targeted extracellular delivery of indoleamine 2,3
TRANSCRIPT

TARGETED EXTRACELLULAR DELIVERY OF INDOLEAMINE 2,3 DIOXYGENASE VIA FUSION WITH GALECTIN 3
By
EVELYN R. BRACHO SANCHEZ
A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY
UNIVERSITY OF FLORIDA
2017

© 2017 Evelyn R. Bracho Sanchez

To my family, near and far, and their unconditional support

4
ACKNOWLEDGMENTS
I would like to express my deepest gratitude to my advisor, Dr. Benjamin
Keselowsky for his guidance, insight and influence during my development as a
scientist and engineer. To my committee and collaborators – Dr. Greg Hudalla, the
fusion proteins presented here were developed in partnership with him and his team,
Dr. Mark Wallet, Dr. Todd Brusko and Dr. Clayton Mathews for their support in
experimental design and guidance through the execution and analysis of immunological
assays, and Dr. Shannon Wallet for her contributions with the periodontal disease
model – your guidance through this process, countless suggestions and positive
feedback continues to be instrumental for my growth.
I would like to express my sincerest appreciation to all my lab mates and close
friends over the years. Thank you, Jamal and Matt for training me in my early career
and welcoming me into the Keselowsky Lab. Thank you, Maigan for explaining
immunology to me countless times, for entertaining my wildest theories, and teaching
me the joys of flow cytometry. Thank you, Antonietta, Maggie, Sabrina, Kevin and Josh
for cheering me on, for being my support system and safety net in the hardest and most
challenging times.
Lastly, I would like to thank my family. You have always been my source of
motivation, faith and energy and I share this accomplishment with you. To my parents,
Rosvel Bracho and Edith Sanchez, your unconditional love, sacrifices and example
continue to be the reason I thrive to be the best version of myself. Thank you for
believing in me even when I doubt myself, thank you for pushing me to always get back
up stronger and wiser. To my siblings, Edith and Rosvel, your courage to pursue your
dreams and remain authentic is inspiring, it is a blessing to have you on my corner. To

5
Jon, thank you for the countless ways you continue to love and support me and all my
goals, both personal and professional. To all my family in Venezuela, thank you for your
love through the distance and continuous encouragement, I am proud of my roots
because they always bring me back to you.

6
TABLE OF CONTENTS page
ACKNOWLEDGMENTS .................................................................................................. 4
LIST OF TABLES ............................................................................................................ 9
LIST OF FIGURES ........................................................................................................ 10
LIST OF ABBREVIATIONS ........................................................................................... 12
ABSTRACT ................................................................................................................... 15
CHAPTER
1 INTRODUCTION .................................................................................................... 17
Micro and Nano Material Carriers for Immunomodulation....................................... 17 Biomaterial-Carries ........................................................................................... 18
Polymeric ................................................................................................... 20 Micro- and nano-particles .......................................................................... 20
Micelles ...................................................................................................... 23 Dendrimers ................................................................................................ 24 Hydrogels ................................................................................................... 25
Lipids ......................................................................................................... 26 Metallic and inorganic ................................................................................ 27
Biologics..................................................................................................... 30
Fusion proteins .......................................................................................... 31
Carrier Properties Influencing Immune Responses .......................................... 33 Biomaterials-Based Immunomodulation of Dendritic Cells ..................................... 35
Innate and Adaptive Immunity Overview .......................................................... 37 Dendritic Cell Subsets ...................................................................................... 41
Inflammatory dendritic cell phenotype ........................................................ 42 Suppressive dendritic cell phenotype ......................................................... 43
Biomaterials-based Immunomodulation ........................................................... 44 Biomaterials-based DC Manipulation Toward an Inflammatory Phenotype ...... 44
Biomaterials as antigen carriers ................................................................. 45 Biomaterials as adjuvants and adjuvant carriers ........................................ 47
Biomaterials-based DC Manipulation Toward a Suppressive Phenotype ......... 50
2 EXTRACELLULAR INDOLEAMINE 2,3 DIOXYGENASE-TREATED DENDRITIC CELLS MAINTAIN AN IMMATURE PHENOTYPE AND SUPPRESS ANTIGEN-SPECIFIC T CELL PROLIFERATION ............................... 59
Background ............................................................................................................. 59 Indoleamine 2,3 Dioxygenase .......................................................................... 59
Tryptophan catabolism and the kynurenine pathway ................................. 59 Tissue and cellular expression ................................................................... 60

7
Role in immune regulation ......................................................................... 61
Materials and Methods............................................................................................ 62
IDO Characteristics and Activity Assay ............................................................ 62 Dendritic Cell Culture and Extracellular Enzyme Treatment ............................. 63 Dendritic Cell Phenotype and Maturation Resistance ...................................... 64 T Cell Isolation and Proliferation Assay ............................................................ 65 Statistical Analysis ............................................................................................ 66
Results .................................................................................................................... 66 IDO-treated DCs Resist LPS-induced Maturation ............................................ 66 IDO-treated DCs Suppress Antigen Specific Proliferation, and Suppression
is Active Enzyme Dependent ........................................................................ 68 Discussion .............................................................................................................. 70
3 FUSION OF GALECTIN 3 WITH A MODEL ENZYME AT THE N-TERMINUS PROLONGS RETENTION TIME IN VIVO .............................................................. 80
Background ............................................................................................................. 80 Galectin 3 ......................................................................................................... 80
NanoLuciferase ................................................................................................ 81 Materials and Methods............................................................................................ 82
Cloning of Galectin 3, Nanoluciferase and NanoLuc-Gal3 ............................... 82 Protein Expression and Purification .................................................................. 83 Mice and Cell Lines .......................................................................................... 84
Galectin 3 and NanoLucGal-3 Binding Affinity .................................................. 84 NanoLuc-Gal3 Binding Affinity to ECM Proteins ............................................... 84
NanoLuc-Gal3 Binding Affinity to Jurkat T Cells ............................................... 85
Jurkat T Cell Agglutination and Viability ........................................................... 85
Quantitative Precipitation Analysis of Galectin 3 and NanoLuc-Gal3 ............... 86 In Vivo Bioluminescence and Imaging .............................................................. 86
NanoLuc-Gal3 Tissue Distribution from Hock Injection .................................... 86 Alternative Injection Sites ................................................................................. 86 Statistical Analysis ............................................................................................ 87
Results .................................................................................................................... 87
NanoLuc-Galectin 3 Retains Binding Affinity for Sugar Moiety when Compared to WT-Gal3 .................................................................................. 87
Galectin 3 Retains Binding Affinity to Sugar Moiety on ECM and Serum Proteins When Fused to an Enzyme on the N-terminus ............................... 87
Galectin 3 Is Not Able to Self-assemble Into Pentamer and Precipitate Asialofetuin When Fused to NanoLuc on the N-terminus .............................. 88
Galectin 3 Binds to Jurkat T Cells but Does Not Induce Agglutination or Apoptosis ...................................................................................................... 88
NanoLuc-Gal3 Prevents Galectin 3-induced Cell Death ................................... 89 NanoLuc-Gal3 Binds Primary Splenocytes in a CRD Dependent Manner,
Binding Does Not Act as a Damage Associated Molecular Pattern .............. 90 Galectin 3 Targeting in NanoLuc-Gal3 Provides Prolonged Retention Time
When Administered Subcutaneously, Intraperitoneally and Intramuscularly .............................................................................................. 90

8
Galectin 3 Targeting in NanoLuc-Gal3 Provides Prolonged Retention Time When Administered Into the Hock with Minimal Drainage to Adjacent Tissues .......................................................................................................... 90
Discussion .............................................................................................................. 91
4 FUSION OF INDOLEAMINE 2,3 DIOXYGENASE WITH GALECTIN 3 RETAINS ENZYME AT INJECTION SITE AND HALTS INFLAMMATION IN VIVO ............. 103
Background ........................................................................................................... 103
Subcutaneous LPS-induced Inflammation...................................................... 103 Periodontal Disease ....................................................................................... 103
Immunopathogenesis ............................................................................... 104 Current treatments ................................................................................... 105
Materials and Methods.......................................................................................... 106
Cloning of IDO-Galectin 3 ............................................................................... 106
Protein Expression and Purification ................................................................ 106 IDO Enzymatic Activity Assay ........................................................................ 107 Galectin 3 and IDO-Gal3 Binding Affinity........................................................ 108
Antibodies ....................................................................................................... 108 Mice and Cell Lines ........................................................................................ 109
DC Maturation and Cytokine Release Profile ................................................. 109 Antigen Specific Co-cultures .......................................................................... 109 LPS Administration at the Hock as a Model of Inflammation .......................... 110
Quantitative PCR ............................................................................................ 110 Mass Spectrometry ........................................................................................ 110
Hock Infiltration ............................................................................................... 111
In vivo Bioluminescence and Imaging ............................................................ 111
Mucosal Induction of Inflammation ................................................................. 111 Subgingival Cytokine Production .................................................................... 112
Results .................................................................................................................. 112 IDO Activity and Gal3 Affinity Are Not Altered Upon Fusion ........................... 112 IDO Retains Immunomodulatory Properties When Fused to Galectin 3 ......... 112 IDO-Gal3 Reduces Inflammatory Cytokine Gene Expression at Various
Time Points. ................................................................................................ 114 IDO-Gal3 Modulates Local Tryptophan Catabolism ....................................... 115 IDO-Gal3 Modulates LPS-induced Inflammation Locally ................................ 116 Gal3 Provides Retention of a Model Enzyme at Subgingival Tissue,
Retention Time Is Not Altered by Infection State ......................................... 117
IDO-Gal3 Suppresses Inflammatory Cytokine and Chemokine Production .... 118
Discussion ............................................................................................................ 119
5 CONCLUSIONS AND FUTURE DIRECTIONS .................................................... 133
LIST OF REFERENCES ............................................................................................. 140
BIOGRAPHICAL SKETCH .......................................................................................... 162

9
LIST OF TABLES
Table page 1-1 Advantages and disadvantages of biomaterials carriers for
immunomodulation ............................................................................................. 19

