lÖsemİ morfolojİ - tphd.org.tr · – sitogenetik. all morfolojİk siniflamasi cd 34 pozitif...
TRANSCRIPT

LÖSEMİ MORFOLOJİSİ
Namık Yaşar ÖZBEKSBÜ Ankara Çocuk Sağlığı ve
Hastalıkları Hematoloji Onkoloji EAH

UYGULAMA
• Posterior superior iliak krestten yapılır
• 18 ay altında tuberositas tibiadan yapılabilir

YAYMA
• Önceden hazırlık yapılmalı (anestezi, vb)• Lamlar isim yazılarak hazır tutulmalı• Birlikte yapılacak incelemeler için yeterli
örnek örnek ayrılmalı• Önce biyopsi sonra aspirasyon yapılmalı

YAYMA/BOYAMA• Uygun yayma tekniği
– İnce yayılmalı– Hücre hasarı oluşturulmamalı
• Uygun boyalarla uygun sürede boyanmalı– Wright boyası yetersiz
• Granülleri yetersiz boyuyor• İmmatür hücre ayrımı zor
– HE veya MGG uygun• Ek boyalar kullanılmalı


MORFOLOJİK TANI BASAMAKLARI
• Önce lösemi tanısı koy• Hasta kliniği• CBC ve PY değerlendirmesi• Kİ değerlendirmesi
• M/E oran• Blast morfolojisi ve yüzdesi
• ALL >%25• AML: WHO >%20; FAB >%30
• Blast dışı hücrelerin özellikleri• Sonra tiplendirme yap

• 164 resimdeki 438 işaretli hücrenin tanımlanması isteniyor
• Tam (> 17/21) konsensus %59.4
MORFOLOJİ ÇOK ÖNEMLİDİR, ANCAK…….
SON TANI DİĞER LABORATUVAR YÖNTEMLERİNİN DE YARDIMIYLA OLUŞTURULMALIDIR



LÖSEMİDE SINIFLAMA
• Neden sınıflama yapıyoruz?– Prognoz hakkında bilgi sahibi olmak– Tedavi düzenlemesini bu prognostik
kriterlere göre oluşturmak• Farklı sınıflamalar mevcut
– Morfolojik– İmmünolojik– Sitogenetik


ALL MORFOLOJİK SINIFLAMASI
CD 34 pozitif hematopoetik kök hücre


MORFOLOJİK SINIFLAMA• 3 subgrup vardır
– L1 tipi:– L2 tipi:– L3 tipi:
• ALL hücreleri PAS (Periodic acid-Schiff) ile boyanır
• Sudan Black veya myeloperoksidaz ile boyanmaz

L1 TİPİ• %85 izlenir• Hücreler ufak,
uniform büyüklükte, dar sitoplazmalı
• Genelde nukleolus içermezler
• Az sayıda vakuol izlenebilir

Hand mirror varyantı

L2 TİPİ• %14 izlenir
(Erişkinde en sık)• Hücreler farklı
büyüklüklerde• Sitoplazması daha
geniş • Nukleolus içerir• Değişik sayılarda
vakuol izlenebilir

L3 TİPİ• %1 bu tip izlenir• Hücreler büyük, koyu
bazofilik sitoplazmalı • Çok sayıda ve belirgin
vakuolizasyon gözlenir • Çoğu B lenfosit kaynaklı• Nadiren pre-B veya T• Burkitt lenfoma
hücrelerine benzer

DİĞER VARYANTLAR• Granuler ALL
• El aynası varyant ALL
• Eozinofiliyle birlikte ALL

