guide to fluorine nmr for organic chemists (dolbier/fluorine nmr) || the single fluorine substituent
TRANSCRIPT

CHAPTER 3
35
THE SINGLE FLUORINE SUBSTITUENT
3.1. INTRODUCTION
The biological activity of a compound can often be affected dramati-cally by the presence of even a single fl uorine substituent that is placed in a particular position within the molecule. There are diverse reasons for this, which have been discussed briefl y in the preface and introduc-tion of this book. A few illustrative examples of bioactive compounds containing a single fl uorine substituent are given in Fig. 3.1 . These include what is probably the fi rst example of enhanced bioactivity due to fl uorine substitution, that of the corticosteroid 3 - 1 below wherein Fried discovered, in 1954, that the enhanced acidity of the fl uorohydrin enhanced the activity of the compound. 1 Also pictured are the antibac-terial β - fl uoro amino acid, FA ( 3 - 2 ), which acts as a suicide substrate enzyme inactivator, and the well - known anti - anthrax drug, CIPRO ( 3 - 3 ).
The information and examples presented in this chapter should enable the reader to predict chemical shift and coupling constant values for a single fl uorine substituent in virtually any possible environment in which it might be encountered.
Guide to Fluorine NMR for Organic Chemists, by William R. Dolbier, Jr.Copyright © 2009 by John Wiley & Sons, Inc.

36 THE SINGLE FLUORINE SUBSTITUENT
3.1.1. Chemical Shifts — General Considerations
As indicated in Chapter 2 , the single fl uorine substituent has an extremely broad range of observed chemical shifts, which include sulfonyl fl uo-rides and acyl fl uorides absorbing downfi eld in the region of +40 and +25 ppm, respectively, all the way up to fl uoromethyltrimethylsilane, with its signal far upfi eld at − 277 ppm.
Even within the different classes of compounds bearing the fl uorine substituent, the ranges of chemical shifts are still quite large, but there are predictable trends, both within each class and connecting the different classes of compounds. For example, the range of chemical shifts for a single fl uorine within a saturated hydrocarbon is − 130 to the value of − 272 ppm observed for methyl fl uoride. Primary fl uorides absorb at the higher fi eld (more negative) end, and tertiary fl uorides absorb at the lower fi eld end of the range. Single vinylic and aromatic fl uorines absorb at an even lower fi eld, within the range of − 95 to − 130 ppm.
3.1.2. Spin – Spin Coupling Constants — General Considerations
It will also be seen that spin – spin coupling constants between fl uorine and hydrogen as well as between fl uorine and fl uorine, and between fl uorine and carbon are quite predictable and thus useful in detailed structure characterization. Compounds containing a single fl uorine substituent exhibit the largest three - bond vicinal F – H coupling among saturated hydrofl uorocarbon systems, such coupling constants ranging from 21 to 27 Hz. However, their two - bond F – H coupling constants
FIGURE 3.1. Examples of bioactive compounds containing a single fl uorine substituent
HN
N N
CO2H
O
F
O
3-1
3-2
3-3
HO
F
O
CH2F
HO2C NH2
H
Antibacterial agent
Ciprof loxacin (CIPRO®)
Anti-inflammatorycorticosteroid

SATURATED HYDROCARBONS 37
(47 – 51 Hz) are smaller than the 56 – 58 Hz coupling constants exhibited in CF 2 H groups.
The one - bond F – C coupling constants observed for – CH 2 F and – CHF - groups, which are generally in the 162 to 170 Hz range, are also much smaller than the 234 to 250 Hz coupling exhibited by – CF 2 H or – CF 2 - groups or the 275 to 285 Hz coupling observed for CF 3 groups. However, both the monofl uoro - and the difl uoromethyl F – C coupling constants will increase signifi cantly when the carbon is also bound to other electronegative substituents. For example, note the large difference between the one - bond F – C coupling constant of methyl fl uoromethyl ether compared to that of 1 - fl uorobutane (219 vs. 165 Hz).
3.2. SATURATED HYDROCARBONS 2,3
Among the large group of compounds represented as monofl uoroal-kanes, primary fl uorine substituents are the most shielded, with the rule governing relative chemical shifts being quite simple:
Shielding of alkyl fl uorides: CH 3 > 1 ° > 2 ° > 3 ° Range of chemical shifts: − 272 → − 130 ppm
This trend is consistent with the trends for both proton and carbon chemical shifts, with the proton on the most highly substituted carbon, and the carbon with the most alkyl substituents being the most highly deshielded.
A corollary rule is that branching at either the β - or γ - position gives rise to increased shielding of 1 ° , 2 ° , or 3 ° fl uorine nuclei.
3.2.1. Primary Alkyl Fluorides
The typical chemical shift for primary n - alkyl fl uorides is − 219, but the values for primary alkyl fl uorides vary between − 212 for ethyl fl uoride and − 226 for 2 - ethyl - 1 - fl uorobutane (Scheme 3.1 ). As mentioned above, alkyl branching leads to shielding of fl uorine nuclei.
The coupling constants given below are typical two - and three - bond H – F values for such systems, with the range of two - bond F – H coupling being about 47 – 49 Hz and that for the three - bond F – H coupling being 21 – 27 Hz. Since the value of the three - bond H – F coupling constant is approximately half that of the two - bond H – F coupling constant, the net result is that the fl uorine signal for an n - alkyl fl uoride generally has the appearance of a septet, as is exemplifi ed for n - butyl fl uoride in Fig. 3.2 .

38 THE SINGLE FLUORINE SUBSTITUENT
When CH 2 F is a substituent on most alicyclic rings, such as a cyclo-hexane ring, the 19 F chemical shift of this group is not signifi cantly altered from that of an acyclic system (Scheme 3.2 ). On the other hand, when it is attached to a cyclopropane ring, a unique deshielding infl uence is observed.
3.2.1.1. 1 H and 13 C NMR Data. The examples in Scheme 3.3 provide insight into expected proton and carbon chemical shift and coupling constant data for primary alkyl fl uorides. It can be seen that the infl uence on both proton and carbon chemical shifts diminishes rapidly as one moves away from the site of fl uorine substitution.
CH3CH2-F –212
F
–226F –219
2JFH = 483JFH = 25
Scheme 3.1
FIGURE 3.2. 19 F NMR spectrum of 1 - fl uorobutane
CH3CH2CH2CH2F
–217.0 –217.5 –218.0 –218.5 –219.0 –219.5 –220.0 –220.5 ppm
CH3 CH2F
–223, 2JFH = 49 –208, 2JFH = 51
H
CH2F
Scheme 3.2
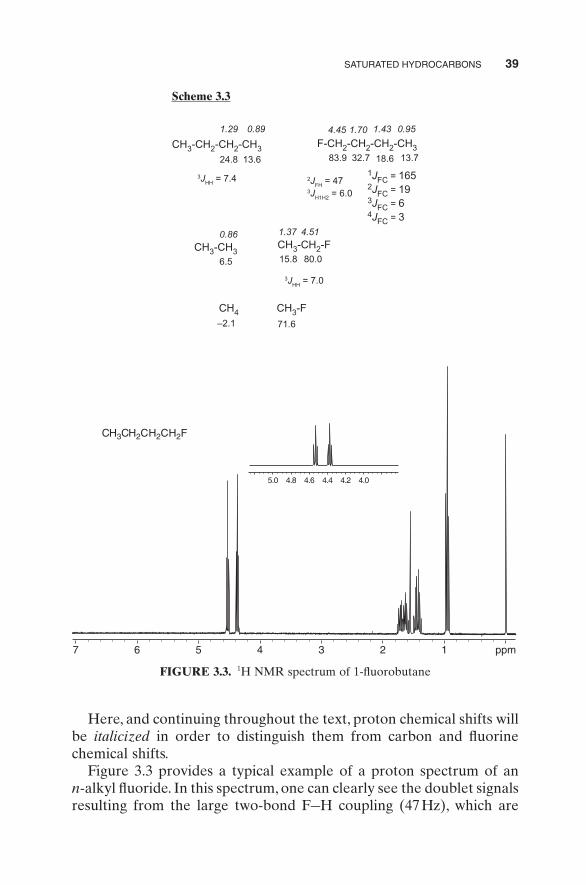
SATURATED HYDROCARBONS 39
Here, and continuing throughout the text, proton chemical shifts will be italicized in order to distinguish them from carbon and fl uorine chemical shifts.
Figure 3.3 provides a typical example of a proton spectrum of an n - alkyl fl uoride. In this spectrum, one can clearly see the doublet signals resulting from the large two - bond F – H coupling (47 Hz), which are
83.9 32.7 18.6 13.7
4.45
CH3-CH2-F80.015.8
CH3-F
71.6
3JHH
= 7.0
1JFC = 1652JFC = 193JFC = 64JFC = 3
CH3-CH2-CH2-CH313.624.8
0.891.29
CH3-CH36.5
0.86
CH4–2.1
F-CH2-CH2-CH2-CH3
3JHH
= 7.4
1.70 1.43 0.95
3JH1H2
= 6.0
2JFH
= 47
1.37 4.51
Scheme 3.3
FIGURE 3.3. 1 H NMR spectrum of 1 - fl uorobutane
7 6 5 4 3 2 1 ppm
CH3CH2CH2CH2F
5.0 4.8 4.6 4.4 4.2 4.0

40 THE SINGLE FLUORINE SUBSTITUENT
themselves split into triplets by the much smaller (6 Hz) three - bond H – H coupling. Chemical shift and coupling constant details for this spectrum are as follows: δ 0.95 ( t , 3 J HH = 7.5 Hz, 3H), 1.43 (sextet, 3 J HH = 7.8 Hz, 2H), 1.70 ( d , pent, 3 J FH = 25, 3 J HH = 7 Hz, 2H), 4.45 ( t , 2 J HF = 47, 3 J HH = 6.0 Hz, 2H).
Figure 3.4 provides the 13 C NMR spectrum of 1 - fl uorobutane, again a typical spectrum of an n - alkyl fl uoride. Examining this spectrum allows one to readily distinguish each carbon with respect to its location relative to the fl uorine substituent. This can be accomplished not only by comparison of their chemical shifts, but even more defi nitively by comparing their F – C coupling constants. In the spectrum, one sees the large (165 Hz) one - bond coupling of the signal at 83.89 ppm, the much smaller (19 Hz) two - bond F – C coupling of the signal at 32.68 ppm, the yet smaller (6.0 Hz) three - bond coupling of the signal at 18.61 ppm, and the still evident 3.1 Hz four - bond coupling constant for the highest fi eld signal at 13.69 ppm. (The multiplet at ∼ 77.2 ppm in most 13 C spectra printed in this book derives from the solvent, CDCl 3 . In those few cases where benzene - d 6 is used, the resultant multiplet will be seen at 126.3 ppm.)
3.2.2. Secondary Alkyl Fluorides
Secondary alkyl fl uorides exhibit a downfi eld (deshielding) shift of about +35 ppm from their primary analogues, their fl uorines typically absorbing at about − 183 ppm (Scheme 3.4 ), and such fl uorines will also experience the usual considerable shielding as a result of branching.
What one can see from examining the fl uorine spectrum of the typical secondary fl uoride, 2 - fl uoropentane, in Fig. 3.5 , is that because
FIGURE 3.4. 13 C NMR spectrum of 1 - fl uorobutane
CH3CH2CH2CH2F
80 70 60 50 40 30 20 10 ppm
19.0 18.8 18.6 18.4
14.5 14.0 13.5

SATURATED HYDROCARBONS 41
of the relatively large (20 – 25 Hz) three - bond H – F coupling to the vicinal hydrogens, one cannot readily distinguish the doublet of 47 Hz deriving from the two - bond H – F coupling within the multiplet at − 173 ppm. The net result is the multiplet seen in Fig. 3.5 . The doublet due to the 47 - Hz coupling is much more clearly seen in the proton spectrum (seen in Fig. 3.6 ) because the potentially complicating three - bond H – H coupling constants are much smaller (6 – 7 Hz).
3.2.2.1. Characteristic 1 H and 13 C NMR Data. The examples in Scheme 3.5 provide insight into expected proton and carbon chemical shift and coupling constant data for secondary alkyl fl uorides.
As mentioned above, the doublet due to the large (48 Hz) two - bond F – H coupling constant can easily be seen in the proton spectrum of 2 - fl uoropentane (Fig. 3.6 ). Note also the nice doublet of doublets centered at 1.31 ppm ( 3 J FH = 24 Hz, 3 J HH = 6 Hz) deriving from the C - 1 methyl group, which exemplifi es the signifi cant difference in magnitude between typical three - bond F – H and three - bond H – H coupling constants.
Figure 3.7 provides a typical 13 C NMR spectrum of a secondary fl uoroalkane, that of 2 - fl uoropentane. In it, one can detect coupling to the fl uorine substituent at all carbons except C - 5.
F
–183–1652J
FH = 47
F
Scheme 3.4
FIGURE 3.5. 19 F NMR of 2 - fl uoropentane
CH3CHFCH2CH2CH3
–168 –169 –170 –171 –172 –173 –174 –175 –176 –177 ppm
–172.6
–172.8
–173.0
–173.2
–173.4

