by an increased dutp/dttp ratio nucleic acids research, 48
TRANSCRIPT

http://www.diva-portal.org
This is the published version of a paper published in Nucleic Acids Research.
Citation for the original published paper (version of record):
Schmidt, T T., Sharma, S., Reyes, G X., Kolodziejczak, A., Wagner, T. et al. (2020)Inactivation of folylpolyglutamate synthetase Met7 results in genome instability drivenby an increased dUTP/dTTP ratioNucleic Acids Research, 48(1): 264-277https://doi.org/10.1093/nar/gkz1006
Access to the published version may require subscription.
N.B. When citing this work, cite the original published paper.
Permanent link to this version:http://urn.kb.se/resolve?urn=urn:nbn:se:umu:diva-170313

264–277 Nucleic Acids Research, 2020, Vol. 48, No. 1 Published online 24 October 2019doi: 10.1093/nar/gkz1006
Inactivation of folylpolyglutamate synthetase Met7results in genome instability driven by an increaseddUTP/dTTP ratioTobias T. Schmidt1,2, Sushma Sharma3, Gloria X. Reyes1, Anna Kolodziejczak1,2,Tina Wagner4, Brian Luke 4,5, Anders Hofer3, Andrei Chabes3,6 and Hans Hombauer 1,*
1DNA Repair Mechanisms and Cancer, German Cancer Research Center (DKFZ), Heidelberg D-69120, Germany,2Faculty of Bioscience, Heidelberg University, Heidelberg D-69120, Germany, 3Department of Medical Biochemistryand Biophysics, Umea University, Umea SE-901 87 Sweden, 4Institute of Developmental Biology and Neurobiology,Johannes Gutenberg Universitat, 55128 Mainz, Germany, 5Institute of Molecular Biology (IMB), 55128 Mainz,Germany and 6Laboratory for Molecular Infection Medicine Sweden (MIMS), Umea University, SE-901 87 Umea,Sweden
Received June 08, 2019; Revised October 11, 2019; Editorial Decision October 14, 2019; Accepted October 16, 2019
ABSTRACT
The accumulation of mutations is frequently asso-ciated with alterations in gene function leading tothe onset of diseases, including cancer. Aiming tofind novel genes that contribute to the stability ofthe genome, we screened the Saccharomyces cere-visiae deletion collection for increased mutator phe-notypes. Among the identified genes, we discov-ered MET7, which encodes folylpolyglutamate syn-thetase (FPGS), an enzyme that facilitates severalfolate-dependent reactions including the synthesisof purines, thymidylate (dTMP) and DNA methylation.Here, we found that Met7-deficient strains show el-evated mutation rates, but also increased levels ofendogenous DNA damage resulting in gross chro-mosomal rearrangements (GCRs). Quantification ofdeoxyribonucleotide (dNTP) pools in cell extractsfrom met7Δ mutant revealed reductions in dTTP anddGTP that cause a constitutively active DNA dam-age checkpoint. In addition, we found that the ab-sence of Met7 leads to dUTP accumulation, at lev-els that allowed its detection in yeast extracts forthe first time. Consequently, a high dUTP/dTTP ratiopromotes uracil incorporation into DNA, followed byfutile repair cycles that compromise both mitochon-drial and nuclear DNA integrity. In summary, this workhighlights the importance of folate polyglutamylationin the maintenance of nucleotide homeostasis andgenome stability.
INTRODUCTION
The one-carbon (1C) cycle is a central metabolic path-way that comprises several modification reactions of folates,which are used as 1C donors in a variety of biosynthetic pro-cesses. Folate cofactors are required for dTMP and purinebiosynthesis (Supplementary Figure S1), glycine/serinehomeostasis, homocysteine remethylation to methionineand the production of formyl-methionyl-tRNA that is nec-essary for the initiation of protein biosynthesis in bacteria,chloroplasts and mitochondria (1,2).
Due to the pivotal role of the 1C metabolism for cellproliferation and growth, drugs that target the 1C cycle(antifolates) have proved beneficial for treatment of can-cer, autoimmune chronic diseases, as well as bacterial andparasite infections (3–7). Antifolates currently in use forcancer treatment inhibit dihydrofolate reductase (DHFR),that converts 7,8-dihydrofolate (DHF) into tetrahydrofolate(THF), the glycinamide ribonucleotide formyltransferase(GARFT) that uses 10-formyl-THF during the synthesisof purines, and thymidylate synthase (TS) that catalyzesthe conversion of 2-deoxyuridine monophosphate (dUMP)into dTMP (8).
Antifolate treatment leads to a reduction in dTMP con-centrations (with a consequent decrease in dTTP levels) andaccumulation of dUMP (4,8). Studies in bacteria, yeast andhuman cells have shown that deprivation of thymine rapidlycompromise cell viability, phenomenon known as thymine-less death (TLD) (9–11). Despite that the underlying mech-anism of TLD is not fully understood (12–14), substan-tial evidence indicates that a high dUTP/dTTP ratio drivesuracil misincorporation into DNA causing genome instabil-ity (12,15). Since eukaryotic DNA replicative polymerasescannot distinguish between dTTP and dUTP (16), an in-
*To whom correspondence should be addressed. Tel: +49 6221 42 3239; Fax: +49 6221 42 3237; Email: [email protected]
C© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), whichpermits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited.
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020

Nucleic Acids Research, 2020, Vol. 48, No. 1 265
creased dUTP/dTTP ratio promotes the incorporation ofuracil (in place of thymine) during DNA synthesis. Misin-corporated uracil triggers base excision repair (BER), thatremoves uracil from DNA; however, high uracil levels leadto reiterative uracil misincorporation/excision or ‘futile re-pair cycles’ resulting in frequent single and double strandbreaks compromising genome integrity (17,18).
Under normal conditions, dUTP level is kept at extremelylow concentrations, as dUTP is efficiently hydrolyzed intodUMP by the dUTP pyrophosphatase (Dut1) enzyme (Sup-plementary Figure S1). Accordingly, previous studies aim-ing to quantify dUTP levels in mammalian cells grown un-der normal conditions, either have failed (18–21), or havereported intracellular dUTP concentrations that differ sev-eral orders of magnitude between reports (11,22).
Complete loss of Dut1 activity in budding yeast causeslethality (23), whereas a dut1 mutant (dut1–1) that retains∼10% of the dUTPase activity shows increased uracil in-corporation and genome instability (24). Up to now, dut1–1together with a hypomorphic thymidylate synthetase allele(cdc21–1) (25), are the only two reported genetic alterationsin budding yeast associated with increased uracil incorpo-ration.
Recently, we performed a genome-wide screen in Sac-charomyces cerevisiae that identified a group of genes thatstrongly enhanced the mutator phenotype of strains ex-pressing DNA polymerase active-site mutant alleles (26).In addition, we also identified 39 single gene deletions (notreported at that time) that confer a mutator phenotype inthe presence of wild-type (WT) DNA polymerases. Withone exception, all identified gene deletions affected well-characterized genes, most of them involved in distinct DNArepair pathways (27,28). The remaining identified hit wasMET7, a gene that has not been previously associated withthe suppression of mutations. MET7 is a non-essential genein S. cerevisiae that encodes for both the cytosolic and themitochondrial folylpolyglutamate synthetase (FPGS) en-zymes (29). In mammals, FPGS also exists as cytosolic andmitochondrial isoforms, but in contrast to Met7, its func-tion is essential for survival of non-transformed proliferat-ing cells (1,30). Met7/FPGS catalyzes the addition of upto eight glutamates (polyglutamyl tail) that are linked tothe first glutamate in folate cofactors (Supplementary Fig-ure S1). The polyglutamylation of folates is important forthe 1C metabolism as it increases folate intracellular re-tention and enhances their affinity to folate-dependent en-zymes (31). Furthermore, polyglutamylation is of clinicalrelevance, as human FPGS not only modifies folates butalso antifolates that are frequently used for cancer treat-ment. Remarkably, a common mechanism of resistance toantifolate treatment in cancer cells occurs through the inac-tivation of human FPGS (5,8).
Previous studies in yeast reported that loss of MET7 re-sults in methionine auxotrophy (32), mitochondrial dys-function (petite phenotype) (33), short telomeres (34–36),imbalanced dNTP pools and a defect in non-homologousend-joining (NHEJ) (36). However, the impact of Met7and folate polyglutamylation on genome stability remainslargely elusive in S. cerevisiae.
This study highlights the importance of Met7 to main-tain nucleotide homeostasis, prevent uracil accumulation
and consequently sustain the stability of both mitochon-drial and nuclear DNA in budding yeast.
MATERIALS AND METHODS
Saccharomyces cerevisiae strains used in this study (Sup-plementary Table S5) are derivatives of the S288c strains:RDKY3686 (MATα ura3–52 leu2Δ1 trp1Δ63 hom3–10his3Δ200 lys2–10A) (37), RDKY5964 (a MATa ver-sion of RDKY3686) (38), RDKY3615 (MATa ura3–52leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl hom3–10 ade2Δ1ade8 yel069c::URA3) (39) or HHY6443 (RDKY5964iYEL072W::hph can1::hisG yel072w::CAN1/URA3bar1::loxP.klLEU2.loxP) (26). To further investigate thephenotype of the dut1–1 mutation we performed some ex-periments (as indicated in Supplementary Figure S4) in theBY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0)/BY4742(MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) background.Strains were cultivated in yeast extract-peptone-dextrose(YPD), yeast extract-peptone-glycerol (YPG) or syntheticmedia (SD) at 30◦C according to standard protocols. Genedeletions and gene-tagging were performed using standardPCR-based recombination methods (40,41), followed byconfirmation by PCR. Tags and junctions were confirmedby sequencing. Yeast strains expressing the dut1–1 allele(dut1-G82S) (24) at the endogenous locus, were generatedby pop-in/pop-out strategy with the integrative vector(pHHB1094). The presence of the dut1-G82S mutation,as well as the absence of additional unwanted mutationsin this gene, was confirmed by sequencing (for details, seeSupplementary Experimental Procedures).
Identification of gene deletions causing mutator phenotypesin S. cerevisiae
We recently reported a genome-wide screen in which weidentified factors that exacerbate the mutator phenotypeof strains expressing active-site mutant DNA polymerases(26) or that result in a mutator phenotype in strains ex-pressing wild-type DNA polymerases. In brief, HHY5298(MATa ura3–52 leu2Δ1 trp1Δ63 his3Δ200 lys2–10A cyh2-Q38K hom3–10.HIS3 pMFA1-klLEU2.hphNT1.lys2–10AMLH2.klURA3 POL1.natNT2) was crossed against thenon-essential gene-deletion collection (MATα his3Δ1leu2Δ0 ura3Δ lys2Δ yfg::kanMX4) using a RoToR robot(Singer Instruments). Strains containing gene deletions inthe presence or absence of DNA polymerase active-site mu-tations were tested for mutator phenotype with two in vivomutational reporters (lys2–10A frameshift reversion assay(37) and CAN1 inactivation assay (42)). Gene deletionsthat have not been previously reported to cause a mutatorphenotype were validated by generating these knockoutsde novo in RDKY5964 and HHY6443 for further analysis.
