disclaimer - dspace.inha.ac.kr · _`mvxyz ' + o & p...
TRANSCRIPT

저 시-비 리- 경 지 20 한민
는 아래 조건 르는 경 에 한하여 게
l 저 물 복제 포 전송 전시 공연 송할 수 습니다
다 과 같 조건 라야 합니다
l 하는 저 물 나 포 경 저 물에 적 된 허락조건 명확하게 나타내어야 합니다
l 저 터 허가를 면 러한 조건들 적 되지 않습니다
저 에 른 리는 내 에 하여 향 지 않습니다
것 허락규약(Legal Code) 해하 쉽게 약한 것 니다
Disclaimer
저 시 하는 원저 를 시하여야 합니다
비 리 하는 저 물 리 목적 할 수 없습니다
경 지 하는 저 물 개 형 또는 가공할 수 없습니다
공학석사학위논문
Random Forest를 이용한 단백질에서의
RNA 결합 부위 예측
Prediction of RNA binding sites in proteins
using a random forest
2012년 2월
인하대학교 대학원
정보공학과
최 혁 진
공학석사학위논문
Random Forest를 이용한 단백질에서의
RNA 결합 부위 예측
Prediciton of RNA binding sites in proteins
using a random forest
2012년 2월
지도교수 한 경 숙
이 논문을 석사학위 논문으로 제출함
인하대학교 대학원
정보공학과
최 혁 진
이 논문을 최혁진의 석사학위논문으로 인정함
2012년 2월 일
주심
부심
위심
- iv -
목 차
그림 목차 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotvi
표 목차 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotvii
국문 요약 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotviii
영문 요약 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot ix
제 1 장 서 론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot1
제 2 장 관련 연구 및 배경 지식 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
21 Protein Data Bank middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
22 Dictionary of protein secondary structure middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
23 Random Forest middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot6
231 결정 트리 분류기 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot6
232 결정 트리의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
233 결정 트리의 설계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
234 결정 트리의 학습 알고리즘 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot11
235 결정 트리의 특성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot13
236 Random Forest의 설계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
24 특징 벡터 기반의 중복 데이터 제거 기법 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot17
25 Ensemble of Under-Sampling middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
제 3 장 데이터 구성 및 특징 선택 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
31 단백질-RNA 결합부위 데이터 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
32 단백질-RNA 결합 부위 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
33 단백질 서열을 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
34 단백질 아미노산 triplet을 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
35 단백질 이차구조를 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot23
36 상대방 RNA를 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
- v -
37 특징 벡터 (feature vector)의 표현 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
38 예측 알고리즘의 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot32
제 4 장 시험 및 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
41 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
42 Random Forest를 이용한 예측 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
43 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot37
44 타 연구 시험과의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot39
45 다른 데이터 셋에서의 테스트 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
제 5 장 결론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot44
참고 문헌 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot45
- vi -
그림 목차
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot4
그림 2 DSSP 데이터베이스의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
그림 3 결정 트리의 개략도 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
그림 4 일반적인 결정 트리 타입 분류기의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
그림 5 결정 트리의 재귀적 트리 성장 프로세스 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot9
그림 6 질문의 판정에 의한 잎 노드의 분할 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot10
그림 7 Random Forest의 모델 구축 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot16
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot19
그림 11 수소 결합의 결합 각도와 거리에 관한 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예 middot31
- vii -
표 목차
표 1 PDB에서 사용된 실험 방법의 통계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot25
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 26
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot27
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 28
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수 middotmiddotmiddotmiddotmiddot 34
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot34
표 8 특징들을 모두 사용하여 10-fold cross validation한 결과 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교 middotmiddotmiddotmiddot 36
표 10 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot38
표 11 Liu et al 논문과 본 논문과의 방법 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot40
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot41
표 13 PR324와 PR102에서의 예측 테스트 성능 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
- viii -
요 약
단백질과 RNA의 상호작용은 단백질 합성 유전자 발현과 복제 바이러스에
의한 감염과 같은 생물학 과정에서 중요한 역할을 하고 있기 때문에 단백질
-RNA 상호작용 부위에 대한 정보는 단백질과 RNA의 기능을 규명하거나 잠재
적으로 질병을 유발하는 단백질과 RNA의 결합을 억제하거나 촉진할 수 있는
신약 개발에 유용하게 사용될 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 그러나 이 기법들은 주어진 단백질과 상호작용하는 상대방
인 RNA를 고려하지 않기 때문에 단백질이 결합하는 RNA가 달라져도 주어진
단백질에 대하여 항상 같은 결합부위를 예측한다
본 연구에서는 단백질 서열뿐만 아니라 RNA 서열을 함께 고려하여 단백질
서열에서 잠재적으로 RNA와 결합하는 부위를 예측하는 Random Forest 분류기
를 논한다 아미노산과 염기 간의 결합 성향은 Protein Data Bank (PDB)에서
단백질-RNA 복합체 분석을 통해 얻었고 Random Forest 분류기의 특징 벡터
에 표현하였다
특징으로 단백질 서열에서 인접한 아미노산 3개의 결합 성향 상대방 RNA
서열의 길이와 염기의 빈도수 단백질 이차 구조 아미노산의 생화학적 특징 등
을 이용하여 RNA와 결합 가능한 아미노산을 예측하는 Random Forest 분류기
를 개발하였다 429개의 단백질-RNA 복합체에서 추출한 3149개의 단백질
-RNA 서열 쌍을 대상으로 한 시험에서 이 Random Forest 분류기는 820의
sensitivity 981의 specificity 962의 accuracy의 예측 성능을 보였다
- ix -
Abstract
Protein-RNA interactions play an important role in a number of biological
processes such as protein synthesis gene expression and replication and viral
infections Information on protein-RNA interaction sites will be useful in
finding the function of proteins and RNA and can facilitate the development of
new drugs against diseases caused by protein-RNA interactions
Recently several machine learning approaches have been attempted to
predict RNA-binding residues in amino acid sequences However none of
these consider interacting partners (ie RNA) for a given protein when
predicting RNA binding amino acids They always predict the same
RNA-binding residues for a given protein even if the protein binds to different
RNA molecules
In this study we present a random forest classifier that takes an RNA
sequence as well as a protein sequence as input and predicts potential RNA
binding residues in the protein sequence The interaction propensity between
an amino acid and RNA nucleotide obtained from the extensive analysis of
protein-RNA complexes in the Protein Data Bank (PDB) was encoded in the
feature vector for the random forest classifier
Various properties of protein and RNA such as interaction propensity of
amino acid triplets RNA sequence length nucleotide frequency in the RNA
sequence protein secondary structure biochemical properties of amino acids
were used to train the random forest classifier On a dataset of 3149
protein-RNA sequence pairs extracted from 429 protein-RNA complexes the
random forest classifier achieved a sensitivity of 820 specificity of 981
and accuracy of 962
- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

공학석사학위논문
Random Forest를 이용한 단백질에서의
RNA 결합 부위 예측
Prediction of RNA binding sites in proteins
using a random forest
2012년 2월
인하대학교 대학원
정보공학과
최 혁 진
공학석사학위논문
Random Forest를 이용한 단백질에서의
RNA 결합 부위 예측
Prediciton of RNA binding sites in proteins
using a random forest
2012년 2월
지도교수 한 경 숙
이 논문을 석사학위 논문으로 제출함
인하대학교 대학원
정보공학과
최 혁 진
이 논문을 최혁진의 석사학위논문으로 인정함
2012년 2월 일
주심
부심
위심
- iv -
목 차
그림 목차 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotvi
표 목차 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotvii
국문 요약 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotviii
영문 요약 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot ix
제 1 장 서 론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot1
제 2 장 관련 연구 및 배경 지식 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
21 Protein Data Bank middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
22 Dictionary of protein secondary structure middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
23 Random Forest middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot6
231 결정 트리 분류기 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot6
232 결정 트리의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
233 결정 트리의 설계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
234 결정 트리의 학습 알고리즘 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot11
235 결정 트리의 특성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot13
236 Random Forest의 설계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
24 특징 벡터 기반의 중복 데이터 제거 기법 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot17
25 Ensemble of Under-Sampling middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
제 3 장 데이터 구성 및 특징 선택 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
31 단백질-RNA 결합부위 데이터 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
32 단백질-RNA 결합 부위 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
33 단백질 서열을 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
34 단백질 아미노산 triplet을 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
35 단백질 이차구조를 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot23
36 상대방 RNA를 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
- v -
37 특징 벡터 (feature vector)의 표현 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
38 예측 알고리즘의 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot32
제 4 장 시험 및 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
41 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
42 Random Forest를 이용한 예측 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
43 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot37
44 타 연구 시험과의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot39
45 다른 데이터 셋에서의 테스트 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
제 5 장 결론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot44
참고 문헌 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot45
- vi -
그림 목차
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot4
그림 2 DSSP 데이터베이스의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
그림 3 결정 트리의 개략도 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
그림 4 일반적인 결정 트리 타입 분류기의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
그림 5 결정 트리의 재귀적 트리 성장 프로세스 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot9
그림 6 질문의 판정에 의한 잎 노드의 분할 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot10
그림 7 Random Forest의 모델 구축 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot16
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot19
그림 11 수소 결합의 결합 각도와 거리에 관한 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예 middot31
- vii -
표 목차
표 1 PDB에서 사용된 실험 방법의 통계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot25
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 26
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot27
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 28
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수 middotmiddotmiddotmiddotmiddot 34
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot34
표 8 특징들을 모두 사용하여 10-fold cross validation한 결과 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교 middotmiddotmiddotmiddot 36
표 10 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot38
표 11 Liu et al 논문과 본 논문과의 방법 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot40
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot41
표 13 PR324와 PR102에서의 예측 테스트 성능 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
- viii -
요 약
단백질과 RNA의 상호작용은 단백질 합성 유전자 발현과 복제 바이러스에