10
LIST OF FIGURES
Figure page 1-1 Interaction of nano- and micro-scale biomaterial carriers with key members of
the innate and adaptive immune system ............................................................ 55
1-2 Simplified immune response to foreign pathogen ............................................... 56
1-3 Dendritic cell phenotype dictates T cell function. ................................................ 57
1-4 Overview of biomaterials-based dendritic cell modulation. ................................. 58
2-1 Tryptophan metabolism through the kynurenine pathway .................................. 74
2-2 Dendritic cells treated with IDO maintain an immature morphology even after LPS challenge .................................................................................................... 75
2-3 Treatment with soluble IDO does not induce dendritic cell death ....................... 76
2-4 Dendritic cells treated with soluble IDO resist LPS maturation. .......................... 77
2-5 Treatment of dendritic cells with exogenous IDO inhibits their IL-12 secretion and maintains IL-10 production. ......................................................................... 77
2-6 IDO-treated DCs suppress antigen specific T cell proliferation, and suppression is active enzyme dependent ........................................................... 78
2-7 Tryptophan depletion and kynurenine accumulation are needed to suppress antigen specific proliferation. .............................................................................. 79
3-1 Classification of galectins ................................................................................... 96
3-2 Fusion protein construct. .................................................................................... 96
3-3 Galectin 3 retains binding affinity to sugar moiety when fused to an enzyme on the N-terminus ............................................................................................... 97
3-4 Biology associated with Galectin 3 is disrupted when fused to an enzyme on the N-terminus .................................................................................................... 98
3-5 NanoLuc-Gal3 fusion prevents wild type Galectin 3-mediated agglutination and apoptosis of Jurkat T cells. .......................................................................... 99
3-6 NanoLuc-Gal3 fusion binds primary splenocytes in a CRD dependent manner, binding to DCs does not act as a damage associated molecular pattern .............................................................................................................. 100

11
3-7 Galectin 3 targeting in NanoLuc-Gal3 provides prolonged retention when administered subcutaneously, intraperitoneally, and intramuscularly. .............. 101
3-8 Galectin 3 targeting provides retention of a model enzyme at the injection site with minimal drainage to adjacent tissues. ....................................................... 102
4-1 Enzymatic activity of IDO is not altered upon fusion with Galectin 3, Galectin 3 retains binding affinity to sugar moiety upon fusion with IDO at N-terminus .. 124
4-2 Indoleamine 2,3-dioxygenase maintains an immature dendritic cell phenotype when fused to Galectin 3. ............................................................... 125
4-3 IDO-Gal3-treated DCs attenuate antigen specific T cell proliferation, suppression is active enzyme dependant. ........................................................ 126
4-4 IDO-Gal3 blocks subcutaneous LPS-induced inflammatory cytokine gene expression. ....................................................................................................... 127
4-5 IDO-Gal3 modulates tryptophan metabolism at the injection site. .................... 128
4-6 IDO-Gal3 blocks subcutaneous LPS-induced inflammatory response, suppression is not systemic .............................................................................. 129
4-7 Galectin 3 provides retention of a model enzyme in the subgingival space; retention profile is not altered by disease state................................................. 130
4-8 Schematic representation of infection model used to induce periodontal disease in mice. ................................................................................................ 131
4-9 Inflammatory cytokine and chemokine production in the subgingival space is significantly reduced by administration of IDO-Gal3 both prophylactically and therapeutically. ................................................................................................. 132
5-1 Proposed mechanism for IDO-related suppression of inflammation by blockage of NF-κβ pathway initiated through binding of LPS to TLR4 on surface of immune cell. ..................................................................................... 138
5-2 Proposed in vivo cytokine network affected by the introduction of IDO. ........... 139

12
LIST OF ABBREVIATIONS
AhR Aryl hydrocarbon receptor
APC Antigen presenting cells
B6 C57BL/6
B-PER Bacterial protein extraction reagent
CD Cluster differentiation
CFSE Carboxyfluorescein succinimidyl ester
CRD Carbohydrate recognition domain
CREB cAMP response element binding protein
DCs Dendritic cells
DNAse Deoxyribonuclease
EDTA Ethylenediaminetetraacetic acid
ELISA Enzyme linked immunosorbent assay
Gal3 Galectin 3
GM-CSF Granulocyte-macrophage colony stimulating factor
H&E Haemotoxylin and Eosin
IACUC Institutional Animal Care and Use Committee
IDO Indoleamine 2,3 dioxygenase
IFNγ Interferon gamma
IΚΚ IκB Kinase
IL-10 Interleukin 10
IL-12 Interleukin 12
IL-1β Interleukin 1 beta
IL-33 Interleukin 33
IL-6 Interleukin 6

13
IM Intramuscular
IP Intraperitoneal
IP10 Interferon gamma-induced protein 10
IPTG isopropyl β-D-1-thiogalactopyranoside
IRAK IL-1 Receptor-Associated Kinases
IV Intravenous
KC Keratinocyte chemoattractant
LacNAc N-Acetyl-D-lactosamine
LATs L-amino acid transporters
LPS Lipopolysaccharide
MAPK Mitogen-Activated Protein Kinase
MCP1 Monocyte chemotactic protein 1
MHC I/II Major histocompatibility complex class I and II
MIP2 Macrophage inflammatory protein 2
MT 1-methyl tryptophan
MyD88 Myeloid Differentiation Primary Response Gene 88
NanoLuc Nanoluciferase
NanoLuc-Gal3 Nanoluciferase-Galectin 3 fusion protein
NF-κβ nuclear factor kappa-light-chain-enhancer of activated B cell
NK cells Natural killer cells
OT-I C57BL/6-Tg(TcraTcrb)1100Mjb/J mice
OT-II B6.Cg-Tg(TcraTcrb)425Cbn/J mice
OVA Ovalbumin
PBS Phosphate-buffered saline
PD Periodontal disease

14
SDS-PAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis
Subq Subcutaneous
TAK1 Transforming growth factor-β-Activated Kinase 1
Teff Effector T cell
TLR4 Toll-like receptor 4
TNFα Tumor necrosis factor alpha
TRAF6 TNF Receptor-Associated Factor
Treg Regulatory T cell

15
Abstract of Dissertation Presented to the Graduate School of the University of Florida in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
TARGETED EXTRACELLULAR DELIVERY OF INDOLEAMINE 2,3 DIOXYGENASE
VIA FUSION WITH GALECTIN 3
By
Evelyn R. Bracho Sanchez
December 2017
Chair: Benjamin Keselowsky Major: Biomedical Engineering
Suppression of inflammatory processes would be beneficial for the treatment of
autoimmune and inflammatory diseases as well as whole organ transplantation. Current
treatments are limited to non-specific systemic immunosuppressant drugs which carry
harmful off-target effects and the potential for opportunistic infections. To address these
limitations, scientists have focused on reprogramming patients own immune systems to
take advantage of naturally occurring tolerance mechanisms such as the upregulation of
indoleamine 2,3 dioxygenase (IDO). Indoleamine 2,3 dioxygenase is an intracellular
enzyme responsible for initiating tryptophan catabolism to kynurenine and downstream
metabolites, and is primarily expressed by lymphoid organs and the placenta.
Overexpression of IDO has been shown to induce T cell tolerance in transplant and
pregnancy. Thus, efforts have primarily focused on increasing intracellular enzyme
expression in antigen presenting cells using pharmacological agents, cytokines and
genetic manipulation. These approaches are constrained by several factors, including
the potentially deleterious biological effects of IDO-inducing agents such as
lipopolysaccharide (LPS) or interferon gamma (IFNγ), or malignancies and even
lethality associated with systemic overexpression. Here we report a new approach for

16
the metabolic programming of immune environments through the local delivery and
retention of IDO, a potent immunomodulator, through fusion with the carbohydrate
binding lectin Galectin 3 (Gal3). This study establishes IDO as an immunomodulator in
the extracellular space and uniquely employs its enzymatic activity for the suppression
of excessive inflammation. Galectin 3 is a member of the lectin family with affinity for N-
Acetyl-D-lactosamine (LacNAc) glycosylation patterns present, for example, on proteins
of the extracellular matrix, such as laminin, and immune cell surfaces. A fusion chimera,
IDO-Gal3, was developed, characterized and demonstrated to maintain function for both
the targeting and enzymatic moieties, in vitro and in vivo. Galectin 3 confers
subcutaneous and oral submucosal retention for 7 and 10 days, respectively, and IDO-
Gal3 suppresses LPS-induced inflammation in a subcutaneous model as well as in a
polymicrobial model of periodontal disease. The findings from this research project will
provide valuable information regarding IDO-Gal3 as a treatment for chronic
inflammation, autoimmune diseases and transplant tolerance.

17
CHAPTER 1 INTRODUCTION
Micro and Nano Material Carriers for Immunomodulation
The immune system is intricately organized, composed of multiple layers that
work in unison to protect the host against foreign invaders and provides homeostatic
regulation of self and non-self. Appropriate immune recognition initiates isolation and
elimination of pathogens, and tolerance to self or benign antigens, such as food
proteins1. Due to its crucial role in health and disease, manipulation of the immune
system by therapeutic interventions is of great interest for the amelioration of
malignancies. Immunostimulation may be sought, as in the case of vaccines and
adjuvants for infectious diseases and cancer. Other times, immunosuppression, or
diminished immune potency, is desired. While systemic immunosuppressants lower the
body’s ability to fight foreign invaders systemic treatments continue to be required for
allergies, autoimmune diseases and transplant rejection.
Cell and whole-organ transplantation has become a standard procedure for the
treatment of numerous conditions including cardiac, hepatic and renal failure2. Donor
tissue is normally derived from an allogenic source, which upon introduction to the
recipient activates a cascade of immune responses. Much progress has been made due
to immunosuppressant therapies, however chronic rejection and dysfunction persist,
with only 47-61% of grafts surviving to the 10 year mark3. Therefore, the induction of
antigen specific tolerance to transplanted tissues remains a primary objective. Antigen
specific therapies aim at preventing the host from rejecting cells or whole-organs while
maintaining complete and functional activity to fight foreign invaders. To achieve
tolerance, key cellular players must be engaged and re-programmed, including antigen

18
presenting cells (APCs,) such as dendritic cells (DCs) and macrophages, T and B
lymphocytes, a strategy currently been explored through the use of nano- and micro-
technologies.
Biomaterials offer unique opportunities to modulate the immune system either
toward a suppressive or stimulatory state by engaging components of the innate and
adaptive immune system (Figure 1-1). Biomaterials are synthetic or naturally-derived
materials suitable for incorporation into the human body, and are meant to perform,
enhance or replace physiological functions4. Given the variety and complexity of signals
that must work together to achieve an immunological outcome, the type of biomaterial,
its structure and properties should be considered when designing nano- and micro-
technologies to ameliorate health concerns. In this chapter, we focus on recent
advancements of biomaterials-based nano- and micro-technologies for
immunomodulation.
Biomaterial-Carries
Many nano and micro-scale biomaterial systems have been described as
platforms for targeting and delivery of therapeutic agents and effective
immunomodulation. These agents can be encapsulated, conjugated, fused or adsorbed
onto the material system and co-delivered with excipients or stabilizers to achieve ideal
release profiles and immunological responses5. Many carriers have been proposed and
can be sorted into polymeric, lipids, metals and inorganics. Recent advances and
limitations of each carrier type are highlighted below and summarized (Table 1).

