hypoglycemia in children philip a. gruppuso and robert ... · pdf filepediatrics in review #...
TRANSCRIPT

DOI: 10.1542/pir.11-4-1171989;11;117Pediatrics in Review
Philip A. Gruppuso and Robert SchwartzHypoglycemia in Children
http://pedsinreview.aappublications.org/content/11/4/117the World Wide Web at:
The online version of this article, along with updated information and services, is located on
Print ISSN: 0191-9601. Village, Illinois, 60007. Copyright © 1989 by the American Academy of Pediatrics. All rights reserved.trademarked by the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elk Grove
andpublication, it has been published continuously since 1979. Pediatrics in Review is owned, published, Pediatrics in Review is the official journal of the American Academy of Pediatrics. A monthly
at UNIV OF CHICAGO on May 21, 2013http://pedsinreview.aappublications.org/Downloaded from

The questions below should helpfocus the reading of this article.
1. How do the manifestations of hy-poglycemia in the neonatal period dif-fer from those in older children?
2. How do the manifestations of post-prandial hypoglycemia differ fromthose of hypoglycemia brought on byprolonged fasting?
3. How can the blood levels of insulin,glucose, ketones, and lactate helpdifferentiate among the causes of hy-poglycemia?
4. What physical findings and labo-ratory data in a newborn infant withhypoglycemia will suggest that thehypoglycemia is due to hypopituitar-ism?
EDUCATIONAL OBJECTIVE
24. The pediatrician should haveknowledge to make an appropriateevaluation of an afebrile 9-month-old girl with seizures and hypogly-cemia, growth hormone deficiency(panhypopituitarism, perinatab as-phyxia), adrenal insufficiency, ke-totic hypoglycemia, organic acidu-na, defect in gluconeogenesis, gly-cogen storage disease, factitioushypoglycemia, and Reye syn-drome, and develop an appropriateplan for management (Topics, 89/90).
Symptoms associated withhypoglycemia are
nonspecific but generallyrelate to adrenergic
responsiveness initially andlater to the central nervous
system.
Neonates and children of allages may have
asymptomatic hypoglycemia(blood glucose concentration<40 mg/dL), especially with
fasting.
pediatrics in review #{149}vol. 11 no. 4 october 1989 PIR 117
Hypoglycemia in ChildrenPhibip A. Gruppuso, MD,* and Robert Schwartz, MDt
Hypoglycemia, although rare inchildhood beyond the newbornperiod, remains a vexing problem forthe pediatrician. First, the symptomsmay be vague and nonspecific, thusmaking diagnosis particularly de-pendent on a high index of suspicion.Second, the pathogenic mechanismsthat result in hypoglycemia are asnumerous and complicated as thephysiologic mechanisms that main-tam euglycemia. Finally, althoughmost patients have no permanent se-quelae, a catastrophic episode of hy-poglycemia can cause neurobogic def-cit and mental retardation. In thisbrief review, the most common causefor hypoglycemia, namely, insulin-de-pendent type I diabetes mebbitus, willnot be considered. Instead, we willfocus on those metabolic disordersthat result directly in a boss of theability to maintain normal serum glu-cose concentrations.
SYMPTOMS
In the olden infant (olden than 2months), child, and adult, a rapid de-
* Associate Professor of Pediatrics.
t Professor of Pediatrics and Medical Science,Dept of Pediatrics, Division of Pediatric Endo-crinology and Metabolism, Rhode Island Hos-pital and Brown University Program in Medi-cine. Address reprint requests to Dr Schwartz,Rhode Island Hospital, 593 Eddy St, Provi-dence, RI 02903.
crease in blood glucose to levels lessthan 40 mg/dL (2.2 mmol/L) may pro-duce hunger and trigger an excessiverelease of epinephnine, causingweakness, anxiety, cold sweat, in-wand trembling and tachycandia.These adrenengic symptoms tend tooccur in persons with postpnandialhypoglycemia. In contrast, fasting hy-poglycemia generally is progressiveand gradual and produces neunogly-copenic symptoms which includeheadache, mental dullness, fatigue,confusion, abnormal behavior or psy-chosis, amnesia on other neunobogicdeficit, seizures, on frank coma.
The definition of hypoglycemia inthe neonate remains controversial.Earlier criteria were based on exten-sive surveys of preterm and termneonates, not all of whom were onoptimal caloric regimens. It was de-tenmined that normal term infants hadblood glucose bevels greaten than 30mg/dL (1 .67 mmob/L), and “normal”pnetenm infants had levels greatenthan 20 mg/dL (1 .1 1 mmob/L) duringthe first 72 hours of life. An alternateview holds that the neonate shouldnot tolerate a glucose bevel less thanthe fetus. Thus, the lowest maternalbevel (less 15% for maternal-fetal gra-dient) is recommended as the cutoffbevel. With this criteria, hypoglycemiaisdefined as a blood glucose concen-tration less than 40 mg/dL (2.2 mmob/L) at any age. It should be noted thatthere have been inadequate long-term neunodevebopmentab studies tohelp choose among these criteria.
Symptoms in the neonate are non-specific and include tremors, jitteri-ness, apnea and cyanosis, hypotonia,irritability, feeding difficulties, convul-sions, coma, high pitched cry, tach-
ypnea, and pallor. These clinical man-ifestations may be seen in the ab-sence of hypoglycemia in infants witha variety of primary central nervoussystem abnormalities including con-genital defects, birth injury, microce-phaby, hemorrhage, and kernicterus.In addition, hypoglycemia may be as-sociated with sepsis, heart disease,severe respiratory distress on as-phyxia, multiple congenital anoma-lies, on endocrine deficiencies.
