pathogenic fungi – genomics, evoluon and …...pathogenic fungi – genomics, evoluon and...
TRANSCRIPT

PathogenicFungi–Genomics,Evolu6onandEpidemiology

PathogenicFungi–Genomics,Evolu6onandEpidemiology
RichardBenne= BrownUniversity,USAGeraldineButler UniversityCollegeDublin,IrelandAntonisRokas VanderbiltUniversity,USALi-JunMa UmassAmherst,USAGuilhemJanbon Ins6tutPasteur,Paris,FranceNormanPavelka A*Star,Singapore

True pathogen Blastomyces dermatitidis
Coccidiodes immitis
Histoplasma capsulatum
Paracoccidioides brasiliensis
25°C 37°C
FUNGI 101
Opportunistic
Candida albicans
Budding yeast
Pseudohyphae
Hyphae
Cryptococcus neoformans
Aspergillus fumigatus
Capsule
Conidia
AdaptedfromRomani,NatRevImmun,2004

True pathogen Blastomyces dermatitidis
Coccidiodes immitis
Histoplasma capsulatum
Paracoccidioides brasiliensis
25°C 37°C
Opportunistic
Candida albicans
Budding yeast
Pseudohyphae
Hyphae
Cryptococcus neoformans
Aspergillus fumigatus
Capsule
Conidia
AdaptedfromRomani,NatRevImmun,2004
FUNGI 101

What is the impact of invasive mycoses?
Worldwide: Life-threatening Mortality rates infections/yr (%) (1) Cryptococcus >1,000,000 20-70
(4) Aspergillus >200,000 30-95
(=2) Pneumocystis >400,000 20-80
Brown et al., Sci. Trans. Med., 2012
(=2) Candida >400,000 46-75

What is the impact of invasive mycoses?
Worldwide: Life-threatening Mortality rates infections/yr (%) (=5) Coccidiodomycosis ~25,000 <1-70
(9) Paracoccidiodomycosis ~4,000 5-27
(8) Penicillosis >8,000 2-75
Brown et al., Sci. Trans. Med., 2012
(=5) Histoplasmosis ~25,000 28-50
(10) Blastomycosis ~3,000 <2-68
(7) Mucormycosis >10,000 30-90

lineages with increased virulence or altered host range (Croll andMcDonald, 2012). This highly adaptive behavior is promoted byan immense fungal genome plasticity (Calo et al., 2013). Besidessexual reproduction, parasexualilty (Pontecorvo, 1956), aneuploidy(Selmecki et al., 2006), transposons, telomere instability (Starneset al., 2012) or horizontal gene transfer (Ma et al., 2010; Richardset al., 2011) amongst others enable adaptive evolution by promot-ing mitotic recombination, independent chromosomal assortment,gain or loss of chromosomes and translocations. Moreover, fungiexhibit morphogenetic plasticity which enables them to colonizeand invade tissue as hyphae, which primarily extend at their tips(Fischer et al., 2008), while often forming differentiated spores,infection structures, fruiting bodies and unicellular, yeast cells,that can aid rapid dispersal (Sudbery et al., 2004).
In this review, we cover an array of fungal species, encompass-ing diverse taxonomic groups, that cover several hundred millionyears of evolution (Fig. 1). We include two major fungal pathogensof humans, Aspergillus fumigatus and Candida albicans, a facultativepathogen of both animals and plants, Fusarium oxysporum, andplant pathogenic species Magnaporthe oryzae, Mycospherella gra-minicola, Ashbya gossypii and Ustilago maydis which display infec-tion-associated dimorphism and cellular differentiation.Collectively, this provides an overview of diverse mechanisms ofpathogenesis and some unifying themes, while also encompassingsome of the best-studied and understood fungal pathosystems.
The purpose of the review is to highlight both specific differ-ences and unifying features of some of the best-studied fungalpathogens and to highlight the significant challenges that remainin developing a deep understanding of fungal pathogenesis.
2. Aspergillus fumigatus
Aspergillus fumigatus is a filamentous fungus that can be isolatedfrom compost soil, where it proliferates in organic debris and plays
an essential role in carbon and nitrogen recycling. Fumigatus is themost important air-borne fungal pathogen and can grow at tem-peratures up to 55 !C, while its spores survive temperatures ofup to 70 !C (Brakhage and Langfelder, 2002). The fungus propa-gates asexually with release of thousands of spores per conidialhead into the atmosphere. Due to their small size (2–3 lm diame-ter), the spores can easily reach lung alveoli (Latge, 2001).
A first description of pulmonary aspergillosis was published in1842 by physician John H. Bennett (Supplementary Fig. 1A), whonoted the presence of a fungus in the lungs of a post mortempatient with pneumothorax. Almost 50 years elapsed, however,before A. fumigatus was recognized as the primary cause of theinfection (Barnes, 2004). Today, A. fumigatus conidia infect millionsof susceptible individuals, causing allergies associated withasthma, allergic sinusitis and bronchoalveolitis (Denning et al.,2013). In cavities in the lungs of tuberculosis patients, A. fumigatusspores germinate and develop into a fungus ball, or non-invasiveaspergilloma (Riscili and Wood, 2009). In addition to these formsof aspergillosis, which are not life-threatening, patients withaltered immune status such as leukemia patients or transplantpatients are at risk to develop invasive aspergillosis (IA), with anestimated number of more than 200,000 cases per year (Brownet al., 2012; Garcia-Vidal et al., 2008). Mortality rates for IA reacharound 50% of the cases when patients are treated and increaseto more than 90% when the diagnosis is missed or delayed(Brown et al., 2012).
2.1. Overview of the A. fumigatus genome
A first draft of the A. fumigatus clinical isolate Af293 genomewas published in 2005 (Nierman et al., 2005). Eight chromosomeswere identified containing about 10,000 genes (Table 2). Threeyears later, the genome sequence of a second A. fumigatus isolate,A1163, was released (Fedorova et al., 2008). Comparative analysisshowed conservation of 98% of the sequence between the two
Fig. 1. Phylogenetic tree. Fungal species phylogeny generated from a concatenated alignment of 49 conserved fungal genes using maximum likelihood. The tree covers 21taxa and 9033 amino acid positions. Bootstrap values for each node are reported as percentages. To generate the tree, protein sequences of the 49 selected conserved singlecopy genes from the 21 fungal species were aligned using Clustal W (Larkin et al., 2007) and the obtained conserved sequence blocks were sampled with G-blocks (Talaveraand Castresana, 2007). All the aligned sequences were then concatenated to one file using Galaxy (Goecks et al., 2010). Finally, PhyML (Guindon et al., 2010) was used togenerate the phylogenetic tree with 100 bootstraps for branch support and LG as the amino acid substitution model as identified by ModelGenerator (Keane et al., 2006)(Keane et al., 2006). The tree was visualized using TreeDyn (Chevenet et al., 2006).
44 E. Perez-Nadales et al. / Fungal Genetics and Biology 70 (2014) 42–67
Perez-Nadalesetal.,FungalGene6csandBiology,2014
Candidaalbicans
Aspergillusfumigatus
Pneumocys7sjirovecii
Phylogeny
Cryptococcusneoformans

Wordle


1976 Bacteriophage
MS2 3.5 kp
1995 H. influenzae
1.8 Mbp
1996 S. cerevisiae
12.1 Mbp
2001 H. sapiens 3.2 Gbp
Improved sequencing technology has pushed forward our understanding of genomes

• what are the dynamics of genome change?
• how often do genomes recombine?
• how does sex impact genome structure?
• how are aneuploidies tolerated?
• how does this impact drug resistance?
• how does host environment impact genomes?
• how does heterozygosity impact mutation?
• can GWAS and QTL mapping be applied?
Questions in fungal genomics