19
Table 1-1. Advantages and disadvantages of biomaterials carriers for immunomodulation
ADVANTAGE DISADVANTAGE
POLYMERIC CARRIERS
MICRO AND NANO PARTICLES
High stability High loading capacity of hydrophilic and hydrophobic molecules Tunable properties Feasibility of various routes of administration
Aggregation leads to difficult handling Fabrication processes are harsh for proteins and enzymes
MICELLES Tunable properties Controlled release Incorporation of poorly soluble molecules
Require stabilizers at low concentrations Difficult polymer synthesis
DENDRIMERS Dimensional length scaling and narrow size distribution Controlled conjugations
Low biocompatibility Material selection may result in increased toxicity High manufacturing cost
HYDROGELS Controlled release Highly biocompatible
Low tensile strength Limited quantity and homogeneity of loads of hydrophobic molecules
LIPID CARRIERS LIPOSOMES Stable encapsulation of both
hydrophilic and hydrophobic molecules Biocompatible, biodegradable Ability to cross lipid bilayer Non-immunogenic
High manufacturing costs Difficult to sterilize Short shelf life and stability
METAL AND INORGANIC
GOLD Controllable synthesis Biocompatible
Non-biodegradable Biodistribution
CARBON Controllable synthesis Multivalent surface conjugation
Toxicity Non-biodegradable
SILICA Ease of fabrication Tunable properties
Non-biodegradable Require surface functionalization for biocompatibility
BIOLOGICS ANTIBODIES High affinity
High bioactivity High doses required High production and distribution cost
PROTEINS Naturally derived Low immunogenicity
Short half-life in vivo Formulation changes upon packaging

20
Polymeric
Polymeric biomaterials have been extensively investigated for the delivery of
drugs, biomolecules and genes. Biocompatibility, low toxicity and biodegradability have
promoted their use as a promising strategy. Additionally, chemical structures and
compositions can be easily tuned to achieve desirable properties such as controlled
release profiles. Examples of polymers widely used for delivery applications include
polyesters (e.g., poly(lactic acid), poly(glycolic acid) and their copolymers),
polyorthoesters, polyanhydrides and polycarbonates. These materials can be fabricated
in the form of particles, micelles, dendrimers and hydrogels, and have each been
extensively studied.
Micro- and nano-particles
The most common form of polymeric carriers are micro and nanoparticles, which
are highly stable can effectively entrap and adsorb hydrophobic as well as hydrophilic
molecules, and are easily administered through various routes. Particle sizes ranging
from nanometers to micrometers can be transported through cellular and subcellular
barriers making them amenable for site specific targeting. In the context of
immunomodulation, for example, polymeric particles can be designed to display
proteins commonly expressed by DCs, thus mimicking APCs and dictating T cell
activation and differentiation. These have been referred to as artificial APCs (aAPCs),
the most widely used form consists of polystyrene beads surface coated with anti-
CD3/CD28 antibodies allowing the delivery of antigen-independent signal to polyclonal
T cells. For antigen-specificity, polystyrene beads have also been coated with MHC-
peptide single chain construct dimers or tetramers and have been used for the ex vivo
expansion of tumor specific T cells.

21
In addition to stimulation of the T cell receptor signaling pathway through
CD3/CD28, co-stimulation and inhibitory signals can be provided by covalently binding
agonistic or antagonistic ligands to the beads. In a recent study, Hippen et al.,
demonstrated anti-CD3 antibody-loaded, aAPCs that displayed CD64 and CD86 on
their surface were able to expand human natural regulatory T cells (nTregs) 6. Upon
original contact nTregs numbers increased 80-fold and a single re-stimulation increased
expansion to 3,000 fold while maintaining Foxp3 expression and suppressor function.
These cells were then infused into an immune-deficient mice and significantly reduced
graft-vs-host lethality, a disease most commonly seen following a bone marrow
transplantation. In another study, Clemente-Casares et al., showed that systemic
delivery of nanoparticles coated with autoimmune-disease relevant peptides bound to
MHC II molecules triggers the generation and expansion of antigen-specific regulatory
CD4+ T cells type 1 (Tr1)-like cells in vivo preventing and reversing type 1 diabetes7.
Tolerance can be introduced not only by transplanting regulatory T cells but by
manipulating existing ones. Low dose IL-2 treatment has been shown to increase the
counts of regulatory T cells ameliorating graft vs host disease and a number of other
autoimmune and inflammatory conditions 8. Biomaterials engineering has incorporated
these findings and increased the functionality of aAPCs by also providing controlled
release of encapsulated cytokines. For example, Steenblock et al., fabricated poly
(lactic co-glycolic) acid (PLGA) microparticles surface-modified with anti-mouse CD3
and CD28 antibodies, and encapsulating IL-2. The authors demonstrated aAPCs
stimulated T cells more strongly than particles with surface ligands alone, and 10-fold

22
higher than soluble supplementation of the cytokine9. Furthermore, they showed the
response of T cells was dependent on the sustained release of IL-2.
In a different approach, polymeric particles can be designed for interactions with
phagocytes and lymphocytes in mind either for the detection or prevention of transplant
rejection 10-12. Early studies aimed to limit immune detection by avoiding protein
adsorption and opsonization, and the use of polyethylene glycol coatings on particles
rapidly emerged, as the polyethylene glycol layer sterically resists protein interactions13.
However, recent efforts have instead focused on actively directing phenotype and
function of immune cells14,15. For example, Shirali et al., fabricated mycophenolic acid
loaded PLGA nanoparticles to prolong murine skin allograft survival by upregulating PD-
L1 on dendritic cells 16; Hlavaty et al., developed PLG antigen-loaded nanoparticles to
promote bone marrow transplant tolerance in sex-mismatched C57BL/6 mice by
interactions with CD4+ and CD8+ T cells17 and Pan Q et al., administered corticosteroid-
loaded PLGA nanoparticles weekly for the prevention of corneal allograft rejection in
rats18. These studies represent an advancement in transplant therapies without the use
of a systemic immunosuppressant. Additionally, Lewis et al. developed a dual
microparticle delivery system using PLGA to encapsulate combinations of immuno-
suppressive factors to condition DCs toward a tolerance-inducing phenotype19.
Following up on this approach, Lewis et al., subcutaneously administered
phagocytosable (encapsulating Vitamin D3 and insulin peptide as antigen) and non-
phagocytosable microparticles (encapsulating TGF-β1 and GM-CSF) to non-obese
diabetic mice and demonstrated 40% protection from Type 1 Diabetes development 20,
representing one of the few microparticle vaccine system to successfully prevent

23
autoimmune diabetes. Furthermore, combinatorial approaches for loading adjuvants
into polymeric carriers are also being investigated, and tested using cellular based
microarrays 21-23
Micelles
Micelles are colloidal particles consisting of self-assembled aggregates of
amphiphilic molecules or surfactants. In aqueous solutions and at low concentrations,
amphiphiles exist as monomers. However, as their concentration increases,
thermodynamic processes drive the formation of aggregates sequestering hydrophobic
regions into a core like structure surrounded by hydrophilic shell24. Micelles have been
used to contain hydrophobic or poorly soluble drugs within its core. Following
administration, dilution occurs rapidly and if the concentration drops below the critical
micelle concentration, the stability can be compromised. However, with the addition of
stabilizers, micelle carriers have successfully been employed by various groups in the
context of immunomodulation. For example, Dane et al., delivered drug-loaded micelles
to lymph nodes and prolonged allograft survival. Specifically, the authors used
poly(ethylene glycol)-bl-poly(propylene sulfide) block copolymers 50 nm micelles to
encapsulate rapamycin and tacrolimus and showed a 2-fold improvement in survival of
MHC-mismatched tail skin allograft in a BALB/c mouse model 25. In another study, Miki
et al., supplemented a dendritic cell vaccine with polymeric nano-micelles comprised of
PEG-polyGlutamate block co-polymer carrying IL-2, and demonstrated enhanced intra-
tumoral accumulation of antigen-specific cytotoxic T lymphocytes in EG7 tumor bearing
mice. Furthermore, this micelle system was able to prolong IL-2 retention in blood
circulation, significantly increasing DC vaccine efficacy against tumors 26.

24
Dendrimers
Dendrimers can be linear, cross-linked, or branched macromolecules forming a
star-like structure. They have been referred to as artificial proteins based on their
dimensional length scaling and narrow size distribution. Dendrimers offer a robust,
covalently fixed, three dimensional structure that can be divided into three domains: (i)
the multivalent surface, containing a large number of potentially reactive sites, (ii) the
interior shell consisting of branches referred to as dendrons, and (iii) the core 27. The
first and most extensively studied dendrimer, poly(amido amine) (PAMAM), is
synthesized by using a step-wise fashion. This results in precisely defined structures
with large functional groups at the surface providing opportunities for controlled
conjugations of drugs and targeting moieties, separating dendrimers from other carriers
by virtue of their well-controlled chemistry. However, dendrimers have shown low
biocompatibility and the material selection may lead to increased toxicity.
Much of the work with dendrimers has focused on the encapsulation in its core
and the covalent attachment of drugs to its surface. For example, carbohydrates
constitute an important class of biological recognition molecules, displaying a wide
variety of spatial structures due to their branching and various occurring isomers. In
order to achieve high binding with cell surfaces, carbohydrates must be presented in a
multivalent or cluster fashion28, and functionalization of dendrimers provides an
excellent platform for such multivalent presentation. For example, Heimburg et al.,
created a PAMAM dendrimer bearing the Thomsen-Friedenreich carbohydrate antigen
(a well-documented antigen for the detection and therapy of carcinomas, particularly
relevant to breast cancer) that was able to raise IgG antibodies against the antigen. This
successfully impeded binding of malignant cells to vascular endothelium, blocking a

25
metastatic step and providing a survival advantage29,30. Although progress has been
made to understand the capability of dendrimers as therapeutics in immune
applications, their clinical translation has been limited by some concerns over
biocompatibility and toxicity. Dendrimers can have affinity for metal ions, lipids, proteins
and nucleic acids sometimes resulting in the disruption of biological processes31.
Additionally, the expense associated with the multi-step synthesis of dendrimers has
also been a concern for translation.
Hydrogels
Hydrogels are three-dimensional, cross-linked networks of highly water-soluble
polymers with high porosity encompassing a wide range of chemical compositions and
bulk properties. They can be formulated in a variety of different forms including micro-
and nano- particles, scaffolds, coatings and films32. Hydrogels can be tuned by
controlling the density of cross-links in the gel matrix and drugs can be loaded with
factors for short-term release at a rate dependent on the diffusion coefficient of the
molecule through the network. Hydrogels can also be highly biocompatible, with a high
water content and similar mechanical properties as the extracellular environment of soft
tissues.
The utilization of hydrogels in transplant applications dates back to the 1980s
when encapsulation of pancreatic islets restricted contact between the donor islets and
the recipients immune cells in diabetic rats33. The pore size of the hydrogel was large
enough to allow small molecules and signaling proteins, including insulin, through but
small enough to block cells and the complement system which resulted in a delayed
rejection. In a more recent study conducted by Neufeld et al., encapsulation of rat islets
within polymer film membranes restored normoglycemia in chemically-induced diabetic