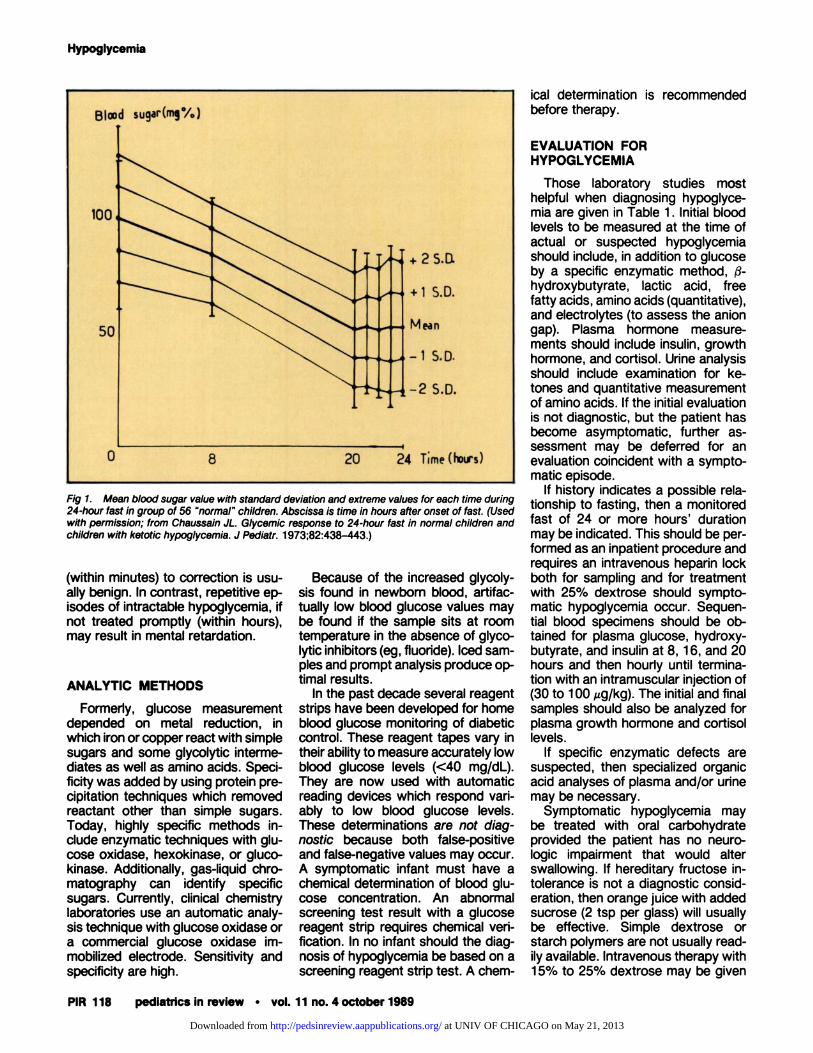
A person of virtually any age mayhave hypoglycemia but be asympto-matic. Infants with type I glycogenstorage disease have tolerated bloodglucose bevels too low to measurewithout obvious symptoms. Normalchildren may tolerate fasts of 24hours without symptoms of hypogly-cemia despite blood glucose levelsless than 40 mg/dL (1.67 mmol/L)
(Fig 1).1 Children with symptomatichypoglycemia may be subject to vari-able bong-term effects. Acute hypo-glycemia which responds rapidly
at UNIV OF CHICAGO on May 21, 2013http://pedsinreview.aappublications.org/Downloaded from
BI�Id sugar(rn��6)
50
+2S.D.
+1 S.D.
Mean
-1 S.D.
0
-2 S.D.
8 20
Fig 1. Mean blood sugar value with standard deviation and extreme values for each time during24-hour fast in group of 56 “normal” children. Abscissa is time in hours after onset of fast. (Usedwith permission; from Chaussain JL. Glycemic response to 24-hour fast in normal children andchildren with ketotic hypoglycemia. J Pediatr. 1973;82:438-443.)
Hypoglycemia
PIR 118 pedIatrics in review #{149} vol. 11 no. 4 october 1989
(within minutes) to correction is usu-ally benign. In contrast, repetitive ep-isodes of intractable hypoglycemia, ifnot treated promptly (within hours),may result in mental retardation.
ANALYTIC METHODS
Formerly, glucose measurementdepended on metal reduction, inwhich iron or copper react with simplesugars and some glycolytic interme-diates as well as amino acids. Speci-ficity was added by using protein pre-cipitation techniques which removedreactant other than simple sugars.Today, highly specific methods in-clude enzymatic techniques with glu-cose oxidase, hexokinase, on gluco-kinase. Additionally, gas-liquid chro-matography can identify specificsugars. Currently, clinical chemistrylaboratories use an automatic analy-sis technique with glucose oxidase ora commercial glucose oxidase im-mobilized electrode. Sensitivity andspecificity are high.
Because of the increased glycoly-sis found in newborn blood, artifac-tually bow blood glucose values maybe found if the sample sits at roomtemperature in the absence of glyco-bytic inhibitors (eg, fluoride). Iced sam-pIes and prompt analysis produce op-timal results.
In the past decade several reagentstrips have been developed for homeblood glucose monitoring of diabeticcontrol. These reagent tapes vary intheir ability to measure accurately lowblood glucose bevels (<40 mg/dL).They are now used with automaticreading devices which respond vari-ably to low blood glucose levels.These determinations are not diag-nostic because both fabse-positiveand false-negative values may occur.A symptomatic infant must have achemical determination of blood glu-cose concentration. An abnormalscreening test result with a glucosereagent strip requires chemical veri-fication. In no infant should the diag-nosis of hypoglycemia be based on ascreening reagent strip test. A chem-
ical determination is recommendedbefore therapy.
