creatine release, potassium-42 content, mechanical...
TRANSCRIPT

Creatine Kinase Release, Potassium-42 Content, andMechanical Performance in Anoxic Rabbit Myocardium
GARYL. CONRAD,EIuc E. RAu, and KENNETHI. SHINE, Departments of Medicineand Physiology, and the American Heart Association, Greater Los AngelesAffiliate Cardiovascular Research Laboratory, University of California atLos Angeles, Centerfor the Health Sciences, Los Angeles, California 90024
A B S T R AC T We studied whether creatine kinaseappearance in venous effluent was specific for, andquantitatively proportional to, the amount of loss offunctioning myocardium. Cell viability was determinedby simultaneously monitoring tissue 42K content andmechanical performance during anoxia and reoxygena-tion in isolated, arterially perfused, interventricularrabbit septa. The septa were paced at 42 beats/minand perfused at 1.8 ml/min per g tissue with a modifiedTyrode solution at 28°C. Net total creatine kinaselosses of 5.3±2.7, 20.6+7.2, 55.3±7.6, and 110.7+27.1IU/g dry wt (mean+SEM) were observed after 20, 30,40, and 60 min of anoxia, respectively. Maximum 42Klosses during the same intervals of anoxia were 16.8±3.4, 38.3+2.9, 47.0±1.4, and 84.3±14.8 mmol K+/kgdry wt and correlated with creatine kinase losses,r = 0.97. Upon reoxygenation, 42K content returned toa new plateau which was expressed as a percentageof decrease from control content. These unrecovered42K losses were -2.7+0.9, 0.7+2.9, 6.6+1.9, and 14.0±6.5% after 20,30,40, and 60 min of anoxia, respectively,and correlated with the creatine kinase loss, r = 0.97.Net loss of developed tension after reoxygenation was9.0±2.3, 26.7+17.9, 31.7+1.1, and 60.7±8.8% of con-trol after these anoxic intervals and correlated withcreatine kinase loss, r = 0.92. The small enzyme lossthat occurred after 20 min anoxia without evidencefor irreversible loss of cell function was _0.1% oftotal tissue enzyme content. The significant correlationof enzyme loss with the irreversible losses of potas-sium content and contractile performance supportedthe hypothesis that creatine kinase appearance in thevenous effluent was the result of cell death.
Address reprint requests to Dr. Conrad at Harvey 402, TheJohns Hopkins Hospital, Baltimore, Md. 21205.
Received for publication 28 August 1978 and in revisedform 26 February 1979.
INTRODUCTION
Creatine kinase (CK)l is used as an enzyme markerfor the identification and quantification of irreversibleloss of cell function and myocardial necrosis (1, 2).This approach is based primarily upon studies correlat-ing CK losses with histological estimates of myocardialnecrosis (1, 3, 4). Yet the specificity of CKappearancefor myocardial cell necrosis has been questioned andstudies in humans and animals have suggested CK re-lease in the absence of overt myocardial cell injuryand without proportionality to the extent of identifiedinjury (5-7). The isolated, arterially perfused, inter-ventricular rabbit septum provides a model for studyingCKappearance in the effluent while monitoring otherparameters of cellular viability including cellular 42Kcontent and mechanical function. Because 98%of myo-cardial potassium is intracellular (8), tissue 42K activityis an excellent real time marker of cellular integrityand viability to follow during stresses. Previous datafrom this laboratory have characterized the mechanicalevents and changes in potassium content during anoxiain this model (9). In this study we will demonstratethe CK appearance in the effluent is proportional tothe amount of irreversible 42K loss and the degree ofinjury to mechanical performance after anoxic stressesin rabbit myocardium.
METHODSExperimental preparations. The experimental prepara-
tions were isolated, arterially perfused, interventricular septaof 1.5-2.0-kg male New Zealand white rabbits, according tothe techniques described by Rau et al. (9). After hepariniza-tion (10,000 U U. S. Pharmacopeia i.v.) and anesthesia (180mg pentobarbital i.v.) the hearts were rapidly removed, theseptal artery branch of the left coronary artery was cannulatedwith a polyethylene cannula, and the septa were perfused
'Abbreviation used in this paper: CK, creatine kinase.