26
pigs for three months with no additional immunosuppression required 34. In principle,
encapsulation decreases the need for systemic immunosuppression, however eventual
loss of glycemic control continues to be observed 35
Hydrogels also have limitations. For instance, low tensile strengths limits their
use in load bearing applications, and can cause premature dissolution. Additionally, due
to their high-water content the quantity and homogeneity of agents loaded into
hydrogels may be limited, particularly in the case of hydrophobic drugs. Their large
water content and high porosity often results in a quick release profile of only a few
hours to a few days, maximum. Clinical administration may also be a concern. Although
many hydrogels can be injected, some must be implanted surgically, giving rise to a
new set of complications.
Lipids
Liposomes are typically assemblies composed of one or more bilayers of
amphipathic lipid molecules enclosing aqueous compartments. Their structure allows
hydrophilic molecules to be incorporated within the inner compartment, while the
hydrophobic compounds will be entrapped in the hydrophobic bilayer. Considering their
biocompatibility, biodegradability and ability to cross lipid bilayer and cell membranes,
liposomes have been widely demonstrated for various delivery platforms for vaccines36,
cancer treatments37, gene therapy38 and transplant39 in various forms including single
layer liposomes, solid lipid nanoparticles and phospholipid micelles. Although lipids tend
to be non-immunogenic, this feature may be intentionally altered by the incorporation of
antigens, and surface ligands.
Liposomes have shown much promise in advanced clinical trials for many years,
with at least two adjuvant systems currently approved for human use: Inflexal®V and

27
Epaxal® both marketed by Crucell (Leiden, Netherlands). More specifically, both
vaccines uses virosomes (unilamellar phospholipid membrane vesicle incorporating
virus derived proteins) to deliver either influenza (Inflexal®V) or hepatitis A (Epaxal®)
antigens to APCs in order to stimulate strong immune responses40,41. Through these
studies it has been hypothesized that the inherent ability of APCs to sequester
nanoscale liposomes more efficiently than larger-sized liposome counterparts may be a
key to enhanced immune responses observed with nano or micro liposome
formulations. More recently, liposomes have been investigated as a novel approach to
induce long-term tolerance in organ transplantation without continuous administration of
immunosuppressants. Hirai et al., at REGiMMUNE Corporation (Santa Clara, CA),
demonstrated donor-specific tolerance can be achieved by induction of mixed
chimerism in various animal models of bone transplantation 42. The authors describe a
novel approach using a ligand (alpha-GalCer) for invariant Natural Killer T cells and
antibody for CD40-CD40L blockage. Treatment resulted in complete acceptance of
transplanted bone marrow as well as cardiac allograft from the same donor.
As with any carrier discussed here, liposomes also have limitations. They are
highly susceptible to chemical and physical degradation resulting in extremely high cost
of manufacturing as conventional cost-effective sterilization techniques may not be
employed. Currently, filtration and aseptic technique are recommended for the
preparation of liposomes to be used in clinical settings. Liposomes also display low
stability decreasing their shelf-life and limiting their widespread application.
Metallic and inorganic
Many inorganic materials have been studied for their use in vaccine and various
immunology related applications. A well-established example is particulates of calcium

28
phosphate, aluminum hydroxide and aluminum phosphate, collectively referred to as
alum. This inorganic material causes antigen aggregation providing a depot and serving
as an adjuvant for many vaccines. Interestingly, alum remains the only material
approved by the U.S. Food and Drug Administration as an adjuvant in human vaccines.
Although inorganic materials may be non-biodegradable, their advantage lies in their
rigid structure and controllable synthesis43. Gold, carbon, and silica particles have all
been studied for their use in vaccine development.
Gold was one of the first metals to be discovered and its use in medical
applications can be traced back to the seventeenth century. Gold particles can easily be
fabricated into different shapes (spherical, rod, cubic, shell), with a size range of 2-150
nm, and can be surface-conjugated to achieve desired outcomes44. Gold nanorods in
particular, have been used as carriers for antigens derived from various viruses such as
influenza45, or as DNA adjuvants for human immunodeficiency virus (HIV)46.
Furthermore, gold nanoparticles have been conjugated with dye-oligonucleotide ( 5′-
Cy5-GAG CTG CAC GCT GCC GTC AAA AAA AAA A-SH-3′) to investigate a non-viral
transfection delivery system for pancreatic islet cell transplantation 47. The authors
demonstrated transfected islets maintained normal mitochondrial function, calcium influx
and insulin release when stimulated by glucose both in vitro and in vivo. This technology
has the potential to facilitate a wide variety of applications, such as the direct
manipulation of factors following transplantation using gold nanoparticles complexed
with siRNA, functional proteins, and pharmacological agents that have previously been
shown to improve outcomes of pancreatic islet transplants. Although widely used in

29
experimental models, the biodistribution, circulation time and toxicity of gold particles
continue to raise concerns and limit their application in clinical settings.
Carbon nanoparticles are another inorganic composition for drug and vaccine
delivery. Carbon nanoparticles are easily synthesized into a variety of shapes
(nanotubes, mesoporous spheres, etc), and can be made to be biocompatible. In
particular, carbon nanotubes offer the possibility of multivalent surface conjugation of
peptide antigens. For example, Villa et al. investigated the delivery of single-wall carbon
nanotubes as antigen carriers to APCs to promote responses to human tumor antigens.
The authors covalently attached a large number of peptide ligands on to carbon
nanotubes. Immunization of mice with the construct along with adjuvant induced specific
IgG responses against the peptide, while in comparison, the peptide with adjuvant
without the vehicle did not induce such a result48.
Although carbon based carriers have attracted much attention because of their
unique physical, chemical and mechanical properties, toxicity data at the molecular,
cellular and whole animal level is often conflicting. During large-scale preparation and
purification procedures impurities, mainly metal catalysts residues, are introduced and
virtually impossible to remove without destroying the structural integrity of the carrier.
These impurities are often released from carbon particles leading to increased oxidative
stress, inflammatory responses, malignant transformation and DNA damage or
mutation49.
Lastly, of the inorganic family, a promising material for immunomodulation is
silica. Silica nanoparticles have properties amenable for various applications including
tumor targeting, real time imaging, and vaccine delivery. They can be prepared with

30
tunable properties including size, shape and porosity which can alter interaction with
immune cells 50. Furthermore, its abundant surface silanol makes silica unique as it
allows for further conjugation and introduction of modulation of cell recognition,
absorption or uptake. For instance, Xia et al. demonstrated that polyethyleneimine
coating of mesoporous silica nanoparticles enhanced pancreatic cancer cellular uptake
and safely delivered siRNA and DNA constructs 51.
Biologics
Given their ubiquitous presence, diverse roles, and importance in the body, it
should not be surprising that proteins dominate the growing list of more than 200
approved biotherapeutic agents used in medicine today52. Protein therapies include the
abundantly present albumin important for osmolarity and volume of blood, vaccines,
enzymes, targeted antibodies, with monoclonal antibodies dominating the market,
fusion proteins and receptors, factors involved in blood clotting, homeostasis and
thrombosis, the potent botulinum neurotoxin, and hormones and cytokines as signaling
and immunomodulating agents, respectively. Protein-based therapies and carriers offer
several advantages over small molecules such as specificity of action and potent
therapeutic efficacy. Additionally, they have predictable behavior following
administration resulting in fewer side effects (e.g. decreased immunogenicity) and faster
regulatory approval time.
Protein-based carriers are often prepared by recombinant DNA technology and
produced in bacterial, yeast, insect or mammalian cells as they are more efficient and
cost-effective means of production. However, the complexity of proteins poses a
challenge as the possibility for heterogeneity due to changes in amino acid sequence,
presence and degree of glycosylation and folding conformation may affect the

31
properties mentioned above. Some proteins have short half-life in serum requiring
frequent parenteral administration leading to poor patient compliance.
Fusion proteins
Chimera fusion proteins, produced through genetic engineering, consist of a
carefully selected and usually short-lived effector domain, generally a peptide, coupled
with a carrier, usually a protein or peptide, that also contributes to the functional
properties of resultant fusion proteins. The linked effector domain can have a wide
variety of functions for example, contributing to recognition, binding and toxicity while its
fused partner can aid in stability or targeting. Effector moieties employed to date have
been limited to the ligand-binding portions of receptors of a few cytokines and growth
factors53,54, extracellular domains of lymphocyte antigens55, coagulation factors56 and
fragments of a toxin57. Three overarching categories drive the successful development
of chimeric proteins: (i) stability or extended half-life (ii) effective targeting and
subsequent binding (iii) capacity to inhibit deleterious processes underlying treated
condition. Most peptides likely to be considered as effectors have a short half-life due to
proteolytic degradation and are usually rapidly cleared, minutes to hours, through the
kidneys. Conjugation to polyethylene glycol (PEG), or pegylation, can extend half-life by
increasing the hydrodynamic radius and decreasing filtration in the kidneys. However,
safety concerns regarding the lack of biodegradability have given pegylation a bad
reputation and have inspired scientists to seek alternative methods.
The Fc region of the human IgG antibody has been the most commonly
employed fusion partner with multiple applications approved for clinical use53,58. The Fc
portion of IgG consist of the CH2-CH3 domains of the immunoglobulin heavy chain, the
hinge region and the two disulfide bridges connecting the heavy chains. In most fusion

32
proteins, the C-terminus of the effector molecule is fused to the N-terminus of the hinge
region59. Receptor-mediated recycling via interaction with the salvage neonatal FcRn
receptor protects Fc-containing molecules from lysosomal degradation. At low pH, FcRn
salvages the Fc fragment by binding, recycling, and releasing the protein in the blood at
neutral pH thus extending the Fc half-life. Furthermore, Fc fusion proteins can also
interact directly with immune cells, dictating the overall outcome of the immunological
response. Although 10 of the 12 currently FDA-approved fusion proteins are Fc derived,
multiple disadvantages have been observed including increased risk for infection,
neurological side effects, and allergic reaction (reviewed in depth in 59) inspiring the
search for alternative fusion constructs to continue.
Human serum albumin fusions have also been employed as partners in
therapeutic chimeric proteins. Albumin, the protein present in the greatest concentration
in plasma, has relative long half-life (14 d) and is normally considered safe due to its low
immunogenic potential. Effector molecules can be attached at either the N- or C-
terminus, and like the Fc fusion proteins, albumin can also engage the FcRn receptor
resulting in recycling by binding at low pH and release at neutral pH. The most
commonly occurring drawback seeing with albumin, however, is the interference of the
carrier with the specific activity of the effector molecule and its target. This has occurred
despite the use of linkers and efforts to orient the relevant domains60. Despite
drawbacks, albumin-based fusions have been successfully approved and implemented
in clinical settings with Eperzan® (dipeptidyl peptidase-4-resistant dimer fused to
albumin) employed for the treatment of type 2 diabetes61, and Idelvion® (factor IX –
albumin fusion protein) for the treatment of hemophilia B62.

