hBACWT gene supports a dominant negative mechanism wherein the mutant protein interferes with normal elastic fiber assembly or function.
Deposition of human WT and CL protein in mouse tissues
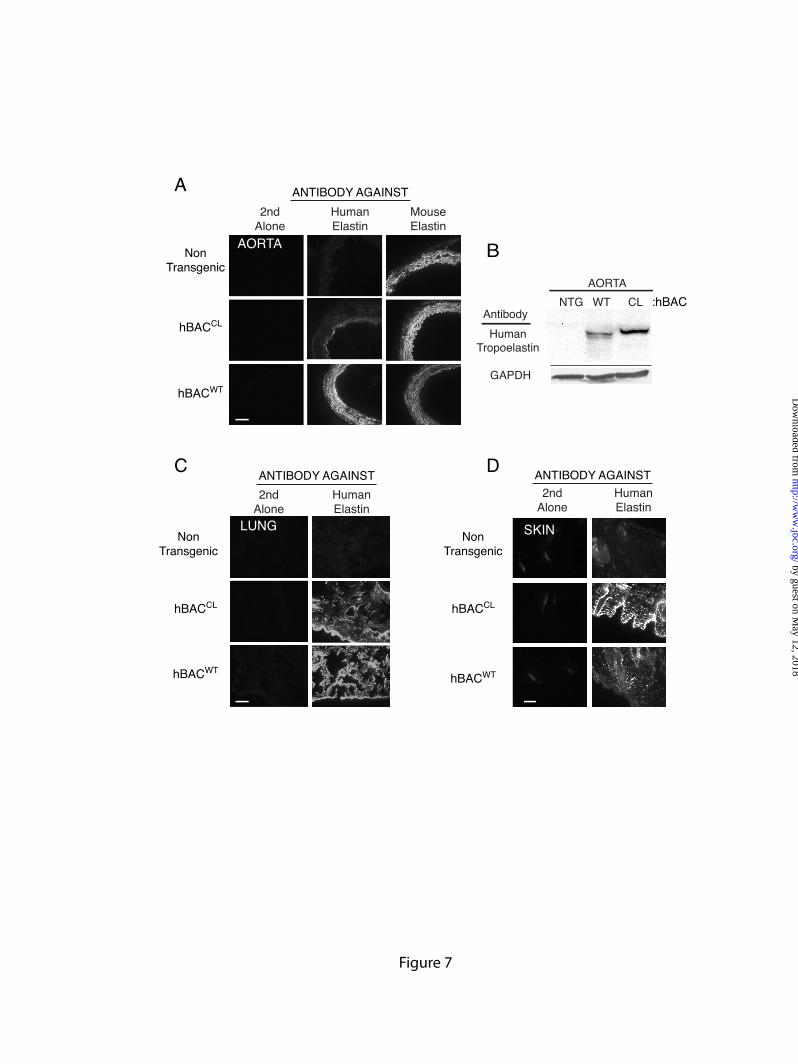
To determine whether tropoelastin containing the CL mutation forms elastic fibers in aorta, lung, and skin, we used human- and mouse-specific elastin antibodies to evaluate the deposition of BAC-transgenic human elastin and endogenous mouse elastin by immunofluorescence microscopy.
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8
Because of the autofluorescence of elastic fibers, it was not possible to utilize double fluorescent labeling techniques to colocalize the human and mouse proteins on the same tissue section. We were able, however, to quench the elastin autofluorescence using the dye pontamine sky blue (27), but this precluded utilizing fluorophores in the red spectrum. Hence, tissue evaluation with the two different antibodies was done on two sections from the same frozen block. It is important to note that, at present, no biochemical method exists that permits the quantification of relative amounts of human versus mouse elastin protein that make up the insoluble fiber.
Figure 7A shows abundant labeling of elastic structures in frozen sections of the aortic wall of non-transgenic animals using the antibody specific for mouse elastin. As expected, the antibody to human elastin showed no reactivity to non-transgenic tissue. These results establish the specificity of each antibody and verify that there is no antibody cross-reactivity between species. Evaluation of aortic sections from hBACWT animals with the antibody to human elastin showed a staining pattern similar to the endogenous WT mouse protein, confirming results from our previous study that the human and mouse proteins interact to form functional elastin fibers (22). Although the anti-human elastin antibody is able to recognize CL mutant elastin (Figure 7B-D), there was little staining with the human-specific antibody in the aorta from hBACCL animals (Figure 7A), suggesting that the frame-shifted hBACCL protein is not incorporated into vascular elastic fibers.
RT-PCR analysis documented abundant mRNA for the CL protein in aorta (Figure 2B) confirming that the mutant transgene is active in this tissue. To determine whether the lack of aortic CL protein is due to postsynthetic degradation, viable aortic segments from both hBACWT and hBACCL animals were extracted and tropoelastin detected by immunoblot using the human-specific elastin antibody. Figure 7B shows that both WT and CL tropoelastins were detectable in the extract at molecular weights appropriate for the full-length proteins (the higher molecular weight of the CL protein is due to the additional missense sequence). Hence, the absence of CL protein in the aortic ECM is not due to selective degradation of the mutant tropoelastin. The absence of a detectable band in the
extract from non-transgenic animals serves as another specificity control for the anti-human elastin antibody.
Different results were obtained when elastin antibody staining was performed on sections from lung and skin. Staining with the human-specific antibody showed abundant deposition of both human WT elastin (from hBACWT) and CL elastin (from hBACCL) in both tissues (Figures 7C and 7D). The pattern of both human WT and CL elastin staining in these two tissues was similar to that of the endogenous mouse elastin, suggesting that both human proteins are able to associate with mouse elastin. The prevalence of immunostaining in the skin was interesting since semi-quantitative RT-PCR showed levels of elastin mRNA in skin to be substantially lower than lung and aorta (Supplemental Figure 3). Hence, incorporation of the mutant protein into skin elastin may be more efficient than in lung.
