chemerin contributes to inflammation by promoting macrophage
TRANSCRIPT

of February 12, 2018.This information is current as
of VLA-4 and VLA-5VCAM-1 and Fibronectin through ClusteringPromoting Macrophage Adhesion to Chemerin Contributes to Inflammation by
Rosie Hart and David R. Greaves
http://www.jimmunol.org/content/185/6/3728doi: 10.4049/jimmunol.0902154August 2010;
2010; 185:3728-3739; Prepublished online 18J Immunol
MaterialSupplementary
4.DC1http://www.jimmunol.org/content/suppl/2010/08/18/jimmunol.090215
average*
4 weeks from acceptance to publicationSpeedy Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
?The JIWhy
Referenceshttp://www.jimmunol.org/content/185/6/3728.full#ref-list-1
, 30 of which you can access for free at: cites 60 articlesThis article
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2010 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Chemerin Contributes to Inflammation by PromotingMacrophage Adhesion to VCAM-1 and Fibronectin throughClustering of VLA-4 and VLA-5
Rosie Hart and David R. Greaves
Chemerin is a potent macrophage chemoattractant protein.We used murine peritoneal exudate cells (PECs) in adhesion, flow cytom-
etry, andconfocalmicroscopyassays to test thehypothesis that chemerincanalso contribute to inflammationbypromotingmacrophage
adhesion. Chemerin stimulated the adhesion of PECs to the extracellular matrix protein fibronectin and to the adhesion molecule
VCAM-1 within a minute, with an EC50 of 322 and 196 pM, respectively. Experiments using pertussis toxin and PECs from
ChemR232/2 mice demonstrated that chemerin stimulated the adhesion of macrophages via the Gi protein-coupled receptor
ChemR23. Blocking Abs against integrin subunits revealed that 89% of chemerin-stimulated adhesion to fibronectin was dependent
on increased avidity of the integrin VLA-5 (a5b1) and that 88% of adhesion to VCAM-1 was dependent on increased avidity of VLA-
4 (a4b1). Although chemerin was unable to induce an increase in integrin affinity as judged by the binding of soluble ligand,
experiments using confocal microscopy revealed an increase in valency resulting from integrin clustering as the mechanism re-
sponsible for chemerin-stimulated macrophage adhesion. PI3K, Akt, and p38 were identified as key signaling mediators in chemerin-
stimulated adhesion. The finding that chemerin can rapidly stimulate macrophage adhesion to extracellular matrix proteins and
adhesion molecules, taken together with its ability to promote chemotaxis, suggests a novel role for chemerin in the recruitment and
retention of macrophages at sites of inflammation. The Journal of Immunology, 2010, 185: 3728–3739.
The coordinated recruitment of leukocytes to sites of tissueinjury is central to the generation of a successful in-flammatory response (1). The directed migration of leu-
kocytes is regulated by a number of inflammatory molecules in-cluding chemokines, cytokines and complement proteins (2).Chemerin was originally isolated from human inflammatory exu-
dates as the protein ligand for the orphan Gi-linked G protein-coupled receptor (GPCR) ChemR23 (3). ChemR23 is structurallyrelated to chemokine receptors, and accordingly, chemerin was dis-covered to possess the chemoattractant abilities of a chemokine (3,4). Chemerin is chemotactic for ChemR23-expressing human mac-rophages, plasmacytoid dendritic cells (pDCs), and NK cells (3, 5–7). ChemR23 is also expressed on human monocytes (6). In the mo-use, macrophages express ChemR23 and migrate toward chemerin(8, 9). Recently, murine pDCs and NK cells were also shown to beChemR23+ (10). Unlike the majority of chemokines, chemerin issecreted in an inactive precursor form (3). It requires cleavage of theC terminus by serine proteases of the coagulation, fibrinolytic, andinflammatory cascades to become active as a chemoattractant forcells expressing ChemR23 (11–13). Moreover, chemerin’s predictedstructure, thought to be the reverse orientation of that of chemokines,distinguishes it from this family of proteins (14).
In addition to mediating chemotaxis, many chemoattractants,including chemokines, are also able to induce the adhesion ofleukocytes to adhesion molecules, such as VCAM-1 and ICAM-1,and to extracellular matrix proteins, such as fibronectin, laminin,and collagen IV (15). They induce adhesion by stimulating anincrease in the affinity and/or the valency of integrin receptorsexpressed on the cell surface (16). Leukocyte adhesion plays a keyrole in inflammation, contributing to leukocyte rolling and arreston the vascular endothelium, migration through the extracellularmatrix to the site of inflammation, retention of leukocytes withinthe inflammatory site, and leukocyte activation (17–22).In this study, we hypothesized that chemerin may contribute to
inflammation not only by mediating macrophage chemotaxis, butalso by promoting the adhesion of macrophages to extracellularmatrix proteins and adhesion molecules.
Materials and MethodsAnimals
All animal experiments were performedwith ethical approval from theDunnSchool of Pathology Local Ethical Review Committee and in accordancewith the U.K. Home Office regulations (Guidance on the Operation ofAnimals, Scientific Procedures Act, 1986).
Reagents
Collagen IV, laminin and fibronectin were obtained Scientific LaboratorySupplies (Nottingham, U.K.). ICAM-1, VCAM-1, and VCAM-1-Fc werefrom R&D Systems (Abingdon, U.K.). The FITC-conjugated GRGDSPpeptidewas purchased fromAnaSpec (Fremont, CA), aswere all the blockingAbs and their isotype controls as follows: rat IgG1 (clone RTK2071), ratIgG2a (cloneRTK2758), rat IgG2b (cloneRTK4530), Armenian hamster IgG(clone HTK888), anti-mouse CD18 (b2; clone M18/2), anti-mouse/ratCD29 (b1; clone HMb1-1), anti-mouse CD49d (a4; clone R1-2), anti-mouseCD49e (a5; clone 5H10-27), anti-mouse CD51 (av; clone RMV-7), and anti-mouse integrin b7 (clone FIB27). Of the inhibitors, Akt inhibitor IV, cyto-chalasin D, LY294002, and SB202190 were purchased from Sigma-Aldrich(Gillingham, U.K.), BAPTA-AM and pertussis toxin (PTX) were from Cal-biochem (Nottingham, U.K.), piceatannol and PP2 were from Tocris Bio-science (Bristol, U.K.), and U0126 was obtained from New England Biolabs
Sir William Dunn School of Pathology, University of Oxford, Oxford, United King-dom
Received for publication July 6, 2009. Accepted for publication July 9, 2010.
This work was supported by British Heart Foundation Grant FS/06/080.
Address correspondence and reprint requests to Dr. David R. Greaves, Sir WilliamDunn School of Pathology, University of Oxford, South Parks Road, Oxford, OxonOX1 3RE, U.K. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this paper: GPCR, G protein-coupled receptor; i.c., isotypecontrols; LPAM, lymphocyte Peyer’s patch adhesion molecule; pDC, plasmacytoiddendritic cell; PEC, peritoneal exudate cell; PTX, pertussis toxin; WT, wild-type.
Copyright� 2010 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0902154
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

(Hitchin,U.K.).Anti-mouseCD16/CD32 (clone FCR4G8), FITC-conjugatedanti-mouse neutrophils (clone 7/4), Alexa Fluor 647-conjugated anti-mouseF4/80 (clone CI:A3-1), FITC-conjugated anti-mouse CD49d (clone MFR5),Pacific blue-conjugated anti-mouse F4/80 (clone CI:A3-1), and Alexa Fluor647-conjugated anti-mouse CD206 (clone MR5D3) were from Serotec(Oxford, U.K.). PE-conjugated anti-mouse ChemR23 (clone BZ194), PE-conjugated rat IgG2a isotype control (clone eBR2a), FITC-conjugated anti-mouse CD49d (clone R1-2), FITC-conjugated rat IgG2a (clone eBR2a) andIgG2b (clone eB149/10H5), and FITC-conjugated anti-mouse CD80 (clone16-10A1) were purchased from eBioscience (Hatfield, U.K.). IFN-g and IL-4were from PeproTech (London, U.K.), and LPS was from Sigma-Aldrich.Murine recombinant chemerin was obtained from R&D Systems, C15 wassynthesized by Biosynthesis (Lewisville, TX), and C9 was a gift fromH. Wang and T. Schall (ChemoCentryx, Mountain View, CA).
Cells
To obtain peritoneal exudate cells (PECs), Bio-Gel P100 polyacrylamidebeads (1 ml 2% w/v in PBS; Bio-Rad, Hemel Hempstead, U.K.) wereinjected into the peritoneal cavities of 8- to 12-wk-old C57BL/6J mice,Sv129 mice, or ChemR232/2 mice on an Sv129 background. Four dayslater, the mice were killed using a Schedule 1 method, and the peritonealcavities were lavaged with 10 ml ice-cold PBS (Lonza, Slough, U.K.) plus 2mM EDTA (Sigma-Aldrich). The resulting cells were used immediately,except for the polarized macrophage experiments in which 23 106 cells/mlPECs in RPMI 1640 (PAA Laboratories, Yeovil, U.K.) supplemented with1% FBS (PAA Laboratories) 0.1% BSA (Sigma-Aldrich), 25 mM HEPES(PAA Laboratories), 50 U/ml penicillin, and 50 mg/ml streptomycin (Invi-trogen, Paisley, U.K.) were incubated overnight in polypropylene tubesat 37˚C. Cells were polarized by the addition of 20 ng/ml IFN-g, 20ng/ml IFN-g plus 100 ng/ml LPS, or 20 ng/ml IL-4 to the media. The nextday the cells were washed twice before use in further experiments.
BaF3 cells stably transfected with the murine ChemR23 receptor(BaF3-ChemR23) were a gift from H. Wang and T. Schall (ChemoCentryx).ChemR23 expression was confirmed by flow cytometry using an anti-mouseChemR23 Ab (data not shown). Wild-type (WT) BaF3 cells and BaF3-ChemR23 cells were cultured in RPMI 1640 supplemented with 10% FBS,10% WEHI conditioned medium, 50 mM 2-ME (Sigma-Aldrich), 50 U/mlpenicillin, and 50 mg/ml streptomycin. BaF3 cells were serum starved inRPMI 1640 supplemented with 0.1% BSA and 25 mM HEPES for 24 hbefore their use in adhesion assays.
Adhesion assay
Ninety-six-well cell culture plates (Corning, Amsterdam, The Netherlands)were coated overnight at 4˚C with 30 mg/ml collagen IV, 30 mg/ml fibro-nectin, 30 mg/ml laminin, 2 mg/ml ICAM-1, or 2 mg/ml VCAM-1. Theplates were washed twice with PBS, and nonspecific binding sites wereblocked with 1% BSA in PBS for 1 h at 37˚C, before an additional threewashes with assay buffer (RPMI 1640 supplemented with 0.1% BSA and25 mM HEPES). PECs or BaF3 cells were resuspended in assay buffer, and50 ml of cell suspension in the absence or presence of chemerin (10 nMunless otherwise stated), C9, or C15 peptide (0.001 nM to 1 mM) was addedto each well of the plate (23 105 cells/well). Plates were incubated at 37˚Cfor 1 min unless otherwise specified, before 20 s of shaking on an orbitalshaker, and three washes with assay buffer to remove nonadherent cells.Adherent cells were quantified using CellTiter-Glo (Promega, South-ampton, U.K.) in a white luminometer plate (Greiner Bio-One, Stonehouse,U.K.), according to the manufacturer’s instructions and a SpectraMax M5microplate reader (Molecular Devices, Wokingham, U.K.).
