photoinduced tautomerism of 2,6-dicarbomethoxyphenol in dmf–water mixtures: perturbation from...
TRANSCRIPT

Photoinduced tautomerism of 2,6-dicarbomethoxyphenolin DMF–water mixtures: Perturbation from intermolecular processes
Abhijit Mandal a,n, Ramprasad Misra b
a Department of Chemistry & Environment, Heritage Institute of Technology, Chowbaga Road, Anandapur, Kolkata 700107, Indiab Department of Physical Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700032, India
a r t i c l e i n f o
Article history:Received 2 October 2013Received in revised form19 December 2013Accepted 7 January 2014Available online 29 January 2014
Keywords:Excited stateProton transferMolecular clusterBinary mixture
a b s t r a c t
In this paper, we report the spectral signatures of photoinduced tautomerism of 4-methyl-2,6-dicarbomethoxyphenol (CMOH) in DMF–water mixtures with varying compositions. Excited stateintramolecular proton transfer (ESIPT) reaction of CMOH has been observed in bulk DMF, indicated bydual fluorescence from its normal and tautomeric forms while only a single emission peak is observed inwater from its anionic species. Binary mixture of a polar aprotic (DMF) and a polar protic (water) solventgives rise to a competition between intramolecular and intermolecular hydrogen bonding (with media)processes of the probe. This competition is found to be largely dependent on the proton affinity of themedia and also on the excitation energy. Solvent separated ion pair and intermolecularly H-bondedCMOH–Solvent complex have been detected in the excited state at specific solvent compositions that areconverted to the anionic form due to the change in excitation wavelengths. The formation of hydrogenbonded 1:1 molecular clusters of different rotamers of CMOH with DMF and water in the ground statehas been investigated using quantum chemical calculations. A combined experimental and theoreticalanalysis indicates that the HOMO to LUMO transitions dictate the electronic absorption profiles of theCMOH–DMF and CMOH–water clusters. These findings are expected to shed light on the mechanism ofacid–base reactions of several hydrogen bonded systems that are part of many biologically relevantprocesses.
& 2014 Elsevier B.V. All rights reserved.
1. Introduction
In recent years, studies of hydrogen bonding and transfer in amolecule in the excited state have been fueled due to theirimmense applicability in chemistry and biology [1–5]. The excitedstate intramolecular proton transfer (ESIPT) is a fundamental acid–base reaction, in which transfer of proton occurs from a protondonor group to a proton acceptor group in the molecule [6,7].Studying proton transfer in different media provides us withunprecedented insight into many chemical and biological pro-cesses as it has similarity with many biological processes, likeorganization of cell membranes, action of ion channels andenzyme catalysis, etc. [8,9]. The molecules that give rise to fluores-cent tautomers through ESIPT are used as laser dyes, in highenergy radiation detectors, in molecular energy storage systems,etc. [10–14]. The ESIPT molecules often exhibit dual emission: anormal emission at lower wavelengths and a large Stokes-shiftedtautomer emission. These emissions have been shown to dependheavily on solvent polarity, pH, hydrogen-bonding character of
solvent, and structure of the molecules in both the groundand excited states [9–11]. 4-methyl-2,6-dicarbomethoxyphenol(CMOH) is a proton transfer probe belonging to ortho-hydro-xybenzaldehyde (OHBA) molecular family [15,16], in which theproton transfer mainly occurs in the excited state. We havereported earlier [17–19] that CMOH can exist in different formsthat have been shown in Scheme 1. The uniqueness of CMOHamong the different OHBA type proton transfer probes is that theemission from the closed conformer has only been observed incase of CMOH (in nonpolar solvents and also in DMF). Out of thetwo rotamers of CMOH (forms A and B; Scheme 1), which arecomparable in energy [17], the ESIPT reaction can occur only forthe rotamer A. So, an incomplete ESIPT taking place in solutions ofCMOH as two different rotamers are present in the ground state inalmost equal proportion. So, it is apparent that some of the formsof CMOH shown in Scheme 1 appear due to intramolecularhydrogen bonding while some of the others appear due tointermolecular interactions (hydrogen bonding or proton abstrac-tion) with the solvents. So, deciphering the competition betweenthe tautomerization of CMOH, an intramolecular phenomenon andthe hydrogen bonding of the probe molecule with the medium, anintermolecular phenomenon are expected to be helpful to under-stand the photophysics of this molecule clearly. It has been
Contents lists available at ScienceDirect
journal homepage: www.elsevier.com/locate/jlumin
Journal of Luminescence
0022-2313/$ - see front matter & 2014 Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.jlumin.2014.01.016
n Corresponding author. Tel.: þ91 92 39458791; fax: þ91 33 24430455.E-mail address: [email protected] (A. Mandal).
Journal of Luminescence 150 (2014) 25–34
reported that the properties of the medium sometimes dictatethe electronic structure and properties of a molecule [20–25].The polarity and the hydrogen bonding properties of a solvent playan important role in controlling the spectral properties of theprobe [26–30]. In many cases, it is not possible to study theimportant various properties of a molecule using pure solventsalone. One should consider the possibility by using binary solventmixtures [31–33]. In recent years, the proton transfer reaction inorganic molecules has been studied in different media, including,micelles, reverse micelles, binary solvent mixtures and ionicliquids [34–40]. We have used mixtures of DMF, a polar aproticsolvent and water, a polar protic solvent with varying compositionas media for studying ESIPT of CMOH. The properties of DMF–water solvent mixtures have been studied by many researchgroups using experimental studies and computer simulations[41,42].
In this contribution, we tried to decipher the competitionbetween the intramolecular and intermolecular hydrogen bondingof CMOH in DMF–water solvent mixtures. The layout of thepaper is as follows. The materials and methodologies used for
the present study have been described in Section 2. In Section 3.1,the spectral properties of CMOH in some pure solvents have beenanalyzed. The results of quantum chemical calculations on mole-cular clusters of CMOH (rotamers A and B) with water and DMFhave been reported in Section 3.2. The spectral response of theprobe in DMF–water binary mixtures with varying compositionhas been described in Section 3.3. Results of time-resolvedexperiments of CMOH in pure (DMF–water) and their binarysolvent mixtures have been discussed in Section 3.4.