GeraldineButler,UCD,Ireland
Genome Diversity of Candida Species
The phylogenetic analysis showed that the C. orthopsilosis mitochondrial genomes belongto four mitotypes (Fig 6B). Isolates from three mitotypes (mt1, mt2 and mt3) correspond tothe nuclear Clades 1, 2 and 3. We designated strain 90–125 as mitotype mt4 because this strainis closely related to nuclear Clade 4 and its mtDNA appears not to be recombinant (see below).There is almost no variation within the mitotypes (0–6 SNPs in the whole genome) whereasdivergence between the mitotypes is significant (up to 222 nucleotide substitutions or 1.1%between mt2 and mt3).
Fig 5. Multiple origins of the species Candida orthopsilosis by independent hybridization events. Eachhybridization involved mating between cells of the same two parental species, A and B, which are 5% different ingenome sequence. We propose that within species A and B, there are multiple distinct lineages. In species B weidentify four lineages corresponding to the nuclear haplotypes B1-B4 found in hybrids. In species A we identify sixlineages comprising the four nuclear haplotypes A1-A4 found in hybrids, and two found in the non-hybrid strains 90–125 and 428. The inferred mitochondrial (mt) mitotypes of each parental lineage are shown where known. The fiveyellow boxes represent the five separate hybridization events we propose, with the genomic contributions fromparents A and B indicated on the horizontal arrows (mitotypes, nuclear haplotypes, and MTL genotypes). See textfor details of the proposed hybridization events.
doi:10.1371/journal.pgen.1006404.g005
Multiple Origins of Candida orthopsilosis
PLOS Genetics | DOI:10.1371/journal.pgen.1006404 November 2, 2016 11 / 25
RESEARCH ARTICLE
Multiple Origins of the Pathogenic YeastCandida orthopsilosis by SeparateHybridizations between Two ParentalSpeciesMarkus S. Schroder1, Kontxi Martinez de San Vicente1, Tamara H. R. Prandini2,Stephen Hammel1, Desmond G. Higgins3, Eduardo Bagagli2, Kenneth H. Wolfe3,Geraldine Butler1
1 School of Biomedical and Biomolecular Science and UCD Conway Institute of Biomolecular andBiomedical Research, Conway Institute, University College Dublin, Belfield, Dublin, Ireland, 2 Instituto deBiocirncias, UNESP - Univ Estadual Paulista, Botucatu, Sao Paulo, Brazil, 3 School of Medicine and UCDConway Institute of Biomolecular and Biomedical Research, University College Dublin, Belfield, Dublin,Ireland
Abstract
Mating between different species produces hybrids that are usually asexual and stuck as
diploids, but can also lead to the formation of new species. Here, we report the genome
sequences of 27 isolates of the pathogenic yeast Candida orthopsilosis. We find that most
isolates are diploid hybrids, products of mating between two unknown parental species (A
and B) that are 5% divergent in sequence. Isolates vary greatly in the extent of homogeni-
zation between A and B, making their genomes a mosaic of highly heterozygous regions
interspersed with homozygous regions. Separate phylogenetic analyses of SNPs in the A-
and B-derived portions of the genome produces almost identical trees of the isolates with
four major clades. However, the presence of two mutually exclusive genotype combinations
at the mating type locus, and recombinant mitochondrial genomes diagnostic of inter-clade
mating, shows that the species C. orthopsilosis does not have a single evolutionary origin
but was created at least four times by separate interspecies hybridizations between parents
A and B. Older hybrids have lost more heterozygosity. We also identify two isolates with
homozygous genomes derived exclusively from parent A, which are pure non-hybrid
strains. The parallel emergence of the same hybrid species from multiple independent
hybridization events is common in plant evolution, but is much less documented in patho-
genic fungi.
Author Summary
The genus Candida is one of the leading causes of fungal morbidity in humans. Manypathogenic Candida species are diploid, and do not have have a full sexual cycle. The
PLOS Genetics | DOI:10.1371/journal.pgen.1006404 November 2, 2016 1 / 25
a11111
23(1$&&(66
Citation: Schroder MS, Martinez de San Vicente K,Prandini THR, Hammel S, Higgins DG, Bagagli E, etal. (2016) Multiple Origins of the Pathogenic YeastCandida orthopsilosis by Separate Hybridizationsbetween Two Parental Species. PLoS Genet12(11): e1006404. doi:10.1371/journal.pgen.1006404
Editor: Gavin Sherlock, Stanford University,UNITED STATES
Received: July 25, 2016
Accepted: October 4, 2016
Published: November 2, 2016
Copyright:� 2016 Schroder et al. This is an openaccess article distributed under the terms of theCreative Commons Attribution License, whichpermits unrestricted use, distribution, andreproduction in any medium, provided the originalauthor and source are credited.
Data Availability Statement: All raw sequencingdata are available in the SRA database usingBioProject number PRJNA322245. Mitochondrialassemblies are available at EMBL (accessionnumbers LT594353-LT594380). The correctassembly of chromosomes 2 and 6 is available atNCBI (BioProject number PRJEA83665).
Funding: This work was supported by theWellcome Trust (097419/Z/11/Z) and ScienceFoundation Ireland (12IA1343). FAPESP (Process2014/04811-6) for the Research Internships
Schroderetal.,2016,PloSGen
The phylogenetic analysis showed that the C. orthopsilosis mitochondrial genomes belongto four mitotypes (Fig 6B). Isolates from three mitotypes (mt1, mt2 and mt3) correspond tothe nuclear Clades 1, 2 and 3. We designated strain 90–125 as mitotype mt4 because this strainis closely related to nuclear Clade 4 and its mtDNA appears not to be recombinant (see below).There is almost no variation within the mitotypes (0–6 SNPs in the whole genome) whereasdivergence between the mitotypes is significant (up to 222 nucleotide substitutions or 1.1%between mt2 and mt3).
Fig 5. Multiple origins of the species Candida orthopsilosis by independent hybridization events. Eachhybridization involved mating between cells of the same two parental species, A and B, which are 5% different ingenome sequence. We propose that within species A and B, there are multiple distinct lineages. In species B weidentify four lineages corresponding to the nuclear haplotypes B1-B4 found in hybrids. In species A we identify sixlineages comprising the four nuclear haplotypes A1-A4 found in hybrids, and two found in the non-hybrid strains 90–125 and 428. The inferred mitochondrial (mt) mitotypes of each parental lineage are shown where known. The fiveyellow boxes represent the five separate hybridization events we propose, with the genomic contributions fromparents A and B indicated on the horizontal arrows (mitotypes, nuclear haplotypes, and MTL genotypes). See textfor details of the proposed hybridization events.
doi:10.1371/journal.pgen.1006404.g005
Multiple Origins of Candida orthopsilosis
PLOS Genetics | DOI:10.1371/journal.pgen.1006404 November 2, 2016 11 / 25

Diego Libkind et al. PNAS, 2011; Walter et al., G3, 2014; Peris et al., PLoS Gen, 2016
The interspecific hybrid S. pastorianus is the most commonly used yeast in brewery fermentations worldwide.
Hybridization drives emergence of new isolates / species : Saccharomyces pastorianus

AntonisRokas,VanderbiltUniversity,Tennessee
Genomicsandthemakingoffungalmetabolicdiversity

Figure 3 The phylogenetic relationships of Saccharomycotina yeasts inferred from the concatenation-based analysis of a 1233 single-copyBUSCO gene amino acid (AA) data matrix. The ML phylogeny was reconstructed based on the concatenation amino acid data matrix (609,899sites) under an unpartitioned LG + GAMMA substitution model using RA·ML version 8.2.3 (Stamatakis 2014). Branch support values nearinternodes are indicated as bootstrap support value (above) and internode certainty (below), respectively. ! indicates bootstrap supportvalues $95%. Thicker branches show conflicts between concatenation-based phylogeny (Figure 3) and coalescence-based phylogeny (Figure 4).Note, branch lengths on the ML tree are given in the inset at the bottom left.
3932 | X.-X. Shen et al.
INVESTIGATION
Reconstructing the Backbone of theSaccharomycotina Yeast Phylogeny UsingGenome-Scale DataXing-Xing Shen,* Xiaofan Zhou,* Jacek Kominek,† Cletus P. Kurtzman,‡,1 Chris Todd Hittinger,†,1
and Antonis Rokas*,1*Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee 37235, †Laboratory of Genetics,Genome Center of Wisconsin, DOE Great Lakes Bioenergy Research Center, Wisconsin Energy Institute, J. F. CrowInstitute for the Study of Evolution, University of Wisconsin-Madison, Madison, Wisconsin 53706, and ‡MycotoxinPrevention and Applied Microbiology Research Unit, National Center for Agricultural Utilization Research, AgriculturalResearch Service, U.S. Department of Agriculture, Peoria, Illinois 61604
ORCID IDs: 0000-0002-2879-6317 (X.Z.); 0000-0002-1916-0122 (J.K.); 0000-0001-5088-7461 (C.T.H.); 0000-0002-7248-6551 (A.R.)
ABSTRACT Understanding the phylogenetic relationships among the yeasts of the subphylum Saccha-romycotina is a prerequisite for understanding the evolution of their metabolisms and ecological lifestyles.In the last two decades, the use of rDNA and multilocus data sets has greatly advanced our understandingof the yeast phylogeny, but many deep relationships remain unsupported. In contrast, phylogenomicanalyses have involved relatively few taxa and lineages that were often selected with limited considerationsfor covering the breadth of yeast biodiversity. Here we used genome sequence data from 86 publiclyavailable yeast genomes representing nine of the 11 known major lineages and 10 nonyeast fungaloutgroups to generate a 1233-gene, 96-taxon data matrix. Species phylogenies reconstructed using twodifferent methods (concatenation and coalescence) and two data matrices (amino acids or the first twocodon positions) yielded identical and highly supported relationships between the nine major lineages.Aside from the lineage comprised by the family Pichiaceae, all other lineages were monophyletic. Mostinterrelationships among yeast species were robust across the two methods and data matrices. However,eight of the 93 internodes conflicted between analyses or data sets, including the placements of: the cladedefined by species that have reassigned the CUG codon to encode serine, instead of leucine; the cladedefined by a whole genome duplication; and the species Ascoidea rubescens. These phylogenomic anal-yses provide a robust roadmap for future comparative work across the yeast subphylum in the disciplines oftaxonomy, molecular genetics, evolutionary biology, ecology, and biotechnology. To further this end, wehave also provided a BLAST server to query the 86 Saccharomycotina genomes, which can be found athttp://y1000plus.org/blast.
KEYWORDSphylogenomicsmaximumlikelihood
incongruencegenomecompleteness
nuclear markers
Molecular phylogenetic analyses show that the fungal phylum As-comycota is comprised of three monophyletic subphyla that share acommon ancestor from !500 MYA (Kurtzman and Robnett 1994;Sugiyama et al. 2006; Taylor and Berbee 2006; James et al. 2006; Liuet al. 2009): the Saccharomycotina (syn. Hemiascomycota; e.g., Sac-charomyces, Pichia, Candida), the Pezizomycotina (syn. Euascomy-cota; e.g., Aspergillus, Neurospora), and the Taphrinomycotina (syn.Archaeascomycota; e.g., Schizosaccharomyces, Pneumocystis).
Yeasts of the fungal subphylumSaccharomycotina exhibit remarkablydiverse heterotrophic metabolisms, which have enabled them to success-fully partition nutrients and ecosystems and inhabit every continent and
every major aquatic and terrestrial biome (Hittinger et al. 2015). Whileyeast species were historically identified by metabolic differences, recentstudies have shown that many of these classic characters are subject torampant homoplasy, convergence, and parallelism (Hittinger et al. 2004;Hall andDietrich 2007;Wenger et al. 2010; Slot and Rokas 2010; Lin andLi 2011; Wolfe et al. 2015). Despite the considerable progress in classi-fying yeasts using multilocus DNA sequence data, critical gaps remain(Kurtzman and Robnett 1998, 2003, 2007, 2013; Nguyen et al. 2006;Kurtzman et al. 2008, 2011; Kurtzman and Suzuki 2010); many generaare paraphyletic or polyphyletic, while circumscriptions at or above thefamily level are often poorly supported (Hittinger et al. 2015).
Volume 6 | December 2016 | 3927
Shenetal.,G3,2016.
“……touncoverthegene.cbasisofyeastmetabolicdiversity….”

