outline - university of cambridge · outline • born-oppenheimer approximation • hohenberg-kohn...
TRANSCRIPT

Outline
• Born-Oppenheimer approximation
• Hohenberg-Kohn theorems
• Kohn-Sham implementation
• The Exc functional
• The local density approximation (LDA)
• Limits of current implementations of DFT

The First Principles Approach
nucleuselectron
it is free of adjustable parametersit treats the electrons explicitlycost of the calculation limits system size and simulation time
eeeNNNeNeN VVVTTH −−− ++++= ˆˆˆˆˆˆ ,where
{ } { }( ) { } { }( )iJeNeNiJeNeN EH rRrR , , ˆ , , , , Ψ=Ψ

Born-Oppenheimer approximation decouples the electronic problemfrom the ionic problem. The electronic problem is:
Born-Oppenheimer Approximation
{ }( ) { }( )ii EH rr ˆ Φ=Φ
Electron mass much smaller than nuclear mass: Timescale associated with nuclear motion much slower than that
associated with electronic motionElectrons follow instantaneously the motion of the nuclei,
remaining always in the same stationary state of the Hamiltonian
{ } { }( ) { }( ) { }( )iJNiJeN rRrR , , ΦΘ=Ψ
{ } { }( ) { } { }( )iJeNeNiJeNeN EH rRrR , , ˆ , , , , Ψ=Ψ
depends only parametrically on {RJ}

Time-independent, non-relativistic Schrödinger equation in theBorn-Oppenheimer approximation [in atomic units: ]:
The Electronic Problem
eNeee VVTH −− ++= ˆˆˆˆ
∑<
− −=
N
ji jieeV
||1ˆ
rr∑∇−=N
iieT 2
21ˆ
∑ −−=−
atN
eNZV
α α
α
||ˆ
Rr
where
1=== heme
{ }( ) { }( )ii EH rr ˆ Φ=Φ
The external potential and the number of electrons, N, completely determine the Hamiltonian
External potential

Quantum States for Many ParticlesWhat happens if we have three particles?
Possibility 1
Number of coefficients = 2 x N (Number of qubits).
Possibility 2
Number of coefficients = 2N
( ) ( ) ( )333322221111 101010 bababa +++
111011101110
001010100000
hgfe
dcba
+++
++++
Its this one

Why Quantum Mechanics is Hard
The mathematical difficulty of solving the Schrödinger equation increases rapidly with the number of particles.
We need a million coefficients to describe N = 20 spins (or qubits).
[OR putting it the other way round if we have a quantum computer with 20 qubits we can do a million calculations at the same time.]
How does density functional theory help?

Density Functional Theory

Density Functional Theory

Theorem 1.
The external potential is uniquely determined by the electronic charge density - n(r) - so the total energy is a unique functional of the density - E[n] !
Theorem 2.
The density which minimises the energy is the ground state density and the minimum energy is the ground state energy:
Density Functional Theory: the Theorems
[ ] 0EnEMinn
=
Hohenberg & Kohn, PRB 136, 864 (1964)
there is a universal functional E[n] which could be minimisedto obtain the exact ground state density and energy.

What is the Functional?
( ).][][][][][][ nEnEnTnEnFnE exteeeext ++=+= −
Eext[n] is trivial:
F (or equivalently T and Ee-e) need to be approximated !
rr dnVnE extext )( ][ ∫∧
=
where F is a unique functional of the electron density
Te[n] is the kinetic energy and Ee-e is the electron-electron interaction.
∑ −−=
atN
extZV
α α
α
||ˆ
Rrwhere

E[n] – The Kohn Sham Approach
Write the density in terms of a set of N non-interacting orbitals:
The non interacting kinetic energy and the classical Coulomb interaction
∑ ∇−=N
iiis nT φφ 2
21][ 21
21
21 )()(21][ rr
rrrr ddnnnEH ∫ −
=
This allows us to recast the energy functional as:
][][][][][ nEnEnEnTnE extxcHs +++=
])[][(])[][(][ nEnEnTnTnE Heesxc −+−= −
where we have introduced the exchange-correlation functional:
∑=N
iin 2)()( rr φ

