experimental observations and mathematical …
TRANSCRIPT

The Pennsylvania State University
The Graduate School
Department of Mathematics
EXPERIMENTAL OBSERVATIONS AND MATHEMATICAL
DESCRIPTION
OF MICELLAR FLUID FLOW
A Thesis in
Mathematics
by
Nestor Z Handzy
c© 2005 Nestor Z Handzy
Submitted in Partial Fulfillmentof the Requirements
for the Degree of
Doctor of Philosophy
May 2005

The thesis of Nestor Z. Handzy has been reviewed and approved* by the following:
Andrew BelmonteAssociate Professor of MathematicsThesis AdvisorChair of the Committee
Diane HendersonAssociate Professor of Mathematics
Anna MazzucatoAssistant Professor of Mathematics
Francesco CostanzoAssociate Professor of Engineering Science and Mechanics
Nigel HigsonProfessor of MathematicsHead of the Department of Mathematics
*Signatures are on file in the Graduate School.

iii
Abstract
We present results from a study of wormlike micellar fluids which includes exper-
imental data and a theoretical mathematical model. Experimentally we examined the
effects of air bubbles rising through solutions of wormlike micelles. A previous study of
this problem reported oscillations in the speed of the rising bubble. Our experiments
revealed two distinct types of oscillations, which we have called “type I” and “type II”.
By mapping the oscillatory instability to a temperature-concentration phase plane we
found that type I oscillations occur when the equilibrium average length of micelles is
larger than a critical value.
Experimental rheology was performed on the same fluids as well, which identified a
transition in equilibrium micellar morphology as concentration increases. This transition
is found to occur in the same concentration range as the transition from type I to type II
oscillations. The rheological results indicate that type I oscillations occur in fluids which
consist of entangled wormlike micelles, while the fluids which give type II oscillations
consist of wormlike micelles in a “fused” or crosslinked network state. The rheological
data also suggest that shear induced structures (SIS) may form in the fluids in which
rising bubbles oscillate, and the oscillatory instability is attributed to the formation and
subsequent destruction of SIS in the wake of a rising bubble. Birefringent images taken
during the free rise of an air bubble support this hypothesis.
The experimental results motivate the inclusion of SIS in a constitutive model for
wormlike micellar fluids. We consider a wormlike micellar fluids to consist of three types

iv
of wormlike micelles: short, long, and “bundles” which represent SIS. The concentra-
tions of these three species are coupled to each other through three ordinary differential
equations. The ODE’s are then coupled to the Maxwell constitutive model for viscoelas-
tic fluids to yield a new “weighted Maxwell model”. With a detailed examination of
the physical meaning of the weighted Maxwell model, we find that further modifica-
tions are necessary in order to remain faithful to the physical properties of wormlike
micelles. These considerations lead us to develop a new “memory kernel” to include in
our weighted Maxwell model. We explain how the modification works and what it means
physically. With numerical simulations, we find that our model is capable of capturing
the rheological properties of wormlike micellar fluids.

v
Table of Contents
List of Tables . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . viii
List of Figures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ix
Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xxi
Chapter 1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
Chapter 2. Rheology and Microscale Architecture of Wormlike Micellar Fluids . 4
2.1 Chemistry and self-assembly . . . . . . . . . . . . . . . . . . . . . . . 4
2.1.1 Alternative chemical components . . . . . . . . . . . . . . . . 10
2.2 Steady shear rheology . . . . . . . . . . . . . . . . . . . . . . . . . . 11
2.3 Transient shear rheology . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.4 Concentration dependence of material parameters . . . . . . . . . . . 27
2.5 Conclusion and suggestions . . . . . . . . . . . . . . . . . . . . . . . 36
Chapter 3. Rising Bubble Oscillations: A New Hydrodynamic Instability Observed
in Wormlike Micellar Fluids . . . . . . . . . . . . . . . . . . . . . . . 38
3.1 Air bubbles rising through liquids . . . . . . . . . . . . . . . . . . . . 38
3.2 Bubbles in wormlike micellar fluids . . . . . . . . . . . . . . . . . . . 40
3.2.1 Preparation of fluids and experimental procedures . . . . . . 44
3.2.2 Concentration and temperature dependence . . . . . . . . . . 47
3.2.3 Inferred length dependence . . . . . . . . . . . . . . . . . . . 52

vi
3.2.4 Topological phase transition . . . . . . . . . . . . . . . . . . . 53
3.2.5 Other types of wormlike micellar fluids . . . . . . . . . . . . . 56
Chapter 4. A New Constitutive Model for Wormlike Micellar Fluids . . . . . . . 60
4.1 Basics of rheology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
4.2 Review of existing models for wormlike micellar fluids. . . . . . . . . 64
4.3 Wormlike micelles as chemical reactants . . . . . . . . . . . . . . . . 82
4.3.1 The 3-species model . . . . . . . . . . . . . . . . . . . . . . . 86
4.4 The law of partial stresses . . . . . . . . . . . . . . . . . . . . . . . . 91
4.5 Steady state solutions to the 2-species model . . . . . . . . . . . . . 94
4.6 Predictions for effective viscosity in constant shear flow to the 3-
species model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100
Chapter 5. Modified Memory: How to Remember to Forget . . . . . . . . . . . . 115
5.1 Modified memory . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
5.2 The time dependent constitutive equation . . . . . . . . . . . . . . . 124
5.3 Predictions for stress in time dependent flow . . . . . . . . . . . . . . 132
5.3.1 Linearly ramping shear . . . . . . . . . . . . . . . . . . . . . 133
5.3.2 Oscillatory shear flow . . . . . . . . . . . . . . . . . . . . . . 141
5.3.3 Thixotropic loop: Linearly ramping up and down . . . . . . . 146
5.3.4 Concluding remarks on time dependent stress predictions . . 151
5.4 Memory integrals and the Fredholm Alternative . . . . . . . . . . . . 152
5.4.1 The idea of an inverse constitutive equation . . . . . . . . . . 153
5.4.2 Integral equations and Fredholm theory . . . . . . . . . . . . 154

vii
Chapter 6. Directions for future research . . . . . . . . . . . . . . . . . . . . . . 161
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164

viii
List of Tables
3.1 Alternate wormlike micellar systems tested which produced no oscillating
bubbles. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
4.1 Chemical-like reactions of the three species of wormlike micelles. On the
left of the arrows are the “reactants” which produce the species to the
right of the arrow. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88

ix
List of Figures
2.1 Drawing of an amphiphile meant to represent a HexadecylPyridinium
molecule. Length scales given are approximate. . . . . . . . . . . . . . . 5
2.2 A spherical micelle. Hydrophilic heads form the surface of the sphere,
while hydrocarbon tails fill the interior . . . . . . . . . . . . . . . . . . . 6
2.3 A cylindrical or wormlike micelle. The micelle terminates in a hemi-
spherical endcap in which amphiphiles may be organized as in a shper-
ical micelle. Length scales are estimateed from experimental data on
womrlike micelles [9, 10] . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
2.4 Steady state effective viscosity as a function of shear rate for 1 mM, 2
mM, and 3 mM CPCl/NaSal at 30C. . . . . . . . . . . . . . . . . . . . 14
2.5 Steady state effective viscosity as a function of shear rate for 3.4 mM,
3.7 mM, and 4 mM CPCl/NaSal at 30C. . . . . . . . . . . . . . . . . . 14
2.6 Steady state effective viscosity as a function of shear rate for 6 mM, 7
mM, 8 mM, and 9 mM CPCl/NaSal at 30C. After shear thinning, each
concentration displays a viscosity increase at high shear rate, shown more
clearly in Figure 2.7. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
2.7 Effective viscosity of 7 mM CPCl/NaSal at 30C. Shown by itself, the
thickening at γ ∼ 50s−1 is more visible. The increase in viscosity from
γ = 40s−1 to γ = 100s−1, is similar to the thickening increase for the
low concentration solutions in Figure 2.5. . . . . . . . . . . . . . . . . . 17

x
2.8 Steady state effective viscosity as a function of shear rate for 10 mM, 20
mM, 25 mM, and 30 mM CPCl/NaSal at 30C. . . . . . . . . . . . . . . 19
2.9 Effective viscosity of 35 mM, 40 mM, 60 mM, and 65 mM CPCl/NaSal
at 30C. The apparent discontinuous drop in viscosity for 60 mM and
65 mM is a spurious result due to a switch from one transducer to a
stronger transducer, which is an automatic response due to an overload
of torque. This is addressed more fully in the text. . . . . . . . . . . . . 19
2.10 Effective viscosity as a function of time for 1 mM CPCl/NaSal at 30C.
The applied shear rate, γ = 25s−1, is in the zero shear viscosity plateau
in Figure 2.4. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.11 Effective viscosity as a function of time for 1 mM CPCl/NaSal at 30C.
The applied shear rate, γ = 40s−1, is in the thickening region in Figure
2.4. The long transient lasting more than 200 seconds could easily be
mistaken for a steady state value. . . . . . . . . . . . . . . . . . . . . . . 22
2.12 Effective viscosity as a function of time for 2 mM CPCl/NaSal, at 30C,
at two different shear rates. Both shear rates are in the thickening region
for 2 mM in Figure 2.4. Like 1 mM in Figure 2.11, at γ = 27s−1 there is
a long transient which appears as a constant, almost steady state value.
At γ = 40s−1, the inception time for thickening is less than 100 seconds.
Both rates produce fluctuating viscosities (stresses). . . . . . . . . . . . 24

xi
2.13 Effective viscosity as a function of time for 6 mM CPCl/NaSal at 30C.
The applied shear rate, γ = 50s−1, is in the thickening region in Figure
2.6. The long inception times of 1 mM and 2 mM are not copied by 6
mM, however there is a roughly constant viscosity of 0.1 after the intial
overshoot, lasting roughly 50 seconds. The viscosity for 6 mM fluctuates
more wildly than for the lower concentrations. . . . . . . . . . . . . . . . 24
2.14 Effective viscosity as a function of time for 8 mM CPCl/NaSal, at 30C,
at two different ahear rates. The lower shear rate, γ = 12s−1 is in the
thinning reagion (see Figure 2.6). The higher rate, γ = 60s−1, is at the
very beginning of the shear thickening region for 8 mM. At γ = 60s−1,
time dependent rheology shows fluctuating viscosity, though there is no
evident inception time for the thickening to begin. . . . . . . . . . . . . 26
2.15 Effective viscosity as a function of time for 10 mM CPCl/NaSal at 30C.
The applied shear rate, γ = 10s−1, is in the shear thinning region in
Figure 2.8. After an initial over shoot, a steady state is quickly achieved. 26
2.16 Effective viscosity as a function of time for 20 mM CPCl/NaSal at 30C.
The applied shear rate, γ = 10s−1, is in the shear thinning region in
Figure 2.8. After an initial over shoot, a steady state is quickly achieved. 28
2.17 Effective viscosity as a function of time for 30 mM CPCl/NaSal at 30C.
The applied shear rate, γ = 10s−1, is at the beginning if the shear
thinning region in Figure 2.8. There is no overshoot at this concnetration
as there is for 10mM and 20 mM (Figures 2.15 and 2.16). . . . . . . . . 28

xii
2.18 Time dependent viscosity for 40 mM at 30C, at shear rate γ = 10s−1.
For shear rates near 10s−1, the viscosity of 40 mM does not change
(Figure 2.9). The shear rate is too modest to activate the non-Newtonian
properties in this experiment. . . . . . . . . . . . . . . . . . . . . . . . . 29
2.19 Zero shear viscosity versus concentration. The line passing through the
data points at 3 mM and 10 mM gives the scaling law obeyed for con-
centrations in this region, the slope of the line is ∼ 5.8, indicating that
stress is relaxed by reptation. . . . . . . . . . . . . . . . . . . . . . . . . 30
2.20 Relaxation time versus concentration. The line gives the scaling law
obeyed for concentraions from 8 mM to 65 mM: λ ∼ ϕ−4.1. . . . . . . . 32
2.21 Elastic modulus G0 versus concentration ϕ of equimolar CPCl/NaSal.
The scaling law holds for all concentrations, whereas the laws for λ and
η0 held for different concentration ranges. . . . . . . . . . . . . . . . . . 33
2.22 The “thickening frequency” or shear rate at which the fluid experiences
an increase in viscosity with increasing shear rate Data in this plot was
obtained from Figures 2.4-2.9. . . . . . . . . . . . . . . . . . . . . . . . . 35
3.1 A rising cusped air bubble in a polymer solution (0.08% carboxymethyl-
cellulose in 50:50 glycerol/water). . . . . . . . . . . . . . . . . . . . . . . 39
3.2 Cusp shape change of an oscillating bubble rising through a 15mM CPCl/NaSal
(weight fraction ϕ = 0.5%) solution at T = 37.5C. The scale at left is
marked in centimeters. Interval between pictures: 0.05 s. . . . . . . . . . 41

xiii
3.3 Height z versus time t of an oscillating bubble in 8mM CPCl/NaSal at
T= 22.4C in a 1.2 m cylinder. The data shown is 40 cm high, and 11.4
seconds in duration. VMin = 2.5cm/s, VMax = 5.2cm/s, VAve = 3.4cm/s. 43
3.4 Temperature and concentration phase diagram for the dynamics of a
rising bubbles in equimolar CPCl/NaSal, showing two distinct regions
of oscillating behavior (shaded) labelled as I and II. The straight line at
low concentrations is an isoline of equation 3.1 and marks a boundary
for type I oscillations. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.5 Oscillating bubble in 30mM CPCl/NaSal at T = 21C. Interval between
pictures : 0.24 s. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
3.6 Rheology of equimolar CPCl/NaSal at T = 30C: a) effective viscosity
versus shear rate for 10, 20, 30, and 40 mM CPCl/NaSal; b) zero shear
viscosity, η0 as a function of concentration. The shaded regions mark the
concentration ranges of bubble oscillations. . . . . . . . . . . . . . . . . 51
3.7 Birefringent images of the wake behind a bubble rising in CPCl/NaSal
at T = 24C: a) 10 mM ; b) 20 mM ; c) 35 mM. Each image is 6.5 cm
high. The diameters of the bubbles are all in the range of 3 mm to 5
mm. Reynolds and Deborah numbers for each image are: a) Re ' 4.72,
De ' 250; b) Re ' 0.02, De ' 6; c) Re ' 1.1, De ' 1.8. . . . . . . . . . 55
3.8 Rising air bubble in 80 mM CPCl - 40 mM NaSal diluted in 500 mM
NaCl. The cusp is stable and no oscillations were observed. . . . . . . . 58

xiv
4.1 Effective viscosity as a function of shear rate at very low concentrations
(dilute regime). Although the viscosity decreases at higher γ, it remains
well above the η0 plateau, indicative of a thickened state. . . . . . . . . 83
4.2 Effective viscosity as a function of shear rate for a semi-dilute solution.
The increase in viscosity near is reminiscent of the thickening at lower
concentrations (Fig.4.1). . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
4.3 Effective viscosity as a function of shear rate. This higher concentration
(30 mM) is no longer in the semi-dilute regime. No thickening occurs in
the range of γ tested. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
4.4 Schematic of micelle reactions in the 3-species model (see Table 4.1). . . 87
4.5 The effective viscosity for the 2-species model (equation 4.30) shows a
zero shear plateau at low shear rates, thinning, and then levelling off to
its asymptotic value. The zero shear viscosity and the asymptotic value
are also depicted (equations 4.31 and 4.32). Parameter choices for ηe are
M = 0.1, V = 1, ηa = 100, ηc = 0.1, k0 = 0.01, k1 = 0.06, m = 0.02,
and n = 0.007. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
4.6 Predictions for the effective viscosity (equation 4.30), with varying flow
dependent reaction rates. Each curve is normalized by its zero shear
viscosity. Both shear thinning and shear thickening are captured by the
model. Parameter choices are M = 0.1, V = 1, ηa = 100, ηc = 0.1,
k0 = 0.01, and k1 = 0.06. . . . . . . . . . . . . . . . . . . . . . . . . . . 98

xv
4.7 Predicted normalized viscosity dependence on shear rate. Parameters
used: k0 = 1, k1 = 100, f0 = f1 = f2 = g = 100γ, ηa = 0.1, ηb = 1,
ηc = 0.01, α = β = 2. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102
4.8 Population dynamics for long a-micelles (circles) and short c-micelles
(triangles) for Fig. 4.7 . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
4.9 Dynamics of bundle population for Fig. 4.7 . . . . . . . . . . . . . . . . 104
4.10 Predicted normalized viscosity dependence on shear rate. Parameters
used: k0 = 1, k1 = 1000, f0 = g = 100γ, f1 = 800γ, f2 = 0.01γ, ηa = 1,
ηb = 1011, ηc = 0.01, α = 0.3, β = 5.4. . . . . . . . . . . . . . . . . . . . 106
4.11 Population dynamics for a-micelles (circles) and c-micelles (triangles) for
Fig. 4.10. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
4.12 Dynamics of bundle population for Fig. 4.10. . . . . . . . . . . . . . . . 107
4.13 Predicted viscosity dependence on shear rate. Parameters used: k0 = 1,
k1 = 1000, f0 = f1 = f2 = 100γ, g = 10−6γ + 1, ηa = 0.1, ηb = 1,
ηc = 0.01, α = β = 2. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
4.14 Population dynamics for a-micelles (circles) and c-micelles (triangles) for
Fig. 4.13 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 110
4.15 Dynamics of bundle population for Fig. 4.13 . . . . . . . . . . . . . . . 110
4.16 Predicted viscosity dependence on shear rate. Parameters used: k0 = 1,
k1 = 100, f0 = 10−6γ, f1 = 10−2γ, f2 = 100γ, g = 10−4γ, ηa = 0.1,
ηb = 1, ηc = 0.01, α = β = 2. . . . . . . . . . . . . . . . . . . . . . . . . 113
4.17 Population dynamics for a-micelles (circles) and c-micelles (triangles) for
Fig. 4.16. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 114

xvi
4.18 Dynamics of bundle population for 4.16. . . . . . . . . . . . . . . . . . . 114
5.1 The concentration a(t) is depicted as the dashed curve, while the solid
curve is the corrected concentration, which excludes the micelles that
grew from t1 = 3π/2 to t2 = 5π/2, and then broke by time t3 = 7π/2. . 119
5.2 The original concentration a(t) is shown as the dashed curve, while the
solid curve is a proposed replacement function for obtaining stress at
time π in equation 5.2. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
5.3 The original concentration, a(t) is shown as the dashed curve. Here the
solid cure is the replacement function for computing stress (equation 5.2
at time 3π. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
5.4 The solid curve is R a(2, t) for the concentration function depicted as the
dashed curve. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
5.5 The solid curve is R a(5, t) for the concentration function depicted as the
dashed curve from Figure 5.4. . . . . . . . . . . . . . . . . . . . . . . . . 123
5.6 Using the same concentration function (dashed curve) as in Figures 5.4-
5.5, the replacement function Pµ(5, t) (solid curve) correctly eliminates
the micelles destroyed from t = 1 to t = 2. For this plot we used µ = 0.02. 126

xvii
5.7 The dynamics of a-micelles is shown as the thick dashed curve. Plots of
P aµ
(0.6, t), P aµ
(1.7, t), and P aµ
(6.5, t) are also shown as solid curves, with
µ = 0.01. The paramaters used for these dynamics are the same for the
stress plots 5.10-5.14 which are: γ(t) = t, k0 = 102, k1 = 103, f0(γ) = 0,
g(γ) = 10−3γ, f1(γ) = 104γ, f2 = 106γ, α = β = 2, ηa = 103, ηb = 10,
ηc = 10−3, and λa = λb = λc = 1. . . . . . . . . . . . . . . . . . . . . . 134
5.8 The dynamics of bundles is shown as the thick dashed curve. A plot of the
modified concentration P bµ
(6.5, t) is shown as the solid curve (µ = 0.01),
which is identical to the concentration function b(t) up to that time since
b(t) is monotonically increasing. Parameter values are the same as for
Figure 5.7. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 134
5.9 The dynamics of c-micelles is shown as the thick dashed curve. Plots
of P cµ
(1.0, t) and P cµ
(3.0, t) are shown as solid curves, both are constant
since the concentration function c(t) is monotonically decreasing. Pa-
rameter values are the same as for Figure 5.7. . . . . . . . . . . . . . . . 135
5.10 Time dependent stress prediction using our model (equation 5.13) for
shear flow with shear rate γ(t) = t. . . . . . . . . . . . . . . . . . . . . . 137
5.11 Time dependent stress prediction of the Maxwell model for the same
shear flow used in Figure 5.10. Parameter values are the same as for
Figure 5.7. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 137
5.12 Time dependent stress with concentration functions inside the Maxwell
memory integral, in the same shear flow used in Figure 5.10.Parameter
values are the same as for Figure 5.7. . . . . . . . . . . . . . . . . . . . . 138

xviii
5.13 Time dependent stress with concentration functions outside the Maxwell
memory integral, in the same shear flow used in Figure 5.10. Parameter
values are the same as for Figure 5.7. . . . . . . . . . . . . . . . . . . . . 138
5.14 The logarithm (base 10) of the difference of stress values between values
from Fig. 5.10 and 5.12, as a percentage of the predicted values in Fig.
5.10 (obtained from equation 5.13). As expected, the values from Fig.
5.12 are consistently greater than those obtained from our model. . . . . 140
5.15 Model prediction (equation 5.13) for shear stress in oscillatory shear flow
plotted against time. Here γ = 0.01 cos(t), and parameter values are:
µ = 0.01, k0 = 1, k1 = 100, f0(γ) = 102|γ|, g(γ) = 10−6|γ|, f1(γ) =
10−2|γ|, f2 = 102|γ|, α = β = 2, ηa = 102, ηb = 104, ηc = 10−2, and
λa = λb = λc = 1. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 142
5.16 Prediction of our model (equation 5.13) in oscillatory shear with shear
rate γ(t) = 0.1 cos(t). All parameter values are the same as in in Figure
5.15. With an increase in the amplitude of shear rate, a slight asymmetry
develops in the oscillating stress. . . . . . . . . . . . . . . . . . . . . . . 142
5.17 Prediction of our model (equation 5.13) in oscillatory shear flow with
γ(t) = cos(t). Parameter values are the same as those in Figures 5.15
and 5.16. At this higher amplitude oscillatory shear rate the asymmetry
is much more pronounced than in Figure 5.16. . . . . . . . . . . . . . . . 143

xix
5.18 Maxwell model prediction for shear stress in oscillatory shear flow with
shear rate γ(t) = cos(t). Values of parameters are identical to those used
in Figures 5.15-5.17. The asymmetry in Figure 5.17 (which uses the same
strain) is absent from the Maxwell prediction. . . . . . . . . . . . . . . . 143
5.19 Prediction for time dependent shear stress in oscillatory shear flow using
concentration functions on the outside of the memory integral. Here
γ(t) = cos(t) and all parameter values are the same as in Figures 5.15-5.18. 144
5.20 Prediction for time dependent shear stress in oscillatory shear flow using
the concentration functions on the inside of the memory integral. Here
γ(t) = cos(t) and all parameter values are the same as in Figures 5.15-5.19. 144
5.21 Difference of stress values between values in Figure 5.17 and 5.20 as a
percentage of the values obtained from our model prediction in Figure
5.17. The occurrence of negative value is explained in the text. . . . . . 145
5.22 Time dependent shear stress prediction of our model equation 5.13 in a
thixotropic loop. Parameter values used are: µ = 0.01, k0 = 102, k1 =
103, f0(γ) = 0, g(γ) = 10−3γ, f1(γ) = 104γ, f2 = 106γ, α = β = 2,
ηa = 103, ηb = 10, ηc = 10−3, and λa = λb = λc = 1. Stress values
obtained while γ is increasing are given as squares, the triangles denote
stress values when the shear rate is decreasing. . . . . . . . . . . . . . . 148

xx
5.23 Maxwell model prediction for shear stress in a thixotropic loop using the
same time dependent shear rate as in Figure 5.22. All paramter values
used to obtain the values in this plot are the same as in Figure 5.22.
The Maxwell model predicts a much greater shear thickening effect than
our model (equation 5.13) shown in Figure 5.22. Stress values obtained
while γ is increasing are given as squares, the triangles denote stress
values when the shear rate is decreasing. . . . . . . . . . . . . . . . . . . 148
5.24 Prediction of shear stress in a thixotropic loop using the concentration
functions outside the memeory integral. The shear rate and all parameter
values used are the same as those used to produce the values in Figure
5.22 and 5.23. Stress values obtained while γ is increasing are given as
squares, the triangles denote stress values when the shear rate is decreasing. 149
5.25 Predicted shear stress values in a thixotropic loop study. The shear rate
and all parameter values used are the same as those used to produce the
values in Figure 5.22-5.24.Stress values obtained while γ is increasing are
given as squares, the triangles denote stress values when the shear rate
is decreasing. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 149
5.26 Logarithm (base 10) of the precent difference of stress values obtained
from Figure 5.24 and 5.22, plotted against shear rate. The squares again-
denote stress values obtained using shear rate values when the rate is
increasing, while triangles are obtained by using shear rates when γ(t) is
decreasing - consistent with thier use in Figures 5.22-5.25. . . . . . . . . 150

xxi
Acknowledgments
During my graduate studies, it has been a joy to work among the undergradu-
ates, graduates, post-docs, and professors associated with the Pritchard Lab. My thesis
advisor, Andrew Belmonte, created an atmosphere of cooperation and friendship in the
lab, which encouraged our interest in our studies and helped us dismiss our intellectual
insecurities. Learning from one another was unavoidable. I enjoyed and benefited from
many discussions with Josh Gladden, Mike Sostarecz, Yuliya Gorb, Jon Jacobsen, Bob
Geist, Linda Smolka, Austin Semerad, Young-Ju Lee, Anand Jayaraman, Thomas Pod-
gorski, Ben Akers, Diane Henderson, Anna Mazzucato, Francesco Costanzo, and Andrew
Belmonte.
I began working with Andrew after a very disappointing period of graduate work
in geometry. His dedication to my education and the value he gave my ideas helped me
to recover and grow. It has been my privilege to study with him, and I have enjoyed our
collaborations. I must also thank the Belmonte family for their warm friendship while
graciously tolerating so many late night visits to meet with Andrew. Let me also say
thanks for feeding an always hungry graduate student.
Many seminar speakers our lab hosted, as well as people I met at conferences, took
time to listen to my experimental and theoretical work. I am inspired by their interest
and grateful for their advice and insight. My sincere thanks to Eric Weeks, Bob Leheny,
Peter Olmsted, Elisha Moses, Ralph Colby, Ranjini Bandyopadhyay, Jim Keener, Lynn
Walker, and Stephen Childres.

xxii
In addition to my studies and research, my graduate duties included teaching and
copious paperwork for which I was born with an inclination to avoid. Unfortunately,
paperwork usually comes equipped with deadlines, and this poses a serious threat to me.
I want to thank the excellent staff in the Mathematics Department, especially Becky
Halpenny, for taking care of things and creating the illusion I live with that “things just
work themselves out”.
I thank my friends Sumant, Stephan, Susan, and my dear friend Paul. My brother
Damian and his wife Renia have often made my life easier by “making things work
themselves out” as well, freeing me to concentrate on my thesis. Their efforts are much
appreciated as is thier love.
Most of all I thank my parents. The love and support my mother and father gave
me directly impacts my work and my life. It is because of their guidance and care that
I have completed my graduate degree. They have my undying love.

1
Chapter 1
Introduction
Imagine holding a pole in the middle of a flowing river. The force you need to
apply to keep the pole upright is a measure of the viscous drag exerted by the river.
Intuition would tell you that the faster the river is flowing, the harder it would be to
keep the pole straight, and after a few minutes you might have a good feel for how much
force the river is exerting. Now suppose that all of a sudden you feel the river pushing
much harder, even though it is not going any faster! Such an unexpected surprise could
find you asking: What is this river made of? The answer would be a fluid made of tiny
objects no bigger than the thickness of a human hair, a wormlike micellar fluid.
This Thesis begins by showing some of the extraordinary behavior of wormlike
micellar fluids, in experiments not so different from the one just described. In highly
controlled situations, the fluid speed can be increased in small increments, and very
precise values of viscous drag (or stress) are recorded. This is the study of rheology, and
in Chapter 2 we show our rheological data after first describing wormlike micellar fluids.
Chapter 3 continues with experimental results, studying the behavior of the fluid
in a more complicated flow. Specifically, we examine the fluid’s response to a constant
stress imposed by the buoyancy of a rising air bubble. To place the results in context we
review aspects of rising bubbles in other liquids, including water and polymer solutions.
The behavior of rising bubbles is studied as a function of the concentration of wormlike

2
micelles in the fluid, and the bubble behavior is able to sense a phase transition in the
fluid microstructure as concentration increases past a certain threshold. These results
are combined with the rheology presented in Chapter 2 to provide a more comprehensive
understanding of the phase transition.
The understanding we develop from the experimental observations (our own and
from published sources) presented in Chapters 2 and 3, helps us to describe the physical
processes taking place inside the fluid which bring about the effects we see. To formalize
such a description, in Chapter 4 we focus on the physical properties we consider most
relevant to the effects seen experimentally, and present these processes mathematically.
Using three coupled ordinary differential equations to describe interactions taking place
among the micelles, we write a model for the stress developed in the fluid during steady
state (time independent) flow. Simulated results are then studied and compared to the
rheological results presented in Chapter 2.
Chapter 5 then pursues a description of fluid flow in the more generic case of
time dependent flows. The physical ideas that led to the mathematics in Chapter 4 are
re-examined, and we find that the time dependent case requires a more sophisticated
mathematical approach. We then write the time dependent model explicitly, and study
how it works in the case of three different types of flow. The results are compared to
similar mathematical equations which we argue are physically inaccurate.
Chapter 5 is concluded with a very interesting connection between our model
and the Fredholm theory of integral equations. We provide results about our model
which show it to be amenable to the powerful techniques of this theory. The possible

3
answers Fredholm theory could provide about our model are discussed and their physical
relevance to known rheological results on wormlike micellar fluids.

4
Chapter 2
Rheology and Microscale Architecture
of Wormlike Micellar Fluids
This chapter is an introduction to wormlike micellar fluids. These fluids are
viscoelastic and form a very interesting subset of non-Newtonian fluids. We start with
a description of how wormlike micelles are formed, their approximate size, and the role
of the different molecules involved in their formation. We then present our results from
numerous rheological tests on certain wormlike micellar systems, and explain what the
data says about the particular system we are studying, and how it relates to known
rheological results.
2.1 Chemistry and self-assembly
Wormlike micelles are an example of what are called “self-assembling” structures.
A micelle is not a molecule, it consists of molecules which have the ability to come
together in aqueous solution to form the micelle. In our experiments we have used a
specific set of molecules to make wormlike micellar fluids, and we begin by describing
the self assembling process of this particular choice of chemicals. Once the process is
explained, it will be easier to discuss how general this process is and other ways wormlike
micellar fluids are made.
The fluids used in our experimental studies were made of three raw components:
CetylPyridiniumChloride (CPCl), Sodium Salicylate (NaSal), and deionized water. The

5
first component, CPCl, is a hydro-carbon chain with a function group (a pyridine ring)
at one end. When mixed with water, the chlorine dissociates as a negatively charged
ion (an anion), leaving a hydrocarbon chain with a positively charged head group [1,
2, 3], sketched in Figure 2.1. The hydrocarbon chain is electrically neutral, and as
such is hydrophobic, while the positively charged head group is hydrophilic. Because of
this ambivalence, this molecule is called an amphiphile. The chemical prefix “cetyl” is
synonymous with “hexadecyl”, which denotes the number 16, referring to the number
of carbon atoms in the hydrocarbon chain. The head group is a pyridinium ring, which
consists of five carbon atoms and one nitrogen atom. The lengths in Figure 2.1 are
estimated by counting atoms and assuming that the distance between bonded atoms is
1-1.5 A[1].
Positively Charged Head Group (hydrophilic)
+
Electrically Neutral Carbon Tail (hydrophobic)
Amphiphilic Molecule
4-5 A
14-16 A
Fig. 2.1. Drawing of an amphiphile meant to represent a HexadecylPyridinium molecule.Length scales given are approximate.

