haploid assembly of diploid genomes
TRANSCRIPT

Haploid Assembly of Diploid Genomes
Challenges, Trials, Tribulations
İnanç Birol
13 October 2011

IEEE InfoVis 2009
Assembly By Short Sequencing
2
3

in Literature
• ~40 citations on tool comparisons
• ~20 citations on using ABySS for a biology study
• Crowded field – 17 teams in Assemblathon 1
4
Overlap-Overlay-Consensus
ARACHNE
CAP3
Celera assembler
MIRA
Newbler
Phred/Phrap
SGA
De Bruijn Graph
Euler
Velvet
ABySS
SOAPdenovo
ALLPATHS
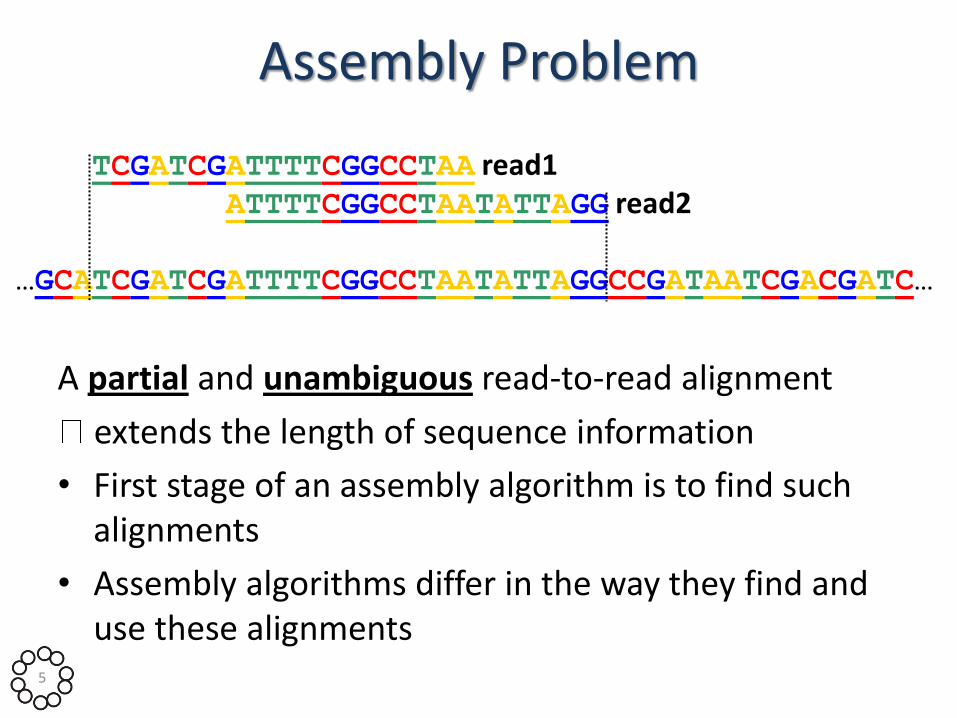
Assembly Problem
A partial and unambiguous read-to-read alignment
extends the length of sequence information
• First stage of an assembly algorithm is to find such alignments
• Assembly algorithms differ in the way they find and use these alignments
5
TCGATCGATTTTCGGCCTAA read1 ATTTTCGGCCTAATATTAGG read2
…GCATCGATCGATTTTCGGCCTAATATTAGGCCGATAATCGACGATC…
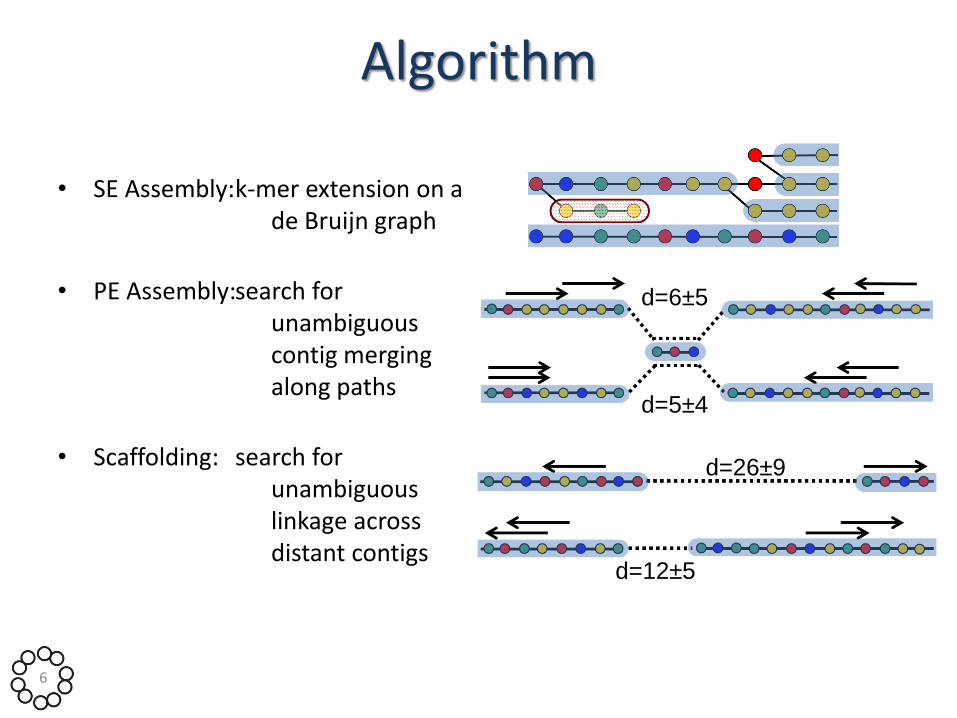
Algorithm
• SE Assembly:
• PE Assembly:
• Scaffolding:
k-mer extension on a de Bruijn graph
search for unambiguous contig merging along paths
search for unambiguous linkage across distant contigs
6
d=6±5
d=5±4
d=26±9
d=12±5
Software
7
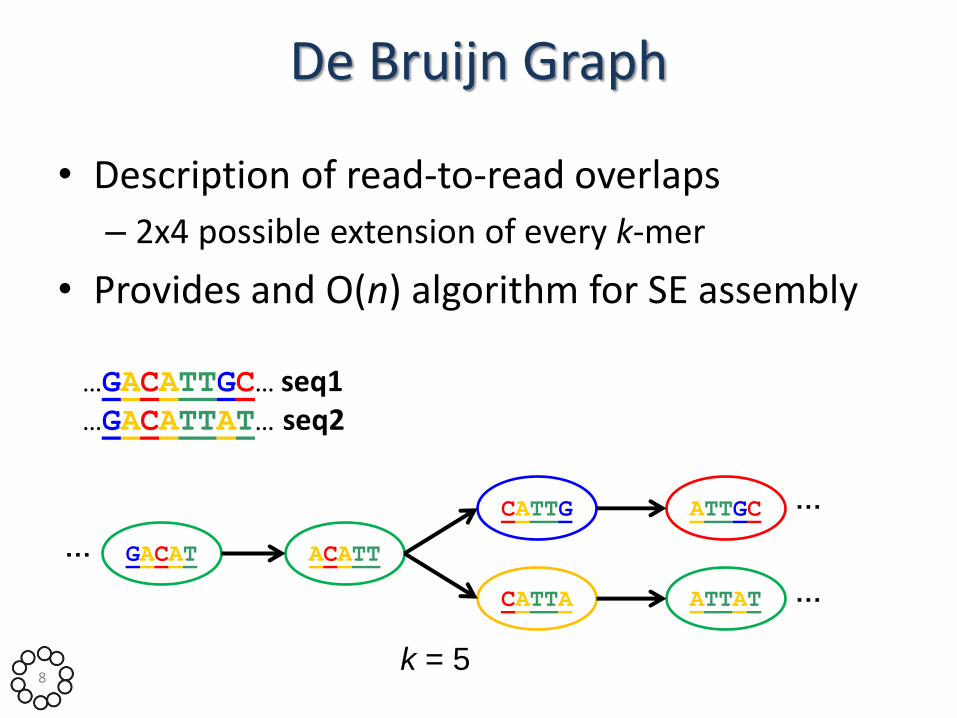
De Bruijn Graph
• Description of read-to-read overlaps
– 2x4 possible extension of every k-mer
• Provides and O(n) algorithm for SE assembly
8
…GACATTGC… seq1 …GACATTAT… seq2
GACAT ACATT
ATTAT CATTA
CATTG ATTGC
…
…
…
k = 5