The Kohn-Sham Equations
Vary the energy with respect to the orbitals and ….
No approximations, So…
If we knew Exc[n] we could solve for the exact ground state energy and density !
)(][)(
rr
nnEV xc
xc ∂∂
=
)()()('')'()(
21 2 rrrr
rrrr iiixcext VdnV φεφ =
⎥⎥⎦
⎤
⎢⎢⎣
⎡+
−++∇− ∫
where:
Kohn & Sham, PRA 140, 1133 (1965)
KS equations are solved via an iterative procedure until self-consistency is reached

The Non-interacting System
There exists an effective mean field potential which, when applied to a system of non-interacting fermions, will generate the exact ground state energy and charge density !!!
,...),(
||1
21 rr
rr
Ψ
− ji)(
)(r
r
i
xcVφ
E[n], n(r)

Density Functional Theory: The History
Thomas-Fermi-Dirac Model (1929):First model where the energy is expressed in terms of the electron density
Kohn-Hohenberg-Sham (1964):Exact unique relationship between groundstate energy and electron density
E. Fermi, Nobel laureate 1938
W. Kohn, Nobel laureate (in chemistry!!) 1998

Exc[n] - Properties
– Does not depend on Vext(the specific system): it is a ‘universal’ functional.
– The exact dependence on n(r) is unknown
Exc must be approximated in applications
])[][(])[][(][ nEnEnTnTnE Heesxc −+−= −

n
εxc
For the homogeneous electron gas the exact dependence of εxc(n) can be computed by Quantum Monte Carlo.
Local Density Approximation (LDA)The idea: build Exc from the knowledge of the exchange-correlation energy per particle, εxc, of the homogeneous electron gas
Kohn & Sham, PRA 140, 1133 (1965)
Ceperley & Alder, PRL 45, 566 (1980)

LDA to Exc: How it is built
nn
( )∫= rrr dnnE xcLDAxc )( )(ε
Picture courtesy of Andreas Savin Kohn & Sham, PRA 140, 1133 (1965)

LDA: Performance
Covalent bondsMetallic bondsIonic bonds
Hydrogen bonds: LDA not adequate
Van der Waals bonds: DFT not adequate – but this is not just a LDA problem…
generally well described(tends to overbind slightly)
Kohn & Sham, PRA 140, 1133 (1965): “We do not expect anaccurate description of chemical bonding”
In reality:
Why does LDA work in most cases ?

The Exchange-Correlation HoleThe exchange correlation hole, Pxc(r1,r2), is the probability of finding an electron at r2 given that there is an electron at r1.
It is the hole the electron at r1 digs for itself in the surrounding electronic density.
There are a number of properties which will be satisfied by the exact exchange correlation hole. For instance it should normalise to exactly one electron:
1),( 221 −=∫ rrr dPxc
LDA satisfies this rule.