Granüler ALL• L1 veya L2 tipinde• En az % 5 blast granüllü
Azurofilik pembe granullerNadiren dev granullu
• Genelde pre-B tipi• Nadiren T (L2) tipi• Prognostik önemi yok
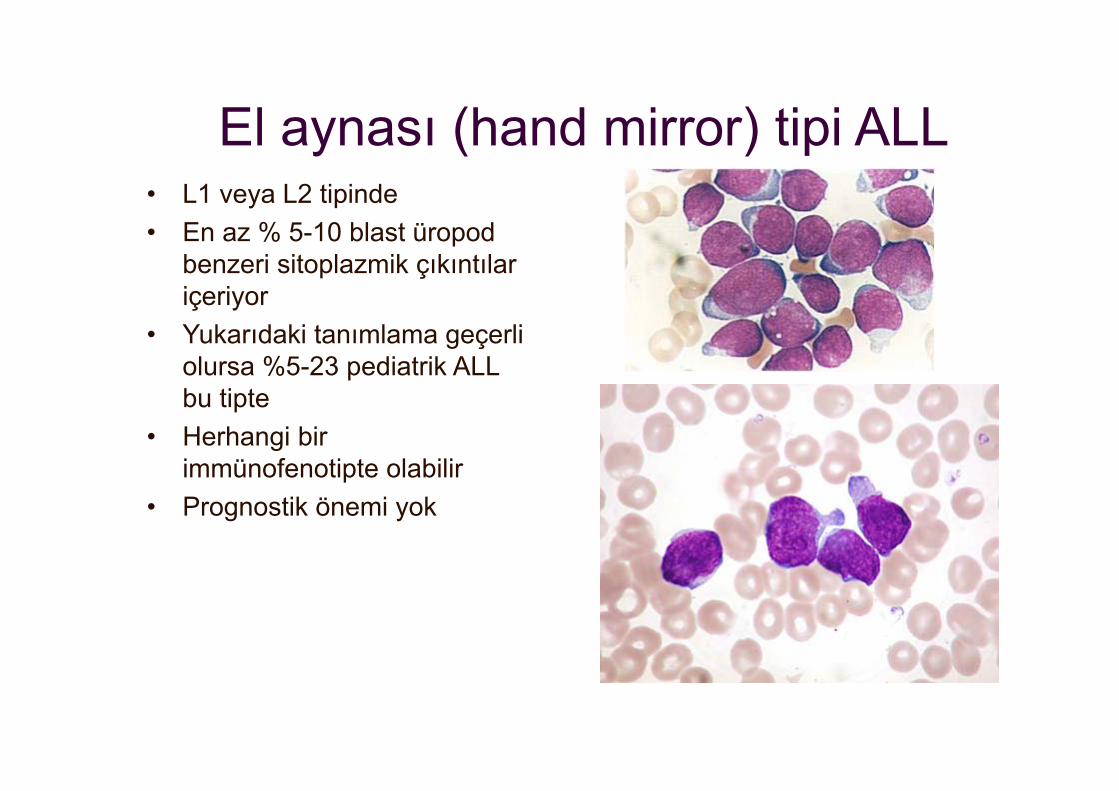
El aynası (hand mirror) tipi ALL• L1 veya L2 tipinde• En az % 5-10 blast üropod