Adjacency Graph
• Description of contig overlaps
– Built during SE assembly
• Overlap = k-1 bp
– Generalized during PE assembly
• Arbitrary overlap
9

Linkage Graph
• Built through read pairs aligned to different contigs
– PE reads from a tight fragment length distribution
• Reliable distance estimates
– MP reads from broader insert length distribution
• Noisy data
• Used in PE assembly (PE) and scaffolding (PE and MP) stages
10

Anchor
• Scrubbing “homozygous” variations
Indel SNPs
11
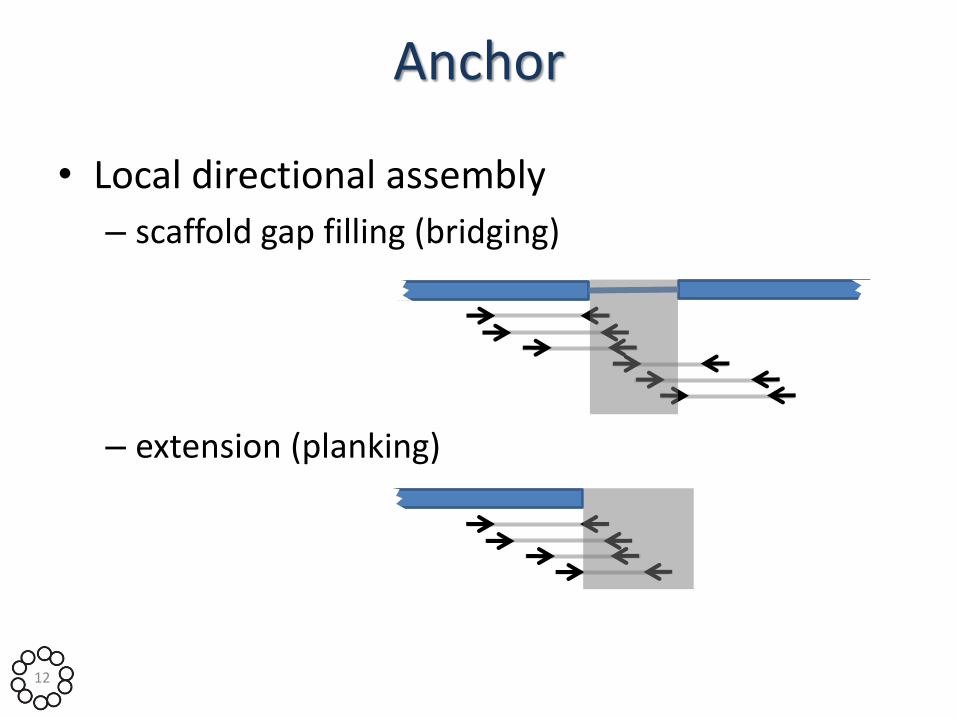
Anchor
• Local directional assembly
– scaffold gap filling (bridging)
– extension (planking)
12

Case Study
Mountain Pine Beetle Genome Assembly
13
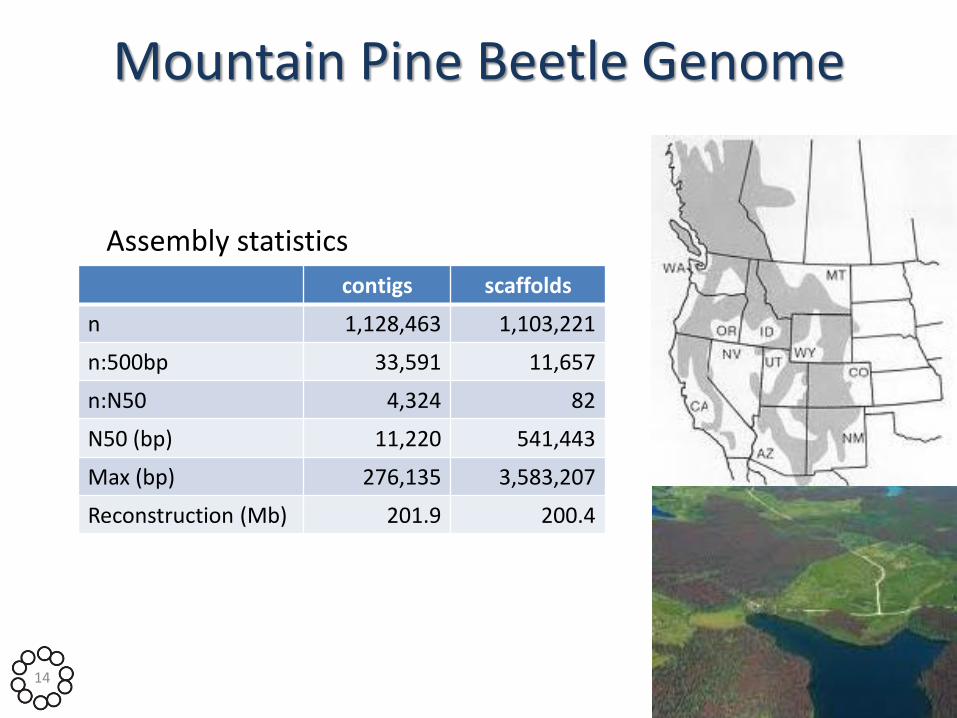
Mountain Pine Beetle Genome
Assembly statistics
contigs scaffolds
n 1,128,463 1,103,221
n:500bp 33,591 11,657
n:N50 4,324 82
N50 (bp) 11,220 541,443
Max (bp) 276,135 3,583,207
Reconstruction (Mb) 201.9 200.4
14