42 THE SINGLE FLUORINE SUBSTITUENT
FIGURE 3.6. 1 H NMR spectrum of 2 - fl uoropentane
CH3CHFCH2CH2CH3
7 6 5 4 3 2 1 ppm
4.9 4.8 4.7 4.6 4.5 4.4 4.3
1.3 1.2 1.1 1.0
9.1 27.6 96.5
3JHH = 7.32JFH = 49
CH3-CH2-CH-CH2-CH3
F
0.96 1.56 4.62CH3-CH-CH3
F
1.33 4.84
3JHH = 7.3
87.322.6
Scheme 3.5
FIGURE 3.7. 13 C NMR spectrum of 2 - fl uoropentane
CH3CH2CH2CHFCH3
90 80 70 60 50 40 30 20 10 ppm
19.0 18.9 18.818.718.6 18.5 18.4 18.3

SATURATED HYDROCARBONS 43
The specifi c chemical shift and coupling constant data for 2 - fl uoropentane is as follows: δ 14.11 (s, C - 5), 18.56 (d, 3 J FC = 5.1 Hz, C - 4), 21.20 (d, 2 J FC = 22.3 Hz, C - 1), 39.24 (d, 2 J FC = 20.4 Hz, C - 3), 91.0 (d, 1 J FC = 164 Hz, C - 2).
3.2.3. Tertiary Alkyl Fluorides
Tertiary alkyl fl uorides exhibit an additional downfi eld shift of about +25 ppm, which is also very sensitive to branching as seen in Scheme 3.6 . The fl uorine spectrum for t - butyl fl uoride is shown in Fig. 3.8 . The signal at − 131 ppm is split into 10 peaks with a three - bond H – F cou-pling constant of 21 Hz.
F
–156–131
3JFH = 21
F
Scheme 3.6
FIGURE 3.8. 19 F NMR spectrum of t - butyl fl uoride
H3CCH3
CH3
F
–129 –130 –131 –132 –133 –134 ppm
3.2.3.1. Characteristic 1 H and 13 C NMR Data. The examples in Scheme 3.7 provide relevant proton and carbon chemical shift data.
The proton and carbon spectra for t - butyl fl uoride are provided in Figs. 3.9 and 3.10 .
The proton spectrum consists of a doublet at δ 1.38 ( 3 J FH = 21), whereas the carbon spectrum exhibits a doublet at 28.7 ppm with 2 J FC = 25 Hz along with a much weaker doublet at 94.1 ppm ( 1 J FC = 162 Hz).

44 THE SINGLE FLUORINE SUBSTITUENT
CH2
CH3
F
1.57, 3JFH = 26
0.87, 3JHH = 7.6
99.528.5
7.3H3C
CH3
CH3F94.1
28.7
1JFC = 1622JFC = 25
1.38 3JFH = 21
Scheme 3.7
FIGURE 3.9. 1 H NMR spectrum of t - butyl fl uoride
H3C CH3
CH3
F
7 6 5 4 3 2 1 ppm
FIGURE 3.10. 13 C NMR spectrum of t - butyl fl uoride
60 50 40 30 20 10 0 ppm
96 95 94 93
A comment about the carbon NMR spectrum of t - butyl fl uoride is appropriate. Because of the signal weakness of carbons such as the tertiary carbon of t - butyl fl uoride, which bear fl uorine but no hydro-gens, many published tabulations of 13 C spectra of compounds that contain such structural features fail to report these crucial signals. They can easily be missed, especially if you do not know what you are looking

SATURATED HYDROCARBONS 45
for. Even with relatively concentrated samples, it is usually necessary to run such spectra overnight in order to accumulate suffi cient FT data to see these weak signals.
3.2.4. Cyclic Alkyl Fluorides
Fluorocycloalkanes exhibit a small downfi eld (+) trend in chemical shifts in going from six - to fi ve - to four - membered rings, but cyclopro-panes are unique in exhibiting a strong upfi eld shift, with fl uorocyclo-propanes having by far the most highly shielded secondary fl uorine at − 213 ppm (Scheme 3.8 ). Notice that a cis methyl group on 1 - fl uoro - 2 - methylcyclopropane shields the fl uorine as compared to the fl uorine of the trans - isomer, an observation that is consistent with the “ branching principle, ” whereby branching at the β - position leads to shielding of fl uorine substituents.
Looking more closely at fl uorocyclohexane systems, it has been observed that a fl uorine substituent in the axial position is much more highly shielded than one in the equatorial position. Of course,
Fcis-isomer, –229trans-isomer, –208
H3C
F F F
F
Fax
Feq
F
CH3 –154t-Bu
F
CH3
t-Bu –127
–1602JFH = 55
–1712JFH = 53
–213, 2JFH = 653JFH(cis) = 223JFH(trans) = 10
–175
–1862JFH = 49
2JFH = 49
–165
FFF –129
–177 –141
Scheme 3.8

46 THE SINGLE FLUORINE SUBSTITUENT
ordinarily one will observe a single, time - averaged 19 F signal for fl uo-rocyclohexanes because of the relatively fast interconversion of the two chair conformations. A chemical shift of − 171 (broad singlet) has been reported for dynamically equilibrated fl uorocyclohexane at room temperature (see Chapter 4 for more details about the conformational dynamics of fl uorocyclohexane systems).
In going from a secondary to a tertiary cycloalkyl fl uoride, one observes the usual deshielding effect as is exemplifi ed by the isomeric 1 - fl uoro - 1 - methyl - 4 - t - butylcyclohexanes. Of course, these two isomers exist essentially in the single conformation given, because of the presence of the 4 - t - butyl substituent.
The chemical shift differences observed for these 1 - fl uoro - 1 - methyl - t - butylcyclohexanes, the cis - and trans - 9 - fl uorodecalins, and for 1 - fl uoroadamantane provide insight regarding the signifi cant infl uence of conformation upon fl uorine chemical shifts in fl uorocyclohexanes. The relative chemical shifts of these various cyclohexyl fl uorides can be rationalized simply on the basis of what is commonly known as an anomeric effect. That is, a vicinal hydrogen that is rigidly anti to a fl uorine substituent will exhibit an “ anomeric ” double bond/no bond resonance (or σ → σ * ) interaction, which will lead to relative shielding of the fl uorine as compared to situations where the fl uorine does not have the anti - hydrogen. Consistent with this explanation is the fact that the highly shielded fl uorine of trans - 9 - fl uorodecalin has three anti - hydrogens, whereas that of the cis - isomer has but one. Likewise the fl uorine of cis - 1 - fl uoro - trans - 1 - methyl - 4 - t - butylcyclohexanes has two antihydrogens, whereas the other isomer has zero. 1 - Fluoroadamantane also has zero hydrogens anti to its fl uorine. Moreover, the absolute values for all of these chemical shifts of tertiary cyclohexyl fl uorides can be directly correlated with the relative number of anti hydrogens present: three = − 177, two = − 154, one = − 141, and zero = − 127 and − 129 ppm.
It should also be noted that the geminal H – F ( 2 J HF ) coupling constants, although normal for cyclohexyl fl uorides ( ∼ 49 Hz), become progressively larger with smaller ring size (cyclopentyl fl uoride, 53 Hz; cyclobutyl fl uoride, 55 Hz), culminating in the characteristically large value ( ∼ 65 Hz) for cyclopropylfl uoride. Such increase is consistent with the presumed increase in the s - character of the carbon orbital used to bond to fl uorine in the series.
3.2.4.1. 1 H and 13 C NMR Data. Pertinent proton and carbon NMR data, in addition to those given above, are provided in Scheme 3.9 . Again, the increase in one - bond F – C coupling constant observed in the

INFLUENCE OF SUBSTITUENTS/FUNCTIONAL GROUPS 47
series, with that for cyclopropyl fl uoride being the largest, is noteworthy and is again consistent with the degree of s - character in the F – C bond. A review of 13 C NMR spectra of fl uorinated cyclopropanes has recently been published. 1
3.3. INFLUENCE OF SUBSTITUENTS/FUNCTIONAL GROUPS
Electronegative substituents, such as halogens and ether functions, deshield fl uorine nuclei when they are bound directly to the carbon bearing the fl uorine substituent. However, these same halogen, ether, and alcohol substituents, as well as similarly electronegative carbonyl functions, when positioned β to the fl uorine substituent, generally give rise to a shielding effect on a fl uorine nucleus.
3.3.1. Halogen Substitution 2
Substitution of a halogen on the same carbon as that bearing the fl uo-rine substituent gives rise to dramatic incremental deshielding effects (Scheme 3.10 ).
The deshielding effects of chlorine and bromine appear to be similar, with the chlorine having a greater deshielding infl uence in the methane examples above but a smaller infl uence in the cyclopropane example in Scheme 3.11 .
F96.5
33.423.1
F91.5
32.23
22.8
25.2
F89.5
31.110.5
HH H
4.572JFH = 49 5.07
2JFH = 53
4.932JF H = 55
H
F
4.322JFH = 65
74.6
1JFC = 2152JFC = 193JFC = 19
1JFC = 2212JFC = 18
1JFC = 1742JFC = 223JFC = 3
1JFC = 1702JFC = 193JFC = 74JFC = 1.5
H2
H3
H4
H5
F
H1
2JFH1 = 55.33JFH2(cis) = 20.63JFH3(trans) = 6.14JFH4(cis) = 8.54JFH5(trans) ~0
H-F Coupling constants in cyclobutyl fluoride
Scheme 3.9

48 THE SINGLE FLUORINE SUBSTITUENT
CH3F CH2ClF CHCl2F CCl3F
H3C CH3
F
H3C CH3
ClF
–268 –169 –82 [0]
–165 –87
CHBr2F
–86
–443JHF = 16.7
F-CCl2-CCl3
–64
F-CCl2-CCl2-F
–69
F-CCl2-CH3
Scheme 3.10
F
H3C
H3C
X X = Cl, –146X = Br, –142
Scheme 3.11
CH3CH2-F Cl-CH2CH2-FFCH2CH2-F
–226 –220
Br-CH2CH2-F
–212–2122JFH = 45
CCl3CH2-FCHCl2CH2-F
–1982JFH = 46
–2082JFH = 463JFH = 8
Cl-CH2CH2-F
–220
RX
FX δF R
HFClBrI
–172–191–182–178–171
n-C4H9n-C11H23n-C9H19n-C10H21n-C10H21
2JFH = 47–49
H3C
Br
F –2102JFH = 473JFH =12.8
Scheme 3.12

INFLUENCE OF SUBSTITUENTS/FUNCTIONAL GROUPS 49
A halogen substituent at the β - position to a fl uorine generally gives rise to shielding of the fl uorine nucleus (Scheme 3.12 ), with fl uorine providing the greatest shielding infl uence and iodine having virtually no effect. Interestingly, the addition of second and third β - chlorine substituents leads to progressive de shielding.
3.3.1.1. 1 H and 13 C NMR Data for Halofl uoroalkanes. Scheme 3.13 provides some pertinent proton and carbon chemical shift and coupling constant data for fl uorochloro - and fl uorobromomethanes, whereas Scheme 3.14 contains relevant data for some typical halo fl uoroalkanes. There does not appear to be anything unusual going on here.
CH2ClF CHCl2F CHBr2F
5.93
2JFH = 49
8.32
2JFH = 50
7.97
2JFH = 54
Scheme 3.13
2JHF3JHF
3JHH
X-CH2-CH2-F12
X δH1 δH2
Cl 4.58 3.67 47 5.7
Br 3.494.61 46 18 4.9
23
I 2.543.88 47 19 6.7
I-CH2-CH2-F
1JFC = 1742JFC = 22
1.2 82.7
Br-CH2-CH2-CH2-F
3.55 4.56
2JHF = 473JHH = 5.7
I-CH2-CH2-CH2-F3.25 2.15 4.48
2JHF = 473JHH = 5.3
3JHH = 6.9
3JHF = 263JHH = 6.9
1.0 33.9 83.01JFC = 1672JFC = 203JFC = 5.7
CH3-CH2-CH-CH2-Br
F
0.97 1.75 4.54 3.45
8.9 26.493.1
33.53JFC = 5.12JFC = 20
1JFC = 176
2JFC = 25
2JHF = 483JHH = 5.33JHH = 7.5
3JHF = 203JHH = 5.3
Scheme 3.14

50 THE SINGLE FLUORINE SUBSTITUENT
3.3.1.2. Vicinal Fluorine Substituents. The examples provided in Scheme 3.15 exemplify the specifi c effect of vicinal fl uorine substituents on both chemical shift and upon spin – spin coupling constants. As indi-cated in Section 3.3.1 (Scheme 3.12 ) each of the fl uorines is somewhat shielded by the presence of the other.
For the aforementioned examples, vicinal F – F coupling constants are in the vicinity of 15 Hz, whereas, as will be seen in Scheme 3.16 , the vicinal F – H coupling constants are larger, generally in the range of 20 – 25 Hz.
In the case of a number of vicinal difl uoro systems, such as 2,3 - difl uoro - 2,3 - diphenylethane or 2,3 - difl uorosuccinic acid derivatives, the coupling systems are AA ′ XX ′ , which means that they will produce second - order spectra (see Chapter 2 , Section 2.3.5 ). A case in point is the fl uorine and proton spectra of 1,2 - difl uoroethane, which have been
CH3F
F
Ph
F
F
CH2CH3
F
F
2JF1,H1 = 482JF2,H2 = 483JFF = 13
2JF1,H1 = 502JF2,H2 = 473JF1,F2 = 16
2JF1,H1 = 482JF2,H2 = 513JFF = 16
–228–183 –232
–192
n-C11H23F
F–231–191
–223–183
H3CCH3 Ph
PhF
F
F
F
Mesod,l
–190–187
–187–183
F
F
–1902JFH = 53
Scheme 3.15