benzeri sitoplazmik çıkıntılar içeriyor
• Yukarıdaki tanımlama geçerli olursa %5-23 pediatrik ALL bu tipte
• Herhangi bir immünofenotipte olabilir
• Prognostik önemi yok
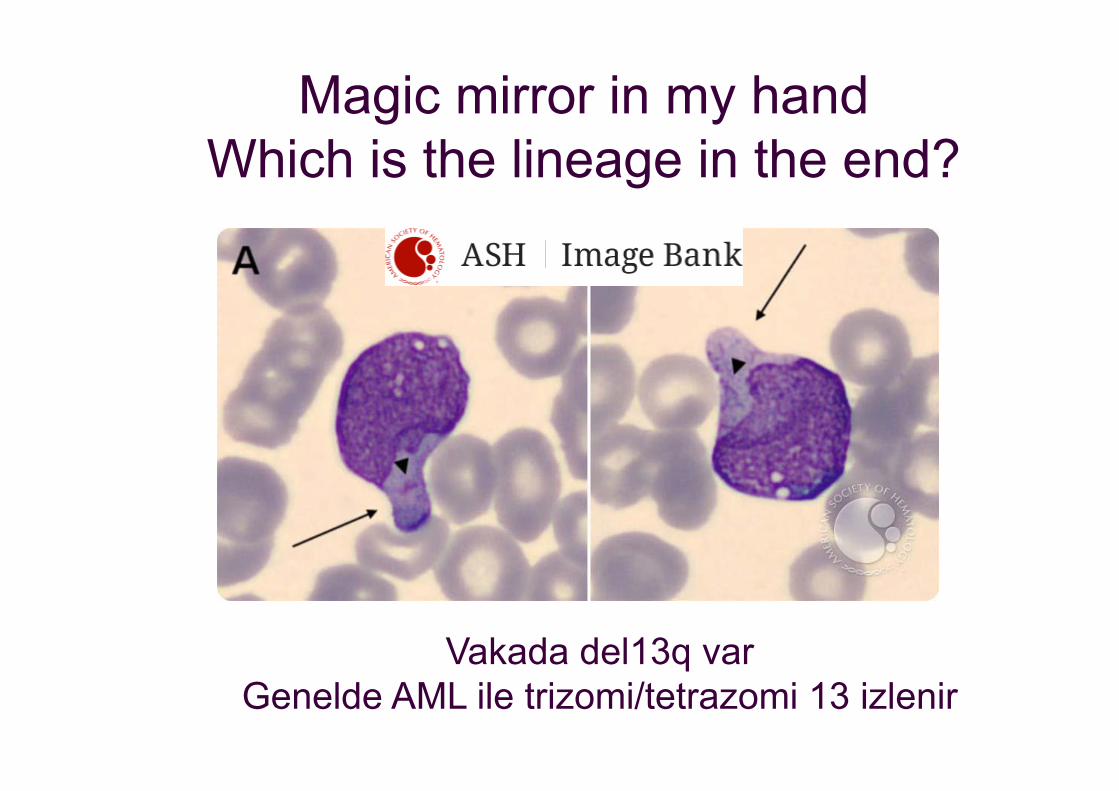
Magic mirror in my handWhich is the lineage in the end?
Vakada del13q varGenelde AML ile trizomi/tetrazomi 13 izlenir