Assembly As a Hairball
• ABySS v1.2.7
– PE/MP information disambiguates short contig extensions
1 2 3 4 5 6+ 1 15822 7354 1882 530 109 1
2 7354 9814 1817 456 72 3
3 1882 1817 1074 238 31 1
4 530 456 238 126 13 1 5 109 72 31 13 10 0 6+ 1 3 1 1 0 0
Node connectivity*
out in
* For contigs 2 kb
15
Scaffolding
16
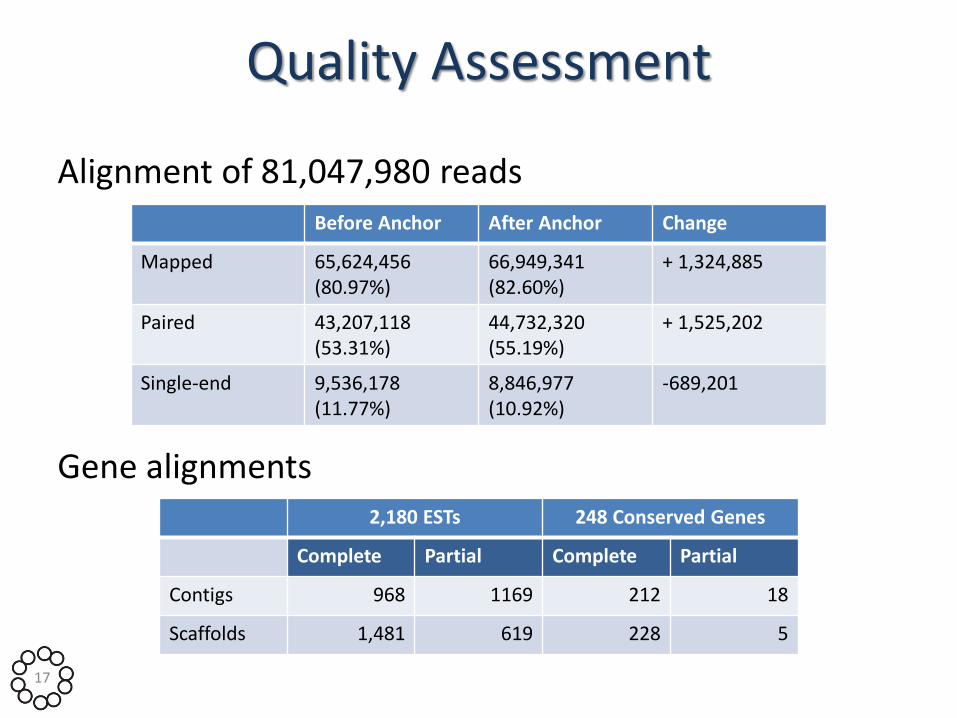
Quality Assessment
Alignment of 81,047,980 reads
Gene alignments
17
Before Anchor After Anchor Change
Mapped 65,624,456 (80.97%)
66,949,341 (82.60%)
+ 1,324,885
Paired 43,207,118 (53.31%)
44,732,320 (55.19%)
+ 1,525,202
Single-end 9,536,178 (11.77%)
8,846,977 (10.92%)
-689,201
2,180 ESTs 248 Conserved Genes
Complete Partial Complete Partial
Contigs 968 1169 212 18
Scaffolds 1,481 619 228 5

Date ABySS Version
Data n:500 N50 Max Sum
August 2009 1.0.11 3x GAiix 81,431 1,526 20,755 107.3e6
November 2009 1.0.15 +2x GAiix 104,958 2,333 55,845 195.8e6
February 2010 1.1.1 +4x GAiix 157,081 2,790 136,637 346.3e6
July 2010 1.2.0 +2x GAiix 146,313 3,354 129,008 376.2e6
November 2010 1.2.4 +1x GAiix +1x GAiix
(MP)
100,690 4,474 294,323 268.8e6
May 2011 1.2.7 -- 18,660 108,158 1,908,773 201.4e6
July 2011 1.2.7 + 1x HiSeq +1x HiSeq
(MP)
11,657 541,443 3,583,207 200.4e6
August 2011 1.2.7 -- 11,523 561,847 3,746,698 206.5e6
18

Transcriptome Assembly
19
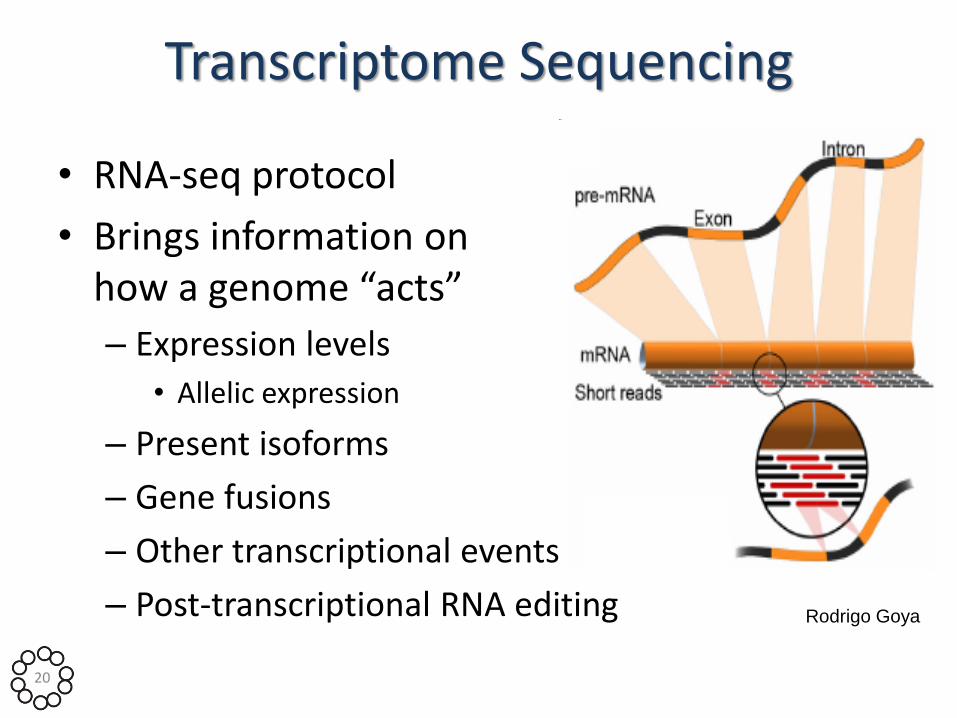
Transcriptome Sequencing
• RNA-seq protocol
• Brings information on how a genome “acts”
– Expression levels
• Allelic expression
– Present isoforms
– Gene fusions
– Other transcriptional events
– Post-transcriptional RNA editing Rodrigo Goya
20
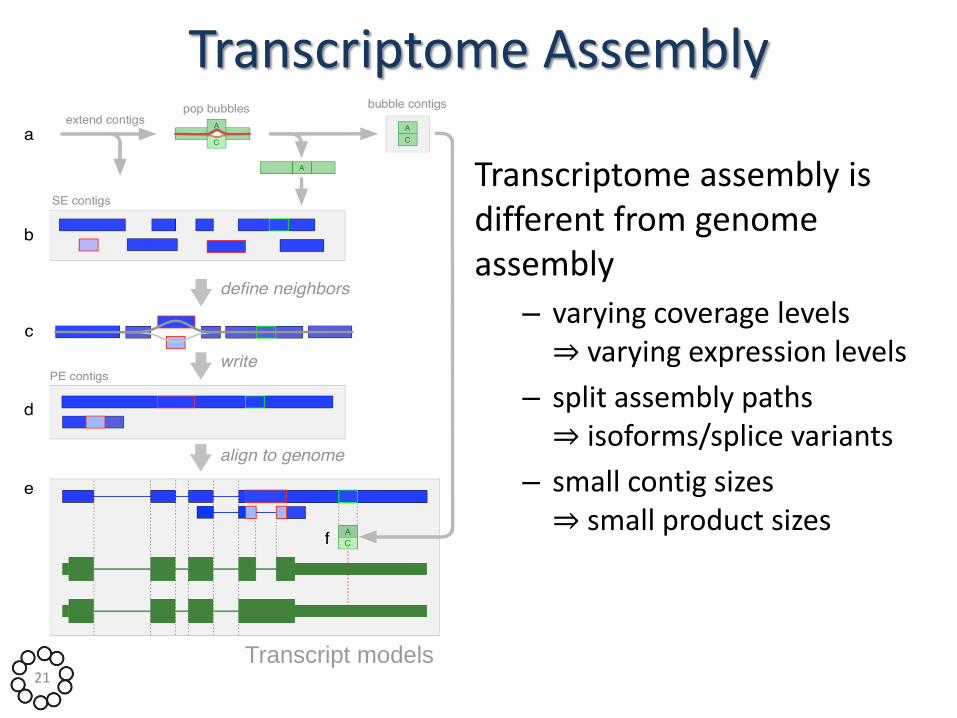
Transcript models
Transcriptome Assembly
Transcriptome assembly is different from genome assembly
– varying coverage levels ⇒ varying expression levels
– split assembly paths ⇒ isoforms/splice variants
– small contig sizes ⇒ small product sizes
21
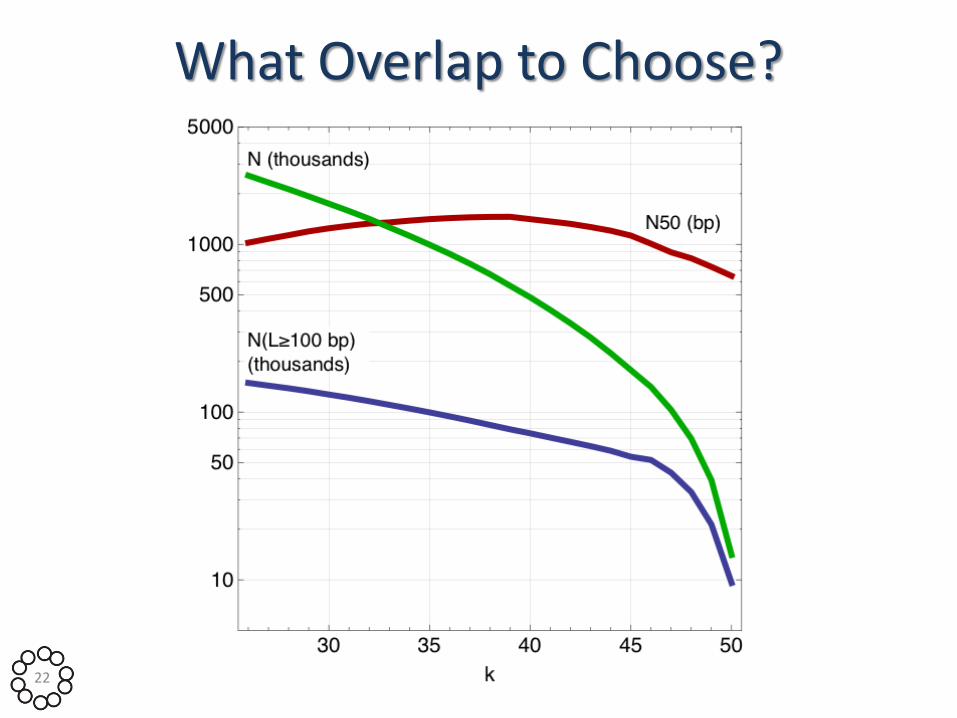
What Overlap to Choose?
22