155J. Clin. Invest. ( The American Society for Clinical Investigation, Inc. 0021-9738/79/07/0155/07 $1.00Volume 64 July 1979 155-161

at a constant flow by a Harvard pump (Harvard ApparatusCo., Inc., Millis, Mass.). A triangular piece of well-perfusedseptum (0.8-1.1 g wet wt) was dissected and a silk sutureused to suspend the apex from a Statham UC4 transducer(Statham Instruments, Inc., Oxnard, Calif.) for tensionmonitoring. The base of the tissue was secured by twoopposing forceps. The transducer recorded only that vectorof tension developed along the axis of the transducer.The proportion of total force represented by this vector re-mained constant throughout each experiment. Resting tensionwas maintained at 5 g, with septa being accepted for studyif they developed a minimum of 15 g tension, 100 g/s positivedT/dt. Perfusate temperature was maintained at 28°C bypassing current through a 200-ohm power resistor placedaround the metal perfusion cannula which was attached tothe polyethylene cannula. Electrodes from a Grass Stimulator(model SD9, Grass Instrument Co., Qunicv, NMass.) attachedto the harmon forceps paced the septa at 42 beats/ruiin. Thetissues were perfused at 1.8 cm3/min per g wet wvt with amodified Tyrode solution containing (in millimolars): NaCl,130; NaHCO3, 12: MgCl2, 1.0; KCl, 5.0; CaCl2, 1.5; NaH2PO4,0.435; dextrose, 5.6 or 20. Solutions were equilibrated with98% 02/2% CO2 or for anoxia, 98% N2 and 2% CO2. Underthese conditions, the tissues reached a steady state formechanical performance, CK release, and ionic and watercontent, after a gain of water and a loss of potassium overthe initial 60-180 min of perfusion (9). 42K (as KCl, NewEngland Nuclear, Boston, Mass.) was added to perfusate tomaintain the final concentration at 5 mMpotassium. The 42Kactivity of the entire muscle was continuously monitoredon-line by a Nal crystal (Nuclear Enterprises, Inc., SanCarlos, Calif.). Details of the isotopic procedure are reportedelsewhere (9). The septa remained stable for 5-7 h, whichallowed for full evaluations of anoxic stresses. It was thuspossible to continuously and simultaneously monitormechanical performance parameters and potassium contentwhile collecting effluent below the septum for enzymeanalysis.
Analytical procedures. 1-cm3 samples of effluent for CKanalysis were collected and stored in 0.01 Mmercaptoethanol,0.01 MEGTA, at 40C. All samples were analyzed 3-7 d aftereach experiment, to allow the 42K to decay, over which timeCK was verified to be 95% stable (10). The amount of CKappearing in the effluent was quantified by integrating thearea within effluent curve minus the extrapolated steady-stateCK appearance. At the onset of anoxia a decrease in the CKappearing in the effluent was usually observed (Fig. 3). Thisamount of CK which did not appear in the effluent duringthe first minutes of anoxia was quantitated and subtractedfrom the CK loss that occurred during the periods of anoxicstress and recovery. This correction decreased the calculatedCK loss that occurred. The necessity for making this subtrac-tion is evidenced in Fig. 1. During quiescence, a transient fallin the level of CK appearing in the effluent was observed.Upon restimulating the septum at 42/min, a transient increasein CK levels appeared, probably because of a direct effectof increased mechanical squeezing. Presumably, this samemechanism operated at the onset of anoxia, as developedtension quickly fell, and at reoxygenation as developedtension increased (Figs. 3 and 4).
By using the modified Rosalki procedure, samples wereanalyzed via coupled enzyme reactions monitoring linearNADHproduction at 340 nm, 30°C, in a Gilford spectro-photometer (Gilford Instruments Laboratories Inc., Oberlin,Ohio). Assay packs were obtained from Calbiochem (Calbio-chem-Behring Corp., American Hoechst Corp., San Diego,Calif.) and CK standards from Hyland (Hyland DiagnosticsDiv., Travenol Laboratories, Inc., Costa Mesa, Calif.). Ef-fluent samples were diluted in a solution containing 0.2%
bovine serum albumin and 0.02 MTris to keep the final CKconcentrations between 10 and 300 IU/liter. Effluent samplesplus reaction mixture showed no NADHproduction in the ab-sence of creatine phosphate. Total tissue CK content wasanalyzed by a method modified from Roberts et al. (11).After being weighed and minced, the tissues were homo-genized in a solution containing 0.25 M sucrose, 0.01 MTris, 0.001 NI EGTA, 0.01 mercaptoethanol, pH 7.4, for three10-s bursts of a Polytron (model PCU-2-110 generator No.PT lOST, Brinkmann Instruments, Inc., Westbury, N. Y.).After centrifugation at 2,000 g for 10 ruin, the supernates wereretained and the pellets were rewashed. More than 75% ofthe total recoverable CK consistentlyv appeared in the firstsupernate, 20% in the second, and <5% in subsequent washeswith this method. CK activity in the first acnd secondl washeswas subse(luently added together as total activity.