Fusariosis is the top emerging opportunistic mycosis in immunocompromised individuals*
* Severe Fungal Infections, 2008. Decision Resources; Montone 2009, AJCP
- Caused by Fusarium species, ubiquitous in environments - Highly invasive, causing disseminated diseases: hepatitis, meningitis
- Mostly refractory to existing antifungals resulting in high mortality rate
Fusariosis: Li-JunMa,UMASSAmherst

Fusariumwiltofbanana,popularlyknownasPanamadisease,isalethalfungaldiseasecausedbythesoil-bornefungusFusariumoxysporumf.sp.cubense(Foc).

TranscriptomedynamicsinC.neoformans
Virulencefactors
Fungalfitness
Transcriptomestructure
GuilhemJanbon,Pasteur
Howistheregula.onofthetranscriptomeachievedinhumanfungalpathogens?And how does this regula.on impact their biologyandvirulence?
Capsule thickness was measured using Indian ink staining of 20randomly selected cells per isolate from 1-week-old colonies thatwere cultured on Littman’s ox gall agar (BD, Franklin Lakes, NJ,U.S.A.) at both 25 and 37 !C. The capsule thickness was calculatedas the ratio between the diameters of the capsule and the cell.
Cutinase, laccase, lipase, phospholipase and protease activitieswere investigated in triplicate by plate assays at 25 and 37 !C.Cutinase activity was detected, according to the method describedby Dantzig et al. (1986), with p-nitrophenyl butyrate (SigmaAldrich, St. Louis, MO, U.S.A.) as the substrate. The yeast isolateswere grown on acetate medium (0.5% yeast extract, 0.5% peptone,0.5% ammonium acetate, 2.0% agar). After incubation for 10 days at25 !C, a loop of cells was transferred onto a glass slide and overlaidwith freshly prepared 0.026% p-nitrophenyl butyrate containing0.011% Triton X-100 (Sigma Aldrich). Distinct yellow stainingwas considered as a positive reading.
Lipase activity was detected in triplicate according to the meth-ods described by Sierra (1957) and Middelhoven (1997), usingTween 40, Tween 60 and Tween 80 as substrates. The presenceof lipase activity was indicated by a diffuse zone of crystalsimmersed around the yeast colony and calculated as the ratiobetween the zone of precipitation and the colony diameter.
Laccase activity was measured in triplicate on L-DOPA and nore-pinephrine media as described before (Petter et al., 2001) at 25 and37 !C. The development of brown pigment was scored daily during10 days, with the following scores: 0 = no melanin production;1 = center of the colony is dull brown, colony edge has no melaninproduction; 2 = colony is dull brown; 3 = center of the colony isdark brown, colony edge is dull brown; 4 = colony is dark browncolored; 5 = colony is black colored.
Phospholipase activity was investigated in triplicate using eggyolk plates as described by Vidotto et al. (1996). The presence ofphospholipase activity was indicated by the occurrence of a clearzone around the yeast colonies and calculated as the ratio betweenthe zone of enzyme activity and the colony diameter.
Protease activity was assessed in triplicate according to themethod described by Braga et al. (1998) using agar plates contain-ing 0.75% casein. The presence of protease activity was indicatedby the occurrence of a clear zone around the yeast colonies and cal-culated as the ratio between zone of enzyme activity and the col-ony diameter.
The activity of 19 pathogenicity associated enzymes was testedby using API-ZYM (BioMerieux, Marcy l’Etoile, France) (Casal andLinares, 1983). Isolates were cultured on MEA medium and incu-bated for 48 h at 37 !C after which the API-ZYM test was carriedout according to the instructions provided by the manufacturer.
2.8. Antifungal susceptibility testing by the broth microdilutionmethod
The in vitro antifungal susceptibility was determined by thebroth microdilution method for amphotericin B (Bristol MyersSquibb, Woerden, The Netherlands), 5-fluorocytosine (ValeantPharmaceuticals, Zoetermeer, The Netherlands), fluconazole andvoriconazole (Pfizer Central Research, Sandwich, Kent, UnitedKingdom), itraconazole (Janssen Cilag, Tilburg, The Netherlands),posaconazole (Schering-Plough Corp., Kenilworth, NJ, U.S.A.), andisavuconazole (Basilea Pharmaceutica, Basel, Switzerland), andminimum inhibiting concentrations calculated according to therecommendations of the National Committee for Clinical
0.0070
CBS8755
B5742
CBS6900
CBS1930
WM779
A47496
WM148
CBS1622
CBS5728
P152
HEC11102
ICB176
RKI-M318/90
A1M-F2866
56A
CBS879
WM179
CBS6886
CBS11687
CBS6998
CBS9172
CBS8710
CBS6289
Bt27
WM721
CBS5758
AMC940441
BD5
CBS10865
CBS918
CBS8336
CBS10089
Bt206
M27055
RV54130
CBS8684
CBS7748
CBS10511
WM161
CBS6993
384C
AMC821330
A1M-F3016
CBS10512
CBS6955
PAH21b
CBS6996
CBS10090
Bt130
C16528
WM178
Bt1
Bt63
WM714
CBS882
WM276
A1M-R265
WM728
C. deuterogattii
C. decagattii
C. gattii
C. tetragattii
C. bacillisporus
C. neoformans
C. deneoformans
BC
A
D
E
F
G
H
I
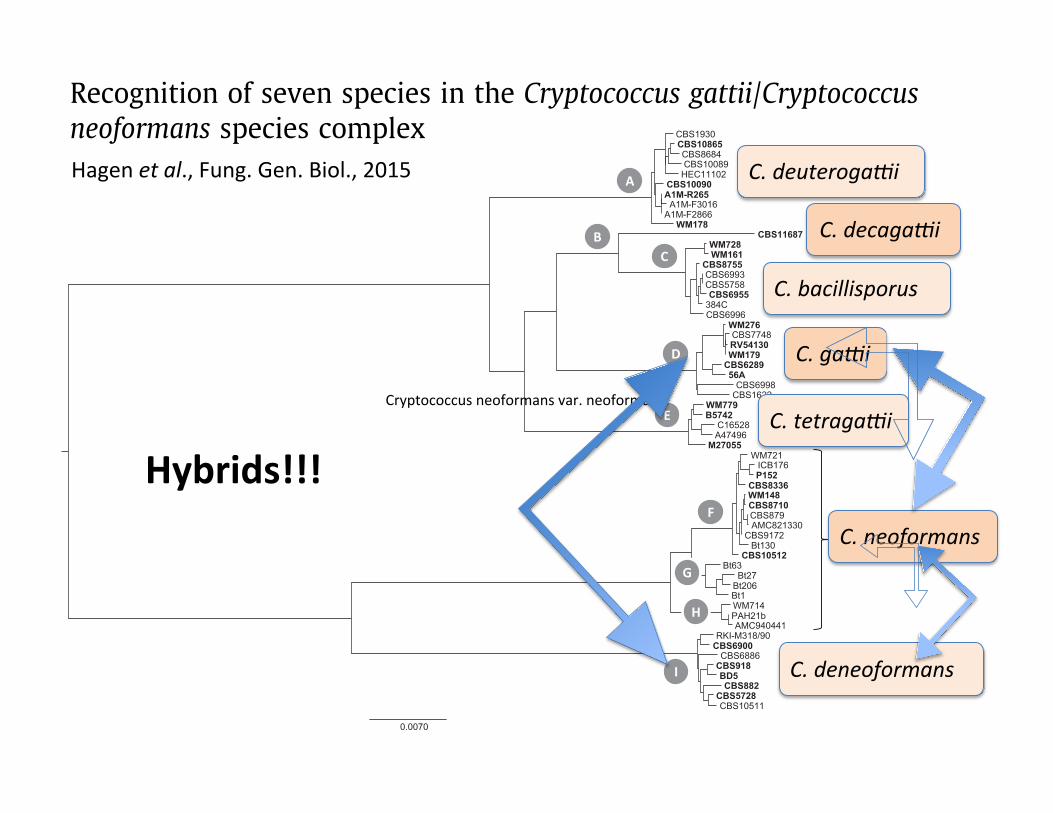
Fig. 1. Diversity in the C. gattii/C. neoformans species complex inferred from a concatenated data set of 11 loci. The clade letters are discussed in the text, isolates indicated inbold were used for describing the species.
20 F. Hagen et al. / Fungal Genetics and Biology 78 (2015) 16–48Recognition of seven species in the Cryptococcus gattii/Cryptococcusneoformans species complex