Eozinofili ile birlikte ALL• L1, L2 tipi• Genelde pre-B tipi• Displastik eozinofiller +• Öncesinde sadece
hipereozinofili olabilir• Hipereozinofilik syn ile
karışabilir• t(5:14) sık (5. kr IL-3 geni)

ALL özel boyamalarBoya SonuçPAS Pozitif
Mpo Negatif
Sudan black Genelde (-)Granular ALL→ Zayıf (+)
Esterazboyamalar
Alfa-naftil bütirat Genelde (-)Granular ALL→ Zayıf (+)
Alfa-naftil asetat Negatif veya zayıf pozitif
Naftol AS-D kloroasetat Negatif veya zayıf pozitif
Asit fosfataz T hücrelilerde pozitifBazı B hücrelilerde pozitif

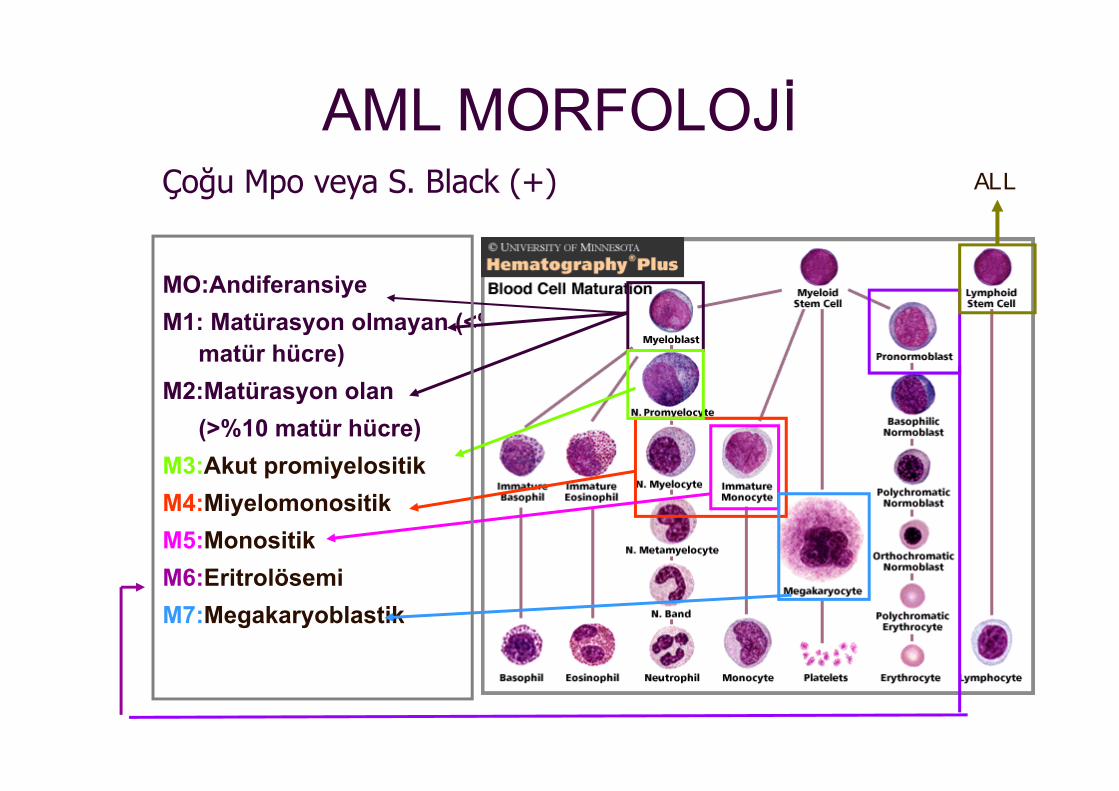
AML MORFOLOJİ
MO:AndiferansiyeM1: Matürasyon olmayan (<%10
matür hücre)M2:Matürasyon olan
(>%10 matür hücre)M3:Akut promiyelositikM4:Miyelomonositik M5:Monositik M6:EritrolösemiM7:Megakaryoblastik
Çoğu Mpo veya S. Black (+) ALL

AML MORFOLOJİ (FAB) MO:Andiferansiye ML
(Mpo sadece e.m. ile +) M1:Matürasyon olmayan ML
(<%10 matür hücre) M2:Matürasyon olan
ML(>%10 matür hücre) M3:Akut promiyelositik L
Auer rod t(15:17)(PML-RAR) DIC
M4:Miyelomonositik L Orbital granulositik sarkom
sıklığı M5:Monositik L
SSS tutulumu -naftil bütirat esteraz (+)
M6:Eritrolösemi PAS (+) Mpo (-)
M7:Megakaryoblastik L Down syn ile Miyelofibrozis CD41 veya CD61 (+)

Mufti et al, Haematologica 2008; 93:1712.

AML-MO• Kİ hiperselüler• Matür hücre yok• Küçük yuvarlak blastlar• Nukleus genelde düzgün• Eksantrik nucleolus• Granül ve Auer rod yok• Mpo negatif• T ve B işaretleyicileri negatif• CD13,14,15,33,34,HLA-DR +