의한 감염과 같은 생물학 과정에서 중요한 역할을 하고 있기 때문에 단백질
-RNA 상호작용 부위에 대한 정보는 단백질과 RNA의 기능을 규명하거나 잠재
적으로 질병을 유발하는 단백질과 RNA의 결합을 억제하거나 촉진할 수 있는
신약 개발에 유용하게 사용될 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 그러나 이 기법들은 주어진 단백질과 상호작용하는 상대방
인 RNA를 고려하지 않기 때문에 단백질이 결합하는 RNA가 달라져도 주어진
단백질에 대하여 항상 같은 결합부위를 예측한다
본 연구에서는 단백질 서열뿐만 아니라 RNA 서열을 함께 고려하여 단백질
서열에서 잠재적으로 RNA와 결합하는 부위를 예측하는 Random Forest 분류기
를 논한다 아미노산과 염기 간의 결합 성향은 Protein Data Bank (PDB)에서
단백질-RNA 복합체 분석을 통해 얻었고 Random Forest 분류기의 특징 벡터
에 표현하였다
특징으로 단백질 서열에서 인접한 아미노산 3개의 결합 성향 상대방 RNA
서열의 길이와 염기의 빈도수 단백질 이차 구조 아미노산의 생화학적 특징 등
을 이용하여 RNA와 결합 가능한 아미노산을 예측하는 Random Forest 분류기
를 개발하였다 429개의 단백질-RNA 복합체에서 추출한 3149개의 단백질
-RNA 서열 쌍을 대상으로 한 시험에서 이 Random Forest 분류기는 820의
sensitivity 981의 specificity 962의 accuracy의 예측 성능을 보였다
- ix -
Abstract
Protein-RNA interactions play an important role in a number of biological
processes such as protein synthesis gene expression and replication and viral
infections Information on protein-RNA interaction sites will be useful in
finding the function of proteins and RNA and can facilitate the development of
new drugs against diseases caused by protein-RNA interactions
Recently several machine learning approaches have been attempted to
predict RNA-binding residues in amino acid sequences However none of
these consider interacting partners (ie RNA) for a given protein when
predicting RNA binding amino acids They always predict the same
RNA-binding residues for a given protein even if the protein binds to different
RNA molecules
In this study we present a random forest classifier that takes an RNA
sequence as well as a protein sequence as input and predicts potential RNA
binding residues in the protein sequence The interaction propensity between
an amino acid and RNA nucleotide obtained from the extensive analysis of
protein-RNA complexes in the Protein Data Bank (PDB) was encoded in the
feature vector for the random forest classifier
Various properties of protein and RNA such as interaction propensity of
amino acid triplets RNA sequence length nucleotide frequency in the RNA
sequence protein secondary structure biochemical properties of amino acids
were used to train the random forest classifier On a dataset of 3149
protein-RNA sequence pairs extracted from 429 protein-RNA complexes the
random forest classifier achieved a sensitivity of 820 specificity of 981
and accuracy of 962
- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

공학석사학위논문
Random Forest를 이용한 단백질에서의
RNA 결합 부위 예측
Prediciton of RNA binding sites in proteins
using a random forest
2012년 2월
지도교수 한 경 숙
이 논문을 석사학위 논문으로 제출함
인하대학교 대학원
정보공학과
최 혁 진
이 논문을 최혁진의 석사학위논문으로 인정함
2012년 2월 일
주심
부심
위심
- iv -
목 차
그림 목차 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotvi
표 목차 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotvii
국문 요약 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotviii
영문 요약 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot ix
제 1 장 서 론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot1
제 2 장 관련 연구 및 배경 지식 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
21 Protein Data Bank middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
22 Dictionary of protein secondary structure middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
23 Random Forest middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot6
231 결정 트리 분류기 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot6
232 결정 트리의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
233 결정 트리의 설계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
234 결정 트리의 학습 알고리즘 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot11
235 결정 트리의 특성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot13
236 Random Forest의 설계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
24 특징 벡터 기반의 중복 데이터 제거 기법 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot17
25 Ensemble of Under-Sampling middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
제 3 장 데이터 구성 및 특징 선택 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
31 단백질-RNA 결합부위 데이터 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
32 단백질-RNA 결합 부위 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
33 단백질 서열을 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
34 단백질 아미노산 triplet을 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
35 단백질 이차구조를 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot23
36 상대방 RNA를 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
- v -
37 특징 벡터 (feature vector)의 표현 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
38 예측 알고리즘의 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot32
제 4 장 시험 및 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
41 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
42 Random Forest를 이용한 예측 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
43 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot37
44 타 연구 시험과의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot39
45 다른 데이터 셋에서의 테스트 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
제 5 장 결론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot44
참고 문헌 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot45
- vi -
그림 목차
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot4
그림 2 DSSP 데이터베이스의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
그림 3 결정 트리의 개략도 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
그림 4 일반적인 결정 트리 타입 분류기의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
그림 5 결정 트리의 재귀적 트리 성장 프로세스 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot9
그림 6 질문의 판정에 의한 잎 노드의 분할 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot10
그림 7 Random Forest의 모델 구축 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot16
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot19
그림 11 수소 결합의 결합 각도와 거리에 관한 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예 middot31
- vii -
표 목차
표 1 PDB에서 사용된 실험 방법의 통계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot25
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 26
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot27
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 28
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수 middotmiddotmiddotmiddotmiddot 34
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot34
표 8 특징들을 모두 사용하여 10-fold cross validation한 결과 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교 middotmiddotmiddotmiddot 36
표 10 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot38
표 11 Liu et al 논문과 본 논문과의 방법 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot40
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot41
표 13 PR324와 PR102에서의 예측 테스트 성능 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
- viii -
요 약
단백질과 RNA의 상호작용은 단백질 합성 유전자 발현과 복제 바이러스에
의한 감염과 같은 생물학 과정에서 중요한 역할을 하고 있기 때문에 단백질
-RNA 상호작용 부위에 대한 정보는 단백질과 RNA의 기능을 규명하거나 잠재
적으로 질병을 유발하는 단백질과 RNA의 결합을 억제하거나 촉진할 수 있는
신약 개발에 유용하게 사용될 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 그러나 이 기법들은 주어진 단백질과 상호작용하는 상대방
인 RNA를 고려하지 않기 때문에 단백질이 결합하는 RNA가 달라져도 주어진
단백질에 대하여 항상 같은 결합부위를 예측한다
본 연구에서는 단백질 서열뿐만 아니라 RNA 서열을 함께 고려하여 단백질
서열에서 잠재적으로 RNA와 결합하는 부위를 예측하는 Random Forest 분류기
를 논한다 아미노산과 염기 간의 결합 성향은 Protein Data Bank (PDB)에서
단백질-RNA 복합체 분석을 통해 얻었고 Random Forest 분류기의 특징 벡터
에 표현하였다
특징으로 단백질 서열에서 인접한 아미노산 3개의 결합 성향 상대방 RNA
서열의 길이와 염기의 빈도수 단백질 이차 구조 아미노산의 생화학적 특징 등
을 이용하여 RNA와 결합 가능한 아미노산을 예측하는 Random Forest 분류기
를 개발하였다 429개의 단백질-RNA 복합체에서 추출한 3149개의 단백질
-RNA 서열 쌍을 대상으로 한 시험에서 이 Random Forest 분류기는 820의
sensitivity 981의 specificity 962의 accuracy의 예측 성능을 보였다
- ix -
Abstract
Protein-RNA interactions play an important role in a number of biological
processes such as protein synthesis gene expression and replication and viral
infections Information on protein-RNA interaction sites will be useful in
finding the function of proteins and RNA and can facilitate the development of
new drugs against diseases caused by protein-RNA interactions
Recently several machine learning approaches have been attempted to
predict RNA-binding residues in amino acid sequences However none of
these consider interacting partners (ie RNA) for a given protein when
predicting RNA binding amino acids They always predict the same
RNA-binding residues for a given protein even if the protein binds to different
RNA molecules
In this study we present a random forest classifier that takes an RNA
sequence as well as a protein sequence as input and predicts potential RNA
binding residues in the protein sequence The interaction propensity between
an amino acid and RNA nucleotide obtained from the extensive analysis of
protein-RNA complexes in the Protein Data Bank (PDB) was encoded in the
feature vector for the random forest classifier
Various properties of protein and RNA such as interaction propensity of
amino acid triplets RNA sequence length nucleotide frequency in the RNA
sequence protein secondary structure biochemical properties of amino acids
were used to train the random forest classifier On a dataset of 3149
protein-RNA sequence pairs extracted from 429 protein-RNA complexes the
random forest classifier achieved a sensitivity of 820 specificity of 981
and accuracy of 962
- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

이 논문을 최혁진의 석사학위논문으로 인정함
2012년 2월 일
주심
부심
위심
- iv -
목 차
그림 목차 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotvi
표 목차 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotvii
국문 요약 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotviii
영문 요약 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot ix
제 1 장 서 론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot1
제 2 장 관련 연구 및 배경 지식 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
21 Protein Data Bank middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
22 Dictionary of protein secondary structure middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
23 Random Forest middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot6
231 결정 트리 분류기 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot6
232 결정 트리의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
233 결정 트리의 설계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
234 결정 트리의 학습 알고리즘 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot11
235 결정 트리의 특성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot13
236 Random Forest의 설계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
24 특징 벡터 기반의 중복 데이터 제거 기법 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot17
25 Ensemble of Under-Sampling middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
제 3 장 데이터 구성 및 특징 선택 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
31 단백질-RNA 결합부위 데이터 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
32 단백질-RNA 결합 부위 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
33 단백질 서열을 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
34 단백질 아미노산 triplet을 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
35 단백질 이차구조를 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot23
36 상대방 RNA를 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
- v -
37 특징 벡터 (feature vector)의 표현 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
38 예측 알고리즘의 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot32
제 4 장 시험 및 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
41 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
42 Random Forest를 이용한 예측 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
43 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot37
44 타 연구 시험과의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot39
45 다른 데이터 셋에서의 테스트 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
제 5 장 결론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot44
