qm/mm calculations and applications to biophysics marcus elstner physical and theoretical chemistry,...
TRANSCRIPT

QM/MM Calculations and Applications to Biophysics
QM/MM Calculations and Applications to Biophysics
Marcus Elstner
Physical and Theoretical Chemistry, Technical University of Braunschweig

Proteins, DNA, lipidsProteins, DNA, lipids
N

Computational challengeComputational challenge
~ 1.000-10.000 atoms in protein
~ ns molecular dynamics simulation
(MD, umbrella sampling)
- chemical reactions: proton transfer
- treatment of excited states
QM

Computational problem I: number of atomsComputational problem I: number of atoms
chemical reaction which needs QM treatment
immediate environment: electrostatic and steric interactions
solution, membrane: polarization and structural effects on protein and reaction!
10.000... - several 100.000 atoms

Computational problem II: sampling with MDComputational problem II: sampling with MD
flexibility: not one global minimum
conformational entropy
solvent relaxation
ps – ns timescale (timestep ~ 1fs)
(folding anyway out of reach!)

Optimal setupOptimal setup
ProteinMembrane: = 10 Membrane: = 10
active
Water: = 80
Water: = 80
= 20
= 20

Combined QM/MMCombined QM/MM
=80
• Computationally efficient
– ~103-5 atoms
• Generally for structural properties
• Bond breaking/formation
• Computationally demanding
– DFT, AI: ~ 50 atoms
– Semi-Empirical: ~102-3 atoms
Quantum mechanical (QM)
Molecular mechanical (MM)
Combined QM/MM• Chemical Rx in macromolecules• DFT (AI) /MM: Reaction path • Semi-Empirical/MM: Potential of mean
force, rate constants
E ˆ H QM ˆ H elQM / MM Evan
QM / MM E MM
•No polarization of MM region!•No charge transfer between QM and MM

Combined QM/MMCombined QM/MM
1976 Warshel and Levitt
1986 Singh and Kollman
1990 Field, Bash and KarplusQM • Semi-empirical• quantum chemistry packages: DFT, HF, MP2, LMP2• DFT plane wave codes: CPMD
MM• CHARMM, AMBER, GROMOS, SIGMA,TINKER, ...
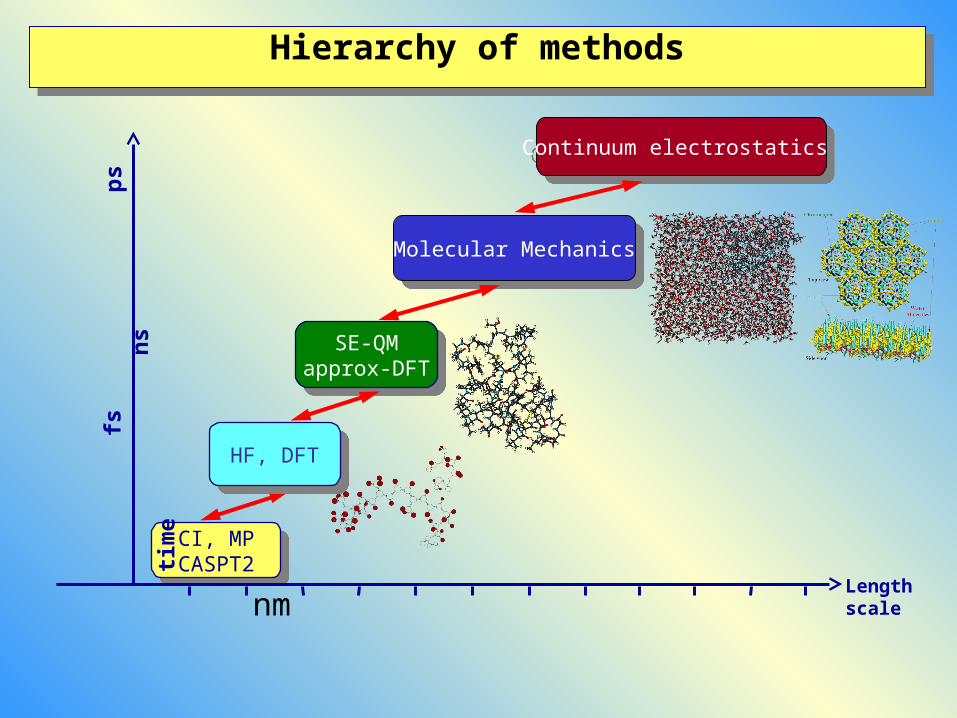
Hierarchy of methodsHierarchy of methods
CI, MPCASPT2
CI, MPCASPT2
Length scale
Continuum electrostatics Continuum electrostatics
Molecular MechanicsMolecular Mechanics
f
s
ps
ns
t i
me
SE-QMapprox-DFT
SE-QMapprox-DFT
HF, DFTHF, DFT
nm