AML-MO

AML-MO
AML-M0Mpo

AML-M1
• 1 yaş altında sık• >%3 Mpo veya SB +• Matür hücre oranı<%10• Granüllü L2 görünümlü• Nukleolus +• Auer rod %50 +• CD13, 14, 15, 33, 34 +• En sık t(9;22) izlenir
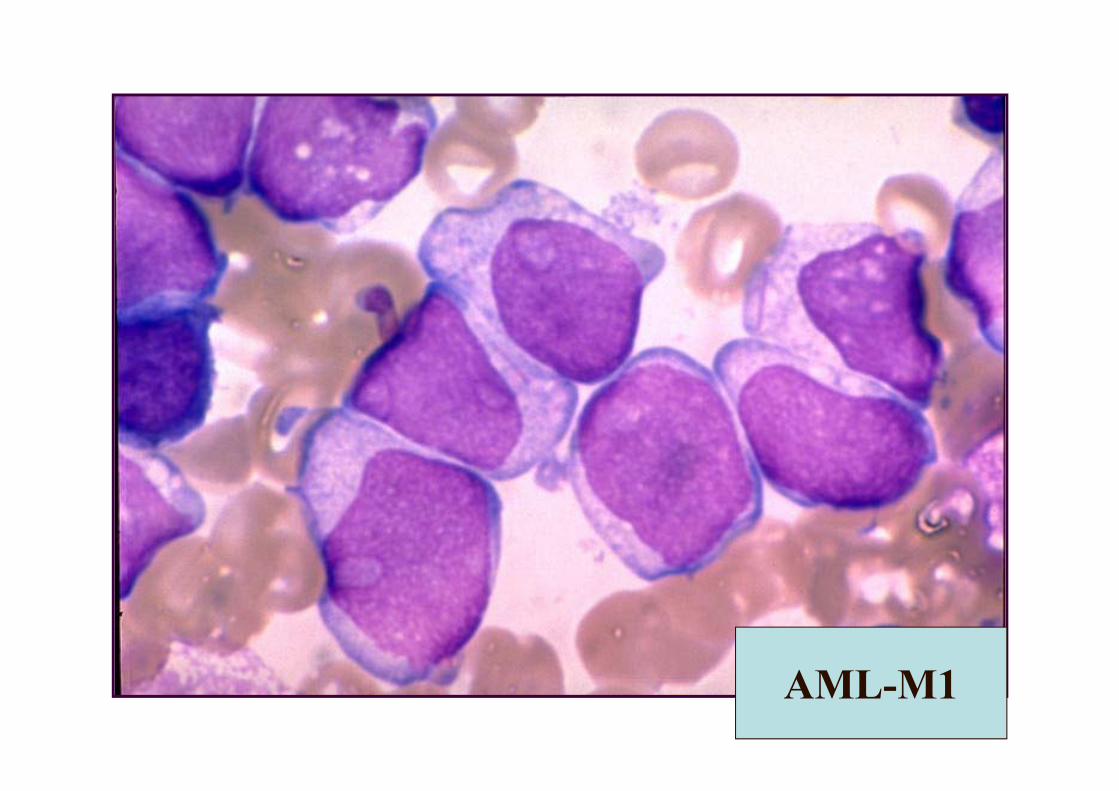
AML-M1
AML-M1


AML-M1Mpo

AML-M1 hand mirror varyant

• PY psödo-Pelgeroid hücreler• >%10 matür hücre• PNL ye kadar her safhada
matürasyon +• <%20 monositik komponent• Eozinofiller %15 e kadar +• Bazen bazofil ↑(M2 bazo)• Mpo + PAS –• t(8;21) sık• CD 13, 15 sık +
AML-M2

AML-M2

AML-M2

AML-M3 (hipergranuler APL)• Kİ genellikle hiperselüler• Granüllü büyük hücreler
– Oval böbrek şekilli veya bilobuleblastlar
– Faggot hücreleri• Mpo +• Genetik:
– t(15;17): Auer rod+– t(11:17)
Auer rod genellikle –Hiperlobule nötrofiller+ATRA cevabı kötü
– t(5:17): Auer rod genellikle -• İmmünfenotip
– CD13,15, 1 ve 33 +– CD 2 veya 19 ±– CD34 ve HLA-DR -

