probing the early temporal and spatial interaction of the sindbis
TRANSCRIPT

Probing the Early Temporal and Spatial Interaction of the SindbisVirus Capsid and E2 Proteins with Reverse Genetics
Jonathan E. Snyder,a Christian J. Berrios,a* Thomas J. Edwards,a Joyce Jose,a Rushika Perera,a and Richard J. Kuhna,b
Markey Center for Structural Biology, Department of Biological Sciences, Purdue University, West Lafayette, Indiana, USA,a and Bindley Bioscience Center, PurdueUniversity, West Lafayette, Indiana, USAb
A 7-Å cryoelectron microscopy-based reconstruction of Sindbis virus (SINV) was recently generated. Fitting the crystal structureof the SINV capsid protein (Cp) into the density map revealed that the F2-G2 loop of the Cp was shifted away from cytoplasmicdomain of E2 (cdE2) in the 7-Å reconstruction relative to its position in the Cp crystal structure. Furthermore, the reconstruc-tion demonstrated that residue E395 in region I of the cytoplasmic domain of the E2 envelope protein (cdE2-RI) and K252 of Cp,part of the Cp F2-G2 loop, formed a putative salt bridge in the virion. We generated amino acid substitutions at residues K250and K252 of the SINV Cp and explored the resulting phenotypes. In the context of cells infected with wild-type or mutant virus,reversing the charge of these two residues resulted in the appearance of Cp aggregates around cytopathic vacuole type I (CPV-I)structures, the absence of nucleocapsid (NC) formation, and a lack of virus particle release in the infected mammalian cell. How-ever, expressing the same Cp mutants in the cell without the envelope proteins or expressing and purifying the mutants from anEscherichia coli expression system and assembling in vitro yielded NC assembly in all cases. In addition, second-site mutationswithin cdE2 restored NC assembly but not release of infectious particles. Our data suggest an early temporal and spatial interac-tion between cdE2-RI and the Cp F2-G2 loop that, when ablated, leads to the absence of NC assembly. This interaction also ap-pears to be important for budding of virus particles.
The alphaviruses are a group of enveloped, positive-sense, sin-gle-stranded RNA viruses in the Togaviridae family. Well-
studied members include the Eastern, Western, and Venezuelanequine encephalitis viruses, Semliki Forest virus, Ross River virus,Chikungunya virus, and the type member, Sindbis virus (SINV)(42). The emergence of Chikungunya virus as an epidemic threathas reemphasized the medical importance of the alphaviruses(47).
The �11.5-kb genome encodes four nonstructural proteinsthat primarily facilitate genome replication and four structuralproteins, capsid, pE2, 6K, and E1, which are initially translatedfrom a subgenomic message as a polyprotein (42). Co- and post-translational processing by viral and host proteases yields the ma-ture structural proteins. The capsid protein (Cp) is an autopro-tease that acts to cotranslationally cleave itself from the growingpolypeptide (30) and later associates with viral genomic RNA toform a nucleocapsid (NC) core. The remaining polyprotein in-serts into the endoplasmic reticulum (ER) in a signal sequence-mediated manner (2). Signalase processing yields pE2 and E1, theenvelope proteins, and 6K, a small membrane-resident proteinwhose function in the virus life cycle remains unclear (26). pE2 islater cleaved by furin to yield the small protein E3 and the largerprotein E2 (52). E1 and E2 are type I integral membrane proteins,with E2 primarily responsible for host receptor engagement (3,24), while E1 contains pH-dependent fusogenic properties re-quired for virus entry (14, 50). The ectodomains of both E1 and E2comprise the bulk of each protein, although both E1 and E2 con-tain some residues, termed cytoplasmic domains, which are foundon the inner face of the envelope and, at least in the case of E2,interact with the NC (15, 17, 28).
The icosahedrally arranged virus particle, released by buddingfrom the host plasma membrane (PM), is comprised of two pro-tein shells (4). In the outer shell, 240 copies each of E1 and E2,arranged as 80 trimers of E1-E2 heterodimers, are embedded in a
plasma membrane-derived lipid bilayer (33). The membranelayer also contains substoichiometric amounts of 6K and sur-rounds the NC core. The NC is comprised of 240 copies of Cp andone copy of genomic RNA and also demonstrates icosahedralsymmetry (4). The icosahedral symmetry of the NC may derivefrom the 1:1 protein-protein interaction between each moleculeof Cp in the NC and a molecule of E2 in the envelope, as theicosahedral arrangement of prebudded, cytoplasmic NCs remainsloosely defined (4, 12, 32).
The alphaviruses have long proven a model system for struc-tural studies of enveloped mammalian viruses owing, in part, tothe T�4 icosahedral arrangement of both the envelope and theNC core of budded virus particles. Genetic and structural evidencesuggests that a hydrophobic pocket on the face of the Cp interactswith the cytoplasmic domain of E2 (cdE2) in the context of abudded virion (36, 38, 44). Specifically, a conserved YxL motif incdE2 is involved in side-chain interactions with residues residentin the Cp hydrophobic pocket, and other evidence suggests inter-actions of C-terminal cdE2 residues, relative to the YxL domain,with Cp (23, 25, 31, 36, 51). Thus, Cp and E2 interact duringbudding, and this interaction is required for budding to occur (43,53). However, details regarding the arrangements of, and the in-teractions between, the Cp and the E1 and E2 envelope proteinsprior to budding events remain unclear.
Received 18 May 2012 Accepted 30 August 2012
Published ahead of print 5 September 2012
Address correspondence to Richard J. Kuhn, [email protected].
* Present address: Christian J. Berrios, Harvard Medical School, Boston,Massachusetts, USA.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01220-12
12372 jvi.asm.org Journal of Virology p. 12372–12383 November 2012 Volume 86 Number 22
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.

It is thought that the trimer of E1-E2 heterodimers, often re-ferred to as spikes, forms a two-dimensional (2D) crystalline arrayat the plasma membrane and probably already arrives at theplasma membrane in a spike-like conformation (20, 41, 49). How-ever, the site of the initial E1-E2 heterodimerization, as well as thesubcellular compartment in which trimerization occurs, is notclear, although these processes likely occur soon after translation(34). Likewise, a detailed understanding of the NC assembly path-way is not available. While Cp homodimers are thought to be keyassembly intermediates, and charge neutralization betweengenomic RNA and the basic Cp likely provides the source for theinitial assembly nucleation, no NC assembly intermediates havebeen identified by either biophysical or microscopy-based tech-niques (27, 46). Additionally, the site of NC assembly is unclear,although NC may assemble at or near the site of viral genomereplication (11, 40). Interestingly, Cp mutants have been de-scribed in which NC formation in the cytoplasm is disrupted butin which wild-type (wt)-like virus particles are nevertheless bud-ded from the cell (9).
While it has long been established that NCs and envelope pro-tein spikes interact during budding at the plasma membrane,emerging data suggest that the interaction of NC and envelopeproteins may occur before either arrives at the plasma membrane,in the form of membranous virus-induced type II cytopathic vac-uoles (CPV-II) (16). These CPV-II molecules have been impli-cated in the traffic of envelope proteins to the plasma membraneand appear to be decorated with NCs (19, 41). The precise originof CPV-II vacuoles and the temporal and spatial aspects of theinitial interaction between NC and envelope proteins on theseCPV-II structures have yet to be detailed. However, the cotraffick-ing of NC and E2 to the site of budding hints at interactions be-tween Cp and the envelope proteins well before virus budding atthe plasma membrane.
A 7-Å cryoelectron microscopy (cryo-EM)-based reconstruc-tion of SINV virus demonstrated several novel molecular linksbetween the E2 envelope protein and the NC in the context of avirus particle (44). Among these data, it was shown that a flexibleloop on the edge of the hydrophobic pocket of Cp, the F2-G2 loopcomposed of amino acids 248-NSKGK-252, was shifted �3.5 Åaway from cdE2 relative to the loop position in the SINV Cp crys-tal structure. Furthermore, a salt bridge between cdE2 E395 andCp K252, part of the F2-G2 loop, was proposed. On the basis ofthose results and the observation that Cp residue K250 was impli-cated in cross-capsomere contacts (45), we performed a muta-tional analysis of Cp residues K250 and K252.
Simultaneous mutation of SINV Cp residues K250 and K252led to production of Cp aggregates around cytopathic type I rep-lication vacuoles (CPV-I) and the complete abrogation of NC for-mation in the cell concomitant with a lack of virus particle release.Surprisingly, though, when the mutant proteins were expressedand assembled in vitro (45), expressed in the absence of the enve-lope proteins, or expressed via a replicon or by a mammalianexpression plasmid, formation of core-like particles (CLPs) wasdetected in all cases. Double SINV mutants were generated inwhich both cdE2-RI (cdE2 391-KARRE-395) and the Cp residuesK250 and K252 were simultaneously mutated. These double mu-tants restored NC formation for the Cp K250D/K252D andK250E/K252E mutant backgrounds but did not restore the releaseof virions.
