structure determination and characterization … · structure determination and characterization of...
TRANSCRIPT

STRUCTURE DETERMINATION AND
CHARACTERIZATION OF THE ENVELOPE PROTEINS
OF SHRIMP WHITE SPOT SYNDROME VIRUS (WSSV)
TANG XUHUA
(B. Sc., Xiamen University, China)
A THESIS SUBMITTED FOR
THE DEGREE OF DOCTOR OF PHILOSOPHY
DEPARTMENT OF BIOLOGICAL SCIENCES
NATIONAL UNIVERSITY OF SINGAPORE
2007

To My Family

i
Acknowledgements
I would like to express my deepest gratitude to my supervisor, Professor Hew Choy Leong, for his invaluable guidance, advice and mentorship. Thanks for giving me an opportunity to commence my research work in Department of Biological Sciences and providing a motivating, enthusiastic, and critical atmosphere for my research.
I am also deeply indebted to Dr. Jayaraman Sivaraman, for his invaluable and
selfless assistance, from whom I obtained the excellent training in the X-ray crystallography. Thanks for his patience and support in my research project and his constructive comments during my thesis time.
I acknowledge Dr. Wu Jinlu for his collaboration on electron microscopy and virus
purification. I am deeply indebted to Dr. Huang Canhua, Dr. Lin Qingsong, Dr. Li Zhengjun, Dr. Song Wenjun and Mr. Shashikant Joshi for their technical guidance molecular biology and mass spectrometry. Thanks for all the insightful comments and constructive criticisms at different stages of my research.
I would like to extend my thanks to all my colleagues and friends for their full
support and help. Thanks specially go to Ms. Chen Jing and Ms. Zhuang Ying for their sincerity and friendship.
Most importantly, none of my achievements is possible without the love and
patience of my family. My family has been a constant source of love, concern, support and strength for me in all my life. I would like to express my heart-felt gratitude to my dear parents for their selfless love and being my spiritual support all the way. Thanks also go to my elder brother, Don, for all the support and encouragement he gave me. I would also like to thank my husband, zhaohua, for his help, understanding and encouragement throughout my graduate studies. My deepest gratitude is for my late grandmother Mdm. Chen Ruiduan who gave me her unconditional love and support. She taught me the good things that really matter in my life. The happy memory of my grandma will provide a persistent inspiration for my journey in life.

ii
Table of Contents
Acknowledgements ............................................................................................................ i
Table of Contents .............................................................................................................. ii
Summary........................................................................................................................... vi
List of Tables ................................................................................................................... vii
List of Figures................................................................................................................. viii
List of Abbreviations ........................................................................................................ x
List of Abbreviations ........................................................................................................ x
1 Literature Review............................................................................................... 1
1.1 Biology of White Spot Syndrome Virus .............................................................. 2
1.1.1 Overview .............................................................................................................. 2
1.1.2 Morphology of WSSV.......................................................................................... 5
1.1.3 The Genome Structure of WSSV ....................................................................... 11
1.1.4 Taxonomy of WSSV .......................................................................................... 13
1.1.5 Envelope Proteins of WSSV .............................................................................. 16
1.1.6 Problems Faced in Study of Envelope Proteins of WSSV................................. 19
1.2 Structural Genomics ........................................................................................... 20
1.2.1 Structural Genomics as a New Research Initiative ............................................ 20
1.2.2 WSSV as a Model for Structural Genomics Study ............................................ 21
1.3 X-ray Crystallography ........................................................................................ 22
1.3.1 Overview ............................................................................................................ 22
1.3.2 Crystallization..................................................................................................... 24
1.3.3 X-ray Diffraction ................................................................................................ 26
1.3.4 Structure Determination ..................................................................................... 27
1.4 Objectives of the Project .................................................................................... 28
1.5 Significance of the Project.................................................................................. 29
1.6 Scope of the Thesis............................................................................................. 29
2 Materials and Methods .................................................................................... 30
2.1 Bacterial Strains, Vectors, Primers and Bacterial Culture ................................. 31
2.2 DNA Manipulation............................................................................................. 32

iii
2.2.1 Amplification of DNA by Polymerase Chain Reaction (PCR) .......................... 32
2.2.2 DNA Digestion and Ligation.............................................................................. 32
2.2.3 Agarose Gel Electrophoresis and DNA Purification.......................................... 33
2.2.4 Preparation of E.coli Competent Cells ............................................................... 33
2.2.5 Transformation of Bacterial Cells ...................................................................... 34
2.2.6 DNA Sequencing................................................................................................ 35
2.3 Virus Treatment and Analysis ............................................................................ 36
2.3.1 Virus Propagation and Purification .................................................................... 36
2.3.2 Treatment of the Intact Virus with Detergent..................................................... 38
2.3.3 Preparation of WSSV Genomic DNA................................................................ 38
2.3.4 Transmission Electron Microscopy.................................................................... 39
2.3.5 Localization Study by Immunoelectron Microscopy ......................................... 39
2.4 Protein Manipulation .......................................................................................... 40
2.4.1 Protein Expression and Solubility Test .............................................................. 40
2.4.2 Expression of Seleno-Methionine Substituted Protein....................................... 40
2.4.3 GST fusion Protein Purification and Removal of GST tag ................................ 41
2.4.4 Purification of Untagged Protein........................................................................ 42
2.4.5 Antibody Preparation.......................................................................................... 42
2.5 Protein Analytical Techniques ........................................................................... 44
2.5.1 SDS-PAGE Gel Electrophoresis ........................................................................ 44
2.5.2 Western blot Analysis......................................................................................... 45
2.5.3 In-Gel Digestion, MALDI-TOF and Tandem MS Sequencing.......................... 46
2.5.4 N-Terminal Sequencing...................................................................................... 47
2.5.5 Circular Dichroism (CD) Spectrum.................................................................... 48
2.5.6 Dynamic Light Scattering (DLS) ....................................................................... 49
2.6 Crystallization..................................................................................................... 51
2.7 Data Collection................................................................................................... 51
2.8 Structure Determination ..................................................................................... 51
3 Characterization of WSSV Envelope Proteins .............................................. 53
3.1 Characterization of Envelope Protein VP300..................................................... 54
3.1.1 Identification of VP300 by Mass Spectrometry ................................................. 54

iv
3.1.2 Expression and Purification of VP300 ............................................................... 54
3.1.3 Localization Study of VP300 ............................................................................. 56
3.1.3.1 Western Analysis of VP300 ............................................................................... 56
3.1.3.2 TEM Immunogold-Labeling Analysis ............................................................... 59
3.1.4 Neutralization Assay of VP300 .......................................................................... 59
3.1.5 Implications ........................................................................................................ 62
3.2 Localization Study of VP26 in WSSV virion..................................................... 64
3.2.1 Immunogold labeling of VP26 in WSSV particles ............................................ 64
3.2.2 Implications ........................................................................................................ 66
3.3 Candidates for Structure Analysis ...................................................................... 67
3.3.1 Solubility Test of Major Envelope Proteins ....................................................... 67
3.3.2 Purification of VP281 Protein and Crystallization Trials................................... 69
3.3.3 Purification of VP466 Protein and Crystallization Trials................................... 71
4 Crystal Study of Two Major Envelope Proteins............................................ 74
4.1 X-ray Structure of Major Envelope Protein VP28 ............................................. 75
4.1.1 Property of rVP28 Protein .................................................................................. 75
4.1.2 Preparation of SeMet rVP28 Protein.................................................................. 78
4.1.3 Crystallization of rVP28..................................................................................... 78
4.1.4 Structure Determination of rVP28...................................................................... 80
4.1.5 Overall Structure of rVP28................................................................................. 82
4.1.6 Oligomerization of rVP28 .................................................................................. 84
4.2 X-ray Structure of Major Envelope Protein VP26 ............................................. 87
4.2.1 Purification of rVP26 and SeMet rVP26 protein ............................................... 87
4.2.2 Crystallization, Data Collection and Structure Determination of rVP26 ........... 89
4.2.3 Overall Structure of rVP26................................................................................. 92
4.2.4 Oligomerization of rVP26 .................................................................................. 94
4.3 Comparison of VP26 and VP28 ......................................................................... 97
4.3.1 Structure Homolog of VP28 and VP26 .............................................................. 97
4.3.2 Comparison with Other Viral Proteins ............................................................. 104
4.3.3 Implications on Gene Duplication.................................................................... 106
4.3.4 Implications on membrane fusion .................................................................... 109

v
5 General Conclusion and Future Studies ...................................................... 112
Bibliography.................................................................................................................. 118
Appendices..................................................................................................................... 130
List of Publications ....................................................................................................... 134

vi
Summary
White spot syndrome virus (WSSV) is a virulent pathogen known to infect
penaeid shrimp and other crustaceans. It has bacilliform morphology with a tail-like
appendage at one end. The envelope consists of four major proteins. Here we report the
localization and crystal structure of two major envelope proteins VP26 and VP28 from
WSSV at 2.2 and 2.0Å respectively. Their structures are being reported for the first time
for WSSV. Both proteins adopt β-barrels architecture with a protruding region.
Furthermore, we reinvestigated the localization of VP26 and VP28 using immunoelectron
microscopy. Our localization study suggests that VP26 and VP28 are on the outer surface
of the virus and resembled as a spike-like structure on the WSSV envelope, this is the
first convincing observation for VP26. The spike-like structure of VP26 and VP28
observed in our immuno-electron microscopy images matches well with the trimeric
shape of the crystal structure. Based on our present studies combined with previous
findings from other groups, we propose that VP26 and VP28 may anchor on the viral
envelope membrane via their predicted N-terminal transmembrane regions, while leaving
the core β-barrel to protrude outside the envelope to interact with the host receptor or to
fuse with the host cell membrane for effective viral infection.

vii
List of Tables
Table 4.1 Summary of Crystallization Conditions and Cryoprotectants .................... 79
Table 4.2 Data collection and refinement statistics of rVP28..................................... 81
Table 4.3 Summary of Crystallization Conditions and Cryoprotectants .................... 90
Table 4.4 Data collection and refinement statistics of rVP26..................................... 91

viii
List of Figures
Figure 1.1 Clinical sign of WSSV in shrimp and crayfish............................................. 4
Figure 1.2 EM of negatively stained intact WSSV virions ............................................ 7
Figure 1.3 Electron micrograph of WSSV particles treated with 1% Triton X-100 ...... 8
Figure 1.4 EM of negatively stained, naked WSSV nucleocapsids ............................... 9
Figure 1.5 Proposed WSSV structure........................................................................... 10
Figure 1.6 Circular representation of the WSSV genome............................................ 12
Figure 1.7 Bootstrap analysis (100 replicates) of an uprooted phylogenetic tree of the DNA polymerase from WSSV.................................................................... 15
Figure 1.8 Synopsis of structure determination by X-ray crystallography .................. 23
Figure 2.1 Banding of purified WSSV Virus in the sucrose gradient.......................... 37
Figure 3.1 1D SDS-PAGE of the WSSV proteins ....................................................... 55
Figure 3.2 Expression of the purified (His)6-VP300 protein........................................ 57
Figure 3.3 Western blot analysis of WSSV with anti-VP300 IgG............................... 58
Figure 3.4 Localization of WSSV VP300 proteins by immunoelectron microscopy .. 60
Figure 3.5 Neutralization Analysis of WSSV infections ............................................. 61
Figure 3.6 localization of WSSV VP26 proteins by immunoelectron microscopy...... 65
Figure 3.7 Solubility study on the envelope proteins of WSSV .................................. 68
Figure 3.8 Expression of the purified VP281 protein .................................................. 70
Figure 3.9 Expression of the purified VP466 protein .................................................. 72
Figure 4.1 DAS transmembrane prediction result for full length VP28 and VP26...... 76
Figure 4.2 Expression of the rVP28 protein in E.coli BL21 DE3 star ......................... 77
Figure 4.3 Ribbon diagram of rVP28 monomer........................................................... 83
Figure 4.4 Ribbon diagram of rVP28 trimer. ............................................................... 85

ix
Figure 4.5 Western blot analysis of WSSV by anti-VP28 ........................................... 86
Figure 4.6 Expression of the rVP26 protein in E.coli BL21 DE3 star ......................... 88
Figure 4.7 Ribbon Diagram of rVP26 monomer.......................................................... 93
Figure 4.8 Ribbon Diagram of rVP26 trimer ............................................................... 95
Figure 4.9 Western blot analysis of WSSV by anti-VP26 ........................................... 96
Figure 4.10 Stereo Cα superposition of rVP28 and rVP26............................................ 98
Figure 4.11 Structure-based sequence alignment of rVP28 and rVP26......................... 99
Figure 4.12 Simulated annealing Fo-Fc omit map showing the conserved region of rVP28 ........................................................................................................ 100
Figure 4.13 Simulated annealing Fo-Fc omit map showing the conserved region of rVP26 ........................................................................................................ 101
Figure 4.14 The molecular surface representation for VP26 and VP28 along with the proposed viral envelope. ........................................................................... 103
Figure 4.15 Sequence Alignment for VP24, VP26 and VP28 ..................................... 107
Figure 4.16 Fusion Proteins of Class I and Class II ..................................................... 110

x
List of Abbreviations
CCP4 collaborative computational project No.4
CD circular dichroism
CNS crystallographic and NMR system
cryo-EM cryo-electron microscopy
DMSO dimethyl sulfoxide
DNase deoxyribonuclease
DLS dynamic light scattering
dsDNA double-stranded DNA
DTT dithiothreitol
dUTPase deoxyuridine-triphosphatase
EDTA ethylenediamine tetraacetic acid
FOM figure of merit
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
HRP horseradish peroxidase
HSQC heteronuclear single-quantum coherence
HIC hydrophobic interaction chromatography
HIV human immunodeficiency virus
Hrs homologous regions
Ig immunoglobulins
IPTG isopropyl-β-D-thiogalactopyranoside
kbp kilo base pair
kDa kilo Dalton
MAD multiple wavelength anomalous dispersion
MALDI matrix assisted laser desorption/ionization
Mbp mega-base pair
MIR multiple isomorphous replacements
MOPS 3-(N-Morpholino)-propanesulfonic acid
MR molecular replacement
MS mass spectrometry

xi
mg milligram
NCS non-crystallographic symmetry
NMR nuclear magnetic resonance
ORF open reading frame
PAGE polyacrylamide gel electrophoresis
PCR polymerase chain reaction
PEG polyethylene glycol
PMSF phenyl-methyl-sulfonyl fluoride
Q-TOF quadrupole-TOF
RMS root mean square
RMSD root-mean-square deviation
SDS sodium dodecyl sulfate
TFA trifluoroacetic acid
TM transmembrane
TOF time-of-flight
Tris tris (hydroxymethyl) aminomethane
UV ultraviolet
WSSV white spot syndrome virus

1
CHAPTER ONE
1 Literature Review

2
1.1 Biology of White Spot Syndrome Virus
1.1.1 Overview
Viruses can only be reproduced by invading and taking over host cells due to their
lack of cellular machinery for reproduction. The term “virus”, which comes from the
Latin word virus, means poison things and usually refers to those particles that infect
eukaryotes including both multi-cellular and many single-cell organisms and prokaryotes
(phages) and archae as well. Typically, these particles carry a small amount of nuclei
acids (either DNA or RNA) surrounded by some forms of protective coat consisting of
proteins, or proteins and lipids (Knipe and Howley 2001).
The discovery that viruses are very abundant in natural waters, surpassing
bacteria by an order of magnitude, has inspired a resurgence of interest in viruses in the
aquatic environment (Wommack and Colwell 2000). The viral pathogen is a major threat,
especially for the aquaculture industry. Intensive cultivation, inadequate sanitation and
worldwide trade of livestock have greatly increased the chance of virus outbreak that
consequently causing catastrophic economic losses. Therefore, effective control and
prevention of viral disease is of upmost important to ensure the long-term operation of
aquaculture.
In the shrimp aquaculture industry, out of more than 20 strains of viruses have
been reported, among which White Spot Syndrome Virus (WSSV) is the most serious

3
pathogen (Chou et al. 1995; Lo et al. 1996; Chen et al. 1997). WSSV first appeared in the
1990s in Taiwan and has since been spread quickly to Southeast Asia, the Indian
continent and the Central- and Latin America (Xie and Yang 2005). The virulence of
WSSV is very high, resulting in a mortality rate of up to 90 to 100% within 3 to 7 days,
which can devastate the regional shrimp culture industry. A major outbreak of WSSV
infection in 1993 resulted in a 70% reduction in production of shrimps in China (Chen et
al. 1997; Cen 1998). The worldwide economic loss caused by WSSV outbreak is
catastrophic and in the range of multi-billion U.S. dollars.
The symptom of the disease caused by this virus include a red color on the entire
body and obvious white spots on the carapace, appendages and the inside surface of body.
That is why scientists named this virus as “white spot syndrome virus”. Most organs and
tissues of the shrimp, except for hepatopancreatocytes and epithelial cells of the midgut,
can be infected by WSSV. Moreover, WSSV has a broad range of hosts, including salt
and brackish water penaeid, crab, spiny lobster, freshwater shrimps and crayfishs (Figure
1.1) (Chou et al. 1995; Wang et al. 1995; Lo et al. 1996; Chen et al. 1997; Huang and
Song 1999; Chen et al. 2000; Huang et al. 2001). As such, WSSV is becoming an
epizootic disease that causes great threat not only to the shrimp aquaculture industry but
also to the marine ecology in general.

4
Figure 1.1 Clinical sign of WSSV in shrimp and crayfish
(A) WSSV infected shrimp (P. monodon) with an apparent presence of white spots on the cuticle. (B) Crayfish (Cherax quadricarinatus) one of WSSV infected hosts, was used in our laboratory for virus propagation and purification.
A
B

5
Prevention and inhibition of infection by this virus, however, is very difficult
largely due to the ability of WSSV to survive for a long period of time in the environment
(around 2 years in a shrimp pond) and also due to a poor understanding of this virus at the
molecular level (Yang et al. 2001). At present, except for the use of nonspecific
immunostimulants such as bacterial glycans (with minimal protection) and low-density
culture conditions, there is no effective treatment protocol for this disease. Therefore
novel control strategies against this virus are highly desirable (Westenberg et al. 2005).
In the following sections, some basic information of the biology on WSSV including its
morphology, genome structure and the taxonomy will be introduced in detail. These
would provide a in-depth and comprehensive understanding of the uniqueness of this
virus and the significance of scientific research on the related disease.
1.1.2 Morphology of WSSV
As mentioned before, WSSV is the major viral pathogen of shrimp with a very
high virulence. In the past decade, WSSV has attracted intensive investigation resulting
in many exciting research progress. The morphology of WSSV has been studied by
electron microscopy technology (Chou et al. 1995; Wang et al. 1995; Wongteerasupaya
et al. 1995; Durand et al. 1997; Lu et al. 1997; Leu et al. 2005). A previous electron
microscopy study showed the that intact WSSV virion is a non-occluded, enveloped
particle with an olive-to-bacilliform shape. The average size of the intact viral particle is

6
approximately 110-130 nm in diameter and 260-350 nm in length. The most prominent
feature of WSSV is the presence of a long tail-like envelope extension at one extremity
that highly resemble to a bacterial flagellum (Huang et al. 2001). The negatively stained
intact WSSV virions observed under EM are shown in Figure 1.2.
The WSSV virion is very sensitive to detergents and the envelope proteins wll be
removed from the virion when treated with 1% Triton X-100 to expose the nucleocapsid
(Figure 1.3). As shown in Figure 1.4, the naked viral nucleocapsid is about 80×350 nm
(Huang et al. 2001). Interestingly, the nucleocapsid is composed of 15 or 16 vertical
segments that are perpendicular to the long axis and about 18 to 20 nm thick. Huang and
coworkers (Huang et al. 2001) have proposed that each segment is composed of double
rows of 14 globular subunits of 8 nm in diameter (Figure 1.5). A “ring” structure could
also be seen in some of the degraded viral nucleocapsid. However, the existence of these
discrete globular subunits is debatable. In a recent paper, Leu et al (2005) have identified
that VP664, the largest viral structural protein ever found, is the major component of the
nucleocapsid. Gold labeling Immunoelectron microscopy showed that the gold particles
indicating the location of VP664 were regularly distributed around the periphery of the
nucleocapsid with a periodicity that matched the characteristic stacked ring subunits
which appear as striations. From this observation, they hypothesized that the stacked,
patterned rings of the nucleocapsid are comprised of the VP664 viral protein.
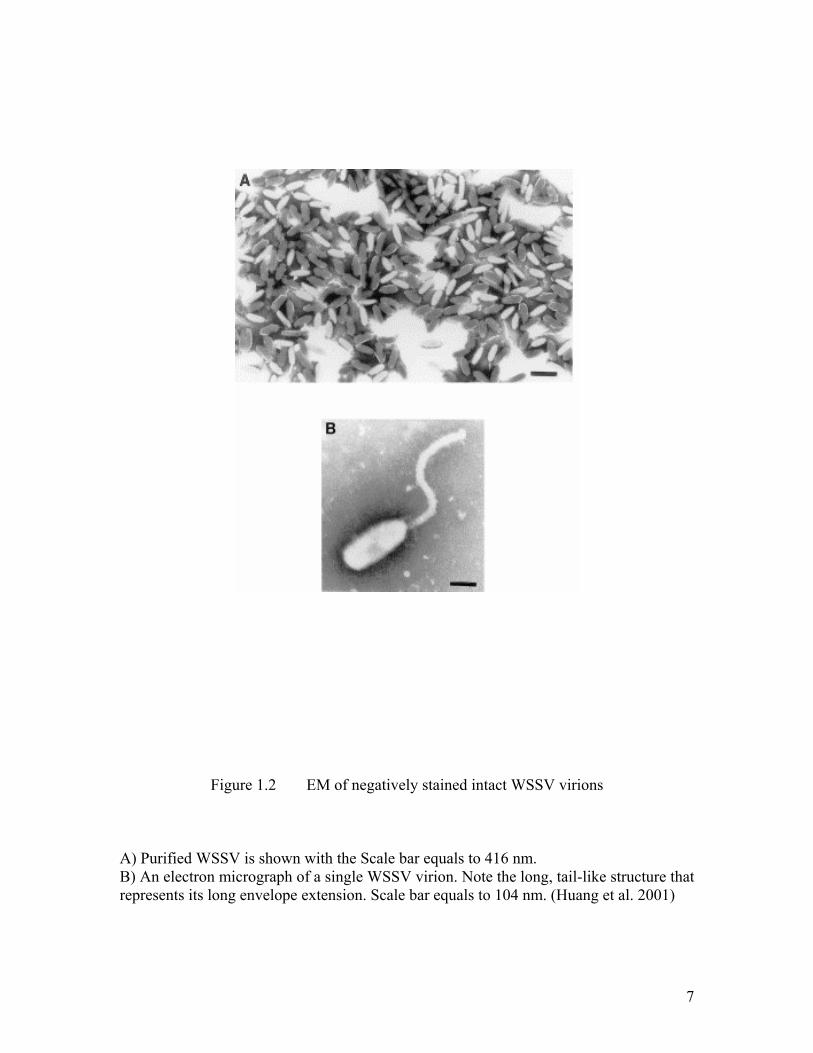
7
Figure 1.2 EM of negatively stained intact WSSV virions
A) Purified WSSV is shown with the Scale bar equals to 416 nm. B) An electron micrograph of a single WSSV virion. Note the long, tail-like structure that represents its long envelope extension. Scale bar equals to 104 nm. (Huang et al. 2001)

8
Figure 1.3 Electron micrograph of WSSV particles treated with 1% Triton X-100
This treatment leads to the distinct separation of the double layered structure of the viral envelop as shown in A (arrow →). The nucleocapsid wrapped inside the double layer envelope shows a "spindle" shape at one extremity (arrow →), while a ‘papilla’ shape structure in the other extremity (arrow →) as shown in B. In A, scale bar equals to 250 nm and in B, scale bar equals to 138 nm. (Huang et al. 2001)

9
Figure 1.4 EM of negatively stained, naked WSSV nucleocapsids
The average size of the nucleocapsid in A is 80 nm in diameter and 350 nm in length with the scale bar equals to 50 nm. Note the 15 conspicuous, vertical helix located horizontally along the long axis of the naked rod-shape nucleocapsid core in B. Each helix has two parallel striations which contain 14 global capsomers or subunit. Scale bar in B equals to 54 nm. (Huang et al. 2001)

10
Figure 1.5 Proposed WSSV structure
(Top) the overall proposed structure of WSSV virion. (Bottom) The average size of the WSSV nucleocapsid encapsidated within the double-layered envelope is 80×350 nm. There are 15 distinct and spiral helices along its long axis. Each spiral helix has two striations composed of seven pairs of globular capsomers, each of which is 8 nm in diameter. The spacing between each spiral helix is 7 nm. A ‘ring-like’ structure is associated at one terminus of the nucleocapsid core. (Huang et al. 2001)

