structural and functional … · circular dichroism spectrometry ... isothermal titration...
TRANSCRIPT

STRUCTURAL AND FUNCTIONAL
CHARACTERIZATION OF TYPE III AND TYPE IV
SECRETION SYSTEM PROTEINS
ABHILASH PADAVANNIL
M. Tech
(Biotechnology and Biochemical Engineering)
A THESIS SUBMITTED FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
Department of Biological Sciences
Faculty of Science
National University of Singapore
2013

Declaration
I hereby declare that the thesis is my original work and it has been
written by me in its entirety. I have duly acknowledged all the sources of
information which have been used in the thesis.
This thesis has also not been submitted for any degree in any university
previously.
Abhilash Padavannil
17th
October 2013

To my dear parents
and to the almighty GOD

iv
Acknowledgements
First and foremost, I would like to thank the greatest teacher of all: God. PhD.
certainly has not been a cakewalk but I thank God for the wonderful opportunity. I
would like to thank anybody and everybody who has anything to do with these
projects.
It is difficult to overstate my gratitude to my PhD. supervisor, Prof. J. Sivaraman
whose passion for perfection and sense of responsibility for his students inspires
my every day. His relentless support and guidance has made, the otherwise
excruciating, Ph.D. research worthwhile. He led us by an example and always
made sure that we understood the process of research. I would like to thank him
for seeing me through the difficult times. Years to come, I will cherish these few
years I spent under his tutelage. Thank you Sir. It is my privilege to know you, to
work with you and to learn from you.
I would like to thank our collaborators Prof. Ilan Rosenshine, Prof. Yu Keung Mok
and Prof. Adrian Velazquez-Campoy for their time and effort in supporting the
research of our common interest. Special thanks to Prof. Ilan Rosenshine for
providing clones and for carefully going through the manuscript time and again
patiently. I would like to thank Dr. Jobichen Chacko for teaching and helping me
solve the structures and for always being there as an elder brother.
My sincere thanks also go to Prof. K. Swaminathan for being such a wonderful
teacher and a great human being. The search for the black cat in the dark room

v
(protein crystals) would not have been possible without the intellectual torch Prof.
Siva and you have lit for us. I would also like to thank Lissa and Tzer Fong for
equipping me with the skills necessary for protein purification and molecular
cloning.
I am also indebted to my lab mates Dr. Kumar, Rajesh, Cherlyn, Veeru, Jermy,
Pankaj, Nilofer, Priyanka, Sarath and Digant for their time and all the help.
Thank you for maintaining an affable environment in the lab. I would also like to
thank Karthik, Kuntal, Siva, Vinod, Sunil, Girish, Vivek, Vijay, Prathiba, Pavitra,
Yang Qinghua, Jack, Kangwei and Sang for sharing their experience and often
suggesting necessary experiments.
To Manjeet, Umar and Thangavelu, thanks for being such a wonderful team.
Thank you for lending me your ears when I needed and raking my brains out when
you needed. I will cherish our friendship and the time we spent in the lab burning
the mid night oil; together we struggled, we stumbled and finally haven’t we
survived?
I thank my mother, father and brother for everything that’s me. They are the
reason I keep going no matter what. Thank you for always being there for me.
Finally, a big thank you to the National University of Singapore for providing me
with an opportunity to pursue Ph.D. and for supporting me with a research
scholarship.

vi
Table of Contents
Acknowledgements ...................................................................................... iv
Summary ........................................................................................................ x
List of Tables ............................................................................................... xii
List of Figures ............................................................................................ xiii
List of Abbreviations ................................................................................ xvii
Publications ..................................................................................................xx
Chapter 1: General Introduction ................................................................. 1
1.1. Bacterial Secretion Systems ............................................................................ 2
1.1.1. Sec pathway .......................................................................................................... 4
1.1.2. Tat pathway .......................................................................................................... 6
1.1.3. Type I Secretion System (T1SS) .......................................................................... 8
1.1.4. Type II Secretion System (T2SS) ....................................................................... 10
1.1.5. Type III Secretion System (T3SS) ...................................................................... 12
1.1.6. Type IV Secretion System (T4SS) ..................................................................... 26
1.1.7. Type V Secretion System (T5SS) ....................................................................... 34
1.1.8. Type VI Secretion System (T6SS) ..................................................................... 36
1.1.9. Type VII Secretion System (T7SS) .................................................................... 39
1.2. Objectives ....................................................................................................... 40

vii
1.2.1. GrlR-GrlA regulatory complex .......................................................................... 40
1.2.2. VirD2 binding protein (VBP) of the VirD4/B T4SS .......................................... 41
Chapter 2: Structure of GrlR-GrlA Complex that Prevents GrlA
Activation of Virulence Genes ....................................................................43
2.1. Introduction ........................................................................................................ 44
2.2. Materials and Methods ...................................................................................... 48
2.2.1. Plasmid and strain construction ................................................................................. 48
2.2.2. GrlR-GrlAΔ complex structure determination .......................................................... 49
2.2.3. Analytical Ultracentrifugation ................................................................................... 51
2.2.4. Circular dichroism spectrometry ............................................................................... 51
2.2.5. Isothermal titration calorimetry ................................................................................. 52
2.2.6. Electrophoretic mobility shift assays ........................................................................ 52
2.2.7. Extracellular protein extraction and detection ........................................................... 53
2.2.8. ler-gfp promoter assay ............................................................................................... 54
2.2.9. Pull down assay and Western blot ............................................................................. 55
2.2.10. Peptide mass finger printing .................................................................................... 55
2.3. Results ................................................................................................................. 59
2.3.1. GrlR-GrlA/GrlAΔ complex purification, characterization and crystallization ......... 59
2.3.2. Overall structure of the GrlR-GrlA complex ............................................................ 62
2.3.3. Sequence and structural homology of GrlA .............................................................. 66
2.3.4. GrlR interacts with the HTH and C-terminal regions of GrlA .................................. 67
2.3.5. ler promoter region and GrlR compete for HTH motif of GrlA ............................... 76
2.3.6. The GrlR-GrlAΔ complex is functional in vivo ........................................................ 81
2.3.7. The key HTH residues are required for GrlA function in vivo.................................. 83
2.3.8. GrlR competes with the regulatory regions of flhDC operon for binding to GrlA ... 85

viii
2.3.9. GrlR competes with the regulatory regions of ehxCABD operon for binding to GrlA
............................................................................................................................................. 88
2.4. Discussion ............................................................................................................ 90
Chapter 3: Dimerization of VirD2 Binding Protein from the Type IV
Secretion System is essential for Agrobacterium induced tumor
formation in plants ......................................................................................94
3.1. Introduction ........................................................................................................ 95
3.2. Materials and Methods ...................................................................................... 97
3.2.1. Plasmid and strain construction ................................................................................. 97
3.2.2. Protein expression and purification ........................................................................... 97
3.2.3. Crystallization and data collection ............................................................................ 98
3.2.4. Analytical Ultracentrifugation ................................................................................... 99
3.2.5. Pull down assay ....................................................................................................... 100
3.2.6. Isothermal titration calorimetry ............................................................................... 100
3.2.7. Circular dichroism spectrometry ............................................................................. 101
3.2.8. Plant virulence Assay .............................................................................................. 101
3.3. Results ............................................................................................................... 102
3.3.1. Overall structure ...................................................................................................... 102
3.3.2. Sequence and structural homology.......................................................................... 106
3.3.3. VBP is a dimer ........................................................................................................ 107
3.3.4. HEPN domain of VBP is the dimerization domain ................................................. 108
3.3.5. Substitution of Asn186 with Asp disrupts the dimerization .................................... 110
3.3.6. VBP functions as a dimer in vivo ............................................................................ 114
3.3.6. Interaction of VBP with ATP and VirD2 ................................................................ 116
3.4. Discussion .......................................................................................................... 119

ix
Chapter 4: Conclusion and future directions .........................................122
4.1. Conclusion ......................................................................................................... 123
4.2. Future directions .............................................................................................. 126
References ................................................................................................................ 128

x
Summary
Bacteria inhabit most environments including the bodies of plants and animals. Protein
secretion plays an important role in modulating the way bacteria interact with their
environment. Bacteria have developed several different secretion systems to secrete the
proteins across their own cell membrane and into the host cell cytoplasm. The secreted
proteins help the bacteria to survive in these harsh environments and facilitate the host-
pathogen interactions to cause the infection. To date seven different secretion systems
(Type I to Type VII) have been identified. This thesis reports the structure and function
of a type III regulatory complex and of a type IV recruitment protein. A detailed
introduction of the bacterial secretion systems is given in Chapter1.
Attaching and effacing (AE) pathogens like the enterohemorrhagic Escherichia
coli (EHEC) and enteropathogenic E. coli (EPEC) possess type III secretion systems
(T3SS) that promote virulence. Most T3SS components and related proteins are encoded
by genes in the locus of enterocyte effacement (LEE). The LEE consists of 41 genes,
clustered in five different operons, termed LEE1-LEE5, and some additional transcription
units. The LEE genes encode type III secretion system (T3SS) proteins and three
associated regulators: Ler, GrlA and GrlR. Ler is a positive regulator for most of the LEE
operons, including grlRA. GrlA controls the expression of ler, ehxCABD and flhDC
operons. GrlR binds to GrlA and suppresses its function.
In chapter II, we report the crystal structure of GrlR-GrlAΔ (aa 1-106) complex
(2:1) and its functional characterization. We show that GrlR interacts with the Helix-
Turn-Helix (HTH) motif of GrlA. Moreover, GrlA binds to the promoter DNA fragments
of ler, ehxCABD and flhDC, and GrlR outcompetes with these promoter DNA sequences

xi
for the HTH motif of GrlA. These findings provide mechanistic insight into a novel
regulatory module for EPEC and EHEC virulence, two important pathogens that cause
devastating diseases.
Type IV secretion system (T4SS) is the only bacterial secretion system known to
translocate DNA in addition to protein substrates. T4SS translocate DNA not only to
other bacteria but also to higher eukaryotic organism in a contact dependent manner.
VirB/D4 system of Agrobacterium tumefaciens is a typical example of T4SS. The
proteins involved in translocation include, the DNA processing and packaging proteins,
the VirB secretion system apparatus proteins and the VirD4 coupling protein. VirD2
binding protein (VBP) is a cytoplasmic protein that plays a key role in recruiting the T-
DNA-protein complex to the VirD4 coupling protein. Thus VBP is an important protein
in the T4SS translocation pathway.
In chapter III, we report the crystal structure of the C-terminal domain of VBP
along with the biophysical and in vivo functional studies. Sequence and structural
analysis shows that the C-terminal domain is homologous to the HEPN domain of Sacsin
protein. Biophysical experiments reveal that VBP is a dimer in solution and HEPN
domain (C-terminal domain) is the dimerization domain of VBP. Furthermore, the in vivo
functional studies with full length VBP have shown that only dimeric VBP can recruit the
T-DNA-protein complex to the VirD4 coupling protein and lead to tumor formation. This
study sheds light on the function of VBP in the recruiting complexes and thus widens the
understanding of T4SS pathway in A.tumefaciens as well as in many pathogenic bacteria
such as Bartonella, Bordetella, Legionella and other species which have homologous
T4SS. The conclusion and future directions are discussed in chapter IV.

xii
List of Tables
Table No. Page No.
Table 1.1 T3SS effectors of pathogens for humans 18,19
Table 2.1 Strains and plasmids 57
Table 2.2 Primers used to amplify the promoter regions 58
Table 2.3 Crystallographic data collection and refinement statistics 64
Table 2.4 Stoichiometry, affinity and favorability of GrlA-GrlR
interactions
73
Table 3.1 Crystallographic data collection and refinement statistics 104
Table 3.2 Structural homologs of HEPN domain as predicted by DALI
search
106
Table 3.3 Stoichiometry, affinity and favorability of VBP-AMPPNP
interactions
118

xiii
List of Figures
Fig. No. Page
no.
Fig. 1.1. Summary of known bacterial secretion systems. 3
Fig. 1.2. a. A schematic representation of the bacterial pre-protein translocase
subunits.
b. A general schematic representation of the secretion process.
5
Fig. 1.3. Ribbon diagram of the E. coli type I secretion system. 9
Fig. 1.4. Model of the Gsp secreton assembly (left), and comparison with a model for
type IV pilus biogenesis in P. aeruginosa (right).
11
Fig. 1.5. Needle complex of S. typhimurium. 13
Fig. 1.6. Model for substrate recognition and delivery of proteins by type III secretion
machines.
14
Fig. 1.7. Genes involved in EHEC pathogenesis. 22
Fig. 1.8. T3SS effector functions of path physiologic importance. 25
Fig. 1.9. Schematic representations of the different type-IV-dependent mechanisms. 27
Fig. 1.10. Topologies of the VirB/D4 subunits of the A. tumefaciens T4SS. 29
Fig. 1.11. Schematic representation of the cellular consequences T4SS. 32
Fig. 1.12. Schematic overview of the type V secretion systems. 35
Fig. 1.13. A model for type VI secretion system assembly and function. 38
Fig. 2.1. Schematic of the regulatory circuits related to the GrlR-GrlA complex. 45

xiv
Fig. 2.2. Structure of GrlR. 47
Fig. 2.3. Size-exclusion chromatography profile of GrlR-GrlA complex. 59
Fig. 2.4. Size-exclusion chromatography profile of GrlR-GrlAΔ complex. 60
Fig. 2.5. AUC profile of GrlR-GrlA protein complex. 61
Fig. 2.6. GrlR-GrlAΔ complex crystals from the screen. 61
Fig. 2.7. Schematic representation of GrlR and GrlA proteins with motifs. 62
Fig. 2.8. Crystal structure of GrlR-GrlAΔ complex. 63
Fig. 2.9. Structure of GrlA. 65
Fig. 2.10. The topology diagram of the GrlA molecule. 66
Fig. 2.11. Structure of the GrlR-GrlAΔ interacting surface. 67
Fig. 2.12. Final 2Fo-Fc electron density map (contoured at 1 σ) for the key residues of
GrlAΔ.
68
Fig. 2.13. Structure of the GrlR-GrlAΔ interactions. 68
Fig. 2.14. Circular dichroism spectroscopic analysis of various MBP-GrlA constructs. 69
Fig. 2.15. Role of the C-terminal region and the Helix-Turn-Helix (HTH) motif region
of GrlA in GrlR-GrlA interactions.
71
Fig. 2.16. Role of the C-terminal region and the Helix-Turn-Helix (HTH) motif region
of GrlA in GrlR-GrlA interactions. (Cont…)
72
Fig. 2.17. Role of the key residues in the Helix-Turn-Helix (HTH) motif region in
GrlR-GrlAΔ interactions.
74
Fig. 2.18. Role of the key residues in the Helix-Turn-Helix (HTH) motif region in
GrlR-GrlAΔ interactions. (Cont…)
75

xv
Fig. 2.19. Interaction between GrlR and different GrlA constructs. 76
Fig. 2.20. Interaction of GrlA with the ler regulatory region in vitro. 77
Fig. 2.21. Competitive EMSA aimed at testing competition between Pler and 6His-GrlR
for binding to MBP-GrlA.
78
Fig. 2.22. Competitive EMSA to study the formation of GrlR-GrlA complex. 79
Fig. 2.23. EMSA to verify the binding of DNA to preformed GrlR-GrlA complex. 80
Fig. 2.24. In vivo functionality of GrlR-GrlAΔ asymmetric complex. 81
Fig. 2.25. In vivo functionality of GrlR-GrlAΔ asymmetric complex. (Cont…) 82
Fig. 2.26. In vivo analysis of the importance of key HTH motif residues in GrlA –DNA
binding.
84
Fig. 2.27. In vivo analysis of the importance of key HTH motif residues in GrlA –DNA
binding. (Cont…)
85
Fig. 2.28. Interaction of MBP-GrlA with the flhDC regulatory region. 86
Fig. 2.29. Interaction of MBP-GrlA mutants with flhDC regulatory region. 87
Fig. 2.30. Competitive EMSA carried out to test the competition between flhDC
regulatory region and 6HisGrlR for binding to MBP-GrlA.
88
Fig. 2.31. Interaction of GrlA with the ehxCABD regulatory region. 89
Fig. 2.32. EMSA with ehxCABD regulatory region and mutant MBP-GrlA proteins. 89
Fig. 2.33. Competitive EMSA carried out to test the competition between ehxCABD
regulatory region and 6HisGrlR for binding to MBP-GrlA.
90
Fig. 3.1. Schematic representation of VBP and its domains. 102
Fig. 3.2. Structure of the HEPN domain of VBP. 103

xvi
Fig. 3.3. Dimer interface of HEPN dimer. 105
Fig. 3.4. A sample 2Fo-Fc electron density map (contoured at 1 σ) of HEPN domain
of VBP.
105
Fig. 3.5. Gel filtration profile of VBP. 107
Fig. 3.6. AUC profile of VBP. 108
Fig. 3.7. Comparison of gel filtration profiles of NT domain and HEPN domain of
VBP.
109
Fig. 3.8. AUC profile of HEPN domain of VBP. 109
Fig. 3.9. AUC profile of NT domain of VBP. 110
Fig. 3.10. Comparison of gel filtration profiles of HEPN domains of VBP. 111
Fig. 3.11. AUC profile of HEPN Asn186Asp domain of VBP. 111
Fig. 3.12 CD spectroscopy of HEPN domain with/without N186D substitution. 112
Fig. 3.13. Comparison of gel filtration profiles of VBP with/without substitution
N186D.
112
Fig. 3.14 AUC profile of VBP Asn186Asp. 113
Fig. 3.15 CD spectroscopy of VBP with/without N186D substitution. 113
Fig. 3.16. The effect of VBP mutations on tumorigenesis. 115
Fig. 3.17. ITC profile for VBP/ VBP N186D / HEPN domain vs. AMPPNP binding. 117
Fig. 3.18. In vitro pull down assay.
118
Fig. 3.19. Schematic representation shows the induction of tumor in plants by
Agrobacterium and the role of VBP.
120

xvii
List of Abbreviations
ABC ATP binding cassette
AE Attaching and Effacing
AMP-PNP Adenylyl-imidodiphosphate
ARF ADP ribosylation factor
AT I Autotransporter-1
AT2 Autotransporter-2
EAEC Enteroaggregative Escherichia coli
EDTA Ethylenediamine tetraacetic acid
EHEC Enterohemorrhagic Escherichia coli
EMSA Electrophoretic mobility shift assay
EPEC Enteropathogenic Escherichia coli
ETEC Enterotoxigenic Escherichia coli
GrlA Global regulator of LEE, activator
GrlR Global regulator of LEE, repressor
HEPN Higher eukaryotes and prokaryote nucleotide binding domain
HTH Helix-turn-helix

xviii
ITC Isothermal titration calorimetry
LEE Locus of enterocyte effacement
NDSB Non-detergent sulfobetaine
NCS Non-crystallographic symmetry
NLS Nuclear localization signal
NTD Nucleotidyl transferase domain
PT Pertusis toxin
rmsd Root mean square deviation
SAD Single Wavelength Anomalous Diffraction
Sec Secretory
SPI-1 Salmonella pathogenicity island-1
SPI-2 Salmonella pathogenicity island -2
SRP Signal recognition particle
T1SS Type I secretion system
T2SS Type II secretion system
T3SS Type III secretion system
T4SS Type IV secretion system
T5SS Type V secretion system

xix
T6SS Type VI secretion system
T7SS Type VII secretion system
Tat Twin arginine translocation
VBP VirD2 binding protein
Vir Virulence

xx
Publications
1. Structure of GrlR-GrlA Complex that Prevents GrlA Activation of Virulence
Genes
Abhilash Padavannil, Chacko Jobichen, Erez Mills, Adrian Velazquez-Campoy,
Mo Li, Ka Yin Leung, Yu Keung Mok, Ilan Rosenshine and J. Sivaraman
Nature Communications 4, Article number: 2546 doi: 10.1038/ncomms3546
2. Dimerization of VirD2 Binding Protein from the Type IV Secretion System is
essential for Agrobacterium induced tumor
Abhilash Padavannil, Chacko Jobichen, Yang Qinghua, Liu Yang, Shen Q. Pan,
J Sivaraman (2013) (Submitted)

1
Chapter 1: General Introduction

2
1.1. Bacterial Secretion Systems
Bacteria are among the first life forms on earth. They inhabit most environments
including the bodies of plants and animals, acidic hot springs, earth's crust, organic
matter and even radioactive waste, providing outstanding examples of mutualism. They
cause a vast number of diseases in humans, plants and animals. Protein secretion plays an
important role in modulating the way the bacteria inhabit in and interact with the
environment. This is more so when it is interacting with a larger host organism (Tseng et
al., 2009). This essential process of protein secretion is responsible for pathogenesis and
symbiosis, the biogenesis of membranes and cell walls, motility, nutrient scavenging and
uptake. The secreted proteins enter the host cell and modify the host physiology to enable
bacterial colonization (Holland, 2010). While several specialized secretion systems have
evolved, especially in Gram-negative bacteria to enable translocation of proteins
(effectors and toxins) across the double membrane, Gram-positive bacteria seem to utilize
rather generalized secretion systems to cater to the need for translocation of virulence
proteins (Papanikou et al., 2007).
Some bacteria can translocate DNA into the host cell which is later incorporated
into the host genome thereby altering not only the physiology but also the genetic
makeup of the host cell. Seven different types of secretion systems have been identified
so far, six of which are predominant in Gram-negative bacteria and one in Gram-positive
bacteria. Fig.1.1 summarizes the known secretion systems (Tseng et al., 2009). In Gram-
negative bacteria the proteins are translocated either in a single step or in a two step
process. Single step translocation involves direct translocation of proteins through the
outer membrane using type I, type III, type IV or type VI pathways. In a two step process

3
the proteins are translocated first into the periplasm via the Sec or Two-arginine (Tat)
pathways and then translocated across the outer membrane via type II, type V or less
commonly, the type I or type IV machinery (Tseng et al., 2009).
Fig. 1.1. Summary of known bacterial secretion systems. HM: Host membrane;
OM:outer membrane; IM:inner membrane; MM: mycomembrane; OMP: outer membrane
protein; MFP: membrane fusion protein. ATPases and chaperones are shown in yellow
(Tseng et al., 2009).
While in Gram- positive bacteria, the proteins are translocated by a single step via
Sec or Tat pathways. Mycobacterium despite being Gram-positive has a hydrophobic
outer cell wall called the mycomemberane. A specialized type VII secretion system helps
in the translocation of protein across the mycomemberane (Tseng et al., 2009). Sec
pathway and Tat translocation pathway are simple and more generalized pathways
present in archaea, prokaryotes and eukaryotes (Tseng et al., 2009). The following
sections briefly discuss these pathways.

4
1.1.1. Sec pathway
The Sec pathway is ubiquitous and essential for viability in all three domains of
life. In addition, the Sec pathway acts as the entry point for many of the other protein
export and sorting pathways (Papanikou et al., 2007). Protein translocation through Sec
pathway is multi-stage reaction that involves post translational modification. This process
can be divided into three distinct stages
a. Protein sorting and targeting. Secretory proteins called the pre-proteins carry
cleavable amino (N)-terminal signal peptides. These signal peptides act as address
tags to be sorted from cytoplasmic proteins. They are recognized directly by
piloting factors, such as the ribonucleoprotein signal-recognition particle (SRP)
(Luirink and Sinning, 2004) or the SecB chaperone (Randall and Hardy, 2002;
Schierle et al., 2003). The resulting SRP-pre-protein or SecB-pre-protein
complexes are targeted to the membrane receptor FtsY and SecA respectively.
SecA does not contribute to the SRP-translocation; however in case of
translocation of long, hydrophilic segments SecA is recruited to catalyse the
export.
b. Translocation. The translocase consists of membrane embedded protein-
conducting channel built of the SecY, SecE and SecG polypeptides and a
molecular motor, the SecA ATPase which drives the translocation at the expense
of metabolic energy or the proton-motive force.
c. Release and maturation. In this last stage the signal peptides are cleaved off by
the signal peptidases converting the pre-proteins to mature proteins. The mature
proteins are folded correctly on the trans side of the membrane. (Gruber et al.,

5
2006; Mogensen and Otzen, 2005; Nakamoto and Bardwell, 2004). SecYEG
heterotrimeric complex forms the core of the translocase (Brundage et al., 1990)
Fig. 1.2.
Fig. 1.2. a. A schematic representation of the bacterial pre-protein translocase
subunits. The translocase consisits of the Sec YEG pre-protein-conducting channel
(yellow) and the ATPase motor SecA (red). b. A general schematic representation of
the secretion process. Secretory pre-proteins (thick orange line) are synthesized with
amino-terminal signal peptides and are targeted to the translocase either by the ribosome
–bound signal-recognition particle (SRP) or by the tetrameric SecB chaperone
(Papanikou et al., 2007).

6
SecA ATPase provides the necessary chemo-mechanical energy conversion for
the translocation (Baud et al., 2002; Karamanou et al., 1999). SecYEG and SecA together
form the active holoenzyme. SecA binds to SecYEG with higher affinity than to the
acidic phospholipids. Cytoplasmic exposed loops of SecY are the possible interaction
sites of SecA. Several auxiliary complexes are also associated to SecYEG translocation
complexes.
1.1.2. Tat pathway
The twin-arginine translocation pathway is one of the two general pathways used
by the bacteria for protein translocation. However, it is unique in that it translocates well
folded proteins (Palmer and Berks, 2012). Translocation of well folded proteins is a
challenging task because they have a much larger cross-section than an unfolded protein
and so require a larger transport pathway. In addition, they adopt diverse range of shapes
and sizes, making it difficult to seal tightly around the protein during transport to
preserve the membrane permeability barrier. Tat pathway is also conserved in plants
where it is present in the thylakoid membranes and plays a key role in photosynthesis
(Mori and Cline, 2002).
The translocation of proteins in Tat pathway is carried out by the integral
membrane proteins TatA (Settles et al., 1997), TatB (Sargent et al., 1998; Settles et al.,
1997) and TatC (Bogsch et al., 1998). TatA and TatB have a single transmembrane helix
followed by an amphipathic helix, whereas TatC has multiple transmembrane helices
(Rollauer et al., 2012; Walther et al., 2010). Proteins to be translocated through the Tat
pathway are targeted to the system by N-terminal signal peptides possessing a twin-
arginine-containing sequence motif that is recognized by the TatC protein within a TatBC

7
complex (Frobel et al., 2012). Once the targeted proteins bind to the TatBC complex, Tat
A is recruited to form a transient TatABC- containing translocation site (Alami et al.,
2003; Mori and Cline, 2002) that facilitates transport by perturbing the membrane bilayer
(Celedon and Cline, 2013; Palmer and Berks, 2012). TatC is the core organizing
component of the Tat pathway, directly and dynamically binding substrate, TatB and
TatA (Fritsch et al., 2012; Frobel et al., 2011), and maintaining interactions during the
transport step (Gerard and Cline, 2006; Mori and Cline, 2002). TatC is the largest and
most conserved element of the Tat translocation machinery.
The presence of an outer membrane in Gram-negative bacteria has forced the
organism to develop a myriad of specialized secretion systems that would enable them to
translocate the protein not only across the double membrane but also sometimes into the
host-cell cytoplasm (Tseng et al., 2009). Some of these secretion systems are
complemented by the more general secretory pathways like the Sec and the Tat pathways
(e.g. type II and type V) and others work independent of the general secretory pathways
(e.g. type I, type III, type IV and type VI pathways).
Different secretion systems from Gram-negative bacteria are discussed below
with special emphasis on type III secretion system (T3SS) and type IV secretion system
(T4SS) which are the main focus of this dissertation.

8
1.1.3. Type I Secretion System (T1SS)
Type I secretion is widespread in gram-negative bacteria. The T1SS allows the
secretion of proteins of various sizes and functions from the cytoplasm to the
extracellular medium in a single step without a stable periplasmic intermediate (Hueck,
1998). The proteins targeted to the T1SS have a signal peptide at the carboxy (C) -
terminus of the protein. The signal peptide is not cleaved during the translocation and
remains intact with the secreted protein. The signal peptide usually contains distinctive
glycine rich repeats (GGXGXDXXX) that specifically bind calcium ions forming
peculiar beta-sandwich or beta-roll structures with calcium ions in the turns. Several
studies have shown that these repeats are necessary for the activity of the secreted
proteins (Delepelaire, 2004).
The translocation machinery is made of three different proteins viz., ATP-binding
cassette protein (ABC), a membrane fusion protein and an outer membrane protein (C.
Wandersman, 1996). The ATP-binding cassette protein consists of a NBD (nucleotide-
binding domain of the ABC class with its conserved features) fused to a membrane
domain (transmembrane domain, TMD) and is localized in the cytoplasmic membrane.
This recognizes the substrate via its C-terminal secretion signal and is responsible for the
specificity of the secretion process. Membrane fusion protein (MFP) or adaptor protein
consists of a short cytoplasmic domain at the N-terminus followed by a membrane anchor
and a large periplasmic domain. The third member of the system is the outer membrane
protein (OMP) of the TolC class. The X-ray structure of OMP is shown in the Fig.1.3.
(Koronakis et al., 2000).
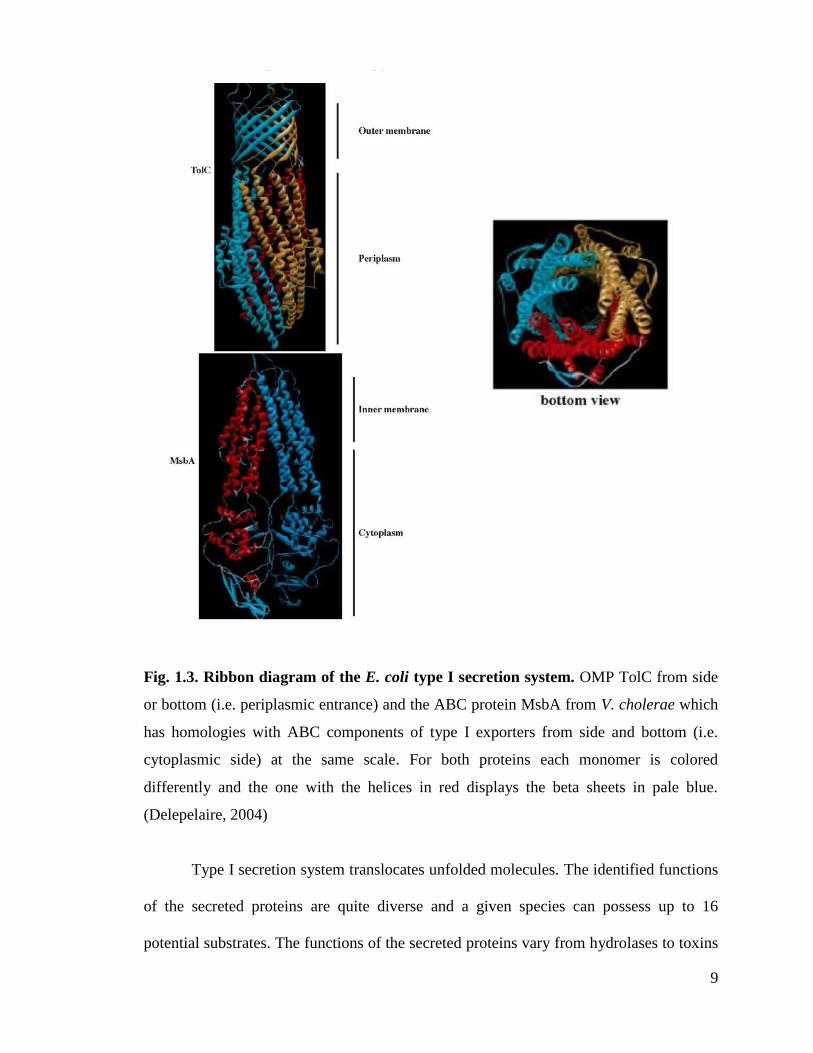
9
Fig. 1.3. Ribbon diagram of the E. coli type I secretion system. OMP TolC from side
or bottom (i.e. periplasmic entrance) and the ABC protein MsbA from V. cholerae which
has homologies with ABC components of type I exporters from side and bottom (i.e.
cytoplasmic side) at the same scale. For both proteins each monomer is colored
differently and the one with the helices in red displays the beta sheets in pale blue.
(Delepelaire, 2004)
Type I secretion system translocates unfolded molecules. The identified functions
of the secreted proteins are quite diverse and a given species can possess up to 16
potential substrates. The functions of the secreted proteins vary from hydrolases to toxins

10
for the host, the first one being the HlyA from uropathogenic Escherichia coli and many
HlyA relatives with different specificities, from bifunctional adenycyclase-hemolysin
from B. pertussis to the tubulin interacting RtxA toxin from V. cholera.
1.1.4. Type II Secretion System (T2SS)
T2SS forms a key component of the general secretory pathway (GSP). GSP is a
two step translocation pathway. The first step involves translocation of the protein across
the cytoplasmic membrane. The targeted proteins are usually synthesized as pro-proteins
with a cleavable N-terminal signal peptide. This pro-protein is targeted and transported
through the inner membrane via Sec pathway (de Keyzer et al., 2003). The signal peptide
is then cleaved by the leader peptidase releasing the mature protein into the periplasm.
These set of events constitute the first step and is known as general export pathway
(GEP). The mature protein is then translocated by machinery, an extension of the GEP, to
assist its translocation across the OM. These set of events constitute the second step and
is called the terminal branch of the GSP (Filloux, 2004).
T2SS is considered the main terminal branch (MTB) of the GSP. T2SS can also
translocate well folded proteins transported into the periplasmic space by the Tat
pathway. The periplasmic form might correspond to an extremely short period or may not
exist, both steps of membrane translocation being then tightly connected. The T2SS is
broadly conserved in Gram-negative bacteria and involves a set of 12–16 different
proteins named GspC-M, GspAB, GspN, GspO, and GspS (Filloux, 2004). The type II
secretion system is highly reminiscent of the type IV piliation assembly. A model of Gsp
secreton assembly is shown along with a comparison with a model for type IV pilus
biogenesis in the Fig. 1.4. Based on findings about the sub cellular localization of the Gsp

11
components, protein–protein interactions between Gsps’ and their multimerisation status,
structural data and electron microscopy observation, a working model has been proposed
that strikingly runs both systems in parallel (Rollauer et al., 2012).
Fig. 1.4. Model of the Gsp secreton assembly (left), and comparison with a model for
type IV pilus biogenesis in P. aeruginosa (right). On the left, the Gsp dependent
exoproteins, shown as grey circles, have initially been exported across the IM via the Sec
or Tat machinery (not shown). The exoproteins are subsequently recognised by the Gsp
machinery and transported across the OM via the secretin, GspDQ. The secretin GspDQ is
shown as a homomultimeric ring forming a channel with a large central opening. The
homologous components of the type IV piliation assembly in P. aeruginosa are shown on
the right for comparision. The major pilin, PilA, is represented in orange, whereas minor
pilins (PilE, PilV-X and FimT-U) involved in the type IV pilus assembly but that are not
found in the extracellular structure are in green. The GspKx homologue, PilX, is
represented in dark green. Type IV pili are retractile appendages. Retraction is promoted
by the PilT NTPase in P.aeruginosa (Filloux, 2004)