EVALUATION FORHYPOGLYCEMIA
Those laboratory studies mosthelpful when diagnosing hypoglyce-mia are given in Table 1 . Initial bloodbevels to be measured at the time ofactual on suspected hypoglycemiashould include, in addition to glucoseby a specific enzymatic method, $-hydnoxybutyrate, lactic acid, freefatty acids, amino acids (quantitative),and electrolytes (to assess the aniongap). Plasma hormone measure-ments should include insulin, growthhormone, and cortisol. Urine analysisshould include examination for ke-tones and quantitative measurementof amino acids. If the initial evaluationis not diagnostic, but the patient hasbecome asymptomatic, further as-sessment may be deferred for anevaluation coincident with a sympto-matic episode.
If history indicates a possible neba-tionship to fasting, then a monitoredfast of 24 on more hours’ durationmay be indicated. This should be per-formed as an inpatient procedure andrequires an intravenous hepanin bockboth for sampling and for treatmentwith 25% dextrose should sympto-matic hypoglycemia occur. Sequen-tial blood specimens should be ob-tamed for plasma glucose, hydnoxy-butynate, and insulin at 8, 16, and 20hours and then hourly until tenmina-tion with an intramuscular injection of(30 to 100 pg/kg). The initial and finalsamples should also be analyzed forplasma growth hormone and contisollevels.
If specific enzymatic defects aresuspected, then specialized organicacid analyses of plasma and/or urinemay be necessary.
Symptomatic hypoglycemia maybe treated with oral carbohydrateprovided the patient has no neuno-logic impairment that would alterswallowing. If hereditary fructose in-tolerance is not a diagnostic consid-eration, then orange juice with addedsucrose (2 tsp per glass) will usuallybe effective. Simple dextrose onstanch polymers are not usually read-ily available. Intravenous therapy with15% to 25% dextrose may be given
at UNIV OF CHICAGO on May 21, 2013http://pedsinreview.aappublications.org/Downloaded from

TABLE 1. Laboratory Studies for the Diagnosis of Hypoglycemia*
At time of symptoms
Substrates Hormones
Blood (plasma) glucoseBlood lactic acidBlood �3-hydnoxybutyr-
atePlasma quantitative
amino acids, Plasma-free fatty acids
Plasma insulinPlasma growth hor-
monePlasma cortisobPlasma glucagonPlasma catecholamines
Plasma electrolytes(anion gap)
Blood gasesUrinary quantitative
amino acidsUrinary quantitative or-
ganic acids
Serial blood specimens,every 2-4 h beforeand after feedings
Blood (plasma) glucose
Prolonged fast 24-30 h,hourly blood speci-mens after 20 h
Plasma insulin
Blood (plasma) glucoseBlood �3-hydroxybutyr-
atePlasma quantitative
amino acidst
Plasma insulinPlasma growth hor-
monetPlasma cortisolt
For suspected defectsin glycogen metabo-lism or gluconeogen-esis
Glucagon tolerance(100-300 pg/kg)
Blood (plasma) glu-coset
Blood lactic acidt
Plasma insulint
*Gkjcose and insulin determinations
according to clinical findings.t Initialand finalsamples.� Every 15 minutes.
are performed on all samples, others
Oral glucose tolerance testcontributes little and should
not be used in evaluatinghypoglycemia in an infant or
child.
METABOLISM
pediatrics in review #{149} vol. 11 no. 4 october 1989 PIR 119
rapidly in a dose of 0.25 to 0.50 g/kg. Because of rebound hypoglyce-mia, a continuous infusion of dex-trose should be maintained at 4 to 6mg/kg per minute until the patient isstabilized with nonmoglycemia.
Hypoglycemia was evaluated pro-spectively by Gutbenlet and Corn-bbath2 from 1971 to 1973. The fre-quency in the neonate was 4.4 pen1000 total live births and 15.6 pen1000 low birth weight infants. Thefrequency figures would be modifiedwith more current criteria. The chang-ing character of patients in specialcane nurseries (ie, more very low birthweight infants) will also change cur-rent incidence data. There are no re-liable incidence data for olden chil-dren; however, estimates indicatetwo to three pen 1000 olden childrenadmitted to hospitals have hypogly-cemia.
PATHOGENESIS ANDMANAGEMENT
Successful management of hypo-glycemia requires an understandingof its pathogenesis. Hypoglycemiacan result from disorders of glycogenmetabolism, gluconeogenesis, lipidoxidation, on amino acid metabolism.A simplified schema of these meta-bolic pathways is presented in Fig 2.