AML-M3

AML-M3
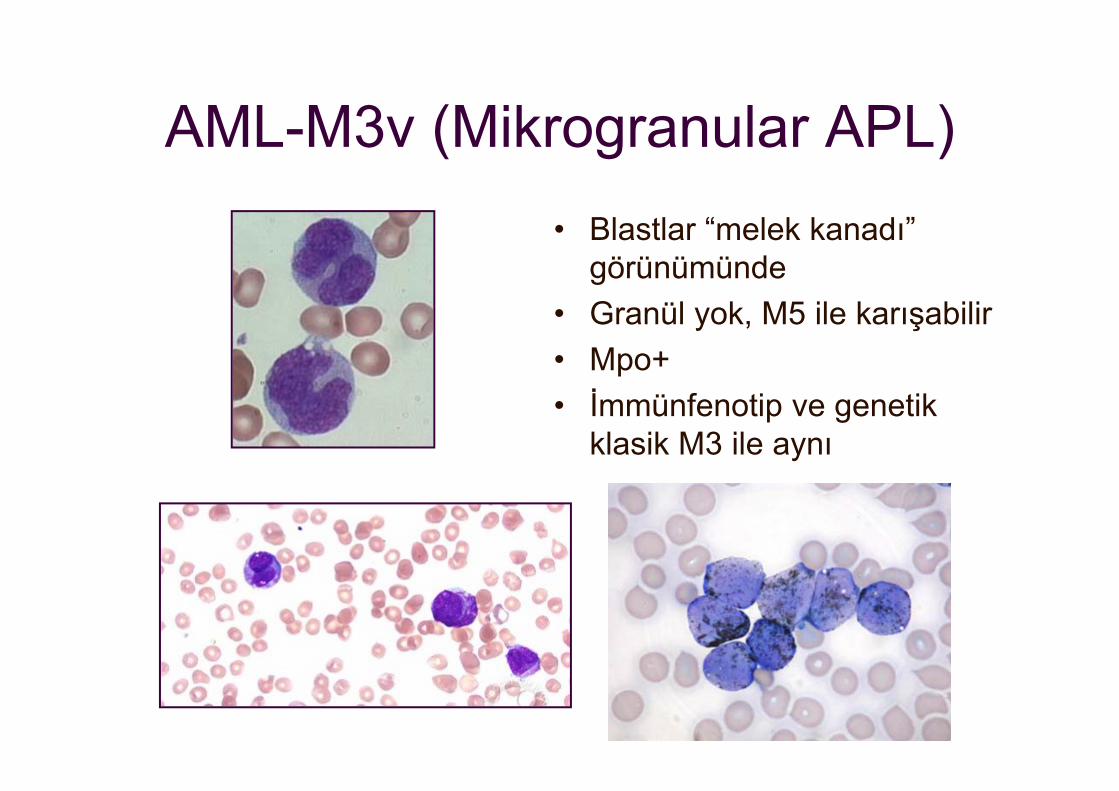
AML-M3v (Mikrogranular APL)• Blastlar “melek kanadı”
görünümünde• Granül yok, M5 ile karışabilir• Mpo+• İmmünfenotip ve genetik
klasik M3 ile aynı



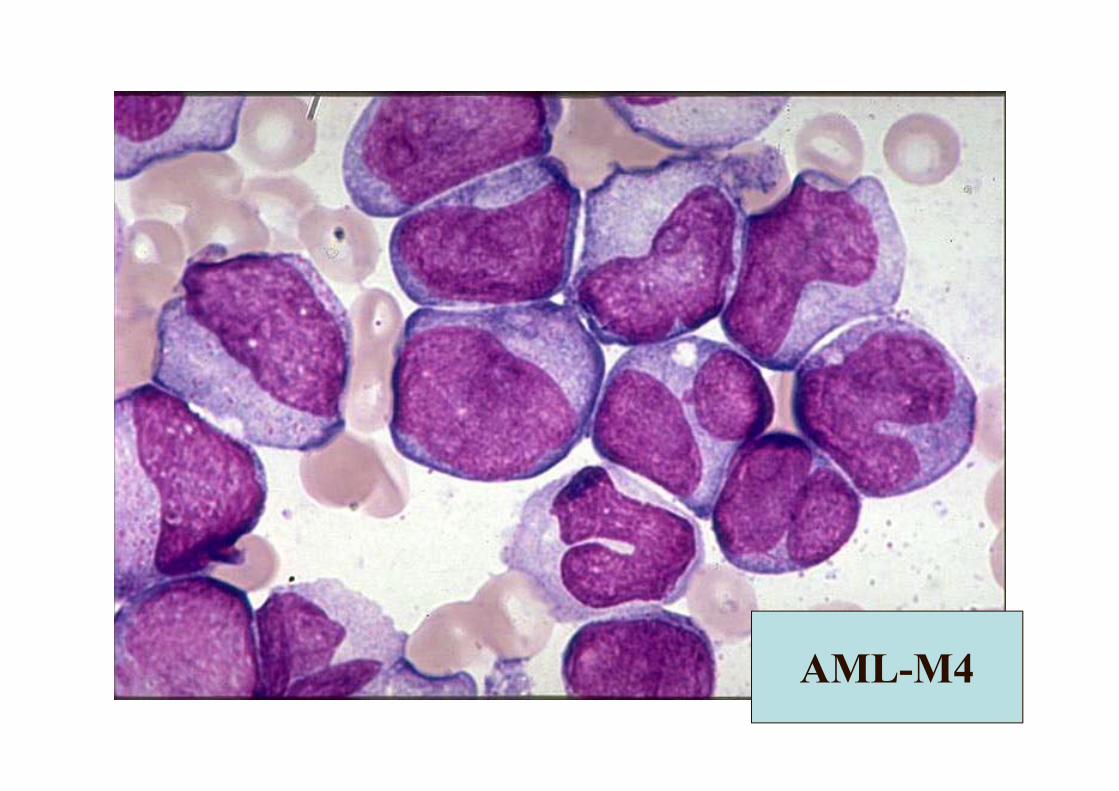
AML-M4(AMML)
• Gingival hiperplazi sık• Serum muramidaz
(lizozim) ↑• PY monosit sayısı
>5x109/L• Kİ <20 monoblast >80
(non eritroid)• İnv(16) del 16q22 +• CD13,33, 11b, 14 +