12
T2SS plays a very important role in bacterial pathogenesis. Genes encoding the
core components of the T2SS are present in many different pathogens. The fact that most
T2SS dependent enzymes are degradative in nature suggests that the system promotes the
damage of host cells and tissue, be it plant or animal. Individual exoenzymes have also
been shown to contribute to virulence. Prominent examples include the ADP-ribosylating
toxins of enterotoxigenic E. coli (heat labile toxin), V. cholerae (cholera toxin) and P.
aeruginosa (exotoxin A). T2SS often works in coordination with other secretion systems
to achieve full virulence; for example T2SS and T4SS are operative in L. pneumophila .
T2SS and T3SS function in X. campestris (da Silva et al., 2002), and T1SS, T2SS, T3SS
and T5SS exist in B. pseudomallei and P. aeruginosa (Holden et al., 2004; Stover et al.,
2000).
1.1.5. Type III Secretion System (T3SS)
T3SS is amongst the bacterial secretion systems that can inject the virulence
factors or the effector proteins into the host organism. They are present in both
pathogenic bacteria as well as endosymbionts. The bacteria inject T3S toxins, called
effectors, through a nano-machine weapon, called injectisome, and involves the assembly
of a pore in the eukaryotic cell membrane formed by two/three type III secreted proteins
called ‘translocators’(Mota and Cornelis, 2005). Proteins are thought to travel this
pathway in a largely unfolded manner, and families of customized cytoplasmic
chaperones, which specifically bind cognate secreted proteins, are essential for secretion
(Akeda and Galan, 2005).
T3SS mainly consists of three groups of proteins: the first group comprises the
secretion system apparatus and is known as structural proteins, the second group which

13
helps in the translocation of proteins is known as translocators and the third group which
is transported using the T3SS is known as effectors (Coburn et al., 2007).
The injectisome was originally discovered in Salmonella typhimurium (Kubori et
al., 1998b), and later identified in several other bacteria. (Blocker et al., 2001; Sekiya et
al., 2001) (Daniell et al., 2001). It consists of a multi-ring base, which anchors the
structure to the bacterial envelope, and a needle-like projection that protrudes several
nanometers from the bacterial surface (Fig. 1.5).
Fig. 1.5. Needle complex of S. typhimurium. a. Electron micrographs of negatively
stained isolated needle complexes. b. Cross-section of the structure of the needle complex
indicating the location of its different substructures. c. Surface rendering of the structure
of the needle complex. Shown here are different views of the structure of a 20-fold
complex with 20-fold symmetry imposed (Kubori et al., 1998a).
The base is traversed by cylindrical substructure that connects the needle to the
basal side of the base substructure. The entire needle complex is traversed by a narrow

14
channel (~28Å in diameter), which functions as the conduit for proteins traveling through
this secretion pathway. The opening of the channel that traverses the needle complex is
so narrow that the proteins can be translocated only in unfolded state (Fig 1.6).
Fig. 1.6. Model for substrate recognition and delivery of proteins by type III
secretion machines. The effector–chaperone complex is recognized by the secretion
machinery, including a type-III-secretion-associated ATPase. The ATPase ‘strips’ the
chaperone from the complex, which remains within the bacterial cell, and mediates the
unfolding and ‘threading’ of the effector protein through the central channel of the needle
complex. A ‘translocator complex’ made up of proteins also secreted by the T3SS is
assembled on the host cell membrane and mediates the passage of the effector proteins
through the target cell membrane. The translocated effectors re-fold within the host cell
to carry out their function (Collazo and Galán, 1996).
Type III secretion machines translocate a selected number of substrate proteins.
Some bacteria encode more than one T3SS simultaneously. Therefore, the mechanisms of
substrate recognition must ensure a level of specificity that helps in targeting the correct

15
substrates to the appropriate machine. Furthermore, studies indicate that the secretion
process follows a hierarchy with a predetermined order in which different proteins are
engaged and secreted by these machines (Collazo and Galán, 1996) (Pettersson et al.,
1996) (Wulff-Strobel et al., 2002). Therefore, it is obvious that the mechanisms of
substrate recognition are complex, involving multiple signals and accessory proteins
(Sorg et al., 2005).
Most proteins targeted by the T3SS posses a secretion signal within the first 20-30
amino acids (Sory et al., 1995) (Schesser et al., 1996). The signals are not cleaved on
secretion and do not seem to have any conserved features. Absence of conserved signal
sequences in the translocated proteins prompts the possibility of some other mechanism
that ensures specificity. First, it is possible that the unstructured flexible segments at the
amino terminus serve as a type III secretion signal. Second, accessory proteins such as a
family of customized cytosolic chaperones that specifically bind at least some of the type
III secreted proteins (Wattiau and Cornelis, 1993) and help in translocation.
The type-III-secretion-associated chaperones are small, acidic, dimeric proteins,
which unlike other chaperones, lack ATP-binding or ATP-hydrolyzing activities
(Feldman and Cornelis, 2003). The T3SS chaperones do not share significant sequence
similarity however the structures are related In general, these chaperones bind a ,50–100
amino acid domain of the secreted protein, located immediately downstream from the N-
terminal secretion signal. The co-crystal structures of the chaperones and their cognate
secreted protein showed that these chaperones maintain the chaperone-binding domain of
their cognate secreted proteins in a non-globular conformation that nevertheless
maintains secondary structure. This observation has led to the proposal that at least one of

16
the functions of these chaperones must be to ‘prime’ the secreted proteins for rapid
unfolding before secretion (Stebbins and Galán, 2003). The chaperones also play a key
role in targeting the secreted protein to the type III secretion apparatus.
The secretion machine, in addition to recognizing secretion signals on the
chaperone – effector complex, must ‘strip’ the chaperone from the effector protein
because T3SS-associated chaperones remain in the bacterial cytosol after delivery of the
effector proteins to the secretion apparatus. Moreover the limitation in size of the
secretion channel (estimated to be, 28Å) dictates that the effector domain present at the
C-terminus of the chaperone binding domain, be unfolded before secretion. Highly
conserved ATPases associated with the T3SS apparatus play a key role in dissociation of
the chaperone-effector complex and the unfolding of the effector domain of the effector
protein (Müller et al., 2006). Furthermore, this unfolding activity may be critical for
energizing the secretion process.
The needle complex alone however, is not capable of mediating protein injection
and needs the activity of a subset of conserved proteins called translocators that are
themselves secreted by the T3SS (Sory et al., 1995) (Håkansson et al., 1996). This group
of proteins inserts itself into the target cell membrane forming a channel through which
the effector proteins can pass on their way to the target cell cytosol (Håkansson et al.,
1996; Sory et al., 1995). A possible scenario is that the needle actually ‘docks’ onto the
pore or channel made up of the translocators thereby allowing the direct delivery of
effector proteins into the target cell. One such structures identified in Yersinia
enterocolitica, is formed by a single protein, LcrV (Mueller et al., 2005). Another more
complex structure has been visualized in the T3SS of enteropathogenic E. coli and some

17
plant pathogenic bacteria (Roine et al., 1997). This structure, which is also formed by a
single protein (for example, EspA in the case of the E.coli T3SS), takes the form of a
long appendage that extends from the tip of the needle and presumably serves as a
‘bridge’ linking the needle with the bacterial translocators on the target cell membrane.
T3SS delivers a unique arsenal of effector proteins, to suit the specific needs of
the bacteria that harbor them. These proteins delivered by different T3SSs can modulate
or interfere with a vast array of cellular functions including actin and tubulin dynamics,
gene expression, vesicular trafficking, programmed cell death and cell cycle progression.
Most of the effector proteins mimic the host cell proteins and thereby modulate the host
cellular functions (Fu and Galan, 1999). For example, the Salmonella SPI-1 T3SS
effector protein SopE is a Rho-family GTPase exchange factor (GEF) that shares no
sequence or structural similarity with eukaryotic GEFs (Hardt et al., 1998) However, the
crystal structure of the complex of SopE with its target Rac1 showed that the interaction
leads to an outcome (that is, conformational changes in the critical switch 1 and switch 2
regions of Rac1) that is nearly indistinguishable from that of the interaction of a bona fide
eukaryotic GEF and the same target (Buchwald et al., 2002).. A review of T3SS effectors
of pathogens for humans is provided in the Table 1.1.
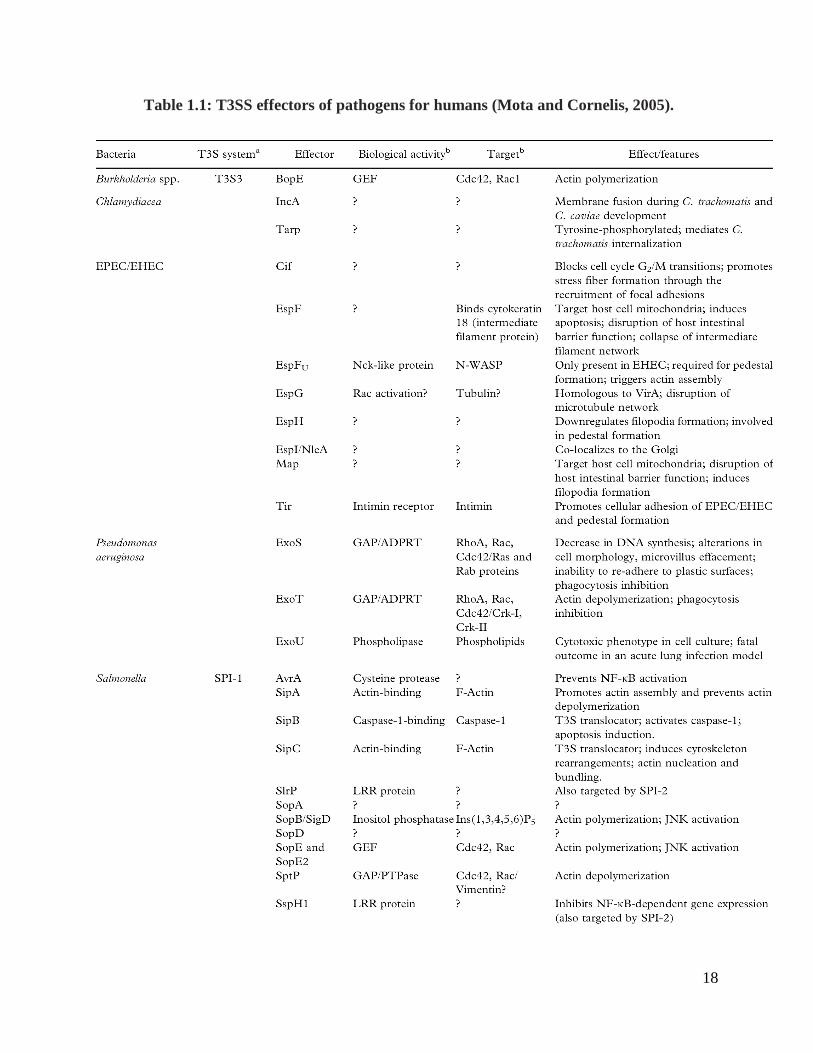
18
Table 1.1: T3SS effectors of pathogens for humans (Mota and Cornelis, 2005).

19
Table 1.1 (continued): T3SS effectors of pathogens for humans
Note: a Some bacteria possess more than one T3S in their genome. The different T3S
systems are identified for each bacterium by their most common names. SPI (Salmonella
Pathogenicity Island)-1 and SPI-2 are pathogenicity islands in the Salmonella
chromosome that encode two distinct T3S systems. b Refs – References. See text for
other relevant references. ?:. Function unknown

20
Major studies on T3SS were conducted in Yersinia (Cornélis, 1987) Shigella
(Lindberg and Pál, 1993), Salmonella (Pang et al., 1995), E. coli (Donnenberg et al.,
1993), Pseudomonas and various plant pathogens like Erwinia, Pseudomonas, Ralstonia,
and Xanthomonas species (Bonas, 1994). A brief description of some of the major
pathogenic bacteria species with T3SS is given below.
Salmonella species Salmonella contain two T3SSs, encoded by two PAIs, namely SPI-1 and SPI-2.
These two T3SSs play different roles during pathogenesis. SPI-1 is required for initial
penetration of the intestinal mucosa and SPI-2 is necessary for subsequent stages of
infection. A broad spectrum of diseases is caused by Salmonella spp. These include
gastroenteritis, bacteremia, and enteric fever. S. enterica serovar typhi causes typhoid
fever in humans (Pang et al., 1995). S. enterica and S. enteritidis are major causative
agents of food poisoning.
Pathogenic E. coli
E. coli belongs to the family Enterobacteriaceae. Most of the E. coli strains are
harmless and are found in the intestines of mammals. The harmless strains are part of the
normal flora of the gut, and can help their hosts by producing vitamin K2, or by
preventing the establishment of pathogenic bacteria within the intestine. However, certain
strains like Enterohemorrhagic E. coli (EHEC), Enteropathogenic E. coli (EPEC),
Enterotoxigenic E. coli (ETEC), Enteroinvasive E. coli (EIEC) and Enteroaggregative E.
coli (EAEC), are virulent and cause a wide variety of diseases ranging from diarrhea to
hemolytic uremic syndrome.

21
AE pathogens
Attaching and Effacing pathogens (AE) pathogens produce shiga toxin
(verotoxin). Certain serotypes cause enteritis, colitis and diarrhea in humans and a
number of different animal species by expressing a virulence factor protein called intimin
which allows intimate attachment of the organism to the microvillus brush border of
enterocyte forming a characteristic attaching and effacing lesion. AE pathogen infection
results in the morphological alteration of tight junctions during natural disease. Tight
junction alteration, characterized by relocalization of the transmembrane tight junction
proteins results in seepage of molecular tracers. Functional junction disruption occurs
with a concomitant increase in colon luminal water content (Guttman et al., 2006).
EHEC, EPEC and Citrobacter rodentium belong to a group of bacterial pathogens known
as AE.
Enterohemorrhagic E. coli (EHEC)
Enterohemorrhagic E. coli (EHEC) is a pathogenic strain of E. coli. EHEC
belongs to the group of diarrheagenic strains of E. coli that include EPEC, EHEC, ETEC
and EAEC. It causes hemorrhagic colitis, acute bloody diarrhea and abdominal cramps.
In children, it can cause hemolytic uremic syndrome, a disease characterized by acute
renal failure, thrombocytopenia, and micro angiopathic hemolytic anemia (Nataro, 1998
). EHEC produces Shiga-like toxin, the key virulence factor responsible for both
hemorrhagic colitis and hemolytic uremic syndrome. The major genes in EHEC
associated with T3SS are shown in Fig. 1.7.

22
Enteropathogenic E. coli (EPEC)
EHEC, EPEC and mouse pathogen Citrobacter rodentium are the three bacterial
species that have the locus of enterocyte effacement (LEE). The genes encoding the T3SS
in all the above species are located in LEE and they are homologous. Moreover, the
studies on EPEC T3SS can be extended to the other two species also. EPEC and EHEC
strains are distinguished from other E. coli strains by their ability to inflict characteristic
lesions in small intestine enterocytes, with gross cytoskeletal damage and loss of brush
border microvilli (Staley et al., 1969).
Fig. 1.7. Genes involved in EHEC pathogenesis. Genes involved in EHEC
pathogenesis are similar to those implicated for EPEC, except for the presence of the Stx
encoding phage on the EHEC chromosome and the presence of the characteristic EHEC
60-Mda plasmid instead of the EAF plasmid of EPEC. The EHEC plasmid is known to
encode the enterohemolysin (ehx) as well as a fimbrial antigen potentially involved in
colonization (Nataro, 1998 ).

23
After the initial adherence to epithelia, these pathogens attach intimately to the
epithelial cell surface and cause effacement of microvilli beneath the bacteria, resulting in
characteristic attaching and effacing (AE) lesions (Moon et al., 1983). In the region of
contact between bacteria and the epithelial cell surface, cup-like pseudopod structures
appear which form progressively elongating pedestals carrying individual bacteria on
their tops (Rosenshine et al., 1996). Intimate attachment, effacing of microvilli and
formation of pedestals require a bacterial adhesin (called intimin) and EPEC type III
secretion. Intimin is not secreted by the T3S pathway, but the encoding gene (eaeA) is
located within the gene cluster that encodes EPEC T3SS (Donnenberg and Kaper, 1991)
(Jerse et al., 1991) and intimin functions in tandem with type III secretion in pedestal and
AE lesion formation. Intimin specifically binds to Tir (translocated intimin receptor),
which is secreted by T3SS and inserted into the eukaryotic membrane (Kenny and Finlay,
1997). EspA and EspB are the other two T3SS secreted proteins which may be
translocated into host cytosol (Donnenberg et al., 1993) (Kenny et al., 1996). These
proteins are required for the membrane insertion of Tir (Kenny and Finlay, 1997). Intimin
directly binds with Tir, thus showing that EPEC strains transfer their own receptor for
intimate attachment into eukaryotic cells. Concomitant with pedestal formation, adherent
EPEC strains induce tyrosine phosphorylation of several proteins in the eukaryotic cell,
including Hsp90/Tir (Rosenshine et al., 1996; Rosenshine et al., 1992) (Rosenshine et al.,
1992; 1996) and phospholipase C-g1 (Kenny and Finlay, 1997) (Kenny and Finlay,
1997). The tyrosine phosphorylation and host cell signaling also depend on the type III
secretion of EspA, EspB, and EspC (Kenny et al., 1996; Rosenshine et al., 1992).

24
T3SSs are highly regulated to ensure that they function at the appropriate time. In
their simplest form, the regulatory mechanisms ensure that the secretion machine is
deployed to the bacterial envelope only when the appropriate cues are present. These
regulatory mechanisms are largely transcriptional and are specific for each T3SS (Francis
et al., 2002) Although the regulatory systems seem specific for each T3SS, a common
mechanism involves the use of regulatory proteins that themselves are substrates of the
T3SS. A detailed description of T3SS regulators and regulatory pathways is given in
Introduction of Chapter II. Previously in our lab, we have determined the structure of
GrlR (global regulator of LEE, repressor), a negative regulator protein of the LEE operon
in EHEC (Jobichen et al., 2009; Jobichen et al., 2007). The chapter II of this thesis
presents the structure of GrlR-GrlA (global regulator of LEE, activator) complex along
with structure based functional studies.
Implication of T3SS proteins in therapeutics
Coburn and co-workers (2007) have reviewed T3SS with special emphasis on the
diseases caused by these proteins and the recent developments in clinical research.
Fig.1.8 shows the details of pathological importance of T3SS. The T3SS proteins are
targeted in different ways for controlling the diseases caused by them. Antibodies
developed against some of the T3SS proteins in Yersinia and Pseudomonas were
successful in mouse models against septic shock as well as bubonic plague (Apodaca et
al., 1995; Goure et al., 2005). Studies using T3SS secreted proteins have shown that they
have the potential to be developed as vaccines for immunizing cattle, the major carriers
of EHEC pathogens (Potter et al., 2004) (Van Donkersgoed et al., 2005). Possibilities are
being explored to develop inhibitors against these proteins and also for using these

25
proteins as diagnostic tools (Kauppi et al., 2003; Li et al., 2005). T3SS helps Gram-
negative bacteria to transport a wide variety of proteins (mainly virulence proteins) into
plant and animal host cells (Hueck, 1998). Recent studies have revealed selected T3SS
proteins to be potential targets for controlling the diseases that are caused by these
organisms by specifically attenuating the causative bacterial pathogens without affecting
the commensal flora. Further developments in this field will eventually aid discovery of
vaccines and other drugs to specifically inhibit T3SS proteins.
Fig. 1.8. T3SS effector functions of path physiologic importance. T3SS effectors have
been implicated in a variety of critical pathogenic behaviors. These virulence strategies
have specific consequences in disease pathogenesis in the infected host. (Coburn et al.,
2007)

26
1.1.6. Type IV Secretion System (T4SS)
T4SS is a unique bacterial secretion system that can translocate DNA into the host
organism. Bacteria use T4SS to serve two of its fundamental objectives – genetic
exchange and delivery of effector molecules to eukaryotic target cells. The T4SSs can be
classified into three different sub-families
a. Conjugation family - This is the largest sub-family of the T4SS, and is found in
most Gram-negative and Gram-positive bacteria. These systems can mediate
DNA transfer both within and between phylogenitically diverse species, and some
systems can even deliver DNA to fungi, plants and human cells (eukaryotic cells).
b. DNA uptake and release family - The second subfamily is the DNA uptake and
release family, which, function independent of contact with the target cell. This
subfamily comprises two (DNA uptake) systems — the Campylobacter jejuni
Cjp/VirB system and the Helicobacter pylori ComB system (Bacon et al., 2000)
(Hofreuter et al., 1998)- and one DNA-release system, an F-plasmid Tra-like
system of N.gonorrhoeae. As with the conjugation machines, these systems
promote genetic exchange and therefore also represent potential mechanisms for
the transfer of survival traits during infection (Chen and Dubnau, 2003).
c. Effector translocator family- This family is indispensable in the infection
processes of several prominent pathogens of plants and mammals (Fig 1.9). These
machines can be viewed as ‘injectisomes’, reminiscent of the type III secretion
(T3S) machines, because they deliver their substrates through direct contact with
the eukaryotic target cell.

27
Fig. 1.9. Schematic representations of the different type-IV-dependent mechanisms.
The three subfamilies of type IV secretion (T4S) systems are shown. Conjugation
machines deliver DNA to recipient bacteria and other cell types by cell-to-cell contact.
DNA-uptake and release systems exchange DNA with the extracellular milieu
independently of contact with target cells. Effector translocators deliver DNA or protein
substrates to eukaryotic cells during infection. The effector translocators contribute in
markedly different way to the infection processes of the bacterial pathogens shown. PT:
pertussis toxin (Cascales and Christie, 2003; Christie et al., 2005)

28
DNA transfer in conjugal systems is enabled by a set of proteins known as the
DNA transfer and replication (Dtr) proteins. The Dtr proteins act on the origin of transfer
(oriT) sequence of mobile DNA elements and process the DNA into single-stranded
DNA and sometimes remains covalently attached to the 5' end of the DNA. One such
protein, the relaxase, generates a strand-specific nick at oriT and remains covalently
bound to the 5' end of the T-strand. The translocation competent form of the DNA
substrate corresponds to a T-strand relaxase nucleoprotein complex (Baron et al., 2002;
Christie, 1997; Zhu et al., 2000). The DNA and protein substrates recruited to the T4S
apparatus, are delivered across one or both membranes by the Mpf structure. The
VirB/D4 system in Agrobacterium is one of the most well studied T4SS. For the A.
tumefaciens VirB/D4 T4S system, the sub cellular locations and topologies of the VirB
Mpf proteins have been defined based on computer predictions and a combination of sub
cellular fractionation and analyses of reporter-protein fusion studies (Fig 1.10).
The conjugation systems of Gram-negative bacteria are an assembly of three
distinct substructures: the coupling protein (CP) homomultimer; a transenvelope-protein
complex; and the conjugative pilus (transfer- or T-pilus). The transenvelope and the
conjugative pilus are assembled from the mating-pore-formation - for example, VirB1–
VirB11 of the A. tumefaciens VirB/D4 T4S system. The CP, transenvelope complex and
the T-pilus act in coordination, as a single, supramolecular organelle, to mediate the
various stages of translocation. These stages include the recruitment of cognate DNA and
protein substrates to the transfer machine, the transfer of substrates across the cell
envelope and the delivery of substrates to target cells.

29
Fig. 1.10. Topologies of the VirB/D4 subunits of the A. tumefaciens T4SS. The
coupling protein (CP) VirD4 and the mating-pore-formation components (VirB1–
VirB11) are represented according to their proposed functions: energetic (blue), channel
(red) or pilus (green) components. Several proteins are post-translationally modified in
the periplasm (Cascales and Christie, 2003; Christie et al., 2005))
The VirB proteins can be divided into three classes according to known or
postulated functions (Baron et al., 2002; Christie, 1997)
i. Channel components which include the inner-membrane proteins VirB6, VirB8
and VirB10, and the outer-membrane proteins VirB3, VirB7 and VirB9.
ii. Two ATPases, VirB4 and VirB11, which are localized at the cytoplasmic face of
the inner membrane, provide energy to drive substrate transfer and, possibly,
biogenesis of the transfer channel and the pilus.

30
iii. The pilin subunit, VirB2, assembles as the T-pilus in association with VirB5 and
the VirB7 lipoprotein (Eisenbrandt et al., 1999; Lai and Kado, 2000; Sagulenko
and Christie, 2001; Schmidt-Eisenlohr et al., 1999).
The VirB4 and VirB11 ATPases are postulated to either mediate VirB/D4 T4SS
machine assembly or to function through dynamic, ATP-driven conformational changes.
Homologues of both ATPases are widely conserved among the T4S system family
members and VirB11-like ATPases constitute a protein super family that extends to the
transport machines of many Gram-negative and Gram-positive bacteria and several
species of the archaea. Conjugation systems have several morphologically distinct pili.
They can be long and flexible like the F-plasmid pilus (Lawley et al., 2003) or short and
rigid like the RP4-plasmid pilus (Eisenbrandt et al., 1999). In case of Agrobacterium
tumifaciens T4SS, the T-pilus resembles the RP4-plasmid pilus and is composed of
VirB2 pilin. These pili help in substrate transfer by promoting mating-pair formation.
The T-DNA integration occurs in an illegitimate recombination, a mechanism that
joins two double-stranded (ds) DNA elements that do not share extensive homology
(Ziemienowicz, 2001). Till date, it has not been possible to target T-DNA to any
particular locus in the genome with any great efficiency. However, one of the major
contributions of A. tumefaciens to genetic engineering research has been the use of T-
DNA as a mutagen to generate the desirable mutant (Valentine, 2003). Moreover, the
molecular mechanisms of the T-DNA integration remain largely elusive. It is likely that
after nuclear import, the ss T-strand is turned into double-stranded DNA (dsDNA) with
the concomitant displacement of VirE2.

31
Unlike the transposons and retroviruses, T-DNA itself does not encode enzymes
that catalyze the integration. Thus, the integration of T-DNA into the plant genome must
be mediated by proteins imported from A. tumifaciens or by host cell factors. The
incorporated T-DNA induce plant cells to synthesize opine food substrates and to induce
proliferation of the transformed plant cells. The outcome of infection is a plant tumor,
known as a crown gall, which for the bacterium represents a good ecological niche as it
acts as a food-producing factory.
T4SS in A. tumifaciens in addition to the T-DNA also translocates three protein
effectors, VirE2 (Ward et al., 2002), VirE3 (Schrammeijer, (2003) ) and VirF (Vergunst,
2000). VirE2 interacts with the T-strand VirD2 particle to form the so-called T-complex,
VirE3 and VirF participate in largely unspecified ways to promote infection. VirD2 and
VirE2 carry nuclear-localization sequences (NLS) that enable interactions with plant
cellular factors and render nuclear targeting, import, and T-DNA integration into the host
genome. Specific interactions between these two bacterial proteins and several eukaryotic
factors have been identified. For example, VirD2 binds three members of the Arabidopsis
cyclophilin chaperone family; these interactions might maintain the proper conformation
of VirD2 in the host-cell cytoplasm or nucleus during T-complex transit (Deng et al.,
1998). Given the large numbers of cellular factors identified so far, it is likely that the T-
DNA and the reported effector proteins represent only a subset of the molecules
translocated by the VirB/D4 T4S system during infection.
Conjugation, competence and other gene-transfer mechanisms help the bacterium
with the capacity to survive changing environments through the acquisition of adaptive
traits. Conversely, T4S effector translocators have evolved for the opposite purpose: to

32
render the harsh environment of the eukaryotic host habitable. This is achieved through
sedition of a plethora of host cellular processes, as illustrated in the Fig 1.11.
Fig. 1.11. Schematic representation of the cellular consequences T4SS. T4S effector
translocation alters various eukaryotic cellular processes, as illustrated for the four
systems in which effector molecules have been identified so far. Agrobacterium
tumefaciens delivery of T-DNA and effector proteins induces synthesis of opine food
substrates and also induces tumour production through modulation of phytohormone
levels. Helicobacter pylori CagA modulates various pathways associated with
eukaryotic-cell differentiation, proliferation and motility. Bordetella pertussis
pertussistoxin (PT) interferes with G-protein-dependent signaling pathways, and
Legionella pneumophila RalF recruits the ARF (ADP ribosylation factor) family of
guanosine triphosphatases to the phagosome to promote intracellular survival (Cascales
and Christie, 2003).

33
Besides A.tumifaciens other pathogens that harbor T4SS for effector translocation
are mentioned below.
Bartonella henselae: The causative agent of cat-scratch disease, a relatively benign
disease that is transmitted to humans by blood-sucking arthropods.
Bordetella pertussis: Responsible for a respiratory disease known as ‘whooping cough’
or pertussis, and transmitted by aerosol droplets.
Brucella spp: The causative agents of brucellosis, or Malta fever, these organisms are
transmitted to humans through direct contact with infected animals, carcasses or milk.
Helicobacter pylori: The causative agent of chronic gastric disorders, and is important in
the development of peptic ulcer and gastric cancers.
Legionella pneumophila: Responsible for pneumonia known as ‘legionnaire’s disease’.
Humans are infected through contact with contaminated water or aerosols.
Significance of T4SS from A.tumifaciens
A.tumefaciens-mediated T-DNA transfer to plant is the most popular method for
the introduction of foreign genes into plant cells and the subsequent regeneration of
transgenic plants. This method has remarkable advantages over other direct
transformation methods such as electroporation, microinjection and particle
bombardment (De la Riva et al., 1998) which include (1) significantly high
transformation efficiency; (2) easy to manipulate; (3) low copy number of the transgene,
usually single copy insertion into the plant genome, potentially resulting in fewer
problems with transgene co-suppression and instability (Hansen et al., 1997); and (4) less
frequent to form mosaic plants (Enriquez-Obregon et al., 1998). A. tumefaciens
represents a major tool for plant molecular breeding and delivery of DNA to other

34
eukaryotic cells. The molecular mechanism by which it genetically transforms the host
cells has been the focus of research for a wide spectrum of biologists, from
bacteriologists to molecular biologists to botanists, for a number of years.
In addition, A. tumefaciens-mediated T-DNA transfer to plant is the only known
example of DNA transport between kingdoms that occurs between kingdoms. The T-
DNA is transferred into eukaryotic cells in the form of nucleoprotein complex. The A.
tumefaciens-mediated T-DNA transfer system can be used as a model system to study the
molecular mechanism of a wide variety of biological processes such as nucleoprotein
trafficking, nuclear targeting of nucleoprotein, and the export of virulence effector
(Christie, 2001). Many of these biological processes are relevant to human pathogen,
human gene therapy, as well as HIV viral infection.
1.1.7. Type V Secretion System (T5SS)
T5SSs are known for their simplicity. There are three different types of T5SSs
viz., the autotransporter system-1 (AT-1 or type Va), the two-partner secretion pathways
(TPS) (type Vb) pathways and autotransporter system-2 (AT-2 or type Vc) (Fig. 1.12).
These three systems are characterized by relatively low number of protein components
involved in the secretion process. They are widely distributed among the pathogenic
bacteria (Henderson and Nataro, 2001; Henderson et al., 1998; Jacob-Dubuisson et al.,
2001; Yen et al., 2002).
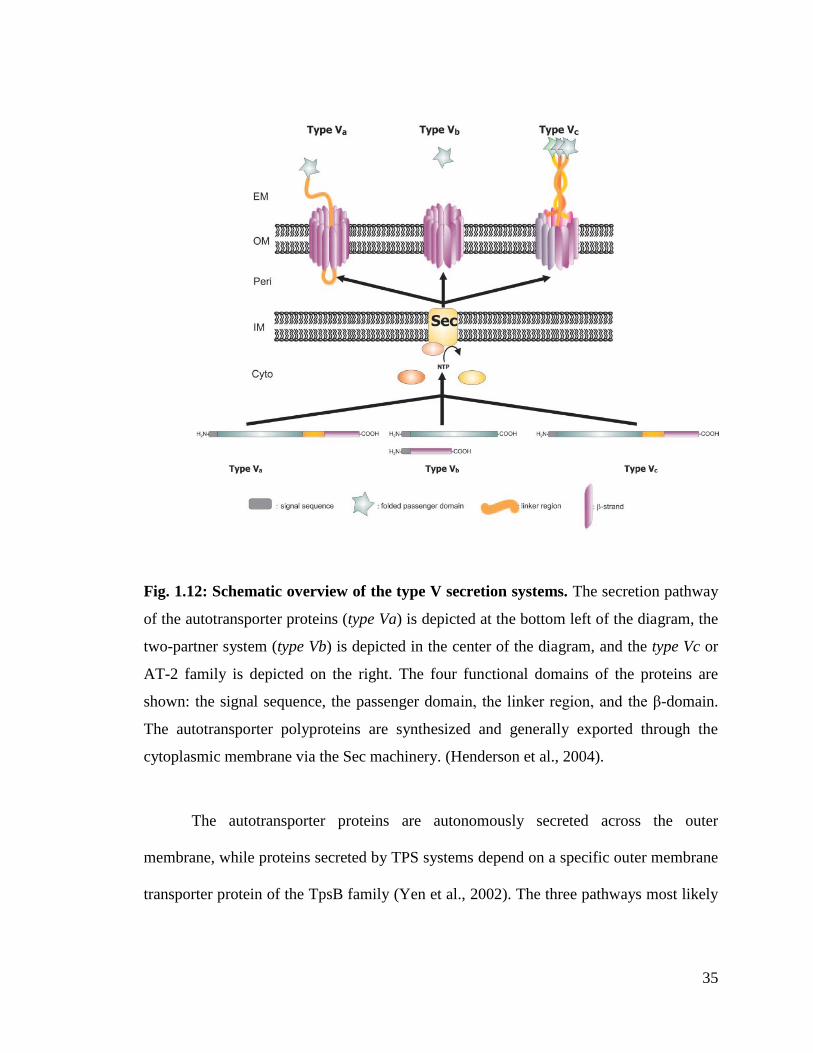
35
Fig. 1.12: Schematic overview of the type V secretion systems. The secretion pathway
of the autotransporter proteins (type Va) is depicted at the bottom left of the diagram, the
two-partner system (type Vb) is depicted in the center of the diagram, and the type Vc or
AT-2 family is depicted on the right. The four functional domains of the proteins are
shown: the signal sequence, the passenger domain, the linker region, and the β-domain.
The autotransporter polyproteins are synthesized and generally exported through the
cytoplasmic membrane via the Sec machinery. (Henderson et al., 2004).
The autotransporter proteins are autonomously secreted across the outer
membrane, while proteins secreted by TPS systems depend on a specific outer membrane
transporter protein of the TpsB family (Yen et al., 2002). The three pathways most likely

36
represent convergent solutions to the secretion of essentially large proteins with certain
folding characteristics.
The proteins targeted by these systems are synthesized with a N-terminal signal
peptide that help their translocation through the inner membrane by the Sec machinery.
Effector proteins with an unusual extended signal sequence, mediates SRP-dependent
export, are found in all three categories of type V secretion (Henderson et al., 1998).
Once through the inner membrane, the signal sequence is cleaved and the β-domain
inserts into the outer membrane in a biophysically favored β-barrel structure that forms a
pore in the outer membrane. AT proteins are modular and in addition to the N-terminal
signal peptide they have a passenger module which carries out the function of the
exoprotein followed by a C-terminal translocation module which serves as a conduit for
the translocation of the passenger domain across the outer membrane (Pugsley, 1993).
TpsA proteins do not have such transporter domain. They instead have cognate protein
partners like TpsB that form β-barrel channels in the outer membrane. TpsB helps in the
translocation of TpsA following a specific recognition event between the two partners
(Yen et al., 2002)
1.1.8. Type VI Secretion System (T6SS)
Type VI secretion is widely distributed in Gram-negative bacteria. T6SS is a key
virulence factor for many pathogenic bacteria and has been implicated in the
translocation of potential effector proteins into eukaryotic cells eg. Rhizobium
leguminosarum, V. cholerae, S. enterica, and P. aeruginosa etc. Studies from our lab
have shown that this particular system is also present in E. tarda and that they play an
important role in pathogenesis (Rao et al., 2004; Zheng et al., 2005).