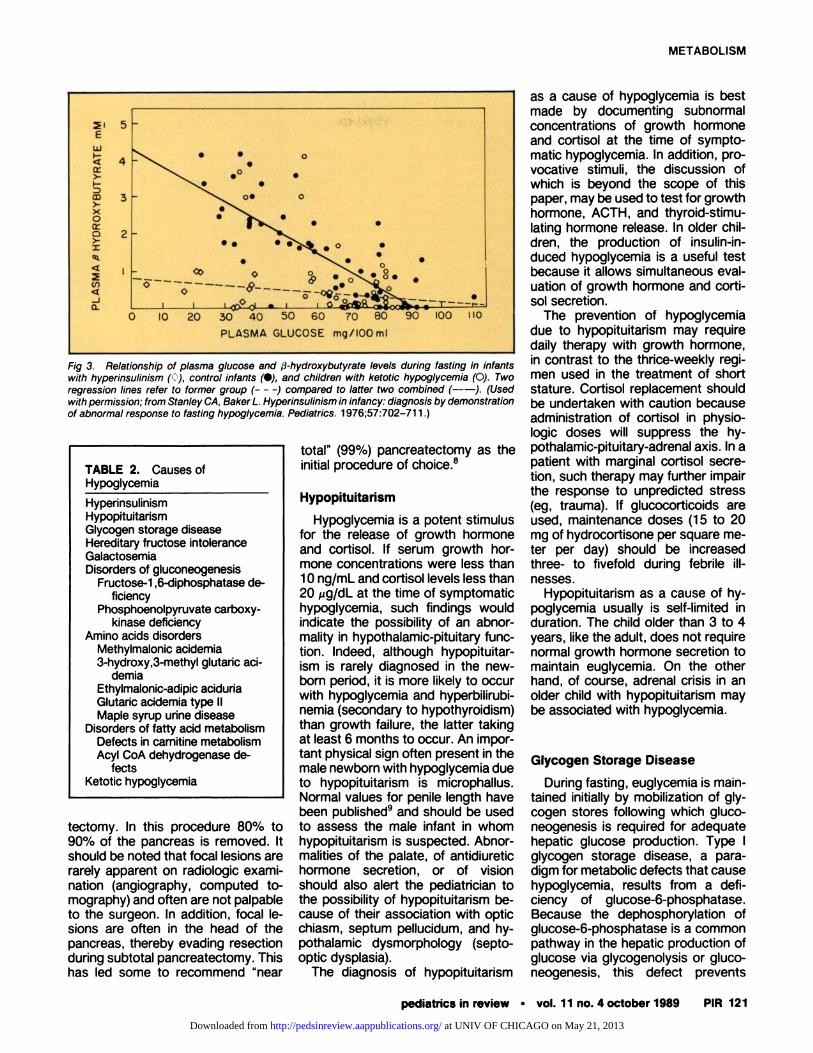
After hypoglycemia has been con-firmed, the cause can be best pur-sued by distinguishing ketosis-asso-ciated hypoglycemia from nonketotichypoglycemia. A prominent metaboliceffect of insulin is suppression of Ii-pobysis and ketogenesis. Becausehypoglycemia suppresses insulin se-cretion, ketosis should result. Inap-propriate secretion of insulin resultsin a relative absence of ketonemia(Fig 3). Measurement of plasma �3-
hydroxybutyrate concentration isuseful in the diagnosis of hypeninsu-linemic hypoglycemia,3 especially inpatients who do not demonstrate ob-vious elevations in circulating insulinconcentration. It should be noted,however, that disorders resulting inimpaired oxidation of fatty acids, in-cluding cannitine deficiency, will alsoresult in nonketotic hypoglycemia.
The ketosis usually associatedwith hypoglycemia in the patient withappropriately suppressed insulin Se-cretion and intact fatty acid oxidationmay not occur in those with glucose6-phosphatase deficiency and hened-itany fructose intolerance. In thesedisorders, lactic acictosis in the ab-sence of ketosis occurs despite pro-found hypoglycemia. However, pa-tients with accelerated starvation(see Ketotic Hypoglycemia), hypopi-tuitanism, and most inborn errors ofmetabolism associated with hypogly-cemia will have associated ketosis.
The most critical diagnostic ma-neuven in the infant with hypoglyce-mia is obtaining a blood sample atthe time of symptoms. The oral glu-cose tolerance test has no role indefining the cause of hypoglycemia inchildren, because reactive hypogly-cemia may well be normal followingglucose loading in children.
The diagnostic wonkup for hypogly-cemia may be presented in the formof an algorithm, as developed by Phil-lip et al.4 Such an approach is likelyto differentiate among specific diag-noses (listed in Table 2), some ofwhich will be discussed here.
Hyperinsulinism
Hypeninsulinemic hypoglycemiacan result from a number of distinctpathologic lesions including diffuse onfocal adenomatosis of the pancreas.5Hyperinsulinemia in infants and chil-dren is often attributed to adenomasor insubinomas, but these lesionsprobably represent adenomatosis.
at UNIV OF CHICAGO on May 21, 2013http://pedsinreview.aappublications.org/Downloaded from

GLYCOGEN
2 3
UDPG
tG4�p
GLUCO� �: G-tL
4
FATTY PC1DS
L�TATE � �. PJ’�W VAlE
�.__�...P�ETYL C0A
V
AMNO’IDS KE�ONE BODIES
Hyperinsulinemia is best
diagnosed by the finding ofan elevated insulin to
glucose ratio in the presenceof low plasma �-
hydroxybutyrateconcentrations.
Fig 2. Interrelationships between glycogen metabolism, gluconeogenesis, glycolysis, and fattyacid metabolism. Numbers depict key enzymes: 1, glucose-6-phosphatase; 2, glycogen synthase;3, phosphorylase; 4, fructose-1,6-diphosphatase; 5, phosphoenolpyruvate carboxykinase; 6,pyruvate dehydrogenase; 7, acetyl CoA carboxylase. Note that arrows represent pathways andnot specific enzymatic reactions. Abbreviations: G-1-P, glucose-i-phosphate; G-6-P, glucose-6-phosphate; UDPG, Uridyl-diphospho-glucose.
Hypoglycemia
PIR 120 pediatrics in review #{149}vol. 11 no. 4 october 1989
Hypeninsulinemia has also long beenattributed to “nesidioblastosis,” anappearance caused by budding of de-veloping islet tissue from ducts. Thisis not a pathologic finding in the infantpancreas, however, and is thereforenot an anatomic correlate of hypen-insubinism. Hypeninsulinemia mayalso be associated with the pan-creatic endocrine hyperplasia foundin infants on diabetic mothers withBeckwith-Wiedemann syndrome andwith hemolytic disease of the new-born (erythnoblastosis fetabis).
The ovensecretion of insulin resultsin augmented glucose utilization,
mostly by skeletal muscle. In additionto the suppression of ketosis, an in-creased rate of glucose utilization isone of the hallmarks of hyperinsuli-nemic hypoglycemia. Whereas a hy-poglycemic infant with a normal rateof glucose uptake (as would be thecase with hypopituitarism or a defectin hepatic glucose production) will re-quire 6 to 8 mg/kg per minute ofglucose (the normal endogenous pro-duction rate during fasting), a hypen-insulinemic infant will often require>12 mg/kg per minute to preventhypoglycemia.