Determination of mutation rates and GCRs
Mutation rates using the CAN1 inactivation assay were de-termined in strains derived from RDKY5964 (38) by fluc-tuation analysis as previously described (43,44). Similarly,GCR rates were measured by fluctuation analysis in strains
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020

266 Nucleic Acids Research, 2020, Vol. 48, No. 1
either derived from RDKY3615 (for the standard GCR as-say) (39) or HHY6443 (for the post-duplication GCR re-porter) (26). Mutation and GCR rates were determinedbased on two biological isolates and at least 14 indepen-dent cultures. 95% confidence intervals were calculated forall fluctuation tests.
Determination of NTP and dNTP pools
NTP and dNTPs were initially measured as described be-fore (45). Briefly, logarithmically growing yeast cells wereharvested by filtration at a density of 0.4 × 107 to 0.5 ×107 cells/ml, disintegrated with a ice-cold mixture of 12%(wt/vol) trichloroacetic acid (TCA) and 15 mM MgCl2,and extracted with an ice-cold freon-trioctylamine mixture[10 ml of freon (1,1,2-trichloro-1,2,2-trifluoroethane); Mil-lipore Sweden AB (>99%) and 2.8 ml of trioctylamine;Sigma–Aldrich Sweden AB (98%)]. 500 �l of the aque-ous phase was treated with or without recombinant humandUTPase (hDut1, ab173062, Abcam) at a concentration of1 ng/�l at 37◦C for 1 h and then analyzed by strong ionexchange (SAX) high-performance liquid chromatography(HPLC) before (for NTP quantification) and after boronatechromatography (Affigel 601, Bio-Rad) (for dNTP quan-tification). Using this SAX-HPLC protocol, we could sep-arate all NTPs and dNTPs except dUTP, which was onlypartially resolved from dTTP (Supplementary Figure S2A).The dTTP peak could still be quantified accurately since itis much larger than dUTP.
To resolve all NTPs and dNTP including dUTP, we de-veloped a HPLC procedure based on reverse phase (RP)chromatography with tetrabutylammonium bromide as ionpairing agent. In this procedure, we introduced an extra stepwhere the cell-containing filters (see above) were washedtwo times (30 ml each) with an ice-cold aqueous solutioncontaining 8 g/l NaCl and 27 g/l glucose before disintegrat-ing the cells with the TCA-MgCl2 solution. After the freon-trioctylamine step, the extracts were purified with OASIS-WAX (46), mixed 1:1 with mobile phase and separated at 0.5ml/min on a 2.1 × 50 mm ACE Excel 2 �m C18-PFP col-umn from Advanced Chromatography Technologies (Ab-erdeen, UK) using a UV-2075 Plus detector (Jasco Inter-national Co. Ltd, Hachioji, Japan) set at 270 nm (STD re-sponse time). A gradient between three aqueous solutionswas used: solution A, B and C. Solution A contained 7%(v/v) methanol and 23 g/l KH2PO4 (HPLC grade fromVWR International, Radnor, PA, USA), and the final solu-tion was pH-adjusted to pH 5.6 with KOH. Solution B con-tained only 7% (v/v) methanol and solution C contained7% (v/v) methanol and 3.52 g/l tetrabutylammonium bro-mide (ion-pair chromatography grade from Merck Group,Darmstadt, Germany). The run started isocratically with10% A, 70% B and 20% C (min 0–10), followed by a lin-ear gradient up to 60% A, 20% B and 20% C (min 10–27),and finally an isocratic step with 60% A, 20% B and 20%C (min 27–35), before returning to the initial conditions.The column was equilibrated for at least 15 min betweenthe runs. Using the RP-HPLC protocol, we could separateall NTPs as well as ADP, dCTP and dUTP (SupplementaryFigure S2B). However, co-purifying metabolites occludedthe analysis of dGTP, and to some extent dTTP (as well as
dATP in the WT extracts), and because of that, we excludedthese peaks from the analyses. The dUTP peak was com-pletely free from interfering peaks, and dCTP was nearlyfree from interference (only a minor peak was close to dCTPin the met7Δ mutant). In Supplementary Table S2, we havegiven the NTP and dNTP pools from the SAX-HPLC pro-tocol and the NTP, dCTP, dUTP and ADP pools from theRP-HPLC protocol, whereas Figure 2A, B and Figure 3Cshow the results obtained from the SAX protocol. The highATP/ADP ratio in all samples (∼20) indicates that the en-ergy status was good and that the cells were not disturbed bythe extra washing steps during harvesting. The quantifica-tion was performed by comparing the peak heights to a nu-cleotide standard for both protocols except for the NTPs inthe RP protocol where areas were used instead. The reasonfor this is that the column needed to be slightly overloadedwith NTPs (and then area is a more accurate measurement)in order to measure the much smaller dNTP peaks accu-rately.
Yeast cell lysates and immunoblotting
Saccharomyces cerevisiae whole-cell protein extracts weregenerated as described (47) and analyzed on SDS-PAGEfollowed by immunoblotting using anti-Rad53 (EL7.E1,Abcam), anti-Rnr3 (AS09574, Agrisera), anti-tubulin/anti-Rnr4 (YL1/2, Sigma), anti-c-Myc (9E10, Millipore), anti-Clb2 (sc-9071, Santa Cruz Biotechnology), anti-Pgk1(22C5D8, Invitrogen) and anti-Sic1 (this study).
DNA content analysis
Logarithmic S. cerevisiae cultures were processed as de-scribed in (47) and analyzed using BD FACS Canto II (BDBiosciences) and FlowJo (v10.1, Tree Star Inc).
Determination of growth rates
Yeast cultures were diluted to an optical density at 595 nm(OD595) of 0.05 by transferring the appropriate volume ofan overnight culture into fresh YPD. Cultures were grownwith shaking at 30◦C for 12 h and OD595 measurementswere taken every 30 min. Doubling times were calculatedbased on measurements obtained from at least two inde-pendent isolates per genotype.
Live-cell imaging of Ddc2-GFP foci
Exponentially growing cells were processed and imaged asdescribed in (38) using a Leica SP5 confocal microscope(Leica) with an Argon laser, a 63x 1.4NA objective and ahigh resonance scanner @8 kHz frequency. Ten 0.4 �m zsections were acquired; image processing such as maximumintensity projections were performed using ImageJ. Threeindependent biological replicates per genotype were ana-lyzed and Mann–Whitney rank sum test was used to com-pare Ddc2-GFP foci abundance in WT and met7Δ strains.Statistical analysis was performed using the SigmaPlot soft-ware.
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020

Nucleic Acids Research, 2020, Vol. 48, No. 1 267
Determination of uracil incorporation into genomic DNA
Uracil accumulation assay was mainly done as described(48). Genomic DNA was isolated from logarithmicallygrowing cells using Puregene Yeast/Bact. Kit B (Qiagen)and further incubated overnight at 37◦C with 10 U uracilDNA glycosylase from E. coli (UDG) and 20 U human APendonuclease 1 (Ape1) (New England Biolabs) in 1× NEB-uffer 4 (50 mM potassium acetate, 20 mM Tris-acetate, 10mM magnesium acetate, 1 mM DTT, pH 7.9). DNA wasprecipitated and loaded on a 0.8% agarose gel stained withGelRed (Biotium). Images were taken using the GelDocsystem (Bio-Rad).
Telomere length analysis by southern blot
Genomic DNA (5 �g) was digested with XhoI for 5 hat 37◦C. The digested DNA was separated on a 0.8%agarose gel overnight at 50 V. DNA in the gel was dena-tured for 1 h (0.4 M NaOH, 0.6 M NaCl) and neutralizedfor 1 h (1 M Trizma Base, 1.5 M NaCl, pH 7.4). DNAwas transferred to a nylon membrane (Hybond NX, GEHealthcare) via capillary transfer in 10X SSC overnightand cross-linked to the membrane with UV light (auto X-link, Stratalinker). The membrane was pre hybridized for5 h at 55◦C in hybridization solution (PerfectHyb™ PlusHybridization Buffer, Sigma). A telomere-specific probewas generated by random primed radioactive labeling withdATP [�-32P] (DECAprime kit II; Thermo Scientific) of adouble-stranded DNA fragment obtained by digestion ofpBL423 (a kind gift from M.P. Longhese (pSP100)) withEcoRI followed by gel extraction. Hybridization was car-ried out overnight at 55◦C. The membrane was washedtwice in 2× SSC with 0.1% SDS for 5 min and twice in 0.5×SSC with 0.1% SDS for 20 min. All washing steps were per-formed at 55◦C. The signal was detected via Typhoon FLA9500 (GE Healthcare).
RESULTS
Inactivation of the folylpolyglutamate synthetase Met7 re-sults in mutator phenotype and GCRs
In a previous study, we identified a group of genes in S.cerevisiae that prevent the accumulation of mutations instrains expressing low-fidelity DNA polymerase alleles (26).As part of that study we also identified 39 genes (not re-ported at that time) that when inactivated in a WT straincaused an increased rate of mutations according to thelys2–10A frameshift reversion reporter and/or the CAN1inactivation assay (Supplementary Table S1). Among thegenes that prevented frameshift mutations, we identifiedknown components of the mismatch repair (MMR) sys-tem (MSH2, MSH6, MSH3, MLH1, MLH3, PMS1 andEXO1) that participate at different steps during the correc-tion of insertions/deletions or base substitutions (49–51).Moreover, we found that inactivation of Elg1, a subunit ofan alternative replication factor C (RFC) complex (52,53)caused a mild increase in frameshift mutations (Supplemen-tary Table S1). Elg1 promotes the unloading of Proliferat-ing Cell Nuclear Antigen (PCNA) from DNA (54) and con-tributes in different aspects to the stability of the genome,including the suppression of frameshift mutations (27).
In addition, we identified 31 genes (e.g. CCS1, CSM2,MET7, MMS2, MPH1, among others) that upon inactiva-tion caused an increased rate of CAN1 inactivation, with-out compromising the repair of frameshift mutations at thelys2–10A reporter. With the exception of MET7, all otheridentified genes have been previously associated with thesuppression of mutations (27,28,55–61). To validate the po-tential role of Met7 in the suppression of mutations we gen-erated de novo met7Δ strains and measured the CAN1 inac-tivation rate by fluctuation analysis. Supporting our initialobservation, loss of Met7 resulted in a 9-fold increase (rel-ative to WT) in the CAN1 inactivation rate (Table 1).
As inactivation of the CAN1 gene can occur due to mi-spaired bases, frameshifts but also as result of GCRs, wetested whether loss of Met7 may also cause chromosomalinstability. For this analysis we used yeast strains that har-bor in the non-essential left arm of chromosome V twocounter-selectable genes (URA3 and CAN1) that confersensitivity to 5-fluoroorotic acid (5-FOA) and canavanine,respectively. By measuring the spontaneous appearance of5-FOA- and canavanine-resistant colonies (5-FOAR/CanR)in multiple independent cultures, it is possible to calculatethe spontaneous inactivation rate of both genes, which ismainly due to GCR events. Quantification of GCR rateswas done using yeast strains either carrying the ‘standard’GCR reporter (39) or the ‘post-duplication GCR reporter’(62). In the latter GCR reporter, GCR events are driven bya 4.2 kb region (HTX13 DSF1, located centromeric to theURA3 and CAN1 genes), that shares high sequence homol-ogy to sequences present in chromosomes XIV, IV and X,mainly resulting in homologous recombination-mediatedGCRs. GCR rates measured in the WT strain using thestandard- and post-duplication-GCR assay, were consistentwith previous reports (39,47,62). Remarkably, Met7 inac-tivation resulted in a 39-fold increase in GCRs using thestandard GCR assay (5.1 × 10–11 and 2.0 × 10–9 CanR 5-FOAR mutations per cell per generation in the WT andmet7Δ strains, respectively) and 177-fold increase in thepost-duplication GCR assay compared to the WT strain(Table 1). Therefore, loss of Met7 not only causes a mutatorphenotype but also results in an increased GCR rate.