Ferry Hagen a,b, Kantarawee Khayhan a,c, Bart Theelen a, Anna Kolecka a, Itzhack Polacheck d,Edward Sionov d,e, Rama Falk d,f, Sittiporn Parnmen g, H. Thorsten Lumbsch h, Teun Boekhout a,i,j,⇑a CBS-KNAW Fungal Biodiversity Centre, Basidiomycete and Yeast Research, Utrecht, The Netherlandsb Department of Medical Microbiology and Infectious Diseases, Canisius-Wilhelmina Hospital, Nijmegen, The Netherlandsc Department of Microbiology and Parasitology, Faculty of Medical Sciences, University of Phayao, Phayao, Thailandd Department of Clinical Microbiology and Infectious Diseases, Hadassah-Hebrew University Medical Center, Ein Kerem, Jerusalem, Israele Department of Food Quality & Safety, Institute for Postharvest and Food Sciences, Agricultural Research Organization, The Volcani Center, Bet Dagan, Israelf Department of Fisheries and Aquaculture, Ministry of Agriculture and Rural Development, Nir-David, Israelg Department of Medical Sciences, Ministry of Public Health, Nonthaburi, Thailandh Science & Education, The Field Museum, Chicago, IL, USAi Shanghai Key Laboratory of Molecular Medical Mycology, Changzheng Hospital, Second Military Medical University, Shanghai, Chinaj Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
a r t i c l e i n f o
Article history:Received 29 October 2014Revised 12 February 2015Accepted 15 February 2015Available online 23 February 2015
Keywords:Cryptic speciesCryptococcus gattiiCryptococcus neoformansYeastPathogenTaxonomy
a b s t r a c t
Phylogenetic analysis of 11 genetic loci and results from many genotyping studies revealed significantgenetic diversity with the pathogenic Cryptococcus gattii/Cryptococcus neoformans species complex.Genealogical concordance, coalescence-based, and species tree approaches supported the presence of dis-tinct and concordant lineages within the complex. Consequently, we propose to recognize the currentC. neoformans var. grubii and C. neoformans var. neoformans as separate species, and five species withinC. gattii. The type strain of C. neoformans CBS132 represents a serotype AD hybrid and is replaced. Thenewly delimited species differ in aspects of pathogenicity, prevalence for patient groups, as well asbiochemical and physiological aspects, such as susceptibility to antifungals. MALDI-TOF mass spectrom-etry readily distinguishes the newly recognized species.! 2015 The Authors. Published by Elsevier Inc. This is an open access article under the CC BY-NC-ND license
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
1. Introduction
Cryptococcus neoformans (Sanfelice) Vuillemin is a clinicallyimportant basidiomycetous yeast that globally causes an estimated1 million new infections and over 625,000 deaths annually.Although this estimate has recently been disputed, the speciesremain a main source of morbidity and mortality (Park et al.,2009, 2014a; Warkentien and Crum-Cianflone, 2010). Accordingto the current classification the species complex comprises twospecies, namely C. neoformans and C. gattii (Vanbreuseghem &Takashio) Kwon-Chung & Boekhout (Kwon-Chung et al., 2002;Kwon-Chung and Varma, 2006) with serotypes A, D and AD forthe former, and B and C for the latter species. Cryptococcus neofor-mans currently consists of two varieties: C. neoformans variety
grubii (serotype A) (Franzot et al., 1999) and C. neoformans varietyneoformans (serotype D) (Kwon-Chung, 2011). With the dualnomenclature that was in use in fungal taxonomy until recently,the teleomorphs (i.e., the sexual stages) were named Filobasidiellaneoformans Kwon-Chung and Filobasidiella bacillisporaKwon-Chung (Franzot et al., 1999; Kwon-Chung, 1975, 1976,2011; Kwon-Chung et al., 2002; Vanbreuseghem and Takashio,1970). A detailed account on the history of our knowledge of thespecies was presented by Drouhet (1997), Barnett (2010) andKwon-Chung et al. (2011) as well as in two books (Casadevalland Perfect, 1998; Heitman et al., 2011). A recent review on C. gattiiinfections highlighted the history, epidemiology and clinicalimpact of that species and its subtypes (or species) (Chen et al.,2014).
During the past two decades, considerable genetic heterogene-ity has been demonstrated to occur in the C. gattii/C. neoformansspecies complex by a plethora of molecular methods, such asmulti-locus enzyme typing (Brandt et al., 1993), amplified frag-ment length polymorphism (AFLP) (Boekhout et al., 2001; Hagen
http://dx.doi.org/10.1016/j.fgb.2015.02.0091087-1845/! 2015 The Authors. Published by Elsevier Inc.This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
⇑ Corresponding author at: CBS-KNAW Fungal Biodiversity Centre, Basidiomycete& Yeast Research, Uppsalalaan 8, NL-3584CT Utrecht, The Netherlands. Tel.: +31(0)30 2122 671; fax: +31 (0)30 2512 097.
E-mail address: [email protected] (T. Boekhout).
Fungal Genetics and Biology 78 (2015) 16–48
Contents lists available at ScienceDirect
Fungal Genetics and Biology
journal homepage: www.elsevier .com/ locate/yfgbi
Hagenetal.,Fung.Gen.Biol.,2015 C.deuteroga;i
C.decaga;i
C.bacillisporus
C.ga;i
C.tetraga;i
C.neoformans
C.deneoformans
Cryptococcusneoformansvar.neoformans
Hybrids!!!
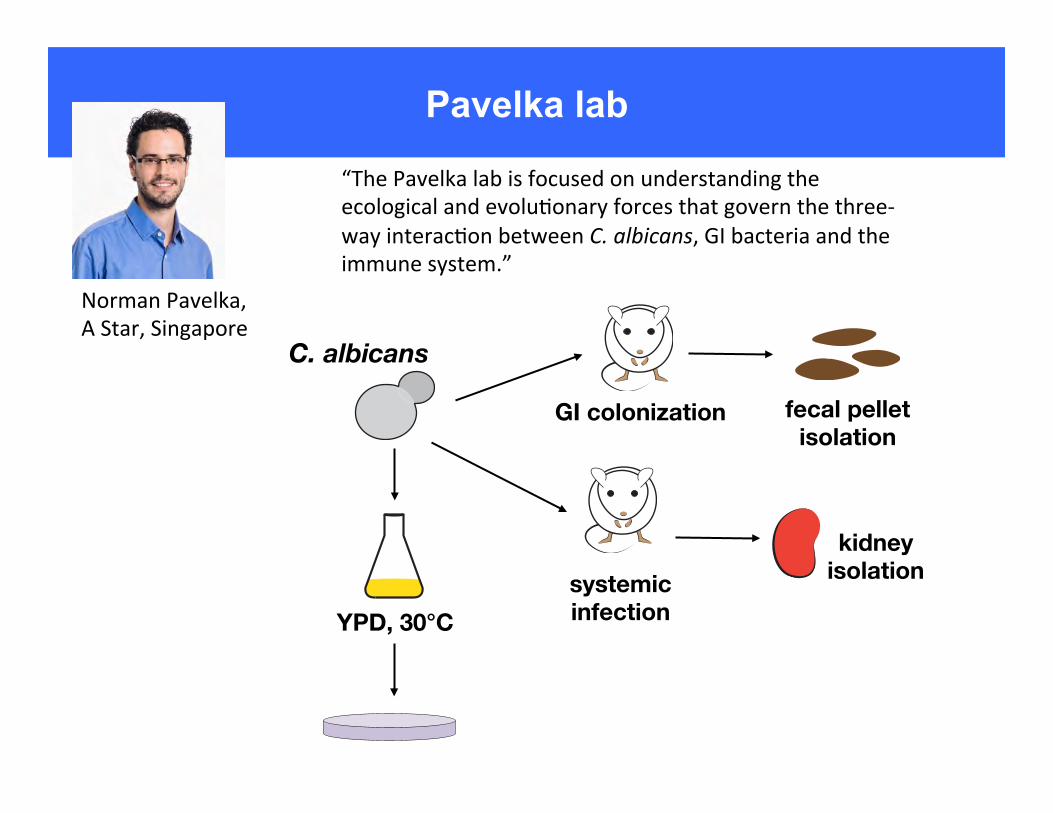
Pavelka lab
NormanPavelka,AStar,Singapore
“ThePavelkalabisfocusedonunderstandingtheecologicalandevolu6onaryforcesthatgovernthethree-wayinterac6onbetweenC.albicans,GIbacteriaandtheimmunesystem.”
C. albicans
YPD, 30°C
GI colonization
systemic infection
fecal pellet isolation
kidney isolation