2. Experimental
2.1. Materials and solutions
CMOH was prepared in the Department of Inorganic Chemistryof this institute using the methodology described elsewhere [43].It was recrystallized from methanol and checked for purity forspectroscopic studies. The solvents used were of spectroscopic gradeand were purchased from commercial sources (Merck/Spectrochem)
CH3
C C
O
O
O
O
OCH3 H
CH3
C C
O
O
O
O
OCH3 H
CH3
C C
O
O
O
O
OCH3
CH3
C C
O
O
O
O
OCH3
CH3
C C
O
O
O
O
OH
CH3
CH3
CH3
CH3
CH3
C C
O
O
O
O
OCH3 CH3H
S
H
CH3
CH31
2
34
5
61
2
34
5
6
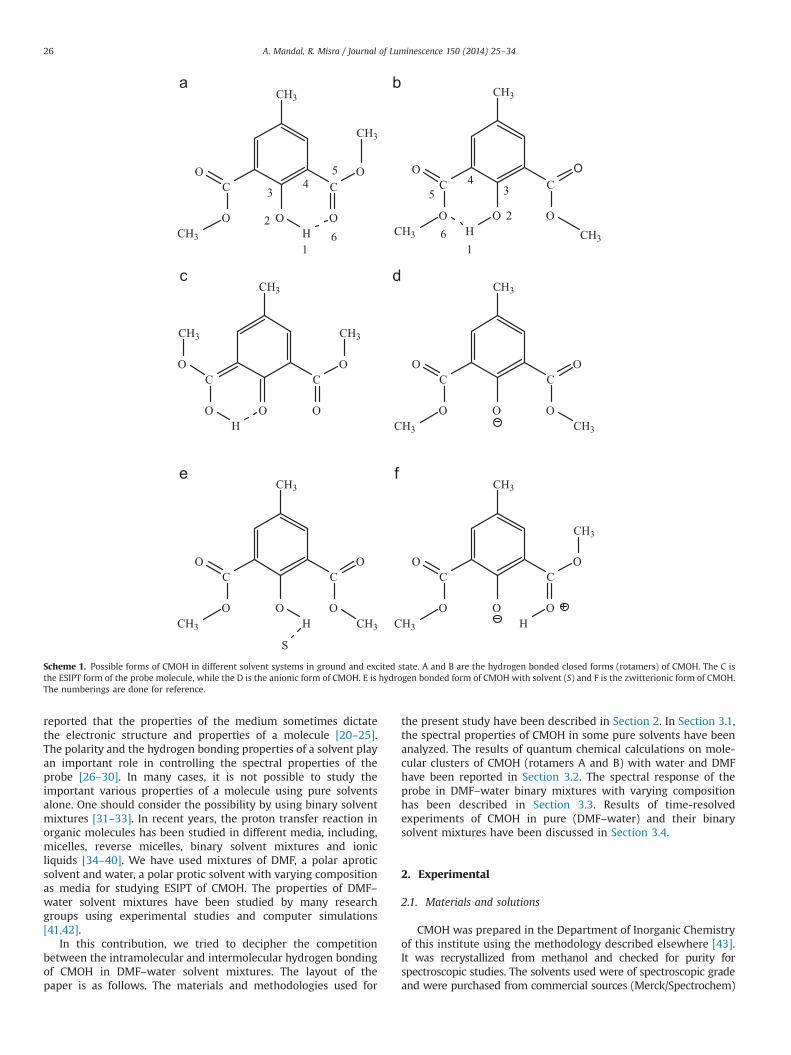
Scheme 1. Possible forms of CMOH in different solvent systems in ground and excited state. A and B are the hydrogen bonded closed forms (rotamers) of CMOH. The C isthe ESIPT form of the probe molecule, while the D is the anionic form of CMOH. E is hydrogen bonded form of CMOH with solvent (S) and F is the zwitterionic form of CMOH.The numberings are done for reference.
A. Mandal, R. Misra / Journal of Luminescence 150 (2014) 25–3426
and checked for residual fluorescence, if any, before use. Triplydistilled water was used through the experiment. Analytical gradeNaOH and triethyl amine (TEA) were used as base. To avoid the effectof concentration, very dilute solutions were used for all spectralmeasurements of the probe (2–3�10�5 mol L�1) and no concentra-tion dependence spectral response of CMOH has been found at thisrange. All the spectral data were collected at an ambient temperatureof 298 K.
2.2. Instruments
Absorption and emission spectra of CMOH were recorded indifferent media using a Shimadzu UV–vis Spectrophotometer,UV-2401 (PC) S220V and Fluoro Max 3 (Horiba Jobin Yvon)fluorescence spectrophotometer, respectively. The excited statelifetime measurements have been performed by using TimeCorrelated Single Photon Counting (TCSPC) technique. The sampleswere excited either using either 340 nm or 375 nm light andthe emissions were monitored at the corresponding steady stateemission maxima. The typical full width at half maximum (fwhm)of the system response was about 86 ps and the repetition rate of1 MHz. The detector used was a Hamamatsu MCP plate photo-multiplier (R3809U). The single photon counting technique com-prised of an Ortec 9327 discriminator (CFD, Tenelec TC 454) andFluoro Hub Single Photon Counting controller. The data werecollected using a DAQ card as a multichannel analyzer. Thefluorescence decays were analyzed by using the IBH DAS6 Soft-ware at Data Station v2.3. The quality of the fit was judged interms of weighted residuals, Durbin–Watson (DW) parameter andreduced χ2 values.
2.3. Quantum chemical calculations
The ground state equilibrium geometries of the 1:1 molecularclusters of CMOH (rotamers A and B) with DMF and water in thegas phase have been optimized using the Hartree–Fock (HF)theory and 6–31 G (d,p) basis set. The uses of diffuse functionsare necessary for accurate determination of energy, especially forthe excited states [39]. The density functional theory (DFT)calculations have been also performed to optimize the geometriesof these clusters. Becke's three parameter-Lee–Yang–Parr (B3LYP)hybrid functional and 6–31 G (d, p) basis set are used for DFTcalculations. The optimizations have been confirmed through thevibrational analyses of the optimized geometries, where nonegative frequencies have been found. Time dependent DFT(TDDFT) calculations have been undertaken to calculate the energy
and oscillator strengths of the first 10 vertical transition of theaforesaid cluster. The level of theory used has been reported to beadequate to investigate these types of systems [39]. All thecalculations were performed using standard quantum chemicalsoftware; Gaussian 03 [44] and visualization of molecular struc-tures are done with Molden [45] and Gausview [46] softwares.
3. Results and discussion
3.1. Steady state absorption and emission studies of CMOH in bulksolvents
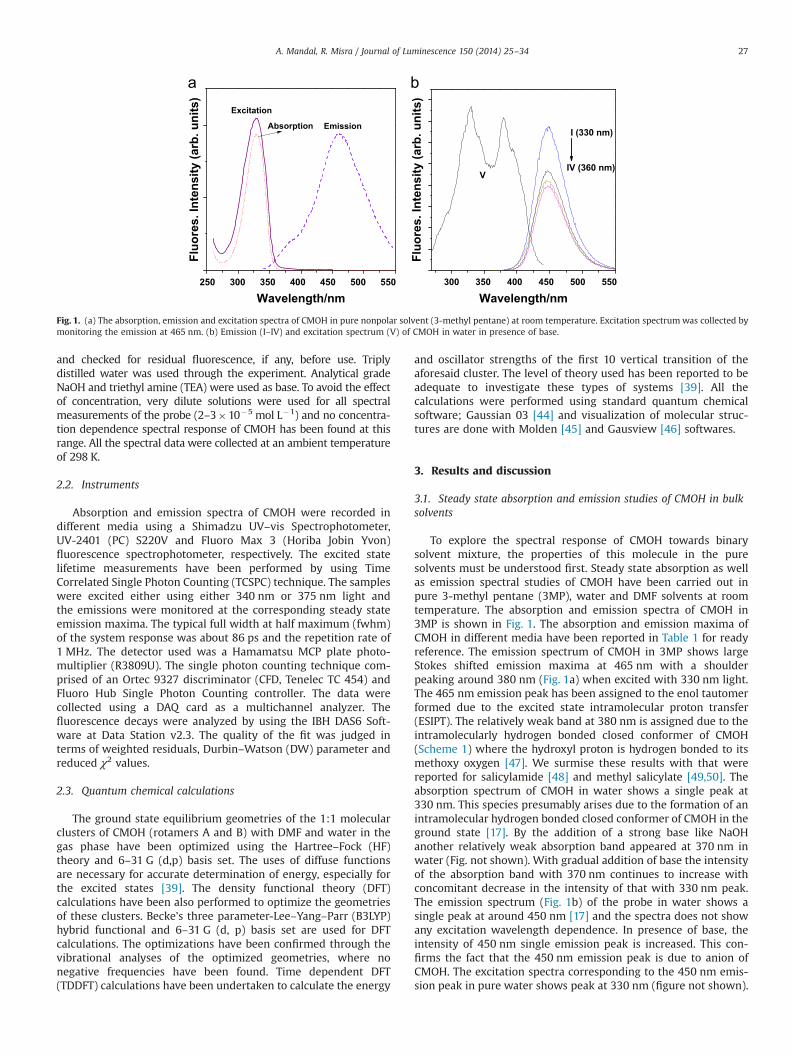
To explore the spectral response of CMOH towards binarysolvent mixture, the properties of this molecule in the puresolvents must be understood first. Steady state absorption as wellas emission spectral studies of CMOH have been carried out inpure 3-methyl pentane (3MP), water and DMF solvents at roomtemperature. The absorption and emission spectra of CMOH in3MP is shown in Fig. 1. The absorption and emission maxima ofCMOH in different media have been reported in Table 1 for readyreference. The emission spectrum of CMOH in 3MP shows largeStokes shifted emission maxima at 465 nm with a shoulderpeaking around 380 nm (Fig. 1a) when excited with 330 nm light.The 465 nm emission peak has been assigned to the enol tautomerformed due to the excited state intramolecular proton transfer(ESIPT). The relatively weak band at 380 nm is assigned due to theintramolecularly hydrogen bonded closed conformer of CMOH(Scheme 1) where the hydroxyl proton is hydrogen bonded to itsmethoxy oxygen [47]. We surmise these results with that werereported for salicylamide [48] and methyl salicylate [49,50]. Theabsorption spectrum of CMOH in water shows a single peak at330 nm. This species presumably arises due to the formation of anintramolecular hydrogen bonded closed conformer of CMOH in theground state [17]. By the addition of a strong base like NaOHanother relatively weak absorption band appeared at 370 nm inwater (Fig. not shown). With gradual addition of base the intensityof the absorption band with 370 nm continues to increase withconcomitant decrease in the intensity of that with 330 nm peak.The emission spectrum (Fig. 1b) of the probe in water shows asingle peak at around 450 nm [17] and the spectra does not showany excitation wavelength dependence. In presence of base, theintensity of 450 nm single emission peak is increased. This con-firms the fact that the 450 nm emission peak is due to anion ofCMOH. The excitation spectra corresponding to the 450 nm emis-sion peak in pure water shows peak at 330 nm (figure not shown).