6
These amphiphilic molecules are sometimes called surfactants. This word is used
because when the amphiphile (surfactant) is in water, it goes to the surface to enable the
hydrophobic carbon tail to escape from the water, leaving the hydrophilic head to remain
in contact with the water. This in turn reduces the surface tension of the liquid. The
point is that the molecule (more correctly the surrounding water) always prefers to have
hydrophobic tail excluded from the water. When a critical concentration of amphiphiles
is reached, another way of excluding or hiding the tails from the water is available. The
molecules can gather together in such a way that the surrounding water does not come
in contact with the tails, and only interacts with the head groups. For example, this
would be achieved if the molecules aggregated into spheres with the heads on the surface
of the sphere, and all tails pointing towards the center as in Figure 2.2.
Spherical Micelle
Fig. 2.2. A spherical micelle. Hydrophilic heads form the surface of the sphere, whilehydrocarbon tails fill the interior

7
This is our first example of a micelle, in this case a spherical micelle, and the
aggregation process is called micellization. The critical concentration of amphiphiles
needed for micellization to occur is called the critical micelle concentration or CMC [2].
The diameter of a spherical micelle would be roughly twice the tail length ∼ 20− 30 A.
Even above the CMC, there is a fixed concentration (equal to the CMC) of amphiphiles
which exist individually in solution, not in a micelle.
Micellization occurs in order to raise the entropy of the surrounding water, thereby
reducing the free energy of the system. However, there are competing forces in micelle,
which push the amphiphiles away from each other when they come too close together.
Since the head groups are positively charged, there is a Coulomb (electrostatic) repulsion
pushing them away from each other. There is also the crowding of the carbon tails
inside the sphere, which lowers their entropy and prevents too many amphiphiles from
occupying a single spherical micelle [2, 5].
This brings us to the second of the three components we listed above, namely
the organic salt NaSal. Dissolved in water, the salt molecule NaSal seperates into Na+
and a salicylate molecule Sal−, which itself has a hydrophobic part to it. If NaSal is
added to a fluid consisting of spherical micelles made from CPCl, the Sal− prefers to
obscure its hydrophobic portion by penetrating into the spherical micelle [2, 4, 5, 6]. It
therefore can serve to screen the Coulomb repulsion of the positively charged head groups
of the amphiphiles. The effect of introducing the Sal− on the geometry of the micelle
is a transition from spherical to rod-like or cylindrical micelles [5, 7, 8]. The cylindrical
micelles terminate at ends which are hemi-spherical, as if the cylinder grew from the
sphere by cutting the sphere into two equal halves and adding a cylinder between them.

8
The sketch in Figure 2.3 shows how the amphiphiles are arranged in the cylin-
drical portion and hemi-spherical endcaps, with approximate length scales. It must be
understood that the rendition in Figure 2.3 is speculation, though the length scales
have been approximated experimentally [3, 9]. The lengths of wormlike micelles can
range from nanometers to microns. The average length of wormlike micelles depends on
factors such as total amphiphile concentration, temperature, and concentration of salt.
Furthermore, the distribution of lengths in a wormlike micellar fluid can be highly dis-
perse [2, 4, 5, 7]. At concentrations sufficiently greater than the CMC, and with enough
salt, the wormlike micelles are long and flexible, resembling something like a worm. The
lengths that wormlike micelles can achieve, together with their flexibility, make them
similar to polymer chains [6]. And like polymer solutions, wormlike micellar fluids are
viscoelastic non-Newtonian fluids.
4nm
100nm - 1µm
Hemi-SphericalEndcap
Fig. 2.3. A cylindrical or wormlike micelle. The micelle terminates in a hemi-sphericalendcap in which amphiphiles may be organized as in a shperical micelle. Length scalesare estimateed from experimental data on womrlike micelles [9, 10]

9
It is important, though, to remember that the micelles are not molecules, and
that there are no chemical bonds between neighboring amphiphiles and organic salt
molecules as there are in polymer chains. The micelles form due to the hydrophobic
effect, and can both break into smaller micelles, or join with other micelles to form
a longer micelle, under the influence of thermal fluctuations. These kinetic reactions
constantly occur in the fluid, even in equilibrium, and individual amphiphiles are also
free to leave a given micelle, enter a new micelle or join the pool of free amphiphiles. In
equilibrium these processes are of course in balance with one another. A well accepted
model for the average length of wormlike micelles [9, 2, 4] in equilibrium predicts that the
average length of micelles, L0, scales with amphiphile concentration ϕ and temperature
T according to the relation:
L0 ∼√
ϕeE/2kT . (2.1)
The constant k is Boltzmann’s constant: k ' 1.4×10−23 Joules/Kelvin. In the exponent,
the E is the energy needed to hold a micelle together, analogous to the bond energy of
a molecule. This “bond” energy is equated with the energy needed to break a micelle
into two micelles, and called the scission or endcap energy. The reason is that it is
thought that the the “bond energy” is due to the high curvature and dense packing
of hydrophobic tails in the hemi-spherical endcaps. The linear cylindrical portion is
believed to have little or no energy cost. Under this assumption, breaking a micelle into
two micelles requires the creation of two hemispherical endcaps, which requires a cost of
energy equal to the “bond energy” of the micelle. Thus the notions of endcap energy,
scission energy, and “bond” energy are identical to one another.

10
The scaling in equation 2.1 is strictly in equilibrium, and in its derivation it is
assumed that there are no interactions among the wormlike micelles [5, 9]. It makes the
most sense, therefore, to use this model in dilute, or perhaps semi-dilute, solutions in
which micelles are on average far enough apart from one another to avoid interactions.
It is likely that motion of the fluid changes the length distribution, and while it is not
known what the non-equilibrium lengths are, predictive models have been hypothesized.
In Chapter 4 we review some of these models and the relevance of their predictions to
experimental rheology.
2.1.1 Alternative chemical components
The description of the self-assembly of amphiphiles (surfactants) into micelles
given in section 2.1 is somewhat generic, not particular to wormlike micelles made from
CPCl and NaSal. Other surfactants that can lead to wormlike micelles in the way we
have described as well [3, 9, 11]. A selection of such surfactants that are commonly found
in experimental literature are listed here: Cetyltrimethylammonium Chloride (CTAB),
Cetyltrimethylammonium Tosylate (CTAT), and tris(2-hydroxyethyl)-tallowalkyl ammo-
nium acetate (TTAA). Each surfactant can be combined with the organic salt Sodium
Tosylate (NaTos), or NaSal [12]. Other non-organic salts sometimes used in addition to
an organic salt include Potassium Bromide (KBr), Sodium Bromide (NaBr), and Sodium
Chloride (NaCl). The organic salts facilitate growth of wormlike micelles because of their
hydrophic carbon chains, which pentrate into the micelle to avoid interactions with the
polar water molecules [2]. Inorganic salts such as KBr, when used with an organic salt,

11
have little or no effect as a catalyst for the sphere to rod transition and subsequent
growth to long, flexible wormlike micelles [7, 9].
Each choice of surfactant and salt will produce micelles with different length
distributions, different degrees of viscosity and elasticity. For a single surfactant, the
amount of salt used can dramatically affect the size distribution as well the rheological
properties of the fluid [9, 5, 6, 8]. For our experiments, we have chosen the combination
of CPCl with organic salt NaSal, and we use them in equal parts (with the exception of
certain experimental results in Chapter 3, where the precise combination is stated). The
NaSal acts a particularly effective counterion, and using equal parts of surfactant and
salt gives us a maximal growth rate of wormlike micelle for the concentration ranges we
use [6].
2.2 Steady shear rheology
Incompressible viscous Newtonian fluids can be characterized mathematically as
those fluids which obey the Navier-Stokes equation
%∂u∂t
+ % (u · ∇)u = −∇p + η∇2u,
in which % is the fluid density, η the viscosity, p is pressure, and u the velocity. For fluids
of constant density there is a single material parameter, η, and for Newtonian fluids it is
not a function of u. Indeed, Newtonian fluids are those for which the stress σ is linearly
related to velocity gradients, with proportionality constant η: σ = η(∇u +∇uT ).

12
Viscoelastic non-Newtonian fluids have more complex material properties in the
sense that they are elastic and their viscosity can depend on the flow. In this section we
examine the material properties of wormlike micellar fluids in motion, specifically in sim-
ple shear flow. In Cartesian coordinates (x1, x2, x3), simple shear flow is a velocity field
u such that u has only one non-zero component, and ∇u has one non-zero component
which is constant in space and orthogonal to the direction of u. Thus if u = (u1, 0, 0),
then
∇u =
0 0 0
γ 0 0
0 0 0
,
in which γ = ∂u2∂x1
. Here, γ is called the shear rate and is spatially constant, but can
depend on time.
The study of the material properties of fluids in shear flow is the subject of shear
rheology [47, 83]. Shear flow is one of two flow types considered in rheology, the other
being extensional flow in which all velocity gradients are parallel to the direction of flow.
Rheology is performed with a rheometer, and we describe now the rheometer we have
used for the data presented here. In shear rheology a fluid sample is loaded into a chamber
which is confined to shearing motion. There are then two possible ways to control the
motion: through applied stress or applied strain rate. In a stress controlled rheometer, a
force of the controller’s design is applied to the walls of the chamber containg the fluid,
inducing shear flow. The rheometer then measures the shear rate and reports this value.
Factors such as duration of measurement, temperature of chamber, and duration of flow

13
before measurement begins are adjustable parameters and can effect the reported values.
In controlled strain rate rheology, a shear rate is applied, and the shear stress is reported.
For all data in this Thesis, we have used a controlled rate of strain rheometer 1, with
circulating temperature bath to control the temperature of the chamber containing the
fluid sample, in a stainless steel Couette geometry.
The data in Figures 2.4-2.9 are “steady shear rate sweep tests.” In these tests,
a constant shear rate is applied to the fluid for as much as 45-500 seconds (called the
delay time), after which the stress is measured for 45-90 seconds (called the measurement
time). The stress having been recorded, the shear rate is increased to a higher constant
value and the measurement is again taken. This procedure is repeated up to a final shear
rate γf which varies slightly for each fluid tested. The fluids used for these experiments
are equimolar concentrations of CPCl and NaSal. (Procedures used to make these fluids
are discussed fully in Chapter 3). The concentrations used vary from 1 mM to 65 mM,
and the exact concentrations used are given in Figures 2.4-2.9. A delay time was chosen
for each concentration based on how long it takes to achieve a steady state stress value
for that concentration. All tests reported here were performed at 30Celsius, accurate
to within 1.0C.
In Figure 2.4, the concentrations used were 1 mM, 2 mM, and 3 mM CPCl/NaSal.
In the data for 1 mM, we start at the smallest rates γ and work our way up. Then the
plot begins with a constant viscosity value for γ ∼ 10s−1 to 30s−1 called the zero-shear
1Rheometrics RFS III with transducer model 100 FRT, Rheometric Scientific is now ownedand operated by TA Instruments, New Castle, Delaware.

14
0 . 0 0 1
0 . 0 1
0 . 1
1
1 0 1 0 0 1 0 0 0
1 mM2 mM3 mM
η eff
(P
oise
)
Shear rate (s- 1)
Effective Viscosity - Shear Thickening
Fig. 2.4. Steady state effective viscosity as a function of shear rate for 1 mM, 2 mM,and 3 mM CPCl/NaSal at 30C.
0 . 0 1
0 . 1
1
1 1 0 1 0 0 1 0 0 0
3.4 mM3.7 mM4 mM
η eff
(P
oise
)
Shear rate (s- 1)
Effective Viscosity - Shear Thickening
Fig. 2.5. Steady state effective viscosity as a function of shear rate for 3.4 mM, 3.7 mM,and 4 mM CPCl/NaSal at 30C.

15
viscosity η0. For 1 mM η0 ∼ 0.01 Poise, which is roughly the viscosity of water, which
means in equilibrium, the fluid seems Newtonian, and there is no evidence that there are
micelles in the fluid. Near γ ∼ 30s−1 however, the viscosity rapidly increases to a value
which is an order of magnitude greater, showing the fluid is certainly non-Newtonian. So
the solution became “thicker” above some critical shear rate γcrit ∼ 30s−1. Clearly then
this fluid is not Newtonian. In fact, this same type of thickening transition is observed
for 2 mM - 4 mM in Figures 2.4 and 2.5.
What we are observing is called “shear thickening”, and is an interesting phe-
nomenon in low viscosity (or dilute) wormlike micellar fluids. In an outstanding ex-
periment, C. Liu and D. Pine [17] observed such thickening in their rheology of dilute
solutions of wormlike micellar fluids made from CTAB/NaSal and performed additional
light scattering experiments to determine length scales of the micelles. What they found
was that the sizes observed were not consistent with wormlike micelles. Rather, they
likened the observed structures to a rope consisting of coiled threads (presumably the
wormlike micelles), so that the micelles “banded” together to form a new larger struc-
ture. This structure has received much attention [35, 38, 39, 40, 84] and has come to be
known as a “shear induced structure” or SIS. The rheology shown in Figures 2.4 and 2.5
are a likely indication of SIS formation in these concentrations. There are two things
to notice about the SIS formation in these Figures: the shear rate at which they begin,
and the zero-shear viscosity of the fluid. Although we will have much more to say about
these quantities in section 2.4, we note that all η0 are within an order of magnitude of
the viscosity of water, and the critical shear rate for thickening is decreasing as concen-
tration increases. We also note that the thickening jump, ηmax − η0 (where ηmax is

16
the maximum viscosity), is decreasing with concentration, and in fact ηmax seems to
plateau.
Increasing concentration further to 6 mM, we see that the shear thickening is
replaced with a decrease in viscosity from the zero shear plateau (Figure 2.6). Fluid
which display a decrease in viscosity with increasing shear rate, like the fluids in Figure
2.6, are called “shear thinning” fluids. Shear thinning in polymer solutions is thought
to happen because polymers become aligned in the direction of flow by the orthogonal
velocity gradient. Aligned in this way, the polymers would transfer less momentum to
their neighbors, resulting in a decreased viscosity. For wormlike micelles, which can
break and reform, thinning could be due to alignment or, possibly, to a decreased size
of micelle from breaking.
Note also that the η0 values for 6 mM, 7 mm, 8 mM, and 9 mM are much higher
than those for the dilute solutions of Figures 2.4 and 2.5. For 6 mM-9 mM, zero shear
viscosities range from 1 to ∼10 Poise, which is 2-3 orders of magnitude greater than the
η0 values for 1-4 mM. So there is a tremendous increase in η0 coincident with the shift
from shear thickening to shear thinning . That these changes occur over such a modest
increase in concentration is poignant. Indeed, by increasing the concentration from 1
mM or 2 mM CPCl to 3 mM or 4 mM, we produce no significant qualitative difference,
but increasing from 4 mM to 6 mM we introduce an entirely new behavior.
Yet at higher shear rates there is an increase in viscosity reminiscent of shear
thickening. It is difficult to see the precise values of viscosity at shear rates γ ∼ 100s−1

17
0 . 0 1
0 . 1
1
1 0
1 0 0
0 . 0 1 0 . 1 1 1 0 1 0 0 1 0 0 0
6 mM7 mM8 mM9 mM
η eff
(P
oise
)
Shear rate (s- 1)
Effective Viscosity - Shear Thinning
Fig. 2.6. Steady state effective viscosity as a function of shear rate for 6 mM, 7 mM, 8mM, and 9 mM CPCl/NaSal at 30C. After shear thinning, each concentration displaysa viscosity increase at high shear rate, shown more clearly in Figure 2.7.
0 . 1
1
1 0
0 . 0 1 0 . 1 1 1 0 1 0 0 1 0 0 0
7 mM
η eff
(P
oise
)
Shear rate (s- 1)
Effective Viscosity
Fig. 2.7. Effective viscosity of 7 mM CPCl/NaSal at 30C. Shown by itself, thethickening at γ ∼ 50s−1 is more visible. The increase in viscosity from γ = 40s−1 toγ = 100s−1, is similar to the thickening increase for the low concentration solutions inFigure 2.5.

18
in Figure 2.6, so we include a plot of a single concentration (7 mM CPCL/NaSal) in
Figure 2.7 to make the effect more visible. We can clearly see that there is a rapid
increase in viscosity starting near γ ∼ 50s−1, but the increase is not as great as the
shear thickening increases in Figures 2.4 and 2.5. Since the shear thickening at low
concentrations is a rheological indication for SIS formation, we can speculate that the
increases we observe in 6 mM -9 mM are due to something similar. Perhaps these more
modest increases are caused by the formation of smaller SIS, or perhaps SIS which have
become aligned from the shear flow and are hence less viscous.
In Figure 2.8 we show the steady shear rheology of CPCl/NaSal for the concen-
trations of 10 mM, 20 mM, 25 mM, and 30 mM. Again there are η0 plateaus at low γ,
and a slight thickening at higher γ as for the concentrations in Figure 2.6. However,
there seems to be an overall qualitative change in behavior. Whereas all the curves in
Figure 2.6 are similar to one another, in Figure 2.8, each concentration seems different,
although the concentration is increased by a larger amount in Figure 2.8 than that in
Figures 2.4-2.6. Nonetheless, this qualitative as well as quantitative change from 10 mM
to 30 mM CPCl/NaSal is interesting, and we will return to this in section 2.4, where
we will examine it in the context of the rheology of all concentrations presented in this
section.
The highest concentrations we tested are 35 mM, 40 mM, 60 mM, and 65 mM, and
their rheology is shown in Figure 2.9. We first note that for both 60 mM and 65 mM, there
is an abrupt drop in reported viscosity near γ ∼ 100s−1. This is a machine induced effect,

19
0 . 1
1
1 0
1 0 0
0 . 0 1 0 . 1 1 1 0 1 0 0 1 0 0 0
10 mM20 mM25 mM30 mM
η eff
(P
oise
)
Shear rate (s- 1)
Effective Viscosity - Shear Thining
Fig. 2.8. Steady state effective viscosity as a function of shear rate for 10 mM, 20 mM,25 mM, and 30 mM CPCl/NaSal at 30C.
0 . 1
1
1 0
0 . 1 1 1 0 1 0 0 1 0 0 0
35 mM40 mM60 mM65 mM
η eff
(P
oise
)
Shear rate (s- 1)
Effective Viscosity
Fig. 2.9. Effective viscosity of 35 mM, 40 mM, 60 mM, and 65 mM CPCl/NaSal at 30C.The apparent discontinuous drop in viscosity for 60 mM and 65 mM is a spurious resultdue to a switch from one transducer to a stronger transducer, which is an automaticresponse due to an overload of torque. This is addressed more fully in the text.

20
meaning the reported values for viscosity were adjusted by an “offset value” starting
precisely at γ = 158s−1 for both 60 mM and 65 mM. The rheometer design is such
that if measured stresses are 80 − 100% of a threshold stress, the machine begins to
record stress with an alternate transducer, right in the middle of the experiment. This
transducer switch is recorded by the rheometer, but the exact offset value is not. While
the offset amount could be estimated, we have chosen to present the raw data because
it is honest and we feel it is an important example of machine induced error. This
does happen in experimental science and acknowledging it is important so that one can
remain wary of spurious effects and identify false data. We can however estimate the
offset value based on calibration parameters for both 60 mM and 65 mM, and we find
that the corrected viscosity values place the data points in Figure 2.9 in line with the
rest of the data for those concentrations.
We observed that the curves for the concentrations in Figure 2.8 were qualitatively
different from each other, and we see that in Figure 2.9 the rheology of 35 mM up to
65 mM all look very similar in terms of zero shear viscosity. Furthermore, they all have
a broad range of shear rates over which they maintain their zero shear plateaus, much
broader than concentrations as low as 6 mM and as high as 20 mM. In addition, there
is no longer a thickening at high γ as there is for each concentration in Figures 2.6 and
2.8.
Although SIS formation is typically associated with, and has been directly ob-
served in, low concentration wormlike micellar fluids [38, 39], there is evidence that
these structures can form at higher concentrations as well. In the same wormlike micel-
lar fluid we use, Wheeler et al. [40] observe SIS formation in equimolar solution of 40

21
mM CPCl/NaSal. While we do not observe any thickening region in our data for 40 mM
in Figure 2.9, it is likely that it is because our experiments are done at 30C, while those
in [40] are performed in the range 19−22C. In fact, we have tested 40 mM CPCl/NaSal
at 20C, and found a thickening region near γ ∼ 60s−1. So even though fluids in the
concentration range 6 mM -30 mM shear thin before they thicken, it is our belief that
SIS form in these fluids as well. In section 2.3, we explore the time dependent rheology
during SIS formation in our low concentration fluids, which will provide more evidence
that the shear thickening we observe in 6 mM -30 mM is a sign of SIS formation.
2.3 Transient shear rheology
The data presented here in Figures 2.10-2.18 represents the results of transient
stress measurements (with the same rheometer and Couette geometry). In each test, a
shear rate is chosen and held fixed, the rate is applied and the resulting stress is recorded
as a function of time. In this way we can gain information about the individual data
points in each of the plots shown in section 2.2. That is to say, by looking at what the
viscosity values are before they reach steady state, we may be able to deduce something
about the micellar interactions or the stability of SIS for example.
The steady state rheology of 1 mM CPCl/NaSal shown in Figure 2.4 shows two
behaviors: zero shear plateau and shear thickened region. Figures 2.10 and 2.11 show
the time dependent viscosity at shear rates in each of these regions. At γ = 25s−1, the
viscosity of 1 mM is still equal to η0, which means the fluid is nearly in equilibrium,

22
-0 .002
0
0 . 0 0 2
0 . 0 0 4
0 . 0 0 6
0 . 0 0 8
0 . 0 1
0 1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 7 0 0
η eff (
Poi
se)
time (seconds)
Transient response in 1 mM CPCl/NaSal
γ = 25 s- 1.
Fig. 2.10. Effective viscosity as a function of time for 1 mM CPCl/NaSal at 30C. Theapplied shear rate, γ = 25s−1, is in the zero shear viscosity plateau in Figure 2.4.
0
0 . 5
1
1 . 5
2
2 . 5
3
0 1 0 0 2 0 0 3 0 0 4 0 0 5 0 0
stre
ss
(d
yne/
cm2)
time (seconds)
1 mM CPCl/NaSal at T = 30 Celsius
γ = 40 s- 1.
Fig. 2.11. Effective viscosity as a function of time for 1 mM CPCl/NaSal at 30C.The applied shear rate, γ = 40s−1, is in the thickening region in Figure 2.4. The longtransient lasting more than 200 seconds could easily be mistaken for a steady state value.

23
so it is not surprising that the time dependent viscosity quickly achieves a steady state
value. The shear thickening for 1 mM CPCl/NaSal begins near γ = 30s−1, so for
shear rates above that, the fluid is far from equilibrium and we could hope to see the
structure formation reflected in the transient response. Figure 2.11 is the transient stress
at γ = 40s−1, in which the stress is constant for roughly 250 seconds, which could easily
be thought to be the steady value. However, the stress begins to dramatically increase
by more than an order of magnitude, whereupon the stress begins to fluctuate. After
400 seconds, it is not easy to judge whether a steady state has been achieved. This time
dependent behavior is consistent with the time dependent observations in [17] in which
the inhomogenous SIS formation is certain [17, 39].
The same type of transient rheology occurs for 2 mM, where again we believe
SIS develop during flow with rates above γ ∼ 25s−1. In Figure 2.12 we show two time
dependent responses at shear rates γ = 27s−1, 40s−1 in the thickening region. Here we
see that at γ = 27s−1, which gives a viscosity less than the maximum in Figure 2.4, the
viscosity rises more slowly than at γ = 40s−1, which is a rate firmly in the thickened
region. The thickening occurs more quickly at γ = 40s−1 than at 27s−1. The shear rates
at which shear thickening begins in fact has an interesting dependence on concentration,
and we will address this in more detail in section 2.4.
The steady rheology for 6 mM in Figure 2.6 shows that this fluid begins to thicken
near γ ∼ 45s−1, at higher γ for which the fluid has shear thinned. The transient rheology
for 6 mM at γ = 50s−1 is shown in Figure 2.13. The inception time for the rise in viscosity

24
-0 .02
0
0 . 0 2
0 . 0 4
0 . 0 6
0 . 0 8
0 . 1
0 1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 7 0 0 8 0 0
ηef
f (P
oise
)
time (seconds)
γ= 27 s- 1.
γ = 40 s- 1.
Transient response in 2 mM CPCl/NaSal
Fig. 2.12. Effective viscosity as a function of time for 2 mM CPCl/NaSal, at 30C,at two different shear rates. Both shear rates are in the thickening region for 2 mM inFigure 2.4. Like 1 mM in Figure 2.11, at γ = 27s−1 there is a long transient whichappears as a constant, almost steady state value. At γ = 40s−1, the inception time forthickening is less than 100 seconds. Both rates produce fluctuating viscosities (stresses).
-0 .05
0
0 . 0 5
0 . 1
0 . 1 5
0 . 2
0 . 2 5
0 . 3
0 5 0 1 0 0 1 5 0 2 0 0 2 5 0 3 0 0 3 5 0
η eff (
Poi
se)
time (seconds)
γ = 50 s- 1.
Transient response in 6 mM CPCl/NaSal
Fig. 2.13. Effective viscosity as a function of time for 6 mM CPCl/NaSal at 30C.The applied shear rate, γ = 50s−1, is in the thickening region in Figure 2.6. The longinception times of 1 mM and 2 mM are not copied by 6 mM, however there is a roughlyconstant viscosity of 0.1 after the intial overshoot, lasting roughly 50 seconds. Theviscosity for 6 mM fluctuates more wildly than for the lower concentrations.

25
to occur is approximately 50 seconds, much shorter than the 250 seconds needed for the
SIS development in 1 and 2 mM (see Figures 2.10 and 2.12). However, at the higher shear
rate of 40s−1, the inception time was less than 100 seconds in 2 mM. Furthermore, the
time dependent rheology for 6 mM shows large fluctuations in viscosity once thickening
has occured, as do 1 mM and 2 mM. The larger fluctuations and shorter inception time
are probably because the shear rate at which thickening occurs is higher in 6 mM than
at the lower concentrations. Note that these fluctuations do not occur at γ = 25s−1 in
1 mM, and thickening is not observed in this fluid (Figure 2.10).
To check if such fluctuations are specific to the thickening shear rate range, we
tested 8 mM at two shear rates (Figure 2.14): γ = 12s−1 which is in the thinning
region, and γ = 60s−1 which is in the thickening region. At a shear rate of γ = 12s−1,
the viscosity quickly achieves its steady value, and remains relatively constant. At γ =
60s−1, the fluctuations are clearly evident, however there is no longer an appreciable
inception time for viscosity growth. The inception time may be reduced because of the
higher shear rates, which could speed the reactions necessary to for the SIS.
The loss of an incepetion time creates doubt that the shear thickening in 6 mM-
35 mM coincides with structure formation. While it seems that, in dilute solutions, an
inception time on the order of hundreds of seconds is typical in SIS formation [17, 84], we
do observe that the inception time decreases from 1 mM to 4 mM CPCl/NaSal (Figures
2.4 and 2.5). It may simply be that the time needed to form SIS is reduced at higher
concentrations because the fluid begins with larger wormlike micelles, making it easier
to form the induced structures.

26
- 0 . 2
0
0 . 2
0 . 4
0 . 6
0 . 8
0 5 0 0 1 0 0 0 1 5 0 0 2 0 0 0
η eff
(P
oise
)
time (seconds)
γ = 12 s - 1.
γ = 60 s- 1.
Transient response in 8 mM CPCL/NaSal
Fig. 2.14. Effective viscosity as a function of time for 8 mM CPCl/NaSal, at 30C, attwo different ahear rates. The lower shear rate, γ = 12s−1 is in the thinning reagion (seeFigure 2.6). The higher rate, γ = 60s−1, is at the very beginning of the shear thickeningregion for 8 mM. At γ = 60s−1, time dependent rheology shows fluctuating viscosity,though there is no evident inception time for the thickening to begin.
- 0 . 5
0
0 . 5
1
1 . 5
2
2 . 5
0 5 0 1 0 0 1 5 0
ηef
f (P
oise
)
time (seconds)
Transient response in 10 mM CPCl/NaSal
γ = 10 s -1.
Fig. 2.15. Effective viscosity as a function of time for 10 mM CPCl/NaSal at 30C.The applied shear rate, γ = 10s−1, is in the shear thinning region in Figure 2.8. Afteran initial over shoot, a steady state is quickly achieved.