ORIGINAL RESEARCHpublished: 22 December 2016
doi: 10.3389/fcimb.2016.00186
Frontiers in Cellular and Infection Microbiology | www.frontiersin.org 1 December 2016 | Volume 6 | Article 186
Edited by:
Brian Wickes,
University of Texas Health Science
Center at San Antonio, USA
Reviewed by:
Karen L. Wozniak,
University of Texas at San Antonio,
USA
Robert T. Wheeler,
University of Maine, USA
*Correspondence:
Norman Pavelka
norman_pavelka@
immunol.a-star.edu.sg
†Present Address:
Marina Yurieva,
The Jackson Laboratory for Genomic
Medicine, Farmington, USA
Francesca Zolezzi,
Galderma R&D, Sophia Antipolis,
France
‡These authors have contributed
equally to this work.
Received: 19 October 2016
Accepted: 30 November 2016
Published: 22 December 2016
Citation:
Sem X, Le GTT, Tan ASM, Tso G,
Yurieva M, Liao WWP, Lum J,
Srinivasan KG, Poidinger M, Zolezzi F
and Pavelka N (2016) β-glucan
Exposure on the Fungal Cell Wall
Tightly Correlates with Competitive
Fitness of Candida Species in the
Mouse Gastrointestinal Tract.
Front. Cell. Infect. Microbiol. 6:186.
doi: 10.3389/fcimb.2016.00186
β-glucan Exposure on the Fungal CellWall Tightly Correlates withCompetitive Fitness of CandidaSpecies in the MouseGastrointestinal TractXiaoHui Sem‡, Giang T. T. Le ‡, Alrina S. M. Tan‡, Gloria Tso, Marina Yurieva †,Webber W. P. Liao, Josephine Lum, Kandhadayar G. Srinivasan, Michael Poidinger,Francesca Zolezzi † and Norman Pavelka*
Singapore Immunology Network, Agency for Science, Technology and Research, Singapore, Singapore
Candida albicans is responsible for ∼400,000 systemic fungal infections annually, with
an associated mortality rate of 46–75%. The human gastrointestinal (GI) tract represents
the largest natural reservoir of Candida species and is a major source of systemic fungal
infections. However, the factors that control GI colonization by Candida species are not
completely understood.We hypothesized that the fungal cell wall would play an important
role in determining the competitive fitness of Candida species in the mammalian GI tract.
To test this hypothesis, we generated a systematic collection of isogenic C. albicans cell
wall mutants and measured their fitness in the mouse GI tract via quantitative competition
assays. Whereas a large variation in competitive fitness was found among mutants,
no correlation was observed between GI fitness and total levels of individual cell wall
components. Similar results were obtained in a set of distantly-related Candida species,
suggesting that total amounts of individual cell wall components do not determine the
ability of fungi to colonize the GI tract. We then subjected this collection ofCandida strains
and species to an extensive quantitative phenotypic profiling in search for features that
might be responsible for their differences in GI fitness, but found no association with
the ability to grow in GI-mimicking and stressful environments or with in vitro and in vivo
virulence. The most significant association with GI fitness was found to be the strength
of signaling through the Dectin-1 receptor. Using a quantitative assay to measure the
amount of exposed β-glucan on the surface of fungal cells, we found this parameter,
unlike total β-glucan levels, to be strongly predictive of competitive fitness in the mouse
GI tract. These data suggest that fungal cell wall architecture, more so than its crude
composition, critically determines the ability of fungi to colonize themammalian GI tract. In
particular, recognition of exposed β-glucan by Dectin-1 receptor appears to severely limit
CandidaGI fitness and hence represents a promising target to reduce fungal colonization
in patients at risks of systemic candidiasis.
Keywords: Candida, Candida albicans, cell wall, competitive fitness, gastrointestinal tract, beta-glucan, dectin-1
ORIGINAL RESEARCHpublished: 22 December 2016
doi: 10.3389/fcimb.2016.00186
Frontiers in Cellular and Infection Microbiology | www.frontiersin.org 1 December 2016 | Volume 6 | Article 186
Edited by:
Brian Wickes,
University of Texas Health Science
Center at San Antonio, USA
Reviewed by:
Karen L. Wozniak,
University of Texas at San Antonio,
USA
Robert T. Wheeler,
University of Maine, USA
*Correspondence:
Norman Pavelka
norman_pavelka@
immunol.a-star.edu.sg
†Present Address:
Marina Yurieva,
The Jackson Laboratory for Genomic
Medicine, Farmington, USA
Francesca Zolezzi,
Galderma R&D, Sophia Antipolis,
France
‡These authors have contributed
equally to this work.
Received: 19 October 2016
Accepted: 30 November 2016
Published: 22 December 2016
Citation:
Sem X, Le GTT, Tan ASM, Tso G,
Yurieva M, Liao WWP, Lum J,
Srinivasan KG, Poidinger M, Zolezzi F
and Pavelka N (2016) β-glucan
Exposure on the Fungal Cell Wall
Tightly Correlates with Competitive
Fitness of Candida Species in the
Mouse Gastrointestinal Tract.
Front. Cell. Infect. Microbiol. 6:186.
doi: 10.3389/fcimb.2016.00186
β-glucan Exposure on the Fungal CellWall Tightly Correlates withCompetitive Fitness of CandidaSpecies in the MouseGastrointestinal TractXiaoHui Sem‡, Giang T. T. Le ‡, Alrina S. M. Tan‡, Gloria Tso, Marina Yurieva †,Webber W. P. Liao, Josephine Lum, Kandhadayar G. Srinivasan, Michael Poidinger,Francesca Zolezzi † and Norman Pavelka*
Singapore Immunology Network, Agency for Science, Technology and Research, Singapore, Singapore
Candida albicans is responsible for ∼400,000 systemic fungal infections annually, with
an associated mortality rate of 46–75%. The human gastrointestinal (GI) tract represents
the largest natural reservoir of Candida species and is a major source of systemic fungal
infections. However, the factors that control GI colonization by Candida species are not
completely understood.We hypothesized that the fungal cell wall would play an important
role in determining the competitive fitness of Candida species in the mammalian GI tract.
To test this hypothesis, we generated a systematic collection of isogenic C. albicans cell
wall mutants and measured their fitness in the mouse GI tract via quantitative competition
assays. Whereas a large variation in competitive fitness was found among mutants,
no correlation was observed between GI fitness and total levels of individual cell wall
components. Similar results were obtained in a set of distantly-related Candida species,
suggesting that total amounts of individual cell wall components do not determine the
ability of fungi to colonize the GI tract. We then subjected this collection ofCandida strains
and species to an extensive quantitative phenotypic profiling in search for features that
might be responsible for their differences in GI fitness, but found no association with
the ability to grow in GI-mimicking and stressful environments or with in vitro and in vivo
virulence. The most significant association with GI fitness was found to be the strength
of signaling through the Dectin-1 receptor. Using a quantitative assay to measure the
amount of exposed β-glucan on the surface of fungal cells, we found this parameter,
unlike total β-glucan levels, to be strongly predictive of competitive fitness in the mouse
GI tract. These data suggest that fungal cell wall architecture, more so than its crude
composition, critically determines the ability of fungi to colonize themammalian GI tract. In
particular, recognition of exposed β-glucan by Dectin-1 receptor appears to severely limit
CandidaGI fitness and hence represents a promising target to reduce fungal colonization
in patients at risks of systemic candidiasis.
Keywords: Candida, Candida albicans, cell wall, competitive fitness, gastrointestinal tract, beta-glucan, dectin-1
Sem et al. Candida Fitness in Mouse Gut
GI Tract Fitness of Candida SpeciesIn order to extend the generality of our findings beyond C.albicans, we then repeated the in vivo competition assays indistantly related Candida species (Figures 3A–E). Interestingly,while C. krusei and C. parapsilosis were significantly less fit,C. glabrata, C. tropicalis, and C. dubliniensis were significantlyfitter than C. albicans in the mouse GI tract (Figure 3F). Inaccordance with the cell wall mutants’ data, compositionalanalysis of the major cell wall polysaccharides did not yieldany significant association with GI tract fitness. In particular,whereas both C. glabrata and C. dubliniensis were significantlyfitter than C. albicans in the mouse GI environment, the formerhad significantly reduced and the latter significantly increasedchitin levels in comparison to C. albicans. Moreover, whereasboth C. tropicalis and C. krusei showed significantly decreasedβ-glucan levels, the former was significantly fitter and the latter
significantly less fit then C. albicans. Hence, despite the existenceof a large variation in both GI tract fitness and cell wallcomposition within the Candida genus, these two traits appearto be largely independent from each other.
Quantitative Phenotypic Profiling RevealsNo Associations with GI Tract FitnessWe next sought to identify factors underlying the variationin mouse GI tract fitness across all our C. albicans cell wallmutants and the other Candida species, employing a systematicprofiling of a large number of quantitative phenotypic parameters(Supplementary Figure 2). Consistent with above findings, nosignificant association was found between competitive fitnessin the mouse GI tract (Supplementary Figure 3A) and totalchitin, mannan, β-glucan, β-1,6-glucan or β-1,3-glucan levels(Supplementary Figures 3B–F). Moreover, no association was
FIGURE 3 | Murine GI tract fitness of Candida spp. strains. Relative frequency of wild-type C. glabrata (A, black), C. tropicalis (B, black), C. dubliniensis (C,
black), C. krusei, (D, black), and C. parapsilosis (E, black) in comparison to SC5314-dTomato (A–E, red) during a competition assay in the mouse GI tract,
represented as in Figures 2A–E. Each pair of lines represents an independent biological assay with a singly-housed mouse, with ≥4 mice per strain and ≥200 CFUs
counted per time point. (F) Murine GI tract fitness of C. albicans (black) and Candida spp. (blue and orange shaded bars) represented as in Figure 2F. (G–I) Cell wall
polysaccharide composition of each Candida species represented as in Figures 2G–I. Asterisks shown represent significant two-tailed p-values obtained from
unpaired Welch’s t-tests between wild-type and mutant strain (* < 0.05; ** < 0.01; *** < 0.001; **** < 0.0001).
Frontiers in Cellular and Infection Microbiology | www.frontiersin.org 7 December 2016 | Volume 6 | Article 186
Sem et al. Candida Fitness in Mouse Gut
GI Tract Fitness of Candida SpeciesIn order to extend the generality of our findings beyond C.albicans, we then repeated the in vivo competition assays indistantly related Candida species (Figures 3A–E). Interestingly,while C. krusei and C. parapsilosis were significantly less fit,C. glabrata, C. tropicalis, and C. dubliniensis were significantlyfitter than C. albicans in the mouse GI tract (Figure 3F). Inaccordance with the cell wall mutants’ data, compositionalanalysis of the major cell wall polysaccharides did not yieldany significant association with GI tract fitness. In particular,whereas both C. glabrata and C. dubliniensis were significantlyfitter than C. albicans in the mouse GI environment, the formerhad significantly reduced and the latter significantly increasedchitin levels in comparison to C. albicans. Moreover, whereasboth C. tropicalis and C. krusei showed significantly decreasedβ-glucan levels, the former was significantly fitter and the latter
significantly less fit then C. albicans. Hence, despite the existenceof a large variation in both GI tract fitness and cell wallcomposition within the Candida genus, these two traits appearto be largely independent from each other.
Quantitative Phenotypic Profiling RevealsNo Associations with GI Tract FitnessWe next sought to identify factors underlying the variationin mouse GI tract fitness across all our C. albicans cell wallmutants and the other Candida species, employing a systematicprofiling of a large number of quantitative phenotypic parameters(Supplementary Figure 2). Consistent with above findings, nosignificant association was found between competitive fitnessin the mouse GI tract (Supplementary Figure 3A) and totalchitin, mannan, β-glucan, β-1,6-glucan or β-1,3-glucan levels(Supplementary Figures 3B–F). Moreover, no association was
FIGURE 3 | Murine GI tract fitness of Candida spp. strains. Relative frequency of wild-type C. glabrata (A, black), C. tropicalis (B, black), C. dubliniensis (C,
black), C. krusei, (D, black), and C. parapsilosis (E, black) in comparison to SC5314-dTomato (A–E, red) during a competition assay in the mouse GI tract,
represented as in Figures 2A–E. Each pair of lines represents an independent biological assay with a singly-housed mouse, with ≥4 mice per strain and ≥200 CFUs
counted per time point. (F) Murine GI tract fitness of C. albicans (black) and Candida spp. (blue and orange shaded bars) represented as in Figure 2F. (G–I) Cell wall
polysaccharide composition of each Candida species represented as in Figures 2G–I. Asterisks shown represent significant two-tailed p-values obtained from
unpaired Welch’s t-tests between wild-type and mutant strain (* < 0.05; ** < 0.01; *** < 0.001; **** < 0.0001).
Frontiers in Cellular and Infection Microbiology | www.frontiersin.org 7 December 2016 | Volume 6 | Article 186
C.albicans
C.glabrata