INFLUENCE OF SUBSTITUENTS/FUNCTIONAL GROUPS 51
analyzed carefully both experimentally and computationally in order to determine details of the conformational distribution of this molecule (Scheme 3.16 ). 3 As is commonly known, the gauche conformation is preferred thermodynamically over the anti conformation .
3.3.1.3. 1 H and 13 C NMR Spectra of Vicinal Difl uoro Systems. Some typical examples of proton and carbon NMR data for vicinal difl uoro - substituted systems are given in Scheme 3.17 .
H
H FF
HHF
H HF
HHH
F HF
HH
0.43 0.430.14
ΔGo = 0.8 kcal mol–1
F-CH2-CH2-F
4.56
–226
Scheme 3.16
3JF1,H2 = 213JF2H1 = 25
3JF1,H2 = 243JF2H1 = 17
F
F4.37
4.35 1.601.00
F
PhF
4.44
5.54
F
F
84.1
91.1
29.9 n-C10H21
27.7
1JFC = 1732JFC = 22
1JFC = 1722JFC = 19
2JFC = 213JFC = 6
3JFC = 4F
F
4.76
1.90
1.70
3JF1,H2 = 123JF1CH2 = 18
Scheme 3.17

52 THE SINGLE FLUORINE SUBSTITUENT
(CF3)2CHF
(CF3)3CF–214
2JFH = 443JFF = 11.53JFH = 5.5
–188
3JFF = 6.1
F2CH-CH2-F CF3CH2-F
–239 –2412JFH = 463JFF = 183JFH = 14, 7.03JHH = 3.5
2JFH = 463JFF = 163JHF = 8.2
CH3CH2-F FCH2CH2-F
–226–212
2JFH = 45
(CH3)2CHF
–165
2JFH = 47
(CH3)3CF
–1313JFH = 21
4.65
–131
5.92 4.46
5.07
4.56
3JFH = 27
2JFH = 48.53JFH = 17
Scheme 3.18
3.3.1.4. More Heavily Fluorinated Compounds. The series of fl uo-rinated ethanes in Scheme 3.18 indicates that the fl uorine nucleus of a CH 2 F group is increasingly shielded as the number of β - fl uorines increases, unlike the situation observed for an increasing number of β - chlorine substituents (Scheme 3.12 ). Note that as one accumulates fl uorines at the β - position, the three - bond H – F coupling constant becomes progressively smaller.
Replacing the hydrogens of CF 3 CH 2 F with CF 3 groups gives the secondary and tertiary fl uoride compounds in Scheme 3.18 , which absorb at progressively lower fi eld. However, in spite of this, the sec-ondary and tertiary fl uorine substituents of these two compounds are the most shielded of any secondary and tertiary fl uorines.
A few pertinent proton chemical shifts are also provided in the aforementioned scheme.
3.3.2. Alcohol, Ether, Ester, Sulfi de, and Sulfone Groups
Compounds with fl uorines bound directly to a carbon bearing a hydroxy group are generally very unstable, although there are exceptions. Hexafl uoroacetone and hexafl uorocyclobutanone both add HF to form stable α - fl uoroalcohols, which release HF quickly in water to form the respective hydrates. The stability of these alcohols derives simply from the relative instability of the respective perfl uoroketones. Fluorine NMR data for the one example available are provided in Scheme 3.19 . Its chemical shift is obviously also infl uenced signifi cantly by the six β - fl uorines.

INFLUENCE OF SUBSTITUENTS/FUNCTIONAL GROUPS 53
An ether oxygen bound directly to a CH 2 F group, as in a fl uoro-methyl ether, deshields the fl uorine more than does a chlorine sub-stituent (Scheme 3.19 ).
Similar sulfur substitution, as in a fl uoromethyl sulfi de, also leads to deshielding, but somewhat less than for the analogous ether.
Again, secondary fl uorides are deshielded considerably compared to the primary systems, with tertiary fl uorides even more so.
Sulfones and sulfoxides bearing a CH 2 F group have similar fl uorine chemical shifts and are quite shielded relative to their unoxidized sulfi de analogues. A CH 2 F group attached to a sulfonium sulfur is slightly deshielded relative to sulfoxides and sulfones (Scheme 3.20 ).
As was the case when one added a β - fl uorine substituent, placement of an ether or alcohol functionality β to a fl uorine substituent leads to modest shielding (Scheme 3.21 ).
A hydroxyl group one or two carbons further removed, γ or δ to the fl uorine substituent, does not infl uence the fl uorine chemical shift sig-nifi cantly (Scheme 3.22 ). In the case of a secondary system, the fl uorine is also unaffected by a γ - hydroxy substituent.
ClCH2F
PhOCH2F
–169
–1492JFH = 54
PhSCH2F
–1802JFH = 54
n-C12H25SCH2F
n-C6H13CHFSCH3
n-C8H17OCH2F
–1522JFH = 57
–1842JFH = 52
CH3CH2F
–212
–112
O OCH2F O2
SO
CH2F–158
–1542JFH = 512JFH = 51
CH3-CH2-S-CHF-CH3
–142
SCH3
CH3
FPh
–108
CH3-S-CH2-F
–1892JFH = 54
2JFH = 593JFH = 21
3JFH = 19
F3C CF3
OHF–126
–83
3JFF = 2
Scheme 3.19

54 THE SINGLE FLUORINE SUBSTITUENT
–2122JFH = 47
–211–213
2JFH = 48
S
CH2F OTf
–2072JFH = 47
PhS
CH2-F
O
Ph-CH2-S-CH2-F
O
O
Ph-S-CH2-F
O
O
Scheme 3.20
CH3OCH2CH2F HOCH2CH2F
–2232JFH = 49
–2272JFH = 483JFH = 32
CH3CH2F
–212
F
–183
F
OH–191
Scheme 3.21
CH3CH2CH2F
–219
HOCH2CH2CH2F
–220
HOCH2CH2CH2CH2F
–216
OH
F
–173 –173
F
Scheme 3.22
3.3.2.1. 1 H and 13 C NMR Data. The examples in Scheme 3.23 provide characteristic proton and carbon chemical shift and coupling constant data for fl uorinated alcohols, ethers, thioethers, sulfoxides, and sul-fones. An ether substituent serves to deshield the carbon of a CH 2 F by about 20 ppm. This can be compared to the 40 - ppm deshielding generally observed in a nonfl uorinated ether system. Thus, the fl uorine substituent seems to have a damping effect on the usual effects of other substituents.

INFLUENCE OF SUBSTITUENTS/FUNCTIONAL GROUPS 55
3.3.2.2. Multiple α - Ether Substituents. There are few examples of such compounds. Three phenoxy substituents do not deshield a C – F fl uorine as much as three chlorine substituents, but a little more than three additional F substituents (Scheme 3.24 ).
O OCH2F
S OCH2F
S
CH2F
OTf
F
OH
5.13
5.07/ 5.042JHH = 8.4
98.01JFC = 220
CH3-O-CH2-F57.3 104.8
3.65 5.45
1JFC = 219
OCH2F
100.51JFC = 217
5.8, 2JFH = 55
CH3-S-CH2-F
SCH2F
83.6
5.625.70 2JFH = 46
R-O-CH2CH2-F4.62
83.6
3.734.37
96.629.2 73.0
1JFC = 168
2JFC = 222JFC = 21
2JFH = 48
88.2 1JFC = 219
6.566.65
2JAB = 9.390.1
1JFC = 242
6.04
93.91JFC = 220
1JFC = 232
5.76
98.3
O
O
CH3CH2-S-CHF-CH3
6.05 1.592.26
Ph-S-CH2-F Ph-S-CH2F
O O
O
Scheme 3.23
(p-CF3-C6H4O)3C-FCl3C-F F3C-F
[0] –53.8 –64.6
Scheme 3.24
3.3.3. Amino and Ammonium Groups
There are no α - amino fl uorides because of the reactivity of this func-tional combination, and for the same reason, there are few β - amino fl uorides. However, when attached to the less reactive nitrogen of a

56 THE SINGLE FLUORINE SUBSTITUENT
benzimidazole, the CH 2 F group is much more stable (Scheme 3.25 ). Note that when attached to a partially positive nitrogen, as in an imid-azolium compound, or a fully positive ammonium nitrogen, the respec-tive fl uorines become progressively more shielded.
Available carbon and proton data are also provided for these compounds in the same scheme.
The example of the β - fl uoroamine given in this scheme indicates that, unlike the effect of a halogen or an alcohol or ether function, a β - amino substituent acts to deshield fl uorine.
An example of a fl uorodiazirene is also provided.
3.3.4. Phosphorous Compounds
There do not appear to be any simple phosphines that bear a CH 2 F group. However, fl uorine NMR spectra of phosphonates, phosphane oxides, and phosphonium compounds with CH 2 F and – CHF - bound to phosphorous have been reported. Examples are given in Scheme 3.26 , including spectral data for the useful Horner – Wadsworth – Emmons reagent, triethyl 2 - fl uoro - 2 - phosphonoacetate.
The fl uoromethyl phosphonium compound is unique. Analogous CHF 2 or CF 3 phosphonium compounds do not appear to have been reported.
3.3.4.1. Proton and Carbon Spectra. Proton and carbon NMR data, including 31 P chemical shift and P – C coupling constants for the above compounds are given in Scheme 3.27 .
N
C2H5
F
–1932JFH = 483JFH = 23
N
N
CH2F –164, 2JFH = 546.14
81.71JFC = 200
N
N
CH2F
CH3
BF4–
6.27
85.3
–171, 2JFH = 49
1JFC = 201N
H3C
CH3
CH2F–188, 2JFH = 45
5.87
98.41JFC = 220
H2N F
4.60
2.91
2JFH = 473JFH = 303JHH = 5
NN
F –133
Scheme 3.25

INFLUENCE OF SUBSTITUENTS/FUNCTIONAL GROUPS 57
3.3.5. Silanes
Whether bound directly to the silicon or on a carbon bound to the silicon, a fl uorine substituent within a silane is highly shielded com-pared to that in a hydrocarbon. For example, the fl uorine of TMS fl uo-ride absorbs more than 25 - ppm upfi eld from that in t - butyl fl uoride (Scheme 3.28 ). (For additional data on Si – F compounds, see Chapter 7 , which deals with compounds that have heteroatom – fl uorine bonds.)
Likewise, a primary – CH 2 F fl uorine adjacent to silicon is shielded by more than 50 ppm compared to the respective hydrocarbon (Scheme 3.29 ), with the value of − 277 ppm observed for fl uoromethyltrimethyl-silane being the largest chemical shift known for a single carbon - bound fl uorine.
Ph3P-C+
–
H2F
BF4
–2442JHF = 452JPF = 58
Ph2P-CH2F
O
EtOP
EtO
O O
OEtF H
–2112JHF = 472JPF = 72
–2422JHF = 472JPF = 49
Ph2P
O
H F
HO
(CH2)10
–2042JHF = 462JPF = 68
EtOP
EtO
O
n-C6H13
F H
–210 2JHF = 472JPF = 76
Scheme 3.26
Ph3P-CH2F
BF4
Ph2P-CH2F
O
EtOP
EtO
O O
OEtF H
Ph2P
O
H F
HO
(CH2)10
5.2084.9
1JFC = 1971JPC = 159
5.18
80.1 1JFC = 1891JPC = 84
5.09
94.7 1JFC = 1991JPC = 82
6.32
76.7
1JFC = 1951JPC = 65
EtOP
EtO
O
n-C6H13
F H 4.62
88.71JFC = 1791JPC = 170
31P, δ 19.0
–
+
Scheme 3.27

58 THE SINGLE FLUORINE SUBSTITUENT
CH3
CH3C
CH3
F
CH3
SiH3C
CH3
F
–131 –158
Scheme 3.28
CH3
CH3C
CH3
CH2F
CH3
SiH3C
CH3
CH2F
–223 –277
4.4
2JFH = 47
Scheme 3.29
3.4. CARBONYL FUNCTIONAL GROUPS
Carbonyl functional groups bound to a carbon - bearing fl uorine give rise to modest shielding of the fl uorine (Scheme 3.30 ), with esters shielding 1 ° CH 2 F somewhat more, but 2 ° CHF somewhat less than ketones. An aldehyde carbonyl appears to have the largest shielding impact.
Carbonyl functions affect the fl uorine chemical shift of secondary systems more greatly than primary systems, shielding their α - fl uorines
1o fluorides:
FH2C CH3
O
FH2C n-alkyl
O
FH2C OCH2CH3
O
–2282JFH = 49
–230–226
H3C
F
CH3
O
–1902JFH = 49
2o f luorides:
n-C3H7
F
n-C3H7
O
–193
FCH2CH2CH3
–219
F
–173
FH2C H
O
–2322JFH = 463JFH = 5.1
F
O
OCl
–1832JFH = 48
3JHF = 24
Scheme 3.30