33
Carrier Properties Influencing Immune Responses
Pre-clinical and clinical evaluations of many types of carriers have demonstrated
that material properties are related to their biological outcome. Much can be learned
from nature as immune cells have evolved to respond to pathogens displaying many
sizes, shapes and surface charges. These same properties are important
considerations when designing carriers for immunomodulation. Carrier size appears to
be a major influence in the cellular uptake and further endocytic pathway directing their
intracellular fate and thus overall biological effect. Carriers may be assimilated by
receptor-mediated endocytosis, which relies on the specific recognition of surface
receptors and their ligands; by receptor-independent endocytosis (pinocytosis) referring
to the invagination of the cell membrane encapsulating liquids from the extracellular
environment; or phagocytosis in which solid factors are engulfed by the cell membrane.
Carriers with diameters larger than 0.5 µm tend to be assimilated through
phagocytosis63, which is carried out by members of the innate immune system (e.g.,
DCs, macrophages, neutrophils and mast cells), and leads to cargo degradation in
lysosomes and presentation on the cell surface for recognition by the adaptive immune
system. Smaller carriers, less than 150 nm, are generally taken by cells via receptor-
mediated endocytosis or pinocytosis which are involved in the uptake of essential
nutrients, downregulation of cell signaling by internalization and degradation of
receptors, and maintaining cellular homeostasis64.
It has also been reported that geometrical shape of particulate carriers influences
cellular uptake and trafficking. While spherical polymeric carriers are quickly
internalized, anisotropic systems are poorly phagocytosed thus increasing their
circulation time and systemic delivery of their cargo65. To demonstrate this concept,

34
Champion et al., used polystyrene particles of various sizes and shapes to study the
phagocytosis of alveolar macrophages. The authors report that all shapes were able to
initiate phagocytosis in at least one direction. However, it was reported that the point of
contact dictated whether macrophages phagocytosed or simply spread on the particles,
concluding this effect is based on the actin structure that must form around the particle
to be internalized66. Further studies elucidating the role of biomaterials shape will not
only allow researchers to understand immune cells interactions with pathogens further
but could inform the design of micro and nano carrier-based therapeutics.
Also important for the design on new technologies to modulate the immune
system, is consideration of carriers’ surface properties, which plays a role in interactions
with innate immune cells. For example, charged gold particles are reported to be more
toxic than their neutral counter parts67, cytotoxicity of PAMAM dendrimers is correlated
with the number of primary amino groups 68, and DCs and macrophages preferentially
interact with cationic molecules69. The surface of biomaterials can also be modified by
protein adsorption, which can direct subsequent cell-protein-material interactions 70-74.
For example, Acharya et al., demonstrated that DC morphology and production of
cytokines is differentially dependent upon adhesive substrates 70. Specifically, DCs
cultured on albumin and serum coated surfaces maintained low levels of stimulatory
and co-stimulatory molecules and produce increased levels of IL-10. Conversely, DCs
cultured on collagen and vitronectin substrates expressed higher stimulatory and co-
stimulatory molecules and generated higher levels of IL-12p40 indicating a suppressive
and inflammatory DC phenotype respectively.

35
Technologies that target the immune system through the use of materials as
nano- and micro carriers have gained traction in recent years. Such biomaterials are
contributing to translation of basic immunology discoveries into therapies for transplant
rejection, autoimmune and infectious diseases, and cancer. They offer many
advantages over current clinical approaches including targeted delivery, controlled
release, and stability. Expanding the implementation of materials-based technologies in
clinical settings is expected to have broad impact.
Biomaterials-Based Immunomodulation of Dendritic Cells
The immune system provides a formidable barrier to the clinical application of
many biomaterials, and controlling interactions with components of the immune system
has recently become a major focus in the field5. The immune system is intricately
organized, composed of multiple layers of protection that work in unison for the defense
of the host against would-be invaders. Historically, biomaterials scientists have
developed materials that simply try to limit chronic aggravation of the immune system.
However, recent biomaterials approaches to actively engage and modulate immune
responses to achieve specific outcomes hold great promise. Key cellular players in
immunological defense and homeostasis, are dendritic cells (DCs), professional antigen
presenting cells that have a crucial role in dictating T cell-mediated immunity. Dendritic
cells naturally encounter, uptake, process and present antigen to naïve T cells, and
appropriately shape the resulting T and B cell responses. These features make DCs a
major target of strategies to manipulate the immune system. Pioneering cellular-based
therapies are ongoing using DCs, where DCs are generated from peripheral blood cells,
exposed to antigen and stimuli ex vivo, and then reintroduced into the patient’s
circulatory system. Successes from these cellular therapy approaches highlight the

36
importance of DC modulation. However, widespread application of DC cellular therapies
is limited for most treatments, as the costs are tremendous. Recent accomplishments
using implanted/injected biomaterials to direct DC function while remaining in situ, are
encouraging, and also address the high costs of cell-based therapies.
Biomaterial based methodologies of modulating DCs can be broadly categorized
as directing DC responses toward either inflammatory or suppressive phenotypes.
Numerous approaches have aimed to augment the inflammatory response of DCs for
their use in infectious diseases and cancer applications. Fewer, but a growing number
of approaches are pursuing suppressive DC phenotypes, which is of interest for
autoimmunity and transplant rejection therapies. Strategies currently employed to direct
DCs through the introduction of biomaterials into the body include the use of both
implanted scaffolds and injected particulates. The modulation of material properties
such as hydrophobicity, surface chemistry and degradation rate, as well as the
biomaterial based delivery of proteins, nucleic acids and small drug molecules, are
approaches actively being investigated for directing immune responses. Suitable
outcomes for strategies promoting inflammatory DCs are typically increased expression
of stimulatory76 and co-stimulatory molecules76, and release of pro-inflammatory
cytokines77. Conversely, desired outcomes of strategies promoting suppressive DCs
are often decreased expression of stimulatory and co-stimulatory molecules19, release
of regulatory cytokines77, and increased expression of inhibitory molecules and
tolerogenic mediators78. The ability to manipulate DC function via biomaterials is proving
to be enabling for numerous immune-related applications.

37
Here, we discuss tools of biomaterials-based DC immunomodulation currently
being developed, and suggest that they are broadly applicable to regenerative
medicine. Given that regenerative medicine applications often require careful balance
between inflammatory and suppressive immunological processes involved in wound
healing and regeneration, the field may substantially benefit from the tools and
principles uncovered from such biomaterial based immunomodulation approaches. To
begin, major principles of immunology will first be introduced, and relevant specifics of
DC biology briefly described.
Innate and Adaptive Immunity Overview
The immune system represents a major hurdle to successful incorporation of
many biomaterial. This complex network of cellular interactions and processes has
evolved as an organized and lethal defense mechanism against invaders, as well as
providing homeostatic regulation of self and non-self. Recognition of foreign entities
initiates a series of events that can lead to isolation and antigen-specific elimination in
the case of a pathogen, or tolerance, as in the case of food proteins. In vertebrates,
immunity can be divided into two main categories: the innate and the adaptive immune
system79.
Innate immunity is considered the first line of defense, consisting of anatomical
barriers such as the skin and mucosal membranes, soluble factors, and key cellular
players80. Members of this system include the complement cascade (made up of a large
number of distinct plasma proteins reacting with one another to opsonize pathogens
and induce a series of inflammatory responses), neutrophils, natural killer (NK) cells,
macrophages and DCs. If a pathogen has penetrated beyond physical skin and
mucosal barriers, then the cellular elements of innate immunity are typically engaged.

38
Innate cellular defense mechanism are triggered by signaling through pattern
recognition receptors (PRRs), which are encoded in the germline and can be secreted
or expressed on the host cellular surfaces. These PRRs can recognize and rapidly
respond to a wide variety of common molecular features known as pathogen associated
molecular patterns79,80, facilitating uptake and clearance of the foreign material. Upon
uptake and degradation of a pathogen, cellular machinery presents peptides from
degraded proteins presented on the cell surface bound to specialized stimulatory
molecules known as human leukocyte antigens (HLA) in humans, and major
histocompatibility complex (MHC) in mice79. This process is known as antigen
presentation, and can be carried out by a number of cells collectively known as antigen
presenting cells (APCs).
The adaptive arm of the immune system provides specific responses to newly
encountered antigens, and is mainly driven by the interactions between APCs and
lymphocytes81. Two major classes of lymphocytes exist, B and T cells, both originating
from a common progenitor in the bone marrow. These lymphocytes patrol the body,
routing through the bloodstream and lymphatic system to travel between peripheral and
lymphatic tissues. The basic functions of B cells include antigen presentation, antibody
production and immunological memory. B lymphocytes are equipped with antigen-
recognizing, membrane-bound immunoglobulins, termed B cell receptors that allow it to
interact with and uptake a vast array of antigens. When a naïve B cell encounters an
antigen with affinity for its B cell receptor, the B cell will then differentiate into either a
plasma or a memory B cell, which proliferate to generate a pool of these antigen-
specific cells to combat the invasion. This process is known as clonal selection and

39
expansion. Plasma cells produce soluble antibodies, which is the secreted form of the
B cell receptor. Antibodies that bind to surface antigens on pathogens will attract the
first components of the complement cascade leading to complement activation. Clonally
expanded memory B cells possess the same antigen-specific B cell receptor and will
persist in vivo for an extended period following the resolution of the infection. These
cells form the basis of an immunological memory response in the event of secondary
invasion by the same pathogen.
Much like B lymphocytes, T lymphocytes express a unique, membrane bound
protein termed the T cell receptor, which recognizes antigenic peptides that are bound
to stimulatory molecules on the surface of APCs. T cells also express associated co-
receptors that coordinate with the T cell receptor. Different types of these co-receptors
(e.g., CD4 or CD8) will correspond to the function of a T cell. Lymphocytes expressing
CD4 are generally categorized as helper T cells. The CD4 molecule recognizes
stimulatory molecules HLA-D/MHC II on the surface of a professional APCs. Main
subtypes of CD4+ helper T cells include Th1, Th2 and Th17. The Th1 T cell subset
supports pro-inflammatory cell-mediated immunity, which is intended to eliminate
intracellular pathogens and inducing B cell production of opsonizing IgG82. Th1 T cells
are characterized by the release of effector cytokines such as IFN-γ, TNF-α and IL-2.
Th2 T cells on the other hand, are associated with less inflammatory immune responses
and B cell production of IgG, IgA, and IgE82. More recently, the Th17 subset, was found
to abundantly produce the pro-inflammatory cytokine IL-1783 and is associated with
antimicrobial responses in epithelial and mucosal tissues. T lymphocytes expressing
CD8 receptors are generally categorized as cytotoxic T cells and recognize intracellular