참고 문헌 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot45
- vi -
그림 목차
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot4
그림 2 DSSP 데이터베이스의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
그림 3 결정 트리의 개략도 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
그림 4 일반적인 결정 트리 타입 분류기의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
그림 5 결정 트리의 재귀적 트리 성장 프로세스 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot9
그림 6 질문의 판정에 의한 잎 노드의 분할 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot10
그림 7 Random Forest의 모델 구축 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot16
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot19
그림 11 수소 결합의 결합 각도와 거리에 관한 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예 middot31
- vii -
표 목차
표 1 PDB에서 사용된 실험 방법의 통계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot25
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 26
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot27
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 28
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수 middotmiddotmiddotmiddotmiddot 34
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot34
표 8 특징들을 모두 사용하여 10-fold cross validation한 결과 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교 middotmiddotmiddotmiddot 36
표 10 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot38
표 11 Liu et al 논문과 본 논문과의 방법 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot40
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot41
표 13 PR324와 PR102에서의 예측 테스트 성능 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
- viii -
요 약
단백질과 RNA의 상호작용은 단백질 합성 유전자 발현과 복제 바이러스에
의한 감염과 같은 생물학 과정에서 중요한 역할을 하고 있기 때문에 단백질
-RNA 상호작용 부위에 대한 정보는 단백질과 RNA의 기능을 규명하거나 잠재
적으로 질병을 유발하는 단백질과 RNA의 결합을 억제하거나 촉진할 수 있는
신약 개발에 유용하게 사용될 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 그러나 이 기법들은 주어진 단백질과 상호작용하는 상대방
인 RNA를 고려하지 않기 때문에 단백질이 결합하는 RNA가 달라져도 주어진
단백질에 대하여 항상 같은 결합부위를 예측한다
본 연구에서는 단백질 서열뿐만 아니라 RNA 서열을 함께 고려하여 단백질
서열에서 잠재적으로 RNA와 결합하는 부위를 예측하는 Random Forest 분류기
를 논한다 아미노산과 염기 간의 결합 성향은 Protein Data Bank (PDB)에서
단백질-RNA 복합체 분석을 통해 얻었고 Random Forest 분류기의 특징 벡터
에 표현하였다
특징으로 단백질 서열에서 인접한 아미노산 3개의 결합 성향 상대방 RNA
서열의 길이와 염기의 빈도수 단백질 이차 구조 아미노산의 생화학적 특징 등
을 이용하여 RNA와 결합 가능한 아미노산을 예측하는 Random Forest 분류기
를 개발하였다 429개의 단백질-RNA 복합체에서 추출한 3149개의 단백질
-RNA 서열 쌍을 대상으로 한 시험에서 이 Random Forest 분류기는 820의
sensitivity 981의 specificity 962의 accuracy의 예측 성능을 보였다
- ix -
Abstract
Protein-RNA interactions play an important role in a number of biological
processes such as protein synthesis gene expression and replication and viral
infections Information on protein-RNA interaction sites will be useful in
finding the function of proteins and RNA and can facilitate the development of
new drugs against diseases caused by protein-RNA interactions
Recently several machine learning approaches have been attempted to
predict RNA-binding residues in amino acid sequences However none of
these consider interacting partners (ie RNA) for a given protein when
predicting RNA binding amino acids They always predict the same
RNA-binding residues for a given protein even if the protein binds to different
RNA molecules
In this study we present a random forest classifier that takes an RNA
sequence as well as a protein sequence as input and predicts potential RNA
binding residues in the protein sequence The interaction propensity between
an amino acid and RNA nucleotide obtained from the extensive analysis of
protein-RNA complexes in the Protein Data Bank (PDB) was encoded in the
feature vector for the random forest classifier
Various properties of protein and RNA such as interaction propensity of
amino acid triplets RNA sequence length nucleotide frequency in the RNA
sequence protein secondary structure biochemical properties of amino acids
were used to train the random forest classifier On a dataset of 3149
protein-RNA sequence pairs extracted from 429 protein-RNA complexes the
random forest classifier achieved a sensitivity of 820 specificity of 981
and accuracy of 962
- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- iv -
목 차
그림 목차 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotvi
표 목차 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotvii
국문 요약 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotviii
영문 요약 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot ix
제 1 장 서 론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot1
제 2 장 관련 연구 및 배경 지식 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
21 Protein Data Bank middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
22 Dictionary of protein secondary structure middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
23 Random Forest middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot6
231 결정 트리 분류기 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot6
232 결정 트리의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
233 결정 트리의 설계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
234 결정 트리의 학습 알고리즘 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot11
235 결정 트리의 특성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot13
236 Random Forest의 설계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
24 특징 벡터 기반의 중복 데이터 제거 기법 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot17
25 Ensemble of Under-Sampling middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
제 3 장 데이터 구성 및 특징 선택 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
31 단백질-RNA 결합부위 데이터 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
32 단백질-RNA 결합 부위 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot21
33 단백질 서열을 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
34 단백질 아미노산 triplet을 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
35 단백질 이차구조를 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot23
36 상대방 RNA를 고려한 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
- v -
37 특징 벡터 (feature vector)의 표현 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
38 예측 알고리즘의 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot32
제 4 장 시험 및 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
41 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
42 Random Forest를 이용한 예측 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
43 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot37
44 타 연구 시험과의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot39
45 다른 데이터 셋에서의 테스트 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
제 5 장 결론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot44
참고 문헌 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot45
- vi -
그림 목차
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot4
그림 2 DSSP 데이터베이스의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
그림 3 결정 트리의 개략도 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
그림 4 일반적인 결정 트리 타입 분류기의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
그림 5 결정 트리의 재귀적 트리 성장 프로세스 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot9
그림 6 질문의 판정에 의한 잎 노드의 분할 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot10
그림 7 Random Forest의 모델 구축 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot16
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot19
그림 11 수소 결합의 결합 각도와 거리에 관한 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예 middot31
- vii -
표 목차
표 1 PDB에서 사용된 실험 방법의 통계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot25
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 26
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot27
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 28
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수 middotmiddotmiddotmiddotmiddot 34
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot34
표 8 특징들을 모두 사용하여 10-fold cross validation한 결과 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교 middotmiddotmiddotmiddot 36
표 10 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot38
표 11 Liu et al 논문과 본 논문과의 방법 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot40
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot41
표 13 PR324와 PR102에서의 예측 테스트 성능 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
- viii -
요 약
단백질과 RNA의 상호작용은 단백질 합성 유전자 발현과 복제 바이러스에
의한 감염과 같은 생물학 과정에서 중요한 역할을 하고 있기 때문에 단백질
-RNA 상호작용 부위에 대한 정보는 단백질과 RNA의 기능을 규명하거나 잠재
적으로 질병을 유발하는 단백질과 RNA의 결합을 억제하거나 촉진할 수 있는
신약 개발에 유용하게 사용될 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 그러나 이 기법들은 주어진 단백질과 상호작용하는 상대방
인 RNA를 고려하지 않기 때문에 단백질이 결합하는 RNA가 달라져도 주어진
단백질에 대하여 항상 같은 결합부위를 예측한다
본 연구에서는 단백질 서열뿐만 아니라 RNA 서열을 함께 고려하여 단백질
서열에서 잠재적으로 RNA와 결합하는 부위를 예측하는 Random Forest 분류기
를 논한다 아미노산과 염기 간의 결합 성향은 Protein Data Bank (PDB)에서
단백질-RNA 복합체 분석을 통해 얻었고 Random Forest 분류기의 특징 벡터
에 표현하였다
특징으로 단백질 서열에서 인접한 아미노산 3개의 결합 성향 상대방 RNA
서열의 길이와 염기의 빈도수 단백질 이차 구조 아미노산의 생화학적 특징 등
을 이용하여 RNA와 결합 가능한 아미노산을 예측하는 Random Forest 분류기
를 개발하였다 429개의 단백질-RNA 복합체에서 추출한 3149개의 단백질
-RNA 서열 쌍을 대상으로 한 시험에서 이 Random Forest 분류기는 820의
sensitivity 981의 specificity 962의 accuracy의 예측 성능을 보였다
- ix -
Abstract
Protein-RNA interactions play an important role in a number of biological
processes such as protein synthesis gene expression and replication and viral
infections Information on protein-RNA interaction sites will be useful in
finding the function of proteins and RNA and can facilitate the development of
new drugs against diseases caused by protein-RNA interactions
Recently several machine learning approaches have been attempted to
predict RNA-binding residues in amino acid sequences However none of
these consider interacting partners (ie RNA) for a given protein when
predicting RNA binding amino acids They always predict the same
RNA-binding residues for a given protein even if the protein binds to different
RNA molecules
In this study we present a random forest classifier that takes an RNA
sequence as well as a protein sequence as input and predicts potential RNA
binding residues in the protein sequence The interaction propensity between
an amino acid and RNA nucleotide obtained from the extensive analysis of
protein-RNA complexes in the Protein Data Bank (PDB) was encoded in the
feature vector for the random forest classifier
Various properties of protein and RNA such as interaction propensity of
amino acid triplets RNA sequence length nucleotide frequency in the RNA
sequence protein secondary structure biochemical properties of amino acids
were used to train the random forest classifier On a dataset of 3149
protein-RNA sequence pairs extracted from 429 protein-RNA complexes the
random forest classifier achieved a sensitivity of 820 specificity of 981
and accuracy of 962
- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- v -
37 특징 벡터 (feature vector)의 표현 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot29
38 예측 알고리즘의 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot32
제 4 장 시험 및 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
41 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot33
42 Random Forest를 이용한 예측 성능 평가 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
43 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot37
44 타 연구 시험과의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot39
45 다른 데이터 셋에서의 테스트 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
제 5 장 결론 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot44
참고 문헌 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot45
- vi -
그림 목차
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot4
그림 2 DSSP 데이터베이스의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
그림 3 결정 트리의 개략도 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
그림 4 일반적인 결정 트리 타입 분류기의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