Experimental procedure. A 150-ruin equilibration periodelapsed before any experimental intervention. When 42Kcounts in the tissue approached asymptotic levels andlmechanical performance was stable, anoxia was indtuced byswitching to identical solution equilibrated with N2/CO2. Afterthe anoxic stress, 42K cou-nts and tension were moniitored forat least 90( min while collecting effluent samples below theseptum at regular intervals for CKanalysis. At the end of eachexperiment the tissue was blotted and dried for 24 h at110°C. The tissue was reweighed to obtain the dry weight,then dissolved in acid and counted for 42K coontenit dleter-minations (9).
Statistics. Regression coefficients were determined by themethod of least squares. Correlation coefficients were cal-culated by standard techniques and used to estimate the pro-portioIn of the variance of one variable that could be at-tributed to its linear regression on the second.
RESULTS
CK loss in the absence of anoxia. As previouslyreported (9), the 42K counts in the perfused septunreached an asymptotic value within 150-180 min (Fig.1). The 42K couints, corrected for decay, and mechanicalparameters remained stable for 5-7 h. After 3 h of per-fusion the CK content of the effluent from septa hadplateaued at 0.4-0.8 IU/irin per g dry wt, or <0.01%/ruin of the total septal CK. This low level of CK ap-pearance corresponds with physiologic levels andlfurther proves the stability of the preparation. In oneexperiment the stimulator was turned off at 264 iuin(Fig. 1). A decline in appearance of CK was observed.At 274 min the stimulation- was resumed. CKcontent ofthe effluent increased abruptly, returning to the pre-(quiescent levels after 6 min.
CK loss during anoxia. Fig. 2 demionstrates thetypical responses to 20 min of anoxia. At 170 min themuscle was perfused with anoxic solution. Tension and42K activity immediately declined. Reoxygenationi at190 min resulted in recovery of tension and 42K contentto new stable levels. The 42K counts always recoveredto a level > 100% of conitrol during recovery from 20 minof anoxia. In the 20-min anoxic studies CKappeared inthe effluent only after reoxygenation. In the experimentillustrated by Fig. 2, the CK loss was calculated to be5 IU/g dry wt.
156 G. L. Conrad, E. E. Rau,, and K. I. Shine

RABBIT42K UPTAKE,CK RELEASE280C, 42 BEATS/ MINCONTROL
TIME (min)
FIGURE 1 CK effluent appearance, 42K counts, and tensionfor a septum with no anoxic intervention. Dots represent42K content (left ordinate) which increased after loading beganat 40 min and plateaued at 180 min. Solid line of effluent CKactivity (right ordinate) fell rapidly and began to level off at230 min. The tension fell slowly as the resting tension de-creased and was restretched at 210 min to maintain a rest-ing tension of 5 g. Stimulator was turned off from 264 to 274min resulting in a fall and then a brief peak in enzyme ap-
pearance.
Fig. 3 demonstrates changes seen during and after30 min of anoxia. In addition to the fall in developedtension during anoxia, the resting tension increasedand did not return to preanoxia levels after reoxygena-
tion. The septum lost 34 mmol K+/kg dry wt duringanoxia and returned to a new plateau level after reoxy-
genation that was 5 mmol K+/kg dry wt less than con-
trol. This potassium loss was termed unrecoveredpotassium. The rate of CK appearance in the effluentdeclined during anoxia, increased upon reoxygenation,and achieved its peak level 20 min after reoxygenation.
The effects of 40 min of anoxia are seen in Fig. 4.
RABBIT42K UPTAKE, CK RELEASE28°C 42 BEATS/MIN
100- 20 MiN ANOXIA
322 mmol K'306 mmdKC 297mmol K* kg Dry Wt
x80- .-0* kg Dry Wt kg Dry Wt
Z i 20 MIN ANOXIA
~60 w
Z 24t 0- 150.
