fisiopatologÍa del hÍgado - albergue de …€¦ · –alteraciones de los microfilamentos...
TRANSCRIPT

FISIOPATOLOGÍA DEL HÍGADO


Enfermedades hepáticas
Motivos de consulta por manifestaciones clínicas (*):
• Ictericia
• Retención hidrosalina (hipertensión portal): ascitis y/o edemas
• Síntomas neuropsiquiátricos: encefalopatía hepática (hipertensión portal + insuficiencia hepática, o insuficiencia hepática aguda grave). Consulta a psiquiatra o a neurólogo
• Hemorragia digestiva (hipertensión portal): Servicio de Urgencias.
(*) Pueden ser la primera manifestación clínica de la enfermedad hepática o aparecer en pacientes con diagnóstico previo de hepatopatía.

Exploración física de un paciente con enfermedad hepática
• Presencia de ictericia (piel, conjuntivas, paladar, etc.)• Adelgazamiento (neoplasia, cirrosis avanzada)• Edemas (retención hidrosalina: ¿Hipertensión portal?• Ingurgitación yugular (hígado cardíaco o de éstasis)• Equímosis (alteración de la coagulación: insuficiencia
hepática)• Señales de rascado (colestasis)• Orientación y alteraciones neuropsiquiátricas
• Estigmas hepáticos:– Arañas vasculares (¿aumento de estrógenos circulantes?) – Eritema palmar
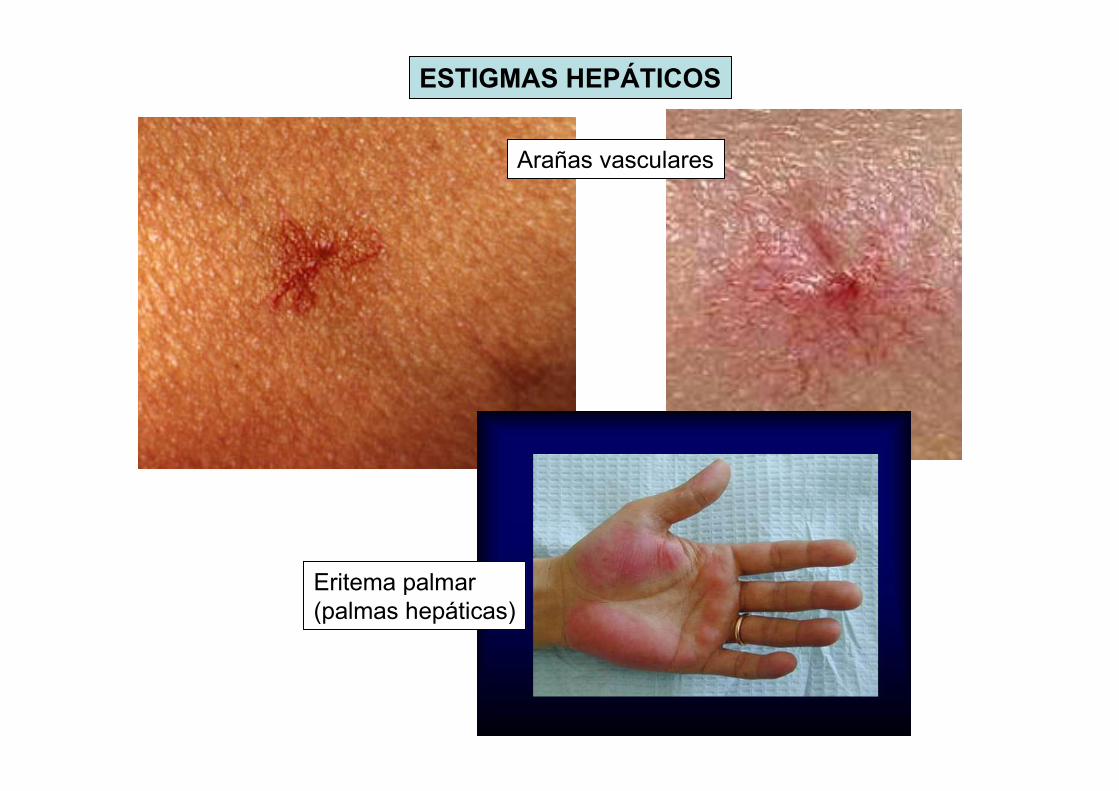
ESTIGMAS HEPÁTICOS
Arañas vasculares
Eritema palmar(palmas hepáticas)