For adhesion assays using blocking Abs, PECs were blocked with 10mg/ml anti-mouse CD16/CD32 for 15 min at 37˚C, followed by a 30-minincubation at 37˚C with 3 mg/ml blocking Ab or its isotype control. Cellswere then added to the coated plate as described previously.
For adhesion assays using inhibitors, PECswere pretreated for 1 h at roomtemperature with 0.015 mg/ml cytochalasin D, 1 mMAkt inhibitor IV, 5 mMBAPTA-AM, 5 mM LY294002, 20 mM piceatannol, 10 mM PP2, 20 mMSB202190, 20mMU0126, or combinations of these inhibitors. For adhesionassays using PTX, PECs were pretreated with 200 ng/ml PTX for 1 hat 37˚C. To ensure that these concentrations of inhibitors were not cytotoxic,CellTiter-Glo was used according to the manufacturer’s instructions tocompare the number of live untreated PECs with the number of live PECsthat had been treated with inhibitors as described previously.
Flow cytometry
To assess ChemR23 expression on PECs, 5 3 105 PECs were resuspendedin assay buffer (PBS supplemented with 5% FBS, 25 mM HEPES, and5 mM EDTA), blocked with 10 mg/ml anti-mouse CD16/CD32 for 15 minon ice, stained with 0.8 mg/ml FITC-conjugated anti-mouse 7/4, 1 mg/ml
Alexa Fluor 647-conjugated anti-mouse F4/80 and either 3 mg/ml PE-conjugated anti-mouse ChemR23 or 3 mg/ml PE-conjugated rat IgG2aisotype control for 30 min on ice, washed with assay buffer, and fixed with2% formaldehyde (Sigma-Aldrich) in PBS. Cells were analyzed on a CyanADP flow cytometer (Dako, Ely, U.K.). Overnight-treated PECs were alsostained with 3 mg/ml FITC-conjugated anti-mouse CD80 or 1 mg/ml AlexaFluor 647-conjugated anti-mouse CD206 in combination with 2 mg/mlPacific blue-conjugated anti-mouse F4/80.
To measure the binding of a fibronectin peptide to macrophages, PECsfrom C57BL/6J mice were resuspended in assay buffer (HBSS [Invit-rogen] with CaCl2 and MgCl2 supplemented with 0.1% BSA and 20 mMHEPES) to a concentration of 5 3 106 cells/ml, blocked with 10 mg/mlanti-mouse CD16/CD32 for 15 min on ice, and stained with 1 mg/ml AlexaFluor 647-conjugated anti-mouse F4/80 for 30 min on ice. FITC-labeledGRGDSP peptide (10 mM) and either assay buffer 10 mM MnCl2 (Sigma-Aldrich) or 10 nM chemerin were added to 100 ml of the stained PECs andincubated at 37˚C for up to 5 min. Tubes were immediately placed on ice tostop the reaction and washed once with assay buffer. Cells were fixed using2% paraformaldehyde (Sigma-Aldrich) in PBS on ice for 1 h. Samples wereanalyzed by flow cytometry on a Cyan ADP flow cytometer.
To measure the binding of VCAM-1-Fc to macrophages, PECs wereresuspended in assay buffer to a concentration of 5 3 106 cells/ml and100 mg/ml VCAM-1-Fc plus either assay buffer 10 mM MnCl2 or 10 nMchemerin were added to 10ml of these cells and incubated at 37˚C for up to 2min. Tubes were immediately placed on ice to stop the reaction and the cellswere fixed with 2% paraformaldehyde in PBS on ice for 1 h. Samples werewashed twice with assay buffer, blocked with 10 mg/ml anti-mouseCD16/CD32 for 15 min on ice, stained with 1 mg/ml Alexa Fluor 647-con-jugated anti-mouse F4/80 and a 1/100 dilution of PE-conjugated anti-humanIgG (Jackson ImmunoResearch Laboratories, Newmarket, U.K.) for 30 minon ice and washed with assay buffer before resuspension in 2% para-formaldehyde in PBS and analysis by flow cytometry on Cyan ADP flowcytometer.
Confocal microscopy
To visualize clustering of the a4 or a5 integrins on macrophages, PECs fromC57BL/6J mice were resuspended in ice-cold RPMI 1640 supplementedwith 0.1% BSA and 25 mM HEPES to a concentration of 83 106 cells/ml.After addition of either media, 100 ng/ml PMA or 10 nM chemerin, the cellswere incubated at 37˚C for 1 min. Tubes were then immediately placed onice to stop the reaction and the cells were fixedwith 2% paraformaldehyde inPBS on ice for 20min. Samples werewashed twicewith assay buffer (HBSSwith CaCl2 and MgCl2 supplemented with 5% FCS, 0.5% BSA, and 20 mMHEPES), blocked with 10 mg/ml anti-mouse CD16/CD32 for 30 min on ice,and stained overnight at 4˚C with 2.5 mg/ml FITC-conjugated anti-mouseCD49d (a4) or CD49e (a5) or their isotype controls (FITC-conjugated ratIgG2b or IgG2a, respectively) in combination with 1 mg/ml Alexa Fluor647-conjugated anti-mouse F4/80. The next day the cells were washed oncein assay buffer then added to wells of a Lab-Tek chamber slide (Cole-Parmer, London, U.K.) that had previously been coated with 100 mg/mlpoly-D-lysine (Sigma-Aldrich) for 1 h at room temperature, washed twotimes with sterile dH2O, and left to air-dry. The cells were allowed to adherefor 1 h at room temperature before thewells werewashed an additional threetimes with assay buffer, and the slides were mounted in Dako fluorescentmounting medium. The cells were analyzed using a Carl Zeiss LSM 510confocal laser scanning systemwith a 633 Plan-Apochromat objective (NA1.4). Macrophages were identified as those cells with an average fluores-cence intensity for F4/80 exceeding a predefined lower limit.
RT-PCR analysis
Total RNA was prepared from freshly obtained PECs using TRIzol (Invi-trogen), isolated using an RNeasy kit (Qiagen, Crawley, U.K.), and reversetranscribed using Moloney murine leukemia virus reverse transcriptase andrandomprimers (Promega).RT-PCRanalysis forGAPDH,ChemR23,GPR1,CCRL2, and BLT1 was performed using SYBRGreen (Qiagen) and thefollowing primers: GAPDH, 59-TGTGTCCGTCGTGGATCTGA-39 (for-ward) and 59-ACCACCTTCTTGATGTCATCATACTT-39 (reverse); Chem-R23, 59-ACACACTGTTCATCAGTAGCAA-39 (forward) and 59-CCTCC-ATGAGGAATGATGTCAA-39 (reverse); GPR1, 59-CTACGTGGCCTTG-AGTTTCCA-39 (forward) and 59-CAGTTGGGCAATGAAGGAATTAA-39 (reverse); CCRL2, 59-ACGGGCGTGTTCCTTTTG-39 (forward) and 59-TCCTGGAAAGCAGACAGGAAA-39 (reverse); and BLT1, 59-TTGCT-CACTGCTCCCTTTTTC-39 (forward) and 59-GCGGCAACCCATCTCT-CTAA-39 (reverse). The number of copies of receptor mRNA per copy ofGAPDH mRNAwas quantified from a standard curve using mouse geno-mic DNA (Promega).
The Journal of Immunology 3729
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Statistical analysis
One-way ANOVA (with Dunnett’s or Bonferroni’s posthoc test) andtwo-way ANOVA (with Bonferonni’s posthoc test) were performed asappropriate using GraphPad Prism.
ResultsChemerin rapidly induces adhesion to fibronectin and VCAM-1
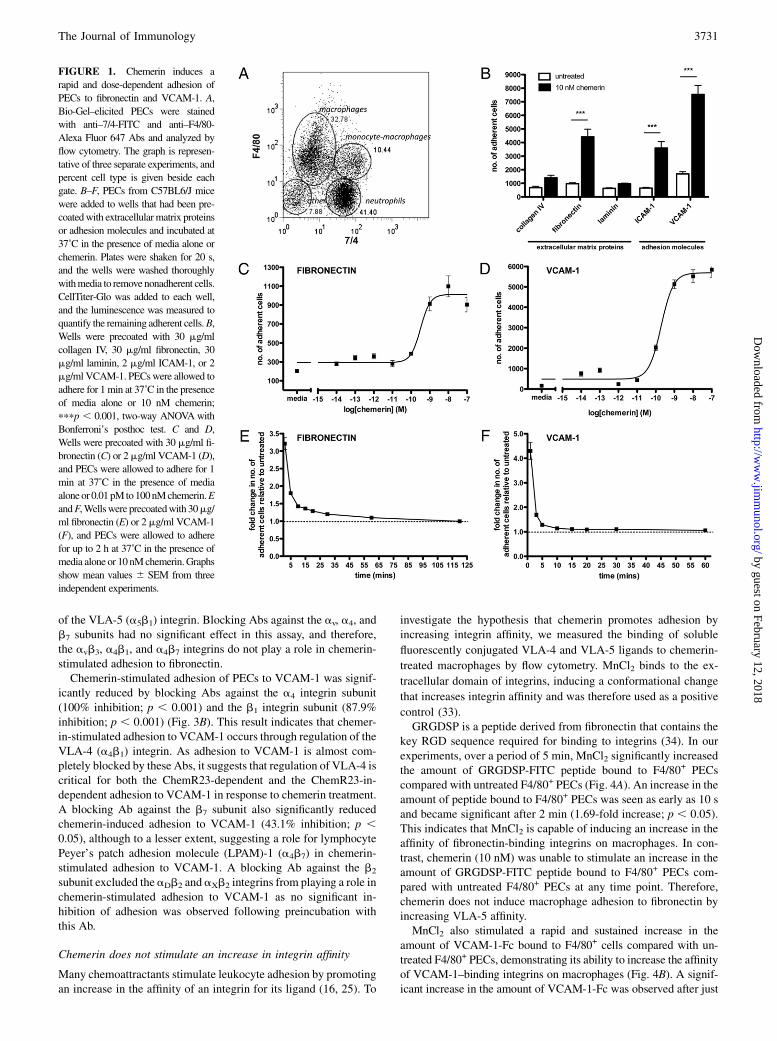
To investigate chemerin’s ability to induce macrophage adhesion toextracellular matrix proteins and recombinant adhesion molecules,we used Bio-Gel–elicited PECs, which are a good source of min-imally activated macrophages, in a static adhesion assay. Bio-Gel–elicited PECs were composed of an average of 33% macrophagesand 10% immature monocyte-macrophages as determined by theirexpression of 7/4 and F4/80 Ags (Fig. 1A).PECs were allowed to adhere to plates coated with collagen IV,
fibronectin, laminin, ICAM-1, or VCAM-1 in the presence or ab-sence of chemerin.Treatment ofPECswith10nMchemerin resultedin a significant increase in thenumberof adherent cells onfibronectin(4.6-fold; p, 0.001), ICAM-1 (5.6-fold; p, 0.001), and VCAM-1(4.5-fold; p , 0.001) (Fig. 1B). Chemerin was not able to inducePEC adhesion to collagen IV or laminin. Chemerin stimulated theadhesion of PECs to fibronectin (Fig. 1C) and VCAM-1 (Fig. 1D) ina dose-dependent manner with maximal effects achieved at 10 nMchemerin. This corresponds well with chemerin’s chemotactic ac-tivity (8). Chemerin had an EC50 value of 322 and 196 pM for celladhesion to fibronectin and VCAM-1, respectively.Chemerin’s effects on adhesion were very rapid. The greatest
increase in the number of adherent cells compared with untreatedoccurred at 1 min, the earliest time point that we could accuratelyquantify, on both fibronectin (Fig. 1E) and VCAM-1 (Fig. 1F).