Taken together, our data suggest (i) that an early temporal and
spatial interaction occurs between Cp and E2 in the infected cell,likely at or near the CPV-I structure, (ii) validation of the model-ing performed in the SINV 7-Å reconstruction with regard to afunctional interaction between cdE2-RI and the Cp F2-G2 loop,and (iii) a new set of cdE2:Cp amino acid interactions, involvingthe N-terminal region of cdE2, that are important for alphavirusassembly and budding.
MATERIALS AND METHODSCell culture. BHK-15 cells obtained from the American Type CultureCollection were maintained in minimal essential medium (MEM) (LifeTechnologies) supplemented with 10% fetal bovine serum (FBS), exceptfor enzyme-linked immunosorbent assay (ELISA) studies in which theFBS was omitted.
Generation of mutants in virus cDNA. All mutants were generated inthe pToto64 SINV cDNA plasmid using standard overlap PCR mutagen-esis with Platinum Pfx DNA polymerase (Invitrogen). Infectious full-length RNA was produced by linearizing the plasmids with SacI followedby SP6-mediated in vitro transcription (Ambion). The Cp-expressing rep-licon was created in the pToto64 background by deleting the nucleotidescorresponding to E3, E2, 6K, and E1 via overlap PCR mutagenesis. Thefirst nine nucleotides of E3 were retained in addition to the stop codonutilized by E1. Plaque diameter analysis was performed by incubatingBHK cells seeded in 6-well culture dishes with 1.5 ml of 0.2 mg/ml DEAEdextran–50 mM Tris (pH 7.4) at 37°C for 1 h, followed by incubation within vitro-transcribed RNA at 25°C for 30 min and subsequent overlay with1% agarose–MEM.
NC accumulation assays. BHK cells were electroporated (1.5 kV, 25�F, 200 �, 0.2-cm-gap cuvette) with 10 �g of in vitro-transcribed RNAsderived from the Toto64 cDNA clones. Infection was allowed to proceedfor 8 h, at which time media were removed and cells washed 3 times withphosphate-buffered saline (PBS) and then scraped off the cell culture dishusing 1 ml PBS. Cells were subjected to pellet formation for 10 min at750 � g and resuspended in hypotonic buffer (10 mM NaCl, 10 mM Tris[pH 7.4], 20 mM EDTA) to swell for 20 min on ice. Triton X-100 (20%)was added to achieve a final concentration of 4% (vol/vol). Lysed celldebris and nuclei were subjected to pellet formation at 2,000 � g for 10min. The solution was then loaded onto a continuous iodixanol gradient(0% to 30%) in TNE-Triton X-100 (50 mM Tris [pH 7.4], 100 mM NaCl,1 mM EDTA, 0.1% Triton X-100) and centrifuged at 32,000 rpm in aBeckman SW-41 rotor for 2.5 h. All steps were carried out at 4°C. Thegradient was fractionated, and each fraction was analyzed by SDS-PAGEon a 12% polyacrylamide gel. Separated proteins were electroeluted andadsorbed to 0.4-�m-pore-size nitrocellulose membranes before beingprobed with a rabbit polyclonal anti-Cp primary antibody and an IRDye680 goat anti-rabbit secondary antibody conjugate (Li-COR). Stainedmembranes were scanned with a Li-COR Odyssey infrared imager. Cpintensities were measured using the Odyssey software and relative inten-sity values plotted as a function of fraction number.
In vitro NC assembly. The wild-type or mutant Cp genes were sub-cloned into pET-11a prokaryotic expression vector (Novagen). Proteinexpression and purification and in vitro assembly of NCs were performedas described previously (45), using an arbitrary single-stranded DNA(ssDNA) 109-mer (5=-GATCCGAAAGCGCGCCGTGAGTGCCTGACGCCATACGCCCTGGCCCCAAACGCCGTAATCCCAGCGGCCGCTGCCGCCGCGTGCTGCGTTAGGTCGGCCAATGCTTAGC-3=).
Transient transfection and expression of SINV Cp or Cp plus enve-lope proteins. The SINV sequences encoding the structural proteins (Cp,pE2, 6K, E1), or the sequences encoding just the Cp, were cloned intopcDNA4/TO/myc/his/A mammalian expression vector (Invitrogen).BHK cells were seeded into 60-mm-diameter culture dishes at �70%confluence and then transiently transfected with 10 �g of plasmid withLipofectamine (Invitrogen) 18 h prior to lysis and gradient analysis asdescribed above.
Early Temporal Interaction of Sindbis Capsid and E2
November 2012 Volume 86 Number 22 jvi.asm.org 12373
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.

Multistep growth curve analysis. BHK cells were infected with stockvirus at a multiplicity of infection (MOI) of 0.5. Virus was allowed toadsorb to cells at 25°C for 1 h followed by incubation at 37°C. Media wereremoved and replaced with fresh media at 30-min intervals for the first 2h and then at 1-h intervals thereafter. The amount of infectious virusrelease was quantified by titration on BHK cells.
Plaque assay. Serial dilutions (10-fold) of culture media or virus weremade in PBS containing 1% FBS plus 10 mM CaCl2 and 10 mM MgCl2.BHK cells at �90% confluence were inoculated with 250 �l of the dilu-tions for 1 h at room temperature and then overlaid with MEM containing5% FBS and 1% agarose. Cells were incubated at 37°C for 48 h and stainedwith neutral red.
ELISA. Posttransfection media (2 ml) were concentrated to 100 �lusing Amicon 100-kDa centrifugal filters and applied to a 96-well Poly-sorp ELISA plate (Sigma) overnight at 4°C. The samples were blockedwith 5% BSA–PBS for 2 h followed by three washes with PBS-T (PBS,0.1% Tween 20). A 1:500 dilution of a monoclonal anti-E2 antibody (202)was applied to the sample for 2 h, followed by three washes with PBS-T. A1:1,500 dilution of a horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody was applied to the sample for 1 h, followed bythree PBS-T washes. A TMB peroxidase detection system (KPL) was usedto develop the ELISA according to the manufacturer’s instructions. Theoptical density (OD) was determined at 450 nm.
Electron microscopy analyses. BHK cells were electroporated with invitro-transcribed RNAs or transfected (Lipofectamine 2000) with plas-mids expressing the viral Cp or structural polyprotein. Cells were fixedand processed at 8 h postelectroporation of RNA or 18 h posttransfectionof plasmid. Samples were fixed in 2% glutaraldehyde– cacodylate buffer(0.1 M sodium cacodylate, 2 mM MgCl2, 2 mM CaCl2, 0.5% NaCl, pH7.4) and then washed in cacodylate buffer followed by water. Sampleswere then postfixed with 2% OsO4 and 1.5% K4Fe(CN)6 and then rinsedwith water. Processing of cells for immunostaining omitted the osmiumtetroxide and potassium ferrocyanide. Cells were subjected to pellet for-mation at 4,000 � g in 1% low-melt agarose. The pellet was dehydrated insequential concentrations of ethanol from 10% to 100%. Pellets were thenembedded in Durcupan resin and hardened at 60°C for 72 h. Sections (50to 70 nm thick) were collected on carbon-coated copper mesh grids (car-bon-coated gold mesh grids for immunostaining) using a diamond knife.Sections received a postembedding stain of 2% uranyl acetate and Sato’slead. Images were collected on a Philips CM-10 Biotwin transmissionelectron microscope (TEM) with film at 80 kV, a FEI CM200F cryoelec-tron microscope with a 1-k charge-coupled-device (CCD) camera at 200kV (FEI Company, Hillsboro, OR), or an FEI Titan Krios TEM at 300 kVwith a 4-k CCD camera. For immunostaining, sections were etched with200 mM NaOH in ethanol and rinsed in ethanol and water followed byincubation in 0.005% Triton X–PBS for 10 min. Grids were then blockedwith 2% bovine serum albumin–1% fish gelatin–PBS for 30 m, followedby exposure to anti-Cp antibody (1:150 in 10% blocking solution–PBS)for 1.5 h. Samples were then rinsed in PBS and blocked with 3% normalgoat serum in PBS for 10 min followed by exposure of anti-rabbit anti-body conjugated to 6-nm-diameter gold (EMS) (1:40 in PBS) for 1.5 h.Samples were rinsed in PBS and cross-linked with 2.5% glutaraldehyde for5 min and then poststained with 2% uranyl acetate and Sato’s lead.
RESULTSPhenotype of the SINV Cp F2-G2 loop mutants. It has previouslybeen suggested that the SINV Cp F2-G2 loop is involved in aputative interaction with cdE2 (44). Furthermore, Cp K250 wasproposed to mediate cross-capsomere contacts within the NC(46) and Cp K252 was postulated to participate in salt bridge con-tacts with cdE2. However, analysis of virus assembly phenotypesafter mutation of these residues has not yet been described. Thus,we generated SINV mutants in which K250 and K252 were simul-taneously mutated to alanine (K250/2A), aspartic acid (K250/2D),or glutamic acid (K250/2E) and a mutant in which the sequence
encoding the tripeptide sequence 250-KGK-252 was deleted(�KGK).