Exploración física de un paciente con enfermedad hepática (cont.)
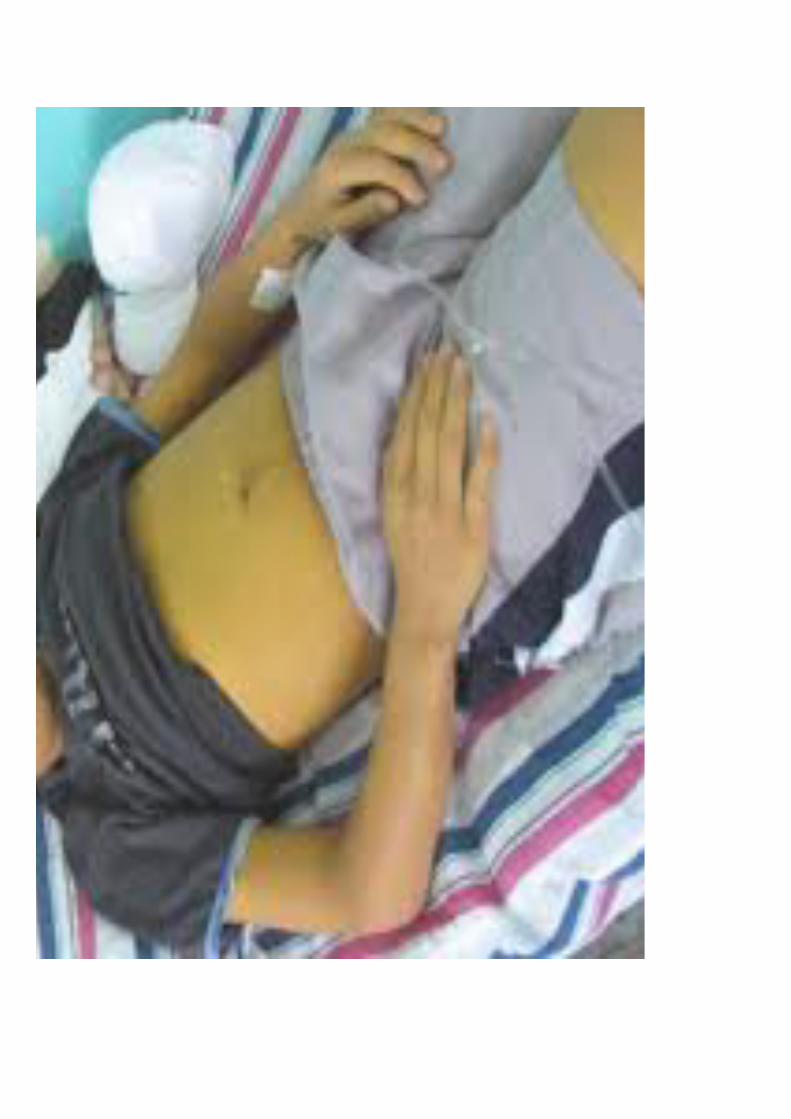
• Ascitis: (acumulación de líquido libre en abdomen)
– Tensa o no tensa
– Matidez cambiante en flancos
– Signo del menisco en bipedestación
– Presencia de hernia umbilical con frecuencia
• Circulación venosa abdominal (hipertensión portal):
– Umbilicofuga
– A veces “cabeza de medusa” (permeabilización de la vena umbilical por hipertensión portal) (síndrome de Cruveilhier-Baumgarten)




Exploración física de un paciente con enfermedad hepática (cont.)
• Hígado (palpación):
– Tamaño: Hepatomegalia (+/-)– Consistencia:
• Blanda: enfermedad aguda (a veces doloroso)• Dura (leñosa): enfermedad crónica• Pétrea: tumor primario o secundario (metastásico)
– Forma:• Lisa• Nodular o irregular: ¿cirrosis?• Abollonada y dura: ¿neoplasia primaria o secundaria?
• Bazo: Esplenomegalia– Hipertensión portal– Algunas hemólisis– Enfermedades de depósito (enf. de Gaucher, etc)

Enfermedades hepáticasDatos de laboratorio indicadores de:
• Citolisis
• Colestasis
• Fallo hepático
• Hipertensión portal

Enfermedades hepáticasDatos de laboratorio indicadores de:
• Citolisis:
Aumento de transaminasas
– GPT (ALT): citosol(T. Glutámico-pirúvica o Alanin amino-transferasa)
Glutámico + Pirúvico α-cetoglutárico + alanina
– GOT (AST): mitocondrias(T. Glutámico-oxalacética o aspartato amino transferasa)Glutámico + Oxalacético α-cetoglutárico + aspártico
(La GOT puede tener origen muscular, miocárdico, renal u otros)
– Aumento de Fe y ferritina

Enfermedades hepáticasDatos de laboratorio indicadores de:
• Colestasis (con o sin hiperbilirrubinemia)
– Aumento de fosfatasa alcalina termoestable a 60ºC (isoenzima hepático)
– Aumento de gamma-GT
– Aumento de colesterol
– Aumento de sales biliares

Enfermedades hepáticasDatos de laboratorio indicadores de:
• Fallo o insuficiencia hepática (disminución crítica de la masa hepática funcionante)
– Disminución del índice de protrombinaNo corregible con vitamina K parenteral. (Se corrige en caso de colestasis, por malabsorción de esta vitamina)
– Disminución de albúmina(menos fiable para valorar un fallo hepático agudo por su larga vida media y por variar según el estado de hemodilución)

Enfermedades hepáticasDatos de laboratorio indicadores de:
• Hipertensión portal (cirrosis)
– Trombopenia moderada, en general no inferior a 50.000/mm3, con o sin neutropenia(hiperesplenismo).
– Aumento de gamma-globulina policlonal, por derivación porto-sistémica de antígenos de procedencia intestinal que estimulan el sistema inmunológico, al no ser captados y destruídos por las células de Kupffer (“filtro hepático”).