그림 5 결정 트리의 재귀적 트리 성장 프로세스 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot9
그림 6 질문의 판정에 의한 잎 노드의 분할 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot10
그림 7 Random Forest의 모델 구축 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot16
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot19
그림 11 수소 결합의 결합 각도와 거리에 관한 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예 middot31
- vii -
표 목차
표 1 PDB에서 사용된 실험 방법의 통계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot25
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 26
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot27
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 28
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수 middotmiddotmiddotmiddotmiddot 34
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot34
표 8 특징들을 모두 사용하여 10-fold cross validation한 결과 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교 middotmiddotmiddotmiddot 36
표 10 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot38
표 11 Liu et al 논문과 본 논문과의 방법 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot40
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot41
표 13 PR324와 PR102에서의 예측 테스트 성능 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
- viii -
요 약
단백질과 RNA의 상호작용은 단백질 합성 유전자 발현과 복제 바이러스에
의한 감염과 같은 생물학 과정에서 중요한 역할을 하고 있기 때문에 단백질
-RNA 상호작용 부위에 대한 정보는 단백질과 RNA의 기능을 규명하거나 잠재
적으로 질병을 유발하는 단백질과 RNA의 결합을 억제하거나 촉진할 수 있는
신약 개발에 유용하게 사용될 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 그러나 이 기법들은 주어진 단백질과 상호작용하는 상대방
인 RNA를 고려하지 않기 때문에 단백질이 결합하는 RNA가 달라져도 주어진
단백질에 대하여 항상 같은 결합부위를 예측한다
본 연구에서는 단백질 서열뿐만 아니라 RNA 서열을 함께 고려하여 단백질
서열에서 잠재적으로 RNA와 결합하는 부위를 예측하는 Random Forest 분류기
를 논한다 아미노산과 염기 간의 결합 성향은 Protein Data Bank (PDB)에서
단백질-RNA 복합체 분석을 통해 얻었고 Random Forest 분류기의 특징 벡터
에 표현하였다
특징으로 단백질 서열에서 인접한 아미노산 3개의 결합 성향 상대방 RNA
서열의 길이와 염기의 빈도수 단백질 이차 구조 아미노산의 생화학적 특징 등
을 이용하여 RNA와 결합 가능한 아미노산을 예측하는 Random Forest 분류기
를 개발하였다 429개의 단백질-RNA 복합체에서 추출한 3149개의 단백질
-RNA 서열 쌍을 대상으로 한 시험에서 이 Random Forest 분류기는 820의
sensitivity 981의 specificity 962의 accuracy의 예측 성능을 보였다
- ix -
Abstract
Protein-RNA interactions play an important role in a number of biological
processes such as protein synthesis gene expression and replication and viral
infections Information on protein-RNA interaction sites will be useful in
finding the function of proteins and RNA and can facilitate the development of
new drugs against diseases caused by protein-RNA interactions
Recently several machine learning approaches have been attempted to
predict RNA-binding residues in amino acid sequences However none of
these consider interacting partners (ie RNA) for a given protein when
predicting RNA binding amino acids They always predict the same
RNA-binding residues for a given protein even if the protein binds to different
RNA molecules
In this study we present a random forest classifier that takes an RNA
sequence as well as a protein sequence as input and predicts potential RNA
binding residues in the protein sequence The interaction propensity between
an amino acid and RNA nucleotide obtained from the extensive analysis of
protein-RNA complexes in the Protein Data Bank (PDB) was encoded in the
feature vector for the random forest classifier
Various properties of protein and RNA such as interaction propensity of
amino acid triplets RNA sequence length nucleotide frequency in the RNA
sequence protein secondary structure biochemical properties of amino acids
were used to train the random forest classifier On a dataset of 3149
protein-RNA sequence pairs extracted from 429 protein-RNA complexes the
random forest classifier achieved a sensitivity of 820 specificity of 981
and accuracy of 962
- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- vi -
그림 목차
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot4
그림 2 DSSP 데이터베이스의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot5
그림 3 결정 트리의 개략도 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot7
그림 4 일반적인 결정 트리 타입 분류기의 구조 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot8
그림 5 결정 트리의 재귀적 트리 성장 프로세스 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot9
그림 6 질문의 판정에 의한 잎 노드의 분할 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot10
그림 7 Random Forest의 모델 구축 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot14
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot16
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot18
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot19
그림 11 수소 결합의 결합 각도와 거리에 관한 정의 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot22
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예 middot31
- vii -
표 목차
표 1 PDB에서 사용된 실험 방법의 통계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot25
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 26
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot27
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 28
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수 middotmiddotmiddotmiddotmiddot 34
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot34
표 8 특징들을 모두 사용하여 10-fold cross validation한 결과 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교 middotmiddotmiddotmiddot 36
표 10 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot38
표 11 Liu et al 논문과 본 논문과의 방법 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot40
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot41
표 13 PR324와 PR102에서의 예측 테스트 성능 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
- viii -
요 약
단백질과 RNA의 상호작용은 단백질 합성 유전자 발현과 복제 바이러스에
의한 감염과 같은 생물학 과정에서 중요한 역할을 하고 있기 때문에 단백질
-RNA 상호작용 부위에 대한 정보는 단백질과 RNA의 기능을 규명하거나 잠재
적으로 질병을 유발하는 단백질과 RNA의 결합을 억제하거나 촉진할 수 있는
신약 개발에 유용하게 사용될 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 그러나 이 기법들은 주어진 단백질과 상호작용하는 상대방
인 RNA를 고려하지 않기 때문에 단백질이 결합하는 RNA가 달라져도 주어진
단백질에 대하여 항상 같은 결합부위를 예측한다
본 연구에서는 단백질 서열뿐만 아니라 RNA 서열을 함께 고려하여 단백질
서열에서 잠재적으로 RNA와 결합하는 부위를 예측하는 Random Forest 분류기
를 논한다 아미노산과 염기 간의 결합 성향은 Protein Data Bank (PDB)에서
단백질-RNA 복합체 분석을 통해 얻었고 Random Forest 분류기의 특징 벡터
에 표현하였다
특징으로 단백질 서열에서 인접한 아미노산 3개의 결합 성향 상대방 RNA
서열의 길이와 염기의 빈도수 단백질 이차 구조 아미노산의 생화학적 특징 등
을 이용하여 RNA와 결합 가능한 아미노산을 예측하는 Random Forest 분류기
를 개발하였다 429개의 단백질-RNA 복합체에서 추출한 3149개의 단백질
-RNA 서열 쌍을 대상으로 한 시험에서 이 Random Forest 분류기는 820의
sensitivity 981의 specificity 962의 accuracy의 예측 성능을 보였다
- ix -
Abstract
Protein-RNA interactions play an important role in a number of biological
processes such as protein synthesis gene expression and replication and viral
infections Information on protein-RNA interaction sites will be useful in
finding the function of proteins and RNA and can facilitate the development of
new drugs against diseases caused by protein-RNA interactions
Recently several machine learning approaches have been attempted to
predict RNA-binding residues in amino acid sequences However none of
these consider interacting partners (ie RNA) for a given protein when
predicting RNA binding amino acids They always predict the same
RNA-binding residues for a given protein even if the protein binds to different
RNA molecules
In this study we present a random forest classifier that takes an RNA
sequence as well as a protein sequence as input and predicts potential RNA
binding residues in the protein sequence The interaction propensity between
an amino acid and RNA nucleotide obtained from the extensive analysis of
protein-RNA complexes in the Protein Data Bank (PDB) was encoded in the
feature vector for the random forest classifier
Various properties of protein and RNA such as interaction propensity of
amino acid triplets RNA sequence length nucleotide frequency in the RNA
sequence protein secondary structure biochemical properties of amino acids
were used to train the random forest classifier On a dataset of 3149
protein-RNA sequence pairs extracted from 429 protein-RNA complexes the
random forest classifier achieved a sensitivity of 820 specificity of 981
and accuracy of 962
- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- vii -
표 목차
표 1 PDB에서 사용된 실험 방법의 통계 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot3
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot25
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 26
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot27
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot 28
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수 middotmiddotmiddotmiddotmiddot 34
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot34
표 8 특징들을 모두 사용하여 10-fold cross validation한 결과 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot35
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교 middotmiddotmiddotmiddot 36
표 10 Random Forest와 SVM의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot38
표 11 Liu et al 논문과 본 논문과의 방법 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot40
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot41
표 13 PR324와 PR102에서의 예측 테스트 성능 middotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddotmiddot42
- viii -
요 약
단백질과 RNA의 상호작용은 단백질 합성 유전자 발현과 복제 바이러스에
의한 감염과 같은 생물학 과정에서 중요한 역할을 하고 있기 때문에 단백질
-RNA 상호작용 부위에 대한 정보는 단백질과 RNA의 기능을 규명하거나 잠재
적으로 질병을 유발하는 단백질과 RNA의 결합을 억제하거나 촉진할 수 있는
신약 개발에 유용하게 사용될 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 그러나 이 기법들은 주어진 단백질과 상호작용하는 상대방
인 RNA를 고려하지 않기 때문에 단백질이 결합하는 RNA가 달라져도 주어진
단백질에 대하여 항상 같은 결합부위를 예측한다
본 연구에서는 단백질 서열뿐만 아니라 RNA 서열을 함께 고려하여 단백질
서열에서 잠재적으로 RNA와 결합하는 부위를 예측하는 Random Forest 분류기
를 논한다 아미노산과 염기 간의 결합 성향은 Protein Data Bank (PDB)에서
단백질-RNA 복합체 분석을 통해 얻었고 Random Forest 분류기의 특징 벡터
에 표현하였다
특징으로 단백질 서열에서 인접한 아미노산 3개의 결합 성향 상대방 RNA
서열의 길이와 염기의 빈도수 단백질 이차 구조 아미노산의 생화학적 특징 등
을 이용하여 RNA와 결합 가능한 아미노산을 예측하는 Random Forest 분류기
를 개발하였다 429개의 단백질-RNA 복합체에서 추출한 3149개의 단백질
-RNA 서열 쌍을 대상으로 한 시험에서 이 Random Forest 분류기는 820의
sensitivity 981의 specificity 962의 accuracy의 예측 성능을 보였다
- ix -
Abstract
Protein-RNA interactions play an important role in a number of biological
processes such as protein synthesis gene expression and replication and viral
infections Information on protein-RNA interaction sites will be useful in
finding the function of proteins and RNA and can facilitate the development of
new drugs against diseases caused by protein-RNA interactions
Recently several machine learning approaches have been attempted to
predict RNA-binding residues in amino acid sequences However none of
these consider interacting partners (ie RNA) for a given protein when
predicting RNA binding amino acids They always predict the same
RNA-binding residues for a given protein even if the protein binds to different
RNA molecules
In this study we present a random forest classifier that takes an RNA
sequence as well as a protein sequence as input and predicts potential RNA
binding residues in the protein sequence The interaction propensity between
an amino acid and RNA nucleotide obtained from the extensive analysis of
protein-RNA complexes in the Protein Data Bank (PDB) was encoded in the
feature vector for the random forest classifier
Various properties of protein and RNA such as interaction propensity of
amino acid triplets RNA sequence length nucleotide frequency in the RNA
sequence protein secondary structure biochemical properties of amino acids
were used to train the random forest classifier On a dataset of 3149
protein-RNA sequence pairs extracted from 429 protein-RNA complexes the
random forest classifier achieved a sensitivity of 820 specificity of 981
and accuracy of 962
- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- viii -
요 약
단백질과 RNA의 상호작용은 단백질 합성 유전자 발현과 복제 바이러스에
의한 감염과 같은 생물학 과정에서 중요한 역할을 하고 있기 때문에 단백질
-RNA 상호작용 부위에 대한 정보는 단백질과 RNA의 기능을 규명하거나 잠재
적으로 질병을 유발하는 단백질과 RNA의 결합을 억제하거나 촉진할 수 있는