After BHK cells were electroporated with in vitro-transcribedviral RNA, infectious virus was recovered from only the wild typeand the K250/2A mutant; no infectious virus was recovered fromthe K250/2D, K250/2E, or �KGK mutant. Growth curve analysisof the wild type and the K250/2A mutant indicated that theK250/2A mutant released �1.5-log-fewer infectious units thanthe wild type throughout the 12-h time course (Fig. 1A).
To ascertain if noninfectious particles were being released inthe K250/2D, K250/2E, or �KGK backgrounds, we collected me-dia 8 h after electroporation of in vitro-transcribed viral RNAs andassayed the amount of infectious particles in the media and theamount of total virus particles in parallel by plaque assay andELISA, respectively (Fig. 1B). The K250/2A mutant showed a de-crease in signal in the ELISA relative to the growth curve, indicat-ing no modulation in specific infectivity in that mutant. TheK250/2D, K250/2E, and �KGK mutants, however, failed to releaseany virus particles as gauged by the ELISA, indicating that, in thesebackgrounds, virus particle release is completely abrogated. Elec-troporating BHK cells with in vitro-transcribed RNA generatedfrom wild-type or mutant cDNAs and growing the electroporatedcells at 30°C, rather than 37°C, did not rescue infectious virus
FIG 1 Analysis of infectious particle release. (A) Growth curve of the wild typeand the K250/2A Sindbis capsid mutant. Infectious stocks of the wild type orthe K250/2A mutant were used to inoculate a monolayer of BHK cells at amultiplicity of infection of 0.5. Media was collected at various time intervalspostinoculation, and the amount of infectious particles released from the cellswas determined via titration on BHK cells. (B) ELISA to detect total Sindbisparticles released after electroporation of in vitro-transcribed viral RNA. BHKcells were electorporated with RNA, and the media were collected at 8 h post-transfection. In parallel, the titer was determined by plaque assay on BHK cellsand the total amount of virions determined by ELISA. The mock sample wasderived from BHK cells electroporated without RNA and processed as de-scribed above. n.p., no plaques; n/a, not applicable.
Snyder et al.
12374 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.

particle release for the K250/2D, K250/2E, and �KGK mutants(data not shown). Thus, when the lysines present at Cp positions250 and 252 were replaced with a small, neutral, and nonpolarresidue, release of infectious virus was attenuated. However, re-versing the charge of these two lysines, as in the K250/2D andK250/2E mutants, or deleting the residues led to a complete dis-ruption of virus particle release from cells.
Reversing the charge of Cp residues K250 and K252 leads toincomplete NC assembly in the mammalian cell. Tang et al. (44)suggested that the Cp F2-G2 loop, which contains the K250 andK252 residues, might shift position when cdE2 inserts into the Cphydrophobic pocket; thus, these Cp residues may be important forbudding. Tellinghuisen and Kuhn suggested that Cp K250 may beinvolved in cross-capsomere contacts in the context of an assem-bled NC core (46). Thus, disruption of the lysine residues in theCp F2-G2 loop could be expected to affect NC assembly or bud-ding or both.
To determine if NC assembly was affected in the F2-G2 loopmutants, we performed a NC accumulation assay in BHK cellsafter electroporation of in vitro-transcribed viral RNAs in a fash-ion similar to that described previously (29). Briefly, 8 h postelec-troporation, lysates of BHK cells were applied to a continuousrate-zonal gradient and centrifuged and the migration of Cp-con-taining complexes was determined by fractionation and densi-
tometry after immunoblot analysis. With the exception of the�KGK mutant, equivalent amounts of total Cp were detected inthe cell (data not shown). The migration of viral genomic RNA-containing complexes was also determined by fractionation andquantitative reverse transcriptase PCR (qRT-PCR).
As shown in Fig. 2, free Cp (45) and free in vitro-transcribedviral genomic RNA (assayed independently of each other) showedonly slight sedimentation into the gradient. Lysates from cellstransfected with the wild type and the K250/2A mutant demon-strated cosedimentation of Cp and viral genomic RNA to fraction7, the position typically occupied by NC (data not shown). TheK250/2D and K250/2E mutants, however, demonstrated sedi-mentation of the bulk of Cp- and genomic RNA-containing com-plexes to fractions 4 and 5, unlike the wild type, suggesting that, inthese mutants, Cp-genomic RNA complexes are formed but areconformationally different from that of authentic NC. In additionto mock-electroporated cells, the �KGK mutant failed to generatefull-length Cp in the cell (data not shown) and was not consideredfurther. We unsuccessfully attempted to visualize the putative ag-gregates isolated from the K2502/2D or K250/2E gradient frac-tions by negative-stain electron microscopy.
To validate the lack of NC formation in the K250/2D andK250/2E mutant backgrounds, we examined transfected BHKcells by electron microscopy. In thin-section EM, we were unable
FIG 2 Rate-zonal gradient analysis of infected cell lysates. BHK cells were electroporated with in vitro-transcribed full-length RNA and hypotonically lysed at 8h posttransfection. Clarified cell lysates were loaded onto an iodoxinol gradient and fractionated from the top of the gradient after 2.5 h. Fraction 1 refers to thetop of the gradient, whereas fraction 11 was collected from the bottom. The amount of capsid protein and genomic RNA in each fraction was determined byimmunoblotting and densitometry and by qRT-PCR, respectively. The y axis represents the normalized intensity in each fraction relative to the fractioncontaining the most abundant signal.
Early Temporal Interaction of Sindbis Capsid and E2
November 2012 Volume 86 Number 22 jvi.asm.org 12375
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.
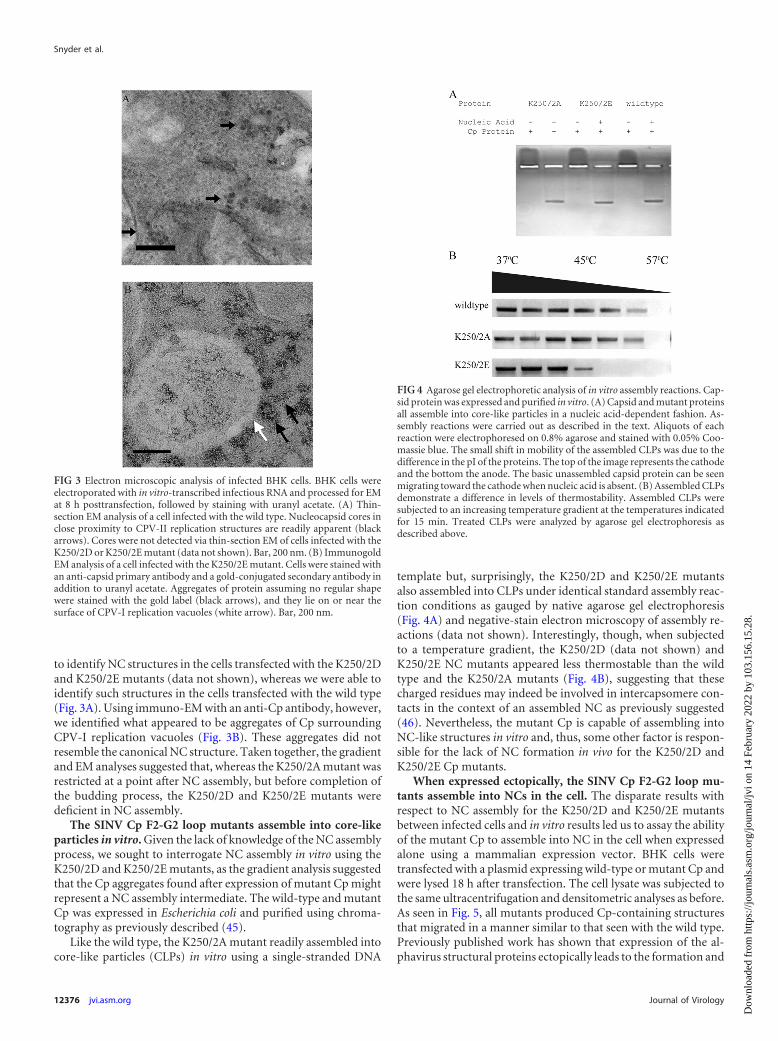
to identify NC structures in the cells transfected with the K250/2Dand K250/2E mutants (data not shown), whereas we were able toidentify such structures in the cells transfected with the wild type(Fig. 3A). Using immuno-EM with an anti-Cp antibody, however,we identified what appeared to be aggregates of Cp surroundingCPV-I replication vacuoles (Fig. 3B). These aggregates did notresemble the canonical NC structure. Taken together, the gradientand EM analyses suggested that, whereas the K250/2A mutant wasrestricted at a point after NC assembly, but before completion ofthe budding process, the K250/2D and K250/2E mutants weredeficient in NC assembly.