250 300 350 400 450 500 550
Absorption
Fluo
res.
Inte
nsity
(arb
. uni
ts)
Wavelength/nm
Excitation
Emission
300 350 400 450 500 550
Fluo
res.
Inte
nsity
(arb
. uni
ts)
Wavelength/nm
I (330 nm)
IV (360 nm)V
Fig. 1. (a) The absorption, emission and excitation spectra of CMOH in pure nonpolar solvent (3-methyl pentane) at room temperature. Excitation spectrumwas collected bymonitoring the emission at 465 nm. (b) Emission (I–IV) and excitation spectrum (V) of CMOH in water in presence of base.
A. Mandal, R. Misra / Journal of Luminescence 150 (2014) 25–34 27

This indicates the formation of anion in the excited state in puresolvents, which was not present in the ground state. This forma-tion of anion in the excited state can be better understood usingthe change in pKa of CMOH (ΔpKa) due to electronic excitation.We can calculate the change in pKa in the excited state (pKn
a) tothat in the ground state (pKG
a ) using ‘Föster cycle’ using thefollowing equation [34]:
ΔpKa ¼ pKn
a�pKGa � �0:00207 νA00�νB00
� �ð1Þ
where, νA00 and νB00 are the electronic transitions (expressed inwavenumbers) of the acid and base, respectively. Using themaximum absorption energies of the normal (A or B form;Scheme 1) and anionic (D) forms of CMOH, which are S0-S1transitions [17], we can calculate the value of ΔpKa as �6.78. So,the decrease in the pKa value, that is increase in acidity in theexcited state facilitates the deprotonation of A or B form to formthe anion (D). The excitation spectrum in basic water shows dualpeaks at 330 and 370 nm when monitored at 450 nm as shown inFig. 1b. The ionization of CMOH in aqueous phase can be attributedto proton abstraction by OH� ions of water, followed by extrastability achieved through proton solvation in water by formingH9O4
þ complex [51]. A hydrogen bonded ‘first solvation shell’ ofthree water molecules is formed beyond the simple hydroxoniumion (H3Oþ). But the stable tetrahedral geometry (like H9O4
þ ion)cannot be formed in case of DMF solvent due to its large size.
The absorption spectra of CMOH in DMF show a strong bandwith a maximum at 330 nm with two shoulders at 375 nm and430–450 nm regions as shown in Fig. S1(a) in Supplementaryinformation. Like 4-methyl-2,6-diformylphenol (MFOH), theabsorption peak of CMOH at 330 nm can be assigned to itsintramolecularly hydrogen bonded closed conformer [17]. Theemission spectrum of CMOH in DMF [Fig. S1(b)] shows a veryinteresting behavior when the sample was excited at 330 nmwavelength. At this excitation wavelength a large Stokes shiftedpeak at around 462 nm, and a shoulder at around 380 nm areobserved and the spectrum is broad [Fig. S1(b)]. So even in purepolar aprotic solvent like DMF, 380 nm emission peak is observedin addition to the ESIPT band. The emission spectra of CMOH inpure DMF show excitation wavelength dependence. The broademission spectrum of CMOH in DMF indicates the presence ofmore than one species in the excited state of CMOH. The 462 nmpeak (λexc¼330 nm) is shifted to 430 nm with increase in excita-tion wavelength (λexc¼360 nm and 370 nm) as shown in Fig. S1(b). This phenomenon also indicates presence of more than one
excited state species of CMOH in DMF. The excitation spectra of462 nm emission show two bands with a strong peak at 330 nmand a relatively weaker peak at around 380 nm. Interestingly, wefound that by the addition of a strong base like TEA, the 375 nmabsorption band [Fig. S1(b)] in DMF increases with concomitantdecrease in the intensity of the 330 nm band. But the band at430–440 nm region in absorption spectrum remains unaffectedeven after the addition of TEA. So 430–440 nm shoulder in theabsorption profile can be attributed to the formation of solventseparated ion pair, in which the proton is transferred to the solventand resulting ions are linked electrostatically.
In presence of a strong base like TEA, the emission spectrum ofCMOH in DMF shows a peak around 450 nm when the solutionwas excited at 330 nm [Fig. S1(c)]. The emission spectra of CMOHin pure DMF and in the presence of TEA show excitation wave-length dependence. The 450 nm emission peak is shifted to430 nm by increasing the excitation wavelength in presence ofbase. By the addition of TEA, a peak at 380 nm in the excitationspectrum appears in addition to the 330 nm one when theemission was monitored at 450 nm. We also found that by gradualaddition of TEA, the intensity of the 380 nm excitation band isincreased. We also observed that if we add an acid like acetic acidto this resulting solution, the excitation spectrum with a max-imum at 380 nm gradually disappears with simultaneous increasein 330 nm band (not shown in Fig.). We can infer from theseobservations that the 380 nm band is obviously due to the anionof CMOH.
3.2. Electronic structure of CMOH-solvent clusters in the groundstate
It has been reported that CMOH forms hydrogen bondedmolecular cluster with solvents [17]. As only one proton can beabstracted from CMOH by the solvent and hydrogen bonding withthe media is the starting point of this, we have performedquantum chemical calculations to investigate the formation ofCMOH–Solvent hydrogen bonded 1:1 molecular clusters in theground state. The optimized structures have been shown in Fig. 2and Figs. (S2–S4) in Supporting Information. The important bondlengths of the aforesaid clusters have been reported in Table 2. Wecomputed the hydrogen bonding distances (rHB) with DMF for therotamers A and B of CMOH and which are 2.648 Å and 2.365 Å,respectively while that with water are 2.135 Å and 2.168 Å,respectively using Hartree–Fock theory. The trend in rHB obtainedfrom DFT calculations follows the similar trend. So, the computed
Table 1The room temperature steady state absorption (λabs) and emission maxima (λemm) of CMOH in DMF (D), water (W) and binary solvent mixtures inpresence and absence of base have been reported. The samples were excited at 330 nm and 370 nm and mentioned as subscripts to correspondingemission maxima. The mole fraction of DMF in the mixture (χP) and the static dielectric constants of the media (ε0) have also been reported.