Chemerin stimulates the adhesion of macrophages throughChemR23
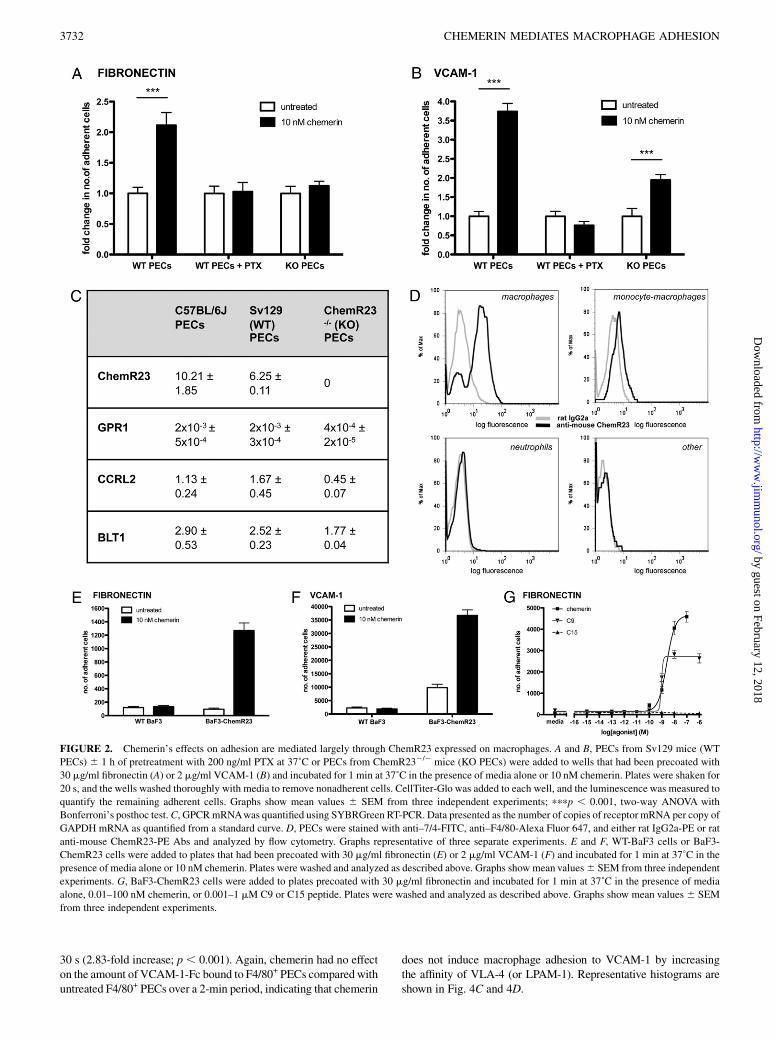
The effects of chemoattractants on adhesion are mediated throughGi-coupled GPCRs (23–26). To determinewhether this was true forchemerin’s effects, PECs from WT Sv129 mice were pretreatedwith PTX. Chemerin induced a significant increase in the adhesionof PECs from WT Sv129 mice to fibronectin (2.1-fold; p , 0.001)(Fig. 2A) and VCAM-1 (3.7-fold; p, 0.001) (Fig. 2B). Chemerin’seffects on cell adhesion to fibronectin and VCAM-1 were com-pletely inhibited by PTX pretreatment, demonstrating the require-ment for a Gi-coupled GPCR (Fig. 2A, 2B).Bio-Gel–elicited peritoneal macrophages from ChemR232/2
mice display no chemotaxis toward chemerin (8). To investigatewhether chemerin-stimulated adhesion is mediated via ChemR23,we assessed the ability of chemerin to stimulate the adhesion ofPECs from Sv129 ChemR232/2 mice (KO PECs) to both fibro-nectin and VCAM-1. Chemerin was unable to promote the adhe-sion of PECs from ChemR232/2 mice to fibronectin (Fig. 2A).Therefore, chemerin-stimulated adhesion to fibronectin is depen-dent on ChemR23 and its coupling to a Gi G protein. However,a significant number of PECs from ChemR232/2 mice were stillcapable of adhering to VCAM-1 (1.9-fold increase; p , 0.001).These results suggest that although chemerin-stimulated adhesionto VCAM-1 is dependent on activation of a Gi-coupled GPCR, justover half of this activity is independent of ChemR23.Quantitative RT-PCR analysis of PECs from C57BL/6J mice,
Sv129 mice, and Sv129 ChemR232/2mice showed minimal levelsof mRNA for GPR1 (0.02–0.03% of ChemR23 mRNA expression),another GPCR shown to bind chemerin (Fig. 2C) (27). Low levelsof mRNA for CCRL2 (11.1–26.7% of ChemR23 mRNA expres-sion), a third nonsignaling receptor for chemerin, were expressed inPECs from C57BL/6J mice, Sv129 mice, and Sv129 ChemR232/2
mice (28). PECs from these three strains of mouse also expressedleukotriene B4 receptor (BLT1) mRNA (28.4–40.3% of ChemR23mRNA expression).Only macrophage and monocyte-macrophage PECs expressed
ChemR23 as determined by flow cytometry (Fig. 2D). Therefore weconcluded that the cells that adhere to fibronectin and VCAM-1 ina ChemR23-dependent manner in response to chemerin treatmentare macrophages and monocyte-macrophages.To test the effect of macrophage polarization on chemerin-
stimulated adhesion, we pretreated PECs with LPS and/or variouscytokines. Macrophages that were treated overnight with IFN-galone expressed increased levels of CD80, a marker of classicallyactivated M1 macrophages (Supplemental Fig. 1A) and the samelevel of ChemR23 expression as untreated cells (Supplemental Fig.1B). Overnight treatment of macrophages with IFN-g plus LPSupregulated CD80 expression to a greater extent than IFN-g alonebut completely downregulated ChemR23 expression. Consistentwith the ChemR23 expression of these macrophage populations,IFN-g–treated cells retained their ability to adhere to fibronectinand VCAM-1 in response to chemerin stimulation, whereas IFN-gplus LPS-stimulated cells lost this activity (Supplemental Fig. 2).Macrophages treated overnight with IL-4 displayed an upregulationin expression of the mannose receptor, the classic marker of al-ternatively activatedM2amacrophages (Supplemental Fig. 1A) andhad the same level of ChemR23 expression as untreated cells(Supplemental Fig. 1B). However, in the adhesion assay, IL-4–treated cells did not adhere to fibronectin or VCAM-1 in response tochemerin stimulation (Supplemental Fig. 2).To provide further evidence that chemerin is capable of promoting
cell adhesion to fibronectin and VCAM-1 solely through ChemR23,we used BaF3 cells (a pro-B cell line) stably transfected withChemR23 in the adhesion assay. Chemerin (10 nM) was capable ofstimulating the adhesion of these cells to both fibronectin (Fig. 2E)and to VCAM-1 (Fig. 2F), but it was not able to induce the adhesionof WT BaF3 cells that did not express the ChemR23 receptor(Fig. 2E, 2F). We also tested whether two peptides derived fromthe C terminus of murine chemerin were able to promote adhesionof the BaF3-ChemR23 cells to fibronectin. These peptides hadboth been previously shown to induce ChemR23-dependent activ-ity (8, 10, 29). C9 (FLPGQFAFS) was a partial agonist, capable ofproducing 58.6% of the adhesive activity of full-length chemerin(Fig. 2G). However, it was slightly more potent than chemerinwith an EC50 of 0.94 nM compared with 2.48 nM for chemerin.Peptide C15 (AGEDPHGYFLPGQFA) was unable to promote theadhesion of BaF3-ChemR23 cells to fibronectin at any concen-tration tested.
Chemerin-stimulated adhesion to fibronectin and VCAM-1 ismediated through the VLA-5 (a5b1) and VLA-4 (a4b1)integrins, respectively
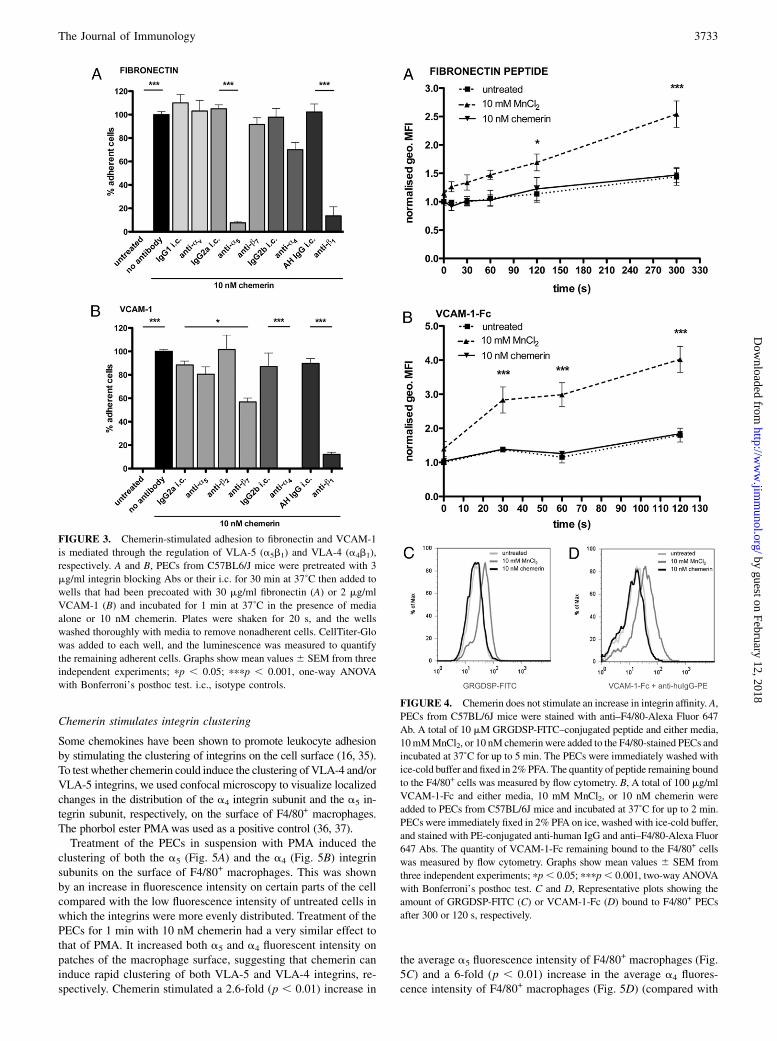
Leukocytes bind to extracellular matrix proteins and adhesionmolecules, such as fibronectin and VCAM-1, through integrinsexpressed on their surface (30). The adhesion of macrophages tofibronectin is mediated via the integrins a4b1, a5b1, avb3, anda4b7, whereas a4b1, aDb2, aXb2, and a4b7 are involved in mac-rophage adhesion to VCAM-1 (31, 32). To identify which of theseintegrins mediates chemerin-induced adhesion to fibronectin andVCAM-1, PECs were pretreated with blocking Abs against theintegrin subunits that constitute the candidate integrins.Chemerin-induced adhesion to fibronectin was significantly
inhibited by Abs that blocked the a5 integrin subunit (92.5% in-hibition; p , 0.001) and the b1 integrin subunit (86.4% inhibition;p , 0.001) (Fig. 3A). This result demonstrates that chemerin stim-ulates the adhesion ofmacrophages to fibronectin through regulation
3730 CHEMERIN MEDIATES MACROPHAGE ADHESION
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
of the VLA-5 (a5b1) integrin. Blocking Abs against the av, a4, andb7 subunits had no significant effect in this assay, and therefore,the avb3, a4b1, and a4b7 integrins do not play a role in chemerin-stimulated adhesion to fibronectin.Chemerin-stimulated adhesion of PECs to VCAM-1 was signif-
icantly reduced by blocking Abs against the a4 integrin subunit(100% inhibition; p , 0.001) and the b1 integrin subunit (87.9%inhibition; p , 0.001) (Fig. 3B). This result indicates that chemer-in-stimulated adhesion to VCAM-1 occurs through regulation of theVLA-4 (a4b1) integrin. As adhesion to VCAM-1 is almost com-pletely blocked by these Abs, it suggests that regulation of VLA-4 iscritical for both the ChemR23-dependent and the ChemR23-in-dependent adhesion to VCAM-1 in response to chemerin treatment.A blocking Ab against the b7 subunit also significantly reducedchemerin-induced adhesion to VCAM-1 (43.1% inhibition; p ,0.05), although to a lesser extent, suggesting a role for lymphocytePeyer’s patch adhesion molecule (LPAM)-1 (a4b7) in chemerin-stimulated adhesion to VCAM-1. A blocking Ab against the b2
subunit excluded theaDb2 and aXb2 integrins from playing a role inchemerin-stimulated adhesion to VCAM-1 as no significant in-hibition of adhesion was observed following preincubation withthis Ab.