The SINV Cp F2-G2 loop mutants assemble into core-likeparticles in vitro. Given the lack of knowledge of the NC assemblyprocess, we sought to interrogate NC assembly in vitro using theK250/2D and K250/2E mutants, as the gradient analysis suggestedthat the Cp aggregates found after expression of mutant Cp mightrepresent a NC assembly intermediate. The wild-type and mutantCp was expressed in Escherichia coli and purified using chroma-tography as previously described (45).
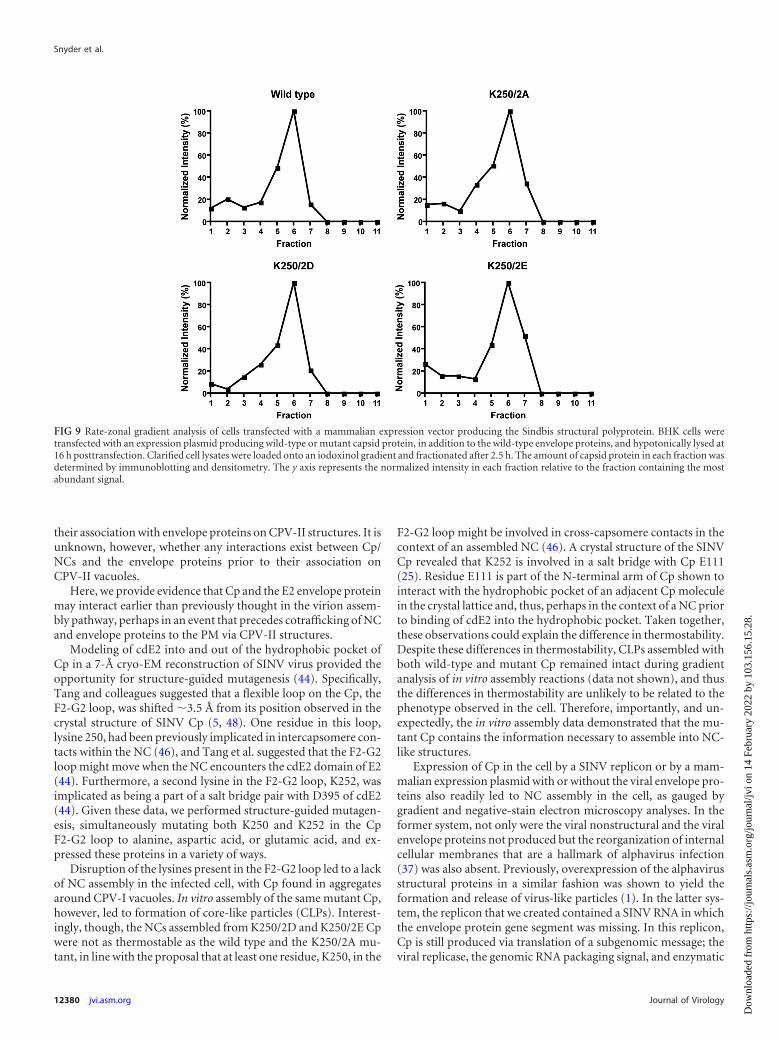
Like the wild type, the K250/2A mutant readily assembled intocore-like particles (CLPs) in vitro using a single-stranded DNA
template but, surprisingly, the K250/2D and K250/2E mutantsalso assembled into CLPs under identical standard assembly reac-tion conditions as gauged by native agarose gel electrophoresis(Fig. 4A) and negative-stain electron microscopy of assembly re-actions (data not shown). Interestingly, though, when subjectedto a temperature gradient, the K250/2D (data not shown) andK250/2E NC mutants appeared less thermostable than the wildtype and the K250/2A mutants (Fig. 4B), suggesting that thesecharged residues may indeed be involved in intercapsomere con-tacts in the context of an assembled NC as previously suggested(46). Nevertheless, the mutant Cp is capable of assembling intoNC-like structures in vitro and, thus, some other factor is respon-sible for the lack of NC formation in vivo for the K250/2D andK250/2E Cp mutants.
When expressed ectopically, the SINV Cp F2-G2 loop mu-tants assemble into NCs in the cell. The disparate results withrespect to NC assembly for the K250/2D and K250/2E mutantsbetween infected cells and in vitro results led us to assay the abilityof the mutant Cp to assemble into NC in the cell when expressedalone using a mammalian expression vector. BHK cells weretransfected with a plasmid expressing wild-type or mutant Cp andwere lysed 18 h after transfection. The cell lysate was subjected tothe same ultracentrifugation and densitometric analyses as before.As seen in Fig. 5, all mutants produced Cp-containing structuresthat migrated in a manner similar to that seen with the wild type.Previously published work has shown that expression of the al-phavirus structural proteins ectopically leads to the formation and
FIG 3 Electron microscopic analysis of infected BHK cells. BHK cells wereelectroporated with in vitro-transcribed infectious RNA and processed for EMat 8 h posttransfection, followed by staining with uranyl acetate. (A) Thin-section EM analysis of a cell infected with the wild type. Nucleocapsid cores inclose proximity to CPV-II replication structures are readily apparent (blackarrows). Cores were not detected via thin-section EM of cells infected with theK250/2D or K250/2E mutant (data not shown). Bar, 200 nm. (B) ImmunogoldEM analysis of a cell infected with the K250/2E mutant. Cells were stained withan anti-capsid primary antibody and a gold-conjugated secondary antibody inaddition to uranyl acetate. Aggregates of protein assuming no regular shapewere stained with the gold label (black arrows), and they lie on or near thesurface of CPV-I replication vacuoles (white arrow). Bar, 200 nm.
FIG 4 Agarose gel electrophoretic analysis of in vitro assembly reactions. Cap-sid protein was expressed and purified in vitro. (A) Capsid and mutant proteinsall assemble into core-like particles in a nucleic acid-dependent fashion. As-sembly reactions were carried out as described in the text. Aliquots of eachreaction were electrophoresed on 0.8% agarose and stained with 0.05% Coo-massie blue. The small shift in mobility of the assembled CLPs was due to thedifference in the pI of the proteins. The top of the image represents the cathodeand the bottom the anode. The basic unassembled capsid protein can be seenmigrating toward the cathode when nucleic acid is absent. (B) Assembled CLPsdemonstrate a difference in levels of thermostability. Assembled CLPs weresubjected to an increasing temperature gradient at the temperatures indicatedfor 15 min. Treated CLPs were analyzed by agarose gel electrophoresis asdescribed above.
Snyder et al.
12376 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.

release of virus-like particles, and alphavirus NC assembly in cellsthat lack nucleic acid has not been described; thus, NCs presum-ably assemble here with the aid of cellular RNAs (1). Therefore,the complexes found in these cell lysates likely represent NCs and,as such, the K250/2D and K250/2E mutant Cp are also capable ofNC assembly in cells. Taken further, there is apparently some fac-tor or environment in the infected cell that prevents authentic NCassembly using K250/2D and K250/2E mutant Cp.
When expressed via a SINV replicon, the SINV Cp F2-G2loop mutants assemble into NCs in the cell. In the infected cell,NCs are found in close proximity to CPV-I structures which com-prise both the site of viral RNA synthesis and the site of NC assem-bly (11). NCs also interact with the envelope proteins (23, 25, 31,36, 51) frequently during the viral life cycle. Thus, in an effort todetermine whether it is the viral replicase, the viral envelope pro-teins, or the environment in an infected cell that is responsible forthe conflicting results seen with infected cells versus the otherexperimental conditions with regard to NC assembly for theK250/2D and K250/2E mutants, we generated a SINV repliconexpressing only the wild-type or mutant Cp. This construct, whichlacked the sequence encoding the viral glycoproteins, was ex-pected to better mimic the environment of an infected cell, giventhe expression of the viral enzymatic proteins and the concurrentestablishment of viral replicases on CPV-I replication vacuoles. Asbefore, BHK cells were electroporated and the cell lysates weresubjected to the same ultracentrifugation and densitometric anal-ysis. Again (Fig. 6A), the mutants all produced Cp-containingstructures that migrated in a manner similar to that seen with thewild type and apparently represented NCs. Indeed, electron mi-croscopy studies validated the presence of NC structures for boththe wild type and the mutants (Fig. 6B). To exclude the possibility
that the shorter length of the replicon nucleic acid, relative to thatof the full-length viral genomic RNA molecule, played a role inrescuing NC assembly, we created a separate construct in whichwe replaced the fourth and fifth codons of E3 with stop codons. Inthis system, only Cp is produced via the subgenomic message, asin the replicon, but the replicated nucleic acid is the same length asin the full-length genome. Both the wt and the K250/2D Cp mu-tant were tested in this background, and we detected NC assemblyin cell lysates after sedimentation analysis in the ultracentrifuge(Fig. 7). Therefore, it appeared that it was the envelope proteinsthat were responsible for the lack of NC assembly in infected cellsexpressing the K250/2D and K250/2E mutant Cp.
