clausthal seminar 030615
TRANSCRIPT

Computer modelling of defects and dopants in LiNbO3, with a look ahead to LiTaO3, and Li(Nb, Ta)O3 solid solutions
Robert A JacksonSchool of Physical & Geographical Sciences
Keele UniversityKeele, Staffordshire ST5 5BG, UK
[email protected]://www.robajackson.com
@robajackson
http://www.slideshare.net/robajackson

Clausthal Seminar: 3 June 2015
Acknowledgements
Mário Valerio, Romel Araujo (Aracaju, Brazil)László Kovács, Krisztián Lengyel (Budapest, Hungary)Bud Bridges (Santa Cruz, USA)Günter Borchardt, Peter Fielitz (Clausthal, Germany)
Günter Borchardt, Holger Fritze, Klaus Dieter Becker… for the invitation!
2

Clausthal Seminar: 3 June 2015
Relevant publications
[1] R A Jackson, M E G Valerio‘A new interatomic potential for the ferroelectric and paraelectric phases of LiNbO3’
Journal of Physics: Condensed Matter, 17, 837-843 (2005)[2] R M Araujo, K Lengyel, R A Jackson, L Kovács, M E G Valerio
‘A computational study of intrinsic and extrinsic defects in LiNbO3’
Journal of Physics: Condensed Matter, 19, 046211 (2007)[3] R M Araujo, M E G Valerio, R A Jackson
‘Computer modelling of trivalent metal dopants in lithium niobate’Journal of Physics: Condensed Matter, 20, 035201 (2008)
[4] R M Araujo, M E G Valerio, R A Jackson‘Computer simulation of metal co-doping in lithium niobate’Proc. R. Soc. A 470, 20140406 (2014)
[5] R M Araujo, M E G Valerio, R A Jackson‘Computer modelling of hafnium doping in lithium niobate’ (http://arxiv.org/abs/1505.01661)
3

Clausthal Seminar: 3 June 2015
Plan of talk
• Summary of our previous work on LiNbO3
– Background– Structural agreement– Intrinsic defects– Dopants
• Modelling LiTaO3
– Potential and structural agreement– Li(Nb, Ta)O3 solid solutions
4

Clausthal Seminar: 3 June 2015
Motivation & background
• The interatomic potential published by Donnerberg and co-workers in 1989-90 was widely used. However:(i) advances in computational software
and(ii) the continued interest in the material and the availability
of new experimental dataprompted us to revisit and re-derive the potential (published in 2005).
5

Clausthal Seminar: 3 June 2015
Potential derivation
• The potential was fitted to simultaneously reproduce the structures of LiNbO3
a, Li2O and Nb2O5 to allow consistency in later defect calculations.
• The GULP codeb was used, employing the free energy option (allowing temperature dependence of the structure to be treated).
a S C Abrahams, P Marsh, Acta Cryst., B 42, 61 (1986)b J Gale, see: http://projects.ivec.org/gulp/
6

Clausthal Seminar: 3 June 2015
Brief details of the potential*
• Full ionic charges on Li, Nb and O.• Buckingham potentials describe the interactions
between Li-O, Nb-O & O-O.• A shell model is employed for O.• A 3-body bond bending potential describes the O-
Nb-O interactions.*R A Jackson, M E G Valerio, J Phys.: Condensed Matter, 17, 837 (2005)
7
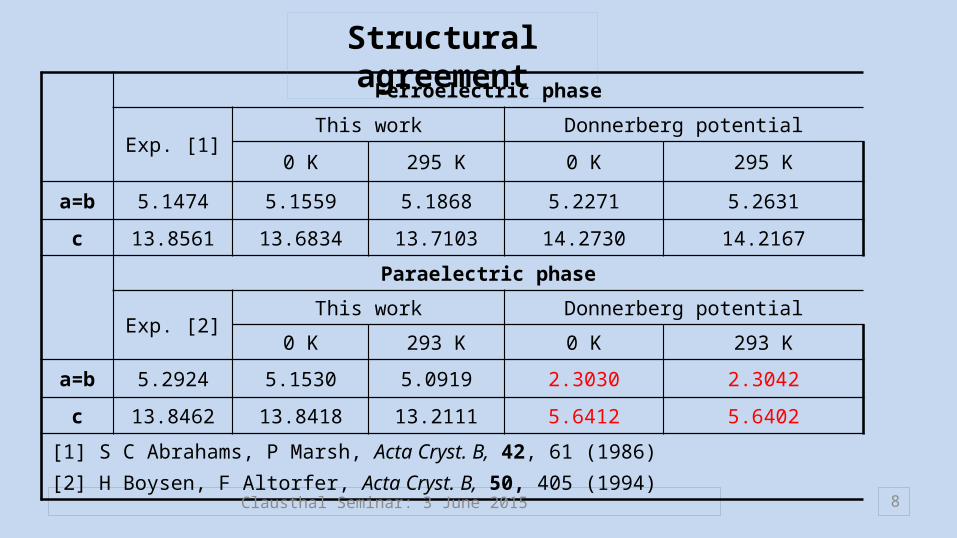
8
Ferroelectric phase
Exp. [1]This work Donnerberg potential
0 K 295 K 0 K 295 K
a=b 5.1474 5.1559 5.1868 5.2271 5.2631
c 13.8561 13.6834 13.7103 14.2730 14.2167
Paraelectric phase
Exp. [2]This work Donnerberg potential
0 K 293 K 0 K 293 K
a=b 5.2924 5.1530 5.0919 2.3030 2.3042
c 13.8462 13.8418 13.2111 5.6412 5.6402
[1] S C Abrahams, P Marsh, Acta Cryst. B, 42, 61 (1986) [2] H Boysen, F Altorfer, Acta Cryst. B, 50, 405 (1994)
Structural agreement
Clausthal Seminar: 3 June 2015