Inactivation of Met7 causes activation of the DNA damageresponse
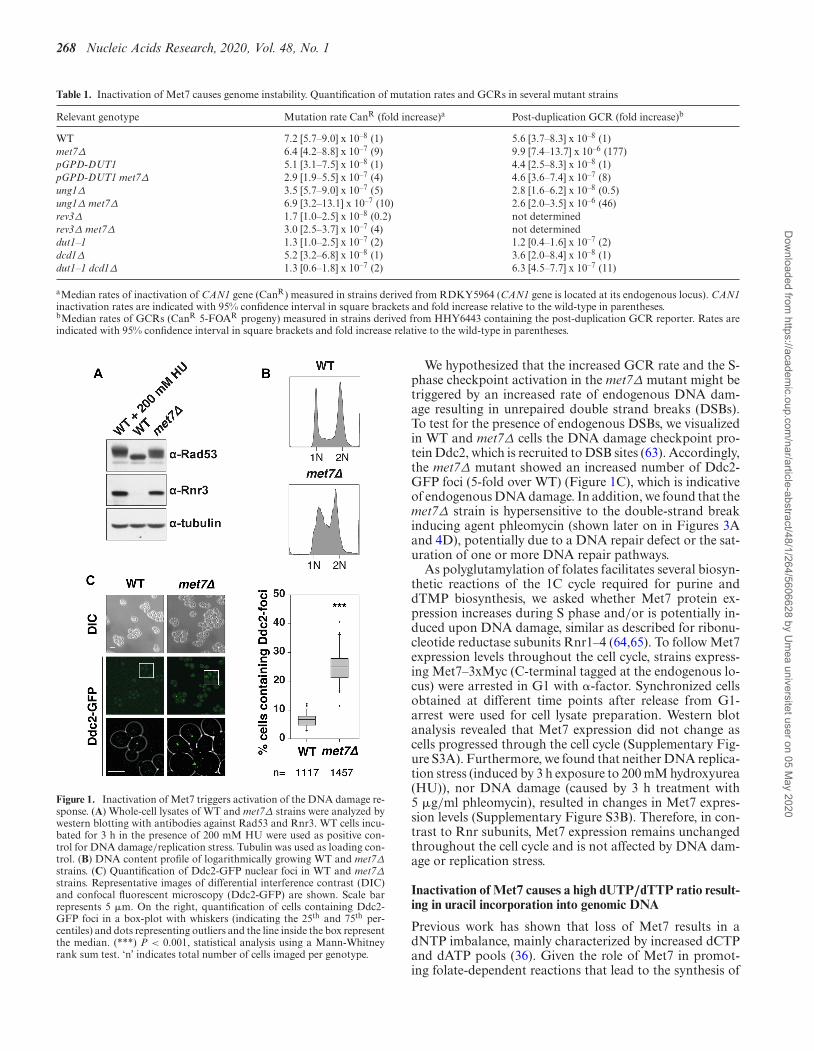
The slow growth phenotype of the met7Δ strain (33) andthe increased genome instability phenotype, prompted usto investigate whether loss of Met7 triggers the activationof the DNA damage response. After analyzing cell lysatesfrom logarithmically growing WT and met7Δ strains bywestern blotting, we found that loss of Met7 resulted inphosphorylation of the DNA damage checkpoint kinaseRad53 (phosphorylated forms of Rad53 give slower elec-trophoretic mobility bands), and up-regulation of the DNAdamage-inducible ribonucleotide reductase (RNR) subunitRnr3 that contributes to dNTP biosynthesis (Figure 1A).These findings, together with the accumulation of cells inS phase observed in the met7Δ mutant (Figure 1B), indi-cate that loss of Met7 results in constitutive S-phase check-point activation, potentially as a consequence of endoge-nous DNA damage and/or DNA replication stress.
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020
268 Nucleic Acids Research, 2020, Vol. 48, No. 1
Table 1. Inactivation of Met7 causes genome instability. Quantification of mutation rates and GCRs in several mutant strains
Relevant genotype Mutation rate CanR (fold increase)a Post-duplication GCR (fold increase)b
WT 7.2 [5.7–9.0] x 10–8 (1) 5.6 [3.7–8.3] x 10–8 (1)met7Δ 6.4 [4.2–8.8] x 10–7 (9) 9.9 [7.4–13.7] x 10–6 (177)pGPD-DUT1 5.1 [3.1–7.5] x 10–8 (1) 4.4 [2.5–8.3] x 10–8 (1)pGPD-DUT1 met7Δ 2.9 [1.9–5.5] x 10–7 (4) 4.6 [3.6–7.4] x 10–7 (8)ung1Δ 3.5 [5.7–9.0] x 10–7 (5) 2.8 [1.6–6.2] x 10–8 (0.5)ung1Δ met7Δ 6.9 [3.2–13.1] x 10–7 (10) 2.6 [2.0–3.5] x 10–6 (46)rev3Δ 1.7 [1.0–2.5] x 10–8 (0.2) not determinedrev3Δ met7Δ 3.0 [2.5–3.7] x 10–7 (4) not determineddut1–1 1.3 [1.0–2.5] x 10–7 (2) 1.2 [0.4–1.6] x 10–7 (2)dcd1Δ 5.2 [3.2–6.8] x 10–8 (1) 3.6 [2.0–8.4] x 10–8 (1)dut1–1 dcd1Δ 1.3 [0.6–1.8] x 10–7 (2) 6.3 [4.5–7.7] x 10–7 (11)
aMedian rates of inactivation of CAN1 gene (CanR) measured in strains derived from RDKY5964 (CAN1 gene is located at its endogenous locus). CAN1inactivation rates are indicated with 95% confidence interval in square brackets and fold increase relative to the wild-type in parentheses.bMedian rates of GCRs (CanR 5-FOAR progeny) measured in strains derived from HHY6443 containing the post-duplication GCR reporter. Rates areindicated with 95% confidence interval in square brackets and fold increase relative to the wild-type in parentheses.
Figure 1. Inactivation of Met7 triggers activation of the DNA damage re-sponse. (A) Whole-cell lysates of WT and met7Δ strains were analyzed bywestern blotting with antibodies against Rad53 and Rnr3. WT cells incu-bated for 3 h in the presence of 200 mM HU were used as positive con-trol for DNA damage/replication stress. Tubulin was used as loading con-trol. (B) DNA content profile of logarithmically growing WT and met7Δ
strains. (C) Quantification of Ddc2-GFP nuclear foci in WT and met7Δ
strains. Representative images of differential interference contrast (DIC)and confocal fluorescent microscopy (Ddc2-GFP) are shown. Scale barrepresents 5 �m. On the right, quantification of cells containing Ddc2-GFP foci in a box-plot with whiskers (indicating the 25th and 75th per-centiles) and dots representing outliers and the line inside the box representthe median. (***) P < 0.001, statistical analysis using a Mann-Whitneyrank sum test. ‘n’ indicates total number of cells imaged per genotype.
We hypothesized that the increased GCR rate and the S-phase checkpoint activation in the met7Δ mutant might betriggered by an increased rate of endogenous DNA dam-age resulting in unrepaired double strand breaks (DSBs).To test for the presence of endogenous DSBs, we visualizedin WT and met7Δ cells the DNA damage checkpoint pro-tein Ddc2, which is recruited to DSB sites (63). Accordingly,the met7Δ mutant showed an increased number of Ddc2-GFP foci (5-fold over WT) (Figure 1C), which is indicativeof endogenous DNA damage. In addition, we found that themet7Δ strain is hypersensitive to the double-strand breakinducing agent phleomycin (shown later on in Figures 3Aand 4D), potentially due to a DNA repair defect or the sat-uration of one or more DNA repair pathways.
As polyglutamylation of folates facilitates several biosyn-thetic reactions of the 1C cycle required for purine anddTMP biosynthesis, we asked whether Met7 protein ex-pression increases during S phase and/or is potentially in-duced upon DNA damage, similar as described for ribonu-cleotide reductase subunits Rnr1–4 (64,65). To follow Met7expression levels throughout the cell cycle, strains express-ing Met7–3xMyc (C-terminal tagged at the endogenous lo-cus) were arrested in G1 with �-factor. Synchronized cellsobtained at different time points after release from G1-arrest were used for cell lysate preparation. Western blotanalysis revealed that Met7 expression did not change ascells progressed through the cell cycle (Supplementary Fig-ure S3A). Furthermore, we found that neither DNA replica-tion stress (induced by 3 h exposure to 200 mM hydroxyurea(HU)), nor DNA damage (caused by 3 h treatment with5 �g/ml phleomycin), resulted in changes in Met7 expres-sion levels (Supplementary Figure S3B). Therefore, in con-trast to Rnr subunits, Met7 expression remains unchangedthroughout the cell cycle and is not affected by DNA dam-age or replication stress.
Inactivation of Met7 causes a high dUTP/dTTP ratio result-ing in uracil incorporation into genomic DNA
Previous work has shown that loss of Met7 results in adNTP imbalance, mainly characterized by increased dCTPand dATP pools (36). Given the role of Met7 in promot-ing folate-dependent reactions that lead to the synthesis of
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020
Nucleic Acids Research, 2020, Vol. 48, No. 1 269
A B
C
ung1∆dut1-1ung1∆
met7∆ung1∆
pGPD-DUT1met7∆ung1∆
- + - + - + - +UDG + Ape1
250
500
7501000
15002000
250
500
7501000
15002000
UA G
AATA
CC
T UG
GC
A GA
ATA
CC
TG
GC
UDG + Ape1
agarose gel electropheresis
DNA fragmentation
dCTP dTTPdATP dGTPdUTP
0
5000
10000
15000
WT met7∆ pGPD-DUT1met7∆
NT
P c
on
cen
trat
ion
in p
mo
l / 1
08 ce
lls CTP UTPATP GTP
1.0
1.0
1.0
1.0
0.9
0.9
0.7
0.7
1.2
1.2
1.2
1.1
dN
TP
co
nce
ntr
atio
n in
pm
ol /
108
cells
hDUT1WT met7∆ pGPD-DUT1 met7∆
- + - + - +
100
200
300
400
500
10
0
1.0
1.0
1.0
1.0 1.
01.0
1.0
0.9
2.7
4.0
2.5
3.9
2.8
2.5
3.7
3.6
0.6
0.6
0.6
0.6 0.