Empirical Force Fields: Molecular Mechanics MM
Empirical Force Fields: Molecular Mechanics MM
models protein + DNA structures quite well
Problem:
- polarization
- charge transfer
- not reactiv in general
ji
ij
ji
ji
ji
ji
ji
ji
ji
dihedrals
N
n impropers
n
bonds anglesb
Dr
rr
knkkbbkV
,,
6
,
,
12
,
,
,
1
2
0
)(2
0
2
0
4
(cos1
kb
k
k
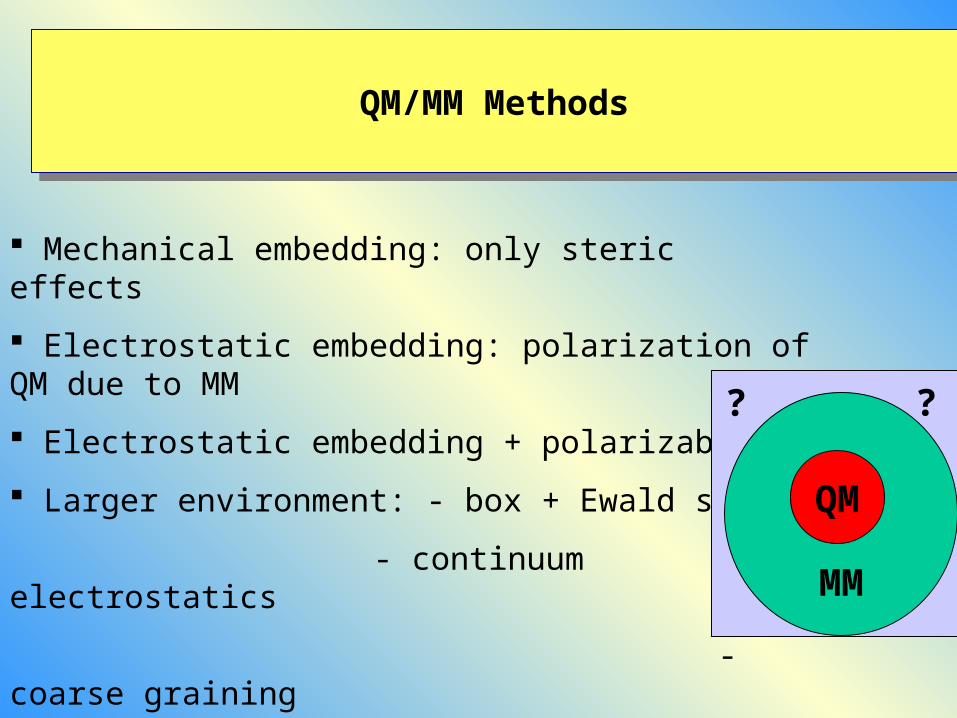
QM/MM MethodsQM/MM Methods
Mechanical embedding: only steric effects
Electrostatic embedding: polarization of QM due to MM
Electrostatic embedding + polarizable MM
Larger environment: - box + Ewald summ.
- continuum electrostatics
- coarse grainingMM
QM
? ?