Chemerin does not stimulate an increase in integrin affinity
Many chemoattractants stimulate leukocyte adhesion by promotingan increase in the affinity of an integrin for its ligand (16, 25). To
investigate the hypothesis that chemerin promotes adhesion byincreasing integrin affinity, we measured the binding of soluble
fluorescently conjugated VLA-4 and VLA-5 ligands to chemerin-
treated macrophages by flow cytometry. MnCl2 binds to the ex-
tracellular domain of integrins, inducing a conformational change
that increases integrin affinity and was therefore used as a positive
control (33).GRGDSP is a peptide derived from fibronectin that contains the
key RGD sequence required for binding to integrins (34). In ourexperiments, over a period of 5 min, MnCl2 significantly increasedthe amount of GRGDSP-FITC peptide bound to F4/80+ PECscompared with untreated F4/80+ PECs (Fig. 4A). An increase in theamount of peptide bound to F4/80+ PECs was seen as early as 10 sand became significant after 2 min (1.69-fold increase; p , 0.05).This indicates that MnCl2 is capable of inducing an increase in theaffinity of fibronectin-binding integrins on macrophages. In con-trast, chemerin (10 nM) was unable to stimulate an increase in theamount of GRGDSP-FITC peptide bound to F4/80+ PECs com-pared with untreated F4/80+ PECs at any time point. Therefore,chemerin does not induce macrophage adhesion to fibronectin byincreasing VLA-5 affinity.MnCl2 also stimulated a rapid and sustained increase in the
amount of VCAM-1-Fc bound to F4/80+ cells compared with un-treated F4/80+ PECs, demonstrating its ability to increase the affinityof VCAM-1–binding integrins on macrophages (Fig. 4B). A signif-icant increase in the amount of VCAM-1-Fc was observed after just
FIGURE 1. Chemerin induces a
rapid and dose-dependent adhesion of
PECs to fibronectin and VCAM-1. A,
Bio-Gel–elicited PECs were stained
with anti–7/4-FITC and anti–F4/80-
Alexa Fluor 647 Abs and analyzed by
flow cytometry. The graph is represen-
tative of three separate experiments, and
percent cell type is given beside each
gate. B–F, PECs from C57BL6/J mice
were added to wells that had been pre-
coatedwith extracellularmatrix proteins
or adhesion molecules and incubated at
37˚C in the presence of media alone or
chemerin. Plates were shaken for 20 s,
and the wells were washed thoroughly
withmedia to removenonadherent cells.
CellTiter-Glo was added to each well,
and the luminescence was measured to
quantify the remaining adherent cells.B,
Wells were precoated with 30 mg/ml
collagen IV, 30 mg/ml fibronectin, 30
mg/ml laminin, 2 mg/ml ICAM-1, or 2
mg/ml VCAM-1. PECswere allowed to
adhere for 1min at 37˚C in the presence
of media alone or 10 nM chemerin;
pppp , 0.001, two-way ANOVAwith
Bonferroni’s posthoc test. C and D,
Wells were precoated with 30 mg/ml fi-
bronectin (C) or 2mg/ml VCAM-1 (D),
and PECs were allowed to adhere for 1
min at 37˚C in the presence of media
aloneor 0.01pMto100nMchemerin.E
andF,Wellswere precoatedwith 30mg/
ml fibronectin (E) or 2mg/ml VCAM-1
(F), and PECs were allowed to adhere
for up to 2 h at 37˚C in the presence of
media alone or 10 nMchemerin.Graphs
show mean values 6 SEM from three
independent experiments.
The Journal of Immunology 3731
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
30 s (2.83-fold increase; p , 0.001). Again, chemerin had no effecton the amount of VCAM-1-Fc bound to F4/80+ PECs compared withuntreated F4/80+ PECs over a 2-min period, indicating that chemerin
does not induce macrophage adhesion to VCAM-1 by increasingthe affinity of VLA-4 (or LPAM-1). Representative histograms areshown in Fig. 4C and 4D.
FIGURE 2. Chemerin’s effects on adhesion are mediated largely through ChemR23 expressed on macrophages. A and B, PECs from Sv129 mice (WT
PECs) 6 1 h of pretreatment with 200 ng/ml PTX at 37˚C or PECs from ChemR232/2 mice (KO PECs) were added to wells that had been precoated with
30 mg/ml fibronectin (A) or 2 mg/ml VCAM-1 (B) and incubated for 1 min at 37˚C in the presence of media alone or 10 nM chemerin. Plates were shaken for
20 s, and the wells washed thoroughly with media to remove nonadherent cells. CellTiter-Glo was added to each well, and the luminescence was measured to
quantify the remaining adherent cells. Graphs show mean values 6 SEM from three independent experiments; pppp , 0.001, two-way ANOVA with
Bonferroni’s posthoc test.C, GPCRmRNAwas quantified using SYBRGreen RT-PCR. Data presented as the number of copies of receptor mRNA per copy of
GAPDH mRNA as quantified from a standard curve. D, PECs were stained with anti–7/4-FITC, anti–F4/80-Alexa Fluor 647, and either rat IgG2a-PE or rat
anti-mouse ChemR23-PE Abs and analyzed by flow cytometry. Graphs representative of three separate experiments. E and F, WT-BaF3 cells or BaF3-
ChemR23 cells were added to plates that had been precoated with 30 mg/ml fibronectin (E) or 2 mg/ml VCAM-1 (F) and incubated for 1 min at 37˚C in the
presence of media alone or 10 nM chemerin. Plates were washed and analyzed as described above. Graphs show mean values6 SEM from three independent
experiments. G, BaF3-ChemR23 cells were added to plates precoated with 30 mg/ml fibronectin and incubated for 1 min at 37˚C in the presence of media
alone, 0.01–100 nM chemerin, or 0.001–1 mM C9 or C15 peptide. Plates were washed and analyzed as described above. Graphs show mean values 6 SEM
from three independent experiments.
3732 CHEMERIN MEDIATES MACROPHAGE ADHESION
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
Chemerin stimulates integrin clustering
Some chemokines have been shown to promote leukocyte adhesionby stimulating the clustering of integrins on the cell surface (16, 35).To test whether chemerin could induce the clustering of VLA-4 and/orVLA-5 integrins, we used confocal microscopy to visualize localizedchanges in the distribution of the a4 integrin subunit and the a5 in-tegrin subunit, respectively, on the surface of F4/80+ macrophages.The phorbol ester PMAwas used as a positive control (36, 37).Treatment of the PECs in suspension with PMA induced the
clustering of both the a5 (Fig. 5A) and the a4 (Fig. 5B) integrinsubunits on the surface of F4/80+ macrophages. This was shownby an increase in fluorescence intensity on certain parts of the cellcompared with the low fluorescence intensity of untreated cells inwhich the integrins were more evenly distributed. Treatment of thePECs for 1 min with 10 nM chemerin had a very similar effect tothat of PMA. It increased both a5 and a4 fluorescent intensity onpatches of the macrophage surface, suggesting that chemerin caninduce rapid clustering of both VLA-5 and VLA-4 integrins, re-spectively. Chemerin stimulated a 2.6-fold (p , 0.01) increase in
the average a5 fluorescence intensity of F4/80+ macrophages (Fig.5C) and a 6-fold (p , 0.01) increase in the average a4 fluores-cence intensity of F4/80+ macrophages (Fig. 5D) (compared with
FIGURE 3. Chemerin-stimulated adhesion to fibronectin and VCAM-1
is mediated through the regulation of VLA-5 (a5b1) and VLA-4 (a4b1),
respectively. A and B, PECs from C57BL6/J mice were pretreated with 3
mg/ml integrin blocking Abs or their i.c. for 30 min at 37˚C then added to
wells that had been precoated with 30 mg/ml fibronectin (A) or 2 mg/ml
VCAM-1 (B) and incubated for 1 min at 37˚C in the presence of media
alone or 10 nM chemerin. Plates were shaken for 20 s, and the wells
washed thoroughly with media to remove nonadherent cells. CellTiter-Glo
was added to each well, and the luminescence was measured to quantify
the remaining adherent cells. Graphs show mean values6 SEM from three
independent experiments; pp , 0.05; pppp , 0.001, one-way ANOVA
with Bonferroni’s posthoc test. i.c., isotype controls.
FIGURE 4. Chemerin does not stimulate an increase in integrin affinity. A,
PECs from C57BL/6J mice were stained with anti–F4/80-Alexa Fluor 647
Ab. A total of 10 mM GRGDSP-FITC–conjugated peptide and either media,
10mMMnCl2, or 10 nM chemerinwere added to the F4/80-stained PECs and
incubated at 37˚C for up to 5 min. The PECs were immediately washed with
ice-cold buffer and fixed in 2%PFA. The quantity of peptide remaining bound
to the F4/80+ cells was measured by flow cytometry. B, A total of 100 mg/ml
VCAM-1-Fc and either media, 10 mM MnCl2, or 10 nM chemerin were
added to PECs from C57BL/6J mice and incubated at 37˚C for up to 2 min.