5
0.5
0.5
0.5
nd nd nd nd nd
D
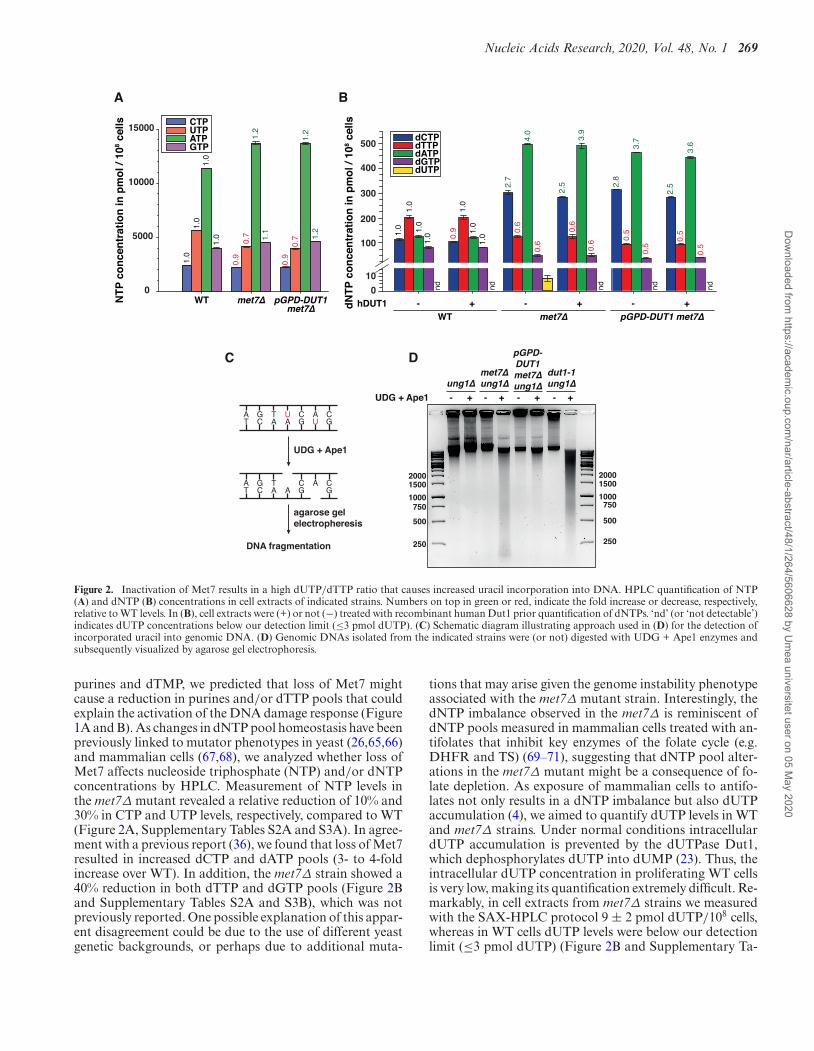
Figure 2. Inactivation of Met7 results in a high dUTP/dTTP ratio that causes increased uracil incorporation into DNA. HPLC quantification of NTP(A) and dNTP (B) concentrations in cell extracts of indicated strains. Numbers on top in green or red, indicate the fold increase or decrease, respectively,relative to WT levels. In (B), cell extracts were (+) or not (−) treated with recombinant human Dut1 prior quantification of dNTPs. ‘nd’ (or ‘not detectable’)indicates dUTP concentrations below our detection limit (≤3 pmol dUTP). (C) Schematic diagram illustrating approach used in (D) for the detection ofincorporated uracil into genomic DNA. (D) Genomic DNAs isolated from the indicated strains were (or not) digested with UDG + Ape1 enzymes andsubsequently visualized by agarose gel electrophoresis.
purines and dTMP, we predicted that loss of Met7 mightcause a reduction in purines and/or dTTP pools that couldexplain the activation of the DNA damage response (Figure1A and B). As changes in dNTP pool homeostasis have beenpreviously linked to mutator phenotypes in yeast (26,65,66)and mammalian cells (67,68), we analyzed whether loss ofMet7 affects nucleoside triphosphate (NTP) and/or dNTPconcentrations by HPLC. Measurement of NTP levels inthe met7Δ mutant revealed a relative reduction of 10% and30% in CTP and UTP levels, respectively, compared to WT(Figure 2A, Supplementary Tables S2A and S3A). In agree-ment with a previous report (36), we found that loss of Met7resulted in increased dCTP and dATP pools (3- to 4-foldincrease over WT). In addition, the met7Δ strain showed a40% reduction in both dTTP and dGTP pools (Figure 2Band Supplementary Tables S2A and S3B), which was notpreviously reported. One possible explanation of this appar-ent disagreement could be due to the use of different yeastgenetic backgrounds, or perhaps due to additional muta-
tions that may arise given the genome instability phenotypeassociated with the met7Δ mutant strain. Interestingly, thedNTP imbalance observed in the met7Δ is reminiscent ofdNTP pools measured in mammalian cells treated with an-tifolates that inhibit key enzymes of the folate cycle (e.g.DHFR and TS) (69–71), suggesting that dNTP pool alter-ations in the met7Δ mutant might be a consequence of fo-late depletion. As exposure of mammalian cells to antifo-lates not only results in a dNTP imbalance but also dUTPaccumulation (4), we aimed to quantify dUTP levels in WTand met7Δ strains. Under normal conditions intracellulardUTP accumulation is prevented by the dUTPase Dut1,which dephosphorylates dUTP into dUMP (23). Thus, theintracellular dUTP concentration in proliferating WT cellsis very low, making its quantification extremely difficult. Re-markably, in cell extracts from met7Δ strains we measuredwith the SAX-HPLC protocol 9 ± 2 pmol dUTP/108 cells,whereas in WT cells dUTP levels were below our detectionlimit (≤3 pmol dUTP) (Figure 2B and Supplementary Ta-
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020

270 Nucleic Acids Research, 2020, Vol. 48, No. 1
ble S2A). To further validate dUTP measurements, cell ex-tracts were treated with recombinant hDut1 prior to dNTPquantification. Strikingly, dUTP was no longer detectablein hDut1-treated met7Δ samples, whereas no major addi-tional changes were observed among other dNTPs (Fig-ure 2B and Supplementary Table S2A). Likewise, met7Δcells overexpressing the DUT1 gene (pGPD-DUT1) underthe control of a strong constitutive promoter showed un-detectable dUTP levels and otherwise identical dNTP con-centrations as measured in the met7Δ strain (Figure 2B andSupplementary Table S2A). To validate the results obtainedby SAX-HPLC, we developed an alternative HPLC proce-dure based on reverse phase chromatography (RP-HPLC)with tetrabutylammonium bromide as an ion-pairing agentthat allowed a more accurate quantification of dUTP pools,corresponding to ≤1 and 11 pmol dUTP/108 cells in WTand met7Δ strains, respectively (Supplementary Table S2B).Otherwise, SAX-HPLC and RP-HPLC methods gave sim-ilar results for NTPs and dNTPs concentrations (Supple-mentary Table S2). The conclusion from these experimentsis that loss of Met7 causes at least a 10-fold increase of in-tracellular dUTP levels.
Previous studies have shown that increased uracil incor-poration observed upon folate depletion (e.g. induced byantifolates) causes genome instability (11,72). Therefore, wehypothesized that the underlying cause of the genome insta-bility phenotype observed in the met7Δ strain might be re-lated to a high dUTP/dTTP ratio that will lead to uracilmisincorporation and futile repair cycles resulting in fre-quent DSBs. In order to test this hypothesis, we treated ge-nomic DNA from WT and met7Δ strains with recombinanturacil DNA glycosylase (UDG) and apurinic/apyrimidinicendonuclease 1 (Ape1), and visualized its potential frag-mentation pattern by agarose gel electrophoresis as previ-ously described (48) (Figure 2C). Since uracil is efficientlyremoved from DNA by the uracil DNA glycosylase (Ung1),all strains used for this analysis were generated in strainslacking UNG1, which encodes for the nuclear and mito-chondrial UDG isoforms (73,74). In addition, we includedin our analysis the dut1–1 mutant that has been previouslyshown to retain <10% dUTPase activity and to cause uracilincorporation into genomic DNA (24). Remarkably, whilethe DNA isolated from ung1Δ remained unchanged af-ter UDG+Ape1 treatment, DNA isolated from the met7Δung1Δ mutant was vastly fragmented by UDG+Ape1 (withDNA fragments as low as 200 bases), indicative of massiveuracil incorporation (Figure 2D). Similar results were ob-tained with DNA isolated from the dut1–1 strain as previ-ously reported (24). Importantly, the DNA fragmentationobserved in the met7Δ ung1Δ strain was largely suppressedby DUT1-overexpression (Figure 2D), which is in line withthe low dUTP levels in this strain (Figure 2B and Supple-mentary Table S2). Thus, inactivation of Met7 causes accu-mulation of intracellular dUTP levels resulting in increaseduracil incorporation into DNA.
Rev3 inactivation or Dut1 overexpression partially suppressesmutations in met7Δ strain
As uracil accumulates in the absence of Met7, we askedwhether the increased uracil incorporation into DNA con-
tributes to genome instability phenotype in the met7Δ mu-tant. The uracil DNA glycosylase Ung1 recognizes and re-moves the uracil moiety leaving behind an abasic site (75).Abasic sites are further processed by apurinic/apyrimidinic(AP) endonucleases, which generate a single strand break,which is subsequently repaired by short- or long-patchBER. If abasic sites are not processed by AP endonucle-ases, they cause stalled DNA replication forks during thenext round of DNA replication, which leads to error-pronerepair via the recruitment of translesion synthesis (TLS)DNA polymerases (75,76). To test the contribution of TLSpolymerase Rev3 to the mutator phenotype observed in theabsence of MET7, we determined the CAN1 inactivationrate in the met7Δ strain in the presence and absence ofREV3. Approximately, 50% of all CAN1 inactivation eventswere REV3-dependent (Table 1). Moreover, DUT1 overex-pression in the met7Δ strain, which prevents dUTP accu-mulation and presumably to a large degree abasic site for-mation, also caused a 2-fold reduction in the CAN1 muta-tion rate (Table 1). A similar reduction in the CAN1 mu-tation rate was not seen when Ung1 was inactivated in themet7Δ strain. This last result suggests that Ung1-dependenturacil removal (and consequently the formation of abasicsites) is not the major source of CAN1 inactivating mu-tations in the met7Δ strain. Moreover, no change in theCAN1 inactivation rate was observed when Dut1 was over-expressed in the WT strain, suggesting that the lower mu-tation rate in the pGPD-DUT1 met7Δ mutant is linked toelevated dUTP pools that are normally not occurring in theWT strain.
To investigate which type of mutations occur in the ab-sence of Met7, we analyzed the CAN1 mutation spectrum inMet7-deficient strains. The mutational spectrum was dom-inated by base substitution mutations (77% of the CAN1-inactivation events), and among those we observed a 1.5-fold increase in the percentage of G-C to A-T mutationscompared to the WT (Supplementary Table S4) (26). Thisincrease in G-C to A-T transitions is likely a consequenceof the reduced dGTP and dTTP, and elevated dATP levelsobserved in the met7Δ mutant (Figure 2B, SupplementaryTables S2A and S3B). This type of dNTP imbalance mayresult in C:A mispairs that in the next DNA replication cy-cle will lead to C to T transitions. No mutational hotspotswere identified, and besides the slight increase in G-C to A-T transitions, the met7Δ mutation spectrum was not signif-icantly different to the WT (Fisher exact test, P = 0.2275).Thus, CAN1 inactivation events in the met7Δ mutant arelikely driven by DNA replication errors due to imbalanceddNTP pools and by potential mutations arising from error-prone repair after uracil excision (75,77)
Dut1 overexpression or Ung1 inactivation suppresses GCRsin the met7Δ mutant
Removal of uracil from genomic DNA can lead to the gen-eration of single and double strand DNA breaks, that couldexplain the chromosomal instability phenotype observed instrains lacking MET7 (Table 1). To test whether the in-creased GCR rate in met7Δ strains is a consequence ofuracil accumulation/repair, we measured GCR rates (post-duplication GCR assay) in strains overexpressing DUT1
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020

Nucleic Acids Research, 2020, Vol. 48, No. 1 271
or lacking UNG1, either in the WT or in the met7Δbackground. Strikingly, the increased GCR rate in themet7Δ strains was suppressed to a large extent by Dut1-overexpression (from 177-fold down to 8-fold over WT lev-els), and to a lesser degree by Ung1 inactivation (from 177-fold down to 46-fold) (Table 1). On the other hand, DUT1-overexpression or Ung1 inactivation in a WT strain did notresult in major changes in GCR rates. Thus, these results in-dicate that the elevated GCR rate in the met7Δ strain is trig-gered by uracil accumulation and can be suppressed eitherby reducing dUTP levels (Figure 2B and Supplementary Ta-ble S2) or by preventing the processing of uracil incorpo-rated into genomic DNA (Figure 2D). Interestingly, theseresults are in agreement with the observation that increaseddUTPase activity or inactivation of Ung1 protects yeastcells against toxic effects of antifolates or 5-fluorouracil ex-posure (48,72).