CARBONYL FUNCTIONAL GROUPS 59
F
–219
FCH2CH2CO2Et
–2202JFH = 473JFH = 26
vs.
Scheme 3.31
to a greater degree. Addition of a second β - carbonyl group gives rise to additional but less shielding than the fi rst carbonyl group.
When the carbonyl function is two carbons removed from the CH 2 F, it has little, if any, infl uence on the fl uorine ’ s chemical shift (Scheme 3.31 ).
3.4.1. 1 H and 13 C NMR Data for Aldehydes, Ketones, and Esters
Typical proton and carbon chemical shift and coupling constant data for α - fl uoroketones and aldehydes are given in Scheme 3.32 , with data for esters being given in Scheme 3.33 .
Two - bond F – H coupling constants for both primary and secondary fl uoroketone systems are always in the range of 47 – 49 Hz, as indicated earlier in Scheme 3.30 . One - bond F – C couplings are in the range of 181 – 183 Hz, whereas two - bond F – C coupling constants can vary between 16 and 25 Hz, as seen in the examples below. A primary CH 2 F bound to a ketone has a carbon chemical shift routinely in the 83 to 85 ppm range, whereas that of a secondary CHF group bound to carbonyl is between 92 and 95 ppm. Proton chemical shifts for a CH 2 F group vary depending on whether the ketone has an aliphatic (4.5 – 4.7 ppm) or an aromatic (5.3 – 5.6 ppm) substituent. Proton chemi-cal shifts for a secondary CHF group adjacent to a ketone are around 4.7 – 4.8 ppm for aliphatic systems.
Both for primary and secondary α - fl uoroesters, the α - protons absorb slightly downfi eld of analogous aliphatic ketones and somewhat upfi eld of analogous aromatic ketones.
H3C
F
OCH2CH3
O
H3C OCH2CH3
O
F
O
–1942JFH = 49
–1852JFH = 48
n-C6H13
F
H
O
–2002JFH = 50
Scheme 3.30 (cont’d)

60 THE SINGLE FLUORINE SUBSTITUENT
OCH3
O
F169.5
89.0
38.5
1JFC = 1882JFC1 = 242JFC3 = 20
O
O
CH2FH3C
OEt OEt
OO
F F
1JFC = 1812JFC1 = 232JFC3 = 22
Et
167.9170.1
61.588.9
85.7
18.335.4
5.04.9
170.5
4.85
1JFC = 1842JFC1 = 242JFC3= 21
1.6 1.9
Scheme 3.33
F FCH2-C N H3C CN
F
–2322JFH = 45
–1822JFH = 46, 3JFH = 233JHH = 7
–219
F
CN
–1672JFH = 47
Scheme 3.34
O
CH2F
O
CH2F
O
CH2F
1JFC = 1851JFC = 1832JFC = 16
84.9
4.46O
CH2F203.5
1JFC = 1812JFC = 18
84.1
4.685.51
5.30
83.5
186.9
193.383.4
OO
F F
4.78 4.72
92.6 208.5 95.9 208.3
1JFC = 1812JFC = 25
1JFC = 1832JFC2 = 262JFC4 = 21
1.41 31.5
H
O
F
31.0
95.5
200.5
1JFC = 1802JFC1 = 352JFC3 = 20
O
F6.24
93.0
183.2
OClH
1JFC = 1882JFC = 24
Scheme 3.32
3.5. NITRILES
A nitrile function behaves much like a carbonyl functionality with respect to its infl uence upon an alkyl fl uorine ’ s chemical shift, acting to shield the fl uorine modestly (Scheme 3.34 ).

ALKENES WITH A SINGLE FLUORINE SUBSTITUENT 61
3.6. ALKENES WITH A SINGLE FLUORINE SUBSTITUENT
Single vinylic fl uorine substituents absorb over quite a wide range of chemical shifts, with fl uoroallene at the high fi eld end ( − 169 ppm) and β - fl uoroacrylate derivatives at the low fi eld end ( − 75 ppm) (Scheme 3.36 ).
TABLE 3.1. 13 C Chemical Shifts for α - Substituted Nitriles
Compound CH 3 CN ClCH 2 CN MeOCH 2 CN FCH 2 CN
δ , CH 2 X 1.5 24.6 59.0 66.7 δ , CN 116.3 114.5 115.6 113.7
H
CNF
6.23
80.3115.3
H3C
F
CN
H
1.72
5.221JFC = 1822JFC = 34
2JFH = 47
2JFH = 473JHH = 6.93JFH = 23
Scheme 3.35
3.5.1. 1 H and 13 C NMR Data for Nitriles
Table 3.1 provides a comparison of the carbon chemical shifts for a number of monosubstituted acetonitriles, 4 whereas Scheme 3.35 provides proton and carbon data for a couple of more highly substituted systems.
C C CHH
F
HRCF=CHCO2H
–1692JFH = 854JFH = 6
–75
toLowfield
Highfield
7.15.5
Scheme 3.36
62 THE SINGLE FLUORINE SUBSTITUENT
C11H23
CH3
F
H
F
F
–1302JFH = 853JFH(cis) = 19
–1312JFH = 853JFH(trans) = 43
F
F
–130
–131
–137
2JFH = 88
F
–113
2JFH = 853JFH(cis) = 203JHF(trans) = 52
Scheme 3.37
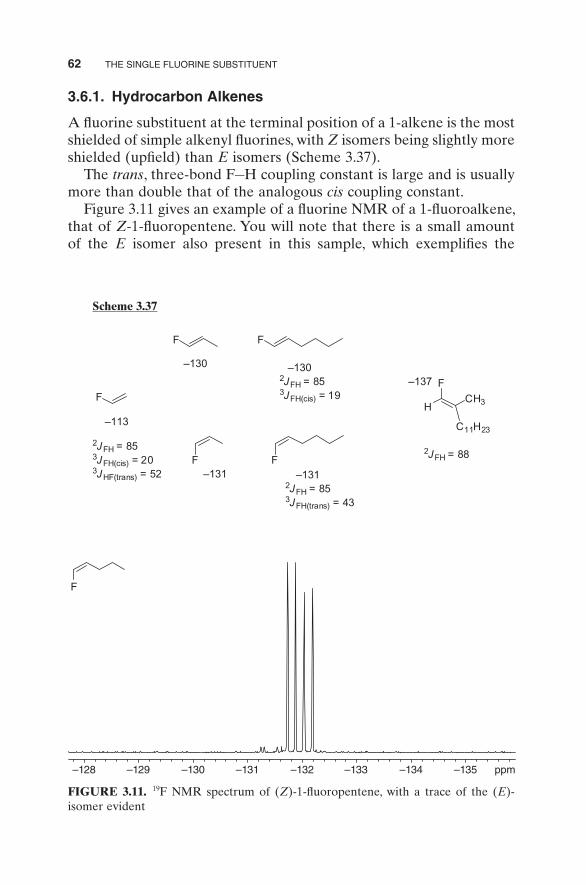
FIGURE 3.11. 19 F NMR spectrum of ( Z ) - 1 - fl uoropentene, with a trace of the ( E ) - isomer evident
F
–128 –129 –130 –131 –132 –133 –134 –135 ppm
3.6.1. Hydrocarbon Alkenes
A fl uorine substituent at the terminal position of a 1 - alkene is the most shielded of simple alkenyl fl uorines, with Z isomers being slightly more shielded (upfi eld) than E isomers (Scheme 3.37 ).
The trans , three - bond F – H coupling constant is large and is usually more than double that of the analogous cis coupling constant.
Figure 3.11 gives an example of a fl uorine NMR of a 1 - fl uoroalkene, that of Z - 1 - fl uoropentene. You will note that there is a small amount of the E isomer also present in this sample, which exemplifi es the

ALKENES WITH A SINGLE FLUORINE SUBSTITUENT 63
signifi cant difference between the cis and trans , three - bond, H – F cou-pling constants. The chemical shifts of the Z and E isomers are − 131.9 and − 131.4, respectively, the two - bond H – F coupling constants of both isomers being 87 Hz, the trans , three - bond H – F coupling constant of the Z isomer being 44 Hz, with the cis , three - bond H – F coupling constant of the E isomer being 18.6 Hz.
The slight shielding of ( Z ) - 1 - fl uoroalkenes vs. ( E ) - 1 - fl uoroalkenes is again worth refl ecting on since, although only a small effect, it is con-sistent with the effect mentioned earlier with respect to chemical shifts of cis - and trans - 2 - methyl - 1 - fl uorocyclopropanes (Section 3.2.4 ), but contrary to the deshielding impact of cis - alkyl groups on the chemical shifts of alkenyl trifl uoromethyl groups, as discussed in Chapter 2 (Section 2.2.1 ).
When the fl uorine substituent is located at the 2 - position or on any alkyl - substituted alkenyl carbon, it experiences the usual deshielding of 30 – 40 ppm (Scheme 3.38 ). Note the interesting variation in the chem-ical shifts and coupling constants for the 1 - fl uorocycloalkenes.
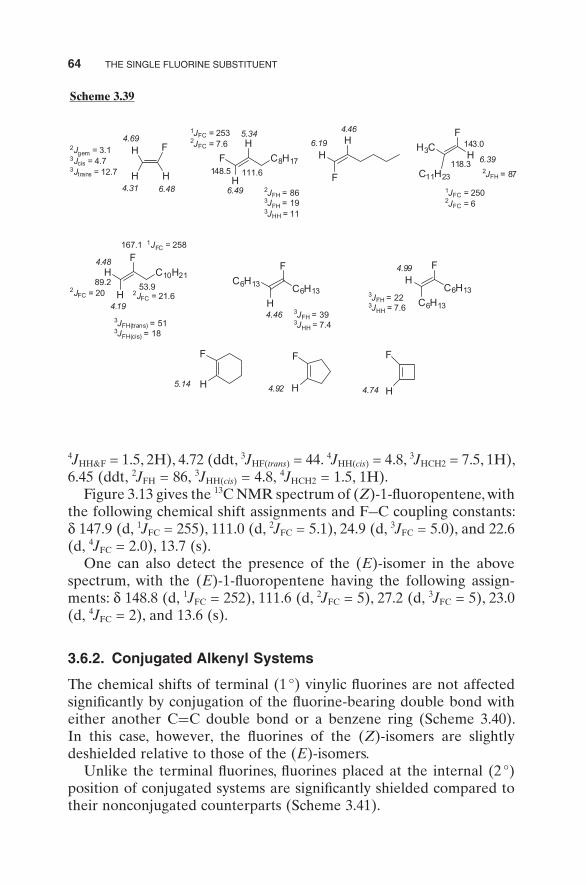
3.6.1.1. 1 H and 13 C NMR Data. The data given in Scheme 3.39 provide some guidelines for proton and carbon NMR chemical shift and cou-pling constant data for fl uoroalkenes. Notice that in all cases, hydrogens that are cis to the fl uorine substituent are deshielded relative to those that are trans .
Figure 3.12 a,b shows the proton NMR spectrum of ( Z ) - 1 - fl uoropen-tene with the following assignments of shift and coupling constant data: δ 0.92 (t, 3 J HH = 7.2, 3H), 1.40 (sextet, 3 J HH = 7.3, 2H), 2.09 (qt, 3 J HH = 7.5,
F
–953JFH(trans) = 513JFH(cis) = 17
F
–88
FFF
–101 –125–85
3JFH = 18.43JFH = 8.83JFH = “small”
C6H13
F
C6H13 C6H13
F
C6H13–111
–1063JFH = 21.5
Scheme 3.38
64 THE SINGLE FLUORINE SUBSTITUENT
F
HH
H4.69
4.31 6.48
2Jgem = 3.13Jcis = 4.73Jt rans = 12.7
1JFC = 2532JFC = 7.6
C8H17F
FH
HH
H5.34
6.49
4.46
6.19
148.5 111.6
2JFH = 863JFH = 193JHH = 11
C11H23
H3C
F
H143.0
118.3
1JFC = 2502JFC = 6
6.392JFH = 87
F FF
C6H13
F
C6H13 C6H13
F
C6H13
C10H21
F
H
H
4.19
4.48
1JFC = 258167.1
89.2 53.9
3JFH(trans) = 513JFH(cis) = 18
2JFC = 21.62JFC = 20
H
H
H H H
4.46
4.99
3JFH = 393JHH = 7.4
3JFH = 223JHH = 7.6
4.925.144.74
Scheme 3.39
4 J HH & F = 1.5, 2H), 4.72 (ddt, 3 J HF( trans ) = 44. 4 J HH( cis ) = 4.8, 3 J HCH2 = 7.5, 1H), 6.45 (ddt, 2 J FH = 86, 3 J HH( cis ) = 4.8, 4 J HCH2 = 1.5, 1H).
Figure 3.13 gives the 13 C NMR spectrum of ( Z ) - 1 - fl uoropentene, with the following chemical shift assignments and F – C coupling constants: δ 147.9 (d, 1 J FC = 255), 111.0 (d, 2 J FC = 5.1), 24.9 (d, 3 J FC = 5.0), and 22.6 (d, 4 J FC = 2.0), 13.7 (s).
One can also detect the presence of the ( E ) - isomer in the above spectrum, with the ( E ) - 1 - fl uoropentene having the following assign-ments: δ 148.8 (d, 1 J FC = 252), 111.6 (d, 2 J FC = 5), 27.2 (d, 3 J FC = 5), 23.0 (d, 4 J FC = 2), and 13.6 (s).
3.6.2. Conjugated Alkenyl Systems
The chemical shifts of terminal (1 ° ) vinylic fl uorines are not affected signifi cantly by conjugation of the fl uorine - bearing double bond with either another C = C double bond or a benzene ring (Scheme 3.40 ). In this case, however, the fl uorines of the ( Z ) - isomers are slightly deshielded relative to those of the ( E ) - isomers.
Unlike the terminal fl uorines, fl uorines placed at the internal (2 ° ) position of conjugated systems are signifi cantly shielded compared to their nonconjugated counterparts (Scheme 3.41 ).
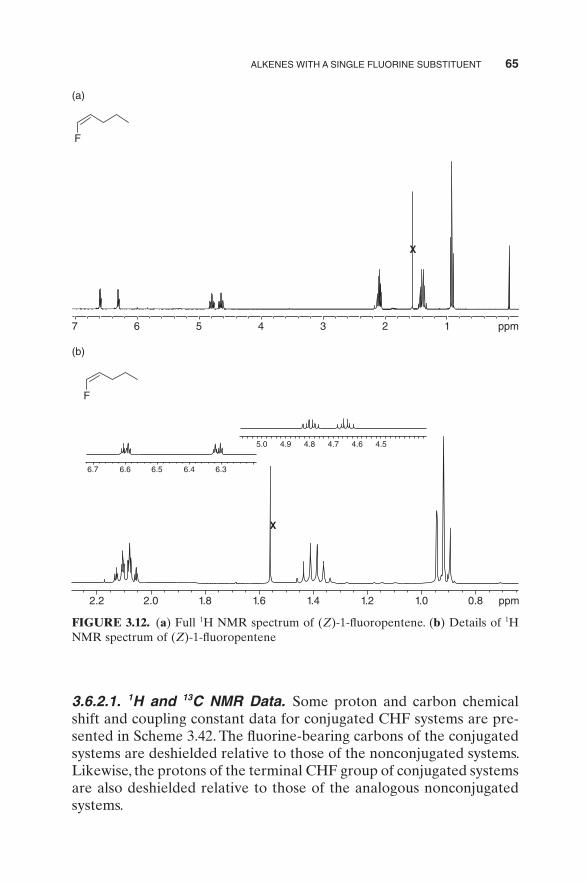
ALKENES WITH A SINGLE FLUORINE SUBSTITUENT 65
FIGURE 3.12. ( a ) Full 1 H NMR spectrum of ( Z ) - 1 - fl uoropentene. ( b ) Details of 1 H NMR spectrum of ( Z ) - 1 - fl uoropentene
F
X
7 6 5 4 3 2 1 ppm
F
X
6.7 6.6 6.5 6.4 6.3
2.2 2.0 1.8 1.6 1.4 1.2 1.0 0.8 ppm
5.0 4.9 4.8 4.7 4.6 4.5
(a)
(b)
3.6.2.1. 1 H and 13 C NMR Data. Some proton and carbon chemical shift and coupling constant data for conjugated CHF systems are pre-sented in Scheme 3.42 . The fl uorine - bearing carbons of the conjugated systems are deshielded relative to those of the nonconjugated systems. Likewise, the protons of the terminal CHF group of conjugated systems are also deshielded relative to those of the analogous nonconjugated systems.