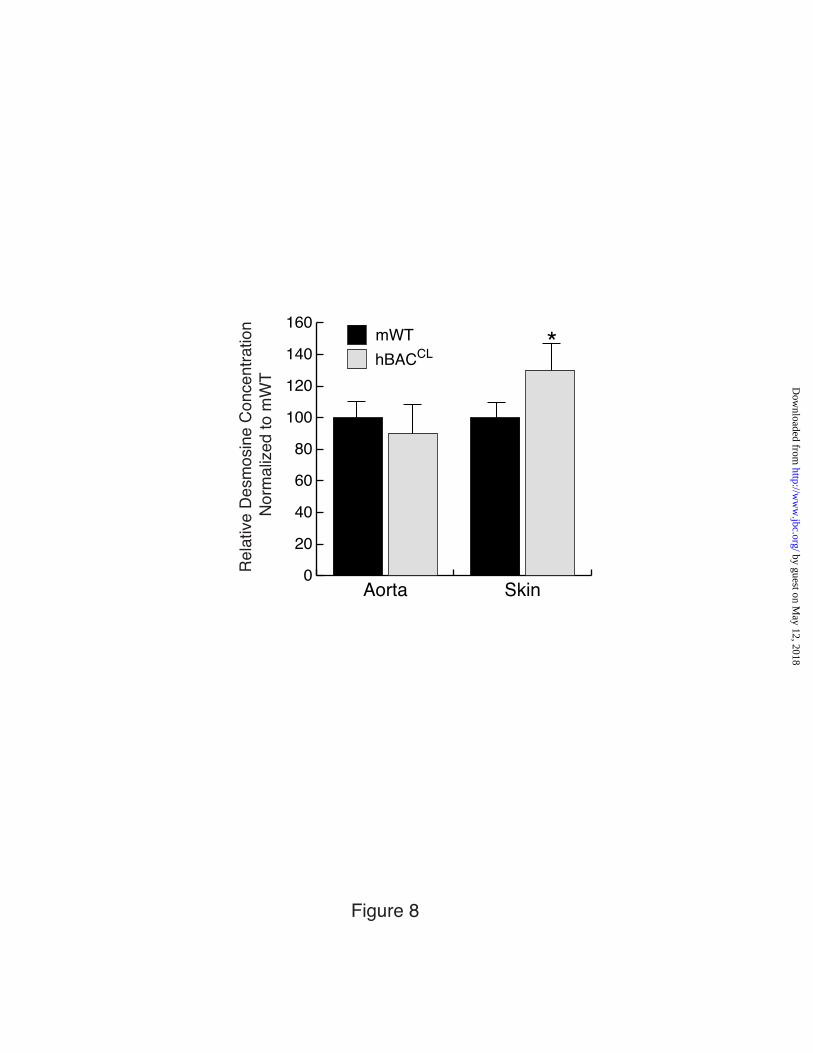
Quantification of desmosine, a crosslinking amino acid specific for mature elastin, showed that the presence of the hBACCL transgene had no effect on total desmosine in the aorta of hBACCL-mWT mice (Figure 8). In the skin, however, desmosine levels were increased significantly in these animals compared to mWT. These results are consistent with the antibody staining data showing little incorporation of the mutant protein into the aortic elastic laminae, whereas there is significant incorporation into skin elastic fibers.
Elastic fiber ultrastructure
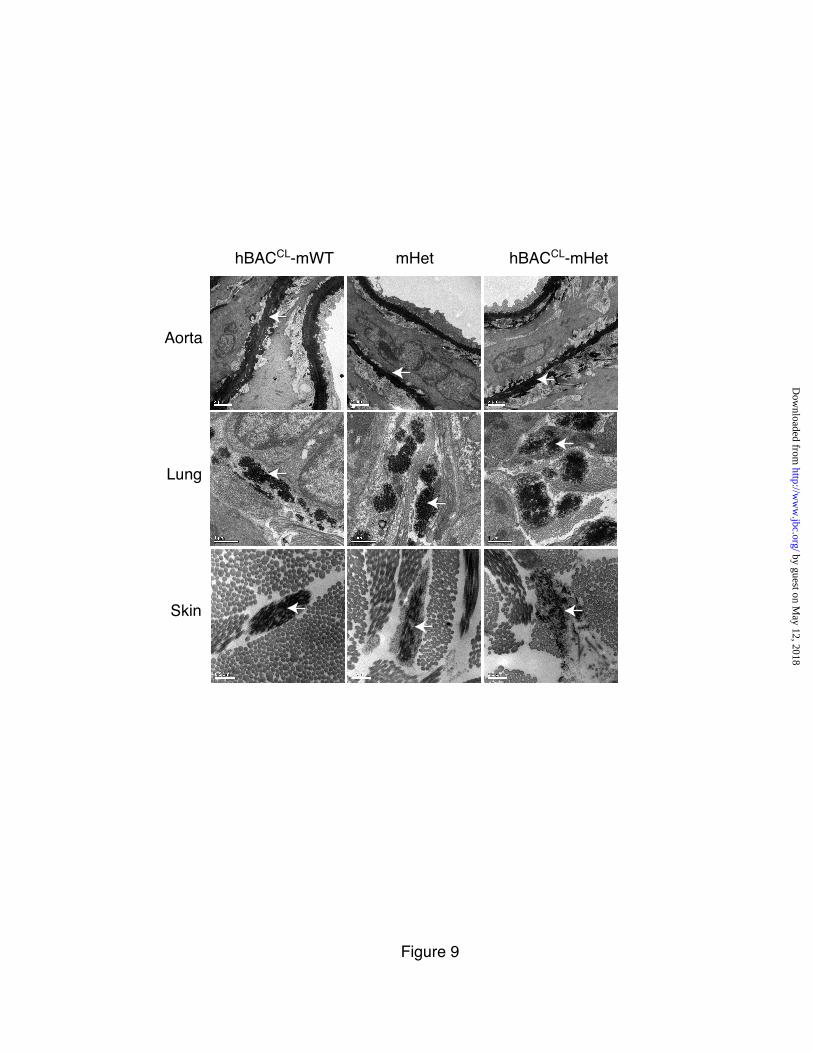
Electron microscopic examination of the aorta, lung and skin in hBACCL-mWT, mHet and hBACCL-mHet mice showed that elastic fibers did assemble in the correct location, in all three tissues, in all three genotypes (Figure 9). Subtle differences, however, were observed. For the hBACCL-mWT samples, a normal distribution of elastic fibers was seen in all of the tissues. In the mHet tissues, as expected due to the production of one-half of the normal amount of elastin, the elastic laminae in the aorta were thinner and less elastin was observed amongst the microfibrils in the other tissues, which was especially evident in the skin. For the hBACCL-mHet tissues, the elastic laminae in the aorta appear slightly ‘ragged’ but were essentially normal. Elastic fibers in the skin, however, were more diffuse and less uniformly dense. In the skin and
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

9
lung of these animals, the elastin within microfibrillar bundles was frequently distributed intermittently within the microfibrils whereas the elastin in mWT, hBACCL-mWT and mHet animals formed a normal, continuous structure. Overall, the changes in hBACCL-containing elastic fibers were subtle, consistent with the mild phenotype (described below) in lung and skin tissues. CL mutations have an adverse effect on skin and lung but not aorta
Elastin loss of function mutations associated with SVAS lead to marked phenotypes in aorta and lung, as characterized by vascular stenosis, pulmonary and systemic hypertension, increased vessel stiffness, increased lung compliance, and, in mice and perhaps humans, a propensity to develop lung disease when exposed to injurious stimuli (11, 29, 37). Interestingly, these mutations do not result in dramatic skin phenotypes in either humans or mice. To determine how the ADCL exon 30 frameshift mutation influences elastin-rich organ systems, we assessed the mechanical function of vascular, lung, and skin tissue in hBACCL-mWT mice. We also determined whether protein from the hBACCL allele could reverse the adverse vascular effects of elastin haploinsufficiency by supplementing mouse elastin in the mHet background, as has been shown for elastin from the hBACWT (22).