infection arising from implanted medical devices (Nobile andMitchell 2006; Finkel and Mitchell 2011). Phenotypic variation isfurther evident in the form ofmorphological plasticity, such as thewhite-opaque switch and the yeast to hyphal transition, which in-volve developmental shifts between distinct cellular states (Slutskyet al. 1985, 1987; Soll 1993; Lohse and Johnson2009; Sudbery 2011).
Genomic variation is also well established for C. albicans andincludes large-scale chromosomal rearrangements such as trans-locations, chromosome truncations, and aneuploidy (Rustchenko2007; Selmecki et al. 2010; Bennett et al. 2014). A number of studieshave demonstrated that karyotypic differences exist between clini-cal isolates, including evidence for both monosomic and trisomicchromosomes (Rustchenko-Bulgac et al. 1990; Rustchenko-Bulgac1991; Barton and Gull 1992; Perepnikhatka et al. 1999; Chibanaet al. 2000; Chen et al. 2004). In one example, the presence of anextra isochromosome composed of the two left arms of Chromo-some 5mediates fluconazole resistance in a subset of clinical strains(Selmecki et al. 2006, 2008). C. albicans strains also exhibit geneticdiversity due to variable tracts of loss of heterozygosity (LOH). MostC. albicans strains are considered to be heterozygous diploids, andyet different strains often contain different LOH tracts, some ofwhich can provide phenotypic advantages. For example, homozy-gosis of hyperactive alleles of ERG11, TAC1, and MRR1 (encodingthe target of azole drugs or transcription factors regulating drug ef-flux pumps) can result in strains with increased azole resistance(White 1997; Coste et al. 2006; Dunkel et al. 2008). Both LOH andkaryotypic changes are induced in response to environmentalstimuli including drug treatment, heat shock, nutrient utilization,and oxidative stress (Rustchenko et al. 1997; Janbon et al. 1998;Forche et al. 2011; Harrison et al. 2014).
Sequencing of C. albicans isolates has provided additionalinsights into the genome structure ofC. albicans. Two isolates havebeen sequenced to date: SC5314, the standard laboratory strain ofC. albicans; and WO-1, in which white-opaque switching was firstobserved (Jones et al. 2004; van het Hoog et al. 2007; Butler et al.2009). The genomes of these strains consisted of eight diploid chro-mosomes, althoughWO-1 had lost one >300-kb region on Chromo-some 3. Both SC5314 and WO-1 contained ;6100 genes and weregenerally heterozygous, although extended LOH regions were ob-served in both isolates (Butler et al. 2009). Recent phasing of thesingle nucleotide polymorphisms (SNPs) in SC5314 included nearlyall of the 69,668 SNPs identified in this strain (Muzzey et al. 2013).
Here, we sequenced the genomes of 21 clinical isolates ofC. albicans selected to represent different clades, different sites ofanatomical origin, and differences in virulence (Wu et al. 2007).We find that genomic variation in C. albicans occurs at the level ofSNPs, inversions, copynumber variation, loss ofheterozygosity (LOH),and aneuploidy. Extensive phenotypic analysis was also performedon the 21 isolates to characterize properties associated with patho-genesis.Wediscuss our findingswith respect to intra-species variationinC. albicans and reveal how a natural isolate has evolved amutationthat promotes the commensal lifestyle at the expense of a morepathogenic one.
Results
Sequencing of 21 clinical isolates of C. albicans
We selected 21 C. albicans strains for phenotypic and genotypiccharacterization (Supplemental Table S1). This is a geographicallydiverse set of strains that originated from bloodstream infections(14 isolates) or superficial infections of oral or vaginal tissues (seven
isolates) and includes the previously sequenced laboratory strainSC5314 for comparison (Jones et al. 2004). With the exception ofSC5314, the virulence of each clinical isolate was previously com-pared using a murine model of disseminated candidiasis (Supple-mental Table S1; Wu et al. 2007).
The set of 21 strainswas typed by fingerprint analysis to cladesI, II, III, SA, and E of C. albicans (Lockhart et al. 1996; Blignaut et al.2002; Pujol et al. 2002). Using variants identified from whole-genome sequence data (Supplemental Table S2; Methods), weevaluated the phylogenetic relationship of these 21 strains (Fig. 1).The major groups corresponded to the previously described finger-print-based clades, with the exception of P94015, which is distantlypositioned relative to other clade I strains.MLSTanalysis was carriedout and compared to a previously typed population (Tavanti et al.2005). The sequenced isolates represent seven of the 17 previouslyidentified clades, including representatives of the five largest clades(1, 2, 3, 4, and 11), as well as two less common clades (6 and 8)(Supplemental Table S3; Supplemental Fig. S1; Odds et al. 2007).
Analysis of the genome-wide variation for these C. albicansisolates revealed they contain, on average, a heterozygous SNP every267 bases. Furthermore, when compared to SC5314, other clade Istrains displayed a homozygous variant every 1404 bases, whereasnon-clade 1 isolates contained a homozygous variant every 235bases. Using these variants, the average genome-wide nucleotidediversity between any twoC. albicans isolates is estimated at 0.37%.
Figure 1. Phylogenetic relationship and clade assignment of studiedisolates. The phylogenetic relationship of the strains was inferred based on112,223 informative SNP positions using maximum parsimony in PAUP*;node labels indicate the support frequency of 1000 bootstrap replicates.Clades were assigned based on multilocus sequence type (MLST) analysisof seven loci or prior work reporting fingerprint clades (FP).
414 Genome Researchwww.genome.org
Hirakawa et al.
Cold Spring Harbor Laboratory Press on July 24, 2015 - Published by genome.cshlp.orgDownloaded from
Research
Genetic and phenotypic intra-species variationin Candida albicansMatthew P. Hirakawa,1 Diego A. Martinez,2,5 Sharadha Sakthikumar,2,5
Matthew Z. Anderson,1,3 Aaron Berlin,2 Sharvari Gujja,2 Qiandong Zeng,2 Ethan Zisson,1
Joshua M. Wang,1 Joshua M. Greenberg,1 Judith Berman,3,4 Richard J. Bennett,1,5
and Christina A. Cuomo2,5
1Department of Molecular Microbiology and Immunology, Brown University, Providence, Rhode Island 02912, USA; 2Broad Institute
of MIT and Harvard, Cambridge, Massachusetts 02142, USA; 3Department of Molecular, Cellular Biology and Genetics, University of
Minnesota, Minneapolis, Minnesota 55455, USA; 4Department of Molecular Microbiology and Biotechnology, Tel Aviv University,
Ramat Aviv 69978, Israel
Candida albicans is a commensal fungus of the human gastrointestinal tract and a prevalent opportunistic pathogen. Toexamine diversity within this species, extensive genomic and phenotypic analyses were performed on 21 clinical C. albicansisolates. Genomic variation was evident in the form of polymorphisms, copy number variations, chromosomal inversions,subtelomeric hypervariation, loss of heterozygosity (LOH), and whole or partial chromosome aneuploidies. All 21 strainswere diploid, although karyotypic changes were present in eight of the 21 isolates, with multiple strains being trisomic forChromosome 4 or Chromosome 7. Aneuploid strains exhibited a general fitness defect relative to euploid strains whengrown under replete conditions. All strains were also heterozygous, yet multiple, distinct LOH tracts were present in eachisolate. Higher overall levels of genome heterozygosity correlated with faster growth rates, consistent with increasedoverall fitness. Genes with the highest rates of amino acid substitutions included many cell wall proteins, implicating fastevolving changes in cell adhesion and host interactions. One clinical isolate, P94015, presented several striking propertiesincluding a novel cellular phenotype, an inability to filament, drug resistance, and decreased virulence. Several of theseproperties were shown to be due to a homozygous nonsense mutation in the EFG1 gene. Furthermore, loss of EFG1 functionresulted in increased fitness of P94015 in a commensal model of infection. Our analysis therefore reveals intra-speciesgenetic and phenotypic differences in C. albicans and delineates a natural mutation that alters the balance between com-mensalism and pathogenicity.
[Supplemental material is available for this article.]
Candida albicans is an important human fungal pathogen, respon-sible for both debilitating mucosal infections and life-threateningsystemic infections (Wey et al. 1988;Gudlaugsson et al. 2003; Pfallerand Diekema 2007). In addition to being an opportunistic patho-gen, C. albicans is a frequent and benign component of the humangastrointestinalmicrobiota (Kleinegger et al. 1996).Many attributeshave been linked to the ability ofC. albicans to successfully colonizeand infect the human host, including its ability to rapidly switchbetween different cell states in response to environmental cues(Slutsky et al. 1987; Lo et al. 1997; Kumamoto and Vinces 2005;Phan et al. 2007; Zhu and Filler 2009).
The majority (97%) of C. albicans isolates are diploid strainsassigned to one of 17 clades, as defined by multilocus sequencetyping (MLST). These clades are enriched for different geographicalareas, with the five largest clades accounting for three-quarters ofclinical isolates (Bougnoux et al. 2004; Tavanti et al. 2005; Oddset al. 2007). The population structure is largely clonal, althoughrecombination events have led to isolates withmixed evolutionaryhistories, consistent with a sexually or parasexually reproducingspecies (Tibayrenc 1997; Odds et al. 2007). C. albicans mating has
been shown to occur betweenMTLa andMTLa diploids, as well aswithin unisexual populations (Hull and Johnson 1999; Hull et al.2000;Magee andMagee 2000; Alby et al. 2009).Mating is regulatedby a phenotypic switch, in which cells must transition from thedefault ‘‘white’’ state to the mating-competent ‘‘opaque’’ state toundergo efficient conjugation (Miller and Johnson 2002). Despiteefficient mating, a conventional meiosis has not been observed inC. albicans; instead, cells shift from the tetraploid state to the diploidstate via a program of random chromosome loss (Bennett andJohnson 2003; Forche et al. 2008; Berman and Hadany 2012). A vi-able haploid state was also recently uncovered, although C. albicanshaploids are unstable, rapidly reverting to diploid, and also exhibitdecreased fitness relative to diploid forms (Hickman et al. 2013).
Natural isolates of C. albicans exhibit a broad spectrum ofphenotypic properties. For example, clinical isolates display strik-ing differences in virulence when compared in the murine modelof systemic infection (Wu et al. 2007; MacCallum et al. 2009).Clinical isolates also show differences in their ability to form bio-films (Li et al. 2003); these are cellular communities that promote
! 2015 Hirakawa et al. This article is distributed exclusively by Cold SpringHarbor Laboratory Press for the first six months after the full-issue publicationdate (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, itis available under a Creative Commons License (Attribution-NonCommercial4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
5These authors contributed equally to this work.Corresponding authors: [email protected], [email protected] published online before print. Article, supplemental material, and pub-lication date are at http://www.genome.org/cgi/doi/10.1101/gr.174623.114.
25:413–425 Published by Cold Spring Harbor Laboratory Press; ISSN 1088-9051/15; www.genome.org Genome Research 413www.genome.org
Cold Spring Harbor Laboratory Press on July 24, 2015 - Published by genome.cshlp.orgDownloaded from
Research
Genetic and phenotypic intra-species variationin Candida albicansMatthew P. Hirakawa,1 Diego A. Martinez,2,5 Sharadha Sakthikumar,2,5
Matthew Z. Anderson,1,3 Aaron Berlin,2 Sharvari Gujja,2 Qiandong Zeng,2 Ethan Zisson,1
Joshua M. Wang,1 Joshua M. Greenberg,1 Judith Berman,3,4 Richard J. Bennett,1,5
and Christina A. Cuomo2,5
1Department of Molecular Microbiology and Immunology, Brown University, Providence, Rhode Island 02912, USA; 2Broad Institute
of MIT and Harvard, Cambridge, Massachusetts 02142, USA; 3Department of Molecular, Cellular Biology and Genetics, University of
Minnesota, Minneapolis, Minnesota 55455, USA; 4Department of Molecular Microbiology and Biotechnology, Tel Aviv University,
Ramat Aviv 69978, Israel
Candida albicans is a commensal fungus of the human gastrointestinal tract and a prevalent opportunistic pathogen. Toexamine diversity within this species, extensive genomic and phenotypic analyses were performed on 21 clinical C. albicansisolates. Genomic variation was evident in the form of polymorphisms, copy number variations, chromosomal inversions,subtelomeric hypervariation, loss of heterozygosity (LOH), and whole or partial chromosome aneuploidies. All 21 strainswere diploid, although karyotypic changes were present in eight of the 21 isolates, with multiple strains being trisomic forChromosome 4 or Chromosome 7. Aneuploid strains exhibited a general fitness defect relative to euploid strains whengrown under replete conditions. All strains were also heterozygous, yet multiple, distinct LOH tracts were present in eachisolate. Higher overall levels of genome heterozygosity correlated with faster growth rates, consistent with increasedoverall fitness. Genes with the highest rates of amino acid substitutions included many cell wall proteins, implicating fastevolving changes in cell adhesion and host interactions. One clinical isolate, P94015, presented several striking propertiesincluding a novel cellular phenotype, an inability to filament, drug resistance, and decreased virulence. Several of theseproperties were shown to be due to a homozygous nonsense mutation in the EFG1 gene. Furthermore, loss of EFG1 functionresulted in increased fitness of P94015 in a commensal model of infection. Our analysis therefore reveals intra-speciesgenetic and phenotypic differences in C. albicans and delineates a natural mutation that alters the balance between com-mensalism and pathogenicity.
[Supplemental material is available for this article.]
Candida albicans is an important human fungal pathogen, respon-sible for both debilitating mucosal infections and life-threateningsystemic infections (Wey et al. 1988;Gudlaugsson et al. 2003; Pfallerand Diekema 2007). In addition to being an opportunistic patho-gen, C. albicans is a frequent and benign component of the humangastrointestinalmicrobiota (Kleinegger et al. 1996).Many attributeshave been linked to the ability ofC. albicans to successfully colonizeand infect the human host, including its ability to rapidly switchbetween different cell states in response to environmental cues(Slutsky et al. 1987; Lo et al. 1997; Kumamoto and Vinces 2005;Phan et al. 2007; Zhu and Filler 2009).
The majority (97%) of C. albicans isolates are diploid strainsassigned to one of 17 clades, as defined by multilocus sequencetyping (MLST). These clades are enriched for different geographicalareas, with the five largest clades accounting for three-quarters ofclinical isolates (Bougnoux et al. 2004; Tavanti et al. 2005; Oddset al. 2007). The population structure is largely clonal, althoughrecombination events have led to isolates withmixed evolutionaryhistories, consistent with a sexually or parasexually reproducingspecies (Tibayrenc 1997; Odds et al. 2007). C. albicans mating has
been shown to occur betweenMTLa andMTLa diploids, as well aswithin unisexual populations (Hull and Johnson 1999; Hull et al.2000;Magee andMagee 2000; Alby et al. 2009).Mating is regulatedby a phenotypic switch, in which cells must transition from thedefault ‘‘white’’ state to the mating-competent ‘‘opaque’’ state toundergo efficient conjugation (Miller and Johnson 2002). Despiteefficient mating, a conventional meiosis has not been observed inC. albicans; instead, cells shift from the tetraploid state to the diploidstate via a program of random chromosome loss (Bennett andJohnson 2003; Forche et al. 2008; Berman and Hadany 2012). A vi-able haploid state was also recently uncovered, although C. albicanshaploids are unstable, rapidly reverting to diploid, and also exhibitdecreased fitness relative to diploid forms (Hickman et al. 2013).
Natural isolates of C. albicans exhibit a broad spectrum ofphenotypic properties. For example, clinical isolates display strik-ing differences in virulence when compared in the murine modelof systemic infection (Wu et al. 2007; MacCallum et al. 2009).Clinical isolates also show differences in their ability to form bio-films (Li et al. 2003); these are cellular communities that promote
! 2015 Hirakawa et al. This article is distributed exclusively by Cold SpringHarbor Laboratory Press for the first six months after the full-issue publicationdate (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, itis available under a Creative Commons License (Attribution-NonCommercial4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
5These authors contributed equally to this work.Corresponding authors: [email protected], [email protected] published online before print. Article, supplemental material, and pub-lication date are at http://www.genome.org/cgi/doi/10.1101/gr.174623.114.
25:413–425 Published by Cold Spring Harbor Laboratory Press; ISSN 1088-9051/15; www.genome.org Genome Research 413www.genome.org
Cold Spring Harbor Laboratory Press on July 24, 2015 - Published by genome.cshlp.orgDownloaded from
21genomes
2015
SNP/indelmuta6ons
*
Aneuploidy
8x
8heterozygouschromosomes
indiploidgenome
Lossofheterozygosity(LOH)
Hirakawaetal.,2015,GenomeResearch