Selection of k
23
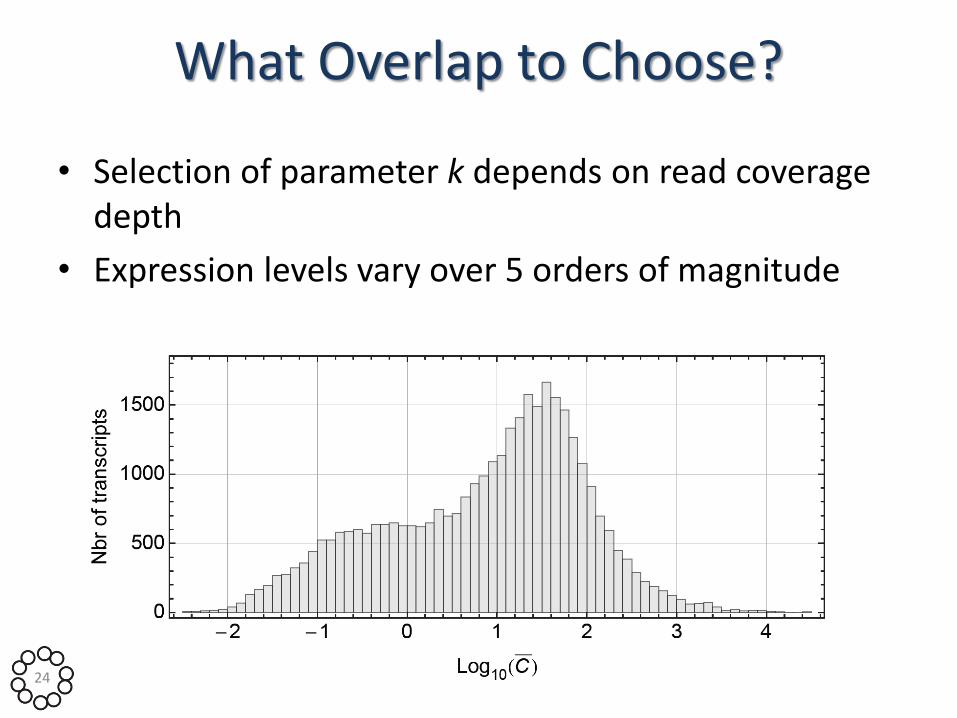
What Overlap to Choose?
• Selection of parameter k depends on read coverage depth
• Expression levels vary over 5 orders of magnitude
24
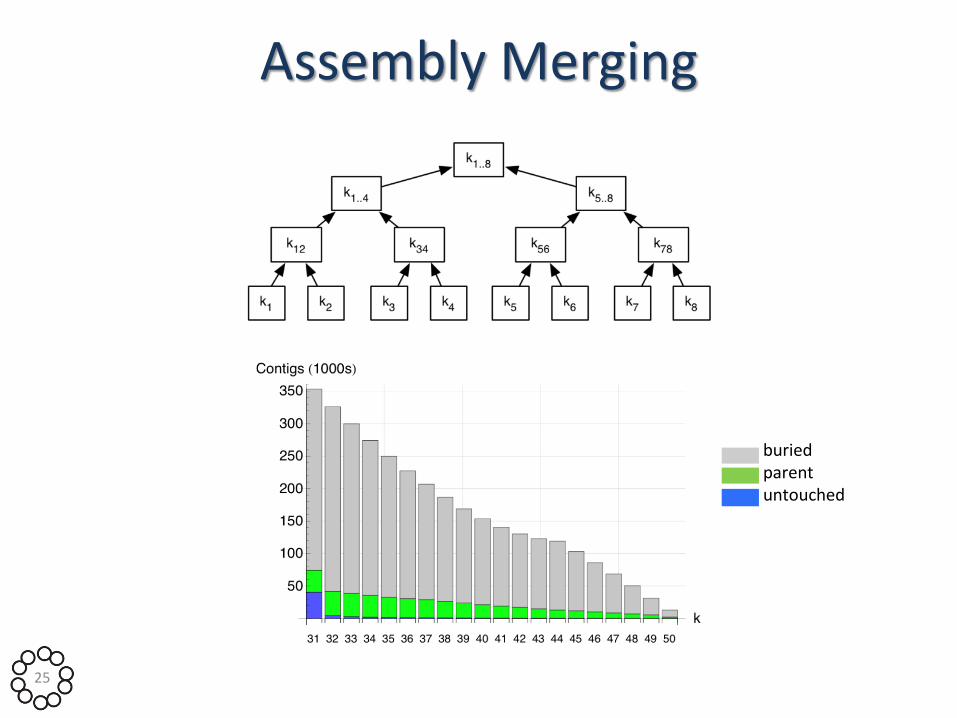
Assembly Merging
25
buried parent untouched
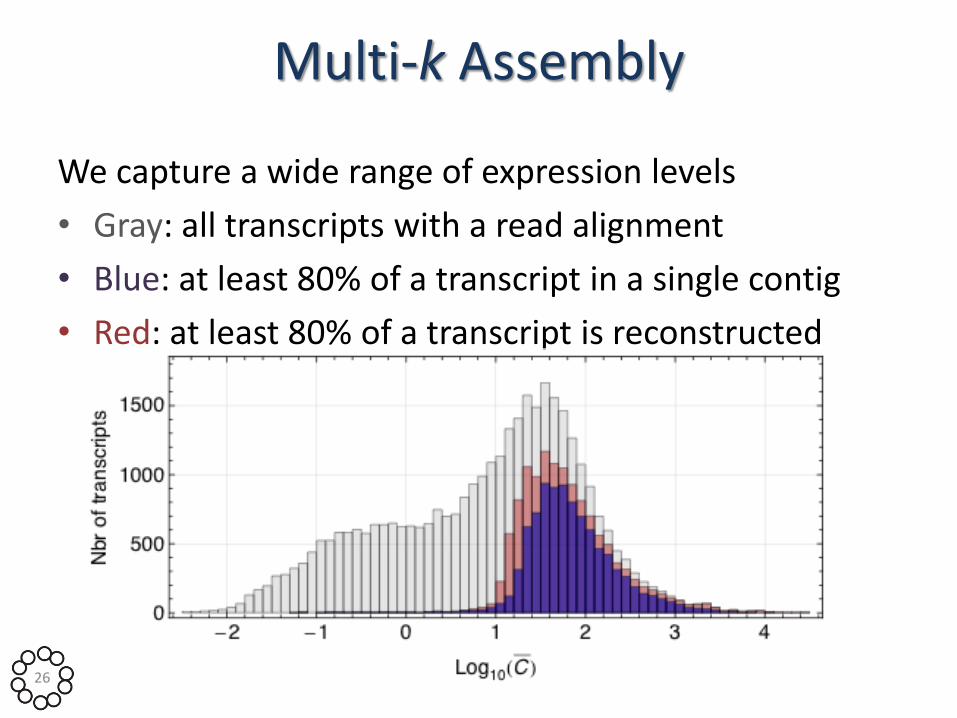
Multi-k Assembly
We capture a wide range of expression levels
• Gray: all transcripts with a read alignment
• Blue: at least 80% of a transcript in a single contig
• Red: at least 80% of a transcript is reconstructed
26
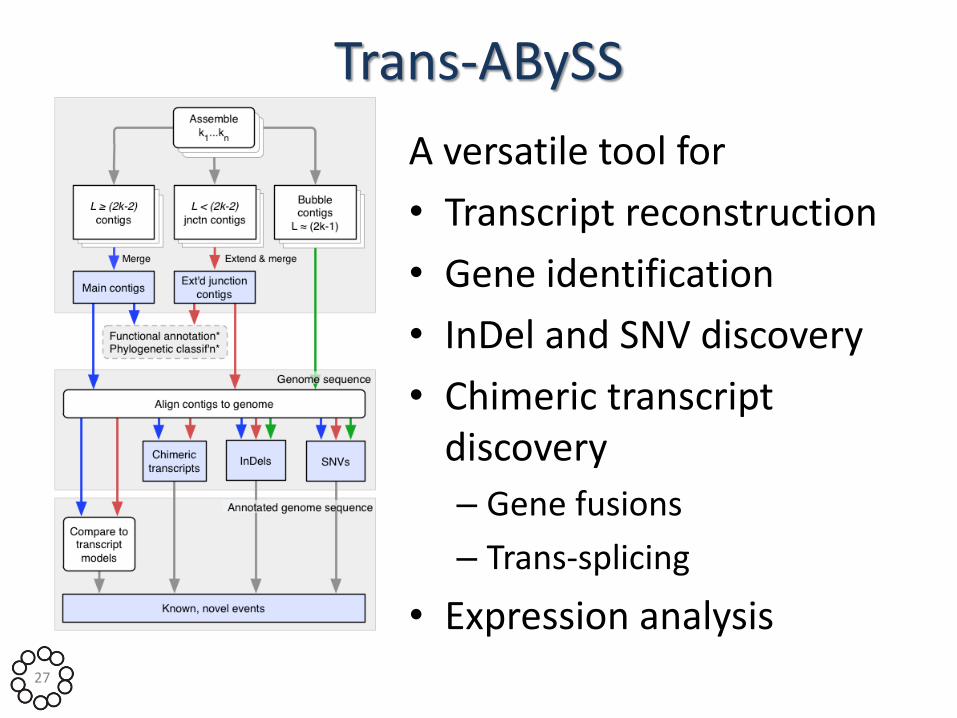
Trans-ABySS
A versatile tool for
• Transcript reconstruction
• Gene identification
• InDel and SNV discovery
• Chimeric transcript discovery
– Gene fusions
– Trans-splicing
• Expression analysis
27
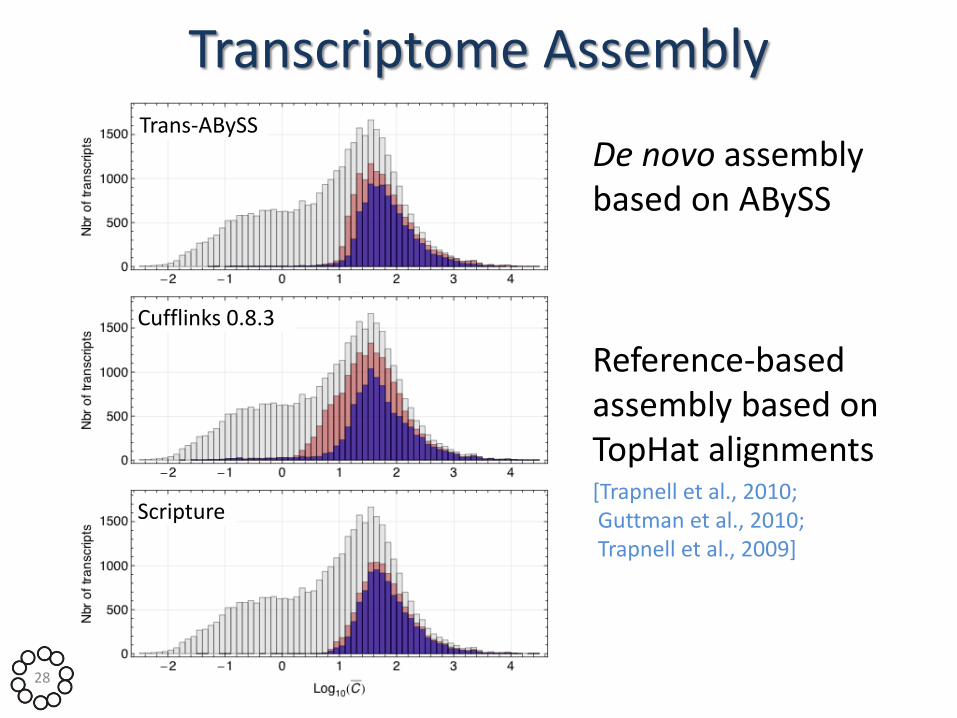
Trans-ABySS
Cufflinks 0.8.3
Scripture
28
Transcriptome Assembly
De novo assembly based on ABySS
Reference-based assembly based on TopHat alignments [Trapnell et al., 2010; Guttman et al., 2010; Trapnell et al., 2009]
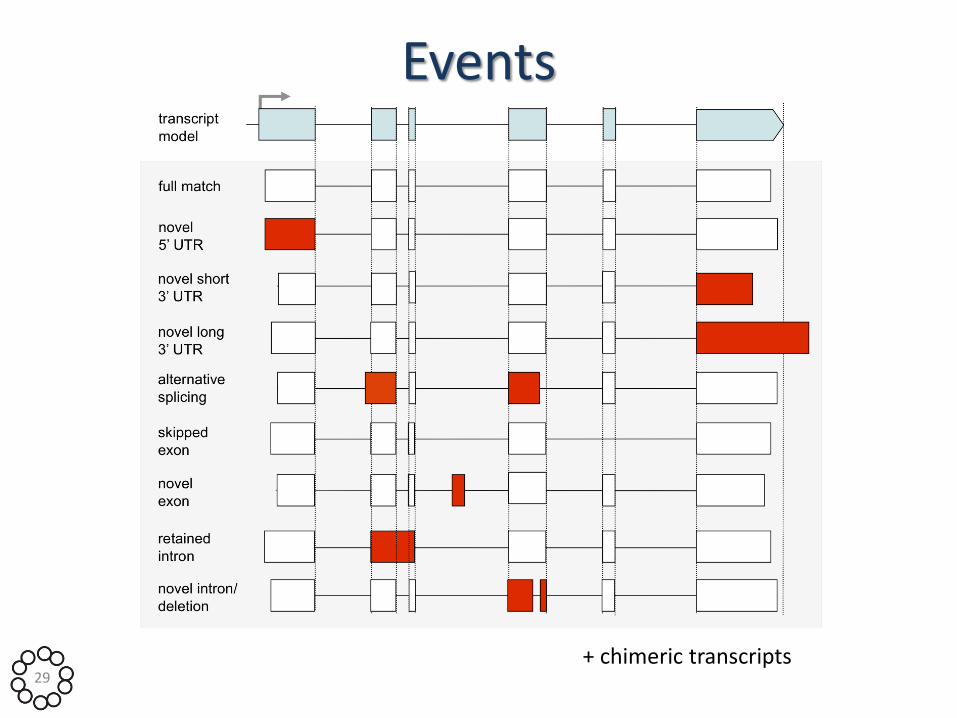
Events
29 + chimeric transcripts
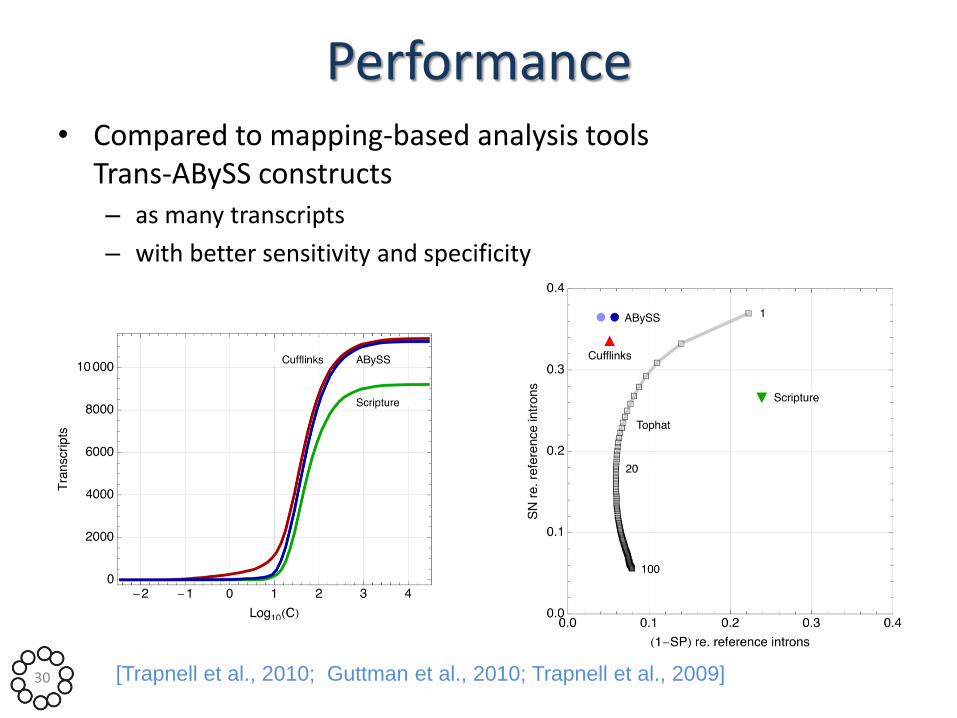
Performance • Compared to mapping-based analysis tools
Trans-ABySS constructs – as many transcripts
– with better sensitivity and specificity
30 [Trapnell et al., 2010; Guttman et al., 2010; Trapnell et al., 2009]