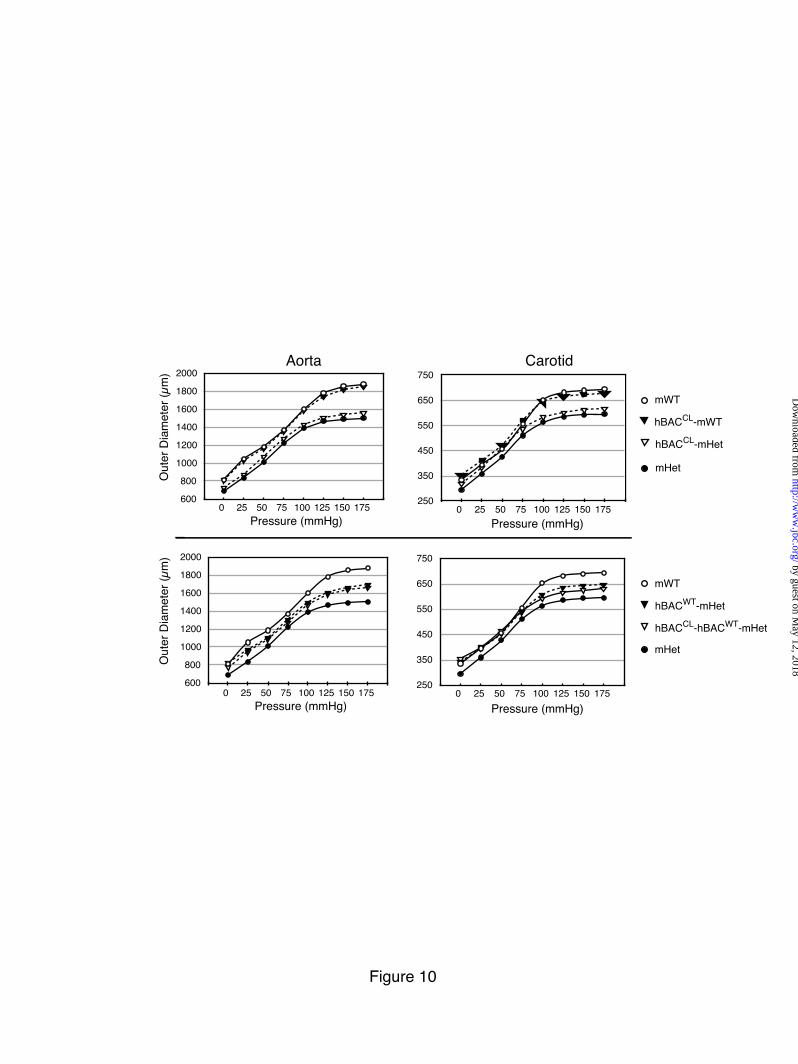
Aorta Figure 10 compares the mechanical properties of
the carotid artery and ascending aorta in hBACWT and hBACCL animals in either the mWT or mHet background. mHet vessels have a smaller diameter at any given pressure and reach maximal distention at lower pressures than similar vessels in mWT animals (29). Consistent with our previous results, expression of human elastin in the mHet background (hBACWT-mHet) was efficacious in restoring normal compliance to the vessel wall (Figure 10). The presence of the hBACCL allele, however, had no such effect, with the pressure-diameter curve for hBACCL-mHet vessels being unchanged from mHet. There was also no adverse effect of the hBACCL allele in the mWT background (Figure 10). Similarly, vessel compliance of both the carotid and ascending aorta was unchanged when the hBACCL allele was bred into the hBACWT-mHet background, resulting in a hBACCL-hBACWT-mHet genotype. Together, the mechanical studies confirm that WT
human and mouse elastin proteins can combine to form a functional fiber. The results also show that the hBACCL protein has neither a positive nor negative effect on vessel compliance, which is consistent with the immunofluorescence findings in Figure 7 and unchanged desmosine levels in Figure 8 showing that only low levels of mutant protein are associated with elastic fibers in the vessel wall.
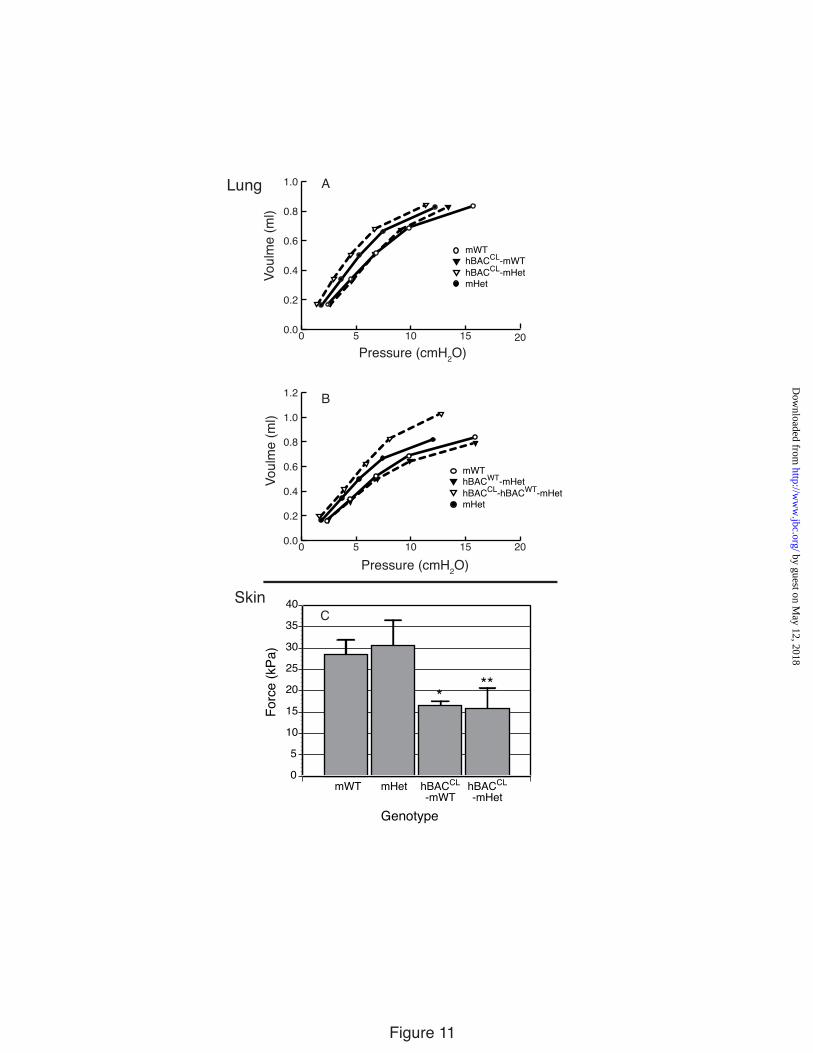
Lung Mice with elastin haploinsufficiency (mHet) have
more compliant lungs than WT animals (37). Representative expiratory quasi-static pressure-volume curves for these animals are shown in Figure 11A, where the curve for the mHet phenotype is shifted to the left, indicating that the lung elastic recoil pressure is reduced. Expression of the hBACCL allele in the mWT background (hBACCL-mWT) had no significant effect on the pressure-volume curve, but there was a trend towards increased compliance in the hBACCL-mHet animals. These results confirm that the ratio of mutant to WT protein is an important modifier of lung dysfunction.
Similar to our previous studies (22), we found a beneficial effect from human WT protein when the hBACWT allele was expressed in the mHet background (hBACWT-mHet), again confirming that the human and mouse WT proteins can combine to form functional elastic fibers. Figure 11B shows that the lung pressure-volume curve in hBACWT-mHet animals shifted to WT values, indicating a positive contribution from human elastin in forming functional elastic fibers. Interestingly, when the hBACCL and hBACWT alleles were combined in the mHet background, a decidedly negative effect was observed, with the lung becoming markedly more compliant, especially at higher pulmonary pressure (Figure 11B). Chord compliance calculated from the slopes of the compliance curves (ΔV/ΔP) indicates that mHet lungs (0.091 ml/cm H2O) have greater compliance than mWT lungs (0.071 ml/cm H2O). The presence of the hBACCL in both the mWT (0.078 ml/cm H2O) and mHet (0.096 ml/cm H2O) backgrounds caused the lung compliance to trend higher, but the differences were not statistically significant. The chord compliance decreased significantly in the hBACWT-mWT animals (0.067 ml/cmH2O) indicating rescue of the compliance phenotype. However, when hBACCL was included with hBACWT to give the genotype
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

10
(hBACCL-hBACWT-mHet), there was a large and significant increase in chord compliance (0.099 ml/cmH2O, p>0.05) indicating a reversal of the beneficial effects of the hBACWT protein on lung stiffness.