Mediaa χP ε0b λabs (nm) λemm (nm)330 λemm (nm)370
DMF 1 39.88 330, 370, 435 462,380 430DMFþTEA – – 330, 370, 435 450 43090% Dþ10% W 0.69 46.21 330, 370, 435 430 43090% Dþ10% WþTEA – – 330, 370, 435 450 43060% Dþ40% W 0.27 63.09 330, 370, 435 460 43060% Dþ40% WþTEA – – 330, 370, 435 450 45050% Dþ50% W 0.20 67.90 330, 370, 435 450 43050% Dþ50% WþTEA – – 370, 435 450 45040% Dþ60% W 0.14 72.12 330, 370, 435 460 43040% Dþ60% WþTEA – – 370, 435 450 45010% Dþ90% W 0.027 76.79 330, 370, 435 450 43010% Dþ90% WþNaOH – – 370, 435 450 450Water 0 79.50 330 450 450Waterþbase – – 330,370 450 450
a TEA stands for triethyamine.b Static dielectric constant of DMF, water and their binary mixtures with varying composition at 25 1C have been taken from reference 39.
A. Mandal, R. Misra / Journal of Luminescence 150 (2014) 25–3428

hydrogen bonding distances between the CMOH and the solventindicate that the rotatmer B forms stronger hydrogen bonds thanrotamer A with the DMF but in case of water the reverse trend isobserved. TDDFT calculations have been performed to calculate theenergies and oscillator strengths of first ten vertical transitions of1:1 clusters of the rotamers A and B of CMOH with DMF and waterand the important results are displayed in Table 3. Comparing thevalues of experimental oscillator strength and absorption energy,it can be said that the absorption of light leads the probe moleculeto the S1 state from the ground state, from which the emissionoccurs. The calculated absorption maxima (λmax) of 1:1 CMOH–DMF and CMOH–water clusters are slightly blue shifted comparedto the experimentally recorded one due to effect of macroscopic
solvation. The values of contribution of dominant electronictransition obtained from TDDFT calculations also indicate thatthe HOMO to LUMO transition is the dominant transition in theCMOH–DMF and CMOH–water clusters and determines the elec-tronic absorption of these clusters. The molecular orbitals of theseclusters are shown in Figs. (3–4). In our previous studies we haveshown that the rotamers A and B of CMOH have comparableenergy and are indistinguishable in the electronic absorptionspectra [17]. Based on the steady state absorption and emissionspectra of CMOH and quantum chemical analysis, the differentforms of the probe observed in DMF and water are shown inScheme 2. In the ground state, hydrogen bonded 1:1 clusters ofCMOH (rotamers A and B) with coexisting DMF and gives rise tothe electronic absorption spectrum of the probe in this solvent.The solvated B form which cannot undergo ESIPT reaction isresponsible for the normal florescence (NF) of the probe. On theother hand, the solvated A form tautomerizes in the excited stateto give proton transferred C form which is responsible for theESIPT emission of CMOH in DMF.
3.3. Steady state absorption and emission studies of CMOHin DMF–water binary mixtures
The absorption spectra of CMOH in DMF–water mixtures withvarying compositions have been reported in Table 1. The volumepercentage of each of the solvents and the mole fractions (χp),which have been calculated using Eq. (2), are also reported in thesame table.
χp ¼nd
nwþndð2Þ
where, nd and nw are the mole numbers of the DMF and watersolvents present in the binary mixture respectively, have beenreported in the same table. The dielectric constants of DMF–watermixtures with varying composition have been studied by Mehro-tra and co-workers [41] using time domain reflectometry (TDF),has also been reported in Table 1. CMOH shows an absorptionmaximum at 330 nm both in DMF and water. The emission spectraof the probe in DMF–water binary mixtures with varying compo-sitions have been recorded and the emission maxima of the probein these media have been displayed in Table 1.
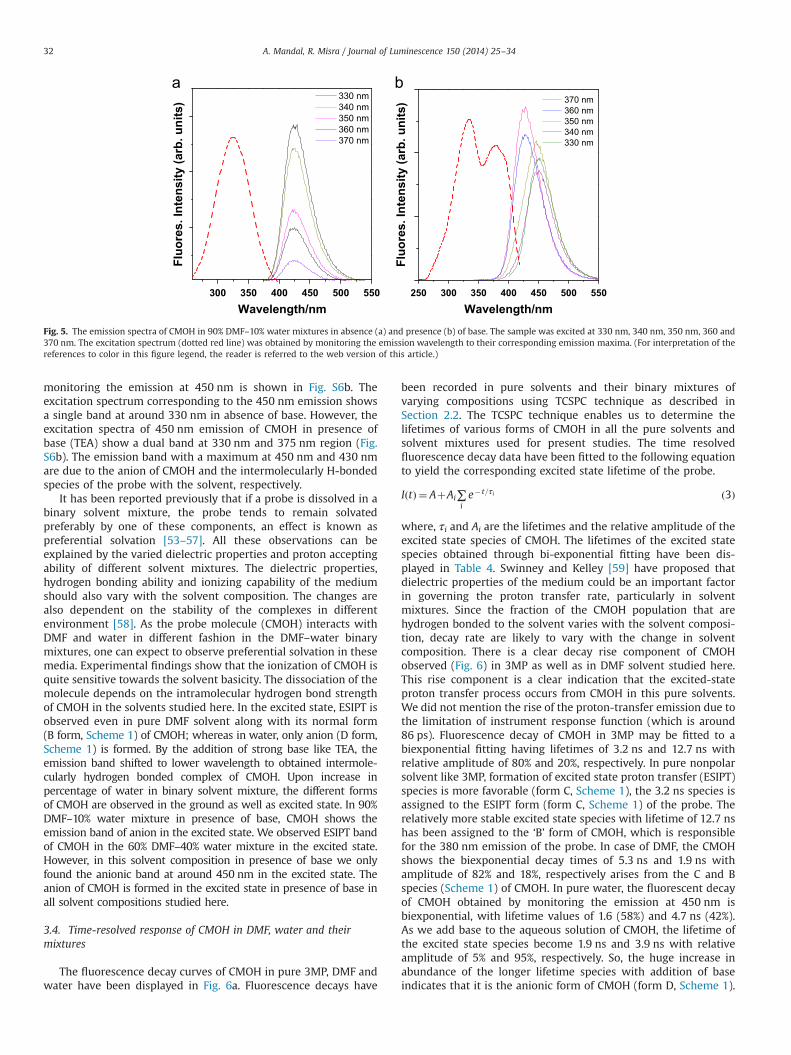
The emission spectra of CMOH in 90% DMF–10% water binarymixture (χ¼0.69) have been recorded and displayed in Fig. 5.Excitation at 330 nm shows a single peak in the emission bandpeak at 430 nm region. We did not find any excitation wavelengthdependency in the emission maxima of CMOH but there is atendency to decrease in emission intensity with increase inexcitation wavelength. We also have observed that even in pre-sence of 10% water the 380 nm emission peak (due to rotamer B;Scheme 1) vanishes. We also recorded the excitation spectrum of
Fig. 2. The ground state optimized geometry of CMOH (Rotamer A) – DMF 1:1molecular cluster as obtained using (a) density functional theory (B3LYP functional)(b) the Hartree–Fock theory and 6–31 G (d,p) basis set.
Table 2The important bond lengths (Angstrom) in 1:1 molecular cluster of CMOH(Rotamer A and Rotamer B) with DMF and water as calculated using DFT (B3LYPfunctional) and HF level of theory and 6–31 G** basis set.