37
The components of the T6SS include IcmF homologue, an ATPase ClpV, a
regulatory forkhead-associated (FHA) protein FHA domain and the secreted proteins
VgrG and Hcp (Bingle et al., 2008). The T6SS translocation apparatus consists of IcmF-
like and ClpV ATPase proteins. ClpV ATP ases constitute a subfamily of the ClpB
family, which comprises hexameric enzymes involved in protein quality control. ClpBs
use ATP energy currency to unfold protein substrates to be degraded. Unlike other
secretion systems, sequence analysis of T6SS proteins predicts a cytoplasmic location for
most of the subunits (Cascales, 2008). Previously from our lab we have reported the
structure and function of EvpC from E.tarda. EvpC is a close homolog of Hcp1 from
Pseudomonas aeruginosa. It forms a hexameric ring with a diameter of 40Å that is
capable of transporting small proteins and ligands (Jobichen et al., 2010).
The genes that are responsible for T6SS are located in the IAHP (IcmF
Associated Homologous Protein) cluster. Two genes encode putative inner membrane
proteins with one (IcmH) or three (IcmF) transmembrane domains. In addition, one
conserved gene encodes a probable outer membrane lipoprotein. With the exception of
the R. leguminosarum RbsB protein, the T6S substrates identified so far lack a canonical
hydrophobic (Sec) or arginine-rich (Tat) N-terminal signal sequence (Cascales, 2008).
Many pathogenic bacteria known to manipulate host-cell physiologies harbor T6SS.
T6SS delivers macromolecules that subvert host-cell defenses such as signaling cascades,
inflammatory responses, intracellular transport, cytoskeleton dynamics or key regulatory
or metabolic pathways. Interestingly, Hcp and VgrG are both secreted and part of the
secretion machine. Furthermore, several subunits share homologies with subunits of the
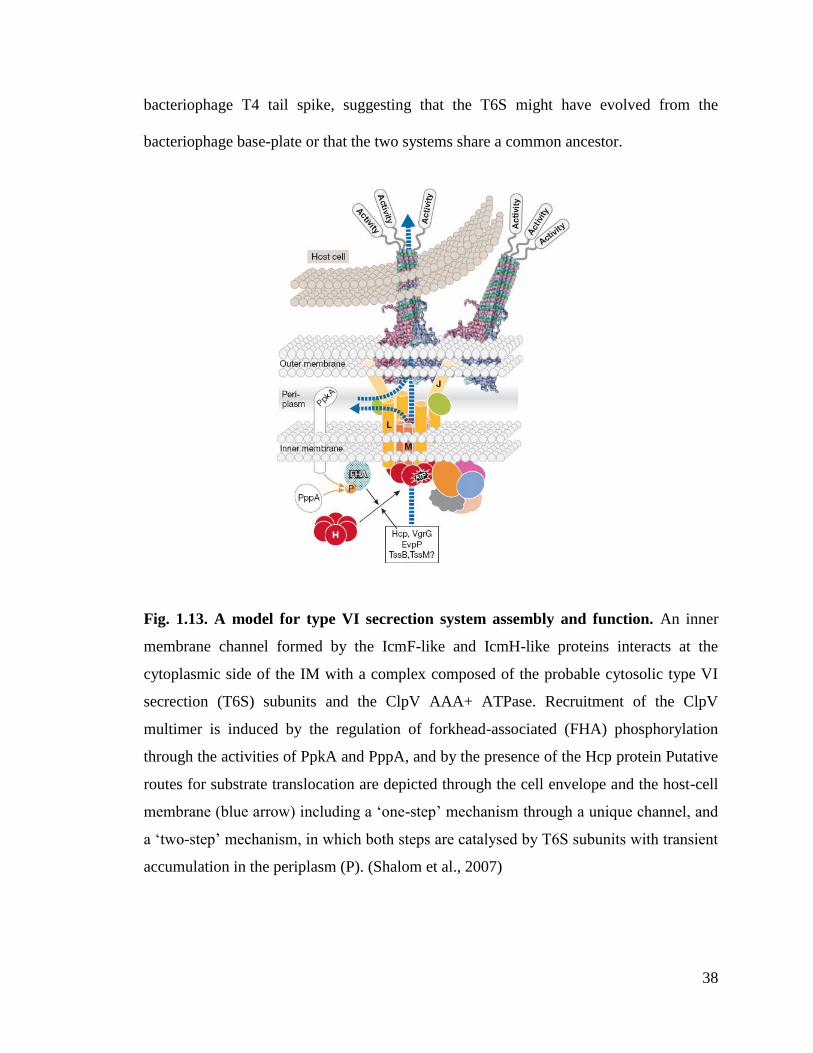
38
bacteriophage T4 tail spike, suggesting that the T6S might have evolved from the
bacteriophage base-plate or that the two systems share a common ancestor.
Fig. 1.13. A model for type VI secrection system assembly and function. An inner
membrane channel formed by the IcmF-like and IcmH-like proteins interacts at the
cytoplasmic side of the IM with a complex composed of the probable cytosolic type VI
secrection (T6S) subunits and the ClpV AAA+ ATPase. Recruitment of the ClpV
multimer is induced by the regulation of forkhead-associated (FHA) phosphorylation
through the activities of PpkA and PppA, and by the presence of the Hcp protein Putative
routes for substrate translocation are depicted through the cell envelope and the host-cell
membrane (blue arrow) including a ‘one-step’ mechanism through a unique channel, and
a ‘two-step’ mechanism, in which both steps are catalysed by T6S subunits with transient
accumulation in the periplasm (P). (Shalom et al., 2007)

39
1.1.9. Type VII Secretion System (T7SS)
Gram-positive bacteria are enclosed by a simple cell membrane without a cell
wall. However some Gram-positive bacteria like the mycobacteria have a complex cell
envelope that contains, in addition to the regular inner membrane, a unique outer
membrane, also called the mycomembrane, made up of covalently attached mycolic acids
intercalated with free (glycol) lipids (Hoffmann et al., 2008). Mycobacteria employ the
most recently discovered transport pathway known as type VII secretion (T7S) for the
secretion of proteins across this complex cell envelope (Abdallah et al., 2007). The
presence of the T7SS was initially predicted based on clustering of genes encoding
secreted proteins that lacked signal sequences with those encoding membrane proteins,
ATPases and/or chaperones (Tseng et al., 2009). Mycobacterial genomes contain up to
five T7SS gene clusters (ESX-1 – ESX-5) that do not functionally complement one
another (Daleke et al., 2012). T7SS gene clusters are also found in the closely related
pathogens Corynebacterium diphtheriae and Nocardia.
The structure and functional components of the T7SS are still being pieced
together. The available studies so far suggest that an integral membrane protein forms the
translocation channel on the inner membrane and a separate channel in the
mycomembrane is formed by an as yet unidentified protein (Abdallah et al., 2007) .
Most of the T7SS substrates contain a unique highly conserved motif, i.e.,
YxxxD/E within the C terminus which is required for translocation. Exchange of these C-
terminal secretion signals between the substrate proteins restored secretion, but each
substrate protein remained secreted via its own ESX secretion system, indicating that an
additional signal(s) provides system specificity (Daleke et al., 2012) .

40
1.2. Objectives
For the present thesis we mainly focus on understanding (1) the regulatory mechanism of
a T3SS regulatory GrlR-GrlA complex from EHEC and (2) the recruiting mechanism of a
T4SS recruiting protein: the VirD2 binding protein (VBP) of the VirD4/B T4SS from
A.tumefaciens
1.2.1. GrlR-GrlA regulatory complex
GrlA is a positive regulator of the LEE operon and forms a positive regulatory
loop with Ler. GrlR binds to GrlA and probably inhibits the positive regulatory loop and
thereby repressing the expression of LEE operon (Barba et al., 2005). Furthermore GrlA
negatively regulates flhDC operon (Iyoda et al., 2006) and positively regulates ehxCABD
operon (Saitoh et al., 2008). We have previously determined the structure of GrlR and
have speculated that a particular EDED motif of GrlR binds to the highly basic C-
terminal region of GrlA (Jobichen et al., 2007). Our objective for the present study is to
understand the mechanism by which GrlR-GrlA complex regulates multiple virulence
operons and enhances pathogenesis in AE pathogens. In particular we want to determine
the competition between GrlR and Pler in binding to GrlA. Once we establish the
competition between Pler and GrlR, we would like to verify the same with other
promoters like the PflhD and Pehx. The following steps are adapted to accomplish our
objectives:
1. Determining the structure of GrlR-GrlA complex for understanding the regulatory
mechanism of the T3SS
2. Structure based functional studies have to be carried out to reveal the role of the
key residues involved in GrlR-GrlA interaction

41
3. Characterizing the interactions using biophysical interaction studies
4. Studying the DNA(Pler) binding property of GrlA
5. Studying the competition between GrlR and DNA for binding to GrlA
6. Validation of the results from the aforementioned studies using in vivo EspB
secretion assay and ler-gfp reporter assay
7. Extending the DNA binding studies to other promoters like the flhDC (Pflh) and
ehxCABD (Pehx)
1.2.2. VirD2 binding protein (VBP) of the VirD4/B T4SS
Recruitment of the VirD2-T-DNA-VirE2 complex (T-complex) is an important
step in the DNA translocation process of the T4SS (Christie, 1997). VirD2 binding
protein (VBP) facilitates the recruitment of bulky VirD2-T-DNA-VirE2 complex to the
VirD4 coupling protein. In addition to VirD2 and VirD4 CP, VBP binds to VirB4 and
VirB11 ATPases of the T4SS. Besides, VBP plays an important role in bacterial
conjugation that is independent of T4SS (Guo et al., 2007a; Guo et al., 2007b).
Understanding the mechanism of recruitment of the T-complex in particular and
translocation of T-DNA in general, is the main objective of this project. As a first step
towards understanding this, the structure of VBP and its interaction with its binding
partner-VirD2 and relevant functional studies have to be carried out. The following steps
are adapted to accomplish our objectives:
1. Determining the crystal structure of VBP and/or its individual domains with its
binding partners to understand the substrate recruiting mechanism of the T4SS.

42
2. Identifying and studying the functional oligomeric form of the key recruiting
complex protein VBP
3. Validation of the importance of the key amino acids of VBP involved in protein-
protein interactions using structure based functional studies
4. Validation of the in vitro findings using in vivo plant virulence assay

43
Chapter 2: Structure of GrlR-GrlA Complex that Prevents
GrlA Activation of Virulence Genes

44
2.1. Introduction
Attaching and effacing (AE) pathogens are a group of enteric pathogens that
include the closely related enterohemorrhagic Escherichia coli (EHEC) and
enteropathogenic E. coli (EPEC). EPEC causes severe diarrhea in young children, while
EHEC is a causative agent of hemorrhagic colitis and hemolytic uremic syndrome (HUS)
(Nataro, 1998 ; Pennington.H, 2010). AE pathogens possess type III secretion systems
(T3SS) that promote virulence. Most components of T3SS and related proteins are
encoded by genes in the locus of enterocyte effacement (LEE). The LEE consists of 41
genes, clustered in five different operons termed LEE1-LEE5 and some additional
transcription units (Deng et al., 2004b).
Under specific conditions such as low temperature, a histone-like nucleoid-
structuring (H-NS) protein binds to the extended regions within the LEE DNA and
silences the expression of entire LEE region. This H-NS-mediated repression, however,
can be countered by Ler (LEE-encoded regulator), a protein encoded by the first gene in
the LEE1 operon that functions as a positive regulator of other LEE operons (LEE2-
LEE5) by counteracting this H-NS mediated repression (Barba et al., 2005). Moreover
Ler is involved in direct or indirect regulation of additional key virulence components of
EPEC and EHEC and is thus considered as the master regulator of the virulence
machineries of these pathogens. Accordingly, activation of LEE1 and ler expression is a
key event in the activation of the entire virulence system of EPEC and EHEC. It is
therefore not surprising that the LEE1 promoter is subjected to tight regulation by
multiple factors (Fig. 2.1), including H-NS and Ler, which function as repressor and
autorepressor, respectively. Expression of the LEE1 operon is strictly dependent on

45
binding of the integration host factor (IHF) protein immediately upstream to the LEE1
promoter (Barba et al., 2005). Besides, activation of LEE1 expression is promoted by
proteins belonging to either one of two classes of redundant positive regulators such as
PerC (Bustamante et al., 2011) (or Pch in EHEC) or GrlA.
Fig. 2.1. Schematic of the regulatory circuits related to the GrlR-GrlA complex. We
propose the following model based on results presented in this report as well as results
previously published by us and other groups. The grlR and grlA genes form a bicistronic
operon located within the LEE. Under non inducing condition the expression of the entire
LEE including grlRA is suppressed by H-NS and the activity of the residual GrlA is
suppressed by GrlR, via direct interaction (repression is indicated by red lines). The
expression of the entire system can be activated by two alternative modes. The first
activation mode involves specific environmental cues that turn on expression of PerC,

46
which in turn activate expression of Ler (activation is indicated by blue lines). The
produced Ler antagonize H-NS and thus stimulate expression of all the other LEE operon
including grlRA. The produced GrlA directly activate ler expression to establish GrlA-
Ler positive feedback loop. Consequently GrlA further promote, indirectly, the
expression of all the LEE genes and in addition it directly activates expression of
the ehxCABD operon and represses the expression of the flagella master
regulator flhDC. The intensity of GrlA-Ler loop is restrained by autorepression activity of
Ler and by GrlR, which directly bind GrlA to inhibit its interaction with target DNA. The
second mode of activation involves environmental signals that trigger the ClpXP protease
to degrade GrlR and thus releasing GrlA to activate ler expression, leading again to
establishing of the Ler-GrlA positive loop described above.
The grlR and grlA genes form a transcriptional unit encoding GrlR and GrlA,
respectively. GrlA is a positive regulator of the LEE1 promoter and thus forms a positive
regulatory loop with Ler. GrlR binds to GrlA and this is thought to account for the
negative effect of GrlR on ler expression (Barba et al., 2005; Deng et al., 2004a; Iyoda et
al., 2006). Since GrlA and GrlR are co-expressed, it is expected that in most cases GrlA
is inhibited by GrlR and that the relative levels of GrlR and GrlA are regulated. For
instance, it was reported that under certain conditions GrlR is degraded by the ClpXP
protease, freeing GrlA to activate the Ler expression (Iyoda and Watanabe, 2005).
Previously, we reported the structure of GrlR (Fig. 2.2) and elucidated its role in
LEE regulation (Jobichen et al., 2009; Jobichen et al., 2007). GrlR forms a stable and
tight dimer in solution as well as in the crystal. The dimeric architecture of GrlR is
maintained by the cluster of hydrophobic interactions as well as numerous hydrogen
bonding contacts at the dimeric interface. GrlA has been functionally characterized
(Barba et al., 2005; Deng et al., 2004a; Jiménez et al., 2010), but no structural

47
information is available to provide insight into the regulatory mechanisms involving
GrlA, GrlR, and Ler.
Fig. 2.2. Structure of GrlR. Ribbon diagram of the GrlR dimer. Monomer A is shown in
green, monomer B is shown in cyan (Jobichen et al., 2007).
In addition to its function as a LEE1 regulator, GrlA negatively regulates
transcription of the flhDC operon and thus controls flagellar gene expression. Kitagawa et
al (Kitagawa et al., 2011) demonstrated that flagellar gene expression in EHEC is strictly
regulated by dual pathways, i.e. (i) post-translational control of the FlhDC master
regulator by protein degradation via ClpXP and (ii) transcriptional control of the flhDC
operon through the GrlR–GrlA system under conditions in which LEE expression is
induced. Moreover, it was reported that deletion of grlR resulted in a non motile
phenotype in the EHEC O157 Sakai strain (Iyoda et al., 2006). In many pathogens,
motility is repressed by negatively regulating flhDC at the same time that T3SS are

48
induced (Kitagawa et al., 2011). GrlA-dependent repression of flagellar regulation is
important for efficient adhesion of EHEC to host cells (Iyoda et al., 2006; Kitagawa et
al., 2011) and perhaps also to avoid detection by the host innate immune sensors TLR5
and NLRP4 (Deretic, 2012; Fujita and Taguchi, 2012).
GrlA is also implicated in the transcriptional activation of the ehxCABD operon in
EHEC (Schmidt et al., 1995; Welch and Pellett, 1988). The ehxA gene encodes
hemolysin, ehxC acts as a modifying factor that converts the inactive hemolysin into an
active form by the addition of a fatty acid group (Issartel et al., 1991), and the specific
secretion machinery required for translocation of EhxA is encoded by ehxB and ehxD
(Wagner et al., 1983).
The in vitro instability of GrlA hindered structural studies and attempts to
elucidate the regulatory mechanism involving Ler, GrlA and GrlR. Here we report the
crystal structure of GrlAΔ (amino acids (aa) 1-106) in a complex with GrlR refined up to
2.7 Å resolution. The structure is asymmetric with a stoichiometry of 2GrlR:1GrlA. In
addition, we identify a novel regulatory mechanism by which GrlR interacts with GrlA at
its Helix-Turn-Helix (HTH) motif, preventing GrlA from binding to its target promoters
DNA.
2.2. Materials and Methods
2.2.1. Plasmid and strain construction
Bacterial strains and plasmids used in this study are listed in Table 2.1.Intact grlR
and grlA genes were PCR-amplified from EHEC EDL933(Hayward et al., 2006)
chromosomal DNA and cloned into MCS1 (with a 6His tag) and MCS2 of the pETDuet-1

49
(Novagen; Madison, WI, USA) vector, respectively. Plasmid pET32-grlR was
constructed by amplifying the grlR gene from EHEC EDL933 chromosomal DNA and
cloning into pET32 vector. Plasmid pGEX-grlA and pMBP-grlA were constructed by
amplifying the grlA DNA fragments from EHEC EDL933 chromosomal DNA and
cloning into pGEX-4T1 (GE Healthcare; Buckinghamshire, UK) and pMAL-c2X (New
England Biolabs; Ipswich, MA, USA), respectively. The EPEC grlRA null mutant
(ΔgrlRA::kn) was constructed by replacing the grlRA gene from an EPEC strain with a
kanamycin cassette by the one-step method using the λ recombinase system(Datsenko
and Wanner, 2000). Site-specific mutations in grlA and grlR were introduced by
overlapping PCR (Ho et al., 1989) which uses complementary oligodeoxyribonucleotide
(oligo) primers and the polymerase chain reaction to generate two DNA fragments having
overlapping ends. These fragments are annealed, allowing the 3' overlap of each strand to
serve as a primer for the 3' extension of the complementary strand. The resulting fusion
product is amplified further by PCR. Specific mutations in the nucleotide sequence were
introduced by incorporating nucleotide changes into the overlapping oligo primers. Each
construct was verified by DNA sequencing. To construct pGY1, a DNA fragment
containing PLEE1 and ler (starting from position -159 compared to the transcriptional start
site) was amplified using specific primers; this amplified fragment was digested by XbaI
and BamHI and cloned into pIR1 (Friedberg et al., 1999) digested by the same enzymes.
The null strain (ΔgrlRA::kn) transformed with pGY1 was named GY2155.
2.2.2. GrlR-GrlAΔ complex structure determination
pETDuet1-grlR-grlAΔ plasmid was transformed into E. coli BL21 cells and
grown in defined M9 medium(Doublié, 1997) supplemented with 25 mg/l L-SeMet at

50
37°C until the optical density reached 0.6 at 600 nm. A 1 L culture was induced with 100
μM IPTG and grown overnight at 20°C. Cells were then harvested by centrifugation and
resuspended in 40 ml of lysis buffer (50 mM Tris-HCl [pH 7.0], 0.2 M NaCl, 1% (w/v)
Triton X-100, 5% (w/v) glycerol, 10mM β-mercaptoethanol) with complete protease
inhibitors (Roche Applied Science; Mannheim, Germany). The GrlA-GrlRΔ complex
was purified in two steps using Ni-NTA (Qiagen; Valencia, CA, USA), followed by gel
filtration (Superdex75, GE Healthcare). The 6His tag remained intact on GrlR. Drops
containing 1 μl of protein solution (7 mg/ml) and 1 μl of reservoir solution were
equilibrated by hanging drop vapour diffusion at 25°C. The best crystals were grown
from 12% PEG 3350, 0.1 M sodium malonate pH 5, 0.4 M non-detergent
sulfobetaines201, with the protein in 30 mM Tris-HCl (pH 7.0), 200 mM NaCl and 5%
(w/v) glycerol. One complex molecule consisted of two GrlR and one GrlAΔ (2:1) in the
asymmetric unit, which accounts for a Matthews coefficient of 2.40 Å3/Da(Matthews,
1968), corresponding to a solvent content of 49%. Crystals were cryo protected in the
reservoir solution supplemented with 25% glycerol and flash cooled at 100K. The
structure was determined using SeMet-labelled protein crystals by single-wavelength
anomalous dispersion (SAD)(Terwilliger and Berendzen, 1997a). X-ray diffraction data
were collected at beamline 13B, National synchrotron radiation research centre (NSRRC,
Taiwan), using a Quantum-315r CCD area detector (ADSC) and processed with
HKL2000(Otwinowski and Minor, 1997). All of the expected eight Se sites of an
asymmetric unit were located using the program Phenix-Autosol. The phases were further
improved by density modification using RESOLVE(Terwilliger, 2003), which gave a
final overall figure of merit of 0.70. Over 50% of the backbone atoms of the model were

51
built by RESOLVE. The remaining residues were manually built using COOT(Emsley
and Cowtan, 2004) and refined with phenix-refine(Adams et al., 2010). Refinement was
continued until the R-value converged to 0.18 (Rfree = 0.23) for reflections I>σ (I) to 2.7
Å resolution (Table 2.3). The model had good stereochemistry, with 99.3% residues
falling within the allowed regions of the Ramachandran plot. Subsequently, the
importance of the key residues in the HTH motif region was validated by structure-based
in vitro studies, such as isothermal titration calorimetry and pull-down assay.
2.2.3. Analytical Ultracentrifugation
The stoichiometric ratio of wild-type GrlR-GrlA complex was investigated by
monitoring their sedimentation properties in sedimentation velocity experiments.
Samples (400 µl) were used at A280 nm of 1.0 in 30 mM Tris-HCl (pH 7.0), 200 mM
NaCl, and 5% glycerol. Sedimentation velocity profiles were collected by monitoring the
absorbance at 280nm. The samples were sedimented at 40,000 rpm at 20°C in a Beckman
Optima XL-I centrifuge (Beckman Coulter Inc., Brea, CA) fitted with a four-hole AN-60
rotor and double-sector aluminium centre pieces and equipped with absorbance optics. A
total of 200 scans were collected and analysed using the Sedfit program(Schuck, 2000).
2.2.4. Circular dichroism spectrometry
Far UV spectra (260–190 nm) of MBP-GrlA/MBP-GrlAΔ and its substituents
were measured using a Jasco J-810 spectropolarimeter (Jasco Europe, MI, Italy) in
phosphate buffer (pH 7.5) at room temperature using a 0.1-cm path length and stoppered
cuvettes. Six scans were recorded, averaged and the baseline subtracted.

52
2.2.5. Isothermal titration calorimetry
MBP-GrlA, MBP-GrlAΔ (with or without substitutions in the HTH motif region)
and GrlR were all purified in gel filtration buffer containing 30 mM Tris-HCl [pH 7.0],
200 mM NaCl and 5% glycerol. ITC experiments were carried out using a VP-ITC
calorimeter (Microcal, LLC; Northampton, MA, USA) at 25°C using 0.01 mM protein
(MBP-GrlA/MBP-GrlAΔ) in the sample cell and 0.15-0.22 mM GrlR in the injector. All
samples were thoroughly degassed and then centrifuged to remove precipitates.
Excluding the first 2-microliter injection, 10-microliter injections were sequentially made
in each experiment. Consecutive injections were separated by 5 min to allow the peak to
return to baseline levels. ITC data were analyzed with a model considering a single class
of binding sites implemented in Origin 7.0 (Origin Lab Corp.; Northampton, MA, USA)
software.
2.2.6. Electrophoretic mobility shift assays
EMSAs were performed by mixing approximately 100 ng of DNA (promoter
region of ler (-275/+217) (numbers indicate the number of base pairs upstream and
downstream from the functional ATG start codon, respectively), flhDC(-455/+223) or
ehxCABD(-261/+22) with increasing concentrations of purified wild-type (WT) or mutant
MBP-GrlA proteins in binding buffer containing 10 mM Tris-HCl [pH 8], 50 mM KCl, 1
mM dithiothreitol (DTT), 0.5mM EDTA, 10 µg/ml bovine serum albumin (BSA) and 5%
glycerol. The primers used to amplify the promoter regions are shown in Table 2.2.
Reaction mixtures were incubated for 30 min at room temperature and then separated by
electrophoresis on 5% polyacrylamide gels in 0.5X Tris-borate-EDTA buffer.DNA bands
were stained with ethidium bromide and visualised with a Syngene transilluminator

53
(Syngene, Frederick, MD, USA). For competitive EMSAs, DNA was incubated with
1µM MBP-GrlA for 30 min, followed by the addition of increasing concentrations of
His-GrlR for an additional 30 min at room temperature. The complexes were visualised
as described above. The formation of GrlR-GrlA complex in the competitive EMSA was
verified by setting up a 300µl amylose-resin bound MBP-GrlA–DNA binding reaction
for 30 min. A 30 µl sample was run as input on EMSA and SDS-PAGE and the rest was
washed several times with excess DNA binding buffer before titrating with GrlR. A 30 µl
sample was run after titrating with GrlR followed by washes. The final beads and all
other washes along with the initial samples were analyzed on both EMSA and SDS-
PAGE gels.
2.2.7. Extracellular protein extraction and detection
Overnight cultures of EPEC strains were grown in DMEM (Invitrogen; Carlsbad,
CA, USA) and supplemented with 30 µg/ml of kanamycin, 40 µg/ml streptomycin,
34µg/ml chloramphenicol until the optical density at 600 nm reached 0.8. The cultures
were diluted 1:50 into fresh DMEM and incubated for 9 h at 37°C in a shaking water bath
at 200 rpm. Bacterial cells were removed from the culture by centrifugation (5,500 x g,
10 min, 4°C), and the supernatant was filtered through a 0.22-µm-pore-size small-protein
binding filter (Millex; Millipore). The Extracellular protein (ECP) fraction was isolated
by trichloroacetic acid precipitation (Shimizu et al., 2002) and the protein pellet was
washed thrice with -20°C acetone and then air dried. ECP pellets were solubilised in
Ready Prep reagent 3 (5 M urea, 2 M thiourea, 2% (w/v) CHAPS, 2% (w/v) SB 3–10, 40
mM Tris, and 0.2% (w/v) Bio-Lyte 3/10 ampholyte (Bio-Rad; Hercules, CA, USA)), and
stored at -80°C, as described(Li et al., 2004). Proteins were transferred to a PVDF

54
membrane. EspB was detected by the addition of diluted anti-EspB (1:2000) polyclonal
antiserum, followed by a 1:5000 dilution of mouse anti-rabbit IgG HRP (1:5000) (Santa
Cruz Biotechnology; Santa Cruz, CA, USA). The PVDF membrane was examined using
the SuperSignal WestPico Chemiluminescent substrate (Pierce Biotechnology; Rockford,
IL, USA) under the conditions recommended by the manufacturer.
2.2.8. ler-gfp promoter assay
The plasmid pGY1 contains a transcriptional fusion of the gfp to the ler gene and
its regulatory region(Yerushalmi et al., 2008). The plasmid was transformed into the
EPEC grlRA::kn mutant strain EM3715 to create strain GY2155. EPEC strain GY2155
was then transformed with a compatible, pACYC184-based plasmid, expressing GrlA or
GrlAΔ and various grlA mutants. The constructed strains were grown in DMEM
overnight as described above. Cells were then harvested by centrifugation at 5,500 xg for
10 min at 4°C and re-suspended in lysis buffer (1X PBS) with complete protease
inhibitors (Roche Applied Science). The cells were lysed and centrifuged down at
30,000xg for 30 min.Protein concentrations in the supernatant were adjusted and
separated on an SDS-PAGE, transferred to a PVDF membrane and subjected to western
blotting. GFP was detected by the addition of diluted anti-GFP (1:2000) rabbit
monoclonal antibody (Invitrogen), followed by a 1:5000 dilution of mouse anti-rabbit
IgG HRP (Santa Cruz Biotechnology). The PVDF membrane was examined using the
SuperSignal WestPico Chemiluminescent substrate under the conditions recommended
by the manufacturer.