Case Study
Acute Myeloid Leukemia Transcriptome Assembly
31
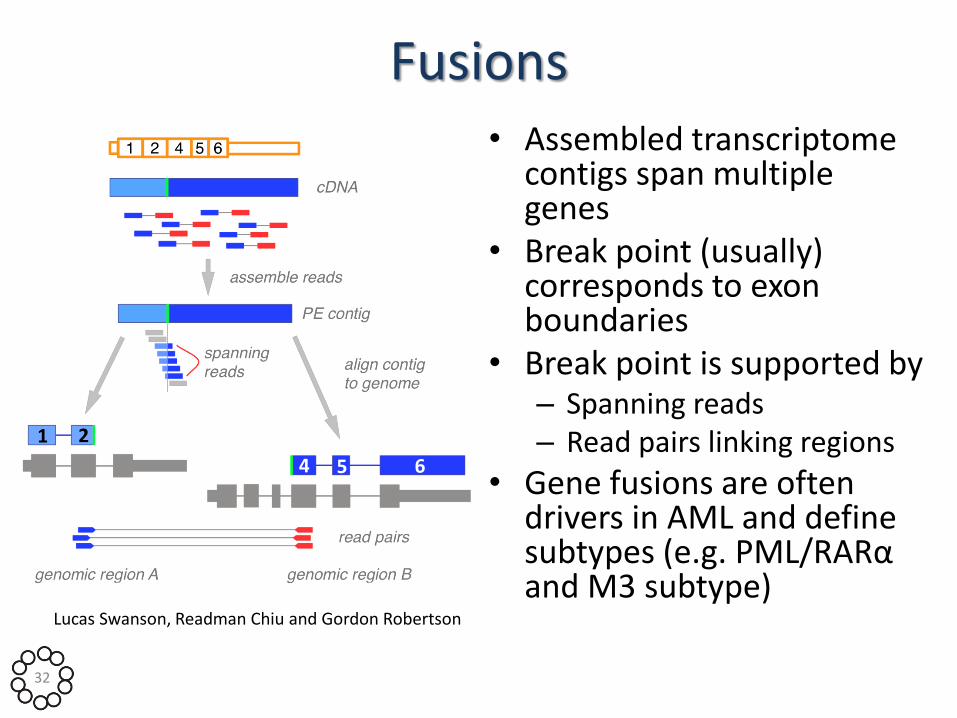
Fusions • Assembled transcriptome
contigs span multiple genes
• Break point (usually) corresponds to exon boundaries
• Break point is supported by – Spanning reads – Read pairs linking regions
• Gene fusions are often drivers in AML and define subtypes (e.g. PML/RARα and M3 subtype)
1 2
4 5 6
Lucas Swanson, Readman Chiu and Gordon Robertson
32