27
In Figure 2.15 the shear rate is γ = 10s−1, for which the steady rheology of 10
mM (Figure 2.8) displays shear thinning. Again the time dependent response shows no
fluctuations of the type we observe in the thickened regions. Here the viscosity quickly
rises in time, and after reaching a maximum, achieves its steady state value where it
remains constant. For 20 mM the transient rheology is very similar in Figure 2.16 for
this same shear rate. According to the steady state rheology in Figure 2.8, 20 mM is
still shear thinning at this shear rate as well.
For both 30 mM and 40 mM, the time dependent response at γ = 10s−1 shows no
fluctuations, and the viscosity rises smoothly to its steady state value with no overshoot-
ing (Figures 2.17 and 2.18). At this shear rate, 30 mM is near the boundary between
the η0 plateau and the thinning region (Figure 2.8). The steady rheology of 40 mM in
Figure 2.9 places this shear rate firmly in the zero shear viscosity range of shear rates.
So the time dependent rheology of both 30 mM and 40 mM, at rates in their zero shear
region, display none of the features we associate with thickening and SIS, as we expect.
2.4 Concentration dependence of material parameters
The data presented in sections 2.2 and 2.3 showed very interesting patterns for
certain concentration ranges. For example, we noted that the steady rheology of concen-
trations in Figure 2.6 looked similar, qualitatively and quantitatively. Again in Figure
28
- 2
0
2
4
6
8
1 0
0 5 0 1 0 0 1 5 0
η eff (
Poi
se)
time (seconds)
Transient response in 20 mM CPCl/NaSal
γ = 10 s -1.
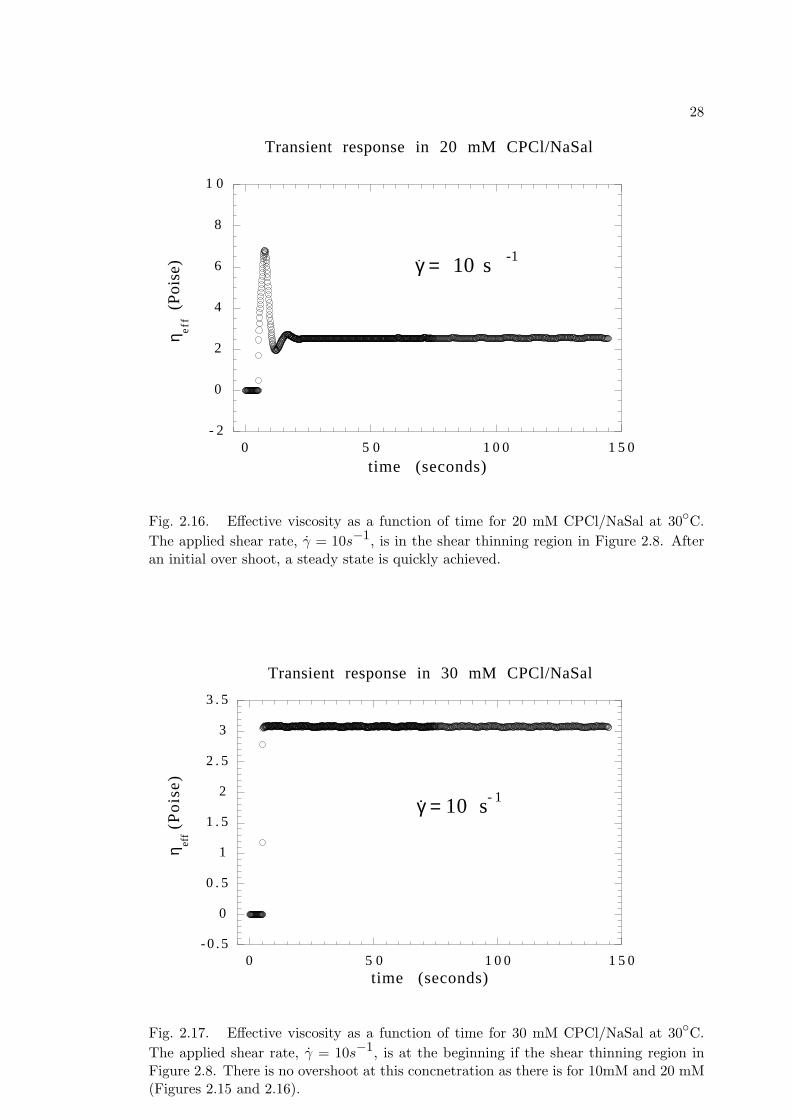
Fig. 2.16. Effective viscosity as a function of time for 20 mM CPCl/NaSal at 30C.The applied shear rate, γ = 10s−1, is in the shear thinning region in Figure 2.8. Afteran initial over shoot, a steady state is quickly achieved.
- 0 . 5
0
0 . 5
1
1 . 5
2
2 . 5
3
3 . 5
0 5 0 1 0 0 1 5 0
ηef
f (P
ois
e)
time (seconds)
Transient response in 30 mM CPCl/NaSal
γ = 10 s- 1.
Fig. 2.17. Effective viscosity as a function of time for 30 mM CPCl/NaSal at 30C.The applied shear rate, γ = 10s−1, is at the beginning if the shear thinning region inFigure 2.8. There is no overshoot at this concnetration as there is for 10mM and 20 mM(Figures 2.15 and 2.16).

29
- 5
0
5
1 0
1 5
2 0
2 5
0 5 0 1 0 0 1 5 0
ηef
f(Po
ise)
time (seconds)
Transient response in 40 mM CPCl/NaSal
γ = 10 s- 1.
Fig. 2.18. Time dependent viscosity for 40 mM at 30C, at shear rate γ = 10s−1. Forshear rates near 10s−1, the viscosity of 40 mM does not change (Figure 2.9). The shearrate is too modest to activate the non-Newtonian properties in this experiment.
2.9 the curves were similar to each other but different from other concentration ranges.
In this section, these observations are put on a firmer footing by studying the behavior
of key material parameters as they vary over the entire concentration range of solutions
we have tested and presented in sections 2.2 and 2.3.
To provide the concentration ranges with a classification scheme, the zero shear
viscosity η0 is shown as a function of concentration in Figure 3.6. The reason for using
η0 to categorize the concentrations is that η0 is a measure of how viscous the fluid is
nearly in equilibrium. Because this value is obtained for low γ, before the fluid responds
with its non-Newtonian character, η0 is a way of probing the structure of the fluid in
steady state with little or no flow, i.e., it is an equilibrium property of the fluids.

30
0 .001
0 .01
0 . 1
1
1 0
1 0 0
1 1 0 1 0 0
η 0
(Poi
se)
Concentration (mM)
Zero Shear Scaling
η0~ϕ5 . 8
Fig. 2.19. Zero shear viscosity versus concentration. The line passing through the datapoints at 3 mM and 10 mM gives the scaling law obeyed for concentrations in this region,the slope of the line is ∼ 5.8, indicating that stress is relaxed by reptation.
In Figure 3.6, the lowest concentrations, 1 mM through 3 mM or 4 mM, have an
η0 comparable to water as we mention in section 2.2. We call this the dilute regime,
since in and near equilibrium the fluid is not very viscous. Following the dilute regime,
we can see a very fast growth in η0 beginning near 3 mM or 4 mM, continuing up to ∼12
mM. The rise in viscosity here is attributed to the growth of small rod like micelles into
longer and flexible wormlike micelles [6, 42]. In this semi-dilute concentration range,
the wormlike micelles are growing to be long enough to give the fluid a much greater
viscosity than in the dilute regime, presumably because the micelles entagle with one
another in their equilibrium configurations. In Figure 3.6 we have fit the data in the
semi-dilute range to a power law model, shown as the solid line. The fit of the this line
to the data is very good, and the slope is found to be 5.8.

31
There are theoretical predictions of how the zero shear viscosity should scale with
concentration in the case of polymers and wormlike micellar fluids [47, 42, 9, 50, 44,
3]. For concentrated solutions of linear polymers, in which stress relaxation occurs by
reptation, the zero shear viscosity η0 is predicted to scale with the concentration ϕ as
ϕ3.4. This prediction is in very good agreement with experiments on polymer solutions
above a critical concentration. Dilute polymer solutions are expected to scale with
concentration linearly [47]. In the case of wormlike micelles, there is the possibility of
relaxing stress by micellar breaking. When micelles break on a much shorter timescale
than the reptative process, a theory by Cates [50, 9] predicts η0 to scale as ϕ3.5. This
exponent value has been seen experimentally in wormlike micellar fluids made with (non-
equimolar) CPCl, NaSal , with NaCl, in which they found η0 ∝ ϕ3.3 [42]. When reptation
is the dominant relaxation mechanism rather than scission reactions, an exponent of
∼ 5.5 is predicted [50, 10]. Our exponent in the semi-dilute region is consistent with this
last prediction and suggests our micelles experience “slow-breaking” reactions.
After the semi-dilute range, there is an unexpected decrease in η0 with concentra-
tion. This is surprising because as we add more amphiphiles to the solution of wormlike
micelles, we would expect the micelles to grow longer and thereby make the fluid more
viscous. However, something else must be happening to reduce η0. If we perceive the
data in Figure 3.6 as a measure of an equilibrium property of the fluid, the decrease in
η0 signals a phase transition in the organization of the wormlike micellar architecture.
Rather than entangled wormlike micelles, a new configuration may be developing, and

32
0 . 0 0 1
0 . 0 1
0 . 1
1
1 0
1 0 0
1 1 0 1 0 0
Rel
axat
ion
ti
me
(se
co
nd
s)
Concentration (mM)
λ ∼ ϕ−4.1
λ Scaling
Fig. 2.20. Relaxation time versus concentration. The line gives the scaling law obeyedfor concentraions from 8 mM to 65 mM: λ ∼ ϕ−4.1.

33
we have suggested in [76] that this is a transition in topology from an entangled state to
a network state with fused junction nodes. This equilibrium phase transition is discussed
fully in Chapter 3, in which additional evidence is provided through experiments on the
fluid in non-equilibrium.
To obtain the relaxation times λ, in Figure 2.20, we used the shear rate at which
the steady state rheology first becomes nonlinear. That is to say, we estimated the
relaxation time by using the relation λ ∼ 1/γc, where γc is defined to be the shear rate
at which shear thinning first begins [3]. We used the standard relation η0 = G0 λ [3] to
obtain estimates the elastic modulus G0 in Figure 2.21. The units of G0 are units of
stress, and it is a measure of how much elasticity the fluid can display [3]. The elastic (or
plateau modulus) is analogous to the spring constant in Hooke’s linear spring force law:
as G0 →∞, we obtain a solid, and if G0 = 0, there is no elastic response to deformation.
0 . 1
1
1 0
1 0 0
1 0 0 0
1 1 0 1 0 0
Ela
stic
m
odu
lus
(d
yne/
sq.
cm)
Concentration (mM)
G0~ϕ3 . 0
G0 Scaling
Fig. 2.21. Elastic modulus G0 versus concentration ϕ of equimolar CPCl/NaSal. Thescaling law holds for all concentrations, whereas the laws for λ and η0 held for differentconcentration ranges.

34
We have seen that the semi-dilute concentration range obeys the zero shear vis-
cosity scaling η0 ∝ ϕ5.8. In Figure 2.20, the fluids with concentration 10 mM (roughly
the end of the semi-dilute regime) and higher obey a law for the scaling of relaxation
time: λ ∝ ϕ−4.1. This alone is interesting, as if each concentration regime obeys a law
for a different material parameter. However, in Figure 2.21 we see a scaling law which is
obeyed by all fluids tested, from 5 mM to 65 mM. The elastic modulus scales with con-
centration as G0 ∝ ϕ3.0, even though there the fluids pass through a phase tranisition
in equilibrium microstructure, the growth of elasticity remains constant.
We note that in the theory of wormlike micelles developed by M. Cates [50], there
is a prediction for the scaling of G0. In the case when kinetic reactions occur faster
than the micelles have time to reptate away their stress, it is expected that G0 ∝ ϕ2.25
[50, 42, 3]. Our observation of a larger exponent for this power law model again implies
that the micelles in our fluid are breaking at rates slower than the reptation time, as
does our observed exponent of 5.8 in our scaling law for the zero shear viscosity.
Finally, we adress the SIS formation and shear thickening in our fluids. In Figure
2.22, we examine the shear rates (called “thickening frequencies” in Figure 2.22) at which
shear thickening begins in the steady state rheology Figures 2.4-2.9. After an intial
decrease from 1 mM to 3 mM, the thickening frequency increases with concentration as
ϕ1.46 . This rule is followed for concentrations ranging from 3 mM to 10 mM, which
defines the semi-dilute regime (Figure 3.6). There is a sudden decrease in thickenning

35
1 0
1 0 0
1 0 0 0
1 1 0 1 0 0
Onset frequency for shear thickening in equimolar CPCl/NaSal
thic
keni
ng
freq
uenc
y (s
-1 )
Concentration (mM)
slope = 1.46
slope=1.28
Fig. 2.22. The “thickening frequency” or shear rate at which the fluid experiencesan increase in viscosity with increasing shear rate Data in this plot was obtained fromFigures 2.4-2.9.

36
frequency between 10 mM and 20 mM, after which it begins to increase at almost the
same rate as the semi-dilute fluids. Although we show only two data points in this
higher concentration range in Figure 2.22, namely 20 mM and 30 mM, if the increase is
obeying a power law, it would have approximately the same exponent as the semi-dilute
concentration range.
So it seems that the thickening frequency is sensitive to the structural transition
taking place in the equilibrium microscale architecture of the fluids. If the viscous thick-
ening in the higher concentration is happening because SIS are forming, as is the case
at low concentrations (1 mM to 4 mM - see Figures 2.4 and 2.5), then the shear induced
structure formation would likely be effected by the change in equilibrium structure oc-
curing between 10 mM and ∼25 mM. We would expect that SIS formation should change
somehow when we increase concentration past the semi-dilute regime to the higher con-
centration phase, and this is precisely what we are observing Figure 2.22.
2.5 Conclusion and suggestions
The rheology presented in this Chapter for equimolar solutions of CPCl/NaSal
identified a phase transition in the equilibrium microscale structure of these fluids. As
concentration increases from 10 mM or 15 mM to ∼25 mM, the behavior of the zero
shear viscosity changes notably from increasing exponentially with concentration (with
exponent 5.8), to decreasing. The inital phase is understood to be a semi-dilute solution,
but the final phase is not as easily identified. We have postulated in [76] that the final
phase is that of a crosslinked network, and have given strong evidence for this conjecture
in [76] which will be discussed thoroughly in Chapter 3.

37
The observed shear thickening rheology at low concentration (1 mM -4 mM) is
nearly identical to that of other systems [17, 39] in which it is known that SIS develop in
the thickenned shear rate range. Our rheology on semi-dilute solutions shows a similar
shear thickening, but only after the fluid has shear thinned. We speculated that this type
of thickening might also be a sign that SIS are forming in the flow, and we performed
transient tests (Figures 2.10-2.18) to address this question. The transients showed that
for rates in the thickened regions, there was a good qualitative match between the rheol-
ogy of dilute solutions (where SIS are known to form) and semi-dilute solutions (where
we speculate SIS form). However, the inception times for viscous thickening between
these concentration ranges are not comparable.
The boundary between these concentration ranges is ∼5 mM. It is obvious that
this concentration must be studied, and concentrations near it, to understand the tran-
sition which replaces shear thickening at low γ with the observed shear thinning, as we
increase concentration and pass into the semi-dilute regime. In fact, we have tested 5
mM CPCl/NaSal, several times, and the data is not reproducible. In some experiments
the fluid thickens much like 4 mM, and in others it shear thins. We feel that future
studies meant to address the possibility of SIS development in the thickening region of
semi-dilute wormlike micellar fluids should include a serious and careful study of fluids
in this middle concentration range centering around 5 mM.

38
Chapter 3
Rising Bubble Oscillations:
A New Hydrodynamic Instability Observed
in Wormlike Micellar Fluids
3.1 Air bubbles rising through liquids
In chapter 2 we explored the rheology of certain wormlike micellar fluids. While
rheological tests provide quantitative information about the fluid (viscosity and relax-
ation time for example), these tests are very limited in that the flow is either pure
extension or pure shear. Rheology attempts to characterize the fluid by its material
paramaters, though it is well known that these properties depend in no small way on the
flow field. For viscoelastic fluids, rheology cannot be expected to lead to a prediction of
the fluid’s response to a general velocity field, since the stress is in general a non-linear
function of the rate of strain. In this chapter we look at a hydrodynamic experiment
that we performed with many wormlike micellar fluids. The experiment is to create an
air bubble at the bottom of a cylinder filled with micellar fluid, and then to examine
such properties as speed and shape of the bubble. This experiment is simple in that
it requires no extraordinary tools, and subjects a fluid initially at rest to the constant
average stress of the buoyancy of the bubble, which causes motion in the fluid.
While the motivation for such an experiment can be pure curiosity, the particular
choice of examining a rising air bubble is linked to previous experiments on rising bubbles
in other viscoelastic fluids, namely polymer solutions, as well as Newtonian fluids. In

39
either type of fluid, several different bubble shapes have been documented as well as
other interesting phenomena [75, 73, 22, 74]. And while bubbles in Newtonian fluids are
always smoothly curved (with the exception of turbulent flows), in polymer solutions
they can have a sharp cusp at their trailing end (Fig. 3.1). Furthermore, this cusp is
non-symmetric [22], that is, if the bubble is seen rotated 90, the sides would not meet
at a point [47]. The effect is a “knife edge”, and this edge itself does not always have
the same shape. An excellent account on the possible cusp shapes is given by Liu, Liao,
& Joseph [22].
Fig. 3.1. A rising cusped air bubble in a polymer solution (0.08% carboxymethylcellulosein 50:50 glycerol/water).
Another effect seen with rising bubbles in non-Newtonian fluids is the so called
“negative wake”, which refers to the direction of the fluid flow in the wake of the bubble.

40
In Newtonian fluids, the fluid follows the bubble in the wake, i.e. it flows up. In polymer
solutions it was first observed by Ole Hassager in 1979 [75] that the flow in the wake of
the bubble is down. A good analogy is with the wind created by a passing car; leaves
on the ground get whipped up behind the car, and then move in the same direction as
the car. Imagine instead the leaves going in the opposite direction and it becomes clear
that this negative wake is extremely counterintuitive and intitiates us to the world of
non-Newtonian fluids.
3.2 Bubbles in wormlike micellar fluids
On a microscale level, Newtonian fluids consist of molecules which can be ap-
proximated by point masses (such as air and water). Non-Newtonian fluids consist of
larger structures, such as polymer chains. This increased size affords a greater number
of degrees of freedom which leads to macroscopic viscoelasticity [47]. Wormlike micelles
are aggregates of amphiphilic molecules, long enough to resemble polymers [6], but with
the added feature that their length distribution is determined by aggregation kinetics
since micelles continually break and reform [2, 4, 16]. The molecular level physics is
more complicated for such fluids, and one can expect this will introduce a new set of
flow properties to the class of non-Newtonian fluids.
In particular, we can ask whether or not micellar fluids will be able to support
a sharp cusp on a rising bubble. The problem is that the stress in the fluid near the
surface of the bubble is proportional to the surface curvature (assuming a constant
surface tension) [47]. Evidently, fluids comprised of large (macro) molecules can support
the large stress at the cusp, but fluids of point particles (Newtonian fluids) do not. If the

41
stress in a micellar fluid is large enough to break the micelles, then they might not be
long enough to accomodate a cusp, resulting in Newtonian-like bubbles. It is interesting
to see the effect of stress and flow on the micellar kinetics, and we can ask the question:
will the impact on micellar kinetics be great enough to be seen macroscopically in the
rise of an air bubble?
Fig. 3.2. Cusp shape change of an oscillating bubble rising through a 15mM CPCl/NaSal(weight fraction ϕ = 0.5%) solution at T = 37.5C. The scale at left is marked incentimeters. Interval between pictures: 0.05 s.
The answer to this question is yes [19]. For certain wormlike micellar fluids, there
is in fact a sharp cusp at the trailing edge of a bubble, but it is unstable in the following
sense. In these wormlike micellar fluids we have observed the cusp extend to a sharp
point and then retract to a blunt edge, as shown in Figure 3.2. This effect repeats itself
during the entire rise of the bubble with no apparent sign of diminishing, as discussed and
shown below. More spectactular is what happens to the rise velocity during the cusp

42
shape change; the bubble velocity rapidly increases when the cusp retracts and then
slows again. Typically, bubbles begin to oscillate within 10 seconds of their formation,
with a similar time between oscillations.
These clearly visible velocity oscillations are apparently not a transient effect; we
have observed their persistence for rise distances larger than one meter, during which
there were more than 30 oscillations in ∼ 35 s (for 8 mM CPCl/NaSal), and velocities
more than doubled during a speed oscillation. Figure 3.3 tracks the position of a rising
bubble. Negative wakes were sometimes observed as well, but the observation was only
possible when a second bubble was in the wake of a higher bubble. In this case the
lower bubble would be pushed downward during the oscillation of the bubble above. It
is unclear whether a negative wake is present when the bubble is not oscillating, and we
could not determine this possibility.
In this chapter we present a detailed experimental survey of the oscillatory motion
of rising bubbles in a wormlike micellar fluid [76]. To study the role of the aggregation
kinetics of the micelles on the observed dynamics, both concentration and fluid tem-
perature were varied over a wide range. As these parameters change, different micellar
architectures are possible - from short linear or branched micelles to crosslinked net-
works [10, 26, 25]. Four types of bubble dynamics were seen: Newtonian behavior at
high temperatures, standard polymeric behavior, and two distinct oscillating responses
occurring in different concentration ranges. Steady rheology experiments were performed
to identify the fluid microstate, and we found that transitions in the equilibrium struc-
ture match transitions in bubble behavior. Critical temperature bounds were also found,

43
Fig. 3.3. Height z versus time t of an oscillating bubble in 8mM CPCl/NaSal at T=22.4C in a 1.2 m cylinder. The data shown is 40 cm high, and 11.4 seconds in duration.VMin = 2.5cm/s, VMax = 5.2cm/s, VAve = 3.4cm/s.

44
which can be interpreted as a minimum length of micelle required for oscillations to oc-
cur, so that for certain concentrations the fluid may be tuned with temperature to make
bubbles oscillate, rise with a stable cusp, or rise as bubbles in Newtonian fluids.
3.2.1 Preparation of fluids and experimental procedures
Non-transient oscillating bubbles were first observed in aqueous solutions of cetyltrimethy-
lammonium bromide (CTAB) and sodium salicylate (NaSal) [19]; here we use another fa-
miliar system, cetylpyridinium chloride (CPCl) and NaSal, with the fixed ratio [NaSal]/[CPCl]
= 1 [6]. The CPCl, NaSal, and NaCl were purchased from Sigma-Aldrich. The surfac-
tant and salts were dissolved in water which was deionized with a Barnstead B-Pure
purification system. Typical conductivity values for deionized water were in the range
16.5− 17.9 MΩ− cm. The procedure for making the fluids was:
1. The surfactant was combined with deionized water and mixed for one hour over a
low heat.
2 The NaSal was dissolved in deionized water and mixed by hand for five to ten
minutes.
3. After mixing the surfactant and water for one hour, the salts (dissolved in water)
were added to the surfactant solution. The heat was turned off and the solution
was allowed to mix for twenty four hours or more.
4. Before any use of the fluid for data collection, the mixed fluid was allowed to sit
undisturbed for three or more days.

45
All experimental results are for a single bubble rising near the center of the tube,
with volumes ranging from 14 mm3 to 110 mm3. The bubble was injected into the fluid
at the bottom of a plexiglas cylinder (31 cm height, 5 cm diameter) using a syringe with
a long stainless steel tube (inner diameter of either 1.0 mm or 1.5 mm). Great care was
taken to record data on a single bubble with the syringe, which was a difficult procedure
because often more than a single bubble was produced at a given time. Many trials
resulted in either multiple bubbles or a single bubble which was near the wall of the
cylinder. The plexiglas cylinder sat in a rectangular plexiglas tank (∼ 8 gallon capacity)
filled with tap water which was temperature controlled by a Neslab RTE-111 circulator
connected to the tank with two hoses. The water level in the tank was controlled with
adjustable clamps on the hoses, and was always at least five centimeters lower than the
top of the cylinder containing the wormlike micellar fluid.
Immediately following each bubble trial, the temperature of the micellar fluid was
measured by inserting a glass mercury thermometer (accurate to 0.1C) directly into the
micellar fluid. The temperature of the fluid was recorded only after it remained constant
for one minute or more. After the temperature was measured, the syringe was removed
and the cylinder was covered with parafilm. Both the syringe and thermometer were
washed by hand with mild soap and tap water, then rinsed first with ethanol (95% pure)
then deionized water and dried with Kimwipes. To allow the fluid to rest and return to
equilibrium, no less than one hour was allowed between succesive bubble trials on a given
fluid. When temperature was changed, no less than two hours was given for the fluid to
equilibrate after the fluid achieved the desired temperature. When a new concentration
was tested, the old fluid was discarded and the cylinder was washed with mild soap and

46
tap water, rinsed with deionized water and dried with compressed nitrogen. The new
fluid was poured into the clean cylinder, covered and allowed to equilibrate at room
temperature for no less than twenty four hours before any experiments were performed.
The ratio of the horizontal diameter of the bubble d to the cylinder diameter D
is d/D ≤ 0.14, and we have checked that bubbles oscillate for d/D ' 0.02. Typical
sizes for the Reynolds number (inertia) are Re ' 10−2−5, while the Deborah number
(elasticity) ranged from De ' 1− 500, similar to values seen for oscillating bubbles and
spheres in CTAB/NaSal [19, 20]. Rheological data were taken with a Rheometrics RFS-
III controlled rate of strain rheometer with circulating fluid temperature bath, using a
stainless steel Couette geometry. Video images were made with a Kodak Ektapro Motion
Analyzer (Model 1012), and a Vision Research, Inc. Phantom v5 CCD camera.
Birefringent images were made by placing the plexiglas cylinder between two
crossed polarizers, the Phantom CCD camera in front and a uniform light source behind.
Optical birefringence is a well-known technique for visualizing stress in non-Newtonian
fluids [34], and is especially effective for wormlike micellar fluids [35]. Before reaching
the micellar fluid in the cylinder, the light is linearly polarized, and then blocked on
its way to the camera by the second polarizer. In this arrangement, when the fluid is
at rest (and is isotropic), no light reaches the camera. The anisotropy (alignment of
micelles) of the fluid caused by the rising bubble rotates the polarization of the light in
the fluid and allows only some light to reach the camera. Birefringence occurs when the
polarizers are either parallel or perpendicular to the alignment of the micelles in order to
obtain birefringence. The result is light regions in the fluid representing different local

47
alignments of micelles, which means the micelles are stretched from their equilibrium
coiled configurations. We therefore associate stress with the light regions in the fluid.
3.2.2 Concentration and temperature dependence
We have studied concentrations from 4 to 40 mM (weight fractions 0.13% ≤ ϕ ≤
1.3%). From 5 to 15 mM, bubbles have a cusp which oscillates in length and changes
shape (Fig. 3.2). While rising, the cusp lengthens (frames 1-4 in Fig.3.2) during which
the velocity as measured at the top of the bubble is nearly constant. At the apex of
the extension, the tail abruptly retracts and the bubble jumps upward (frames 4 and
5). After this it slows to a nearly constant velocity until the cycle repeats. We call this
behavior type I oscillations, which occurs in the temperature ranges shown in Fig.3.4
and is consistent with the oscillations previously observed in CTAB/NaSal [19].
Above a critical temperature in the type I range (Fig.3.4), bubbles have a sharp
cusp which does not change in length (“steady cusp” in Fig.3.4) and velocities smoothly
reach a steady state as in polymer solutions, i.e, the oscillatory instability vanishes. By
60C, the solutions appear Newtonian (“no cusp” in Fig.3.4): large enough bubbles are
ellipsoidal and undergo the well-known side-to-side oscillations [27]. In this temperature
range, the micelles must be predominantly spherical or more like rigid rods [2] neither
of which are not stretched by the passing bubble.
Upon increasing the concentration from 15 mM to 16 mM (ϕ = 0.5% to 0.53%),
all oscillations cease. Bubbles rise steadily with a stable cusp, for concentrations up
to 20 mM in temperature ranges shown in Fig.3.4, but there are no temperatures at
which oscillations occur. This is puzzling, as these fluids are still viscoelastic, consist

48
20 25 30 35 40 45 50 55 60
4
6
810
30
no cuspsteady cusposcillations
Temperature [°C]
Con
cent
ratio
n [
mM
]
I
II
Fig. 3.4. Temperature and concentration phase diagram for the dynamics of a risingbubbles in equimolar CPCl/NaSal, showing two distinct regions of oscillating behavior(shaded) labelled as I and II. The straight line at low concentrations is an isoline ofequation 3.1 and marks a boundary for type I oscillations.

49
of wormlike micelles, and have a rheology similar to type I fluids (discussed below).
Note that the transition temperature from polymeric to Newtonian behavior reaches a
maximum at this concentration (Fig.3.4).
More surprising is the re-emergence of oscillations for concentrations from 25 mM
to 40 mM (0.8% ≤ ϕ ≤ 1.3%). Yet here the oscillations, which we call type II, are
visibly different from those at lower concentrations (Fig. 3.5); the shape change involves
the entire bubble, whereas in type I it seems restricted to the tail and cusp. A type
II oscillation begins with a constriction in width near the top of the bubble, while the
whole bubble lengthens. This constriction then travels downward, as if the bubble were
squeezing through a hoop. We define wmax to be the maximum (relaxed) width (at
its waist), wmin to be its most constricted width, and ∆w = wmax − wmin, we found
∆w/wmax ' 0.13 in type II fluids and 0.04 in type I. Length extensions, however, were
∆l/lmin ' 0.25 in type II and 0.27 in type I; for previously observed bubbles rising
in CTAB/NaSal, ∆l/lmin ' 0.26 [19]. This previous study revealed only one type of
oscillation, which due to the shape dynamics we identify as type I [19].
Although wormlike micelles are comprised of surfactants, it is unlikely that surface
tension plays a role in the mechanism of bubble oscillations; falling rigid spheres have also
been observed to oscillate in wormlike micellar fluids [20, 36]. Figure 1 in the article by
Jayaraman and Belmonte [20] is an overlaid time series of a solid sphere in a wormlike
micellar fluid made from 9mM CTAB/NaSal. The time interval between images is
constant, though the spacing between succesive sphere positions is not, and indicating
speed oscillations. Furthermore, our observation of two types of oscillation suggests that
the viscoelastic character of the bulk fluid is changing with concentration. We address

50
Fig. 3.5. Oscillating bubble in 30mM CPCl/NaSal at T = 21C. Interval betweenpictures : 0.24 s.
this with rheology measurements, controlling the shear strain rate γ and recording the
effective viscosity, ηeff = σxy/γ, where σxy is the steady shear stress. Transient tests
were also performed, to ensure that steady state was achieved (see chapter 2). Shown in
Fig.3.6a is ηeff as a function of γ at 30C, for concentrations in both oscillation regions,
as well as the non-oscillating range between them. All fluids are shear thinning: ηeff
decreases at high enough γ. While there is a noticable difference in the rheology upon
increasing from 20 to 30 mM, 30 and 40 mM (type II region) are strikingly similar;
they have a constant viscosity over a broader range of γ than the other fluids. More
interestingly, the zero shear viscosity, η0 (defined as the plateau value at low γ [47]),
decreases with concentration. As concentration increased, there is more material to form
micelles and we would expect them to thus be longer, which would increase viscosity.
The observed decrease is therefore another counterintuitive aspect of wormlike micellar
fluids.

51
0.001
0.01
0.1
1
1 0
1 0 0
1 1 0
η 0 (
Poi
se)
Concentration (mM)
I II
0.1
1
1 0
1 0 0
0.01 0.1 1 1 0 1 0 0
10 mM20 mM30 mM40 mM
Shear Rate (s- 1
)
η eff (
Poi
se)
a )
b )
Fig. 3.6. Rheology of equimolar CPCl/NaSal at T = 30C: a) effective viscosity versusshear rate for 10, 20, 30, and 40 mM CPCl/NaSal; b) zero shear viscosity, η0 as afunction of concentration. The shaded regions mark the concentration ranges of bubbleoscillations.

52
3.2.3 Inferred length dependence
The changes in temperature and concentration introduce variations in the fluid
microstructure, among which is the micellar length. For low concentrations in equilib-
rium, the average length of a wormlike micelle L0, can be described within a mean field
approximation [9, 2, 5]:
L0(φ, T ) ∼√
φ eE/2kT (3.1)
where φ is the total amphiphile concentration, k is Boltzmann’s constant, T is tem-
perature, and E is the scission energy required to break one wormlike micelle into two
wormlike micelles. The scission energy is directly related to the energy required to sus-
tain an endcap of a micelle. To break a micelle in two requires the formation of two new
endcaps, so the scission energy is exactly twice the endcap energy. Estimates based on
light scattering measurements for E are on the order of 10kT [9, 37, 25] - much lower
than the covalent bonds of polymers, yet large enough for some micelles to reach appre-
ciable lengths against thermal fluctuations [9]. The flow near the bubble may increase
the equilibrium length, and there is evidence that shear-induced structures (SIS) much
larger than individual micelles form in such flow in both CTAB/NaSal [17, 38, 39] and
CPCl/NaSal [40, 41]. We assume simply that for the type I region, L0 is the relevant
quantity determining whether or not the bubble oscillates. Specifically, we attribute the
oscillations to a breaking instability of micelles or SIS in the bubble wake, occurring only
if L0 exceeds some critical value Lc. Using equation.3.1, the solid line in Figure 3.4 is
an isoline of L0 corresponding to a scission energy of E = 1.01× 10−19 Joules, which is
about 24kT .