66 THE SINGLE FLUORINE SUBSTITUENT
FIGURE 3.13. 13 C NMR spectrum of ( Z ) - 1 - fl uoropentene
F
140 120 100 80 60 40 20 ppm
112.0 111.5 111.0 110.5 110.0
23.0
22.9
22.8
22.722.622.5
22.4
26.5
26.0
25.5
25.0
24.5
24.0
F
F–127
2JFH = 833JFH(cis) = 17
–1262JFH = 833JFH(trans) = 41
PhF Ph
F–130
2JFH = 843JFH(cis) = 19
–1232JFH(cis) = 813JFH(trans) = 45
Scheme 3.40
F
–1203JFH(trans) = 503JFH(cis) = 163JFCH2 = 26
Ph
F
–1203JFH(trans) = 503JFH(cis) = 17
F
–95
Scheme 3.41

ALKENES WITH A SINGLE FLUORINE SUBSTITUENT 67
3.6.3. Allylic Alcohols, Ethers, and Halides
Oxygen functionalities, such as alcohols, ethers, and acetate groups, and halogens at the allylic position, deshield when the fl uorine is at the terminal position and shield when it is at the internal 2 - position (Scheme 3.43 ).
The F – H spin – spin coupling constants of these compounds remain much the same as those of the simple alkenes.
3.6.3.1. Proton and Carbon NMR Data. Some characteristic 13 C and 1 H NMR data for fl uorinated allylic alcohols and bromide are provided in Scheme 3.44 .
3.6.4. Halofl uoroalkenes and Fluorovinyl Ethers
Geminal chlorine or bromine substituents deshield vinylic fl uorine sig-nifi cantly, whereas a vicinal chlorine substituent shields the fl uorine, much as was the case for the saturated systems. Again similarly, a second vicinal chlorine substituent reverses the trend and shifts the fl uorine signal downfi eld (Scheme 3.45 ).
Few monofl uoro vinyl ethers have been reported in the literature. The NMR data for one example is given. It can be seen that the β - ether substituent shields the fl uorine much more than does a β - chlorine substituent.
Note the impact of the geminal chlorine or bromine substituent to diminish the cis and trans F – H coupling constants in these systems.
H
F
H
150.1
3JFH = 193JHH = 12
1JFC = 258
7.17
6.40
113.8
F
H
H
F6.89
152.9
6.49
148.5
1JFC = 261 1JFC = 280
H
F
H
148.9
1JFC = 256
7.09
6.35
113.8
H
H
F
146.9
1JFC = 267
6.60
5.55
H3CO H3CO
Scheme 3.42

68 THE SINGLE FLUORINE SUBSTITUENT
BrFBr
FCl
F
–1242JFH = 813JFH(cis) = 15
–1252JFH = 823JFH(trans) = 38
–1013JFH(trans) = 473JFH(cis) = 14
OAcF
–1232JFH = 823JFH(cis) = 16
OAc
F
–1252JFH = 833JFH(trans) = 40
OF
–1252JFH = 833JFH(cis) = 17
CH2PhO
F
–1262JFH = 843JFH(trans) = 42
CH2Ph
OHPh
F
–1183JFH(trans) = 35
OH
F
–1063JFH(trans) = 373JF,CH2 = 17
OH
F
I
F
–1083JFH(trans) = 493JFH(cis) = 17
–973JFH(trans) = 463JFH(cis) = 15
OH
FOH
F
Ph
H
H
Ph–114 –1093JFH(trans) = 393JF,CH2 = 14
3JFH(trans) = 203JF,CH2 = 22
OH
Ph
F
–983JFH(cis) = 20
Scheme 3.43
Br
F
OHPh
F
OH
F
OH
F
OH
FPh
H
H
Ph4.26
4.37
158.2
5.78
6.41
159.51JFC = 267 1JFC = 254
H
H
4.09
4.54
4.66
3JFH = 122JHH = 3
3JFH = 49
3JFH = 1790.9
164.5
60.3 1JFC = 2602JFCH2 = 332JFC = 15
26.994.3
161.2
1JFC = 2542JFCH2 = 332JFC = 20
OH
Ph
FH H
5.45
4.05
3JFH = 20
3JHH = 7.5
5.40
4.25
3JFH = 35
3JHH = 8
Scheme 3.44

ALKENES WITH A SINGLE FLUORINE SUBSTITUENT 69
Cl
FF F
Cl
FCl
Cl
Cl
F
–1132JFH = 853JFH(trans) = 523JFH(cis) = 20
–683JFH(trans) = 393JFH(cis) = 7
–1312JFH = 793JFH(cis) = 9
–1282JFH = 773JFH(trans) = 28
–1222JFH = 76
Cl F
ClCl
–80
Br
F
–613JFH(trans) = 423JFH(cis) = 10
Br
F
Br
F
–66–68
3JFH(cis) = 153JFH(trans) = 33
Br
F–71
H
H
Cl
F
Cl
F
Cl
FCl
F H3C
H3C
–72
–74
3JFH(cis) = 12.9
3JFH(trans) = 30.7
–81.5
–82.1
H3CO
H3CO
4JFH = 4.5
FCl –128FOvs.
–1562JFH = 753JFH(trans) = 26
Scheme 3.45
3.6.4.1. Proton and Carbon NMR Data. Some selected chemical shift and coupling constant data from proton and carbon spectra of chloro - and bromofl uoroethylenes are presented in Scheme 3.46 .
3.6.5. Multifl uoroalkenes
3.6.5.1. Vicinal Difl uoroalkenes. Each of the two vicinal fl uorine substituents is signifi cantly shielded by the presence of the other (Scheme 3.47 ). However, it can be seen that when the vicinal fl uorines are cis to each other, both of the fl uorines appear at a much lower fi eld than when they are trans to each other.

70 THE SINGLE FLUORINE SUBSTITUENT
Cl
F
Cl
F
Cl
F
Cl
F H3C
H3CH3C
H3C
Cl
FF F
Cl
FCl
Br
F
Br
F
Br
FH
H5.34
4.91
2JHH = 4.4
H
H H
H H H
H
H
HH
H4.69
4.31 6.48
2Jgem = 3.13Jcis = 4.73Jtrans = 12.7
4.78
4.54
2Jgem = 4
6.11
6.87
3Jtrans = 10.6
5.51 6.79
6.65
5.97
I
IH
H
6.30
5.74
3JHF = 12.3
3JHF = 30.2
145.3106.8
142.1106.4
15.1
16.4
1.92
1.95
4JFH = 4.5
4JFH = 3.2
1JFC = 2992JFC = 29
1JFC = 3132JFC = 9.2
FO
H H 6.726.54
3JHH = 2.4
Scheme 3.46
The observed trans F – F coupling constants are very large ( > 130 Hz), whereas the analogous cis couplings are much smaller ( < 15 Hz). Both the trans - and cis - three - bond F – H couplings are much smaller than those observed for monofl uoroalkenes, being affected more by the vicinal fl uorine than they were by a vicinal chlorine.
3.6.5.1.1. Proton and Carbon NMR Data. Some representative chem-ical shift and coupling constant data are provided in Scheme 3.48 for alkenes with vicinal fl uorines.
Comparing geometric isomers of the type CHF = CFR, a proton cis to fl uorine is more deshielded than one that is trans .
3.6.5.2. Trifl uorovinyl Groups. Trifl uorovinyl groups have charac-teristic chemical shifts and coupling constants that are exemplifi ed in Scheme 3.49 (see Chapter 6 for more details and examples).
3.6.6. α , β - Unsaturated Carbonyl Compounds
The usual deshielding that is observed at the β - position of α , β - unsaturated carbonyl compounds in both proton and carbon NMR

ALKENES WITH A SINGLE FLUORINE SUBSTITUENT 71
FH
F
–1842JF,H = 773JF,F(trans) = 128
–1602JF,H = 773JFF(trans) = 1303JF,H(cis) = 33JF,CH2 = 23
F F
F
F
–165, 2JFH = 723JFH (trans) = 21
–186, 2JFH = 753JFH (cis) = 3.4
F
F
F F
Cl Cl
H
H
–105–156
–130
–174
2JFH = 743JFF(trans) = 1333JFH(cis) =1.2 2JFH = 73
3JFF(cis) = 11.83JFH(trans) =12.4
F F
I H
–109 –136
2JFH = 75
F
FI
H–134
–159
2JFH = 763JFF(trans) = 145
F
FO
H F F
O HF3C F3C
–132
–188
–105 –180
–63–62
2JFH = 723JFF(trans) = 1233JFH(cis) = 4.3
2JFH = 703JFH(trans) = 11.73JFF(cis) = 24.2
F
FCl
Cl F F
Cl Cl
–120 –106
3JFF(trans) = 129 3JFF(cis) = 36
CH2OH
FH3C
F
CH2OH
F
H3C
F
CH2OH
FCl
FCH2OH
F
Cl
F
–1453JFF(trans) = 134
–1343JF,CH2 = 25
–1273JFF(cis) = 8
–1483JF,CH2 = 24
–1223JFF(trans) = 138
–1523JF,CH2 = 22
–1043JFF(cis) = 14
–1413JF,CH2 = 22
H
F F
I
F F–165 –143
3JFF(cis) = 11.82JFH = 73
3JFF(cis) = 5.1
–101 –109
F
H F
–175
–167
3JFF(trans) = 1252JFH = 75
Scheme 3.47