55
2.2.9. Pull down assay and Western blot
MBP fusions of GrlA protein with or without substitutions in the HTH motif
region were bound to amylose beads, incubated with His-GrlR overnight, washed and
eluted with 20 mM maltose. The eluted proteins were separated by 12.5% SDS-PAGE,
followed by gel staining with coomassie brilliant blue and destained. For western blot
analysis the proteins were transferred to a PVDF membrane. The membrane was cut
carefully according to size difference between MBP-GrlA and His GrlR and treated
separately for detection of the proteins. MBP-GrlA fusion was detected by the addition of
diluted anti-MBP antiserum (1:2000), (New England Biolabs, UK) followed by a 1:5000
dilution of mouse anti-rabbit IgG HRP (1:5000) (Santa Cruz Biotechnology; Santa Cruz,
CA, USA). 6His-GrlR was detected by the addition of anti-His monoclonal antibody
(1:10000) (Santa Cruz Biotechnology; Santa Cruz, CA, USA). The PVDF membrane was
examined using the SuperSignal WestPico Chemiluminescent substrate (Pierce
Biotechnology; Rockford, IL, USA) under the conditions recommended by the
manufacturer.
2.2.10. Peptide mass finger printing
Protein bands were excised and washed with 25 mM ammonium bicarbonate
(ABB) in 50% acetonitrile (ACN) buffer thrice. The proteins in the gel were reduced with
10 mM DTT in 25 mM ABB buffer, alkylated with 5 mM iodoacetamide, dehydrated and
digested with trypsin overnight. After in-gel digestion, the solution was transferred to a
clean tube and sonicated for 30 min in the presence of 50ml 50% ACN and 5% acetic
acid for protein extraction. This extraction procedure was repeated three times; the

56
pooled extracts were dried with a vacuum concentrator. The samples were processed and
analysed using a LTQ-FT ultra mass spectrometer.
For each experiment, MS/MS (dta) spectra were extracted from the raw data files
using the extract_msn program in Biowork 3.3 (ThermoFinnigan). The extracted dta files
were combined into a single file in the Mascot generic file (mgf) format. Except for the
conversion of precursor mass from MH+ in dta to m/z in mgf, the fragment ion m/z and
intensity values were used as determined. Proteins were identified by searching the
combined data against the entire database via an in-house Mascot server (version 2.2.07).
Two missing cleavages were allowed. Precursor ion and MS/MS fragment ion error
tolerances were set to < 10 p.p.m. and < 0.8 Da, respectively. A protein was accepted as a
true positive if it had a significant score (P< 0.05) and at least two unique peptides.
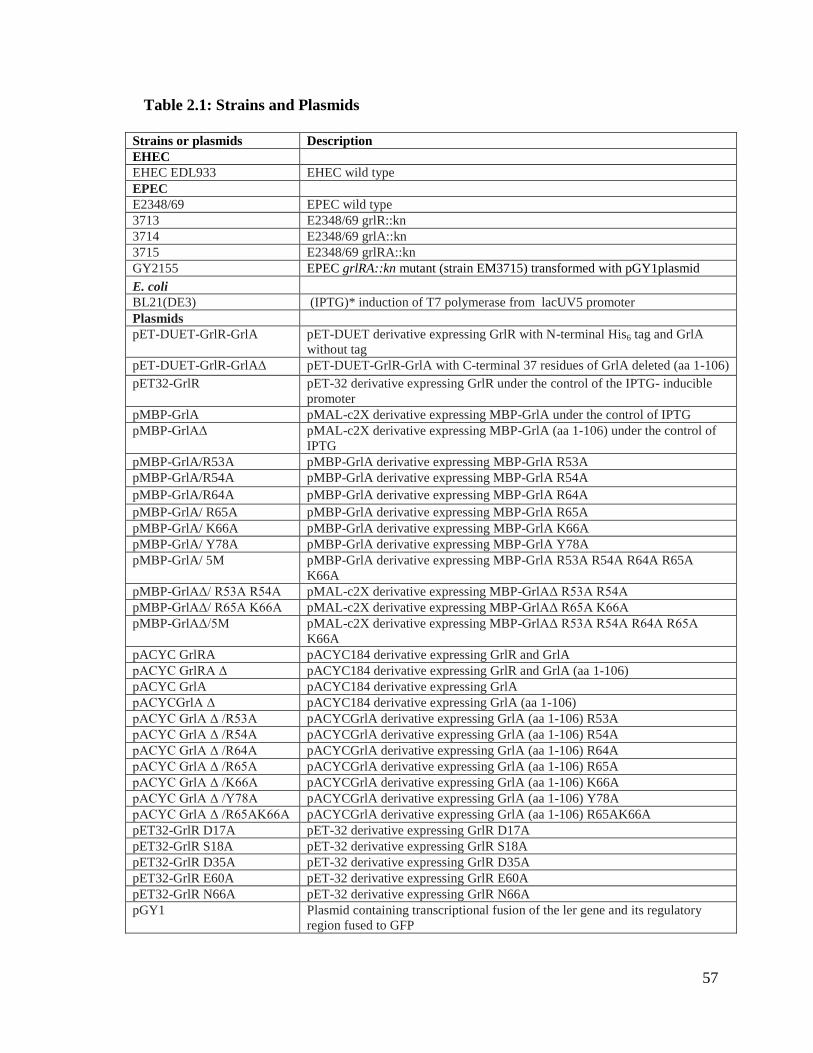
57
Table 2.1: Strains and Plasmids
Strains or plasmids Description
EHEC
EHEC EDL933 EHEC wild type
EPEC
E2348/69 EPEC wild type
3713 E2348/69 grlR::kn
3714 E2348/69 grlA::kn
3715 E2348/69 grlRA::kn
GY2155 EPEC grlRA::kn mutant (strain EM3715) transformed with pGY1plasmid
E. coli
BL21(DE3) (IPTG)* induction of T7 polymerase from lacUV5 promoter
Plasmids
pET-DUET-GrlR-GrlA pET-DUET derivative expressing GrlR with N-terminal His6 tag and GrlA
without tag
pET-DUET-GrlR-GrlAΔ pET-DUET-GrlR-GrlA with C-terminal 37 residues of GrlA deleted (aa 1-106)
pET32-GrlR pET-32 derivative expressing GrlR under the control of the IPTG- inducible
promoter
pMBP-GrlA pMAL-c2X derivative expressing MBP-GrlA under the control of IPTG
pMBP-GrlAΔ pMAL-c2X derivative expressing MBP-GrlA (aa 1-106) under the control of
IPTG
pMBP-GrlA/R53A pMBP-GrlA derivative expressing MBP-GrlA R53A
pMBP-GrlA/R54A pMBP-GrlA derivative expressing MBP-GrlA R54A
pMBP-GrlA/R64A pMBP-GrlA derivative expressing MBP-GrlA R64A
pMBP-GrlA/ R65A pMBP-GrlA derivative expressing MBP-GrlA R65A
pMBP-GrlA/ K66A pMBP-GrlA derivative expressing MBP-GrlA K66A
pMBP-GrlA/ Y78A pMBP-GrlA derivative expressing MBP-GrlA Y78A
pMBP-GrlA/ 5M pMBP-GrlA derivative expressing MBP-GrlA R53A R54A R64A R65A
K66A
pMBP-GrlAΔ/ R53A R54A pMAL-c2X derivative expressing MBP-GrlAΔ R53A R54A
pMBP-GrlAΔ/ R65A K66A pMAL-c2X derivative expressing MBP-GrlAΔ R65A K66A
pMBP-GrlAΔ/5M pMAL-c2X derivative expressing MBP-GrlAΔ R53A R54A R64A R65A
K66A
pACYC GrlRA pACYC184 derivative expressing GrlR and GrlA
pACYC GrlRA Δ pACYC184 derivative expressing GrlR and GrlA (aa 1-106)
pACYC GrlA pACYC184 derivative expressing GrlA
pACYCGrlA Δ pACYC184 derivative expressing GrlA (aa 1-106)
pACYC GrlA Δ /R53A pACYCGrlA derivative expressing GrlA (aa 1-106) R53A
pACYC GrlA Δ /R54A pACYCGrlA derivative expressing GrlA (aa 1-106) R54A
pACYC GrlA Δ /R64A pACYCGrlA derivative expressing GrlA (aa 1-106) R64A
pACYC GrlA Δ /R65A pACYCGrlA derivative expressing GrlA (aa 1-106) R65A
pACYC GrlA Δ /K66A pACYCGrlA derivative expressing GrlA (aa 1-106) K66A
pACYC GrlA Δ /Y78A pACYCGrlA derivative expressing GrlA (aa 1-106) Y78A
pACYC GrlA Δ /R65AK66A pACYCGrlA derivative expressing GrlA (aa 1-106) R65AK66A
pET32-GrlR D17A pET-32 derivative expressing GrlR D17A
pET32-GrlR S18A pET-32 derivative expressing GrlR S18A
pET32-GrlR D35A pET-32 derivative expressing GrlR D35A
pET32-GrlR E60A pET-32 derivative expressing GrlR E60A
pET32-GrlR N66A pET-32 derivative expressing GrlR N66A
pGY1 Plasmid containing transcriptional fusion of the ler gene and its regulatory
region fused to GFP

58
Table 2.2: Primers used to amplify the promoter regions
Primers
-275 For primer
ler promoter 5' CGT TTG TTA ACG AGA TGA TTT TCT TCT ATA TCA TTG ATT TT 3'
217 Rev primer
ler promoter 5' TTC CGG CGA GCG AGT CCA TCA TCA GGC AC 3'
-261 For primer
hlyC promoter 5' CATTTGTCACGTGGCTATTCATATGAAAATCATACGT 3'
22 Rev primer
hlyC promoter 5' CGTCAAAAGCATTAGATTTCATAATGTTTAAATAAATAAGA 3'
-455 For primer
flhD promoter
5' ATG AAA GTG ATT ATT TAT AGC AGA TGA TTA TTT ACG GTG AGT TAT TTT
AAC TGT GC 3'
223 Rev primer
flhD promoter
5' GGC TGT CAA AAC GGA AGT GAC AAA CCA GCT GAT TGG 3'
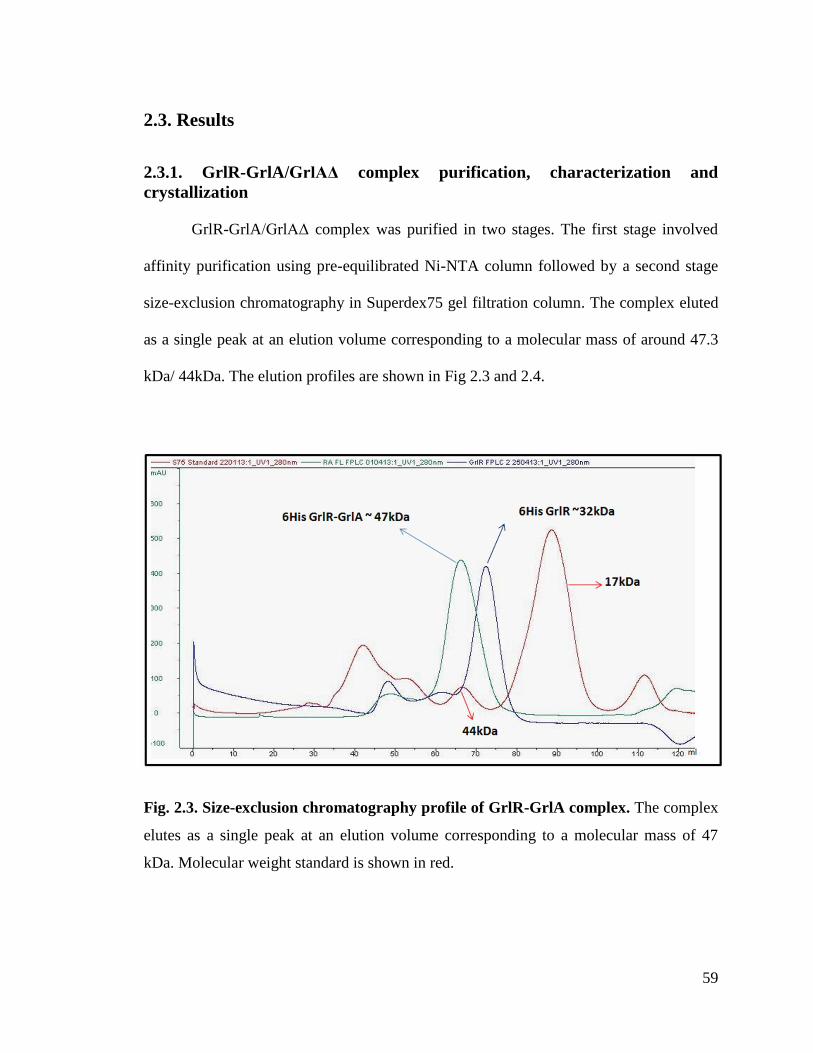
59
2.3. Results
2.3.1. GrlR-GrlA/GrlAΔ complex purification, characterization and
crystallization
GrlR-GrlA/GrlAΔ complex was purified in two stages. The first stage involved
affinity purification using pre-equilibrated Ni-NTA column followed by a second stage
size-exclusion chromatography in Superdex75 gel filtration column. The complex eluted
as a single peak at an elution volume corresponding to a molecular mass of around 47.3
kDa/ 44kDa. The elution profiles are shown in Fig 2.3 and 2.4.
Fig. 2.3. Size-exclusion chromatography profile of GrlR-GrlA complex. The complex
elutes as a single peak at an elution volume corresponding to a molecular mass of 47
kDa. Molecular weight standard is shown in red.
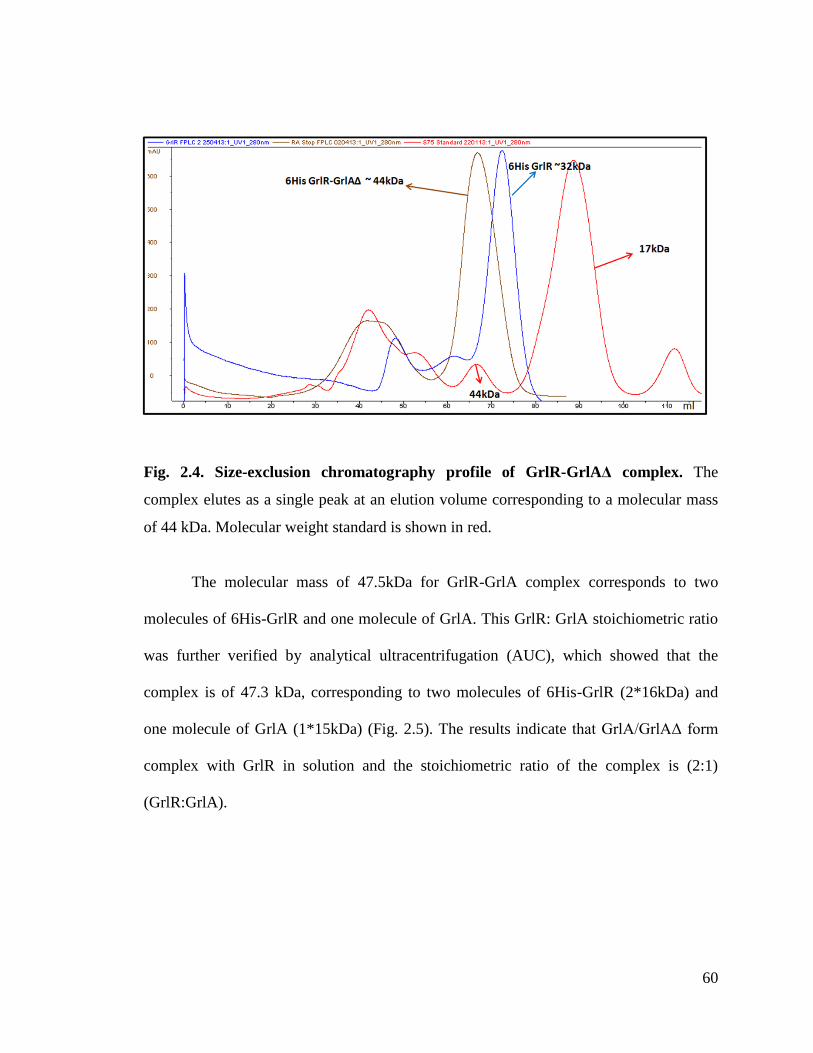
60
Fig. 2.4. Size-exclusion chromatography profile of GrlR-GrlAΔ complex. The
complex elutes as a single peak at an elution volume corresponding to a molecular mass
of 44 kDa. Molecular weight standard is shown in red.
The molecular mass of 47.5kDa for GrlR-GrlA complex corresponds to two
molecules of 6His-GrlR and one molecule of GrlA. This GrlR: GrlA stoichiometric ratio
was further verified by analytical ultracentrifugation (AUC), which showed that the
complex is of 47.3 kDa, corresponding to two molecules of 6His-GrlR (2*16kDa) and
one molecule of GrlA (1*15kDa) (Fig. 2.5). The results indicate that GrlA/GrlAΔ form
complex with GrlR in solution and the stoichiometric ratio of the complex is (2:1)
(GrlR:GrlA).
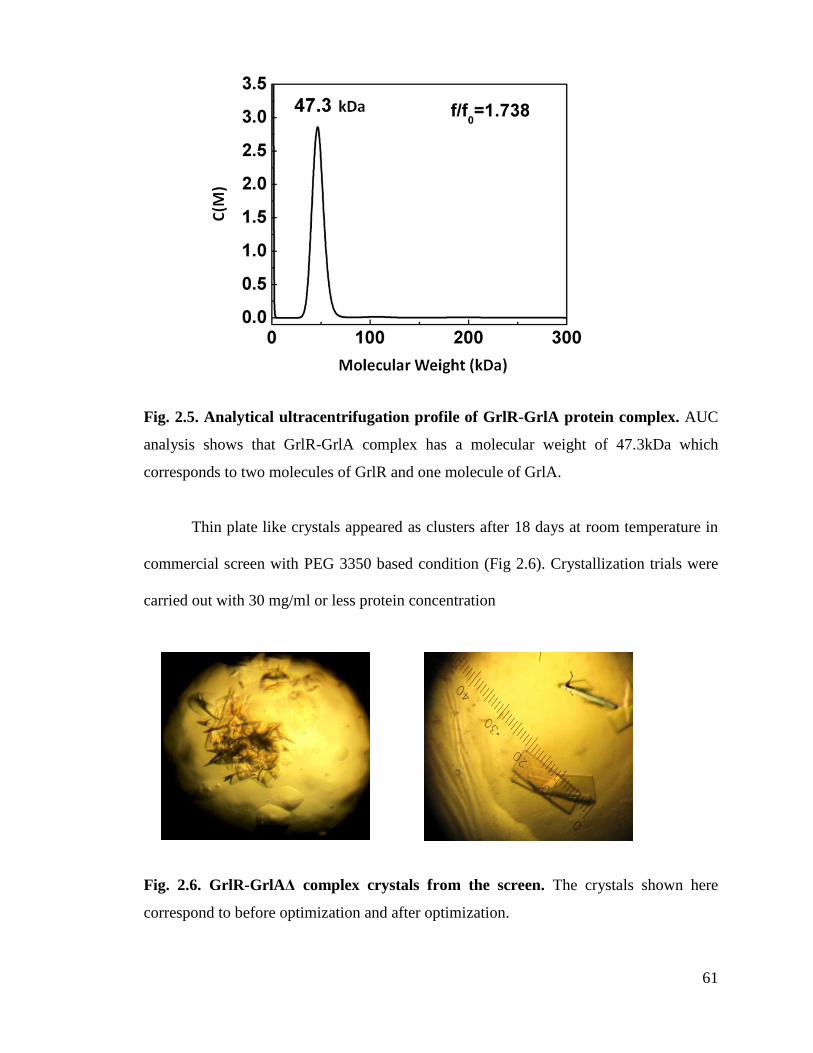
61
Fig. 2.5. Analytical ultracentrifugation profile of GrlR-GrlA protein complex. AUC
analysis shows that GrlR-GrlA complex has a molecular weight of 47.3kDa which
corresponds to two molecules of GrlR and one molecule of GrlA.
Thin plate like crystals appeared as clusters after 18 days at room temperature in
commercial screen with PEG 3350 based condition (Fig 2.6). Crystallization trials were
carried out with 30 mg/ml or less protein concentration
Fig. 2.6. GrlR-GrlAΔ complex crystals from the screen. The crystals shown here
correspond to before optimization and after optimization.

62
Grid screening and additive screening were carried out to improve the quality of
the crystal. However the crystals were not of good quality. Single, plate like crystals
were obtained with GrlR-GrlAΔ complex within 3 days in the same condition when 2.0
M NDSB-201 was added as an additive (Fig 2.6).
2.3.2. Overall structure of the GrlR-GrlA complex
We initially attempted to determine the structure of full-length GrlA in complex
with GrlR. However, the full-length GrlR-GrlA complex did not yield diffraction-quality
crystals, presumably because of the unstable nature of GrlA. As a result, we generated
GrlR-GrlA complexes with varying lengths of GrlA. A complex of GrlR with GrlAΔ (aa
1-106) yielded diffraction quality crystals (Fig. 2.7).
Fig. 2.7. Schematic representation of GrlR and GrlA proteins with motifs.
The structure was determined and refined up to 2.7 Å resolution (Fig. 2.8). The
first 8 residues and last 11 residues of GrlA were not well defined in the electron density
map and were not included in the model (Fig. 2.8; Table 2.3).

63
Fig. 2.8. Crystal structure of GrlR-GrlAΔ complex. The two monomers of GrlR are
shown in green (monomer A) and cyan (monomer B). GrlA is shown in magenta. The
ligand Triton-X 100 is shown as a stick model.

64
Table 2.3: Crystallographic statistics and refinement details
SelMet SAD
Data collection
Space group C2221
Cell dimensions
a, b, c (Å) a = 83.19 b = 121.21 c = 84.83
α, β, γ () 90
Wavelength 0.97893
Resolution (Å) 50.0-2.62(2.67-2.62)
Rsym or Rmerge 0.12 (0.39)
I / σI 16.23 (3.56)
Completeness (%) 98.0(82.8)
Redundancy 13.9(10.6)
Refinement
Resolution (Å) 15.0-2.7
No. reflections 22108
Rwork / Rfree 0.18/0.23
No. atoms
Protein 2543
Ligand/ion 26
Water 15
B-factors
Protein 53.9
Ligand/ion 54.80
Water 45.7
R.m.s deviations
Bond lengths (Å) 0.01
Bond angles () 1.283
a Rsym = |Ii -<I>|/ |Ii| where Ii is the intensity of the i
th measurement, and <I> is the mean
intensity for that reflection.
bRwork = | Fobs - Fcalc|/ |Fobs| where Fcalc and Fobs are the calculated and observed
structure factor amplitudes, respectively.
cRfree = as for Rwork, but for 10.0% of the total reflections chosen at random and omitted
from refinement.
Individual B-factor refinement was carried out.
*Values in the parenthesis are the highest resolution bin values.

65
GrlR is a β-barrel protein, structurally similar to our previously determined GrlR
structures (rmsd of 0.87Å for all Cα atoms) (Jobichen et al., 2009; Jobichen et al., 2007),
whereas GrlAΔ mainly comprises a Helix-Turn-Helix (HTH) motif (aa 39-66) and an
anti-parallel β-sheet in the C-terminus (Fig. 2.9 and 2.10). The complex consists of one
dimer of GrlR bound to a monomer of GrlA (2:1 stoichiometric ratio). There are 15
hydrogen bonding contacts (<3.2Å) between GrlR and GrlAΔ with a buried area of
1043.8 Å2.
Fig. 2.9. Structure of GrlA. The ribbon diagram of GrlA

66
Fig. 2.10. The topology diagram of the GrlA molecule.
2.3.3. Sequence and structural homology of GrlA
GrlA homologs are present in over 100 species of bacteria including EPEC,
EHEC, Citrobacter rodentium, Shigella sp. and Salmonella sp. GrlA shares 33%
sequence identity (46% similarity) with CaiF, a potential transcriptional activator of
carnitine metabolism in E. coli. GrlA belongs to the PFAM family of DUF1401
(http://pfam.sanger.ac.uk/family/DUF1401). Despite a very low sequence identity (<7%),
the DALI (Holm and Sander, 1998) search for structural homologs of GrlA identified
several regulatory proteins. The closest homologue are also HTH motif containing
proteins such as a transcriptional regulator from Methanosarcina mazei (PDB 3R0A;
RMSD 2.8 Å for 72 Cα atoms), followed by a DNA-binding protein from Pyrococcus
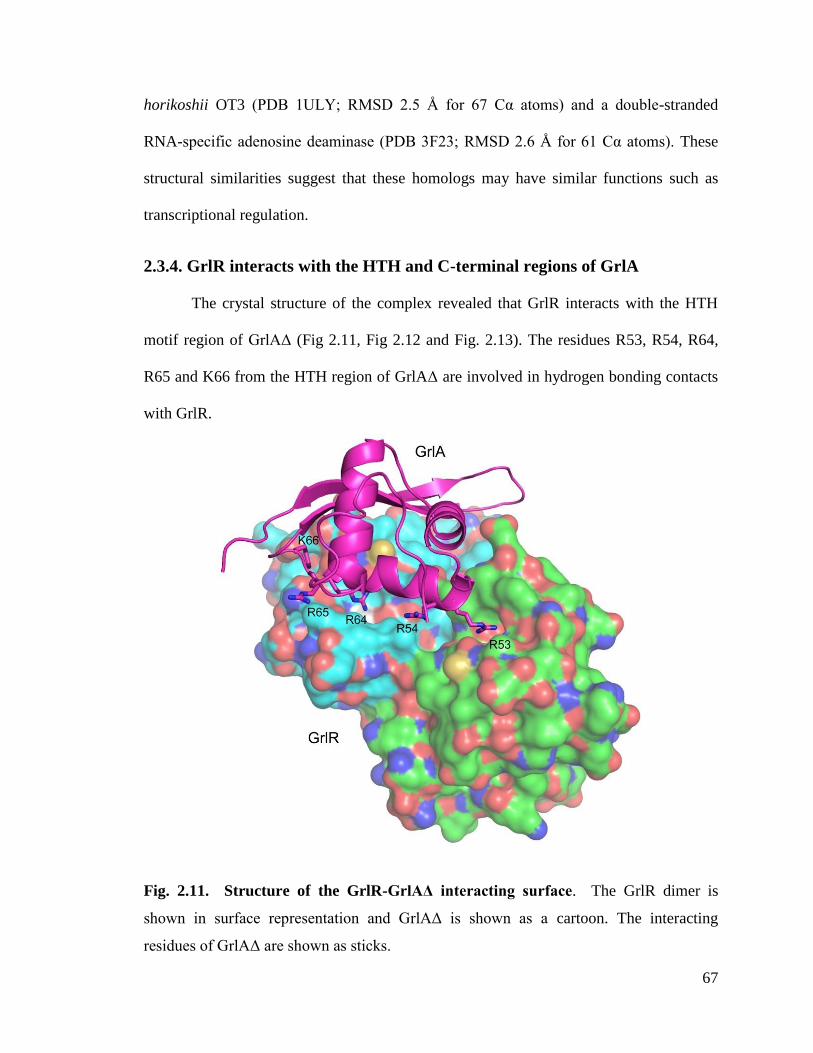
67
horikoshii OT3 (PDB 1ULY; RMSD 2.5 Å for 67 Cα atoms) and a double-stranded
RNA-specific adenosine deaminase (PDB 3F23; RMSD 2.6 Å for 61 Cα atoms). These
structural similarities suggest that these homologs may have similar functions such as
transcriptional regulation.
2.3.4. GrlR interacts with the HTH and C-terminal regions of GrlA
The crystal structure of the complex revealed that GrlR interacts with the HTH
motif region of GrlAΔ (Fig 2.11, Fig 2.12 and Fig. 2.13). The residues R53, R54, R64,
R65 and K66 from the HTH region of GrlAΔ are involved in hydrogen bonding contacts
with GrlR.
Fig. 2.11. Structure of the GrlR-GrlAΔ interacting surface. The GrlR dimer is
shown in surface representation and GrlAΔ is shown as a cartoon. The interacting
residues of GrlAΔ are shown as sticks.

68
Fig. 2.12. Final 2Fo-Fc electron density map (contoured at 1 σ) for the key residues of
GrlAΔ.
Fig. 2.13. Structure of the GrlR-GrlAΔ interactions. (a) The interacting residues of
GrlR (monomer A) in green and GrlAΔ in magenta shown as a stick model. (b) The
interacting residues of GrlR (monomer B) in cyan.
a b

69
Our previous studies with GrlA (Jobichen et al., 2007) suggested that the C-
terminal region of GrlA might be involved in binding to GrlR. Therefore, we sought to
independently verify the contribution of the N-terminal HTH motif and the C-terminus of
GrlA in its interaction with GrlR. To verify this, we created three constructs by
substituting the above mentioned five interacting amino acids of the HTH motif region
with alanine (denoted as GrlA5M), truncating the C-terminal region of GrlA (GrlAΔ), or
both mutations (GrlAΔ5M). Thus, the various GrlA constructs used in these interaction
studies include MBP-GrlA, MBP-GrlA5M, MBP-GrlAΔ and MBP-GrlAΔ5M. The
circular dichroism (CD) spectrum of the wild-type and mutants of GrlA suggest that the
mutants have the same fold as the wild-type GrlA (Fig. 2.14)
Fig. 2.14. Circular dichroism spectroscopic analysis of various MBP-GrlA
constructs. Readout from Circular Dichroism (CD) spectroscopy analysis showing that
MBP-GrlA, MBP-GrlAΔ, MBP-GrlA5M and MBP-GrlAΔ5M are all well folded and
have the same secondary structure with no global structural changes. The profiles are
color coded.

70
The Kd for the interaction between GrlR and MBP-GrlA was determined by
isothermal titration calorimetry (ITC). GrlR interacts with MBP-GrlA with an
approximate 35-fold higher affinity (Kd= 0.031 µM) than with MBP-GrlAΔ (Kd=1.1 µM)
(Fig. 2.15; Table 2.4). Besides the 35- and 31-fold reductions in affinity respectively,
MBP-GrlA and MBP-GrlA5M also showed thermodynamic profiles distinct from the
wild-type MBP-GrlA. While the MBP-GrlA interaction with GrlR is enthalpically and
entropically favourable (H = -4.8 kcal/mol, -TS = -5.4 kcal/mol), the other two protein
variants (GrlAΔ and GrlA5M) showed an enthalpically driven interaction, with
unfavorable entropic contributions (H = -21.0 kcal/mol, -TS = 12.9 kcal/mol, for
MBP-GrlA; H = -9.6 kcal/mol, -TS = 1.3 kcal/mol, for GrlA5M) (Table 2.4).

71
Fig. 2.15. Role of the C-terminal region and the Helix-Turn-Helix (HTH) motif
region of GrlA in GrlR-GrlA interactions. The binding affinities of MBP-GrlA/MBP-
GrlAΔ to GrlR were determined using isothermal titration calorimetry (ITC).
Representative ITC profiles are shown.