53
Type II oscillations have a much simpler dependence on temperature; they occur
only for T . 36C (Figure 3.4). If crosslinks have indeed formed, then the mean field
length is no longer an appropriate quantity. The transition temperature Tc = 36C
suggests a critical energy condition (Ec ' 4.3 × 10−21 Joules), much smaller than the
endcap (scission) energy, which may describe the transition from junctions to endcaps.
Thus for T > Tc, endcaps would dominate and the fluid is comprised of individual
micelles which can entangle like polymers. For T < Tc, the microscale structure remains
a crosslinked network of micelles fused at junction nodes. Though this is at present only
speculation, it is consistent with both the rheology and the rising bubble dynamics.
3.2.4 Topological phase transition
The overall dependence of η0 on ϕ is complicated, and indicates transitions in
micellar microstructure. This is shown in Fig.3.6b, in which the different oscillation
regimes are shaded. The transition at low concentration from constant η0 to rapid
growth marks the overlap concentration ϕ∗ ' 3 mM, below which the viscosity is similar
to the viscosity of the solvent, and this is therefore called the dilute regime. Above ϕ∗
the fluid viscosities begin rapid growth. The region above ϕ∗ is clearly distinguished
from the dilute, and we call this the semi-dilute regime. In the semi-dilute regime it is
reasonable to assume that micelles exist as individual worms in a disordered entangled
network [42], which would account for the higher viscosities. Here we find η0 ∼ ϕ5.5,
close to the value 5.4 associated with the relaxation of stress by reptation [10, 29]. The
semi-dilute regime ends near ϕ ' 12 − 15 mM, and is followed by an extraordinary

54
decrease of η0 with concentration, which continues to approximately ϕ ' 30 mM, after
which η0 varies weakly.
The dramatic change in the concentration dependence of η0 indicates a transition
in the equilibrium fluid structure, coinciding with the loss of type I oscillations. It may be
that the entangled micelles in the semi-dilute range have begun to fuse at entanglement
points, forming a crosslinked network [10, 29, 31, 32]. The crosslinked network state of
micellar structure was also proposed for another wormlike micellar system which shares
rheological properties with our type II fluids [10], among which are the broad zero shear
viscosity plateaus. The junction nodes are free to slide along the micelles in such a
formation, which would account for the decrease in viscosity. We assume that the ratio
of crosslinks to entanglement points would grow as η0 decreases, until the new state is
fully formed and η0 stabilizes near ∼ 30 mM = 1%. A crosslinked network state was
also proposed by Lequeux and Candau [33] for systems of CTAB/NaSal at ϕ ' 1%, and
it is noteworthy that CTAB/NaSal solutions also give rise to oscillating bubbles [19].
Transitions in micellar morphology would naturally lead to transitions in the
mechanisms for stress relaxation, which should be observable in the stress field around
the rising bubble. These transitions are made evident through birefringent visualization,
shown in Fig.3.7 for the three concentration regimes, in which bright areas correspond
to anisotropic fluid (see Chapter 2 section 1). Fig.3.7a (10 mM) shows the localization of
stress in the wake of a type I oscillating bubble. This birefringent tail mirrors the dynam-
ics of the bubble’s cusp (Fig.3.2), and when the cusp retracts upward, the birefringent
tail disperses to the sides. At 20 mM a similar - but shorter - tail is seen (Fig.3.7b), whose
length remains constant, like the bubble tail. Fig.3.7c corresponds to type II oscillations

55
Fig. 3.7. Birefringent images of the wake behind a bubble rising in CPCl/NaSal atT = 24C: a) 10 mM ; b) 20 mM ; c) 35 mM. Each image is 6.5 cm high. The diametersof the bubbles are all in the range of 3 mm to 5 mm. Reynolds and Deborah numbers foreach image are: a) Re ' 4.72, De ' 250; b) Re ' 0.02, De ' 6; c) Re ' 1.1, De ' 1.8.

56
(35 mM), in which three equally spaced birefringent bands or wings indicate a broader
distribution of stress. This pattern is slightly altered during the course of an oscillation:
the lateral birefringent wings move down a little the bubble during an oscillation and
rise again afterwards. These images clearly show that each type of oscillatory motion is
matched by a unique stress response in the fluid.
3.2.5 Other types of wormlike micellar fluids
The different oscillations we have observed appear linked to two different mi-
croscale architectures as we have shown. We extended our study to include the well
characterized ternary system CPCl-NaSal diluted in concentrated NaCl, with a ratio
[NaSal]/[CPCl] = 0.5 [42, 43, 16]. We tested several concentrations of these fluids for
oscillating bubbles in our apparatus (Table 3.1) at T = 30C, a temperature central to
both type I and II oscillations (Fig.3.4).
Experiment [CPCl] [NaSal] [NaCl]
1 30mM 15mM 500mM
2 40mM 20mM 500mM
3 50mM 25mM 500mM
4 60mM 30mM 500mM
5 70mM 35mM 500mM
6 80mM 40mM 500mM
Table 3.1. Alternate wormlike micellar systems tested which produced no oscillatingbubbles.

57
A typical bubble shape is shown in Figure 3.8. We observed no oscillations or
any behavior different from steadily rising bubbles in conventional polymeric fluids [22].
This ternary system is known to consist of entangled wormlike micelles, which when
taken with the observed scaling η0 ∼ ϕ3.3 [30], indicates that micellar breaking is the
dominant stress relaxation mechanism (occurring on a shorter timescale than reptation)
[9, 42]. The observed scaling law for η0 for our fluids in the semi-dilute regime indicates
stress relaxation by reptation rather than micellar scission kinetics. This difference could
account for the absence of oscillations in the ternary system with added NaCl. If so, the
type I oscillatory motion of rising bubbles would not be a characteristic of fluids in this
“fast-breaking” limit [9, 30].
We may still ask why this ternary system could not support type II oscillations,
and a clue is offered through our conjecture of a crosslinked network. The ternary
systems we tested that failed to produce oscillating bubbles (given in Table 3.1) have
been well studied and no decrease in zero shear viscosity has been reported as surfactant
concentration was increased [42]. Using similar techniques, it was found that in other
wormlike micellar systems the size of micelle decreased as concentration was increased
[44, 45] and it was again proposed that these systems were in a crosslinked network state.
The data collected by Berret et al. [42] for the ternary sytem we have tested led experts
in the field to conclude that these ternary systems “are prototypes for entangled but
not branched wormlike micellar systems” [42]. Perhaps then the crosslinks (branches)
are essential in producing type II oscillations. There is at present not enough data on
bubble oscillations to put this conjecture on firm ground, though all of the evidence

58
collected so far (here and elsewhere) on wormlike micellar fluids either supports it or
fails to contradict it.
Fig. 3.8. Rising air bubble in 80 mM CPCl - 40 mM NaSal diluted in 500 mM NaCl.The cusp is stable and no oscillations were observed.
Wormlike micelles are far more delicate structures than polymers as constituents
of complex fluids, and the interplay of their microscopic dynamics with macroscopic flow
promises to include novel instabilities to the class of viscoelastic fluids. Indeed, the os-
cillatory motion of a rising bubble through a wormlike micellar fluid illustrates some of
the dramatic possibilities of this coupling. Lengths of micelles can change locally with
flow, and micelles may themselves aggregate into SIS. We have shown that this response
to flow can in fact be tuned by the choice of temperature and concentration, and the
effects can produce dynamics in which the forces never balance - no steady state is ob-
served, despite viscous damping. Beyond bubble dynamics, this ‘tunable fluid’ provides
a wide variety of different dynamics (Newtonian, polymeric, time-dependent) which may

59
be useful as either a biomolecular medium or as a versatile industrial fluid. The dynamic
behavior of a rising bubbles demonstrates the possible variety of hydrodynamic effects
and their sensitivity to physical microstructure in wormlike micellar fluids.

60
Chapter 4
A New Constitutive Model
for Wormlike Micellar Fluids
4.1 Basics of rheology
As was seen in Chapters 1 and 2, non-Newtonian fluids have very different flow
properties from viscous Newtonian fluids. Chapter 1 gave examples of the elastic nature
of polymer liquids as well as mathematical descriptions of both types of fluids, and Chap-
ter 2 introduced wormlike micellar fluids and examined ways they differ from Newtonian
and other non-Newtonian fluids through experimental rheology. Chapter 3 examined an
interesting instability, and differentiated wormlike micellar fluids from classical polymer
solutions. With this as background, we have enough information about these fluids to
begin to consider them from a mathematical perspective. In this chapter we consider
what physical aspects of wormlike micellar fluids can account for their unusual flow prop-
erties, and how we can begin to model them. To this end, we briefly review concepts
from previous chapters which will be relevant to modelling.
For Newtonian fluids, we know that σ = 2ηD, in which η is a constant known as
the viscosity and D is the symmetric part of the velocity gradient: D = 12(∇u +∇uT ).

61
If the flow is pure shear, then in Cartesian coordinates
D =12
0 γ 0
γ 0 0
0 0 0
,
where γ is known as the shear rate. In the case of steady shear flow, γ is a constant, and
we can view the shear component of stress σxy as a function of γ. Note that σxy = σyx
are the only non-zero components of stress in pure shear flow; in extensional flows the
diagonal components of D would not be zero. For any Newtonian fluid, the steady
state value of σxy grows linearly with γ, and the slope is simply the viscosity η. For
viscoelastic fluids, typically there is a range of small values of γ for which σxy depends
linearly on γ [3]. This is because in equilibrium the macromolecule (or micelle) will be
“coiled” up, and for small enough shear gradients, it will not unravel, but move through
the fluid like a colloidal sphere. The internal degrees of freedom not being activated, the
macromolecules behave like large point particles, and the fluid seems Newtonian. In this
“Newtonian” region the linear proportionality constant is called the zero shear viscosity
and denoted by η0.
At higher shear rates, deviations are often experimentally observed [3]: if σxy
grows slower than η0γ, we say the fluid is shear thinning, and if σxy grows faster than
η0γ, it is shear thickening. Since for non-Newtonian fluids, σxy is often no longer linearly
proportional to γ, it is inappropriate to speak of a single viscosity, and the effective

62
viscosity is defined as ηe = σ/γ in analogy to Newtonian fluids [47]. Thus at low shear
rates, ηe = η0, and at higher γ we would have ηe < η0 for shear thinning fluids.
In general, any constitutive equation used to model a fluid should capture, at least
qualitatively, those material parameters which play a role in the effects under study;
if elasticity is important for example, then the constitutive equation should predict a
relaxation time. Unlike Newtonian fluids, there is no single constitutive equation which
succesfully describes all viscoelastic fluids, even for a given type of flow [77]. Instead,
various models are used depending on particular circumstances of the fluid, the flow and
the phenomena under investigation. A first approach to modelling viscoelasticity came
from J.C. Maxwell in 1867 [48]:
σ + λ∂tσ = 2η0D. (4.1)
The Maxwell equation introduces a new material parameter in λ, which is a time con-
stant, while η0 is the zero shear viscosity (a constant). The λ ∂tσ term is the elastic
contribution, the other terms give the Newtonian constitutive relation if λ = 0. The
meaning of λ becomes more clear when we solve this first order linear ordinary differen-
tial equation using the integrating factor et/λ:
σ =2η0λ
∫ t
0e(t′−t)/λD(t′)dt′. (4.2)
In this form it is easy to see that elasticity has been accounted for in the form of memory.
That is to say, stress is determined not only by what the current velocity gradient is,

63
but the velocity gradient at all previous times. The decaying exponential term can be
thought of as a weight, so that D(t′) contributes less the farther back in time one goes,
and λ is a measure of the amount “forgotten” about D as we look back in time. This
model is sometimes referred to as having “fading memory” for this reason [47].
The Maxwell model (equation 4.1) gives a plausible description of elasticity, but
some underlying physics is not entirely correct. The problem is that when we consider
an equation for fluid motion, we must remember that we are describing an object (a fluid
“packet”) which is moving. If we use spatial variables (x1, x2, x3) and a time variable
t then, since the position of a fluid particle is moving, xi = xi(t). It is well known [78]
that it is not physically correct to use a partial time derivative in the Maxwell equation,
but rather the total time derivative of σ should be used:
d
dtσ(x(t), t) =
∑
i
∂σ
∂xi
∂xi∂t
+∂σ
∂t
=∑
i
ui∂σ
∂xi+
∂σ
∂t
= u · ∇σ +∂σ
∂t(u is the velocity)
The total time derivative (sometimes denoted with capital letters DDt = u · ∇ + ∂
∂t) is
called the convective or material derivative. The alteration to 4.1 using this derivative
is commonly called the convected Maxwell model:
σ + λ (u · ∇σ +∂σ
∂t) = 2η0 D.

64
The convected Maxwell model differs from equation 4.1 in the nonlinear term
u ·∇σ. For the remainder of this thesis we assume our flow to be homogenous, for which
u · ∇σ = 0, so that the two forms of the Maxwell model are identical.
Although the Maxwell model includes a term to account for relaxation, in steady
shear flow it predicts σxy = η0γ, so that ηe is constant equal to the zero shear viscosity,
and no thinning or thickening is possible. One can easily write models which allow for
the nonlinear viscoelasticity, for instance a power law model such as σ = η0γk, gives
ηe = σγ = η0γk−1, which is no longer constant with γ as long as k 6= 1. The White-
Metzner model [47] incorporates nonlinear viscoelasticity directly in the Maxwell model:
σ +η(γ)G
∂tσ = 2 η(γ) γ.
In this model η(γ) can be any function such as a power law, or it can be determined by
fitting to data. Note that here we restrict ourselves to pure shear flow.
In our endeavor to model wormlike micellar fluids, we choose to start with the
Maxwell model for its elegant way of including elasticity through memory and describing
fading memory with a decaying exponential kernel. Our alterations to the Maxwell model
will be motivated by the idea of memory in wormlike micellar fluids and the particular
microscale physics these fluids posess.
4.2 Review of existing models for wormlike micellar fluids.
Wormlike micellar fluids began receiving increased attention in the early 1980’s
after several experimental results were published showing that these fluids have unusual

65
and often unprecedented responses to flow [49, 6]. The experimental and theoretical
challenge was to understand what had been been observed as well as predict how the
fluids would react to flow. The general problem facing the theoretician is to understand
what are the prominent properties a system posesses, and which are relevant or respon-
sible for specific observations. This is only the first step though, since there can be
many such properties and they in general depend on each other. So there is the prob-
lem of understanding the physical processes, followed by or simultaneously developed
with a mathematical description, of which there may be many. This is the problem of
modelling. In this section we provide a brief overview of previous models put forth to
describe wormlike micellar fluids.
In developing a theory, a standard mindset is to compare the given problem to
an analogous problem. In the case of wormlike micelles, which may be long and flexible,
there is an obvious comparison with polymers. A polymer solution is in some sense the
standard when describing macromolecular or non-Newtonian fluids, though there are
many non-Newtonian fluids which consist of particles very different from polymers (e.g.
dense colloidal suspensions, clays, corn starch). Many constitutive equations have been
written to describe the fluid properties of polymer solutions [47, 77, 3], and this offers
a starting point in modelling wormlike micellar fluids. The next step is to identify key
differences from the analogous system and then adapt the equations to include these new
effects. When we compare wormlike micelles to polymers, one salient difference is the
reversible scission reactions of the micelles. So the idea would be to model this aspect
and incorporate it into a pre-existing constitutive equation. This is a common technique
in modelling wormlike micellar fluids [50, 51, 52, 53].

66
The ability of wormlike micelles to break and also combine affects the fluid in
many ways, and one example is the distribution of micelle lengths. While classical
polymer solutions may be polydisperse (i.e. there are polymers of varying lengths in a
given fluid), once the polymers are formed, the size distribution is fixed. In the case
of wormlike micelles, the size distribution is in thermal equilibrium (more precisely,
chemical equilibrium), since breaking and recombining events persist when the fluid
is not moving [9, 2]; recall chapter 3 in which a mean field model prediction for the
average length of micelles and the length distribution in equilibrium. It is very sensible
to imagine that flow will increase the average number of collisions between micelles and
it becomes obvious to ask how this will affect the length distribution. This question has
received a good deal of attention in the fluids community because of some astounding
rheological effects, such as shear banded flow and shear thickening [17, 11, 54, 39] in which
the micelles aggregate to form larger structures, sometimes referred to as “bundling”,
forming a shear induced phase (SIP) or shear induced structure (SIS). Modelling this
“gelation” has provided some insight into the mechanism at work, but the reasons why
it occurs is still not understood.
While we review papers in this section to give the reader an idea of what has been
done in modelling micellar fluids or related subjects, there are many other articles which
have had an impact on the theory of these fluids. We list some of these models here and
give a brief description of their approach:

67
1. M. E. Cates (1987) [50] and M. E. Cates (1990) [51] - an adaptation of deGennes’
reptation model is presented (known as the “reaction-reptation model), which in-
cludes the effects of reversible scission reactions.
2. N. A. Spenley, M. E. Cates, & T. C. B. McLeish (1993) [81] - a simulation of
the reaction-reptation model [50] to predict the nonlinear rheology of wormlike
micelles, including a stress plateau associated with shear banding.
3. V. Schmitt, C. M. Marques, & F. Lequeux (1995) [55] - a constitutive equation is
presented which includes effects of flow on concentration, creating spatial concen-
tration gradients to describe shear induced phase separations.
4. B. H. A. A. van den Brule & P. J. Hoogerbrugge (1995) [56] - a simulation of rheo-
logical properties, including Brownian motion of a crosslinked network of molecules
which can detach and reattach themselves.
5. I. Jeon (1997) [57] - solutions to coagulation-fragmentation equations are approxi-
mated by Markov chains, and their qualitative behavior is studied.
6. A. Vaccaro & G. Marrucci (2000) [58] - a model describing a network (or “web”) of
polymers which may detach themselves and reattach elsewhere, much like micelles.
7. J. L. Goveas & P. D. Olmsted (2001) [59] - micelles are treated as chemical reactants
and a reaction-diffusion equation is written to model to study phase separation due
to flow.
In addition, we now review four other important papers in more detail.

68
8. R. Bruinsma, W. Gelbart, & A. Ben-Shaul (1992) [60]. This model describes
the effect of flow on the micellar length distribution with a “coagulation-fragmentation”
model to count the number of micelles of a given length. Bruinsma et al. use theory first
developed by Paine [61] in 1912 and von Smoluchowski [62] in 1916, for the coagulation
seen in colloidal suspensions under shear flow. Colloidal spheres were experimentally
seen to aggregate in shear flow [61] due to hydrodynamic interactions. This is called
orthokinetic coagulation, in contrast to perikinetic coagulation which occurs under the
influence of Brownian motion. The authors argue that we may expect orthokinetic
coagulation to begin to dominate over perikinetic at a critical shear rate γc = Dt/R2,
where Dt is the translational diffusion coefficient, and R is the radius of the sphere. The
derivation of this value for γc comes from the comparison of particle fluxes due to flow
and Brownian motion. Assuming the reaction cross section for coagulation is just the
particle radius, the flux due to shear is Js ∝ v A, where v is the impact velocity and A is
the reaction cross section. Assuming A = R2 and v = γ R, gives Js = ϕ(R)γR3 where
ϕ(R) is the concentration of particles with radius R. A similar argument for the flux
due to Brownian motion gives the relation Jb ∼ DtϕR R. The ratio of these quantities
gives the Peclet number for the flow: Pe = Js/Jb = γR2/Dt. When Pe > 1 orthokinetic
coagulation dominates, and γc is defined for Pe = 1. When applied to wormlike micelles,
R is replaced with the length L, so that the shear rate at which “gelation” or thickening
occurs grows like γc ∝ 1/L2. This is a nice result because it follows from very simple
and seemingly reasonable ideas, and the prediction makes sense: for longer micelles,
gelling will occur at more modest rates, that is, it is easier to induce coagulation since
the micelles are so long to begin with.

69
The problem with this idea is that shear flow will stretch the coiled micelles,
weaking the analogy with a colloidal sphere, and it will align the micelles as well. The
authors postulate that stretching of micelles will begin when γ = Dr, where Dr is the
rotational diffusion constant. An approximate relation between Dt and Dr is given in
[63] as Dr ∼ Dt/L2. So that γ/Dr = 1 = γL2/Dt occurs exactly at γc, i.e. alignment
begins at Pe = 1 when flow induced coagulation begin. It is also assumed by the authors
that alignment will reduce the number of collisions between micelles, so the condition
Pe = 1 for orthokinetic coagulation to dominate is questionable.
The basic equation used in the model of Bruinsma et al. is an integral-differential
equation for the function N(L, Ω) representing the number density of micelles with length
L and angular orientation Ω. When the micelles combine or break slowly enough (the
“slow reaction” regime) to re-equilibrate their orientation before another scission (or
recombination) reaction, this function decomposes as N(L, Ω) = N(L) fL(Ω), where
N(L) satisfies:
∂
∂tN(L) = −N(L)
∫ L
0dL′kb(L|L′) + 2
∫ ∞L
dL′N(L′)kb(L′|L) (4.3)
− N(L)∫ ∞0
dL′N(L′)kc(L|L′) +∫ L/2
0dL′N(L′)N(L− L′)kc(L
′|L− L′).
Equation 4.3 is an example of a coagulation-fragmentation model [64, 65, 57]. Here kb
and kc are reaction rates: kc(L|L′) is the rate that micelles of length L and L′ combine
into a micelle of length L + L′, and kb(L′|L) is the rate at which a micelle of length L′
breaks into two micelles of length L and L− L′. Thus the four terms on the right hand
side of equation 4.3 represent the ways of gaining and losing micelles of length L due to

70
coagulation (combining) and fragmentation (breaking). When collisions occur quickly,
the authors argue that the many collisions randomize the orientation of micelles before
coagulation or fragmentation, so that equation 4.3 again holds in this ”fast reaction”
regime.
In thermal equilibrium, the reversible scission reactions of micelles are expected
to obey the law of detailed balance, which says that for each breaking event there is a
recombining event, which gives a steady state length distribution N(L). Detailed balance
requires that
N(L′) N(L− L′) kc(L′|L− L′) = N(L) kb(L|L′) + N(L) kb(L|L− L′). (4.4)
The breaking rates on the right hand side of equation 4.4 must be equal by symmetry
of the breaking event: a micelle of length L which breaks to give a micelle of length L′
necessarily makes a micelle of length L−L′ as well, so that kb(L|L′) = kb(L|L−L′). This
does not mean however, that these breaking reactions are identical; they are considered
distinct events because they involve different angular orientations. The authors solve
equation 4.4 in the case where L = 2L′ to obtain solutions of the form:
N(L) ∝ 1
g(L)1/ln 2e−L/L, (4.5)
in which L represents the average length. An exact solution to equation 4.3 is found
in equilibrium which has the form of the detailed balance solution (equation 4.5) with
g(L) = constant, with an average length L depending on concentration, temperature,

71
and scission energy in precisely the same way predicted by the mean field equilibrium
length model presented in chapter 2, i.e. L0 ∝ ϕ1/2eE/2kT . Hence equilibrium requires
that g(L) be constant in equation 4.5.
Although equations 4.4 and 4.5 hold only in equilibrium, and may not hold for
γ 6= 0, the authors assume that length distributions N(L) with flow are still of the form
of equation 4.5. For this model, the effect of flow is restricted to the ratio of rates g(L),
i.e. g(L) is allowed to be non-constant and L is solved for self-consistently. This is a
perfect example of the techniques of modelling we described at the beginning of this
section. So far, certain assumptions have been made, but they have all been reasonable
and physically motivated, and only solutions in equilibrium have been discussed, for
which there are existing models. The coagulation-fragmentation model given in equation
4.3 seeks to extend those results, and it has been shown to at least recover the accepted
equilibrium results. Now we come to the heart of the matter, namely, how does flow affect
the equilibrium distribution? By assuming that non-equilibrium solutions to equation
4.3 have the same general form as the equilibrium solutions, except that g(L) is flow
dependent, the authors have chosen to model the influence of flow in a specific way: the
effects of flow enter the equations only through the reaction rates kb and kc.
The remaining task is then to describe the ratio g(L). Starting from the analogy
to colloidal spheres, different forms for g(L) are assumed, depending on flow rate and
collision rate. Parameter space is partitioned into four cases: Peclet number Pe À 1,
or Pe ¿ 1, and the “slow reaction” versus the “fast reaction” regime. Bruinsma et al.
obtain the distribution N(L) in analytic form for these cases, which are used to obtain

72
expressions describing L. Using the relation
dL
dt= α(L)
∫ ∞0
∂N(L)∂t
dL (4.6)
in which α(L) is a different function of L for the cases Pe À 1 and Pe ¿ 1, together
with the coagulation-fragmentation equation (4.3), an ordinary differential equation is
obtained for L in each of the four cases. Rather than attempting to solve for analytic
forms for L, its steady state value can be obtained by minimizing the function v(L)
defined by
− d v(L)dL
≡ dL
dt.
It is natural to think of v(L) as a sort of potential for L. The results for the four cases
are [60]:
i) Pe ¿ 1, slow reaction regime:−L is shifted to only a slightly larger value than
L0, specifically, L ' L0(1 + Pe). We must remember that Pe ∼ γ and assuming
R2/Dt is O(1), we may conclude that γ is small in this formula.
ii) Pe À 1, slow reaction regime: L ∼ L0 Pe1/3, so that while Pe is much greater
than in case i), L grows even more slowly in this regime. Physically, we can say
that the stronger flow increases alignment and decreases reaction cross sections.
iii) Pe ¿ 1, fast reaction regime: In this case the “potential” v(L) has a local minimum
near L0 followed by a local maximum, i.e. there is a potential “well” and “barrier”.
This means that if the flow is strong enough (still with Pe ¿ 1) or the reaction

73
rates for coagulation are large enough, it is more favorable for micelles to continue
growing longer.
iv) Pe À 1, fast reaction regime: Here the potential v(L) has no minimum, but is
monotonically decreasing, i.e. this is the gelation regime.
The authors make a connection between the slow and fast reaction settings; slow
reactions are expected in dilute solutions, while semi-dilute solutions experience fast
reactions. Hence we can understand their findings as a continuous progression as we
increase concentration and flow rate (Peclet number Pe). In each of these cases it is
proposed that there is an “energy barrier” to be overcome for gelation to occur (L →∞),
and that it is too large for dilute solutions to overcome. As we increase flow rate in the
dilute regime, L grows, but there is a well defined value depending on flow rate. As
concentration is increased and collisions are much more frequent, the collisions decrease
the effect of alignment due to flow, and coagulation events are more frequent. The
stronger the flow, the more easily the barrier can be overcome, until a “runaway” gelation
process occurs [60].
9. M. E. Cates & M. S. Turner (1990) [66]. The next model we consider focusses
again on the length distribution of micelles, but here extensional flow is considered rather
than shear flow. In extensional flows, the velocity gradient at a point is parallel to the
velocity itself at the point. In this sense, the flow “elongates” or extends a fluid packet
in the direction it is travelling. In Cartesian coordinates, the rate of strain tensor D

74
would be diagonal, for example in elongational extension we have [47]:
D =
− ε2 0 0
0 − ε2 0
0 0 ε
(4.7)
In this matrix form for D, the extensional rate ε has units of inverse seconds and may be
constant or time dependent, and for inhomogenous flows may also be spatially dependent.
It is completely analogous to the shear rate γ in shear flows. Notice that extensional
flows have no shear component to them, and are examples of shear-free flows.
Because micelles are thought to be highly aligned in such flows, the model given
by Cates and Turner [66] assumes that micellar reactions (combining and breaking)
involve only collinear micelles. These authors also seek a mechanism for gelation to
occur, and the rough idea is that alignment will increase collisions and reactions because
all collisions here are for collinear micelles, making micelles longer. The longer micelles
will then have less angular mobility and will therefore be more easily aligned, which in
turn enhances growth again, and a gelation is predicted at a critical flow rate ε.
In this paper, the concentration of rod like micelles with length L and angular
orientation u is denoted by ψ(L,u), which is implicitly time dependent. The goal is to
find steady state solutions, i.e. solutions dψdt (L,u) = 0. Turner and Cates decompose dψ
dt
into the sum of two contributions: dψdt = F1 + F2, with F1(ψ) describing rotations and
angular diffusion, and F2(ψ) accounting for micellar scission and recombination. While
the functional form for F2(ψ) is a coagulation-fragmentation equation very similar to

75
that found in Bruinsma et al. [60], F1 is given as
F1 = R · [D(L)Rψ(L,u) − f(u) ψ(L,u)], (4.8)
in whichR = u×∂/∂u, f(u) = u×∇v·u (v is the velocity field), and D(L) is the angular
diffusion constant. It is assumed that angular diffusion is of the form D(L) = D0 L−ζ ,
with ζ a free parameter. The reasons for this choice are discussed below.
Recall that in Bruinsma et al. [60], a solution was first obtained with no flow (and
in equilibrium), and the general solution to the length distribution function was related
to the equilibrium solution, making the problem more tractable. Here too, Turner and
Cates use equilibrium as a stepping stone in finding solutions to dψdt (L,u) = 0, in a
slightly different manner. The authors define a “fast-reaction” regime in which collisions
occur much faster than angular diffusion, and argue that micelles rapidly reach a chemical
(thermal) equilibrium for each angle u. This then requires that for each given u, ψ(L,u)
must be a solution F2 = 0. A solution to F2 = 0 is obtained as:
ψ(L,u) = e−E−L/λ(u), (4.9)
where λ(u) is to be solved for using F1(ψ) and E is a constant. Note the similarity
of this model with the Bruinsma approach. Equation 4.9 is a solution when there is a
non-zero flow, and in the case of equilibrium λ(u) = λ0 the average equilibrium length.
Thus the same equilibrium solution is obtained, for each angular orientation u, as in the
equilibrium solution in Bruinsma’s model. This is also means that Cates and Turner

76
are saying that the effect of flow on ψ(L,u) is to alter the average micellar length. In
solving for λ(u) the authors restrict their consideration to flows v such that there exists
a “potential” function V (u) satisfying RV (u) = −f(u). This is by no means a crippling
assumption; the elongational flow described earlier by 4.7 is an example of such a flow.
In this case the authors find that, using F1,
λ(u) = (c/ζ)1/ζ (V (u)/D0)−1/ζ . (4.10)
Here both D0 and ζ come from the angular diffusion constant D(L). In the particular
case of elongational flow with extensional rate ε, Cates and Turner use a conservation
of mass equation to find that equation 4.10 predicts that λ(u) remains bounded as long
as ζ < 2. For ζ ≥ 2, λ becomes unbounded at a critical flow rate εc ∝ D0λ−ζ0 , where
λ0 is the equilibrium average length of micelle. It is this unbounded solution that the
authors identify as a “gelation”. A natural question to ask is whether values of ζ near 2
are reasonable physically. For the case of a dilute solution of stiff rods, D(L) ∼ D0L−3,
while for entangled rods (which do not break) ζ ∼ 5 [63]. It is reasonable then that
η may be larger or smaller than 2, yet a theory for the angular diffusion constant for
rod like micelles has not been introduced, and it may be quite different from the form
assumed in this article by Cates and Turner [66].
10. F. Bautista, J. F. A. Soltero, J. H. Perez-Lopez, J. E. Puig, & O. Manero
(2000) [67] and O. Manero, F. Bautista, J. F. A. Soltero, & J. E. Puig (2002) [53]. Unlike
the papers we reviewed above [60, 66], these authors are interested not in describing the
length distribution and average length in flow, but a constitutive relation for wormlike