신약 개발에 유용하게 사용될 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 그러나 이 기법들은 주어진 단백질과 상호작용하는 상대방
인 RNA를 고려하지 않기 때문에 단백질이 결합하는 RNA가 달라져도 주어진
단백질에 대하여 항상 같은 결합부위를 예측한다
본 연구에서는 단백질 서열뿐만 아니라 RNA 서열을 함께 고려하여 단백질
서열에서 잠재적으로 RNA와 결합하는 부위를 예측하는 Random Forest 분류기
를 논한다 아미노산과 염기 간의 결합 성향은 Protein Data Bank (PDB)에서
단백질-RNA 복합체 분석을 통해 얻었고 Random Forest 분류기의 특징 벡터
에 표현하였다
특징으로 단백질 서열에서 인접한 아미노산 3개의 결합 성향 상대방 RNA
서열의 길이와 염기의 빈도수 단백질 이차 구조 아미노산의 생화학적 특징 등
을 이용하여 RNA와 결합 가능한 아미노산을 예측하는 Random Forest 분류기
를 개발하였다 429개의 단백질-RNA 복합체에서 추출한 3149개의 단백질
-RNA 서열 쌍을 대상으로 한 시험에서 이 Random Forest 분류기는 820의
sensitivity 981의 specificity 962의 accuracy의 예측 성능을 보였다
- ix -
Abstract
Protein-RNA interactions play an important role in a number of biological
processes such as protein synthesis gene expression and replication and viral
infections Information on protein-RNA interaction sites will be useful in
finding the function of proteins and RNA and can facilitate the development of
new drugs against diseases caused by protein-RNA interactions
Recently several machine learning approaches have been attempted to
predict RNA-binding residues in amino acid sequences However none of
these consider interacting partners (ie RNA) for a given protein when
predicting RNA binding amino acids They always predict the same
RNA-binding residues for a given protein even if the protein binds to different
RNA molecules
In this study we present a random forest classifier that takes an RNA
sequence as well as a protein sequence as input and predicts potential RNA
binding residues in the protein sequence The interaction propensity between
an amino acid and RNA nucleotide obtained from the extensive analysis of
protein-RNA complexes in the Protein Data Bank (PDB) was encoded in the
feature vector for the random forest classifier
Various properties of protein and RNA such as interaction propensity of
amino acid triplets RNA sequence length nucleotide frequency in the RNA
sequence protein secondary structure biochemical properties of amino acids
were used to train the random forest classifier On a dataset of 3149
protein-RNA sequence pairs extracted from 429 protein-RNA complexes the
random forest classifier achieved a sensitivity of 820 specificity of 981
and accuracy of 962
- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- ix -
Abstract
Protein-RNA interactions play an important role in a number of biological
processes such as protein synthesis gene expression and replication and viral
infections Information on protein-RNA interaction sites will be useful in
finding the function of proteins and RNA and can facilitate the development of
new drugs against diseases caused by protein-RNA interactions
Recently several machine learning approaches have been attempted to
predict RNA-binding residues in amino acid sequences However none of
these consider interacting partners (ie RNA) for a given protein when
predicting RNA binding amino acids They always predict the same
RNA-binding residues for a given protein even if the protein binds to different
RNA molecules
In this study we present a random forest classifier that takes an RNA
sequence as well as a protein sequence as input and predicts potential RNA
binding residues in the protein sequence The interaction propensity between
an amino acid and RNA nucleotide obtained from the extensive analysis of
protein-RNA complexes in the Protein Data Bank (PDB) was encoded in the
feature vector for the random forest classifier
Various properties of protein and RNA such as interaction propensity of
amino acid triplets RNA sequence length nucleotide frequency in the RNA
sequence protein secondary structure biochemical properties of amino acids
were used to train the random forest classifier On a dataset of 3149
protein-RNA sequence pairs extracted from 429 protein-RNA complexes the
random forest classifier achieved a sensitivity of 820 specificity of 981
and accuracy of 962
- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 1 -
제 1 장 서 론
세포내의 단백질은 다른 단백질과의 상호작용뿐 아니라 RNA 또는 DNA 분
자와의 상호작용을 통하여 그 기능을 수행하기 때문에 단백질-RNA 상호작용
과 단백질-DNA 상호작용이 단백질의 기능 수행을 위해서 중요하다 예를 들
면 transcription factor는 promoter에 있는 DNA 모티브와 결합함으로써 전사
(transcription)를 활성 또는 억제한다 [1]
단백질과 RNA의 상호작용은 라이보솜의 assembly와 생물학적 기능 진핵생
물의 splicesome에 필수적인 역할을 하는 것으로 알려져 있다 [2 3]
pre-mRNA processing splicing polyadenylation mRNA 운반 및 번역 등의
다양한 과정을 포함하는 전사 후 조절은 특정 RNA-결합 단백질 또는 RNA-결
합 단백질이 다른 단백질과의 상호작용을 통하여 가능하다 [4 5]
단백질과 RNA가 생명 현상 유지 및 유전자의 형질 발현 조절 그리고 바이
러스 감염 등과 같은 과정에 상호작용을 통해 이루어진다는 사실이 점차 밝혀
지면서 단백질과 RNA 상호작용에 대한 연구가 활발히 이루어지고 있다 단백
질과 RNA의 상호작용을 연구하는 것은 질병 치료와 신약 개발에 매우 유용하
게 활용될 수 있다 일반적으로 신약개발을 하기 위해서는 무수히 반복되는 실
험이 수반되어야 하며 그 과정에서 막대한 비용과 시간이 소모된다 본 연구에
서 예측한 단백질-RNA 결합부위를 활용하여 표적 단백질을 한정시킴으로써 실
험 횟수를 현저하게 줄일 수 있다
최근 여러 기계학습 기법이 단백질 서열에서 RNA와 결합하는 잔기를 예측
하는데 사용되었다 대표적인 연구로서 Support Vector Machine (SVM)을 이
용한 BindN [6]과 Naive Bayes Classifier를 이용한 RNABindR [7]은 단백질
서열에서 RNA와 결합하는 아미노산을 예측한다
본 연구에서는 단백질 서열뿐만 아니라 상호작용하는 상대방인 RNA 서열과
도 함께 고려하며 단백질 이차구조의 특징을 개발하여 단백질 서열에서 잠재적
으로 RNA와 결합하는 부위를 예측하는 Random Forest 모델 [8]을 개발하였다
이 Random Forest 모델은 특징 벡터로서 실험적으로 밝혀진 아미노산의 생화
- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 2 -
학적 특징 normalized position 단백질 서열에서 인접한 아미노산 3개의 결합
성향 단백질의 이차구조 상대방 RNA 서열의 길이와 염기의 빈도수 등을 이용
하고 특징 벡터에 기반을 두어 데이터 중복이 없는 학습 데이터를 생성하였다
[14] 그 결과 429개의 단백질-RNA 복합체 데이터에서 sensitivity = 820
specificity = 981 accuracy = 962 예측 성능을 가지고 결합부위를 예측하
였다
본 논문은 Protein Data Bank (PDB) [9]로부터 단백질-RNA 결합부위를 추
출하는 방법 학습 데이터를 생성하는 과정 예측에 사용된 단백질과 RNA의 특
징 예측 시험 결과 등을 논한다
- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 3 -
Exp Method Molecule Type
Protein RNAProteinRNA
ComplexesOther Total
X-ray 62152 453 948 2926 66479
NMR 7888 400 68 675 9031
Microscopy 258 21 92 5 376
Other 215 4 6 18 243
Total 70513 878 1114 3624 76129
제 2 장 관련 연구 및 배경 지식
21 Protein Data Bank
Protein Data Bank (PDB) [9]는 실험적으로 밝혀진 생체내의 거대 분자들의
구조들에 대한 정보들을 모아 놓은 데이터베이스로서 1971년에 미국의
Brookhaven National Laboratory (BNL)에서 처음 구축하여 현재 전 세계적으
로 이용되고 있다
PDB에 저장된 각각의 분자 데이터들은 PDB ID에 의해 구분되고 X-ray
crystallography NMR (Nuclear Magnetic Resonance) Electron Microscopy 등
과 같은 방법을 이용하여 밝혀진 실험 데이터들로 구성되어있다 그리고 이 데
이터들을 기반으로 하여 얻어진 원자들의 삼차원 좌표 데이터 분자 서열정보
삼차원 구조 영상 등을 제공하고 있다
표 1 PDB에서 사용된 실험 방법의 통계
표 1은 PDB 웹 사이트에서 2011년 9월을 기준으로 실험적으로 밝혀진 분자
구조 데이터의 개수를 실험 방법 별로 구성한 표이다 PDB 파일 형식은 text로
된 파일 형식으로서 PDB에 저장되어 있는 분자들의 3차원 구조를 설명하는데
- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 4 -
사용된다 이 파일은 원자 위치정보 서열 정보 그리고 구조를 정하고 주석으로
첨부한 연구자들에 대한 정보를 포함하며 추가적으로 정보가 수록될 수도 있
다
그림 1 PDB에서 제공하는 시각화 프로그램인 Jmol을 이용한 PDB ID 2GJW
의 삼차원 구조 영상
- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 5 -
22 Dictionary of protein secondary structure
단백질의 이차구조는 backbone chain 간의 수소 결합에 의하여 생기는 구조
이다 DSSP 프로그램 [12]은 단백질 이차 구조 분류의 표준을 위해 Wolfgang
Kabsch와 Chris Sander에 의해서 설계되었다
DSSP는 Protein Data Bank (PDB)에 있는 구조가 알려져 있는 모든 단백질
항목에 대한 이차 구조를 계산해 준다 DSSP 프로그램은 단백질의 이차 구조
를 여덟 가지 (H alpha helix B residue in isolated beta-bridge E extended
strand and participates in beta ladder G 3-helix(310 helix) I 5 helix(pi
helix) T hydrogen bonded turn S bend X otherwise)로 분류한다
본 연구에서는 여덟 가지의 분류를 Helix Sheet Turn Coil의 네 가지 이차
구조 유형으로 줄여서 사용한다 (IGH rarr Helix EB rarr Sheet T rarr Turn
SX rarr Coil)
그림 2 DSSP 데이터베이스의 구조
- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 6 -
23 Random Forest
기계 학습 분류 기법의 하나인 Random Forest [8]는 Breiman에 의해 개발
된 것으로 트리 (tree) 타입의 분류기들을 이용한 앙상블 (ensemble) 분류 기법
이다 [17 18] 앙상블 분류 기법은 경험적인 객체들로 몇 개의 분류기를 훈련시
키고 그 분류기들의 결정을 결합하여 가중 투표를 통해 비경험적인 객체를 분
류하는 방법이다 앙상블 기법은 트리들의 결과를 합산하여 더욱 신뢰도 높고
안정적인 분류 결과를 내놓는다는 장점이 있다 [18]
Random Forest는 동일한 뿌리 노드를 갖는 여러 결정 트리 (decision tree)
들로 구성된다 각각의 결정 트리는 마지막 노드에서 최종 결정을 하고 그 결정
들로 Random Forest의 결정이 이루어진다 따라서 Random Forest의 최종 결
정은 Forest를 구성하는 결정 트리들의 결정에 귀속되므로 결정 트리의 생성방
법과 결정 트리의 수가 알고리즘의 정확도와 처리속도를 결정하는 변수가 된다
Random Forest는 학습 샘플들로 결정 트리 분류기를 생성하는 과정에서 학
습 샘플들이 갖는 방대한 데이터 안에서 효과적으로 작용하여 데이터 가공 삭
제 등의 과정 없이 데이터를 다룬다 이 과정에서 분류에 유리한 데이터를 선정
하여 어떤 데이터가 중요한지 보여준다 또한 트리 기법이 갖는 특징으로 학습
속도가 빠르고 과적합 (over fitting)하지 않는다는 장점이 있다
과도하게 복합한 분류시스템은 훈련 샘플들에 대해서는 완벽한 분류가 가능
할지 몰라도 새로운 패턴들에 대해서는 제대로 동작하지 않는 경향이 있다 랜
덤 포레스트는 크고 복잡한 시스템에서 중요한 데이터를 선택하여 분류기준을
삼으므로 빠르면서도 정확한 평가를 한다는 평을 받는다 또한 결정트리 수가
늘어날수록 정확도가 높아지다가 수렴한다는 특징을 갖는다
231 결정 트리 분류기
결정 트리의 분류방법은 뿌리 노드 (root node)에서 잎 노드 (leaf node)까지
결정 결정이 가능한 질문을 통해 최종 의사결정을 하는 방법이다 이때 결정
트리는 대개 일련의 간단한 질문을 사용한다
- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 7 -
그림 3은 결정 트리 분류기를 묘사한 그림이다 화살표는 질문에 의한 경호
는 뜻한다 그림 3에 입력된 객체는 네 가지 질문에 의해 최종 결과 판정을 받
았다 이와 같이 결정 트리는 트리의 해당 잎 노드로의 경로 판정들로 특정 패
턴에 대한 판정을 쉽게 해석할 수 있다는 장점이 있다
결정 트리는 입력된 객체의 모든 데이터를 분류 속성들과 한꺼번에 비교하
지 않고 순차적인 방법으로 분류된 서브데이터를 다른 질문을 통해 또다시 분
류하는 방법을 사용하기 때문에 빠르면서도 정확한 분류를 한다 [18]
그림 3 결정 트리의 개략도
232 결정 트리의 구조
결정 트리는 최상위 뿌리 노드에서부터 분지 (branch)를 시작하여 각 노드의
하위 노드에 연결된다 이들은 더 이상 경로가 없는 노드 또는 마지막 잎 노드
에 도달할 때까지 연결된다 트리의 각 노드에는 분류 되어야 할 데이터 공간을
둘로 나눌 수 있는 간단한 판정 (test) 조건이 들어 있다
특정 패턴의 분류는 패턴의 특정 특성의 값을 요구하는 뿌리 노드에서 시작
한다 패턴이 트리에 특징벡터의 형태로 입력되었을 때 입력된 벡터의 특정 요
소의 값을 판정하여 경로를 결정한다 이 방법으로 마지막 노드까지 경로를 결
정한다 마지막 노드의 결과 값으로 입력 벡터가 속할 것으로 추정되는 최종 클
- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 8 -
래스를 결정한다
그림 4는 앞서 설명한 결정 트리 타입 분류기의 구조를 묘사한 것이다 트리
t (Tree t)는 뿌리 노드에서 질문1 (test 1)에서 입력된 특징벡터의 특정 요소를
판정하여 하위 노드로의 경로를 결정한다 입력된 특징벡터는 트리의 깊이만큼
판정하여 잎 노드에 도달한다 그림 4의 잎 노드에는 각 클래스별 최종 확률 값
이 들어 있다
그림 4 일반적인 결정 트리 타입 분류기의 구조
233 결정 트리의 설계
트리는 사전 지식인 학습 샘플들로부터 판정 질문을 결정하여 자연스러운
분류 방법을 제공한다 뿌리 노드에서 잎 노드까지 생성되는 서로 다른 경로는
판정에서 서로 다른 가능한 값을 갖는다는 것을 의미한다 상위 노드의 판정에
의한 결정 결정에 의하여 왼쪽과 오른쪽 경로로 하위 노드로 간다 이 방법을
- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 9 -
더 이상 질문을 갖지 않는 잎 노드에 도달할 때까지 계속하여 트리가 생성된다
그림 5는 결정 트리의 재귀적 트리의 성장 프로세스를 나타낸다 클래스가
레이블링 (labeling)된 객체들이 담긴 현재노드 (current node)에서 결정 분할
(decision splitting)을 할 것인지를 결정한다 현재 노드에 담긴 객체들의 레이블
의 분포가 또 다른 분할을 요구한다면 분할하여 하위 노드 둘을 생성한다 만
약 더 이상 분할이 필요치 않다면 루프 (loop)를 종료한다 이때 분할이 종료된
현재 노드는 잎 노드가 된다 뿌리노드에서 시작한 성장은 최종 노드가 잎 노드
가 될 때까지 분할하며 분할이 종료되어 모든 가지가 잎 노드에 도달하면 하나
의 결정 트리가 완성된다
그림 5 결정 트리의 재귀적 트리 성장 프로세스
트리 설계 작업의 많은 부분은 각 노드에서 어떤 질문이 수행되어야 하는지
를 결정하는데 있다 질문에 따른 판정에 의하여 훈련 샘플들은 더욱 더 작은
부분 집합으로 나누어진다
그림 6은 test라는 질문에 의하여 Ss집합이 no와 yes 판정을 받은 Ss1과 Ss2
의 부분집합으로 나누어 분류되는 모습이다 보통 각 부분 집합에는 서로 다른
클래스의 샘플들이 섞여 있다 따라서 각 분지에 대해 분할을 중단하고 불완전
한 판정을 받아들이거나 또는 그 대신 또 다른 질문을 선택해서 트리를 더 키
울 것인지를 결정해야 한다
- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 10 -
ne
(1)
에서에속한샘플의수 (2)
그림 6 질문의 판정에 의한 잎 노드의 분할
트리 생성의 밑에 깔려 있는 근본적 원칙은 단순성 원칙이다 우리는 적은
수의 노드를 갖는 단순하고 간결한 트리로 이끄는 판정을 선호한다 이 때문에
우리는 바로 아래 하위 노드들에 도달하는 데이터를 가능한 순수하게 만드는
특성 질문을 각 노드에서 모색한다 이 개념을 공식화 하는데 노드의 순수성보
다는 불순도를 정의하는게 더 편리한 것으로 밝혀졌다 불순도 (impurity)에 대
한 몇 가지 수학적 척도가 제안되어 왔으며 그들 모두가 똑같은 작용을 가진다
고 알려지고 있다 [18]
불순도는 여러 방법으로 정의할 수 있는데 Random Forest는 지니 불순도
(Gini impurity)를 측정하는 방법을 사용하여 결정 트리의 잎 노드의 분할을 수
행한다 지니 불순도 (Gini impurity)는 식 1과 같이 정의된다
식 1에서 ωi는 M개의 부류 중에 i번째를 나타내며 P(ωi|T)는 노드 T에서 ωi
가 발생할 확률이다 불순도 측정은 노드 T에서 ωi가 발생할 확률인 P(ωi|T)를
사용하는데 식 2에 의해 정의된다
- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 11 -
입력 훈련 집합 X=(x1 t1)⋯(xN tN)
출력 결정 트리 R
알고리즘
1 노드 하나를 생성하고 그것을 R이라 한다 이것이 루트 노드
2 T = R
3 XT = X
4 split_node(T XT) 루트 노드를 시작점으로 하여 순환 함수 호출
5 split_node(T XT) 순환 함수
6 노드 T에서 후보 질문을 생성한다
7 모든 후보 질문의 불순도 감소량을 측정한다 지니 불순도 이용
8 불순도 감소량이 최대인 질문 q를 선택한다
9 if (T가 멈춤 조건을 만족)
식 2의 XT는 원래 훈련 집합 X의 부분 집합을 의미하고 |XT|는 집합 XT의
크기를 의미한다 만일 노드에 도달하는 패턴 모두가 똑같은 클래스 레이블을
가지고 있다면 그 값이 0으로 되길 원하며 클래스가 동등하게 나타나면 값이
크게 나타난다
234 결정 트리의 학습 알고리즘
알고리즘 1은 결정 트리의 학습 알고리즘이다 결정 트리와 관련된 알고리즘
은 대부분 순환 (recursive) 함수로 작성된다 라인 6~8은 앞서 설명한 지니 불
순도 (Gini impurity)를 측정하는 방법을 사용하여 후보 질문을 생성하고 그들
중 가장 좋은 것을 고른다 그런 후 자식 노드를 생성할 필요가 있는지 검사한
다 이때 사용하는 조건이 멈춤 조건이다 만일 멈춤 조건을 만족하면 그 노드