72 THE SINGLE FLUORINE SUBSTITUENT
F
FO
H F F
O HF3C F3C
F
F
F F
Cl
Cl
H
H 6.39
7.26
F
FI
H F F
I H
7.50
6.32
104.1 146.2
1JFC = 3162JFC = 57
1JFC = 2482JFC = 56 97.9 138.5
1JFC = 3312JFC = 19
1JFC = 2732JFC = 9
H
F F
6.90
134.1148.51JFC = 2562JFC = 15.6
1JFC = 2472JFC = 10.2
F
H F7.45 3JFH = 6
3JFH = 17
4.59
4.86
F
FH
H 7.54
F F
H H 6.62
3JHH = 9.5
3JHH = 2.0
Scheme 3.48
FF
F
2JFF = 903JFH(trans) = 114
–174
2JFF = 903JFF (cis) = 32
–126
–104
Scheme 3.49
αβ
O
Fluorinedeshielding
Fluorineshielding
Scheme 3.50

FLUOROAROMATICS 73
is also observed for a fl uorine substituted at this position (Scheme 3.50 ).
Fluorines in the β - position are deshielded by as much as 20 ppm relative to a simple fl uoroalkene, whereas those at the α - position are shielded by about 20 ppm, similar to fl uorines at the 2 - position of a 1,3 - diene (Scheme 3.51 ). Generally, in pairs of geometric isomers, fl uo-rines that are cis to the carbonyl function appear at higher fi elds than those that are trans to the carbonyl function.
3.6.6.1. 1 H and 13 C NMR Data. The carbons at the β - position of α , β - unsaturated carbonyl compounds are also deshielded relative to ordinary terminal fl uoroalkenes (Scheme 3.52 ).
C6H13F
H
–1122JFH = 83
OCH3
O
H
H
F
OC8H17
O
–1172JFH = 83
Hn–Bu
F–823JFH(trans) = 403JF,CH2 = 18
C2H5
O
H
n–Bu
F
–783JFH(cis) = 213JF,CH2 = 26
C2H5
O
Hn-Bu
F
–793JFH(trans) = 333JF,CH2 = 16
OCH3
O
H
n–Bu
F
OCH3
O
–763JFH(cis) = 203JF,CH2 = 26
H
F
Ph
OCH3
O
–973JFH(trans) = 33
H
C2H 5
F
OHO
–733JFH(cis) = 193JF,CH2 = 24
FH
H
–1163JFH(trans) = 463JFH(cis) = 17
CH3
O
F
H
H –1173JFH(trans) = 463JFH(cis) = 17
O-n-BuO
Scheme 3.51

74 THE SINGLE FLUORINE SUBSTITUENT
C6H13F
H OCH3
O
H
H
F
OC8H17
O7.56
5.78
7.53165.3
162.9 106.8158.0 118.6
167.22JFH = 793JFH = 153JHH = 11
2JFH = 82
1JFC = 2802JFC = 153JFC = 23
1JFC = 2762JFC = 113JFC = 19
H3CH2CH2C
F
OHO
H5.60179.3
31.6
100.8
172.3
1JFC = 276
2JFC = 30
2JFC = 22 3JFC = 27
3JFH = 19.3
FPh
H OEtO6.93
3JFH = 35
Fn-C7H15
H O-t-BuO6.05
3JFH = 34
F
n-C7H15
H
O-t-BuO
5.85
3JFH = 21
147.01JFC = 268
FH
H OCH2PhO5.683JFH = 43
151.3102.8
160.2
1JFC = 2652JFC = 153JFCO = 35
5.323JFH = 13
Scheme 3.52
F
H OEtO
–1263JFH(trans) = 35
FH
OEtO
–118
3JFH(cis) = 21
F
H
n-C7H15
O-t-BuO
–130.63JFH(trans) = 34
FH
n-C7H15 O-t-BuO
–121.33JFH(cis) = 21
Scheme 3.51 (cont’d)

FLUOROAROMATICS 75
3.7. ACETYLENIC FLUORINE
There has been but one report of a fl uorine NMR spectrum of a fl uo-roacetylene, that of the parent fl uoroacetylene . 5 It was reported to have a fl uorine chemical shift of − 210 ppm.
3.8. ALLYLIC, PROPARGYLIC, AND BENZYLIC FLUORIDES
The proximity of carbon – carbon double or triple bonds or a phenyl substituent, as in allylic, propargylic, or benzylic systems, has very little impact upon a fl uorine substituent ’ s chemical shift (Scheme 3.53 ). Note that one would not expect allyl fl uoride and methallyl fl uoride to have the same chemical shift.
3.8.1. 1 H and 13 C NMR Data
Some typical proton and carbon chemical shift and coupling constant data for allylic and benzylic systems are given in Scheme 3.54 . An alkenyl substituent or a phenyl substituent on either a CH 2 F or a – CHF - group has virtually no effect upon that carbon ’ s chemical shift, and they also only affect the proton chemical shift by about 0.5 ppm.
3.9. FLUOROAROMATICS
Ring current (anisotropic) effects do not play a signifi cant role in fl uorine NMR. Therefore, fl uorine substituents on a benzene ring absorb in the general region of fl uoroalkenes, with fl uorobenzene and 1 - fl uoronaphthalene having chemical shifts of − 113.5 and − 123.9 ppm, respectively. The fl uorine NMR of fl uorobenzene is shown in Fig. 3.14 .
3.9.1. Monofl uoroaromatics
Table 3.2 provides chemical shift data for various substituted fl uoro-benzenes. 6 The chemical shifts of para - substituted fl uorobenzenes have a reasonable correlation with the σ p values of the substituents, the more electron - withdrawing substituents leading to greater deshielding of the p - fl uorine. The chemical shifts of ortho - substituted fl uorobenzenes also exhibit a rough correlation, but there are some signifi cant aberrations. The chemical shifts of meta - substituted fl uorobenzenes exhibit no cor-relation and vary over a much smaller range.
It should be noted (and can be seen from Table 3.2 ) that there can be signifi cant solvent effects upon the chemical shifts of fl uorobenzenes.

76 THE SINGLE FLUORINE SUBSTITUENT
CH2F CH2F Ph-CH2F
–2162JFH = 48
–2182JFH = 48
–206
H3C
F
CH3 Ph
F
CH3 Ph
F
Ph
1o
2o
–165 –167 –167
3o Ph
Ph
F
PhH3C
CH3
F
CH3
–130 –127
F
–218
CH2FH3C
CH2FPh
–208 –2112JFH = 48
CH3
F–170
2JFH = 48
CH3
F
Ph
–166
CH3
F
–216
Scheme 3.53
1JFC = 166 1JFC = 167
CH2-FCH3
F
84.6
5.355.6
90.9
F
4.852JFH = 46
H3C F
H
H4.75
5.27
5.6583.5
125.7
132.5
17 .6
Ph F
H
H
6.36
5.01
6.69
123.0
133.7
83.4
1JFC = 1612JFC = 163JFC = 12.3
1JFC = 1602JFC = 16.33JFC = 12.34JFC = 2.5
CH3
F
CH3
F
Ph
HH5.02
5.26
1.541.3720.9
89.9
137.7
116.0
21.5
90.2
128.8
131.81JFC = 1632JFC = 18.9, 25(C1)3JFC = 11.81JFC = 163
2JFC = 16.3, 24(C1)3JFC = 11.7
CH3
F
4.73
1.76
4.98
Scheme 3.54

FLUOROAROMATICS 77
FIGURE 3.14. 19 F NMR of fl uorobenzene
F
–113.2 –113.3 –113.4 –113.5 –113.6 –113.7 –113.8 –113.9–113.1
3.9.1.1. Interplay of 19 F , 13 C , and 1 H NMR Spectra for Fluoroaromatics. The two - dimensional character of the 1 H NMR spectrum (Fig. 3.15 ) makes analysis by examination impossible.
A fl uorine substituent on benzene has a characteristic effect upon the 13 C spectrum of benzene, and it couples in a distinctive and highly consistent manner with the ipso , ortho , meta , and para carbons (Scheme 3.55 ).
The 13 C NMR of fl uorobenzene itself, shown in Fig. 3.16 , exemplifi es this nicely with four doublets being clearly observable. The chemical shifts seen in this spectrum are slightly different from those given in Scheme 3.55 because of the choice of solvent (C 6 D 6 ).
3.9.1.2. Complete NMR Analysis of o - , m - , and p - Nitrofl uorobenzenes. The complete set of NMR data for one series of o - , m - , and p - disubstituted fl uorobenzene compounds, that of the nitrofl uoroben-zenes, will serve to further exemplify the interplay of 19 F, 13 C, and 1 H chemical shifts and coupling constants that provide unique insight into the structures of disubstituted fl uorobenzenes. These data are given in Tables 3.3 – 3.5 .
3.9.1.3. Coupling Constants. The usual three - bond H – H coupling constant in fl uorobenzenes is about 8 Hz, whereas the four - bond cou-pling constant is between 1 and 3 Hz. Five - bond coupling is usually not observed. Likewise, the three - bond F – H coupling constant is about 8 Hz, the four - bond value 5 – 6 Hz, and the fi ve - bond coupling constant about 1 Hz.

78 THE SINGLE FLUORINE SUBSTITUENT
TAB
LE
3.2
. 19
F C
hem
ical
Shi
fts
for
Flu
orob
enze
nes 6
Subs
titu
ent
orth
o
met
a
para
σ p
val
ue
Ace
tone
- d 6
D
MSO
A
ceto
ne - d
6
DM
SO
Ace
tone
- d 6
D
MSO
CO
Cl
− 109
.5
− 113
.6
− 101
.8
0.61
N
O 2
− 1
19.7
− 1
19.0
− 1
10.0
− 1
09.5
− 1
03.0
− 1
02.4
0.
78
CN
− 1
08.6
− 1
07.9
− 1
10.9
− 1
10.0
− 1
04.0
− 1
02.8
0.
66
CH
O
− 122
.4
− 120
.7
− 112
.6
− 111
.6
− 104
.3
− 103
.2
0.42
C
OC
H 3
− 1
10.6
− 1
10.0
− 1
13.1
− 1
12.0
− 1
07.1
− 1
05.9
0.
50
CO
2 H
− 110
.0
− 110
.1
− 113
.3
− 112
.0
− 107
.2
− 106
.5
0.45
C
F 3
− 1
15.8
− 1
15.4
− 1
11.4
− 1
10.3
− 1
08.0
− 1
06.8
0.
54
CO
NH
2
− 113
.6
− 113
.3
− 113
.4
− 112
.6
− 109
.8
− 109
.2
0.36
H
− 1
13.8
− 1
12.6
0.
0 I
− 94.
4 − 1
06.2
− 1
10.9
− 1
10.3
− 1
14.8
− 1
14.2
0.
18
Br
− 108
.1
− 107
.7
− 110
.8
− 110
.0
− 115
.6
− 114
.7
0.23
C
l − 1
16.3
− 1
15.9
− 1
11.2
− 1
10.3
− 1
16.7
− 1
15.2
0.
23
F
− 139
.7
− 138
.8
− 110
.6
− 109
.5
− 120
.0
− 119
.4
0.06
C
H 3
− 1
18.4
− 1
17.3
− 1
14.9
− 1
13.7
− 1
19.2
− 1
18.0
− 0
.17
NH
Ac
− 125
.6
− 124
.6
− 112
.8
− 111
.8
− 120
.3
− 119
.4
0.0
OC
H 3
− 1
36.1
− 1
35.3
− 1
12.6
− 1
11.4
− 1
25.2
− 1
24.0
− 0
.27
OH
− 1
38.0
− 1
36.3
− 1
13.2
− 1
12.1
− 1
26.8
− 1
25.0
− 0
.37
NH
2
− 136
.3
− 134
.9
− 115
.6
− 113
.5
− 129
.7
− 129
.4
− 0.6
6

FIGURE 3.15. 1 H NMR spectrum of fl uorobenzene (benzene - d 6 )
F
6.95 6.90 6.85 6.80 6.75 6.70 ppm
F 245.3 Hz
21.0 Hz
7.7 Hz
3.3 Hz
F–C benzenespin–spincouplingconstants
F13CChemical shifts
offluorobenzene
162.8115.2
130.0
124.0
compared to
128.4
(in CDCl3)
Scheme 3.55
FIGURE 3.16. 13 C NMR of fl uorobenzene (benzene - d 6 )
F
160 150 140 130 120 ppm
131.
013
0.8
130.
613
0.4
130.
2
124.
612
4.5
124.
412
4.3