11
1.1.3 The Genome Structure of WSSV
The complete genome sequence of three different isolates of WSSV has been
published (van Hulten et al. 2001a; Yang et al. 2001; Chen et al. 2002a) (Genebank
Accession Nos. AF332093, AF369029, AF440570 for viruses isolated from China,
Thailand and Taiwan, respectively). WSSV is the first marine invertebrate virus that has
its genome completely sequenced and it is also at present the largest animal virus whose
complete genome has been sequenced.
The complete WSSV genome is a double-stranded circular DNA of 305,107 bp,
very close to the previous estimate of 290 kb (Yang et al. 1997). Since the origin of
replication was unknown, the start of the largest BamHI fragment was chosen to be base
1. Three percent of the WSSV genome is made up of nine homologous regions (hrs),
while the remaining 97% of the sequences are unique (see description below). The
genome has a total G+C content of 41% that are uniformly distributed (Yang et al. 2001).
A total of 531 putative open reading frames (ORFs) were identified by sequence
analysis, among which 181 ORFs encoding more than 50 amino acids are likely for
functional proteins. This corresponds to an average gene density of one gene per 1.7kb.
The relative positions of the ORFs and homologous regions (hrs) in the genome are
shown in Figure 1.6. There is a potential polyadenylation site (AATAAA) downstream of
the ORFs for 80% of the 181 putative ORFs. The putative proteins encoded by 18 ORFs
show 40 to 68% identity to known proteins from other viruses or organisms and contain
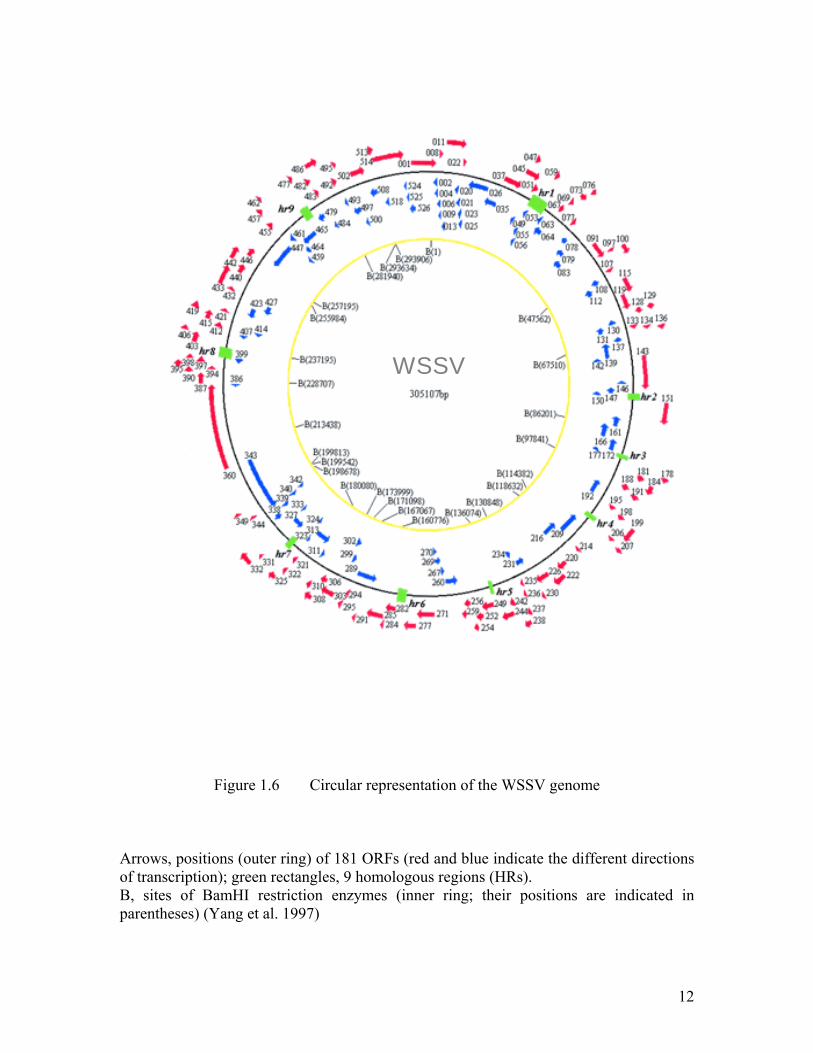
12
Figure 1.6 Circular representation of the WSSV genome
Arrows, positions (outer ring) of 181 ORFs (red and blue indicate the different directions of transcription); green rectangles, 9 homologous regions (HRs). B, sites of BamHI restriction enzymes (inner ring; their positions are indicated in parentheses) (Yang et al. 1997)
WSSV

13
an identifiable functional domain. These proteins include enzymes involved in nucleic
acid metabolism and DNA replication, a collagen-like protein, and three viral structural
proteins. Proteins predicted from thirty ORFs also show partial homology (20 to 39%
identity) to known proteins or contain one or two sequence motifs (versus a real
functional domain). However, the remaining 133 ORFs encode novel proteins with no
homology to any known proteins or motifs (Yang et al. 2001).
The most unique feature of WSSV is the presence of an collagen gene, a gene
encodes an extracellular matrix protein of animal cells that has never been found in any
other viruses (Yang et al. 2001). The genome information provides us the molecular basis
of viral replication and infection.
The analysis of the genome showed that WSSV differs from all known viruses,
suggesting that it represents a novel class of viruses and perhaps implying a significant
evolutionary distance between marine and terrestrial viruses. The next section will
introduce studies on the taxonomy of WSSV during the past decades.
1.1.4 Taxonomy of WSSV
WSSV was classified in the Baculoviridae family and the subfamily of the non-
occluded baculoviruses with its rod-shaped and enveloped morphology that are similar to
insect baculovirus (Francki et al. 1991). However, more in-depth studies on the general

14
and molecular characteristics of WSSV (Inouye et al. 1994; Momoyama et al. 1994;
Wongteerasupaya et al. 1995; Lo et al. 1997) and analysis of the complete genome
sequence (van Hulten et al. 2001a; Yang et al. 2001; Chen et al. 2002a) suggest that
WSSV is not related to any virus of the Baculoviridae family or any other family of
viruses. Looking at the phylogenetic tree of the genes for WSSV DNA polymerase
(Figure 1.7), ribonucleotide reductase large (rr1) and small (rr2) subunits, protein kinase,
thymidine-thymidylate kinase and endonuclease, no obvious relationship was revealed
between WSSV and other established family of large dsDNA virus (Chen et al. 2002b).
Based on the uniqueness of WSSV, many scientists have suggested that it should
be excluded from the Baculovirus family and be classified as a new virus family.
Recently, the International Committee on Taxonomy of Viruses approved a proposal to
elect WSSV as the type species of the genus Whispovirus, family Nimaviridae
(www.ncbi.nlm.nih.gov/ICTVdb/Ictv/index.htm). The family name reflects the most
notable physical feature of the virus, the tail-like projection extending from one end of
the WSSV virion. The uniqueness of WSSV has attracted extensive research into its
evolution and infection mechanism. In the protein level, most researches are focused on
the envelope proteins that play important roles in the infection and maturation
mechanisms of the virus.

15
Figure 1.7 Bootstrap analysis (100 replicates) of an uprooted phylogenetic tree of the
DNA polymerase from WSSV.
The numbers at the branches indicate frequency of the clusters and frequencies over 70% are indicated by thick lines.
(http://www.danforthcenter.org/iltab/ictvnet/images/paris/Nimaviridae.pdf)

16
1.1.5 Envelope Proteins of WSSV
In general, viral proteins are divided into three temporal classes: early proteins
synthesized prior to the replication of the DNA, the intermediate and late proteins
synthesized after the onset of DNA replication (Jensen et al. 1996). The late proteins that
synthesized from 5 to 6 hours after infection are assumed to be virion-associated proteins.
In characterizing any virus, its virion-associated proteins are particularly important for
understanding the mechanisms of viral infection and morphogenesis.
A virion is a complex assembly of macromolecules exquisitely suited for the
protection and delivery of viral genomes (Tsai et al. 2006). As a double-layer enveloped
virus, virion-associated proteins of WSSV include proteins of both the envelope and
nucleocapsid. Thirty nine virion-associated proteins have been identified due to the gel
based mass spectrometry techniques (Huang et al. 2002a; Huang et al. 2002b; Li et al.
2004; Tsai et al. 2004). Shotgun proteomic studies have identified fifty five virion-
associated proteins in our laboratory recently (Li et al, unpublished data).
Among these virion-associated proteins, the envelope proteins are of extremely
importance because they are believed to be the first molecules to interact with the host
and hence play critical roles in targeting the host cell and triggering host defenses.
Determining the localization of structural proteins in the virion is important to elucidate
their roles in both virus assembly and infection. Up to date, four major envelope proteins
(VP19, VP24, VP26 and VP28) and several minor envelope proteins (VP31, VP39,

17
VP124, VP187, VP281, VP292, VP466) were detected by Western blotting technique
and localization study under electron microscope (Huang et al. 2002a; Huang et al. 2002b;
Zhang et al. 2002a; Zhang et al. 2002b; Zhang et al. 2004; Li et al. 2005b; Li et al. 2006;
Xie et al. 2006; Zhu et al. 2006).
There are different classes of membranes proteins: Integral proteins interact with
hydrophobic parts of the bilayer’s phospholipids, and they are not easy to remove. Most
span the bilayer, with their hydrophilic domains extending past both of its surfaces.
Peripheral proteins are positioned at the membrane surface, not in the bilayer. Weak
interactions, including hydrogen bonds, allow them to associate with integral proteins and
with polar heads of the membrane lipids. For major envelope proteins of WSSV such as
VP26 and VP28, they both have a hydrophobic transmembrane domain that serves to
anchor them to the surface of the lipid bilayer.
Using SDS-PAGE profiles of envelope fractions from WSSV, VP19, VP24, VP26
and VP28 have been shown to constitute the major part of the entire envelope.
Interestingly, there are striking similarities between VP24, VP26 and VP28 at the amino
acid and the nucleotide sequence level. VP26 and VP28 share a sequence similarity of
41%, whereas VP24 shares significant sequence similarities of 41% and 46% with VP26
and VP28 respectively. These three proteins have the same size (~ 206 aa) and all are
predicted to have a transmembrane region of approximately 30 amino acid in length at
the N-terminus. This strongly suggests that the genes of these three envelope protein may
have evloved by gene duplication. Another major envelope protein, VP19, has no

18
sequence homology with other WSSV or other known viral proteins. The transmembrane
region of VP19 is predicted to be situated at the middle of the protein sequence.
As far as functional study is concerned, VP28, VP68, VP281, and VP466 were
believed to play an important role in the systemic WSSV infection in shrimp by using an
in vivo neutralization assay (van Hulten et al. 2001b; Li et al. 2005a; Xu et al. 2005).
VP28 is often used for detecting the WSSV in crustaceans because of its abundance in
the proteins profile (Yoganandhan et al. 2004). VP26 is reported to be capable of binding
to actin or actin-associated proteins (Xie and Yang 2005). Another envelope protein,
VP281, which contains a cell attachment RGD motif, is also supposed to play an
important role in mediating infection by WSSV (Huang et al. 2002a; Liang et al. 2005).

19
1.1.6 Problems Faced in Study of Envelope Proteins of WSSV
Envelope proteins have been investigated extensively because of their importance
in viral infection and this project will also focus on the characterization of these envelope
proteins. There are, however, many well-known technical difficulties that hinder in-depth
studies to elucidate the function of the envelope proteins from WSSV. The lack of
established shrimp cell lines for in vitro reproduction of WSSV is one of the major
obstacles. The function of these envelope proteins are also hard to predict because of
their lack of homology with other known proteins. Therefore, we will utilize structural
genomics approaches to elucidate the structure of the WSSV proteins to understand their
function towards the drug design.
Among the 55 structural proteins identified by mass spectrometry, 11 and 3 of
them have been assigned as the envelope and the nucleocapsid proteins respectively. The
localizations of the other 40 structural proteins are still unknown. At present, only VP28
has been confirmed to be involved in the attachment and penetration of the WSSV into
shrimp cells (Yi et al. 2004). The roles of other envelope proteins in the life cycle of the
virus remain to be defined. Moreover, up to now, tertiary structures are not available for
any WSSV proteins. This project will therefore focus on the characterization and
elucidation of the three dimensional structure of the envelope proteins of WSSV.

20
1.2 Structural Genomics
1.2.1 Structural Genomics as a New Research Initiative
New research initiatives in biology are being driven by complete sequencing of
the genomes from various organisms (Green 2001; Lander et al. 2001; Aparicio et al.
2002; Yu et al. 2002), which gave us the protein repertoires of diverse organisms from all
kingdoms (Marsden et al. 2006). The genome-sequence projects have accumulated lots of
sequence data providing us nearly complete lists of macromolecules present in an
organism. However, the knowledge of the components relatively little about the function
and organization of biological systems (Bravo and Aloy 2006). The challenge to
understand these gene products has led to the development of functional genomics
approaches, which collectively aim to improve the biological understanding of the
genetic sequence. Structural genomics is one of such approaches, with an unique promise
to reveal the molecular function of protein domains (Ashburner et al. 2000). A good
understanding of molecular functions & interactions can come from high-resolution
three-dimensional (3D) structures, as they provide key atomic details on the binding
surfaces. Structural genomics projects, together with computational structure prediction
methods (Bradley et al. 2005), are likely to provide 3D structures for most of the proteins
within an organism.
Worldwide structural genomics initiatives, advanced by the development of
improved technologies in X-ray crystallography and NMR, are expanding our knowledge
on structural families with increasing number of novel protein folds. Methods for

21
detecting remote sequence similarities have also been improved in sensitivity and enabled
us to map domains from these structural families onto genome sequences directly to
understand how these families are distributed throughout the genomes. This in turn reveal
how these proteins might influence the functional repertoires and biological complexities
of the organisms (Marsden et al. 2006).
1.2.2 WSSV as a Model for Structural Genomics Study
WSSV provides an important and interesting model for structural genomics study.
It has a relatively small genome with only about 181 open reading frames, much smaller
than herpes virus and other viruses with large genomes. Many of the WSSV putative
ORFs do not have match with any known proteins and functions. Structural genomics
therefore offers an opportunity to elucidate the function of these proteins through
structure determination and prediction. Structure elucidation of WSSV will provide
insights on the mechanisms of host-pathogen interaction, the maturation and assembly of
the virus particles and many other important biological processes. More importantly, it
will provide a wealth of information for structure based drug design.

22
1.3 X-ray Crystallography
1.3.1 Overview
It is necessary to know the precise arrangement of atoms in biological
macromolecules in order to understand how they function at the molecular level. In
addition, this information is required for the rational design of drugs that target a specific
activity of the molecule or prevent the molecule from binding to other molecules with
which it normally interacts (Yaffe 2005). X-ray crystallography is one of the most
powerful techniques that are used to obtain atomic information about molecular structure.
In brief, structure determination of a protein by X-ray crystallography requires the
growth of high-quality crystals from the purified protein, measurement of the directions
and intensities of X-ray beams diffracted from the crystals, and the use of a computer to
calculate the electron density map after the phase problem is being solved. Finally, the
protein backbone is fitted into the electron density map and refined to give the structure
coordinates (Rhodes 2000).
In Figure 1.8, the synopsis of structure determination by X-ray crystallography is
clearly shown. The macromolecule of interest is crystallized, and a single crystal that
contains thousands of copies of the macromolecule is placed in an X-ray beam. The
crystal is slowly rotated in the X-ray beam and diffraction patterns similar to the one
shown are collected for every 0.5–1 degree of rotation until almost all points in the

23
Figure 1.8 Synopsis of structure determination by X-ray crystallography
The macromolecule of interest is crystallized, and a single crystal containing thousands of copies of the macromolecule is placed into an X-ray beam. The crystal is slowly rotated in the X-ray beam and diffraction images similar to the one shown are collected every 0.5–1 degree of rotation until all points in the crystal have been sampled. The intensity of the diffracted X-ray, together with information about their phases, is collected and converted by a Fourier transforming to an electron density map of the protein. A model of the protein structure is then built into the electron density map (Yaffe, 2005).

24
reciprocal space have been sampled. Structure determination of a molecule requires
knowing both the amplitude and the phase of the photon wave being diffracted from the
crystalline sample. Although a detector can measure the intensity of a wave, which is
proportional to its amplitude, there is no way to measure its phase. The phase angel is the
difference of the angles between the combined X-ray scattered by all atoms of this
molecule and the original X-ray. Thus, it is common to hear that one has "lost the phase"
when measuring a diffraction pattern. Reconstructing the phase of the diffracted wave is
the crux of the phase problem. The intensity of the diffracted X-ray, together with
information about their phases, is converted by Fourier transformation into an electron
density map of the protein. A model of the protein structure is then built into the electron
density map (Yaffe 2005).
1.3.2 Crystallization
X-ray crystallography is totally dependent on highly ordered crystals. Obtaining
such crystals is the rate-limiting step in structure determination and the pressure is to
produce crystals. As a result, the science of crystallization is gathering a new momentum
and is becoming a rapidly developing field (Chayen 2004).
Crystallization is a phase transition phenomenon. Crystals grow from an aqueous
protein solution when the solution is brought into supersaturation (Ataka 1993; Ducruix
and Giege 1999; McPherson 1999). Supersaturation is achieved by varying the

25
concentration of precipitant, protein and additives, pH, temperature and other parameters.
The task to produce suitable crystals can be tackled using two different approaches. The
first relies on empirical techniques that is based mainly on trial and error, and what is
perceived to be the “art” of crystallization. The second approach is aimed at gaining an
understanding of the fundamental principles that govern crystallization; this knowledge
may be applied to design experimental methodology for producing high-quality crystals
of medical and industrial interest (Chayen 2004).
The past few years have seen some of the greatest achievements in the field of
protein crystallization e.g. automation and miniaturization of crystallization trials (Kuhn
et al. 2002; Walter et al. 2005). The high-throughput approach has reduced the time
needed to set up a series of experiments. The subsequent phases of different pattern
capture and analysis of the data is also progressing at high speed (Luft et al. 2003).
However, the high-throughput has not led to high output. Production of suitable crystals
still remains as a rate-limiting step. It is still not understood why some proteins crystallize
easily while others stubbornly refuse to produce suitable crystals. Obviously there is an
urgent need to solve the problem by a more systematic & scientific approach (Chayen
2004).
The success rate to obtain high-quality crystals is improving rapidly and it will
improve further as we progress to the more sophisticated techniques, which will play a
major role in crystallization and structural biology.

26
1.3.3 X-ray Diffraction
X-ray is an electromagnetic radiation with wavelengths in the range from 0.1 to
1000 Å. X-ray rays are generated when electrons (or other charged particles) undergo
severe acceleration (or deceleration). There are two types of X-ray sources used for
macromolecular crystallography, those from rotating anode and the synchrotron radiation.
The rotating anode type X-ray generator is the laboratory scale sources. An anode
of appropriate target metal (copper, molybdenum etc.), a cathode and a tungsten filament
are kept inside an evacuated tube. When a high voltage difference is maintained between
the anode and cathode, electrons, produced by the filament by thermionic emission, hit
the anode and X-rays are produced. Depending on the target and transition, the radiation
is named as Cu Kα, Cu Kβ etc. The anode is rotated by a motor for efficient distribution
and dissipation of heat and a high intensity beam is produced. The X-rays produced at the
impact point are distributed over a wide angular range and cover a wide range of energies
up to a maximum energy determined by the voltage at which the tube is operated. In a
synchrotron, electrons travel in a storage ring at a high speed and are emitted as radiation
and it is possible to change the wavelength of synchrotron radiation. Here bunches of
very high energy electrons (several GeV) move in a closed loop. At each bending point
an intense and very narrowly defined pencil beam of radiation is emitted. The beam
contains radiation of a wide range of energies - UV through to X-rays. Synchrotrons are
very large central facilities that have their own operating procedures and safety systems
that visitors and users must follow.

27
1.3.4 Structure Determination
To determine the structure of a protein means that position of all atoms of the
protein in the unitcell represented by the electron density map are known. There is a
direct relationship between the electron density map of the unitcell and the structure
factors of the diffraction pattern as related by the Fourier transformation. However, in
order to calculate the electron density map of the unitcell, one needs the experimental
structure amplitudes and their phase angles.
As the phase angle is an immeasurable quantity, the information can only be
derived by one of the following methods. In the molecular replacement (MR) method, an
available similar structure is used as a model to probe the experimental data to identify
the structure of interest. Direct methods are based on the positivity and atomicity of
electron density that leads to phase relationships between structure factors. The phase
angle is obtained by mathematical means. In the multiple isomorphous replacement (MIR)
method, the data set for a native crystal and data sets for two derivative crystals, obtained
by soaking the native crystal in two different heavy metal containing solutions, are
collected. In the multiwavelength anomalous dispersion method (MAD), a crystal
containing an anomalously diffracting atom is used for data collection at three different
wavelengths using a synchrotron. Positions of these anomalous atoms are first
determined and the electron density map is made. SAD (single-wavelength anomalous
diffraction) method is becoming increasingly possible to collect data at just a single

28
wavelength, typically at the absorption peak, and use density-modification protocols to
break the phase ambiguity and provide interpretable maps.
The electron density map is calculated after the phase problem is being solved.
The protein model is refined until the difference between the experimental and calculated
(using the model) structure factors (R-factor) reaches at an acceptable minimum. The
most used refinement program is “CNS” (Crystallographic and NMR System). CNS
adopts the algorithm of energy minimization for the refinement
1.4 Objectives of the Project
The characterization of viral structural proteins especially the envelope proteins is
of significant importance to study the virus taxonomy, infection assembly. Due to the
lack of a shrimp cell line for in vitro reproduction of this virus and low homology with
other proteins of known function, we have decided to take a structural proteomics
approach to study the envelope proteins of WSSV. The objectives of this project are:
1) To identify more envelope proteins of WSSV. Among the 55 structural proteins
identified by mass spectrometry, only 11 have been assigned as envelope proteins and 3
as nucleocapsid proteins. There are 40 structural proteins unassigned. We will use
western blotting and immuno-gold-labeling electron microscopy to study the localization
of these envelope proteins. Neutralization assay that can determine whether the envelope
proteins are involved in the virus entry during infection will also be performed.

29
2) To elucidate the three dimensional structure of the viral envelope proteins. The
study attempts to determine the structures of these envelope proteins to understand their
function towards the drug design.
1.5 Significance of the Project
Once the 3D structures of major envelope proteins are solved, their structural
features should provide us an unique opportunity to postulate the function of these
envelope proteins. Also, the structure of these envelope proteins and their interactions
explain the unique morphological features of WSSV. The structural elucidation of WSSV
envelope proteins will provide us much useful information for further research on host-
virus interaction, the maturation and assembly of the virus particles and many other
important questions. It will also help us to better understand the infection mechanism and
life cycle of WSSV. Finally, it will provide a wealth of information for structure based
drug design that can be used to develop specific inhibitors to control or even prevent the
outbreak of WSSV.
1.6 Scope of the Thesis
X-ray crystallography techniques will be used to determine the structures of the
envelope proteins. The major envelope proteins like VP19, VP24, VP26 and VP28 are the
primary candidates for crystallography and functional study.