77
micellar fluids. In [53], Manero et al. formulate a constitutive equation for the stress in
a wormlike micellar fluid which is based on a variation of the upper convected Maxwell
model which we saw in Chapter 1. Manero et al. [53] couple an equation describing
micellar kinetics to the Oldroyd-B constitutive equation. The Oldroyd-B constitutive
law is essentially the upper convected Maxwell model [47], with a term linear in rate of
strain, meant to include viscous effects of the water surrounding the micelles in a micellar
fluid.
The equations comprising the model for the stress σ in a wormlike micellar fluid
with rate of deformation D in [53] are:
σ +η
G0
Oσ = 2η(D + λJ
OD), (4.11)
dη−1
dt=
1λ
(1η0− 1
η) + k(
1η∞
− 1η) σ : D (4.12)
In equation 4.11, η, G0, and λJ are constants representing respectively the fluid
viscosity, elasticity, and a retardation time, respectively. The symbol O positioned over
σ and D indicates the upper convected derivative of these terms (see chapter 1). The
term last term in equation 4.12 can be defined in terms of components:
σ : D =∑
i,j
σijDji.
Equation 4.12 is the authors’ contribution to the constitutive law which is based
on the model by Vaccaro & Marrucci [58] (#6 in the list above). In this equation, λ
is the relaxation time, η0 is the zero shear viscosity, and η∞ is the limiting value of

78
viscosity at high shear rates, all of which are constants. The constant k is meant to
describe “structure breakdown”, though it is not a scission rate, in fact it has units of
1/stress. Under certain assumptions, equations 4.11 and 4.12 lead to a single equation
in which the kinetic constant has been chosen as k = G−10 :
1 + (λ/G0)(λ0/λ∞)(σ : D)1 + (λ/G0)(σ : D)
σ + λ0Oσ = 2G0λ0(D + λJ
OD). (4.13)
Manero et al. [53] compare the predictions of their model (equation 4.13) to
experimental data in steady state shear flow and small amplitude oscillatory shear. Of
primary interest is a comparison between the viscosity η in steady shear and the complex
viscosity η∗ in oscillatory shear [47]. The Cox-Merz rule [68] states that these two
viscosities should coincide as functions of shear rate. And while polydisperse systems of
polymers adhere to the Cox-Merz rule [68], it has been observed that in some wormlike
micellar fluids η and η∗ do not agree [69, 70].
In steady shear flow, an analytic expression for the shear stress (σ12) is obtained
from equation 4.13 in steady state:
σ12 = G0(λ0λγ2 − 1) +
√(λ0λγ2 − 1)2 + (λ/λ∞)λ2
0γ2
2(λ/λ∞)λ0γ. (4.14)
This solution predicts linear increase of σ with shear rate for small shear rates γ, followed
by σ = G0√
λ∞/λ =constant beginning at γ = λ−10 . The constant stress ends at
γ = λ−1∞ , after which σ again increases linearly, with a different proportionality constant
than at low γ. The significance of this is that wormlike micellar fluids often display a

79
“stress plateau” in which, for a finite range of γ, the stress is at least nearly independent
of shear rate. This phenomenon has been associated with a shear banding instability, and
is predicted by other models as well such as the convected Jeffreys and Johnson-Segalman
models [47].
In small amplitude oscillatory shear flow, the model of Manero et al. 4.13 reduces
to the convected Jeffreys model in the linear viscoelastic limit. Under the assumption
that 1 + λ0λJ
≈ λ0λJ
, this model gives the same expression for the storage modulus G′ as
the Maxwell model: G′ = G0λ20ω2
1+λ20ω2 . The loss modulus, however, is given by
G′′ = G0λ0ω
1 + λ20ω2 + G0λJω, (4.15)
whereas the Maxwell model predicts equation 4.15 without the G0λJω term. This extra
term gives G′′ a second critical point that the Maxwell model does not have, which is
a local minimum and has been seen experimentally in many wormlike micellar fluids.
This minimum has gained importance because it has been linked with sizes of wormlike
micelles and under certain conditions can give a measure of their entanglement [44].
In addition to a model with predictions, the authors report experimental data they
obtained from testing two different wormlike micellar systems: erucyl bis(hydroxyethyl)-
methylammonium chloride (EHAC) with potassium chloride (KCl) and cetyltrimethyl-
ammonium tosilate (CTAT). The results for these systems and model predictions are
quite impressive. In oscillatory shear experiments, both systems showed storage and
loss moduli which agreed very well with model predictions [53]. Moreover, the model
predicts that the viscosity η in steady shear will not coincide with the complex viscosity

80
η∗ through an oscillatory shear experiment, and the model predictions for both η and
η∗ agree beautifully with data for both the EHAC and CTAT systems. These results
are quantitatively correct for the experiments shown by the authors, and shows that for
these systems the Cox-Merz rule is not obeyed, though no clear explanation is given.
The authors of [67], many of whom authored [53], use essentially the same model
as 4.11 and 4.12, and focus on the prediction of the stress plateau in steady shear
experiments. In this paper, the kinetic constant k is assumed to be linear in γ instead
of setting k = G−10 as in [53]. Following the ideas of Acierno et al. [71], Bautista et al.
[67] choose to let k = k(γ) = k0(1 + µγ), where k0 and µ are constants. In this model,
the magnitude of “structure breakdown” is controlled by µ γ. Since the experimental
rheology that the authors attempt to reproduce with their model is performed across a
range of shear rates, the effective control parameter for structure breakdown is simply
µ.
The authors give their model predictions for steady shear flow, and obtain what
is classically observed for many wormlike micellar fluids; stress increases linearly with
γ until some critical rate γc after which stress “plateaus” and becomes constant until a
second finite value of γ, after which it increases linearly with γ again. Additionally, the
authors simulate their model in transient experiments, examining the time dependence
of stress at fixed shear rate. Experimentally, this is a very important consideration to
ensure that one knows how long to run an experiment before steady state values are
obtained. In wormlike micellar fluids which exhibit stress plateaus (those which shear
band), different and extraordinary transients have been observed [30, 15, 17] at rates for
which stress is constant. Most spectacular are the very long transient stresses [15] which

81
can last for thousands of seconds and more (also see Fig. in chapter 2) without reaching
steady state.
The model of Bautista et al. [67] qualitatively matches certain experimental
transient rheology such as Maxwell growth [6] and stress overshoots [30]. While the
authors of [67] argue that they do achieve such transients with their model, the magnitude
of deviation from a steady state stress value is unclear; the authors offer no numerical
data to support their claim that they have long timescale transients. From the graphs
presented in the article, the stress does in fact seem to achieve a steady state value in a
much shorter time than what is seen experimentally. The failure to address this point
leaves their last result open to question.
We have listed several papers which model wormlike micellar fluids (or closely
related topics) and have given a detailed review to some of these. The equations and
methods these authors used should provide an idea of what the issues are in the theoreti-
cal side of wormlike micellar fluids. Persistent themes include a non-equilibrium micellar
length distribution, restriction to a single type of flow (usually shear flow), and the use
of existing models or theories for guidance. We also note the many assumptions used in
modelling, which usually but not always, have at least some physical motivation.
The models reviewed in section 4.2 were written specifically to capture certain as-
pects of wormlike micellar fluids in motion (e.g. length distribution and stress plateaus).
Each model included the ability of micelles to break and reform, though the way this
was described varied considerably. This physical process is highlighted since it is per-
haps the most distinguishing property of wormlike micelles from classical polymers, and
we also base our model on this single property. While the molecular level physics of a

82
solution of wormlike micelles may indeed be complex, in the next section we narrow our
focus on the ability of micelles to exchange mass in order to determine what aspects of
these fluids are caused by this particular property. The nodel we present endeavors to
avoid the phenomenological approach (e.g Manero et al. [53]) as well as the detailed
considerations of the more physical models from this section (e.g. Bruinsma et al. [60]
and Cates [66]), while remaining faithful to the physics.
4.3 Wormlike micelles as chemical reactants
Since wormlike micelles exchange matter with one another, even with no flow [9],
their microscale physics is more complex than conventional polymers; the kinetic inter-
actions influence and are influenced by the internal stresses developed from the motion
of the micelles themselves. This dynamic coupling manifests itself macroscopically, for
example in the non-linear rheology of the fluid, or the oscillatory instability of rising
bubbles and falling spheres [19, 20, 76].
In Figs. 4.1-4.3, our experimental results from shear rheology tests are shown
for equimolar solutions of CPCl/NaSal (see also Chapter 2). The concentrations repre-
sented were chosen to show the variety of rheology seen in wormlike micellar fluids, and
correspond to the three concentration regimes relevant to the oscillatory instability in
rising bubbles, as described in Chapter 2.
Each data point in Figures 4.1-4.3 represents the steady state value of the effective
viscosity for that shear rate. The rheology of a dilute wormlike micellar solution is shown
in Figure 4.1 in which the effective viscosity shows a plateau at low γ, which gives the

83
0 . 0 0 1
0 . 0 1
0 . 1
1 0 1 0 0 1 0 0 0
η eff
(P
oise
)
Shear rate (seconds- 1)
1 mM CPCl/NaSal T=30 oC
Fig. 4.1. Effective viscosity as a function of shear rate at very low concentrations(dilute regime). Although the viscosity decreases at higher γ, it remains well above theη0 plateau, indicative of a thickened state.
value of η0. Note that the viscosity is constant up to γ ' 30s−1, where a sudden
thickening begins which is sustained for the range of the test.
This rheology is the signature of SIS or bundle formation [17], also described in
Chapter 2. Figure 4.2 again shows an η0 plateau for a 8 mM micellar solution, though the
value is more than two orders of magnitude greater than that of the 1mM solution shown
in Fig. 4.1. The plateau is quickly followed by shear thinning during which ηe decreases
by an order of magnitude. This is followed by an interesting thickening, during which the
viscosity roughly doubles, and then decreases again. This is typical of our experimental
rheology for semi-dilute solutions of CPCl/NaSal, and it is interesting that there is still
a type of thickening transition after the departure from dilute solutions. The highest
concentration shown is 30mM CPCl/NaSal, in Fig. 4.3. For this higher concentration,
there is a wide range of γ for η0. This implies a much shorter relaxation time λ than at

84
0 . 1
1
1 0
0 . 0 1 0 . 1 1 1 0 1 0 0 1 0 0 0
8 mM CPCl/NaSal at T = 30 Celsius
η
Shear Rate (s- 1)
ef
f(P
ois
e)
Fig. 4.2. Effective viscosity as a function of shear rate for a semi-dilute solution.The increase in viscosity near is reminiscent of the thickening at lower concentrations(Fig.4.1).
concentrations in the semi-dilute regime. The fluid then undergoes thinning, decreasing
modestly over the tested range of γ.
While extensive experimental rheology data was presented in Chapter 2, we have
recalled these plots as a reminder of what sort of rheology we are trying to reproduce
with a constitutive equation. What the data in Figures 4.1-4.3 show is that wormlike
micellar fluids can both shear thin and thicken, even a single concentration can do both
at different shear rates. Our goal is to write a model for wormlike micelles which produces
this rheology while capturing the molecular level physics of the micelles. When we first
consider this modelling problem, it may seem to involve many different aspects of the
fluid: the motion and interactions of many (∼ 1023) non-rigid particles (the micelles)
which are not point masses as in the Newtonian case, their ability to change size and
possibly their topology (recall the phase transitions seen in Chapter 3), and the effect

85
1
1 0
0 . 1 1 1 0 1 0 0
η eff
(P
oise
)
Shear rate (seconds- 1)
30 mM CPCl/NaSal T=30 oC
Fig. 4.3. Effective viscosity as a function of shear rate. This higher concentration(30mM) is no longer in the semi-dilute regime. No thickening occurs in the range of γtested.
of the Newtonian solvent on the micelles. The first task is to recognize which of these
properties are common to other fluids and to use them as an analogy. We will use models
designed for those systems as a starting point for our problem, so that the problem is
reduced to studying only those properties which distinguish wormlike micellar fluids
from the analogue, and creatively adjusting the analogous model to account for the new
features.
We have a viscoelastic fluid consisting of self-assembling wormlike micelles, as
opposed to covalently bonded ploymers. However, both systems are viscoelastic, and
we can use models which are meant to describe this commonality as our starting point.
Namely, we may start modelling wormlike micellar fluids with the Maxwell model 4.2.
The novel aspect we still need to include is the reversible scission reactions which occur
among micelles. A principle we adopt in modelling this is feature is simplicity. We

86
will alter the Maxwell equation to accomodate breaking and reforming, but the fewer
new terms we introduce, the more understanding we gain about these terms. More
than that, if predictions from the model match even approximately what is physically
observed, then we can understand how the physical process enters into the equation.
4.3.1 The 3-species model
We also model the mass exchange property in flow using these ideas. While there
is in reality a large spectrum of micelle lengths, we consider a simplified situation in
which there are only two sizes of micelles, described as short and long, together with a
third species representing SIS or “bundles”, which will represent the structures believed
responsible for the shear thickening observed in rheological studies:
short micelles in concentration C(t)
long micelles in concentration A(t)
bundles of micelles in concentration B(t)
The ability of micelles to exchange mass is accounted for by having long and short
micelles; instead of describing the process of mass exchange, we allow the concentrations
of long or short micelles to increase with a simultaneous decrease in the other concen-
tration. Clearly the minimum number of species needed in such a description is two,
and the third concentration of bundles is included specifically to model the formation of
bundles. To make this “3-species” model more specific, we let long micelles be precisely
twice as long as short micelles, in this way we are allowing long micelles to break into 2

87
shorter micelles, as well as the reverse reaction. It is not necessary to make this choice of
size difference for A and C; we could let long micelles break into three shorter micelles,
increasing the value of C. We consider these species analogously to chemical reactants,
and the reactions are listed with their respective rates in Table 1 and depicted in Fig.
4.4:
A
B
Ck1 + f1(γ)
f2(γ)g(γ) g(γ)
k0 + f0(γ)
~
~
~
~ ~
~ ~
Fig. 4.4. Schematic of micelle reactions in the 3-species model (see Table 4.1).
Both k0 and k1 are kinetic constants which describe the rate of exchange of mass
with no flow. The parameters f0(γ), f1(γ), f2(γ), and g(γ) are functions of γ, and
satisfy fi(0) ≡ 0 ≡ g(0) for i = 0, 1, 2. That is to say that these parameters describe the
flow induced mass exchange.
These reactions lead us to a system of three coupled ordinary differential equations
(ODE’s) for the concentrations of species A, B, and C:

88
Reaction Rate
A → 2C (k0 + f0(γ))A
A + A + A → B f2(γ) A3
C + C → A (k1 + f1(γ))C2
B → αA + βC g(γ) B
Table 4.1. Chemical-like reactions of the three species of wormlike micelles. On the leftof the arrows are the “reactants” which produce the species to the right of the arrow.
dA
dt= −(k0 + f0)A− 3f2A3 + (k1 + f1)C2 + α g B (4.16)
dB
dt= βf2A3 − gB (4.17)
dC
dt= 2(k0 + f0)A− 2(k1 + f1)C2 + β g B (4.18)
There are two parameters in equations 4.16 that we have not yet defined: α and β,
which appear as prefactors to terms representing populations of micelles arising from the
destruction of bundles. A bundle is created from three a-micelles, but it can break into
three a-micelles or six c-micelles, since a-micelles consist of two c-micelles. In fact, four
combinations are possible for a single bundle breaking up, which occur with probabilities

89
r1, r2, r3, and r4. The constraint on α and β is then:
α = 3r1 + 2r2 + r3
β = 2r2 + 4r3 + 6r4.
We can choose any three of the probabilities arbitrarily, which means one of α or β is
arbitrary and the other determined by α + β/2 = 3.
In this 3-species model, the concentration functions have no spatial dependence,
only time dependence, which means that we consider the fluid to be homogeneous and no
concentration gradients are possible. While there is experimental evidence for inhomo-
geneities developing during shear banding flows [38, 39], our approach does not include
this aspect explicitly, rather it rests on the existence of bundles as sufficient to produce
the thickening phase. Built into eqns. 4.16- 4.18 is a conservation of mass equation.
Since a long micelle is twice as long as a short micelle, it is exactly twice the mass of
a short micelle, while a bundle is 6 times more massive than a short micelle. Thus we
expect
Conservation of Mass 2A + 6B + C = M = constant. (4.19)
We note from eqns 4.16 - 4.18 that 2dAdt + 6dB
dt + dCdt = 0. The constant M representing
mass, is actually a concentration with units of molesliter . The reaction rates k0, f0, and
g all have unit of (seconds)−1, while k1 and f1 have units of (liters/mole)(seconds)−1,
and f2 has units of (liters/mole)2 (seconds)−1. In order to nondimensionalize eqns 4.16
- 4.18 we need to choose a timescale τ in units of seconds s and a volume scale V in

90
units of litersmole . Then each parameter in Table 2, as well as the concentrations A, B,
and C, and time t, has a nondimensional counterpart: k0 = τ k0, f0 = τ f0, g = τ g,
k1 = (τ/V ) k1, f1 = (τ/V ) f1, f2 = (τ/V 2) f2, a = V A, b = V B, c = V C, t = t/τ .
Using these non-dimensional parameters, ODE’s 4.16 - 4.18 then become:
dA
dt=
1τV
d a
dt= −(
k0τ
+f0τ
)a
V− 3
V 2
τf2
a3
V 3 + (V
τk1 +
V
τf1)
c2
V 2 + αg
τ
b
V
=1
τV(k0 + f0)− 3
1τV
f2a3 +1
τV(k1 + f1)c2 +
1τV
α g b
Multiplying both sides by τV we obtain the non-dimensional ODE for a, unaltered from
the original ODE for A. The computation for the non-dimensionalization of B and C
are identical, giving
da
dt= −(k0 + f0(γ)) a− 3f2(γ) a3 + (k1 + f1(γ)) c2 + α g(γ) b (4.20)
db
dt= f2(γ) a3 − g(γ) b (4.21)
dc
dt= 2(k0 + f0(γ)) a− 2(k1 + f1(γ)) c2 + β g(γ) b (4.22)
While we have a mathematical description of the process of mass transfer, the
ODE’s (4.20-4.22) do not describe the internal stresses developed during fluid flow. Now
that we have three coexisting species, we need to describe how each species contributes
to stress in the fluid, as well as how the stress due to each species combine to give the
total stress in the fluid.

91
4.4 The law of partial stresses
With our 3-species model, we have separated the fluid into different species each
of which can create stress in the fluid and we must relate these individual stresses to the
total stress in the fluid. To this end, we assume a simple relation with the total stress
in the fluid, σ, and the stress contributions of each species (σa, σb, and σc); we let each
contribute equally σ = σa +σb +σc. This assumption is not such an original idea, it has
been used in other models for wormlike micellar fluids [59], and is similar to the law of
partial pressures for gasses. The law of partial pressures is also an assumption which says
that for example in a confined space of fixed volume with 2 gasses which exert pressures
P1 and P2, the total pressure exerted is the sum P = P1 + P2 [82]. This law is exact if
the equation of state is given by the Ideal Gas Law:
PV = nRT,
for a gas with pressure P , volume V , at temperature T , where n is the number of
molecules, and R is the universal gas constant. If we solve for pressure to obtain P =
nRT/V , then the law of partial pressures says that if for example 2 gasses with numbers
of molecules n1 and n2 (both at the same temperature and collectively occupying the
same volume), the total pressure is P = P1+P2 = n1RT/V +n2RT/V = (n1+n2) RT/V
which is perfectly self-consistent. Pressure is an isotropic stress, and we use the same
idea of additive forces (the principle of superposition) to decompose arbitrary stress
contributions from diffrent micellar species.

92
In our model, the partial stresses are each given by the Maxwell equation, but
weighted by the concentration of each species. It can be thought of as saying that the
fluid is a mixture of three fluids of different viscosities, each of which contributes to the
total stress proportional to the amount of that type of fluid (which is the “weight” of
that constituent fluid). Of course, in addition to this idea, the ODE’s 4.20-4.22 describe
the way these three constituent fluids interact or “mix”. In the case of steady shear flow,
we define the steady state partial stresses by
σa = limt→∞ a(t)
2ηaλa
∫ t
0e(t′−t)/λaD(t′)dt′ (4.23)
Both σb and σc are defined analogously. Then we can compute the effective viscosity:
ηe = limt→∞
σ
γ(4.24)
= limt→∞
1γ
ηaλa
a(t)∫ t
0e(t′−t)/λa γ(t′)dt′
(continued) +1γ
ηbλb
b(t)∫ t
0e(t′−t)/λb γ(t′)dt′
(continued) +1γ
ηcλc
c(t)∫ t
0e(t′−t)/λc γ(t′)dt′)
= limt→∞ [ηa a(t)(1− e−t/λa) + ηb b(t)(1− e−t/λb)
(continued) + ηc c(t)(1− e−t/λc)]
= ηa as + ηb bs + ηc cs,
where as = limt→∞ a(t), and both bs and cs are similarly defined. Since the reaction
rates in equations 4.16-4.18 depend on γ, the concentrations will as well. This means

93
that ηe is not constant, but depends on γ. This is a significant point: the Maxwell model
predicts a constant ηe, and while we have only added the “weight” of each species, we
already have the possibility of a non-linear stress dependence on the rate of strain. This
moves our focus to steady state solutions of the 3-species model.
So the model in equations 4.20-4.22 and 4.24 for wormlike micellar fluids in-
troduces breaking and reforming into the Maxwell model. In the steady state case this
involves adding Maxwell stresses each weighted by a population (a number), and we have
written O.D.E.’s for coupling of these weights. We note that this is not a unique strategy
in that other models are also variants of the Maxwell model, adjusted to “correct” pre-
dictions in extensional flows (where the Maxwell model develops infinite stresses). The
finite extensible non-linear elastic (“FENE”) family of models are examples [47, 79, 80].
Since the Maxwell model describes macromolecules as Gaussian chains, or equivalently
Hookean springs, they allow for an infinite extension in the spring. The FENE models
replace the linear spring force Fs ∝ R (where R is the extension vector, which is end to
end vector of the molecule, see chapter 1) with the more physical law given by Warner
[72]:
Fs ∝R
1− (‖R‖2 /L2), (4.25)
in which L is the maximum extendable length of the spring, i.e. the total length of
the macromolecule. This spring force diverges to infinity as R → L, as the molecule
“straightens out” and the ends approach their maximum separation. Our modification
of the Maxwell model is similar to the FENE approach in that both begin with the

94
Maxwell equation and include a single new idea, though the ideas and the methods are
quite different.
In the next section we will consider a reduced version of our model 4.20-4.22 and
equation 4.24. As a first step in simulating solutions to the model, we restrict to the
case where we have only a and c-micelles, using only the ODE’s 4.20 and 4.22, and refer
to this reduced model as the “2-species” model. There are two reasons for this: when
we simulate the full 3-species model in steady state (in section 4.6) we will compare the
results to the 2-species model to understand better the effect of introducing bundles.
Also, for the 2-species model we are able to analytically solve for the steady state stress,
providing exact solutions rather than numerical approximations.
4.5 Steady state solutions to the 2-species model
The ODE’s 4.16 - 4.18 describe the interactions between the different species
under flow as functions of time, and it is natural to ask whether for a given shear rate γ,
the populations of a, b, and c ever achieve steady state values, or do they always depend
on time for certain values of γ. We begin to address this question by first simplifying
the ODE’s to the case where only a and c are interacting; that is, we set b ≡ 0 and the
rates to and from b (f2 and g - see Figure 4.4) are set to 0 as well. This reduced system
of ODE’s (our “2-species” model) becomes (in non-dimensional form):
da
dt= −(k0 + f0)a + (k1 + f1)c2 (4.26)
dc
dt= 2(k0 + f0)a− 2(k1 + f1)c2, (4.27)

95
while the non-dimensional conservation of mass equation is 2a + c = MV . In steady
state dadt ≡ 0 ≡ dc
dt , so 4.26 becomes equivalent to 4.27, which gives a in terms of c:
a = k1+f1k0+f0
c2. Using conservation of mass gives a quadratic equation for cs (the steady
state value for c) with two real solutions:
cs = − k0 + f04(k1 + f1)
± k0 + f04(k1 + f1)
√1 +
8MV (k1 + f1)k0 + f0
.
Since k0, k1, f0, and f1 are all positive,√
1 + 8MV (k1+f1)k0+f0
> 1, so there is a positive
and a negative solution for cs. Since c represents a concentration, only the positive root
is physically relevant:
cs = − k0 + f04(k1 + f1)
+k0 + f0
4(k1 + f1)
√1 +
8MV (k1 + f1)k0 + f0
> 0 (4.28)
as =k0 + f0
8(k1 + f1)+
MV
2− k0 + f0
8(k1 + f1)
√1 +
8MV (k1 + f1)k0 + f0
> 0 (4.29)
For steady shear flow, the equation for the steady state effective viscosity is
ηe = ηaas + ηccs. (4.30)
By introducing flow dependent reaction rates in ODE’s coupled to the Maxwell model,
our ηe is a non-constant function of γ (unlike the Maxwell model) even for just two
species. A first means of examining the dependence of ηe on γ is by examining the
limiting cases of γ → 0 and γ →∞.

96
η0 = limγ→0
= ηa( k08k1
+MV
2− k0
8k1
√1 +
8MV k1k0
) − ηc( k04k1
− k04k1
√1 +
8MV k1k0
)
(4.31)
To compute the formula for limγ→∞ ηe, we assume a linear asymptotic dependence
f0 = mγ and f1 = nγ, where m,n ∈ R+.
η∞ = limγ→∞
ηe = ηe(γ →∞) = ηa (m
8n+
MV
2−m
8n
√1 +
8MV n
m)−ηc (
m
4n−m
4n
√1 +
8MV n
m)
(4.32)
A sample of the predictions for ηe = ηaas + ηccs is given in Figures 4.5 - 4.6. For
each both figures below (and in this section), we have chosen explicit functions for the
flow rates, namely: f0 = mγ and f1 = nγ . In Fig. 4.5, three curves are shown: the
zero shear viscosity prediction η0, the asymptotic viscosity value η∞, and the effective
viscosity prediction ηe. The choices for parameters to produce ηe are: M = 0.1, V = 1,
ηa = 100, ηc = 0.1, k0 = 0.01, k1 = 0.06, m = 0.02, and n = 0.007. The parameters for
η0 and η∞ are the same as for ηe in Fig. 4.5. Notice that the range of shear rates in
this prediction is similar to those in used in the experimental data in Figs. 4.1, 4.2, and
4.3. Also note the similar values of η0 in Fig. 4.3 with the zero shear viscosity in Fig.
4.1 for 30 mM CPCl / NaSal. While this predicted ηe is not identical to the rheology
shown in Figs. 4.1-4.3, it is qualitatively correct in the sense that it has a well defined η0
observable at shear rates consistent with experiement, and predicts thinning to a value
∼ 0.1 Poise, which is a very reasonable value of viscosity for these fluids.

97
10−2
10−1
100
101
102
100
Shear rate
Eff
ectiv
e V
isco
sitie
s
η0
η∞
Fig. 4.5. The effective viscosity for the 2-species model (equation 4.30) shows a zeroshear plateau at low shear rates, thinning, and then levelling off to its asymptotic value.The zero shear viscosity and the asymptotic value are also depicted (equations 4.31 and4.32). Parameter choices for ηe are M = 0.1, V = 1, ηa = 100, ηc = 0.1, k0 = 0.01, k1 =0.06, m = 0.02, and n = 0.007.

98
10−2
10−1
100
101
102
10−2
10−1
100
101
Shear rate
η e / η 0
m=0.05, n=0.001
m=0.005,n=0.001
m=0.0005,n=0.0001
m=0.0001,n=0.005
m=0.01, n=1
Fig. 4.6. Predictions for the effective viscosity (equation 4.30), with varying flow de-pendent reaction rates. Each curve is normalized by its zero shear viscosity. Bothshear thinning and shear thickening are captured by the model. Parameter choices areM = 0.1, V = 1, ηa = 100, ηc = 0.1, k0 = 0.01, and k1 = 0.06.

99
Depicted in Figure 4.6 is a series of plots for ηe (equation 4.24) normalized by
their respective values of η0 so that each has a zero shear viscosity value of η0 = 1. The
only parameters which vary for these curves are m and n, which are the “strengths”
of the flow induced reaction rates f0 = mγ and f1 = nγ (see Figure 4.4). The other
parameters are the same as those used in producing the curve in figure 4.5: M = 0.1,
V = 1, ηa = 100, ηc = 0.1, k0 = 0.01, and k1 = 0.06.
Figure 4.6 shows the surprisingly rich steady shear rheology for the 2-species
model. When the ratio of flow dependant reaction rates is m/n = 50, the effective
viscosity shows shear thinning beginning at γ ∼ 0.01s−1, and as this ratio decreases the
rate at which thinning begins occurs at higher rates until m/n = 0.02 at which the fluid
shear thickens instead, and thickens at a lower shear rate when m/n = 0.01. This makes
sense in our model; the ratio m/n describes the relative strength of flow induced breaking
(a → c) to flow induced coagulation (c → a), so that as coagulation dominates breaking,
the fluid begins to thicken because the longer a-micelles contribute to viscosity more
than the shorter c-micelles. Again the shear rates for these curves are consistent with
experimental data. In particular notice that for m = 0.0005 and n = 0.0001, thinning
occurs at γ ∼ 10s−1 as does 30 mM CPCl / NaSal (Fig. 4.3), and a similar amount over
a decade of shear rates.
Perhaps the greatest significance of Figs.4.5 and 4.6 is that they show non-constant
viscosities for our model, even with only 2 species and even though we are considering
steady shear. Furthermore, this single model easily predicts both shear thinning and
shear thickening in a way which makes physical sense. By doing nothing other than
introducing two interacting species each with its own “intrinsic” viscosity and summing

100
their Maxwell stresses, we have changed the effective viscosity of the Maxwell model
to give physically realistic predictions. To our knowledge this modification has never
been studied, and it is a very reasonable modification to make for a fluid consisting
of wormlike micelles. Furthermore, it shows that considerations of angular orientation
or other specialized assumptions of fast or slow reaction regimes as in the papers we
reviewed in section 4.2, are not necessary effects to explain the observed rheology of
wormlike micellar fluids. If nonlinear rheology is sought, it is enough to assume that
there exist different viscous species within the fluid whose reactions are coupled to flow
in the nonlinear way described by equations 4.20-4.22.
4.6 Predictions for effective viscosity in constant shear flow to the 3-
species model
In the previous section, steady state solutions (with a and c only) were obtained
analytically to the 2-species model . While these solutions showed non-trivial predictions
for rheology, the interactions of the bundle species was ignored. The predicted rheology of
the 2-species model did show shear thinning similar to the experimental rheology depicted
in Figure 4.3. The shear thickening prediction of the 2-species model did not, however,
subsequently thin at higher shear rates. The actual rheology of wormlike micellar fluids
often includes regions of thinning and thickening in a single experiment, which the 2-
species model failed to produce.
In this section, we examine the 3-species model rheology including a wider range
of reaction rates as well. The hope is that by introducing the bundle population, we will
be able to produce the both thinning and thickening for a single choice of parameters.