Ho to study reactions and (rare) dynamical eventsHo to study reactions and (rare) dynamical events
direct MD
accelerated MD
- hyperdynamics (Voter)
- chemical flooding (Grubmüller)
- metadynamics (Parinello)
reaction path methods
- NEB (nudged elastic band, Jonsson)
- CPR (conjugate peak refinement, Fischer, Karplus)
- dimer method (Jonsson)
free energy sampling techniques
- umbrella sampling
- free energy perturbation
- transition path sampling

Ho to study reactions and (rare) dynamical eventsHo to study reactions and (rare) dynamical events
accelerated MD
- metadynamics
reaction path methods- CPR
free energy sampling techniques
- umbrella sampling

QM/MM MethodsQM/MM Methods

Subtractive vs. additive modelsSubtractive vs. additive models
- subtractive: several layers: QM-MM
doublecounting on the regions is subtracted
- additive: different methods in different regions +
interaction between the regions
MM
QM

Additive QM/MMAdditive QM/MM
total energy QM= +
QM+
MM
MM
interaction

Subtractive QM/MM: ONIOM Morokuma and co.: GAUSSIAN
Subtractive QM/MM: ONIOM Morokuma and co.: GAUSSIAN
total energy
QM
MM=
-+ MM

The ONIOM Method (an ONION-like method)First LayerBond-formation/breaking takes place. Use the "High level" (H) method.
Second LayerElectronic effect on the first layer. Use the "Medium level" (M) method.
Third LayerEnvironmental effects on the first layer. Use the "Low level" (L) method.
Sm
all
Mo
del
S
yste
m (
SM
)
Inte
rmed
iate
M
od
el S
yste
m (
IM)
Rea
l S
yste
m (
R)
Example: The binding energy of 3C-C 3HPE)
C C
Hexaphenylethane
C
(HPE)
2
Triphenylmethyl radical
(TPMR)
ONIOM: 16.4 kcal/mol
from S. Irle
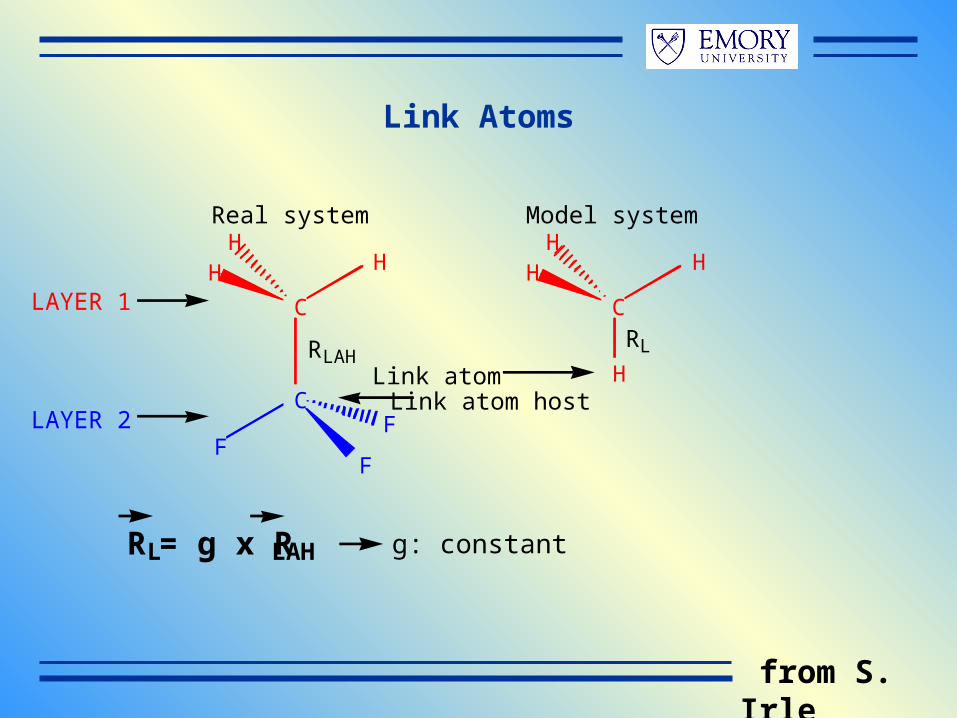
Link Atoms
C
C
FF
F
HHH
C
H
HHH
Link atomLink atom host
RL = g x RLAH
RLAH
LAYER 1
LAYER 2
Real system Model system
RL
g: constant
from S. Irle
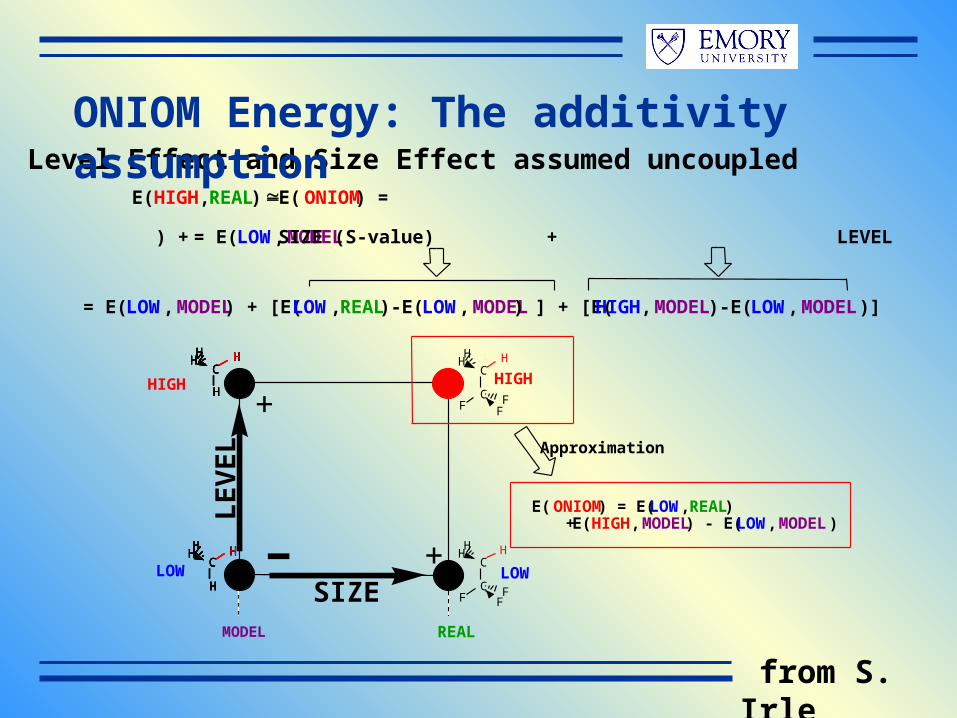
E(ONIOM) = E(LOW,REAL) + E(HIGH,MODEL) - E(LOW,MODEL)
C
CF F
F
HHH
C
CF F
F
HHH
C
H
HHH
C
H
HHH
C
H
HHH
C
H
HHH
C
H
HHH
C
H
HHH
C
H
HHH
C
H
HHH
C
H
HHH
C
H
HHH
C
H
HHH
C
H
H
HIGH HIGHHH
LOW LOW
SIZE
LE
VE
LE(HIGH,REAL) E(ONIOM) =
= E(LOW,MODEL) + SIZE (S-value) + LEVEL
Level Effect and Size Effect assumed uncoupled
Approximation
+
+-MODEL REAL
= E(LOW,MODEL) + [E(LOW,REAL)-E(LOW,MODEL) ] + [E(HIGH,MODEL)-E(LOW,MODEL)]
ONIOM Energy: The additivity assumption
from S. Irle