30
CHAPTER TWO
2 Materials and Methods

31
2.1 Bacterial Strains, Vectors, Primers and Bacterial Culture
Bacterial Strain DH5α (Invitrogen) was used in this study as host for cloning
experiments and BL21 Star (DE3) strain (Invitrogen) for protein expression. Vector
pGEX6p-1 (Amersham Bioscience) was used for GST fusion protein expression and
pET32a(+) (Novagen) for His-tag protein expression. Escherichia coli starins were
grown at 37 °C in LB (Luria broth) liquid media (10g Tryptone, 5g yeast extract, 10g
NaCl, pH7.5) or 1.5 % agar medium (Sambrook and Russell 2001). For long-term storage,
all bacterial strains were stored in LB with 50% glycerol at -80 °C freezer.
The sequences of some primers used in this study are shown as following:
N-terminus truncated VP28 (residues 31-204)
CGC GGA TCC AAC ACT GTG ACC AGG ACC ATC GAA
CCG GAA TTC TTA CTC GGT CTC AGT GCC AGA
N-terminus truncated VP26 (residues 35-204)
CGC GGA TCC ATG AAC ACA CGT GTT GGA AGA
CCG GAA TTC TTA CTT CTT CTT GAT TTC GTC CTT
Full-length VP300
CGC GGA TCC ATG GGA GAT AAG CAA AAG GTG GAA
CGC GAA TTC TTA GGA GCA TGT GCA TGT GAT CCT
Full-length VP281
CGC GGA TCC ATG GCG GTA AAC TTG GAT AAT
CGC GAA TTC TTA TGT CCA ACA ATT TAA AAA GAA

32
2.2 DNA Manipulation
DNA manipulations were carried out following standard molecular protocols
cited from Molecular Cloning (Sambrook and Russell 2001) with some modifications.
2.2.1 Amplification of DNA by Polymerase Chain Reaction (PCR)
DNA fragments and related genes were amplified by the basic polymerase chain
reaction (PCR). High fidelity Pfu DNA polymerase (Promega) was used to amplify the
genes from WSSV genomic DNA. The composition of PCR reaction mixture was shown
as following: 5 µl 10 × PCR buffer (without MgCl2), 5 µl MgCl2 (25 mM), 1 µl 5’ Primer,
1 µl 3’ Primer, 1 µ dNTPs (25 mM) l, 1 µl Template DNA (20-100 ng/ µl), 1 µl Tag DNA
polymerase (1 unit/ µl), and distilled water to a final volume of 50 µl.
The PCR was run on an iCycler Thermal Cycler (Bio-rad) with the following
program. 1 cycle of 95 °C for 1 ~ 2 minutes, 30 cycles of each at 95 °C for 30 seconds,
50 °C for 30 seconds and 72 °C for 1 minute per 1kb DNA, and 1 cycle of 72 °C for 15
minutes.
2.2.2 DNA Digestion and Ligation
DNA digestion and ligation were conducted according to the instruction manual
by the supplier. The DNA digestion mixture comprised of 10 µl DNA, 1 µl restriction
enzymes, 2 µl 10×BSA, 2 µl reaction buffer and 4 µl sterile H2O. The mixture was

33
incubated at 37 °C for 2 hours. For single-digestion vectors, dephosphorylation is
required for the subsequent ligation. In this case, the vector mixture was treated
additionally with alkaline phophatase (Promega) at 37 °C for 1 hour, followed by
inactivation of the alkaline phophatase at 75 °C for 10 minutes. The digested DNA
products were further purified using agarose gel electrophoresis and QIAquick Gel
Extraction Kit (QIAGEN). Bacteriophage T4 DNA ligase was used to ligate the digested
DNA fragments to the digested vectors. The ligation was performed at room temperature
for 3 ~ 4 hours or at 4 °C for overnight.
2.2.3 Agarose Gel Electrophoresis and DNA Purification
DNA fragments were separated by agarose gel electrophoresis in TAE buffer
(0.04M Tris-acetate, 0.001 M EDTA, pH 8.0) along with a standard DNA ladder
(Fermentas). Normally, the concentration of agarose gel is 1% and ethidium bromide (EB)
was directly added to the agarose gel to a final concentration of 0.5 µg/ml. The separated
DNA fragments were recovered from the agarose gel by using the QIAquick Gel
Extraction Kit (QIAGEN) according to the manufacturer’s instructions.
2.2.4 Preparation of E.coli Competent Cells
Highly efficient competent cells of E. coli DH5α and BL21 (DE3) strains were
prepared by the rubidium chloride method. Freshly growing E. coli colonies from LB

34
agar plates were inoculated into 100 ml of Psi broth medium (5g yeast extract, 20g
tryptone, 5g magnesium sulfate, pH7.6) in a 1-liter conical flask and then allowed to
grow at 37 °C with vigorous shaking (200 rpm). After the cells grew to an OD550 of
0.45~0.5, the culture was chilled on ice for 15 min and then the cells were collected by
centrifugation at 5,000 rpm (Beckman J2-21 ) for 5 min at 4 °C. The cell pellet was
resuspended in 40 ml of ice-cold TfbI buffer (30 mM potassium acetate, 100 mM
rubidium chloride, 10 mM CaCl2, 50 mM MgCl2, 15% v/v glycerol, pH 5.8) and
incubated on ice for 15 min. After centrifugation, the cell pellet was resuspended by 4 ml
TfbII buffer (10 mM MOPS, 10 mM rubidium chloride, 75 mM CaCl2, 15% v/v glycerol,
pH 6.5). The competent cells can be either used immediately or quickly frozen as 0.25 ~
0.5 ml aliquots in liquid nitrogen prior to storage in a -80 °C freezer.
(http://130.15.90.245/e__coli_competent_cells.htm)
2.2.5 Transformation of Bacterial Cells
In most operations, DNA was transferred into E. coli cells by heat-shock
following the standard protocol (Sambrook and Russell 2001). Frozen competent cells
(50 µl) were thawed on ice. The transforming DNA (up to 25 ng per 50 µl of competent
cells), including plasmid or ligation product, was added to the competent cell suspension
in a volume not exceeding 5 % of that of the competent cells. The competent cells were
mixed with the transforming DNA by swirling gently and then incubated on ice for 30
minutes. The mixture of cells and DNA were heat-shocked at 42 °C for 1 minute and
immediately chilled on ice for 2 min. Eight hundred microliters of fresh LB medium were

35
added to the transformed cells. The transformed cells were incubated at 37 °C for 45 min
with shaking to allow the bacteria to recover and to express the antibiotic resistance
marker encoded by the plasmid vector. The cells were collected by centrifugation and
spread onto LB agar plates containing the appropriate antibiotic. Colonies usually
appeared after overnight incubation at 37 °C.
2.2.6 DNA Sequencing
The sequencing PCR run mixture was 10 µl and contained: 2 µl BigDyeTM Ready
Mix (v 3.0), 0.5 µl primer, 2 µl template DNA (purified plasmids or PCR products,
100~200 ng µl-1) and 5.5 µl deionized water. The sequencing cycle was normally
performed using the following program: 1 cycle at 96 °C for 0.5 ~ 1 min and then 25
cycles each at 96 °C for 15 seconds, annealing at 50 °C for 15 seconds and extension at
60 °C for 4 min. The sequence product was kept at 4 °C until ready to purify. After the
cycle sequencing was finished, the contents in the PCR tube were spun down and then
transferred into a clear 1.5-ml tube. 80 µl of acetate-ethanol mixture (3 µl of 3M sodium
acetate, 62.5 µl 95% ethanol and 14.5 µl water) was added to the tube. The contents in
the tube were mixed by vortexing and the tube was allowed to stand at room temperature
for 15 minutes to precipitate the extension products. The pellet was collected by
centrifugation at 14,000 rpm (Eppendorf 5417C) for 20 min and then washed twice with
70% ethanol before centrifugation at 14,000 rpm (Eppendorf 5417C) for 10 min. After
the supernatant was carefully aspirated from the tube, the pellet was dried and stored at –
20 °C. This pellet is ready to be submitted for running electrophoresis.

36
2.3 Virus Treatment and Analysis
2.3.1 Virus Propagation and Purification
The WSSV isolate used in this study was originated from WSSV infected
Penaeus chinensis (Ningbo, China) and was propagated in an alternate host, red claw
crayfish, Cherax quadricarinatus. Preparation of WSSV virions was carried out based on
the method described previously (Huang et al. 2001) with some modifications. The
haemolymph collected from infected crayfish were diluted in PBS buffer and stored in -
80 oC as the stock. One tube of the stock haemolymph was thawed and after
centrifugation at 1000 × g for 10 minutes, the supernatant was filtered (0.45 µm filter)
and diluted 10 times in PBS buffer as the virus inoculum. Then we injected 0.3-0.4ml of
the virus inoculant was injected intramuscularly into the healthy red claw crayfish in the
lateral area of the fourth abdominal segment. After four to five days later, hemolymph
freshly extracted from moribund crayfish was first centrifuged at 2,000 × g to get rid of
the tissue debris. The supernatant was layered on the top of a 30 – 60% (w/v) stepwise
sucrose gradient and centrifuged at 20,000 rpm using a SW28 rotor in a Beckman
ultracentrifuge (XL-90; Beckman Coulter) for 2 hours at 4°C. Virus bands (Figure 2.1)
were collected and then diluted two or three times with TN buffer (20mM Tris-HCl and
400mM NaCl, pH7.4) before centrifugation again at 20,000 rpm using a SW28 rotor for 1
hour. The pellets were washed with TN buffer followed by a centrifuge at 12,000 × g for
30 minutes at 4 °C. The wash step was repeated to remove all the sucrose. The resulting
pellets were resuspended in TN buffer and the intact virion samples were stored at -80 oC.

37
Figure 2.1 Banding of purified WSSV Virus in the sucrose gradient
The stepwise sucrose gradient used was 30%, 40%, 50%, 60% (w/v) from the top. The
virus band showed in the interface between 40% and 50% gradient after
ultracentrifugation at 20,000rpm for 1 hour.
WSSV

38
2.3.2 Treatment of the Intact Virus with Detergent
The virus envelope was separated from the virus particles by treatment with Triton
X-100. Generally, the purified WSSV was mixed with an equal volume of 0.2% Triton
X-100 and incubated for 1 h at room temperature. The nucleocapsids were purified by
centrifugation at 20,000 × g for 20 min at 4 °C. The envelope fraction was collected in
the supernatant. The nucleocapsid fraction was subjected to a second round of Triton X-
100 extraction to ensure the envelopes were removed completely. The degree of purity of
the intact virions isolated and the nucleocapsid fractions were evaluated by negative-
staining transmission electron microscopy.
2.3.3 Preparation of WSSV Genomic DNA
WSSV genomic DNA from gradient-purified virions was extracted by protease K
and cetyltrimethylammonium bromide (CTAB) treatments, followed by phenol-
chloroform extraction and ethanol precipitation (Lo et al. 1996). Briefly, protease K and
N-laurysarcosine were added to the purified viral particles suspended in TNE buffer to a
final concentration of 0.5 mg/ml and 0.5%, respectively. After 1 hour incubation at 65 °C,
the solution was then treated with 1% CTAB for 15 minutes at 65 °C. This was followed
by successive extractions with an equal volume of phenol once, an equal volume of
phenol:chloroform:isoamyl alcohol (25:24:1) for 3 ~ 4 times, and then an equal volume
of chloroform:isoamyl alcohol once. Viral DNA was recovered by ethanol precipitation,
dried and dissolved in a small volume of 0.1 × TE buffer (50 mM Tris, 1 mM EDTA,

39
pH 8.0) containing RNAse A (10ug/ml) before quantified by spectrophotometry with the
Shimadzu spectrophotometer (Model UV-300).
2.3.4 Transmission Electron Microscopy
For transmission electron microscopy (TEM) examination, virus samples were
deposited onto formvar-coated, carbon-stabilized copper grids (200-mesh, Electron
Microscopy Sciences) before they were negatively stained with 2% (w/v) sodium
phosphotungastate (PTA, pH 7.0). The grids were dried and examined under electron
microscope (JEOL JEM 2010F or PHILIPS CM 10).
2.3.5 Localization Study by Immunoelectron Microscopy
WSSV intact virion and nuclocapsid suspension were mounted on carbon-coated
nickel grids (300 meshs, Electron Microscopy Sciences) and kept for 5 minutes before
blocking in 5% BSA (in 20 mM phosphate buffer) for 30 minutes at room temperature.
The first antibody (diluted with 5% BSA buffer, concentration of 40 mg/ml) was added
and kept at 4°C overnight. After washing at least three times for 10 minutes, the gold
labeled second antibody (goat-anti-mouse/rabbit IgG, 1:40 dilution, Electron Microscopy
Sciences) was added and incubated for 2 ~3 hour at room temperature. The grids were
washed by 5% BSA buffer twice and followed by water once. Negative stain by 2% PTA
was performed for 2 ~3 minutes and the grids were dried before examining under TEM.

40
2.4 Protein Manipulation
2.4.1 Protein Expression and Solubility Test
The constructs encoding WSSV envelope proteins were introduced into E. coli.
The bacterial cells were grown to a concentration of approximately OD600 = 0.5 ~ 0.6 at
37 °C. IPTG was added to the cultures in a final concentration of 0.1 mM to induce the
expression of fusion proteins. The bacterial cultures were allowed to grow for additional
5 ~6 hours at 25 °C or overnight at 18°C. All the subsequent operations were performed
at 4 °C. The cells from 500 ml of culture were harvested by centrifugation and
resuspended in 25 ml lysis buffer (20 mM Tris, 500 mM NaCl; 10 mM EDTA, 2mM
DTT, 100 µM PMSF, 5% Glycerol, pH 8.0). After treatment with chicken egg lysozyme
(final concentration of 1 mg/ml) for 30 minutes, the cell suspension was sonicated to near
clarity and centrifuged at 20,000 rpm for 1 hour using a J6-HC centrifuge (Beckman
Coulter). The supernatant and pellets sample were loaded into SDS-PAGE to determine
the solubility of the fusion proteins expressed.
2.4.2 Expression of Seleno-Methionine Substituted Protein
The selenomethionine substituted protein was expressed in defined LeMaster
medium using the methionine auxotrophic strain (DL41). The LeMaster medium amino
acid mixture for 1L medium were prepared as following: 0.5 g Alanin, 0.58 g
Arginine.HCL, 0.4 g Aspartic acid, 0.03 g Cystine, 0.67 g Glutamic acid, 0.33 g

41
Glutamine, 0.54 g Glycine, 0.06 g Histidine, 0.23 g Isoleucine, 0.23 g Leucine, 0.42 g
Lysine.HCL, 0.13 g Phenylalanine, 0.1 g Proline, 2.08 g Serine, 0.23 g Valine, 0.5 g
Adenine, 0.67 g Adenine, 0.67 g Guanosine, 0.17 g Thymine, 0.5 g Uracil, 1.5 g Sodium
acetate, 1.5 g Succinic acid, 0.75 g Ammonium chloride, 1.08 g Sodium hydroxide, 10.5
g Dibasic K2HPO4. This autoclaved mixture should be stored dry at -20°C.
Dissolve 24.1 g of the amino acid mixture to 1 Liter of deionized water and
autoclave for 20 minutes. Dissolve 10 g D-glucose, 0.25 g MgSO4.7H2O and 4.18 mg
FeSO4.7H2O in 90ml deionized water. 8.3 µl of concentrated H2SO4 and 10 ml Kao and
Michayluk vitamen solution (Sigma) were added and filter sterilize. All these are added
into the above autoclaved medium. Add 25 mg L-selenomethionine (Sigma) and 1 ml 0.1
g/ml amipicillin immdediately prior to inoculation. Inoculate the methionine auxotrophic
strain (DL41) containing the plasmid of interest in defined LeMaster medium and grow at
37 °C until the reading of OD600 is above 0.6. IPTG was then added into the culture
medium for the protein over expression.
2.4.3 GST fusion Protein Purification and Removal of GST tag
All the subsequent operations were performed at 4 °C. The soluble fusion proteins
were present in the supernatant of cell lysate from above steps. The supernatant was
filtered through 0.45 µm filter (Millpore) and loaded onto a Glutathione Sepharose 4B
column (Amersham Biosciences), followed by washing with at least 10 column volume
of lysis buffer. The protein was eluted with 20 mM reduced glutathione freshly prepared

42
in lysis buffer. The concentration of eluted GST fusion protein was determined with
Coomassie Protein Assay kit (Pierce) according to the manufacturer’s instructions.
ProScission™ protease (Amersham Biosciences) was added to the eluted protein solution
and kept at 4°C overnight for reaction. After digestion, the solution was loaded onto a
HiPrep Desalting column (Amersham Biosciences) with the desalting buffer (20 mM Tris,
100 mM NaCl, 2 mM DTT, pH8.0) to get rid of the reduced glutathione. After desalting,
the protein solution was passed through the Glutathione Sepharose 4B column to remove
the GST cleaved away from the fusion protein.
2.4.4 Purification of Untagged Protein
After removal of GST, the solution of untagged protein was further purified by
Superdex 75 size exclusion chromatography (Amersham Biosciences) using gel filtration
buffer. The purified protein fractions were collected and concentrated to a final
concentration of 8 mg/ml with a VIVASPIN 20 (Vivascience) concentrator. The purity of
the protein was examined by SDS-PAGE as well as by native gel electrophoresis.
2.4.5 Antibody Preparation
To obtain the polyclonal anti-rabbit antibody, New Zealand wild type female
rabbits were used in this study. A small amount (about 5 ml) of preimmune blood was
drawn from each rabbit prior to any injections as negative controls. 0.5 ml of purified

43
protein (0.1 mg) was mixed and emulsified with an equal volume of complete Freund’s
Adjuvant. The freshly prepared emulsion was immediately injected into one limb of the
rabbit. The first boost was done four weeks later on another limb. The dosage for each
booster was 50 µg, which was emulsified in 0.5 ml of Incomplete Freund’s Adjuvant
(IFA) emulsion and injected into another limb. After the first booster, the rabbit was
injected every week and the limbs were used in rotation during the subsequent boost
injections.
To obtain the polyclonal anti-mouse antibody, the purified protein was used to
immunize 3- to 4- week-old Swiss Albino mice once every 2 weeks by intradermal
injection over an 8-week period. Antigen (20 µg) was mixed with an equal volume of
Freund’s complete adjuvant (Sigma) for the first injection. Subsequent injections were
conducted using 20 µg of protein mixed with an equal volume of Freund’s incomplete
adjuvant (Sigma).
The blood collected from rabbit or mouse was allowed to clot at room
temperature for overnight. The antiserum was collected from the clot by centrifugation at
1,500 × g for 10 minutes at room temperature. Titers of the antiserum was determined by
ELISA (Harlow and Lane 1988). Protein A Sepharose CL-4B was used to isolate anti-
mouse antibody according to the manufacturer’s instructions (Amersham Biosciences).
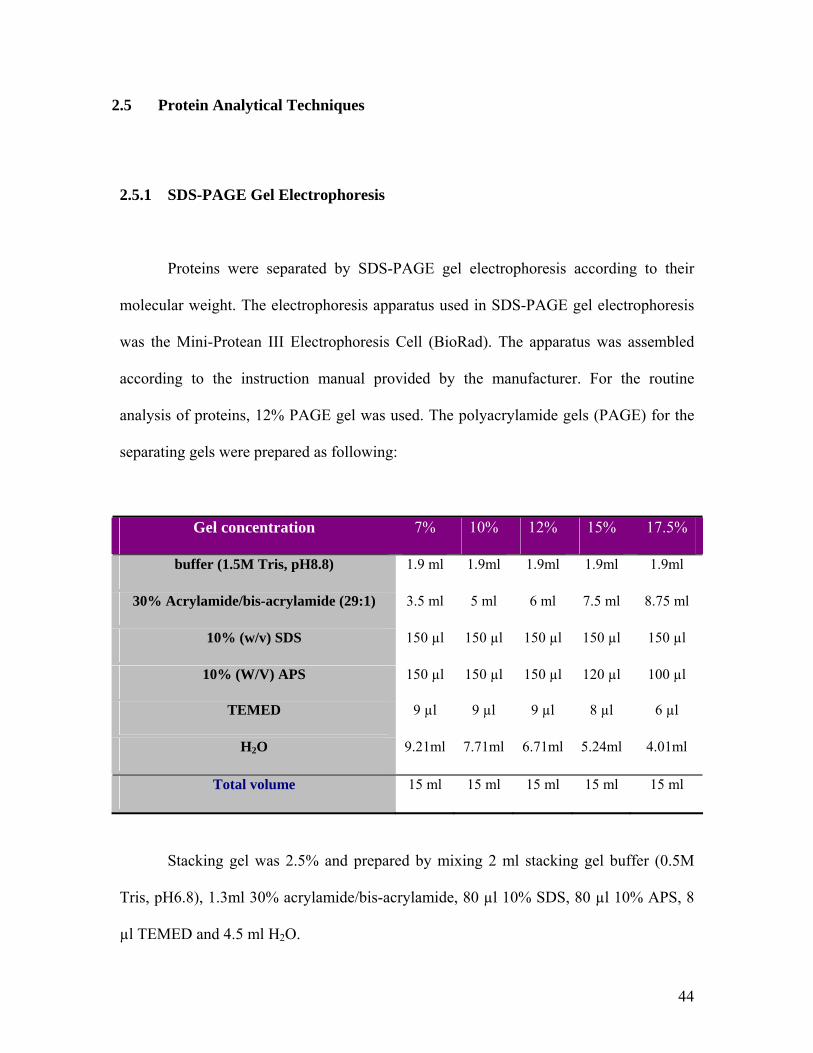
44
2.5 Protein Analytical Techniques
2.5.1 SDS-PAGE Gel Electrophoresis
Proteins were separated by SDS-PAGE gel electrophoresis according to their
molecular weight. The electrophoresis apparatus used in SDS-PAGE gel electrophoresis
was the Mini-Protean III Electrophoresis Cell (BioRad). The apparatus was assembled
according to the instruction manual provided by the manufacturer. For the routine
analysis of proteins, 12% PAGE gel was used. The polyacrylamide gels (PAGE) for the
separating gels were prepared as following:
Gel concentration 7% 10% 12% 15% 17.5%
buffer (1.5M Tris, pH8.8) 1.9 ml 1.9ml 1.9ml 1.9ml 1.9ml
30% Acrylamide/bis-acrylamide (29:1) 3.5 ml 5 ml 6 ml 7.5 ml 8.75 ml
10% (w/v) SDS 150 µl 150 µl 150 µl 150 µl 150 µl
10% (W/V) APS 150 µl 150 µl 150 µl 120 µl 100 µl
TEMED 9 µl 9 µl 9 µl 8 µl 6 µl
H2O 9.21ml 7.71ml 6.71ml 5.24ml 4.01ml
Total volume 15 ml 15 ml 15 ml 15 ml 15 ml
Stacking gel was 2.5% and prepared by mixing 2 ml stacking gel buffer (0.5M
Tris, pH6.8), 1.3ml 30% acrylamide/bis-acrylamide, 80 µl 10% SDS, 80 µl 10% APS, 8
µl TEMED and 4.5 ml H2O.

45
Before loading, proteins sample was mixed with 2 × loading buffer (100mM Tris
pH6.8, 200mM DTT, 4% SDS, 0.2% bromophenol blue, 20% Glycerol) and boiled for 3
~ 5 minutes to fully denature the protein and reduce disulfide bonds. The gel was run at a
constant voltage of 100 V until the bromophenol blue reached the bottom of the gel. After
SDS-PAGE, the gel was stained in Coomassie staining solution (45% methanol, 10%
acetic acid and 0.25% coomassie brilliant blue R-250) for 2 ~4 hours and then destained
with destain solution containing 5% methanol and 7.5% acetic acid. The destained gel
was washed in water to remove the organic solvents before drying.
2.5.2 Western blot Analysis
For Western blot analysis, proteins separated by SDS-PAGE gel electrophoresis
were transferred electrophoretically onto a nitrocellular membrane with Mini Trans-Blot
Cell (Bio-rad). The transfer buffer used in this study was Towbin buffer (3.03g/l Tris,
14.4 g/l Glycine, 20% V/V Methanol). Transfer was performed either at 75 mA for 2~3
hours or at 30 mA for overnight at 4°C. After the transfer was completed, the membrane
was removed from the sandwich and then incubated with 5% non-fat milk in TBST
buffer (20 mM Tris, 150 mM NaCl, 0.05% V/V Tween-20, pH 7.4) to block the
remaining protein-binding sites in the membrane. The blocking reaction was carried out
for 1 hours at room temperature on an orbital shaker. After blocking reaction was
completed, the membrane was washed at least three times with TBST buffer for ten
minutes each on a rocking platform. Then, the membrane was incubated with the TBST-
diluted primary antibody (usually use 100ug/ml as final concentration) for 1 hour at room

46
temperature while continuously rocking the sample. To remove the unbound primary
antibody, the membrane must be washed at least three times as aforementioned. After the
washing was completed, the membrane was incubated with the TBST-diluted second
antibody (usually in a 1:5000 dilution). The unbound second antibodies were washed
away by three extensive TBST washes. The fluorescence produced by the HRP
(horseradish peroxidase) -conjugated second antibody was detected with the SuperSignal
West Pico System (Pierce).
2.5.3 In-Gel Digestion, MALDI-TOF and Tandem MS Sequencing
Protein bands were carefully excised from the gel, cut into small pieces, and then
destained with several washes of 50 mM ammonium bicarbonate in 50% aqueous
acetonitrile. The gel pieces were dehydrated with 100% acetonitrile and dried in Savant
Speed-vac. The dried gel pieces were rehydrated with a solution containing 10 mM DTT
and 100 mM ammonium bicarbonate and incubated at 57°C for 60 min to reduce the
disulfide bonds. The reduced sulfhydryl group was alkylated by 55 mM iodoacetamide in
100 mM ammonium bicarbonate at room temperature for 60 minutes. After alkylation,
the gel pieces were first washed with 100 mM ammonium bicarbonate solution, then
dehydrated with 100% acetonitrile at least 6 times, and then dried in Savant Speed-vac
again. Sequencing grade modified trypsin (12.5 mg l-1
in 50 mM ammonium bicarbonate,
pH 8.0) was added to the dried gel pieces and incubated overnight at 37°C. The resulting
trypsinized peptides were extracted with 20 mM ammonium bicarbonate solution, 5%
formic acid in 50% aqueous acetonitrile, and 100% acetonitrile, respectively. The