40
antigen presented on stimulatory molecules HLA-A/B/C (human) or MHC I (mouse),
which are expressed by all nucleated cells including both professional APCs and non-
professional APCs. CD8+ T cells directly bind to infected cells and eliminate them by
releasing cytotoxins.
Professional APCs (DCs, macrophages, B cells) serve as the bridge between the
innate and adaptive immune system. These cells patrol the body and can capture
pathogens in their immature state via several uptake mechanisms. Once engulfed by an
APC, antigen is loaded into acidic vesicles where proteins are degraded into peptide
fragments, and presented in the context of the stimulatory HLA/MHC molecules. The
binding of a T cell receptor to antigen-loaded HLA/MHC molecules is commonly termed
signal 1 of the T cell-APC interaction. Once bound to a T cell receptor, APCs can also
provide signal 2, by presenting co-stimulatory molecules, which bind cognate T cell
receptors that activate the T cell and trigger clonal T cell expansion. Furthermore, APCs
can produce a signal 3, which refers to the cytokines released by APCs. Through these
mechanisms APCs shape T cell responses. Antigen presenting cells can also
sometimes engage in the secondary mechanism of cross presentation. During this
process, an extracellularly-supplied antigen is loaded on to HLA-A/B/C or MHC I
molecules (instead of the primary route via HLA-D/MHC II) and presented to CD8+ T
cells. The role of macrophages as APCs is primarily to stimulate immunity to previously
encountered antigens, and the role of B cells as APCs is primarily to engage helper T
cell support to cognate antigen. Dendritic cells, in contrast, are of particular interest due
to their unique ability to initiate and tune naïve lymphocyte responses to new antigens.
As such, DCs are a central regulator of immunity and have become a major target for

41
immunomodulation. In summary, Figure 1-2 depicts a simplified schematic of the typical
immune response to a foreign pathogen.
The successful application of immunological principles has been most widely
implemented through the use of vaccines. Vaccination engages innate and adaptive
immunity and has been able to manipulate immunological responses for the treatment
of numerous pathologies, particularly infectious diseases, and more recently cancer.
Vaccines have been primarily successful in eliciting humoral (antibody-mediated)
responses, but have had limited ability to induce cellular immunity (which provides
clearance of intracellular pathogens in infected cells). Incorporation of biomaterials tools
into vaccine strategies holds potential to address this and other limitations, and will be
discussed further.
Dendritic Cell Subsets
Two main subsets of dendritic cells are plasmacytoid dendritic cells (pDCs) and
conventional DCs. Both cell types express low levels of stimulatory HLA-D/MHC II and
co-stimulatory molecules (e.g., CD40, CD80 and CD86) at homeostatic resting state,
which are upregulated upon activation, and are distinguished by high expression of the
integrin receptor αXβ2 (the alpha subunit of which is also known as CD11c).
Plasmacytoid dendritic cells arise from lymphoid committed precursors and constitute a
small subset of DCs mainly found in the blood and lymphoid tissue, and enter the lymph
nodes through peripheral blood circulation79. Upon activation, pDCs are characterized
by the copious release of type 1 interferon (IFN-α and IFN-β), particularly in response to
viral infections. Plasmacytoid DCs, however, do not play a crucial role in the activation
of naïve T lymphocytes, which is in contrast to conventional DCs. Conventional DCs
originate from myeloid precursors and form a small subset of hematopoietic cells that

42
populate most lymphatic and non-lymphatic tissue. Conventional DCs are the most
potent antigen presenting cells of the immune system as they have an enhanced ability
to sense injury, capture antigen, process and present it to T lymphocytes. Conventional
DCs can also further differentiate into phenotypes that are either inflammatory or
tolerogenic84 (Figure 1-3). Another major type of DC, the Langerhans cell, resides in the
skin and mucosa, and unlike conventional DCs, during homeostasis they originate from
a unique pool of skin-localized precursors85. While these (and more) distinct DC
subsets have been identified, the field of biomaterials based immune modulation has
primarily focused on conventional DCs due to their ability to initiate de novo antigen
response and their relative abundance.
Inflammatory dendritic cell phenotype
In the immature state, DCs are constantly monitoring their microenvironment,
sampling, processing and presenting antigens, and ready to respond to signals of
danger. They capture antigens through several mechanisms including pinocytosis79 (an
actin-driven mechanism in which the plasma membrane ruffles to form a vesicle
incorporating foreign material), receptor mediated endocytosis79 (either via C-type lectin
receptors, or CD64 and CD32), and phagocytosis79 (internalization of larger particulates
usually aided by specific membrane receptors). Once immunogenic antigen is captured
in conjunction with activation signals (e.g., from PRRs), DCs undergo phenotypical
changes rendering their mature state. These changes are characterized by morphology
alteration (e.g., loss of cell surface adhesion molecules, cytoskeleton reorganization and
increased cellular motility79), increased expression of HLA/MHC stimulatory molecules,
upregulation of co-stimulatory molecules (such as CD40, CD80 and CD86), and release
of pro-inflammatory cytokines (e.g., IL-12). Dendritic cell maturation is closely linked

43
with migration from peripheral tissue to the secondary lymphoid organs (spleen, lymph
nodes, tonsils, appendix and Peyer’s patches) where they interact with T cells and
influence their function.
Suppressive dendritic cell phenotype
In the periphery, DCs encounter self-derived antigens and present them to T
cells as a normal process of homeostasis. Rarely, this presentation results in the
pathological activation of the adaptive immune system and can result in autoimmunity
against self-antigen. Typically, the normal homeostatic reaction involves DC initiation of
suppressive immune networks that can lead to T cell anergy, deletion, or expansion of a
regulatory phenotype. For instance, transforming growth factor-β1 (TGF-β1) and/or
interleukin-10 (IL-10) secreted by suppressive DCs can induce regulatory T cells
(Tregs). These Tregs suppress inflammatory immune responses, and are characterized
by the expression of surface receptors CD4 and CD25, and the transcription factor,
forkhead box P3 (Foxp3). Subset of Tregs can also secrete IL-10 and have suppressive
roles when in contact with effector T cells. Generally, characteristics of tolerance-
promoting DCs have been reported to include low expression of stimulatory and co-
stimulatory molecules, low production of inflammatory cytokines (e.g., IL-12), increase
production of regulatory cytokines (e.g., IL-10, TGF-β1), high levels of inhibitory
molecules (e.g., programmed death ligand 1; PD-L1), as well as high expression of
tolerogenic mediators such as indoleamine 2,3-dioxygenase (IDO)84,86. There is
tremendous potential in targeting DCs for therapeutic applications across the spectrum
of immune diseases.

44
Biomaterials-based Immunomodulation
Numerous synthetic polymers, have been investigated for their use as
biomaterials in regenerative medicine applications, including polyesters (e.g., poly(lactic
acid), poly(glycolic acid) and their copolymers), polyorthoesters, polyanhydrides and
polycarbonates. Of these, polymers fabricated using biodegradable copolymer
poly(lactic co-glycolic) acid (PLGA) have been the most researched for the delivery and
controlled release of immunotherapeutics, and their interaction with DCs well
characterized10. By altering the composition of the PLGA copolymer, microparticles can
be fabricated to provide an initial burst of encapsulated immunomodulatory molecule
followed by sustained release. This feature has notably been identified as enabling the
development of therapeutics which can administer both prime and boost doses in a
single injection19. This section details efforts in the manipulation of immune responses
through the use of polymeric systems as controlled release carriers of
immunomodulatory molecules, as well as the potential for biomaterials to provide direct
interactions with DCs, for example, through proteins adsorbed onto the biomaterial
surface. A conceptual summary of biomaterials-based immunomodulation of DCs is
illustrated in Figure 1-4.
Biomaterials-based DC Manipulation Toward an Inflammatory Phenotype
The quintessential scenario in which an inflammatory DC phenotype would be
desired is vaccine applications. Vaccines are generally composed of an antigen,
adjuvant, and a delivery system (e.g., a liquid solvent able to be injected). Adjuvants
can be materials or molecules which stimulate innate immunity by activating APCs. This
engagement of the innate branch of immunity results in the development of long-lasting,
antigen-specific immune responses toward the co-delivered antigen87. The only FDA

45
approved agents for use as adjuvants in the US, interestingly, are materials consisting
of inorganic particulates of calcium phosphate, aluminum hydroxide and aluminum
phosphate, which are collectively referred to as alum87. Alum causes aggregation of
antigen onto the particulates, providing an antigen depot, and eliciting a strong
inflammatory response87. Although these materials have been used for decades, their
mechanism of action is still being uncovered. Recent reports have shown that alum
engages the NALP (NACHT, LRR and pyrin domain-containing protein) -3
inflammasome, resulting in the production of pro-inflammatory cytokines IL-1 and IL-
1888. However, alum has limitations, in that it is not optimal for different types of
antigens, and is ineffective at inducing cellular immunity. This motivates development of
new adjuvant technologies where biomaterials can be implemented as both antigen
carriers, and serve as adjuvants or adjuvant carriers.
Biomaterials as antigen carriers
The use of biomaterials is an attractive approach as they can be formulated to
provide an antigen depot with long term protection, persistence, and controlled release
of antigens, potentially eliminating the need for booster doses, while increasing the
likelihood for antigen uptake and processing by APCs. In contrast to alum, synthetic
biodegradable polymers can be used to provide a depot for more antigens than just
proteins, including, for example, small antigenic peptides. Antigens can be loaded into,
or conjugated onto biomaterial particles or scaffolds, protecting the cargo from
enzymatic degradation. For example, studies conducted by Ali et. al, sought to create
an inflammatory microenvironment through the use of implanted porous PLGA scaffolds
as a cancer vaccine89. This approach aimed at not only using biomaterials as a delivery
vehicle but also as a structural support in which DCs are attracted through release of a

46
recruiting molecule, granulocyte macrophage colony stimulating factor (GM-CSF), and
in which the DCs can reside while receiving co-encapsulated antigen (tumor lysate) and
activation signal (CpG). The investigators reported that after 14 days in vivo, the cellular
infiltration of the scaffolds containing GM-CSF was significantly higher when compared
to the matrix containing no recruiting agent. Not only were the cells primed in a
controlled microenvironment, activated DCs were also able to leave the PLGA matrix
and travel to draining lymph nodes for further interaction with T cells89. Notably, these
immune modulating materials were able to develop specific and protective anti-tumor
immunity reporting a 90% survival rate in melanoma mouse models.
Biomaterials in particulate form can promote targeted delivery of antigen to DCs
and intracellular DC compartments, facilitating specific processing and presentation on
either MHC I or MHC II complexes. For example, Lewis et. al. demonstrated that DC
uptake was markedly increased when PLGA microparticles were surface conjugated
with either anti-DEC-205 antibody, anti-CD11c antibody or the CD11c-binding peptide,
P-D272. Notably, the PD-2 peptide conjugation greatly improved DC MHC II-antigen
presentation compared to controls, by prolonging presentation to a 4 day time span, in
vitro. It was reported that none of the modified or unmodified PLGA particles (50:50
composition) activated DCs(activating factors were not included). The P-D2 peptide
surface modification also greatly promoted in vivo DC uptake and trafficking of
microparticles to lymph nodes72. In contrast to delivering exogenous antigen for MHC II
loading, promotion of cross-presentation, the exogenous delivery of antigen to induce
immunity through the MHC I pathway, is known to be challenging. This is due to the fact
that internalized particulates are quickly trafficked to acidic endosomes where they are