72
Fig. 2.16. Role of the C-terminal region and the Helix-Turn-Helix (HTH) motif
region of GrlA in GrlR-GrlA interactions (Cont…). The binding affinities of GrlR to
MBP-GrlA/MBP-GrlAΔ with substitutions in the HTH motif region (5M) were
determined using ITC. (5M refers to substitutions at R53A R54A R64A R65A K66A.)
Similarly, the Kd for the interaction between GrlR and MBP-GrlA5M was also
higher (Kd= 0.96 µM) than the wild-type interaction (Fig 2.16; Table 2.4). Probably, the
C-terminal region in MBP-GrlA, predicted to be mainly unstructured, contributes
significantly to the desolvation entropy gain associated to the binding, and this
contribution is absent in MBP-GrlA yielding a less favorable entropy (-TS = 18.3
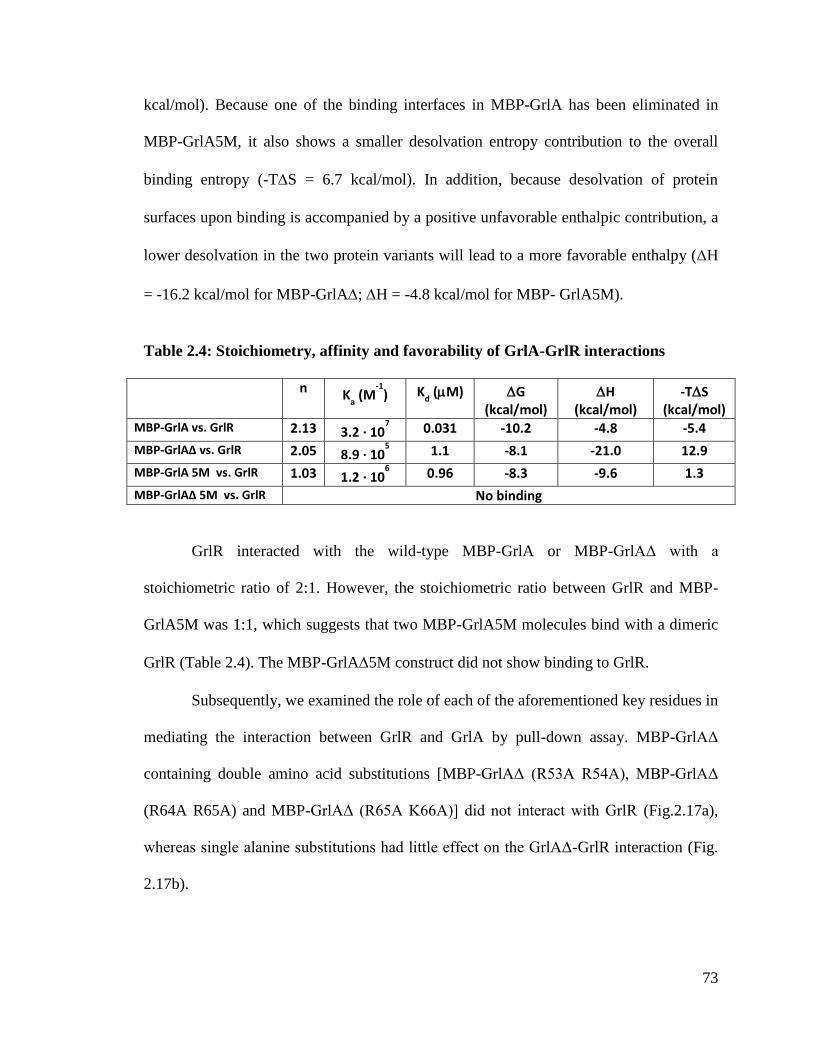
73
kcal/mol). Because one of the binding interfaces in MBP-GrlA has been eliminated in
MBP-GrlA5M, it also shows a smaller desolvation entropy contribution to the overall
binding entropy (-TS = 6.7 kcal/mol). In addition, because desolvation of protein
surfaces upon binding is accompanied by a positive unfavorable enthalpic contribution, a
lower desolvation in the two protein variants will lead to a more favorable enthalpy (H
= -16.2 kcal/mol for MBP-GrlA; H = -4.8 kcal/mol for MBP- GrlA5M).
Table 2.4: Stoichiometry, affinity and favorability of GrlA-GrlR interactions
n Ka (M
-1) K
d (M) G
(kcal/mol) H
(kcal/mol) -TS
(kcal/mol) MBP-GrlA vs. GrlR 2.13 3.2 · 10
7 0.031 -10.2 -4.8 -5.4
MBP-GrlAΔ vs. GrlR 2.05 8.9 · 105 1.1 -8.1 -21.0 12.9
MBP-GrlA 5M vs. GrlR 1.03 1.2 · 106 0.96 -8.3 -9.6 1.3
MBP-GrlAΔ 5M vs. GrlR No binding
GrlR interacted with the wild-type MBP-GrlA or MBP-GrlAΔ with a
stoichiometric ratio of 2:1. However, the stoichiometric ratio between GrlR and MBP-
GrlA5M was 1:1, which suggests that two MBP-GrlA5M molecules bind with a dimeric
GrlR (Table 2.4). The MBP-GrlA5M construct did not show binding to GrlR.
Subsequently, we examined the role of each of the aforementioned key residues in
mediating the interaction between GrlR and GrlA by pull-down assay. MBP-GrlAΔ
containing double amino acid substitutions [MBP-GrlAΔ (R53A R54A), MBP-GrlAΔ
(R64A R65A) and MBP-GrlAΔ (R65A K66A)] did not interact with GrlR (Fig.2.17a),
whereas single alanine substitutions had little effect on the GrlAΔ-GrlR interaction (Fig.
2.17b).
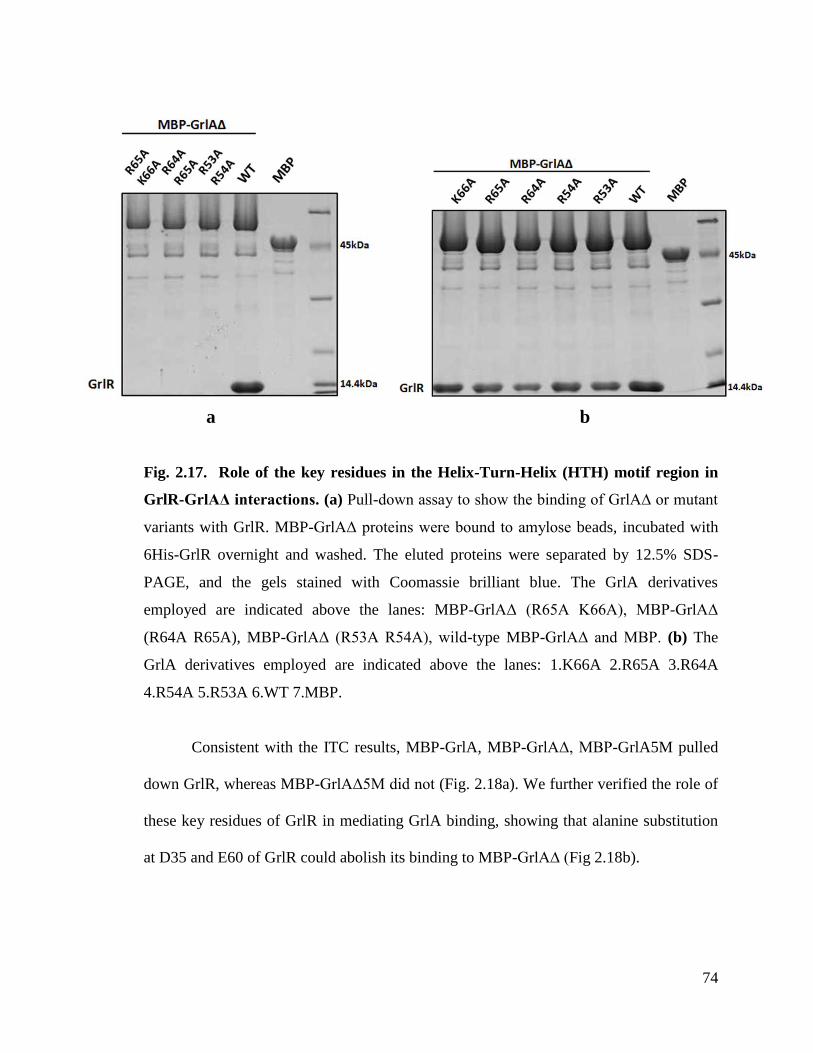
74
Fig. 2.17. Role of the key residues in the Helix-Turn-Helix (HTH) motif region in
GrlR-GrlAΔ interactions. (a) Pull-down assay to show the binding of GrlA∆ or mutant
variants with GrlR. MBP-GrlAΔ proteins were bound to amylose beads, incubated with
6His-GrlR overnight and washed. The eluted proteins were separated by 12.5% SDS-
PAGE, and the gels stained with Coomassie brilliant blue. The GrlA derivatives
employed are indicated above the lanes: MBP-GrlAΔ (R65A K66A), MBP-GrlAΔ
(R64A R65A), MBP-GrlAΔ (R53A R54A), wild-type MBP-GrlAΔ and MBP. (b) The
GrlA derivatives employed are indicated above the lanes: 1.K66A 2.R65A 3.R64A
4.R54A 5.R53A 6.WT 7.MBP.
Consistent with the ITC results, MBP-GrlA, MBP-GrlAΔ, MBP-GrlA5M pulled
down GrlR, whereas MBP-GrlAΔ5M did not (Fig. 2.18a). We further verified the role of
these key residues of GrlR in mediating GrlA binding, showing that alanine substitution
at D35 and E60 of GrlR could abolish its binding to MBP-GrlA∆ (Fig 2.18b).
a b
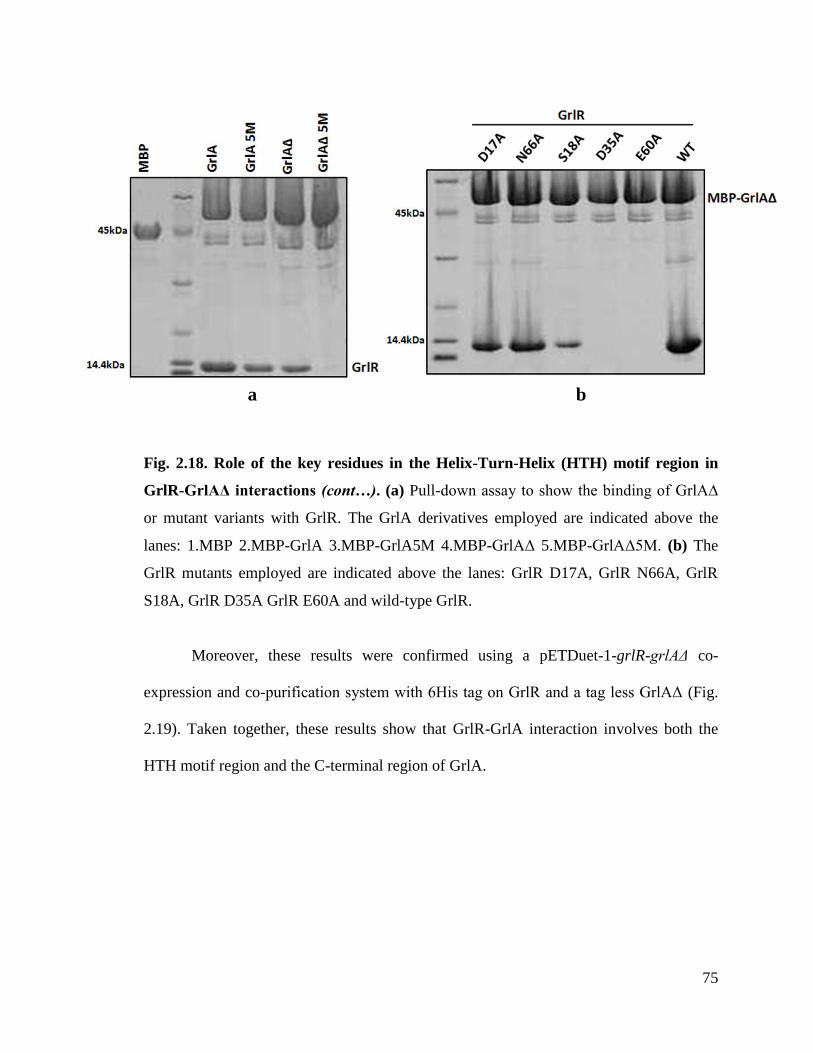
75
Fig. 2.18. Role of the key residues in the Helix-Turn-Helix (HTH) motif region in
GrlR-GrlAΔ interactions (cont…). (a) Pull-down assay to show the binding of GrlA∆
or mutant variants with GrlR. The GrlA derivatives employed are indicated above the
lanes: 1.MBP 2.MBP-GrlA 3.MBP-GrlA5M 4.MBP-GrlAΔ 5.MBP-GrlAΔ5M. (b) The
GrlR mutants employed are indicated above the lanes: GrlR D17A, GrlR N66A, GrlR
S18A, GrlR D35A GrlR E60A and wild-type GrlR.
Moreover, these results were confirmed using a pETDuet-1-grlR-grlAΔ co-
expression and co-purification system with 6His tag on GrlR and a tag less GrlAΔ (Fig.
2.19). Taken together, these results show that GrlR-GrlA interaction involves both the
HTH motif region and the C-terminal region of GrlA.
a b
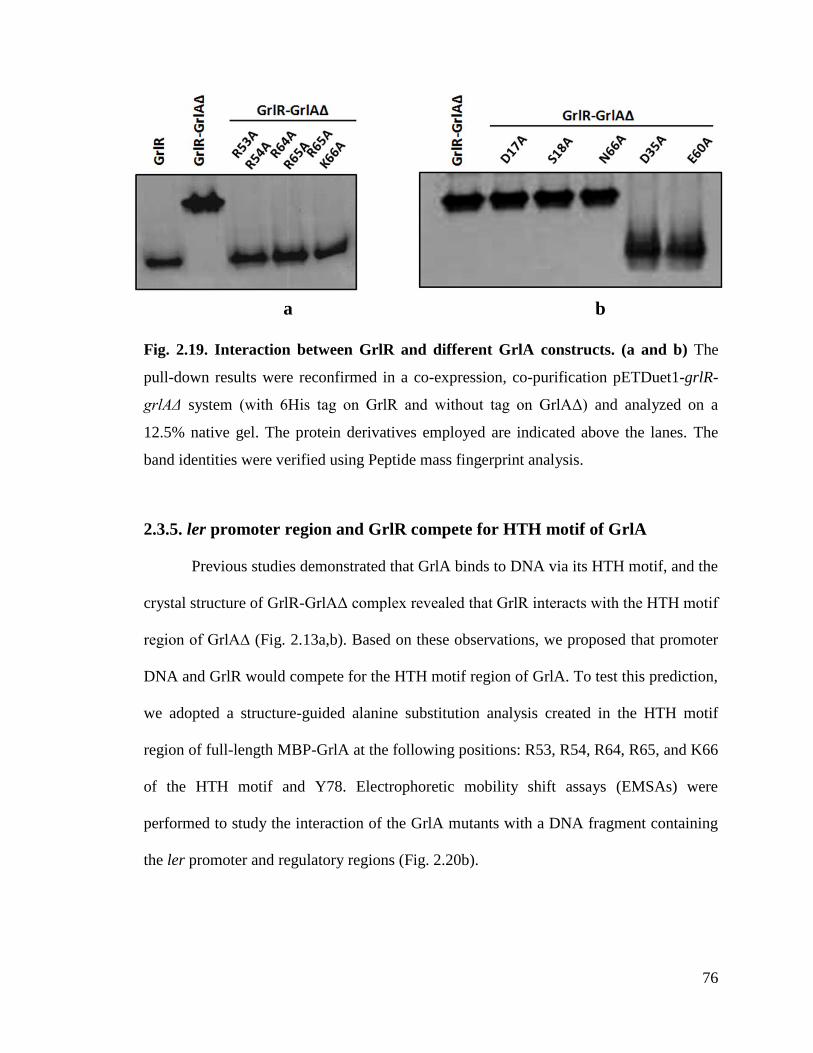
76
a b
Fig. 2.19. Interaction between GrlR and different GrlA constructs. (a and b) The
pull-down results were reconfirmed in a co-expression, co-purification pETDuet1-grlR-
grlAΔ system (with 6His tag on GrlR and without tag on GrlAΔ) and analyzed on a
12.5% native gel. The protein derivatives employed are indicated above the lanes. The
band identities were verified using Peptide mass fingerprint analysis.
2.3.5. ler promoter region and GrlR compete for HTH motif of GrlA
Previous studies demonstrated that GrlA binds to DNA via its HTH motif, and the
crystal structure of GrlR-GrlAΔ complex revealed that GrlR interacts with the HTH motif
region of GrlA∆ (Fig. 2.13a,b). Based on these observations, we proposed that promoter
DNA and GrlR would compete for the HTH motif region of GrlA. To test this prediction,
we adopted a structure-guided alanine substitution analysis created in the HTH motif
region of full-length MBP-GrlA at the following positions: R53, R54, R64, R65, and K66
of the HTH motif and Y78. Electrophoretic mobility shift assays (EMSAs) were
performed to study the interaction of the GrlA mutants with a DNA fragment containing
the ler promoter and regulatory regions (Fig. 2.20b).
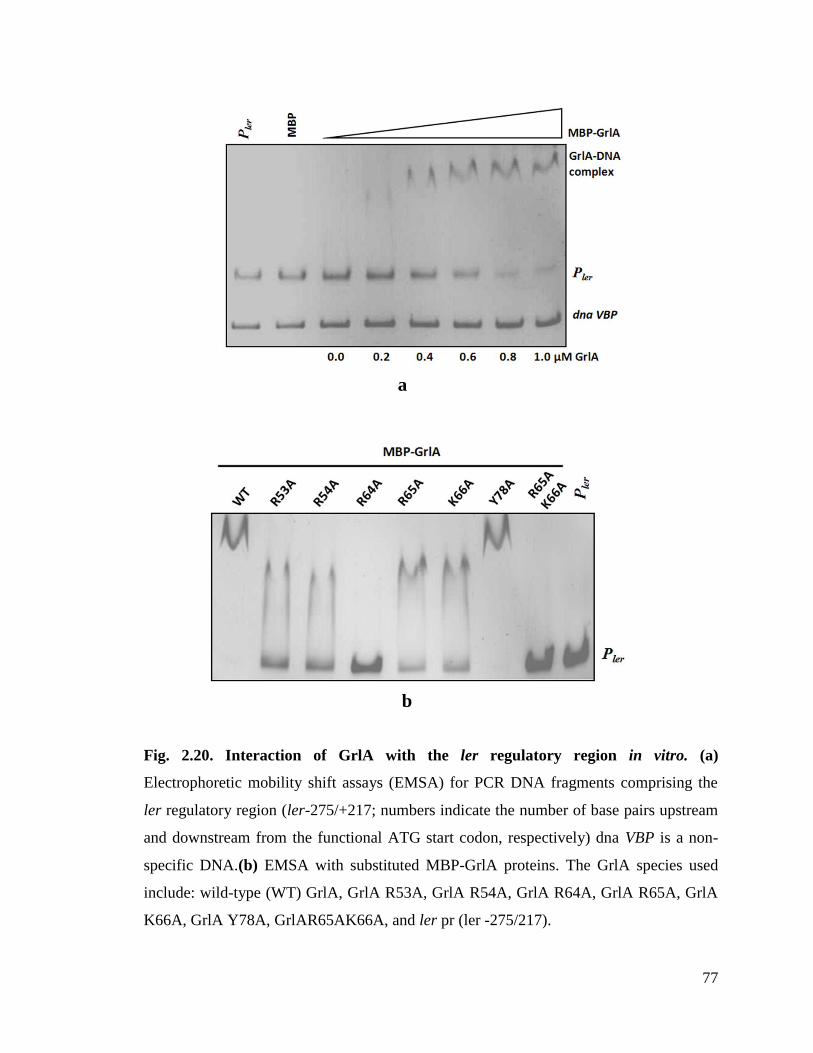
77
Fig. 2.20. Interaction of GrlA with the ler regulatory region in vitro. (a)
Electrophoretic mobility shift assays (EMSA) for PCR DNA fragments comprising the
ler regulatory region (ler-275/+217; numbers indicate the number of base pairs upstream
and downstream from the functional ATG start codon, respectively) dna VBP is a non-
specific DNA.(b) EMSA with substituted MBP-GrlA proteins. The GrlA species used
include: wild-type (WT) GrlA, GrlA R53A, GrlA R54A, GrlA R64A, GrlA R65A, GrlA
K66A, GrlA Y78A, GrlAR65AK66A, and ler pr (ler -275/217).
a
b

78
Mutants with single alanine substitutions at R53, R54, R65 and K66 exhibited
mobility shifts that were less than that observed for the wild-type GrlA, indicating a
reduced affinity for DNA. The MBP-GrlA R64A mutant and a double mutant of MBP-
GrlA (R65A K66A) failed to interact with DNA (Fig. 2.20b, lanes 4 and 8). Furthermore,
the Y78A mutant formed a shifted complex that was comparable to the wild-type GrlA-
DNA complex (Fig. 2.20b, lane 7).
To determine whether the addition of GrlR could dissociate MBP-GrlA from the
ler promoter, a competitive EMSA was conducted (Fig. 2.21). Under the given
experimental conditions, when the concentration of GrlR was increased to 0.3 µM, the
MBP-GrlA-DNA complex dissociated. No mobility shift of the DNA was detected at
higher GrlR concentrations (>0.3 µM).
Fig. 2.21. Competitive EMSA aimed at testing competition between Pler and 6His-
GrlR for binding to MBP-GrlA. PCR DNA fragments comprising the regulatory region
(ler-275/+217) were mixed and incubated with 1 µM purified MBP-GrlA for 15 min,
then combined with increasing concentrations (0.0, 0.15, 0.3, 0.45, and 0.6 µM) of 6His-
GrlR for an additional 15 min. The complexes and free DNA were separated on a 5%
native polyacrylamide gel and stained with ethidium bromide (ler pr: ler -275/+217).

79
In order to verify the formation of GrlR-GrlA complex, a pre-formed MBP-GrlA-
DNA complex bound to amylose beads was titrated against GrlR and washed with DNA
binding buffer. Beads from each of these stages and washes were simultaneously
analysed using SDS-PAGE and EMSA (Fig. 2.22)
Fig. 2.22. Competitive EMSA to study the formation of GrlR-GrlA complex. A
300µl MBP-GrlA-DNA binding reaction was set up by incubating DNA with amylose-
resin bound MBP-GrlA for 30 min. 30 µl sample was run as input on EMSA and SDS-
PAGE and the rest was washed carefully with excess DNA binding buffer before
incubating with GrlR. 30 µl sample was run after titrating with GrlR and the rest was
washed with excess DNA binding buffer. The final beads and all other washes along with
the initial samples were analyzed on (a) EMSA and (b) SDS-PAGE gels. The final beads
run in lane 9 show only MBP-GrlA and GrlR on SDS-PAGE and so is marked
accordingly. EMSA gel showed no apparent shift in the DNA compared to DNA alone
after the MBP-GrlA-DNA complex was titrated with GrlR (lane 6 and lane 1). SDS-
PAGE showed bands corresponding to MBP-GrlA and GrlR in the final beads (lane 9).
a b

80
The EMSA gel analysis showed no apparent shift in the DNA as compared with
DNA alone after the MBP-GrlA-DNA complex was titrated with GrlR (Fig. 2.22a). SDS-
PAGE showed bands corresponding to MBP-GrlA and GrlR in the final beads (Fig.
2.22b).These results indicate that DNA bound to MBP-GrlA was replaced by GrlR.
Besides, these findings show that GrlR outcompetes with DNA to bind to the GrlA HTH
motif. Next, we sought to verify the ability of DNA to pull out GrlA from the GrlR-GrlA
complex. A pre-formed GrlA-GrlR complex was incubated with increasing
concentrations of DNA (Pler) for about 30 min and then analysed using EMSA.
Fig. 2.23. EMSA to verify the binding of DNA to preformed GrlR-GrlA complex. A
preformed MBP-GrlA-GrlR complex was incubated with increasing concentrations of
DNA for 30 min in DNA binding buffer. The samples were run on 5% EMSA gel and
stained with ethidium bromide. There is no apparent shift in the DNA incubated with
preformed MBP-GrlA-GrlR complex compared to DNA alone clearly indicating that
DNA is not capable of displacing GrlR from the MBP-GrlA-GrlR complex and forming
complex with MBP-GrlA.

81
There was no apparent shift in the DNA incubated with pre-formed GrlA-GrlR
complex compared with the control DNA fragment (Fig. 2.23). These results clearly
show the inability of DNA to displace GrlR from the GrlR-GrlA complex.
2.3.6. The GrlR-GrlAΔ complex is functional in vivo
We next determined whether the GrlAΔ could form a functional regulatory
complex with DNA in vivo. To this end, we employed the EPEC null strain (ΔgrlRA::kn),
which contains a ler-gfp transcriptional fusion (GY2155), and for which the GFP
expression levels report the activity of the ler promoter. This strain was transformed with
a plasmid expressing GrlA or GrlAΔ and the GFP levels were compared by western blot
analysis using an antibody directed against GFP.
Fig. 2.24. In vivo functionality of GrlR-GrlAΔ asymmetric complex. Immunoblot
analysis using anti-GFP antibodies to compare the expression of GFP via the ler
promoter. The used strains include a deletion mutant of grlRA (control) (indicated as
GrlR-A
-); this mutant complemented with plasmids expressing GrlA, GrlAΔ, GrlRA and
GlRAΔ, as indicted above the lanes. All strains contained a compatible GFP-expressing
plasmid via the ler promoter.
The results show that the activity of the ler promoter in the strain expressing the
full-length GrlA and GrlAΔ is almost the same, indicating that GrlAΔ is functional in

82
vivo. This finding also suggests a negligible role of the last 31 amino acids of GrlA in
terms of ler-promoter binding. The strain expressing both GrlR and GrlA showed less
activity as compared with the strain expressing GrlA alone, illustrating the repressive
activity of GrlR. Repression by GrlR in the GrlAΔ-expressing strain was comparable
with that expressing both GrlR and full-length GrlA (Fig.2.24).
An additional readout for the functionality of GrlA∆ is the secretion of EspB,
which reflects Ler expression and functionality (Mellies et al., 1999). EspB is a major
T3SS effector protein and EspB secretion acts as an indicator for the formation of a
functional T3SS (Yerushalmi et al., 2008).
Fig. 2.25. In vivo functionality of GrlR-GrlAΔ asymmetric complex (Cont…).
Secreted proteins were concentrated from supernatants of bacterial culture grown in
DMEM and resolved using 12% SDS-PAGE. These samples were then transferred to a
PVDF membrane and analysed using a monoclonal antibody against representative
secretory protein EspB. The strains used are as in Fig.2.24, but all lack the GFP-
expressing plasmid. Strains are indicated above the lanes as in Fig.2.24.
Our measurements of EspB secretion is in agreement with the data obtained with
the ler-gfp promoter assay. The secretion assay showed comparable EspB secretion
between ΔgrlRA::kn null strains supplemented with a plasmid expressing either GrlA∆ or

83
full-length GrlA (Fig. 2.25). Similarly, a comparable amount of EspB was secreted from
the mutant expressing GrlRA or GrlRAΔ. Although deletion of the C-terminal region of
GrlA resulted in a substantial decrease in the affinity between GrlA and GrlR (Fig. 2.15),
we did not observe significant phenotypic consequences under the experimental
conditions used in this study (Fig. 2.24 and Fig 2.25). This suggests that, as long as the
N-terminal region of GrlA is intact, the protein remains functional. The role of the
additional interaction from the C-terminal region of GrlA thus remains to be identified.
2.3.7. The key HTH residues are required for GrlA function in vivo
EspB secretion was further used to examine the role of the key HTH residues in
the regulatory functions of GrlA. To eliminate the negative regulatory effect of GrlR, we
transformed the ΔgrlRA::kn null strain with a plasmid expressing GrlA∆. Bacteria were
grown under conditions that favoured positive regulation of the T3SS by GrlA
(Bustamante et al., 2011), and the amount of EspB secretion was determined by western
blot analysis (Fig. 2.26). Secretion of EspB was elevated when the ΔgrlRA::kn null strain
was supplemented with GrlA∆, and GrlA with alanine substitutions at R65 or K66
induced efficient EspB secretion (Fig. 2.26). In contrast, the GrlAΔ R64A mutant failed
to induce EspB secretion (Fig. 2.26) as did the GrlAΔ (R65A K66A) double mutant.
Taken together, the results show that substitutions in key residues in the GrlA HTH
region, which hamper binding to the ler regulatory region, elicit a profound effect on the
ability of the bacteria to assemble a functional T3SS and thus on the bacterial virulence.
These effects are presumably related to reduced activity of the ler promoter and
consequently reduced expression of some or all of the LEE operons.
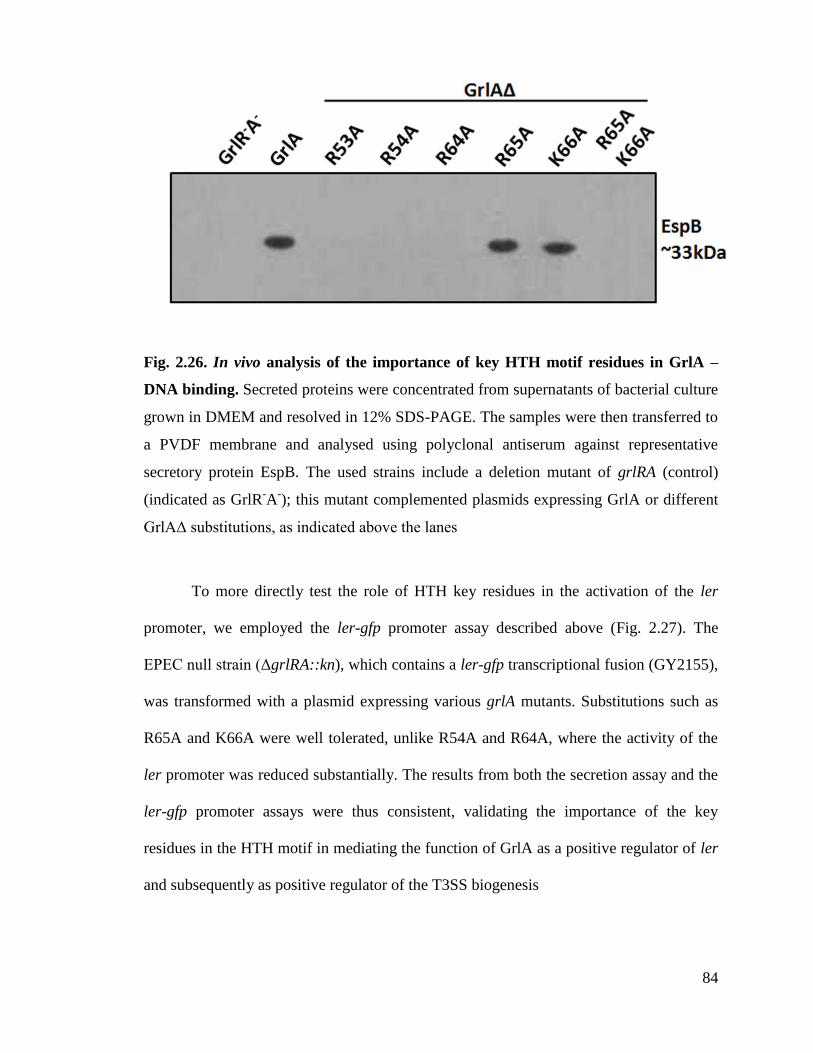
84
Fig. 2.26. In vivo analysis of the importance of key HTH motif residues in GrlA –
DNA binding. Secreted proteins were concentrated from supernatants of bacterial culture
grown in DMEM and resolved in 12% SDS-PAGE. The samples were then transferred to
a PVDF membrane and analysed using polyclonal antiserum against representative
secretory protein EspB. The used strains include a deletion mutant of grlRA (control)
(indicated as GrlR-A
-); this mutant complemented plasmids expressing GrlA or different
GrlAΔ substitutions, as indicated above the lanes
To more directly test the role of HTH key residues in the activation of the ler
promoter, we employed the ler-gfp promoter assay described above (Fig. 2.27). The
EPEC null strain (ΔgrlRA::kn), which contains a ler-gfp transcriptional fusion (GY2155),
was transformed with a plasmid expressing various grlA mutants. Substitutions such as
R65A and K66A were well tolerated, unlike R54A and R64A, where the activity of the
ler promoter was reduced substantially. The results from both the secretion assay and the
ler-gfp promoter assays were thus consistent, validating the importance of the key
residues in the HTH motif in mediating the function of GrlA as a positive regulator of ler
and subsequently as positive regulator of the T3SS biogenesis

85
Fig. 2.27. In vivo analysis of the importance of key HTH motif residues in GrlA –
DNA binding (Cont…) Immunoblot analysis using anti-GFP antibodies to compare the
expression of GFP via the ler promoter. The used strains are similar to those described in
(Fig. 8a), with the exception that all contain a compatible GFP-expression plasmid via the
ler promoter. Strains are indicated above the lanes.
2.3.8. GrlR competes with the regulatory regions of flhDC operon for binding
to GrlA
We next examined whether competition between a promoter region and GrlR for
binding to GrlA was unique to the ler promoter or a more general mode of operation of
the GrlR-GrlA system. To this end, we examined two additional GrlA targets: the flhDC
and ehxCABD promoters. We performed EMSAs by mixing MBP-GrlA with a DNA
fragment containing the flhDC promoter region (-455/+223). Addition of GrlA shifted the
DNA, indicating that MBP-GrlA bound to the promoter region of the flhDC operon (-
455/+223) (Fig. 2.28).
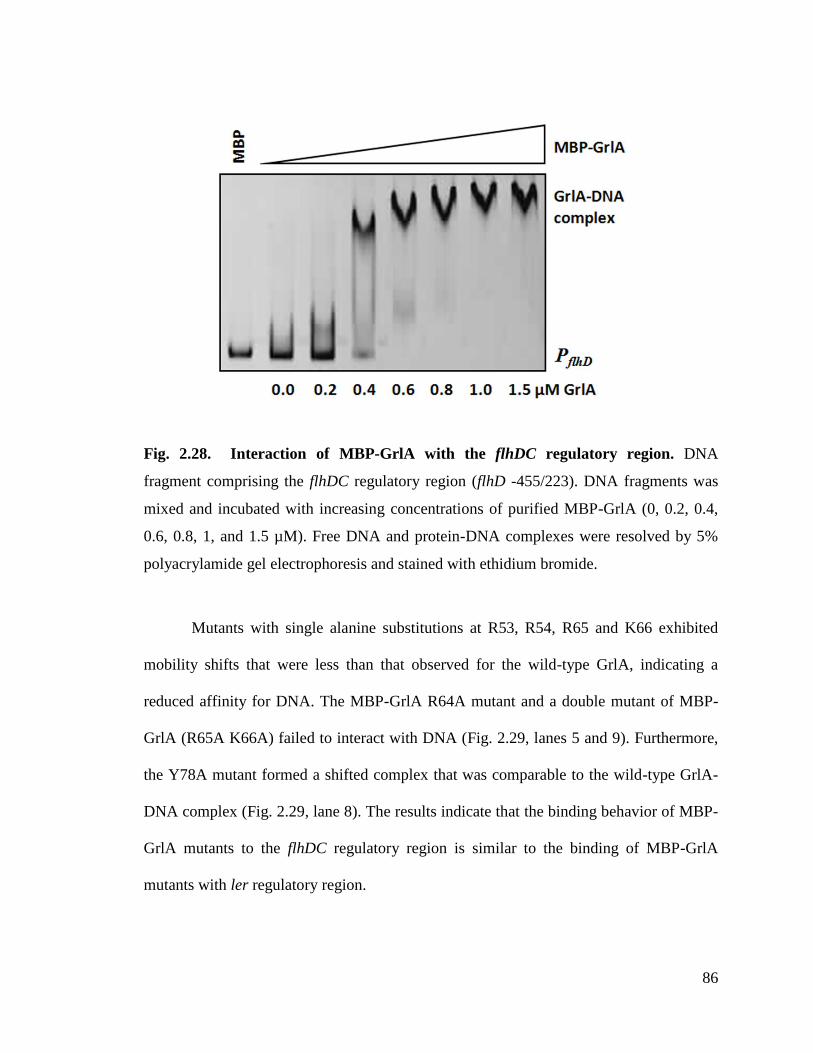
86
Fig. 2.28. Interaction of MBP-GrlA with the flhDC regulatory region. DNA
fragment comprising the flhDC regulatory region (flhD -455/223). DNA fragments was
mixed and incubated with increasing concentrations of purified MBP-GrlA (0, 0.2, 0.4,
0.6, 0.8, 1, and 1.5 µM). Free DNA and protein-DNA complexes were resolved by 5%
polyacrylamide gel electrophoresis and stained with ethidium bromide.
Mutants with single alanine substitutions at R53, R54, R65 and K66 exhibited
mobility shifts that were less than that observed for the wild-type GrlA, indicating a
reduced affinity for DNA. The MBP-GrlA R64A mutant and a double mutant of MBP-
GrlA (R65A K66A) failed to interact with DNA (Fig. 2.29, lanes 5 and 9). Furthermore,
the Y78A mutant formed a shifted complex that was comparable to the wild-type GrlA-
DNA complex (Fig. 2.29, lane 8). The results indicate that the binding behavior of MBP-
GrlA mutants to the flhDC regulatory region is similar to the binding of MBP-GrlA
mutants with ler regulatory region.
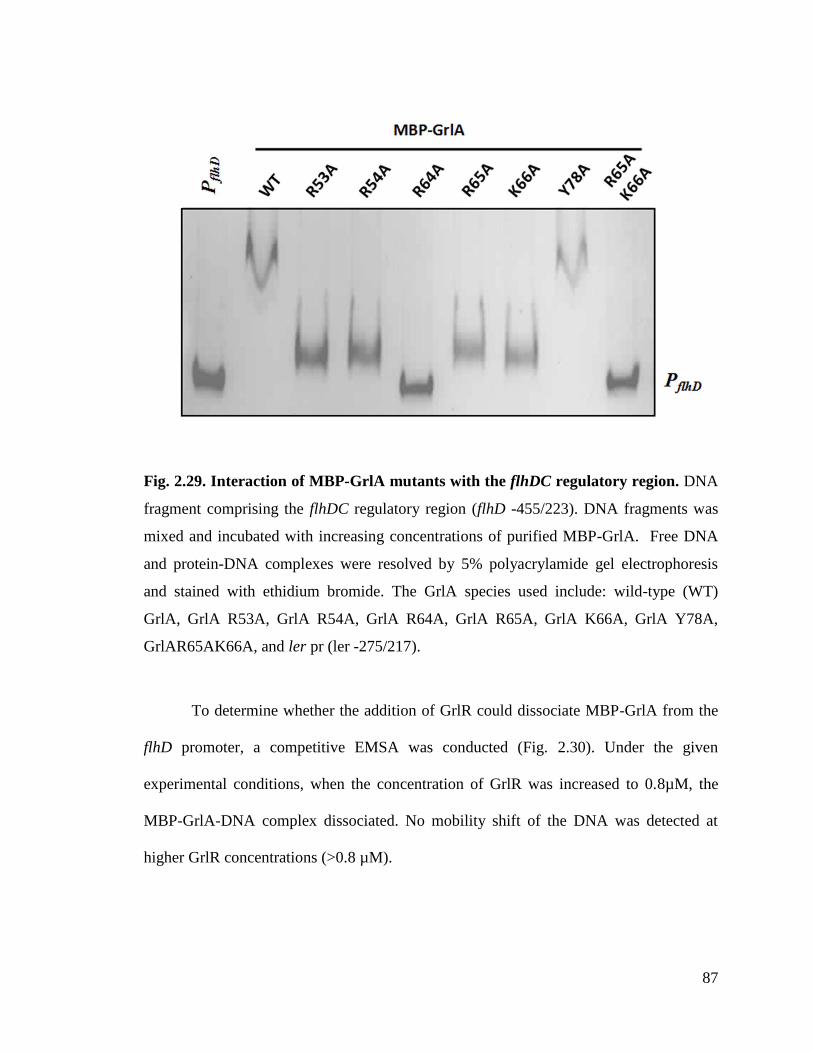
87
Fig. 2.29. Interaction of MBP-GrlA mutants with the flhDC regulatory region. DNA
fragment comprising the flhDC regulatory region (flhD -455/223). DNA fragments was
mixed and incubated with increasing concentrations of purified MBP-GrlA. Free DNA
and protein-DNA complexes were resolved by 5% polyacrylamide gel electrophoresis
and stained with ethidium bromide. The GrlA species used include: wild-type (WT)
GrlA, GrlA R53A, GrlA R54A, GrlA R64A, GrlA R65A, GrlA K66A, GrlA Y78A,
GrlAR65AK66A, and ler pr (ler -275/217).
To determine whether the addition of GrlR could dissociate MBP-GrlA from the
flhD promoter, a competitive EMSA was conducted (Fig. 2.30). Under the given
experimental conditions, when the concentration of GrlR was increased to 0.8µM, the
MBP-GrlA-DNA complex dissociated. No mobility shift of the DNA was detected at
higher GrlR concentrations (>0.8 µM).