80 THE SINGLE FLUORINE SUBSTITUENT
The F coupling to carbon can vary considerably for the carbon directly substituted (ipso), depending on its substitution environment, but it is always very large, 250 Hz or larger. F coupling to the ortho - position is usually about 20 – 26 Hz, to the meta - position about 8 – 10 Hz, and to the para - position about 4 Hz.
TABLE 3.3. NMR Analysis of ortho - Nitrofl uorobenzene
Coupling Constants to Carbon (Hz)
Chemical Shifts Coupling Constants (Hz)
F H3 H4 H5 H6 F/H H/H
C1 − 262.6 − 8.1 − 1.9 − 11.5 − 5.0 F
NO2
–119.6
H
H
H
H
8.14
7.48
7.83
7.50
156.1
138.2126.8
125.8
136.9
119.1
4 J FH3 = 7.9 3 J H3H4 = 8.1
C2 8.8 — — — — 5 J FH4 = − 0.9 3 J H4H5 = 7.5 C3, − 2.6 169.4 2.6 9.0 1.0 4 J FH5 = 4.6 3 J H5H6 = 8.5 C4 4.0 0.9 167.8 0.9 8.7 3 J FH6 = 11.5 4 J H3H5 = 1.7 C5 8.8 9.2 1.5 165.5 0.5 4 J H4H6 = 1.2 C6 20.7 1.3 8.3 1.3 167.2
TABLE 3.4. NMR Analysis of meta - Nitrofl uorobenzene
Coupling Constants to Carbon (Hz)
Chemical Shifts Coupling Constants (Hz)
F H2 H4 H5 H6 F/H H/H
C1 − 249.0 − 6.0 − 1.4 − 11.9 − 4.5 F
H
–110.2
H
H
H
NO2
7.65
7.76
8.12
8.00163.2
111.7150.0
120.3
132.2
122.8
3 J FH2 = 8.9 3 J H4H5 = 8.3
C2 26.5 171.1 − 5.2 − 1.5 − 4.3 5 J FH4 = − 1.0 3 J H5H6 = 8.3 C3, 8.8 − 3.9 − 1.5 − 11.4 − 2.6 4 J FH5 = 5.7 4 J H2H4 = 2.2 C4 3.1 − 4.2 171.3 − 1.7 − 8.0 3 J FH6 = 8.3 4 J H2H6 = 2.6 C5 8.4 0.0 0.0 167.9 0.0 4 J H4H6 = 0.9 C6 21.6 4.0 8.0 − 2.4 168.1
TABLE 3.5. NMR Analysis of para - Nitrofl uorobenzene
Coupling Constants to Carbon (Hz)
Chemical Shifts Coupling Constants (Hz)
F H2 H3 H5 H6 F/H H/H
C1 − 255.7 4.6 10.9 − 10.9 − 4.6 F
H
–103.5
H
H
NO2
H8.35
7.43167.2
117.3127.2
145.5
3 J FH2 = 8.2 3 J H2H3 = 9.2
C2 24.1 168.9 0.0 0.05 4.5 4 J FH3 = 4.8 C3, 10.2 0.0 171.4 5.4 0.0 C4 4.5 10.2 5.8 5.8 10.2

FLUOROAROMATICS 81
Ipso (one - bond) coupling of H to C is consistently between 165 and 172 Hz, whereas two - bond H – C coupling constants (0 – 5 Hz) are usually much smaller than three - bond H – C couplings (4 – 10 Hz).
Usually, a careful analysis of the combination of fl uorine, proton, and carbon NMR chemical shifts and spin – spin coupling constants will provide defi nitive information regarding the structure of disubstituted fl uoroaromatics.
3.9.2. Fluoropolycyclic Aromatics: Fluoronaphthalenes
The isomeric 1 - and 2 - fl uoronaphthalenes have fl uorine chemical shifts of − 124 and − 116 ppm, respectively. A full analysis of the proton and carbon spectra of 1 - fl uoronaphthalene is given in Scheme 3.56 . NMR data for a number of other fl uoropolycyclic aromatic compounds are available. 7
3.9.2.1. Steric Deshielding of Fluorine Nucleus. As can be seen from the data in Table 3.2 and for 4 - methyl - 1 - fl uoronaphthalene (Scheme 3.57 ), ordinarily a methyl group on a fl uoroaromatic will give rise to shielding of the fl uorine nucleus. However, a methyl group in the peri - 8 - position of 1 - fl uoronaphthalene provides a rare example of steric deshielding of a fl uorine atom. 8 Other groups in this position, such as ethyl and acetyl, give rise to similar deshielding effects, with
F
H
H
HH
H
H
H7.17 (3JFH = 10.7)
7.42 (4JFH = 5.4)
7.65 (5JFH ~ 0.5)7.88(7JFH = 2.3)
8.13(4JFH ~ 0.6)
7.56 (5JFH ~ 0)
7.55 (6JFH ~ 0)
F158.8 (1JFC = 252)
109.4 (2JFC = 19.8)
125.6 (3JFC = 8.4)
123.6(4JFC = 4.1)
134.9(5JFC = 4.8)
127.5(6JFC = 3.2)
126.8 (5JFC = 0.9)
126.2 (4JFC = 1.8)
120.6(3JFC = 5.2)
123.72JFC = 16.5)
Scheme 3.56

82 THE SINGLE FLUORINE SUBSTITUENT
the t - butyl group providing the largest observed effect, deshielding the fl uorine by approximately 28 ppm (to − 96 ppm ). Further discussion of such deshielding effects has been provided in Chapter 2 , Section 2.2.1 .
One should also note the signifi cant 7.5 Hz F – H coupling constant between the methyl hydrogens of the 8 - methyl - 1 - fl uoronaphthalene and its fl uorine substituent. This likely derives, at least in part, from through - space F – H coupling (see Chapter 2 , Section 2.3.1 ).
3.9.3. Polyfl uoroaromatics
A second fl uorine substituent shields in the ortho - and especially in the para - position, but one in the meta - position deshields, with 1,3 - 5 - trifl uorobenzene having the most deshielded fl uorines in a polyfl uoro-aromatic system (Scheme 3.58 ). On the other hand, hexafl uorobenzene has highly shielded fl uorines. The fl uorine spectra of these multifl uoro-benzenes are second order in nature and their appearance is thus not generally predicable on the basis of fi rst - order logic.
FFF CH3
CH3
–124 –127 –1135JFCH3 = 7.5
Scheme 3.57
F F
F
F
F
F
F
F
FF
F
F
F
F
F
F
–113 –119 –110–139
–101
–162
Scheme 3.58
FLUOROHETEROCYCLES 83
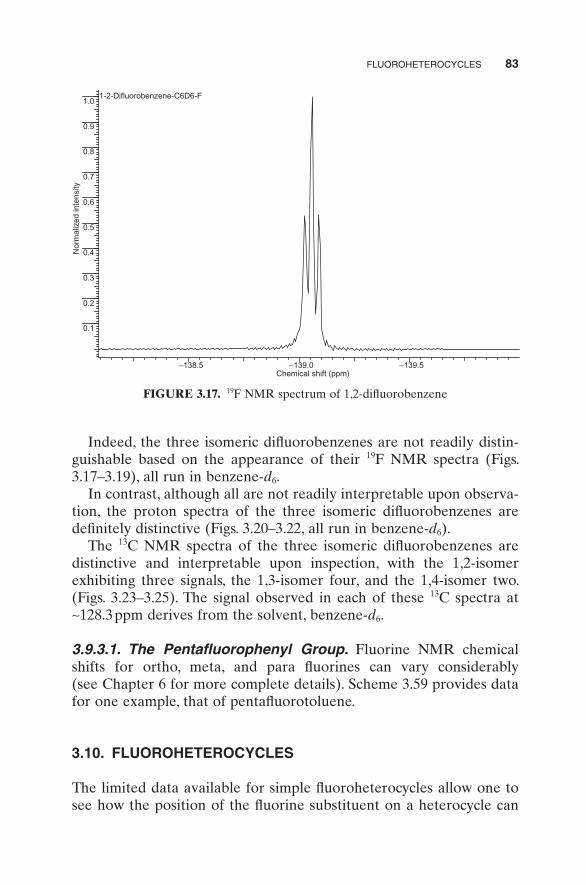
FIGURE 3.17. 19 F NMR spectrum of 1,2 - difl uorobenzene
1-2-Difluorobenzene-C6D6-F
–138.5 –139.0 –139.5Chemical shift (ppm)
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Nor
mal
ized
inte
nsity
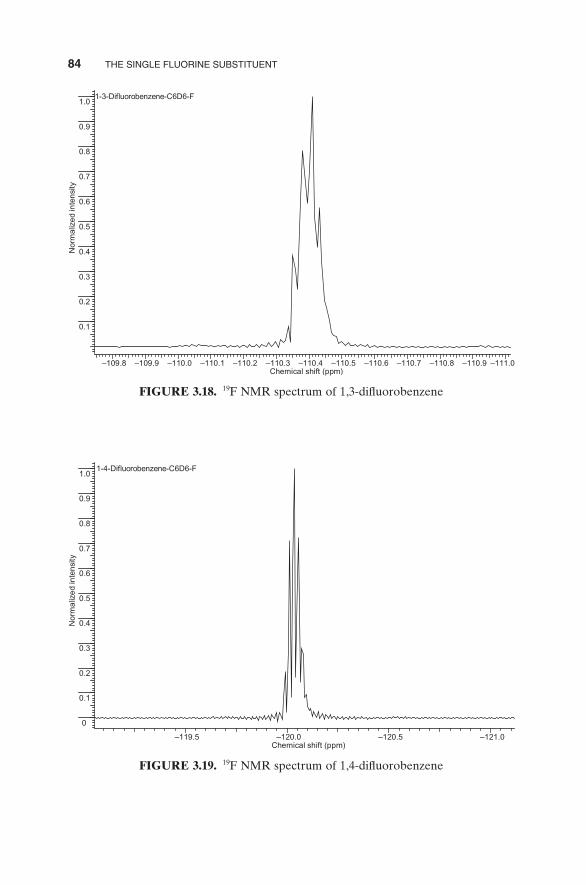
Indeed, the three isomeric difl uorobenzenes are not readily distin-guishable based on the appearance of their 19 F NMR spectra (Figs. 3.17 – 3.19 ), all run in benzene - d 6 .
In contrast, although all are not readily interpretable upon observa-tion, the proton spectra of the three isomeric difl uorobenzenes are defi nitely distinctive (Figs. 3.20 – 3.22 , all run in benzene - d 6 ).
The 13 C NMR spectra of the three isomeric difl uorobenzenes are distinctive and interpretable upon inspection, with the 1,2 - isomer exhibiting three signals, the 1,3 - isomer four, and the 1,4 - isomer two. (Figs. 3.23 – 3.25 ). The signal observed in each of these 13 C spectra at ∼ 128.3 ppm derives from the solvent, benzene - d 6 .
3.9.3.1. The Pentafl uorophenyl Group. Fluorine NMR chemical shifts for ortho, meta, and para fl uorines can vary considerably (see Chapter 6 for more complete details). Scheme 3.59 provides data for one example, that of pentafl uorotoluene.
3.10. FLUOROHETEROCYCLES
The limited data available for simple fl uoroheterocycles allow one to see how the position of the fl uorine substituent on a heterocycle can
84 THE SINGLE FLUORINE SUBSTITUENT
FIGURE 3.18. 19 F NMR spectrum of 1,3 - difl uorobenzene
1-3-Difluorobenzene-C6D6-F
–109.8 –109.9 –110.0 –110.1 –110.2 –110.3 –110.4 –110.5 –110.6 –110.7 –110.8 –110.9 –111.0Chemical shift (ppm)
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Nor
mal
ize
d in
ten
sity
FIGURE 3.19. 19 F NMR spectrum of 1,4 - difl uorobenzene
1-4-Difluorobenzene-C6D6-F
–119.5 –120.0 –120.5 –121.0Chemical shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Nor
mal
ize
d in
ten
sity

FLUOROHETEROCYCLES 85
FIGURE 3.20. 1 H NMR spectrum of 1,2 - difl uorobenzene
1-2-Difluorobenzene-C6D6-H
7.0 6.9 6.8 6.7 6.6 6.5 6.4 6.3 6.2Chemical shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Nor
mal
ize
d in
ten
sity
FIGURE 3.21. 1 H NMR spectrum of 1,3 - difl uorobenzene
1-3-Difluorobenzene-C6D6-H
6.90 6.85 6.80 6.75 6.70 6.65 6.60 6.55 6.50 6.45 6.40 6.35 6.30 6.25 6.20Chemical shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Nor
ma
lized
inte
nsity

86 THE SINGLE FLUORINE SUBSTITUENT
FIGURE 3.22. 1 H NMR spectrum of 1,4 - difl uorobenzene
1-4-Difluorobenzene-C6D6-H
6.90 6.85 6.80 6.75 6.70 6.65 6.60 6.55 6.50 6.45 6.40 6.35 6.30 6.25 6.20 6.15Chemical shift (ppm)
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Nor
mal
ize
d in
tens
ity
FIGURE 3.23. 13 C NMR spectrum of 1,2 - difl uorobenzene
F
F
150 145 140 135 130 125 120 115 ppm
126.0
118.5 118.0 117.5
125.5 125.0 124.5
signifi cantly affect its chemical shift. For all nitrogen and oxygen het-erocycles, a fl uorine substituent on a carbon bound to the nitrogen or oxygen will be deshielded compared to a fl uorine at any other position. The situation is reversed for sulfur heterocycles, although the observed differences are small.