Cluster and theory 1-2 2-3 3-4 4-5 5-6 6-1 HBa
CMOH(A)–DMF/HF 0.995 1.319 1.406 1.478 1.200 1.835 2.648CMOH(A)–DMF/DFT 0.986 1.331 1.425 1.471 1.230 1.788 2.334CMOH(B)–DMF/HF 0.950 1.324 1.404 1.488 1.326 1.861 2.365CMOH(B)–DMF/DFT 0.980 1.339 1.422 1.487 1.359 1.773 2.309CMOH(A)–water/HF 0.950 1.319 1.406 1.475 1.205 1.878 2.135CMOH(A)–water/DFT 0.982 1.333 1.425 1.470 1.232 1.826 2.048CMOH(B)–water/HF 0.947 1.332 1.402 1.488 1.335 1.802 2.168CMOH(B)–water/DFT 0.978 1.347 1.420 1.485 1.372 1.714 2.087
a The hydrogen bonding lengths of the cluster.
A. Mandal, R. Misra / Journal of Luminescence 150 (2014) 25–34 29

the probe in 90% DMF–10% water binary mixture by monitoringthe 430 nm emission and has been shown in the same figure (Fig.5a). The recorded excitation spectrum show peak at 330 nm,which indicates that all the species responsible for the emissionspectrum of the probe at χ¼0.69 originated in the ground state[52]. In 90% DMF–10% water binary mixture and in presence ofbase the probe shows emission peaks at 450 nm when the probewas excited at 330 nm. Upon excitation of the probe at 370 nm theemission maxima shifted to 430 nm with increase in emissionintensity compared to that of the spectra obtained at 330 nmexcitation. We have measured the excitation spectra of the probein 90% DMF–10% water binary mixture (in presence of base) bymonitoring the emission at 450 nm and the spectra is shown inFig. 5b. However, the excitation spectra of 450 nm emission ofCMOH in presence of base (TEA) show a dual band at 330 nm and380 nm region. The 450 nm emission is due to the anion of CMOHand the 430 nm peak is due to the intermolecularly hydrogenbonded species of the probe with the solvent.
The emission spectra of CMOH at 50% DMF–50% water binarymixture (χ¼0.20) is measured by exciting the solution mixturewith varying excitation wavelengths (Fig. S5 in Supplementary
information). At excitation wavelength of 330 nm at χ¼0.20, theemission spectrum of CMOH shows a maxima at 450 nm. Withincrease in excitation wavelength to 370 nm, the 450 nm emissionpeak gets blue-shifted to 430 nm. On the other hand, CMOH in 50%DMF–50% water binary mixture and in presence of base (TEA)shows emission peaks at 450 nm when the probe was excited at330 nm. We did not find any excitation wavelength dependencehere, except for only decreases in fluorescence intensity withincrease in the excitation wavelength in presence of base. We alsohave recorded the excitation spectrum of the probe in 50% DMF–50% water binary mixture by monitoring the emission at 450 nm.The excitation spectrum of 450 nm emission shows a single bandwith a maximum at 330 nm (Fig. S5a). On the other hand, inpresence of base at χ¼0.20, CMOH shows peak at around 380 nmalong with 330 nm in its excitation spectrum, when the emissionwas monitored at 450 nm (Fig. S5b). The emission peak at 450 nmregion is due to the anionic form of CMOH; whereas the 430 nmpeak is assigned to the intermolecularly H-bonded species ofCMOH (Scheme 1) with the solvent molecules.
The emission spectral behavior of the probe in 10% DMF–90%water binary mixtures (χ¼0.027) have been recorded and are
Table 3Electronic transition energy, oscillator strength and dominant transitions of 1:1 molecular cluster of CMOH (Rotamer A and Rotamer B) with DMF andwater as calculated using TD-DFT (B3LYP functional) and 6–31 G** basis set.
Cluster Transition λabs (eV) λabs (nm) f Dominant transitionsa
1. CMOH(A)–DMF S0-S1 3.954 313.5 0.14 [(H�3)�(Lþ1)]1, [H-L]2
S0-S2 4.680 265.0 0.00 [(H�1)�L]3
2. CMOH(B)–DMF S0-S1 4.084 303.5 0.12 [(H�2)–(Lþ1)]4, [H–L]5
S0-S2 4.794 259.0 0.0005 [(H�3)–L]6
3. CMOH(A)–water S0-S1 3.965 313.5 0.14 [(H�1)–(Lþ1)]7, [H–L]8
S0-S2 4.651 266.5 0.0005 [(H�2)–L]9, [(H�1)–L]10
4. CMOH(B)–water S0-S1 4.130 300.0 0.11 [(H�1)–(Lþ1)]11, [H–L]12
S0-S2 4.719 263.0 0.0002 [(H�3)–L]13, [(H�2)–L]14
a H¼HOMO and L¼LUMO. Contributions of transitions: 1: 0.17, 2: 0.64, 3: 0.70, 4: 0.16, 5: 0.65, 6: 0.63, 7: 0.15, 8: 0.65, 9: 0.65, 10: 0.37, 11: 0.21,12: 0.64, 13: -0.37 and 14: 0.52.
Fig. 3. The Molecular Orbital pictures of 1:1 molecular cluster of (a) Rotamer A and (b) Rotamer B of CMOH with DMF as obtained using DFT and 6–31 G (d,p) basis set.
A. Mandal, R. Misra / Journal of Luminescence 150 (2014) 25–3430
shown in Fig. S6a in Supplementary information. At excitationwavelength of 330 nm, the emission spectrum of CMOH at thiscomposition shows a peak at 450 nm along with a small shoulderat around 380 nm. This 380 nm band is due to another rotamer ofCMOH (form B, Scheme 1) which exists at this solvent composition[17]. Increasing the excitation wavelength to 370 nm shows a blue-shift of the emission peak at 450 nm to 430 nm. The excitationspectrum of CMOH at χ¼0.027 shows peaks at 330 nm, indicating
that all the species detected in the excited state at this composi-tion have been originated at the ground state. We also haveinvestigated the effect of base on the emission behavior of CMOHat χ¼0.027. The recorded spectra have been shown in Fig. S6b. Theemission spectrum shows a peak at around 450 nm when thesample was excited at 330 nm. At this stage, CMOH does not showany excitation wavelength dependent emission spectra. Excitationspectrum of the probe in 10% DMF–90% water binary mixture by
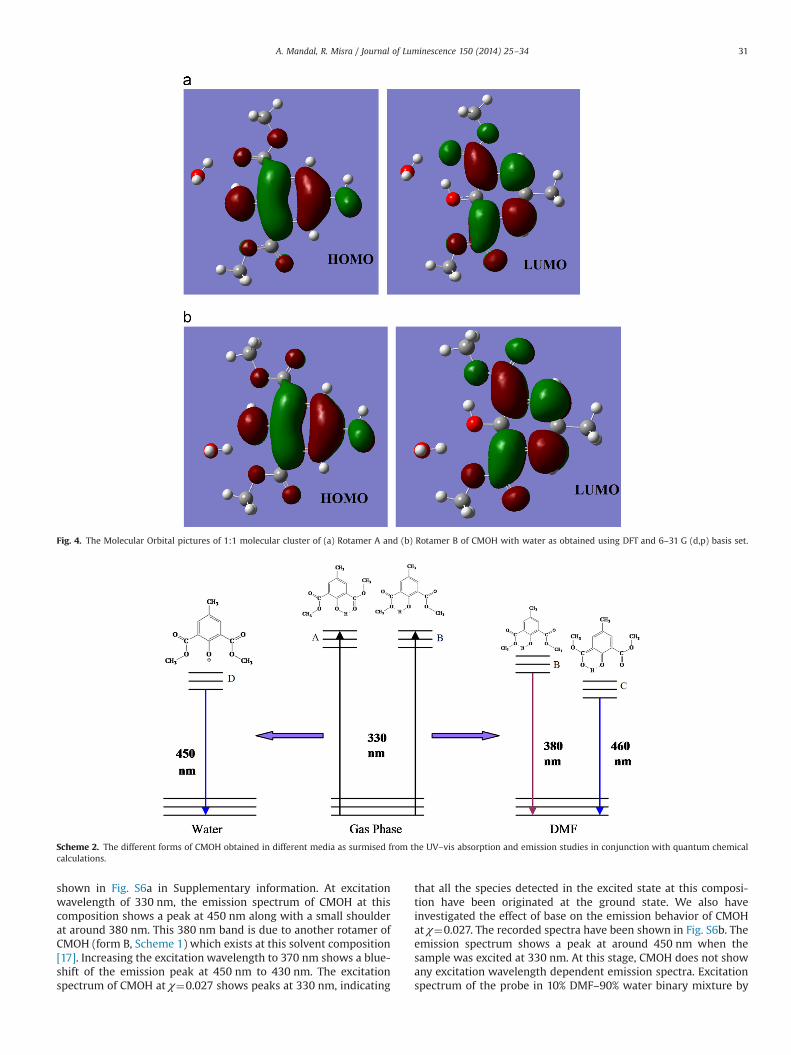
Fig. 4. The Molecular Orbital pictures of 1:1 molecular cluster of (a) Rotamer A and (b) Rotamer B of CMOH with water as obtained using DFT and 6–31 G (d,p) basis set.