AML Gene Fusions
0
2
4
6
8
10
12
14
16
Nu
mb
er
of
pat
ien
ts
Candidate fusion events
9%
5%
4% MLL fusions
Known AML fusion events (12) Known polymorphism (1) Novel fusion event (17)
Low frequency (<1%)
71 events in 65/173 (38%) patients 30 different gene fusions identified ≥94% validation by RT-PCR sequencing
Karen Mungall 33
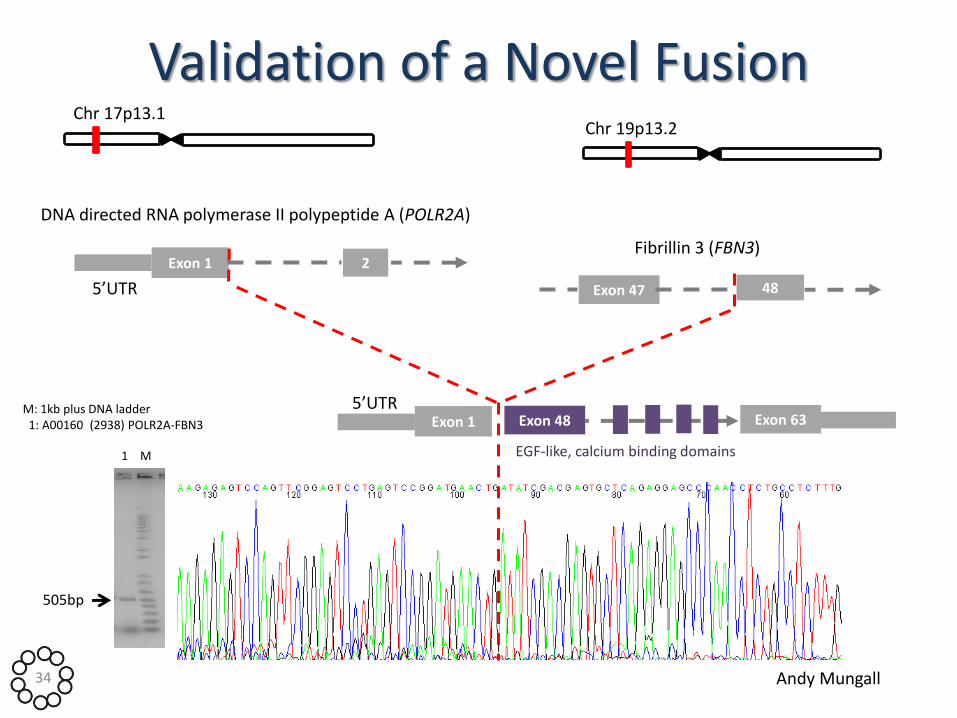
Validation of a Novel Fusion
M: 1kb plus DNA ladder 1: A00160 (2938) POLR2A-FBN3
505bp
Chr 17p13.1
DNA directed RNA polymerase II polypeptide A (POLR2A)
Exon 1 2
5’UTR
Fibrillin 3 (FBN3)
Chr 19p13.2
Exon 47 48
Exon 1 5’UTR
Exon 48 Exon 63
EGF-like, calcium binding domains 1 M
Andy Mungall 34