Choice of combination of levels is critical
S(ubstituent)-Value test: Does the low level work?
MODEL REAL
S(LOW)
LE
VE
L
SIZE
S(HIGH)If S(HIGH) = S(LOW)
E(ONIOM) = E( HIGH,REAL)
LOW
HIGH
Low level describes substituent effect as good as high level does!
Must be as close to zero as possible
S(HIGH) - S(LOW)
Combinations can be investigated using the S-Value test
Several E(HIGH,REAL) calculations necessary
S(LEVEL) = E(LEVEL,REAL) - E(LEVEL,MODEL)
from S. Irle

ONIOM Potential Energy Surface and PropertiesONIOM energy E(ONIOM, Real) = E(Low,Real) + E(High,Model) - E(Low,Model)
Potential energy surface well defined, and also derivatives are available.
ONIOM gradient G(ONIOM, Real) = G(Low,Real) + G(High,Model) x J - E(Low,Model) x J
J = (Real coord.)/ (Model coord.) is the Jacobian that converts the model system coordinate to the real system coordinate
ONIOM Hessian H(ONIOM,Real) = H(Low,Real) + JT x H(High,Model) x J - JT x H(Low,Model) x J
Scale each Hessian by s(Low)**2 or s(High)**2 to get scaled H(ONIOM)
ONIOM density (ONIOM, Real) = (Low,Real) + (High,Model) - (Low,Model)
ONIOM properties < o (ONIOM, Real)> = < o (Low,Real) > + < o (High,Model) > - < o (Low,Model) >
from S. Irle

Three-layer ONIOM (ONIOM3)
SIZE
LE
VE
L
MODEL REALINTERMEDIATE
LOW
MEDIUM
HIGH
E(ONIOM)= E(LOW,REAL)
- E(MEDIUM,MODEL)
+ E(HIGH,MODEL)
+ E(MEDIUM,INTERMEDIATE)
- E(LOW,INTERMEDIATE) +
+
+-
-MO:MO:MOMO:MO:MM
Target
from S. Irle

Additive QM/MM: linkingAdditive QM/MM: linking

Additive QM/MMAdditive QM/MM
total energy QM= +
QM+
MM
MM
interaction

/ˆ ˆ ˆ ˆ
QM MM QM MMH H H H
M
coorMMQM
M
M
M
M
Mi M M
M
iM
MMMQM H
R
B
R
A
R
qZ
r
qH
,
.int/612
, ,/
ˆˆ
Additive QM/MM: Additive QM/MM:
Elecrostatic mechanical embedding

Combined QM/MMCombined QM/MM
Amaro , Field , Chem Acc. 2003Bonds:
a) take force field terms
b) - link atom
- pseudo atoms
- frontier bonds
Nonbonding:
- VdW
- electrostatics
M
coorMMQM
M
M
M
M
Mi M M
M
iM
MMMQM H
R
B
R
A
R
qZ
r
qH
,
.int/612
, ,/
ˆˆ

Combined QM/MMCombined QM/MM
Reuter et al, JPCA 2000
Bonds:
a)
from force field

Combined QM/MM: link atomCombined QM/MM: link atom
Amaro & Field , T. Chem Acc. 2003
a) constrain or not?
(artificial forces)
relevant for MD
b) Electrostatics
- LA included – excluded
(include!)
- QM-MM:
exclude MM-host
exclude MM-hostgroup
- DFT, HF: gaussian broadening of MM point charges, pseudopotentails (e spill out)

Combined QM/MM: frozen orbitalsCombined QM/MM: frozen orbitals
Warshel, Levitt 1976
Rivail + co. 1996-2002
Gao et al 1998
M
coorMMQM
M
M
M
M
Mi M M
M
iM
MMMQM H
R
B
R
A
R
qZ
r
qH
,
.int/612
, ,/
ˆˆ
Reuter et al, JPCA 2000