TENSIONIg) -5
20;: .1
0.5 D
0 Qo~~~~~~~~~~~~~~~~~80 160 240 320
TIME (min)
FIGURE 2 42K counts, (dots), CK release (solid line), and ten-sion with 20 min of anoxia. There was a small fall in 42Kcounts during anoxia with the characteristic rise to a higherplateau after reoxygenation.
TIME (min)
FIGURE 3 42K counts, CKappearance (solid line), and tensionwith 30 min of anoxia. Resting tension rose during anoxiawith incomplete return to preanoxia levels after reoxygena-tion. Note that CKappearance decreased initially during theanoxia stress as developed tension fell.
Spontaneous premature contractions occurred at160-170 min and resulted in a loss of developed ten-sion. Anoxia was initiated at 210 min. After 40 min a
significant rise in resting tension was observed. Uponreoxygenation developed tension recovered to a new
level for a net loss of 39% from the preanoxia level.Potassium content declined from 244 mmol K+/kg drywt before anoxia to a postreoxygenation level of 235mmol K+/kg dry wt, representing an unrecovered lossof 4%of the control content. The rate of CKappearancein the effluent began to increase before reoxygenationin this and all anoxic stresses longer than 30 min. TheCKappearance rate was accelerated by reoxygenationand the peak level occurred 20 min later. The rate of
0
z
"I
Hfz
0
yN
It
210 270 330 390
TIME (min)
2.0
- 1.5
*0
-1.0 .
DY
-0.5
FIGURE 4 42K counts, CKappearance (solid line), and tensionwith 40 min of anoxia. Fall in tension at 165 min resultedfrom spontaneous ectopy. Note that CK appearance initiallydecreased during anoxia then began increasing at 30 min, be-fore reoxygenation.
Creatine Kinase in Anoxic Rabbit Myocardium
X-0
Z
z
0C-)
53
0
>cV -
- C)
E Z>%:D
e 00
o >-
5;>-0
yNl
157
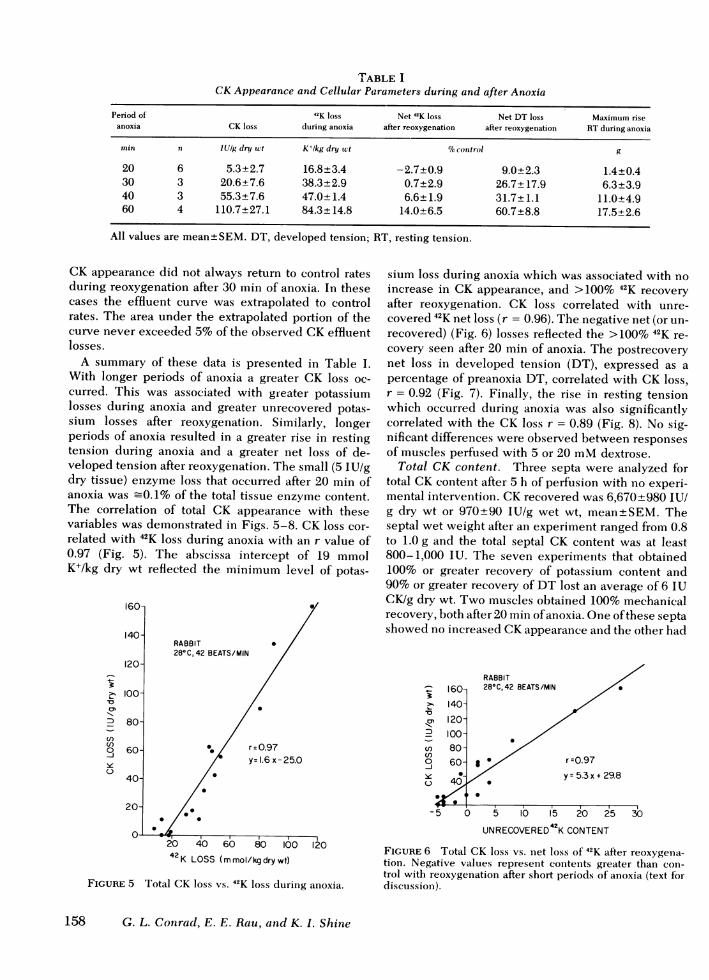
TABLE ICKAppearance and Cellular Parameters during and after Anoxia
Period of 42K loss Net 42K loss Net DT loss Maximum riseanoxia CK loss during anoxia after reoxygenation after reoxygenation RTduring anoxia
min n lUig dry wct K+lkg dry wt %ocontrol g
20 6 5.3±2.7 16.8±3.4 -2.7±0.9 9.0±2.3 1.4±0.430 3 20.6±7.6 38.3±2.9 0.7±2.9 26.7± 17.9 6.3±3.940 3 55.3±7.6 47.0±1.4 6.6±1.9 31.7±1.1 11.0±4.960 4 110.7±27.1 84.3±+14.8 14.0±6.5 60.7+8.8 17.5±2.6
All values are mean±SEM. DT, developed tension; RT, resting tension.