88
Fig. 2.30. Competitive EMSA carried out to test the competition between flhDC
regulatory region and 6HisGrlR for binding to MBP-GrlA. PCR DNA fragments
comprising the regulatory region (flhD -455/+223) were mixed and incubated with
purified MBP-GrlA 1 µM for 15 min, then increasing concentrations (0 0.2 0.4 0.6 0.8
1.0) µM of HisGrlR were added and incubated for additional 15 min. The complexes and
free DNA were run in 5% polyacrylamide gel and stained with ethidium bromide. flhD
pr: (flhD -455/+223).
2.3.9. GrlR competes with the regulatory regions of ehxCABD operon for
binding to GrlA
We next investigated a direct role of the GrlR-GrlA system in controlling the
ehxCABD operon by EMSA assays. MBP-GrlA bound to the ehx promoter region (-
261/22). Subsequent EMSA experiments using mutant MBP-GrlA showed various
degrees of reduction in the mobility shifts, consistent with those seen for GrlA binding to
ler and the flhDC promoter regions. Cumulatively, our findings suggest that the general
mode of operation of the GrlR-GrlA system involves mutually exclusive binding of the
GrlA HTH motif with either GrlR or its target DNA.
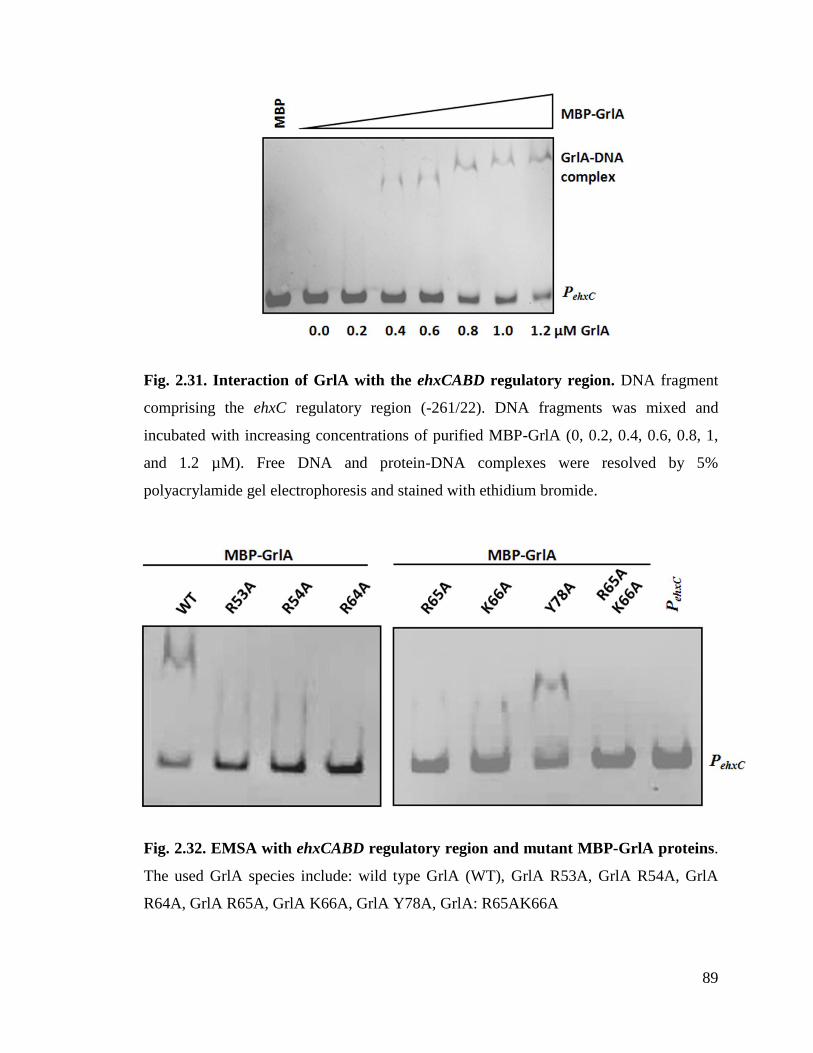
89
Fig. 2.31. Interaction of GrlA with the ehxCABD regulatory region. DNA fragment
comprising the ehxC regulatory region (-261/22). DNA fragments was mixed and
incubated with increasing concentrations of purified MBP-GrlA (0, 0.2, 0.4, 0.6, 0.8, 1,
and 1.2 µM). Free DNA and protein-DNA complexes were resolved by 5%
polyacrylamide gel electrophoresis and stained with ethidium bromide.
Fig. 2.32. EMSA with ehxCABD regulatory region and mutant MBP-GrlA proteins.
The used GrlA species include: wild type GrlA (WT), GrlA R53A, GrlA R54A, GrlA
R64A, GrlA R65A, GrlA K66A, GrlA Y78A, GrlA: R65AK66A

90
Fig. 2.33. Competitive EMSA carried out to test the competition between ehxCABD
regulatory region and 6HisGrlR for binding to MBP-GrlA. PCR DNA fragments
comprising the regulatory region (ehxC (-261/22)) were mixed and incubated with
purified MBP-GrlA 1 µM for 15 min, then increasing concentrations (0 0.2 0.4 0.6 0.8
1.0) µM of HisGrlR were added and incubated for additional 15 min. The complexes and
free DNA were run in 5% polyacrylamide gel and stained with ethidium bromide. ehxc
pr: (ehxC (-261/22)).
2.4. Discussion
Extensive functional analyses have shown that the GrlR-GrlA is a key regulatory
complex involved in the direct or indirect regulation of most virulence genes in AE
pathogens, including EPEC, EHEC and C. rodentium (Barba et al., 2005; Deng et al.,
2004a; Iyoda et al., 2006). These studies include demonstration of the regulatory role of
the GrlR-GrlA system in three independent operons: LEE1, flhDC and ehxCABD (Barba
et al., 2005; Kitagawa et al., 2011; Saitoh et al., 2008). GrlA positively regulates the
expression of ler, the first gene in the LEE1 operon, which in turn regulates the
expression of various downstream genes involved in T3SS biogenesis (Barba et al.,

91
2005). In addition to LEE1, GrlA is also involved in the positive regulation of ehxCABD
operons and the negative regulation of the flhDC operon (Fig. 2.1). GrlR binds directly to
GrlA and functions as an anti-regulator (Barba et al., 2005). However, the molecular
details of the regulation of the GrlR-GrlA complex have, until now, remained elusive
because of the lack of structural details. Here, we close this gap, reporting the crystal
structure of the GrlR-GrlAΔ complex, along with supportive functional studies.
In a previous study, we reported the binding between GrlR and GrlA mediated by
the possible interaction of an46
EDED49
motif of GrlR with the C-terminal region of GrlA
(Jobichen et al., 2007). However, we could not confirm this finding in the present
complex structure because of the absence of the C-terminal region (aa107-137) of GrlA
in the crystallized construct, wherein a majority of GrlAΔ interactions occur with one
monomer of the dimeric GrlR. Despite this, we were able to verify the binding of full-
length GrlA to GrlR using ITC and pull down experiments (Fig.15; Fig.18a; Table.2.4).
The ITC experiments indicated that full-length GrlA binds to GrlR with a ~35-fold higher
affinity than GrlAΔ. These results suggest the role of the GrlA C-terminus in its
interaction with GrlR. Despite this difference in affinity, under the employed
experimental conditions, GrlAΔ exhibited an activity comparable to full-length GrlA
(Fig. 2.24 and Fig 2.25).
Previous studies of the GrlR-GrlA interaction were performed with GST-tagged
proteins (Jiménez et al., 2010; Jobichen et al., 2007). These studies suggest that the GST
tag might interfere with the interaction, and could be the reason for the complete
disruption of GrlA binding with GrlR following alanine substitution in the
46EDED
49motif in our previous study (Jobichen et al., 2007). To avoid this potential

92
pitfall, the current experiments were carried out using MBP-tagged GrlA. We and others
have independently verified that MBP is not involved in the GrlA-GrlR interaction
(Jiménez et al., 2010).
Here, we show that substitutions in the HTH motif of GrlA∆ interfered with GrlR
binding, while the full-length GrlA with substitutions in the HTH region was able to bind
to GrlR probably through its C-terminal region (Fig. 16; Fig. 18a; Table 2.4).Jimenez et
al. show that GrlA∆1(aa1-100) was able to pull down GrlR (Jiménez et al., 2010), and our
observation of two binding sites agrees with this finding. Thus, even though the binding
site located to the GrlA C-terminus was deleted; these deletion constructs can still pull
down GrlR via this alternative binding site located at the HTH region.
Our data shows that GrlR interacts with GrlA to prevent its binding to target
DNA. The fact that grlR and grlA are co-transcribed raises the question of under what
conditions GrlA can escape suppression by GrlR and interact with its target DNA. Given
the tight GrlR-GrlA mode of interaction and the observation that two GrlR molecules are
needed to suppress one GrlA molecule, it can be assumed that GrlA will function as a
regulator only when its steady state level reaches more than half of that for GrlR. The
relative steady state level of the two proteins is controlled by their relative translation
rates and stability. While virtually nothing is known about the first, it is expected that the
translation rate would be equal in generating both GrlR-bound, as well as free, GrlA. The
differential stability and specific GrlR degradation by ClpXP has been previously
reported (Iyoda et al., 2006). An additional point of view on this system is that GrlR in
complex with GrlA stabilises GrlA and maintains it in an inactive state that can become

93
functional and rapidly released following an appropriate signal and ClpXP-mediated
GrlR degradation.
In conclusion, the present study revealed the molecular structure and mechanism
of the GrlR-GrlA complex. GrlR and the promoter regions of ler, flhDC and ehxCABD
compete for the HTH motif region of GrlA. GrlR outcompetes with these promoter DNA
sequences for GrlA. Regulation of multiple virulence operons by a central hetero-trimeric
GrlR-GrlA complex would help the pathogen to precisely control the expression of
various genes involved in its pathogenesis. By differentially regulating the ler and ehx
operons positively and the flhDC operon negatively, GrlR and GrlA coordinate and
optimize gene expression by the pathogen during the infection process.
The next chapter discusses the structural and functional characterization of VirD2
binding protein (VBP), a key recruiting protein of VirB/D4 T4SS of Agrobacterium
tumefaciens.

94
Chapter 3: Dimerization of VirD2 Binding Protein from the
Type IV Secretion System is essential for Agrobacterium
induced tumor formation in plants

95
3.1. Introduction
The Type IV Secretion System (T4SS) has an unmatched versatility among the
seven different secretion systems known in bacteria. T4SS can translocate not only
proteins but also DNA to phylogenitically diverse taxa, including many bacterial species
and different types of eukaryotic cells, as well as import and export DNA from the
extracellular milieu. The T4SS shares a common ancestry with bacterial conjugation
systems (Cascales and Christie, 2003). Three types of T4SS have been described: (1)
conjugation systems, defined as machines that translocate DNA substrates to recipient
cells by a contact-dependent process; (2) effector translocation systems, functioning to
deliver proteins or other effector molecules to eukaryotic target cells; (3) DNA release or
uptake systems that translocate DNA to or from the extracellular milieu (Alvarez-
Martinez and Christie, 2009).
Conjugation, similar to other gene transfer mechanisms, endows bacterium
with the capacity to survive changing environments through the acquisition of adaptive
traits. The proteins involved in these conjugation systems are classified into three
functionally distinct classes (Christie et al., 2005). For the VirB/D4 (virulence B/D4)
conjugation system of Agrobacterium tumefaciens, Class I constitutes proteins involved
in the processing and packing of transferred DNA intermediates (VirD2 relaxase, VirD1,
VirC proteins, VirE2, VirE1 and VirF) (Tzfira and Citovsky, 2000). Class II comprises
11 VirB proteins that form the T4SS apparatus, which functions to translocate the VirD2-
T-DNA-VirE2 complex (T-DNA-protein complex) into plant cells (Tzfira and Citovsky,
2000). Class III constitutes the coupling proteins (CP) that mediate the interaction
between the substrate (T-DNA-protein complex) and the transport apparatus (Christie,

96
1997). In the A. tumefaciens VirB/D4 system, the coupling protein is VirD4, an inner
membrane protein with a large cytoplasmic domain that is required for the transfer of
both T-strand and VirE2 to host cells (Hamilton et al., 2000; Kumar and Das, 2002;
Okamoto et al., 1991).
The T-DNA-protein complex forms in the cytoplasm, but there is no evidence to
suggest that VirD4 CP can recruit the bulky T-DNA-protein complex to the T4SS
apparatus (Guo et al., 2007a). In 2007, Guo et al. reported the existence of a subset of
proteins defined as ‘recruiting proteins’ that help recruit the nucleoprotein substrate
complex to the VirD4 CP. The VirD2 binding protein (VBP) was identified as a key
protein belonging to this subset and was shown to recruit the T-DNA-protein complex to
the VirD4 CP (Guo et al., 2007b). Site-directed mutagenesis has shown that the VBP-
VirD2 interaction is important for T-DNA transfer (Guo et al., 2007b), with VBP
interacting with both the T-DNA-protein complex and independently with several T4SS
components, including VirD4, VirB4 and VirB11. However, the molecular mechanism(s)
by which VBP recruits the complex to VirD4 coupling protein remains unknown.
Here we report the crystal structure of the C-terminal domain of VBP along with
its functional studies. The two monomers of the asymmetric unit form a tight anti-parallel
dimer and structural analysis confirms that this domain adopts a HEPN (higher
eukaryotes and prokaryotes nucleotide-binding) fold. Solution studies with gel filtration
and analytical ultracentrifugation confirm the dimeric nature of HEPN and full-length
VBP. Furthermore, the HEPN domain is revealed as the dimerization domain of VBP.
Using plant virulence assays, we show that only full-length dimeric VBP is capable of

97
inducing tumors in plants. These studies broaden the understanding the role of VBP in
the T-DNA-protein transfer from A. tumefaciens to the plant cell.
3.2. Materials and Methods
3.2.1. Plasmid and strain construction
The strains and plasmids used are given in Table S3. Intact vbp and vird2 genes
were amplified from A. tumefaciens C58 plasmid and Ti plasmid, respectively. These
genes were then cloned to pET32a (Novagen; Madison, WI, USA) and pRSET
(Invitrogen, Carlsbad, CA, USA) vectors, respectively. N-terminal nucleotidyltransferase
(NT) domain and C-terminal HEPN domain vbp constructs were created using specific
primers that amplify these regions and were cloned into pGEX-6p1 (GE Healthcare;
Buckinghamshire, UK). Site-specific mutations in vbp were introduced by overlapping
PCR, as described previously (Ho et al., 1989). Each construct was verified by DNA
sequencing. A fragment of virF cassette cloned from pTiBo542 (GenBank: DQ058764.1)
was inserted into the SphI–ApaI site on pCB301 (Oliver et al., 1999). The virF coding
sequence was substituted with a multiple cloning site, resulting in pQH300.
3.2.2. Protein expression and purification
The plasmid pET32a-vbp was transformed into E. coli BL21 (DE3) cells and was
grown in LB broth at 37°C overnight. The overnight culture was transferred into 1 L of
LB broth and the protein expression was induced at an absorbance of 0.6 with 350 μM
IPTG for 20 h at 20°C. Cells were harvested and lysed in lysis buffer (20 mM Tris-HCl,
pH 8.0, 200 mM NaCl and 1 mM PMSF). Cell lysates were centrifuged and the

98
supernatants transferred to affinity columns containing Ni-NTA agarose (Qiagen;
Valencia, CA, USA), pre-equilibrated with the lysis buffer. The His6-VBP bound to Ni-
NTA was eluted with 400 mM imidazole following three wash steps to remove non-
specific proteins. The eluted protein was purified through size-exclusion chromatography
using a HiLoad 16/12075 Superdex75 gel filtration column (AKTA FPLC UPC-900
system, GE Healthcare) containing buffer (20 mM Tris-HCl, pH 8.0, 200 mM NaCl, and
5% glycerol). The GST fusion proteins (GST-HEPN and GST-NTD) were expressed as
described above using M9 media (Doublié, 1997). The fusion proteins were purified by
affinity chromatography on GST-Sepharose resin, and the tags were removed by cleavage
with precession proteases (GE Healthcare; Buckinghamshire, UK). The HEPN domain
was additionally purified by size-exclusion chromatography in gel-filtration buffer (30
mM CHES pH 9.0, NaCl 200 mM, 5% glycerol). The NT domain was purified in the
same way but using a buffer containing 30 mM Tris-HCl, pH 7.5 and 200 mM NaCl
buffer.
3.2.3. Crystallization and data collection
Initial crystallization conditions were identified by hanging drop vapor diffusion
method using an index screen (Hampton Research, Aliso Viejo, CA, USA). Diffraction-
quality crystals were obtained by equilibrating a l.0 µl drop of protein (4 mg/ml) in 30
mM CHES, pH 9.0, 200 mM NaCl and 5% glycerol mixed with 1.0 µl of reservoir
solution (8% (w/v) PEG 3350, 2% v/v tacsimate, 5% v/v 2-proponal, and 0.1 M
imidazole) suspended over 1 ml of reservoir solution. Crystals grew in 1–3 days at 16°C.
For data collection, 15% glycerol was added as a cryo-protectant and the crystals were
flash-cooled in an N2 cold stream.

99
A complete single wavelength anomalous diffraction (SAD) (Terwilliger and
Berendzen, 1997b) dataset was collected to 2.7 Å resolution at the synchrotron beamline
X6A (National Synchrotron Light Source, Brookhaven National Laboratory, Upton, NY)
using a Quantum4-CCD detector (Area Detector Systems Corp., Poway, CA). The
datasets were processed and scaled using HKL2000 (Otwinowski and Minor, 1997). The
crystals belonged to a P212121 space group. There were two monomers in the asymmetric
unit corresponding to Vm =2.49 Å3 Da
-1 and a solvent content of 50.6%. The position of
the selenium atoms were determined using the program Phenix Autosol (Terwilliger,
2003). The obtained phases were further improved by density modification using
RESOLVE (Terwilliger, 2003). Over 50% of the backbone atoms of the model were built
by RESOLVE. The remaining residues were manually built using Coot (Emsley and
Cowtan, 2004) and subsequently refined using Phenix-refine (Adams et al., 2010).
Refinement was continued until the R-value converged to 0.22 (Rfree = 0.27) for
reflections I>σ (I) to 2.7 Å resolution (Table 1). The model had good stereochemistry,
with 99.3% residues within the allowed regions of the Ramachandran plot. Subsequently,
the importance of the key residues at the dimeric interface was validated by structure-
based in vitro studies, such as analytical ultracentrifugation and pull down assays, and in
vivo plant virulence studies.
3.2.4. Analytical Ultracentrifugation
The oligomeric state of full-length VBP, HEPN and NTD domain of VBP and
their mutants was investigated by monitoring the sedimentation properties of each protein
in sedimentation velocity experiments. For these experiments, 400 μl of samples at 1
mg/ml in appropriate buffer were used, with the experiments carried out in duplicate. The

100
sedimentation velocity profiles were collected by monitoring the absorbance at 280 nm.
The samples were centrifuged at 40,000 rpm at 24°C in a Beckman Optima XL-I
centrifuge fitted with a four-hole AN-60 rotor and double-sector aluminum centerpieces
and equipped with absorbance optics. 80 scans were collected and analyzed using Sedfit
program (Brown and Schuck, 2006).
3.2.5. Pull down assay
MBP-VirD2 bound to amylose resin (New England Biolabs, Ipswich, MA, USA)
was incubated with purified 6His-VBP, with or without substitution at Asn186Asp. The
beads were washed several times before resolving the eluent on 12.5% SDS-PAGE. For
western blot analysis, the proteins were transferred to a PVDF membrane. 6His-VBP was
detected by the addition of diluted anti-His antibody (Santa Cruz Biotechnology; Santa
Cruz, CA, USA). The signal was detected using the SuperSignal WestPico
Chemiluminescent substrate (Pierce Biotechnology; Rockford, IL, USA) under the
conditions recommended by the manufacturer.
3.2.6. Isothermal titration calorimetry
His-VBP, with or without substitution at Asn186Asp, were purified in gel
filtration buffer containing 30 mM Tris-HCl, pH 8.0, 200 mM NaCl and 5% glycerol.
ITC experiments were carried out using a VP-ITC calorimeter (Microcal, LLC,
Northampton, MA, USA) at 25°C using 0.01 mM VBP protein in the sample cell and
0.25 mM AMP-PNP in the injector. All samples were thoroughly degassed and then
centrifuged to remove precipitates. With the exception of the first experiment, 10 μl
volumes per injection were used for different experiments. Consecutive injections were
separated by 5 min to allow the peak to return to baseline levels. ITC data were analyzed

101
with a single-site fitting model using Origin 7.0 (Origin Lab Corp., Northampton, MA,
USA) software.
3.2.7. Circular dichroism spectrometry
Far UV spectra (260–190 nm) of VBP, HEPN, NTD domains and their mutants
were measured using a Jasco J-810 spectropolarimeter (Jasco Europe, MI, Italy) in
phosphate buffer (pH 7.5) at room temperature using a 0.1-cm path length-stoppered
cuvette. Six scans were recorded, averaged and then baseline subtracted to obtain values.
3.2.8. Plant virulence Assay
A. tumefaciens strains were grown in MG/L liquid medium overnight at 28°C
supplemented with appropriate antibiotics. The bacterial cells were collected by
centrifugation and re-suspended in a buffer solution consisting of 10 mM MgCl2 and 10
mM MES, pH5.5. Cell concentrations were adjusted to OD600 = 0.1. The leaves
of Kalanchoe were wounded with a hypodermic syringe needle and 5 μl of bacterial cell
suspension was inoculated onto each wound area. The tumors were photographed 25 and
35 days after inoculation.

102
3.3. Results
3.3.1. Overall structure
We initially attempted to crystallize full-length VBP with intact N- and C-
terminal domains (Fig. 3.1).
Fig. 3.1. Schematic representation of VBP and its domains. Based on the sequences
analysis, the N-terminal (aa 10-133) is predicted to be a Nucleotidyltransferase (NT)
domain and the C-terminal domain is predicted to be Higher Eukaryotes and Prokaryotes
Nucleotide (HEPN) (aa 159-279) binding domain.
Peptide-mass finger printing analysis on the crystals showed that only the C-
terminal domain of VBP had crystallized. The boundaries of the crystallized protein were
determined using N-terminal sequencing and mass spectrometric analysis. Subsequently,
we generated a new construct that consisted of the C-terminal domain, and obtained
crystals that diffracted up to 2.7 Å resolution. The structure was determined using the
SAD method. The asymmetric unit consists of two monomers forming a tight dimer.
Each monomer contains a five-helix bundle with three long anti-parallel α helices (α1,
α2, and α6) that forms the major part of this domain, and three shorter helices (α3, α4 and
α5) that stack at an angle to the long helices (Fig. 3.2 and Table 3.1). The monomers are
arranged as anti-parallel dimers with a large buried surface area of 2255.6 Å2
(or 13% of
the total surface area of each monomer). The dimer is held together by tight interactions
between α1, α2, and α6 helices from both monomers (Fig. 3.3).
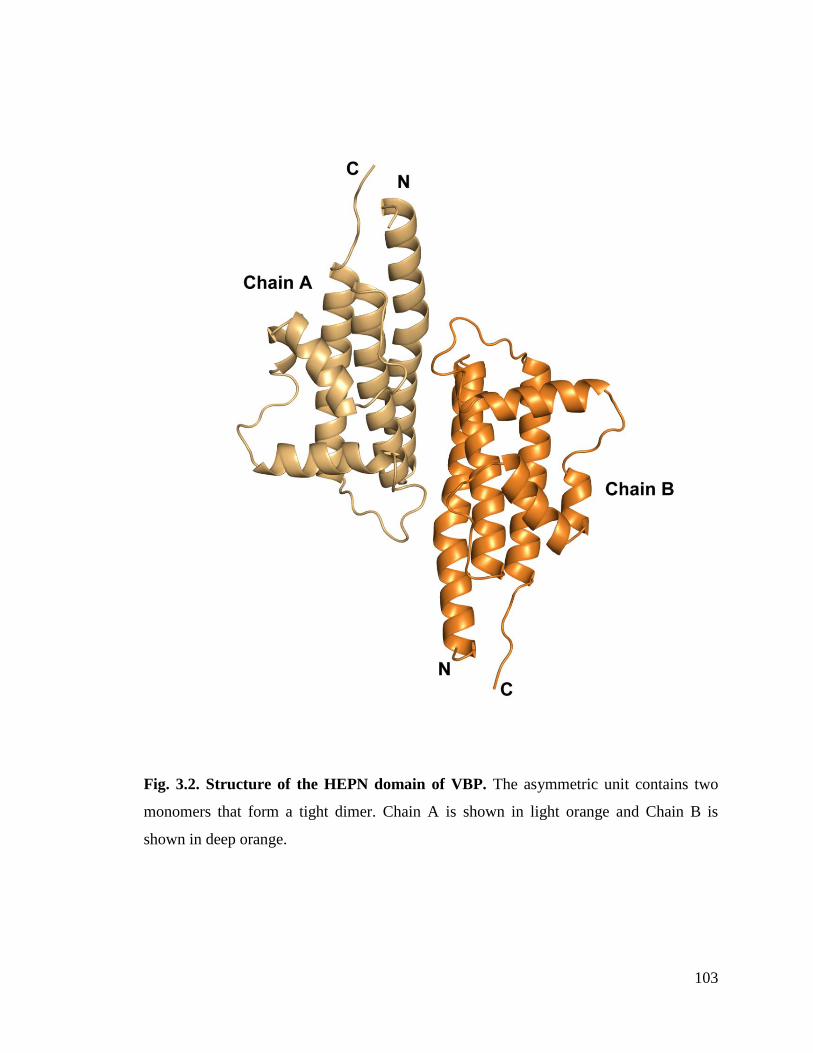
103
Fig. 3.2. Structure of the HEPN domain of VBP. The asymmetric unit contains two
monomers that form a tight dimer. Chain A is shown in light orange and Chain B is
shown in deep orange.

104
Table 3.1: Crystallographic statistics and refinement details
SelMet SAD
Data collection
Space group P212121
Cell dimensions
a, b, c (Å) a = 63.10 b = 71.57 c = 83.42
α, β, γ () 90
Wavelength 0.97945
Resolution (Å) 50.0-2.7(2.75-2.7)
Rsym or Rmerge 0.05 (0.15)
I / σI 22.4 (5.5)
Completeness (%) 99.7(99.3)
Redundancy 7.5 (7.4)
Refinement
Resolution (Å) 14.8-2.7
No. reflections 10677
Rwork / Rfree 0.22/0.27
No. atoms
Protein 2248
Water 13
B-factors
Protein 63
Water 57
R.m.s deviations
Bond lengths (Å) 0.04
Bond angles () 0.897
a Rsym = |Ii -<I>|/ |Ii| where Ii is the intensity of the i
th measurement, and <I> is the mean
intensity for that reflection.
bRwork = | Fobs - Fcalc|/ |Fobs| where Fcalc and Fobs are the calculated and observed
structure factor amplitudes, respectively.
cRfree = as for Rwork, but for 10.0% of the total reflections chosen at random and omitted
from refinement.
Individual B-factor refinement was carried out.
*Values in the parenthesis are the highest resolution bin values.

105
Fig. 3.3. Dimer interface of HEPN dimer. The contacts at the core of the HEPN dimer
are shown.
Fig. 3.4. A sample 2Fo-Fc electron density map (contoured at 1 σ) of HEPN domain of
VBP.

106
3.3.2. Sequence and structural homology
PSI–BLAST searches of the non-redundant protein database using the full length
VBP (gi|159141484) resulted in several hits of nucleotidyl transferase proteins from
Rhizobium, Sinorhizobium and other species of Agrobacterium. Sequence based
predictions revealed that VBP has two domains such as an NT_KNTase_like domain at
the N-terminus and a HEPN domain at the C-terminus. The NT domain is implicated in
nucleotidyl transfersae function while HEPN domain is implicated in nucleotide binding
(Altschul et al., 1997). A DALI search for the structural homologs of VBP C-terminal
domain identified several proteins with a HEPN domain (Table 3.2); this confirmed that
the C-terminal domain of VBP adopts a HEPN fold (hereafter referred to as HEPN
domain).
Table 3.2: Structural homologs of HEPN domain as predicted by DALI search
No PDB z rmsd aa Total
residues
%id Protein name/detail
1: 3o10 15.0 2.0 121 136 16 SACSIN
2: 1ufb 13.5 2.2 116 127 16 TT1696 PROTEIN
3: 2q00 10.5 2.7 106 122 14 ORF C02003 PROTEIN
4: 3jyy 8.9 3.0 103 266 7 LINCOSAMIDE NUCLEOTIDYLTRANSFERASE
5: 1kny 7.6 3.3 108 253 7 KANAMYCIN NUCLEOTIDYLTRANSFERASE
6: 3agt 7.0 3.0 100 133 5 HEMERYTHRIN-LIKE DOMAIN PROTEIN
Notably, the HEPN domain of VBP aligns with the C-terminal domain of kanamycin
nucleotidyl transferase from Staphylococcus aureus (PDB code: 1KNY) with an rmsd of
2.9 Ǻ for the 104 Cα atoms. Although VBP has very low sequence identity (14 to 16%)
with its structural homologs, it might have similar nucleotide binding and transfer
function.
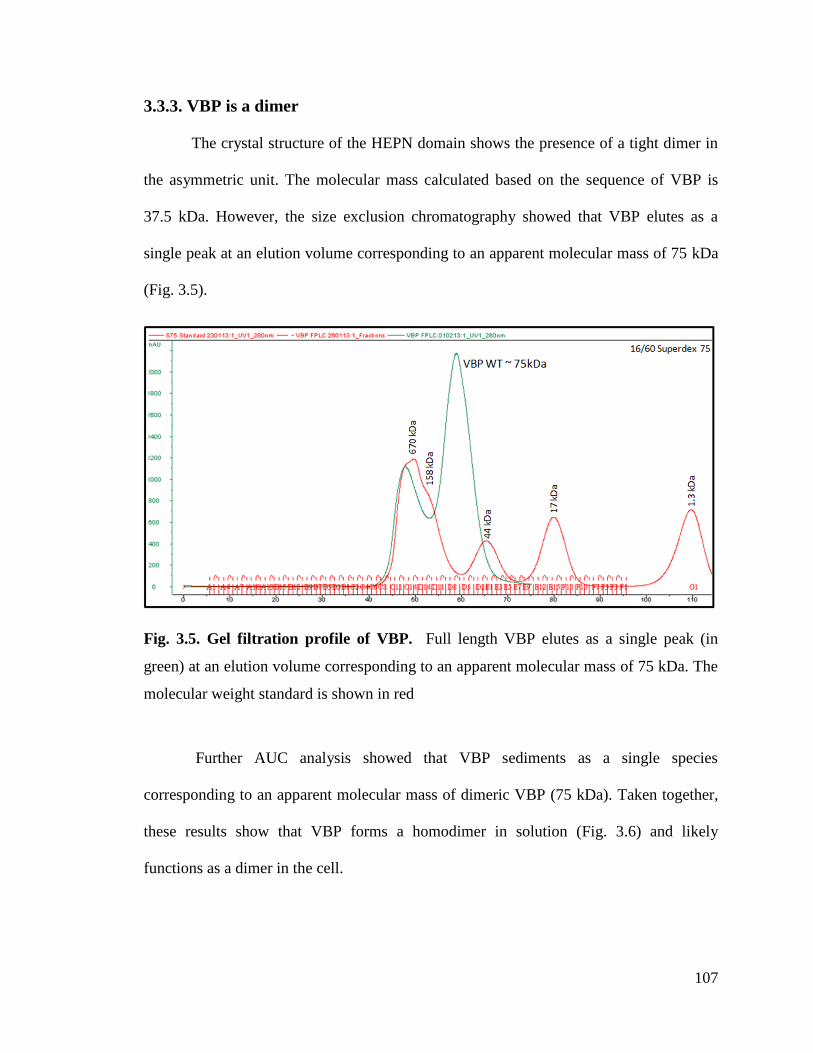
107
3.3.3. VBP is a dimer
The crystal structure of the HEPN domain shows the presence of a tight dimer in
the asymmetric unit. The molecular mass calculated based on the sequence of VBP is
37.5 kDa. However, the size exclusion chromatography showed that VBP elutes as a
single peak at an elution volume corresponding to an apparent molecular mass of 75 kDa
(Fig. 3.5).
Fig. 3.5. Gel filtration profile of VBP. Full length VBP elutes as a single peak (in
green) at an elution volume corresponding to an apparent molecular mass of 75 kDa. The
molecular weight standard is shown in red
Further AUC analysis showed that VBP sediments as a single species
corresponding to an apparent molecular mass of dimeric VBP (75 kDa). Taken together,
these results show that VBP forms a homodimer in solution (Fig. 3.6) and likely
functions as a dimer in the cell.

108
Fig. 3.6. AUC profile of VBP. Full length VBP sediments as a single species at an
apparent molecular mass of 75kDa.
3.3.4. HEPN domain of VBP is the dimerization domain
We examined the role of the HEPN domain in VBP oligomerization by generating
individual constructs for the N-terminal (NT) and C-terminal (HEPN) domains of VBP.
The size-exclusion chromatography of the purified proteins showed that the NT domain
elutes as a single peak at an elution volume corresponding to an apparent molecular mass
of 16.8 kDa (NT as monomer), whereas the HEPN domain elutes as a single peak at an
elution volume corresponding to an apparent molecular mass of 37 kDa (HEPN as a
dimer) (Fig. 3.7). These results were further confirmed using analytical
ultracentrifugation experiments (Fig. 3.8 and Fig. 3.9), and are consistent with the crystal
structure findings that show the presence of a tight dimeric HEPN domains in the
asymmetric unit. Taken together, these results suggest that HEPN domain is responsible
for the dimerization of the full-length VBP.
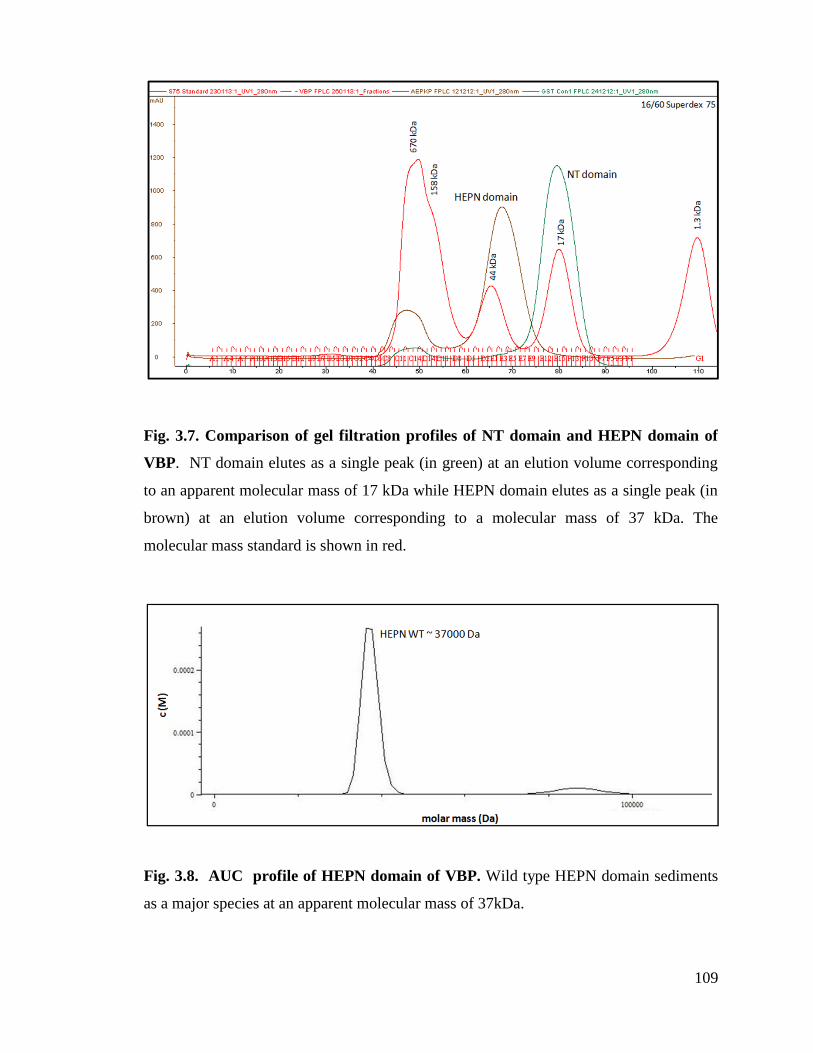
109
Fig. 3.7. Comparison of gel filtration profiles of NT domain and HEPN domain of
VBP. NT domain elutes as a single peak (in green) at an elution volume corresponding
to an apparent molecular mass of 17 kDa while HEPN domain elutes as a single peak (in
brown) at an elution volume corresponding to a molecular mass of 37 kDa. The
molecular mass standard is shown in red.
Fig. 3.8. AUC profile of HEPN domain of VBP. Wild type HEPN domain sediments
as a major species at an apparent molecular mass of 37kDa.