Combined QM/MM: PseudoatomsCombined QM/MM: Pseudoatoms
Amaro & Field ,T Chem Acc. 2003
Pseudobond- connection atom
Zhang, Lee, Yang, JCP 110, 46
Antes&Thiel, JPCA 103 9290
No link atom: parametrize C H2 as pseudoatom
X
M
coorMMQM
M
M
M
M
Mi M M
M
iM
MMMQM H
R
B
R
A
R
qZ
r
qH
,
.int/612
, ,/
ˆˆ

Nonbonding terms:
VdW
- take from force field
- reoptimize for QM level
Coulomb:
which charges?
M
coorMMQM
M
M
M
M
Mi M M
M
iM
MMMQM H
R
B
R
A
R
qZ
r
qH
,
.int/612
, ,/
ˆˆ
Combined QM/MMCombined QM/MM
Amaro & Field ,T Chem Acc. 2003

Tests:
- C-C bond lengths, vib. frequencies
- C-C torsional barrier
- H-bonding complexes
- proton affinities, deprotonation
energies
Combined QM/MMCombined QM/MM

Subtractive vs. additive QM/MMSubtractive vs. additive QM/MM
- parametrization of methods for all regions required
e.g. MM for Ligands
SE for metals
+ QM/QM/MM conceptionally simple and applicable

Local Orbital vs. plane wave approaches:Local Orbital vs. plane wave approaches:
PW implementations
(most implementations in LCAO)
- periodic boundary conditions and large box! lots of empty space in unit cell
- hybride functionals have better accuracy: B3LYP, PBE0 etc.
+ no BSSE
+ parallelization (e.g. DNA with ~1000 Atoms)

• QM and MM accuracy
• QM/MM coupling
• model setup: solvent, restraints
• PES vs. FES: importance of sampling
All these factors CAN introduce errors in similar magnitude
ProblemsProblems

Modelling StratgiesModelling Stratgies

How much can we treat ? =How much can we afford
How much can we treat ? =How much can we afford
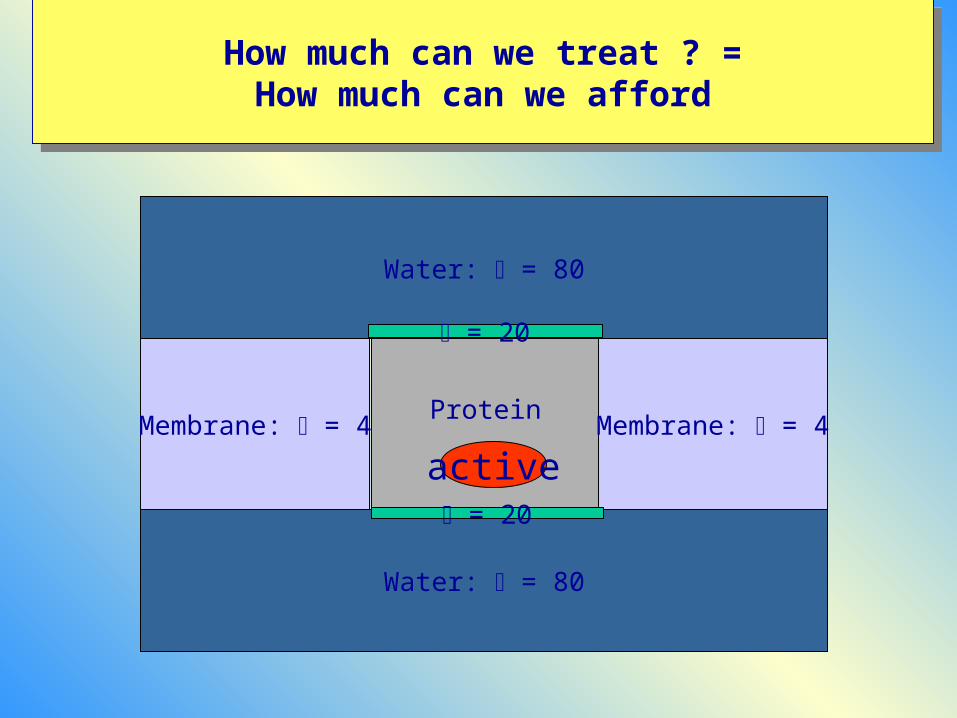
ProteinMembrane: = 4 Membrane: = 4
active
Water: = 80
Water: = 80
= 20
= 20