Internal Tandem Duplications • Contig alignments result in
– Query gaps – Contiguous target blocks
• Read support on break point(s) • Aberrant read pair distances • Known AML ITDs:
– 29/173 (17%) harbour partial FLT3 exon 14 duplication
– 6/173 (3.5%) harbour partial WT1 exon 7 duplication
– Nakao et al., Leukemia 1996; Christiansen et al., Leukemia 2001 2 2’
2’
2
35
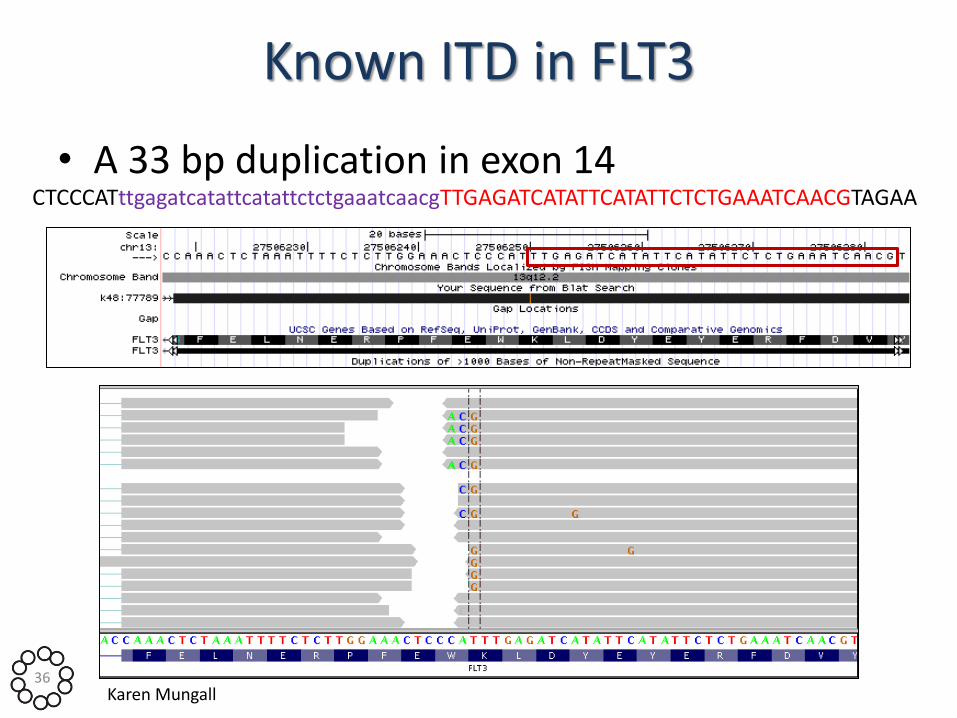
Known ITD in FLT3
• A 33 bp duplication in exon 14 CTCCCATttgagatcatattcatattctctgaaatcaacgTTGAGATCATATTCATATTCTCTGAAATCAACGTAGAA
Karen Mungall 36
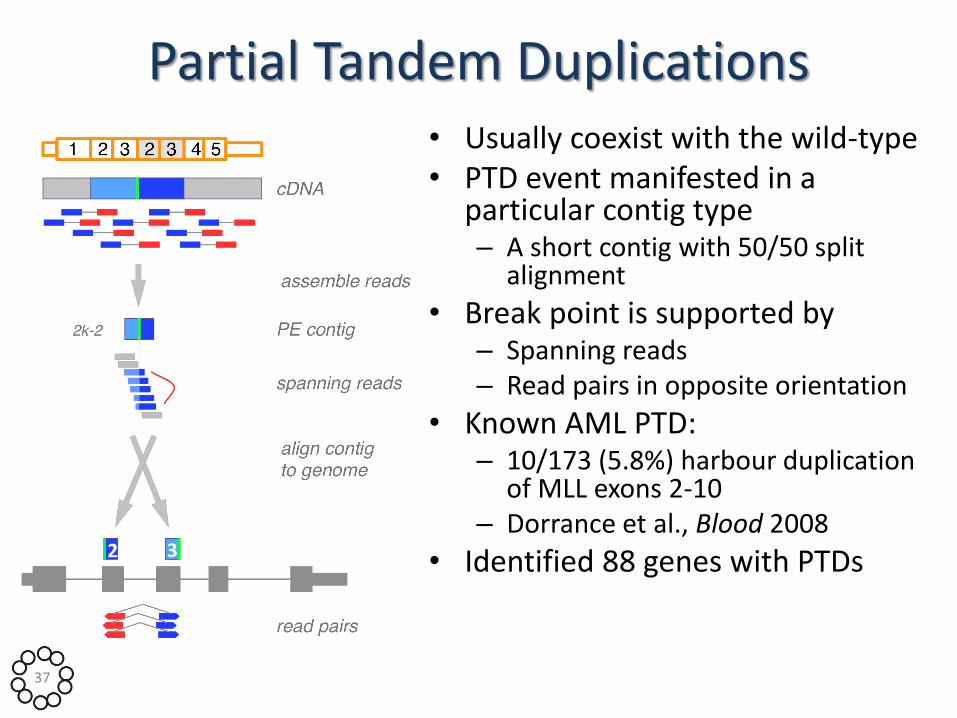
Partial Tandem Duplications • Usually coexist with the wild-type • PTD event manifested in a
particular contig type – A short contig with 50/50 split
alignment
• Break point is supported by – Spanning reads – Read pairs in opposite orientation
• Known AML PTD: – 10/173 (5.8%) harbour duplication
of MLL exons 2-10 – Dorrance et al., Blood 2008
• Identified 88 genes with PTDs 2 3
37