110
Fig. 3.9. AUC profile of NT domain of VBP. NT domain sediments as a major species
at an apparent molecular mass of 17kDa.
3.3.5. Substitution of Asn186 with Asp disrupts the dimerization
The structural analysis indicates that the HEPN dimer is held together by the
contacts maintained throughout the helices α1 and α2; in particular Asp173, Lys184 and
Asn186 of the HEPN domain play an important role in holding the dimer. Asn186 is
located at the edge of a loop which links the α1 and α2 helices of the dimer interface (Fig.
3.3). Asn186, Lys184 and Asp173 of one monomer form hydrogen bonding contact with
Asp173 and Asn186, Lys184 of the second monomer respectively. We substituted
Asn186 with Asp in the HEPN domain and verified its oligomerization state by size
exclusion chromatography and analytical ultracentrifugation experiments (Fig. 3.10 and
Fig. 3.11).Both experiments show that HEPN Asn186Asp exists as a monomer in
solution. These results indicate that substitution of Asn186Asp breaks the dimer. The
secondary structure of HEPN and HEPN mutant was compared using circular dichroism
spectroscopy (Fig. 3.12)

111
Fig. 3.10. Comparison of gel filtration profiles of HEPN domain of VBP3. HEPN
domain elutes as a single peak (in brown) at an elution volume corresponding to an
apparent molecular mass of 37 kDa while HEPN domain with Asn186Asp elutes as a
single peak (in blue) at an elution volume corresponding to a molecular mass of 18.5
kDa. The molecular weight standard is shown in red.
Fig. 3.11. AUC profile of HEPN N186D domain of VBP. HEPN domain with
Asn186Asp sediments as a single species at an apparent molecular mass of 18.5kDa.
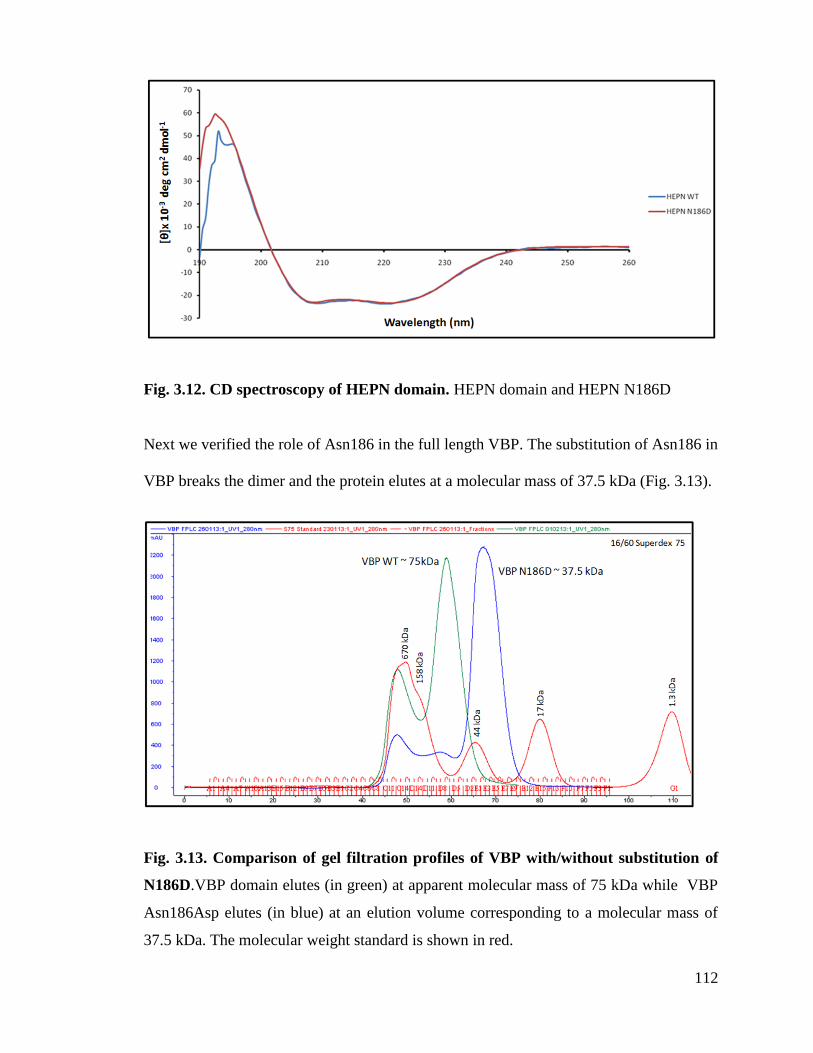
112
Fig. 3.12. CD spectroscopy of HEPN domain. HEPN domain and HEPN N186D
Next we verified the role of Asn186 in the full length VBP. The substitution of Asn186 in
VBP breaks the dimer and the protein elutes at a molecular mass of 37.5 kDa (Fig. 3.13).
Fig. 3.13. Comparison of gel filtration profiles of VBP with/without substitution of
N186D.VBP domain elutes (in green) at apparent molecular mass of 75 kDa while VBP
Asn186Asp elutes (in blue) at an elution volume corresponding to a molecular mass of
37.5 kDa. The molecular weight standard is shown in red.
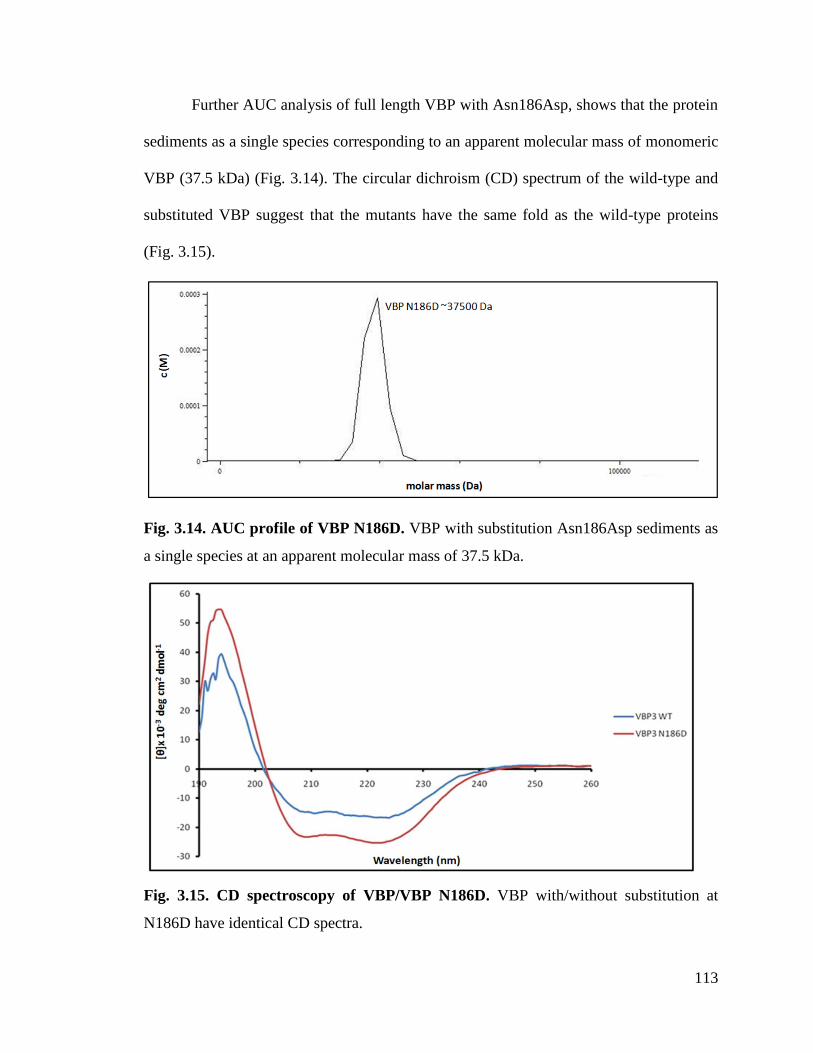
113
Further AUC analysis of full length VBP with Asn186Asp, shows that the protein
sediments as a single species corresponding to an apparent molecular mass of monomeric
VBP (37.5 kDa) (Fig. 3.14). The circular dichroism (CD) spectrum of the wild-type and
substituted VBP suggest that the mutants have the same fold as the wild-type proteins
(Fig. 3.15).
Fig. 3.14. AUC profile of VBP N186D. VBP with substitution Asn186Asp sediments as
a single species at an apparent molecular mass of 37.5 kDa.
Fig. 3.15. CD spectroscopy of VBP/VBP N186D. VBP with/without substitution at
N186D have identical CD spectra.

114
These results indicate that the two hydrogen bonds present at the core of the dimeric
interface play a key role in holding the dimeric HEPN intact. Moreover these
observations reiterate that HEPN domain is the dimerization domain of VBP.
3.3.6. VBP functions as a dimer in vivo
Recruitment of the T-DNA complex to the CP is a key step in the T4SS, for
which VBP plays a central role. The observed dimeric nature of VBP in solution and in
the crystal structure prompted us to verify the functional state of VBP inside the cell
using a plant virulence assay. A triple vbp null mutant strain GMI9017Δvbp2Δvbp3 (for
which all three existing vbp genes were knocked out) was complemented with pQH300-
17 plasmid harboring substituted constructs for a functional vbp gene, a gene expressing
the HEPN domain, and a gene expressing the NT domain. We found that null mutants
transformed with a plasmid expressing VBP could cause a tumor-like phenotype in the
wild-type plants (Fig. 3.16). Strains expressing a substituted VBP (VBPAsn186Asp) or one
of the other deletion mutants (pQH-NTD or pQH-HEPN) did not cause tumors. These
results indicate that VBP functions as a dimer in the cells and that full-length VBP is
required for tumor formation in plants.

115
Fig. 3.16. The effect of VBP mutations on tumorigenesis. Agrobacterium. tumefaciens strains were grown in MG/L medium at
28°C overnight. The cell density was adjusted to 108
cells per milliliter. Then 5µl of cell suspension was inoculated onto each wound
site on the leaves of Kalanchoe plants. The tumors were photographed (a) 25 days after inoculation and (b) 35 days after inoculation.
a b

116
3.3.6. Interaction of VBP with ATP and VirD2
Proteins homologous to VBP are predicted to bind ATP (Kozlov et al., 2011). A
previously reported structure of kanamycin nucleotidyl transferase (PDB code: 1KNY), a
structural homolog of VBP, in complex with an ATP analog and kanamycin, shows that
the nucleotide binding pocket involves residues from the N-terminal domain of one
monomer and the C-terminal domain of the second monomer. We verified the ATP
binding property of VBP using ITC (Fig. 3.16 a, b Table 3.3). The ITC analysis shows
that VBP binds to the ATP analog (AMPPNP) (Kd= 1.8 µM), whereas VBP with an
Asn186Asp substitution does not bind to AMPPNP. These results indicate that only
dimeric VBP binds to ATP. Further, we sought to verify whether VBP-HEPN domain
alone can bind nucleotides using ITC experiments (Fig. 3.16c). Our results indicate that
the HEPN domain alone cannot bind nucleotides. We infer that, similar to the kanamycin
nucleotidyl transferase, the ATP binding in VBP might involve both N-terminal and C-
terminal domains. Notably, the structure of the kanamycin nucleotidyl transferase (PDB
code: 1KNY) complexed with a nucleotide analog and kanamycin shows that the two
monomers of the dimer interact in an anti-parallel fashion to form the ATP binding
pocket. Similarly, the structure of the HEPN domain from VBP shows that the two HEPN
monomers form a tight dimer in which the monomers run in anti-parallel. Although the
relative orientation of monomers in the dimers of both proteins is not the same, both
proteins might have a similar ATP-binding mechanism.

117
Fig. 3.17. ITC profile for VBP/ VBP N186D / HEPN domain vs. AMPPNP binding. The binding affinities of VBP/VBP
N186D/HEPN domain to AMPPNP were determined using isothermal titration calorimetry (ITC). Representative ITC profiles are
shown.
a b c

118
Table 3.3: Stoichiometry, affinity and favorability of VBP vs. AMPPNP interactions
VBP is the key recruiting protein that binds to VirD2-T-DNA-VirE2 complex and
recruits it to the VirD4 CP for subsequent translocation to the host (Guo et al., 2007b).
Thus, we finally sought to verify the VirD2 binding property of VBP using pull-down
assays. We showed that VBP binds to VirD2, whereas VBP with an Asn186Asp
substitution does not bind VirD2 (Fig. 3.17).
Fig. 3.18. In vitro pull down assay. MBP/MBP-VirD2 bound to amylose resin was
incubated overnight with 6His-VBP, followed by washes. The final beads were resolved
in a 12.5% SDS-PAGE. The protein was transferred to a PVDF membrane and treated
with Anti-His monoclonal antibody (1:10000). The PVDF membrane was examined
using the SuperSignal WestPico Chemiluminescent substrate (Pierce Biotechnology;
Rockford, IL, USA) under the conditions recommended by the manufacturer.6His-VBP
is loaded in the last lane for reference.
n Ka (M
-1) K
d (M) G
(kcal/mol) H
(kcal/mol) -TS
(kcal/mol)
VBP vs. AMPPNP 2.157 5.6 X 105 1.8 -7.97 -0.521 -7.45
VBP N186D vs. AMPPNP No binding
HEPN vs. AMPPNP No binding

119
The results indicate that, similar to the nucleotide binding property, the VirD2 binding
property also requires the dimeric nature of VBP, and thus the dimerization of VBP is
essential for its function.
3.4. Discussion
A. tumefaciens causes crown gall disease in over 140 species of dicots (Moore et
al., 1997), instigating infection through the efficient translocation of the T-DNA-protein
complex (Alvarez-Martinez and Christie, 2009), a prerequisite for tumor formation in
plants (Pitzschke and Hirt, 2010). T-DNA is a segment of the Ti plasmid between the
right and left border sequences, and the Ti plasmid encodes most of the proteins that are
involved in the T-DNA-protein complex, with each protein catering to a particular stage
of the translocation process. The VirD4 CP is known to couple the T-DNA-protein
complex (VBP-VirD2-VirE2) as a recruiting complex to the T4SS secretion apparatus.
Of these three components, the VBP (VirD2 binding protein) is the key component of the
recruiting complex (Guo et al., 2007).
VBP belongs to the class of HEPN domain–containing proteins that have a N-
terminal NT-domain and a C-terminal HEPN domain (Grynberg et al., 2003). Here we
sought to analyze the structural and functional aspects of the HEPN domain of VBP. We
show that HEPN domain facilitates dimerization of VBP, forming tight anti-parallel
dimers. The structural similarity observed between the HEPN domain of VBP and
kanamycin nucleotidyl transferase, in addition to the results from our ITC experiments,
suggest that a nucleotide binding pocket is formed by the dimeric interface in this protein.
Further, our pull-down assays show that only dimeric VBP can bind VirD2 and that
dimeric VBP is essential for its virulence in in vivo (Fig. 3.18, Fig 3.19.).

120
Fig. 3.19. Schematic representation shows the induction of tumor in plants by Agrobacterium and the role of VBP. Our
experiments show that only dimeric VBP can bind to VirD2. Once VBP binds to VirD2 (steps 1 and 2), it recruits the VirD2-T-DNA-
VirE2 complex to the T4SS apparatus which constitutes the 11 VirB proteins (3). Guo et al have shown that VBP interacts with
VirD4, VirB4 and VirB11 proteins independently. Once recruited to the T4SS apparatus the VirD2-T-DNA-VirE2 complex is
translocated into the host cell cytoplasm (4). It is yet unclear whether VBP is translocated or not (Guo et al., 2007b). Inside the host
cell, certain host cytoplasmic proteins recognize and bind to the nuclear localization signals on VirD2 and VirE2 and translocate the
VirD2-T-DNA-VirE2 complex to the nucleus(5) (Zupan and Zambryski, 1995). Inside the nucleus VirD2 and VirE2 along with a
plethora of host proteins help the T-DNA to integrate with the host DNA (6) (Citovsky et al., 2007). The integrated T-DNA modulates
the host cell process to enable the bacterial colonization and growth leading to the formation of tumor (7).

121
Based on the sequence analysis, it has been predicted that the NT domain of VBP
belongs to the DNA polymerase β superfamily of proteins (Aravind and Koonin, 1999).
This superfamily includes nucleotidyl transferases that catalyze nucleotidylation of
proteins in yet unidentified pathways (Aravind and Koonin, 1999). VBP binds to VirD2,
VirD4, VirB4 and VirB11 energizing components of the T4SS (Guo et al., 2007b), which
makes it difficult to predict the exact nucleotidylation site of the protein. However, based
on our results, we propose that nucleotide binding to the dimeric VBP and its transfer to
another as yet unknown protein in the translocation machinery are two essential steps in
T-DNA translocation in this system. Furthermore, we are tempted to speculate that VBP
acts as an ATP-driven shuttle that recruits the T-DNA-protein complex to the VirD4 CP.
The present study sheds light on the role of VBP in the T-DNA-protein complex
translocation in VirB/D4 T4SS of A. tumefaciens and other bacteria that use the Type IV
secretion conjugation systems.

122
Chapter 4: Conclusion and future directions

123
4.1. Conclusion
This thesis reports the structural and functional characterization of two key
secretion system components viz., GrlR-GrlA complex from the T3SS and HEPN domain
of VBP from the T4SS.
A major infection mechanism employed by the AE pathogens like the EHEC and
EPEC is the type III secretion system. T3SS is a syringe-like apparatus composed of
approximately 20 proteins that serve to transfer virulence proteins from the bacteria
directly into the host cytoplasm. The genes encoding for the T3SS components and
related proteins are organized in several operons that are clustered in the locus of
enterocyte effacement (LEE). GrlR-GrlA is a major regulatory complex of the T3SS.
GrlA is a positive regulator of the LEE1 promoter and forms a positive regulatory loop
with Ler. GrlR binds to GrlA and this is thought to account for the negative effect of
GrlR on ler expression (Barba et al., 2005; Deng et al., 2004a; Iyoda et al., 2006). In
addition to its function as a LEE1 regulator, GrlA negatively regulates the transcription of
the flhDC operon and thus controls flagellar gene expression. Besides GrlA is implicated
in the transcriptional activation of the ehxCABD operon in EHEC (Schmidt et al., 1995;
Welch and Pellett, 1988).
We reported the crystal structure of GrlAΔ (aa1-106) in complex with GrlR,
refined to 2.7 Å resolution, along with the functional studies in the second chapter. The
complex structure is asymmetric with a stoichiometry of 2 GrlR: 1 GrlAΔ. The crystal
structure shows that GrlR binds to the HTH motif region of GrlA. ITC experiments with
various constructs of GrlA /GrlAΔ with GrlR have shown that full length GrlA binds to
GrlR with high affinity than does GrlAΔ clearly indicating the presence of a second

124
region on GrlA that involves in binding GrlR. We verified the role of the key residues
from the HTH motif in DNA binding using in vivo EspB secretion assay and ler-gfp
promoter assay. Competitive EMSA studies revealed a novel regulatory mechanism by
which GrlR interacts with GrlA at its Helix-Turn-Helix (HTH) motif, preventing GrlA
from binding to its target promoter DNA. We also carried out EMSA with flhDC and
ehxCABD promoters. Results from these assays show that GrlA binds with these
promoters like it does with ler promoter. Furthermore GrlR can replace this promoter
DNA from GrlA-DNA complex. Regulation of multiple virulence operons by a central
heterotrimeric GrlR-GrlA complex would help the pathogen to precisely control the
expression of various genes involved in its pathogenesis. By differentially regulating the
ler and ehx operons positively and the flhDC operon negatively, GrlR and GrlA
coordinate and optimize gene expression by the pathogen during the infection process.
Further we studied the VirD2 binding protein (VBP) from Agrobacterium
tumefaciens. A.tumefaciens harbors a T4SS. T4SS has unparalleled versatility among the
known bacterial translocation systems. These systems export both DNA and protein
substrates by cell-contact-dependent and cell-contact-independent mechanisms, and also
import DNA from the extracellular milieu. The T4SS apparatus of A.tumefaciens
comprise of the 11 VirB proteins which form the T4SS apparatus that translocates the
VirD2-T-DNA-VirE2 complex to plant cells. The coupling proteins (CP) mediate the
interaction between the substrate and the transport apparatus. VirD4 acts as a coupling
protein in A.tumefaciens. It could recruit VirE2 to the bacterial cell poles. VirD2 binding
protein (VBP) was identified as a key protein that recruits the VirD2- T-DNA complex to

125
the VirD4 coupling protein. However, the molecular mechanism by which VBP recruits
the complex is unknown.
The crystal structure of the C-terminal domain of VBP determined to 2.8 Å along
with the functional studies is reported in the third chapter. Sequence and structural
analysis of the C-terminal domain have shown that it is homologous to the HEPN domain
of Sacsin. HEPN domain forms dimer in solution as well as in the crystal consistent with
VBP which forms dimer in solution. The NT domain however, is a monomer in solution
clearly indicating that HEPN domain of VBP is the dimerization domain. The crystal
structure showed that HEPN is a α-helical domain. The dimeric interface is kept intact by
a two tethered contact between Asp-173 of one protomer and Asn-186 of the second
protomer. Using site-directed mutagenesis and Analytical ultracentrifugation we showed
that substitution of Asn-186 with Asp, HEPN domain becomes monomer both in the
HEPN domain and in the full length VBP. Moreover, virulence assay was performed to
verify the functional form of VBP in Agrobacterium. Our results indicated that VBP is a
functional dimer and that HEPN domain or the NT domain alone cannot cause tumor in
the plants. This study sheds light into the molecular mechanism of T-DNA transfer to
plants and this will lead to an effective treatment of crown gall disease in plants.
In conclusion, the GrlR-GrlA structure and the DNA binding studies have
demonstrated the regulatory mechanism involving Ler, GrlR and GrlA. Moreover, it
revealed the unique synchronization the GrlR-GrlA complex brings into the regulation of
multiple virulence operons thereby enhancing pathogenesis. The structure of HEPN
domain of VBP and the structure based functional studies have revealed the importance
of dimeric nature of VBP in binding to VirD2 and inducing tumorigenesis. In the

126
following section, the future direction in which these projects could be pursued is
discussed.
4.2. Future directions
ITC analysis of binding between GrlA/GrlAΔ and GrlR has shown that GrlA has
more affinity to GrlR than does GrlAΔ, clearly indicating that the additional region from
the C-terminus of GrlA contributes to increased affinity. Crystal structure of full length
GrlR-GrlA complex will help to understand this additional contribution.
Our EMSA analysis has shown that GrlA binds to the ler, flhDC and ehxCABD
promoters in a similar fashion and involves the same residues from the HTH motif of
GrlA; however, it positively regulates ler and ehxCABD promoters and negatively
regulates flhDC. Crystal structure of GrlA with the promoter DNA can vividly explain
these differences in modes of regulation of various promoters.
Although we have verified the binding of GrlA with flhDC and ehxCABD using
EMSA, an in vivo study like the motility assay and a toxin study would warrant a clear
understanding of the phenotypic effects of the substitutions on GrlA. The plethora of
proteins and the intricate networks involved make the system all the more complex to
understand. An mRNA based quantitative RT-PCR to compare the mRNA levels of ler,
flhDC and ehxCABD genes will help us understand the mode and the stage of regulation
involved.
The details from these studies will help to design drugs or vaccines that can target
the protein secretion system. Since these drugs target the secretion system, they have a
great potential to reduce mortality associated with infection without generating resistant
pathogenic strains.

127
VBP is a key missing link in the T4SS recruitment pathway and is also involved
in binding various key proteins that constitutes T-DNA-protein complex. The
prerequisite in understanding the molecular details of recognition is the establishment of
interaction among the member proteins of the T4SS. The interaction of VirD2 with VBP
has been established using co-immunoprecipitation. Previously, the physiological
relevance of VirD4 CP (Coupling Protein) –VirD2 interaction has been verified by
transfer DNA immunoprecipitation. As a continuation of our studies, the role of VBP in
establishing the interaction between VirD4 CP and VirD2-T-DNA can be verified by
forming a tertiary complex of the three proteins involved and determine the structure
using X-ray crystallography and / or cryo-electron microscopy.
Notably, studies in other systems of T4SS have shown that the CP translocates
promiscuous plasmid clearly depicting that there is an underlying recognition mechanism
that is conserved(Cascales, 2008). This mechanism can be well established only with the
structure of the tertiary complex. Further, the interaction of VBP with the channel
proteins can be verified by co-immunoprecipitation.
VBP has independent binding with various other molecules of the T4SS. Structure
determination, of various molecular (e.g. with VirD2, VirD4 etc.,) complexes with VBP
can be carried out.
Results from our current studies, in combination with those planned for the future,
will provide a better understanding of the type III and type IV secretion systems. This
will lead to the design of molecules which can block the secretion of virulence proteins.
Inability to secrete the virulence proteins will considerably reduce the infectivity of these
pathogenic bacteria.

128
References
Abdallah, A.M., Gey van Pittius, N.C., Champion, P.A., Cox, J., Luirink, J.,
Vandenbroucke-Grauls, C.M., Appelmelk, B.J., Bitter, W., 2007. Type VII
secretion--mycobacteria show the way. Nat Rev Microbiol 5, 883-891.
Adams, P.D., Afonine, P.V., Bunkóczi, G., Chen, V.B., Davis, I.W., Echols, N., Headd,
J.J., Hung, L.W., Kapral, G.J., Grosse-Kunstleve, R.W., McCoy, A.J., Moriarty,
N.W., Oeffner, R., Read, R.J., Richardson, D.C., Richardson, J.S., Terwilliger,
T.C., Zwart, P.H., 2010. PHENIX: a comprehensive Python-based system for
macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213-
221.
Akeda, Y., Galan, J.E., 2005. Chaperone release and unfolding of substrates in type III
secretion. Nature 437, 911-915.
Alami, M., Luke, I., Deitermann, S., Eisner, G., Koch, H.G., Brunner, J., Muller, M.,
2003. Differential interactions between a twin-arginine signal peptide and its
translocase in Escherichia coli. Mol Cell 12, 937-946.
Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., Lipman,
D.J., 1997. Gapped BLAST and PSI-BLAST: a new generation of protein
database search programs. Nucleic Acids Res 25, 3389-3402.
Alvarez-Martinez, C.E., Christie, P.J., 2009. Biological diversity of prokaryotic type IV
secretion systems. Microbiol Mol Biol Rev 73, 775-808.
Apodaca, G., Bomsel, M., Lindstedt, R., Engel, J., Frank, D., Mostov, K.E., Wiener-
Kronish, J., 1995. Characterization of Pseudomonas aeruginosa-induced MDCK
cell injury: glycosylation-defective host cells are resistant to bacterial killing.
Infect Immun 63, 1541-1551.
Aravind, L., Koonin, E.V., 1999. DNA polymerase beta-like nucleotidyltransferase
superfamily: identification of three new families, classification and evolutionary
history. Nucleic Acids Res 27, 1609-1618.
Bacon, D.J., Alm, R.A., Burr, D.H., Hu, L., Kopecko, D.J., Ewing, C.P., Trust, T.J.,
Guerry, P., 2000. Involvement of a plasmid in virulence of Campylobacter jejuni
81-176. Infect Immun 68, 4384-4390.
Barba, J., Bustamante, V., Flores-Valdez, M., Deng, W., Finlay, B., Puente, J., 2005. A
positive regulatory loop controls expression of the locus of enterocyte effacement-
encoded regulators Ler and GrlA. J Bacteriol 187, 7918-7930.
Baron, C., D, O.C., Lanka, E., 2002. Bacterial secrets of secretion: EuroConference on
the biology of type IV secretion processes. Mol Microbiol 43, 1359-1365.

129
Baud, C., Karamanou, S., Sianidis, G., Vrontou, E., Politou, A.S., Economou, A., 2002.
Allosteric communication between signal peptides and the SecA protein DEAD
motor ATPase domain. J Biol Chem 277, 13724-13731.
Bingle, L.E., Bailey, C.M., Pallen, M.J., 2008. Type VI secretion: a beginner's guide.
Curr Opin Microbiol 11, 3-8.
Blocker, A., Jouihri, N., Larquet, E., Gounon, P., Ebel, F., Parsot, C., Sansonetti, P.,
Allaoui, A., 2001. Structure and composition of the Shigella flexneri "needle
complex", a part of its type III secreton. Molecular microbiology 39, 652-663.
Bogsch, E.G., Sargent, F., Stanley, N.R., Berks, B.C., Robinson, C., Palmer, T., 1998. An
essential component of a novel bacterial protein export system with homologues
in plastids and mitochondria. J Biol Chem 273, 18003-18006.
Bonas, U., 1994. hrp genes of phytopathogenic bacteria. Curr Top Microbiol Immunol
192, 79-98.
Brown, P.H., Schuck, P., 2006. Macromolecular size-and-shape distributions by
sedimentation velocity analytical ultracentrifugation. Biophysical journal 90,
4651-4661.
Brundage, L., Hendrick, J.P., Schiebel, E., Driessen, A.J., Wickner, W., 1990. The
purified E. coli integral membrane protein SecY/E is sufficient for reconstitution
of SecA-dependent precursor protein translocation. Cell 62, 649-657.
Buchwald, G., Friebel, A., Galan, J.E., Hardt, W.D., Wittinghofer, A., Scheffzek, K.,
2002. Structural basis for the reversible activation of a Rho protein by the
bacterial toxin SopE. EMBO J 21, 3286-3295.
Bustamante, V.H., Villalba, M.I., Garcia-Angulo, V.A., Vazquez, A., Martinez, L.C.,
Jimenez, R., Puente, J.L., 2011. PerC and GrlA independently regulate Ler
expression in enteropathogenic Escherichia coli. Mol Microbiol 82, 398-415.
C. Wandersman, i.F.C.N.E., 1996. Escherichia coli and Salmonella tiphymurium
Cellular and Molecular Biology, ASM Press,Washington, DC,, 955–967.
Cascales, E., 2008. The type VI secretion toolkit. EMBO Rep 9, 735-741.
Cascales, E., Christie, P.J., 2003. The versatile bacterial type IV secretion systems. Nat
Rev Microbiol 1, 137-149.
Celedon, J.M., Cline, K., 2013. Intra-plastid protein trafficking: How plant cells adapted
prokaryotic mechanisms to the eukaryotic condition. Biochim Biophys Acta 1833,
341-351.

130
Chen, I., Dubnau, D., 2003. DNA transport during transformation. Front Biosci 8, s544-
556.
Christie, P.J., 1997. Agrobacterium tumefaciens T-complex transport apparatus: a
paradigm for a new family of multifunctional transporters in eubacteria. J
Bacteriol 179, 3085-3094.
Christie, P.J., 2001. Type IV secretion: intercellular transfer of macromolecules by
systems ancestrally related to conjugation machines. Mol Microbiol 40, 294-305.
Christie, P.J., Atmakuri, K., Krishnamoorthy, V., Jakubowski, S., Cascales, E., 2005.
Biogenesis, architecture, and function of bacterial type IV secretion systems.
Annu Rev Microbiol 59, 451-485.
Citovsky, V., Kozlovsky, S.V., Lacroix, B., Zaltsman, A., Dafny-Yelin, M., Vyas, S.,
Tovkach, A., Tzfira, T., 2007. Biological systems of the host cell involved in
Agrobacterium infection. Cell Microbiol 9, 9-20.
Coburn, B., Sekirov, I., Finlay, B.B., 2007. Type III secretion systems and disease.
Clinical microbiology reviews 20, 535-549.
Collazo, C.M., Galán, J.E., 1996. Requirement for exported proteins in secretion through
the invasion-associated type III system of Salmonella typhimurium. Infection and
immunity 64, 3524-3531.
Cornélis, G., 1987. [Yersinia enterocolitica, an excellent model for the molecular study of
the pathogenicity of invasive bacteria]. Bull Mem Acad R Med Belg 142, 126-
136.
da Silva, A.C., Ferro, J.A., Reinach, F.C., Farah, C.S., Furlan, L.R., Quaggio, R.B.,
Monteiro-Vitorello, C.B., Van Sluys, M.A., Almeida, N.F., Alves, L.M., do
Amaral, A.M., Bertolini, M.C., Camargo, L.E., Camarotte, G., Cannavan, F.,
Cardozo, J., Chambergo, F., Ciapina, L.P., Cicarelli, R.M., Coutinho, L.L.,
Cursino-Santos, J.R., El-Dorry, H., Faria, J.B., Ferreira, A.J., Ferreira, R.C.,
Ferro, M.I., Formighieri, E.F., Franco, M.C., Greggio, C.C., Gruber, A.,
Katsuyama, A.M., Kishi, L.T., Leite, R.P., Lemos, E.G., Lemos, M.V., Locali,
E.C., Machado, M.A., Madeira, A.M., Martinez-Rossi, N.M., Martins, E.C.,
Meidanis, J., Menck, C.F., Miyaki, C.Y., Moon, D.H., Moreira, L.M., Novo,
M.T., Okura, V.K., Oliveira, M.C., Oliveira, V.R., Pereira, H.A., Rossi, A., Sena,
J.A., Silva, C., de Souza, R.F., Spinola, L.A., Takita, M.A., Tamura, R.E.,
Teixeira, E.C., Tezza, R.I., Trindade dos Santos, M., Truffi, D., Tsai, S.M.,
White, F.F., Setubal, J.C., Kitajima, J.P., 2002. Comparison of the genomes of
two Xanthomonas pathogens with differing host specificities. Nature 417, 459-
463.