ICTERICIAS

Ictericia• Coloración amarilla de la piel y mucosas visibles
por depósito de pigmento biliar (bilirrubina).
• Para que aparezca ictericia se requieren niveles séricos de bilirrubina > 3 mg/dl (normal: 1 mg/dl). (Depende de textura, grado de pigmentación melánica y riego sanguíneo de piel y mucosas).
• Se detecta sobretodo en zonas ricas en fibras elásticas (conjuntiva, paladar, etc.)


SRE1.- Aumento producción (hemolisis)
Br. no conjugada (indirecta) + albúmina
2.- Disminución captación________________________________________ Membrana sinusoidal
Ligandos Y y Z
Microsomas (REL): UDP-GT 1 (UGT 1) 3.- Disminución conjugación(a. glucurónico)
- S. de Gilbert- S. de Criggler-Najjar
-----------------------------------------------------------------------------------------------------------Br. conjugada (directa) 4.- Disminución almacenamiento
- S. de Rotor
5.- Disminución transporte y secreción biliar
- S. de Dubin-Johnson6.- CITOLISIS (ictericia hepatocelular o parenquimatosa)
Membrana canalicular7.- COLESTASIS (ictericia colestática intra o extrahepática)
ICTERICIAS

Ictericias por hiperbilirrubinemiano conjugadano conjugada o indirecta
1. Aumento de producción:• Hemolisis (ictericia hemolítica)• Reabsorción de grandes hematomas• Eritropoyesis ineficaz (hemolisis intramedular)
2. Disminución de la captación• Competición con fármacos (rifampicina) o
contrastes radiológicos
3. Disminución de la conjugación (por disminución hereditaria de la UGT-1)
• Síndrome de Gilbert• Síndrome de Criggler-Najjar

Ictericias por hiperbilirrubinemiano conjugadano conjugada o indirecta (hemolíticas)
• No coluria (Br. no hidrosoluble)• Urobilinuria (aumento de Br. en intestino)• Heces coloreadas (pleiocrómicas)• Tendencia a litiasis biliar pigmentaria
(bilirrubinato cálcico) (por bilis espesa por exceso de eliminación de bilirrubina)
• Encefalopatía por bilirrubina (ictericia nuclear, “kernicterus”), sobretodo en el recién nacido (por ser Br. liposoluble).

Ictericias por hiperbilirrubinemia de predominio conjugadaconjugada o directa
1. HEPATOCELULAR O PARENQUIMATOSA• Por citolisis o necrosis hepatocelular
• Hepatitis víricas o tóxicas (alcohol, fármacos…)
• Isquemia (shock, obstrucción arteria hepática, etc)
• Congestión
• etc.
• Aumentan ambas fracciones, con predominio de la br.conjugada (que compite con la no conjugada en presencia de lesión hepatocelular, ya que hay disminución de la masa hepática funcionante)
• Coluria e hipocolia
• Urobilinuria (fallo de recaptación hepática)(experimenta circulación entero-hepática)
• Aumento de transaminasas (GPT o ALT y GOT o AST)

Ictericias por hiperbilirrubinemia de predominio conjugadaconjugada o directa
2.- COLESTÁTICAS • Alteración excreción biliar de algunos o todos los componentes
orgánicos de la bilis (colesterol, sales biliares, fosfolípidos y bilirrubina) (*)
• Aumento casi exclusivo de fracción DIRECTA (muy poco o nada de no conjugada o indirecta).
• Acolia o hipocolia y coluria• Ausencia de urobilinuria (apenas pasa br. al intestino y la
recaptación hepática es normal)• Aumento en sangre de colesterol (xantomas) y sales biliares• Prurito• Aumento de fosfatasa alcalina y γ-glutamiltranspeptidasa
(GGT)
(*) Retención de componentes de la bilis, pero no de bilirrubina (o sea, ausencia de ictericia): colestasis disociada.