How to model the environmentHow to model the environment
1) Only QM (implicit solvent)
2) QM/MM w/wo MM polarization
3) Truncated systems and charge scaling
System in water with periodic boundary conditions: pbc and Ewald summation
Truncated system and implicit solvent models

How much can we treat ? =How much can we afford
How much can we treat ? =How much can we afford
Don‘t have or don‘t trust QM/MM or too complicated Only active site models
= ??
active

How much can we treat ? =How much can we afford
How much can we treat ? =How much can we afford
Protein
active
Small protein
Simple QM/MM: - fix most of the protein
- neglect polarization of environment

• solvation charge scaling
• freezing vs. stochastic boundary
• size of movable MM?
• size of QM?
First approximations: First approximations:
How much can we treat ? =How much can we afford
How much can we treat ? =How much can we afford
Protein: polarizable
active
Small protein
Simple QM/MM: - fix most of the protein
- include polarization from environment

Absolute excitation energies
S1 excitation energy (eV)
exp TD-B3LYP1
TD-DFTB
OM2/
CIS
CASSCF2 OM2/
MRCI
SORCI
vacuum 2.42 2.14 2.34 2.86 2.13 1.86
bR (QM:RET) 2.18
2.53 2.21 2.54 3.94 2.53 2.341Vreven[2003] 2 Hayashi[2000]
• TDDFT nearly zero
• CIS shifts still too small ~50%
• SORCI, CASPT2
• OM2/MRCI compares very well
0.1 0.2 1.0 0.5

Polarizable force field for environment
• MM charges
• MM polarization
RESP charges for residues in gas phase
atomic polarizabilities: = E
Polarization red shift of about 0.1 eV:

How much can we treat ? =How much can we afford
How much can we treat ? =How much can we afford
Explicit Watermolecules
pbc
Protein
active
How much can we treat ? =How much can we afford
How much can we treat ? =How much can we afford
ProteinMembrane: = 4 Membrane: = 4
active
Water: = 80
Water: = 80
= 20
= 20

Ion channelsIon channels
Membrane: = 4 Membrane: = 4
Water: = 80
Water: = 80
Explicit water
Explicit water

Implicit solvent: Generalized Solvent Boundary Potential (GSBP, B. Roux)
Implicit solvent: Generalized Solvent Boundary Potential (GSBP, B. Roux)
•Drawback of conventional implicit solvation: e.g. specific water molecules important
•Compromise: 2 layers, one explicit solvent layer before implicit solvation model.
• inner region: MD, geomopt
• outer region: fixed QM/MM
explicit MM
implicit

GSBPGSBP
Solvation free energy of point charges
rf s v

GSBPGSBP
Depends on inner coordinates!
Basis set expansion of inner density calculate reaction field for basis set
QM/MM DFTB implementation by Cui group (Madison)

Water structure in AquaporinWater structure in Aquaporin
Water structure only in agreement with full solvent simulations when GSBP is used!

-differences in protein
conformations
Problems with the PES: CPR, NEB etc.Problems with the PES: CPR, NEB etc.
Zhang et al JPCB 107 (2003) 44459

- differences in protein conformations
(starting the reaction path calculation)
- problems along the reaction pathway
* flipping of water molecules
* size of movable MM region
different H-bonding pattern
average over these effects:
potential of mean force/free energy
Problems with the PES: complex energy landscape
Problems with the PES: complex energy landscape

Ion channelsIon channels