CK appearance did not always return to control ratesduring reoxygenation after 30 min of anoxia. In thesecases the effluent curve was extrapolated to controlrates. The area under the extrapolated portion of thecurve never exceeded 5%of the observed CKeffluentlosses.
A summary of these data is presented in Table I.With longer periods of anoxia a greater CK loss oc-curred. This was associated with greater potassiumlosses during anoxia and greater unrecovered potas-sium losses after reoxygenation. Similarly, longerperiods of anoxia resulted in a greater rise in restingtension during anoxia and a greater net loss of de-veloped tension after reoxygenation. The small (5 IU/gdry tissue) enzyme loss that occurred after 20 min ofanoxia was -0.1% of the total tissue enzyme content.The correlation of total CK appearance with thesevariables was demonstrated in Figs. 5-8. CK loss cor-related with 42K loss during anoxia with an r value of0.97 (Fig. 5). The abscissa intercept of 19 mmolK+/kg dry wt reflected the minimum level of potas-
160
140-RABBIT *28°C, 42 BEATS/MIN
120-
> 100-C0D 80-
C')
O 60- r0.97y=1.6x-25.0
40-
20- *
O- *20 40 60 80 100 12042K LOSS (mmol/kg dry wt)
FIGURE 5 Total CK loss vs. 42K loss during anoxia.
sium loss during anoxia which was associated with noincrease in CK appearance, and >100% 42K recoveryafter reoxygenation. CK loss correlated with unre-covered 42K net loss (r = 0.96). The negative net (or un-recovered) (Fig. 6) losses reflected the >100% 42K re-covery seen after 20 min of anoxia. The postrecoverynet loss in developed tension (DT), expressed as apercentage of preanoxia DT, correlated with CK loss,r = 0.92 (Fig. 7). Finally, the rise in resting tensionwhich occurred during anoxia was also significantlycorrelated with the CK loss r = 0.89 (Fig. 8). No sig-nificant differences were observed between responsesof muscles perfused with 5 or 20 mMdextrose.
Total CK content. Three septa were analyzed fortotal CK content after 5 h of perfusion with no experi-mental intervention. CK recovered was 6,670+980 IU/g dry wt or 970±90 IU/g wet wt, mean±SEM. Theseptal wet weight after an experiment ranged from 0.8to 1.0 g and the total septal CK content was at least800-1,000 IU. The seven experiments that obtained100% or greater recovery of potassium content and90% or greater recovery of DT lost an average of 6 IUCK/g dry wt. Two muscles obtained 100% mechanicalrecovery, both after 20 min of anoxia. One of these septashowed no increased CKappearance anid the other had
0l
C')C')
-JU)
160-140-
,,120-100-80-60-
I '
RABBIT/28°C,42 BEATS/MIN
J r =0.97
/ ~ ~~~~~~y=5.3 x +29.8
-5 ) 5 10 15 20 25
UNRECOVERED42K CONTENT
30
FIGURE 6 Total CK loss vs. net loss Of 42K after reoxygena-tion. Negative values represent contents greater than coni-trol with reoxygenation after short periods of anoxia (text fordiscussioni).
158 G. L. Conrad, E. E. Rau, and K. I. Shine

1I00-
D 80-cn0-J 60-y0
40-
20-
RABBIT28° C, 42 BEATS/MIN
0
0
r=0.92y= 1.9 x-12.9
0
20 40 60 80 100NET LOSS DEVELOPEDTENSION
(% CONTROL)
FIGURE 7 Total CK loss vs. net loss of developed tensionafter recovery.
a total of CK appearance of 12 IU/g dry wt, =0.2% ofthe average total CKcontent.