Acolia
Orina normal Orina colúrica

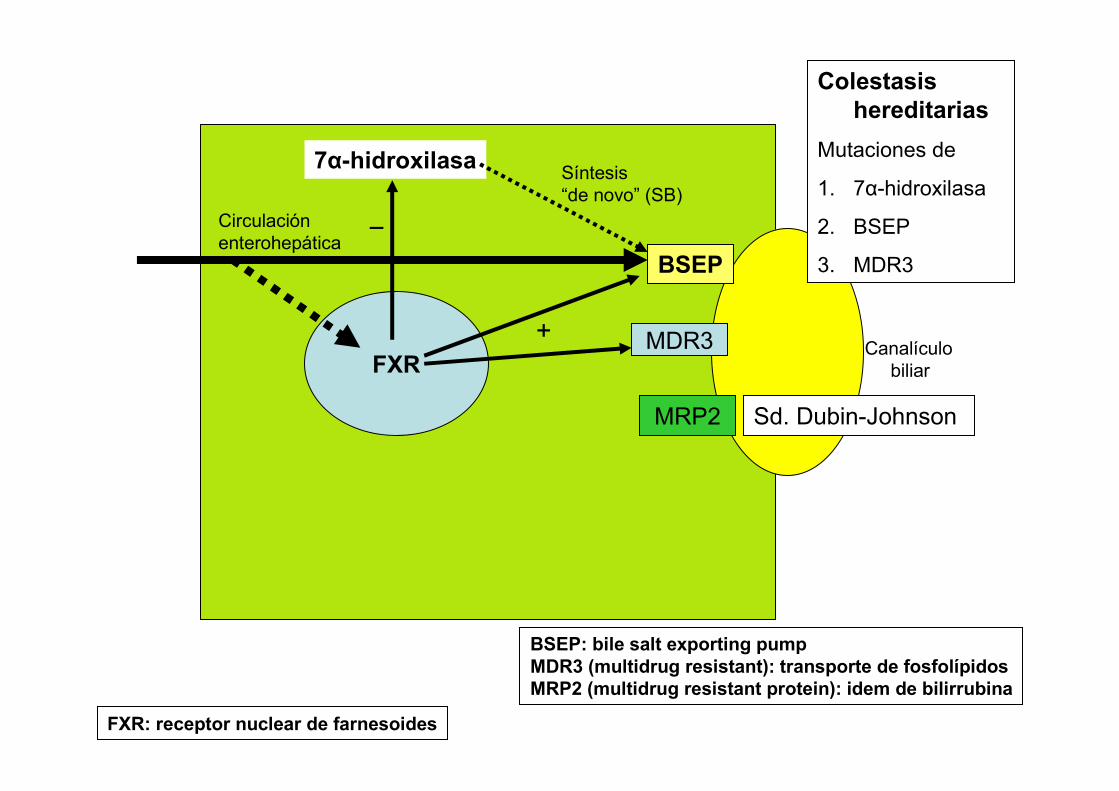
FXR
7α-hidroxilasa
_
BSEP
BSEP: bile salt exporting pumpMDR3 (multidrug resistant): transporte de fosfolípidosMRP2 (multidrug resistant protein): idem de bilirrubina
FXR: receptor nuclear de farnesoides
Canalículobiliar
Síntesis“de novo” (SB)
MDR3
MRP2
+
Circulaciónenterohepática
Colestasishereditarias
Mutaciones de
1. 7α-hidroxilasa
2. BSEP
3. MDR3
Sd. Dubin-Johnson

Ictericias por hiperbilirrubinemia de predominio conjugadaconjugada o directa
2.- Colestáticas (cont.)– Intrahepáticas
• Intracelulares (hereditarias)• Canaliculares: p.ej. por estrógenos.
– Alteraciones de la membrana canalicular– Alteraciones de los microfilamentos pericanaliculares
• Ductales: p.ej. Cirrosis biliar primaria (autoinmune)
– Extrahepáticas u obstructivas (p.ej. Cálculo en colédoco, tumor de cabeza de páncreas, etc.):
• Dilatación de las vías biliares extrahepáticas. • A veces distensión indolora de la vesícula (signo de
Courvoisier-Terrier, sospecha de causa tumoral, no litiásica)

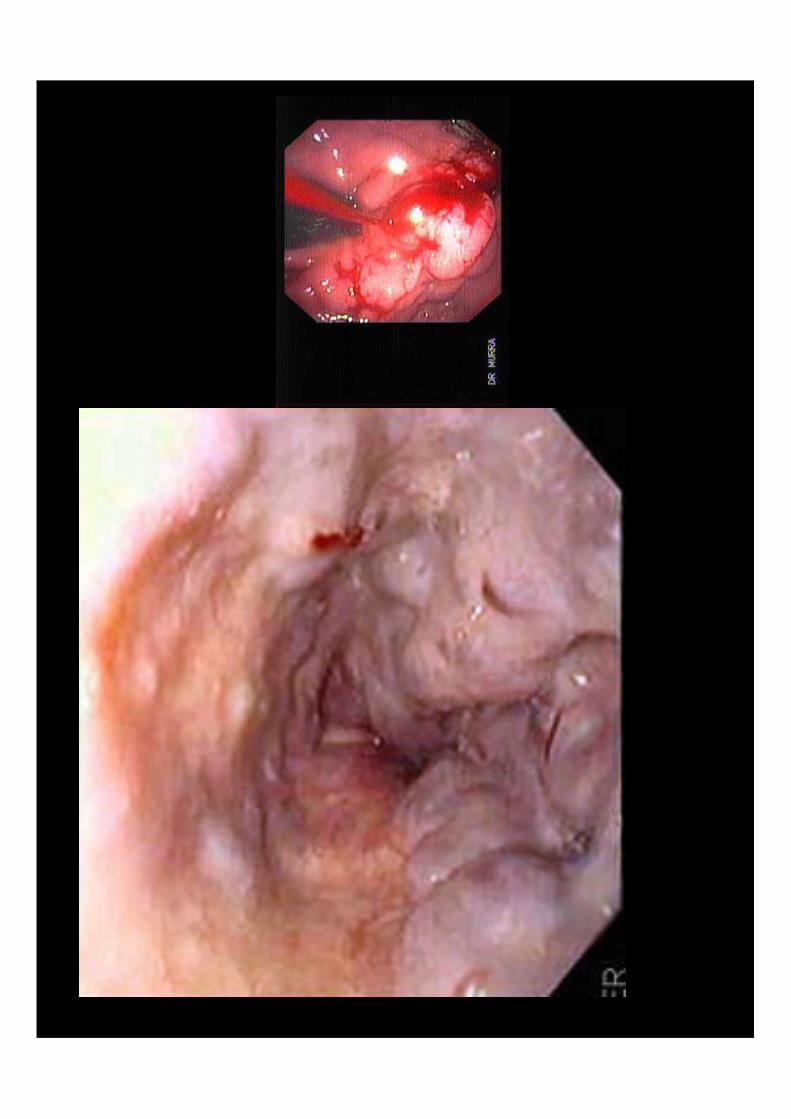
COLEDOCOLITIASIS

Circulación enterohepática de las sales biliares
Enterocito(íleon terminal)
FXR
SB conjugada
7α-hidroxilasa
_
BSEP*
*BSEP:bile salt exporting pump
FXR: receptor nuclear de farnesoides
Canalículobiliar
Síntesis“de novo”(0,5 gr/día)
Enterocito(yeyuno, etc)
SB desconjugada
v. porta
“pool” de SB: 4 gr