Skin Measurements of skin elasticity in mWT, mHet,
hBACCL-mHet and hBACCL-mWT animals were obtained using the DermaLab suction cup (38). Figure 11C shows that skin from animals with the hBACCL allele is more lax than mWT animals, in agreement with the lax skin phenotype seen in humans with this mutation. After four conditioning cycles, approximately equivalent force was required to displace skin by 2 mm in mWT and mHet animals, whereas only about half as much force was required to displace skin from hBACCL-mWT and hBACCL-mHet animals. These results show that skin in the CL mutants have different physical properties than animals without the mutant allele, even when compared to animals having half the normal elastin levels (mHet). DISCUSSION
As a model organism, the mouse has been valuble for exploring the relationship between gene mutation and human disease. The ability to introduce a human gene mutation into the mouse ortholog using conventional gene targeting is a powerful tool for understanding disease causality. This approach is useful when the mouse and human genes are similar, but may not be physiologically relevant if the genes are substantially different. Such is the case for the elastin gene, which has undergone considerable alteration in the primate lineage such that the human gene differs from the mouse gene in both structure and splicing of the gene transcript (23). Most human elastin gene mutations that are associated with dominant cutis laxa-type phenotypes are +1 or -1 frameshift mutations that adversely affect exons that encode critical assembly domains (e.g., exons 30 and 36) in the 3’ end of the gene. This 3’ region also undergoes extensive alternative splicing in humans where splicing influences the types and amounts of product that contribute to disease. Splicing out the exon containing the mutation, or splicing out downstream exons, can have the effect of silencing the mutation or modifying the nature of the RNA products when the mutant allele is transcribed. For example, the impact of transcript splicing on mutant
protein is illustrated by types of missense sequence produced by the ∆2012G mutation described in this study (Figure 3). In the human gene, missense sequence downstream of the exon 30 mutation ends at a premature termination codon in exon 32. When exon 32 is deleted, which occurs >50% of the time in human skin fibroblasts and in fibroblasts from the transgenic BAC mouse, missense sequence continues into the 3’UTR until a new stop codon is encountered downstream of the normal translation termination site, resulting in a 25 amino acid extension. Transcripts from the mouse elastin gene, in contrast, show no splicing of these 3’exons (22) and, hence, cannot model the variability in missense sequence associated with the human mutations.
In a previous study we showed that the hBACWT transgene in mice has a temporal and tissue expression pattern similar to the endogenous mouse elastin gene. The human transgene also retains the human splice pattern and does not alter splicing of the endogenous mouse gene (22). These characteristics are also true for the hBACCL transgene, with the notable difference that transcript levels from hBACCL are ~50% lower than those from hBACWT. This decrease was correlated with alternative mRNA splicing, with substantial, but not complete, degradation of the hBACCLexon32in transcript through nonsense mediated decay. It is interesting that nonsense mediated decay did not degrade the hBACCLexon32out transcript, indicating that this splicing product, with significant missense sequence, is stable and capable of producing mutant protein. How the two protein products of hBACCLexon32in and exon32out combine to alter normal elastic fiber assembly is unknown. It is intriguing to speculate, however, that the variable severity and phenotypic heterogeneity of ADCL may be related to the amount of mutant exon32in transcript that survives NMD.
Functional studies comparing the mechanical properties of skin and lung establishes that the hBACCL protein has a negative effect on mechanical function, providing direct evidence for a dominant-negative mechanism. Electron microscopic analysis of tissues from hBACCL mice showed some suggestion of disorganized bundles of elastin in the lung and skin, but the difference from mWT were subtle. This suggests that the CL protein does not dramatically change the organization or packing of elastin (at least in ways that are obvious by electron microscopy), but changes the functional properties
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

11
of the fiber as was evident from the tissue mechanical studies.
In contrast to lung and skin, the CL mutant protein incorporates inefficiently in aorta, consistent with the absence of a vascular phenotype in the hBACCL mice and in ADCL individuals with the mutation modeled in this report. Similar results were obtained by Hu et al. who expressed a cDNA for tropoelastin containing a 25bp deletion (2114-2138 del) in exon 30 as a transgene in mice (17). Like the mutant tropoelastin described in our study, the exon 30(2114-2138 del) protein was found to incorporate into lung but not aorta.
The reason why elastin incorporation is different in the aorta is not known. Because the extent of exon 32 splicing is not substantially different between aorta and lung or skin, the loss of C- terminal 'assembly sequences' is more consequential for assembly of elastic fibers in the aorta as compared to skin/lung, suggesting either different mechanisms or a more lax restriction on assembly in skin as compared to aorta. It is important to note, however, that alternative splicing of elastin gene transcripts is developmentally regulated (39-41), such that the splice pattern we observe in adult animals may be different from what occurs during development when elastin deposition is at its highest level. Developmental changes in the ratio of hBACCLexon32in and exon32out splicing could impact the mutant protein isoforms and be an important modifier of disease pathogenesis.
The C-terminal region of tropoelastin that is altered in ADCL (exons 30 through 36 in this study) contains important sequences for fiber assembly and it is easy to envision how modification of this region has negative consequences for proper fiber
formation. In vitro studies and studies with transgenic mice showed that tropoelastin with altered or deleted sequences in this region are still capable of interacting with microfibrils to form an elastic fiber, although with abnormal crosslinking (7, 42, 43). How this region of tropoelastin facilitates fiber assembly is still unknown, although recent studies have identified binding sites for fibulin-4 and fibulin-5—two proteins that facilitate elastin organization (44, 45). Inactivation of these two fibulins in mice results in abnormal elastic fiber formation, and, particularly relevant to our study, mutations in the human genes for fibulin-4 and fibulin-5 have been linked to an autosomal recessive form of cutis laxa (ARCL Type I) (46-50). The fact that reduced fibulin-4 levels achieved through gene inactivation (51) or RNA knockdown (44) result in increased desmosine crosslinks—similar to our findings in hBACCL mice—suggests that the critical interaction disrupted by the CL-frameshift is likely between tropoelastin and fibulin-4.
In humans, the 2012∆G mutation results in a phenotype that includes loose skin, redundant mitral and tricuspid valves, mild aortic dilation, and umbilical and inguinal hernias. Pulmonary traits include mild right ventricular hypertrophy and reduced expiratory flow indicative of upper airway obstruction. Although we did not study the heart or ligaments in the hBACCL mice, the phenotypic traits that we did describe show significant overlap with the human phenotype. Thus, the hBACCL mouse provides an excellent model to develop mechanistic insight into the human disease and to advance our understanding of elastic fiber assembly and function in vivo.