PECs were immediately fixed in 2% PFA on ice, washed with ice-cold buffer,
and stained with PE-conjugated anti-human IgG and anti–F4/80-Alexa Fluor
647 Abs. The quantity of VCAM-1-Fc remaining bound to the F4/80+ cells
was measured by flow cytometry. Graphs show mean values 6 SEM from
three independent experiments; pp, 0.05; pppp, 0.001, two-way ANOVA
with Bonferroni’s posthoc test. C and D, Representative plots showing the
amount of GRGDSP-FITC (C) or VCAM-1-Fc (D) bound to F4/80+ PECs
after 300 or 120 s, respectively.
The Journal of Immunology 3733
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
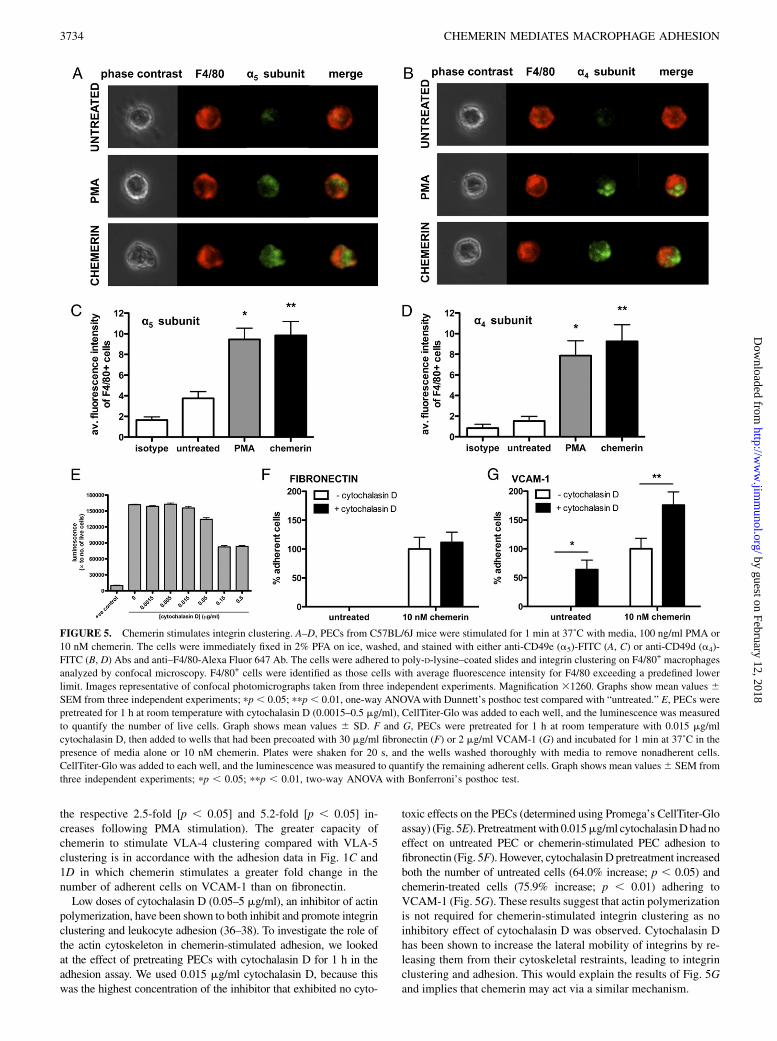
the respective 2.5-fold [p , 0.05] and 5.2-fold [p , 0.05] in-creases following PMA stimulation). The greater capacity ofchemerin to stimulate VLA-4 clustering compared with VLA-5clustering is in accordance with the adhesion data in Fig. 1C and1D in which chemerin stimulates a greater fold change in thenumber of adherent cells on VCAM-1 than on fibronectin.Low doses of cytochalasin D (0.05–5 mg/ml), an inhibitor of actin
polymerization, have been shown to both inhibit and promote integrinclustering and leukocyte adhesion (36–38). To investigate the role ofthe actin cytoskeleton in chemerin-stimulated adhesion, we lookedat the effect of pretreating PECs with cytochalasin D for 1 h in theadhesion assay. We used 0.015 mg/ml cytochalasin D, because thiswas the highest concentration of the inhibitor that exhibited no cyto-
toxic effects on the PECs (determined using Promega’s CellTiter-Gloassay) (Fig. 5E). Pretreatmentwith 0.015mg/ml cytochalasinDhadnoeffect on untreated PEC or chemerin-stimulated PEC adhesion tofibronectin (Fig. 5F). However, cytochalasinD pretreatment increasedboth the number of untreated cells (64.0% increase; p , 0.05) andchemerin-treated cells (75.9% increase; p , 0.01) adhering toVCAM-1 (Fig. 5G). These results suggest that actin polymerizationis not required for chemerin-stimulated integrin clustering as noinhibitory effect of cytochalasin D was observed. Cytochalasin Dhas been shown to increase the lateral mobility of integrins by re-leasing them from their cytoskeletal restraints, leading to integrinclustering and adhesion. This would explain the results of Fig. 5Gand implies that chemerin may act via a similar mechanism.
FIGURE 5. Chemerin stimulates integrin clustering. A–D, PECs from C57BL/6J mice were stimulated for 1 min at 37˚C with media, 100 ng/ml PMA or
10 nM chemerin. The cells were immediately fixed in 2% PFA on ice, washed, and stained with either anti-CD49e (a5)-FITC (A, C) or anti-CD49d (a4)-
FITC (B, D) Abs and anti–F4/80-Alexa Fluor 647 Ab. The cells were adhered to poly-D-lysine–coated slides and integrin clustering on F4/80+ macrophages
analyzed by confocal microscopy. F4/80+ cells were identified as those cells with average fluorescence intensity for F4/80 exceeding a predefined lower
limit. Images representative of confocal photomicrographs taken from three independent experiments. Magnification 31260. Graphs show mean values 6SEM from three independent experiments; pp, 0.05; ppp, 0.01, one-way ANOVAwith Dunnett’s posthoc test compared with “untreated.” E, PECs were
pretreated for 1 h at room temperature with cytochalasin D (0.0015–0.5 mg/ml), CellTiter-Glo was added to each well, and the luminescence was measured
to quantify the number of live cells. Graph shows mean values 6 SD. F and G, PECs were pretreated for 1 h at room temperature with 0.015 mg/ml
cytochalasin D, then added to wells that had been precoated with 30 mg/ml fibronectin (F) or 2 mg/ml VCAM-1 (G) and incubated for 1 min at 37˚C in the
presence of media alone or 10 nM chemerin. Plates were shaken for 20 s, and the wells washed thoroughly with media to remove nonadherent cells.
CellTiter-Glo was added to each well, and the luminescence was measured to quantify the remaining adherent cells. Graph shows mean values6 SEM from
three independent experiments; pp , 0.05; ppp , 0.01, two-way ANOVA with Bonferroni’s posthoc test.
3734 CHEMERIN MEDIATES MACROPHAGE ADHESION
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
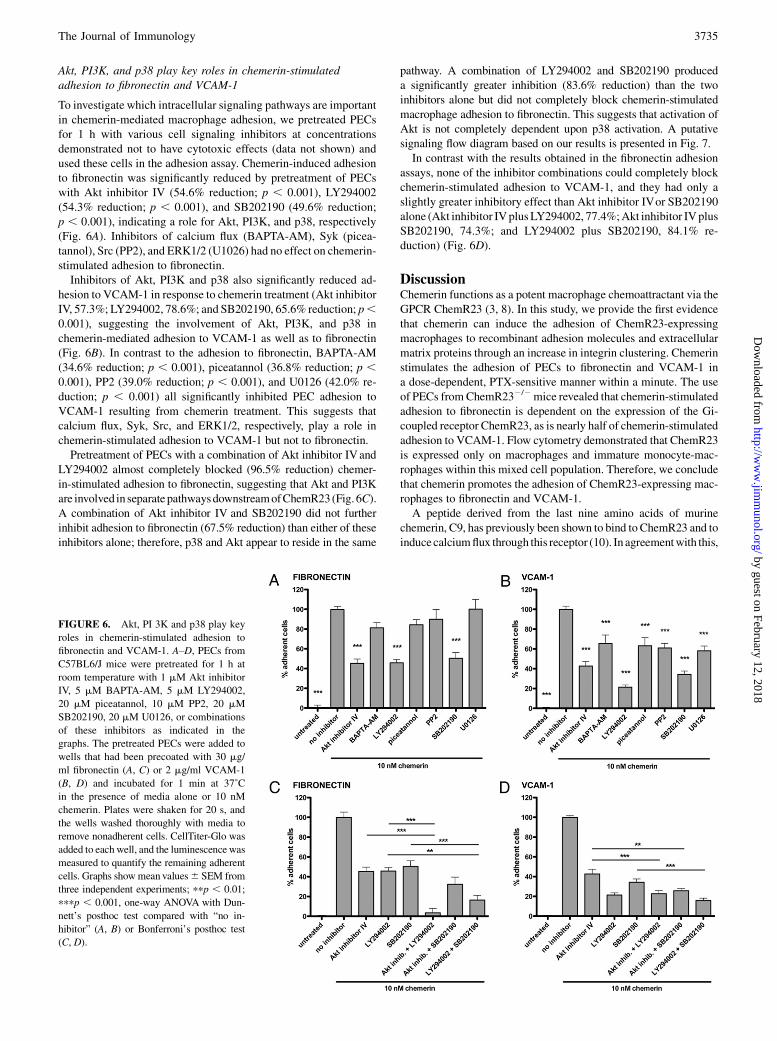
Akt, PI3K, and p38 play key roles in chemerin-stimulatedadhesion to fibronectin and VCAM-1
To investigate which intracellular signaling pathways are importantin chemerin-mediated macrophage adhesion, we pretreated PECsfor 1 h with various cell signaling inhibitors at concentrationsdemonstrated not to have cytotoxic effects (data not shown) andused these cells in the adhesion assay. Chemerin-induced adhesionto fibronectin was significantly reduced by pretreatment of PECswith Akt inhibitor IV (54.6% reduction; p , 0.001), LY294002(54.3% reduction; p , 0.001), and SB202190 (49.6% reduction;p , 0.001), indicating a role for Akt, PI3K, and p38, respectively(Fig. 6A). Inhibitors of calcium flux (BAPTA-AM), Syk (picea-tannol), Src (PP2), and ERK1/2 (U1026) had no effect on chemerin-stimulated adhesion to fibronectin.Inhibitors of Akt, PI3K and p38 also significantly reduced ad-
hesion to VCAM-1 in response to chemerin treatment (Akt inhibitorIV, 57.3%; LY294002, 78.6%; and SB202190, 65.6% reduction; p,0.001), suggesting the involvement of Akt, PI3K, and p38 inchemerin-mediated adhesion to VCAM-1 as well as to fibronectin(Fig. 6B). In contrast to the adhesion to fibronectin, BAPTA-AM(34.6% reduction; p , 0.001), piceatannol (36.8% reduction; p ,0.001), PP2 (39.0% reduction; p , 0.001), and U0126 (42.0% re-duction; p , 0.001) all significantly inhibited PEC adhesion toVCAM-1 resulting from chemerin treatment. This suggests thatcalcium flux, Syk, Src, and ERK1/2, respectively, play a role inchemerin-stimulated adhesion to VCAM-1 but not to fibronectin.Pretreatment of PECs with a combination of Akt inhibitor IVand
LY294002 almost completely blocked (96.5% reduction) chemer-in-stimulated adhesion to fibronectin, suggesting that Akt and PI3Kare involved in separatepathwaysdownstreamofChemR23(Fig. 6C).A combination of Akt inhibitor IV and SB202190 did not furtherinhibit adhesion to fibronectin (67.5% reduction) than either of theseinhibitors alone; therefore, p38 and Akt appear to reside in the same
pathway. A combination of LY294002 and SB202190 produceda significantly greater inhibition (83.6% reduction) than the twoinhibitors alone but did not completely block chemerin-stimulatedmacrophage adhesion to fibronectin. This suggests that activation ofAkt is not completely dependent upon p38 activation. A putativesignaling flow diagram based on our results is presented in Fig. 7.In contrast with the results obtained in the fibronectin adhesion
assays, none of the inhibitor combinations could completely blockchemerin-stimulated adhesion to VCAM-1, and they had only aslightly greater inhibitory effect than Akt inhibitor IVor SB202190alone (Akt inhibitor IVplusLY294002, 77.4%;Akt inhibitor IVplusSB202190, 74.3%; and LY294002 plus SB202190, 84.1% re-duction) (Fig. 6D).