47
extracted peptides were combined, lyophilized, and then resuspended in 0.1%
trifluoroacetic acid (TFA). An aliquot of the resuspended peptides was spotted onto the
stainless steel MALDI sample plate and overlaid with equal volume of matrix solution
(20 g l-1 α-cyano-4-hydroxycinnamic acid in 0.1% TFA; 50% aqueous acetonitrile). The
sample (matrix mixture) was allowed to air dry. An Applied Biosystems Voyager-DE
STR MALDI mass spectrometer was used to acquire the MALDI-time of flight (TOF)
spectra. The instrument was operated in the positive reflector delayed extraction mode.
Peptide mass spectra were searched against theoretically spectra derived from proteins in
the non-redundant databases of National Center for Biotechnology Information (NCBI) to
match peptide mass fingerprints. Partial amino acid sequence of a single peptide was
obtained by quadrupole-time of flight (Q-TOF) hybrid tandem mass (MS/MS)
spectrometer. For Q-TOF MS/MS analysis, the trypsinized peptide samples were
concentrated and desalted by the Zip-Tip method. Based on the mass spectra acquired by
MALDI-TOF, dominant peptides were selected for MS/MS analysis. After peptides were
selected, the Q-TOF mass spectrometer was automatically switched from MS to MS/MS
mode and the Q-TOF MS/MS spectra were collected. The amino acid sequences were
determined by searching the Q-TOF MS/MS spectra against the NCBI non-redundant
protein databases (Westermeier 2002).
.
2.5.4 N-Terminal Sequencing
Purified protein sample (1 ~ 2 mg/ml) was loaded into the ABI Procise 494
Protein Sequencer for N-terminal sequencing analysis. Principally, in this reaction

48
phenylisothiocyanate (PITC) reacts with the amino acid residue at the amino terminus
under basic conditions (provided by n-methylpiperidine/methanol/water) to form a
phenylthiocarbamyl derivative (PTC-protein). Trifluoroacetic acid was then used to
cleave off the first amino acid as its anilinothialinone derivative (ATZ-amino acid) and
leave the new amino terminus for the next degradation cycle. The ATZ amino acid is then
removed by extraction with N-butyl chloride and converted to a phenylthiohydantoin
derivative (PTH-amino acid) with 25% TFA/water. The PTH-amino acid is transferred to
a reverse-phase C-18 column for detection at 270 nm. This chromatogram provided
standard retention times of the amino acids for comparison with each Edman degradation
cycle chromatogram. The HPLC chromatograms were collected using a computer data
analysis system. To determine the amino acid present at a particular residue number, the
chromatogram from the residues of interest was compared with the chromatogram from
the previous residue by overlaying one on top of the other. From this, the amino acid for
the particular residue can be determined. This process was repeated sequentially to
provide the N-terminal sequence of the protein.
(http://www.protein.iastate.edu/nsequence494.html)
2.5.5 Circular Dichroism (CD) Spectrum
Circular Dichroism (CD) spectroscopy is a type of absorption spectroscopy that
can provide information on the secondary & tertiary structures of many types of
biological macromolecules. Circular Dichroism is the difference between the absorption
of left and right handed circularly-polarised light and is scanned as a function of

49
wavelength. Biological macromolecules, such as proteins, carbohydrates and nucleic
acids, are composed of many optically active or chiral units that exhibit large CD signals.
CD spectroscopy has therefore been used extensively in the study of proteins, where
asymmetric carbon atoms in their amino-acid 'backbone' give rise to a CD spectrum. The
shape of the spectrum depends on the protein's secondary structure content and allows the
proportions of helix, beta structure, turns and random coil to be determined.
Purified protein was diluted in phosphate buffer to final concentration of 100
µg/ml. 200 µl protein sample was loaded and use water as blank control.
(http://www.cryst.bbk.ac.uk/BBS/whatis/cd_website.html)
2.5.6 Dynamic Light Scattering (DLS)
The dynamic light scattering (DLS) is a well established technique for measuring
particle size over the size range from a few nanometers to a few microns. The technique
uses the idea that small particles in a suspension move in a random pattern. When a beam
of light passes through a colloidal dispersion, the particles or droplets scatter some of the

50
light in all directions. When the particles are very small compared with the wavelength of
the light, the intensity of the scattered light is uniform in all directions; for larger particles
(above approximately 250 nm diameter), the intensity is angle dependent. If the light is
coherent and monochromatic, as from a laser for example, it is possible to observe time-
dependent fluctuations in the scattered intensity using a suitable detector such as a
photomultiplier capable of operating in photon counting mode.
(http://www.brookhaven.co.uk/dynamic-light-scattering.html). Observation of larger
particles compared to smaller particles will show that the larger particles tumble more
slowly than the smaller ones if the temperature is the same. According to Einstein's
developments in his Kinetic Molecular Theory (applied to heat), molecules that are much
smaller than the particles can impart a change to the direction of the particle and its
velocity. Thus water molecules (0.00033 microns) can move polystyrene particles as
large as a couple of microns. The combination of these effects is observed as vibrations
and an overall movement of the particle. Concentrated and purified protein prior to
crystallization was tested for homogeneity using the Dynamic Light Scattering (DynaPro,
Protein Solutions). A few minutes are required for the sample and cell to equilibrate with
the actively controlled temperature environment. The cuvette must be washed with sterile
water until the counts are below 10. Then 50 µl concentrated sample was loaded into the
cuvette and any bubbles should be avoided. At least 15 times of counting should be
collected. (http://www.microtrac.com/dynamicscattering.cfm)

51
2.6 Crystallization
Crystallization screens were performed using the hanging-drop method with
Hampton 24-well plastic plates at 20 °C. The initial screens were carried out using
Hampton crystallization screen kits (crystal screen 1&2, index). A drop composed of a
mixture of 1 µl protein solution and 1µl reservoir solution is placed in vapor equilibration
with a liquid reservoir of regent. The initial conditions were optimized to get diffraction-
quality crystals. The crystals were soaked into the cryoprotectant buffer before flash-
frozen in liquid nitrogen for data collection.
2.7 Data Collection
X-ray diffraction data were collected at 100°K using synchrotron radiation. A
three wavelength experimental data set for the rVP28 crystals was collected at beamline
X29, NSLS, USA while that of rVP26 crystals was collected at beamline X26C, NSLS,
USA. Data for rVP26 and rVP28 were processed and scaled using the HKL suite
(Otwinowski and Minor 1997).
2.8 Structure Determination
The structure of rVP28 and rVP26 were determined using the MAD method. The
position of the seleno-methionine were determined and phases computed with SOLVE
(Terwilliger and Berendzen 1999). Density modification was effected by using

52
RESOLVE (Terwilliger 2000). The resulting electron density map was not good enough
and the model was built manually by using the program O (Jones et al. 1991). The initial
model was refined by rigid body, individual B-factor and energy minimization refinement
using CNS (Brunger et al. 1998). For rVP28, the restrained non-crystallographic
symmetry (NCS) was applied to increase the observation to parameter ratio throughout
the CNS refinement. Also, the test set (Rfree) was introduced into the refinement to
monitor and validate the model refinement. At each round of refinement, the geometry of
the structure was checked with PROCHECK using the Ramachandran plot (Laskowski et
al. 1993). Water molecules are added in the final stage of refinement. Structural
superposition was performed with the program O (Jones et al. 1991).

53
CHAPTER THREE
3 Characterization of WSSV Envelope Proteins

54
3.1 Characterization of Envelope Protein VP300
3.1.1 Identification of VP300 by Mass Spectrometry
Purified WSSV virions were separated by SDS-PAGE (Figure 3.1). More than 30
bands were visualized using Coomassie brilliant blue R-250 staining. Many major bands
have been identified previously (such as VP19, VP24, VP26 and VP28 as envelope
proteins and VP15 as nucleocapsid) however many minor bands are still undefined. The
band highlighted by red arrow was excised and digested by trypsin and analyzed by
MALDI-TOF mass spectrometry. Search the WSSV ORF database with the masses of a
list of trypsin-treated peptides has identified a protein as the product of the vp300 gene.
The ORF is located between nucleotide position 132994-133893 on the genome (Yang et
al. 2001). This 900 bp ORF presumably encoded a 300 aa protein (hence termed the
vp300 gene, GenBank accession number AF403003, and the resulting protein termed
VP300). One more interesting finding from the mass spectrometry result is that VP292 is
also identified by MALDI and Q-TOF from the same band. The mass spectrometry result
is consistent with previous proteomic research (Tsai et al. 2004; Zhang et al. 2004). It
suggests that VP300 might be a WSSV virion-associated protein.
3.1.2 Expression and Purification of VP300
The VP300 gene was amplified by PCR from the WSSV genome and inserted
into the vector pET-32(a) to express the (His)6-VP300. The recombinant protein was

55
Figure 3.1 1D SDS-PAGE of the WSSV proteins Viral proteins were purified and separated via one-dimensional SDS-PAGE. Left lane is the protein marker and the right lane is WSSV proteins. Four major structural proteins (VP19, VP24, VP26 and VP28) are indicated by black arrow. The band highlighted by red arrow was excised and digested by trypsin and then analyzed by MALDI-TOF mass spectrometry. The search of WSSV ORF database with a list of trypsin-treated peptide masses identified one of the proteins as the product of the vp300 gene. .
25
15
37
50
75
200150100
kDa
VP300
VP28 VP26 VP24
VP19

56
expressed in the BL21 (DE3) cell and purified by Ni-NTA under denaturing condition.
Figure 3.2 shows that SDS-PAGE gel of the purified VP300 protein. The purified protein
was injected into mouse to raise the mouse anti-(His)6-VP300 antibody as mentioned in
Chapter Two.
3.1.3 Localization Study of VP300
3.1.3.1 Western Analysis of VP300
To test specificity of the anti-VP300 antibody, western blot analysis was
performed. From Figure 3.3A, the purified mouse anti-(His)6-VP300 IgGs was used and
it can detect the VP300 protein among a mixture of all viral proteins as well as the
positive control of purified (His)6-VP300 protein. As a negative control, the antibody did
not react with purified VP281, another envelope protein of WSSV. It showed that the
purified mouse anti-(His)6-VP300 IgGs reacted specifically with VP300. Also it proved
that VP300 was of the WSSV origin rather than a contaminating protein linked
superficially to the WSSV virion. To further identify the location of VP300 in the virion,
the virus sample was fractionated into envelope and nucleocapsid fractions by the
treatment with 2% Triton X-100. As in Figure 3.3B, the western blot result shows that
VP300 was clearly detected in the WSSV whole virion and the purified viral envelope
fraction. However, the anti-VP300 antibodies did not react with the proteins of WSSV
nucleocapsid. This indicates that VP300 belongs to viral envelope, instead of
nucleocapsid.

57
Figure 3.2 Expression of the purified (His)6-VP300 protein
Shown are Coomassie Brilliant Blue –stained 12% SDS-PAGE gels. Lane 1, protein marker; Lane 2, recombinant pET-32(a) plasmid containing vp300 gene, non-induced; Lane 3, recombinant pET-32(a) plasmid containing vp300 gene, induced; Lane 4, purified (His)6-VP300 protein by Ni-NTA resin.
14.4
21.5
31.0
45.0
66.0
200.0
kDa
1 2 3 4
(His)6-VP300

58
Figure 3.3 Western blot analysis of WSSV with anti-VP300 IgG
Figure 3.3A is Coomassie Brilliant Blue-stained SDS-PAGE gels and the corresponding western blot analysis with anti-VP300 IgG. Lane 1, purified (His)6-VP300 protein as positive control; Lane 2, purified VP281 protein as negative control; Lane 3, protein marker; Lane 4, WSSV virion. Figure 3.3B is the western blot analysis of the different fractions of WSSV with anti-VP300 IgG. Lane 1, WSSV whole virion; Lane 2, WSSV viral envelope; Lane 3, WSSV nucleocapsid.
VP300
Western blot with anti-VP300 antibody Coomassie Brilliant Blue-stained gel
1 2 3 4 1 2 3 4
A
B
1 2 3

59
3.1.3.2 TEM Immunogold-Labeling Analysis
TEM immunogold-labeling method was further used to confirm the location of
VP300. The results shown in Figure 3.4 demonstrated that high-density gold particles
were located specifically at the viral envelope, while no apparent signals could be
observed in the nucleocapsid. The mouse anti-(His)6 antibody was also used as control to
replace the anti-(His)6-VP300 IgGs but no gold labeling signals were observed in the
viral envelope. From the Western blotting assay and TEM observations, it is concluded
that VP300 is a WSSV envelope protein.
3.1.4 Neutralization Assay of VP300
Virus envelope proteins play important roles in viral infection, such as recognition
and attachment to receptors on the surface of host cell. So being an envelope protein, is
VP300 involved in viral entry during infection? In an attempt to identify the envelope
protein’s involvement in WSSV infection on shrimp, the neutralization assay of infection
by WSSV virion with antibodies against VP300 was performed. First, the suitable dosage
of WSSV for shrimp infection was determined by performing an in vivo virus titration.
As expected, the mortality varied among groups of shrimp injected with different amount
of viruses. Delay in shrimp mortality was observed with the serial dilution of virus. When
the WSSV virion was diluted in PBS buffer with a ratio of 1:106, a mortality of 100%
was observed in a period of 11 days (Figure 3.5B). Based on the in vivo virus titration,
the WSSV virion concentration of 106 dilutions was selected for later experiments

60
Figure 3.4 Localization of WSSV VP300 proteins by immunoelectron microscopy
A, gold particles localize in virus envelope (arrow indicated); B, No gold particles was found in the nucleocapsid. The primary antibody is the purified mouse anti-(His)6-VP300 IgG, and the second antibody is goat anti-mouse IgG conjugated with 15 nm of gold.
A
B

61
Neutralization
0
0.2
0.4
0.6
0.8
1
1.2
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
day
mor
talit
y 0.9%NaClWSSV+Anti-GSTWSSV(1:1000000)WSSV + Anti-VP300
0.00%
20.00%
40.00%
60.00%
80.00%
100.00%
120.00%
1 2 3 4 5 6 7 8 9 10 11
1 / 1001 /10001 / 100001 /1000001 /1000000
Figure 3.5 Neutralization Analysis of WSSV infections
A, Neutralization of WSSV infection in crayfish by antibody against VP300. B, Dosage of WSSV for shrimp infection. Days post-infection were shown on the abscissa and accumulated mortality on the ordinate.
B
A

62
because it is expected to give the optimal response to the neutralization in terms of
mortality. After we selected the suitable dosage of WSSV, we performed the
neutralization assay by adding specific antibodies. The antibodies against VP300 and
WSSV virion, as well as the anti-GST antibodies as control, were used in the in vivo
neutralization assays. After incubation of WSSV with each antibody, the mixtures were
injected into shrimp. The results are shown in Figure 3.5A. It showed that the shrimp
mortalities were very low, almost zero for the negative control (0.9% NaCl), whereas the
shrimp from the positive control (WSSV only) displayed 100% mortality in 15 days post-
infection. However, when the mixture of WSSV and anti-VP300 IgGs was injected, the
shrimp mortality fell significantly to almost zero, indicating that the infection of WSSV
could be delayed or neutralized by anti-VP300 IgGs.
3.1.5 Implications
Proteomic study by mass spectrometry had identified the existence of VP300
protein in the WSSV virion. From the SDS gel, the observed molecular mass of VP300 is
around 38 KDa, which is greater than the theoretical molecular mass (34.4 KDa) of the
vp300 gene product. This difference indicates that some posttranslational modifications
might have occurred. Computer assisted analysis revealed that there are three possible
phoshporylation sites and one potential N-linked glycosylation site in VP300.
Furthermore, VP292, an envelope protein that previously confirmed by Huang et al
(2002), was also detected by mass spectrometry from the same SDS PAGE band. When
we compared the protein sequence of VP300 and VP292, there is no significant

63
homology. Since the functional characterization of VP292 is still in progress, we cannot
confirm any functional relationships between these two proteins.
Besides the mass spectrometry results, the observations from Western blot and
immuno-gold labeling TEM further confirmed that VP300 is a WSSV envelope protein
but not a nucleocapsid protein. Similar to many known membrane proteins, VP300 has a
transmembrane region predicted by sequence analysis using the DAS server (Stockholm
University). It suggests that this hydrophobic region was responsible for insertion into
membrane lipid bilayer and VP300 may be a membrane anchor protein.
To date, several envelope proteins have been identified (VP19, VP31, VP24,
VP26, VP28, VP124, VP281, VP292, VP466). Previous neutralization study of six
WSSV envelope proteins indicates that some of the envelope proteins had function on
viral infection while some do not. In this study, neutralization assay showed that the anti-
VP300 IgGs functions to delay or neutralize the infection of shrimp by WSSV. The
antibody against VP300 could bind to envelope spikes on the virion and prevent the
attachment of the virus to the cell surface or uncoating of the virus (Burton et al. 2000;
Wu et al. 2005). VP300 might be a key protein in viral invasion and could be a good
candidate for screening of receptors on the cell surface. Further study on VP300 would
help to understand the mechanism of the virus infection. Up until now, no efficient
method has been found to prevent the infection of shrimp by WSSV. Antibody against
VP300 could be used directly as a passive immune strategy to control the WSSV
infection.

64
3.2 Localization Study of VP26 in WSSV virion
3.2.1 Immunogold labeling of VP26 in WSSV particles
(This part of experiments was done by Dr. Jinlu Wu)
Compared with the confirmed envelope protein of VP28, the location of VP26 in
the virion is still unclear. VP26 was first reported as a nucleocapsid protein (van Hulten
et al. 2001b) and later reported as an envelope protein (Zhang et al. 2002b). However, the
most recent publications claimed that VP26 is a linker (Xie and Yang 2005) or a
tegument protein (Tsai et al. 2006). Clarification of the location of VP26 should improve
our understanding on function of the protein.
To study the location of VP26 in the virion, the virus was treated with Tween 20
to get partial and complete separation of viral envelope from the nucleocapsid so that we
can distinguish the location of gold particles. Figure3.6 shows that the detergent
treatment separated the envelope from the nucleocapsid. Interestingly, some completely
separated envelope formed vesicles of different sizes and shapes. The treatment also
resulted in visible surface projections and spike-like structures, which were not observed
on the envelope of an intact virion. Figure 3.6A-3.6D show immunogold-labeled VP26,
and Figure 3.6E show immunogold-labeled VP28. All gold particles were found to bind
at the outer surface of the envelope, while none bind to the inner surface of envelope,
nucleocapsid or the space between them. Some gold particles were clearly localized on
the spike-like structures when using anti-VP26 for immunogold labeling (insert in Figure
3.6D). The negative control (Figure 3.6F) shows very low level of non-specific binding
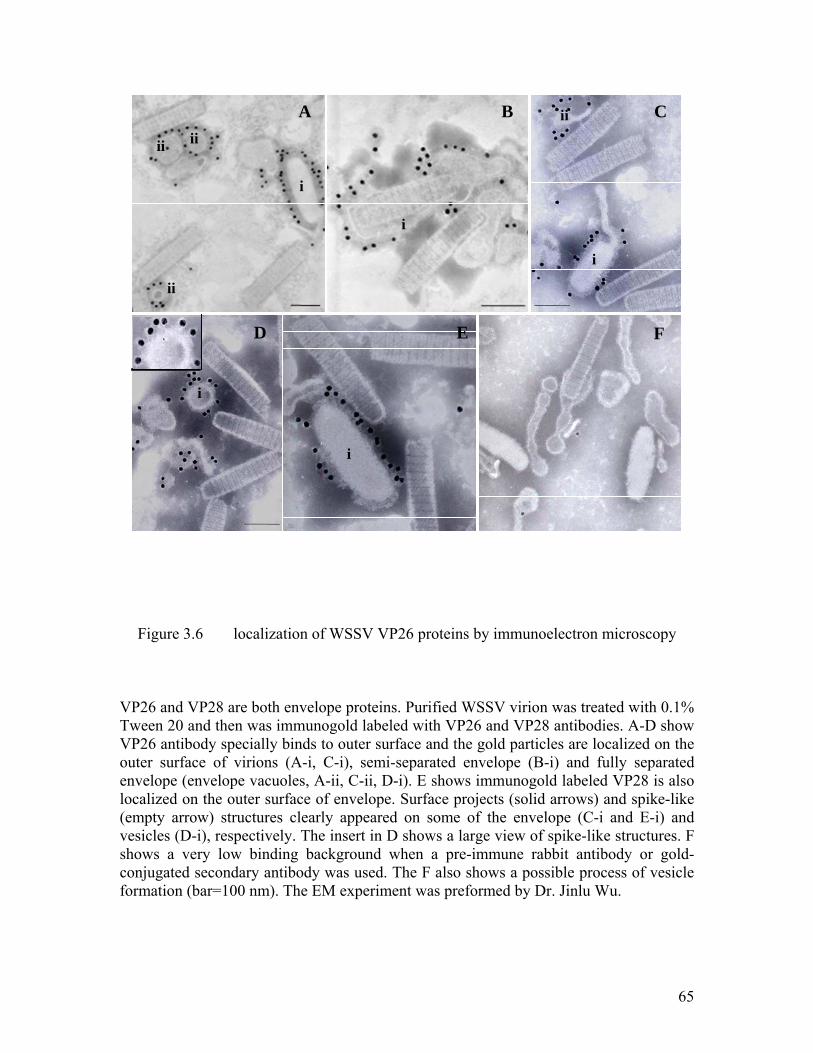
65
A CB
i
iiii
ii
i
i
D E
i
F
ii
i
A CB
i
iiii
ii
i
i
D E
i
F
ii
i
Figure 3.6 localization of WSSV VP26 proteins by immunoelectron microscopy
VP26 and VP28 are both envelope proteins. Purified WSSV virion was treated with 0.1% Tween 20 and then was immunogold labeled with VP26 and VP28 antibodies. A-D show VP26 antibody specially binds to outer surface and the gold particles are localized on the outer surface of virions (A-i, C-i), semi-separated envelope (B-i) and fully separated envelope (envelope vacuoles, A-ii, C-ii, D-i). E shows immunogold labeled VP28 is also localized on the outer surface of envelope. Surface projects (solid arrows) and spike-like (empty arrow) structures clearly appeared on some of the envelope (C-i and E-i) and vesicles (D-i), respectively. The insert in D shows a large view of spike-like structures. F shows a very low binding background when a pre-immune rabbit antibody or gold-conjugated secondary antibody was used. The F also shows a possible process of vesicle formation (bar=100 nm). The EM experiment was preformed by Dr. Jinlu Wu.

66
and a possible mechanism for the formation of vesicles. On the other hand, if the virion
was over treated with Tween 20, VP26 can only be scarcely labeled. We also tried to treat
the virion with 0.1% Triton X-100, but such treatment always resulted in complete loss of
viral envelope, only nucleocapsid remained after the subsequent immunogold labeling
was finished.
3.2.2 Implications
Due to the dispute over the location of VP26, we have carried out an examination
on VP26 by immunoelectron microscopy in this study. In this study we chose to use
Triton X-100 and Tween 20 to treat the virion to expose antibody binding sites and to
separate the envelope from the nucleocapsid. Our result showed that 0.1% Tween 20
could effectively separate the envelope from the nucleocapsid, but the treatment with
0.1% Triton X-100 was too harsh and resulted in complete loss of envelope during
subsequent steps of immunogold labeling. More interestingly, surface projections and
spike-like structures could be observed in the outer surface of the envelope after
treatment with Tween 20. It suggests that the envelope structure became loosely packed,
thus more antibody binding sites for both VP26 and VP28 were exposed resulting in
higher labeling efficiency. Even though, no gold particles were observed in the space
between the inner surfaces of envelope or the nucleocapsid, all the gold particles were
localized on the outer surface of the semi or fully separated envelope. Taken together, we
proposed that VP26 itself is an envelope protein and is anchored at the viral lipid bilayer
by its N-terminal transmembrane region.