101
There is strong physical evidence that bundles (which are more viscous than normal
wormlike micelles) are the cause for the shear thickening in these fluids [17, 38, 39, 40].
So we hope to see that any predicted shear thickening should be concomitant to a rise
in bundle population. The ultimate goal is to see that the 3-species model is capable
of producing rheology similar to the rheology shown at the start of this chapter (Figs.
4.1-4.3).
When we wrote the 3-species model 4.20-4.22 and 4.24, we included the bundles
because of the shear thickening rheology that wormlike micellar fluids display ([17, 39]
and our own data in Figs. 4.1 and 4.2). Our idea is that the short c-micelles have the
lowest viscosity or contribute the least amount of stress of the 3 populations. That is
to say, a fluid of given concentration consisting of only c-micelles would be less viscous,
at any shear rate, than a fluid with the same concentration but consisting of a micelles.
Likewise, bundles are presumed to be more viscous than long a-micelles. With this
notion, the explanation for thinning or thickening is simply that the dynamics between
the three interacting species causes one population to increase at the expense of another
species; shear thinning occurs when a solution decreases its population of long micelles
by beaking them into c-micelles. Similarly, if thickening is observed, it is believed to be
due to SIS or bundle formation, which means we expect that the b population will grow
at the expense of either or both a and c.
We emphasize that the following case studies are results for steady state rheology
(t → ∞), and represent normalized viscosities ηe/η0. To obtain steady state values,
we numerically simulated up to t = 1000 in equation 4.24, after checking that the
populations a, c, and b achieve steady state in equations 4.20-4.22. For each ηe we

102
Fig. 4.7. Predicted normalized viscosity dependence on shear rate. Parameters used:k0 = 1, k1 = 100, f0 = f1 = f2 = g = 100γ, ηa = 0.1, ηb = 1, ηc = 0.01, α = β = 2.

103
use a different set of parameters in the non-dimensional equation 4.24, however for all
of the figures we have used τ = 100, V = 1 for non-dimensionalization, and M = 0.01,
and λa = λb = λc = 1. We note that the predictions for the effective viscosities did not
seem to be sensitive to the paramters α and β in simulations made, and the examples
shown here do not include a wide variety of choices for these parameters.
Case I
For Fig. 4.7-4.9 the choices of the remaining parameters are: k0 = 1, k1 = 100,
f0 = f1 = f2 = g = 100γ, ηa = 0.1, ηb = 1, ηc = 0.01, and α = β = 2. Figure 4.7
shows a zero shear viscosity at low γ followed by modest thinning (less than an order
of magnitude) to a final value. Although thinning occurs at physically small values γ
(typical rheological tests in steady shear use shear rates no lower than ∼ 0.01 s−1),
this has also been observed in certain concentrations of wormlike micellar fluids at low
temperatures (∼ 20− 25C). Because the flow dependent reaction rates are equal, and
the scission term is linear in a in equation 4.22 while the combination rate is quadratic
in c in equation 4.20 (with c < 1), the equations favor scission when f0 = f1, so that the
fluid thins. The effect of bundle population is negligible; the initial population for the
bundles is zero, and only slight growth occurs at a restricted range of shear rates (Fig.
4.9). This effect could be made more influencial however if the bundle population had a
much higher intrinsic viscosity than we assigned for these plots (µb = 1).
The population dynamics that yield the viscosity curve in Fig. 4.7 are shown in
Figs. 4.8. From Figure 4.8 the reason for thinning is clear; the population of a-micelles
decreases and feeds the c-micelle population which is less viscous at the same shear rate
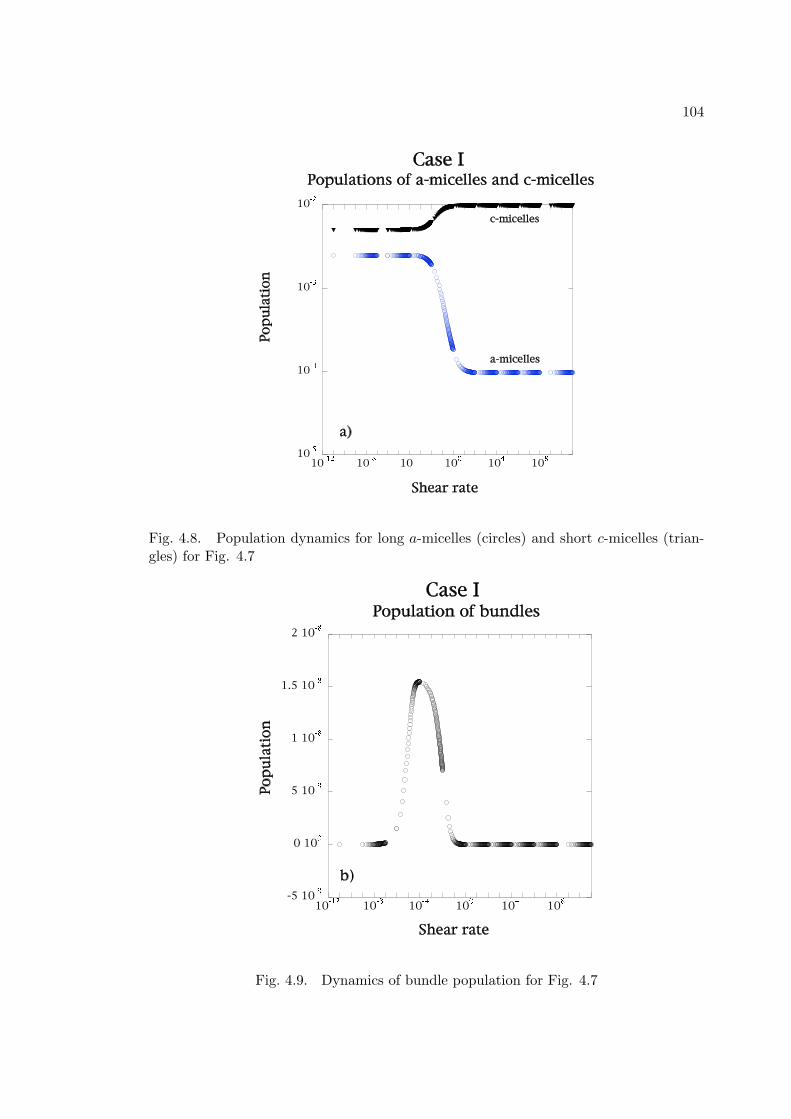
104
Fig. 4.8. Population dynamics for long a-micelles (circles) and short c-micelles (trian-gles) for Fig. 4.7
Fig. 4.9. Dynamics of bundle population for Fig. 4.7

105
at which thinning begins. The flow induced reaction rates for scission f0 and combination
f1 are equal, but the equilibrium rates are not, k0 < k1.
Case II The parameters chosen for the predicted viscosity in Fig.4.10 are: k0 = 1,
k1 = 1000, f0 = g = 100γ, f1 = 800γ, f2 = 0.01γ, α = 0.3, β = 5.4, ηa = 1, ηb = 1011,
and ηc = 0.1. Compared to Fig. 4.7, we have greatly increased the intrinsic bundle
viscosity µb, slightly increased the flow induced coagulation rate f1, and decreased the
breaking up of bundles in f2. The effect of changing k1 is to change initial conditions
(see equation 4.28 and 4.29) and we decreased f2 with the hope that it would prevent
the bundles from being destroyed until at least higher shear rates from those in Fig 4.7.
As we see in Fig. 4.10, the fluid is now predicted to shear thicken, again after an inital
zero shear plateau, but at very low shear rates γ ∼ 10−8 − 1 s−1, after which it thins
to roughly its zero shear viscosity value. From the population graphs (Figs. 4.11 and
4.12), it is evident that the thickening is due to bundle formation as is physically the
case [17] but the a and c-micelles are interacting very much like as in Fig. 4.7. This is
not surprising since we essentailly only altered the viscosity µb. Apparently, the effect of
reducing f2, the flow induced breaking of bundles, is to delay the destruction of bundles
until a higher shear rate.
Case III
In the next plot, Fig 4.13, the parameters are: k0 = 1, k1 = 100, f0 = 100γ =
f1 = f2, g = 10−6γ + 1, α = 2 = β, ηa = 0.1, ηb = 1, and ηc = 0.01. This simulation is
interesting because it predicts first shear thinning and then shear thickening, as in our
experimental data Fig. 4.2. Although it does not agree quantitatively with Fig. 4.2,

106
Fig. 4.10. Predicted normalized viscosity dependence on shear rate. Parameters used:k0 = 1, k1 = 1000, f0 = g = 100γ, f1 = 800γ, f2 = 0.01γ, ηa = 1, ηb = 1011, ηc = 0.01,α = 0.3, β = 5.4.

107
Fig. 4.11. Population dynamics for a-micelles (circles) and c-micelles (triangles) for Fig.4.10.
Fig. 4.12. Dynamics of bundle population for Fig. 4.10.

108
it is in some sense at the opposite end of a spectrum from Fig. 4.7 i.e. both Figs. 4.7
and 4.13 predict both thinning and thickening but in reverse order. There is only one
change in parameters for these figures in the breaking rate of bundles: g = 100γ for Fig.
4.7 whereas g = 10−6γ + 1 for Fig. 4.13. So by decreasing the breaking rate further
from the value used in producing Fig. 4.7, we have pushed the formation of bundles
to a much higher shear rate (Fig. 4.15), and we have stabilized bundle existence by
decreasing the flow dependent breaking rate adding a constant to g. It is not clear that
the added constant has a specific and identifiable role in the population dynamics for
this case. Fig. 4.14 shows that the dynamics of the a and c-micelles have not changed
qualitatively from the previous dynamics with different choices of parameters. So this
is a first approximation to the rheology data for 3.5 mM CPCl / NaSal in Fig. 4.2. As
expected the rise in bundle population is mirrored by a decline in a-micelle population,
visible in Fig. 4.14 near γ ∼ 103 s−1.
We note that although the shear rates at which the thickening occurs and sub-
sequent thinning, this plot (Fig. 4.10) is in amazingly good qualitative agreement with
physically observed results for low concentration wormlike micellar fluids (Figure 1 in
[17]). The increase of viscosity in this plot is over two orders of magnitude, and experi-
mentally thickening is less than a single order of magnitude. Viscosities increase twofold
or threefold in typical experiments on dilute solutions of wormlike micelles, so that by de-
creasing ηb in our simulations we would both recover a more physically accurate rheology
profile, and have a more reasonable value for ηb.
Case IV

109
Fig. 4.13. Predicted viscosity dependence on shear rate. Parameters used: k0 = 1,k1 = 1000, f0 = f1 = f2 = 100γ, g = 10−6γ +1, ηa = 0.1, ηb = 1, ηc = 0.01, α = β = 2.

110
Fig. 4.14. Population dynamics for a-micelles (circles) and c-micelles (triangles) for Fig.4.13
Fig. 4.15. Dynamics of bundle population for Fig. 4.13

111
Finally, we include a plot in which only shear thickening occurs. For Fig. 4.16
the choice of parameters is: k0 = 1, k1 = 100, f0 = 10−6γ, f1 = 10−2γ, f2 = 100γ,
g = 10−4γ, α = 2 = β, ηa = 0.1, ηb = 1, and ηc = 0.01. This choice of parameters a
priori seems like it should give the thickest fluid yet, since the flow-induced coagulation of
c into a is greater than a → c scission and the bundle formation from a is at a greater rate
f2 than either f0 or f1, and the breaking rate of bundles is much lower than the bundle
formation rate. And as we see in Fig. 4.16 the fluid has a constant zero shear viscosity
at low γ followed by shear thickening at a shear rate which is physically reasonable:
γ ∼ 0.1 s−1. Moreover, the value of viscosity increase is perfectly sensible, and the curve
agrees qualitatively with the experimental data shown in Fig. 4.1 for a dilute solution of
1 mM CPCl / NaSal. Here again our choices of parameters result in a bundle formation
which does not decay over a large range of shear rates, but interestingly, after the initial
rise of b near γ ∼ 1000 s−1, there is a secondary increase in b with a coincident decrease
in c-micelle population. In Fig. 4.13 there is also a secondary increase in b population
but there it was fed by a-micelles. This increase in b from c is noticeable in Fig. 4.16
near γ ∼ 104 s−1. Also, both a and c micelle concentration feed into the b population
at the same shear rate, which is not surprising because our choice of parameters is one
that favors a growth at the expense of c, and b growth over a.
While the 2-species model was able to capture shear thickening, it is the rise
and subsequent decrease in viscosity that we associate with SIS formation. Physically
we expect that at high enough shear rates, the bundles will break apart, decreasing
the effective viscosity of the fluid. With the inclusion of the bundle species, the 3-
species model was able to produce the rheology of SIS formation but the 2-species model

112
was not. The sample predictions we have shown are not necessarily characteristic of
all possible predictions, rather they show that the model is able to adress the various
rheological features of wormlike micellar fluids. The 3-species model has therefore been
successful, qualitatively and to a large extent quantitatively, in the sense of capturing
the rheology we set out to reproduce. So far, though, we have examined only steady
state behaviour and in the next chapter time dependence will be included. As we will
see, this will introduce additional and subtle mathematical complexities, which have a
very nice physical interpretation.

113
Fig. 4.16. Predicted viscosity dependence on shear rate. Parameters used: k0 = 1,k1 = 100, f0 = 10−6γ, f1 = 10−2γ, f2 = 100γ, g = 10−4γ, ηa = 0.1, ηb = 1, ηc = 0.01,α = β = 2.

114
Fig. 4.17. Population dynamics for a-micelles (circles) and c-micelles (triangles) for Fig.4.16.
Fig. 4.18. Dynamics of bundle population for 4.16.

115
Chapter 5
Modified Memory:
How to Remember to Forget
5.1 Modified memory
In equation 4.24 for the steady state stresses of the 3-species model, the concen-
tration functions a(t), b(t), and c(t) appear on the outside of the memory integrals, as
weights to the Maxwell stress for each species. Since we were only concerned with the
steady state, concentrations were treated as functions of γ, without time dependence
since we were using limiting values (e.g. limt→∞ a(t)). The point is that we could
regard the concentrations simply as weights, placing them outside the integrals in equa-
tions 4.24. As steady state values, the concentrations could be placed inside or outside
the integrals equally, there was no reason to consider the mathematical impact or phys-
ical difference of this placement. In this chapter, however, we consider time dependent
stresses. As we will see, the two choices of placing the concentrations inside or outside
the memory integral have different physical meanings, both of which are contradicted by
the physical process we model.
We can consider the following cases, which expose an interesting connection be-
tween memory and the breaking and joining of micelles. These kinetic processes are
identified in the concentration functions as a decrease (breaking) or an increase (join-
ing). The cases to consider are the various combinations of increasing or decreasing

116
concentrations, placed inside or outside the memory integral for stress. First suppose
that a(t) is strictly increasing, for example a(t) = t. Then if we write
σa(t0) = a(t0)2η
λ
∫ t0
0e(t′−t0)/λD(t′)dt′, (5.1)
we are effectively remembering that there were a(t0) = t0 micelles at time t0 and at
all previous times. Because the concentration function is not under the integral, we are
weighting the Maxwell stress by a(t0) even as we integrate over the past history of the
rate of strain when there were fewer a-micelles. In this case we are over-counting since
a(t′) < a(t0) for all times t′ < t0.
Next suppose that a(t) is decreasing (due to breaking), say from t = 0 to t0,
and the concentration is placed again outside the memory integral as in equation 5.1.
Computing stress at t0 would use a(t0), and exclude the larger values of a at previous
times. There is no memory of the micelles present in the fluid at earlier times, which
seems like under-counting. However, we are excluding the contribution of micelles which
have converted to another species - equation 5.1 is the stress from a-micelles. Should
we allow micelles, which existed in the past but no longer exist, to still contribute to the
stress?
To answer this let us recall that the way polymers and micelles are believed to
supply stress to the fluid is through their extension, or uncoiling. When their ends
are separated, they act like stretched springs, storing energy and exerting a force on
the nearby fluid. Now imagine instantaneously removing a stretched spring, is there

117
any force left from it? If an a-micelle breaks (presumably instantaneously) into two c-
micelles, then any stress contribution would come from the c-micelles, but the a-micelle
no longer exists, so it cannot continue to stress the fluid. So when any micelle (or
bundle) disappears due to breaking or joining, any stress it was carrying disappears as
well. Therefore, there is no contradiction with placing a decreasing concentration outside
the integral. But a general concentration function (solution to the ODE’s 4.20-4.22) can
decrease over certain times, and increase as well. Putting the concentration outside the
integral would then be physically inaccurate in the general case.
It is certainly possible that when a-micelles break, the new c-micelles (or bundles)
born from a converted a-micelle may carry stress, if the a-micelle broke in a way to create
stretched c-micelles. We do not exclude this possibility, in fact we depend on it. We
assume that some c-micelles will be stretched when they come from broken a-micelles,
and some will be less stretched. Each species in flow will carry stress from the deformation
caused by the flow, and we can consider each member of a species to carry the average
amount of stress caused by flow. Our assumption is that the average amount of stress
in a newly born micelle (or bundle) is equal to the average amount of stress caused
by flow. This means that we need only count how much of a species is present in the
fluid to compute the stress, and we do not need to keep track of how many members of
the species came about by a breaking or joining event. The only stress we exclude are
the stress contributions from micelles which are no longer present in the fluid, but were
perhaps at one time.

118
We next suppose that a(t) should appear under the integral sign. Let a(t) be
increasing from t0 to t1. To compute stress at t1 we would use
σa(t1) =2ηaλa
∫ t1
0a(t) e(t−t1)/λaD(t)dt.
As we integrate over the past history, we are using the past concentration as well. The
integral is “remembering” that we had fewer micelles in the past, which is appropriate
since these micelles still exist at the time we compute stress at t1. So there is no problem
with this scenario.
However, let us take a(t) to be decreasing from say time t1 to t2, with t1 < t2.
With the concentration inside the integral, the formula for stress at t2 would be
σa(t2) =2ηaλa
∫ t2
0a(t′) e(t′−t2)/λaD(t′)dt′.
In this case, as we integrate from 0 to t2, we remember the micelles which existed at
times t < t2, but which have vanished by the time we compute stress at t2! We have
just argued that when they vanish, the stress they were carrying also vanishes, so these
micelles should be excluded from the stress computation.
In general then, it is not correct to have a(t) either outside or inside the integral.
We could resolve this problem if we kept something like the concentration inside the
integral sign but somehow excluded the micelles that broke. So we need to find a function
of time which is almost the concentration but makes our model for the memory of stress
more physically accurate. So rather than use weighted Maxwell stresses, as we could in

119
the steady state case, we now propose stresses of the form
σa(t) =ηaλa
∫ t
0(“Replacement for the concentration a(t)”) e(t′−t)/λaD(t′)dt′, (5.2)
with similar expressions for σb(t) and σc(t).
3 Π
25 Π
27 Π
2
time
0.2
0.4
0.6
0.8
1
Forgetting Broken Micelles
Fig. 5.1. The concentration a(t) is depicted as the dashed curve, while the solid curveis the corrected concentration, which excludes the micelles that grew from t1 = 3π/2 tot2 = 5π/2, and then broke by time t3 = 7π/2.
This raises an interesting point about our course of modelling. That is, we first set
out to model the ability of micelles to exchange mass by having three species of micelle,
and allowing thier concentrations to interact throught the ODE’s 4.20-4.22. We then
used the law of partial stresses to combine the species specific stresses, each given by
the Maxwell equation appropriately weighted with concentration. But we are now faced
again with the process we have already modelled, and we see that this physical property

120
of micelles cannot be decoupled from the stress equation as we have attempted to do by
writing separate ODE’s for it. We must again model breaking and reforming. Whereas
we first used the ODE’s 4.20-4.22 to model what species was in the fluid, we now need
to model how the ability to convert from one species to another effects memory.
Let us consider some examples to motivate our first attempt of determing a physi-
cally relevant “replacement function.” For example, say a(t) increases from t1 = 3π/2 to
t2 = 5π/2, and decreases from t2 to t3 = 7π/2, as in Figure 5.1. If we wish to compute
stress at time t3 = 7π/2, then we need to replace a(t) with a similar function, but which
completely ignores the “bump” ranging from t1 to t3. This means forgetting that the
long micelles created during (t1, t2) ever existed, because they subsequently died over
(t2, t3). To get the stress at time t3 = 7π/2, we should use as a replacement function
the solid curve depicted in Figure 5.1. This curve is only depicted up to 7π/2, since that
is all that is relevant for the memory integral in equation 5.2.
If we had asked for the stress at time t2 = 5π/2, then the micelles (in the previous
example of Figure 5.1) born from 3π/2 to 5π/2 would not yet have been destroyed.
Clearly the solid curve in Figure 5.1 would be unsuitable, and we see immediately that
the “replacement concentration function” depends on the time at which we compute
stress. To make this point more clear we use the following simple examples. Suppose
the concentration a(t) is as depicted by the dashed curve in Figure 5.2. To compute
stress at time π, we would have to ignore the amount of a-micelles that was destroyed
before time π, so the proper replacement concentration would be given by the solid curve
in Figure 5.2. Let us denote this replacement function by R a(π, t), the superscript a

121
Π 3 Πtime
0.2
0.4
0.6
0.8
1Replacement function for ΣaHΠL
Fig. 5.2. The original concentration a(t) is shown as the dashed curve, while the solidcurve is a proposed replacement function for obtaining stress at time π in equation 5.2.
Π 3 Πtime
0.2
0.4
0.6
0.8
1Replacement function for ΣaH3ΠL
Fig. 5.3. The original concentration, a(t) is shown as the dashed curve. Here the solidcure is the replacement function for computing stress (equation 5.2 at time 3π.

122
denoting a-micelles. The stress at π would then be given by
σa(π) =ηaλa
∫ π
0R a(π, t′)e(t′−t)/λaD(t′)dt′. (5.3)
Now suppose, using the same concentration function a(t), that we would like the
stress at time 3π. In this case we would need the replacement concentration shown as
the solid curve in Figure 5.3, which we denote by R a(3π, t). Notice that R a(π, t′) 6=
R a(3π, t′) for all t′ < π.
The three example given in Figures 5.1-5.3 imply that the replacement function
should somehow compare minima of the concentration at different times. More precisely,
to compute stress at time s, we could use the replacement function R a(s, t) defined by
R a(s, t) =
mina(s), a(t) t ≤ s
a(t) t > s,
(5.4)
with similar definitions for bundles and c-micelles. The choice of making R a(s, t) = a(t)
for t > s seems somewhat arbitrary since it does not enter into the stress equation, but
we make this choice to be consistent with a function which we define in the next section.
To compute stress at time s we would then have
σa(s) =ηaλa
∫ s
0R a(s, t)e(t−s)/λaD(t)dt. (5.5)
Equation 5.4 is our first attempt at the replacement function. This definition for Ra(s, t)
was in fact used to produce each of the solid curves in Figure 5.1-5.3.

123
1 2 3 4 5time
1
3
5
RaH2,tL: Replacement function for ΣaH2L
Fig. 5.4. The solid curve is R a(2, t) for the concentration function depicted as thedashed curve.
1 2 3 4 5time
1
3
5
RaH5,tL: Replacement function for ΣaH5L
Fig. 5.5. The solid curve is R a(5, t) for the concentration function depicted as thedashed curve from Figure 5.4.

124
Given the definition in equation 5.4, R a(s, t) is clearly bounded by the actual
concentration function a(t). If it is to eliminate all events in which members of a species
disappear, then the replacement function should also be non-decreasing. Indeed, the way
we identify that an amount of one species has converted to another species is when the
concentration decreases. However, our first attempt in equation 5.4 unfortunately fails
in this respect. Consider a concentration function such as the dashed curve in Figure
5.4. While R a(2, t) is physically accurate (the solid curve in Figure 5.4) in the sense that
it “forgot” about the destroyed micelle population, R a(5, t) is no longer non-decreasing
as shown by the solid curve in Figure 5.5. The replacement function correctly forgot
the micelles destroyed from t = 1 to t = 2 when we compute stress at s = 2, but at
a later time (s = 5) it “forgot to forget” these same destroyed micelles. We need a
new replacement function, one which correctly forgets destroyed micelles, and which will
never again “re-remember” them.
Although equation 5.4 is not suitable to compute stress with, it was presented
for three reasons. First, it is correct for some choices of concentration functions, and
those for which it fails reveal also what exactly needs to be fixed. Second, it serves as
a stepping stone and will make it easier to understand the more complex definition we
present in the next section. Finally, R a(s, t) was our actual first attempt on our way to
defining a final replacement function.
5.2 The time dependent constitutive equation
The task now is to write a physically correct time dependent function which
replaces the concentration functions a(t), b(t), and c(t). The previous section gave us

125
a good start, but we have at least to correct the flaw in equation 5.4 for R a(s, t). In
fact, it is the view that R a(s, t) “forgot to forget” that we use as inspiration for the next
definition for the replacement function. What this phrase refers to is the problem we
exposed with R a(s, t) in Figure 5.5. The problem can be stated by saying that while
R a(2, t) eliminated the micelles that were destroyed from t = 1 to t = 2, R a(5, t) did
not “remember” to eliminate this same amount of micelles. What we need is to use the
information from R a(2, t) when we compute R a(5, t). Indeed, if we can make R a(s, t)
always “remember to forget” that which it forgot before, we will have our non-decreasing,
bounded replacement function. We do this in two steps: first we do it approximately by
asking R a(s, t) to check itself against R a(s− ε, t), then we let ε → 0.
A definition for the replacement function for the concentration a(t) which checks
itself in this way is given by
P aµ
(s, t) =
mina(t), a(s), P aµ
(s− µ, t) 0 ≤ t < s, µ < s
mina(t), a(s) 0 ≤ t < s, µ ≥ s
a(t) t ≥ s ≥ 0.
(5.6)
This function is only defined for s ≥ 0, t ≥ 0, and for all cases we use only µ > 0. The
superscript a denotes the concentration a(t), and again, there are analogous definitions
for P bµ
(s, t) and P cµ
(s, t). The notation is cumbersome so in the sequel we may suppress
the superscript and refer to this simply as Pµ.
Our new function Pµ(s, t) has two variable (and the parameter µ), both of which
represent time, which we now discuss. Recall that the first variable s is regarded as
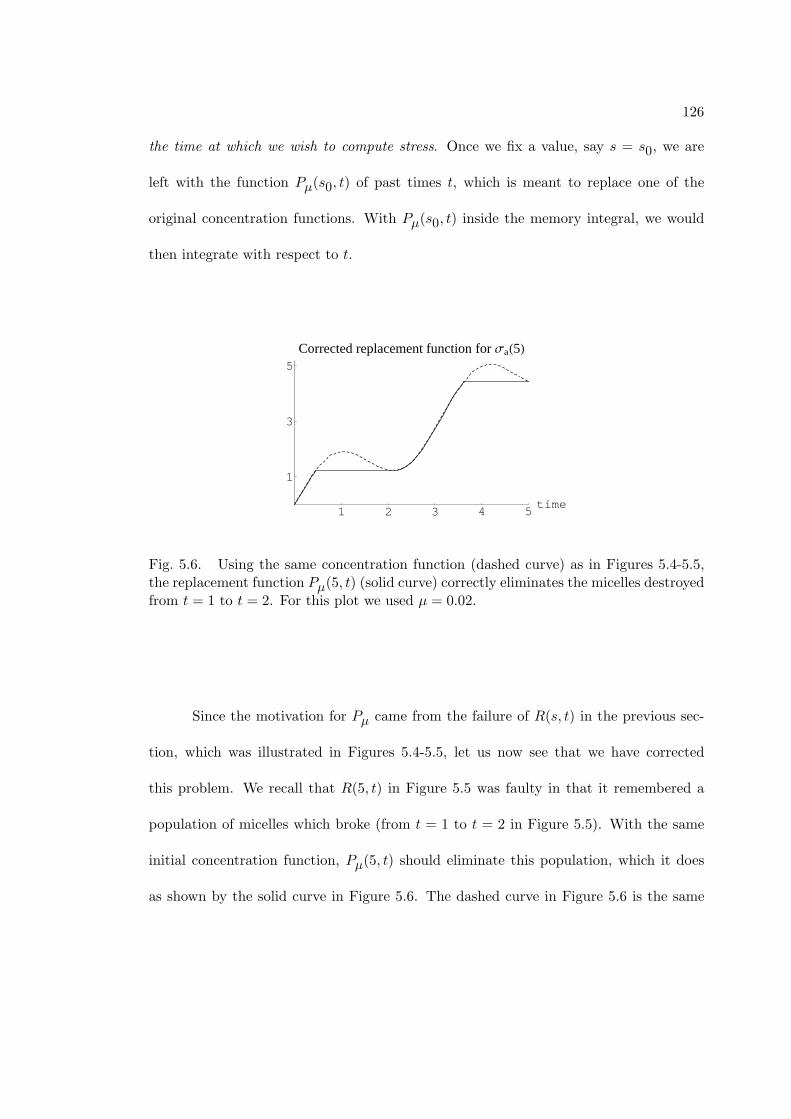
126
the time at which we wish to compute stress. Once we fix a value, say s = s0, we are
left with the function Pµ(s0, t) of past times t, which is meant to replace one of the
original concentration functions. With Pµ(s0, t) inside the memory integral, we would
then integrate with respect to t.
1 2 3 4 5time
1
3
5
Corrected replacement function for ΣaH5L
Fig. 5.6. Using the same concentration function (dashed curve) as in Figures 5.4-5.5,the replacement function Pµ(5, t) (solid curve) correctly eliminates the micelles destroyedfrom t = 1 to t = 2. For this plot we used µ = 0.02.
Since the motivation for Pµ came from the failure of R(s, t) in the previous sec-
tion, which was illustrated in Figures 5.4-5.5, let us now see that we have corrected
this problem. We recall that R(5, t) in Figure 5.5 was faulty in that it remembered a
population of micelles which broke (from t = 1 to t = 2 in Figure 5.5). With the same
initial concentration function, Pµ(5, t) should eliminate this population, which it does
as shown by the solid curve in Figure 5.6. The dashed curve in Figure 5.6 is the same

127
concentration function which was used in Figures 5.4-5.5. The value of the parameter µ
to produce this plot was µ = 0.02.
Before taking the limit as µ → 0, we address the role of the parameter µ. The
difference between equation 5.4 for R(s, t) and equation 5.6 for Pµ is the extra term
Pµ(s − µ, t) in the case when t ≤ s and µ < s. This term is therefore responsible for
correcting the problem with R(s, t), and it serves to iteratively compare Pµ with earlier
versions of itself. So that Pµ(s, t) checks itself against Pµ(s − µ, t), which checks itself
again Pµ(s− 2µ, t), and so on until (s− kµ) ≤ µ. This is how Pµ “remembers to forget”
what it had forgotten in “previous versions of itself.” As soon as (s − kµ) ≤ µ, the
definition of Pµ in equation 5.6 toggles into the second case because we can no longer
subtract µ from the first argument (s−kµ) (both arguments of Pµ must be non-negative).
Toggling into the second case means that we are using exactly our definition of R(s, t)
(equation 5.4). So the function Pµ is not completely correct; if we ask for stress at times
s such that s ≤ µ, we could have a flawed replacement function. As we stated at the
beginning of this section, equation 5.6 is approximate. In the limit as µ → 0, there will
be no values of s for which Pµ toggles into the second case in definition 5.6; this is the
function we actually want. So our next task is to show that the limit of Pµ as µ → 0
exists.
PROPOSITION 5.2.1. The function defined by
P (s, t) = limµ→0
Pµ(s, t)
exists for each point (s, t) such that s ≥ 0 and t ≥ 0.