를 잎 노드로 만들고 적절한 부류를 할당한다 그렇지 않으면 왼쪽과 오른쪽 자
식 노드를 생성하고 각각에 대해 순환 함수를 호출하여 계속 진행한다
알고리즘 1 결정 트리 학습 알고리즘
- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 12 -
10 T에 부류를 할당한다
11 return
12
13 else
14 q로 XT를 XTleft와 XTright로 나눈다
15 새로운 노드 Tleft와 Tright를 생성한다
16 split_node(Tleft XTleft)
17 split_node(Tright XTright)
18
19
알고리즘 1에서 부연 설명할 곳이 두 군데 있다 첫 번째는 라인 9의 멈춤
조건이다 노드 T에서 더 분기할 것인지 아니면 분기를 멈추고 잎 노드로 할
것인지 결정해야 한다 기본 원칙은 XT의 불순도가 충분히 낮을 때 멈추게 하
면 된다 하지만 어느 정도가 충분한지 애매할 수 있다 따라서 아래와 같은 여
러 조건을 and 또는 or로 결합하여 사용하는 것이 보통이다
조건 1 불순도가 0이다 이 경우는 더 이상 분기할 필요가 없다
조건 2 XT의 샘플 개수가 임계값 이하이다
조건 3 라인 8에서 결정한 q의 불순도 감소량이 임계값 이하이다
만일 조건 1만 가지고 멈춤을 결정하면 자칫 과적합 (overfitting)이 발생할
수 있다 심한 경우 대부분 잎 노드가 단 하나의 샘플만을 가지게 될 수도 있
다 너무 늦게 멈추는 셈이다 이런 결정 트리는 일반화 능력이 낮을 수밖에 없
다 반대로 조건 2와 조건 3에서 임계값을 너무 크게 하여 느슨한 조건을 사용
하면 설익은 수렴 (premature convergence)에 도달할 수 있다 너무 빨리 멈추
는 꼴이다 따라서 적절한 임계값 설정이 매우 중요하다
라인 10은 잎 노드가 발생했을 때 그것에 부류를 할당하는 작업이다 이 작
- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 13 -
업은 매우 간단하다 그 잎 노드의 샘플 집합 XT를 조사하여 가장 큰 빈도를
갖는 부류를 선정하면 된다
235 결정 트리의 특성
결정 트리는 아주 다양한 형태를 띤다 예를 들어 불순도 측정 방법을 바꾸
면 다른 트리가 만들어 진다 또한 멈춤 조건에 따라서 아주 큰 트리가 만들어
질 수도 있고 반대로 아주 작은 트리가 될 수도 있다 결정 트리의 몇 가지 특
성을 살펴보기로 한다
1 결정 트리는 계량 값을 갖는 특징뿐 아니라 비계량 특징도 다룰 수 있고 이
둘이 혼합되어 있는 패턴도 다룰 수 있다 또한 특징 값들을 크기 정규화하
는 등의 전처리 과정이 필요 없다 반면 신경망이나 SVM은 계량 데이터만
다룰 수 있다 결정 트리가 갖는 중요한 장점 중의 하나이다
2 결정 트리는 분류 결과가 lsquo해석 가능하다rsquo는 장점을 갖는다 신경망이나 SVM
등에서는 샘플이 어떤 부류로 분류되었을 때 왜 그런 결정이 내려졌는지 이
유를 대기가 매우 어렵다 하지만 결정 트리에서는 그 샘플이 루트부터 잎
노드까지 거쳐온 경로를 보면 왜 그런 결정이 내려졌는지 해석할 수가 있다
3 학습이 끝난 결정 트리를 가지고 인식하는 작업은 매우 빠르다 루트 노드에
서 간단한 비교 연산을 통하여 예아니오 결정을 하고 그 결과에 따라 자식
노드로 이동한다 이 과정을 잎 노드를 만날 때까지 순환적으로 반복하면 된
다 트리의 깊이가 h라면 최대 h-1 번의 노드만 거치면 된다 보통 트리의
깊이가 몇 십 정도이므로 매우 빠르게 인식을 마칠 수 있다
4 트리의 크기를 줄이기 위해 가지치기(pruning) 방법을 사용할 수 있다 멈춤
조건에서 임계값 설정을 낮게 허용하거나 불순도 감소량이 0이 될 때까지 분
기를 허용하여 충분히 큰 트리를 만든다 그런 후 반대로 밑에서부터 잎 노
드를 합치는 연산을 수행한다 이런 연산을 노드 가지치기라 한다 두 자식
노드를 조사하여 그것을 합쳤을 때 불순도 증가가 충분히 적으면 그것들을
하나로 합친다 노드 가지치기는 트리의 크기를 줄여준다
5 결정 트리는 불안정성 (instability)을 보이기도 한다 불안정성이란 단지 한두
- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 14 -
개 샘플의 값이 조금 바뀌었는데 전체 트리 모양이 크게 바뀌는 현상을 뜻한
다 일반적으로 단점으로 볼 수 있다 하지만 혼합 모델의 배깅과 같은 기법
에서는 오히려 불안정성을 이용하여 좋은 효과를 얻기도 한다
236 Random Forest의 설계
Random Forest는 의사결정 트리 분류기를 위해 특별히 설계된 앙상블 기법
으로 다수의 의사결정 트리에 의한 예측을 종합한다 각각의 트리는 그림 7과
같이 임의의 벡터들이 독립적인 집합 값들에 기초하여 생성된다
그림 7 Random Forest의 모델 구축 과정
Random Forest는 학습 샘플의 배깅 (bagging)을 사용하여 생성된다 배깅이
란 분류를 향상시키기 위해 데이터를 재사용하거나 선택하는 것을 가리키는 말
- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 15 -
일반화오류le
(3)
max (4)
로 부트스트랩 (bootstrap) 집합 (aggregation)에서 온 이름이다 배깅은
Random Forest의 특별한 경우로써 원래의 훈련 집합에서 교체와 함께 N개의
샘플을 임의로 선택하는 방법을 통해 무작위성이 모델 구축 과정에 포함되었다
트리의 수가 충분히 많은 경우 Random Forest의 일반화 오류의 상한선은 다음
의 식 3으로 수렴된다
식 3에서의 p는 트리 (tree)들 사이의 평균 상관관계이고 s는 트리 분류기의
강도를 측정하는 수치이다
분류기 집합의 강도는 분류기들의 평균 성능을 말하는데 여기서 성능은 분
류기의 margin 관점에서 확률적으로 측정된다
식 4에서 Yθ는 무작위 벡터 θ로부터 구축된 분류기에 의해 예측된 X클래
스이고 margin 값이 클수록 분류기가 주어진 사례 X를 정확히 예측할 가능성
이 크다 트리 사이의 상관관계가 커지거나 앙상블의 강도가 감소하면 일반화
오류의 한계치는 증가하는 경향이 있다 무작위성은 의사결정 트리 사이의 상관
관계를 줄여줌으로써 앙상블의 일반화 오류를 개선할 수 있다 [27]
배깅은 학습 집합 (training set)의 여러 버전을 사용하는데 각 버전은 학습
집합들로부터 전체 학습 샘플 수 보다 적은 수의 샘플을 뽑아 생성된다 여기
서 샘플 뽑는 방법은 뽑힌 것을 확인하고 다시 넣고 뽑는 방식이다 이 부트스
트랩 훈련 집합의 각각은 다른 트리 분류기를 훈련시키는 데 사용되며 최종 클
래스 판정은 각 트리 분류기의 다수결 투표에 기반 한다 [18]
어떤 분류기의 훈련 데이터에 작은 변화가 다른 분류기들과 비교적 큰 정확
도의 변화로 이끌면 비공식적으로 불안정하다고 불린다 단일 훈련 점의 위치
의 작은 변화가 완전히 다른 트리를 생성할 수 있기 때문이다 일반적으로 배깅
- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 16 -
(5)
은 최종 클래스 판정의 불연속들에 대해 효과적으로 평균을 내기 때문에 불안
정한 분류기들의 인식률을 향상 시킨다 [8 17 18]
그림 8 Random Forest의 최종 결과 값을 얻어내는 과정
그림 8은 Random Forest 분류기가 최종 결과를 얻어내는 과정이다 학습에
의하여 구축된 Random Forest 분류기에 분류하고자하는 특징벡터를 입력한다
각 트리에서 특징벡터를 분류하여 마지막 노드에 속한 특징벡터가 각 클래스로
판명될 확률을 얻는다 각 트리의 결과는 경로 선정에 따른 판정 과정에 의해
최종 선택되는 확률 분포이므로 판정 과정에 따라 다른 확률 분포를 가질 수
있다 각 트리에서 얻은 최종 확률 값을 합하여 트리의 개수로 나누면 Random
Forest의 최종 결과인 평균 확률이 된다 m개의 트리에서 특징 벡터 v가 클래
스 c에 속하는 Random Forest의 최종 확률 값은 식 5에 의해 얻어진다
본 연구에서는 Weka [13]에 있는 Random Forest 패키지를 사용하여 예측
모델을 생성하였고 특징 벡터는 Weka의 입력 포맷 중 하나인 Arff 포맷으로
- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 17 -
표현하였다
24 특징 벡터 기반의 중복 데이터 제거 기법
많은 관련 연구들의 지도학습 기법에서 학습될 패턴들의 다양성을 높이기
위해 일반적으로 슬라이딩 윈도우 (Sliding window) 기법을 사용하고 있다 슬
라이딩 윈도우 기법이란 개별적인 아미노산 또는 염기를 하나의 학습 패턴으로
보는 것이 아닌 양 옆의 이웃되어 있는 연속된 아미노산을 패턴화 한 후 이를
하나의 학습 패턴으로 구성하는 기법으로서 단백질 서열에서 보다 다양한 학습
패턴을 생성할 수 있으며 이러한 학습 패턴으로 학습된 예측 모델의 성능을 높
일 수 있다고 알려져 있다 하지만 슬라이딩 윈도우 기법과 서열간 유사성에 기
반을 둔 단백질 서열 데이터 중복을 제거하게 되면 제거된 서열에서 부분적으
로 존재할 수 있는 중요한 결합 패턴 정보를 함께 제거하게 되며 이렇게 손실
된 패턴 정보들은 이후 예측 모델 학습 과정에서 전혀 고려되지 못하게 된다
이러한 문제점을 보완하기 위하여 본 연구에서는 특징 벡터 기반의 중복 데이
터 제거 기법을 사용하여 학습 데이터 생성에 사용 하였다 [14]
특징 벡터 기반의 중복 데이터 제거 기법은 데이터 셋에서 서열간 유사한
서열을 제거하여 슬라이딩 윈도우 기법으로 패턴화시키는 방법이 아닌 데이터
셋에서 슬라이딩 윈도우 기법으로 먼저 패턴화 시킨 후 각 패턴이 예측 모델
학습에 필요한 형태인 특징 벡터로 변환할 때 특징 벡터와 해당 패턴이 속하는
클래스를 고려하여 해당 패턴이 다른 패턴들과 중복적인 데이터인지 아닌지에
대한 판단을 하게 되며 동일하다고 판단된 패턴 중 한 패턴만을 학습 데이터에
포함시킴으로써 원래의 단백질 서열에 존재하는 중복적인 패턴들을 제거하며
또한 원래의 서열들로부터 가능한 모든 패턴 정보가 학습 데이터로 사용된다
패턴을 구성하는 아미노산의 종류와 순서가 동일하다고 하여도 해당 패턴들이
속하는 클래스 (Positive 또는 Negative)가 다르면 다른 패턴으로 사용되었다
그림 9에서는 매우 유사한 두 개의 단백질 서열에 대하여 원도우 크기를 3
으로 하였을 때 유사한 서열 기반의 중복 데이터 제거 기법과 특징 벡터 기반
의 중복 데이터 제거 기법을 비교하고 있다
유사한 서열 기반의 중복 데이터 제거 기법은 두 번째 단백질 서열을 제거
- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 18 -
함으로써 제거된 서열이 보유하고 있는 패턴들 역시 제거 되지만 특징 벡터 기
반의 중복 데이터 제거 기법에서는 가능한 모든 부분적인 아미노산 패턴 정보
를 학습 데이터에 포함시키고 있어 보다 완전하며 중복 데이터를 포함하지 않
는 학습 데이터 생성을 보인다
그림 9 서열 간 유사성 기반의 중복 데이터 제거 기법과 특징 벡터 기반의 중
복 데이터 기법을 통한 학습 데이터 생성 과정의 비교
본 연구에서는 상대방 RNA를 고려한 결합 성향도 특징 벡터에 포함되어 단
백질 서열에서 중복적인 패턴을 가져도 상대방 RNA의 서열에 따라 다른 특징
벡터로 표현된다
25 Ensemble of Under-Sampling
단백질-RNA 복합체의 단백질 서열과 RNA 서열에서 결합 잔기 (positive
data)의 개수가 비 결합 잔기 (negative data)보다 훨씬 적기 때문에 positive 클
래스와 negative 클래스 사이에 심각한 불균형이 존재 한다 이러한 불균형은
결합 부위의 예측 기법의 성능을 저하시키는 요소로 관련 선행 연구에서 지적
- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 19 -
된 바가 있다 [28] 이는 적은 개수의 데이터 또는 다양성이 낮은 데이터들로
학습된 SVM 모델의 정확도를 낮추는 요인이 된다 불균형 문제를 해소하기 위
해 상호작용하지 않는 RNA-단백질 쌍의 일부를 무작위로 선택하여 균형을 맞
출 수 있지만 이렇게 선택된 데이터는 본래의 데이터 군을 모두 표현할 수 없
기 때문에 선택되지 않은 데이터에 대한 정보를 놓치게 된다 [29]
이러한 데이터 불균형을 해소하기 위하여 Under-Sampling 기반 앙상블 결
정 트리 기법을 사용하였다 데이터 불균형 문제의 관련 선행 연구 [30]에서
Ensemble of Under-Sampling은 데이터의 불균형이 심하고 소수 범주에 속하
는 데이터의 수가 매우 적은 경우 Under-Sampling이 가지는 단점을 극복하기
위하여 Ensemble of Under-Sampled SVMs (EUS SVMs)를 제안하였다 본 논
문은 SVM (Support vector machine) 대신 결정 트리에 이 기법을 접목시킴으
로써 Under-Sampling 기법을 가지는 Random Forest 분류기를 구현하게 되었
다
그림 10 Ensemble of Under-Sampling 기법의 학습 데이터 생성
그림 10은 Under-Sampling 기반 앙상블 결정 트리 기법에서 사용된 학습
데이터 생성 방법을 나타내었다 첫 번째 단계에서는 전체 학습 데이터를 소수
범주 데이터와 다수 범주 데이터로 분류한다 두 번째 단계에서는 다수 범주 데
이터에서 소수 범주 데이터의 수만큼 비 복원 랜덤 추출을 하여 하나의 데이터
- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 20 -
집합을 생성한다 하나의 데이터 집합이 생성되면 다시 전체 다수 범주의 데이
터를 사용하여 새로운 데이터 집합을 생성한다 이 방법을 사용함으로써 복원
추출시의 데이터의 중복을 방지 하는 효과와 비 복원 추출을 할 경우 소수 범
주 데이터의 수에 앙상블의 크기가 영향을 받는 단점도 극복할 수 있다 마지막
으로 앙상블의 크기만큼 다수 범주에서의 데이터 집합 생성이 완료되면 각각의
데이터 집합과 소수 범주 데이터를 결합하여 범주 비율이 동일한 학습 데이터
를 생성한다 이러한 절차를 통하여 앙상블 학습 데이터를 구성함으로써 소수
범주에 의해 데이터의 분포가 왜곡되지 않으면서 높은 성능을 나타내는 분류기
를 구축할 수 있게 된다
- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 21 -
제 3 장 데이터 구성 및 특징 선택
31 단백질-RNA 결합 부위 데이터
단백질-RNA 결합부위 정보를 위하여 Protein Data Bank (PDB) [9]에서 단
백질-RNA 복합체 구조 데이터를 수집하였다 PDB는 단백질이나 DNA 또는
RNA와 같은 거대분자의 구조정보를 담고 있으며 모든 원자의 3차원 좌표 값을
포함하고 있기 때문에 원자 간의 거리와 각도를 계산하면 원자 간의 결합을 추
출할 수 있다
NMR (Nuclear Magnetic Resonance) Electron Microscopy 실험 기법 등
정확성이 부족한 방법에 의해 얻어진 데이터는 제외하고 신뢰도가 높은 X-ray
crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å 보다 높은 해상
도를 갖는 구조 데이터를 사용하였다 DNA 또는 DNARNA 하이브리드의 서
열은 포함하지 않는 데이터들을 검색하여 429개의 단백질-RNA 복합체 구조 데
이터를 추출하였으며 총 3149개의 단백질-RNA 서열 쌍을 분석 데이터로 수집
하였다 분석 데이터는 2663개의 단백질 서열과 812개의 RNA 서열 정보를 가
진다
32 단백질-RNA 결합 부위 정의
본 연구에서 가장 근본이 되는 개념인 lsquo결합rsquo에 관한 여부를 판단하기 위해
결합에 관한 정의가 필요하다 생물학적으로 단백질-RNA 결합 부위 정의는 수
소결합 (H-bond) 또는 원자 사이의 거리 등으로 정의할 수 있는데 그 중 가장
강력하고 많이 발생하는 결합이 바로 수소결합이기 때문에 본 연구에서는 아미
노산과 염기 사이의 수소결합 정의를 채택하였다 각 연구에서 사용하는 단백질
과 RNA의 수소 결합에 대한 정의가 조금씩 차이가 있다 [10 11] 본 논문에서
사용한 수소 결합은 수소 원자 (H hydrogen atom) 수소 공여자 (D donor)
수소 수용자 (A acceptor) acceptor antecedent (AA)에 해당하는 원자들이 다
- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 22 -
(6)
음과 같은 기하학적 조건을 만족해야 한다 D-A 거리 lt 39Å H-A 거리 lt
27Å D-H-A 각도 ge 90deg H-A-AA 각도 ge 90deg
그림 11 수소 결합의 결합 각도와 거리에 관한 정의
33 단백질 서열을 고려한 결합 성향
실험적으로 밝혀진 아미노산의 생화학적 특징으로서 accesible surface area
(ASA) molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다
normalized position은 각 아미노산의 서열에서의 위치를 단백질 서열 길이로
나눈 값이다 (식 6 참조)
34 단백질 아미노산 triplet을 고려한 결합 성향
각 아미노산 또는 염기가 독립적으로 결합에 관여하기 보다는 동일한 서열
에서 주변의 아미노산 또는 염기들이 함께 상호작용에 관여한다는 연구 결과
[15 16]에 착안하여 아미노산 triplet을 고려한 결합 성향 함수를 사용하였다
- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 23 -
sum
summiddotcossummiddotsum
middot (7)
연속한 아미노산 3개를 의미하는 아미노산 triplet t가 RNA 염기 a와 결합하는
성향 IPta는 식 7에 의하여 정의하였다
식 7에서 sum middot cos 는 단백질-RNA 결합 데이터 셋에 존재하
는 아미노산 triplet t의 중앙에 있는 아미노산과 염기 a 사이의 수소 결합에서
H-A가 D-A로 투영된 값의 역수를 모두 합한 것이다 sum 는 데이터 셋에 있
는 모든 아미노산 triplet과 염기 쌍의 개수 는 데이터 셋에 있는 아미노산
triplet t의 개수 는 데이터 셋에 있는 염기 a의 개수 sum 와 sum는 각각 데
이터 셋에 있는 아미노산 triplet의 총 개수와 염기의 총 개수를 의미한다
식 7을 적용하여 429개의 단백질-RNA 복합체 구조 데이터에서 203=8000인
아미노산 triplet들과 4개의 염기사이에 존재하는 8000times4=32000개의 IPta값이
계산된다 전체 IPta값에서 572에 해당하는 18301개의 IPta값만 0이 아니며
triplet (Arginine-Histidine-Proline R-H-P)과 염기 (Cytosine C) 사이의 IPta
값이 4789796으로 가장 높은 결합 성향을 보이고 있다
35 단백질 이차구조를 고려한 결합 성향
본 연구에서는 DSSP 프로그램 [12]의 분류법에 따라서 단백질 이차구조를
Helix Sheet Turn Coil의 네 가지 유형으로 분류하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 결합부위 예측에 사용하기 위한
통계적 특징을 개발하였다 특히 단순한 결합회수가 아닌 성향성을 구해야 정확
한 예측이 이루어질 수 있다 어떠한 아미노산 a와 염기가 빈번히 결합하는 것
이 발견된다고 하여도 원래 단백질을 이루는 성분의 대부분이 그 아미노산 a라
면 성향성이 높다고 말하는 것은 잘못된 것이기 때문이다 가령 매우 적은 양
이 존재하는 아미노산 b가 비록 앞서 예를 들었던 아미노산 a 보다는 결합했다
고 발견된 개수는 적을지라도 존재하는 아미노산 b의 대부분이 결합에 참여하
- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 24 -
middot summiddotsum
sum (8)
였다면 성향성은 높다고 해야 할 것이다 이러한 판단을 하기 위해 수소 결합
정의에 의하여 이차구조 유형별로 아미노산 a와 염기 r의 결합 성향 IPar을 식
8에 의하여 사용하였다 [19 20]
식 8에서 은 염기 r과 상호작용하는 이차구조 아미노산 a의 개수 sum 는
임의의 염기와 결합하는 이차구조 아미노산의 총 개수 는 모든 아미노산 a의
개수 은 모든 염기 r의 개수 sum 는 모든 아미노산 총 개수의 합 sum는 모
든 염기 총 개수의 합을 의미한다
표 2-5는 IPar을 사용하여 Helix Sheet Turn Coil의 이차구조 유형별로 계
산한 결합 성향을 나타낸다 Interaction propensity는 IPar에 의해 계산된 20개
의 아미노산과 4개의 염기 사이의 결합 성향 값을 나타내고 있다 Amino acid
Average는 각 아미노산의 결합 성향 평균값을 나타내고 RNA Average는 각
염기의 결합 성향 평균값을 나타내고 있다 본 연구에서 이차구조 유형별 아미
노산과 염기 사이의 특징 값은 각각의 20개 아미노산 별 RNA Average를 기준
으로 사용하였으며 IPar로 정의하겠다
식 8의 단백질 이차구조를 고려한 결합 성향은 1을 기준으로 1보다 큰 값은
결합 성향이 강한 것을 의미하고 1보다 작은 것은 결합 성향이 약함을 의미한
다
- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 25 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340822 0317793 0374597 0327005 0340054
C 0029169 000307 0023028 0001535 0014201
D 0379203 0095184 0459035 0308582 0310501
E 0376132 0244102 1034747 0343892 0499718
F 000307 0 0076762 0010747 0022645
G 0274807 0429865 0534261 0194975 0358477
H 0563431 0743053 0466711 0287089 0515071
I 0007676 0024564 0018423 0112072 0040684
K 1467683 220613 2846322 1628882 2037254
L 0210327 0121283 0021493 0029169 0095568
M 0066015 0027634 0500486 0004606 0149685
N 0302441 0445218 0931886 0802927 0620618
P 0095184 0 0090579 0001535 0046825
Q 0677038 0822885 1019395 0531191 0762627
R 4855942 4835984 4736194 3052043 4370041
S 0563431 0950309 1065452 0302441 0720408
T 0568036 0591065 1191341 0634051 0746123
V 0007676 0102861 0093649 0007676 0052966
W 0092114 0118213 016427 0038381 0103244
Y 0144312 0214933 0558825 016427 0270585
RNA Average 0551225 0614707 0810373 0439153
표 2 이차 구조가 Helix인 아미노산이 각 염기와 결합하는 결합 성향