Clausthal Seminar: 3 June 2015 9
Lattice parameter as a function of temperature
T/K aexp (Å) acalc (Å) a () cexp (Å) ccalc (Å) c ()
0 - 5.1559 - - 13.6834 -
10 - 5.1745 - - 13.7035 -
295 5.1474 5.1868 0.77 13.8561 13.7103 -1.06
297 5.1483 5.1864 0.74 13.8631 13.7101 -1.10
523 5.1700 5.2133 0.84 13.8700 13.7200 -1.08

Clausthal Seminar: 3 June 2015 10
Modelling defect properties
• Using the Mott-Littleton method, energies of formation of the intrinsic defects in LiNbO3 were calculated.
• These allow predictions to be made about the defect chemistry of the material.
(See Araujo et al: Journal of Physics: Condensed Matter, 19, 046211 (2007))

Mott-Littleton approximation
Region IIons are strongly perturbed by the defect and are relaxed explicitly with respect to their Cartesian coordinates.
Region IIIons are weakly perturbed and therefore their displacements, with the associated energy of relaxation, can be approximated.
© Mark Read
11Clausthal Seminar: 3 June 2015
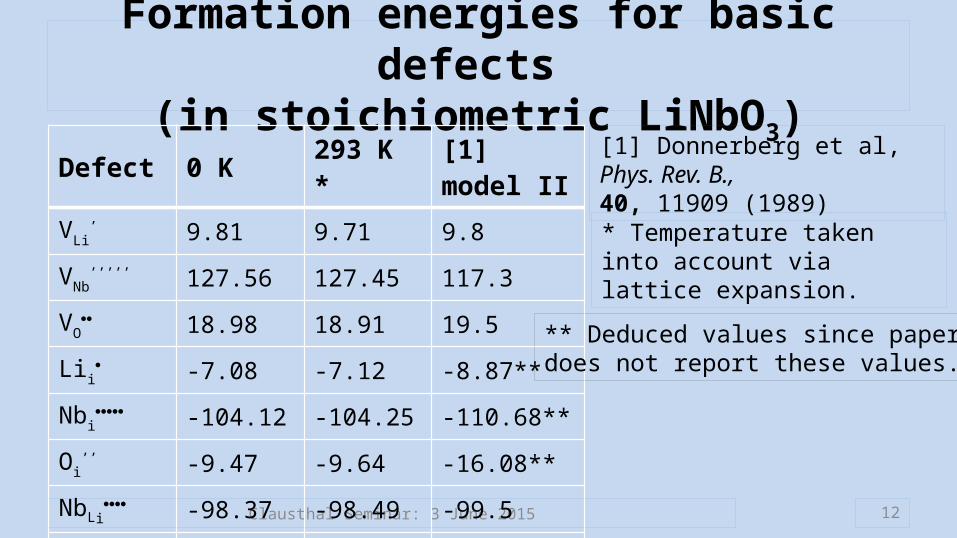
Formation energies for basic defects (in stoichiometric LiNbO3)
Clausthal Seminar: 3 June 2015 12
Defect 0 K 293 K * [1] model II
VLi’ 9.81 9.71 9.8
VNb’’’’’ 127.56 127.45 117.3
VO 18.98 18.91 19.5
Lii -7.08 -7.12 -8.87**
Nbi -104.12 -104.25 -110.68**
Oi’’ -9.47 -9.64 -16.08**
NbLi -98.37 -98.49 -99.5
LiNb’’’’ -113.99
[1] Donnerberg et al, Phys. Rev. B.,40, 11909 (1989)
* Temperature taken into account via lattice expansion.
** Deduced values since paper does not report these values.