REFERENCES 1. Liem, K. F. (1988) Amer. Zool. 28, 739-759 2. Li, D. Y., Brooke, B., Davis, E. C., Mecham, R. P., Sorensen, L. K., Boak, B. B., Eichwald, E. and
Keating, M. T. (1998) Nature 393, 276-280 3. Kozel, B. A., Mecham, R. P. and Rosenbloom, J. (2011) in The Extracellular Matrix: an Overview
(Mecham, R. P., ed.) pp. 267-301, Springer Berlin Heidelberg, Berlin, Heidelberg 4. Foster, J. A. and Curtiss, S. W. (1990) Am. J. Physiol. 259, L13-L23 5. Barrineau, L. L., Rich, C. B., Przybyla, A. and Foster, J. A. (1981) Dev Biol 87, 46-51 6. Wrenn, D. S., Parks, W. C., Whitehouse, L. A., Crouch, E. C., Kucich, U., Rosenbloom, J. and
Mecham, R. P. (1987) J. Biol. Chem. 262, 2244-2249 7. Kozel, B. A., Wachi, H., Davis, E. C. and Mecham, R. P. (2003) J. Biol. Chem. 278, 18491-18498
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

12
8. Curran, M. E., Atkinson, D. L., Ewart, A. K., Morris, C. A., Leppert, M. F. and Keating, M. T. (1993) Cell 73, 159-168
9. Li, D. Y., Toland, A. E., Boak, B. B., Atkinson, D. L., Ensing, G. J., Morris, C. A. and Keating, M. R. (1997) Hum. Molec. Gen. 6, 1021-1028
10. Olson, T. M., Michels, V. V., Urban, Z., Csiszar, K., Christiano, A. M., Driscoll, D. J., Feldt, R. H., Boyd, C. D. and Thibodeau, S. N. (1995) Hum. Mol. Genet. 4, 1677-1679
11. Pober, B. R., Johnson, M. and Urban, Z. (2008) J. Clin. Invest. 118, 1606-1615 12. Urban, Z., Michels, V. V., Thibodeau, S. N., Davis, E. C., Bonnefont, J.-P., Munnich, A., Eyskens, B.,
Gewillig, M., Devriendt, K. and Boyd, C. D. (2000) Hum. Genet. 106, 577-588 13. Graul-Neumann, L. M., Hausser, I., Essayie, M., Rauch, A. and Kraus, C. (2008) Am. J. Med. Genet. A
146A, 977-983 14. Rodriguez-Revenga, L., Iranzo, P., Badenas, C., Puig, S., Carrio, A. and Mila, M. (2004) Arch.
Dermatol. 140, 1135-1139 15. Tassabehji, M., Metcalfe, K., Hurst, J., Ashcroft, G. S., Kielty, C., Wilmot, C., Donnai, D., Read, A. P.
and Jones, C. J. P. (1998) Hum. Molec. Gen. 7, 1021-1028 16. Zhang, M. C., He, L., Giro, M., Yong, S. L., Tiller, G. E. and Davidson, J. M. (1999) J. Biol. Chem.
274, 981-986 17. Hu, Q., Shifren, A., Sens, C., Choi, J., Szabo, Z., Starcher, B. C., Knutsen, R. H., Shipley, J. M.,
Davis, E. C., Mecham, R. P. and Urban, Z. (2010) Matrix Biol 29, 621-628 18. Callewaert, B., Renard, M., Hucthagowder, V., Albrecht, B., Hausser, I., Blair, E., Dias, C., Albino,
A., Wachi, H., Sato, F., Mecham, R. P., Loeys, B., Coucke, P. J., De Paepe, A. and Urban, Z. (2011) Hum Mutat 32, 445-455
19. Damkier, A., Brandrup, F. and Starklint, H. (1991) Clin Genet 39, 321-329 20. Szabo, Z., Crepeau, M. W., Mitchell, A. L., Stephan, M. J., Puntel, R. A., Yin Loke, K., Kirk, R. C.
and Urban, Z. (2006) J. Med. Genet. 43, 255-258 21. Urban, Z., Gao, J., Pope, F. M. and Davis, E. C. (2005) J. Invest. Dermatol. 124, 1193-1199 22. Hirano, E., Knutsen, R. H., Sugitani, H., Ciliberto, C. H. and Mecham, R. P. (2007) Circ. Res. 101,
523-531 23. Szabo, Z., Levi-Minzi, S. A., Christiano, A. M., Struminger, C., Stoneking, M., Batzer, M. A. and
Boyd, C. D. (1999) J. Mol. Evol. 49, 664-671 24. Warming, S., Costantino, N., Court, D. L., Jenkins, N. A. and Copeland, N. G. (2005) Nucleic Acids
Res. 33, e36 25. Lalioti, M. and Heath, J. (2001) Nucleic Acids Res. 29, E14 26. Livak, K. J. and Schmittgen, T. D. (2001) Methods 25, 402-408 27. Cowen, T., Haven, A. J. and Burnstock, G. (1985) Histochemistry 82, 205-208 28. Davis, E. C. (1993) Lab. Invest. 68, 89-99 29. Faury, G., Pezet, M., Knutsen, R. H., Boyle, W. A., Heximer, S. P., McLean, S. E., Minkes, R. K.,
Blumer, K. J., Kovacs, A., Kelly, D. P., Li, D. Y., Starcher, B. and Mecham, R. P. (2003) J. Clin. Invest. 112, 1419-1428
30. Wagenseil, J. E., Nerurkar, N. L., Knutsen, R. H., Okamoto, R. J., Li, D. Y. and Mecham, R. P. (2005) Am. J. Physiol. Heart Circ. Physiol. 289, H1209-1217
31. Vanoirbeek, J. A., Rinaldi, M., De Vooght, V., Haenen, S., Bobic, S., Gayan-Ramirez, G., Hoet, P. H., Verbeken, E., Decramer, M., Nemery, B. and Janssens, W. (2010) Am J Respir Cell Mol Biol 42, 96-104
32. Brown-Augsburger, P., Tisdale, C., Broekelmann, T., Sloan, C. and Mecham, R. P. (1995) J. Biol. Chem. 270, 17778-17783
33. Copeland, N. G., Jenkins, N. A. and Court, D. L. (2001) Nat. Rev. Genet. 2, 769-779 34. Muyrers, J. P., Zhang, Y., Benes, V., Testa, G., Ansorge, W. and Stewart, A. F. (2000) EMBO Rep. 1,
239-243 35. Indik, Z., Yoon, K., Morrow, S. D., Cicila, G., Rosenbloom, J., Rosenbloom, J. and Ornstein-
Goldstein, N. (1987) Connect. Tissue Res. 16, 197-211
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

13
36. Noensie, E. N. and Dietz, H. C. (2001) Nat. Biotechnol. 19, 434-439 37. Shifren, A., Durmowicz, A. G., Knutsen, R. H., Hirano, E. and Mecham, R. P. (2006) Am. J. Physiol.
Lung Cell Mol. Physiol. 292, L778-787 38. Pedersen, L., Hansen, B. and Jemec, G. B. (2003) Skin Res. Technol. 9, 111-115 39. Baule, V. J. and Foster, J. A. (1988) Biochem. Biophys. Res. Commun. 154, 1054-1060 40. Yeh, H., Anderson, N., Ornstein-Goldstein, N., Bashir, M. M., Rosenbloom, J. C., Abrams, W., Indik,
Z., Yoon, K., Parks, W., Mecham, R. and Rosenbloom, J. (1989) Biochemistry 28, 2365-2370 41. Heim, R. A., Pierce, R. A., Deak, S. B., Riley, D. J., Boyd, C. D. and Stolle, C. A. (1991) Matrix 11,
359-366 42. Kozel, B. A., Ciliberto, C. H. and Mecham, R. P. (2004) Matrix Biol. 23, 23-34 43. Hsiao, H., Stone, P. J., Toselli, P., Rosenbloom, J., Franzblau, C. and Schreiber, B. M. (1999) Connect.