Increased uracil incorporation in the dut1-1 mutant has nomajor consequences on genome stability
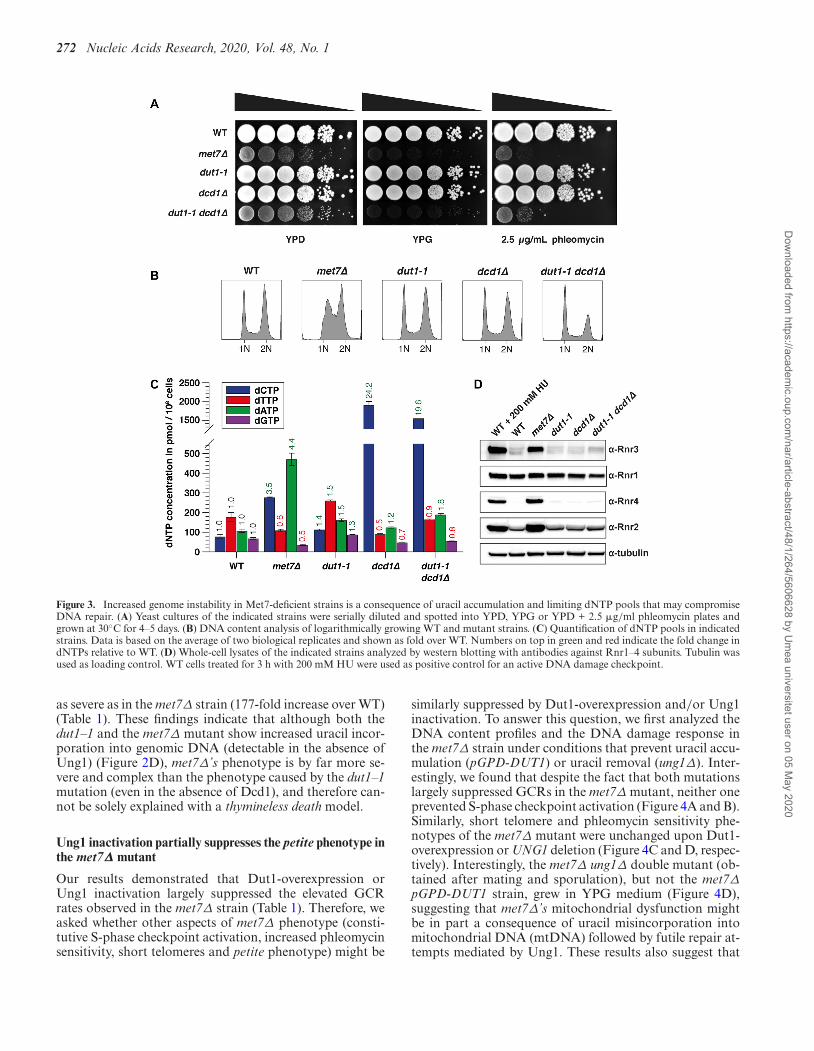
Our results indicate that the genome instability phenotypeof the met7Δ mutant is, at least in part, a consequenceof a high dUTP/dTTP ratio that leads to uracil incorpo-ration followed by futile repair cycles. We asked whetheruracil incorporation is solely sufficient to drive elevatedGCR rates, or if other additional factors may contributeto met7Δ’s genome instability phenotype. To answer thisquestion, we measured mutation rates and GCR rates inthe dut1–1 strain, which like the met7Δ mutant showed in-creased uracil incorporation (Figure 2D). Interestingly, incontrast to the findings reported by (24), the dut1–1 mu-tant despite its increased uracil incorporation phenotype,showed only a minor increase (2-fold) in both, the CAN1inactivation and GCR assays (Table 1).
To exclude the possibility of potential suppressor mu-tations that may have arisen during the generation ofthe dut1–1 mutant, we re-introduced the dut1–1 muta-tion in our S288c strain background, and also in an al-ternative yeast strain (BY4741/BY4742), and performedmating/sporulation and tetrad dissection analysis, followedby characterization of the obtained dut1–1 mutants. Tetraddissection analysis revealed that the dut1–1 mutation didnot cause an apparent growth defect in our S288c back-ground, although it consistently resulted in colonies of re-duced size in the BY-strain (Supplementary Figure S4A).These initial observations were complemented with growthrate analysis (Supplementary Figure S5), revealing in bothyeast backgrounds slightly reduced growth rates in thedut1–1 mutants, reflected on extended doubling times rel-ative to the WTs (97 ± 0.7 versus 86 ± 0.6 min in theS288c background, and 95 ± 1.3 versus 86 ± 0.5 min inthe BY-background). These results are in agreement withthe growth defect previously described in the dut1–1 mutant(24).
On the other hand, the qualitative mutator analysis indut1–1 mutant strains obtained after tetrad dissection, re-vealed no mutator phenotype in both tested genetic back-grounds (Supplementary Figure S4B and C), which sup-ports our previous measurements (Table 1), and arguesagainst the presence of unwanted mutations that may sup-
press the expected dut1–1 genome instability phenotype(24). There are at least two possibilities that could explainthis discrepancy: (i) The previously characterized dut1–1strain might contain an additional mutation that in combi-nation with the dut1–1 allele could result in a strong mutatorphenotype. (ii) Alternatively, the dut1–1 mutation may re-sult in a mutator phenotype in specific genetic backgrounds,perhaps as result of differences in DNA replication fidelityand/or DNA repair efficiency.
In contrast to the met7Δ mutant, the dut1–1 strain isneither a petite (indicated by the ability to use glycerolas carbon source), nor it is hypersensitive to DNA dam-age induced by phleomycin (Figure 3A). Furthermore, thedut1–1 mutant, despite its slightly increased doubling time,showed a DNA content profile and intracellular dNTPpools, almost indistinguishable from the WT strain (Fig-ure 3B-C and Supplementary Table S3). In agreement withthese observations, cell lysates of the dut1–1 strain ana-lyzed by western blotting, showed WT-like levels of theDNA damage-inducible RNR subunits (Rnr3, Rnr2 andRnr4), indicating that the DNA damage checkpoint is notactivated (Figure 3D). Taken together, these results sug-gest that uracil incorporation alone might not be suffi-cient to cause increased mutagenesis or GCRs in buddingyeast.
One difference between the dut1–1 and the met7Δ strainsis that the former one showed no reduction in dTTP levels.We speculate that the more severe dUTP/dTTP ratio in themet7Δ mutant might be responsible for the genome insta-bility phenotype. In budding yeast, dTMP is synthesized bythymidylate synthase (Cdc21) (Supplementary Figure S1)that transfers a methyl group from 5,10-methylene-THFto the C5 position of dUMP to generate dTMP. Impor-tantly, 60% of the dUMP pool is supplied by Dut1 throughhydrolysis of dUTP, and 40% by deamination of deoxy-cytidine monophosphate (dCMP), a reaction catalyzed bythe dCMP deaminase Dcd1 (24,78) (Supplementary Fig-ure S1). For this reason, we asked whether a dcd1Δ dut1–1 double mutant, if viable, might recapitulate the genomeinstability phenotype observed in the met7Δ strain. Inter-estingly, dcd1Δ dut1–1 double mutant, similar to the met7Δstrain, grew poorly, showed phleomycin sensitivity and a pe-tite phenotype, indicated by the inability to grow on mediacontaining glycerol as carbon source (YPG plates) due tomitochondrial dysfunction (Figure 3A), but did not showan S phase delay or an activated DNA damage response(Figure 3B and D). Quantification of dNTP pools in thedcd1Δ single mutant revealed reduced dTTP and increaseddCTP levels (Figure 3C and Supplementary Table S3B),which is in agreement with a previous report (78). However,in contrast to our expectations, the dcd1Δ dut1–1 doublemutant did not show lower but rather increased dTTP lev-els compared to the dcd1Δ single mutant (1.8-fold higher).In general, dNTPs in the dcd1Δ dut1–1 double mutant ap-proximate to the average of the dcd1Δ and the dut1–1 singlemutant dNTP concentrations. As the dut1–1 single mutantshows slightly increased dNTPs (∼1.5-fold over WT), wepropose that the dut1–1 mutation causes an overall increasein dNTP levels in the dcd1Δ strain.
In addition, we found that the dcd1Δ dut1–1 double mu-tant showed elevated GCR rates (11-fold over WT), but not
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020
272 Nucleic Acids Research, 2020, Vol. 48, No. 1
Figure 3. Increased genome instability in Met7-deficient strains is a consequence of uracil accumulation and limiting dNTP pools that may compromiseDNA repair. (A) Yeast cultures of the indicated strains were serially diluted and spotted into YPD, YPG or YPD + 2.5 �g/ml phleomycin plates andgrown at 30◦C for 4–5 days. (B) DNA content analysis of logarithmically growing WT and mutant strains. (C) Quantification of dNTP pools in indicatedstrains. Data is based on the average of two biological replicates and shown as fold over WT. Numbers on top in green and red indicate the fold change indNTPs relative to WT. (D) Whole-cell lysates of the indicated strains analyzed by western blotting with antibodies against Rnr1–4 subunits. Tubulin wasused as loading control. WT cells treated for 3 h with 200 mM HU were used as positive control for an active DNA damage checkpoint.
as severe as in the met7Δ strain (177-fold increase over WT)(Table 1). These findings indicate that although both thedut1–1 and the met7Δ mutant show increased uracil incor-poration into genomic DNA (detectable in the absence ofUng1) (Figure 2D), met7Δ’s phenotype is by far more se-vere and complex than the phenotype caused by the dut1–1mutation (even in the absence of Dcd1), and therefore can-not be solely explained with a thymineless death model.