DISCUSSION
The appearance of CK in serum is used as a bio-chemical marker for the identification and quantifica-tion of myocardial necrosis (1, 2). However conflictingevidence exists in the literature concerning the inter-pretation of elevated CK levels. Evidence supporting
>%11
_
cn
d3)0"03
60- RABBIT28C,42 BEATS/min
40-0
20-r =0.89
00- y= 5.9 x-5.5
80-~~~~~~~30~~~~~~~~~
60- 0
40- -0
20-
4 8 12 16 20MAX RISE RESTING TENSION (g)
FIGuRE 8 Total CK loss vs. maximum rise in resting tensionduring anoxia.
the use of serum CK levels as an index of cell deathrests largely on data from canine experiments andperfused whole heart experiments and suggests thatmembrane disruption associated with cell death allowsthe appearance of CK in serum. Hearse and Humphrey(12) used nonbeating, anoxic whole heart preparationsfrom several species to study CK release and showedclose correlations between CKappearance, the falls inATP and creatine phosphate levels, and disruption ofcellular ultrastructure. Using a canine preparationAhmed et al. (3) evaluated serum CKand histologicalchanges after ischemia and found that large CK ap-pearances were highly correlated with histochemicallyproven cell death. Evidence against the use of CK ap-pearances as a marker of cell death argue that smallincreases in serum enzyme levels could be related tothe state of cellular energy levels or cellular membraneintegrity, and not to cell death and disruption of the cel-lular membrane (13). Small rises in serum CK levelshave been observed with no histochemical evidence ofcell death in ischemic dog hearts (3) or loss of mechan-ical performance in ischemic guinea pig hearts (7).Similarly, Klein et al. (6) observed increases in serumlevels of the MBisoenzyme of CKafter cardiovascularsurgery when electrocardiogram and radionucleotidescans failed to detect myocardial cell necrosis.
Similar conflicting information exists concerning theuse of elevated serum CK levels as an index of the sizeor amount of cell necrosis. Shell et al. (1) found thattotal CKappearance in the serum was proportional tothe amount of CK depletion in the area of experi-mental infarction, and Kjekshus and Sobel (4) showedthat CK losses correlated with histologically deter-mined sizes of infarction. Contraindicating the use ofserum CK levels as an index of the amount of cellnecrosis are the results of studies by Jarmakani et al.(5) and Maroko and Vatner (14), which showed a largeeffect of reperfusion on the amount of CK releasedand would therefore allow prediction of infarct sizefrom CKdata only if the reperfusion effect was knownand quantified. Despite these discrepancies, theamount of CK appearing after a myocardial infarctionor intervention is being used as a measure of theamount of cell necrosis (15). Use of an isolated, con-trolled, experimental preparation allowed us to directlyand simultaneously evaluate CK release, cellular via-bility as measured by the ability to maintain or re-cover a potassium gradient, and mechanical perform-ance in the heart.
We used the isolated, arterially perfused, beating,intraventricular rabbit septum as the preparation forthese studies. This preparation allowed complete con-trol of the entire tissue being stressed by anoxia in-cluding control of: temperature, beating rate, enddiastolic tension (rest tension), perfusate content,and rate of perfusion, and permitted simultaneous,
Creatine Kinase in Anoxic Rabbit Myocardium 159

real time monitoring of contractile performance, tissuepotassium content (42K), and appearance of CK in theeffluent. Because 98% of tissue 42K is intracellular (8),monitoring tissue counts was a sensitive index of cel-lular 42K content and therefore of the cellular sarcolem-mal membrane integrity and performance (9). Post-anoxic recovery of tissue 42K counts served as an indica-tion of cell viability as it has previously been shownthat irreversibly injured cells do not recover theirpotassium content while reversibly injured ones can(16). CKappearance in the effluent was substantial for150 min after cannulation of the isolated septum (Fig.1). This initial CK appearance averaged 80 IU andrepresented losses up to 13% of the total tissue CKcontent. These losses apparently resulted from injuryto cells during dissection and cannulation. ThereafterCK loss diminished to a level <0.01%o/min of total tis-sue CK and the septa remained mechanically stable,and maintained their 42K content for 5-7 h. During thisstable period we followed enzyme appearance, 42Kcounts, and tension during anoxia and after reoxygena-tion.