Scheme 2. The different forms of CMOH obtained in different media as surmised from the UV–vis absorption and emission studies in conjunction with quantum chemicalcalculations.
A. Mandal, R. Misra / Journal of Luminescence 150 (2014) 25–34 31
monitoring the emission at 450 nm is shown in Fig. S6b. Theexcitation spectrum corresponding to the 450 nm emission showsa single band at around 330 nm in absence of base. However, theexcitation spectra of 450 nm emission of CMOH in presence ofbase (TEA) show a dual band at 330 nm and 375 nm region (Fig.S6b). The emission band with a maximum at 450 nm and 430 nmare due to the anion of CMOH and the intermolecularly H-bondedspecies of the probe with the solvent, respectively.
It has been reported previously that if a probe is dissolved in abinary solvent mixture, the probe tends to remain solvatedpreferably by one of these components, an effect is known aspreferential solvation [53–57]. All these observations can beexplained by the varied dielectric properties and proton acceptingability of different solvent mixtures. The dielectric properties,hydrogen bonding ability and ionizing capability of the mediumshould also vary with the solvent composition. The changes arealso dependent on the stability of the complexes in differentenvironment [58]. As the probe molecule (CMOH) interacts withDMF and water in different fashion in the DMF–water binarymixtures, one can expect to observe preferential solvation in thesemedia. Experimental findings show that the ionization of CMOH isquite sensitive towards the solvent basicity. The dissociation of themolecule depends on the intramolecular hydrogen bond strengthof CMOH in the solvents studied here. In the excited state, ESIPT isobserved even in pure DMF solvent along with its normal form(B form, Scheme 1) of CMOH; whereas in water, only anion (D form,Scheme 1) is formed. By the addition of strong base like TEA, theemission band shifted to lower wavelength to obtained intermole-cularly hydrogen bonded complex of CMOH. Upon increase inpercentage of water in binary solvent mixture, the different formsof CMOH are observed in the ground as well as excited state. In 90%DMF–10% water mixture in presence of base, CMOH shows theemission band of anion in the excited state. We observed ESIPT bandof CMOH in the 60% DMF–40% water mixture in the excited state.However, in this solvent composition in presence of base we onlyfound the anionic band at around 450 nm in the excited state. Theanion of CMOH is formed in the excited state in presence of base inall solvent compositions studied here.
3.4. Time-resolved response of CMOH in DMF, water and theirmixtures
The fluorescence decay curves of CMOH in pure 3MP, DMF andwater have been displayed in Fig. 6a. Fluorescence decays have
been recorded in pure solvents and their binary mixtures ofvarying compositions using TCSPC technique as described inSection 2.2. The TCSPC technique enables us to determine thelifetimes of various forms of CMOH in all the pure solvents andsolvent mixtures used for present studies. The time resolvedfluorescence decay data have been fitted to the following equationto yield the corresponding excited state lifetime of the probe.
IðtÞ ¼ AþAi∑ie� t=τi ð3Þ
where, τi and Ai are the lifetimes and the relative amplitude of theexcited state species of CMOH. The lifetimes of the excited statespecies obtained through bi-exponential fitting have been dis-played in Table 4. Swinney and Kelley [59] have proposed thatdielectric properties of the medium could be an important factorin governing the proton transfer rate, particularly in solventmixtures. Since the fraction of the CMOH population that arehydrogen bonded to the solvent varies with the solvent composi-tion, decay rate are likely to vary with the change in solventcomposition. There is a clear decay rise component of CMOHobserved (Fig. 6) in 3MP as well as in DMF solvent studied here.This rise component is a clear indication that the excited-stateproton transfer process occurs from CMOH in this pure solvents.We did not mention the rise of the proton-transfer emission due tothe limitation of instrument response function (which is around86 ps). Fluorescence decay of CMOH in 3MP may be fitted to abiexponential fitting having lifetimes of 3.2 ns and 12.7 ns withrelative amplitude of 80% and 20%, respectively. In pure nonpolarsolvent like 3MP, formation of excited state proton transfer (ESIPT)species is more favorable (form C, Scheme 1), the 3.2 ns species isassigned to the ESIPT form (form C, Scheme 1) of the probe. Therelatively more stable excited state species with lifetime of 12.7 nshas been assigned to the ‘B’ form of CMOH, which is responsiblefor the 380 nm emission of the probe. In case of DMF, the CMOHshows the biexponential decay times of 5.3 ns and 1.9 ns withamplitude of 82% and 18%, respectively arises from the C and Bspecies (Scheme 1) of CMOH. In pure water, the fluorescent decayof CMOH obtained by monitoring the emission at 450 nm isbiexponential, with lifetime values of 1.6 (58%) and 4.7 ns (42%).As we add base to the aqueous solution of CMOH, the lifetime ofthe excited state species become 1.9 ns and 3.9 ns with relativeamplitude of 5% and 95%, respectively. So, the huge increase inabundance of the longer lifetime species with addition of baseindicates that it is the anionic form of CMOH (form D, Scheme 1).
300 350 400 450 500 550
Fluo
res.
Inte
nsity
(arb
. uni
ts)
Wavelength/nm
330 nm 340 nm 350 nm 360 nm 370 nm
250 300 350 400 450 500 550
Fluo
res.
Inte
nsity
(arb
. uni
ts)
Wavelength/nm
370 nm 360 nm 350 nm 340 nm 330 nm
Fig. 5. The emission spectra of CMOH in 90% DMF–10% water mixtures in absence (a) and presence (b) of base. The sample was excited at 330 nm, 340 nm, 350 nm, 360 and370 nm. The excitation spectrum (dotted red line) was obtained by monitoring the emission wavelength to their corresponding emission maxima. (For interpretation of thereferences to color in this figure legend, the reader is referred to the web version of this article.)