131
Daleke, M.H., Ummels, R., Bawono, P., Heringa, J., Vandenbroucke-Grauls, C.M.,
Luirink, J., Bitter, W., 2012. General secretion signal for the mycobacterial type
VII secretion pathway. Proc Natl Acad Sci U S A 109, 11342-11347.
Daniell, S.J., Delahay, R.M., Shaw, R.K., Hartland, E.L., Pallen, M.J., Booy, F., Ebel, F.,
Knutton, S., Frankel, G., 2001. Coiled-coil domain of enteropathogenic
Escherichia coli type III secreted protein EspD is involved in EspA filament-
mediated cell attachment and hemolysis. Infect Immun 69, 4055-4064.
Datsenko, K.A., Wanner, B.L., 2000. One-step inactivation of chromosomal genes in
Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97, 6640-
6645.
de Keyzer, J., van der Does, C., Driessen, A.J., 2003. The bacterial translocase: a
dynamic protein channel complex. Cell Mol Life Sci 60, 2034-2052.
Delepelaire, P., 2004. Type I secretion in gram-negative bacteria. Biochim Biophys Acta
1694, 149-161.
Deng, W., Chen, L., Wood, D.W., Metcalfe, T., Liang, X., Gordon, M.P., Comai, L.,
Nester, E.W., 1998. Agrobacterium VirD2 protein interacts with plant host
cyclophilins. Proc Natl Acad Sci U S A 95, 7040-7045.
Deng, W., Puente, J., Gruenheid, S., Li, Y., Vallance, B., Vázquez, A., Barba, J., Ibarra,
J., O'Donnell, P., Metalnikov, P., Ashman, K., Lee, S., Goode, D., Pawson, T.,
Finlay, B., 2004a. Dissecting virulence: systematic and functional analyses of a
pathogenicity island. Proc Natl Acad Sci U S A 101, 3597-3602.
Deng, W., Puente, J.L., Gruenheid, S., Li, Y., Vallance, B.A., Vazquez, A., Barba, J.,
Ibarra, J.A., O'Donnell, P., Metalnikov, P., Ashman, K., Lee, S., Goode, D.,
Pawson, T., Finlay, B.B., 2004b. Dissecting virulence: systematic and functional
analyses of a pathogenicity island. Proceedings of the National Academy of
Sciences of the United States of America 101, 3597-3602.
Deretic, V., 2012. Autophagy as an innate immunity paradigm: expanding the scope and
repertoire of pattern recognition receptors. Curr Opin Immunol 24, 21-31.
Donnenberg, M.S., Kaper, J.B., 1991. Construction of an eae deletion mutant of
enteropathogenic Escherichia coli by using a positive-selection suicide vector.
Infection and immunity 59, 4310-4317.
Donnenberg, M.S., Tzipori, S., McKee, M.L., O'Brien, A.D., Alroy, J., Kaper, J.B., 1993.
The role of the eae gene of enterohemorrhagic Escherichia coli in intimate
attachment in vitro and in a porcine model. J Clin Invest 92, 1418-1424.

132
Doublié, S., 1997. Preparation of selenomethionyl proteins for phase determination.
Methods Enzymol 276, 523-530.
Eisenbrandt, R., Kalkum, M., Lai, E.M., Lurz, R., Kado, C.I., Lanka, E., 1999.
Conjugative pili of IncP plasmids, and the Ti plasmid T pilus are composed of
cyclic subunits. The Journal of biological chemistry 274, 22548-22555.
Emsley, P., Cowtan, K., 2004. Coot: model-building tools for molecular graphics. Acta
Crystallogr D Biol Crystallogr 60, 2126-2132.
Feldman, M.F., Cornelis, G.R., 2003. The multitalented type III chaperones: all you can
do with 15 kDa. FEMS Microbiol Lett 219, 151-158.
Filloux, A., 2004. The underlying mechanisms of type II protein secretion. Biochim
Biophys Acta 1694, 163-179.
Francis, M.S., Wolf-Watz, H., Forsberg, A., 2002. Regulation of type III secretion
systems. Curr Opin Microbiol 5, 166-172.
Friedberg, D., Umanski, T., Fang, Y., Rosenshine, I., 1999. Hierarchy in the expression
of the locus of enterocyte effacement genes of enteropathogenic Escherichia coli.
Mol Microbiol 34, 941-952.
Fritsch, M.J., Krehenbrink, M., Tarry, M.J., Berks, B.C., Palmer, T., 2012. Processing by
rhomboid protease is required for Providencia stuartii TatA to interact with TatC
and to form functional homo-oligomeric complexes. Mol Microbiol 84, 1108-
1123.
Frobel, J., Rose, P., Muller, M., 2011. Early contacts between substrate proteins and TatA
translocase component in twin-arginine translocation. J Biol Chem 286, 43679-
43689.
Frobel, J., Rose, P., Lausberg, F., Blummel, A.S., Freudl, R., Muller, M., 2012.
Transmembrane insertion of twin-arginine signal peptides is driven by TatC and
regulated by TatB. Nature communications 3, 1311.
Fu, Y., Galan, J.E., 1999. A salmonella protein antagonizes Rac-1 and Cdc42 to mediate
host-cell recovery after bacterial invasion. Nature 401, 293-297.
Fujita, Y., Taguchi, H., 2012. Overview and outlook of Toll-like receptor ligand-antigen
conjugate vaccines. Ther Deliv 3, 749-760.
Gerard, F., Cline, K., 2006. Efficient twin arginine translocation (Tat) pathway transport
of a precursor protein covalently anchored to its initial cpTatC binding site. J Biol
Chem 281, 6130-6135.

133
Goure, J., Broz, P., Attree, O., Cornelis, G.R., Attree, I., 2005. Protective anti-V
antibodies inhibit Pseudomonas and Yersinia translocon assembly within host
membranes. J Infect Dis 192, 218-225.
Gruber, C.W., Cemazar, M., Heras, B., Martin, J.L., Craik, D.J., 2006. Protein disulfide
isomerase: the structure of oxidative folding. Trends Biochem Sci 31, 455-464.
Grynberg, M., Erlandsen, H., Godzik, A., 2003. HEPN: a common domain in bacterial
drug resistance and human neurodegenerative proteins. Trends Biochem Sci 28,
224-226.
Guo, M., Hou, Q., Hew, C.L., Pan, S.Q., 2007a. Agrobacterium VirD2-binding protein is
involved in tumorigenesis and redundantly encoded in conjugative transfer gene
clusters. Mol Plant Microbe Interact 20, 1201-1212.
Guo, M., Jin, S., Sun, D., Hew, C.L., Pan, S.Q., 2007b. Recruitment of conjugative DNA
transfer substrate to Agrobacterium type IV secretion apparatus. Proc Natl Acad
Sci U S A 104, 20019-20024.
Guttman, J.A., Li, Y., Wickham, M.E., Deng, W., Vogl, A.W., Finlay, B.B., 2006.
Attaching and effacing pathogen-induced tight junction disruption in vivo. Cell
Microbiol 8, 634-645.
Håkansson, S., Schesser, K., Persson, C., Galyov, E.E., Rosqvist, R., Homblé, F., Wolf-
Watz, H., 1996. The YopB protein of Yersinia pseudotuberculosis is essential for
the translocation of Yop effector proteins across the target cell plasma membrane
and displays a contact-dependent membrane disrupting activity. The EMBO
journal 15, 5812-5823.
Hamilton, C.M., Lee, H., Li, P.L., Cook, D.M., Piper, K.R., von Bodman, S.B., Lanka,
E., Ream, W., Farrand, S.K., 2000. TraG from RP4 and TraG and VirD4 from Ti
plasmids confer relaxosome specificity to the conjugal transfer system of pTiC58.
J Bacteriol 182, 1541-1548.
Hansen, G., Shillito, R.D., Chilton, M.D., 1997. T-strand integration in maize protoplasts
after codelivery of a T-DNA substrate and virulence genes. Proc Natl Acad Sci U
S A 94, 11726-11730.
Hardt, W.D., Chen, L.M., Schuebel, K.E., Bustelo, X.R., Galán, J.E., 1998. S.
typhimurium encodes an activator of Rho GTPases that induces membrane
ruffling and nuclear responses in host cells. Cell 93, 815-826.
Hayward, R.D., Leong, J.M., Koronakis, V., Campellone, K.G., 2006. Exploiting
pathogenic Escherichia coli to model transmembrane receptor signalling. Nat Rev
Microbiol 4, 358-370.

134
Henderson, I.R., Nataro, J.P., 2001. Virulence functions of autotransporter proteins.
Infect Immun 69, 1231-1243.
Henderson, I.R., Navarro-Garcia, F., Nataro, J.P., 1998. The great escape: structure and
function of the autotransporter proteins. Trends Microbiol 6, 370-378.
Henderson, I.R., Navarro-Garcia, F., Desvaux, M., Fernandez, R.C., Ala'Aldeen, D.,
2004. Type V protein secretion pathway: the autotransporter story. Microbiol Mol
Biol Rev 68, 692-744.
Ho, S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., Pease, L.R., 1989. Site-directed
mutagenesis by overlap extension using the polymerase chain reaction. Gene 77,
51-59.
Hoffmann, C., Leis, A., Niederweis, M., Plitzko, J.M., Engelhardt, H., 2008. Disclosure
of the mycobacterial outer membrane: cryo-electron tomography and vitreous
sections reveal the lipid bilayer structure. Proc Natl Acad Sci U S A 105, 3963-
3967.
Hofreuter, D., Odenbreit, S., Henke, G., Haas, R., 1998. Natural competence for DNA
transformation in Helicobacter pylori: identification and genetic characterization
of the comB locus. Mol Microbiol 28, 1027-1038.
Holden, M.T., Titball, R.W., Peacock, S.J., Cerdeno-Tarraga, A.M., Atkins, T.,
Crossman, L.C., Pitt, T., Churcher, C., Mungall, K., Bentley, S.D., Sebaihia, M.,
Thomson, N.R., Bason, N., Beacham, I.R., Brooks, K., Brown, K.A., Brown,
N.F., Challis, G.L., Cherevach, I., Chillingworth, T., Cronin, A., Crossett, B.,
Davis, P., DeShazer, D., Feltwell, T., Fraser, A., Hance, Z., Hauser, H., Holroyd,
S., Jagels, K., Keith, K.E., Maddison, M., Moule, S., Price, C., Quail, M.A.,
Rabbinowitsch, E., Rutherford, K., Sanders, M., Simmonds, M., Songsivilai, S.,
Stevens, K., Tumapa, S., Vesaratchavest, M., Whitehead, S., Yeats, C., Barrell,
B.G., Oyston, P.C., Parkhill, J., 2004. Genomic plasticity of the causative agent of
melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci U S A 101, 14240-
14245.
Holland, I.B., 2010. The extraordinary diversity of bacterial protein secretion
mechanisms. Methods Mol Biol 619, 1-20.
Holm, L., Sander, C., 1998. Touring protein fold space with Dali/FSSP. Nucleic Acids
Res 26, 316-319.
Hueck, C.J., 1998. Type III protein secretion systems in bacterial pathogens of animals
and plants. Microbiol Mol Biol Rev 62, 379-433.

135
Issartel, J.P., Koronakis, V., Hughes, C., 1991. Activation of Escherichia coli
prohaemolysin to the mature toxin by acyl carrier protein-dependent fatty
acylation. Nature 351, 759-761.
Iyoda, S., Watanabe, H., 2005. ClpXP protease controls expression of the type III protein
secretion system through regulation of RpoS and GrlR levels in
enterohemorrhagic Escherichia coli. J Bacteriol 187, 4086-4094.
Iyoda, S., Koizumi, N., Satou, H., Lu, Y., Saitoh, T., Ohnishi, M., Watanabe, H., 2006.
The GrlR-GrlA regulatory system coordinately controls the expression of flagellar
and LEE-encoded type III protein secretion systems in enterohemorrhagic
Escherichia coli. J Bacteriol 188, 5682-5692.
Jacob-Dubuisson, F., Locht, C., Antoine, R., 2001. Two-partner secretion in Gram-
negative bacteria: a thrifty, specific pathway for large virulence proteins. Mol
Microbiol 40, 306-313.
Jerse, A.E., Gicquelais, K.G., Kaper, J.B., 1991. Plasmid and chromosomal elements
involved in the pathogenesis of attaching and effacing Escherichia coli. Infect
Immun 59, 3869-3875.
Jiménez, R., Cruz-Migoni, S.B., Huerta-Saquero, A., Bustamante, V.H., Puente, J.L.,
2010. Molecular characterization of GrlA, a specific positive regulator of ler
expression in enteropathogenic Escherichia coli. J Bacteriol 192, 4627-4642.
Jobichen, C., Fernandis, A.Z., Velazquez-Campoy, A., Leung, K.Y., Mok, Y.K., Wenk,
M.R., Sivaraman, J., 2009. Identification and characterization of the lipid-binding
property of GrlR, a locus of enterocyte effacement regulator. Biochem J 420, 191-
199.
Jobichen, C., Li, M., Yerushalmi, G., Tan, Y., Mok, Y., Rosenshine, I., Leung, K.,
Sivaraman, J., 2007. Structure of GrlR and the implication of its EDED motif in
mediating the regulation of type III secretion system in EHEC. PLoS pathogens 3,
e69.
Jobichen, C., Chakraborty, S., Li, M., Zheng, J., Joseph, L., Mok, Y.K., Leung, K.Y.,
Sivaraman, J., 2010. Structural basis for the secretion of EvpC: a key type VI
secretion system protein from Edwardsiella tarda. PLoS One 5, e12910.
Karamanou, S., Vrontou, E., Sianidis, G., Baud, C., Roos, T., Kuhn, A., Politou, A.S.,
Economou, A., 1999. A molecular switch in SecA protein couples ATP
hydrolysis to protein translocation. Mol Microbiol 34, 1133-1145.
Kauppi, A.M., Nordfelth, R., Hagglund, U., Wolf-Watz, H., Elofsson, M., 2003.
Salicylanilides are potent inhibitors of type III secretion in Yersinia. Adv Exp
Med Biol 529, 97-100.

136
Kenny, B., Finlay, B.B., 1997. Intimin-dependent binding of enteropathogenic
Escherichia coli to host cells triggers novel signaling events, including tyrosine
phosphorylation of phospholipase C-gamma1. Infect Immun 65, 2528-2536.
Kenny, B., Lai, L.C., Finlay, B.B., Donnenberg, M.S., 1996. EspA, a protein secreted by
enteropathogenic Escherichia coli, is required to induce signals in epithelial cells.
Mol Microbiol 20, 313-323.
Kitagawa, R., Takaya, A., Yamamoto, T., 2011. Dual regulatory pathways of flagellar
gene expression by ClpXP protease in enterohaemorrhagic Escherichia coli.
Microbiology 157, 3094-3103.
Koronakis, V., Sharff, A., Koronakis, E., Luisi, B., Hughes, C., 2000. Crystal structure of
the bacterial membrane protein TolC central to multidrug efflux and protein
export. Nature 405, 914-919.
Kozlov, G., Denisov, A.Y., Girard, M., Dicaire, M.J., Hamlin, J., McPherson, P.S., Brais,
B., Gehring, K., 2011. Structural basis of defects in the sacsin HEPN domain
responsible for autosomal recessive spastic ataxia of Charlevoix-Saguenay
(ARSACS). J Biol Chem 286, 20407-20412.
Kubori, T., Matsushima, Y., Nakamura, D., Uralil, J., Lara-Tejero, M., Sukhan, A.,
Galan, J.E., Aizawa, S.I., 1998a. Supramolecular structure of the Salmonella
typhimurium type III protein secretion system. Science 280, 602-605.
Kubori, T., Matsushima, Y., Nakamura, D., Uralil, J., Lara-Tejero, M., Sukhan, A.,
Galán, J.E., Aizawa, S.I., 1998b. Supramolecular structure of the Salmonella
typhimurium type III protein secretion system. Science 280, 602-605.
Kumar, R.B., Das, A., 2002. Polar location and functional domains of the Agrobacterium
tumefaciens DNA transfer protein VirD4. Mol Microbiol 43, 1523-1532.
Lai, E.M., Kado, C.I., 2000. The T-pilus of Agrobacterium tumefaciens. Trends
Microbiol 8, 361-369.
Lawley, T.D., Klimke, W.A., Gubbins, M.J., Frost, L.S., 2003. F factor conjugation is a
true type IV secretion system. FEMS Microbiol Lett 224, 1-15.
Li, L., Ledizet, M., Kar, K., Koski, R.A., Kazmierczak, B.I., 2005. An indirect enzyme-
linked immunosorbent assay for rapid and quantitative assessment of Type III
virulence phenotypes of Pseudomonas aeruginosa isolates. Ann Clin Microbiol
Antimicrob 4, 22.
Li, M., Rosenshine, I., Tung, S.L., Wang, X.H., Friedberg, D., Hew, C.L., Leung, K.Y.,
2004. Comparative proteomic analysis of extracellular proteins of

137
enterohemorrhagic and enteropathogenic Escherichia coli strains and their ihf and
ler mutants. Applied and environmental microbiology 70, 5274-5282.
Lindberg, A.A., Pál, T., 1993. Strategies for development of potential candidate Shigella
vaccines. Vaccine 11, 168-179.
Luirink, J., Sinning, I., 2004. SRP-mediated protein targeting: structure and function
revisited. Biochim Biophys Acta 1694, 17-35.
Matthews, B., 1968. Solvent content of protein crystals. J Mol Biol 33, 491-497.
Mellies, J., Elliott, S., Sperandio, V., Donnenberg, M., Kaper, J., 1999. The Per regulon
of enteropathogenic Escherichia coli : identification of a regulatory cascade and a
novel transcriptional activator, the locus of enterocyte effacement (LEE)-encoded
regulator (Ler). Mol Microbiol 33, 296-306.
Mogensen, J.E., Otzen, D.E., 2005. Interactions between folding factors and bacterial
outer membrane proteins. Mol Microbiol 57, 326-346.
Moon, H.W., Baetz, A.L., Giannella, R.A., 1983. Immunization of swine with heat-stable
Escherichia coli enterotoxin coupled to a carrier protein does not protect suckling
pigs against an Escherichia coli strain that produces heat-stable enterotoxin. Infect
Immun 39, 990-992.
Moore, L.W., Chilton, W.S., Canfield, M.L., 1997. Diversity of opines and opine-
catabolizing bacteria isolated from naturally occurring crown gall tumors. Appl
Environ Microbiol 63, 201-207.
Mori, H., Cline, K., 2002. A twin arginine signal peptide and the pH gradient trigger
reversible assembly of the thylakoid [Delta]pH/Tat translocase. J Cell Biol 157,
205-210.
Mota, L.J., Cornelis, G.R., 2005. The bacterial injection kit: type III secretion systems.
Ann Med 37, 234-249.
Mueller, C.A., Broz, P., Müller, S.A., Ringler, P., Erne-Brand, F., Sorg, I., Kuhn, M.,
Engel, A., Cornelis, G.R., 2005. The V-antigen of Yersinia forms a distinct
structure at the tip of injectisome needles. Science 310, 674-676.
Müller, S.A., Pozidis, C., Stone, R., Meesters, C., Chami, M., Engel, A., Economou, A.,
Stahlberg, H., 2006. Double hexameric ring assembly of the type III protein
translocase ATPase HrcN. Molecular microbiology 61, 119-125.
Nakamoto, H., Bardwell, J.C., 2004. Catalysis of disulfide bond formation and
isomerization in the Escherichia coli periplasm. Biochim Biophys Acta 1694,
111-119.

138
Nataro, J.a.K., J. , 1998 Diarrheagenic Escherichia coli. . Clin Microbiol Rev. 11, , 142-
201.
Okamoto, S., Toyoda-Yamamoto, A., Ito, K., Takebe, I., Machida, Y., 1991. Localization
and orientation of the VirD4 protein of Agrobacterium tumefaciens in the cell
membrane. Molecular & general genetics : MGG 228, 24-32.
Oliver, D.J., Xiang, C.B., Han, P., Lutziger, I., Wang, K., 1999. A mini binary vector
series for plant transformation. Plant Mol Biol 40, 711-717.
Otwinowski, Z., Minor, W., 1997. Processing of X-ray diffraction data collected in
oscillation mode, p. 307-326, Methods in enzymology, Academic press.
Palmer, T., Berks, B.C., 2012. The twin-arginine translocation (Tat) protein export
pathway. Nat Rev Microbiol 10, 483-496.
Pang, T., Bhutta, Z.A., Finlay, B.B., Altwegg, M., 1995. Typhoid fever and other
salmonellosis: a continuing challenge. Trends Microbiol 3, 253-255.
Papanikou, E., Karamanou, S., Economou, A., 2007. Bacterial protein secretion through
the translocase nanomachine. Nat Rev Microbiol 5, 839-851.
Pennington.H, 2010. Escherichia coli O157. . Lancet 376 1428-1435.
Pettersson, J., Nordfelth, R., Dubinina, E., Bergman, T., Gustafsson, M., Magnusson,
K.E., Wolf-Watz, H., 1996. Modulation of virulence factor expression by
pathogen target cell contact. Science 273, 1231-1233.
Pitzschke, A., Hirt, H., 2010. New insights into an old story: Agrobacterium-induced
tumour formation in plants by plant transformation. EMBO J 29, 1021-1032.
Potter, A.A., Klashinsky, S., Li, Y., Frey, E., Townsend, H., Rogan, D., Erickson, G.,
Hinkley, S., Klopfenstein, T., Moxley, R.A., Smith, D.R., Finlay, B.B., 2004.
Decreased shedding of Escherichia coli O157:H7 by cattle following vaccination
with type III secreted proteins. Vaccine 22, 362-369.
Pugsley, A.P., 1993. The complete general secretory pathway in gram-negative bacteria.
Microbiol Rev 57, 50-108.
Randall, L.L., Hardy, S.J., 2002. SecB, one small chaperone in the complex milieu of the
cell. Cell Mol Life Sci 59, 1617-1623.
Roine, E., Wei, W., Yuan, J., Nurmiaho-Lassila, E.L., Kalkkinen, N., Romantschuk, M.,
He, S.Y., 1997. Hrp pilus: an hrp-dependent bacterial surface appendage
produced by Pseudomonas syringae pv. tomato DC3000. Proceedings of the
National Academy of Sciences of the United States of America 94, 3459-3464.

139
Rollauer, S.E., Tarry, M.J., Graham, J.E., Jaaskelainen, M., Jager, F., Johnson, S.,
Krehenbrink, M., Liu, S.M., Lukey, M.J., Marcoux, J., McDowell, M.A.,
Rodriguez, F., Roversi, P., Stansfeld, P.J., Robinson, C.V., Sansom, M.S., Palmer,
T., Hogbom, M., Berks, B.C., Lea, S.M., 2012. Structure of the TatC core of the
twin-arginine protein transport system. Nature 492, 210-214.
Rosenshine, I., Ruschkowski, S., Finlay, B.B., 1996. Expression of attaching/effacing
activity by enteropathogenic Escherichia coli depends on growth phase,
temperature, and protein synthesis upon contact with epithelial cells. Infect
Immun 64, 966-973.
Rosenshine, I., Donnenberg, M.S., Kaper, J.B., Finlay, B.B., 1992. Signal transduction
between enteropathogenic Escherichia coli (EPEC) and epithelial cells: EPEC
induces tyrosine phosphorylation of host cell proteins to initiate cytoskeletal
rearrangement and bacterial uptake. EMBO J 11, 3551-3560.
Sagulenko, V., Sagulenko, E., Jakubowski, S., Spudich, E. &, Christie, P.J., 2001. VirB7
lipoprotein is exocellular and associates with the Agrobacterium tumefaciens T
pilus. . J. Bacteriol. 183 3642–3651.
Saitoh, T., Iyoda, S., Yamamoto, S., Lu, Y., Shimuta, K., Ohnishi, M., Terajima, J.,
Watanabe, H., 2008. Transcription of the ehx enterohemolysin gene is positively
regulated by GrlA, a global regulator encoded within the locus of enterocyte
effacement in enterohemorrhagic Escherichia coli. J Bacteriol 190, 4822-4830.
Sargent, F., Bogsch, E.G., Stanley, N.R., Wexler, M., Robinson, C., Berks, B.C., Palmer,
T., 1998. Overlapping functions of components of a bacterial Sec-independent
protein export pathway. EMBO J 17, 3640-3650.
Schesser, K., Frithz-Lindsten, E., Wolf-Watz, H., 1996. Delineation and mutational
analysis of the Yersinia pseudotuberculosis YopE domains which mediate
translocation across bacterial and eukaryotic cellular membranes. Journal of
bacteriology 178, 7227-7233.
Schierle, C.F., Berkmen, M., Huber, D., Kumamoto, C., Boyd, D., Beckwith, J., 2003.
The DsbA signal sequence directs efficient, cotranslational export of passenger
proteins to the Escherichia coli periplasm via the signal recognition particle
pathway. J Bacteriol 185, 5706-5713.
Schmidt-Eisenlohr, H., Domke, N., Angerer, C., Wanner, G., Zambryski, P.C., Baron, C.,
1999. Vir proteins stabilize VirB5 and mediate its association with the T pilus of
Agrobacterium tumefaciens. J Bacteriol 181, 7485-7492.
Schmidt, H., Beutin, L., Karch, H., 1995. Molecular analysis of the plasmid-encoded
hemolysin of Escherichia coli O157:H7 strain EDL 933. Infection and immunity
63, 1055-1061.

140
Schrammeijer, B., Dulk-Ras, A., Vergunst, A.C., Jacome, E.J., and Hooykaas, P.J.J. ,
(2003) Analysis of Vir protein translocation from Agrobacterium tumefaciens
using Saccharomyces cerevisiae as a model: evidence for transport of a novel
effector protein VirE3. . Nucleic Acids Research 31, 860-868.
Schuck, P., 2000. Size-distribution analysis of macromolecules by sedimentation velocity
ultracentrifugation and lamm equation modeling. Biophysical journal 78, 1606-
1619.
Sekiya, K., Ohishi, M., Ogino, T., Tamano, K., Sasakawa, C., Abe, A., 2001.
Supermolecular structure of the enteropathogenic Escherichia coli type III
secretion system and its direct interaction with the EspA-sheath-like structure.
Proceedings of the National Academy of Sciences of the United States of America
98, 11638-11643.
Settles, A.M., Yonetani, A., Baron, A., Bush, D.R., Cline, K., Martienssen, R., 1997. Sec-
independent protein translocation by the maize Hcf106 protein. Science 278,
1467-1470.
Shalom, G., Shaw, J.G., Thomas, M.S., 2007. In vivo expression technology identifies a
type VI secretion system locus in Burkholderia pseudomallei that is induced upon
invasion of macrophages. Microbiology 153, 2689-2699.
Shimizu, T., Shima, K., Yoshino, K., Yonezawa, K., Shimizu, T., Hayashi, H., 2002.
Proteome and transcriptome analysis of the virulence genes regulated by the
VirR/VirS system in Clostridium perfringens. J Bacteriol 184, 2587-2594.
Sorg, J.A., Miller, N.C., Schneewind, O., 2005. Substrate recognition of type III secretion
machines--testing the RNA signal hypothesis. Cell Microbiol 7, 1217-1225.
Sory, M.P., Boland, A., Lambermont, I., Cornelis, G.R., 1995. Identification of the YopE
and YopH domains required for secretion and internalization into the cytosol of
macrophages, using the cyaA gene fusion approach. Proceedings of the National
Academy of Sciences of the United States of America 92, 11998-12002.
Staley, T.E., Jones, E.W., Corley, L.D., 1969. Attachment and penetration of Escherichia
coli into intestinal epithelium of the ileum in newborn pigs. Am J Pathol 56, 371-
392.
Stebbins, C.E., Galán, J.E., 2003. Priming virulence factors for delivery into the host. Nat
Rev Mol Cell Biol 4, 738-743.
Stover, C.K., Pham, X.Q., Erwin, A.L., Mizoguchi, S.D., Warrener, P., Hickey, M.J.,
Brinkman, F.S., Hufnagle, W.O., Kowalik, D.J., Lagrou, M., Garber, R.L., Goltry,
L., Tolentino, E., Westbrock-Wadman, S., Yuan, Y., Brody, L.L., Coulter, S.N.,
Folger, K.R., Kas, A., Larbig, K., Lim, R., Smith, K., Spencer, D., Wong, G.K.,

141
Wu, Z., Paulsen, I.T., Reizer, J., Saier, M.H., Hancock, R.E., Lory, S., Olson,
M.V., 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an
opportunistic pathogen. Nature 406, 959-964.
Terwilliger, T., 2003. SOLVE and RESOLVE: automated structure solution and density
modification. Methods Enzymol 374, 22-37.
Terwilliger, T., Berendzen, J., 1997a. Bayesian correlated MAD phasing. Acta
Crystallogr D Biol Crystallogr 53, 571-579.
Terwilliger, T.C., Berendzen, J., 1997b. Bayesian correlated MAD phasing. Acta
Crystallogr D Biol Crystallogr 53, 571-579.
Tseng, T.T., Tyler, B.M., Setubal, J.C., 2009. Protein secretion systems in bacterial-host
associations, and their description in the Gene Ontology. BMC Microbiol 9 Suppl
1, S2.
Tzfira, T., Citovsky, V., 2000. From host recognition to T-DNA integration: the function
of bacterial and plant genes in the Agrobacterium-plant cell interaction. Molecular
plant pathology 1, 201-212.
Valentine, L., 2003. Agrobacterium tumefaciens and the plant: the David and Goliath of
modern genetics. Plant physiology 133, 948-955.
Van Donkersgoed, J., Hancock, D., Rogan, D., Potter, A.A., 2005. Escherichia coli
O157:H7 vaccine field trial in 9 feedlots in Alberta and Saskatchewan. Can Vet J
46, 724-728.
Vergunst, A.C., Schrammeijer, B., Dulk-Ras, A., Vlaam, C.M.T., Regensburg-Tuink,
T.J.G., and Hooykaas, P.J.J. , 2000. VirB/D4-dependent protein translocation
from Agrobacterium into plant cells. . Science 290, 979-982.
Wagner, W., Vogel, M., Goebel, W., 1983. Transport of hemolysin across the outer
membrane of Escherichia coli requires two functions. Journal of bacteriology 154,
200-210.
Walther, T.H., Grage, S.L., Roth, N., Ulrich, A.S., 2010. Membrane alignment of the
pore-forming component TatA(d) of the twin-arginine translocase from Bacillus
subtilis resolved by solid-state NMR spectroscopy. J Am Chem Soc 132, 15945-
15956.
Ward, D.V., Draper, O., Zupan, J.R., Zambryski, P.C., 2002. Peptide linkage mapping of
the Agrobacterium tumefaciens vir-encoded type IV secretion system reveals
protein subassemblies. Proceedings of the National Academy of Sciences of the
United States of America 99, 11493-11500.

142
Wattiau, P., Cornelis, G.R., 1993. SycE, a chaperone-like protein of Yersinia
enterocolitica involved in Ohe secretion of YopE. Molecular microbiology 8, 123-
131.
Welch, R.A., Pellett, S., 1988. Transcriptional organization of the Escherichia coli
hemolysin genes. Journal of bacteriology 170, 1622-1630.
Wulff-Strobel, C.R., Williams, A.W., Straley, S.C., 2002. LcrQ and SycH function
together at the Ysc type III secretion system in Yersinia pestis to impose a
hierarchy of secretion. Molecular microbiology 43, 411-423.
Yen, M.R., Peabody, C.R., Partovi, S.M., Zhai, Y., Tseng, Y.H., Saier, M.H., 2002.
Protein-translocating outer membrane porins of Gram-negative bacteria. Biochim
Biophys Acta 1562, 6-31.
Yerushalmi, G., Nadler, C., Berdichevski, T., Rosenshine, I., 2008. Mutational analysis
of the locus of enterocyte effacement-encoded regulator (Ler) of enteropathogenic
Escherichia coli. J Bacteriol 190, 7808-7818.
Zhu, J., Oger, P.M., Schrammeijer, B., Hooykaas, P.J., Farrand, S.K., Winans, S.C.,
2000. The bases of crown gall tumorigenesis. Journal of bacteriology 182, 3885-
3895.
Ziemienowicz, A., Merkle, T., Schoumacher, F., Hohn, B., and Rossi, L. , 2001. Import
of Agrobacterium T-DNA into plant nuclei: two distinct functions of VirD2 and
VirE2 proteins. . The Plant Cell 13, 369-383.
Zupan, J.R., Zambryski, P., 1995. Transfer of T-DNA from Agrobacterium to the plant
cell. Plant Physiol 107, 1041-1047.

ARTICLE
Received 22 Feb 2013 | Accepted 3 Sep 2013 | Published 4 Oct 2013
Structure of GrlR–GrlA complex that preventsGrlA activation of virulence genesAbhilash Padavannil1, Chacko Jobichen1, Erez Mills2, Adrian Velazquez-Campoy3,4,5, Mo Li1, Ka Yin Leung6,7,
Yu Keung Mok1, Ilan Rosenshine2 & J. Sivaraman1
The locus of enterocyte effacement (LEE) is essential for virulence of enterohaemorrhagic
Escherichia coli (EHEC) and enteropathogenic E. coli (EPEC). The 41 genes of the LEE encode
type III secretion system proteins and three associated regulators: Ler, GrlA and GrlR. Ler is a
positive regulator for most of the LEE operons, including grlRA. GrlA controls the expression
of ler, ehxCABD and flhDC operons. GrlR binds to GrlA and suppresses its function. Here we
report the crystal structure of GrlR–GrlAD (aa 1–106) complex (2:1) and its functional
characterization. We show that GrlR interacts with the Helix-Turn-Helix motif of GrlA.
Moreover, GrlA binds to the promoter DNA fragments of ler, ehxCABD and flhDC, and GrlR
outcompetes with these promoter DNA sequences for the Helix-Turn-Helix motif of GrlA.
These findings provide mechanistic insight into a regulatory module for the virulence of EPEC
and EHEC, two important pathogens that cause devastating diseases.
DOI: 10.1038/ncomms3546
1 Department of Biological Sciences, National University of Singapore, 14 Science Drive 4, Singapore 117543, Singapore. 2 Faculty of Medicine, Department ofMicrobiology and Molecular Genetics, IMRIC, The Hebrew University, Jerusalem 91120, Israel. 3 Institute of Biocomputation and Physics of Complex Systems(BIFI), Joint-Unit IQFR-CSIC-BIFI, Zaragoza 50018, Spain. 4 Department of Biochemistry and Molecular and Cell Biology, University of Zaragoza, Zaragoza50009, Spain. 5 Fundacion ARAID, Government of Aragon, Zaragoza 50018, Spain. 6 Department of Biology, Trinity Western University, 7600 Glover Road,Langley, British Columbia, Canada V2Y 1Y1. 7 State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai200237, China. Correspondence and requests for materials should be addressed to J.S. (email: [email protected]).
NATURE COMMUNICATIONS | 4:2546 | DOI: 10.1038/ncomms3546 | www.nature.com/naturecommunications 1
& 2013 Macmillan Publishers Limited. All rights reserved.