Tiss. Res. 40, 83-95 44. Choudhury, R., McGovern, A., Ridley, C., Cain, S. A., Baldwin, A., Wang, M. C., Guo, C., Mironov
Jnr, A., Drymoussi, Z., Trump, D., Shuttleworth, A., Baldock, C. and Kielty, C. M. (2009) J. Biol. Chem. 284, 24553-24567
45. Wachi, H., Nonaka, R., Sato, F., Shibata-Sato, K., Ishida, M., Iketani, S., Maeda, I., Okamoto, K., Urban, Z., Onoue, S. and Seyama, Y. (2008) J. Biochem. 143, 633-639
46. Claus, S., Fischer, J., Megarbane, H., Megarbane, A., Jobard, F., Debret, R., Peyrol, S., Saker, S., Devillers, M., Sommer, P. and Damour, O. (2008) J. Invest. Dermatol. 128, 1442-1450
47. Dasouki, M., Markova, D., Garola, R., Sasaki, T., Charbonneau, N. L., Sakai, L. Y. and Chu, M. L. (2007) Am. J. Med. Genet. A 143, 2635-2641
48. Hu, Q., Loeys, B. L., Coucke, P. J., De Paepe, A., Mecham, R. P., Choi, J., Davis, E. C. and Urban, Z. (2006) Hum. Mol. Genet. 15, 3379-3386
49. Hucthagowder, V., Sausgruber, N., Kim, K. H., Angle, B., Marmorstein, L. Y. and Urban, Z. (2006) Am. J. Hum. Genet. 78, 1075-1080
50. Loeys, B., Van Maldergem, L., Mortier, G., Coucke, P., Gerniers, S., Naeyaert, J. M. and De Paepe, A. (2002) Hum. Mol. Genet. 11, 2113-2118
51. McLaughlin, P. J., Chen, Q., Horiguchi, M., Starcher, B. C., Stanton, J. B., Broekelmann, T. J., Marmorstein, A. D., McKay, B., Mecham, R., Nakamura, T. and Marmorstein, L. Y. (2006) Mol. Cell Biol. 26, 1700-1709
Acknowledgments– We thank Ron McCarthy of the Washington University Pulmonary Mouse Core (supported by HL295994) for oocyte injection and production of the hBACCL mice. We also thank Terese Hall for administrative support. FOOTNOTES *This work was supported by NIH grants HL53325, HL74138, HL105314 and HL084922 to RPM, K12 HL089968 to AS, HL087563 to JEW, the Canadian Institutes of Heath Research (MOP86713) to ECD, and by a grant from the National Marfan Foundation (RPM). ECD is a Canada Research Chair.
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

14
FIGURE LEGENDS Figure 1: Confirmation of Ex30ΔG mutation in the human elastin BAC. A single base deletion in exon 30 of the human elastin gene was engineered using two-step recombineering and DY380 cells. DNA was isolated from the bacteria following each recombination (steps 4 & 6, Supplementary Data Figure 1) and digested with Bgl1. Panel A is an ethidium bromide gel of the restriction fragments with molecular weight markers on the left (lane M). A 3.9 kb fragment (lane 1) corresponds to wild-type and mutant fragments without the sacB-Kan cassette (see schematic on the right of the panel). This band shifted in size to 7.5 kb in intermediate clones (lanes 2) after kanamycin selection, which is the expected size of the fragment containing the sacB-Kan selection cassette. After the second recombination step, the size of the band returned to 3.9 kb (lanes 3), confirming removal of the sacB-Kan cassette. Southern blot analysis of the Bgl1 digests with probes located outside of the homologous recombination arms confirmed the correct size of the constructs (Panel B). Figure 2: Characterization of hBACCL transgenic mice. A) Southern blot identifying the presence of the hBACCL transgene (arrow) in three founder lines (the band in Line 2 is not evident at this exposure). WT is DNA from a nontransgenic animal. Transgenic line 1 was selected for subsequent studies. B) RT-PCR analysis of mRNA from 4 week-old transgenic animals comparing expression of the WT and CL transgenes in aorta and lung. Shown is mean ± Std. Dev. n=6. C): RT-PCR analysis of elastin mRNA levels in transgenic mouse aorta documenting a down regulation of elastin production with age in both hBACWT and hBACCL animals. * = indicates that hBACCL expression is significantly lower (p<0.01) than hBACWT in each tissue. Figure 3: Comparison of exon structure of human and mouse elastin mRNA and assessment of alternative splicing. A) Functional coding of exons in mouse and human elastin genes. Exons encoding hydrophobic domains are green, K-P crosslinking domains yellow, and K-A crosslinking domains orange. Exons known to undergo alternative splicing in the human gene are indicated by dotted lines. B) Possible splice products from the hBACWT and hBACCL transgenes. The full-length hBAC transcript is 688 amino acids (hBACWT32in, light blue). Deletion of exon 32 (hBACWT32out, dark blue) maintains a normal reading frame through to the stop codon in exon 36 (670 amino acids). The Ex30ΔG mutation (arrow) in the hBACCL gene (hBACCL32in, light green) produces missense sequence downstream of the mutation with a premature stop codon in exon 32 (transcript of 667 amino acids). When exon 32 is spliced out (hBACCL32out, dark green), missense sequence continues into the 3’ UTR (transcript of 713 amino acids). Exon 22 is deleted in all human transcripts. C) The extent of exon 32 splicing was assessed from total RNA isolated from hBACCL and hBACWT lung tissue using RT-PCR with human-specific primers that span exons 25-36 (brown line). The agarose gel shows that exon32in and exon32out transcripts are both produced by the hBACWT and hBACCL genes, although at different ratios. Figure 4: Levels of exon32in transcript are reduced by nonsense-mediated decay. A) Semiquantitative RT-PCR with human-specific primers that span exons 25-36 at 21 and 23 cycles showed that the hBACWT-exon32in and -exon32out products were of approximately equal abundance, whereas the hBACCL-exon32in transcript was about half as abundant as the RNA with exon 32 spliced out (hBACCL-exon32out). B) The abundance of the hBACCL-exon32in transcript is increased following treatment of cells with the protein synthesis inhibitors puromycin or anisomycin compared to vehicle treatment (DMSO), suggesting that the reduced levels of this transcript are the result of nonsense-mediated decay. The graph at the bottom represents data from densitometric scans of the hBACWT and hBACCL data ± puromycin shown in B. Values are mean ±Std. Dev., n=4 scans. Data in panels A and B are from two separate experiments. Figure 5: Comparison of exon32 splicing in aorta, lung and skin.