A. Mandal, R. Misra / Journal of Luminescence 150 (2014) 25–3432
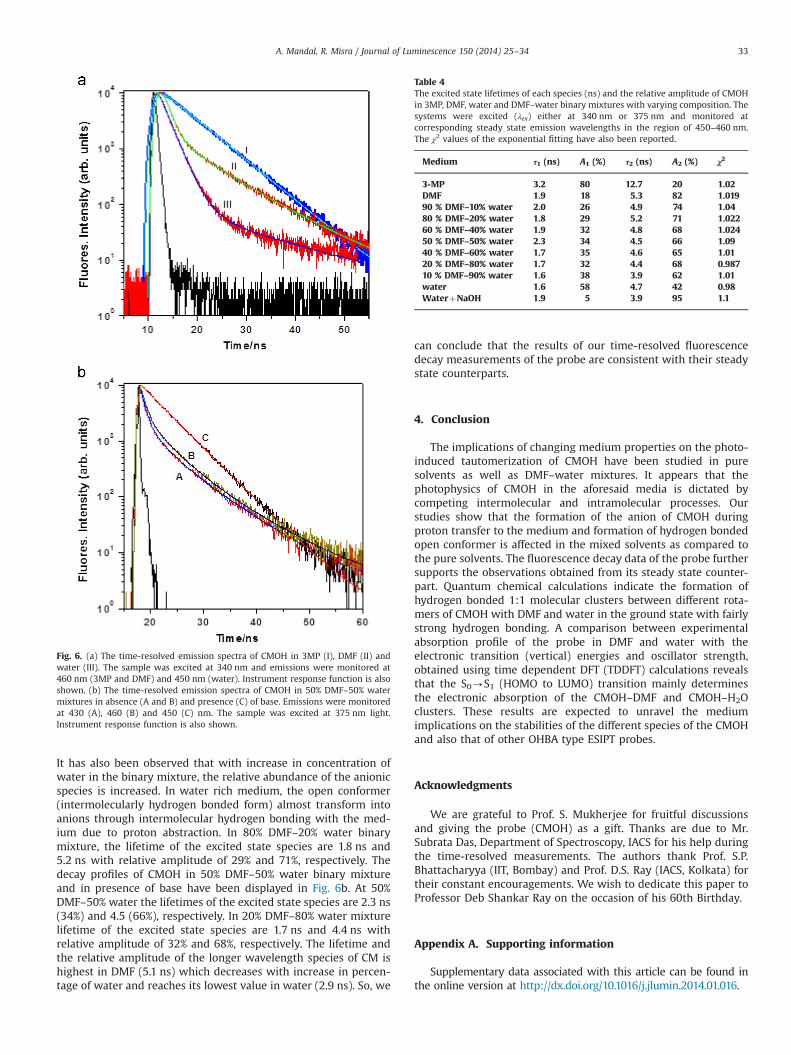
It has also been observed that with increase in concentration ofwater in the binary mixture, the relative abundance of the anionicspecies is increased. In water rich medium, the open conformer(intermolecularly hydrogen bonded form) almost transform intoanions through intermolecular hydrogen bonding with the med-ium due to proton abstraction. In 80% DMF–20% water binarymixture, the lifetime of the excited state species are 1.8 ns and5.2 ns with relative amplitude of 29% and 71%, respectively. Thedecay profiles of CMOH in 50% DMF–50% water binary mixtureand in presence of base have been displayed in Fig. 6b. At 50%DMF–50% water the lifetimes of the excited state species are 2.3 ns(34%) and 4.5 (66%), respectively. In 20% DMF–80% water mixturelifetime of the excited state species are 1.7 ns and 4.4 ns withrelative amplitude of 32% and 68%, respectively. The lifetime andthe relative amplitude of the longer wavelength species of CM ishighest in DMF (5.1 ns) which decreases with increase in percen-tage of water and reaches its lowest value in water (2.9 ns). So, we
can conclude that the results of our time-resolved fluorescencedecay measurements of the probe are consistent with their steadystate counterparts.
4. Conclusion
The implications of changing medium properties on the photo-induced tautomerization of CMOH have been studied in puresolvents as well as DMF–water mixtures. It appears that thephotophysics of CMOH in the aforesaid media is dictated bycompeting intermolecular and intramolecular processes. Ourstudies show that the formation of the anion of CMOH duringproton transfer to the medium and formation of hydrogen bondedopen conformer is affected in the mixed solvents as compared tothe pure solvents. The fluorescence decay data of the probe furthersupports the observations obtained from its steady state counter-part. Quantum chemical calculations indicate the formation ofhydrogen bonded 1:1 molecular clusters between different rota-mers of CMOH with DMF and water in the ground state with fairlystrong hydrogen bonding. A comparison between experimentalabsorption profile of the probe in DMF and water with theelectronic transition (vertical) energies and oscillator strength,obtained using time dependent DFT (TDDFT) calculations revealsthat the S0-S1 (HOMO to LUMO) transition mainly determinesthe electronic absorption of the CMOH–DMF and CMOH–H2Oclusters. These results are expected to unravel the mediumimplications on the stabilities of the different species of the CMOHand also that of other OHBA type ESIPT probes.
Acknowledgments
We are grateful to Prof. S. Mukherjee for fruitful discussionsand giving the probe (CMOH) as a gift. Thanks are due to Mr.Subrata Das, Department of Spectroscopy, IACS for his help duringthe time-resolved measurements. The authors thank Prof. S.P.Bhattacharyya (IIT, Bombay) and Prof. D.S. Ray (IACS, Kolkata) fortheir constant encouragements. We wish to dedicate this paper toProfessor Deb Shankar Ray on the occasion of his 60th Birthday.
Appendix A. Supporting information
Supplementary data associated with this article can be found inthe online version at http://dx.doi.org/10.1016/j.jlumin.2014.01.016.
Fig. 6. (a) The time-resolved emission spectra of CMOH in 3MP (I), DMF (II) andwater (III). The sample was excited at 340 nm and emissions were monitored at460 nm (3MP and DMF) and 450 nm (water). Instrument response function is alsoshown. (b) The time-resolved emission spectra of CMOH in 50% DMF–50% watermixtures in absence (A and B) and presence (C) of base. Emissions were monitoredat 430 (A), 460 (B) and 450 (C) nm. The sample was excited at 375 nm light.Instrument response function is also shown.
Table 4The excited state lifetimes of each species (ns) and the relative amplitude of CMOHin 3MP, DMF, water and DMF–water binary mixtures with varying composition. Thesystems were excited (λex) either at 340 nm or 375 nm and monitored atcorresponding steady state emission wavelengths in the region of 450–460 nm.The χ2 values of the exponential fitting have also been reported.
Medium τ1 (ns) A1 (%) τ2 (ns) A2 (%) χ2
3-MP 3.2 80 12.7 20 1.02DMF 1.9 18 5.3 82 1.01990 % DMF–10% water 2.0 26 4.9 74 1.0480 % DMF–20% water 1.8 29 5.2 71 1.02260 % DMF–40% water 1.9 32 4.8 68 1.02450 % DMF–50% water 2.3 34 4.5 66 1.0940 % DMF–60% water 1.7 35 4.6 65 1.0120 % DMF–80% water 1.7 32 4.4 68 0.98710 % DMF–90% water 1.6 38 3.9 62 1.01water 1.6 58 4.7 42 0.98WaterþNaOH 1.9 5 3.9 95 1.1
A. Mandal, R. Misra / Journal of Luminescence 150 (2014) 25–34 33

References
[1] C. Reichardt, T. Welton, Solvents and Solvent Effects in Organic Chemistry,4th Ed., Wiley, 2011.
[2] A. Migani, L. Blancafort, M.A. Robb, A.D. DeBellis, J. Am. Chem. Soc. 130 (2008)6932.
[3] W.S. Yu, C.C. Cheng, Y.M. Cheng, P.C. Wu, Y.H. Song, Y. Chi, P.T. Chou, J. Am.Chem. Soc. 125 (2003) 10800.