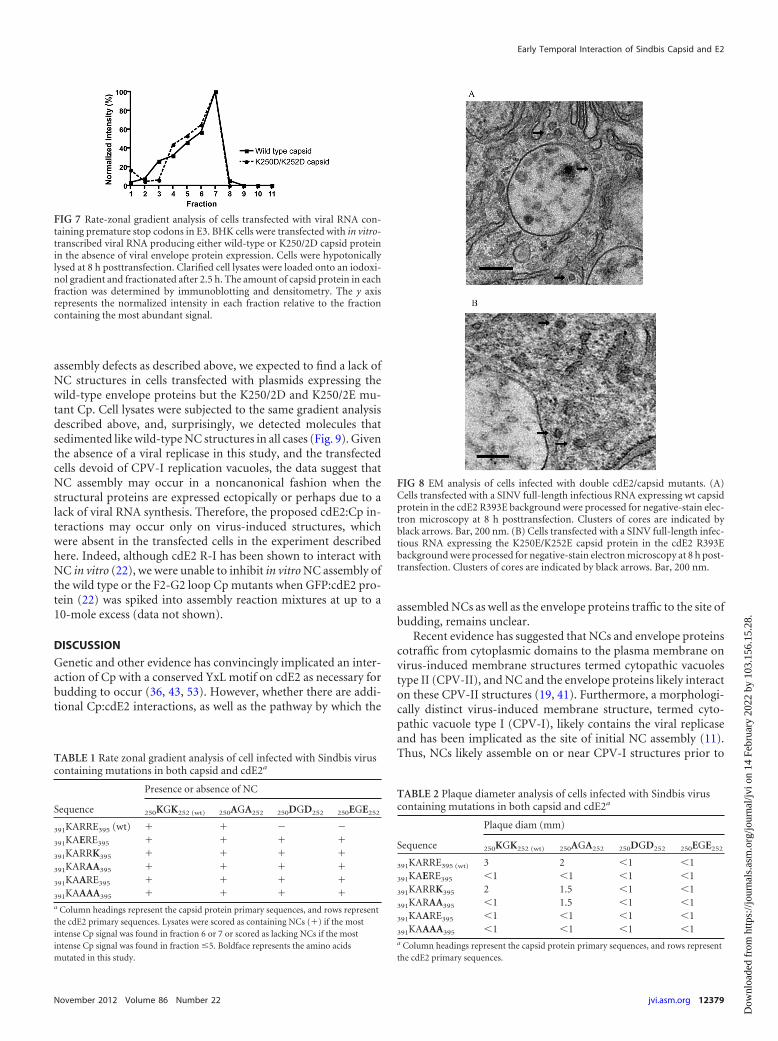
Double mutants containing mutations in both the Cp F2-G2loop and cdE2 region I rescue NC assembly but not virus particlerelease. Given the 7-Å SINV reconstruction, as well as previouslypublished biophysical analyses of residues involved in Cp:E2 in-teractions (22, 44), we speculated that region I of the cytoplasmicdomain of E2 (cdE2-RI), when present, may be responsible for thelack of NC assembly in the K250/2D and K250/2E mutant Cpbackgrounds. We therefore created a set of double mutants inwhich cdE2-RI (cdE2 391-KARRE-395) and Cp residues K250and K252 were simultaneously changed. Interestingly, gradientanalysis of the double mutants demonstrated restoration of NCassembly in the K250/2D and K250/2E Cp backgrounds (Table 1).Lysates were scored as containing NCs (�) if the most intense Cpsignal was found in fraction 6 or 7 or scored as lacking NCs if themost intense Cp signal was found in fraction 1, 2, 3, 4, or 5. Neg-ative-stain electron microscopy analyses of a subset of the doublemutants validated the gradient findings (Fig. 8). Specifically, weidentified electron-dense NC structures adjacent to CPV-I struc-tures for both the wt and K250/2E Cp mutant backgrounds con-
FIG 5 Rate-zonal gradient analysis of cells transfected with a mammalian expression vector producing capsid protein. BHK cells were transfected with anexpression plasmid producing wild-type or mutant capsid proteins and hypotonically lysed at 16 h posttransfection. Clarified cell lysates were loaded onto aniodoxinol gradient and fractionated after 2.5 h. The amount of capsid protein in each fraction was determined by immunoblotting and densitometry. The y axisrepresents the normalized intensity in each fraction relative to the fraction containing the most abundant signal.
Early Temporal Interaction of Sindbis Capsid and E2
November 2012 Volume 86 Number 22 jvi.asm.org 12377
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.

currently with a lack of aggregates of Cp around these CPV-Istructures using immuno-EM (data not shown). However, recov-ery of infectious particles as determined by plaque titration ofposttransfected BHK cell media, or immediate agarose overlayafter transfection of infectious RNA, was not restored for theK250/2D or K250/2E Cp background despite the formation ofpinpoint-sized syncytium-derived plaques (Table 2). Further-more, modulation of the sequence of cdE2 R-I residues 393-RRE-395 also impaired the ability of virus with a wild-type or K250/2ACp background to release infectious particles. Taken together,
these data suggest that cdE2-RI is important for efficient buddingbut also that cdE2-RI may interact with Cp at a prebudding stage,perhaps before or during NC assembly.
Ectopic expression of the entire structural polyprotein withCp F2-G2 loop mutants yields NC assembly in the mammaliancell. Next, we generated a set of mammalian expression plasmidsin which only the structural polyprotein, containing the wild-typeenvelope proteins, with either the wild-type Cp or the F2-G2 loopmutant Cp, was produced from a single message. Given the pres-ence of the wild-type cdE2 R-I sequence, and its implication in NC
FIG 6 Analysis of cells transfected with Sindbis replicon expressing the capsid. BHK cells were electroporated with in vitro-transcribed replicon RNA. (A) Cellswere hypotonically lysed at 8 h posttransfection. Clarified cell lysates were loaded onto an iodoxinol gradient and fractionated after 2.5 h. The amount of capsidprotein in each fraction was determined by immunoblotting and densitometry. The y axis represents the normalized intensity in each fraction relative to thefraction containing the most abundant signal. (B) Cells transfected with a SINV replicon expressing wt capsid protein were processed for negative-stain electronmicroscopy at 8 h posttransfection. Clusters of cores are indicated by black arrows. Bar, 200 nm. (C) Cells transfected with a SINV replicon expressing theK250E/K252E capsid protein were processed for negative stain and EM at 8 h posttransfection. Clusters of cores are indicated by black arrows. Bar, 200 nm.
Snyder et al.
12378 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.
assembly defects as described above, we expected to find a lack ofNC structures in cells transfected with plasmids expressing thewild-type envelope proteins but the K250/2D and K250/2E mu-tant Cp. Cell lysates were subjected to the same gradient analysisdescribed above, and, surprisingly, we detected molecules thatsedimented like wild-type NC structures in all cases (Fig. 9). Giventhe absence of a viral replicase in this study, and the transfectedcells devoid of CPV-I replication vacuoles, the data suggest thatNC assembly may occur in a noncanonical fashion when thestructural proteins are expressed ectopically or perhaps due to alack of viral RNA synthesis. Therefore, the proposed cdE2:Cp in-teractions may occur only on virus-induced structures, whichwere absent in the transfected cells in the experiment describedhere. Indeed, although cdE2 R-I has been shown to interact withNC in vitro (22), we were unable to inhibit in vitro NC assembly ofthe wild type or the F2-G2 loop Cp mutants when GFP:cdE2 pro-tein (22) was spiked into assembly reaction mixtures at up to a10-mole excess (data not shown).
DISCUSSION
Genetic and other evidence has convincingly implicated an inter-action of Cp with a conserved YxL motif on cdE2 as necessary forbudding to occur (36, 43, 53). However, whether there are addi-tional Cp:cdE2 interactions, as well as the pathway by which the
assembled NCs as well as the envelope proteins traffic to the site ofbudding, remains unclear.
Recent evidence has suggested that NCs and envelope proteinscotraffic from cytoplasmic domains to the plasma membrane onvirus-induced membrane structures termed cytopathic vacuolestype II (CPV-II), and NC and the envelope proteins likely interacton these CPV-II structures (19, 41). Furthermore, a morphologi-cally distinct virus-induced membrane structure, termed cyto-pathic vacuole type I (CPV-I), likely contains the viral replicaseand has been implicated as the site of initial NC assembly (11).Thus, NCs likely assemble on or near CPV-I structures prior to
FIG 7 Rate-zonal gradient analysis of cells transfected with viral RNA con-taining premature stop codons in E3. BHK cells were transfected with in vitro-transcribed viral RNA producing either wild-type or K250/2D capsid proteinin the absence of viral envelope protein expression. Cells were hypotonicallylysed at 8 h posttransfection. Clarified cell lysates were loaded onto an iodoxi-nol gradient and fractionated after 2.5 h. The amount of capsid protein in eachfraction was determined by immunoblotting and densitometry. The y axisrepresents the normalized intensity in each fraction relative to the fractioncontaining the most abundant signal.