Our results showed a significant correlation be-tween CK appearance and the loss of potassium andtension after anoxic stress. Mean changes of mechanicalfunction, CK appearance in the effluent, and 42K con-tent after four different time periods of anoxia weresummarized in Table I. CK appearance always fellduring anoxia as the developed tension decreased.Upon reoxygenation the effluent enzyme concentrationincreased and peaked at 20 min. With longer periodsof anoxia CK appearance began to increase at 30 minbut further increased upon reoxygenation. This ac-celeration in rate of appearance upon reoxygenationcould have resulted from a mechanical squeeze as thedeveloped tension increased (17), in addition to thedirect cellular effects of oxygen described in the studiesof Hearse and Humphrey (12). Potassium losses duringanoxia have been shown to be due to increased effluxof ions from cells despite the presence of high energyphosphate levels sufficient to maintain the Na-K pump(9). The potassium lost during the period of anoxiawas highly correlated with the CK appearance in theeffluent (Fig. 5). The recovery of potassium contentwas a direct measurement of cellular viability becauseirreversibly injured cells cannot regain their potassiumcontent. Recovery of potassium content to levelsslightly higher than control values after 20 min ofanoxia has been previously observed and was due to adiminished efflux of potassium after reoxygenation (9).This observation explains the ordinate intercept seenin Fig. 6. With longer periods of anoxia the potas-sium content did not return to preanoxia levels afterreoxygenation. During recovery after 60 min anoxia,four septa recovered an average of 86% of the controlpotassium content, representing a net loss of 14%. This
evidence for loss of cellular viability was associatedwith a proportional increase in CKeffluent appearance(Fig. 6). The mean CK losses after the 60 min of anoxiawas _2%of the total septal CKcontent, or one-seventhof the observed net potassium loss. A similar propor-tionality of 1:7 has been observed in experimentalmodels using CK depletion from infarcted myocar-dium, where only one-seventh of the depleted CKis ac-counted for in the serum (2). This disappearance ofenzyme was presumed to be due to degradation or inac-tivation and could explain the discrepancy we ob-served between the total CK losses and potassiumlosses after the various periods of anoxia. The sig-nificant correlation between unrecovered potassiumcontent and CKappearance was direct evidence for theappearance of the enzyme correlating with the amountof loss of nonfunctioning myocardium.
Mechanical parameters have previously been used tofollow cellular viability and function (9). Loss of de-veloped tension, after anoxia, although not as a directa measurement of irreversible cell damage as potas-sium loss, correlated well with total CK loss. Rest-ing tension rises during anoxia, because irreversiblydamaged myocardium goes into contracture. Thisparameter was also proportional and well correlatedwith the total CK loss.
Seven septa obtained 100% or greater recovery ofpotassium and 90% or greater recovery of developedtension. Concerning the question of specificity of CKappearance for cell necrosis, a small increase in CKappearance occurred in several of these septa, averag-ing 6±3 IU/g dry wt, or 0.1% of the total tissue levelsof CK. After correcting for the estimated 1:7 propor-tionality of CK appearance:CK loss, the quantity ofenzyme lost by these tissues obtaining 90-100%recovery was < 1%of the total CKcontent of the tissue.This small loss may possibly have reflected the greatersensitivity of the effluent enzyme appearance for a verysmall quantity of cellular necrosis. We could not dif-ferentiate a loss of CKdue to irreversible damage to a1% fraction of less well perfused cells from a smallCK "leak" from viable cells with 100% recovery, butthe contribution of such a leak would have been <1%.
This experimental model, involving a stable beatingventricular septum with physiologic perfusion rates,allows several further observations on CKrelease. Ourstudies confirmed the importance of contractile func-tion, probably occurring through increased mechanicalsqueezing, influencing enzyme appearance in the ef-fluent perfusate. This has been previously describedfor lactate dehydrogenase (17). Also, previous studiesof CKrelease during graded hypoxia may be influencednot only by mechanical effects but also by the strikinginhomogeneity of the hypoxic insult which mayactually be anoxic in some areas with adequate oxy-genation in others (18). The degree of mechanical im-
160 G. L. Conrad, E. E. Rau, and K. I. Shine

pairment and the rate of recovery of contractile func-tion during or after tissue injury significantly in-fluenced the time course of appearance of the enzyme.
Using the postanoxic recovery of 42K content as amarker of cellular viability while measuring mechan-ical performance and CK losses, we obtained a signifi-cant correlation between the net total CK loss and boththe loss of 42K content and loss of contractile perform-ance. The amount of CK that appeared in the venouseffluent, in several experiments in the absence of loss ofcell function, was a very small percentage of the totaltissue content of CK. CK appeared to be a quantita-tive marker for cell death.