INSUFICIENCIA HEPÁTICA

INSUFICIENCIA HEPÁTICA
CONCEPTO: Síndrome clínico originado por la pérdida brusca o progresiva de las funciones del hígado:– Alteración de la síntesis y secreción de los componentes
biliares (función digestiva) y– trastorno metabólico múltiple (función metabólica), sobretodo
de la síntesis proteica (casi todas las proteinas del plasma) y– alteración de la función detoxificante (NH3, biotransformación
de xenobióticos, etc)
CLASIFICACIÓN:• Aguda o fulminante. (En días o semanas) necrosis masiva
aguda del hígado: virus, fármacos, tóxicos (setas, p.ej), etc
• Crónica progresiva. (Variable, en general al cabo de años)cirrosis hepática de cualquier etiología (alcohol, virus, etc)

INSUFICIENCIA HEPÁTICASíndrome metabólico-clínico
• Funciones con reserva elevada (se conservan hasta fases muy avanzadas o incluso terminales):– Glucuronización (conjugación) de la bilirrubina– Síntesis de fibrinógeno
• Funciones con reserva limitada (se alteran precozmente):– Captación y secreción de sales biliares– Síntesis de otros factores de la coagulación
(*) Variabilidad de las alteraciones metabólicas y su expresividad clínica de unos enfermos a otros.

INSUFICIENCIA HEPÁTICASíndrome metabólico-clínico
• Metabolismo de la bilirrubina: alteración de la excreción de bilirrubina conjugada (y de la captación): – Ictericia mixta (bilirr. de predominio directa: coluria e hipocolia).
• Metabolismo de los carbohidratos: alteración de la glucogénesis, gluconeogénesis y glucogenolisis: – Hipoglucemia (sólo en la forma aguda, no en la progresiva)
• Metabolismo de los lípidos:– Disminución de colesterol esterificado (déficit de síntesis
hepática de LCAT)– Disminución de síntesis y recaptación de sales biliares:
• Disminución de S.B. en bilis: propensión a litiasis biliar.• Id. en intestino: propensión a malabsorción y esteatorrea
– Alteración del metabolismo de los AGL de cadena corta• ¿Encefalopatía hepática?

INSUFICIENCIA HEPÁTICASíndrome metabólico-clínico
• Metabolismo nitrogenado (1)– 1.- Alteración del balance de aminoácidos en plasma:
• Aumento de aa. aromáticos y metionina (utilizados por el hígado para formar amonio, urea y para la síntesis proteica intracelular y del plasma)
• Disminución de los aa. ramificados (utilizados por el músculo esquelético)
Barrera hemato-encefálica S.N.C.
aa. aromáticos neurotransmisores falsosaa. ramificados (¿encefalopatía hepática?)
• Hiperaminoacidemia con aminoaciduria por rebosamiento en insuficiencia hepática aguda.

INSUFICIENCIA HEPÁTICA (I.H.) Síndrome metabólico-clínico
• Metabolismo nitrogenado (2)– 2.- Aumento de amonio en sangre:
• Procedencia:– Intestinal (colon): acción de la flora sobre las proteinas de la dieta.
– Desaminación de aminoácidos (en el catabolismo proteico)
– Renal (amoniopoyesis en túbulos renales por acción de la glutaminasa)
– Intestino delgado (glutaminasa)
• Destino: sobre todo ciclo de la urea en hígado: síntesis de urea
• I.H. Aguda: aumenta amonio en sangre y disminuye síntesis de urea (aunque por ins. renal se enmascara el descenso de urea)
• I.H. Crónica progresiva: idem, y por la hipertensión portal (HTP) de la cirrosis (a través de las colaterales portosistémicas)
• El amonio es el principal compuesto relacionado con la encefalopatía hepática en ambos tipos de ins. hepática.

INSUFICIENCIA HEPÁTICASíndrome metabólico-clínico
• Metabolismo nitrogenado (3)– 3.- Disminución de la SÍNTESIS PROTEICA
• Descenso de albúmina sérica (contribuye al edema) (tardío)
• Idem Factores de la coagulación: muy precoz, dada su corta semivida.
– La alteración de la coagulación ocasiona tendencia hemorrágica, y no se corrige con vitamina K parenteral.
– 4.- Alteración de neurotransmisores en SNC (encefalopatía hepática)• Síntesis en intestino, llegan al SNC sin pasar por hígado:
Neurotransmisores falsos o débiles (octopamina, fenil-etanolamina, que desplazan a los normales: dopamina, nAd)
• Síntesis en el propio SNC: disbalance aa aromáticos/ramificados
• Aumento de neurotransmisores inhibidores:– Gamma-aminobutírico (probable origen bacteriano intestinal)– Serotonina (a partir del aa aromático triptófano)– Benzodiazepinas (BZ) endógenas (origen incierto): hipersensibilidad cerebral a
las BZ.