[4] L.M. Tolbert, K.M. Solntsev, Acc. Chem. Res. 35 (2002) 19.[5] R. Misra, D.K. Maity, S.P. Bhattacharyya, Chem. Phys. 425 (2013) 148.[6] R. Yang, S.G. Schulman, Luminescence 20 (2005) 358.[7] A. Sytnik, M. Kasha, Proc. Nat. Acad. Sci. USA 91 (1994) 8627.[8] A. Jarczewski, C.D. Hubbard, J. Mol. Struct. 649 (2003) 287.[9] M. Eigen, Angew. Chem. Int. Ed. 3 (1994) 1.[10] M.J. Cox, R.L.A. Timmer, H.J. Bakker, S. Park, N. Agmon, J. Phys. Chem. A
113 (2009) 6599.[11] E.L. Roberts, J. Dey, I.M. Warner, J. Phys. Chem. A 101 (1997) 5296.[12] B. Bhattacharya, A. Samanta, J. Phys. Chem. B 112 (2008) 10101.[13] D. Stoner-Ma, A.A. Jaye, K.L. Ronayne, J. Nappa, S.R. Meech, P.J. Tonge, J. Am.
Chem. Soc. 130 (2008) 1227.[14] J. Qian, A.M. Brouwer, Phys. Chem. Chem. Phys. 12 (2010) 12562.[15] D.C. Luhrs, R. Knochenmuss, I. Fischer, Phys. Chem. Chem. Phys. 2 (2000) 4335.[16] M. Barbatti, A.J.A. Aquino, H. Lischka, C. Schriever, S. Lochbrunner, E. Riedle,
Phys. Chem. Chem. Phys. 11 (2009) 1406.[17] R. Misra, S.P. Bhattacharyya, A. Mandal, J. Phys. Chem. B 115 (2011) 11840.[18] A. Mandal, D. Guha, S. Mukherjee, J. Mol. Spectrosc. 210 (2001) 183.[19] R. Misra, A. Mandal, M. Mukhopadhyay, D.K. Maity, S.P. Bhattacharyya, J. Phys.
Chem. B 113 (2009) 10779.[20] M. Maroncelli, J. MacInnis, G.R. Fleming, Science 243 (1989) 1674.[21] A. Weigel, N.P. Ernsting, J. Phys. Chem. B 114 (2010) 7879.[22] J.R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd Ed., Springer,
New York, 2006.[23] S.I. Druzhinin, S.A. Kovalenko, T.A. Senyushkina, A. Demeter, K.A. Zachariasse,
J. Phys. Chem. A 114 (2010) 1621.[24] S. Gao, L. Hao, J. Li, P. Lin, D. Li, S. Shuang, C. Dong, Luminescence 28 (2013)
412.[25] R. Misra, D.K. Maity, S.P. Bhattacharyya, Chem. Phys. 402 (2012) 96.[26] R. Misra, S. Kar, Chem. Phys. 397 (2012) 65.[27] P. Dahiya, M. Kumbhakar, D.K. Maity, T. Mukherjee, A.B.R. Tripathy,
N. Chattopadhyay, H. Pal, J. Photochem. Photobiol. 181 (2006) 338.[28] A. Sarkar, T.K. Mandal, D.K. Rana, S. Dhar, S. Chall, S.C. Bhattacharya, J. Lumin.
130 (2010) 2271.[29] J.M. Lee, J.M. Prausnitz, Chem. Phys. Lett. 492 (2010) 55.[30] S.K. Biring, R. Sharma, R. Misra, P. Chaudhury, J. Clust. Sci. 24 (2013) 715.[31] D. Chakrabarty, A. Chakraborty, P. Hazra, D. Seth, N. Sarkar, Chem. Phys. Lett.
397 (2004) 216.[32] A.K. Laha, P.K. Das, S. Bagchi, J. Phys. Chem. A 106 (2002) 3230.
[33] D. Mandal, S.K. Pal, K. Bhattacharyya, J. Phys. Chem. A 102 (1998) 9710.[34] A. Kyrychenko, J. Herbich, M. Izydorzak, F. Wu, R.P. Thummel, J. Waluk, J. Am.
Chem. Soc. 121 (1999) 11179.[35] A. Mironczyk, A. Jankowski, J. Photochem. Photobiol. A 153 (2002) 89.[36] D. Banerjee, S.K. Pal, J. Phys. Chem. B 112 (2008) 7314.[37] F. Gao, X. Ye, H. Li, X. Zhong, Q. Wang, Chem. Phys. Chem. 13 (2012) 1313.[38] M.K. Nayak, B.-H. Kim, J.E. Kwon, S. Park, J. Seo, J.W. Chung, S.Y. Park, Chem. –
A Eur. J. 16 (2010) 7437.[39] M.K. Nayak, J. Photochem. Photobiol. A 241 (2012) 26.[40] K. Suda, M. Terazima, Y. Kimura, Chem. Phys. Lett. 531 (2012) 70.[41] A.C. Kumbhakarne, S.M. Puranik, S.C. Mehrotra, J. Solun. Chem 23 (1993) 219.[42] G.-Z. Jia, K.-M. Huang, L.-J. Yang, X.-Q. Yang, Int. J. Mol. Sci. 10 (2009) 1590.[43] S.K. Mandal, K. Nag, J. Org. Chem. 91 (1986) 3900.[44] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman
, J.A. Montgomery, T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar,J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega,G.A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda,J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene,X. Li, J.E. Knox, H.P. Hratchian, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo,R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli,J.W. Ochterski, P.Y. Ayala, K. Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg,V.G. Zakrzewski, S. Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick,A.D. Rabuck, K. Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul,S. Clifford, J. Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz,I. Komaromi, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng,A. Nanayakkara, M. Challacombe, P.M. Gill, B. Johnson, W. Chen, M.W. Wong,C. Gonzalez, J.A. Pople, Gaussian 03, Revision E.01, Gaussian, Inc., WallingfordCT, 2004.
[45] G. Schaftenaar, J.H. Noordik, J. Comp. – Aided. Mol. Des. 14 (2000) 123.[46] Roy Dennington, Todd Keith, John Millam, GaussView, Version 5, Semichem
Inc., Shawnee Mission, KS, 2009.[47] A. Mandal, D.N. Nath, S. Mukherjee, S. Mitra, R. Das, J. Chem. Phys. 117 (2002)
5280.[48] A.U. Acuna, A. Costela, J.M. Munoj, J. Phys. Chem. 90 (1986) 2807.[49] A.U. Acuna, F. Amat, J. Catalan, F. Gonzalez, J. Phys. Chem. 84 (1980) 629.[50] K.K. Smith, K.J. Kaufmann, J. Phys. Chem. 85 (1981) 2895.[51] R.A. Moore, J. Lee, G.W. Robinson, J. Phys. Chem. 89 (1985) 3648.[52] S. Mitra, R. Das, D. Guha, S. Mukherjee, Spectrochim. Acta A 54 (1998) 1073.[53] A. Chandra, B. Bagchi, J. Mol. Liq. 57 (1993) 39.[54] T. Molotsky, D. Huppert, J. Phys. Chem. A 107 (2003) 8449.[55] N. Agmon, J. Phys. Chem. A 106 (2002) 7256.[56] B.B. Raju, S.M.B. Costa, Phys. Chem. Chem. Phys. 1 (1999) 3539.[57] A. Henseler, M. von Raumer, P. Suppan, J. Chem. Soc. Faraday Trans. 92 (1996)
391.[58] N. Agmon, D. Huppert, A. Masad, E. Pines, J. Phys. Chem. 95 (1991) 10407.[59] T.C. Swinney, D.F. Kelley, J. Phys. Chem. 95 (1991) 10369.
A. Mandal, R. Misra / Journal of Luminescence 150 (2014) 25–3434