47
enzymatically degraded. However, several groups have made the attempt, with varying
degrees of success. For example, Sneh-Edri et. al, investigated PLGA nanoparticles
decorated with peptides with an endoplasmic reticulum localizing motif and loaded with
antigenic materials. Although enhanced localization to the endoplasmic reticulum was
not achieved, the particle modifications did increase endocytosis, and influenced
intracellular trafficking, which provided low levels of cross-presentation of the antigenic
peptide, for a modestly prolonged time of 2 day, in vitro90. More successful strategies
have overcome the challenge of inducing cross-presentation through the development
of pH-responsive polymers91 that disrupt endosomal membranes, thereby delivering
antigen to the cytoplasm (where processing occurs for MHC I loading). For example,
Flanary et. al, investigated the use of poly(propylacrylic acid; PPPA) as a protein
vaccine carrier, and reported enhanced delivery of ovalbumin (as a model antigen) into
the MHC I pathway92. In the ionized state PPAA maintains aqueous solubility, but when
the environment becomes acidic, as is the case of endosomes, the pendant carboxyl
groups become protonated and the polymer transitions to an insoluble state that is
membrane destabilizing, delivering cargo to the cytosol. This cytosolic delivery resulted
in an increased MHC I presentation and antigen specific CD8+ lymphocyte activation,
promoting cellular immunity92. These studies illustrate the use of biomaterials for the
delivery of antigen to cellular and subcellular compartments.
Biomaterials as adjuvants and adjuvant carriers
In addition to being able to provide an antigen depot, biomaterials can also serve
adjuvant functions by either delivering adjuvant molecules93, or by providing direct
activation through cell-material interactions94. Several experimental adjuvant molecules,
such as unmethylated bacterial CpG DNA (CpG), polyinosinic:polycytidylic acid

48
(PolyI:C), lipopolysaccharide (LPS), and monophosphoryl lipid A (MPLA) are known to
activate APCs directly. The mechanism involves the binding of toll-like receptors on
DCs, resulting in the upreguation of MHC II, co-stimulatory molecules CD80, CD86 and
CD40 and secretion of pro-inflammatory cytokines such as IL-6. Studies by Schlosser
et. al showed that CpG and PolyI:C encapsulated along with ovalbumin (model antigen)
in a PLGA microparticle stimulated a potent cytotoxic T lymphocyte response. A single
immunization was provided and significant levels of antigen-specific tetramer
(SIINFEKL/H-2K(b)) -positive cytotoxic T lymphocytes were detected, as was production
of IFN-α, efficient cytolysis, and protection from vaccinia virus infection93. Similarly,
Beaudette et. al, reported that the co-encapsulation of ovalbumin and CpG resulted in
higher cytotoxic T lymphocyte activity when compared to particles co-administered with
the CpG adjuvant in the free form95. In another example, Elamanchili et. al,
demonstrated that DCs pulsed with MPLA-encapsulating PLGA nanoparticles showed
upregulation of MHC II and CD86 molecules resulting in the activation of naïve T
lymphocytes96. Extending upon their previous work89 (described above), Mooney and
colleagues further experimented with CpG and tumor lysate loaded PLGA scaffold
materials, implanted as a cancer vaccine, this time comparing co-encapsulation with
different recruiting factors GM-CSF, Flt3L, or CCL2097. The authors demonstrated a
significant increase in recruited and activated DC numbers (with high levels of MHC II
and CD86) infiltrated into the scaffold, leading to potent anti-tumor T cell responses.
These vaccine formulations resulted in long-term survival in melanoma mice models97.
Combination approaches such as the co-delivery of adjuvants, particularly of PRR-
activating molecules, along with the immune modulating nucleotides such as silencing

49
RNA (siRNA) are promising98,99. Specifically, studies conducted by Pradhan et. al,
conjugated IL-10 siRNA and CpG onto the surface of a PLGA microparticle loaded with
pDNA antigen. Their studies demonstrated uptake, high expression of stimulatory and
co-stimulatory molecules (CD80, CD86 and MHC II), low expression of inhibitory
cytokines (IL-10) and high expression of pro-inflammatory cytokines (IL-12 and TNF-α)
by DCs treated with microparticles when compared to its soluble formulation. This DC
profile shifted the T cell activation to a Th1 subset which resulted in increased survival
of animal models of lymphoma99.
In contrast to approaches where biomaterials serve as an adjuvant carrier,
another strategy is designing materials that can directly act as adjuvants themselves.
Studies conducted by Hudalla et. al, in the Collier group developed peptides bearing p-
nitrophenyl phosphonate ligands that self-assembled into nanofibers displaying properly
folded, biologically active protein antigens100. These nanofibers were able to
successfully present controlled amount of a model protein antigen (green fluorescent
protein) and acted as a self-adjuvanting vaccine material, in mice100. Additionally,
Narasimhan and colleagues demonstrated the ability of a family of polyanhydride
nanoparticles to differentially act as self-adjuvants, depending on the polymer
composition, inducing potent immunomodulatory activity101-103. Along these lines, it is
has been observed that various PLGA compositions influence DC activation differently.
Studies conducted by Fischer et. al have shown microparticles with a 50:50 ratio of
lactide to glycolide maintain DC immaturity104 while the more hydrophobic ratio of 75:25
has been reported to induce DC activation94. Guiding principles for materials chemistry-
based immune modulation are still being explored, but surface immobilized adhesive

50
proteins may play a role by directing DC adhesion71,73,105. Additionally, like alum and
other particulate materials, PLGA microparticles are capable of engaging the NALP3
inflammasome88.Altogether, this literature supports the concept that biomaterials have
tremendous potential to improve current adjuvant technologies by either direct activation
of APCs, or encapsulation of stimulating agents.
Biomaterials-based DC Manipulation Toward a Suppressive Phenotype
So far, discussion has centered on the modulation of DCs to initiate an
inflammatory immunological response, and this has been the largest focus in the field.
However, biomaterials immune modulation of DCs for suppressive applications is
gaining momentum, and hinges on the fact that DCs can also support suppressive
pathways that maintain homeostasis and dictate key regulators of peripheral tolerance.
It has been reported that suppressive DCs can elicit T cell anergy, expansion of
regulatory T cells and repression of B cell differentiation and expansion106. T cell anergy
is a tolerogenic mechanism in which lymphocytes become hypo-responsive following
the incomplete delivery of co-stimulatory signals107. In addition to inducing T cell anergy,
DCs can also expand the formation of CD4+CD25+FoxP3+ regulatory T cells. This Treg
subset can directly halt effector T cells, and by inhibiting antigen presentation from
DCs108. Therefore, guidance of DC phenotype toward a suppressive or tolerogenic state
holds great promise, particularly for the mitigation of autoimmune diseases and
transplant rejection. While the list of works to date is shorter compared to inflammatory
applications, biomaterial approaches also offer opportunities for targeting, controlled
release and payload protection to deliver modulatory molecules and nucleotides for
suppressive applications. These biomaterial strategies can thereby overcome barriers of

51
limited bioavailability that often accompanies administration of these types of
therapeutic molecules.
Numerous pharmacological methods have been reported to inhibit DC
maturation, including the use of anti-inflammatory (e.g., corticosteroids109, salicylates110)
or immunosuppressive (e.g., mycophenolate mofetil111, 1α,25-dihydroxyvitamin D3112,113)
agents, as well as certain cytokines (transforming growth factor β [TGF-β]114, IL-
10115).The application of pharmacological agents is often limited when administered
systemically in soluble form. Targeted delivery of these agents to specific cells in vivo
can be critical, given the pleiotropic nature of many of them possess, with potential off
target effects and toxicity on bystander cells and tissues. This limitation provides
another window of opportunity for the use of biomaterials to provide targeted delivery
and controlled release of these types of factors. For example, the systemic
administration of the widely used immunosuppressant rapamycin, in vivo, decreases DC
surface expression of CD40, CD80, CD86 and MHC II molecules116. Taking advantage
of this, Bryant et. al, investigated the use of PLGA particles for the delivery of donor
antigens to induce transplant tolerance in allogeneic islet transplantation model117.
Infusion of the particles mediated tolerance in approximately 20% of recipients, and
combination with a low dose rapamycin at the time of transplant significantly improved
tolerance to approximately 60% efficacy. This method offered a biomaterials-based
approach to inducing long term donor graft-specific tolerance117. However, long term
systemic administration of rapamycin and other such immunosuppressants at normal
therapeutic levels results in systemic immunosuppression, by affecting off-target
lymphocytes118. This has motivated the use rapamycin encapsulating particles. Work by

52
the Little group examined the effects of rapamycin loaded PLGA microparticles on
DCs113 This study demonstrated low expression of stimulatory and co-stimulatory
molecules (MHC II, CD86, CD40) and decreased T cell activation113. Additionally,
Maldonado et. al, demonstrated that biodegradable PLGA nanoparticles carrying either
protein or peptide antigen, co-formulated with rapamycin induced a durable antigen-
specific tolerogenic effect119. Specifically, treated animals showed reduced allergic
hypersensitivity disorders, protection from disease relapse in a multiple sclerosis model,
and prevention of antidrug antibodies responses in a hemophilia A model. Treatment
with these nanoparticles resulted in the inhibition of CD4+ and CD8+ T cell activation, an
increase in regulatory T cells and durable B cell tolerance resistant to multiple
immunogenic challenges with protein or peptide antigen. Importantly, the authors
demonstrated that only the encapsulated rapamycin, and not the soluble form could
induce immunological tolerance at the doses used119. Altogether, these works show
how immunomodulation can be improved through the use of biomaterials encapsulating
small drug molecules.
Dendritic cell adhesive interactions with biomaterials and biomaterials-adsorbed
proteins have also been investigated, and these interactions have been reported to be
able differentially promote suppressive DC phenotypes71,120. For example, studies
performed by Acharya et. al, indicate that DC morphology and production of cytokines is
differentially dependent upon adhesive substrates70. Specifically, DCs cultured on
albumin and serum-coated tissue cultured-treated substrates maintained low levels of
CD80, CD86 and MHC II, and produced increased levels of IL-1070. Using a different
approach, Leclerc et. al, demonstrated that DCs can receive biological cues from