67
3.3 Candidates for Structure Analysis
3.3.1 Solubility Test of Major Envelope Proteins
Although many structural genomic programs have used different criteria or
priorities for the selection of target proteins (Brenner 2001), the common theme is to
restrict the selection to candidates to proteins of unknown structure, expected interest and
accessibility (yields, solubility and ease of labeling etc). In the present proposal, we have
focused on the envelope proteins what are important for viral infection, assembly and
morphogenesis. These candidates were expressed in a bacterial system. The yield and
solubility of these proteins were examined and protocols will be developed for protein
purifications and characterization. To express and purify all these envelope proteins, the
encoding DNA fragment was cloned into different kinds of vectors, including pET15b,
pET32(a), pGEX-4T-2 and pGEX-6P-1 and then transformed into the BL21 (DE3) cell
for expression as mentioned in Chapter Two. All envelope proteins, except for VP281,
are predicted to contain a transmembrane domain. So the first step is to check the
solubility of these full length proteins. Initial trials were performed by expression using
0.1 mM IPTG at 30 °C for 4 hours. The expression condition was optimized by changing
the final concentration of IPTG and culture temperature. As shown in Figure 3.7, when
expressed as GST fusion proteins, VP281 and VP466 were partially soluble when the
cells with the recombinant plasmid were cultured at the optimal condition. But for VP19,
VP24, VP28 and VP68, they were all expressed as insoluble proteins. VP281 and VP466
were thus first selected as candidates for X-ray crystallography study because of their
higher solubilities.
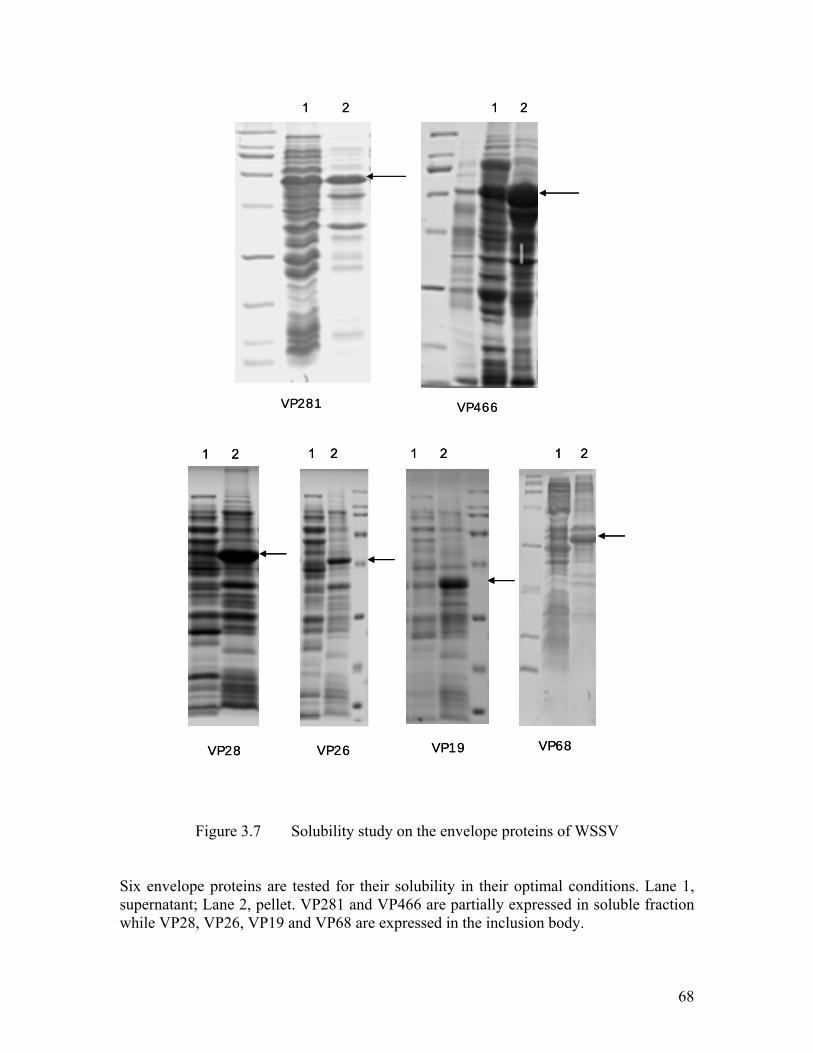
68
VP281 VP466
1 2 1 2
VP281 VP466
1 2 1 2
VP28 VP26 VP19 VP68
1 2 1 21 21 2
VP28 VP26 VP19 VP68
1 2 1 21 21 2
Figure 3.7 Solubility study on the envelope proteins of WSSV
Six envelope proteins are tested for their solubility in their optimal conditions. Lane 1, supernatant; Lane 2, pellet. VP281 and VP466 are partially expressed in soluble fraction while VP28, VP26, VP19 and VP68 are expressed in the inclusion body.

69
3.3.2 Purification of VP281 Protein and Crystallization Trials
The solubility test showed that VP281 and VP466 were expressed as soluble
proteins in a bacterial system. Thus we performed the large scale expression and
purification of these two proteins. The vp281 gene was cloned into pGEX-6P-1 using the
restriction enzyme sites, BamHI and EcoRI. The VP281-GST fusion protein was first
purified using glutathione sepharose and then the GST tag was removed by the cleavage
using ProScission protease as mentioned in Chapter Two. The expressed protein is 286 aa
in size with predicted MW of 31970.80 Da and pI of 4.47. There are 4 cysteine residues
in the VP281 sequence indicating that disulfide bonds might form inside VP281 to
stabilize its fold or/and to form linkage between two VP281 molecules.
The SDS-PAGE gel of purified VP281 is shown in Figure 3.8. Further
purification of VP281 was performed by ion exchange, hydrophobic interaction
chromatography (HIC) and size exclusion chromatography. MALDI-TOF MS and Q-
TOF MS as well as N-terminal sequencing were carried out to confirm the identity of the
purified protein. MALDI-TOF MS result showed that the amino acid sequence coverage
is about 42% (120 of 281). And the Q-TOF MS result showed that the MW of VP281 is
31973.80 Da, almost the same as the theoretical MW of 31970.80 Da. The N-terminal
sequence of 12 amino acids also confirmed identity of the purified VP281 protein. A CD
spectrum was also taken to study the secondary structure of this protein and from the
result we interpreted that VP281 consisted mainly of α-helical secondary structure.

70
GST-VP281
VP281
GST
1 2 3 4 5 6 7 8
14.4
21.5
31.0
45.0
66.0
200.0
kDa
GST-VP281
VP281
GST
1 2 3 4 5 6 7 8
14.4
21.5
31.0
45.0
66.0
200.0
kDa
14.4
21.5
31.0
45.0
66.0
200.0
kDa
Figure 3.8 Expression of the purified VP281 protein
Shown is Coomassie Brilliant Blue –stained 12% SDS-PAGE gel. Lane 1, protein marker; Lane 2, pGEX-6P-1 (vector only as control)-pLysS, non-induced; Lane 3, pGEX-6P-1-pLysS, induced; Lane 4, recombinant pGEX-6P-1 plasmid containing vp281 gene, non-induced; Lane 5, recombinant pGEX-6P-1 plasmid containing vp281 gene, induced; Lane 6, purified GST-VP281 protein; Lane 7, GST-VP281 fusion protein cleavage with ProScission™ protease; Lane 8, purified VP281 protein.

71
Homogeneity of the purified protein is the key to grow a well-diffracting crystal.
Single band purity on native PAGE is neither a sufficient nor a necessary prerequisite to
obtaining crystals but has always been regarded as a good starting point for crystal
growth. The native gel and SDS-gel together will give some hints about the extent of
protein aggregation. The purified VP281 protein was concentrated to 10 mg/ml and ran in
native gel. The gel filtration profile showed that the protein was eluted as a dimeric but
when running in the native gel we could clearly see several bands, indicating that there
was significant aggregation happened in VP281 protein. The most frequent cause of non-
specific aggregation is suboptimal protein solvent conditions. We have tried to change
the buffer condition, such as the concentrations of NaCl, glycerol and the reducing agent,
but to no avoid. However, there was no apparent difference of all in the variations in the
native gel. It is impossible to obtain any diffraction-quality crystals from this poor quality
protein. Further purification steps or truncation treatments are required for this protein for
crystallization.
3.3.3 Purification of VP466 Protein and Crystallization Trials
The vp466 gene was cloned into pGEX-4T-1 between the restriction enzyme sites,
BamHI and XhoI. VP466 was first expressed as a GST fusion protein using Glutathione
sepharose 4B beads and then the GST tag was removed by thrombin protease cleavage.
The SDS-PAGE gel of purified VP466 is shown in Figure 3.9. The corresponding bands
were excised from the gel, digested by trypsin and then portion of tryptic peptide
fragments was studied by MALDI-TOF MS. The fitted amino acids residues covered

72
14.4
21.5
31.0
45.0
66.0
200.0
kDa
GST-VP466
VP466
GST
1 2 3 4 5 6 7 8
14.4
21.5
31.0
45.0
66.0
200.0
kDa
14.4
21.5
31.0
45.0
66.0
200.0
kDa
GST-VP466
VP466
GST
1 2 3 4 5 6 7 8
Figure 3.9 Expression of the purified VP466 protein
Shown is Coomassie Brilliant Blue –stained 12% SDS-PAGE gel. Lane 1, protein marker; Lane 2, pGEX-4T-1 (vector only as control)-pLysS, non-induced; Lane 3, pGEX-4T-1-pLysS, induced; Lane 4, recombinant pGEX-4T-1 plasmid containing vp466 gene, non-induced; Lane 5, recombinant pGEX-4T-1 plasmid containing vp466 gene, induced; Lane 6, purified GST-VP466 protein; Lane 7, GST-VP466 fusion protein cleavage with thrombin protease; Lane 8, purified VP466 protein.

73
42% of the whole sequence (198 of 466). And the Q-TOF MS result showed that the MW
of VP466 was 52053.20 Da, slightly larger than the predicted M.W. of 52052.30 Da, but
the difference was within the allowable error range. Furthermore, the N-terminal
sequence of 14 amino acids also confirmed the identity of the purified VP466 protein. A
CD spectrum showed that VP466 has a typical α-helical secondary structure.
The single band of purified VP466 protein in native gel indicated that the protein
was homogenous. However we were unable to get any crystals from the crystallization
screening. More crystallization trials on purified VP466 will be performed under different
conditions. Different crystal growth techniques like sitting drop vapor diffusion or under
oil will also be attempted.

74
CHAPTER FOUR
4 Crystal Study of Two Major Envelope Proteins

75
4.1 X-ray Structure of Major Envelope Protein VP28
4.1.1 Property of rVP28 Protein
VP28 and VP26 are two most abundant envelope proteins that consist 60% of the
envelope. Previous experiments have shown VP28 play important role in WSSV
infection and VP26 was able to interact with actin. As their importance and abundance in
viral envelope, we selected these two proteins for the structure determination. As we
mentioned before that the expression and purification trials on full-length VP28 and
VP26 resulted in a very low level of expression and the production of an insoluble
protein. Extensive detergent screen to solubilize and crystallize the protein was
unsuccessful. Sequence analysis using DAS server (Stockholm University) indicated the
presence of a transmembrane region at the N-terminal region of VP28 and VP26 (Figure
4.1). We hypothesized that removal of the transmembrane region would increase the
solubility and the yield of crystals. Thus we designed a construct encoding the major part
(Asn31-Glu204) of VP28 without the N-terminal transmembrane domain (Met01-His30).
Theoretical pI and MW of rVP28, the truncated protein, is 4.63 and 19258.47 Da
respectively. The SDS-PAGE electrophoresis is shown in Figure 4.2. The homogeneity of
purified rVP28 at different concentration was tested by native gel and dynamic light
scattering experiment respectively. The gel filtration profile as well as the dynamic light
scattering data showed that rVP28 is a monomer in solution. rVP28 at a concentration of
8-10 mg/ml was homogenous and suitable for crystallization trials.
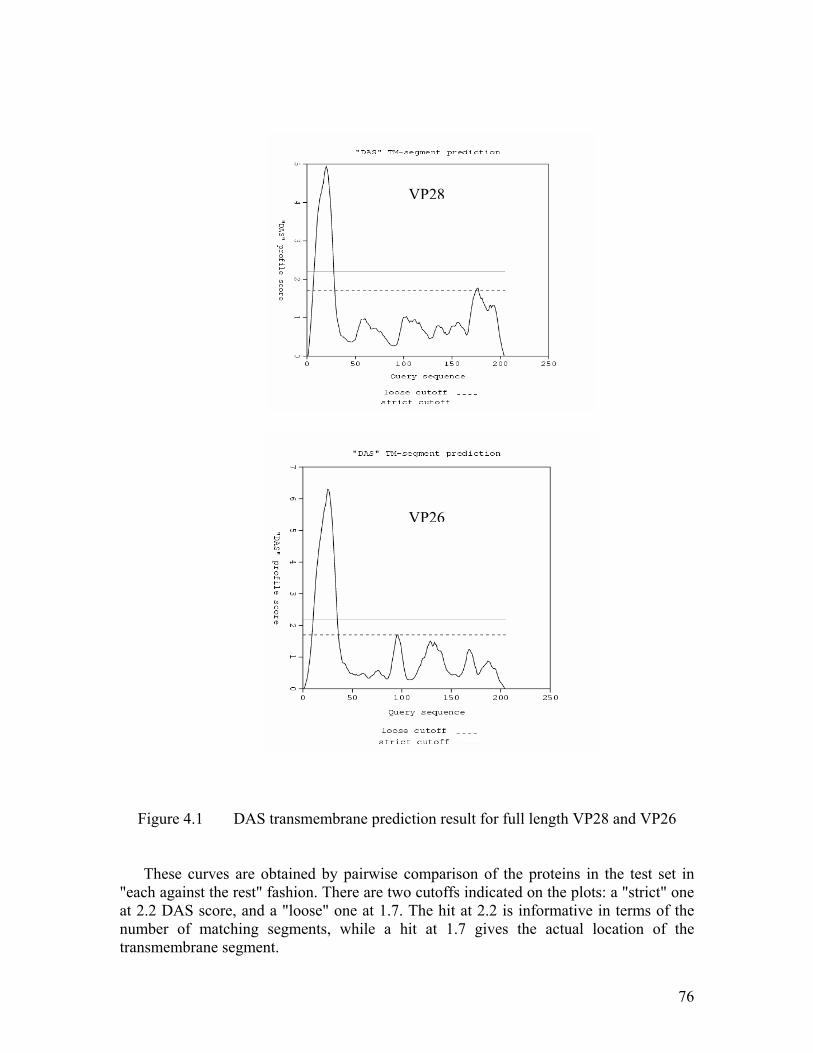
76
Figure 4.1 DAS transmembrane prediction result for full length VP28 and VP26
These curves are obtained by pairwise comparison of the proteins in the test set in
"each against the rest" fashion. There are two cutoffs indicated on the plots: a "strict" one at 2.2 DAS score, and a "loose" one at 1.7. The hit at 2.2 is informative in terms of the number of matching segments, while a hit at 1.7 gives the actual location of the transmembrane segment.
VP28
VP26

77
GST-rVP28
GST
rVP28
1 2 3 4 5 6
15
20
25
37
50
75100
Figure 4.2 Expression of the rVP28 protein in E.coli BL21 DE3 star
Shown is Coomassie Brilliant Blue –stained 12% SDS-PAGE gel Lane 1, protein marker; Lane 2, recombinant pGEX-6P-1 plasmid containing vp28 gene, non-induced; Lane 3, recombinant pGEX-6P-1 plasmid containing vp28 gene, induced; Lane 4, purified GST-rVP28 protein; Lane 5, GST-rVP28 fusion protein cleavage with ProScission™ protease; Lane 6, purified rVP28 protein.
KDa

78
4.1.2 Preparation of SeMet rVP28 Protein
As VP28 lacks homology with any other known structure of viral proteins, the
MAD method was used to solve the structure of rVP28. The seleno-methionine rVP28
was expressed in defined LeMaster medium using the methionine auxotrophic strain
(DL41). The purification procedure was the same as that for the native protein. The
SeMet rVP28 was purified by Superdex-75 gel filtration chromatography using a gel
filtration buffer containing 20 mM Tris-Cl (pH 7.4), 150 mM NaCl, 10 mM DTT and 5%
Glycerol. The purified SeMet rVP28 was concentrated to around 10 mg/ml for
crystallization. To confirm the substitution status, the MW of seleno-methionine rVP28
and the native rVP28 were determined by MALDI TOF MS. The MW difference of
native and seleno-methionine rVP28 was around 271 Dalton, while the MW difference of
selenium and sulfur is 46.895 Dalton, indicating that all of the six methionine residues of
rVP28 were fully substituted by seleno-methionine.
4.1.3 Crystallization of rVP28
The initial crystals were obtained using under oil method. The condition was
later optimized using the hanging drop method to obtain diffraction-quality crystals.
Glycerol was used as the cryoprotectant for flash-frozen in liquid nitrogen. The details of
the conditions were listed in Table 4.1.

79
Table 4.1 Summary of Crystallization Conditions and Cryoprotectants
for rVP28 Crystals
Initial crystallization condition
20% PEG8K
0.2 M Ca acetate
0.1 M Na HEPES (pH 7.5)
Optimized Crystallization condition
25% PEG8K
0.2 M Ca acetate
0.1 M Na HEPES (pH 7.5)
1.5% w/v 1,2,3-heptanetriol
Cyroprotectant
25% PEG8K
0.2 M Ca acetate
0.1 M Na HEPES (pH 7.5)
20% glycerol

80
4.1.4 Structure Determination of rVP28
The structure of the recombinant SeMet VP28 was solved using the MAD method.
Three data sets in different wavelength were collected from the synchrotron beam line
X29 at the Brookhaven National Laboratories. Data were processed and scaled using the
HKL suite. BNP was used to locate the Se sites for VP28.
The phases were further developed using RESOLVE which improved the overall
FOM (figure of merit) to 0.75. The initial models were built automatically by RESOLVE
containing ~50% of the total residues. The rest of the model was manually built using
program O and refined using the program CNS interspersed with several rounds of
manual refitting. All the measured reflections were used in the refinement. The
asymmetric unit of VP28 consists of twelve molecules. Throughout the refinement the
Non Crystallographic Symmetry (NCS) restraints were used for VP28. The final R-
factors for VP28 is 0.24 (Rfree=0.28) at 2.0Å resolutions. The model was refined with
good stereochemical parameters. Statistics of the Ramachandran plot from an analysis by
PROCHECK gave an overall 89.7% of non-glycine residues in the most favored region
and only Asp166 in the generously allowed regions. This residue belongs to a less well-
ordered segment connecting two β strands. The details of data collection and refinement
are listed in Table 4.2.

81
Table 4.2 Data collection and refinement statistics of rVP28
Experiment
Cell parameters and angles a=105.33, b=106.71, c=200.37, α=β=γ=90°
Space group P212121
Data collection Peak Inflection Remote
Resolution range (Å) 50-2.0 50-2.1 50-2.1
Wavelength (Å) 0.9796 0.9799 0.9600
Observed Reflections>1 1818612 1608124 774774
Unique Reflections 152547 132247 131772
Overall (I/σI) 15.3 15.9 15.5
Redundancya 11.9(9.7) 12.2(11.2) 5.9(5.4)
Completenessa (%) 100(99.9) 100(100) 99.7(99.5)
Rsyma,b (%) 0.08(0.209) 0.072(0.179) 0.073(0.181)
Refinement Resolution range (Å) 45.0-2.0
Number of Reflections 294116
R factorc (%) 24.82
Rfreed (%) 28.12
RMSD bond lengths (Å) 0.008
RMSD bond angles ( °) 1.24
Average B-factors (Å2) 60.00
Water molecules 1422
Ramachandran Plot
Most favored region (%) 89.7
Additional allowed regions (%) 9.7
Generously allowed regions (%) 0.6
Disallowed regions (%) 0.0
a Last shell values givn in parentheses. b.Rsym = |Ii -<I>|/|Ii| where Ii is the intensity of the ith measurement, and <I> is the mean intensity for that reflection. b Rwork = | Fobs - Fcalc|/|Fobs| where Fcalc and Fobs are the calculated and observed structure factor amplitudes, respectively. c Rfree is calculated using the same equation as that for R work but 3% of reflections where chosen randomly and omitted from the refinement.

82
4.1.5 Overall Structure of rVP28
The full-length rVP28 exhibits single β-barrel architecture and an α-helix is
protruding from the β-barrel. The N-terminal extended (protruding) α-helix is 15 residues
(Thr32-Asn47) of approximately 23 Å in length from the surface of the β-barrel. A total
of nine β-strands (β1↓β2↑β3↓β4↓ß5↑β6↓β7↑β8↓β9↑), mostly anti-parallel, form the
barrel. The N-terminal protruding α-helix is linked by 2 residues to the core β-barrel. The
overall dimension of the β-barrel is approximately 35 Å (height) x 15 Å (diameter)
(Figure 4.3).
The pore of the β-barrel is highly hydrophobic in nature with 25 hydrophobic side
chains lining the inner surface, contributed by side chains from seven Phe, eight Ile, one
Leu, four Met and five Val residues. Taken together the size and the hydrophobic nature
of the pore, the latter may help to recognize small, specific, hydrophobic ligands/ions
which could potentially establish hydrophobic or van der Waals interactions with the side
chains of the pore residues. The exact role of this pore for the function of VP28 is yet to
be established.

83
Figure 4.3 Ribbon diagram of rVP28 monomer. The N and C termini are labled. Figure was prepared using the programs MOLSCRIPT (Kraulis 1991) and Raster3D (Merrit 1997).
N
C

84
4.1.6 Oligomerization of rVP28
In the crystal, an asymmetric unit consists of twelve copies of rVP28 molecules,
each with 170 residues from Thr32-Thr201 assembled into four trimers (Figure 4.4).
However, Gel filtration, SDS-PAGE and dynamic light scattering results showed that
rVP28 is a monomer in solution. Purified WSSV was treated with RIPA buffer and
analyzed by non-reducing SDS-PAGE and Western blotting. The bands detected by anti-
VP28 were excised from the corresponding gel and analyzed by MALDI TOF TOF MS.
There were three bands with different M.W. that matched to masses of a list of trypsin-
treated peptide of VP28. The bands correspond to a trimer (~75KD), dimer (~50 KD) and
monomer (~25KD, the most abundant species) (shown in Figure 4.5). This suggests that
VP28 naturally forms trimers in the viral envelope but the trimer species may be very
fragile during virus purification and lysis treatment, hence it becomes a monomer in
solution. However, during crystal formation the monomers tend to associate as trimers,
the most natural oligomerization state of VP28.
In the crystal of rVP28, a channel is formed by the N-terminal α-helix and the
wall of the β-barrel from each monomer of the trimeric rVP28. The length and weight of
the channel are approximately 58 X 12 Å. No salt bridges are apparent between any of
these residues. There are 10 hydrogen bonding contacts (<3.2 Å) and several hydrophobic
interactions between each monomers of the trimeric rVP28 to keep the integrity of the
channel. The surface area of a monomer is around 9024 Å2, whereas around 1170 Å2
(around 13%) is buried at the monomer interface of the trimer.
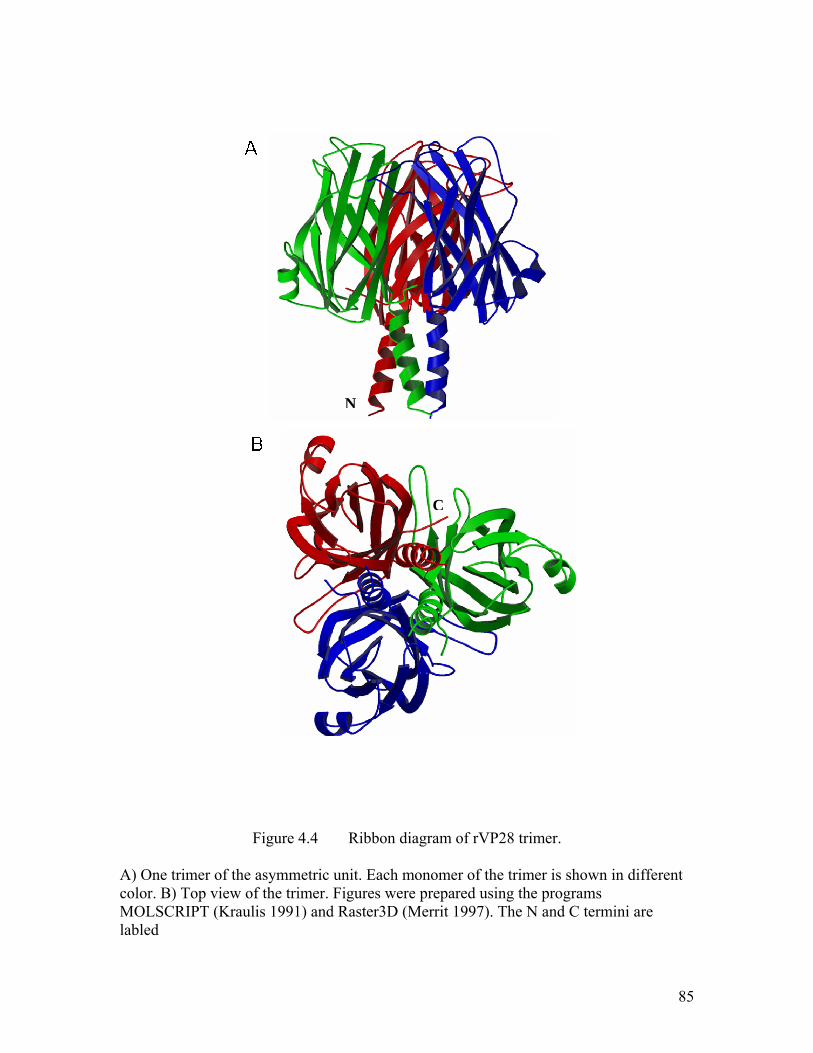
85
Figure 4.4 Ribbon diagram of rVP28 trimer. A) One trimer of the asymmetric unit. Each monomer of the trimer is shown in different color. B) Top view of the trimer. Figures were prepared using the programs MOLSCRIPT (Kraulis 1991) and Raster3D (Merrit 1997). The N and C termini are labled
N
C

86
Figure 4.5 Western blot analysis of WSSV by anti-VP28
The bands that were detected by anti-VP28 IgGs were excised for MALDI TOF TOF MS. The bands indicated by arrows match to VP28 of WSSV.
.