Novel PTD in Arid1a
• Exons 2-4 tandemly repeated in 5 AML libraries
• Recurrent across tissues and species
WT CT
Source Observations
AML 5/173 Libraries
LBC 5/54 Libraries
Normal mouse 3/7 Libraries
NCBI EST colon_ins , placenta_normal
38

Summary
39

ABySS Team: Shaun Jackman Tony Raymond Rod Docking Beetle Project: Joerg Bohlmann Chris Keeling Nancy Liao Greg Taylor Simon Chan Diana Palmquist
Trans-ABySS Team: Readman Chiu Karen Mungall Gordon Robertson Ka Ming Nip Jenny Qian Rong She Lucas Swanson AML Project: Richard Moore Yongjun Zhao Andy Mungall Aly Karsan
GSC: Sequencing Team Library Core Systems Team Steven Jones Marco Marra

Final Hairball
• ABySS v1.2.7
– Read pairs and inferred distances allow for scaffolding
41
contigs scaffolds
n 1,128,463 1,103,221
n:500bp 33,591 11,657
n:N50 4,324 82
N50 (bp) 11,220 541,443
Max (bp) 276,135 3,583,207
Reconstruction (Gb) 201.9 200.4
Biotin Read-Through
circularized insert
42

43
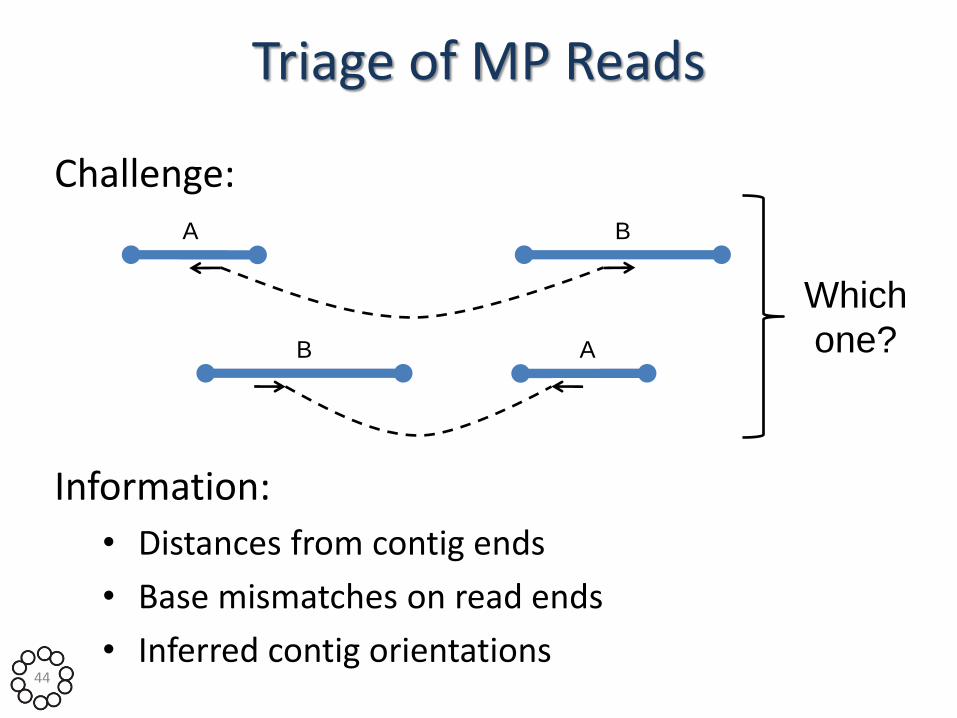
Triage of MP Reads
Challenge: A B
A B
Which
one?
Information:
• Distances from contig ends
• Base mismatches on read ends
• Inferred contig orientations 44
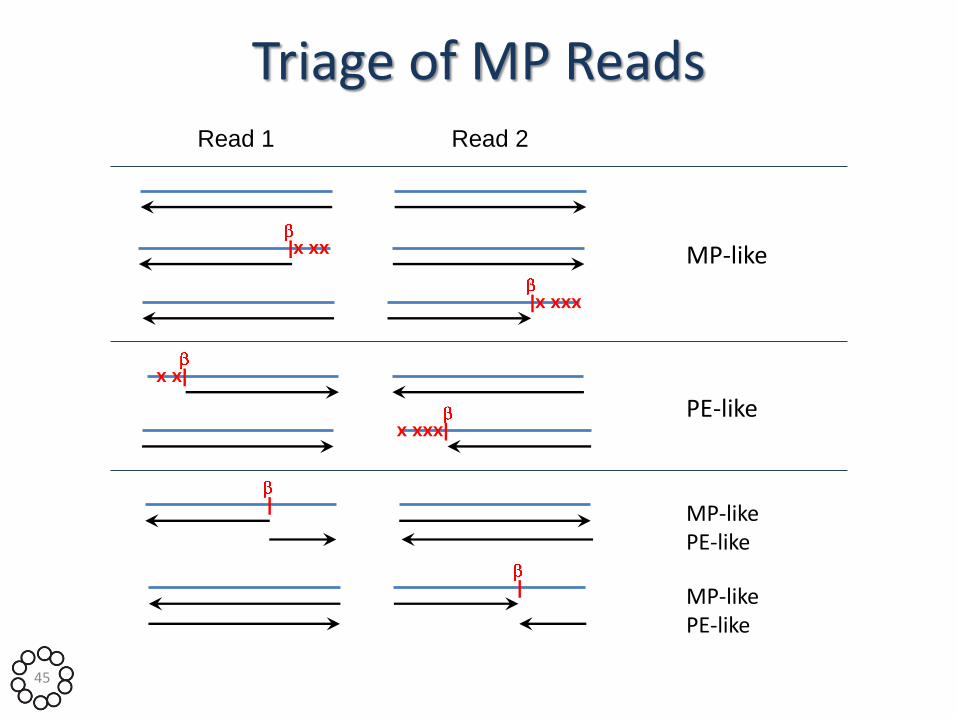
Triage of MP Reads Read 1 Read 2
MP-like
PE-like
MP-like PE-like
MP-like PE-like
|x xx
|x xxx
x x|
x xxx|
|
|
45