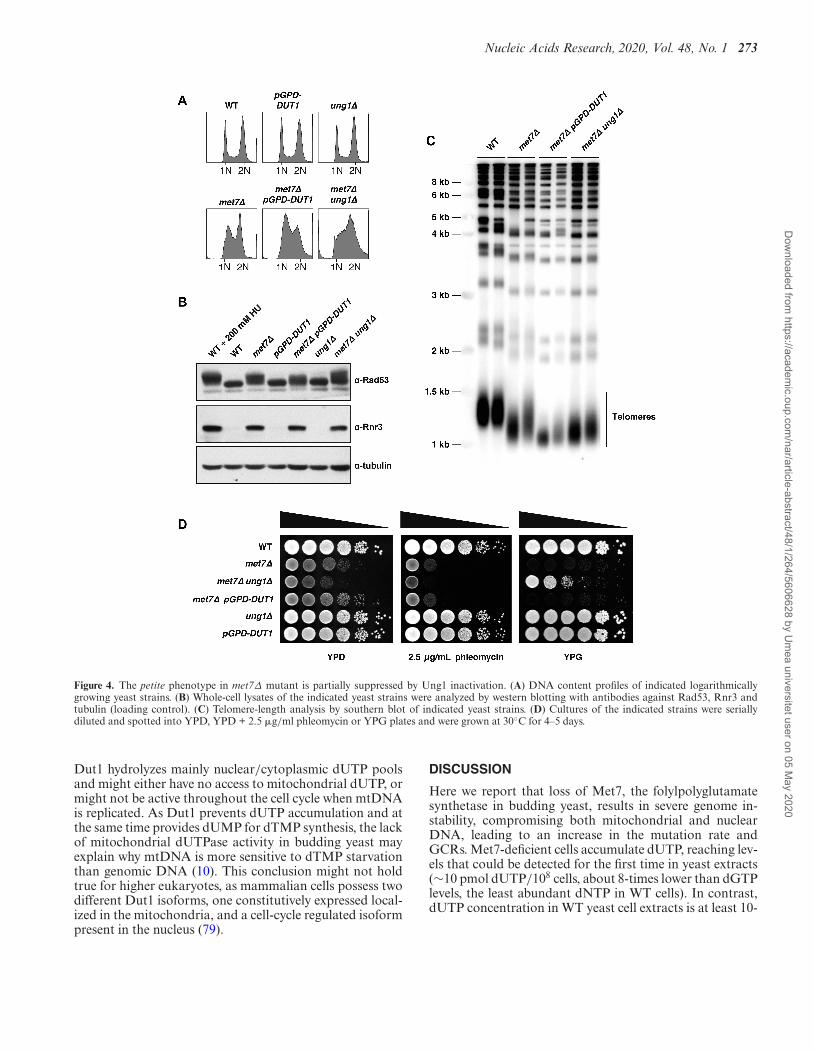
Ung1 inactivation partially suppresses the petite phenotype inthe met7Δ mutant
Our results demonstrated that Dut1-overexpression orUng1 inactivation largely suppressed the elevated GCRrates observed in the met7Δ strain (Table 1). Therefore, weasked whether other aspects of met7Δ phenotype (consti-tutive S-phase checkpoint activation, increased phleomycinsensitivity, short telomeres and petite phenotype) might be
similarly suppressed by Dut1-overexpression and/or Ung1inactivation. To answer this question, we first analyzed theDNA content profiles and the DNA damage response inthe met7Δ strain under conditions that prevent uracil accu-mulation (pGPD-DUT1) or uracil removal (ung1Δ). Inter-estingly, we found that despite the fact that both mutationslargely suppressed GCRs in the met7Δ mutant, neither oneprevented S-phase checkpoint activation (Figure 4A and B).Similarly, short telomere and phleomycin sensitivity phe-notypes of the met7Δ mutant were unchanged upon Dut1-overexpression or UNG1 deletion (Figure 4C and D, respec-tively). Interestingly, the met7Δ ung1Δ double mutant (ob-tained after mating and sporulation), but not the met7ΔpGPD-DUT1 strain, grew in YPG medium (Figure 4D),suggesting that met7Δ’s mitochondrial dysfunction mightbe in part a consequence of uracil misincorporation intomitochondrial DNA (mtDNA) followed by futile repair at-tempts mediated by Ung1. These results also suggest that
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020
Nucleic Acids Research, 2020, Vol. 48, No. 1 273
Figure 4. The petite phenotype in met7Δ mutant is partially suppressed by Ung1 inactivation. (A) DNA content profiles of indicated logarithmicallygrowing yeast strains. (B) Whole-cell lysates of the indicated yeast strains were analyzed by western blotting with antibodies against Rad53, Rnr3 andtubulin (loading control). (C) Telomere-length analysis by southern blot of indicated yeast strains. (D) Cultures of the indicated strains were seriallydiluted and spotted into YPD, YPD + 2.5 �g/ml phleomycin or YPG plates and were grown at 30◦C for 4–5 days.
Dut1 hydrolyzes mainly nuclear/cytoplasmic dUTP poolsand might either have no access to mitochondrial dUTP, ormight not be active throughout the cell cycle when mtDNAis replicated. As Dut1 prevents dUTP accumulation and atthe same time provides dUMP for dTMP synthesis, the lackof mitochondrial dUTPase activity in budding yeast mayexplain why mtDNA is more sensitive to dTMP starvationthan genomic DNA (10). This conclusion might not holdtrue for higher eukaryotes, as mammalian cells possess twodifferent Dut1 isoforms, one constitutively expressed local-ized in the mitochondria, and a cell-cycle regulated isoformpresent in the nucleus (79).
DISCUSSION
Here we report that loss of Met7, the folylpolyglutamatesynthetase in budding yeast, results in severe genome in-stability, compromising both mitochondrial and nuclearDNA, leading to an increase in the mutation rate andGCRs. Met7-deficient cells accumulate dUTP, reaching lev-els that could be detected for the first time in yeast extracts(∼10 pmol dUTP/108 cells, about 8-times lower than dGTPlevels, the least abundant dNTP in WT cells). In contrast,dUTP concentration in WT yeast cell extracts is at least 10-
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020

274 Nucleic Acids Research, 2020, Vol. 48, No. 1
lack of folate polyglutamylation
DSB
increase in dUTP / dTTP ratio
dUTP incorporation
into DNA
excisionby Ung1
abasicsites
ung1∆
DUT1
futile-repaircycles
Loss of Met7 function
folate depletion
reduced dTMP biosynthesis
dUTP accumulation
low dTTP & dGTP
limiting dNTPs
checkpointactivation
inability to increase
dTTP and dGTP pools
DNA repairdefect
dNTP poolimbalance
GCRs
genome instability
mutations
+
?
Figure 5. Met7 contributes to the stability of the genome. Inactivation ofMet7 leads to folate depletion causing limiting dTTP and dGTP and accu-mulation of dUTP. A high dUTP/dTTP ratio promotes uracil incorpora-tion into DNA. Uracil is removed from DNA by Ung1, however the highdUTP/dTTP ratio leads to futile uracil excision/incorporation cycles re-sulting in abasic sites and DSBs. Limiting dNTPs (dTTP and dGTP) mayalso affect DNA repair capacity and increase the frequency of mutations.
times lower (≤1 pmol dUTP/108 cells) and therefore is ex-tremely difficult to quantify. This result is in agreement withseveral studies that could not determine intracellular dUTPlevels in WT yeast or mammalian cells grown under normalconditions, either by HPLC, DNA polymerase-based assaysor mass spectrometry (18–21).
Our experiments also demonstrated that genomic DNAisolated from the met7Δ mutant contained a substantialfraction of uracil indicated by the DNA fragmentation ob-served upon digestion with recombinant UDG+Ape1.
In general, the presence of uracil in DNA can beexplained by two not mutually exclusive mechanisms:an elevated dUTP/dTTP ratio that favors uracilmisincorporation during DNA replication, or thespontaneous/enzymatic deamination of cytosine basesin DNA (77,80). The observation that the met7Δ mutantshowed increased accumulation of intracellular dUTP andreduced dTTP levels, and the fact that Dut1-overexpressionlargely suppressed dUTP accumulation and the presenceof uracil in genomic DNA, strongly indicates that uracildetected in the met7Δ strain is mainly the result of dUTPmisincorporation and not due to cytosine deaminationevents. Moreover, the elevated GCR rates in the met7Δstrain are linked to the removal of misincorporated uracil
from genomic DNA. This is supported by the observa-tion that the increased GCR rates can be suppressed bypreventing uracil accumulation or its excision from DNA.
Given that the met7Δ and the dut1–1 mutants both haveincreased uracil incorporation in DNA but only the formerone showed constitutive activation of the DNA damage re-sponse, it is likely that the reduced dTTP and dGTP con-centrations found in the met7Δ mutant result in the acti-vation of the DNA damage response, rather than uracil in-corporation (or its excision). This hypothesis is further sup-ported by the observation that although Dut1 overexpres-sion in met7Δ prevents dUTP accumulation and GCRs, itneither suppresses the S phase delay nor the DNA damagecheckpoint activation. The fact that the dut1–1 strain, de-spite its massive uracil incorporation, lacks a genome insta-bility phenotype, indicates that uracil incorporation in thedut1–1 mutant is not sufficient to drive genome instability,at least in the two genetic backgrounds tested in this study.
Previous reports in budding yeast have indicated thatMet7 is necessary for the maintenance of an intact mi-tochondrial genome (33); however, given the pleiotropicconsequences caused by the loss of Met7, it remained un-clear how Met7 contributes to mtDNA stability. As Met7supports the production of formyl-methionyl-tRNA re-quired for the initiation of protein biosynthesis in the mito-chondria (1,2), one possible scenario is that mitochondrialgenome instability might arise as a consequence of a defectin mitochondrial protein synthesis. However, our observa-tions in the met7Δ mutant, including the increased uracilincorporation into DNA, together with the partial suppres-sion of its petite phenotype upon inactivation of Ung1,strongly argues that mitochondrial dysfunction is rathercaused by elevated levels of uracil incorporation into DNA.Yet, is the mitochondrial dysfunction a direct consequenceof increased uracil incorporation into mtDNA or might bean indirect effect caused by an unstable nuclear genome?The previous characterization of a thymidylate synthasemutant (cdc21–1), that similar to the met7Δ mutation, pre-vents dTMP biosynthesis and shows increased frequency ofpetite formation (81) supports the first of these two hypothe-ses. The authors found that cdc21–1 mutants accumulateuracil in both genomic and mitochondrial DNA, and thatthe appearance of petites was dependent on the mitochon-drial but not the nuclear isoform of Ung1. Thus, based onthis previous study and our findings, we propose that themitochondrial dysfunction in met7Δ mutant is rather a di-rect consequence of Ung1-dependent uracil excision repaircycles compromising mtDNA integrity.
The reduced telomere length in met7Δ strain is un-likely to be the consequence of DNA damage triggered bythe incorporation of uracil into DNA, as neither Dut1-overexpression nor Ung1 deletion suppress the short telom-ere phenotype. Instead, this phenotype is most likely asso-ciated with limiting dGTP concentrations in this mutantstrain. Previous studies reported a positive correlation be-tween intracellular dGTP levels and both telomere lengthand telomerase processivity in vivo (82,83). Thus, our find-ings showing reduced dGTP levels in the met7Δ mutant thatcorrelate with its short telomere phenotype support the hy-pothesis that telomerase activity is positively regulated invivo by dGTP levels.
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020

Nucleic Acids Research, 2020, Vol. 48, No. 1 275
In this study, we also found that the met7Δ mutantshows a higher percentage of cells containing Ddc2-fociand increased sensitivity to the double-strand break induc-ing agent phleomycin. Together, these observations indi-cate that the met7Δ mutant accumulates endogenous DNAdamage that may overload the DNA repair machinery orpotentially compromise its function in an unrelated man-ner. The increased sensitivity to phleomycin in the met7Δmutant seems not to be related to uracil accumulation, as itis not suppressed by the Dut1-overexpression. Instead, wepropose that phleomycin sensitivity and the DNA repair de-fect (36) in the absence of Met7 are caused by the inabilityto increase dTTP and dGTP levels to facilitate DNA repair(84).