TABLE 1 Rate zonal gradient analysis of cell infected with Sindbis viruscontaining mutations in both capsid and cdE2a
Sequence
Presence or absence of NC
250KGK252 (wt) 250AGA252 250DGD252 250EGE252
391KARRE395 (wt) � � � �
391KAERE395 � � � �
391KARRK395 � � � �
391KARAA395 � � � �
391KAARE395 � � � �
391KAAAA395 � � � �a Column headings represent the capsid protein primary sequences, and rows representthe cdE2 primary sequences. Lysates were scored as containing NCs (�) if the mostintense Cp signal was found in fraction 6 or 7 or scored as lacking NCs if the mostintense Cp signal was found in fraction �5. Boldface represents the amino acidsmutated in this study.
FIG 8 EM analysis of cells infected with double cdE2/capsid mutants. (A)Cells transfected with a SINV full-length infectious RNA expressing wt capsidprotein in the cdE2 R393E background were processed for negative-stain elec-tron microscopy at 8 h posttransfection. Clusters of cores are indicated byblack arrows. Bar, 200 nm. (B) Cells transfected with a SINV full-length infec-tious RNA expressing the K250E/K252E capsid protein in the cdE2 R393Ebackground were processed for negative-stain electron microscopy at 8 h post-transfection. Clusters of cores are indicated by black arrows. Bar, 200 nm.
TABLE 2 Plaque diameter analysis of cells infected with Sindbis viruscontaining mutations in both capsid and cdE2a
Sequence
Plaque diam (mm)
250KGK252 (wt) 250AGA252 250DGD252 250EGE252
391KARRE395 (wt) 3 2 1 1
391KAERE395 1 1 1 1
391KARRK395 2 1.5 1 1
391KARAA395 1 1.5 1 1
391KAARE395 1 1 1 1
391KAAAA395 1 1 1 1a Column headings represent the capsid protein primary sequences, and rows representthe cdE2 primary sequences.
Early Temporal Interaction of Sindbis Capsid and E2
November 2012 Volume 86 Number 22 jvi.asm.org 12379
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.
their association with envelope proteins on CPV-II structures. It isunknown, however, whether any interactions exist between Cp/NCs and the envelope proteins prior to their association onCPV-II vacuoles.
Here, we provide evidence that Cp and the E2 envelope proteinmay interact earlier than previously thought in the virion assem-bly pathway, perhaps in an event that precedes cotrafficking of NCand envelope proteins to the PM via CPV-II structures.
Modeling of cdE2 into and out of the hydrophobic pocket ofCp in a 7-Å cryo-EM reconstruction of SINV virus provided theopportunity for structure-guided mutagenesis (44). Specifically,Tang and colleagues suggested that a flexible loop on the Cp, theF2-G2 loop, was shifted �3.5 Å from its position observed in thecrystal structure of SINV Cp (5, 48). One residue in this loop,lysine 250, had been previously implicated in intercapsomere con-tacts within the NC (46), and Tang et al. suggested that the F2-G2loop might move when the NC encounters the cdE2 domain of E2(44). Furthermore, a second lysine in the F2-G2 loop, K252, wasimplicated as being a part of a salt bridge pair with D395 of cdE2(44). Given these data, we performed structure-guided mutagen-esis, simultaneously mutating both K250 and K252 in the CpF2-G2 loop to alanine, aspartic acid, or glutamic acid, and ex-pressed these proteins in a variety of ways.
Disruption of the lysines present in the F2-G2 loop led to a lackof NC assembly in the infected cell, with Cp found in aggregatesaround CPV-I vacuoles. In vitro assembly of the same mutant Cp,however, led to formation of core-like particles (CLPs). Interest-ingly, though, the NCs assembled from K250/2D and K250/2E Cpwere not as thermostable as the wild type and the K250/2A mu-tant, in line with the proposal that at least one residue, K250, in the
F2-G2 loop might be involved in cross-capsomere contacts in thecontext of an assembled NC (46). A crystal structure of the SINVCp revealed that K252 is involved in a salt bridge with Cp E111(25). Residue E111 is part of the N-terminal arm of Cp shown tointeract with the hydrophobic pocket of an adjacent Cp moleculein the crystal lattice and, thus, perhaps in the context of a NC priorto binding of cdE2 into the hydrophobic pocket. Taken together,these observations could explain the difference in thermostability.Despite these differences in thermostability, CLPs assembled withboth wild-type and mutant Cp remained intact during gradientanalysis of in vitro assembly reactions (data not shown), and thusthe differences in thermostability are unlikely to be related to thephenotype observed in the cell. Therefore, importantly, and un-expectedly, the in vitro assembly data demonstrated that the mu-tant Cp contains the information necessary to assemble into NC-like structures.
Expression of Cp in the cell by a SINV replicon or by a mam-malian expression plasmid with or without the viral envelope pro-teins also readily led to NC assembly in the cell, as gauged bygradient and negative-stain electron microscopy analyses. In theformer system, not only were the viral nonstructural and the viralenvelope proteins not produced but the reorganization of internalcellular membranes that are a hallmark of alphavirus infection(37) was also absent. Previously, overexpression of the alphavirusstructural proteins in a similar fashion was shown to yield theformation and release of virus-like particles (1). In the latter sys-tem, the replicon that we created contained a SINV RNA in whichthe envelope protein gene segment was missing. In this replicon,Cp is still produced via translation of a subgenomic message; theviral replicase, the genomic RNA packaging signal, and enzymatic
FIG 9 Rate-zonal gradient analysis of cells transfected with a mammalian expression vector producing the Sindbis structural polyprotein. BHK cells weretransfected with an expression plasmid producing wild-type or mutant capsid protein, in addition to the wild-type envelope proteins, and hypotonically lysed at16 h posttransfection. Clarified cell lysates were loaded onto an iodoxinol gradient and fractionated after 2.5 h. The amount of capsid protein in each fraction wasdetermined by immunoblotting and densitometry. The y axis represents the normalized intensity in each fraction relative to the fraction containing the mostabundant signal.
Snyder et al.
12380 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.

proteins are present and CPV-I vacuoles are observed in the trans-fected cell (data not shown), but the envelope proteins are notproduced and CPV-II vacuoles are not observed (data not shown).Additionally, in a separate experiment, we excluded the length ofthe capsid-expressing nucleic acid as a factor in rescuing NC as-sembly. Taken together, the data suggested that the envelope pro-teins were responsible for the NC assembly defect seen in the in-fected cell.
Previously published genetic and structural data strongly sug-gested that cdE2 interacts closely with a hydrophobic pocket onthe surface of Cp (36, 38, 44). Jose and colleagues recently dem-onstrated that region I of cdE2 (cdE2-RI), comprised of E2 391-KARRE-395, specifically interacts with both NCs isolated fromcells infected with SINV, as well as in vitro-assembled CLPs (22).cdE2 E395 has been specifically implicated in virus assembly (13),and Tang et al. proposed that cdE2 residue E395, part of region I,formed a salt bridge with Cp K252, a residue in the F2-G2 loopunder investigation here (44). Thus, we speculated that cdE2-RI,when present, was responsible for the lack of NC assembly in theK250/2D and K250/2E mutant Cp backgrounds.
Double mutants in which the primary sequences of both theCp F2-G2 loop and cdE2-RI were mutated showed that multipledifferent alterations of the cdE2 sequence 393-RRE-395 were ca-pable of restoring the ability of the K250/2D and K250/2E mutantCp to incorporate into NCs in the cell. The fact that multipledifferent substitutions in this region of cdE2 rescued the NC as-sembly defect suggests that it is not necessarily one particular Cp:cdE2 side-chain interaction that is responsible for the K250/2Dand K250/2E mutant phenotypes but perhaps more likely theshape or conformation of cdE2. Indeed, it has been suggested that,given the conservation of the positions of prolines and cysteines incdE2 across the alphavirus genus, cdE2 might adopt a conservedconformation important for Cp binding (42).
While mutation of cdE2-RI restored the capacity for NC for-mation in the K250/2D and K250/2E backgrounds, it did not re-store the release of infectious particles. Except for the E2 E395Kmutant, all other cdE2-RI mutants prevented release of infectiousparticles by virus containing wild-type Cp. That cdE2 region Imight be important in virus assembly was also recently proposedby Jose et al. (22). Thus, cdE2-RI may have a multifaceted role inthe virus assembly process through interaction with the Cp. Inter-estingly, as previously noted, some degree of covariance existsbetween cdE2-RI and the F2-G2 loop of Cp (22). Specifically, inmost alphaviruses sequenced to date, when the Cp residue corre-sponding to SINV K250 is basic, the cdE2 residue correspondingto E395 is acidic, and when the Cp residue corresponding to K250is acidic, the cdE2 residue is basic. Note that it is Cp residue K252,not K250, that is reported to form a salt bridge with cdE2 residueE395 (44); thus, modeling based on static images of virus particlesand isolated NC may underscore the true fluidity of the cdE2 andCp domains that apparently interact with one another.