The diagnosis of hyperinsulinemia
based on circulating plasma insulinconcentrations alone is a problem.Serum on plasma insulin concentra-tions must be intenpneted in relationto serum glucose, because insulinconcentrations at times of hypogly-cemia may be in the “normal” range.In general, insulin is appropriatelysuppressed if the ratio of insulin toglucose (in microunits pen milliliter tomilligrams pen deciliter) is less than0.3. Hypeninsulinism should be asso-ciated with the expected secondaryeffects: suppressed bevels of plasmaf�-hydnoxybutyrate relative to glu-cose, a similar suppression of totalbranch chain amino acids (beucine +isoleucine + vabine),6 increased glu-cose requirements, and an excessiveglycemic response to glucagon dueto increased glycogen stones.7
The demonstration of hypeninsubi-nemia on the biochemical conse-quences of hypeninsulinism is an in-dication for therapy aimed at sup-pressing insulin secretion. This isbest accomplished with diazoxide,given in three divided doses for a totalof 5 to 10 mg/kg per day, and nomore than 20 to 25 mg/kg pen day.This may be augmented by the useof corticostenoids (for example, withprednisone, starting at 2 mg/kg penday [60 mg/m2 pen day]) which impairresponsiveness to insulin. In addition,after intnavenous dextrose infusionsare no longer necessary, frequentfeedings (every 3 to 4 hours) shouldbe given until resolution of the hyper-insulinism. Most recently, feedings ofraw cornstarch, which is slowly ab-sorbed, have been used to maintaina safe level of glycemia.
If medical therapy is not adequate(ie, there is persistent hypoglycemiaand/on excessive weight gain), sur-gical removal of the pancreas is indi-cated. The initial procedure mostoften advocated is subtotal pancrea-
at UNIV OF CHICAGO on May 21, 2013http://pedsinreview.aappublications.org/Downloaded from
5
4
3
2
Ew
>-I.-
0
>.
Cr,
-Ja.
30 40 50 60 70
PLASMA GLUCOSE mg/lOOml
Fig 3. Relationship of plasma glucose and �3-hydroxybutyrate levels during fasting in infantswith hyperinsuilnism (<)), control infants (#{149}),and children with ketotic hypoglycemia (0). Tworegression lines refer to former group (- - -) compared to latter two combined (--). (Usedwith permission; from Stanley CA, Baker L. Hyperinsulinism in infancy: diagnosis by demonstrationof abnormal response to fasting hypoglycemia. Pediatrics. 1976;57:702-71 1.)
TABLE 2. Causes ofHypoglycemia
HypeninsubinismHypopituitanismGlycogen storage diseaseHereditary fructose intoleranceGalactosemiaDisorders of gluconeogenesis
Fructose-i ,6-diphosphatase de-ficiency
Phosphoenolpynuvate carboxy-kinase deficiency
Amino acids disordersMethylmabonic acidemia3-hydroxy,3-methyl glutanc aci-
demiaEthylmabonic-adipic acidunaGlutaric acidemia type lbMaple syrup urine disease
Disorders of fatty acid metabolismDefects in camitine metabolismAcyl CoA dehydrogenase de-
fectsKetotic hypoglycemia
METABOLISM
pediatrics in review #{149} vol. 11 no. 4 october 1989 PIR 121
tectomy. In this procedure 80% to
90% of the pancreas is removed. Itshould be noted that focal lesions arerarely apparent on nadiobogic exami-nation (angiognaphy, computed to-mography) and often are not palpableto the surgeon. In addition, focal le-sions are often in the head of thepancreas, thereby evading resectiondunng subtotal pancreatectomy. Thishas led some to recommend “near
total” (99%) pancreatectomy as theinitial procedure of choice.8
Hypopituitarism
Hypoglycemia is a potent stimulusfor the release of growth hormoneand cortisob. If serum growth hon-mone concentrations were less than10 ng/mL and cortisol levels less than20 zg/dL at the time of symptomatichypoglycemia, such findings wouldindicate the possibility of an abnor-mabity in hypothalamic-pituitary func-tion. Indeed, although hypopituitan-ism is rarely diagnosed in the new-born period, it is more likely to occurwith hypoglycemia and hypenbilinubi-nemia (secondary to hypothyroidism)than growth failure, the latter takingat beast 6 months to occur. An impor-tant physical sign often present in themale newborn with hypoglycemia dueto hypopituitanism is microphallus.Normal values for penile length havebeen published9 and should be usedto assess the male infant in whomhypopituitanism is suspected. Abnor-malities of the palate, of antidiunetichormone secretion, or of visionshould also alert the pediatrician tothe possibility of hypopituitanism be-cause of their association with opticchiasm, septum pellucidum, and hy-pothalamic dysmorphobogy (septo-optic dysplasia).
The diagnosis of hypopituitanism
as a cause of hypoglycemia is bestmade by documenting subnormalconcentrations of growth hormoneand cortisol at the time of sympto-matic hypoglycemia. In addition, pro-vocative stimuli, the discussion ofwhich is beyond the scope of thispaper, may be used to test for growthhormone, ACTH, and thynoid-stimu-bating hormone release. In olden chil-dren, the production of insulin-in-duced hypoglycemia is a useful testbecause it allows simultaneous evab-uation of growth hormone and corti-sol secretion.
The prevention of hypoglycemiadue to hypopituitanism may requiredaily therapy with growth hormone,in contrast to the thrice-weekly negi-men used in the treatment of shortstature. Cortisol replacement shouldbe undertaken with caution becauseadministration of cortisob in physio-logic doses will suppress the hy-pothalamic-pituitary-adnenal axis. In apatient with marginal cortisol secre-tion, such therapy may further impairthe response to unpredicted stress(eg, trauma). If glucocorticoids areused, maintenance doses (1 5 to 20mg of hydnocortisone pen square me-ten per day) should be increasedthree- to fivefold during febnile ill-nesses.