87
4.2 X-ray Structure of Major Envelope Protein VP26
4.2.1 Purification of rVP26 and SeMet rVP26 protein
The expression and purification trials on full-length VP26 had resulted in a very
low expression and the production of an insoluble protein as we have mentioned before.
We hypothesize that the low solubility was due to the N-terminal strong hydrophobic
region which was predicted as a transmembrane region by sequence analysis. The
transmembrane truncated form of VP26 (Asn34-Lys204) were constructed, with the
theoretical pI and M.W. of 9.57 and 19038.8 Da respectively. The analysis of the
expressed protein by SDS-PAGE is shown in Figure 4.6.
Similar to the native protein, SeMet substituted rVP26 was purified by Superdex-
75 gel filtration chromatography using a gel filtration buffer containing 20 mM Tris-Cl
(pH 7.4), 200 mM NaCl, 10 mM DT and 5% Glycerol. It is a monomer in solution as
confirmed by the gel filtration profile and the native gel. MALDI TOF mass spectrometry
was used for the measurement of the molecular weight of purified SeMet rVP26 as well
as native rVP26, to make sure that all of the nine methionine residues of rVP26 were
fully substituted by seleno-methionine. The purified SeMet rVP26 protein was
concentrated. The DLS result showed that the purified rVP26 protein was homogenous
up to a concentration of 10 mg/ml. However aggregation will start to appear in solutions
with higher concentration. Thus a sample of concentration of 10 mg/ml was used later for
crystallization trials.

88
15
20
25
37
50
75
kDa
100
GST-rVP26
GST
rVP26
1 2 3 4 5 6
15
20
25
37
50
75
kDa
100
15
20
25
37
50
75
kDa
100
GST-rVP26
GST
rVP26
1 2 3 4 5 6
Figure 4.6 Expression of the rVP26 protein in E.coli BL21 DE3 star
Shown is Coomassie Brilliant Blue –stained 12% SDS-PAGE gel Lane 1, protein marker; Lane 2, recombinant pGEX-6P-1 plasmid containing vp26 gene, non-induced; Lane 3, recombinant pGEX-6P-1 plasmid containing vp26 gene, induced; Lane 4, purified GST-rVP26 protein; Lane 5, GST-rVP26 fusion protein cleavage with ProScission™ protease; Lane 6, purified rVP26 protein.

89
4.2.2 Crystallization, Data Collection and Structure Determination of rVP26
Crystals of rVP26 were grown using the hanging drop vapor diffusion method at
room temperature. Extensive additives screening was performed to optimize the
crystallization condition to obtain best diffraction-quality crystals. Glycerol was used as
the cryoprotectant for flash-frozen the crystals in liquid nitrogen. The details of the
crystallization conditions and cryoprotectants are listed in Table 4.3.
The structure of the recombinant SeMet rVP26 protein was solved using a MAD
data set collected from the synchrotron beam line X26 at the Brookhaven National
Laboratories. Data were processed and scaled using the HKL suite. SOLVE was used to
locate the Se sited and the phases were further developed using RESOLVE which
improved the overall FOM (figure of merit) to 0.67. The initial models were built
automatically by RESOLVE containing ~50% of the total residues. The rest of the model
was manually built using program O and refined with all the measured reflections using
the program CNS interspersed with several rounds of manual refitting. The structure was
refined at 2.1 Å resolution, to a final R-factor of 0.21 (Rfree=0.27). The model has been
refined with good stereochemical parameters. Statistics for the Ramachandran plot from
an analysis using PROCHECK (Laskowski et al., 1993) gave an overall 85.3% of non-
glycine residues in the most favored region, with only Ala 58 in the generously allowed
regions. This residue belongs to a poorly ordered peptide segment connecting two β
strands. No residue was observed in the disallowed region. The details of data collection
and refinement are listed in Table 4.4.

90
Table 4.3 Summary of Crystallization Conditions and Cryoprotectants
for rVP26 Crystals
Initial crystallization condition
0.1 M citric acid (pH 3.5)
3.0 M sodium chloride
Optimized Crystallization condition
0.1 M citric acid (pH 3.5)
3.0 M sodium chloride
1% w/v polyethylene glycol 3350
Cyroprotectant
0.1 M citric acid (pH 3.5)
3.0 M sodium chloride
30% glycerol
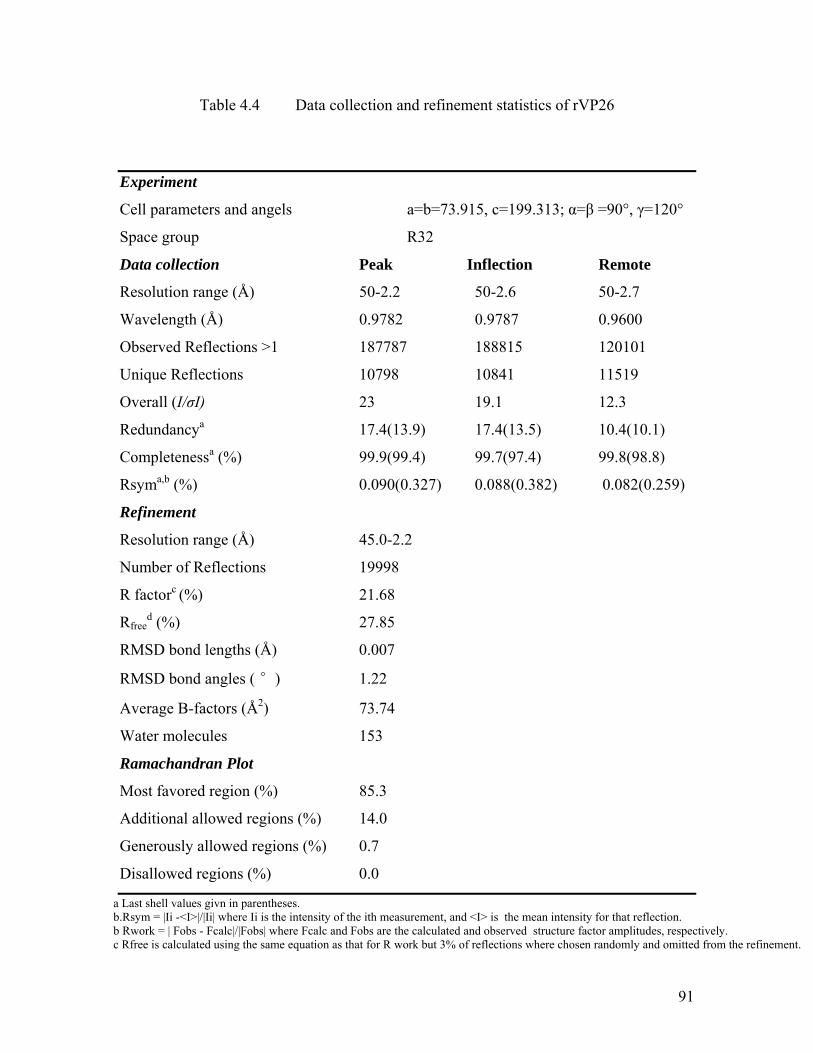
91
Table 4.4 Data collection and refinement statistics of rVP26
Experiment
Cell parameters and angels a=b=73.915, c=199.313; α=β =90°, γ=120°
Space group R32
Data collection Peak Inflection Remote
Resolution range (Å) 50-2.2 50-2.6 50-2.7
Wavelength (Å) 0.9782 0.9787 0.9600
Observed Reflections >1 187787 188815 120101
Unique Reflections 10798 10841 11519
Overall (I/σI) 23 19.1 12.3
Redundancya 17.4(13.9) 17.4(13.5) 10.4(10.1)
Completenessa (%) 99.9(99.4) 99.7(97.4) 99.8(98.8)
Rsyma,b (%) 0.090(0.327) 0.088(0.382) 0.082(0.259)
Refinement
Resolution range (Å) 45.0-2.2
Number of Reflections 19998
R factorc (%) 21.68
Rfreed (%) 27.85
RMSD bond lengths (Å) 0.007
RMSD bond angles ( °) 1.22
Average B-factors (Å2) 73.74
Water molecules 153
Ramachandran Plot
Most favored region (%) 85.3
Additional allowed regions (%) 14.0
Generously allowed regions (%) 0.7
Disallowed regions (%) 0.0
a Last shell values givn in parentheses. b.Rsym = |Ii -<I>|/|Ii| where Ii is the intensity of the ith measurement, and <I> is the mean intensity for that reflection. b Rwork = | Fobs - Fcalc|/|Fobs| where Fcalc and Fobs are the calculated and observed structure factor amplitudes, respectively. c Rfree is calculated using the same equation as that for R work but 3% of reflections where chosen randomly and omitted from the refinement.

92
4.2.3 Overall Structure of rVP26
Figure 4.7 shows the crystal structure of rVP26. The asymmetric unit contains one
VP26 molecule comprising 161 residues from Ser41 to Ile201 and a total of 153 water
molecules. Residues 34-40 and 202-204 from part of the N-terminal/C-terminal region
and had no interpretable density and were not modeled. The full-length rVP26 consists of
a nine stranded β-barrel with mostly anti-parallel β-strands in the orientation of
β1↓β2↑β3↓β4↓ß5↑β6↓β7↑β8↓β9↑. In addition, a two strand β-sheet (β1↓β9↑) protrudes
out of the β-barrel parallel to the barrel axis. Besides, there is a two turn α-helix hanging
outside of the β-barrel structure. In the protruding β-sheet, one strand is the N-terminal
part of the molecule, whereas the second strand is the C-terminal strand of VP26. The N-
and C-terminal of the molecule comes together to form this β-sheet. The N-terminal β-
strand is the longest in the molecule, having around 50Å in length; this β-strand is the
part of the β-barrel as well as the protruding β-sheet. The pore of the β-barrel is highly
hydrophobic in nature with side chains from three Phe, seven Ile, two Leu, five Met and
four Val, a total of 21 hydrophobic side chains lining the inner pore surface. The
approximate dimensions of the β-barrel are 38.5 Å (height) and 15.5 Å (diameter). We
suggest that this pore, similar to the β-barrel pore of rVP28, may allow the binding of
small, specific ligand/ions. The exact role of this pore for the function of rVP26 is yet to
be established.

93
Figure 4.7 Ribbon Diagram of rVP26 monomer
The β strand is colored green, while α helix and loop are colored as red and blue respectively. The N and C termini are labled. Figure was prepared using the programs MOLSCRIPT (Kraulis 1991) and Raster3D (Merrit 1997).
N
C

94
4.2.4 Oligomerization of rVP26
The oligomerization of rVP26 was investigated by gel filtration chromatography,
SDS-PAGE and native gel. All the experiments showed an apparent molecular mass of
20 KDa that corresponded to a monomer in solution. Only one molecule of rVP26 is
observed in an asymmetric unit. Interestingly, rVP26 crystallized in R32 space group
which has a three fold axis. The crystal packing showed a trimeric arrangement within
the crystal lattices from the symmetry related molecules (Figure 4.8).
Non-reduced SDS-PAGE and western blot analysis on the purified WSSV in
Figure 4.9 showed the presence of trimer (~66KD), dimer (~44 KD) and monomer
(~22KD, the most abundant species) of VP26. The arrow indicates the result
corresponding to MALDI TOF TOF MS. Similar to the rVP28 trimer, we speculate that
formation of the trimer may be the desirable aggregated form of this protein in the viral
envelope. The monomers of the trimer may be held together by very weak interactions
that are but not sufficiently strong to retain its oligomerization state in solution. The
immuno-electron images of VP26 that are shown in Chapter three indicate that the
observed spike-like structure should be a trimer.
Similar to the rVP28 trimer, a channel is formed by the protruding N terminal
region and the wall of the β-barrel from each monomer of the rVP26 trimer. There are
several water molecules present in the channel. The height and diameter are
approximately 58 × 12 Å. The channel may allow the passage of ions or small molecules,
but the exact role of these channels remains to be established.

95
Figure 4.8 Ribbon Diagram of rVP26 trimer
A) The rVP26 trimer (Crystallographic symmetry related molecules). Each monomer is shown in different color. B) Top view of the VP26 trimer. Figures were prepared using the programs MOLSCRIPT (Kraulis 1991) and Raster3D (Merrit 1997). The N and C termini are labled.
C
N
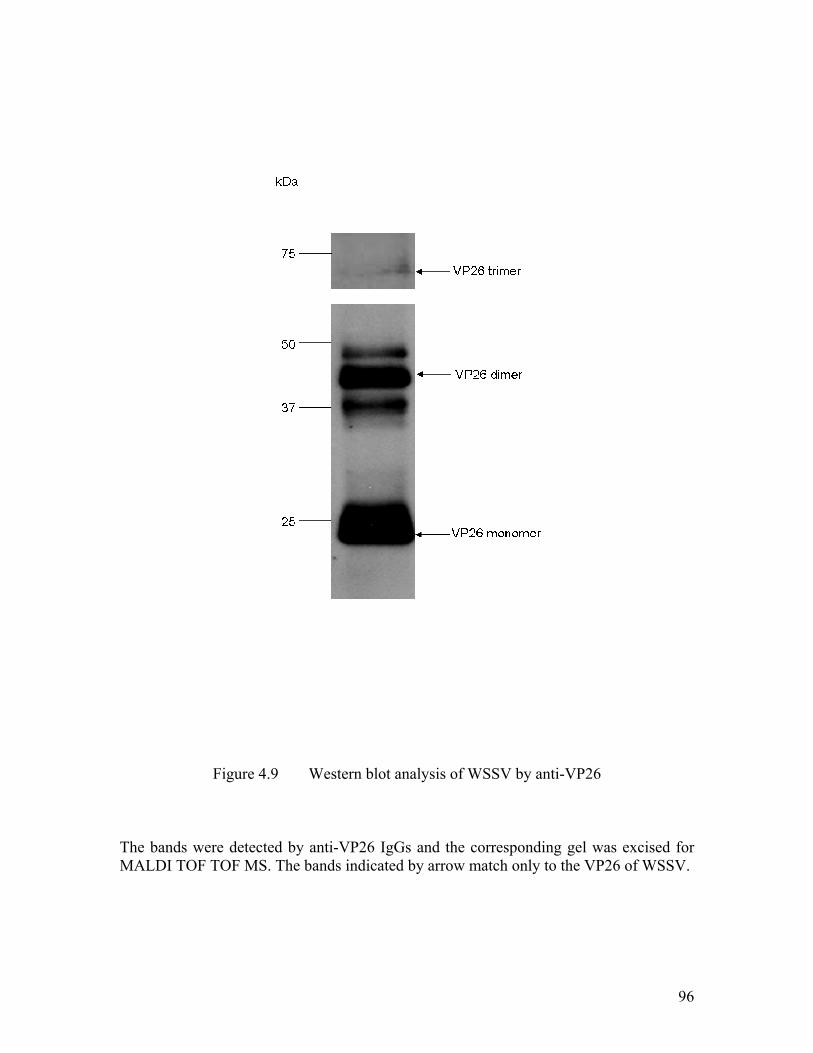
96
Figure 4.9 Western blot analysis of WSSV by anti-VP26
The bands were detected by anti-VP26 IgGs and the corresponding gel was excised for MALDI TOF TOF MS. The bands indicated by arrow match only to the VP26 of WSSV.

97
4.3 Comparison of VP26 and VP28
4.3.1 Structure Homolog of VP28 and VP26
A search of structural similarities for both full length rVP26 and rVP28 using the
DALI (Holm and Sander 1995) database failed to recognize significant matches to any
known structure. Interestingly, rVP26 and rVP28 display strong structural as well as
sequence homology among themselves. The structural superposition of rVP26 on rVP28
gave an rmsd of 2.6Å for 144 Cα atoms, as shown in Figure 4.10, with a sequence
similarity of ~36%. The structure based sequence alignment revealed that most of the
structurally invariant residues are located at β1, β3, α1 (equivalent of α2 in rVP28), β4
and β5. The rVP26 and rVP28 monomers possess a pore of similar size that is very
hydrophobic in nature. The hydrophobic side chain residues lining inside the pore are
quite conserved in these two proteins (Figure 4.11, Figure 4.12 and Figure 4.13). The
most significant difference between them is an extended β-strand in rVP26 and an α-
helix in rVP28 as their protruding parts. In the case of rVP26, the N-terminal part and the
C-termial strand come together to form a two stranded ß-sheet (β1↓β9↑) protruding out
of the β-barrel and parallel to the barrel axis. Whereas in rVP28, a long α-helix is
hanging outside the barrel at the N-terminus.

98
Figure 4.10 Stereo Cα superposition of rVP28 and rVP26
Cα of rVP28 in blue and rVP26 in green is shown. The superposition was generated using the program O (Jones et al. 1991), using the consered residues as the starting point. This figure was prepared using the program MOLSCRIPT (Kraulis 1991).
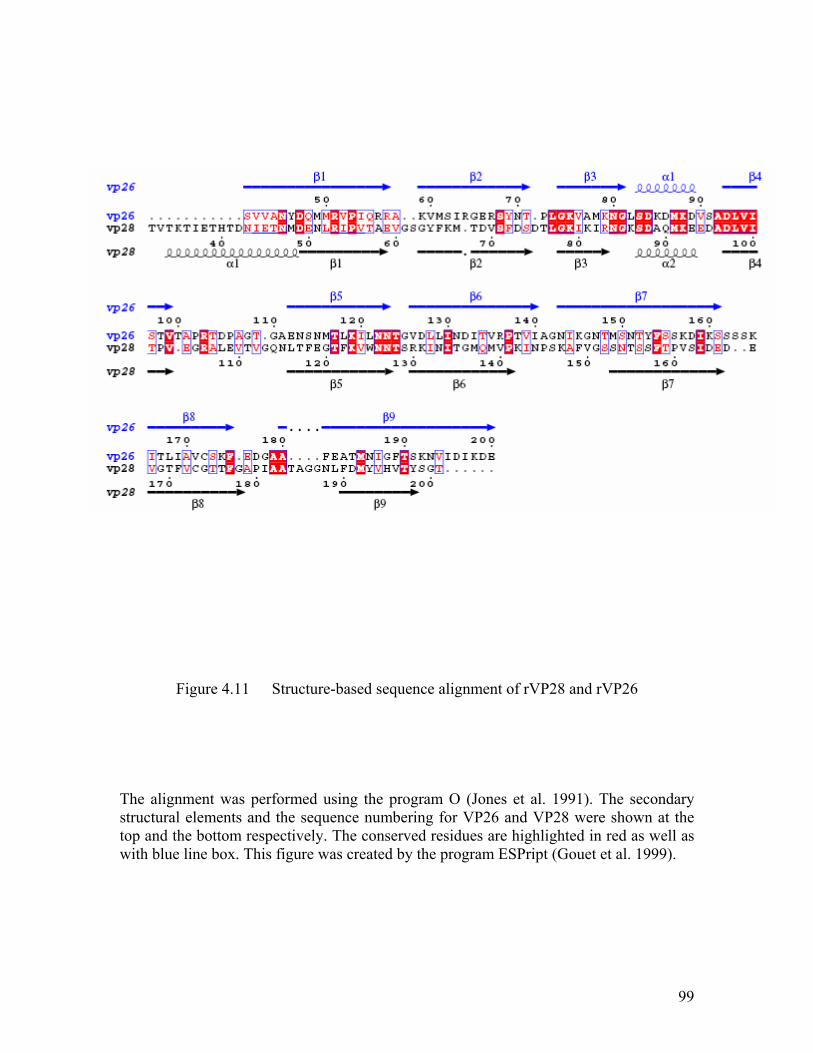
99
Figure 4.11 Structure-based sequence alignment of rVP28 and rVP26
The alignment was performed using the program O (Jones et al. 1991). The secondary structural elements and the sequence numbering for VP26 and VP28 were shown at the top and the bottom respectively. The conserved residues are highlighted in red as well as with blue line box. This figure was created by the program ESPript (Gouet et al. 1999).
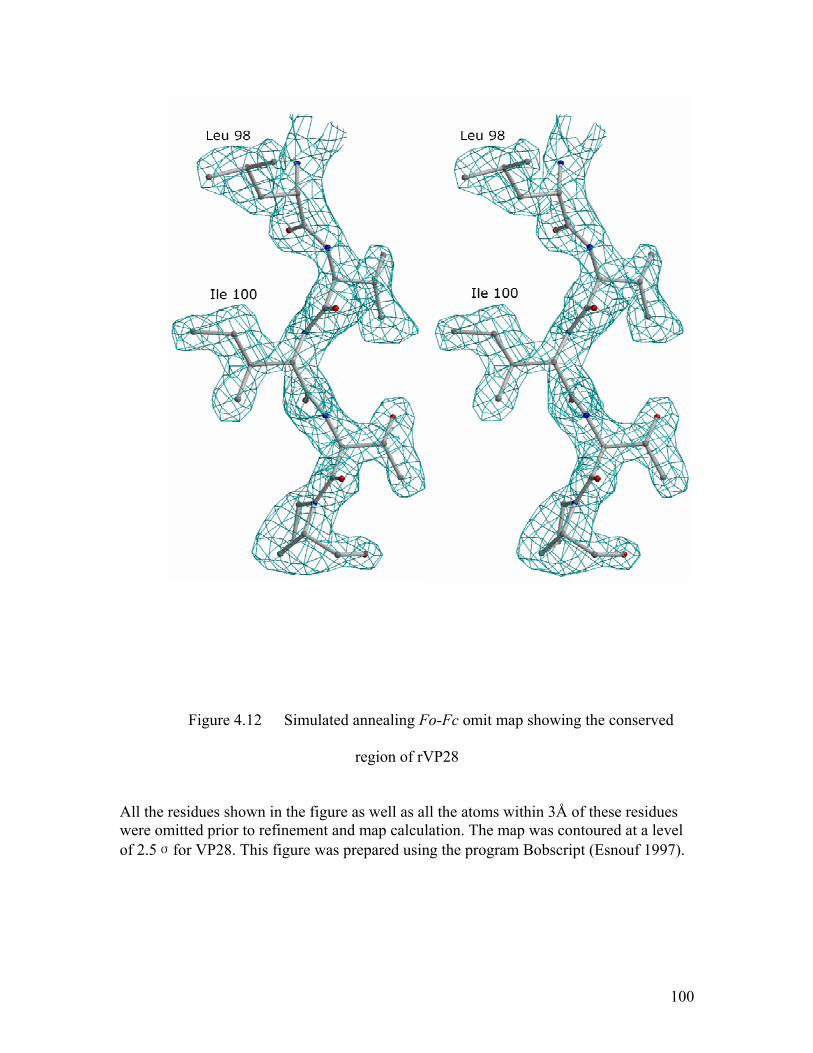
100
Figure 4.12 Simulated annealing Fo-Fc omit map showing the conserved
region of rVP28
All the residues shown in the figure as well as all the atoms within 3Å of these residues were omitted prior to refinement and map calculation. The map was contoured at a level of 2.5σfor VP28. This figure was prepared using the program Bobscript (Esnouf 1997).

101
Figure 4.13 Simulated annealing Fo-Fc omit map showing the conserved
region of rVP26
All the residues shown in the figure as well as all the atoms within 3Å of these residues were omitted prior to refinement and map calculation. The map was contoured at a level of 2σfor rVP26. This figure was prepared using the program Bobscript (Esnouf 1997).