Frenkel, Schottky and pseudo-Schottky energies* (per defect)
Clausthal Seminar: 3 June 2015 13
Defect 0 K 293 K [1]Li Frenkel 1.37 1.30 0.93
Nb Frenkel 11.72 11.60 6.26
O Frenkel 4.76 4.64 3.42
Schottky LiNbO3
3.95 3.85 3.91
Pseudo-Schottky Li2O
1.81 1.80 1.94
Pseudo-Schottky Nb2O5
5.09 5.07 2.85
* Calculated for information only since observed defects are more complex.
Expected trends in values are observed.

Clausthal Seminar: 3 June 2015 14
Models to explain the observed experimental data
• The simple Frenkel and Schottky models do not explain the observed behaviour in LiNbO3.
• For example, the NbLi + 4VLi
’ defect cluster has a formation energy of –63.61 eV.
• We needed to consider possible reactions that give rise to such defects.

Clausthal Seminar: 3 June 2015 15
Explaining the observed non-stoichiometry
• Following the work of Kovács and Polgár*, we considered models based on antisite or interstitial Nb compensated by Li or Nb vacancies.
• 3 possible reactions were considered (see next slide):
* L Kovács and K Polgár, Crystal Research and Technology, 21, K101 (1986)

Clausthal Seminar: 3 June 2015 16
Possible defect reactions that give rise to Li deficiency
Antisite Nb compensated by Li vacancies5LiLi + ½Nb2O5 ® 4V’Li + NbLi
···· + 5/2Li2O
E(reaction) = -0.98 (-2.52*) eV per Li2O formula unit
Antisite Nb compensated by Nb vacancies5LiLi + 4NbNb + ½Nb2O5 ® 5NbLi
···· + 4VNb’’’’’ + 5/2Li2O E(reaction) = 29.8 eV per Li2O formula unit
Interstitial Nb compensated by Li vacancies5LiLi + ½Nb2O5 ® 5VLi’ + Nbi
····· + 5/2Li2O E(reaction) = 0.49 eV per Li2O formula unit
* ‘Bound’ defect configuration

Clausthal Seminar: 3 June 2015 17
Conclusions from the reactions
• If the reaction energies are calculated, using the basic defect energies already obtained, we concluded that:– only the antisite Nb/Li vacancy model is energetically
favourable.– of the other two mechanisms, the interstitial Nb/Li
vacancy model is more favourable than the antisite Nb/Nb vacancy model.

Clausthal Seminar: 3 June 2015 18
Divalent and trivalent dopants
• The incorporation of a range of dopant ions in LiNbO3 was modelled.
• Divalent, trivalent and tetravalent ion substitution was considered.
• Charge compensation is needed for substitution at either the Li+ or Nb5+ site.

Clausthal Seminar: 3 June 2015 19
Dopant ions considered
• Reference [2]: M2+ dopants Mg, Mn, Fe, Co, Ni, Zn, Sr, Cd, Ba & Pb, and M3+ lanthanide dopants Ce-Lu.
• Reference [3] focused on M3+ dopants: Sc, Cr, Fe and In.• References [4] and [5] consider co-doping and Hf doping
respectively.[2] Journal of Physics: Condensed Matter, 19, 046211 (2007)[3] Journal of Physics: Condensed Matter, 20, 035201 (2008)[4] Proc. R. Soc. A 470, 20140406 (2014)[5] http://arxiv.org/abs/1505.01661

Clausthal Seminar: 3 June 2015 20
Summary of modelling procedure
• The GULP code is used to calculate the substitution energies, e.g. M2+ at the Li+ site, denoted by MLi
in Kroger-Vink notation.
• The substitution energies are then converted into solution energies, which give the total energy involved in the process:

Clausthal Seminar: 3 June 2015 21
Solution energies
• Assuming M2+ substitution at the Li+ site, a possible scheme could be (using Kröger-Vink notation):MO + 2 LiLi → MLi
+ VLi’ + Li2O• This assumes charge compensation by Li vacancies, but
other possibilities are considered.• The same idea is applied to M3+ dopants.