Hypopituitanism as a cause of hy-poglycemia usually is self-limited induration. The child olden than 3 to 4years, bike the adult, does not nequinenormal growth hormone secretion tomaintain euglycemia. On the otherhand, of course, adrenal crisis in anolder child with hypopituitanism maybe associated with hypoglycemia.
Glycogen Storage Disease
During fasting, euglycemia is main-tamed initially by mobilization of gly-cogen stones following which gluco-neogenesis is required for adequatehepatic glucose production. Type Iglycogen storage disease, a para-digm for metabolic defects that causehypoglycemia, results from a defi-ciency of glucose-6-phosphatase.Because the dephosphonylation ofglucose-6-phosphatase is a commonpathway in the hepatic production ofglucose via glycogenolysis on gluco-neogenesis, this defect pnevents
at UNIV OF CHICAGO on May 21, 2013http://pedsinreview.aappublications.org/Downloaded from

Raw, uncooked cornstarchgiven orally provides a slow-release glucose mechanism
from the intestine whichprevents hypoglycemia
during brief fasting as in typeI glycogen storage disease
(gbucose-6-phosphatasedeficiency).
Response to glucagon,following a prolonged fast,
may provide clues as to thecause of hypoglycemia.
Hypoglycemia
PIR 122 pediatrics in review #{149}vol. 11 no. 4 october 1989
maintenance of glycemia postabsorp-tion. Furthermore, the mobilization ofglycogen and induction of gluconeo-genesis result in lactate rather thanglucose production. This metabolicerror also leads to accumulation ofhepatic glycogen and fatty infiltrationof the liver, the batten being responsi-ble for marked hepatomegaly.
The diagnosis of glucose-6-phos-phatase deficiency should be sus-pected in patients with fasting hypo-glycemia, metabolic acidosis with anincreased anion gap (due to lactateaccumulation), and hepatomegaly.Despite appropriate suppression ofinsulin secretion, such patients havea relative absence of ketosis, prob-ably due to the shunting of glycolyticintermediates toward fatty acid syn-thesis. A hallmark of this disorder isthe absence of a glycemic responseto glucagon, a potent glycogenobytichormone. The administration of 30pg/kg of glucagon intramuscularly (onas much as 1 00 zg/kg in a newborn)should result in a greater than 50%increase in serum glucose concentra-tion within 30 minutes. In patientswith glucose-6-phosphatase defi-ciency, this increase in glucose doesnot occur, whereas blood lactate in-creases markedly.
When suspected, the diagnosis ofgbucose-6-phosphatase deficiencymay be confirmed by directdetermi-nation of enzyme activity in a liverbiopsy specimen. It should be noted,however, that type Ia glycogen stor-age disease is associated with defi-cient in vitro activity of this enzyme,and type lb has normal in vitro activ-ity. In the latter condition, the in vivoactivityof glucose-6-phosphatase isimpaired because of a defect in glu-cose-6-phosphate transport.In addi-tion, three other types of glycogenstorage disease may occur ininfancywith findings similar to those in infantswith glucose-6-phosphatase defi-ciency. They are amybo-l ,6-glucosi-
dase deficiency (type Ill), hepaticphosphonylase deficiency (type VI),and phosphorylase kinase deficiency(type IX).
The treatment of glycogen storagedisease is based on avoiding thepostabsorptive state, thereby elimi-nating hepatic metabolic flux towardgbucose-6-phosphate. In the past,this was accomplished through theuse of frequent feedings, more ne-cently in concert with continuousnighttime enteral infusions. Today,therapy has been considerably sim-plifled by the use of raw, uncooked
cornstarch, given by mouth.1#{176}Conn-stanch is slowly hydrolyzed in the in-testine, allowing prolonged, steadyabsorption of carbohydrate. It istaken every 4 to 5 hours as a slurryto provide sufficient carbohydrate (6to 10 mg/kg pen minute) to suppresshepatic lactate production.
Hereditary Fructose Intolerance
This disorder (deficiency of hepatic1-phosphofructose abdolase) is char-actenized by vomiting, diaphonesis,tremor, convulsions, and/on coma fob-lowing the oral ingestion of fructose.Itmay occur in infancy with failure tothrive accompanied by recurrentvomiting, hypoglycemia, and hepato-megaby. In childhood, the aforemen-tioned symptoms occur in concertwith hypoglycemia and metabolic aci-dosis. Patients with this disorderhave an extraordinary aversion tosweets which may lead one to sus-pect the diagnosis.Confirmation maybe obtained through the controlledadministration of panentenab on oralfructose with monitoring of serumglucose, lactate, and phosphate con-centrations and acid-base status.Treatment is the avoidance of fruc-
tose. This is extremely difficult giventhe ubiquitous presence of fructosein prepared foods, some vegetables,all fruits, and many medicines.Chronic intake of small amounts offructose will lead to a renal tubulo-pathy and a continued impairment ofgrowth.
Galactosemia
The deficiency of gabactose-1 -
phosphate unidyl transfenase (“clas-sic” gabactosemia) is a serious dison-den that often occurs in the newbornperiod. Because galactose is a majorconstituent of the diet of most new-bonns, hepatic dysfunction due to the
� inability to use galactose may occur� early in life. In fact, galactose of ma-
ternal origin may result in hepaticdamage in the affected fetus.
Hypoglycemia in this disorder is� rare but is presumably due to accu-� mulation of galactose-l -phosphate;� however, the precise mechanism by� which glucose production by the liven� is impaired is unknown. The diagno-� sis should be suspected in the infant
with hepatomegaby, jaundice, vomit-ing, irritability, and failure to thrive.Preliminary diagnosis is often basedon the presence of nonglucose-ne-ducing substances in urine speci-mens collected while the patient isreceivinggalactose. Glucose oxidaseurine dipstickswillnot detect galac-tose. The enzymatic defect can beconfirmed by the analysisof enythro-cyte galactose-1-phosphate unidyltransfenase activity.Neither livenbi-opsy non galactose challenge are re-quired to confirm the diagnosis.Treatment consists of the eliminationof galactose fnom the diet.Ifthe di-agnosis is made before the develop-ment of hepatic cirrhosis and mentalretardation, the prognosis is favona-ble.