AML-M4
AML-M4

AML-M4Mpo

AML-M4-NAFTİL BÜTİRAT ESTERAZ

AML-M4Eo
• >%5 eozinofil• 16. kromozom
anomalileri ↑• Daha iyi prognoz


Orbital granulositik sarkom

AML-M5 (AMoL)• Ekstramedüller infiltrasyon sık• Serum muramidaz (lizozim) ↑• M5a: Maturasyon<%4
Monosit seri>%80Monoblast >%80Granulositik seri<%20
• M5b: Maturasyon>%4Monosit seri>%80Tüm maturasyon evreleri varGranulositik seri<%20
• CD11b ve 14 +• 11q23 bozukluğu sık

AML-M5 (AMoL)
• M5a (promonositik)
• M5b

AML-M5a

AML-M5a

AML-M5a

AML-M5b

AML-M5bMpo

AML-M5bGlikoforin

• NSE pozitif• Alfa naftil bütirat/asetat
esteraz ile daha iyi sonuç elde edilir
• Mpo negatif

AML-M5-NAFTİL BÜTİRAT ESTERAZ

AML-M6• PY anemi, anizositoz,
poikilositoz• Eritrolösemik hücrelerde
– ileri derecede displazi – multinuklearite – vakuolizasyon– sitoplazmik çıkıntılar – megaloblastoid değişiklikler
• Kİ tanı– %50 ↑ eritroid seri – Noneritroidlerde blast>%30

AML M6
• Pür eritroid tip– >%80 eritroid prekürsörler– CD235a (Glycophorin A), Hemoglobin A + – CD117+/-– CD34, HLADR, Mpo-
• Eritroid/miyeloid tip– >%50 eritroid prekürsörler– CD235a, Hb A + , CD71 dim/-, MPO-– > 20 miyeloblast (Eritroidler hücreler hariç)– CD34, CD117+, MPO-/+


AML-M6
• PAS +• CD13, 33, CD71,
Glikoforin +• CD34, HLA-DR
negatif• 8+, -5, del(5q), ve -7
izlenebilir

AML-M6

AML-M6

AML-M6PAS

AML-M7 • Kİ : “dry tap” fibrozis• Binükleer blast olabilir• PAS +Mpo –• Ekstrameduller tutulum:
– k.c.; KCFT bozuk, fibrozis– cilt
• CD41,42,61 +• Genetik subtipler
– Down syn ile AMKL– t(1;22) ile AMKL– Her ikisini de taşımayan AMKL