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

15
Expression levels of exon32 splice variants in mRNA from hBACWT and hBACCL aorta, lung, and skin was determined by qPCR and normalized to cyclophilin as an endogenous control. The fold difference between exon32in and exon32out was used to calculate the percent of 32 spliced out for each tissue and each genotype. Values are mean ±Std. Dev., n=3. Figure 6: hBACCL transgene cannot rescue the elastin-null phenotype and antagonizes the rescue potential of the hBACWT allele. Percentage of animals that survive to two months of age normalized to WT (100%). Expression of the hBACWT transgene in the mouse null background (hBACWT -mNull) rescues the lethality of the null phenotype. Expression of the hBACCL in the mouse null background (hBACCL –mNull) in contrast, does not rescue the lethality of the null phenotype. When expressed with the hBACWT allele in the null background (hBACWT-hBACCL-mNull), hBACCL reduces the survival of hBACWT-rescued animals. Figure 7: Incorporation of CL-mutant tropoelastin into elastic fibers in lung and skin but not aorta. A) Elastin in frozen sections of aorta from non-transgenic, hBACWT and hBACCL mice detected with antibodies specific for the human or mouse elastin protein. Abundant human elastin was detected in hBACWT tissue, but only low levels of the CL mutant protein were incorporated into aortic elastin fibers in hBACCL animals. Secondary antibody alone served as a staining control. B) Western blot of aortic extracts from non-transgenic (NTG), hBACWT (WB), and hBACCL (CB) animals confirming the production of wild-type and mutant human tropoelastin in each tissue. The higher molecular weight of the CB protein is due to the additional missense sequence. GAPDH, detected by western blot, served as a loading control. C & D) In contrast to the aorta, immunofluorescence localization shows abundant incorporation of hBACCL elastin (as well as hBACWT protein) in the lung and skin. Bar = 100 µm and applies to all figures in each panel. Figure 8: Desmosine levels in mWT and hBACCL tissues. Relative desmosine levels in aorta and skin from mWT and hBACCL-mWT animals. Values are normalized to mWT (defined as 100%) in each tissue. Values are mean ±Std. Dev., n=6 independent samples. The asterisk indicates p<0.5. Figure 9: Ultrastructure of elastic fibers Electron microscopic images of aorta, lung, and skin sections from hBACCL-mWT, mHet and hBACCL-mHet. Elastic fibers (white arrow) appear normal in the hBACCL-mWT tissues. In the mHet, as expected, there are thinner elastic laminae in the aorta and less elastin, exposing more microfibrils, in the lung and skin. The aorta of the hBACCL-mHet also has thin elastic laminae but there is more elastin loosely associated with the laminae and cell surface. The elastic fibers in the lung of the hBACCL-mHet appeared similar to the mHet, whereas the fibers in the skin were often quite diffusely assembled as shown. Bars = 2 µm (aortas), 1 µm (lung) and 0.5 µm (skin). Figure 10: Compliance of aorta and carotid artery Pressure-diameter curves assessing the mechanical properties of the carotid artery and ascending aorta in hBACCL (top panels) and hBACWT (bottom panels) animals in either the Eln+/+(mWT) or Eln+/- (mHet) backgrounds. Expression of wild-type human elastin from the hBACWT in the mHet background (hBACWT-mHet) was efficacious in restoring normal compliance to the vessel wall. Pressure-diameter curves were unchanged from mHet values by expression of the hBACCL allele (hBACCL-mHet). There was also no adverse effect from the hBACCL allele in the mWT background (hBACCL-mWT) nor when bred into the hBACWT-mHet background (hBACCL-hBACWT-mHet). Shown are mean values for each genotype. Standard deviations are not shown so as to enhance figure clarity but can be found in Table I in supplementary material.
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

16