Pxc is very poorly estimated the LDA
Jones & Gunnarsson, Rev. Mod. Phys. 61, 689 (1979)
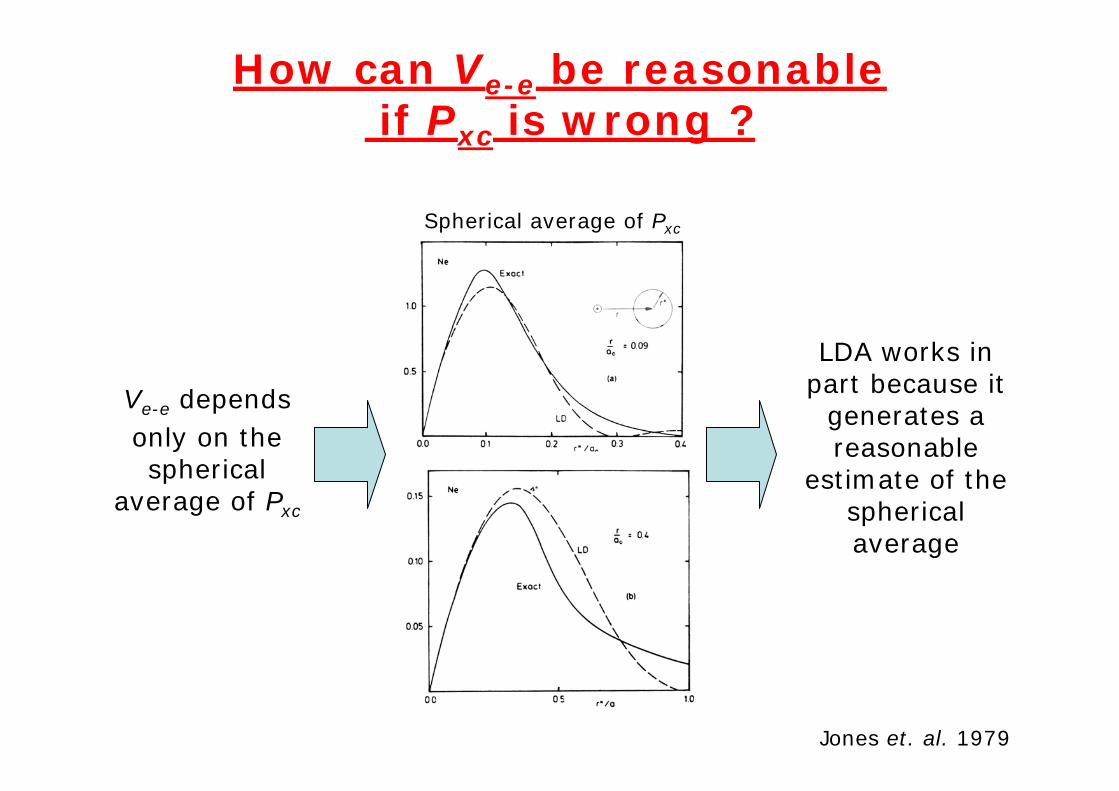
How can Ve-e be reasonableif Pxc is wrong ?
Ve-e depends only on the spherical
average of Pxc
Jones et. al. 1979
LDA works in part because it
generates a reasonable
estimate of the spherical average
Spherical average of Pxc

The LDA energy densities
The difference between the exact (V-QMC) and LDA energy density in bulk silicon (au)
Exchange Correlation
Hood et al PRB 57 8972 (1998)

So Why does the LDA work ?
• Exact properties of the xc-hole maintained
• The electron-electron interaction depends only on the spherical average of the xc-hole – this is reasonably well reproduced
• The errors in the exchange and correlation energy densities tend to cancel

Some things that do not workin this approach
• Van der Waals interactions: due to mutual dynamical charge polarisation of the atoms not properly included in any existing approximations to Exc
• Excited states: DFT is a ground state theory (ways forward: time-dependent DFT, GW, …)
• Non Born-Oppenheimer processes (i.e, non-radiativetransitions between electronic states)
• Self-interaction problem: each electron lives in the field created by all electrons including itself ( ways forward: SIC, hybrid DFT)

�
Disadvantages of DFT
It only applies to the electronic groundstate (or an electronic system in thermal equilibrium).Have to use approximations to the true density functional.Not possible to predict error in the value of any particular property.Not possible to systematically improve accuracy of calculation.

�
Advantages of DFTKohn was awarded the Nobel prize for DFT - this has endowed DFT with a prestige that makes it hard to criticise!It offers very good scaling of computational cost with system size.It allows calculations to be performed on large and complex systems.Given the very large number of DFT calculations the likely accuracy of property prediction for many properties/systems is known.

�
Some myths and half-truths about DFT
DFT tells you nothing about the excited states.Although the bandgap is wrong, the wavefunctions of excited states are meaningful.Density functional theory can fail.The true density functional gets all groundstate energies and densities correct, but it only manages this by psychopathic behaviour. Infinitely small changes in the electron density produce large changes in the XC potential infinitely far away.

Useful References
• Hohenberg & Kohn, PRB 136, 864 (1964)
• Kohn & Sham, PRA 140, 1133 (1965)
• R. O. Jones and O. Gunnarsson, Rev. Mod. Phys. 61, 689 (1989)
• M. C. Payne et al., Rev. Mod. Phys 64, 1045 (1992)
• R. M. Martin, “Electronic Structure: basic theory and practical methods”, Cambridge University Press (2004)
• J. Kohanoff, “Electronic Structure Calculations for Solids and Molecules: Theory and Computational Methods”, Cambridge University Press (2006)