INSUFICIENCIA HEPÁTICASíndrome metabólico-clínico
• 5.- Metabolismo hidrosalino:Vasodilatación esplácnica + vasoconstricción renal
(idem que en hipertensión portal postsinusoidal)
– Retención isotónica de Na y agua, debido a:• Reducción del filtrado glomerular• Aldosterona
– Retención de agua libre debido a:• Liberación de vasopresina (ADH)
• Consecuencias:• Edemas/ascitis (retención hidrosalina)• Hiponatremia dilucional (puede agravar encefalopatía,
favoreciendo además el edema cerebral)

INSUFICIENCIA HEPÁTICASíndrome metabólico-clínico
Encefalopatía hepática
CONCEPTO:– Síndrome neuropsíquico asociado al fallo o insuficiencia
hepática aguda grave (pérdida prácticamente total de las funciones hepáticas),
– o a una insuficiencia hepática crónica progresiva asociada a una enfermedad hepática crónica (cirrosis), en la que participan colaterales porto-sistémicas desarrolladas como consecuencia de la hipertensión portal: encefalopatía hepática porto-sistémica.
– En cualquier caso falla la detoxificación hepática de sustancias de procedencia sobre todo intestinal (especialmente amonio), y que entran directamente al SNC, donde ejercen su efecto tóxico.

INSUFICIENCIA HEPÁTICASíndrome metabólico-clínico
Encefalopatía hepática
Síndrome clínico• Alteraciones psíquicas (agitación, desorientación, trastornos del
comportamiento)
• Inversión del ritmo del sueño
• Lenguaje lento, disartria
• Apraxia constructiva (dibujo, escritura, etc)
• Asterixis (temblor aleteante o “flapping tremor”)
• A veces trastornos neurológicos (piramidales o extrapiramidales)
• Estupor….Coma

Insuficiencia hepáticaENCEFALOPATÍA HEPÁTICA (compuestos responsables)
• 1.- AMONIO:– Detoxificación:
• Hígado (ciclo de la urea, Zn como cofactor) (un déficit de Zn puede favorecer la encefalopatía)
• Músculo esquelético (formación de glutamina) (la atrofia muscular, frecuente en enfermos cirróticos, puede favorecer la encefalopatía)
• SNC (formación de glutamina, que se acumula en los astrocitos)
– Efectos• Inhibe metabolismo energético cerebral:
– Deprime el ciclo de Krebs– Descenso de ATP y consumo de O2
– Dumenta la glicolisis anaerobia ciclo : á. láctico)
• Aumenta la permeabilidad de la barrera hematoencefálica para él mismo y para otras sustancias tóxicas para el SNC.
• Aumenta la glutamina en astrocitos: edema cerebral.• Aumenta la afinidad de receptores neuronales GABA
(hipersensibilidad a benzadiazepinas)

Insuficiencia hepáticaENCEFALOPATÍA HEPÁTICA
(compuestos responsables) (cont)• 2.- Neurotransmisores falsos o débiles (de procedencia intestinal o
sintetizados en el propio SNC a partir de los aa aromáticos)
• 3.- Descenso de neurotransmisores excitadores– Dopamina– Noradrenalina– Glutamato
• 4.- Aumento de neurotransmisores inhibidores (GABA, serotonina).
• 5.- Manganeso (parece aumentar en la activación de los receptoresGABA y probablemente relacionado con sínd. extrapiramidal)
• 6.- Mercaptanos (procedente del metabolismo de la metionina por laflora del colon)(responsables del fetor hepático)
• 7.- Ácidos grasos de cadena corta (procedentes del colon)
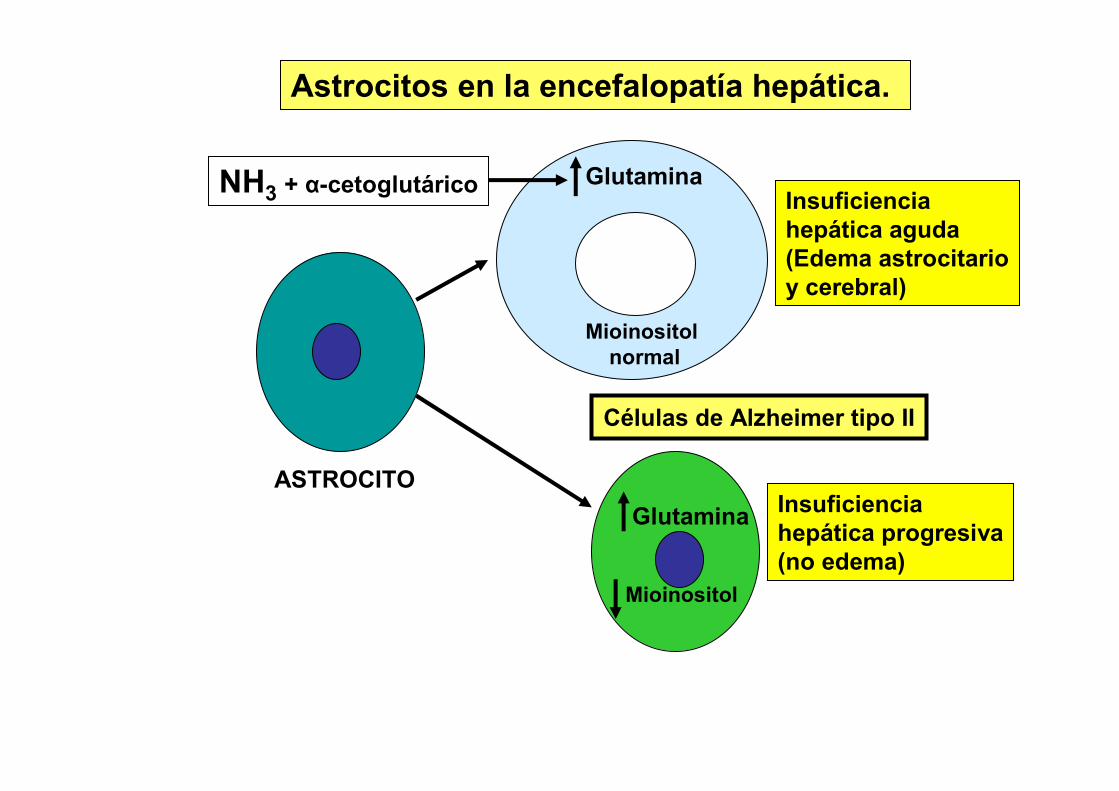
Glutamina
Mioinositolnormal
NH3 + α-cetoglutárico
Glutamina
Mioinositol
Insuficienciahepática aguda(Edema astrocitarioy cerebral)
Insuficienciahepática progresiva(no edema)
Células de Alzheimer tipo II
ASTROCITO
Astrocitos en la encefalopatía hepática.