This study sheds light on the understanding of met7Δ’spleiotropic phenotype. Based on our results, we proposethat dNTP limitations observed in the met7Δ mutant giverise to the S-phase delay, the DNA damage checkpoint acti-vation and potentially also for the short telomere phenotypepreviously reported for this mutant (34–36). Our results arein agreement with a model in which the absence of Met7causes reduced folate pools, preventing the conversion ofdUMP into dTMP and consequently the synthesis of dTTP(Figure 5). The severe genome instability phenotype in themet7Δ mutant occurs presumably as a combinatorial effectof imbalanced dNTP pools, that may interfere with DNArepair transactions, and increased accumulation of dUTP,resulting in a high dUTP/dTTP ratio that will cause fre-quent uracil incorporation into DNA, compromising bothmitochondrial and nuclear DNA integrity.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Richard D. Kolodner and Gislene Pereira forstrains and reagents. We are grateful to Annette Kopp-Schneider for advice on statistical analysis. We also thankthe Antibody Unit of the DKFZ Genomics and ProteomicsCore Facility for antibody generation, and Michael Knopand Matthias Meurer for support with the tetrad dissectionanalysis. We are grateful to Frank Exner and Kerstin Griesfor technical assistance.
FUNDING
German Cancer Research Center (DKFZ); Marie Curie In-tegration Grant ‘iMMR’ (both granted to H.H.); SwedishCancer Society, the Knut and Alice Wallenberg Foundationand the Swedish Research Council (granted to A.C.). Fund-ing for open access charge: Deutsches Krebsforschungszen-trum.Conflict of interest statement. None declared.
REFERENCES1. Ducker,G.S. and Rabinowitz,J.D. (2017) One-Carbon metabolism in
health and disease. Cell Metab., 25, 27–42.2. Appling,D.R. (1991) Compartmentation of folate-mediated
one-carbon metabolism in eukaryotes. FASEB J., 5, 2645–2651.
3. Chattopadhyay,S., Moran,R.G. and Goldman,I.D. (2007)Pemetrexed: biochemical and cellular pharmacology, mechanisms,and clinical applications. Mol. Cancer Ther., 6, 404–417.
4. Van Triest,B., Pinedo,H.M., Giaccone,G. and Peters,G.J. (2000)Downstream molecular determinants of response to 5-fluorouraciland antifolate thymidylate synthase inhibitors. Ann. Oncol., 11,385–391.
5. Visentin,M., Zhao,R. and Goldman,I.D. (2012) The antifolates.Hematol. Oncol. Clin. North Am., 26, 629–648.
6. Nzila,A. (2006) The past, present and future of antifolates in thetreatment of Plasmodium falciparum infection. J. Antimicrob.Chemother., 57, 1043–1054.
7. Murima,P., McKinney,J.D. and Pethe,K. (2014) Targeting bacterialcentral metabolism for drug development. Chem. Biol., 21,1423–1432.
8. Gonen,N. and Assaraf,Y.G. (2012) Antifolates in cancer therapy:structure, activity and mechanisms of drug resistance. Drug Resist.Updat., 15, 183–210.
9. Barner,H.D. and Cohen,S.S. (1954) The induction of thyminesynthesis by T2 infection of a thymine requiring mutant ofEscherichia coli. J. Bacteriol., 68, 80–88.
10. Barclay,B.J. and Little,J.G. (1977) Selection of yeast auxotrophs bythymidylate starvation. J. Bacteriol., 132, 1036–1037.
11. Goulian,M., Bleile,B. and Tseng,B.Y. (1980) Methotrexate-inducedmisincorporation of uracil into DNA. Proc. Natl. Acad. Sci. U.S.A.,77, 1956–1960.
12. Khodursky,A., Guzman,E.C. and Hanawalt,P.C. (2015) Thyminelessdeath lives on: new insights into a classic phenomenon. Annu. Rev.Microbiol., 69, 247–263.
13. Guzman,E.C. and Martin,C.M. (2015) Thymineless death, at theorigin. Front Microbiol., 6, 499.
14. Khan,S.R. and Kuzminov,A. (2019) Thymineless death in escherichiacoli is unaffected by chromosomal replication complexity. J.Bacteriol., 201, e00797-18.
15. Goulian,M., Bleile,B.M., Dickey,L.M., Grafstrom,R.H.,Ingraham,H.A., Neynaber,S.A., Peterson,M.S. and Tseng,B.Y. (1986)Mechanism of thymineless death. Adv. Exp. Med. Biol., 195, 89–95.
16. Bessman,M.J., Lehman,I.R., Adler,J., Zimmerman,S.B., Simms,E.S.and Kornberg,A. (1958) Enzymatic synthesis of deoxyribonucleicacid. Iii. The incorporation of pyrimidine and purine analogues intodeoxyribonucleic acid. Proc. Natl. Acad. Sci. U.S.A., 44, 633–640.
17. Berger,S.H., Pittman,D.L. and Wyatt,M.D. (2008) Uracil in DNA:consequences for carcinogenesis and chemotherapy. Biochem.Pharmacol., 76, 697–706.
18. Goulian,M., Bleile,B. and Tseng,B.Y. (1980) The effect ofmethotrexate on levels of dUTP in animal cells. J. Biol. Chem., 255,10630–10637.
19. Zhang,W., Tan,S., Paintsil,E., Dutschman,G.E., Gullen,E.A., Chu,E.and Cheng,Y.C. (2011) Analysis of deoxyribonucleotide pools inhuman cancer cell lines using a liquid chromatography coupled withtandem mass spectrometry technique. Biochem. Pharmacol., 82,411–417.
20. Chen,C.W., Tsao,N., Huang,L.Y., Yen,Y., Liu,X., Lehman,C.,Wang,Y.H., Tseng,M.C., Chen,Y.J., Ho,Y.C. et al. (2016) The impactof dUTPase on ribonucleotide reductase-induced genome instabilityin cancer cells. Cell Rep., 16, 1287–1299.
21. Wilson,P.M., Labonte,M.J., Russell,J., Louie,S., Ghobrial,A.A. andLadner,R.D. (2011) A novel fluorescence-based assay for the rapiddetection and quantification of cellular deoxyribonucleosidetriphosphates. Nucleic Acids Res., 39, e112.
22. Horowitz,R.W., Zhang,H., Schwartz,E.L., Ladner,R.D. andWadler,S. (1997) Measurement of deoxyuridine triphosphate andthymidine triphosphate in the extracts of thymidylatesynthase-inhibited cells using a modified DNA polymerase assay.Biochem. Pharmacol., 54, 635–638.
23. Gadsden,M.H., McIntosh,E.M., Game,J.C., Wilson,P.J. andHaynes,R.H. (1993) dUTP pyrophosphatase is an essential enzyme inSaccharomyces cerevisiae. EMBO J., 12, 4425–4431.
24. Guillet,M., Van Der Kemp,P.A. and Boiteux,S. (2006) dUTPaseactivity is critical to maintain genetic stability in Saccharomycescerevisiae. Nucleic Acids Res., 34, 2056–2066.
25. Taylor,G.R., Lagosky,P.A., Storms,R.K. and Haynes,R.H. (1987)Molecular characterization of the cell cycle-regulated thymidylate
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020

276 Nucleic Acids Research, 2020, Vol. 48, No. 1
synthase gene of Saccharomyces cerevisiae. J. Biol. Chem., 262,5298–5307.
26. Schmidt,T.T., Reyes,G., Gries,K., Ceylan,C.U., Sharma,S.,Meurer,M., Knop,M., Chabes,A. and Hombauer,H. (2017)Alterations in cellular metabolism triggered by URA7 or GLN3inactivation cause imbalanced dNTP pools and increasedmutagenesis. Proc. Natl. Acad. Sci. U.S.A., 114, E4442–E4451.
27. Huang,M.E., Rio,A.G., Nicolas,A. and Kolodner,R.D. (2003) Agenomewide screen in Saccharomyces cerevisiae for genes thatsuppress the accumulation of mutations. Proc. Natl. Acad. Sci.U.S.A., 100, 11529–11534.
28. Smith,S., Hwang,J.Y., Banerjee,S., Majeed,A., Gupta,A. andMyung,K. (2004) Mutator genes for suppression of grosschromosomal rearrangements identified by a genome-wide screeningin Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A., 101,9039–9044.
29. DeSouza,L., Shen,Y. and Bognar,A.L. (2000) Disruption ofcytoplasmic and mitochondrial folylpolyglutamate synthetase activityin Saccharomyces cerevisiae. Arch. Biochem. Biophys., 376, 299–312.
30. McBurney,M.W. and Whitmore,G.F. (1974) Isolation andbiochemical characterization of folate deficient mutants of Chinesehamster cells. Cell, 2, 173–182.
31. Schirch,V. and Strong,W.B. (1989) Interaction of folylpolyglutamateswith enzymes in one-carbon metabolism. Arch. Biochem. Biophys.,269, 371–380.
32. Masselot,M. and De Robichon-Szulmajster,H. (1975) Methioninebiosynthesis in Saccharomyces cerevisiae. I. Genetical analysis ofauxotrophic mutants. Mol. Gen. Genet.: MGG, 139, 121–132.
33. Cherest,H., Thomas,D. and Surdin-Kerjan,Y. (2000)Polyglutamylation of folate coenzymes is necessary for methioninebiosynthesis and maintenance of intact mitochondrial genome inSaccharomyces cerevisiae. J. Biol. Chem., 275, 14056–14063.
34. Askree,S.H., Yehuda,T., Smolikov,S., Gurevich,R., Hawk,J.,Coker,C., Krauskopf,A., Kupiec,M. and McEachern,M.J. (2004) Agenome-wide screen for Saccharomyces cerevisiae deletion mutantsthat affect telomere length. Proc. Natl. Acad. Sci. U.S.A., 101,8658–8663.
35. Gatbonton,T., Imbesi,M., Nelson,M., Akey,J.M., Ruderfer,D.M.,Kruglyak,L., Simon,J.A. and Bedalov,A. (2006) Telomere length as aquantitative trait: genome-wide survey and genetic mapping oftelomere length-control genes in yeast. PLos Genet., 2, e35.
36. Rubinstein,L., Ungar,L., Harari,Y., Babin,V., Ben-Aroya,S.,Merenyi,G., Marjavaara,L., Chabes,A. and Kupiec,M. (2014)Telomere length kinetics assay (TELKA) sorts the telomere lengthmaintenance (tlm) mutants into functional groups. Nucleic AcidsRes., 42, 6314–6325.
37. Amin,N.S., Nguyen,M.N., Oh,S. and Kolodner,R.D. (2001)exo1-Dependent mutator mutations: model system for studyingfunctional interactions in mismatch repair. Mol. Cell Biol., 21,5142–5155.
38. Hombauer,H., Campbell,C.S., Smith,C.E., Desai,A. andKolodner,R.D. (2011) Visualization of eukaryotic DNA mismatchrepair reveals distinct recognition and repair intermediates. Cell, 147,1040–1053.
39. Chen,C. and Kolodner,R.D. (1999) Gross chromosomalrearrangements in Saccharomyces cerevisiae replication andrecombination defective mutants. Nat. Genet., 23, 81–85.
40. Sikorski,R.S. and Hieter,P. (1989) A system of shuttle vectors andyeast host strains designed for efficient manipulation of DNA inSaccharomyces cerevisiae. Genetics, 122, 19–27.