Wuetal.,MolMicro,2007
L26 GC75 P37005 P57072 P60002
19FP9712CP94015*P787048
Survival(%
)Survival(%
)
Time(Days)
Howdoesintra-speciesdiversityaffectphenotypessuchasvirulence?

The role of EFG1 in P94015?
Increased Filamentation SC5314 P94015
P94015+EFG1 (1) P94015+EFG1 (2)
A
1 2 3 4 5 6 7 8 9 10 11 12 13 140
50
100
Days Post Infection
Rel
ativ
e S
train
Fre
quen
cy
in F
eces
(%)
P94015 P94015+EFG1 (1)
P94015 P94015+EFG1 (2)
vs.
vs.
E
0 5 100
20
40
60
80
100
Days Post Infection
% S
urvi
val
P94015P94015+EFG1 (1)P94015+EFG1 (2)
C
P9401
5
P9401
5+EFG1
(1)
P9401
5+EFG1
(2)
0
5
10
15
% F
ilam
ento
us C
ells
B
*
*
Inocu
lum
Stomac
h
Small In
testin
e
Cecum
Colon
0
50
100
Rel
ativ
e S
train
Fre
quen
cy
in O
rgan
s (%
)
P94015P94015+EFG1
F
0 70
50
100
Rel
ativ
e S
train
Fre
quen
cy
in K
idne
y (%
)
P94015 P94015+EFG1 (1)
P94015 P94015+EFG1 (2)
vs.
vs.
D
0
Days Post Infection
SC5314 P94015
P94015+EFG1 (1) P94015+EFG1 (2)
A
1 2 3 4 5 6 7 8 9 10 11 12 13 140
50
100
Days Post Infection
Rel
ativ
e S
train
Fre
quen
cy
in F
eces
(%)
P94015 P94015+EFG1 (1)
P94015 P94015+EFG1 (2)
vs.
vs.
E
0 5 100
20
40
60
80
100
Days Post Infection
% S
urvi
val
P94015P94015+EFG1 (1)P94015+EFG1 (2)
C
P9401
5
P9401
5+EFG1
(1)
P9401
5+EFG1
(2)
0
5
10
15
% F
ilam
ento
us C
ells
B
*
*
Inocu
lum
Stomac
h
Small In
testin
e
Cecum
Colon
0
50
100
Rel
ativ
e S
train
Fre
quen
cy
in O
rgan
s (%
)
P94015P94015+EFG1
F
0 70
50
100
Rel
ativ
e S
train
Fre
quen
cy
in K
idne
y (%
)
P94015 P94015+EFG1 (1)
P94015 P94015+EFG1 (2)
vs.
vs.
D
0
Days Post Infection
Increased Virulence
SC5314 P94015
P94015+EFG1 (1) P94015+EFG1 (2)
A
1 2 3 4 5 6 7 8 9 10 11 12 13 140
50
100
Days Post Infection
Re
lativ
e S
train
Fre
qu
en
cy
in F
ece
s (%
)
P94015 P94015+EFG1 (1)
P94015 P94015+EFG1 (2)
vs.
vs.
E
0 5 100
20
40
60
80
100
Days Post Infection%
Su
rviv
al
P94015P94015+EFG1 (1)P94015+EFG1 (2)
C
P9401
5
P9401
5+EFG1
(1)
P9401
5+EFG1
(2)
0
5
10
15
% F
ilam
en
tou
s C
ells
B
*
*
Inocu
lum
Stomac
h
Small In
testin
e
Cecum
Colon
0
50
100
Rel
ativ
e S
train
Fre
quen
cy
in O
rgan
s (%
)
P94015P94015+EFG1
F
0 70
50
100
Rel
ativ
e S
train
Fre
quen
cy
in K
idne
y (%
)
P94015 P94015+EFG1 (1)
P94015 P94015+EFG1 (2)
vs.
vs.
D
0
Days Post Infection
SC5314 P94015
P94015+EFG1 (1) P94015+EFG1 (2)
A
1 2 3 4 5 6 7 8 9 10 11 12 13 140
50
100
Days Post Infection
Rel
ativ
e S
train
Fre
quen
cy
in F
eces
(%)
P94015 P94015+EFG1 (1)
P94015 P94015+EFG1 (2)
vs.
vs.
E
0 5 100
20
40
60
80
100
Days Post Infection
% S
urvi
val
P94015P94015+EFG1 (1)P94015+EFG1 (2)
C
P9401
5
P9401
5+EFG1
(1)
P9401
5+EFG1
(2)
0
5
10
15
% F
ilam
ento
us C
ells
B
*
*
Inocu
lum
Stomac
h
Small In
testin
e
Cecum
Colon
0
50
100
Rel
ativ
e S
train
Fre
quen
cy
in O
rgan
s (%
)
P94015P94015+EFG1
F
0 70
50
100
Rel
ativ
e S
train
Fre
quen
cy
in K
idne
y (%
)
P94015 P94015+EFG1 (1)
P94015 P94015+EFG1 (2)
vs.
vs.
D
0
Days Post InfectionDays Post Infection
Restoring EFG1 increased filamentation and virulence
Contains a homozygous, premature stop codon in EFG1
* aa80 1 525
APSES DNA-binding domain
Hirakawaetal.,2015,GenomeResearch