DiscussionChemerin functions as a potent macrophage chemoattractant via theGPCR ChemR23 (3, 8). In this study, we provide the first evidencethat chemerin can induce the adhesion of ChemR23-expressingmacrophages to recombinant adhesion molecules and extracellularmatrix proteins through an increase in integrin clustering. Chemerinstimulates the adhesion of PECs to fibronectin and VCAM-1 ina dose-dependent, PTX-sensitive manner within a minute. The useof PECs fromChemR232/2mice revealed that chemerin-stimulatedadhesion to fibronectin is dependent on the expression of the Gi-coupled receptor ChemR23, as is nearly half of chemerin-stimulatedadhesion to VCAM-1. Flow cytometry demonstrated that ChemR23is expressed only on macrophages and immature monocyte-mac-rophages within this mixed cell population. Therefore, we concludethat chemerin promotes the adhesion of ChemR23-expressing mac-rophages to fibronectin and VCAM-1.A peptide derived from the last nine amino acids of murine
chemerin, C9, has previously been shown to bind to ChemR23 and toinducecalciumflux through this receptor (10). In agreementwith this,
FIGURE 6. Akt, PI 3K and p38 play key
roles in chemerin-stimulated adhesion to
fibronectin and VCAM-1. A–D, PECs from
C57BL6/J mice were pretreated for 1 h at
room temperature with 1 mM Akt inhibitor
IV, 5 mM BAPTA-AM, 5 mM LY294002,
20 mM piceatannol, 10 mM PP2, 20 mM
SB202190, 20 mM U0126, or combinations
of these inhibitors as indicated in the
graphs. The pretreated PECs were added to
wells that had been precoated with 30 mg/
ml fibronectin (A, C) or 2 mg/ml VCAM-1
(B, D) and incubated for 1 min at 37˚C
in the presence of media alone or 10 nM
chemerin. Plates were shaken for 20 s, and
the wells washed thoroughly with media to
remove nonadherent cells. CellTiter-Glo was
added to each well, and the luminescencewas
measured to quantify the remaining adherent
cells. Graphs showmean values6 SEM from
three independent experiments; ppp , 0.01;
pppp , 0.001, one-way ANOVA with Dun-
nett’s posthoc test compared with “no in-
hibitor” (A, B) or Bonferroni’s posthoc test
(C, D).
The Journal of Immunology 3735
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

we found that C9 is capable of stimulating the adhesion ofChemR23-transfected cells to fibronectin and is in fact slightly morepotent than chemerin in this assay, although it is a partial agonist thatis unable to achieve the maximal cell adhesion induced by chemerin.C15, another peptide derived from the C terminus of chemerin, hasbeen shown to have anti-inflammatory and prophagocytic effects onmacrophages that are dependent on expression of the ChemR23receptor (8, 29). We were unable to see an effect of C15 on the ad-hesion of ChemR23-transfected cells to fibronectin. This also agreeswith the data of Luangsay et al. (10) who were unable to detectcalcium release in ChemR23-transfected cells in response to C15treatment. This does not, however, rule out the possibility that C15activates alternative signaling pathways downstream of ChemR23onmacrophages by acting as an allosteric modulator or by binding toa heterodimer of ChemR23 and another unknown GPCR.
Chemerin, macrophages, and inflammation
Leukocyte adhesion and chemotaxis are important for maintainingtissue homeostasis as well as in generating a successful inflammatoryresponse (2). Given that chemerin is activated by inflammatoryproteases and that ChemR23 is predominantly expressed by mac-rophages in the mouse, chemerin is most likely to contribute to thedevelopment of inflammatory diseases in which macrophages playa central role (e.g., atherosclerosis or arthritis [9, 11]). Taking ath-erosclerosis as an example, the migration of tissue and newly dif-ferentiated macrophages from the surrounding intima and adventitiato the atherosclerotic lesion requires adhesion via integrins to ex-tracellular matrix proteins, such as fibronectin (22, 39, 40). The ad-hesion of macrophages to the extracellular matrix and to VCAM-1expressed on smooth muscle cells that have migrated into the sub-endothelial space, are thought to contribute to macrophage retentionwithin the atherosclerotic plaque, thereby potentiating the pathogenicprocess (41, 42).Macrophage adhesion to fibronectin has been shownto increase the expression of proinflammatory molecules, such asmatrix metalloproteinases, which have a deleterious effect on plaquestability (21, 43, 44). Our findings suggest that chemerin couldpromote any one of the above processes, thereby contributing to theprogression of an inflammatory disease, such as atherosclerosis.Macrophages that have been activated by a combination of IFN-g
plus TNF-a (LPS) are known as classically activated M1 macro-phages, whereas macrophages activated by IL-4 are known as al-ternatively activated M2a macrophages (45). These two proinflam-matory macrophage types have distinct functional properties. Ourresults suggest that polarized macrophages no longer adhere to fi-bronectin or VCAM-1 in response to chemerin stimulation, eitherbecause of a complete downregulation in ChemR23 expressionin the case of M1 macrophages or, in the case of M2a macrophagesthat retain their ChemR23 expression, for another reason thatmay involve the functionality of the integrins themselves. (M1
macrophages resulting from IFN-g stimulation alone retain theirChemR23 expression and their ability to adhere to fibronectin andVCAM-1 in response to chemerin.) This suggests that chemerin-stimulated adhesion may only be relevant in the very earlyinitiating stages of inflammation. However, M1 and M2a macro-phages represent the extreme phenotypes and in inflammatoryenvironments, where a number of stimulatory ligands are present,macrophages exhibit plasticity and may display a spectrum of func-tionalities for which it is hard to predict their responsiveness tochemerin (46).Although we have focused on macrophages, ChemR23 is addi-
tionally expressed onmonocytes, pDCs, andNK cells in humans andwas also recently shown to be expressed by pDCs andNKcells in themouse (5–7, 10). Because we have shown that chemerin rapidlyincreases murine macrophage adhesion to VCAM-1, it is possiblethat chemerin can induce the rapid adhesion of monocytes, NK cells,or pDCs to VCAM-1 presented on activated vascular endotheliumas well as to fibronectin, thereby recruiting these cells to sites ofinflammation. Chemerin has an overall positive charge that wouldallow it to be presented on negatively charged heparin or sulfatedglycosaminoglycans, making monocyte, NK cell, and pDC re-cruitment from the bloodstream a feasible hypothesis (14). Indeed,chemerin immunoreactivity has been detected on endothelial cellslining blood vessels in inflamed tissues and chemerin expression hasbeen shown to correlate with ChemR23+ pDC recruitment in pso-riatic skin biopsies (6, 47–49).
Chemerin-stimulated integrin regulation
Integrins are the receptors expressed on leukocytes that bind to ex-tracellular matrix proteins and adhesion molecules and are conse-quently responsible for leukocyte adhesion (30). The binding of manychemoattractants to their GPCRs can initiate a process termed insi-de-out signaling that results in an increased capacity for the integrinsto bind their ligands (16, 50). Although the regulation of integrinactivity has been well studied in many leukocyte subsets, such asneutrophils, monocytes and lymphocytes, this process in primarymacrophages remains relatively unexplored.We used blocking Abs toidentify VLA-5 and VLA-4 as the integrins responsible for chemerin-stimulated macrophage adhesion to fibronectin and VCAM-1, re-spectively. LPAM-1 was also partially responsible for chemerin-stimulated adhesion to VCAM-1.Chemerin’s effects on adhesion are extremely rapid and so we
considered it unlikely that an upregulation in integrin expression onthe surface of macrophages was responsible for the increase inadhesion to fibronectin and VCAM-1. GPCR agonist-stimulatedinside-out signaling increases integrin avidity and subsequentlyleukocyte adhesion (16). An increase in integrin avidity can resultfrom either an increase in integrin affinity and/or an increase inintegrin valency (51). Many chemoattractants are capable of al-tering the conformational state of integrins, thereby increasing theiraffinity. For example, CXCL12 promotes a rapid increase in theaffinity of VLA-4 on monocytes for VCAM-1 (25). However, ourexperiments using soluble ligands revealed that chemerin is unableto increase the affinity of either VLA-4 or VLA-5 (or LPAM-1) onmacrophages.Increased integrin valency results from cytoskeletal rearrange-
ments that result in the redistribution of integrins on the cell surface(51, 52). This increases the cooperative binding of the integrins,thereby increasing the overall bond strength and allowing leukocyteadhesion (51, 53). Using confocal microscopy, we demonstrated thatchemerin stimulates the ligand-independent clustering of VLA-4and VLA-5, thereby providing a mechanism for chemerin-stimu-lated adhesion to VCAM-1 and fibronectin, respectively. Somechemokines have also been shown to stimulate integrin clustering
FIGURE 7. A diagram to illustrate the putative signaling pathway
downstream of ChemR23 that leads to adhesion to fibronectin.