표 2는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Helix인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Helix 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 26 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0475014 0010735 012345 0279104 0222076
C 0010735 0048306 0005367 0029521 0023482
D 037035 052332 0566259 0080511 038511
E 0448177 0466963 0848047 0260318 0505876
F 0053674 0147603 0504535 0037572 0185846
G 007246 0064409 0045623 0008051 0047636
H 1274755 1360633 0756802 0566259 0989612
I 0222747 0193226 0037572 005099 0126134
K 1771238 3338516 2090598 1392837 2148297
L 0163705 0155654 0335462 0037572 0173098
M 0002684 0034888 0131501 0069776 0059712
N 039987 0351564 0466963 0389136 0401883
P 0201277 0005367 0 0032204 0059712
Q 0241532 1022487 1186193 1368684 0954724
R 1175458 4127522 3700814 2039608 276085
S 1183509 0853415 1556543 0233481 0956737
T 1827596 1532389 0831945 0912456 1276097
V 0024153 0091246 0426707 0185175 018182
W 0040255 0016102 0061725 0077827 0048977
Y 0700444 0678975 0413289 0485749 0569614
RNA Average 0532982 0751166 070447 0426842
표 3 이차 구조가 Sheet인 아미노산이 각 염기와 결합하는 결합 성향
표 3은 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Sheet인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Sheet 이
차 구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향
이 가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이
에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Cytosine C)는 모
든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-
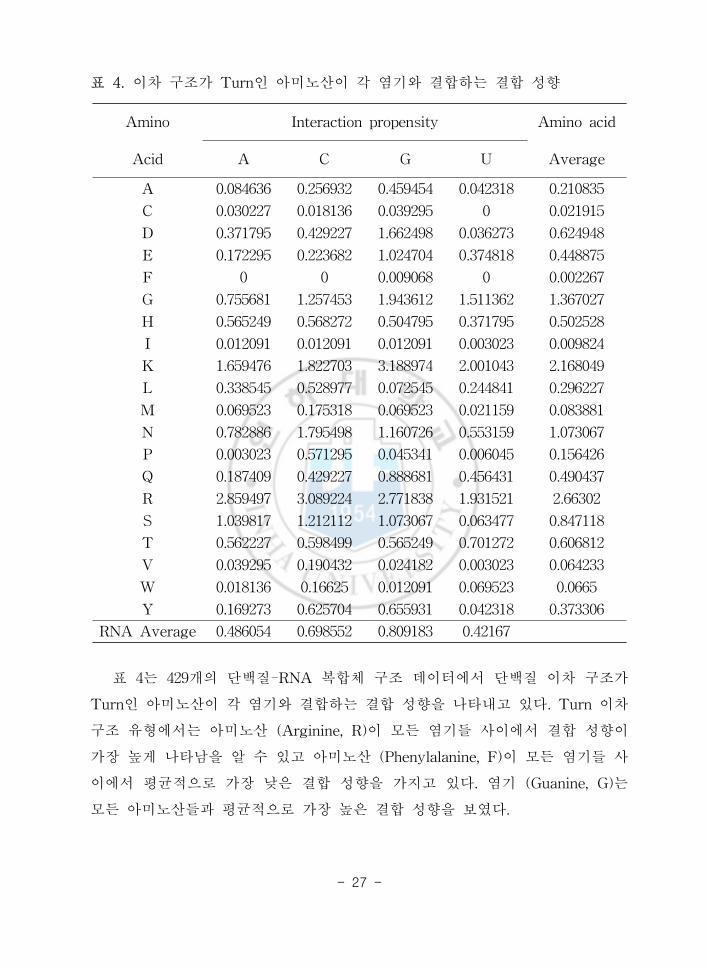
- 27 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0084636 0256932 0459454 0042318 0210835
C 0030227 0018136 0039295 0 0021915
D 0371795 0429227 1662498 0036273 0624948
E 0172295 0223682 1024704 0374818 0448875
F 0 0 0009068 0 0002267
G 0755681 1257453 1943612 1511362 1367027
H 0565249 0568272 0504795 0371795 0502528
I 0012091 0012091 0012091 0003023 0009824
K 1659476 1822703 3188974 2001043 2168049
L 0338545 0528977 0072545 0244841 0296227
M 0069523 0175318 0069523 0021159 0083881
N 0782886 1795498 1160726 0553159 1073067
P 0003023 0571295 0045341 0006045 0156426
Q 0187409 0429227 0888681 0456431 0490437
R 2859497 3089224 2771838 1931521 266302
S 1039817 1212112 1073067 0063477 0847118
T 0562227 0598499 0565249 0701272 0606812
V 0039295 0190432 0024182 0003023 0064233
W 0018136 016625 0012091 0069523 00665
Y 0169273 0625704 0655931 0042318 0373306
RNA Average 0486054 0698552 0809183 042167
표 4 이차 구조가 Turn인 아미노산이 각 염기와 결합하는 결합 성향
표 4는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Turn인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Turn 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Phenylalanine F)이 모든 염기들 사
이에서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는
모든 아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 28 -
Amino Interaction propensity Amino acid
Acid A C G U Average
A 0340257 0418537 0331907 0185784 0319121
C 0021918 0002087 0039662 0017743 0020353
D 0152385 0476986 092266 0407056 0489772
E 0105417 0254671 0372613 0231709 0241102
F 0134642 0001044 0074105 0072018 0070452
G 1250392 1328672 1164806 0632502 1094093
H 0496817 0673208 0560485 0864211 064868
I 0 0015656 0092892 0017743 0031573
K 224507 2227326 2197058 1162718 1958043
L 0080367 0221271 0096023 0028181 0106461
M 0033399 0269283 013986 0107504 0137512
N 0684689 0703476 0927879 0642939 0739746
P 0041749 0076193 008663 0056362 0065233
Q 0665902 0779688 0482205 0524998 0613193
R 2979858 3443275 3685421 1668929 2944371
S 1534287 1361028 1185681 0598059 1169764
T 0913266 051456 157708 0714957 0929966
V 0328776 0049055 0038618 0024006 0110114
W 0163866 0038618 0198309 0010437 0102808
Y 0475942 0171172 0450893 0041749 0284939
RNA Average 063245 0651289 0731239 040048
표 5 이차 구조가 Coil인 아미노산이 각 염기와 결합하는 결합 성향
표 5는 429개의 단백질-RNA 복합체 구조 데이터에서 단백질 이차 구조가
Coil인 아미노산이 각 염기와 결합하는 결합 성향을 나타내고 있다 Coil 이차
구조 유형에서는 아미노산 (Arginine R)이 모든 염기들 사이에서 결합 성향이
가장 높게 나타남을 알 수 있고 아미노산 (Cysteine C)이 모든 염기들 사이에
서 평균적으로 가장 낮은 결합 성향을 가지고 있다 염기 (Guanine G)는 모든
아미노산들과 평균적으로 가장 높은 결합 성향을 보였다
- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 29 -
middotmiddot (9)
36 상대방 RNA를 고려한 결합 성향
BindN [6]이나 RNABindR [7]과 같이 단백질 서열에서 RNA와 결합하는 잔
기를 예측하기 위하여 개발된 기존 기법들의 문제점 중의 하나는 공통적으로
단백질의 결합 상대방인 RNA와 관련하여 서열 정보조차 입력 받지 않고 RNA
와 결합하는 아미노산을 예측한다는 것이다 RNA를 전혀 고려하고 있지 않기
때문에 결합하는 RNA가 바뀌어도 특정 단백질 서열에 대한 결합부위는 항상
동일하게 예측한다 그러나 단백질과 RNA는 상호작용을 위하여 단백질 자체의
구조가 조금 변형되기도 하며 RNA 자체의 구조도 변형될 수 있다 따라서 본
연구에서는 결합 상대방인 RNA에 따라 단백질의 결합부위도 달라진다는 중요
한 사실을 파악하고 단백질에서의 RNA 결합 부위를 예측할 때 단백질 서열뿐
만 아니라 해당 서열의 상호작용 상대방으로 RNA 서열도 함께 고려한 결합 성
향 함수를 개발하여 단백질과 RNA간 결합부위 예측 알고리즘의 성능을 보완하
였다 상대방 RNA를 고려한 결합 성향은 IPtr을 식 9에 의하여 정의된다
식 9는 해당 단백질 서열에서 연속한 아미노산 3개를 의미하는 아미노산
triplet t와 상대방 RNA를 고려한 결합 성향 함수이다 IPta는 식 7에 정의된 함
수이며 Nucleotide frequency는 상대방 RNA 서열에서 각각의 염기 (A C G
U)에 대한 빈도수 RNA length는 상대방 RNA 서열의 길이를 의미한다
37 특징 벡터 (feature vector)의 표현
대부분의 지도학습 (supervised learning) 기법이 학습될 패턴을 수치적으로
표현한 특징 벡터(feature vector)를 요구하듯이 random forest 모델도 입력 데
이터를 특징 벡터로 변환하는 과정이 필요하다 본 연구에서는 단백질-RNA 상
호작용에 참여하는 단백질 서열의 특징을 다음 네 가지로 나누어 특징 벡터에
표현하였다
- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 30 -
sect 단백질 서열을 고려한 결합 성향 실험적으로 밝혀진 각 아미노산의 생화학
적 특징을 사용하였으며 사용된 특징은 accesible surface area (ASA)
molecular mass (Mass) side chain Pka (Pka) hydropathy 이외에
normalized position [14]을 단백질 서열을 고려한 특징으로 사용하였다 (식
4 참조)
sect 단백질 아미노산 Triplet을 고려한 결합 성향 IPta (식 7 참조)
sect 단백질 이차 구조를 고려한 결합 성향 IPar (식 8 참조)
sect 상대방 RNA를 고려한 결합 성향 IPtr (식 9 참조)
Random Forest에 의해 학습될 데이터 셋에서 해당 아미노산의 특징을 추출
하기 위해 슬라이딩 윈도우 기법을 적용하였다 기존의 연구에서 Target 아미노
산을 중심으로 양 옆의 이웃되어 있는 아미노산들은 비슷한 상호작용 성향을
띄고 있음이 밝혀졌고 슬라이딩 윈도우 크기의 조정에 따라 예측 결과가 바뀌
는 것을 볼 수 있다
단백질-RNA 상호작용에 참여하는 단백질 서열의 특징을 특징 벡터에 표현
하였다 그림 12는 슬라이딩 윈도우 크기를 7로 적용하였을 때 표적 아미노산이
수치화된 특징인 아미노산의 생화학적 특징 normalized position 단백질 아미
노산 triplet을 고려한 결합 성향 (IPta) 단백질 이차 구조를 고려한 결합 성향
(IPar) 상대방 RNA를 고려한 결합 성향 (IPtr)을 사용하였을 경우 생성되는 특
징 벡터의 예를 보인다
아미노산 패턴의 i번째 아미노산 ai를 표현하는 특징벡터는 5개의 겹치는
triplet ta(ai-3 ai-2 ai-1) ta(ai-2 ai-1 ai) ta(ai-1 ai ai+1) ta(ai ai+1 ai+2) ta(ai+1
ai+2 ai+3) 정보를 포함한다 각 아미노산에 대하여 생화학적 특징들과
normalized position을 포함하고 각 triplet은 단백질 아미노산 triplet을 고려한
결합 성향 함수를 사용하여 IPta_A IPta_C IPta_G IPta_U 값들을 포함한다 단
백질 이차구조를 고려한 결합 성향 함수를 사용하여 각 아미노산에 대하여
IPar_ACGU (Helix) IPar_ACGU (Sheet) IPar_ACGU (Turn) IPar_ACGU
- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 31 -
(Coil) 값들을 생성한다 상호작용하는 상대방 RNA 서열에서 서열의 길이와 각
염기 (A C G U)의 빈도수를 추출하여 수치화한 후 상대방 RNA를 고려한
결합 성향 함수를 통해 IPtr_A IPtr_C IPtr_G IPtr_U 값들을 생성한다 결론적으
로 7+7+7+7+1+(7times4)+(5times4)+(5times4)=97개의 값으로 구성된 특징 벡터가 생성된다
그림 12 윈도우 크기=7로 했을 때 아미노산 서열을 특징 벡터로 변환한 예
- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 32 -
(10)
(11)
(12)
(13)
timestimes (14)
timestimes (15)
38 예측 알고리즘의 성능 평가
Random Forest 모델의 예측 결과를 검증하기 위해 실제 데이터와 비교하여
True positive (TP) True negative (TN) False positive (FP) 그리고 False
negative (FN)를 수집하였다 TP는 결합 부위로 바르게 예측된 실제의 결합 부
위 TN은 비결합 부위로 바르게 예측된 실제의 비결합 부위 FP는 결합 부위로
잘못 예측된 비결합 부위 FN은 비결합 부위로 잘못 예측된 결합 부위를 의미
한다
예측 모델의 성능은 다음과 같은 6개의 기준으로 평가하였다 Sensitivity
(SN) Specificity (SP) Accuracy (ACC) Net prediction (NP) F-measure
(FM) Correlation coefficient (CC) 6개의 평가 기준은 다음 식 10-15와 같은
함수로 정의된다
- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 33 -
제 4 장 시험 및 평가
본 연구는 PDB로부터 2009년 11월에 X-ray crystallography 기법에 의해
밝혀진 복합체 중 해상도가 30Å 보다 좋은 해상도를 갖는 429개의 단백질
-RNA 복합체 구조 데이터를 구성하였다 단백질-RNA 결합 부위 정의는 수소
결합을 채택하여 결합 부위를 갖는 3149개의 단백질-RNA 서열 쌍을 데이터
셋으로 구성하였다 데이터 셋은 2663개의 단백질 서열과 812개의 RNA 서열로
구성된다 모든 예측 시험 및 평가는 데이터 마이닝 프로그램인 Weka [13]에
포함되어 있는 Random Forest 분류기에 의해 시험되었으며 파라미터로 120개
의 결정 트리 (Decision tree)와 특징 벡터의 크기 개수만큼의 random feature
selection이 사용되었다
41 학습 데이터 생성
단백질 서열 정보와 상호작용 상대방인 RNA 서열의 정보를 함께 고려하여
단백질에서의 RNA 결합 부위를 예측하기 위하여 3149개의 단백질-RNA 서열
쌍 결합 데이터 셋으로부터 특징 벡터 기반 중복 제거 기법을 사용한 데이터를
여러 가지 슬라이딩 윈도우 크기 별로 생성하였다 특징 벡터로 변환하기 위해
사용된 특징은 아미노산의 생화학적 특징 (ASA Mass Pka hydropathy)
normalized position IPta IPar IPtr이다
표 6은 상대방 RNA 서열 정보를 고려하지 않은 패턴으로 단백질 서열에서
만 윈도우 크기별로 생성한 feature vector들의 통계이고 표 7은 단백질 서열에
서 표적 아미노산 패턴과 함께 상대방 RNA 서열의 길이 염기 (A C G U)의
빈도수를 고려하여 생성한 feature vector들의 통계를 보인다 표 6과 표 7 공통
적으로 앞에서 기술한 특징 벡터 기반의 중복 데이터 제거 기법을 사용하여
posive 데이터와 negative 데이터에서 독립적으로 중복되는 특징 벡터를 제거하
였다 positive feature vector들은 상호작용하는 RNA와 결합하는 표적 아미노
산을 패턴화 시킨 개수를 나타내며 negative feature vector들은 상호작용하는
- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 34 -
Window positive negative total common
3 19451 84203 103654 2403
5 23445 160749 184194 2888
7 23508 162006 185514 2819
9 23539 162226 185765 2820
11 23562 162410 185972 2821
13 23581 162549 186130 2823
15 23606 162673 186279 2832
Window positive negative total common
3 31403 232457 263860 7272
5 33152 258750 291902 2956
7 33172 259155 292327 2933
9 33184 259263 292447 2936
11 33195 259356 292551 2936
13 33205 259430 292635 2937
15 33215 259501 292716 2940
RNA와 결합하지 않는 표적 아미노산을 패턴화 시킨 개수이다
표 6 단백질 서열 정보만 고려하여 구성한 데이터에서 특징 벡터의 개수
표 7 단백질 서열과 RNA 서열 정보를 함께 고려하여 구성한 데이터에서 특징
벡터의 개수
- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 35 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
3 6401 9572 9194 7987 7672 0609
5 8167 9810 9624 8989 8914 0810
7 8177 9813 9629 8996 8921 0812
9 8189 9814 9630 9002 8929 0813
11 8191 9814 9630 9003 8930 0813
13 8176 9814 9628 8995 8920 0812
15 8146 9818 9629 8982 8904 0812
특징 벡터 제거 기법을 사용하여 각각 positive 패턴에서 중복되는 패턴을
제거하였고 negative 패턴에서 중복되는 패턴을 제거하였다 total vector는 중
복이 제거된 positive feature vector들과 negative feature vector들의 합이고
common vector는 중복이 제거된 positive feature vector들과 negative feature
vector들 사이에서 공통으로 중복되는 패턴의 개수를 의미하며 positive 패턴과
negative 패턴에서 공통으로 중복되는 패턴은 서로 다른 패턴으로 간주하여 제
거하지 않았다
42 Random Forest를 이용한 예측 성능 평가
표 8은 수치화된 모든 특징을 사용하고 각 슬라이딩 윈도우 크기 별로 구성
된 표 7의 데이터를 5-fold cross validation 기법을 통하여 상호작용 상대방인
RNA 서열 정보를 고려한 단백질에서의 RNA 결합 부위 예측 성능을 평가한
결과를 보이고 있다
표 8 특징들을 모두 사용하여 5-fold cross validation한 결과
- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 36 -
FeatureSN
()
SP
()
ACC
()
NP
()
FM
()CC
PF 648 950 912 799 770 060
IPta 707 927 900 817 802 059
IPar 712 919 893 816 803 057
IPtr 810 979 960 895 887 079
PF+IPta 734 962 933 848 833 070
PF+IPar 735 961 932 848 833 069
PF+IPtr 814 979 960 897 889 080
PF+IPta+IPar 737 961 932 849 834 070
PF+IPta+IPtr 817 979 961 898 891 080
PF+IPta+IPar+IPtr 819 981 963 900 893 081
표 8에서 윈도우 크기가 가장 작은 3일 경우에 모든 평가지표에서 가장 저
조한 성능을 보이는데 학습 데이터에서 positive와 negative 데이터에 공통된
feature vector가 많을수록 지도학습 기법의 성능이 낮아지는 것을 반영하는 것
이라고 할 수 있다 윈도우 크기가 5-15인 경우에는 서로 비슷한 학습 데이터
개수와 공통 feature vector를 포함하고 있고 (표 7 참조) 이에 따라 예측 성능
도 비슷한 결과를 보이는데 윈도우 크기가 11일 경우가 근소한 차이이지만 가
장 좋은 성능을 보인다
표 9 사용한 특징들로 단백질에서의 RNA 결합 부위를 예측한 성능 비교
표 9는 윈도우 크기를 11로 고정하고 5-fold cross validation으로 사용한 특
징별로 예측 성능을 평가한 결과이다 PF는 아미노산의 생화학적 특징인 ASA
Mass Pka hydropathy와 normalized position을 모두 사용함을 의미한다 단백
질 서열 정보만 고려하는 PF IPta IPar의 특징 벡터는 표 6의 데이터에서 생성
- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 37 -
하였고 단백질과 RNA 서열 정보를 함께 고려하는 IPtr을 사용한 특징 벡터는
표 7의 데이터에서 생성하였다
단일 특징으로서는 많은 데이터 개수를 가지고 있는 IPtr을 사용하였을 때
가장 좋은 성능을 보였고 IPtr을 포함하여 두 개 이상의 특징을 사용하는 것이
IPtr을 제외한 복수의 특징들을 사용하는 것보다 sensitivity (SN)와 specificity
(SP)를 모두 향상시켰다 결과적으로 IPtr을 포함하여 모든 특징들
(PF+IPta+IPar+IPtr)을 사용하였을 때 평균적으로 가장 좋은 결과를 보임을 알 수
있다
43 Random Forest와 SVM의 예측 성능 비교
Random Forest 분류기 [8]는 질적 (qualitatively)인 측면에서 여러 구간으로
구분하는 비계량 (nonmetric) 데이터를 다루며 질적 분류기 (qualitative
classifier)라 불리고 SVM (support vector machine) 분류기 [21]는 범주형 데이
터가 아닌 어떤 사물 또는 개념을 양적(quantitatively)으로 표시한 계량
(metric) 데이터를 다루는 양적 분류기 (quantitative classifier)라 불려진다
Random Forest는 여러 트리 (tree)들의 앙상블 기법으로 각각의 트리가 판
정한 결과에 다수결 투표를 따라 최종적인 해를 구하고 SVM은 벡터공간에서
임의의 경계를 찾아 두 개의 집합을 분류하는 방법으로 주어진 조건하에서 수
학적으로 최적의 해를 찾을 수 있다고 알려져 있다
본 논문은 이러한 특징을 가지는 Random Forest와 SVM 분류기의 실제 예