Clausthal Seminar: 3 June 2015 22
Predicted doping schemes: M2+ ions
• From the calculations, the following predictions are made based on lowest energies:
• Co-doping at both Li+ and Nb5+ sites, except for Fe2+ and Cd2+ for which substitution at the Nb5+ site with charge compensation by Nb - Li anti-site substitution is preferred.

Clausthal Seminar: 3 June 2015 23
Predicted doping schemes: M3+ ions
• The predicted scheme for all the lanthanide ions and Sc, Cr and Fe is self-compensation:M2O3 + LiLi + NbNb → MLi
+ MNb’’ + LiNbO3
• For In, the preferred scheme involves doping at the Nb5+ site with charge compensation by Nb-Li anti-sites.

Clausthal Seminar: 3 June 2015 24
Some relevant experimental data
• Studies of M2+ & M3+ dopants in LiNbO3 have included:Mn2+ - LiNbO3: Darwish et al, NIMB, 141, 679-683 (1998)
Supports the idea of Mn2+ self compensation; does not give dopant concentration.Mg2+ - LiNbO3: González-Martínez et al, Opt. Comm., 282, 1212-1219 (2009)
Dopant concentration 0.0714-0.2422 mol%; suggests that self compensation occurs ‘after a certain dopant concentration level’.
Er3+, Cr3+ - LiNbO3: Dierolf & Sandmann, J. Lum., 125, 67-79 (2007) Mainly assumes Li site occupancy, but dopant concentration is unclear as several
samples have been used.

Clausthal Seminar: 3 June 2015 25
‘EXAFS evidence for a primary ZnLi dopant in LiNbO3’
(F Bridges et al, Phys. Rev. B 85 064107 (2012))
• Doesn’t find Zn at the Nb site, but may not be directly comparable with the calculations (concentration effects, stoichiometry of sample?)– Measurements on a ‘stoichiometric’ sample give same result.– EXAFS measurements on In have also been performed, and
preliminary results suggest InLi dopants.

Clausthal Seminar: 3 June 2015 26
General comments on comparison with experimental data
• The calculation results reported are at infinite dilution, so no concentration effects are considered.– Now we are looking at finite dopant concentrations in other materials,
and this could be done for dopants in LiNbO3 (needs persons and €€€).
• There may be issues with the stoichiometry of the older crystal samples (i.e. are we comparing ‘like with like’?)– But recently EXAFS was done on a stoichiometric sample and we are
comparing results.

Clausthal Seminar: 3 June 2015 27
Hf4+ doping
• The lowest energy scheme again involves self-compensation:
4HfO2 + LiLi + 3NbNb → HfLi + 3HfNb’ + ½ Li2O + 3/2 Nb2O5
• These results are supported by experimental studies:Marques J G, Kling A , de Jesus C M , Soares J C , da Silva M F, Dieguez E and Agulló-Lopez F 1998 Nuclear Instruments and Methods in Physics Research B 141 326-331Li S, Liu S, Kong Y, Deng D, Gao G, Li Y, Gao H, Zhang L, Hang Z, Chen S and Xu J 2006 J. Phys.: Condens. Matter 18 3527–3534

Clausthal Seminar: 3 June 2015 28
Co-doping by transition metal ions
• TM co-doped LiNbO3 finds applications in holographic recording devices.
• We have modelled co-doping by Fe3+Cu+, Ce3+Cu+, Ce4+Mn2+, Rh3+Fe3+ and Ru4+Fe3+.
• In most cases the solution energy is reduced compared to doping with single ions.– Has implications for device development.

Clausthal Seminar: 3 June 2015 29
Modelling LiTaO3: obtaining an (initial) Ta-O potential
Structure from: Abrahams & Bernstein, J. Phys. Chem. Sol. 28 (1967) 1685-1692
Structure after refitting of Ta-O potential.
Based on Ta-O potential from KTaO3 paper (Exner)
Phys. Rev. B 52(6) (1995) 3930-3940
Exp. Calc. %
a=b 5.15428 5.18092 0.52
c 13.7835 13.7852 0.01
=β 90 90 0 120 120 0

Clausthal Seminar: 3 June 2015 30
Modelling Li(Nb, Ta)O3 solid solutions
• Two approaches:– Mean field calculation: take LiNbO3 and treat the Nb site as
an averaged (Nb, Ta) site.• With good potentials, can predict structure-composition
relationship.
– Supercell calculation: model a supercell of LiNbO3 with Ta substituted for the Nb as required.• Takes more time, but gives more detailed results.

Clausthal Seminar: 3 June 2015 31
Photos from my last visit to Clausthal (August 2001)

Thank you!32Clausthal Seminar: 3 June 2015