102
Furthermore, an interesting feature of these two proteins is their extreme opposite
charges on the surface, although the inner pore region of the β-barrel maintains a strong
hydrophobic environment with highly conserved residues lining the pore. The rVP26
trimer (crystallographic symmetry related molecules) depicts patches of positively
charged residues on the surfaces, whereas the rVP28 trimer depicts patches of negative
charged regions (Figure 4.14). It has been reported that VP28 will interact with VP26 by
far western blot analysis and pulldown assay (Xie et al. 2006; Xie and Yang 2006).
However, it is unclear that what these kinds of charge distribution contribute to the
function of the proteins such as the interaction between these envelope proteins or with
receptors on host cells.
Here we propose the trimeric VP26 and VP28 models along with the viral
envelope. The trimer arrangement of VP26 and VP28 matches well with the shape of the
spike-like structure in EM images after the treatment of Tween 20 (sees Figure 3.6 A-D,
as well as inserts Figure 3.6 D). It has been proven that viral structural proteins can exert
multiple functions by adopting different conformations (Hartlieb & Weissenhorn, 2006)
such as the Ebola virus matix protein VP40 was monomers when targeted to cellular
membranes but change its conformation to form hexamer via specific interactions of the
N-terminus (Nguyen et al., 2005). We propose that VP28/VP26 of WSSV could also
adopt a different conformation for different function, in which the trimer of VP28/VP26
may play an important role in membrane fusion during viral infection. However, the
questions that what is the factor to trigger the trimerization of these two proteins and
what is the role of monomeric protein are to be answered by further investigations.
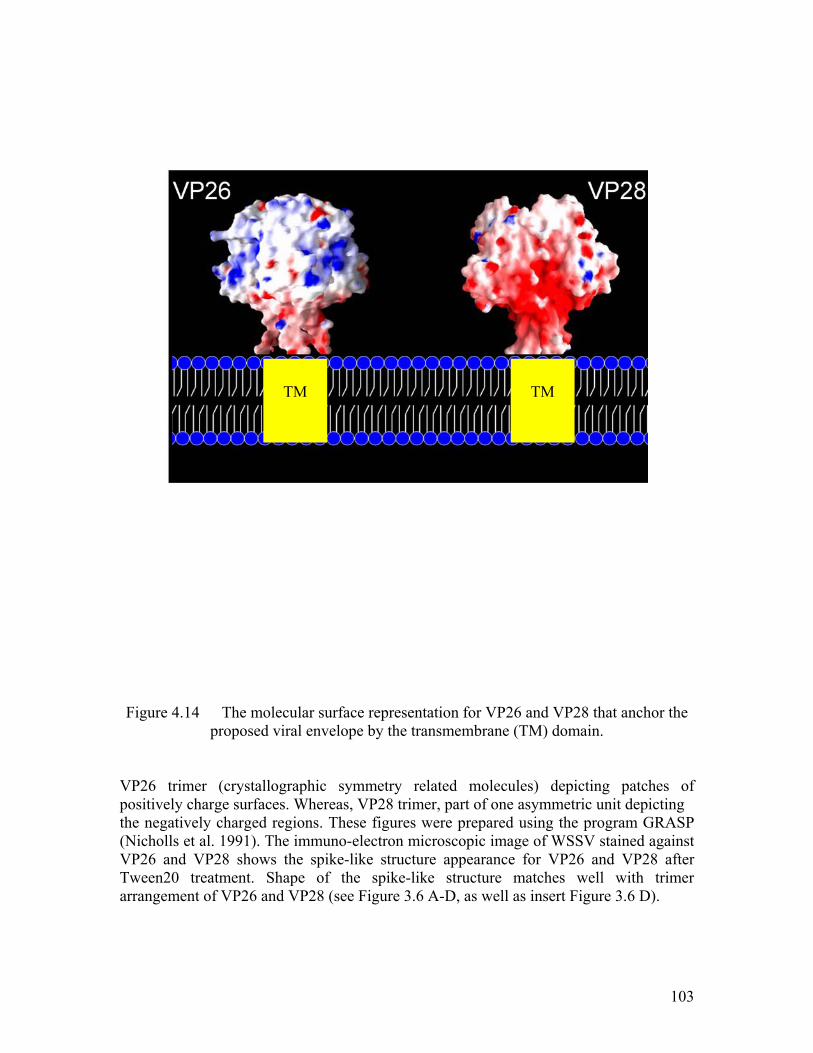
103
Figure 4.14 The molecular surface representation for VP26 and VP28 that anchor the proposed viral envelope by the transmembrane (TM) domain.
VP26 trimer (crystallographic symmetry related molecules) depicting patches of positively charge surfaces. Whereas, VP28 trimer, part of one asymmetric unit depicting the negatively charged regions. These figures were prepared using the program GRASP (Nicholls et al. 1991). The immuno-electron microscopic image of WSSV stained against VP26 and VP28 shows the spike-like structure appearance for VP26 and VP28 after Tween20 treatment. Shape of the spike-like structure matches well with trimer arrangement of VP26 and VP28 (see Figure 3.6 A-D, as well as insert Figure 3.6 D).
TM
TM

104
4.3.2 Comparison with Other Viral Proteins
The β structure is one of the most common structures for viral structural protein
and has been found in various viral proteins in RNA viruses from plant, insect and animal
origins ((Rossmann and Johnson 1989), in double-stranded DNA viruses such as
adenovirus (Roberts et al. 1986), PRD1 (Benson et al. 1999) and SV40 (Stehle et al.
1996), and in dsRNA viruses such as blue tongue virus (Grimes et al. 1998) and
orthoreovirus (Reinisch et al. 2000). The β structures of VP26 and VP28 reveal
interesting structural similarities between these two proteins as well as with several
apparently unrelated virus types, although neither VP28 nor VP26 shares significant
sequence homology with known structural proteins from any other viruses.
However, a search of structural similarities for both full length rVP26 and rVP28
using the DALI (Holm and Sander 1995) database failed to recognize any significant
matches to any known protein structure. The DALI search shows that the highest
structural homology is with the gamma COP appendage domain of an intracellular
trafficking pathway protein (pdb code 1 pzd; rmsd is 3.3 Å for 87 Cα atoms of rVP28 and
89 Cα atoms of rVP26). The only viral protein structures that show partial structural
similarity were VP7 protein of Bluetongue virus (pdb code 1bvp; rmsd is 3.6 Å for 82 Cα
atoms of rVP26), A21 fragment of human coxsackievirus (pdb code 1z7z; rmsd is 3.6 Å
only for 72 Cα atoms of rVP28) and the capsid protein of carnation mottle virus (pdb
code 1opo; rmsd is 3.6 Å for only 64 Cα atoms of rVP28 and 3.7 Å for only 63 Cα atoms
of rVP26). Structurally the most common feature among these proteins is the presence of

105
a β-barrel structure, which is exposed on the outer surface. The topologies of these
proteins are, however, differenced from each other.
Unlike the predominant eight-stranded jelly roll topology, VP26 and VP28 has a
nine stranded ß-barrel that is folded into a novel topology and has not been seen in any
other virus up to date. The novelty of the structure of the major envelope proteins of
WSSV from any other virus to date therefore places WSSV outside of any of the
structurally linked groupings that have been identified so far. This unique feature of
WSSV is consistent with the phylogenetic tree of the WSSV DNA polymerase genes,
showing that WSSV is not related to any of the larger dsDNA virus family (Chen et al.
2002b). This further supports the proposition that WSSV should be a representative of a
new family (Nimavirida) under a new viral genus (Whispovirus).

106
4.3.3 Implications on Gene Duplication
VP24 is another important envelope protein of WSSV and interestingly it is also
highly homologous to VP26 and VP28 with 41% and 46% sequence similarity
respectively (Figure 4.15). Similar to VP26 and VP28, a highly hydrophobic region in the
N-terminus of VP24 might represent a transmembrane region. From the homolog of
VP26 and VP28 and the conservation of the sequence, we can hypothesize that VP24
adopts a nine-stranded β-barrel fold with a hydrophobic pore similar to that observed for
VP26 and VP28. However, the expression and purification trials on full-length VP24 or
the truncated form invariably resulted in the production of an insoluble protein.
Sequence comparison of VP24, VP26 and VP28 revealed that these three proteins
are highly related and most likely arise from by gene duplication. The structure similarity
of VP26 and VP28 further confirms this view. As the β sheet angle reflects packing of
the hydrophobic pore, it should also be conserved upon gene duplication. Conversely,
horizontal gene transfer would be less likely to yield a virus in which all three proteins
would possess the same β sheet angle. Thus we believe that the structures of VP26 and
VP28 bear evidence that support their evolution by gene duplication within the viral
genome. A similar example can be seen in the outer coat proteins P3-V1, P3-V2 and P5
of Bacteriophage PRD1. All these proteins have similar β barrel structure of same β
sheet angles and are believed to be evolved by gene duplication (Merckel et al. 2005).

107
Figure 4.15 Sequence Alignment for VP24, VP26 and VP28
The sequence alignment of VP24, VP26 and VP28 were analyzed by DNAman. The identity residues are highlighted in black. The residues with similar properties are highlighted in blue.
40VP28 40VP26 37VP24
Consensus
MM.
DE.
LF.
SGM
FNH
TLM
LTW
SNG
VLV
VDY
SVAa
AAAi
III
LILa
AAA
IIG
TLL
AST
VIL
IAI
AIL
VIV
FAV
ILI
VIS
IVI
FIV
RMV
YVT
HIN
NMI
TIE
VVL
TFN
KNK
TTK
IRL
EVD
TGK
HRK
80VP28 76VP26 77VP24
Consensus
T.D
D.K
NSD
IVA
EVY
TAP
NNV
MYE
DDS
EQE
NMI
LMI
RRN
IVL
PPT
VII
TQN
ARG
ERV
VAA
GKR
SVG
GMN
YSH
FIF
KRN
MGF
T.V
DEN
VRG
SST
FYL
DNQ
S.T
DTR
TPN
LLYg
GGGk
KKK
IVV
116VP28 112VP26 117VP24
Consensus
KAY
IMV
RKA
.
.G
NNQg
GGG
KLTs
SSSd
DDD
AKS
QDE
MML
KKV
EDK
EVK
DSK
AAGd
DDD
LLI
VVI
IIL
TST
PTS
VVL
ETL
GAG
RPD
.
.G
.
.D
.
.H
ART
LTL
EDN
VPV
TAN
VGK
GTA
QGE
NAS
LEK
155VP28 151VP26 157VP24
Consensus
TNE
FSL
ENE
GML
TTY
FLA
KKR
VIV
WLYn
NNNn
NNNt
TTT
SGK
RVR
KDD
ILI
NLT
IIV
TND
GDS
MIV
QTS
MVL
VRSp
PPP
KTG
IVL
NIN
PAA
.
.T
SGG
KNR
AIE
FKF
VGS
GNA
STN
SMK
NSF
TNV
193VP28 191VP26 195VP24
Consensus
STL
SYYf
FFF
TSK
PSP
VKT
SDV
IIL
DKK
ESK
DSN
.S.
.S.
EKR
VII
GTN
TLT
FIL
VDV
CVF
GCG
TSA
TKTf
FFF
GED
ADE
PGD
IAI
AAD
AFD
TET
AAN
GTR
GMH
NNY
LIL
FGL
DFS
MTM
YSR
204VP28 203VP26 207VP24
Consensus
VKF
HNS
VVP
TIG
YDN
SID
GKL
TDF
EEK
TIV
EKG
.KE

108
It is surprised that WSSV encodes three most abundant envelope proteins of
similar structure. One possible explanation is that these membrane anchor proteins can
aggregate on the surface of membrane with β-barrels in which there are no "edge" with
unfulfilled hydrogen bonding. The presence of similar hydrophobic barrel indicated that
these three proteins most probably are capable of forming homo- and heteromultimers. It
is recently reported that VP28 interacts with VP26 and VP24 to form a complex (Xie et al.
2006; Xie and Yang 2006). Intermolecular interactions between thee similar structures
may play a very important role in virus morphogenesis.
Another way to explain the highly similar structure to assume that these proteins
have been evolved with different functions in the WSSV virion such as VP26 was
previously reported to be capable of binding to actin or actin-associated proteins (Xie and
Yang 2005). Or they maybe related with infection to different host cell as the virus has a
broad range of hosts including salt and brackish water penaeid, crab, spiny lobster,
freshwater shrimps and crayfishes. However, whether these three proteins are just a
redundant copy of each other for backup or tave been evolved with functional divergence
is still unclear and needs further investigations.

109
4.3.4 Implications on membrane fusion
Viral structural proteins can exert multiple functions by adopting different
conformations (Hartlieb & Weissenhorn, 2006). For example, the Ebola virus matix
protein VP40 was shown to form monomers when targeted to cellular membranes but
change its conformation to form hexamer via specific interactions of the N-terminal
domain (Nguyen et al., 2005). We propose that VP28/VP26 of WSSV could also adopt a
different conformation for different function, in which the trimer of VP28/VP26 may
play an important role in membrane fusion during viral infection.
Enveloped viruses gain entry into the cytoplasm by the fusion of their lipid
envelope with the host cell membrane and the virus–induced membrane fusion is
triggered by conformational changes of viral envelope proteins (Da Poian et al. 2005). To
date, two classes of virus membrane-fusion proteins have been defined based on their key
structural features (Kielian 2006; Kielian and Rey 2006). Viruses that are currently
assigned to Class I or Class II are listed in Figure 4.16A. The Class I membrane-fusion
protein is mediated by the refolding of the fusion protein to a highly stable rod-like
structure with a central trimeric α-helical coiled coil. The Class II membrane-fusion
protein undergoes an oligomeric rearrangement during fusion, converting from the
metastable prefusion dimer to a considerably more stable homotrimer conformation. β
structure is the predominant secondary structure of Class II fusion protein. The details of
the structure features of the proteins in these two classes are list in Figure 4.16B. The
structures of some examples of these fusion proteins are shown in Figure 4.16C.

110
Figure 4.16 Fusion Proteins of Class I and Class II
A) Class I or Class II virus fusion proteins B) Comparison of Class I & Class II virus membrane fusion proteins. C) Structures of some fusion proteins. (Kielian and Rey 2006)
A
C
B

111
Though, VP26 and VP28 are not highly homologous to any of the viral envelope
fusion proteins, it retains the ß-barrel as well as trimer architecture similar to other viral
envelope fusion proteins such as E1 structural protein of Semliki Forest Virus (SFV)
(Gibbons et al. 2004). Previous study has shown that VP28 plays an important role in the
systemic WSSV infection in shrimp (Li et al. 2005a) and VP28 can bind to shrimp cells
as an attachment protein only in a low pH condition and can help the virus enter to the
cytoplasm (Yi et al. 2004). It has been well established that some of the enveloped
viruses enter the cells by endocytic pathway and this fusion depends on the acidification
of the endosomal compartment. More recently it has been shown that the interaction
between the host and WSSV is mediated through the host protein PmRab7 and VP28
(Sritunyalucksana et al. 2006). Rab proteins act as molecular switches to control
trafficking of endocytic vesicles within cells, as well as their subsequent fusion to
endosomes. Considering this we suggest that the fusion of WSSV viral envelope and the
host membrane through VP26 and VP28 is triggered by the exposure to low pH in the
endocytic pathway or the combination of receptor binding and low pH.
This completely different fusion mechanism also indicates that WSSV is a very
unique virus that is not closely related to any other virus, underscoring the distinct
taxonomic position of WSSV among invertebrate viruses. The many questions on the
fusion of WSSV will be very important topics to be answered in the future.

112
CHAPTER FIVE
5 General Conclusion and Future Studies

113
The characterization of viral structural proteins especially the envelope proteins is
of significant importance to study the taxonomy of the virus, its mechanism of infection
and also the assembly process of the mature virus. Due to the lack of a suitable shrimp
cell line and a low homology with other proteins of known function, we took the
structural approach to study WSSV envelope proteins.
A novel envelope protein, VP300, was identified by mass spectrometry. A
localization study using Western blotting and immuno-gold-labeling electron microscopy
are performed to confirm that VP300 localizes in WSSV envelope, not in the
nucleocapsids. The neutralization assay showed that anti-VP300 IgGs functions to delay
or neutralize the infection of WSSV to shrimp. Perhaps the antibody of VP300 could bind
to envelope spikes on the virion and prevent virus uncoating or the attachment of the
virus to the cell surface or virus uncoating. Further study on VP300 would be helpful to
realize the mechanism of the viral infection. Up to now, no efficient method has been
found to prevent the infection of shrimp by WSSV. Antibodies against VP300 could be
used directly as a passive immune strategy to control the WSSV infection and also might
be a choice for the virus control in the shrimp industry.
In this study, localization of VP26 with immuno-electron microscopy has also
been investigated. Compared with the highly confirmed envelope protein of VP28, the
location of VP26 in the virion is still unclear. VP26 was first reported as a nucleocapsid
protein (van Hulten et al. 2000), and later reported as an envelope protein (Zhang et al.
2002b). However, most recent publications claimed that VP26 is a linker (Xie and Yang

114
2005), or a tegument protein (Tsai et al. 2006). Clarification of the location of VP26
should improve our understanding on function of the protein. In this study we have first
used Tween 20 treatment to separate viral envelope from the nucleocapsid and then
localized VP26 by immunogold labeling. Both the VP26 and VP28 were clearly observed
on the outer surface of the virus envelope by electron microscopy and that is the very first
time we observed the spike-like structure in WSSV envelope.
As the continuation of our effort to understand the structure and function of
WSSV proteins, we determinate the crystal structure of two major envelop proteins VP26
and VP28 from WSSV, the first two structural envelop proteins to be reported from
WSSV for the first time. We have analyzed the three dimensional structures of VP26 and
VP28 and postulated that VP26 and VP28 might fuse with the envelop membrane by
their N-terminal transmembrane domains. A search for full length VP26 and VP28
structural similarities using the DALI (Holm and Sander 1995) database failed to
recognize any significant matches. Interestingly, VP26 and VP28 display strong
structural as well as significant sequence homology. Both proteins adopt the β-barrel
architecture with a protruding N-terminal region. The mushroom-like shape of the
trimeric VP26 and VP28 matches well with the spike structure we have observed in the
localization study. They are highly likely function to interact with host receptor proteins
or fuse with host cell membrane, representing a different model from the fusion proteins
of class I and class II.

115
This is the first report that structures of WSSV major envelope proteins. It greatly
inspires us to further determine the structure of other envelope proteins or nucleocapsid
proteins as they have important role in mechanism of viral morphogenesis and the
infection mechanism.
There are four major envelope proteins (VP19, VP24, VP26 and VP28) proteins
that consist almost 80-90% of the entire envelope. We have speculated that VP24 may
adopt a β-barrel structure with a hydrophobic pore similar to that of VP26 and VP28
because of their very high sequence homology. However, VP19, which almost constitutes
30% of the envelope, has no sequence similarities to proteins from WSSV or other
known viruses. Structural elucidation of VP19 will be extremely important for studying
the evolution of WSSV and also for the infection mechanism of this unique virus. A brief
attempt at the expression and purification trials of full length VP19 resulted in an
insoluble protein. Considering a predicted transmembrane region at the middle of
sequence, we will try to express the two sides of the fragment that may represent two
domains that are located outside and inside of the membrane respectively.
VP281, with a cell attachment RGD motif, is supposed to play an important role
in mediating WSSV infectivity (Huang et al. 2002a; Liang et al. 2005). As another
candidate for a structural genomics study, this protein has been successfully expressed
and purified as a soluble protein in the bacterial system as we mentioned in Chapter three.
However, we are still unable to grow any crystal of this protein from the crystallization
trials. We suspect that the failure of crystal growth is because of a probable floppy

116
domain in the protein that might cause aggregation. So in the future work, the following
approachs can be used to remove this floppy region. The first one is to construct different
truncated forms of VP281, such as to remove the N-terminus or C-terminus. The second
approach is to remove the flexible region by performing limited proteolysis. An
experiment using intact proteins and small amounts of exogenously added proteases can
be performed to isolate a fragment that will be more amenable to crystallization. The
ratio of protein to protease and the reaction time can be varied for limited proteolysis.
Once identified, the protease resistant fragment can be purified for crystallization.
More recently it has been shown that the interaction between the host and WSSV
is mediated through the host protein PmRab7 and VP28. Rab proteins are known to act as
molecular switches to control trafficking of endocytic vesicles within cells, as well as
their subsequent fusion to endosomes. It has been reported that once the interaction of
these two proteins was blocked by their specific antibodies the efficiency of the WSSV
infection were greatly reduced. Thus we can try to solve the complex structure of VP28
and PmRab7 to figure out the key residues which are involved in their interaction. Based
on these residues, peptides or small molecules can be designed to inhibit their interaction.
Besides the structure determination, an infection assay can be done to figure out
the key factors affecting the viral infection. Such as this assay can be done at various
different pH to prove the proposal that acidic pH is essential for viral infection of WSSV.
These experiments can also lead to the question of why pH will affect viral infection and
whether this is somehow related to the charge distribution on VP26 and VP28. Similar to

117
the neutralization study by antibodies, an experiment can be set up to use mutant VP26 or
VP28 to map out the host receptor binding region of these proteins. Purified wild type
protein should be able to neutralize WSSV infection as it should compete with the virus
to the host cell receptor. By mutating residues on the surface of the protein, the mutant
may lose the ability to inhibit WSSV infection. Those mutated residues can be identified
to be essential for interaction with the host receptor. However, the neutralization assay is
not that easy to be carried out because that it is very hard for us to select out the WSSV-
free shrimps or crayfishes for the assay. Majority of the shrimps or crayfishes we brought
for experiments are tested to be the carrier of the WSSV and unable to be used in the
neutralization assay. Therefore, we will continue to screen out the shrimp (crayfish/crab)
cell lines for in vitro reproduction of WSSV what is suitable for the infection assay.
In a conclusion, after the 3D structures of these envelope and nucleocapsid
proteins are solved, we can obtain more comprehensive information for further research
on host-virus interaction, the maturation and assembly of the virus particles and answer
many other important questions. Besides that, infection assays can be done to figure out
the key factors that affect viral infectivity of WSSV and select the inhibitors to block the
interaction to the host cell receptor that can greatly reduce the infection. Finally, our
future study should also extend towards drug design and vaccine development against
WSSV.

118
6 Bibliography
Abbott, A. (2002). "The society of proteins." Nature 417(6892): 894-6.
Aloy, P. and R. B. Russell (2002). "The third dimension for protein interactions and
complexes." Trends Biochem Sci 27(12): 633-8.
Aparicio, S., J. Chapman, E. Stupka, N. Putnam, J. M. Chia, P. Dehal, A.
Christoffels, S. Rash, S. Hoon, et al. (2002). "Whole-genome shotgun assembly and
analysis of the genome of Fugu rubripes." Science 297(5585): 1301-10.
Ashburner, M., C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P.
Davis, K. Dolinski, S. S. Dwight, et al. (2000). "Gene ontology: tool for the unification
of biology. The Gene Ontology Consortium." Nat Genet 25(1): 25-9.
Ataka, M. (1993). "Protein crystal growth: ab approach based on phase diagram
determination." Phase Transitions 45: 205-219.
Ban, N., P. Nissen, J. Hansen, P. B. Moore and T. A. Steitz (2000). "The complete
atomic structure of the large ribosomal subunit at 2.4 A resolution." Science 289(5481):
905-20.
Ben-Shem, A., F. Frolow and N. Nelson (2003). "Crystal structure of plant photosystem
I." Nature 426(6967): 630-5.
Benson, S. D., J. K. Bamford, D. H. Bamford and R. M. Burnett (1999). "Viral
evolution revealed by bacteriophage PRD1 and human adenovirus coat protein
structures." Cell 98(6): 825-33.

119
Bradley, P., K. M. Misura and D. Baker (2005). "Toward high-resolution de novo
structure prediction for small proteins." Science 309(5742): 1868-71.
Bravo, J. and P. Aloy (2006). "Target selection for complex structural genomics." Curr
Opin Struct Biol.
Brenner, S. E. (2001). "A tour of structural genomics." Nat Rev Genet 2(10): 801-9.
Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-
Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, et al. (1998). "Crystallography &
NMR system: A new software suite for macromolecular structure determination." Acta
Crystallogr D Biol Crystallogr 54(Pt 5): 905-21.
Burton, D. R., R. A. Williamson and P. W. Parren (2000). "Antibody and virus:
binding and neutralization." Virology 270(1): 1-3.
Cen, F. (1998). "The existing condition and development strategy of shrimp culture
industry in China " In Y.Q.Su(ed.), The health culture of shrimps. China Ocean Press,
Beijing, People's Republic of China 32-38.
Chance, M. R., A. R. Bresnick, S. K. Burley, J. S. Jiang, C. D. Lima, A. Sali, S. C.
Almo, J. B. Bonanno, J. A. Buglino, et al. (2002). "Structural genomics: a pipeline for
providing structures for the biologist." Protein Sci 11(4): 723-38.
Chayen, N. E. (2004). "Turning protein crystallisation from an art into a science." Curr
Opin Struct Biol 14(5): 577-83.
Chen, L. L., J. H. Leu, C. J. Huang, C. M. Chou, S. M. Chen, C. H. Wang, C. F. Lo
and G. H. Kou (2002a). "Identification of a nucleocapsid protein (VP35) gene of shrimp
white spot syndrome virus and characterization of the motif important for targeting VP35
to the nuclei of transfected insect cells." Virology 293(1): 44-53.

120
Chen, L. L., C. F. Lo, Y. L. Chiu, C. F. Chang and G. H. Kou (2000). "Natural and
experimental infection of white spot syndrome virus (WSSV) in benthic larvae of mud
crab Scylla serrata." Dis Aquat Organ 40(2): 157-61.
Chen, L. L., H. C. Wang, C. J. Huang, S. E. Peng, Y. G. Chen, S. J. Lin, W. Y. Chen,
C. F. Dai, H. T. Yu, et al. (2002b). "Transcriptional analysis of the DNA polymerase
gene of shrimp white spot syndrome virus." Virology 301(1): 136-47.
Chen, X. F., C. Chen, D. H. Wu, H. Huai and X. C. Chi (1997). "A new baculovirus of
cultured shrimp." Sci. China Ser 40: 630-635.
Chou, H. Y., C. Y. Huang, C. H. Wang, H. C. Chiang and C. F. Lo (1995).
"Pathogenicity of a baculovirus infection causing white spot syndrome in cultured
penaeid shrimp in Taiwan." Dis. Aquat. Org. 23: 165-173.
Cramer, P., D. A. Bushnell and R. D. Kornberg (2001). "Structural basis of
transcription: RNA polymerase II at 2.8 angstrom resolution." Science 292(5523): 1863-
76.
Da Poian, A. T., F. A. Carneiro and F. Stauffer (2005). "Viral membrane fusion: is
glycoprotein G of rhabdoviruses a representative of a new class of viral fusion proteins?"
Braz J Med Biol Res 38(6): 813-23.
Ducruix, A. and R. Giege (1999). "Crystallisation of Nucleic Acids and Proteins. A
practical Approach, edn 2. Oxford: Oxford University Press."
Durand, S., D. V. Lightner, R. M. Redman and J. R. Bonami (1997). "Ultrastructure
and morphogenesis of white spot syndrome baculovirus (WSSV)." Dis Aquat Organ 29:
205-211.