When we assayed the ability of the K250/2D and K250/2E Cpmutants to assemble when they were ectopically expressed as partof the complete structural polyprotein, we expected that theK250/2D and K250/2E Cp mutants would not assemble into NCs.However, gradient analysis demonstrated the presence of Cp-con-taining structures that sedimented just like the wild type. Thesedata suggest that, while cdE2 does play a role in the NC assemblydefect phenotypes, the ultrastructure of the infected cell does aswell. Indeed, in this system, CPV-I vacuoles are absent, and the
polyprotein is likely translated in a canonical fashion rather thanproximal to CPV-I structures. The nonstructural proteins, likelycompartmentalized at CPV-I vacuoles in a regular infection, havebeen shown to interact with numerous host proteins (6, 18, 35). Inshort, the NC assembly defects described here may not be able tobe recapitulated via ectopic expression of the structural polypro-tein because of the inherent differences in the organization orarrangement of viral proteins in the uninfected versus infectedcell. Indeed, when GFP:cdE2 protein was added at up to a 10-moleexcess into in vitro assembly reactions using wild-type or mutantF2-G2 loop Cp, assembly of CLPs was unaffected, again demon-strating that the presence of cdE2 alone is not sufficient to recon-stitute the Cp:cdE2 interactions that occur in the infected cell.
The results described here provide additional insights into thetemporal and spatial interactions between SINV Cp and cdE2. Wedemonstrated that Cp containing mutations in the F2-G2 loopstill retained the information necessary to assemble into NCs.CPV-I vacuoles are proposed to be the site of normal NC assembly(11), but in cells transfected with in vitro transcripts containingthe Cp mutation K250/2D or K250/2E, an accumulation of aggre-gates of Cp and viral genomic RNA complex at CPV-I vacuoleswas detected. Given that this phenotype can be rescued by modu-lation of the primary sequence of cdE2-RI, a region already impli-cated in Cp:E2 interactions (44), and that, in the absence of thesesecond-site mutations, K250/2D and K250/2E Cp form aggregateson CPV-I vacuoles, our data suggest that Cp and E2 may interacton or near CPV-I vacuoles, the site of NC assembly (11), soon afterCp synthesis.
It is tempting to speculate that cdE2 might act as a chaperone,aiding the assembly of NCs. Other viruses employ such a mecha-nism (7); however, additional data are required to interrogate thishypothesis. Another mechanistic possibility, and perhaps one thatis more likely, is that changing the local charge of the Cp F2-G2loop in the K250/2D and K2502/E mutants increases the affinity ofCp for cdE2. This could lead to an altered rate of nucleation be-tween Cp and cdE2 and the formation of aggregates. In the in-fected cell, the K250/2D Cp and the K2502/E Cp may becomekinetically trapped by this interaction, unable to continue downthe NC assembly pathway. Computational studies with otherRNA viruses have demonstrated, counterintuitively, that increas-ing the affinity of a viral Cp for its cognate RNA leads to an aber-rant and incorrect NC assembly pathway by altering the rate ofnucleation (8, 21).
Nevertheless, the data presented here have provided additionalknowledge concerning the interactions of Cp and E2 in the in-fected cell and, coupled with recent additions to the literature,have provided a new level of insight into the interactions andtrafficking of the alphavirus NC and envelope proteins and allowus to propose a more detailed model regarding the NC assemblypathway. Wilkinson et al. proposed that Cp and RNA may initiallyassociate on the cytoplasmic faces of intracellular membranes(51). Here, we show that Cp and cdE2 may interact on CPV-Ivacuoles, although direct evidence of the presence of E2 in CPV-Istructures has yet to be published. However, the analysis of alpha-virus chimeras with respect to genome replication kinetics as wellas work with an anti-idiotypic antibody in which the original an-tigen was cdE2 have led to speculation that molecules of E2 mightindeed be present in the replicase complex (42, 43). Given thatCPV-I vacuoles are derived from the plasma membrane (10), the
Early Temporal Interaction of Sindbis Capsid and E2
November 2012 Volume 86 Number 22 jvi.asm.org 12381
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.

ultimate destination for E2, the presence of the envelope proteinsin or near the viral replicase may not actually be that surprising.
Soonsawad and colleagues showed that CPV-II vacuoles con-tain envelope proteins spikes, are decorated with NCs, and trafficfrom sites internal to the cell to the plasma membrane (41). Un-published electron tomography data (T. J. Edwards and R. J.Kuhn) have demonstrated the movement of assembled NCs fromCPV-I vacuoles to CPV-II vacuoles, and recent studies using mi-croinjection of viral RNA into envelope protein-expressing cellsdemonstrated a preference for newly synthesized envelope pro-teins during envelopment of NCs (39).
Taking these data together, we can propose a more detailedmodel regarding the fate of Cp and envelope proteins in the cell.Specifically, newly translated Cp may associate with CPV-I mem-branes, interact with viral genomic RNA, and assemble into NCstructures. Molecules of cdE2 resident in the CPV-I structuresmay temporarily interact with Cp before or during NC assembly.Assembled NCs may then traffic from CPV-I vacuoles to CPV-IIvacuoles, where they interact with additional molecules of cdE2.Presumably, NC and cdE2 interact strongly here, perhaps via theconserved cdE2 YxL motif and the hydrophobic pocket on Cp.The C-terminal domain of cdE2 (cdE2-CTD) is reported to ini-tially transverse the membrane, flipping back and exposing cdE2-CTD to the cytosol well after translation in a phosphorylation-dependent manner (28). Thus, cdE2 may assume differentconformations within the cell, providing scaffolds of different lev-els of affinity for Cp when embedded in CPV-I versus CPV-IImembranous structures. A strong interaction between NC andcdE2 at CPV-II may tether NCs to CPV-II, and NCs and envelopeproteins could cotraffic to the plasma membrane (41). The struc-ture, or perhaps composition, of CPV-II is such that prematurebudding is inefficient (41). Only when CPV-II reaches and fuseswith the PM do the envelope proteins form a 2D lattice that isplanar enough to envelop the NC, providing the free energy to buda virus particle through the PM. This model, while attractive, isalmost certainly overly simplistic and requires further studies beperformed to completely elucidate the alphavirus virion assemblypathway.
The work presented here correlates with previously publishedgenetic and structural analyses and expands our knowledge ofalphavirus assembly. Taken together, our data (i) suggest that anearly temporal and spatial interaction occurs between Cp and E2at or near the CPV-I replication structures and (ii) provide a de-scription of a new set of cdE2:Cp amino acid interactions, involv-ing the N-terminal region of cdE2, that are important for alpha-virus NC assembly as well as budding.
ACKNOWLEDGMENTS
We thank Anita Robinson for clerical assistance as well as the Life ScienceMicroscopy Facility and Biological Electron Microscopy Facility at Pur-due University.
We acknowledge support from the NIH through NIGMS awardGM56279 to R.J.K. and NIH Biophysics Training Grants 5T32GM008296-21and 5T32GM008296-22 to J.E.S.
REFERENCES1. Akahata W, et al. 2010. A virus-like particle vaccine for epidemic Chi-
kungunya virus protects nonhuman primates against infection. Nat. Med.16:334 –338.
2. Bonatti S, Migliaccio G, Blobel G, Walter P. 1984. Role of signal recog-nition particle in the membrane assembly of Sindbis viral glycoproteins.Eur. J. Biochem. 140:499 –502.
3. Byrnes AP, Griffin DE. 1998. Binding of Sindbis virus to cell surfaceheparan sulfate. J. Virol. 72:7349 –7356.
4. Cheng RH, et al. 1995. Nucleocapsid and glycoprotein organization in anenveloped virus. Cell 80:621– 630.
5. Choi HK, et al. 1991. Structure of Sindbis virus core protein reveals achymotrypsin-like serine proteinase and the organization of the virion.Nature 354:37– 43.
6. Cristea IM, et al. 2010. Host factors associated with the Sindbis virusRNA-dependent RNA polymerase: role for G3BP1 and G3BP2 in virusreplication. J. Virol. 84:6720 – 6732.
7. Dokland T. 1999. Scaffolding proteins and their role in viral assembly.Cell. Mol. Life Sci. 56:580 – 603.
8. Elrad OM, Hagan MF. 2010. Encapsulation of a polymer by an icosa-hedral virus. Phys Biol. 7:045003. doi:10.1088/1478-3975/7/4/045003.
9. Forsell K, Griffiths G, Garoff H. 1996. Preformed cytoplasmic nucleo-capsids are not necessary for alphavirus budding. EMBO J. 15:6495– 6505.