측 성능을 비교해 보기 위해 동일한 학습 데이터를 통해 비교 시험해 보았다
표 10은 각 슬라이딩 윈도우 크기 별로 구성된 [표 7]의 데이터를 5-fold
cross validation 기법을 통하여 Random Forest 분류기와 SVM (support
vector machine) 분류기에서 상호작용 상대방인 RNA 서열 정보를 고려한 단백
질에서의 RNA 결합 부위 예측 성능을 비교한 결과를 보이고 있다
Random Forest 분류기는 파라미터로 10개의 결정 트리 (Decision tree)와
log2(특징 벡터의 크기+1)개의 random feature selection이 사용되었고 SVM 분
류기는 libSVM 패키지를 사용하였고 RBF 커널을 사용하여 파라미터로 C=10
(gamma)=1특징 벡터의 크기 학습 데이터의 negative 클래스의 크기가 N이
- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 38 -
WindowSN
()
SP
()
ACC
()
NP
()
FM
()CC
Random Forest
3 6437 9541 9172 7989 7688 0602
5 8200 9801 9619 9001 8930 0809
7 8183 9808 9624 8995 8922 0810
9 8193 9807 9624 9000 8928 0811
11 8205 9807 9625 9006 8935 0812
13 8194 9807 9624 9001 8928 0811
15 8195 9809 9626 9002 8930 0812
SVM
3 5597 8521 8219 7059 6756 0319
5 6452 9557 9204 8005 7704 0603
7 6656 9590 9269 8123 7858 0625
9 6808 9589 9285 8199 7963 0635
11 6934 9594 9303 8264 8050 0646
13 7028 9597 9316 8313 8114 0654
15 7067 9581 9295 8324 8134 0655
고 positive 클래스의 크기가 P인 경우 positive 클래스에 대한 weight 값=NP
negative 클래스의 weight 값=1이 입력되었다
표 10 Random Forest와 SVM의 예측 성능 비교
- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 39 -
5-fold cross validation한 Random Forest 분류기에서 윈도우 크기가 15일
때 다른 윈도우 크기보다 좋은 평가를 받고 있었으며 SVM 분류기의 경우 윈
도우 크기가 13일 때와 15일 때 가장 좋은 성능을 보이고 있었다 한편
Random Forest 분류기가 SVM 분류기 보다 모든 평가 지표에서 뛰어난 예측
성능을 보이고 있었고 특히 sensitivity와 correlation coefficient의 평가 지표에
서 월등한 차이를 보이고 있다
사용된 학습 데이터들의 크기는 평균 80MB 정도를 보이고 있는데 SVM의
cross validation 수행 시간은 평균 3일이 소요되고 Random Forest는 평균 15
분이 소요되므로 Random Forest가 SVM에 비해 훨씬 수행 속도가 빠르면서
정확한 예측 결과를 보이고 있음을 알 수 있었다
44 타 연구 시험과의 비교
단백질에서의 RNA 결합 부위 예측 성능 평가에서 타 연구 시험과의 비교를
위해 Random Forest를 사용하고 최근에 발표되었던 Liu et al [22] 연구진의
논문을 참조하였다
Liu et al [22]가 Random Forest 기법을 사용하여 단백질에서의 RNA 결합
부위를 예측한 논문에서는 본 논문과 근본적으로 차이가 있는데 표 11은 본 논
문과의 방법의 차이를 비교하여 정리한 표이다
타 연구 시험과의 비교에 앞서 예측 성능 비교의 공평성을 가지기 위해 결
합부위 데이터 결합부위 정의 특징벡터의 표현방법들은 Liu et al 논문에서 공
개된 데이터 구성과 표현방법을 그대로 따랐고 나머지 사용된 특징만 달리하였
다
Nucleic Acid 데이터베이스 [24]에 있는 RsiteDB [25]로부터 수집한 339개의
단백질-RNA 복합체 구조 데이터 안에서 PSI-BLAST 프로그램 [26]을 이용하
여 각각 단백질 서열에서 25가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하였고 RNA 서열에서 60가 넘는 서로 유사한 상동성을 가지는 서열들을
제거하여 유사한 서열 중복이 없는 164개의 단백질-RNA 복합체 구조 데이터로
구성된다 결합부위에 대한 정의는 ENTANGLE 프로그램 [23]을 이용하여
hydrogen bonding electrostatic hydrophobic van der Waals의 결합 기준 중
- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 40 -
Liu et al 본 논문
결합부위
데이터
164개의 단백질-RNA
복합체 구조 데이터
429개의 단백질-RNA
복합체 구조 데이터
결합부위
정의
hydrogen bonding
electrostatic
hydrophobic
van der Waals [23]
hydrogen bonding
특징벡터의
표현방법유사한 서열 중복제거 특징벡터 기반 중복제거
사용된
특징
Mutual interaction propensity
Physicochemical characteristics
Hydrophobicity
PSSM value
Accessible surface
Secondary structure
Side chain Pka
Accessible surface
molecular mass
Side chain Pka
Hydrophobicity
normalized position
IPta
IPar
IPtr
하나만 성립이 되어도 결합부위로 인정이 되었다 Random Forest를 사용해
5-fold cross validation 기법으로 시험을 하였다
표 11 Liu et al 논문과 본 논문과의 방법 비교
- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 41 -
Window SN () SP () ACC () FM ()
Our method
3 690 968 936 806
5 745 985 957 848
7 747 986 958 850
9 757 987 960 857
11 756 987 960 856
13 755 988 960 856
Method of Liu et al
3 831 866 834 848
5 844 858 845 851
7 849 850 849 850
9 850 845 849 850
11 825 860 828 842
13 838 840 839 840
표 12 Liu et al 논문과 본 논문과의 예측 성능 비교
표 12는 Liu et al 논문에서 사용한 164개의 단백질-RNA 복합체 구조 데이
터를 대상으로 Liu et al 논문과 본 논문과의 예측 성능을 각각의 윈도우 크기
별로 평가 지표별로 나타낸다 Liu et al 논문에서 사용한 특징들보다 본 논문
에서 사용한 특징들이 sensitivity이 모두 낮게 나오고 있지만 specificity와
accuracy는 모두 높게 나오고 있음을 확인하였다 F-measure 성능 평가 지표에
서는 윈도우 크기가 7이상 일 때부터 계속 크기가 커질수록 Liu et al 논문의
예측 성능을 추월하고 있음을 알 수 있었다
- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 42 -
테스트 데이터 SN () SP () ACC () NP ()
PR324 537 986 954 762
PR102 354 772 732 563
Liu et al 논문은 단백질과 상호작용하는 상대방 RNA 서열 정보를 고려하
지 않고 있어 본 논문에서 사용한 IPtr의 특징이 예측 성능을 높여주는데 기여
도가 크다고 고려된다 또한 특징 벡터 기반의 중복 제거 기법이 아닌 유사한
서열 중복 제거 기법을 통해서도 본 논문이 사용한 특징들은 뛰어난 예측 성능
평가를 받고 있다
45 다른 데이터 셋에서의 테스트
본 논문에서 개발되어진 Random Forest 모델을 테스트하기 위해 테스트 데
이터 집합을 PR324와 PR102로 구성하였다 PR324는 PDB로부터 2009년 11월
이후에 X-ray crystallography 기법에 의해 밝혀진 복합체 중 해상도가 30Å
보다 좋은 해상도를 갖는 324개의 단백질-RNA 복합체 서열 쌍 데이터를 의미
한다 PR102는 PSI-BLAST [26]를 사용하여 PR324 데이터 안에서 학습 데이
터의 서열 정보와 비교를 통해 단백질 서열과 RNA 서열 별로 각각 서열 간 유
사도가 60 이상인 서열의 중복을 제거하여 구성한 102개의 단백질-RNA 복합
체 서열 쌍 데이터이다 PR324와 PR102 모두 학습 데이터와의 PDB ID는 중복
되지 않는다
새로운 데이터에 대한 테스트를 위해 Under-Sampling 기법을 가지는
Random Forest 분류기가 사용되었고 학습 데이터의 subset은 7개로 구성되어
지며 결정 트리는 CART [31]를 사용하였다 결론적으로 7개의 CART를 통해
판정된 결과를 종합하여 다수결 투표 방식으로 최종 결정을 내리도록 구현되었
다
표 13 PR324와 PR102에서의 예측 테스트 성능
- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 43 -
PR324와 PR102의 테스트는 개발된 Random Forest 모델의 결합부위 예측
테스트를 수행한 후 검증을 위해 실제 결합부위 정보와의 비교를 통해 True
positive (TP) True negative (TN) False positive (FP) 그리고 False negative
(FN)를 수집하였다
표 13은 PR324와 PR102의 테스트 데이터에서 결합부위 예측 테스트 성능을
나타낸다 PR324가 PR102보다 모든 성능 평가 지표에서 결합부위 예측을 잘하
고 있었는데 PR324의 테스트 데이터는 학습 데이터에 구성된 서열 정보와 유사
한 서열 중복 제거를 하지 않았기 때문에 서로 다른 단백질-RNA 복합체 구조
데이터라 하더라도 결합부위와 비 결합부위를 잘 예측하고 있었다 반면 PR102
의 테스트는 학습 데이터와 유사한 서열을 가지는 서열 정보를 제거하였기 때
문에 PR324에 비해 예측 테스트 성능이 저조함을 알 수 있었다
- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 44 -
제 5 장 결 론
본 연구에서는 단백질에서의 RNA 결합 부위를 예측하기 위해 단백질 서열
과 상호작용 상대방인 RNA 서열 정보를 함께 고려하였다 단백질 서열에서 이
차구조 유형별로 결합 성향을 분석하고 이를 예측에 사용하기 위한 특징으로
사용하였다 441개의 단백질-RNA 복합체 구조 데이터로부터 추출한 3149개의
단백질-RNA 서열 쌍 데이터 셋에서 특징 벡터 기반의 중복 데이터 제거 기법
을 적용하여 학습 데이터를 생성하였다 단백질 서열 정보만 고려한 학습 데이
터보다 상호작용하는 상대방 RNA 정보가 포함된 데이터가 학습될 수 있는 패
턴의 수가 많아져서 예측 성능의 향상을 가져왔다
윈도우 크기 별로 아미노산의 생화학적 특징 normalized position IPta IPar
IPtr의 모든 특징들을 특징 벡터로 변환하고 특징 벡터 기반의 중복 제거 과정
을 거쳐 Random Forest 분류기로 시험한 결과 윈도우 크기가 11일 경우
820의 sensitivity 981의 specificity 962의 accuracy로 가장 좋은 성능을
보였다 단일 특징 중에서는 상대방 RNA를 고려한 아미노산 triplet의 결합성
향인 IPtr이 가장 높은 예측 성능을 보였다
윈도우 크기 별로 Random Forest와 SVM의 단백질에서의 RNA 결합부위
예측 성능 비교를 통해 SVM 기법보다는 Radom Forest 기법이 수행 속도 면
에서나 예측 성능 평가 면에서나 모두 뛰어나다는 사실을 입증했다 또한 특징
벡터 기반의 중복 제거 기법이 아닌 유사한 서열 중복 제거 기법을 사용한 타
연구 시험과의 예측 성능 비교에서도 개선된 평가를 받고 있어 타 연구 시험보
다 본 논문이 사용한 특징들의 우수함을 보였다
개발되어진 Random Forest 모델의 검증을 위해 Under-Sampling 기법을 가
지는 Random Forest 분류기를 학습 데이터에 적용시켜 새로운 서열 정보를 가
지는 테스트 데이터에 결합부위 예측을 시험하였다
본 연구 결과는 단백질-RNA 상호작용 연구나 단백질과 RNA와 관련된 신
약개발에 많은 도움이 될 것으로 기대하며 현재 예측 성능을 추가로 향상시키
기 위한 연구를 진행하고 있다
- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 45 -
참고 문헌
[1] Ptashne M ldquoRegulation of transcription from lambda to eukaryotesrdquo
Trends Biochem Sci vol 30 pp 275-279 2005
[2] Noller H F ldquoRNA structure reading the ribosomerdquo Science vol 309
pp 1508-1514 2005
[3] Hertel K J Graveley B R ldquoRS domains contact the pre-mRNA
throughout splicesome assemblyrdquo Trends Biochem Sci vol 30 pp
115-118 2005
[4] Dreyfuss G Matunis M J Pinol-Roma S Burd C G ldquoHnRNP
proteins and the biogenesis of mRNArdquo Annu Rev Biochem vol 62 pp
289-321 1993
[5] Simpson G G Filipowicz W ldquoSplicing of precursors to mRNA in
higher plants mechanism regulation and sub-nuclear organization of the
spliceosomal machineryrdquo Plant Mol Biol vol 32 pp 1-41 1996
[6] Wang L Brown S J ldquoBindN a web-based tool for efficient prediction
of DNA and RNA binding sites in amino acid sequencesrdquo Nucleic Acids
Res vol 34 pp 243-248 2006
[7] Terribilini M Sander J D et al ldquoRNABindR a server for analyzing
and predicting RNA-binding sites in proteinsrdquo Nucleic Acids Res vol
35 pp 578-584 2007
[8] Breiman L ldquoRandom forestsrdquo Mach Learning vol 45 pp 5-32 2001
[9] Berman H M Westbrook J et al ldquoThe Protein Data Bankrdquo Nucleic
Acids Res vol 28 pp 235-242 2000
[10] Ellis J J Jones S ldquoProtein-RNA interactions structural analysis and
functional classesrdquo Proteins vol 66 pp 903ndash911 2007
[11] Torshine I Y Weber I T Harrison R W ldquoGeometric criteria of
hydrogen bonds in proteins and identification of lsquobifurcatedrsquo hydrogen
bondsrdquo Protein Eng vol 15 pp 359-363 2002
- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 46 -
[12] Kabsch W Sander C ldquoDictionary of protein secondary structure
pattern recognition of hydrogen-bonded and geometrical featuresrdquo
Biopolymers vol 22 pp 2577-2637 1983
[13] Bouckaert R R Frank E et al ldquoWEKA Experiences with a java
open-source projectrdquo J Machine Learning Res vol 11 pp 2533-2541
2010
[14] Choi S Han K ldquoPrediction of RNA-binding amino acids from protein
and RNA sequencesrdquo BMC Bioinformatics vol 12 2011 (in press)
[15] Xia X Xie Z ldquoProtein Structure Neighbor Effect and a New Index
of Amino Acid Dissimilaritiesrdquo Mol Biol Evol vol 19 pp 58-67 2002
[16] Rose J R Turkett W H et al ldquoCorrelation of amino acid preference
and mammalian viral genome typerdquo Bioinformatics vol 21 pp
1349-1357 2005
[17] Lepetit V Fua P Keypoint recognition using randomized trees IEEE
PAMI vol 28 pp 1465-1479 2006
[18] Duda R O Hart P E Stork D G ldquoPattern Classificationrdquo Wiley
2002
[19] Moodie SL Mitchell JB and Thornton JM ldquoProtein recognition of
adenylate an example of a fuzzy recognition templaterdquo J Mol Biol vol
263 pp 486ndash500 1996
[20] Kim H Jeong E Lee SW and Han K ldquoComputational analysis of
hydrogen bonds in proteinndashRNA complexes for interaction patternsrdquo
FEBS Lett vol 552 pp 231ndash239 2003
[21] Cortes C Vapnik V ldquoSupport-Vector Networksrdquo Mach Learning vol
20 pp 273-297 1995
[22] Liu ZP Wu LY Wang Y Zhang XS Chen L ldquoPrediction of
protein-RNA binding sites by a random forest method with combined
featuresrdquo Bioinformatics vol 26 pp 1616-1622 2010
[23] Allers J Shamoo Y ldquoStructure-based analysis of protein-RNA
interactions using the program ENTANGLErdquo J Mol Biol vol 311 pp
- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-

- 47 -
75ndash86 2001
[24] Berman HM et al ldquoThe nucleic acid database A comprehensive
relational database of three-dimensional structures of nucleic acidsrdquo
Biophys J vol 63 pp 751ndash759 1992
[25] Shulman-Peleg A Shatsky M Nussinov R Wolfson HJ ldquoPrediction
of interacting single-stranded RNA bases by protein-binding patternsrdquo
J Mol Biol vol 379 pp 299ndash316 2008
[26] Altschul SF et al ldquoGapped BLAST and PSI-BLAST a new
generation of protein database search programsrdquo Nucleic Acids Res vol
25 pp 3389ndash3402 1997
[27] Pang NT Micheal S Vipin K ldquoIntroduction to Data Miningrdquo
Addison-Wesley pp 283-285 2007
[28] Kim J Park B Han K ldquoPrediction of Interacting Motif Pairs
using Stochastic Boostingrdquo Proc FBIT pp 95-100 2007
[29] Brown M P S Grundy W N Lin D Cristianini N Sugnet C
W Furey T S Ares M and Haussler D ldquoKnowledge-based
analysis of microarray gene expression data by using support vector
machinesrdquo Proc Natl Acad Sci vol 97 pp 262-267 2000
[30] 강필성 조성준 ldquo데이터 불균형 해결을 위한 Under-Sampling 기반 앙상블
SVMsrdquo 한국데이터마이닝학회 2006
[31] Breiman L Friedman J Olshen R Stone C ldquoClassification and
regression treesrdquo Wadsworth Pacific Grove 1984
- 1 서론
- 2 관련 연구 및 배경 지식
-
- 21 Protein Data Bank
- 22 Dictionary of protein secondary structure
- 23 Random Forest
-
- 231 결정 트리 분류기
- 232 결정 트리의 구조
- 233 결정 트리의 설계
- 234 결정 트리의 학습 알고리즘
- 235 결정 트리의 특성
- 236 Random Forest의 설계
-
- 24 특징 벡터 기반의 중복 데이터 제거 기법
- 25 Ensemble of Under-Sampling
-
- 3 데이터 구성 및 특징 선택
-
- 31 단백질-RNA 결합부위 데이터
- 32 단백질-RNA 결합 부위 정의
- 33 단백질 서열을 고려한 결합 성향
- 34 단백질 아미노산 triplet을 고려한 결합 성향
- 35 단백질 이차구조를 고려한 결합 성향
- 36 상대방 RNA를 고려한 결합 성향
- 37 특징 벡터 (feature vector)의 표현
- 38 예측 알고리즘의 성능 평가
-
- 4 시험 및 평가
-
- 41 학습 데이터 생성
- 42 Random Forest를 이용한 예측 성능 평가
- 43 Random Forest와 SVM의 예측 성능 비교
- 44 타 연구 시험과의 비교
- 45 다른 데이터 셋에서의 테스트
-
- 5 결론
-