121
Egea, P. F., S. O. Shan, J. Napetschnig, D. F. Savage, P. Walter and R. M. Stroud
(2004). "Substrate twinning activates the signal recognition particle and its receptor."
Nature 427(6971): 215-21.
Francki, R. I. B., C. M. Fanquet, D. L. Knudson and F. Brown (1991). "Classification
and nomenclature of viruses. Fifth report of the International Committee on Taxonomy of
Viruses." Arch Virol Suppl. 2: 1-450.
Gouet, P., E. Courcelle, D. I. Stuart and F. Metoz (1999). "ESPript: analysis of
multiple sequence alignments in PostScript." Bioinformatics 15(4): 305-8.
Green, E. D. (2001). "Strategies for the systematic sequencing of complex genomes."
Nat Rev Genet 2(8): 573-83.
Grimes, J. M., J. N. Burroughs, P. Gouet, J. M. Diprose, R. Malby, S. Zientara, P. P.
Mertens and D. I. Stuart (1998). "The atomic structure of the bluetongue virus core."
Nature 395(6701): 470-8.
Harlow, E. and D. Lane (1988). "Antibodies: A laboratory Manual." Cold Spring
Harbor, NY: Cold Spring Harbor Laboratory.: 553-612.
Holm, L. and C. Sander (1995). "Dali: a network tool for protein structure comparison."
Trends Biochem Sci 20(11): 478-80.
Huang, C., L. Zhang, J. Zhang, L. Xiao, Q. Wu, D. Chen and J. K. Li (2001).
"Purification and characterization of White Spot syndrome virus (WSSV) produced in an
alternate host: crayfish, Cambarus clarkii." Virus Res 76(2): 115-25.
Huang, C., X. Zhang, Q. Lin, X. Xu and C. L. Hew (2002a). "Characterization of a
novel envelope protein (VP281) of shrimp white spot syndrome virus by mass
spectrometry." J Gen Virol 83(Pt 10): 2385-92.

122
Huang, C., X. Zhang, Q. Lin, X. Xu, Z. Hu and C. L. Hew (2002b). "Proteomic
analysis of shrimp white spot syndrome viral proteins and characterization of a novel
envelope protein VP466." Mol Cell Proteomics 1(3): 223-31.
Huang, C. C. and Y. L. Song (1999). "Maternal transmission of immunity to white spot
syndrome associated virus (WSSV) in shrimp (Penaeus monodon)." Dev Comp Immunol
23(7-8): 545-52.
Inouye, K., S. Miwa, N. Oseko, H. Nakano and T. Kimura (1994). "Mass mortalities
of cultured kuruma shrimp, Penaeus japonicus, in Japan in 1993: electron microscopic
evidence of the causative virus." Fish Pathol. 29: 149-158.
Jensen, O. N., T. Houthaeve, A. Shevchenko, S. Cudmore, T. Ashford, M. Mann, G.
Griffiths and J. Krijnse Locker (1996). "Identification of the major membrane and core
proteins of vaccinia virus by two-dimensional electrophoresis." J Virol 70: 7485-7497.
Jones, T. A., J. Y. Zou, S. W. Cowan and M. Kjeldgaard (1991). "Improved methods
for building protein models in electron-density maps and the location of errors in these
models." Acta Crystallogr A 47: 110-119.
Kielian, M. (2006). "Class II virus membrane fusion proteins." Virology 344(1): 38-47.
Kielian, M. and F. A. Rey (2006). "Virus membrane-fusion proteins: more than one way
to make a hairpin." Nat Rev Microbiol 4(1): 67-76.
Knipe, D. M. and P. M. Howley (2001). "Fundamental Virology 4th Edition."
Lippincott Williams and Wilkins Press.
Koppers-Lalic, D., E. A. Reits, M. E. Ressing, A. D. Lipinska, R. Abele, J. Koch, M.
Marcondes Rezende, P. Admiraal, D. van Leeuwen, et al. (2005). "Varicelloviruses

123
avoid T cell recognition by UL49.5-mediated inactivation of the transporter associated
with antigen processing." Proc Natl Acad Sci U S A 102(14): 5144-9.
Kraulis, P. J. (1991). "MOLSCRIPT: a program to produce both detailed and schematic
plots of protein structures." J. Appl. Cryst. 24: 946-950.
Kuhn, P., K. Wilson, M. G. Patch and R. C. Stevens (2002). "The genesis of high-
throughput structure-based drug discovery using protein crystallography." Curr Opin
Chem Biol 6(5): 704-10.
Lander, E. S., L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K.
Devon, K. Dewar, M. Doyle, et al. (2001). "Initial sequencing and analysis of the human
genome." Nature 409(6822): 860-921.
Laskowski, R. A., D. S. Moss and J. M. Thornton (1993). "Main-chain bond lengths
and bond angles in protein structures." J. Mol. Biol. 231: 1049-1067.
Leu, J. H., J. M. Tsai, H. C. Wang, A. H. Wang, C. H. Wang, G. H. Kou and C. F.
Lo (2005). "The unique stacked rings in the nucleocapsid of the white spot syndrome
virus virion are formed by the major structural protein VP664, the largest viral structural
protein ever found." J Virol 79(1): 140-9.
Li, H., Y. Zhu, X. Xie and F. Yang (2006). "Identification of a novel envelope protein
(VP187) gene from shrimp white spot syndrome virus." Virus Res 115(1): 76-84.
Li, H. X., X. L. Meng, J. P. Xu, W. Lu and J. Wang (2005a). "Protection of crayfish,
Cambarus clarkii, from white spot syndrome virus by polyclonal antibodies against a
viral envelope fusion protein." J Fish Dis 28(5): 285-91.

124
Li, L., X. Xie and F. Yang (2005b). "Identification and characterization of a prawn
white spot syndrome virus gene that encodes an envelope protein VP31." Virology
340(1): 125-32.
Li, Q., Y. Chen and F. Yang (2004). "Identification of a collagen-like protein gene from
white spot syndrome virus." Arch Virol 149(2): 215-23.
Liang, X., B. Chow, C. Raggo and L. A. Babiuk (1996). "Bovine herpesvirus 1 UL49.5
homolog gene encodes a novel viral envelope protein that forms a disulfide-linked
complex with a second virion structural protein." J Virol 70(3): 1448-54.
Liang, Y., J. Huang, X. L. Song, P. J. Zhang and H. S. Xu (2005). "Four viral proteins
of white spot syndrome virus (WSSV) that attach to shrimp cell membranes." Dis Aquat
Organ 66(1): 81-5.
Liu, Z., H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An and W. Chang (2004).
"Crystal structure of spinach major light-harvesting complex at 2.72 A resolution."
Nature 428(6980): 287-92.
Lo, C. F., C. H. Ho, C. H. Chen, K. F. Liu, Y. L. Chiu, P. Y. Yeh, S. E. Peng, H. E.
Hsu, H. C. Liu, et al. (1997). "Detection and tissue tropism of white spot syndrome
baculovirus (WSBV) in captured brooders of Penaeus monodon with a special emphasis
on reproductive organs." Dis Aquat Organ 30: 53-72.
Lo, C. F., C. H. Ho, S. E. Peng, C. H. Chen, H. E. Hsu, Y. L. Chiu, C. F. Chang, K. F.
Liu, M. S. Su, et al. (1996). "White spot syndrome associated virus (WSBV) detected in
cultured and captured shrimp, crabs and other arthropods." Dis. Aquat. Org. 27: 215-225.
Lu, C. P., S. Zhu, F. S. Guo and S. Y. Wu (1997). "Electron microscopic observation
on a non-occluded baculo-like virus in shrimps." Arch Virol 142(10): 2073-8.

125
Luft, J. R., R. J. Collins, N. A. Fehrman, A. M. Lauricella, C. K. Veatch and G. T.
DeTitta (2003). "A deliberate approach to screening for initial crystallization conditions
of biological macromolecules." J Struct Biol 142(1): 170-9.
Marsden, R. L., J. A. Ranea, A. Sillero, O. Redfern, C. Yeats, M. Maibaum, D. Lee,
S. Addou, G. A. Reeves, et al. (2006). "Exploiting protein structure data to explore the
evolution of protein function and biological complexity." Philos Trans R Soc Lond B
Biol Sci 361(1467): 425-40.
McPherson, A. (1999). "Crystallization of biological Macromolecules. Cold Spring
Harbor: Cold Spring Harbor Laboratory press."
Merckel, M. C., J. T. Huiskonen, D. H. Bamford, A. Goldman and R. Tuma (2005).
"The structure of the bacteriophage PRD1 spike sheds light on the evolution of viral
capsid architecture." Mol Cell 18(2): 161-70.
Merrit, E. A., and Bacon, D. J. (1997). Methods Enzymol. 277: 505-524.
Momoyama, K., M. Hiraoka, H. Nakano, H. Koube, K. Inouye and N. Oseko (1994).
"Mass mortalities of cultured kuruma shrimp, Penaeus japonicus, in Japan in 1993:
histopathological study." Fish Pathol. 29: 141-148.
Otwinowski, Z. and W. Minor (1997). "Processing of X-ray diffraction data collected in
oscillation mode." Methods Enzymol. 276: 307-326.
Reinisch, K. M., M. L. Nibert and S. C. Harrison (2000). "Structure of the reovirus
core at 3.6 A resolution." Nature 404(6781): 960-7.
Rhodes, G. (2000). "crystallography made crystal clear, second edition. Academic
Press."

126
Roberts, M. M., J. L. White, M. G. Grutter and R. M. Burnett (1986). "Three-
dimensional structure of the adenovirus major coat protein hexon." Science 232(4754):
1148-51.
Rossmann, M. G. and J. E. Johnson (1989). "Icosahedral RNA virus structure." Annu
Rev Biochem 58: 533-73.
Sambrook, J. and D. W. Russell (2001). "Molecular Cloning: A Laboratory Manual
(3rd Edition)." Cold Spring Harbor Laboratory.
Spahn, C. M., R. Beckmann, N. Eswar, P. A. Penczek, A. Sali, G. Blobel and J.
Frank (2001). "Structure of the 80S ribosome from Saccharomyces cerevisiae--tRNA-
ribosome and subunit-subunit interactions." Cell 107(3): 373-86.
Stehle, T., S. J. Gamblin, Y. Yan and S. C. Harrison (1996). "The structure of simian
virus 40 refined at 3.1 A resolution." Structure 4(2): 165-82.
Terwilliger, T. C. (2000). "Maximum-likelihood density modification." Acta Crystallogr
D Biol Crystallogr 56(Pt 8): 965-72.
Terwilliger, T. C. and J. Berendzen (1999). "Automated MAD and MIR structure
solution." Acta Crystallogr D Biol Crystallogr 55(Pt 4): 849-61.
Tsai, J. M., H. C. Wang, J. H. Leu, H. H. Hsiao, A. H. Wang, G. H. Kou and C. F.
Lo (2004). "Genomic and proteomic analysis of thirty-nine structural proteins of shrimp
white spot syndrome virus." J Virol 78(20): 11360-70.
Tsai, J. M., H. C. Wang, J. H. Leu, A. H. Wang, Y. Zhuang, P. J. Walker, G. H. Kou
and C. F. Lo (2006). "Identification of the nucleocapsid, tegument, and envelope
proteins of the shrimp white spot syndrome virus virion." J Virol 80(6): 3021-9.

127
van Hulten, M. C., M. Westenberg, S. D. Goodall and J. M. Vlak (2000).
"Identification of two major virion protein genes of white spot syndrome virus of
shrimp." Virology 266(2): 227-36.
van Hulten, M. C., J. Witteveldt, S. Peters, N. Kloosterboer, R. Tarchini, M. Fiers,
H. Sandbrink, R. K. Lankhorst and J. M. Vlak (2001a). "The white spot syndrome
virus DNA genome sequence." Virology 286(1): 7-22.
van Hulten, M. C., J. Witteveldt, M. Snippe and J. M. Vlak (2001b). "White spot
syndrome virus envelope protein VP28 is involved in the systemic infection of shrimp."
Virology 285(2): 228-33.
Walter, T. S., J. M. Diprose, C. J. Mayo, C. Siebold, M. G. Pickford, L. Carter, G. C.
Sutton, N. S. Berrow, J. Brown, et al. (2005). "A procedure for setting up high-
throughput nanolitre crystallization experiments. Crystallization workflow for initial
screening, automated storage, imaging and optimization." Acta Crystallogr D Biol
Crystallogr 61(Pt 6): 651-7.
Wang, C. H., C. F. Lo, J. H. Leu, C. M. Chou, P. Y. Yeh, M. C. Tung, C. F. Chang,
M. S. Su and G. H. Kou (1995). "Purification and genomic analysis of baculovirus
associated with white spot syndrome (WSBV) of Penaeus monodon." Dis Aquat Organ
23: 239-242.
Westenberg, M., B. Heinhuis, D. Zuidema and J. M. Vlak (2005). "siRNA injection
induces sequence-independent protection in Penaeus monodon against white spot
syndrome virus." Virus Res 114(1-2): 133-9.
Westermeier, R. N., Tom (2002). Proteomics in Practice. A Laboratory Manual of
Proteome Analysis, Wiley-VCH, Weinheim.

128
Wommack, K. E. and R. R. Colwell (2000). "Virioplankton: viruses in aquatic
ecosystems." Microbiol Mol Biol Rev 64(1): 69-114.
Wongteerasupaya, C., J. E. Vickers, S. Sriurairatana, G. L. Nash, A. Akarajamorn,
V. Boonsaeng, S. Panyim, A. Tassanakajon, B. Withyachumnarnkul, et al. (1995).
"A non-occluded, systemic baculovirus that occurs in cells of ectodermal and
mesodermal origin and causes high mortality in black tiger prawn Penaeus monodon "
Dis Aquat Organ 21: 69-77.
Wu, W., L. Wang and X. Zhang (2005). "Identification of white spot syndrome virus
(WSSV) envelope proteins involved in shrimp infection." Virology 332(2): 578-83.
Xie, X., L. Xu and F. Yang (2006). "Proteomic analysis of the major envelope and
nucleocapsid proteins of white spot syndrome virus (WSSV)." J Virol.
Xie, X. and F. Yang (2005). "Interaction of white spot syndrome virus VP26 protein
with actin." Virology 336(1): 93-9.
Xie, X. and F. Yang (2006). "White spot syndrome virus VP24 interacts with VP28 and
is involved in virus infection." J Gen Virol 87(Pt 7): 1903-8.
Xu, Y. X., X. F. Wu, Y. F. Zhu, Z. R. Xu and W. D. Shen (2005). "[The expression of
VP19 gene from prawn white spot syndrome virus in silkworm, Bombyx mori using host
range-expanded HyNPV]." Sheng Wu Gong Cheng Xue Bao 21(5): 837-9.
Yaffe, M. B. (2005). "X-ray crystallography and structural biology." Crit Care Med
33(12 Suppl): S435-40.
Yang, F., J. He, X. Lin, Q. Li, D. Pan, X. Zhang and X. Xu (2001). "Complete genome
sequence of the shrimp white spot bacilliform virus." J Virol 75(23): 11811-20.

129
Yang, F., W. Wang, R. Z. Chen and X. Xu (1997). "A simple and efficient method for
purification of prawn baculovirus DNA." J Virol Methods 67(1): 1-4.
Yi, G., Z. Wang, Y. Qi, L. Yao, J. Qian and L. Hu (2004). "Vp28 of shrimp white spot
syndrome virus is involved in the attachment and penetration into shrimp cells." J
Biochem Mol Biol 37(6): 726-34.
Yoganandhan, K., S. Syed Musthaq, R. B. Narayanan and A. S. Sahul Hameed
(2004). "Production of polyclonal antiserum against recombinant VP28 protein and its
application for the detection of white spot syndrome virus in crustaceans." J Fish Dis
27(9): 517-22.
Yu, J., S. Hu, J. Wang, G. K. Wong, S. Li, B. Liu, Y. Deng, L. Dai, Y. Zhou, et al.
(2002). "A draft sequence of the rice genome (Oryza sativa L. ssp. indica)." Science
296(5565): 79-92.
Zhang, X., C. Huang, X. Tang, Y. Zhuang and C. L. Hew (2004). "Identification of
structural proteins from shrimp white spot syndrome virus (WSSV) by 2DE-MS."
Proteins 55(2): 229-35.
Zhang, X., C. Huang, X. Xu and C. L. Hew (2002a). "Identification and localization of
a prawn white spot syndrome virus gene that encodes an envelope protein." J Gen Virol
83(Pt 5): 1069-74.
Zhang, X., C. Huang, X. Xu and C. L. Hew (2002b). "Transcription and identification
of an envelope protein gene (p22) from shrimp white spot syndrome virus." J Gen Virol
83(Pt 2): 471-7.
Zhu, Y. B., H. Y. Li and F. Yang (2006). "Identification of an envelope protein (VP39)
gene from shrimp white spot syndrome virus." Arch Virol 151(1): 71-82.

130
7 Appendices
A. Media and solutions
LB broth (per liter) To 950 mL of MQ H2O, add: Bacto-tryptone 10 g Bacto-yeast extract 5 g NaCl 10 g Dissolve solutes. Adjust pH to 7.0 with 1 M NaOH. Adjust volume to 1L with
MQ H2O. Sterilize by autoclaving. 2 × YT medium (per liter) To 950 mL of MQ H2O, add: Bacto-tryptone 16 g Bacto-yeast extract 10 g NaCl 5 g Dissolve solutes. Adjust pH to 7.0 with 1 M NaOH. Adjust volume to 1L with
MQ H2O. Sterilize by autoclaving. SOC medium (per liter) To 950 mL of MQ H2O, add: Bacto-tryptone 20 g Bacto-yeast extract 5 g NaCl 0.58 g Dissolve solutes. Add 10 mL of 250 mM KCl. Adjust pH to 7.0 with 1 M NaOH.
Adjust volume to 980 mL with MQ water. Sterilize by autoclaving. Allow to cool to 60°C or less, and then add 20 mL of sterile 1M glucose. Add 10 mL of sterile 1 M MgCl2 just prior to use.
Tfb I (per liter) To 950 mL of MQ H2O, add: CH3COOK 2.94 g RbCl 12.1 g

131
CaCl2·2H2O 14.7 g MnCl2·4H2O 9.9 g Glycerol 150 mL Adjust pH to 5.8 with dilute acetic acid, add MQ H2O to 1 L and autoclave. Tfb II (per liter) To 950 mL of MQ H2O, add: MOPS 2.1 g CaCl2 11.1 g RbCl 1.21 g Glycerol 150 mL Adjust pH to 6.5 with 1 M NaOH, add MQ H2O to 1 L and autoclave. 10 × TNE buffer (per liter) To 950 mL of MQ H2O, add: Tris 6.05 g NaCl 5.85 g EDTA 0.292 g Adjust pH to 7.4 with 1 M HCl and add MQ H2O to 1 L. 10 × TE buffer (per liter) To 950 mL of MQ H2O, add: Tris 6.05 g EDTA 0.292 g Adjust pH to 7.4 with 1 M HCl and add MQ H2O to 1 L. 10 × TN buffer (per liter) To 950 mL of MQ H2O, add: Tris 6.05 g NaCl 9 g Adjust pH to 7.4 with 1 M HCl and add MQ H2O to 1 L.

132
B. Restriction enzymes All restriction enzymes were bought from New England Biolabs (NEB). BamH I NEB, #R0136L EcoR I NEB, #R0101L Hind III NEB, #R0104L Xho I NEB, #R0146L C. Other enzymes Protease K Sigma, #P2308 T4 DNA polymerase NEB, #M0203S T4 DNA ligase NEB, #M0202L T4 polynucleotide kinase Promega, #M4103 DNA Taq polymerase Promega, #M1665 RNase-free DNase I QIAGEN, #79254 Sequencing grade modified trypsin Promega, #V511A Trypsin-EDTA GIBCO, #25200-072 D. Antibiotics Ampicillin Sigma, #A-6140 E. Kits High pure PCR product purification kit Roche, #11732668001 Rapid DNA ligation kit Roche, #11635379001 QIAprep spin miniprep kit QIAGEN, #27106 QIAquick gel extraction kit QIAGEN, #28706 RNeasy mini kit QIAGEN, #74106 OneStep RT-PCR kit QIAGEN, #210212 BigDye® terminator v3.1 cycle sequencing kit Applied Biosystems, #4337455 Hi-Di formamide Applied Biosystems, #4311320 RC DC protein assay kit Bio-Rad, #500-0119 F. Crystallization screen kits Index Hampton Research, HR2-144 PEG/Ion Screen Hampton Research, HR2-126 Crystal Screen Kit Hampton Research, HR2-110 Crystal Screen 2 Hampton Research, HR2-112

133
Natrix Hampton Research, HR2-116 MembFac kit Hampton Research, HR2-114 Additive Screen Hampton Research, HR2-428 G. Equipments High Speed Centrifuge Beckman, J2-21 Ultracentrifuge Beckman, XL-90 MicroPulser® Electroporator Bio-Rad Thermal Cycler Bio-Rad ABI PRISM™ 3100 Genetic Analyzer Applied Biosystems Voyager-DE™ STR BioSpectrometry Workstation Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF™ optics Applied Biosystems Ultimate™ LC system Dionex LC Packings

134
8 List of Publications Publications: Xiaobo Zhang, Canhua Huang, Xuhua Tang, Ying Zhuang, Choy Leong Hew
Identification of structural proteins from shrimp white spot syndrome virus (WSSV) by 2DE-MS. Proteins: Structure, Function, and Bioinformatics, Volume 55, Issue 2, Pages 229-235 (1 May 2004)
Xuhua Tang, Jinlu Wu, Jayaraman Sivaraman, Choy Leong Hew
Crystal Structure of two major envelope proteins from shrimp white spot syndrome virus (WSSV). Journal of Virology.(accepted, under revision)
Xuhua Tang, Jayaraman Sivaraman, Choy Leong Hew
Expression, purification and crystallization of two major envelope proteins from white spot syndrome virus at 2.0 and 2.2Å resolution (submitted)
Abstract Publications: Xuhua Tang, Jayaraman Sivaraman, choy-leong Hew
X-ray crystal structural studies of VP26 of WSSV. FASEB Journal, 20(4), Part 1 (2006): A489.
Xuhua Tang, Jayaraman Sivaraman, choy-leong Hew Structural studies of envelop proteins of white spot syndrome virus. FASEB Journal, 19(4), Part 1 Suppl. S (2005): A312-A313.
Poster Presentations on Conferences:
1. X-ray Crystal Structural Studies of VP26 of WSSV Xuhua Tang, Jayaraman Sivaraman, choy-leong Hew
Experimental Biology 2006, April 1-5, 2006, San Francisco, U.S.A **Graduate Travel Award sponsored by ASBMB (American Society for Biochemistry and
Molecular Biology) 2. Structural studies of envelope proteins of white spot syndrome virus
Xuhua Tang, Jayaraman Sivaraman, choy-leong Hew Experimental Biology 2005, April 2-6, 2005, San Diego, U.S.A **Graduate Travel Award sponsored by ASBMB (American Society for Biochemistry and
Molecular Biology)

135
3. Crystal Structure of envelope protein VP26 of white spot syndrome virus
Xuhua Tang, Jayaraman Sivaraman, choy-leong Hew Biology in Asia International Conference, Dec 7-10, 2004, Nanyang Technological
University, Singapore **Graduate Conference Fellowship sponsored by Department of Biological Sciences, NUS
4. Identification and Structure Determination of envelope proteins of WSSV Xuhua Tang, Canhua Huang, choy-leong Hew
The 10th SCBA (International Symposium, July 18 -23, 2005, Beijing, China
5. Structural studies of envelope proteins of whit spot syndrome virus Xuhua Tang, Jayaraman Sivaraman, choy-leong Hew Third International Conference on Structural Biology & Functional Genomics, Dec 2004, Singapore
6. Functional and structural Genomics Studies of Marine Viral pathogens.
Xuhua Tang, Wenjun Song, Yang Liu, Jing Chen, Ying Zhuang and Choy Leong Hew International Conference on Marine Science and Technology, May 13-14, 2004, Taiwan
7. Structural Genomics Studies of Shrimp White Spot Syndrome Virus. Choy Leong Hew, Yang Liu, Xuhua Tang, Ying Zhuang, Canhua Huang, Zhengjun Li, Jianxing Song,
World Aquaculture Society, Hawaii, U.S.A. March 2004
8. Structural Genomics Studies of Shrimp White Spot Syndrome Virus. Yang Liu, Xuhua Tang, Ying Zhuang, Canhua Huang, Zhengjun Li, Jianxing Song, Choy-Leong Hew
Asian Pacific Aquaculture, Bangkok, September 2003
9. Structural Determination of the envelope proteins from the shrimp white spot syndrome virus.
Xuhua Tang and Choy Leong Hew The 4th Sino-Singapore Conference in Biotechnology, 11-13 Nov 2003, Singapore
10. Structural Determination and characterization of the envelope proteins from the shrimp white spot syndrome virus.
Xuhua Tang and Choy Leong Hew 8th Biological Science Graduate Congress, Department of Biological Sciences, NUS,
3-6 Dec, 2003

136