AML-M7



AML-M7CD41 BOYAMA

AML-M7FVIII BOYAMA

AML özel boyamalarBoya Miyeloblast Monoblast Eritroblast Megakaryoblast
PAS Negatif Genelde (-)Bazen ince/kalın granulasyon
Kuvvetli pozitifKaba granulasyonpaterni
Negatif veya ince granular pozitif
Mpo Pozitif Pozitif/Negatif Negatif Negatif
Sudan black Pozitif Pozitif/Negatif Negatif Negatif
Esteraz Alfa-naftilbütirat
Negatif Pozitif Negatif Negatif
Alfa-naftilasetat
Negatif/Pozitif Diffüz pozitifFloridle inh (+)
PozitifFloridle inh (-)
Lokalize pozitifFloridle inh (parsiyel)
Naftol AS-D kloroasetat
Pozitif Pozitif/Negatif Negatif Negatif
Asit fosfataz
Negatif Negatif Negatif Lokalize pozitif

AML MORFOLOJİ (WHO)1. REKÜREN GENETİK ANOMALİ OLANLAR• t(8:21) AML• İnv (16) veya t(16:16) AML• Akut promiyelositik L • t (9:11) veya11q23 (MLL)
translokasyonlu AML• İnv 3 veya t(3:3) AML• t (1:22) AML• Gen mutasyonlu AML
NPM1 mutasyonlu AML CEBPA mutasyonlu AML
2. MİYELODİSPLAZİYLE BİRLİKTE AML3. TEDAVİYE SEKONDER AML4. SPESİFİYE EDİLMEMİŞ AML• Minimal diferansiyasyonlu• Maturasyonsuz• Maturasyonlu
•Miyelomonositik• Monoblastik• Eritroid
EritrolösemiPür eritroid lösemi
• Megakaryoblastik• Bazofilik • Miyelofibrozisle birlikte panmiyelozis5.MİYELOİD SARKOM6.DOWN SYN İLİŞKİLİ MİYELOİD PROLİFERASYON•Geçici anormal miyelopoez• AML7.BLASTİK PLASMOSİTOİD DENDRİTİK HÜCRELİ AML

t(8:21) AML • Çocukluk AML %15’i• RUNX1T1:RUNX1 → Oncoprotein görevi yapar• FAB: %70-80 AML-M2, kalanı AML-M4• Auer rod sık izlenir• Dismiyelopoetik bulgular olabilir
Psödo Pelger Huet, Chediac Higashi benzeri granüller Megakaryositlerde ve eritroid seride daha az belirgin
• Diğer miyeloid elemanlarda hiperplazi olabilirEn sık eozinofil, daha az mastosit artışı


İnv (16) veya t(6:6) AML • Genellikle AMMoL-Eo tipi• Nadiren promonositik, M4 veya M5 tipi• M1 ve M2 tipi de izlenebilir• Kİ genellikle atipik eozinofilisi mevcut

t(9:11) veya 11q23 translokasyonlu AML
• AML’lerin %15-20 si• Genellikle monositer komponentli (M4, M5)• T(9:11) genellikle M5• MLL taşıyanlar her tip olabilir

NPM1 mutasyonlu AML • NPM1 (nukleofosmin) 5q üzerinde• M3 dışı tüm tiplerde olabilir• %75 M2 veya M4• %60 vakada “fincansı” (cup like) blast• İyi klinik gidişle ilişkili
BLOOD, 1 SEPTEMBER 2006 VOLUME 108, NUMBER 5, pp1784

CEBPA mutasyonlu AML
BLOOD, 1 SEPTEMBER 2006 VOLUME 108, NUMBER 5, pp1784
• CCAAT/enhancer binding protein α (CEBPA)• Germline mutasyon üzerinde duruluyor (ailevi AML)• Kemik iliği hiperselüler• Genelde M1 ve M2 sık• M6 dışı tüm tipler olabilir• Prognoz
• Tek allelde mutasyon→ Diğer AML pleriyle aynı• Biallelik mutasyon → Diğer AML plerinden iyi!
• Aberan CD7 ekspresyonu %50 vakada izleniyor