3736 CHEMERIN MEDIATES MACROPHAGE ADHESION
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

without stimulating an increase in integrin affinity. For example,CXCL12 promotes the rapid clustering of VLA-4 on lymphocytes toachieve adhesion to VCAM-1 while having no effect on VLA-4affinity (35). Integrin clustering alone is sufficient to support celladhesion under the flow conditions that model the environmentfound within blood vessels (54).It is generally thought that release of integrins from cytoskeletal
restraints increases integrin lateral mobility and allows integrinclustering and leukocyte adhesion to takeplace (52). Evidence for thiscomes from the stimulatory effect of cytochalasin D, an inhibitor ofactin polymerization on integrin clustering and the inhibitory effectof integrin mutations on integrin clustering arising from an increasein the cytoskeletal anchoring of the integrin tail (37, 38, 55). Inagreement with this, cytochalasin D pretreatment increased un-treated and chemerin-stimulated adhesion to VCAM-1. It did notinhibit chemerin-stimulated adhesion to VCAM-1 or fibronectin,arguing against an active recruitment of integrins into clustersthrough actin polymerization (36). This implies that chemerin mayfunction through a similar mechanism, acting to release cytoskeletalrestraints holding VLA-4 and VLA-5 in place, thereby allowingthem to passively diffuse into clusters. There is some evidence tosuggest that movement of integrins into lipid rafts allows focusedclustering to take place and is required for cell adhesion (56).The lack of effect of cytochalasinD on cell adhesion to fibronectin
may be explained by a low expression of VLA-5 on the macrophagesurface. The number of untreated PECs adhering to fibronectin was3.4-fold lower than the binding of these cells to VCAM-1 (data notshown). This may result from a lower expression of VLA-5 on thesurface, and together with the low dose used of this inhibitor to avoidcytotoxic effects, cytochalasin D may not be able to sufficiently re-lease enough VLA-5 integrins from the cytoskeleton to cause clus-tering and adhesion, unlike 10 nM chemerin.
Signaling pathways mediating chemerin-stimulated adhesion
In common with other chemoattractants, chemerin’s ability to in-duce adhesion to fibronectin and VCAM-1 is dependent on theactivation of a Gi G protein (23–26). Many studies have looked atthe signaling requirements downstream of the Gi G protein that arerequired for stimulation of leukocyte adhesion (16, 57). However, ithas become clear that no single pathway is responsible, rather thesignaling pathway depends on the specific leukocyte subset, theagonist, GPCR, and specific integrin involved. The inside-outpathways responsible for increased integrin avidity in macro-phages, in particular, have not been studied in detail, and the same istrue of the pathways responsible for increases in integrin valency inother leukocyte subsets. Agonists at Gi-coupled GPCRs are capableof activating a multitude of signaling pathways involving PI3K,phospholipase C, tyrosine kinases, MAPKs, and small GTPases(58, 59). Chemerin was previously demonstrated to induce calciumflux and activate ERK through ChemR23 (3).In this study, we have clearly demonstrated that PI3K, Akt, and
p38 are involved in chemerin-stimulated adhesion to fibronectin andtherefore in the increase in VLA-5 clustering on the macrophagesurface. Combinations of inhibitors revealed that Akt and PI3K areinvolved in separate pathways downstream of ChemR23, whereasp38 and Akt reside in the same pathway (Fig. 7). Similarly, PI3K,Akt, and p38 play a key role in chemerin-stimulated adhesion toVCAM-1 and therefore in the increase in the clustering of VLA-4.However, combinations of inhibitors against these signaling com-ponents are unable to completely block chemerin-stimulated ad-hesion to VCAM-1, and instead, calcium flux, Syk, Src, and ERKplay a role. Taken together, our data suggest that chemerin activatesat least one additional separate signaling pathway that leads toVLA-4 clustering and adhesion to VCAM-1.
Many studies have found a role for PI3K in integrin activation. Forexample, clustering of LFA-1 in lymphocytes was shown to bedependent on PI3K activity, but PI3K was not involved in increasesin LFA-1 affinity in these cells (60). It was suggested that PI3K,through its ability to promote cytoskeletal remodeling, might re-lease LFA-1 from its cytoskeletal restraints, resulting in integrinclustering and lymphocyte adhesion. The calcium-dependent pro-tease calpain has also been implicated in the cleavage of a cyto-skeletal component, triggering the release of LFA-1 from the cyto-skeleton and integrin clustering (61). A similar mechanism may bepartly responsible for chemerin-stimulated VLA-4 clustering, be-cause inhibition of calcium flux partially inhibited cell adhesion toVCAM-1
ChemR23-independent adhesion
Chemerin-stimulated adhesion of macrophages to fibronectin iscompletelydependenton theexpressionofChemR23.Unexpectedly,chemerin is still capable of promoting the adhesion of PECsfrom ChemR232/2 mice to VCAM-1, revealing that just under halfof chemerin-stimulated adhesion to VCAM-1 is dependent onChemR23 expression. Chemerin was originally thought to bind onlyto ChemR23, thereby forming a highly specific receptor-ligand pair,but recent studies have identified two additional high-affinity re-ceptors for chemerin. CCRL2 is a nonsignaling GPCR expressedon mast cells and macrophages that is proposed to bind chemerin,increasing its local concentration and presenting it for interactionwith ChemR23 expressed on adjacent cells (28). Low levels ofCCRL2 mRNA are expressed by the PECs used in our experiments;however, CCRL2’s reported inability to initiate intracellular sig-naling would seem to rule it out as the additional receptor mediatingchemerin-stimulated adhesion to VCAM-1 (28). Chemerin alsobinds to the GPCR GPR1, initiating an increase in intracellularcalcium that is also seen in cells expressing ChemR23 (27). How-ever, negligible levels of GPR1 mRNA are expressed by PECs,suggesting the existence of a fourth unknown chemerin receptor.Pretreatment with PTX completely ablates chemerin-stimulatedadhesion to VCAM-1, implying that the unknown receptor is cou-pled to a Gi G protein. A possible candidate for the unknown re-ceptor is the leukotriene B4 receptor (BLT1), which is expressed atthe mRNA level by PECs. The lipid resolvin E1 has been reported tobe a ligand for both ChemR23 and BLT1 (62, 63).In summary, we have shown for the first time that the macrophage
chemoattractant protein chemerin, which is generated at sites of in-flammation, rapidly stimulates the adhesion of macrophages to fi-bronectin and VCAM-1 in a ChemR23-dependent manner, by pro-moting clusteringof the integrinsVLA-5andVLA-4, respectively.Wehave also identified some of the key signaling components involved inthe regulation of VLA-5 and VLA-4 valency on macrophages in re-sponse to chemerin treatment. Chemerin-stimulated adhesion mayplay an important role in recruiting and retainingmacrophages at sitesof inflammation and in their subsequent activation.
AcknowledgmentsWe thank Linda Randall, Denise Jelfs, Jenna Cash, Thomas Tan, and
Gemma White for advice and support.
DisclosuresThe authors have no financial conflicts of interest.
References1. Majno, G., and I. Joris. 2004. Cells, Tissues, and Disease: Principles of General
Pathology. Oxford University Press, New York, Oxford.2. Sallusto, F., and C. R. Mackay. 2004. Chemoattractants and their receptors in
homeostasis and inflammation. Curr. Opin. Immunol. 16: 724–731.
The Journal of Immunology 3737
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

3. Wittamer, V., J. D. Franssen, M. Vulcano, J. F. Mirjolet, E. Le Poul, I. Migeotte,S. Brezillon, R. Tyldesley, C. Blanpain, M. Detheux, et al. 2003. Specific re-cruitment of antigen-presenting cells by chemerin, a novel processed ligand fromhuman inflammatory fluids. J. Exp. Med. 198: 977–985.
4. Samson, M., A. L. Edinger, P. Stordeur, J. Rucker, V. Verhasselt, M. Sharron,C. Govaerts, C. Mollereau, G. Vassart, R. W. Doms, and M. Parmentier. 1998.ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and someprimary HIV-1 strains. Eur. J. Immunol. 28: 1689–1700.
5. Zabel, B. A., A. M. Silverio, and E. C. Butcher. 2005. Chemokine-like receptor 1expression and chemerin-directed chemotaxis distinguish plasmacytoid frommyeloid dendritic cells in human blood. J. Immunol. 174: 244–251.
6. Vermi, W., E. Riboldi, V. Wittamer, F. Gentili, W. Luini, S. Marrelli, A. Vecchi,J. D. Franssen, D. Communi, L. Massardi, et al. 2005. Role of ChemR23 indirecting the migration of myeloid and plasmacytoid dendritic cells to lymphoidorgans and inflamed skin. J. Exp. Med. 201: 509–515.
7. Parolini, S., A. Santoro, E. Marcenaro, W. Luini, L. Massardi, F. Facchetti,D. Communi, M. Parmentier, A. Majorana, M. Sironi, et al. 2007. The role ofchemerin in the colocalization of NK and dendritic cell subsets into inflamedtissues. Blood 109: 3625–3632.
8. Cash, J. L., R. Hart, A. Russ, J. P. Dixon, W. H. Colledge, J. Doran, A. G. Hendrick,M. B. Carlton, and D. R. Greaves. 2008. Synthetic chemerin-derived peptidessuppress inflammation through ChemR23. J. Exp. Med. 205: 767–775.
9. Zabel, B. A., T. Ohyama, L. Zuniga, J. Y. Kim, B. Johnston, S. J. Allen,D. G. Guido, T. M. Handel, and E. C. Butcher. 2006. Chemokine-like receptor 1expression by macrophages in vivo: regulation by TGF-b and TLR ligands. Exp.Hematol. 34: 1106–1114.
10. Luangsay, S., V. Wittamer, B. Bondue, O. De Henau, L. Rouger, M. Brait,J. D. Franssen, P. de Nadai, F. Huaux, and M. Parmentier. 2009. MouseChemR23 is expressed in dendritic cell subsets and macrophages, and mediatesan anti-inflammatory activity of chemerin in a lung disease model. J. Immunol.183: 6489–6499.
11. Zabel, B. A., S. J. Allen, P. Kulig, J. A. Allen, J. Cichy, T. M. Handel, andE. C. Butcher. 2005. Chemerin activation by serine proteases of the coagulation,fibrinolytic, and inflammatory cascades. J. Biol. Chem. 280: 34661–34666.
12. Wittamer, V., B. Bondue, A. Guillabert, G. Vassart, M. Parmentier, andD. Communi. 2005. Neutrophil-mediated maturation of chemerin: a link be-tween innate and adaptive immunity. J. Immunol. 175: 487–493.
13. Du, X. Y., B. A. Zabel, T. Myles, S. J. Allen, T. M. Handel, P. P. Lee,E. C. Butcher, and L. L. Leung. 2009. Regulation of chemerin bioactivity byplasma carboxypeptidase N, carboxypeptidase B (activated thrombin-activablefibrinolysis inhibitor), and platelets. J. Biol. Chem. 284: 751–758.
14. Zabel, B. A., L. Zuniga, T. Ohyama, S. J. Allen, J. Cichy, T. M. Handel, andE. C. Butcher. 2006. Chemoattractants, extracellular proteases, and the in-tegrated host defense response. Exp. Hematol. 34: 1021–1032.
15. Diamond, M. S., and T. A. Springer. 1994. The dynamic regulation of integrinadhesiveness. Curr. Biol. 4: 506–517.
16. Laudanna, C., J. Y. Kim, G. Constantin, and E. Butcher. 2002. Rapid leukocyteintegrin activation by chemokines. Immunol. Rev. 186: 37–46.
17. Ley, K., C. Laudanna, M. I. Cybulsky, and S. Nourshargh. 2007. Getting to the siteof inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7:678–689.
18. Jia, W., H. Li, and Y. W. He. 2005. The extracellular matrix protein mindinserves as an integrin ligand and is critical for inflammatory cell recruitment.Blood 106: 3854–3859.