ACKNOWLEDGMENTS
The research for this paper was supported by grant HL11351-11 from the U. S. Public Health Service and grant 570from the American Heart Association, Greater Los AngelesAffiliate.
REFERENCES
1. Shell, W. E., J. K. Kjekshus, and B. E. Sobel. 1971.Quantitative assessment of the extent of myocardial in-farction in the conscious dog by means of analysis ofserial changes in serum creatine phosphokinase activity.
J. Clin. Invest. 50: 2614-2625.2. Sobel, B. E., R. Roberts, and K. B. Larson. 1976. Con-
siderations in the use of biochemical markers of ischemicinjury. Circ. Res. 38: I-99-I-108.
3. Ahmed, S. A., J. R. Williamson, R. Roberts, R. E. Clark,and B. E. Sobel. 1976. The association of increased plasmaMB CPK activity and irreversible ischemic myocardialinjury in the dog. Circulation. 54: 187-193.
4. Kjekshus, J. K., and B. E. Sobel, 1970. Depressed myo-cardial creatine phosphokinase activity following experi-mental myocardial infarction in rabbit. Circ. Res. 27:403-414.
5. Jarmakani, J. M., L. Limbird, T. C. Graham, and R. A.Marks. 1976. Effect of reperfusion on myocardial infarct,and the accuracy of estimating infaret size from serum
creatine phosphokinase in the dog. Cardiovasc. Res. 10:245-253.
6. Klein, M. S., R. E. Coleman, C. S. Weldon, B. E. Sobel, andR. Roberts. 1976. Concordance of electrocardiographicand scintigraphic criteria of myocardial injury aftercardiac surgery. J. Thorac. Cardiovasc. Surg. 71: 934-937.
7. Sakai, K., M. M. Gebhard, P. G. Spieckermann, and J. H.Bretschneider. 1975. Enzyme release resulting from totalischemia and reperfusion in the isolated, perfused guineapig heart. J. Mol. Cell. Cardiol. 7: 827-840.
8. Blesa, E. S., G. A. Langer, A. J. Brady, and S. D. Serena.1970. Potassium exchange in rat ventricular myocardium:its relation to rate of stimulation. Am. J. Physiol. 219:747-754.
9. Rau, E. E., K. I. Shine, and G. A. Langer. 1977. Potas-sium exchange and mechanical performance in anoxicmammalian myocardium. Am. J. Physiol. 232(1): H85-H94.
10. Morin, L. G. 1977. Creatine kinase: stability, inactivation,reactivation. Clin. Chem. 23: 646-652.
11. Roberts, R., K. S. Gowda, P. A. Ludbrook, and B. E.Sobel. 1975. Specificity of elevated serum MB creatinephosphokinase activity in the diagnosis of acute myo-cardial infarction. Am. J. Cardiol. 36: 433-437.
12. Hearse, D. J., and S. M. Humphrey. 1975. Enzymerelease during myocardial anoxia: a study of metabolicprotection.J. Mol. Cell. Cardiol. 7: 463-482.
13. Gebhard, M. M., H. Denhaus, K. Sakai, and P. G. Spiecker-mann. 1977. Energy metabolism and enzyme release. J.Mol. Med. 2: 271-283.
14. Maroko, P. R., and S. F. Vatner. 1977. Altered relation-ship between phosphokinase and infarct size with reper-fusion in conscious dogs. J. Mol. Med. 2: 309-315.
15. Sobel, B. E., and W. E. Shell. 1975. Diagnostic and prog-nostic value of serum enzyme changes in patients withacute myocardial infarction. Prog. Cardiol. 8: 165-198.
16. Jennings, R. B., H. M. Sommers, J. P. Kaltenbach, andJ. J. West. 1964. Electrolyte alterations in acute myo-cardial ischemic injury. Circ. Res. 14: 260-269.
17. DeLeiris, J., and D. Feuvray. 1973. Factors affecting therelease of lactate dehydrogenase from isolated rat heartafter calcium and magnesium free perfusion. Cardiovasc.Res. 7: 383-390.
18. Steenbergen, C., G. Deleeuw, C. Barlow, B. Change, andJ. R. Williamson. 1977. Heterogeneity of the hypoxicstate in perfused rat heart. Circ. Res. 41: 606-615.
Creatine Kinase in Anoxic Rabbit Myocardium 161