Does reintroduction of a functional EFG1 allele into P94015 alter commensal fitness?
P94015 outcompetes P94015-EFG1 in a commensal model of GI colonization
Restoring EFG1 reduced fitness in the commensal GI model

Natural isolates show preferential specificity for different host niches
Strains biased towards a more commensal lifestyle
Strains biased towards a more pathogenic lifestyle
A natural polymorphism impacts commensal fitness and virulence
Loss of EFG1 function
Hirakawa et al., Genome Res, 2015

A note of caution with genomes!
INVESTIGATION
Finding a Missing Gene: EFG1 RegulatesMorphogenesis in Candida tropicalisEugenio Mancera,* Allison M. Porman,‡ Christina A. Cuomo,§ Richard J. Bennett,‡
and Alexander D. Johnson*,†*Department of Microbiology and Immunology and †Department of Biochemistry and Biophysics, University of CaliforniaSan Francisco, San Francisco, California 94158, ‡Department of Microbiology and Immunology, Brown University,Providence, Rhode Island 02912, and §Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142
ORCID ID: 0000-0003-0146-8732 (E.M.)
ABSTRACT Fungi from the genus Candida are common members of the human microbiota; however, theyare also important opportunistic pathogens in immunocompromised hosts. Several morphological transitionshave been linked to the ability of these fungi to occupy the different ecological niches in the human body. Thetranscription factor Efg1 from the APSES family plays a central role in the transcription circuits underlying severalof these morphological changes. In Candida albicans, for example, Efg1 is a central regulator of filamentation,biofilm formation, and white-opaque switching, processes associated with survival in the human host. Ortho-logs of Efg1 are present throughout the Candida clade but, surprisingly, the genome sequence of Candidatropicalis failed to uncover a gene coding for Efg1. One possibility was that the paralog of Efg1, Efh1, hadassumed the function of Efg1 in C. tropicalis. However, we show that this gene has only a minor role in themorphological transitions mentioned above. Instead, we report here that C. tropicalis does have an ortholog ofthe EFG1 gene found in other Candida species. The gene is located in a different genomic position than EFG1in C. albicans, in a region that contains a gap in the current genome assembly of C. tropicalis. We show that thenewly identified C. tropicalis EFG1 gene regulates filamentation, biofilm formation, and white-opaque switch-ing. Our results highlight the conserved role of Efg1 in controlling morphogenesis in Candida species andremind us that published genome sequences are drafts that require continuous curation and careful scrutiny.
KEYWORDSCandidamorphogenesis
biofilmfilamentationphenotypicswitch
transcriptionalregulation
Several species of the Candida genus belong to a monophyletic clade ofascomycetous fungi that translate the CTG codon as serine instead ofleucine (Butler et al. 2009). Members of this CTG clade include speciesthat are commensals of the human microbiota with no known envi-ronmental reservoirs outside of animals. However, these commensalscan also cause mucosal disease in healthy individuals as well as sys-
temic infections in immunocompromised hosts (Calderone 2002).Members of the CTG clade, therefore, are important fungal humanpathogens, both in terms of their prevalence and their mortality rate.
Although they are most commonly encountered in the yeast form(unicellular spherical cells), most CTG species are able to undergoa variety of changes in cell and colony morphology. The best studiedof these is the ability to switch between the yeast and hyphal(filamentous) forms, a transition that is closely linked to pathogenesisand biofilm formation (Sudbery 2011). Biofilms are communities ofcells associated with a biologic or inert surface. In the case of Candidaspecies, these communities are typically composed of several layers ofyeast cells and hyphae that are embedded within an extracellularmatrix (Finkel and Mitchell 2011; Nobile et al. 2012). Other morpho-logical transitions include the phenomenon of heritable phenotypicswitching. In C. albicans, this includes the white-opaque switch, inwhich cells transition between two different cell types that generateeither shiny and dome-shaped colonies (white) or dull and flat colo-nies (opaque) (Slutsky et al. 1987; Lohse and Johnson 2009). Whiteand opaque cells are morphologically distinct, have different metabolic
Copyright © 2015 Mancera et al.doi: 10.1534/g3.115.017566Manuscript received January 8, 2015; accepted for publication March 5, 2015;published Early Online March 9, 2015.This is an open-access article distributed under the terms of the CreativeCommons Attribution Unported License (http://creativecommons.org/licenses/by/3.0/), which permits unrestricted use, distribution, and reproduction in anymedium, provided the original work is properly cited.Supporting information is available online at http://www.g3journal.org/lookup/suppl/doi:10.1534/g3.115.017566/-/DC11Corresponding author: University of California, San Francisco, Department ofMicrobiology & Immunology, N372 Genentech Hall, Mission Bay, 600 16th Street,Box 2200, San Francisco, CA 94143-2200. E-mail: [email protected] [email protected]
Volume 5 | May 2015 | 849
Figure 1 Genomic and phylogenetic position of EFG1 in C. tropicalis. (A) Schematic depiction of the genomic location of EFG1 in C. albicans andthe corresponding genomic location in C. tropicalis. (B) As (A), but for C. tropicalis EFG1 and the corresponding location in C. albicans. In (A and B)
852 | E. Mancera et al.
“MydatasuggeststhatthegenesislocatedinSupercon6g_3.1inthegapspanningfrom887,643to888,943”
Figure 1 Genomic and phylogenetic position of EFG1 in C. tropicalis. (A) Schematic depiction of the genomic location of EFG1 in C. albicans andthe corresponding genomic location in C. tropicalis. (B) As (A), but for C. tropicalis EFG1 and the corresponding location in C. albicans. In (A and B)
852 | E. Mancera et al.

opaque
a α
opaque
2N 2N
2N
white
a α
white
2Na/α4N
Recombinantparasexualproducts
αa
a α
a α
2N2N
2N+1
2N+22N+2
2N+1
Spo11/DSBs
C.albicans
opaque
a α
opaque
2N 2N
2N
white
a α
white
2Na/α4N
Recombinantparasexualproducts
αa
a α
a α
2N2N
2N+1
2N+22N+2
2N+1
Spo11/DSBs
What about sex?

More genomes…… and genome resources…. and tools
h=p://1000.fungalgenomes.org/home/about/

PathogenicFungi–Genomics,Evolu6onandEpidemiology
GeraldineButler UniversityCollegeDublin,IrelandAntonisRokas VanderbiltUniversity,USALi-JunMa UmassAmherst,USAGuilhemJanbon Ins6tutPasteur,Paris,FranceNormanPavelka A*Star,Singapore