HIPERTENSIÓN PORTAL

HIPERTENSIÓN PORTALCONCEPTO: aumento de presión en la vena porta y sus tributarias, que son venas de baja presión y bajo flujo (normal 5-10 mmHg).
• La causa más frecuente es la cirrosis hepática.• Clasificación:
– Presinusoidal– Postsinusoidal– La presinusoidal puede ser segmentaria (sólo afecta
a una tributaria, por ejemplo, la vena esplénica)
• Puede tener consecuencias graves:– Hemorragias– Retención hidrosalina (ascitis, etc)– Encefalopatía hepática porto-sistémica

Hipertensión portal: Clasificación
• Presinusoidal (presión sinusoidal normal)– Prehepática o extrahepática:
• Trombosis portal
• Compresiones extrínsecas de vena porta (adenopatías, tumores pancreáticos, hiperplasia nodular focal del hilio hepático,…)
– Intrahepática (afectación difusa a nivel de espacios porta)
CONSECUENCIAS:– Hiperesplenismo (sobre todo trombopenia moderada)
– Circulación colateral (varices esofágicas, que pueden sangrar)
– NO suele dar ascitis ni encefalopatía.


Hipertensión portal: Clasificación
• Postsinusoidal (Presión sinusoidal aumentada)
– Intrahepática
• CIRROSIS HEPÁTICA (componente sinusoidal, por colagenización del espacio de Disse: capilarización)
– Extrahepática o posthepática• Síndrome de Budd-Chiari
–Trombosis suprahepática–Compresiones de v. suprahepáticas
(tumores hepáticos, hipernefroma del riñón derecho)
• Insuficiencia cardíaca derecha– Valvulopatías derechas (estenosis tricuspídea)– Pericarditis constrictiva

Hipertensión portal postsinusoidalpostsinusoidal
• CONSECUENCIAS:– Hiperesplenismo (sobre todo trombopenia moderada)– Circulación colateral porto-cava (varices esofágicas).
• Hemorragia por rotura de las varices• Estimulación inmunológica por paso directo de antígenos de
procedencia intestinal a la circulación sistémica (aumento de gamma-globulinas)
• Propensión a infecciones (fallo del “filtro bacteriano hepático”: células de Kupffer)
• Intolerancia a la glucosa (hiperinsulinismo e hiperglucagonemia, al no metabolizarse las hormonas insulares en el hígado)
– Retención hidrosalina (ascitis, edemas, etc).– Encefalopatía (en caso de cirrosis, ya que debe existir
un grado mayor o menor de deterioro de la función hepática: insuficiencia hepática crónica progresiva).

HIGADO CIRRÓTICO


Capilarización sinusoidal(colagenización del espacio de Disse)
Normal
Cirrosis

HIPERTENSIÓN PORTAL EN LA CIRROSIS
–– COMPONENTE ANATCOMPONENTE ANATÓÓMICO POSTSINUSOIDAL MICO POSTSINUSOIDAL ((prácticamente irreversible).Compresión de los vasos, fundamentalmente de las venas de drenaje del lobulillo (centrolobulillares), por la fibrosisfibrosis y por el crecimiento de los nnóódulos.dulos.
– COMPONENTE SINUSOIDAL (parcialmente reversible). Disminución de elasticidad sinusoidal (colagenizacióndel espacio de Disse o capilarizacicapilarizacióónn sinusoidal)sinusoidal)
– COMPONENTE ACTIVO, FUNCIONAL (reversible) Contracción de miofibroblastos (procedentes de la transformación de células estrelladas o perisinusoidales).