FLUOROHETEROCYCLES 87
FIGURE 3.24. 13 C NMR spectrum of 1,3 - difl uorobenzene
FF
160 150 140 130 120 110 ppm
166 165 164 163 162
132.0 131.5 131.0 130.5
112.5112.0111.5
111.0
110.5110.0
105.5105.0104.5104.0103.5103.0
FIGURE 3.25. 13 C NMR spectrum of 1,4 - difl uorobenzene
F
F
160 150 140 130 120 110 ppm
118.0
117.5
117.0
116.5116.0
CH3
F
F
F
F
F –144
–164
–159
3J23 = 20.43J34 = 18.95J25 = 8.6
Scheme 3.59

88 THE SINGLE FLUORINE SUBSTITUENT
3.10.1. Fluoropyridines and Quinolines
In the case of pyridine, large differences in chemical shift are observed for fl uorines at the 2 - , 3 - , and 4 - positions, with fl uorines at the 2 - position of pyridines and quinolines being the most deshielded, and those at the 3 - position being the most shielded. (Scheme 3.60 ).
The chemical shifts for fl uoropyrimidines, a quinoxoline, and for 5 - fl uorouracil are also provided in Scheme 3.61 .
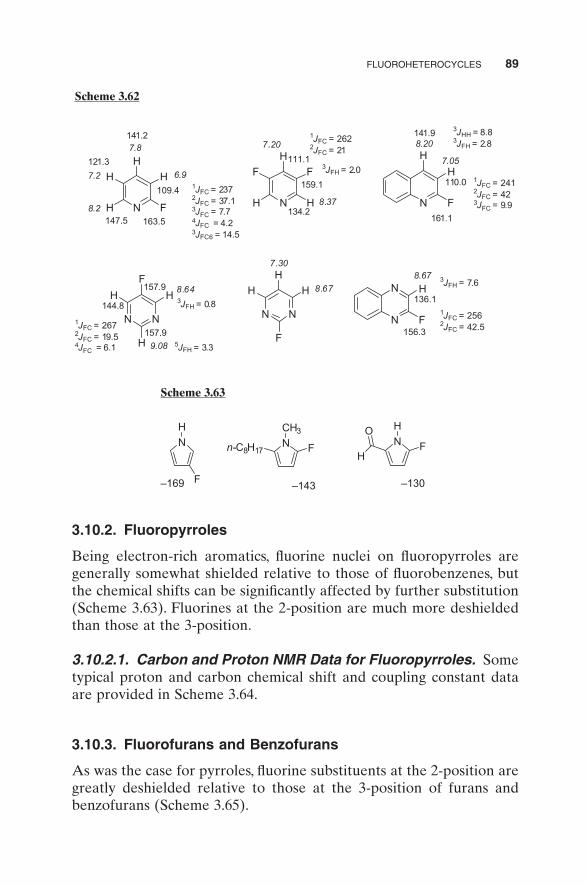
3.10.1.1. Carbon and Proton NMR Data. 13 C and 1 H NMR chemical shift and coupling constant data for 2 - fl uoropyridine, 2 - fl uoroquino-lines, and 2 - fl uoroquinoxoline are provided in Scheme 3.62 .
N N NF
F
F
–68
–126
–103
N–63
N
FF–124
N
F
HN F
H –79
–114
NN
–123 F
F–126
F
3JHF = 9.44JHF = 5.6
3JHF = 10.34JHF = 4.7
N
F
–114
Scheme 3.60
N
N
F–74
N N
N N
N N
F
F
–44
–63
–137F
N
N
O
O
H
H
F
–171
5JHF = 3.3
H3C
CH3
N N
F
CH3Cl
–44
N N
F
CH3
–142
Scheme 3.61
FLUOROHETEROCYCLES 89
N N
F
HH
H
N F N F
N
N
F
H
H
H
H
H
H
H
6.9
7.8
7.2
8.2
163.5
109.4
141.2
121.3
147.5
1JFC = 2372JFC = 37.13JFC = 7.74JFC = 4.23JFC6 = 14.5
7.05
8.20
161.1
110.0
141.9
1JFC = 2412JFC = 423JFC = 9.9
8.67
3JHH = 8.83JFH = 2.8
3JFH = 7.6
156.3
136.11JFC = 2562JFC = 42.5
9.08
8.64
5JFH = 3.3
3JFH = 0.8N N
F
H
H
H 8.67
7.30
157.9
144.8
157.9
1JFC = 2672JFC = 19.54JFC = 6.1
N H
F
H
F
H 8.37
7.20
111.1
159.1
134.2
1JFC = 2622JFC = 21
3JFH = 2.0
Scheme 3.62
3.10.2. Fluoropyrroles
Being electron - rich aromatics, fl uorine nuclei on fl uoropyrroles are generally somewhat shielded relative to those of fl uorobenzenes, but the chemical shifts can be signifi cantly affected by further substitution (Scheme 3.63 ). Fluorines at the 2 - position are much more deshielded than those at the 3 - position.
3.10.2.1. Carbon and Proton NMR Data for Fluoropyrroles. Some typical proton and carbon chemical shift and coupling constant data are provided in Scheme 3.64 .
3.10.3. Fluorofurans and Benzofurans
As was the case for pyrroles, fl uorine substituents at the 2 - position are greatly deshielded relative to those at the 3 - position of furans and benzofurans (Scheme 3.65 ).
N N
H CH3N
H
F
Fn-C8H17 FH
O
–169 –143 –130
Scheme 3.63

90 THE SINGLE FLUORINE SUBSTITUENT
NN
CH3CH3
F
Fn-C8H17HH
HH H
N
CH3
H
FH
H
6.51
5.79
6.44
5.395.82
6.23
146.4
82.7101.2
124.2
1JFC = 2572J
FC = 10.33JFC = 4.84JFC = 1.5
5.275.56
Scheme 3.64
OO O
F
Fn-C8H17 n-C8H17
–165–118 –176
Ph
F
OF
n-Bu
–120
Scheme 3.65
OF
FO
FF
–123 –182
H
H
HH
3JFH = 4.63JFH = 8
Scheme 3.66
The pair of difl uorofurans given in Scheme 3.66 also exhibits the signifi cant difference in fl uorine chemical shift between the 2 - and the 3 - positions of furan.
3.10.3.1. Carbon and Proton NMR Data. Some typical proton and carbon chemical shift and coupling constant data for fl uorofurans are provided in Scheme 3.67 .
3.10.3.2. Fluorodibenzofurans. Three fl uorodibenzofurans have been reported. Their fl uorine NMR data are given in Scheme 3.68 .
3.10.4. Fluorothiophene and Benzothiophene
Unlike the nitrogen of pyrroles and the oxygen of furans, the sulfur of thiophenes does not signifi cantly affect fl uorine chemical shifts either inductively or as an electron donor. Thus, the fl uorine chemical

FLUOROHETEROCYCLES 91
OF
n-Bu
OF
C8H17O
FH
H
C8H17
HH5.26
5.87
7.05
6.23
138.6
148.6102.8
138.5146.2
105.6115.26
156.5 1JFC = 2732J
FC = 12.73JFC = 04JFC = 1.2
1JFC = 2452J
FCH = 20.42JFC = 25.63JFC = 8.8
OF
FO
FF
H
H
HH
7.13
5.28
3JFH = 4.6
3JFH = 8
157.1
90.6
1JFC = 2782J
FC = 12
Scheme 3.67
O OO
F
FF–114 –121 –119
Scheme 3.68
shifts of fl uorothiophenes are generally in the region of electron - rich fl uorobenzenes (Scheme 3.69 ). Moreover, in this heterocycle, fl uorines at the 2 - position are slightly shielded compared to those at the 3 - position.
3.10.4.1. Carbon and Proton NMR Data. Some typical carbon and proton chemical shift and coupling constant data for fl uorothiophenes are given in Scheme 3.70 . Note that the two - , three - , and four - bond F – C
SF
n-Bu
S FH3C
–134
S F
–135
SH3C
F
–128
–131
S
F
S
F
C8H17
–137 –133
Scheme 3.69

92 THE SINGLE FLUORINE SUBSTITUENT
coupling constants are dampened when the fl uorine is bound to the carbon - bearing sulfur.
3.10.5. Fluoroimidazoles and Pyrazoles
Fluorine, proton, and carbon NMR spectra for 4 - fl uoro - and 4,5 - difl uoroimidazole, for 4 - fl uoropyrazole, and for substituted 3 - fl uoropyrazoles have been reported (Scheme 3.71 ).
Comparing the monofl uoropyrazoles with the difl uoropyrazoles, notice the signifi cant effect of the second fl uorine on the chemical shifts of both fl uorines!
3.11. OTHER COMMON GROUPS WITH A SINGLE FLUORINE SUBSTITUENT
Two other functional groups that contain a single fl uorine substituent are acyl fl uorides and sulfonyl fl uorides. Such fl uorines are among the rare few that absorb in the highly deshielded region downfi eld of CFCl 3 .
3.11.1. Acyl Fluorides
Acyl fl uorides are one of the few examples of a single fl uorine absorb-ing at lower fi eld than CFCl 3 (Scheme 3.72 ).
3.11.1.1. 13 C NMR Data. Some typical carbon NMR data are given in Scheme 3.73 for acid fl uorides and for carbonyl fl uoride.
1JFC = 2892JFC = 10
159.2
115.5
SF
n-Bu
S FC8H17S F
S
F
S
F
C8H17
6.356.54
6.74
H
H H
H H
H
H H
H
H
6.69
7.17
6.83
6.426.53
6.74
6.93
119.5
116.7 136.6
121.4
163.3
105.7119.4
142.8
1JFC = 2872JFC = 10.23JFC = 3.9
1JFC = 2532JFCH = 27.32JFC = 18.73JFC = 10.1
3JHH = 5.4 3JFH = 3.4
158.5
124.8 117.2
103.1
2JFC = 26.9
2JFC = 21.1
3JFC = 9.1
1JFC = 258
3JFH = 3.4
S
F
H CO2CH3
H7.42
6.86118.5
160.2112.5
130.1
2JFC = 24.9
1JFC = 2762JFC = 12.5
3JFC = 10.5 160.9
3JFC = 3.8
3JFH = 3.8
3JHH = 5.5
Scheme 3.70

OTHER COMMON GROUPS WITH A SINGLE FLUORINE SUBSTITUENT 93
N
HN
FN
HN
F
F
H
HH 6.97
118.83JFC = 11
–160
130.91JFC = 245
12.5
7.26
3JFC = 16
128.9
94.4
157.01JFC = 245
2JFC = 38
6.563JFH = 8
–140
N
HNH
FH
7.4410.45
–181 116.6144.6
1JFC = 248
2JFC = 25
3JFH = 4.5
N
HNPh
n-BuF
–139
163.8N
NF
n-BuCH3
CH3
–133
152.0
1JFC = 244
1JFC = 275
NNH
FH
Ph
–176 151.2
1JFC = 249
NNH
HF
CH3
NNH
FH
CH3
NNF
HH
CH3
–129 –175
–136
5.76
7.30
7.28
7.28
5.727.36
4JFH = 2.5
3JFH = 4.63JFH = 6.0
3JFH = 2.3
3JFH = 6.0
3JFH = 4.6
3JHH = 2.3
3JHH = 2.0
NNH
FF
CH3
NNF
FH
CH3
NNF
HF
CH3
–139 –185
–124
–185
3JFF = 9
3JFF = 0
3JFF = 5.5
–143
–120
Some difluoropyrazoles:
Scheme 3.71
F F
OCarbonylfluoride
–23
H3C F
O
H3CH2CH2C F
OF
O
F3C F
O
+493JFH = 7
+42+21
+623JFF = 6
Scheme 3.72
94 THE SINGLE FLUORINE SUBSTITUENT
H3C F
O
F
O160.8, 1JFC = 354
18.7, 1JFC = 354
157.3, 1JFC = 344
F F
O
133.61JFC = 308
Scheme 3.73
F O
O
CH3
–19
Scheme 3.74
SF
SO2FO
+6 +65
Scheme 3.75
3.11.3. Sulfi nyl and Sulfonyl Fluorides
Chapter 6 will provide a more thorough coverage of compounds with fl uorine bound directly to sulfur, but typical examples of sulfi nyl and sulfonyl fl uorides are given in Scheme 3.75 .
3.11.2. Fluoroformates
This class of compounds is exemplifi ed by the data for methyl fl uoro-formate (Scheme 3.74 ).
REFERENCES
1. Brey , W. S. Magn. Res. Chem. 2008 , 46 , 480 – 492 . 2. Weigert , F. J. J. Fluorine Chem. 1990 , 46 , 375 – 384 . 3. Hirano , T. ; Nonogama , S. ; Miyajima , T. ; Kurita , Y. ; Kawamura , T. ; Sato , H.
J. Chem. Soc. Chem. Commun. 1986 , 606 – 607 . 4. Butt , G. ; Cilmi , J. ; Hoobin , P. M. ; Topsom , R. D. Spectrochim. Acta Part A
1980 , 36A , 521 – 524 .

REFERENCES 95
5. Simonnin , M. - P. J. Organometal. Chem. 1966 , 5 , 155 – 165 . 6. Fifolt , M. J. ; Sojka , S. A. ; Wolfe , R. A. ; Hojnicki , D. S. ; Bieron , J. F. ; Dinon ,
F. J. J. Org. Chem. 1989 , 54 , 3019 – 3023 . 7. Lutnaes , B. F. ; Luthe , G. ; Brinkman , U. A. T. ; Johansen , J. E. ; Krane , J. Magn.
Reson. Chem. 2005 , 43 , 588 – 594 . 8. Gribble , G. W. ; Keavy , D. J. ; Olson , E. R. ; Rae , I. D. ; Staffa , A. ; Herr , T. E. ;
Ferrara , M. B. ; Contreras , R. H. Magn. Reson. Chem. 1991 , 29 , 422 – 432 .