Figure 11. Lung and skin compliance A) Pressure-volume curves assessing the mechanical properties of lungs from hBACCL animals in either the Eln+/+ (mWT) or Eln+/- (mHet) backgrounds. mHet mice demonstrate increased lung compliance compared with WT mice. There was no significant effect on lung compliance when the hBACCL allele was expressed in the in the mWT background (hBACCL-mWT). Expression of the hBACCL allele in the Eln+/- background (hBACCL-mHet) resulted in an increase in lung compliance compared with mHet mice. B) Pressure-volume curves assessing the mechanical properties of lungs from hBACCL and hBACWT animals in the mWT or mHet backgrounds. Expression of wild-type elastin from the human BAC (hBACWT) in the mHet background (hBACWT-mHet) restores lung compliance to WT levels. This effect is lost when the hBACCL allele is bred into the hBACWT-mHet background (hBACCL-hBACWT-mHet). Standard deviations are not shown to enhance figure clarity but can be found in Table I in supplementary material. C) Skin compliance, as determined by the force required to displace skin 2 mm using a vacuum suction cup, shows that equivalent force was required to displace skin in mWT and mHet animals whereas only about half as much force was required to displace skin from hBACCL-mWT and hBACCL-mHet animals. Values are mean ±Std. Dev., n=4. The single asterisk indicates p <0.05 and the double asterisk indicates p <0.01.
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
7.5 kb
3.9 kb
25
BglI BglI
29 30
DNA ladder data
Figure 1
M 1 2 3
10 8 6 5 4 3
Southern Blot
1 2 3
7.5 kb
3.9 kb
A
B
25
BglI BglI
29 30sacB-Kankb
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
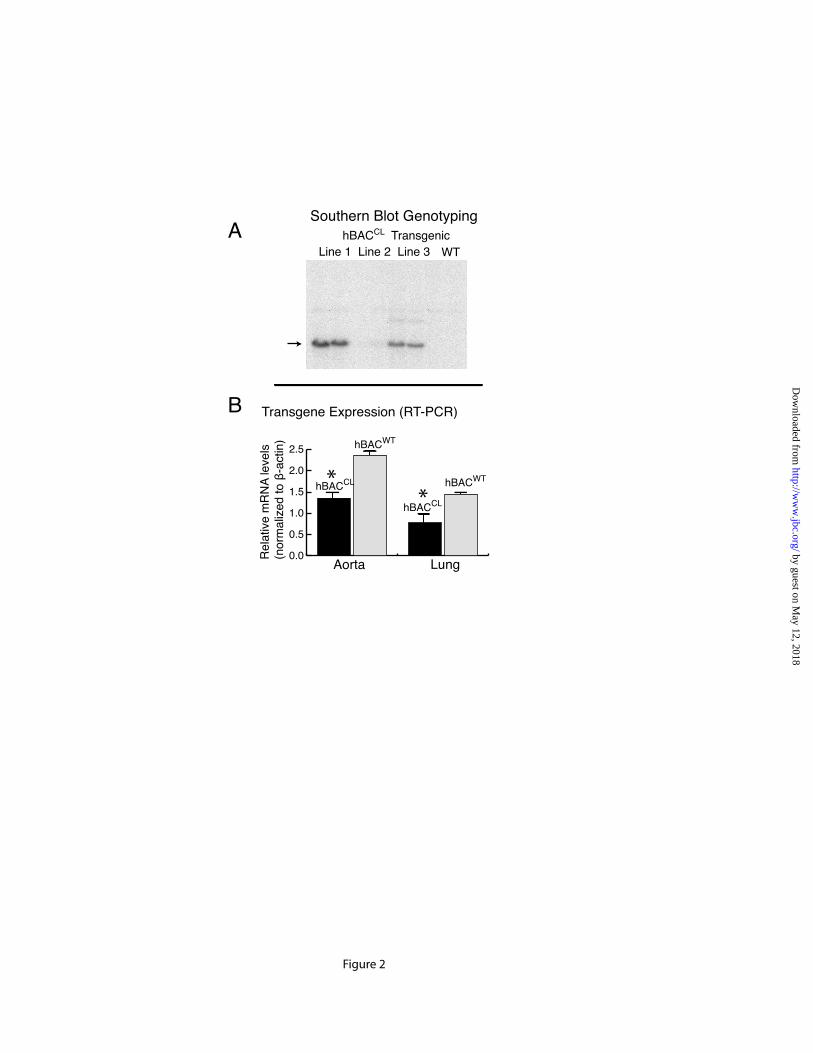
Figure 2
Southern Blot Genotyping
Transgene Expression (RT-PCR)
hBACCL TransgenicWTLine 1 Line 2 Line 3
0.0
0.5
1.0
1.5
2.0
2.5
hBACCL
hBACWT
Aorta LungRel
ativ
e m
RN
A le
vels
(nor
mal
ized
to β
-act
in)
hBACCL
hBACWT
A
B
**
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36
ATG STOP3’ UTR
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 36
ATG STOP3’ UTR
STOP
STOP
STOPMS
MS25 36
Exon 32inExon 32out
hBACWT hBACCL
A
B
ΔG
Mouse
Human
hBACWT32inhBACWT32out
hBACCL32inhBACCL32out
STOP
50%
Exon #:
688670
667713
amino acids
C
Figure 3
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

WT CL WT CLExon 32inExon 32out
CL WTDMSO Puromycin Anisomycin
Figure 4
WT CL WT CL21 cycles 23 cycles
Exon 32inExon 32out
A
B
hBAC:
hBAC:
hBAC genotype: WT CL WT CLtreatment: control puromycin
Scan
Inte
nsity
(rela
tive
units
)
25000
20000
15000
10000
5000
0
Densitometry Scan of Bandsin Gel B (exon 32in)
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

100
75
50
25
0Aorta Lung Skin
* *
% E
last
in m
RN
A w
ith
Exon
32 S
plic
ed O
ut
Figure 5
hBACWT
hBACCL
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
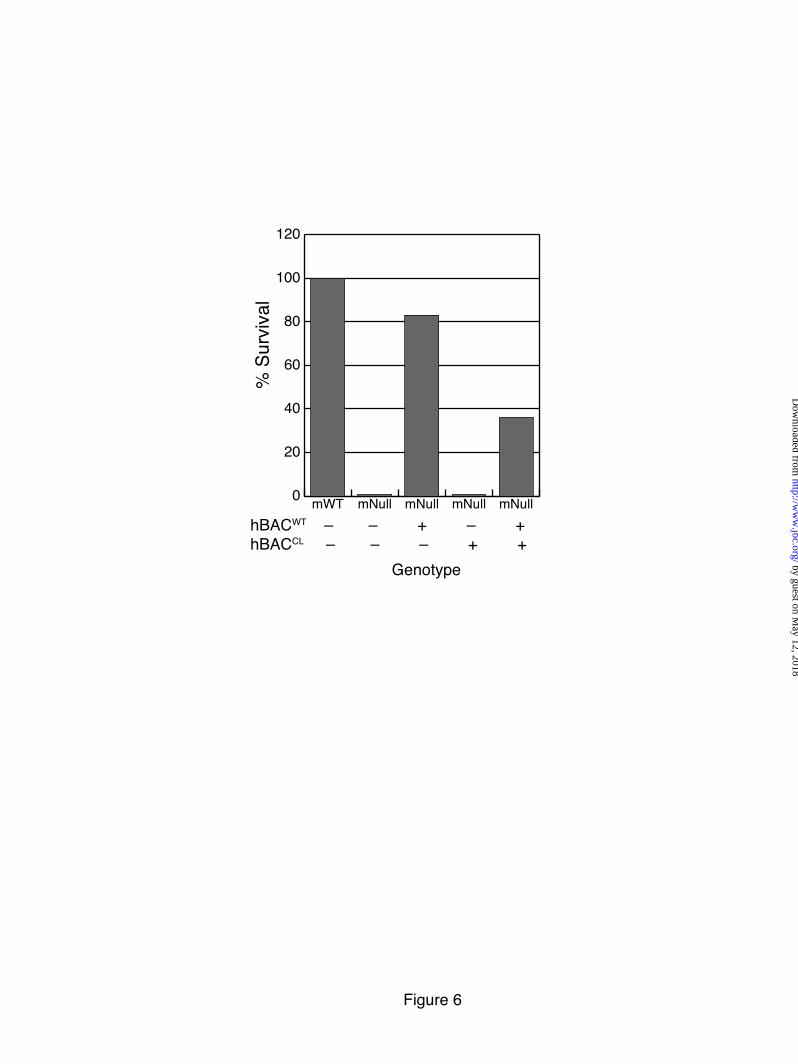
Figure 6
mNullmNullmNullmNullmWThBACWT – – + – + hBACCL – – – + +
% S
urvi
val
Genotype
0
20
40
60
80
100
120
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
2ndAlone
HumanElastin
MouseElastin
ANTIBODY AGAINST
NonTransgenic
HumanElastin
HumanElastin
2ndAlone
2ndAlone
NTG WT CL
HumanTropoelastin
GAPDH
AORTA
Antibody
ANTIBODY AGAINST
NonTransgenic
NonTransgenic
A
B
C D
Figure 7
hBACCL
hBACCL hBACCL
hBACWT
hBACWThBACWT
AORTA
LUNG SKIN
:hBAC
ANTIBODY AGAINST
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Rel
ativ
e D
esm
osin
e C
once
ntra
tion
Nor
mal
ized
to m
WT
Figure 8
0
20
40
60
80
100
120
140
160
SkinAorta
mWThBACCL *
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
hBACCL-mWT mHet hBACCL-mHet
Aorta
Lung
Skin
Figure 9
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
600
800
1000
1200
1400
1600
1800
2000
0 25 50 75 100 125 150 175
250
350
450
550
650
750
0 25 50 75 100 125 150 175600
800
1000
1200
1400
1600
1800
2000
0 25 50 75 100 125 150 175
250
350
450
550
650
750
0 25 50 75 100 125 150 175
mWT
mHet
hBACCL-mWT
hBACCL-mHet
mWT
mHet
hBACWT-mHet
hBACCL-hBACWT-mHet
Pressure (mmHg) Pressure (mmHg)
Pressure (mmHg) Pressure (mmHg)
Out
er D
iam
eter
(µm
)O
uter
Dia
met
er (µ
m)
Aorta Carotid
Figure 10
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
0 5 10 15 200.0
0.2
0.4
0.6
0.8
1.0
0 5 10 15 200.0
0.2
0.4
0.6
0.8
1.0
1.2
mWT
mHet
hBACCL-mWThBACCL-mHet
mWT
mHet
hBACWT-mHethBACCL-hBACWT-mHet
Figure 11
A
B
Voul
me
(ml)
Pressure (cmH2O)
Pressure (cmH2O)
Voul
me
(ml)
0
5
10
15
20
25
30
35
40
Forc
e (k
Pa)
mWT hBACCL
-mWThBACCL
-mHetmHet
Genotype
***
Lung
SkinC
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Broekelmann and Robert P. MechamChristopher Ciliberto, Beth A. Kozel, Zsolt Urban, Elaine C. Davis, Thomas J.
Hideki Sugitani, Eiichi Hirano, Russell H. Knutsen, Adrian Shifren, Jessica E. Wagenseil,of human elastin gene frame shift mutations associated with dominant cutis laxa
Alternative splicing and tissue-specific elastin misassembly act as biological modifiers
published online May 9, 2012J. Biol. Chem.
10.1074/jbc.M111.327940Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2012/05/09/M111.327940.DC1
by guest on May 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from