Disorders of Gluconeogenesis
Disorders of gluconeogenesis inwhich specific enzymes are affectedinclude fructose-i,6-diphosphatasedeficiencyand phosphoenolpyruvatecanboxykinase (PEPCK) deficiency.
Patients with fructose-i ,6-diphos-phatase deficiencyhave hepatomeg-aby,lacticacidosis,ketonunia,and hy-poglycemia. Conversion of galactoseto glucose is normal but conversion
at UNIV OF CHICAGO on May 21, 2013http://pedsinreview.aappublications.org/Downloaded from

METABOLISM
pediatrics in review #{149}vol. 11 no. 4 october 1989 PIR 123
of fructose to glucose is blocked. Theelimination of fructose from the dietand avoidance of prolonged fasts areeffective therapy. In contrast to per-sons with hereditary fructose intolen-ance (in which fasting hypoglycemiadoes not occur), these patients donot have an aversion to sweet foods.
Deficiency of PEPCK is associatedwith feeding problems, failure tothrive, metabolic acidosis, and hypo-glycemia. Hepatomegaly may occursecondary to fatty infiltration of theliven. This is a rare disorder and littleis known about therapy; frequentfeeding is indicated to prevent stim-ulation of hepatic gluconeogensis.
Disorders of Amino AcidMetabolism
A number of metabolic defects ne-suIting in hypoglycemia and the ex-cretion of abnormal organic acids inthe urine have been characterized. Ingeneral, children with these disordershave failure to thrive, poor feeding,vomiting, and hepatomegaly. Labo-natony data often demonstrate hypen-ammonemia and hypochlonemic (in-creased anion gap) metabolic aci-dosis in conjunction with hypogly-cemia. Specific diagnoses are oftenbased on identification of specific on-ganic acids in the urine by gas chro-matognaphy/mass spectnoscopy. Inmany cases, definitive diagnosis isbased either on in vitro analyses ofliven biopsy specimens or on investi-gation of specific metabolic pathwaysin cultured fibroblasts.
Specific metabolic defects associ-ated with organic acidunia and caus-ing hypoglycemia include methylma-Ionic acidemia, 3-hydnoxy-3-methylglutanic acidemia, ethylmabonicadipicacidunia,and glutanicacidemia typelb.The enzymatic defects inthe battentwo disorders also affect fatty acidoxidation.
Maple syrup urine disease is a dis-order of branched chain amino acidmetabolism which may occur in thenewborn period and manifest as poorfeeding, severe metabolic acidosis,and hypoglycemia. Progressive neu-nobogic deterioration beading to deathin infancy occurs in untreated pa-tients. Blood and urine contain exces-sive bevels of the three branchedchain amino acids, leucine, isoleu-
cine, and vabine, and their respectiveketoacids. The enzymatic defect,which involves decanboxylation ofthese ketoacids, can be demon-strated in leukocytes.
Treatment of the aforementioneddisorders involves limiting the intakeof the offending amino acids. Thisrequires a limited protein intake withsupplementation of essential aminoacids where applicable.
Disorders of Fatty AcidMetabolism
A recently described class of dis-orders that can cause hypoglycemiainvolves defects in the $-oxidation offatty acids. The various forms of can-nitine deficiency as well as distinct oncombined abnormalities of short-,medium-, or long-chain acyl CoA de-hydnogenases result in accumulationof abnormal metabolites of fattyacids. These defects are associatedwith impaired ketogenesis due to im-pained mitochondnial fatty acid oxi-dation. Diagnosis may be ascertainedby the detection of the products ofextramitochondnial oxidation (dicar-boxybic acids) and acylcannitine inurine. The avoidance of prolongedfasts through the use of frequent can-bohydrate-nich meals is currently theonly available form of therapy for pni-mary enzymatic defects in �3-oxida-tion. Dietary carnitine supplementa-tion is succesful in treating disordersof cannitine synthesis on excessiverenal carnitine loss and may be mdi-cated in other defects in fatty acidmetabolism in which secondary can-nitine deficiency occurs.
Ketotic Hypoglycemia(Accelerated Starvation)
This entity should not be consid-ered a pathologic disorder resultingin hypoglycemia. Rather, it is a non-specific designation applied to pa-tients in whom fasting beads to phys-iologic hypoglycemia (Fig 1) accom-panied by hypoglycemic symptoms.Age of onset is typicallybefore 18months. Episodes occur aftera pro-longed fast, usually in the morning.Hypoglycemia is accompanied by ap-propriate suppression of plasma in-sulin concentrations, elevated serumgrowth hormone and cortisol concen-
trations, acetonunia, and increasedplasma $-hydnoxybutynate concen-tnations (Fig 2). Lactic acidosis doesnot occur. Diagnosis is based on ebic-iting ketosis with hypoglycemia dun-ing a monitored fast; other specificdiagnoses must be excluded. Thecondition is self-limiting and treatedeffectively by the avoidance of pro-longed fasts.
Factitious Causes ofHypoglycemia
A variety of drugs and toxins can
lead to hypoglycemia. Most promi-nent among these is insulin. The di-agnosis of surreptitious insulin ad-ministration may be made by docu-menting hypeninsulmnemia in thepresence of suppressed plasma C-peptide concentrations. Because C-peptide is produced during the proc-essing of proinsubin, its appearancein the circulation parallels insulin se-cnetion. Conversely, suppression ofendogenous insulin secretion by ex-ogenous administration will diminishsecretion of C-peptide. A secondcause of factitious insulin secretion isthe ingestion of oral hypoglycemicagents. These drugs can be detectedin urine.