HIPERTENSIÓN PORTAL
ASCITIS CONCEPTO: Acumulación de líquido libre en la cavidad peritoneal, como consecuencia de la hipertensión portal postsinusoidal (y sinusoidal en el caso de cirrosis) que induce retención de Na y agua.
• Es un trasudado- Concentración de proteinas < 2,5 gr/dl - Gradiente de albúmina suero/ascitis > 1,1
• A veces se acompaña de hidrotórax (derecho) y frecuentemente edemas (que muchas veces preceden a la ascitis).
• Inicialmente es frecuente la aparición de hernia umbilical
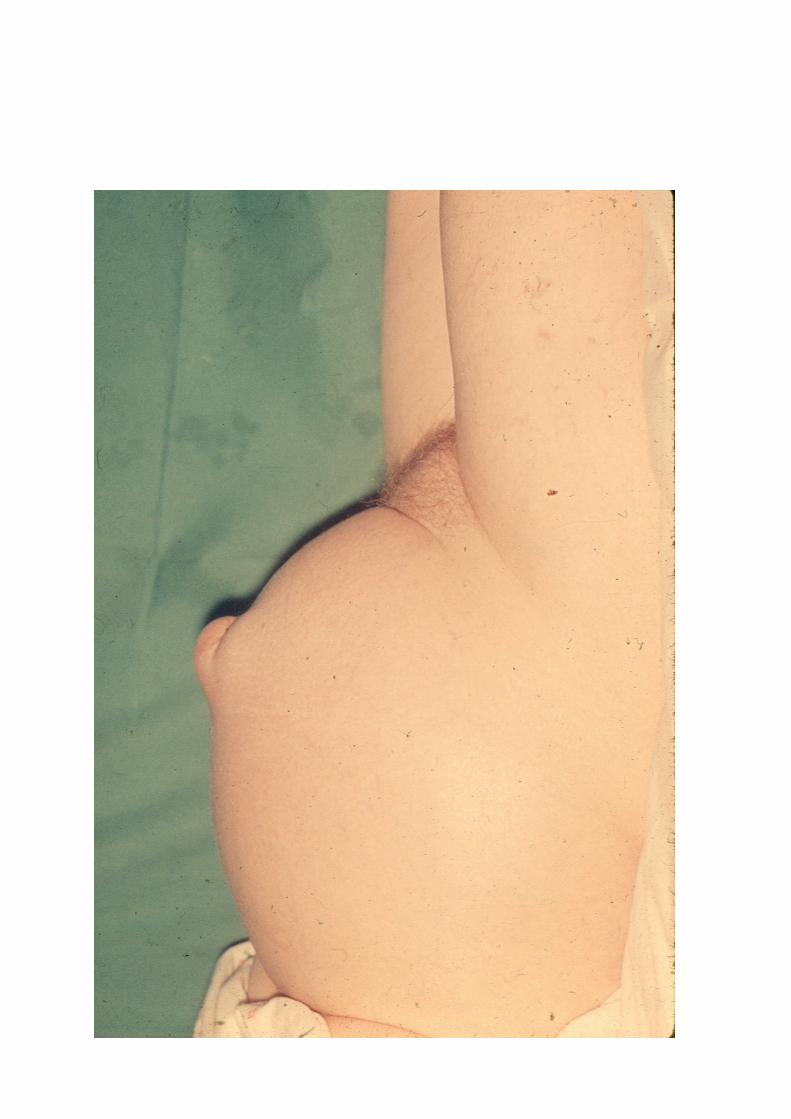

HIPERTENSIÓN PORTAL en la CIRROSIS
ASCITISMecanismos fisiopatológicos de la formación de ascitis
• Aumento de presión hidrostática en el territorio capilar mesentérico (ultrafiltrado, sin proteinas)
• Hipoalbuminemia, por disminución de la presión oncótica (en caso de insuficiencia hepática)
• Derrame linfático en peritoneo (sólo en HT postsinusoidal): casi ultrafiltrado en caso de cirrosis (por la capilarización sinusoidal) (pobre en proteinas)
• Retención renal de Na y agua (sólo en HT postsinusoidal): (por vasodilatación esplácnica e hipovolemia efectiva o por redistribución)
RESULTADO: El líquido ascítico es un trasudado (proteinas < 2,5 gr/dl)

CIRROSIS ASCITIS ASCITIS FisiopatologFisiopatologíía Teora Teoríía linfa linfááticatica

CIRROSIS ASCITIS. Teoría de la vasodilatación---------------------------------------------------------------------------------------------------
AUMENTO DE LA PRESIÓN SINUSOIDALNOx, etc (?)
VASODILATACION ESPLÁCNICA y sistémica (hipovolemia efectiva)
Liberación de sustancias presoras
S.N.Simpático (nAd) Renina---Angiotensina
VASOCONSTRICCIÓN RENAL
F.Gl. Filtrado Glomerular ALDOSTERONA
Resorción isotónica proximal de Na Id. distal UreaCreatinina Tienden a mantener la situación hemodinámica, pero
PGI2(locales) persiste la vasodilatación esplácnica(AINEs, p.ej)
SIND.HEPATO-RENAL Liberación de ADH HIPONATREMIA(Insuficiencia renal funcional)