41. Janke,C., Magiera,M.M., Rathfelder,N., Taxis,C., Reber,S.,Maekawa,H., Moreno-Borchart,A., Doenges,G., Schwob,E.,Schiebel,E. et al. (2004) A versatile toolbox for PCR-based tagging ofyeast genes: new fluorescent proteins, more markers and promotersubstitution cassettes. Yeast, 21, 947–962.
42. Reenan,R.A. and Kolodner,R.D. (1992) Characterization of insertionmutations in the Saccharomyces cerevisiae MSH1 and MSH2 genes:evidence for separate mitochondrial and nuclear functions. Genetics,132, 975–985.
43. Marsischky,G.T., Filosi,N., Kane,M.F. and Kolodner,R. (1996)Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 inMSH2-dependent mismatch repair. Genes Dev., 10, 407–420.
44. Putnam,C.D. and Kolodner,R.D. (2010) Determination of grosschromosomal rearrangement rates. Cold Spring Harb. Protoc., 2010,pdb prot5492.
45. Watt,D.L., Buckland,R.J., Lujan,S.A., Kunkel,T.A. and Chabes,A.(2016) Genome-wide analysis of the specificity and mechanisms ofreplication infidelity driven by imbalanced dNTP pools. Nucleic AcidsRes., 44, 1669–1680.
46. Kong,Z., Jia,S., Chabes,A.L., Appelblad,P., Lundmark,R., Moritz,T.and Chabes,A. (2018) Simultaneous determination of ribonucleosideand deoxyribonucleoside triphosphates in biological samples byhydrophilic interaction liquid chromatography coupled with tandemmass spectrometry. Nucleic Acids Res., 46, e66.
47. Hombauer,H., Srivatsan,A., Putnam,C.D. and Kolodner,R.D. (2011)Mismatch repair, but not heteroduplex rejection, is temporallycoupled to DNA replication. Science, 334, 1713–1716.
48. Seiple,L., Jaruga,P., Dizdaroglu,M. and Stivers,J.T. (2006) Linkinguracil base excision repair and 5-fluorouracil toxicity in yeast. NucleicAcids Res., 34, 140–151.
49. Goellner,E.M., Putnam,C.D. and Kolodner,R.D. (2015) Exonuclease1-dependent and independent mismatch repair. DNA Repair (Amst.),32, 24–32.
50. Reyes,G.X., Schmidt,T.T., Kolodner,R.D. and Hombauer,H. (2015)New insights into the mechanism of DNA mismatch repair.Chromosoma, 124, 443–462.
51. Kunkel,T.A. and Erie,D.A. (2015) Eukaryotic mismatch repair inrelation to DNA replication. Annu. Rev. Genet., 49, 291–313.
52. Ben-Aroya,S., Koren,A., Liefshitz,B., Steinlauf,R. and Kupiec,M.(2003) ELG1, a yeast gene required for genome stability, forms acomplex related to replication factor C. Proc. Natl. Acad. Sci. U.S.A.,100, 9906–9911.
53. Kanellis,P., Agyei,R. and Durocher,D. (2003) Elg1 forms analternative PCNA-interacting RFC complex required to maintaingenome stability. Curr. Biol., 13, 1583–1595.
54. Kubota,T., Nishimura,K., Kanemaki,M.T. and Donaldson,A.D.(2013) The Elg1 replication factor C-like complex functions in PCNAunloading during DNA replication. Mol. Cell, 50, 273–280.
55. Howlett,N.G. and Schiestl,R.H. (2004) Nucleotide excision repairdeficiency causes elevated levels of chromosome gain inSaccharomyces cerevisiae. DNA Repair (Amst.), 3, 127–134.
56. Tishkoff,D.X., Boerger,A.L., Bertrand,P., Filosi,N., Gaida,G.M.,Kane,M.F. and Kolodner,R.D. (1997) Identification andcharacterization of Saccharomyces cerevisiae EXO1, a gene encodingan exonuclease that interacts with MSH2. Proc. Natl. Acad. Sci.U.S.A., 94, 7487–7492.
57. Flores-Rozas,H. and Kolodner,R.D. (1998) The Saccharomycescerevisiae MLH3 gene functions in MSH3-dependent suppression offrameshift mutations. Proc. Natl. Acad. Sci. U.S.A., 95, 12404–12409.
58. Bertrand,P., Tishkoff,D.X., Filosi,N., Dasgupta,R. andKolodner,R.D. (1998) Physical interaction between components ofDNA mismatch repair and nucleotide excision repair. Proc. Natl.Acad. Sci. U.S.A., 95, 14278–14283.
59. Scott,A.D., Neishabury,M., Jones,D.H., Reed,S.H., Boiteux,S. andWaters,R. (1999) Spontaneous mutation, oxidative DNA damage,and the roles of base and nucleotide excision repair in the yeastSaccharomyces cerevisiae. Yeast, 15, 205–218.
60. Collura,A., Kemp,P.A. and Boiteux,S. (2012) Abasic sites linked todUTP incorporation in DNA are a major cause of spontaneousmutations in absence of base excision repair and Rad17-Mec3-Ddc1(9-1-1) DNA damage checkpoint clamp in Saccharomyces cerevisiae.DNA Repair (Amst.), 11, 294–303.
61. Brusky,J., Zhu,Y. and Xiao,W. (2000) UBC13, aDNA-damage-inducible gene, is a member of the error-freepostreplication repair pathway in Saccharomyces cerevisiae. Curr.Genet., 37, 168–174.
62. Putnam,C.D., Hayes,T.K. and Kolodner,R.D. (2009) Specificpathways prevent duplication-mediated genome rearrangements.Nature, 460, 984–989.
63. Lisby,M., Barlow,J.H., Burgess,R.C. and Rothstein,R. (2004)Choreography of the DNA damage response: spatiotemporalrelationships among checkpoint and repair proteins. Cell, 118,699–713.
64. Elledge,S.J. and Davis,R.W. (1990) Two genes differentially regulatedin the cell cycle and by DNA-damaging agents encode alternative
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020

Nucleic Acids Research, 2020, Vol. 48, No. 1 277
regulatory subunits of ribonucleotide reductase. Genes Dev., 4,740–751.
65. Kumar,D., Viberg,J., Nilsson,A.K. and Chabes,A. (2010) Highlymutagenic and severely imbalanced dNTP pools can escape detectionby the S-phase checkpoint. Nucleic Acids Res., 38, 3975–3983.
66. Schmidt,T.T., Sharma,S., Reyes,G.X., Gries,K., Gross,M., Zhao,B.,Yuan,J.H., Wade,R., Chabes,A. and Hombauer,H. (2019) A geneticscreen pinpoints ribonucleotide reductase residues that sustain dNTPhomeostasis and specifies a highly mutagenic type of dNTPimbalance. Nucleic Acids Res., 47, 237–252.
67. Trudel,M., Van Genechten,T. and Meuth,M. (1984) Biochemicalcharacterization of the hamster thy mutator gene and its revertants. J.Biol. Chem., 259, 2355–2359.
68. Weinberg,G., Ullman,B. and Martin,D.W. Jr (1981) Mutatorphenotypes in mammalian cell mutants with distinct biochemicaldefects and abnormal deoxyribonucleoside triphosphate pools. Proc.Natl. Acad. Sci. U.S.A., 78, 2447–2451.
69. Tattersall,M.H. and Harrap,K.R. (1973) Changes in thedeoxyribonucleoside triphosphate pools of mouse 5178Y lymphomacells following exposure to methotrexate or 5-fluorouracil. CancerRes., 33, 3086–3090.
70. Ritter,E.J., Scott,W.J., Wilson,J.G., Lampkin,B.C. and Neely,J.E.(1980) Effect of 5-fluoro-2′-deoxyuridine on deoxyribonucleotidepools in vivo. J. Natl. Cancer Inst., 65, 603–605.
71. Yoshioka,A., Tanaka,S., Hiraoka,O., Koyama,Y., Hirota,Y.,Ayusawa,D., Seno,T., Garrett,C. and Wataya,Y. (1987)Deoxyribonucleoside triphosphate imbalance.5-Fluorodeoxyuridine-induced DNA double strand breaks in mouseFM3A cells and the mechanism of cell death. J. Biol. Chem., 262,8235–8241.
72. Tinkelenberg,B.A., Hansbury,M.J. and Ladner,R.D. (2002) dUTPaseand uracil-DNA glycosylase are central modulators of antifolatetoxicity in Saccharomyces cerevisiae. Cancer Res., 62, 4909–4915.
73. Chatterjee,A. and Singh,K.K. (2001) Uracil-DNAglycosylase-deficient yeast exhibit a mitochondrial mutatorphenotype. Nucleic Acids Res., 29, 4935–4940.
74. Burgers,P.M. and Klein,M.B. (1986) Selection by genetictransformation of a Saccharomyces cerevisiae mutant defective forthe nuclear uracil-DNA-glycosylase. J. Bacteriol., 166, 905–913.
75. Boiteux,S. and Jinks-Robertson,S. (2013) DNA repair mechanismsand the bypass of DNA damage in Saccharomyces cerevisiae.Genetics, 193, 1025–1064.
76. Sharma,S., Helchowski,C.M. and Canman,C.E. (2013) The roles ofDNA polymerase zeta and the Y family DNA polymerases inpromoting or preventing genome instability. Mutat. Res., 743-744,97–110.
77. Owiti,N., Stokdyk,K. and Kim,N. (2019) The etiology of uracilresidues in the Saccharomyces cerevisiae genomic DNA. Curr. Genet.,65, 393–399.
78. Kohalmi,S.E., Glattke,M., McIntosh,E.M. and Kunz,B.A. (1991)Mutational specificity of DNA precursor pool imbalances in yeastarising from deoxycytidylate deaminase deficiency or treatment withthymidylate. J. Mol. Biol., 220, 933–946.
79. Ladner,R.D., McNulty,D.E., Carr,S.A., Roberts,G.D. andCaradonna,S.J. (1996) Characterization of distinct nuclear andmitochondrial forms of human deoxyuridine triphosphatenucleotidohydrolase. J. Biol. Chem., 271, 7745–7751.
80. Chon,J., Field,M.S. and Stover,P.J. (2019) Deoxyuracil in DNA anddisease: Genomic signal or managed situation? DNA Repair (Amst.),77, 36–44.
81. Chien,C.Y., Chou,C.K. and Su,J.Y. (2009) Ung1p-mediateduracil-base excision repair in mitochondria is responsible for thepetite formation in thymidylate deficient yeast. FEBS Lett., 583,1499–1504.
82. Gupta,A., Sharma,S., Reichenbach,P., Marjavaara,L., Nilsson,A.K.,Lingner,J., Chabes,A., Rothstein,R. and Chang,M. (2013) Telomerelength homeostasis responds to changes in intracellular dNTP pools.Genetics, 193, 1095–1105.
83. Maicher,A., Gazy,I., Sharma,S., Marjavaara,L., Grinberg,G.,Shemesh,K., Chabes,A. and Kupiec,M. (2017) Rnr1, but not Rnr3,facilitates the sustained telomerase-dependent elongation oftelomeres. PLos Genet., 13, e1007082.
84. Chabes,A., Georgieva,B., Domkin,V., Zhao,X., Rothstein,R. andThelander,L. (2003) Survival of DNA damage in yeast directlydepends on increased dNTP levels allowed by relaxed feedbackinhibition of ribonucleotide reductase. Cell, 112, 391–401.
Dow
nloaded from https://academ
ic.oup.com/nar/article-abstract/48/1/264/5606628 by U
mea universitet user on 05 M
ay 2020