Jamaican vomiting sickness is thename given to the acute illness, ac-companied by hypoglycemia, whichfollows the ingestion of unripe akeefruit. The offending substance, hy-poglycin, probably interferes withfatty acid oxidation, thus producing aconditioning similar in its pathogen-esis to the errors in fatty acid metab-obism. More familiar toxins that causehypoglycemia are aspirin and alcohol.Both result in impaired hepatic glu-cose production.
CONCLUSION
Prompt recognition and treatmentof hypoglycemia isimportant to pre-vention of central nervous system im-pairment. Diagnostic evaluation isaimed at specific physiologic and bio-chemical defects. Appropriate inter-vention can be initiated after a diag-nosis is established. Hypoglycemiamust be viewed as a symptom com-plex that requires detailed assess-ment.
at UNIV OF CHICAGO on May 21, 2013http://pedsinreview.aappublications.org/Downloaded from

Hypoglycemia
PIR 124 pediatrics in review #{149} vol. 11 no.4 october 1989
REFERENCES
1 . Chaussain JL. Glycemic response to 24hour fast in normal children and childrenwith ketotic hypoglycemia. J Pediatr.1973;82:438-443
2. Gutberlet AL, Comblath M. Neonatal hy-poglycemia revisited, 1975. Pediatrics.1976;58:1 0-17
3. Stanley CA, Baker L. Hyperinsulinism ininfancy: diagnosis by demonstration of ab-normal response to fasting hypoglycemia.Pediatrics. 1976;57:702-71 1
4. Phillip M, Bashan N, Smith CP, Moses SW.An algorithmic approach to diagnosis ofhypoglycemia. J Pediatr. 1987;1 10:387-390
5. Witte DP, Greider MH, DeSchryver-Kec-skemeti K, Kissane JM, White NH. Thejuvenile human endocrine pancreas: nor-mal v idiopathic hypennsulinemic hypogly-cemia. Semin Diag Pathol. 1984;1 :30-42
6. Chaussain J-L, Georges P, Gendrel D,Donnadieu M, Job J-C. Serum branched-chain amino acids in the diagnosis of hy-perinsulinism in infancy. J Pediatr.1980;96:923-926
7. Finegold DN, Stanley CA, Baker L. Gly-cemic response to glucagon during fastinghypoglycemia: an aid in the diagnosis ofhyperinsulinism. J pediatr. 1980;96:257-259
8. Kramer JL, Bell MJ, DeSchryver K, BowerRJ, Temberg JL, White NH. Clinical andhistologic indications for extensive pan-creatic resection in nesidioblastosis. Am J
Surg. 1982;143:1 16-1199. Lee PA, Mazur T, Danish A, et al. Micro-
penis, I. criteria, etiologies and classifica-tion. Johns Hopkins Med J. 1980;146:156-164
10. Chen Y-T, Comblath M, Sidbury JB. Corn-starch therapy in type I glycogen storagedisease. N EngI J Med. 1984;310:171-175
SUGGESTED READING
Aynsley-Green A, Soltesz G. Hypoglycaemiain Infancy and Childhood. London, England:Churchill-Livingstone; 1985
Comblath M, Schwartz A, eds. Disorders ofCarbohydrate Metabolism in Infancy. Phila-delphia, PA: WB Saunders; 1976
Self-Evaluation Quiz
10. In the evaluation of hypoglycemia in ayoung child, the least likely helpful diagnos-tic test among the following would be:
A. Oral glucose tolerance test.B. Serum ketones level.C. Serum lacticacad level.D. Plasma insulin level.E. Plasma level of growth hormone.
11. In an infant with hypoglycemia, simul-taneous measurement of serum glucoseand growth hormone levels 4 hours after a
feeding, at a time when the infant wassymptmatic, found both levels to be abner-mally low. These findings suggest that theprimary defect is most likely to be:
A. Hyperinsullnism.B. Hypopituitansm.C. Glycogen storage disease, type I (von
Gierke).D. Accelerated fasting.E. Phosphoenolpyruvate carboxykinase de-
ficiency.
12. In an infant or child with postprandlalhypoglycemia, among the following findingsthe least likely is:A. Weakness.B. Tremor.
C. Pallor.0. Bradycardia.E. Cold sweat.
13. An infant with hypoglycemia has appar-ently suppressed serum levels of insulin andappropriately elevated levels of ketones ata time when she is symptomatic as a resuftof hypoglycemia. These findings suggestthat among the following her primary con-dition is most likely to be:
A. A disorder of fatty acid metabolism.B. Camitine deficiency.C. Glycogen storage disease, type I.D. Hypennsulinism.E. Hypopituitarism.
Department of Corrections
In the article by Dewitt in the July 1989 issue of Pediatrics in Review, “AcuteDiarrhea in Children,” itisunfortunatethatinTable 3 on page lithe spelling of therehydration solution “Rehydralyte” was printed as “Rydrolute.” In addition, Hydra-Lyteand Infalyte are no longer marketed.
at UNIV OF CHICAGO on May 21, 2013http://pedsinreview.aappublications.org/Downloaded from

DOI: 10.1542/pir.11-4-1171989;11;117Pediatrics in Review
Philip A. Gruppuso and Robert SchwartzHypoglycemia in Children
ServicesUpdated Information &
http://pedsinreview.aappublications.org/content/11/4/117including high resolution figures, can be found at:
Permissions & Licensing
/site/misc/Permissions.xhtmlits entirety can be found online at: Information about reproducing this article in parts (figures, tables) or in
Reprints/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
at UNIV OF CHICAGO on May 21, 2013http://pedsinreview.aappublications.org/Downloaded from