128
Proof:
We will show that for each point (s, t) with s ≥ 0 and t ≥ 0, and any convergent
sequence µn → 0, the sequence Pµn(s, t) is Cauchy.
Case i) Both s and t are fixed, and 0 ≤ t < s. First we will go out far enough in
the sequence of µn to have Pµntoggle into the first case in definition 5.6 at (s, t) and
(s − µ, t). So let N1 be such that ∀ n > N1 we have 3µn < s and 2µn < (s − t), so
that µn < (s − 2µn) < (s − µn) and t < (s − 2µn) < (s − µn). For each n > N1, the
Archimedean Order Property and Completeness of R, guarantees that we can find an
integer kn such that
s− (kn + 1)µn ≤ t ≤ (s− knµn). (5.7)
For each n > N1, we can also find jn ∈ Z+ with
s− (jn + 1)µn ≤ µn ≤ (s− jnµn). (5.8)
So as long as n > N1, we have the first case in the definition of Pµ, i.e. Pµn(s, t) =
mina(t), a(s), Pµn(s − µn, t) as well as Pµn
(s − µn, t) = mina(t), a(s), Pµn(s −
2µn, t).
If jn < kn, then Pµn(s − (jn + 1)µn, t) = mina(t), a(s). If kn ≤ jn, then
Pµn(s − (kn + 1)µn, t) = a(t). Without loss of generality, we may assume kn ≤ jn, to

129
get
Pµn(s, t) = mina(t), a(s), Pµn
(s− µn, t)
Pµn(s− µn, t) = mina(t), a(s− µn), Pµn
(s− 2µn, t)
·:
Pµn(s− knµn, t) = mina(t), a(s− knµn), Pµn
(s− (kn + 1)µn, t)
= mina(t), a(s− knµn),
and hence
Pµn(s, t) = mina(t), a(s), a(s− µn), a(s− 2µn), ..., a(s− knµn). (5.9)
If it were the case that jn < kn, then we would still end up with equation 5.9 for
Pµn(s, t), but with jn replacing kn.
Note that the concentration functions a(t), b(t), and c(t) are solutions to ODE’s
4.20-4.22, so we may assume that they are each continuous. Since the interval [t, s] ⊂ R
is compact, the concentrations are uniformly continuous on [t, s]. Then for any given ε,
find δ1 such that for x, y ∈ [t, s],
‖x− y‖< δ1 =⇒ ‖a(x)− a(y)‖< ε/2.
Before proceeding, we will need the following lemma.

130
LEMMA 5.2.2. “Cramping the µn” Suppose we are given µn, µm ∈ (0, δ1/4), and
0 ≤ t < s. Let kn and km be as defined as in equation 5.7. If α ∈ Z with 0 ≤ α ≤ kn,
then there exists a γ ∈ Z with 0 ≤ γ ≤ km such that ‖(s− γµm)− (s− αµn)‖< δ1/2.
Proof:
If α = 0, then choose γ = 0 and we are done. We next assume that s− αµn < s.
Find the largest γ ∈ Z, γ ≥ 0, such that (s−αµn ) < s−γµm. If (s−γµm)−(s−αµn) ≥
δ1/4, then s− (γ + 1)µm− (s−αµn) = (s− γµm)− (s−αµn)−µm ≥ δ1/4 > 0, which
contradicts the definition of γ. So we conclude that (s − γµm) − (s − αµn) < δ1/2.
Finally, since (s− γµm) > (s− αµn) > t, then by the definition of km (equation 5.7),
it must be that γ ≤ km. Lemma 5.2.2 is now proved.
We are now ready to prove Pµn(s, t) is Cauchy. So let ε > 0 be given. We need to
find an N ∈ Z+ such that whenever n, m > N , we have ‖Pµn(s, t)−Pµm
(s, t)‖< ε. Take
N > N1 (defined above) such that ∀ n > N , we have ‖µn ‖< δ1/4, using the Lemma.
Using equation 5.9, we know it may happen that for some n, m > N , either one or
both of Pµn, Pµm
may be equal to a(t). If both are equal to a(t), then clearly we have
‖Pµn(s, t) − Pµm
(s, t) ‖< ε. Suppose Pµn(s, t) = a(t), while Pµm
(s, t) = a(s − βµm),
with β ∈ Z and 0 ≤ β ≤ km, with km as defined above. If ‖ a(t) − a(s − βµm) ‖> ε,
then we will obtain a contradiction. Since Pµm(s, t) = a(s − βµm), we know from
equation 5.9 that a(s − βµm) ≤ a(t). From Lemma 5.2.2, we can find γ such that
‖(s− γµn)− (s− βµm)‖< δ1/2, with 0 ≤ γ ≤ kn. Since Pµn(s, t) = a(t), we know that
a(t) < a(s− γµn). Hence
‖a(t)− a(s− βµm)‖= a(t)− a(s− βµm) < a(s− γµn)− a(s− βµm) < ε/2 < ε.

131
But then this contradicts our assumption. Therefore, for all n,m > N , if Pµn(s, t) = a(t),
and Pµm(s, t) = a(s− βµm), then ‖Pµn
(s, t)− Pµm(s, t)‖< ε.
We next suppose that with n, m > N , we have Pµn(s, t) = a(s − αµn) with
0 ≤ α ≤ kn, and Pµm(s, t) = a(s− βµm) with 0 ≤ β ≤ km. Without loss of generality,
we may assume that
a(s− αµn) < a(s− βµm).
So find γ ∈ Z, 0 ≤ γ ≤ km, such that ‖ (s − αµn) − (s − γµm) ‖< δ1/2. Then
since Pµm(s, t) = a(s− βµm), we know that a(s− γµm) ≥ a(s− βµm). Therefore,
‖a(s−βµm)−a(s−αµn)‖= a(s−βµm)−a(s−αµn) ≤ a(s−γµm)−a(s−βµn) < ε/2.
This concludes the proof of propostion 5.2.1 for case i) in which 0 ≤ t < s.
Case ii) Both s and t are fixed, with t ≥ s. This case is trivial since Pµn(s, t) =
Pµm(s, t) = a(t).
Since we are taking the limit as µ → 0, we do not have to check the case where
0 ≤ t < s and µ ≥ s. For a fixed s, we just go far enough out in the sequence that we
are guaranteed all µn < s. This concludes the proof to proposition 5.2.1.
So for each concentration function a(t), b(t), and c(t), our model uses a replace-
ment concentration given by the limit functions we now know exist. We denote these

132
functions by
P a(s, t) = limµ→0
P aµ
(s, t) (5.10)
P b(s, t) = limµ→0
P bµ
(s, t) (5.11)
P c(s, t) = limµ→0
P cµ
(s, t). (5.12)
The time dependent constitutive equation is then a sum of three terms using the functions
5.10-5.12:
σ(s) = σa(s) + σb(s) + σc(s) (5.13)
=2ηaλa
∫ s
0P a(s, t) e(t−s)/λa D(t) dt +
2ηbλb
∫ s
0P b(s, t) e(t−s)/λb D(t) dt
+2ηcλc
∫ s
0P c(s, t) e(t−s)/λc D(t) dt.
In the next section of this chapter, we return to the “discrete” replacement func-
tion Pµ(s, t) to obtain time dependent numerical simulations of our model. In section 5.4
we will examine some interesting mathematics of the function P (s, t). We will consider
our model equation 5.13 in a more general context in that section, and show how tools
from functional analysis might be used to explore the types of solutions our model could
bear.
5.3 Predictions for stress in time dependent flow
Using now the function Pµ(s, t) inside the memory integral for stress, in this sec-
tion we present numerical simulations for stress in various time dependent rheological

133
flows. Our model accounts for the reversible scission reactions in the stress computation,
and here we show how it works in live examples. In the first choice of flow, in addition
to stress predictions we show the dynamics of the concentration functions and the corre-
sponding replacement functions Pµ, as an initiation to the details of the model. We also
show how using Pµ differs from the models which do not include the scission reactions of
micelles, namely the approach of using the original concentration functions placed inside
or outside the integral, as well as the Maxwell model. Parameter choices differ for the
different flows we test, but for all of the figures in this section some paramters are held
fixed. These parameters are: τ = 100, V = 1, and M = 0.01.
5.3.1 Linearly ramping shear
All of the flows we consider are time dependent simple shear flows, and the first
that we consider is given by γ(t) = t. For this flow we show several plots (Figures
5.7-5.14), for which we use the same parameter values: γ(t) = t, µ = 0.01, k0 = 102,
k1 = 103, f0(γ) = 0, g(γ) = 10−3γ, f1(γ) = 104γ, f2 = 106γ, α = β = 2, ηa = 103,
ηb = 10, ηc = 10−3, and λa = λb = λc = 1. Since this is our first time dependent flow, it
is the first time we can see how our model (equations 4.20-4.22 with 5.13) works. Since
it differs from the Maxwell model in its modification of the concentration functions, we
begin by examining the dynamics of the three species in Figures 5.7-5.9. Shown also in
each of these plots are samples of our modified memory function Pµ.
In Figure 5.7 we see that the population of a-micelles peaks quickly and then
decays to a plateau value of roughly limt→∞ a(t) ∼ 0.00011. When we compute stress
at times near s ∼ 10 − 20, the modified concentration function P aµ
will be constant

134
0 5 10 15 20
0.00025
0.0005
0.00075
0.001
0.00125
0.0015
Dynamics of a-micelles with PΜaHs,tL
Fig. 5.7. The dynamics of a-micelles is shown as the thick dashed curve. Plots ofP a
µ(0.6, t), P a
µ(1.7, t), and P a
µ(6.5, t) are also shown as solid curves, with µ = 0.01. The
paramaters used for these dynamics are the same for the stress plots 5.10-5.14 whichare: γ(t) = t, k0 = 102, k1 = 103, f0(γ) = 0, g(γ) = 10−3γ, f1(γ) = 104γ, f2 = 106γ,α = β = 2, ηa = 103, ηb = 10, ηc = 10−3, and λa = λb = λc = 1.
0 5 10 15 200
0.00025
0.0005
0.00075
0.001
0.00125
0.0015
Dynamics of bundles with PΜbHs,tL
Fig. 5.8. The dynamics of bundles is shown as the thick dashed curve. A plot ofthe modified concentration P b
µ(6.5, t) is shown as the solid curve (µ = 0.01), which is
identical to the concentration function b(t) up to that time since b(t) is monotonicallyincreasing. Parameter values are the same as for Figure 5.7.

135
and approximately equal to the plateau value limt→∞ a(t), such as shown by the plot
of P aµ
(6.5, t). At smaller times, before any a-micelles convert to bundles or break into
c-micelles, P aµ
will be equal to a(t). When a(t) starts decreasing, P aµ
correctly excludes
the amount of a-micelles lost to another species, as shown by the plots of P aµ
(0.6, t) and
P aµ
(1.7, t).
0 5 10 15 200
0.002
0.004
0.006
0.008
0.01Dynamics of c-micelles with PΜ
cHs,tL
Fig. 5.9. The dynamics of c-micelles is shown as the thick dashed curve. Plots ofP c
µ(1.0, t) and P c
µ(3.0, t) are shown as solid curves, both are constant since the concen-
tration function c(t) is monotonically decreasing. Parameter values are the same as forFigure 5.7.
Evidently, the a-micelle decrease is feeding the bundle population b(t), shown in
Figure 5.8 (since the c-micelle concentration is decreasing for all times in Figure 5.9).
For bundles, which begin at b(0) = 0, there is a monotonic increase, so that for all s and
t we have P bµ
(s, t) = b(t). A sample of the modified concentration is given in Figure 5.8
by P bµ
(6.5, t) as the solid curve, which follows b(t) perfectly from t = 0 to t = 6.5 as it

136
should. The c-micelle decrease in Figure 5.9 must be feeding the early a-micelle increase.
This makes sense since we have chosen k0 < k1, f0(γ) = 0 while f1(γ) = 104γ (see the
schematic of reaction rates in Figure 4.4). The plateau value for c is similar to that of
a: limt→∞ c(t) ∼ 0.00015. We must remember that these values are mole fractions, and
with our choices of M = 0.01 and V = 1, they are each always less than M · V = 0.01,
obeying the non-dimensional conservation of mass (equation 4.19 multiplied by V on
both sides).
With these as population dynamics for the shear flow γ(t) = t, the stress given
by equation 5.13 is shown in Figure 5.10. In this Figure, in which stress is plotted
against time, the stress increases as we expected since γ is increasing. The effect of
the replacement function is best seen when we compare Figure 5.10 with the stress
prediction of the Maxwell model for the same flow (γ(t) = t), given in Figure 5.11. We
also compare our prediction with predictions of two other flawed models: the first model
comes from adding the Maxwell stresses for each of the three species with the respective
concentration function placed outside the memeory integral, the second model places the
concentration functions inside the integral. In other words, the first model is the sum of
equation 5.1 for the three species, this gives the “outside the integral” prediction (Figure
5.12). Moving the concentration functions to the inside of equation 5.1, and summing
the contribution of each species gives the “inside the integral” prediction seen in Figure
5.13.
Each of the Figures 5.10-5.13 shows an increasing stress. Our model prediction
in Figure 5.10 gives a significantly different prediciton from the Maxwell model, but is
close to the “inside” and even closer to the “outside” prediction. From the resolution

137
0 2 4 6 80
0.2
0.4
0.6
0.8
1
1.2
Time dependent shear stress, Γ =t
Fig. 5.10. Time dependent stress prediction using our model (equation 5.13) for shearflow with shear rate γ(t) = t.
0 2 4 6 80
2000
4000
6000
8000
Time dependent Maxwell stress, Γ =t
Fig. 5.11. Time dependent stress prediction of the Maxwell model for the same shearflow used in Figure 5.10. Parameter values are the same as for Figure 5.7.

138
0 2 4 6 80
0.2
0.4
0.6
0.8
1
1.2
Outside the integral, Γ =t
Fig. 5.12. Time dependent stress with concentration functions inside the Maxwellmemory integral, in the same shear flow used in Figure 5.10.Parameter values are thesame as for Figure 5.7.
0 2 4 6 80
0.2
0.4
0.6
0.8
1
1.2
Inside the integral, Γ =t
Fig. 5.13. Time dependent stress with concentration functions outside the Maxwellmemory integral, in the same shear flow used in Figure 5.10. Parameter values are thesame as for Figure 5.7.

139
of the plots, it seems Figures 5.10 and 5.12 are identical. However, let us understand if
they should be the same or not from the dynamics of the concentration functions and
our understanding of the function Pµ. Since the function c(t) for this flow is strictly
decreasing, for any s0 we have P cµ
(s0, t) = c(s0). Since it is constant it can be moved to
the outside of the integral, so that σc(s0) is the same whether we use P cµ
or simply use
c(s0) outside the integral.
The concentration of a-micelles is also decreasing except very early where it is
increasing. When a(t) has decreased past a(0), P aµ
(s0, t) = a(s0) is constant for the
stress computation and can be pulled out of the integral as well. Before it decreases past
a(0) though, using the concentration outside the integral will be overcounting, so it the
should report values larger than our model. Since the bundle population is increasing
for all time, placing b(s) outside the integral will again overcount and will give a larger
contribution to stress than σb(s) in our model using P bµ
(s, t). So we should expect that
placing the concentrations outside the integral is actually giving larger stresses than our
model. We have plotted the difference of the stresses for these two approaches in Figure
5.14. More precisely, Figure 5.14 is the logarithm (base 10) of the percentage that the
“outside the integral” approach is greater than the corrected prediction given by our
model.
The fact we could use a logarithmic scale means that all stress values obtained
from the “outside” approach are larger than those we obtain with our model. This
confirms our expectation that we overcount by placing the concentrations outside the
memory integral. We also note that the difference is as great as ∼ 11%! The error is
greatest for small times, which makes it more significant. The reason for this is that

140
0 2 4 6 8
-1
-0.5
0
0.5
1
1.5Log of the Percentage Difference, Γ =t
Fig. 5.14. The logarithm (base 10) of the difference of stress values between values fromFig. 5.10 and 5.12, as a percentage of the predicted values in Fig. 5.10 (obtained fromequation 5.13). As expected, the values from Fig. 5.12 are consistently greater thanthose obtained from our model.
in transient rheology tests on non-Newtonian fluids, there is sometimes a “stress over-
shoot” [30, 3], meaning stress peaks and then decreases to its steady value, much like
the population of a-micelles in Figure 5.7. However, in the case of wormlike micelles,
a second transient (“sigmoidal decay”) is sometimes seen in which stress decays more
slowly and does not begin at the high peak of a typical stress overshoot [30]. Our
model is predicting a smaller stress than when the concentrations are placed outside
the integrals, especially where stress overshoots occur, and could therefore be avoiding a
spurious report of a stress overshoot that a weighted Maxwell model would predict. The
inclusion of Pµ in the integral is allowing a second mechanism of stress relaxtion (due
to breaking), whereas a weighted Maxwell model can relax stress only through fading
memory.

141
5.3.2 Oscillatory shear flow
The next flow we consider is an oscillatory shear flow, a standard rheological
test [47]. For each of Figures 5.15-5.21 the parameters values are: µ = 0.01, k0 = 1,
k1 = 100, f0(γ) = 102|γ|, g(γ) = 10−6|γ|, f1(γ) = 10−2|γ|, f2 = 102|γ|, α = β = 2,
ηa = 102, ηb = 104, ηc = 10−2, and λa = λb = λc = 1. The shear rate for Figure 5.15
is γ(t) = 0.01 cos(t), and for Figure 5.16 we use γ(t) = 0.1 cos(t). For the remaining
Figures 5.17-5.21 the shear rate is γ(t) = cos(t).
The first three Figures 5.15-5.17 are predictions of our model for the time depen-
dent stress in shear flow with increasing amplitude of oscillation in the shear rate. It is
not surprising that the stress is oscillating since we are applying an oscillating flow. It is
very interesting that as we increase the strain - which is the amplitude of the shear rate
- the symmetry breaks. The Maxwell model predicts no such effect, as we see in Figure
5.18 for the same strain as used in Figure 5.17. So for this flow too our model is showing
considerable difference with the Maxwell model.
Whereas in the previous section, with flow γ(t) = t, our model predictions in
Figure 5.10 were similar to those obtained using the “outside the integral” model (Figure
5.12), for oscillatory shear the “outside the integral” approach predicts stress which
is extremely different from the other model predictions (Figure 5.19). However, the
“inside the integral” model predicts a stress, shown in Figure 5.20, which is apparently
very similar to our model predictions in Figure 5.17. We again provide a plot of the
difference between the values in these figures to reveal quantitatively the error of using
the actual concentration functions inside the integral. The values shown in Figure 5.21

142
0 5 10 15 20
-0.1
-0.05
0
0.05
0.1
Time dependent shear stress, Strain=0.01
Fig. 5.15. Model prediction (equation 5.13) for shear stress in oscillatory shear flowplotted against time. Here γ = 0.01 cos(t), and parameter values are: µ = 0.01, k0 = 1,k1 = 100, f0(γ) = 102|γ|, g(γ) = 10−6|γ|, f1(γ) = 10−2|γ|, f2 = 102|γ|, α = β = 2,ηa = 102, ηb = 104, ηc = 10−2, and λa = λb = λc = 1.
0 5 10 15 20
-0.04
-0.02
0
0.02
0.04
0.06Time dependent shear stress, Strain=0.1
Fig. 5.16. Prediction of our model (equation 5.13) in oscillatory shear with shear rateγ(t) = 0.1 cos(t). All parameter values are the same as in in Figure 5.15. With anincrease in the amplitude of shear rate, a slight asymmetry develops in the oscillatingstress.

143
0 5 10 15 20
-0.005
0
0.005
0.01Time dependent shear stress, Strain=1
Fig. 5.17. Prediction of our model (equation 5.13) in oscillatory shear flow with γ(t) =cos(t). Parameter values are the same as those in Figures 5.15 and 5.16. At this higheramplitude oscillatory shear rate the asymmetry is much more pronounced than in Figure5.16.
0 5 10 15 20
-75
-50
-25
0
25
50
75
100Time dependent Maxwell stress, Strain=1
Fig. 5.18. Maxwell model prediction for shear stress in oscillatory shear flow with shearrate γ(t) = cos(t). Values of parameters are identical to those used in Figures 5.15-5.17.The asymmetry in Figure 5.17 (which uses the same strain) is absent from the Maxwellprediction.

144
0 5 10 15 20
-0.04
-0.02
0
0.02
0.04
Outside time dependent stress, Strain=1
Fig. 5.19. Prediction for time dependent shear stress in oscillatory shear flow usingconcentration functions on the outside of the memory integral. Here γ(t) = cos(t) andall parameter values are the same as in Figures 5.15-5.18.
0 5 10 15 20
-0.005
0
0.005
0.01Inside time dependent stress, Strain=1
Fig. 5.20. Prediction for time dependent shear stress in oscillatory shear flow using theconcentration functions on the inside of the memory integral. Here γ(t) = cos(t) and allparameter values are the same as in Figures 5.15-5.19.
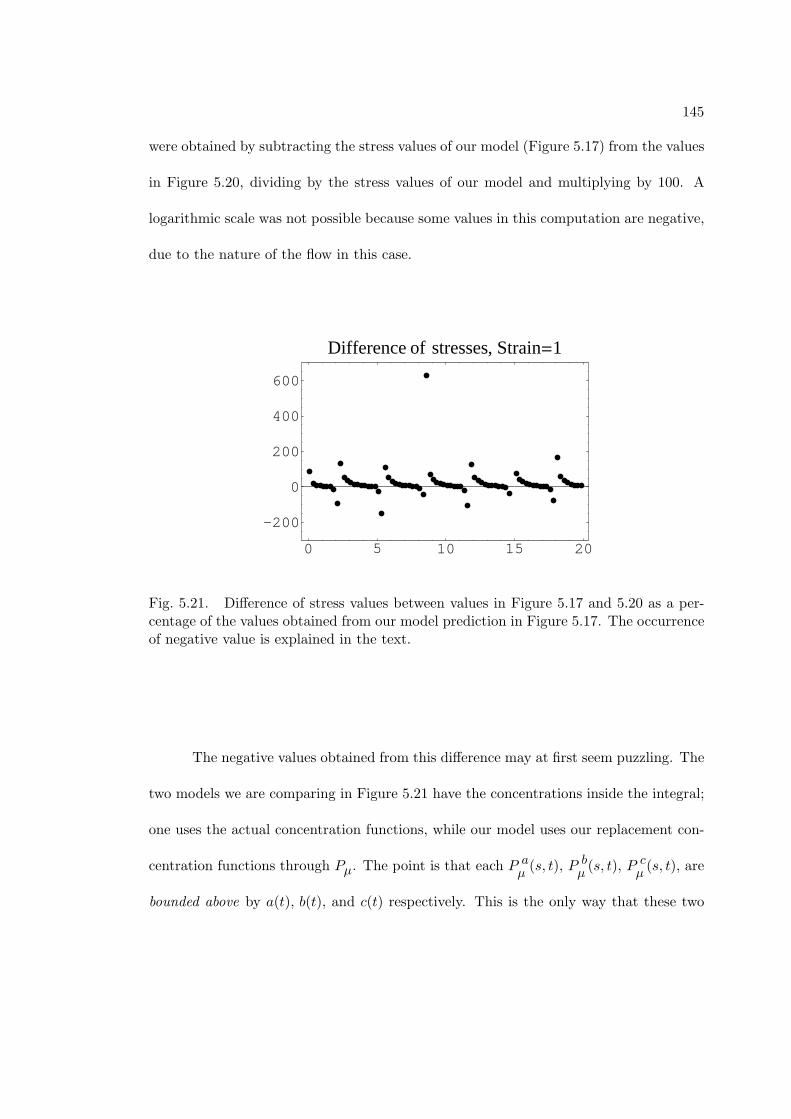
145
were obtained by subtracting the stress values of our model (Figure 5.17) from the values
in Figure 5.20, dividing by the stress values of our model and multiplying by 100. A
logarithmic scale was not possible because some values in this computation are negative,
due to the nature of the flow in this case.
0 5 10 15 20
-200
0
200
400
600
Difference of stresses, Strain=1
Fig. 5.21. Difference of stress values between values in Figure 5.17 and 5.20 as a per-centage of the values obtained from our model prediction in Figure 5.17. The occurrenceof negative value is explained in the text.
The negative values obtained from this difference may at first seem puzzling. The
two models we are comparing in Figure 5.21 have the concentrations inside the integral;
one uses the actual concentration functions, while our model uses our replacement con-
centration functions through Pµ. The point is that each P aµ
(s, t), P bµ
(s, t), P cµ
(s, t), are
bounded above by a(t), b(t), and c(t) respectively. This is the only way that these two

146
models differ, so how could using the functions Pµ yield a stress greater than we ob-
tain using the actual concentrations? The answer is in the flow we have chosen, namely
γ(t) = cos(t), which becomes negative periodically. In the computation of stress, the
memory integral includes the values of the flow for all times less than or equal to the
time at which stress is computed. It will therefore include the negative values of γ(t),
and since the actual concentration, for example a(t), is greater than or equal to its coun-
terpart P aµ
(s, t), we will obtain values of stress which are smaller in magnitude by using
the “inside the integral” model. So even when both models predict a positive stress,
our model can predict a smaller positive stress than that obtained by using the actual
concentration functions.
The error reported in Figure 5.21 is substantial, sometimes over 100% , and more
than 600% near t = 10. While the stress prediction from using the “inside” approach
is qualitatively similar to the predictions of our model, the values in Figure 5.21 give a
clear indication that if we simply place the concentrations inside the Maxwell memory
integral we will obtain false values for stress when using a population model for wormlike
micellar fluids.
5.3.3 Thixotropic loop: Linearly ramping up and down
The flow we consider here is one used in a very interesting rheological test, called
a thixotropic loop [83]. The viscoelasticity in complex fluids is often used synonymously
with “memory”, meaning the fluids remember that they were recently stressed or ex-
perienced a strain. A thixotropic loop study investigates the effect of this macroscopic

147
memory in simple shear flow in an important way. While Newtonian fluids have a con-
stant viscosity, non-Newtonian fluids can have a viscosity which changes, for example
with shear rate in simple shear flow, as in Figures 4.1-4.3 in Chapter 4. It is commonly
said that a non-Newtonian fluid has a viscosity which depends on shear rate, but even
more is true. There is no single viscosity value at a given shear rate, but rather it can
depend on the prior history of deformation. This is known as hysteresis and can occur
in non-Newtonian fluids [83].
In this section we use a simple shear flow with a first increasing then decreasing
shear rate: γ(t) = t if t ≤ 5, and γ(t) = 5 − t if 5 ≤ t ≤ 10. When the shear rate is
decreasing from γ = 5 to γ = 0, we can compare to the values of stress when the shear
rate was increasing from γ = 0 to γ = 5. In this way we obtain a prediction of the
hysteresis in reported values of stress. This test is an example of a “thixotropic loop”, so
called because it is typical to see higher stress at a given shear rate when γ is decreasing
than when it was increasing.
The parameter values used to produce Figures 5.22-5.26 are: µ = 0.01, k0 = 102,
k1 = 103, f0(γ) = 0, g(γ) = 10−3γ, f1(γ) = 104γ, f2 = 106γ, α = β = 2, ηa = 103,
ηb = 10, ηc = 10−3, and λa = λb = λc = 1. In Figure 5.22 the squares denote stress
values at a given shear rate when γ is (linearly) increasing. Upon reaching a shear rate of
γ = 5, the rate decreases linearly back to γ = 0, with stress values given by the triangles
in Figure 5.22. The fact that the stresses at a given shear rate do not coincide is a
prediction of history dependence. The shape of the curve in Figure 5.22 makes physical
sense. By increasing the flow rate, the fluid becomes more stressed, and when the shear
rate begins to decrease, the built up stress needs time to dissipate. The values of stress

148
0 1 2 3 4 50
0.01
0.02
0.03
0.04Thixotropic loop
Fig. 5.22. Time dependent shear stress prediction of our model equation 5.13 in athixotropic loop. Parameter values used are: µ = 0.01, k0 = 102, k1 = 103, f0(γ) = 0,g(γ) = 10−3γ, f1(γ) = 104γ, f2 = 106γ, α = β = 2, ηa = 103, ηb = 10, ηc = 10−3, andλa = λb = λc = 1. Stress values obtained while γ is increasing are given as squares, thetriangles denote stress values when the shear rate is decreasing.
0 1 2 3 4 50
50
100
150
200
250
300Maxwell thixotropic loop
Fig. 5.23. Maxwell model prediction for shear stress in a thixotropic loop using thesame time dependent shear rate as in Figure 5.22. All paramter values used to obtainthe values in this plot are the same as in Figure 5.22. The Maxwell model predicts amuch greater shear thickening effect than our model (equation 5.13) shown in Figure5.22. Stress values obtained while γ is increasing are given as squares, the trianglesdenote stress values when the shear rate is decreasing.

149
0 1 2 3 4 50
0.01
0.02
0.03
0.04Outside the integral thixotropic loop
Fig. 5.24. Prediction of shear stress in a thixotropic loop using the concentrationfunctions outside the memeory integral. The shear rate and all parameter values usedare the same as those used to produce the values in Figure 5.22 and 5.23. Stress valuesobtained while γ is increasing are given as squares, the triangles denote stress valueswhen the shear rate is decreasing.
0 1 2 3 4 50
0.02
0.04
0.06
0.08
0.1Inside the integral thixotropic loop
Fig. 5.25. Predicted shear stress values in a thixotropic loop study. The shear rate andall parameter values used are the same as those used to produce the values in Figure5.22-5.24.Stress values obtained while γ is increasing are given as squares, the trianglesdenote stress values when the shear rate is decreasing.

150
on the “ramp up” in shear rate would naturally be lower than on the “ramp down”
because the fluid begin with no initial stress. On the “ramp down”, however, there is an
initial pre-stress, which causes the higher values.
While the values of stress at γ(10) = 0 is non-zero, it is still much lower than the
predicted value of stress from the Maxwell model at this point, shown in Figure 5.23.
The Maxwell model predicts an increasing stress for all time in this case, even though the
flow rate is decreasing down to zero. The other models, “outside” and “inside”, predict
stress values shown in Figures 5.24 and 5.25 respectively. The “inside the integral” model
predicts a more modest history dependence than the Maxwell model at low shear rates.
However, placing the concentration functions on the ouside of the memory integral seems
more reasonable, and resembles the prediction of our model in Figure 5.22. The exact
difference between Figures 5.22 and 5.24 is given in Figure 5.26.
0 1 2 3 4 5
-0.5
0
0.5
1
1.5
2Log of the percentage difference
Fig. 5.26. Logarithm (base 10) of the precent difference of stress values obtained fromFigure 5.24 and 5.22, plotted against shear rate. The squares againdenote stress valuesobtained using shear rate values when the rate is increasing, while triangles are obtainedby using shear rates when γ(t) is decreasing - consistent with thier use in Figures 5.22-5.25.