10. Frolova EI, Gorchakov R, Pereboeva L, Atasheva S, Frolov I. 2010.Functional Sindbis virus replicative complexes are formed at the plasmamembrane. J. Virol. 84:11679 –11695.
11. Froshauer S, Kartenbeck J, Helenius A. 1988. Alphavirus RNA replicaseis located on the cytoplasmic surface of endosomes and lysosomes. J. CellBiol. 107:2075–2086.
12. Fuller SD, Berriman JA, Butcher SJ, Gowen BE. 1995. Low pH inducesswiveling of the glycoprotein heterodimers in the Semliki Forest virusspike complex. Cell 81:715–725.
13. Gaedigk-Nitschko K, Schlesinger MJ. 1991. Site-directed mutations inSindbis virus E2 glycoprotein’s cytoplasmic domain and the 6K proteinlead to similar defects in virus assembly and budding. Virology 183:206 –214.
14. Garoff H, Frischauf AM, Simons K, Lehrach H, Delius H. 1980. Nucle-otide sequence of cdna coding for Semliki Forest virus membrane glyco-proteins. Nature 288:236 –241.
15. Garoff H, Simons K. 1974. Location of the spike glycoproteins in theSemliki Forest virus membrane. Proc. Natl. Acad. Sci. U. S. A. 71:3988 –3992.
16. Garoff H, Sjoberg M, Cheng RH. 2004. Budding of alphaviruses. VirusRes. 106:103–116.
17. Garoff H, Soderlund H. 1978. The amphiphilic membrane glycoproteinsof Semliki Forest virus are attached to the lipid bilayer by their COOH-terminal ends. J. Mol. Biol. 124:535–549.
18. Gorchakov R, Garmashova N, Frolova E, Frolov I. 2008. Different typesof nsP3-containing protein complexes in Sindbis virus-infected cells. J.Virol. 82:10088 –10101.
19. Griffiths G, et al. 1989. The dynamic nature of the Golgi complex. J. CellBiol. 108:277–297.
20. Gualtieri EJ, et al. 2011. Detection of membrane protein two-dimensional crystals in living cells. Biophys. J. 100:207–214.
21. Hagan MF, Elrad OM. 2010. Understanding the concentration depen-dence of viral capsid assembly kinetics—the origin of the lag time andidentifying the critical nucleus size. Biophys. J. 98:1065–1074.
22. Jose J, et al. 2012. Interactions of the cytoplasmic domain of Sindbis virusE2 with nucleocapsid cores promote alphavirus budding. J. Virol. 86:2585–2599.
23. Kail M, et al. 1991. The cytoplasmic domain of alphavirus E2 glycopro-tein contains a short linear recognition signal required for viral budding.EMBO J. 10:2343–2351.
24. Klimstra WB, Ryman KD, Johnston RE. 1998. Adaptation of Sindbisvirus to BHK cells selects for use of heparan sulfate as an attachmentreceptor. J. Virol. 72:7357–7366.
25. Lee S, et al. 1996. Identification of a protein binding site on the surface ofthe alphavirus nucleocapsid and its implication in virus assembly. Struc-ture 4:531–541.
26. Liljeström P, Garoff H. 1991. Internally located cleavable signal se-quences direct the formation of Semliki Forest virus membrane proteinsfrom a polyprotein precursor. J. Virol. 65:147–154.
27. Linger BR, Kunovska L, Kuhn RJ, Golden BL. 2004. Sindbis virusnucleocapsid assembly: RNA folding promotes capsid protein dimeriza-tion. RNA 10:128 –138.
28. Liu N, Brown DT. 1993. Transient translocation of the cytoplasmic(endo) domain of a type I membrane glycoprotein into cellular mem-branes. J. Cell Biol. 120:877– 883.
29. Lopez S, Yao JS, Kuhn RJ, Strauss EG, Strauss JH. 1994. Nucleocapsid-
Snyder et al.
12382 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.

glycoprotein interactions required for assembly of alphaviruses. J. Virol.68:1316 –1323.
30. Melancon P, Garoff H. 1987. Processing of the Semliki Forest virusstructural polyprotein: role of the capsid protease. J. Virol. 61:1301–1309.
31. Metsikkö K, Garoff H. 1990. Oligomers of the cytoplasmic domain of thep62/E2 membrane protein of Semliki Forest virus bind to the nucleocap-sid in vitro. J. Virol. 64:4678 – 4683.
32. Mukhopadhyay S, Chipman PR, Hong EM, Kuhn RJ, Rossmann MG.2002. In vitro-assembled alphavirus core-like particles maintain a struc-ture similar to that of nucleocapsid cores in mature virus. J. Virol. 76:11128 –11132.
33. Mukhopadhyay S, et al. 2006. Mapping the structure and function of theE1 and E2 glycoproteins in alphaviruses. Structure 14:63–73.
34. Mulvey M, Brown DT. 1996. Assembly of the Sindbis virus spike proteincomplex. Virology 219:125–132.
35. Neuvonen M, et al. 2011. SH3 domain-mediated recruitment of host cellamphiphysins by alphavirus nsP3 promotes viral RNA replication. PLoSPathog. 7:e1002383. doi:10.1371/journal.ppat.1002383.
36. Owen KE, Kuhn RJ. 1997. Alphavirus budding is dependent on theinteraction between the nucleocapsid and hydrophobic amino acids onthe cytoplasmic domain of the E2 envelope glycoprotein. Virology 230:187–196.
37. Schwartz M, Chen J, Lee WM, Janda M, Ahlquist P. 2004. Alternate,virus-induced membrane rearrangements support positive-strand RNAvirus genome replication. Proc. Natl. Acad. Sci. U. S. A. 101:11263–11268.
38. Skoging U, Vihinen M, Nilsson L, Liljestrom P. 1996. Aromatic inter-actions define the binding of the alphavirus spike to its nucleocapsid.Structure 4:519 –529.
39. Snyder JE, et al. 2011. Rescue of infectious particles from preassembledalphavirus nucleocapsid cores. J. Virol. 85:5773–5781.
40. Söderlund H. 1973. Kinetics of formation of the Semliki Forest virusnucleocapsid. Intervirology 1:354 –361.
41. Soonsawad P, et al. 2010. Structural evidence of glycoprotein assembly in
cellular membrane compartments prior to alphavirus budding. J. Virol.84:11145–11151.
42. Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, repli-cation, and evolution. Microbiol. Rev. 58:491–562.
43. Suomalainen M, Liljestrom P, Garoff H. 1992. Spike protein-nucleocapsid interactions drive the budding of alphaviruses. J. Virol. 66:4737– 4747.
44. Tang J, et al. 2011. Molecular links between the E2 envelope glycoproteinand nucleocapsid core in Sindbis virus. J. Mol. Biol. 414:442– 459.
45. Tellinghuisen TL, Hamburger AE, Fisher BR, Ostendorp R, Kuhn RJ.1999. In vitro assembly of alphavirus cores by using nucleocapsid proteinexpressed in Escherichia coli. J. Virol. 73:5309 –5319.
46. Tellinghuisen TL, Kuhn RJ. 2000. Nucleic acid-dependent cross-linkingof the nucleocapsid protein of Sindbis virus. J. Virol. 74:4302– 4309.
47. Thiboutot MM, et al. 2010. Chikungunya: a potentially emerging epi-demic? PLoS Negl. Trop. Dis. 4:e623. doi:10.1371/journal.pntd.0000623.
48. Tong L, Choi HK, Minor W, Rossmann MG. 1992. The structuredetermination of Sindbis virus core protein using isomorphous replace-ment and molecular replacement averaging between two crystal forms.Acta Crystallogr. A 48(Pt 4):430 – 442.
49. von Bonsdorff CH, Harrison SC. 1978. Hexagonal glycoprotein arraysfrom Sindbis virus membranes. J. Virol. 28:578 –583.
50. Wahlberg JM, Bron R, Wilschut J, Garoff H. 1992. Membrane fusion ofSemliki Forest virus involves homotrimers of the fusion protein. J. Virol.66:7309 –7318.
51. Wilkinson TA, Tellinghuisen TL, Kuhn RJ, Post CB. 2005. Associationof Sindbis virus capsid protein with phospholipid membranes and the E2glycoprotein: implications for alphavirus assembly. Biochemistry 44:2800 –2810.
52. Zhang X, Fugere M, Day R, Kielian M. 2003. Furin processing andproteolytic activation of Semliki Forest virus. J. Virol. 77:2981–2989.
53. Zhao H, Lindqvist B, Garoff H, von Bonsdorff CH, Liljestrom P. 1994.A tyrosine-based motif in the cytoplasmic domain of the alphavirus enve-lope protein is essential for budding. EMBO J. 13:4204 – 4211.
Early Temporal Interaction of Sindbis Capsid and E2
November 2012 Volume 86 Number 22 jvi.asm.org 12383
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 14
Feb
ruar
y 20
22 b
y 10
3.15
6.15
.28.