19. Lindbom, L., and J. Werr. 2002. Integrin-dependent neutrophil migration inextravascular tissue. Semin. Immunol. 14: 115–121.
20. Sixt, M., M. Bauer, T. Lammermann, and R. Fassler. 2006. b1 Integrins: zipcodes and signaling relay for blood cells. Curr. Opin. Cell Biol. 18: 482–490.
21. Lin, T. H., C. Rosales, K. Mondal, J. B. Bolen, S. Haskill, and R. L. Juliano.1995. Integrin-mediated tyrosine phosphorylation and cytokine message in-duction in monocytic cells: a possible signaling role for the Syk tyrosine kinase.J. Biol. Chem. 270: 16189–16197.
22. Lauffenburger, D. A., and A. F. Horwitz. 1996. Cell migration: a physicallyintegrated molecular process. Cell 84: 359–369.
23. Campbell, J. J., J. Hedrick, A. Zlotnik, M. A. Siani, D. A. Thompson, andE. C. Butcher. 1998. Chemokines and the arrest of lymphocytes rolling underflow conditions. Science 279: 381–384.
24. Goda, S., T. Imai, O. Yoshie, O. Yoneda, H. Inoue, Y. Nagano, T. Okazaki,H. Imai, E. T. Bloom, N. Domae, and H. Umehara. 2000. CX3C-chemokine,fractalkine-enhanced adhesion of THP-1 cells to endothelial cells throughintegrin-dependent and -independent mechanisms. J. Immunol. 164: 4313–4320.
25. Chan, J. R., S. J. Hyduk, and M. I. Cybulsky. 2001. Chemoattractants inducea rapid and transient upregulation of monocyte alpha4 integrin affinity for vas-cular cell adhesion molecule 1 which mediates arrest: an early step in the processof emigration. J. Exp. Med. 193: 1149–1158.
26. Campbell, J. J., S. Qin, K. B. Bacon, C. R. Mackay, and E. C. Butcher. 1996.Biology of chemokine and classical chemoattractant receptors: differentialrequirements for adhesion-triggering versus chemotactic responses in lymphoidcells. J. Cell Biol. 134: 255–266.
27. Barnea, G., W. Strapps, G. Herrada, Y. Berman, J. Ong, B. Kloss, R. Axel, andK. J. Lee. 2008. The genetic design of signaling cascades to record receptoractivation. Proc. Natl. Acad. Sci. USA 105: 64–69.
28. Zabel, B. A., S. Nakae, L. Zuniga, J. Y. Kim, T. Ohyama, C. Alt, J. Pan, H. Suto,D. Soler, S. J. Allen, et al. 2008. Mast cell-expressed orphan receptor CCRL2binds chemerin and is required for optimal induction of IgE-mediated passivecutaneous anaphylaxis. J. Exp. Med. 205: 2207–2220.
29. Cash, J. L., A. R. Christian, and D. R. Greaves. 2010. Chemerin peptides pro-mote phagocytosis in a ChemR23- and Syk-dependent manner. J. Immunol. 184:5315–5324.
30. Hynes, R. O. 1987. Integrins: a family of cell surface receptors. Cell 48: 549–554.31. Gordon, S. 2003. The Macrophage as Therapeutic Target. Springer, Berlin,
London.32. Smith, C. W. 2008. 3. Adhesion molecules and receptors. J. Allergy Clin.
Immunol. 121(2, Suppl.)S375–S379, quiz S414.33. Takagi, J., B. M. Petre, T. Walz, and T. A. Springer. 2002. Global conformational
rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110: 599–611.
34. Danen, E. H., S. Aota, A. A. van Kraats, K. M. Yamada, D. J. Ruiter, andG. N. van Muijen. 1995. Requirement for the synergy site for cell adhesion tofibronectin depends on the activation state of integrin alpha 5 beta 1. J. Biol.Chem. 270: 21612–21618.
35. Grabovsky, V., S. Feigelson, C. Chen, D. A. Bleijs, A. Peled, G. Cinamon,F. Baleux, F. Arenzana-Seisdedos, T. Lapidot, R. R. Lobb, R. Alon; van Kooyk Y.2000. Subsecond induction of a4 integrin clustering by immobilized chemokinesstimulates leukocyte tethering and rolling on endothelial vascular cell adhesionmolecule 1 under flow conditions. J. Exp. Med. 192: 495–506.
36. Haverstick, D. M., H. Sakai, and L. S. Gray. 1992. Lymphocyte adhesion can beregulated by cytoskeleton-associated, PMA-induced capping of surface receptors.Am. J. Physiol. 262: C916–C926.
37. Kucik, D. F., M. L. Dustin, J. M. Miller, and E. J. Brown. 1996. Adhesion-activating phorbol ester increases the mobility of leukocyte integrin LFA-1in cultured lymphocytes. J. Clin. Invest. 97: 2139–2144.
38. Lub, M., Y. van Kooyk, S. J. van Vliet, and C. G. Figdor. 1997. Dual role of theactin cytoskeleton in regulating cell adhesion mediated by the integrin lym-phocyte function-associated molecule-1. Mol. Biol. Cell 8: 341–351.
39. Katsuda, S., and T. Kaji. 2003. Atherosclerosis and extracellular matrix. J.Atheroscler. Thromb. 10: 267–274.
40. Maiellaro, K., and W. R. Taylor. 2007. The role of the adventitia in vascularinflammation. Cardiovasc. Res. 75: 640–648.
41. Santiago-Garcıa, J., T. Kodama, and R. E. Pitas. 2003. The class A scavengerreceptor binds to proteoglycans and mediates adhesion of macrophages to theextracellular matrix. J. Biol. Chem. 278: 6942–6946.
42. Cai, Q., L. Lanting, and R. Natarajan. 2004. Growth factors induce monocytebinding to vascular smooth muscle cells: implications for monocyte retention inatherosclerosis. Am. J. Physiol. Cell Physiol. 287: C707–C714.
43. Madri, J. A., and D. Graesser. 2000. Cell migration in the immune system: theevolving inter-related roles of adhesion molecules and proteinases. Dev. Immu-nol. 7: 103–116.
44. Xie, B., A. Laouar, and E. Huberman. 1998. Fibronectin-mediated cell adhesionis required for induction of 92-kDa type IV collagenase/gelatinase (MMP-9)gene expression during macrophage differentiation: the signaling roleof protein kinase C-b. J. Biol. Chem. 273: 11576–11582.
45. Mantovani, A., A. Sica, S. Sozzani, P. Allavena, A. Vecchi, and M. Locati. 2004.The chemokine system in diverse forms of macrophage activation and polari-zation. Trends Immunol. 25: 677–686.
46. Stout, R. D., C. Jiang, B. Matta, I. Tietzel, S. K. Watkins, and J. Suttles. 2005.Macrophages sequentially change their functional phenotype in response tochanges in microenvironmental influences. J. Immunol. 175: 342–349.
47. Albanesi, C., C. Scarponi, S. Pallotta, R. Daniele, D. Bosisio, S. Madonna,P. Fortugno, S. Gonzalvo-Feo, J. D. Franssen, M. Parmentier, et al. 2009.Chemerin expression marks early psoriatic skin lesions and correlates withplasmacytoid dendritic cell recruitment. J. Exp. Med. 206: 249–258.
48. Skrzeczynska-Moncznik, J., K. Wawro, A. Stefanska, E. Oleszycka, P. Kulig,B. A. Zabel, M. Sułkowski, M. Kapinska-Mrowiecka, M. Czubak-Macugowska,E. C. Butcher, and J. Cichy. 2009. Potential role of chemerin in recruitment ofplasmacytoid dendritic cells to diseased skin. Biochem. Biophys. Res. Commun.380: 323–327.
49. Vermi, W., S. Lonardi, M. Morassi, C. Rossini, R. Tardanico, M. Venturini,R. Sala, A. Tincani, P. L. Poliani, P. G. Calzavara-Pinton, et al. 2009. Cutaneousdistribution of plasmacytoid dendritic cells in lupus erythematosus: selectivetropism at the site of epithelial apoptotic damage. Immunobiology 214: 877–886.
50. Dustin, M. L., and T. A. Springer. 1989. T-cell receptor cross-linking transientlystimulates adhesiveness through LFA-1. Nature 341: 619–624.
51. Carman, C. V., and T. A. Springer. 2003. Integrin avidity regulation: are changesin affinity and conformation underemphasized? Curr. Opin. Cell Biol. 15: 547–556.
52. Kucik, D. F. 2002. Rearrangement of integrins in avidity regulation by leuko-cytes. Immunol. Res. 26: 199–206.
53. Chen, A., and V. T. Moy. 2000. Cross-linking of cell surface receptors enhancescooperativity of molecular adhesion. Biophys. J. 78: 2814–2820.
54. Yu, T., X. Wu, K. B. Gupta, and D. F. Kucik. Affinity, lateral mobility andclustering contribute independently to b2 integrin-mediated adhesion. Am JPhysiol Cell Physiol. In press.
55. Yauch, R. L., D. P. Felsenfeld, S. K. Kraeft, L. B. Chen, M. P. Sheetz, andM. E. Hemler. 1997. Mutational evidence for control of cell adhesion throughintegrin diffusion/clustering, independent of ligand binding. J. Exp. Med. 186:1347–1355.
56. Leitinger, B., and N. Hogg. 2002. The involvement of lipid rafts in the regulationof integrin function. J. Cell Sci. 115: 963–972.
57. Laudanna, C., and R. Alon. 2006. Right on the spot. Chemokine triggering ofintegrin-mediated arrest of rolling leukocytes. Thromb. Haemost. 95: 5–11.
58. Premack, B. A., and T. J. Schall. 1996. Chemokine receptors: gateways to in-flammation and infection. Nat. Med. 2: 1174–1178.
59. Thelen, M. 2001. Dancing to the tune of chemokines. Nat. Immunol. 2: 129–134.
3738 CHEMERIN MEDIATES MACROPHAGE ADHESION
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

60. Constantin, G., M. Majeed, C. Giagulli, L. Piccio, J. Y. Kim, E. C. Butcher, andC. Laudanna. 2000. Chemokines trigger immediate b2 integrin affinity andmobility changes: differential regulation and roles in lymphocyte arrest underflow. Immunity 13: 759–769.
61. Stewart, M. P., A. McDowall, and N. Hogg. 1998. LFA-1–mediated adhesion isregulated by cytoskeletal restraint and by a Ca2+-dependent protease, calpain. J.Cell Biol. 140: 699–707.
62. Arita, M., F. Bianchini, J. Aliberti, A. Sher, N. Chiang, S. Hong, R. Yang,N. A. Petasis, and C. N. Serhan. 2005. Stereochemical assignment, antiin-flammatory properties, and receptor for the omega-3 lipid mediator resolvin E1.J. Exp. Med. 201: 713–722.
63. Arita, M., T. Ohira, Y. P. Sun, S. Elangovan, N. Chiang, and C. N. Serhan. 2007.Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 andChemR23 to regulate inflammation. J. Immunol. 178: 3912–3917.
The Journal of Immunology 3739
by guest on February 12, 2018http://w
ww
.jimm
unol.org/D
ownloaded from