151
Shown in Figure 5.26 is the logarithm (base 10) of the percentage that the “out-
side” approach is greater than the values obtained by our model. Our use of the logarithm
means that the “outside” stress predictions are always greater than those given by our
model using the modified concentration functions. The difference in this case is greatest
at the lowest shear rates. The error here again is not at all negligible; it is more than
10% at small shear rates.
5.3.4 Concluding remarks on time dependent stress predictions
We began to build our model with the idea of modifying the Maxwell model
with small changes but sufficient to accomodate the physics of wormlike micelles. The
results of the three time dependent simple shear flows we have considered show a definite
difference between our model and the Maxwell model. The model we have put forth
in equation 5.13 and the ODE’s 4.20-4.22, corrects the Maxwell model in the case of
wormlike micelles, by adjusting the memory kernel to forget “broken micelles”. It is
physically inaccurate to place the concentrations inside or outside the memory integral,
and we have solved this problem by defining the functions Pµ. By including the ability
to exchange mass among species, and the effect this has on memory, into the Maxwell
model, we have produced a new model, for both steady and time dependent shear flow.
Summarizing the results for the different flow types, we have found that for both
the linear ramp and the thixotropic loop tests, the “outside the integral” model had
predictions closest to our model with Pµ. For the oscillatory shear flow, however, it
was the “inside the integral” model which came closest. Placing the concentrations
on the outisde can account for breaking events if the concentration is monotonically

152
decreasing. For monotincally increasing concentrations, our function Pµ reduces to the
actual concentration. This suggests that in both the linear ramp and thixotropic loop,
the breaking events occurred on a timescale comparable to the relaxation time, so that
our model could be approximated by having the concentrations outside the integral. For
oscillatory shear, the populations must be oscillating, so that there are both combing
and breaking events. With the concentrations inside the integral, we are overcounting
as shown in Figure 5.21, but evidently the frequent breaking events must be diminishing
this effect, so that the prediction is qualitatively similar to our model.
Although the constitutive equation we have written (equation 5.13) is meant only
for the shear component of stress, we can propose the same model for the full stress
tensor as well. Allowing σ to represent the full stress tensor, the constitutive equation
would still be given by equation 5.13, with the understanding that to compute the ijth
component of σ one must use the ijth component of the rate of strain tensor D.
5.4 Memory integrals and the Fredholm Alternative
In this final section we present material and results which are somewhat specu-
lative. We attempt to exploit an interesting application of ideas in functional analysis
to our model equation 5.13 coupled to the ODE’s 4.20-4.22. After first describing the
problem to be solved, we present the mathematical tools that might be capable of find-
ing a solution. We then give some inital results we have obtained toward this end. The
problems with this approach are also addressed, and we suggest methods in which they
might be handled.

153
5.4.1 The idea of an inverse constitutive equation
While constitutive equations are often thought of as a way to solve determine the
stress in a particular flow field, in this section we consider equation 5.13 from another
point of view. In the following sections we consider the inverse problem, i.e. given a
stress σ, is there a flow u with gradient D which satisfies our model given by equation
5.13 with the given σ.
The motivation for this question comes mainly from interesting rheology of worm-
like micellar fluids. During simple shear flow, wormlike micellar fluids can develop in-
homogeneities and structures larger than wormlike micelles [17, 40, 85, 86]. During a
rheology experiment, these new structures can cause the fluid to “band” [86] into two
seperate fluids moving at different speeds within the chamber holding the fluid sample.
Structures forming in regular patterns have also been observed, which move through
the fluid during experiments [40]. Such phenomena change the velocity gradients in the
fluid, turning an intial homogeneous, viscometric flow such as simple shear flow, into
something more complicated which is no longer pure shear.
The question we ask is whether for a given stress σ(t), there exists a solution
to equation 5.13, i.e. a flow D, which is smooth for only a finite amount of time, and
eventually develops singularities. More ambitious projects would include a classification
of those stresses σ for which C∞ solutions exist, those for which there are no smooth or
even continuous solutions.
We do not answer these questions here, rather we show how useful tools from
functional analysis might yield answers. We next include a review of theorems and

154
definitions if only to establish notation and nomenclature. We then present the results
we have obtained that show our model in equation 5.13 enjoys some key properties that
make it amenable to some of the powerful tools of Fredholm Theory.
5.4.2 Integral equations and Fredholm theory
Let K be a linear operator acting on a Hilbert space H, K : H → H. Recall that
the norm of K is defined as ‖K ‖= inf‖f‖=1 ‖Kf ‖, in which the norms of f and Kf use
the norm on H. In other words, since K is bounded, ‖Kf ‖≤ M ‖ f ‖ for each f ∈ H,
and then ‖K ‖ is the infimum of all such M . An operator is compact if it maps bounded
sets to sets which are sequentially compact. A set is sequentially compact if any sequence
of points, which all belong to the set, has a convergent subsequence. A standard text
containing definitions and standard proofs is Keener [87].
The operators we will be interested in are integral operators acting on functions
in the Hilbert space L2[0, T ], where a ∈ R is a (large) constant. If u ∈ L2[0, T ], then for
an integral operator on L2[0, T ] of the form:
Ku (x) =∫ T
0k(x, y)u(y)dy, (5.14)
is said to have kernel k(x, y). Given a kernel k(x, y), there is an associated integral
operator on L2[0, T ] is simply given by equation 5.14.
We would now like to construct an integral operator using a kernel generated by
P (s, t), dropping the superscript which denotes one of the concentration functions a(t),
b(t), or c(t). For the present discussion, we limit ourselves to any single concentration

155
function, and consider the stress due to that species alone. In the end, we will simply
add the stresses according to our law of partial stresses from section 4.4. We begin by
defining the function H(x, y) by:
H(x, y) =
0 x < y
1 x ≥ y
The kernel of our integral operator is then k(x, y) = H(x, y)P (x, y) e(x−y)/λ, and
the associated integral operator L is:
Lf(x) =∫ T
0k(x, y) f(y) dy =
∫ x
0P (x, y) e(x−y)/λ f(y) dy. (5.15)
We first of all need to check that L is well defined on L2[0, T ].
PROPOSITION 5.4.1. The operator L defined in equation 5.15 is a well defined op-
erator L : L2[0, T ] → L2[0, T ]. Furthermore, L is linear and bounded.
Proof: The proof is simple and straightforward. We first check that if ‖ f ‖< ∞, then
‖Lf ‖< ∞ too. We can as well assume that ‖f ‖= 1. Note that P (x, y) is always bounded
by the concentration function it replaces, which is bounded by the total mass in the
system, so by the non-dimensional form of equation 4.19, we have P (x, y) ≤ MV . The
exponential factor in equation 5.15 is also bounded by 1, i.e., for a fixed x, e(x−y)/λ < 1

156
for any y ∈ [0, x]. Using these bounds we find for norm of Lf :
‖Lf(x)‖ =(∫ T
0(Lf(x))2dx
)1/2
=
(∫ T
0
(∫ x
0P (x, y)e(x−y)/λf(y)dy
)2dx
)1/2
≤ MV
(∫ T
0
(∫ x
0f(y)dy
)2dx
)1/2
≤ MV
(∫ T
0
(∫ x
0|f(y)|dy
)2dx
)1/2
≤ MV
(∫ T
0
(∫ T
0|f(y)|dy
)2dx
)1/2.
Using the inner product on L2[0, T ], and the fact that the function 1 which takes
the constant value 1 ∈ R is in L2[0, T ], we can write < f, 1 >=∫ T0 |f(y)|dy. So by
Cauchy-Schwarz we get
(∫ T
0|f(y)|dy
)2= < |f |, 1 >2 ≤ ‖f ‖2‖1‖2
=(∫ T
0|f(y)|2dy
) (∫ T
012dy
)
=(∫ T
0|f(y)|2dy
)T.
We therefore get
(∫ T
0
(∫ T
0|f(y)|dy
)2dx
)1/2≤
(∫ T
0
(∫ T
0|f(y)|2dy
)(T ) dx
)1/2
=(∫ T
0|f(y)|2dy
)1/2 (∫ T
0T dx
)1/2= T ‖f ‖,

157
and hence ‖Lf ‖≤ MV T ‖f ‖. Since ‖f ‖< ∞, we have also that ‖Lf ‖ is finite. Hence
L is an operator on L2[0, T ]. This also shows that L is a bounded operator. Finally, from
the definition of L in equation 5.15 we can tell that L is clearly linear. This concludes
the proof of proposition 5.4.1.
A kernel k(x, y) is a Hilbert-Schmidt kernel if it satisfies
∫ b
a
∫ b
ak2(x, y) dx dy < ∞.
In this case, the associated integral operator is called Hilbert-Schmidt as well. Integral
operators which are Hilbert-Schmidt are particularly nice because they are compact [87].
PROPOSITION 5.4.2. The integral operator L defined by equation 5.15 is a Hilbert-
Schmidt operator.
Proof: First observe that H(x, y) P (x, y) = 0 whenever y > x. So
Lf(x) =∫ x
0H(x, y) P (x, y)e(x−y)/λ dx dy =
∫ T
0H(x, y)P (x, y)e(x−y)/λ dx dy.
The kernel k(x, y) = H(x, y)P (x, y)e(x−y)/λ satisfies
∫ T
0
∫ T
0H(x, y) P 2(x, y)e2(x−y)/λ dx dy ≤
∫ T
0
∫ T
0(1)2e2(x−y)/λ dx dy
=λ2
4(e2T/λ − 1)(1− e−2T/λ) < ∞,
which is the defining condition for the associated operator to be Hilbert-Schmidt, so the
proposition is verified. In particular this says that the kernel k(x, y) is itself an element

158
of L2([0, T ] × [0, T ]). While our operator is compact, which allows us to apply some
key theorems from functional analysis, we note that it is not self-adjoint (unless it is
identically 0). This is easy to see since our kernel has the property that k(x, y) = 0
whenever y > x. The proofs of these theorems are straightforward and can be found in
most textbooks on the subject, for example [87, 88].
The Fredholm Alternative: If K is a bounded linear operator on a Hilbert
space H such that the range of K is closed, then the equation Kf = g has a solution g
if and only if < g, v >= 0 for every v ∈ ker(K∗).
The theorem of main interest to us is: If L is a compact linear operator, then
(I + νL)u = f has a solution u if and only if f is orthogonal to ker(I + νL)∗, in which ν
is a scalar. It is not hard to prove that if L is compact, then (I + νL) has closed range,
for instance see [87].
The connection to our model 5.13 is given by fixing σ, the stress in the fluid, and
then looking for solutions D, the rate of strain, which satisfy (I − νL)D = σ. The left
hand side is slightly more complicated than our memory integral in equation 5.13, we
write this out fully so that we may comment on it more easily:
D(s) + ν
∫ T
0H(s, t)P (s, t)e(s−t)/λD(t)dt = σ(s). (5.16)
This equation would be our model if not for the first term D and the constant
“ν”. We can interpret the first term D as a Newtonian contribution to the stress, since

159
the Newtonian stress is simply 2ηD. In the second term, which constitutes the non-
Newtonian stress contribution, we need to set ν = 1/2λD. Then σ would not represent
stress σ, but rather σ = σ/2λD.
So fixing a stress σ, the theorem can tell us when a flow D exists, which causes that
stress. This presents an interesting opportunity for identifying “Fredholm instabilities”
in fluid flow. For a given σ, if the Fredholm Alternative provides a solution in L2[0, T ]
which is not smooth. Such a solution would correspond to a non-viscometric flow. By
workling in L2[0, T ], we can find non-smooth solutions, or solutions which are smooth
for only a portion of time. Note that if no solution is found for a given σ, it does not
mean that a solution in L2[0, T ] does not exist, but only that it can be found with the
Fredholm technique. Approximate solutions can in fact be obtained by such methods as
Neumann
There is a problem with this approach, however. The function P (s, t) itself de-
pends on D. In the theorems it is implicit that the kernel itself is known, whereas we
must solve for it and σ simultaneously. Nonetheless, the framework of the Fredholm
theorem, we feel is powerful enough to merit the pursuit of its use in this context. To
that end, we offer some thoughts on how one might first determine the kernel before
seeking solutions to equation 5.16.
The concentration functions can be assumed to be of a certain form, say decreasing
or increasing, with a given functional form. Such a choice could be made based on
rheological results, which suggest the dominance of one species over another. Another
possibility is to seek solutions of equation 5.16 without giving the kernel an explicit
functional form. If it could be determined under what conditions on the kernel a solution

160
would or would not exist, this itself would be a useful result. Our view is that the
theory of integral equations should at least be considered in obtaining information on
the predictions of any integral model. We have started along that path and shown not
only how to connect the Fredholm theorem to our constitutive equation 5.13, but that
our replacement function P (s, t) has the necessary properties to implement the theory.

161
Chapter 6
Directions for future research
Both experimental observations on wormlike micellar fluids and a mathematical
description of the fluid flow have been considered. Experimentally we have observed
shear thickening in dilute solutions of equimolar CPCl/NaSal. In the semi-dilute regime,
there is a again a thickening range of shear rates, but at lower shear rates in the semi-
dilute range, the fluids shear thin. It is well established that the thickening in dilute
solutions is due to the formation of SIS, though the mechanism of this formation is
not understood. Studying the thickening process for dilute and semi-dilute fluids may
give insight to this process. The fluids that shear thin probably consist of longer, more
flexible micelles than the dilute fluids, so if it could be determined wether SIS form in
the thickening range of shear rates, such factors as inception time for growth, maximum
viscosity reached and magnitude of fluctuations could be correlated with micellar length.
This could prove very helpful in understanding the processes leading to SIS formation.
The dependence of thickening and SIS formation on micellar length could also be studied
through temperature.
We have presented data suggesting that the micellar scission reactions in equimo-
lar solutions of CPCl/NaSal are in the “slow-breaking” regime. A natural question to
ask is whether SIS form in the “fast-breaking” limit, and if so how they compare to those
formed in equimolar CPCl/NaSal. We have also suggested that SIS play a role in the

162
oscillations of a rising bubble in equimolar CPCl/NaSal, and we saw that bubbles do not
oscillate in the fluids we tested which are in the “fast-breaking” limit. The dependence
of SIS formation on the micellar scission rates would also provide information about the
oscillatory instability in rising air bubbles.
The formation of SIS or “bundles” is central to the experiments in rheology and
rising bubbles, but it also plays a role in our model. The prediction of shear thickening
was made by the 2-species (Figure 4.6) model which excluded bundle formation, but it
failed to predict a subsequent thinning. With the inclusion of the bundle population
b(t), the model was able to capture shear thickening followed by shear thining (Figure
4.10). What we would like to see in our model, is a predicted rheology which shows a
zero shear plateau, shear thinning, a modest shear thickening followed by thining again,
as for 7 mM CPCl/NaSal in Figure 2.7. A better understanding of SIS formation could
lead to such a prediction for the model. The model parameters in the ODE’s 4.20-4.22
could then be chosen to represent what experiments suggest are responsible for observed
rheology.
The integral equation 5.13 offers a variety of directions for future research. The
application of Fredholm theory could be explored to answer fundamental questions on
the predictions of the model. Assuming functional forms for the concentration functions
to determine the replacement functions P aµ
, P bµ
, and P cµ
, one can seek solutions D to
prescribed stresses σ(t). As we mentioned in Chapter 5, it would be interesting to see
solutions which have singularities. It may be possible to find approximate solutions
through the use of Neumann iterates.

163
Coupling the model we have written (equation 5.13 with the ODE’s 4.20-4.22)
with the equation for conservation of momentum, would give an equation of motion
for the fluid. Then through numerical simulations we could see if our model predicts
oscillations in velocity for flow past a rising bubble or solid sphere. A first step in that
direction would be through the ODE’s 4.20-4.22 themselves. It seems likely that the
populations would need to be time dependent for all time if oscillations in velocity are
to be seen. This is because once the concentration functions reach a steady state, the
replacement functions Pµ also reach a steady state, and then as we go further into future
times our model would become more like a weighted Maxwell model.
So a first step in modelling should be an analysis of the types of solutions we
can obtain through our reaction ODE’s 4.20-4.22. It may be that certain reaction rates
need to be changed to see periodic solutions. Other physical ideas for the reaction rates
could change them from functions of flow rate γ to functions of stress. We also suggest
investigating the properties of the ODE’s using reaction rates which depend on stress
gradients.
The model we have developed is physically accurate and novel in the way it
accounts for the effect of micellar kinetics on memory. The ODE’s represent these kinetic
reactions, and they offer a means of investigating what quantities control these reactions.
Investigating their behavior through the functional forms of the reaction rates, and
the subsequent behavior of the reaplcement functions Pµ, promises a very interesting
approach to the study of wormlike micellar fluid dynamics.

164
References
[1] L. Pauling, General Chemistry, 3rd edition, (W. H. Freeman, San Francisco, 1970).
[2] J. Israelachvili, Intermolecular and Surface Forces, 2nd edition, (Academic Press,
New York, 1991).
[3] R. G. Larson, The Structure and Rheology of Complex Fluids
[4] Micelles, Membranes, Microemulsions, and Monolayers, W. Gelbart, et al., editors,
(Springer, New York, 1994).
[5] A. Ben-Shaul & W. Gelbart, in [4].
[6] H. Rehage & H. Hoffmann, Rheological Properties of Viscoelastic Surfactant Sys-
tems. J. Phys. Chem. 92, 4712 (1988).
[7] Z. Lin, J. J. Cai, L. E. Scriven, & H. T. Davis, Spherical to Wormlike Micelle
Transition in CTAB Solutions, J. Phys. Chem 98, 5984 (1994).
[8] I. A. Kadoma, C. Ylitalo, & J. W. van Egmond, Structural Tranisitions in Wormlike
Micelles, Rheol. Acta 36, 1 (1997).
[9] M. E. Cates & S. Candau, Statics and Dynamics of Worm-Like Surfactant Micelles.
J. Phys.: Condens. Matter 2, 6869 (1990).
[10] J. Appell, G. Porte, A. Khatory, F. Kern, & S. Candau, Static and Dynamic Prop-
erties of a Network of Wormlike Surfactant Micelles (CetylPyridinium Chlorate in
Sodium-Chlorate Brine). J. de Physique II 2, 1045 (1992).

165
[11] H. Rehage & H. Hoffmann, Viscoelastic Surfactant Solutions - Model Systems for
Rheological Research. Molecular Physics 74, 933 (1991).
[12] V. Hartmann & R. Cressely, Shear Thickening of an Aqueous Micellar Solution of
Cetyltrimethylammonium Bromide and Sodium Tosylate, J. Phys. II France 7, 1087
(1997).
[13] V. Degiorgio & M. Corti, in Physics of Amphiphiles: Micelles, Vesicles, and Mi-
croemulsions, (Elsevier, New York, 1985).
[14] T. Shikata, H. Hirata, E. Takatori, & K Osaki, Nonlinear Viscoelastic Behavior of
Aqueous Detergent Solutions. J. Non-Newtonian Fluid Mech. 28, 171 (1988).
[15] C. Grand, J. Arrault, & M. E. Cates, Slow Transients and Metastability in Wormlike
Micellar Rheology. J. de Physique II 7, 1071 (1997).
[16] S. Lerouge, J-P. Decruppe, J-F. Berret, Correlations Between Rheological and Op-
tical Properties of a Micellar Solution Under Shear Banding Flow. Langmuir 16,
6464 (2000).
[17] C. Liu & D. Pine, Shear-Induced Gelation and Fracture in Micellar Solutions. Phys.
Rev. Lett. 77, 2121 (1996).
[18] L. Smolka & A. Belmonte, Drop Pinch-Off and Filament Dynamics of Wormlike
Micellar Fluids. J. Non-Newtonian Fluid Mech. 115, 1 (2003).
[19] A. Belmonte, Self-Oscillations of a Cusped Bubble Rising Through a Micellar Solu-
tion. Rheol. Acta 39, 554 (2000).

166
[20] A. Jayaraman & A. Belmonte, Oscillations of a Solid Sphere Falling Through a
Wormlike Micellar Fluid. Phys. Rev. E 67, 65301 (2003).
[21] J. Rothstein, Transient Extensional Rheology of Wormlike Micellar Solutions. J.
Rheol. 47, 1227 (2003).
[22] Y. Liu, T. Liao, & D. Joseph, A Two-Dimensional Cusp at the Trailing Edge of an
Air Bubble Rising in a Viscoelastic Liquid. J. Fluid Mech. 304, 321 (1995).
[23] A. Mollinger, E. Cornelissen, & B. van den Brule, An Unexpected Phenomenon
Observed in Particle Settling: Oscillating Falling Spheres. J. Non-Newtonian Fluid
Mech. 86, 389 (1999).
[24] P. Weidman, B. Roberts, & S. Eisen, Progress in Nonlinear Science, Proceedings
III, V.D. Shalfeev ed., (Nizhny Novgorod, 2002), 103.
[25] M. In, G. Warr, & R. Zana, Dynamics of Branched Threadlike Micelles. Phys. Rev.
Lett. 83, 2278 (1999).
[26] V. Schmitt, F. Lequeux, A. Pousse, & D. Roux, Flow Behavior and Shear Induced
Transition Near an Isotropic-Nematic Transition in Equilibrium Polymers. Langmuir
10, 955 (1994).
[27] R. Hartunian & W. Sears, J. Fluid Mech. 3, 27 (1957).
[28] D. Rodrigue, D. De Kee, & C. Chan Man Fong, An Experimental Study of the
Effect of Surfactants on the Free Rise Velocity of Gas Bubbles. J. Non-Newtonian
Fluid Mech. 66, 213 (1996).

167
[29] S. Candau, A. Khatory, F. Lequeux, & F. Kern, Rheological Behavior of Wormlike
Micelles - Effect of Salt Content. J. de Physique IV 3, 197 (1993).
[30] J.-F. Berret, Transient Rheology of Wormlike Micelles. Langmuir 13, 2227 (1997).
[31] T. Drye & M. E. Cates, Living Networks - The Role of Cross-Links in Entangled
Surfactant Solutions. J. Chem. Phys. 96, 1367 (1992).
[32] P. Pincus, Phase Tranisitions - Y’s and Ends. Science 290, 1307 (2000).
[33] F. Lequeux & S. Candau, in [46].
[34] G. Fuller, Optical Rheometry. Ann. Rev. Fluid Mech. 22, 387 (1990).
[35] Y. Hu, S. Wang, & A. Jamieson, Rheological and Flow Birefringence Studies of a
Shear Thickening Complex Fluid - a Surfactant Model System. J. Rheol. 37, 531
(1993).
[36] S. Chen & J. P. Rothstein, Flow of a Wormlike Micellar System Past a Falling
Sphere. J. Non-Newtonian Fluid Mech. 116, 205 (2004).
[37] S. Kwon & M. Kim, Topological Transition in Aqueous Micellar Solutions. Phys.
Rev. Lett. 89, 258302 (2002).
[38] P. Boltenhagen, Y. Hu, E. Matthys, & D. Pine, Inhomogenous Structure Formation
and Shear Thickening in Worm-Like Micellar Solutions. Europhys. Lett. 38, 389
(1997).

168
[39] S. Keller, P. Boltenhagen, D. Pine, & J. Zasadzinski, Direct Observation of Shear
Induced Structures in Wormlike Micellar Solutions by Freeze Fracture Electron Mi-
croscopy. Phys. Rev. Lett. 80, 2725 (1998).
[40] E. Wheeler, P. Fischer, & G. Fuller, Time-Periodic Flow Induced Structures and
Instabilites in a Viscoelastic Surfactant Solution. J. Non-Newtonian Fluid Mech.
75, 193 (1998).
[41] P. Fischer, Time Dependent Flow in Equimolar Micellar Solutions: Transient Be-
haviour of the Shear Stress and First Normal Stress Difference in Shear Induced
Structures Coupled with Flow Instabilites. Rheol. Acta 39, 234 (2000).
[42] J.-F. Berret, J. Appell, & G. Porte, Linear Rheology of Entangled Wormlike Mi-
celles. Langmuir 9, 2851 (1993).
[43] J.-F. Berret, D. C. Roux, & G. Porte, Isotropic-to-Nematic Transition in Wormlike
Micelles Under Shear. J. de Physique II 4, 1261 (1994).
[44] R. Granek & M. E. Cates, Stress Relaxation in Living Polymers-Results from a
Poisson Renewal Model. J. Chem. Phys. 96, 4758 (1992).
[45] A. Khatory, F. Lequeux, F. Kern, & S. J. Candau, Linear and Nonlinear Viscoelas-
ticity of Semidilute Solutions of Wormilike Micelles at High Salt Content. Langmuir
9, 1456 (1993).
[46] Theoretical Challenges in the Dynamics of Complex Fluids, T. McLeish, ed.,
(Kluwer, Boston, 1997)

169
[47] R. B. Bird, R. C. Armstrong, & O. Hassager, Dynamics of Polymeric Liquids, 2nd
ed. (Wiley, New York, 1987), vol. 1.
[48] James Clerk Maxwell, On the Dynamical Theory of Gasses. Phil. Trans. Roy. Soc.
A157, 49 (1867).
[49] H. Rehage & H. Hoffmann, Viscoelastic Detergent Solutions. Faraday Discuss.
Chem. Soc. 76, 363 (1983).
[50] M. E. Cates, Reptation of Living Polymers - Dynamics of Entangled Polymers in
the Presence of Reversible Chain-Scission. Macromolecules 20, 2289 (1987).
[51] M. E. Cates, Nonlinear Viscoelasticity of Wormlike Micelles. J. Phys. Chem. 94,
371 (1990).
[52] F. Lequeux, Linear Response of Self Assembling Systems - Mean Field Solution. J.
de Physique II 1, 195 (1991).
[53] O. Manero, F. Bautista, J. F. A. Soltero, & J. E. Puig, Dynamics of Wormlike
Micelles: The Cox-Merz Rule. J. Non-Newtonian Fluid Mech. 106, 1 (2002).
[54] E. Cappelaere, J.-F. Berret, J.-P. Decruppe, R. Cressely, & P. Lindner, Rheology,
Birefringence, and Small Angle Neutron Scattering in a Charged Micellar System:
Evidence of a Shear Induced Phase Transition. Phys. Rev. E 56, 1869 (1997).
[55] V. Schmitt, C. M. Marques, & F. Lequeux, Shear - Induced Phase-Separation of
Complex Fluids-The Role of Flow Concentration Coupling. Phys. Rev. E 52, 4009
(1995).

170
[56] B. H. A. A. van den Brule & P. J. Hoogerbrugge, Brownian Dynamics Simulation
of Reversible Polymeric Networks. J. Non-Netonian Fluid Mech. 60, 303 (1995).
[57] J. Intae, Existence of Gelling Solutions for Coagulation-Fragmentation Equations.
Comm. Math. Phys. 194, 541 (1998).
[58] A. Vaccaro & G. Marrucci, A Model for the Nonlinear Rheology of Associating
Polymers. J. Non-Newtonian Fluid Mech. 92, 261 (2000).
[59] J. L. Goveas & P. D. Olmsted, A Minimal Model for Vorticity and Gradient Banding
in Complex Fluids. Eur. Phys. J. E 6, 79 (2001).
[60] R. Bruinsma, W. Gelbart, & A. Ben-Shaul, Flow-Induced Gelation of Living (Mi-
cellar) Polymers. J. Chem. Phys. 96, 15 (1992).
[61] H. Paine, Koll. Z 11, 2115 (1912).
[62] M. von Smoluchowski, Z. Phys. Chem. 112, 1 (1916).
[63] M. Doi & S. F. Edwards, The Theory of Polymer Dynamics, (Oxford University
Press, New York, 1986).
[64] M. S. Turner & M. E. Cates, The Relaxation Spectrum of Polymer Length Distri-
butions. J. Phys. France 51, 307 (1990).
[65] C. M. Marques & M. E. Cates, Nonlinear Thermodynamic Relaxation in Living
Polymer Systems. J. Phys. II 1, 489 (1991).
[66] M. E. Cates & M. S. Turner, Flow-Induced Gelation of Rodlike Micelles. Europhys.
Lett. 11, 681 (1990).

171
[67] F. Bautista, J. F. A. Soltero, J. H. Perez-Lopez, J. E. Puig, & O. Manero, On
the Shear Banding Flow of Elongated Micellar Solutions. J. Non-Newtonian Fluid
Mech. 94, 57 (2000).
[68] J. D. Ferry, Viscoelastic Properties of Polymers, (Wiley, New York, 1980).
[69] J. F. A. Soltero, J. E. Puig, O. Manero, Rheology of the CetyltrimethylAmmonium
Tosilate-Water System.2. Linear Viscoelastic Regime. Langmuir 12, 2654 (1996).
[70] J. F. A. Soltero, F. Bautista, J. E. Puig, O. Manero, Rheology of CetyltrimethylAm-
monium p-Toluenesulfonate-Water System.3. Nonlinear Viscoelasticity. Langmuir
15, 1604 (1999).
[71] D. Acierno, F. P. La Mantia, G. Marucci, G. Titomanlio, J. Non-Newtonian Fluid
Mech. 1, 125 (1976).
[72] H. R. Warner Jr., IE & C Fundam. 11, 379 (1972).
[73] H. A. Stone, Dynamics of Drop Deformation and Breakup in Viscous Fluids. Annual
Review of Fluid Mechanics 26, 65 (1994).
[74] A. Prosperetti, Bubbles. Phys. Fluids 16, 1852 (2004).
[75] O. Hassager, Nature 279, 402 (1979).
[76] N. Z. Handzy & A. Belmonte, Oscillatory Rise of Bubbles in Wormlike Micellar
Fluids with Different Microstructures. Phys. Rev. Lett. 92, 124501 (2204).
[77] M. Renardy, Mathematical Analysis of Viscoelastic Flows, (Society for Industrial
and Applied Mathematics, Philadelphia, 2000).

172
[78] G. K. Batchelor, An Introduction to Fluid Dynamics, (Cambridge University Press,
Cambridge, 1967).
[79] M. D. Chilcott & J. M. Rallison, Creeping Flow of Dilute Polymer Solutions Past
Cylinders and Spheres. J. Non-Newtonian Fluid Mech. 29, 381 (1988).
[80] L. E. Wedgewood, D. N. Ostrov, & R. B. Bird, A Finitely Extensible Bead-Spring
Chain Model for Dilute Polymer Solutions. J. Non-Newtonian Fluid Mech. 40, 119
(1991).
[81] N. A. Spenley, M. E. Cates, & T. C. B. McLeish, Nonlinear Rheology of Wormlike
Micelles. Phys. Rev. Lett. 71, 939 (1993).
[82] F. Reif, Fundamentals of Statistical and Thermal Physics, (McGraw-Hill, New York,
1965).
[83] R. I. Tanner, Engineering Rheology, (Oxford University Press, New York, 1985).
[84] R. Gamez-Corrales, J.-F. Berret, L. M. Walker, & J. Oberdisse, Shear Thicken-
ing Dilute Surfactant Solutions: Equilibrium Structure as Studied by Small-Angle
Neutron Scattering. Langmuir 15, 6755 (1999).
[85] G. Porte, J.-F. Berret, & J. L. Harden, Inhomogeneous Flows of Complex Fluids:
Mechanical Instability versus Non-Equilibrium Phase Transition, J. Phys. II 7, 459
(1997).
[86] O. Radulescu & P. D. Olmsted, Matched Asymptotic Solutions for the Steady
Banded Flow of the Diffusive Johnson-Segalman Model in Various Geometries, J.
Non-Newtonian Fluid Mech. 91, 143 (2000).

173
[87] J. P. Keener, Principles of Applied Mathematics, (Perseus Books, Reading, Mas-
sachusetts, 1988).
[88] F. Riesz & B. Sz.-Nagy, Functional Analysis, (Dover Publications, New York, 1990).

Vita
Nestor Zenon Handzy was born to Jerry Nestor Handzy and Alexandra Ann
Handzy, in New York City. He received a B.A. in mathematics with a minor in physics
from the University of Pennsylvania in Philadelphia. After receiving his Ph.D. in math-
ematics from The Pennsylvania State University, he will work in the Department of
Complex Systems at The Weizmann Institute in Rehovot, Israel. American by birth,
Nestor’s ancestry is Ukrainian, and he appreciates and enjoys his Ukrainian heritage
through language, arts, history, and religion.