53
tethered signaling molecules121. The authors reported covalently immobilized GM-CSF
onto planar silane and polystyrene surfaces resulted in increased DC differentiation in
response to GM-CSF tethering121.
Delivery of immune modulating nucleotides also has the potential to modulate
DC responses and further dictate the outcome of an immunological response.
Biomaterials can serve as delivery vehicles for silencing RNA99, gene vectors122,
messenger RNA123, and antisense oligonucleotides124. Biomaterial carriers can provide
a vehicle to protect nucleotides from systemic elimination, and provide intact delivery to
subcellular compartments125. For example, Giannoukakis and colleagues demonstrated
that non-obese diabetic mouse (NOD; a well-established animal model for type 1
diabetes)-derived DCs treated ex vivo with antisense oligonucleotides resulted in
diminished expression of costimulatory molecules (CD40, CD80 and CD86) and a
significant delay in the incidence of autoimmune diabetes in NOD mice124. Furthermore,
phase I safety clinical trials concluded positively, where antisense-treated autologous
DCs were manipulated ex vivo and administered as a cellular therapy to established
type 1 diabetes adult patients126. Nevertheless, the limiting high cost of cellular
therapies has motivated the group to pursue a biomaterials microparticle-based
approach. Along this line, encapsulation of antisense oligonucleotides in a polymeric
microparticle was able to effect gene knockdown in DCs127. The subcutaneously
injected microparticles were trafficked to pancreatic draining lymph nodes, augmented
Tregs and provoked hypo-responsiveness to β-cell antigen without compromising global
immunity127. The studies presented in this section highlight the delivery of small
molecules and nucleotides through the use of biomaterials as well as surface

54
interactions specifically targeting DCs for suppressive applications, and are directly
relevant to regenerative medicine.
Biomaterials can be engineered to fulfill many functions in the context of
regenerative medicine. They have long been studied to perform, augment or replace a
natural function, historically attempting to avoid provoking the immune system.
However, more recently biomaterials have gained much interest as a tool with which to
actively engage the immune system, particularly through the targeting of DCs. Dendritic
cells play a crucial role in dictating the fate of an immune response and maintaining
homeostasis and have therefore become the focus of numerous experimental
approaches. This chapter has outlined key concepts for the understanding of immune
responses, described relevant DC subsets and functions, and detailed illustrative
biomaterials-based technologies directing DC phenotype toward either an inflammatory
or a suppressive role. Techniques being developed include the use of biomaterials as
antigen carriers, biomaterials as adjuvants and adjuvant carriers, the delivery of small
molecules and nucleotides, and adhesive interactions with biomaterials. The field of
biomaterials-driven immunotherapies will continue to grow as critical mechanisms are
uncovered and more advanced material designs emerge. Notably, the concepts
encompassing the field of regenerative medicine have grown over the years to include
fields such as tissue engineering and wound healing. Looking forward, through the use
of new tools such as biomaterials-based immunomodulation of dendritic cells, it is
conceivable that the range of regenerative medicine will be further expanded to include
related disciplines such as cancer, transplant rejection, autoimmune diseases, and
infectious diseases.
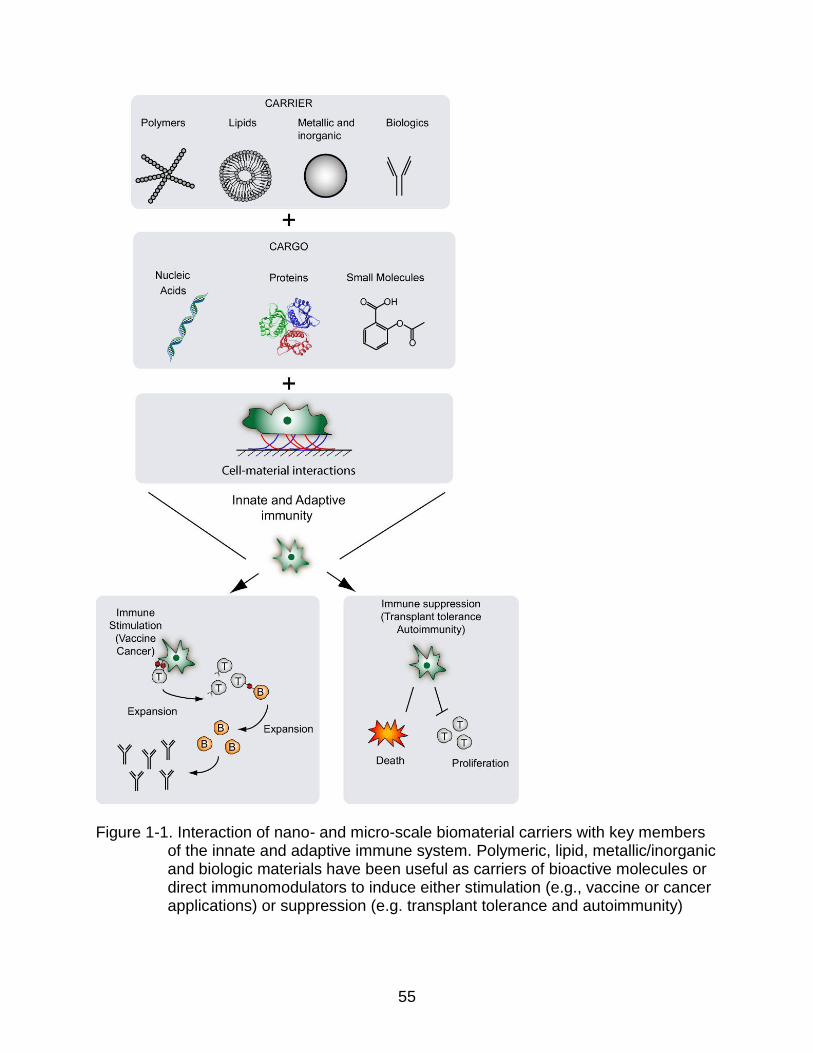
55
Figure 1-1. Interaction of nano- and micro-scale biomaterial carriers with key members
of the innate and adaptive immune system. Polymeric, lipid, metallic/inorganic and biologic materials have been useful as carriers of bioactive molecules or direct immunomodulators to induce either stimulation (e.g., vaccine or cancer applications) or suppression (e.g. transplant tolerance and autoimmunity)

56
Figure 1-2. Simplified immune response to foreign pathogen. 1) Pathogen encounters
epithelial barrier which serves as first line of defense. 2) If the barrier is broken, the innate immune system will attempt to clear it by attacks mediated largely by macrophages, neutrophils and NK cells. 3) To initiate adaptive immune system, dendritic cells (DCs) encounter antigen, undergo maturation during the antigen processing and present it on their surface for recognition by T cells. 4) CD8+ T cells then undergo clonal expansion and become antigen specific cytotoxic T cells. 5) CD4+ T cells also undergo clonal expansion and interact with B cells leading to, 6) the production of pathogen specific antibodies.

57
Figure 1-3. Dendritic cell phenotype dictates T cell function. Immature dendritic cells can
be derived from monocytes in the presence of IL-4 and GM-CSF. Dendritic cells can then become tolerogenic or inflammatory depending on the environmental stimulus, and subsequently direct T cell polarization. Inflammatory danger signals and pathogen associated molecular patterns (PAMPs) can activate iDCs into classically mature (inflammatory) DCs which express high levels of MHC class II and co-stimulatory molecules, release pro-inflammatory cytokines, migrate to lymph nodes and stimulate naïve T cells to differentiate into Th1, Th2 or Th17 effector T cells. Suppressive DCs can be induced in response to signals such as apoptotic cells and TGF-β1. Suppressive DCs can induce antigen-specific T cell apoptosis, anergy and Treg expansion.

58
Figure 1-4 Overview of biomaterials-based dendritic cell modulation. Common
strategies can be broadly divided into those employing biomaterials as antigen carriers, for targeted delivery of small molecules and nucleotides, and controlled release of adjuvants. Biomaterials can furthermore directly function as an adjuvant, possibly through dendritic cell (DC) interactions with surface-adsorbed proteins.

59
CHAPTER 2 EXTRACELLULAR INDOLEAMINE 2,3 DIOXYGENASE-TREATED DENDRITIC CELLS MAINTAIN AN IMMATURE PHENOTYPE AND SUPPRESS ANTIGEN-
SPECIFIC T CELL PROLIFERATION
Background
Indoleamine 2,3 Dioxygenase
Tryptophan catabolism and the kynurenine pathway
Indoleamine 2,3-dioxygenase (IDO) is a non-secreted heme containing enzyme
responsible for the catalysis of the least-abundant amino acid, tryptophan, along the
kynurenine pathway128. IDO catalyzes the conversation of L-tryptophan into N-formyl
kynurenine, the rate limiting step, by oxidative cleavage of the indole ring present in
tryptophan. N-formyl-kynurenine is further converted into formic acid and the stable end-
product kynurenine either spontaneously under acidotic conditions or enzymatically via
formidase129,130. Depending on the cell type and expression level of IDO and other
kynurenine pathway enzymes, a cascade of reactions occurs yielding various
biologically active metabolites collectively known as kynurenines including kynunrenine,
formic acid, anthranilic acid, kynurenic acid, xanthurenic acid, 3-hydroxykynurenine, 3-
hydroxyanthralinic acid, quinolinic acid and picolinic acid (Figure 2-1). Approximately
95% of free tryptophan is metabolized through the kynurenine pathway with the
remaining 4% consumed through the methoxyindole pathway for conversion into
serotonin and melatonin, and the remainder used for protein synthesis. Intracellular
expression of IDO can act as a sink for extracellular tryptophan resulting in a reduction
of local levels of the amino acid. Early studies suggest tryptophan is transported into
IDO-expressing cells from the extracellular space via system L amino acid transporters
(LATs) which have affinity for l-tryptophan and other larger neutral amino acids131. Other

60
studies have demonstrated uptake of tryptophan can also be enhanced by stimulation
with interferon gamma (IFNγ)132-135, lipopolysaccharide (LPS)136 and aryl hydrocarbon
receptor (AhR) agonists, which include kynurenine128,130,137,138. Metabolites generated
during catabolism of tryptophan can be released into the extracellular environment for
uptake by nearby cells via LATs or binding to the AhR. This represents a positive
feedback loop for cell signaling which can influence the function and phenotype of
surrounding cells that do not necessarily express IDO themselves.
Tissue and cellular expression
Indoleamine 2,3-dioxygenase is expressed in a wide range of tissues and cell
types either constitutively or upon stimulation. IDO expression under normal
physiological conditions in mice occurs primarily in mucosal tissues including the
placenta, epidermis, prostate, adipose tissue, in lymphoid organs such as the thymus,
spleen, the central nervous system, kidney, pancreas, lungs and regions of the eye.
Expression of IDO in the small and large intestines, as well as lymph nodes is
dependent on IFNγ, and is thought to play a role in the homeostasis of mucosal
immunity in response to the local microbiota128
In many cell types, IDO is not expressed under normal physiological conditions
but instead requires inflammatory stimuli. IDO expression is induced in antigen
presenting cells, such as monocyte-derived macrophages and dendritic cell subsets,
neutrophils and eosinophils, and non-haematopoietic including endothelial cells,
epithelial cells, fibroblasts and smooth muscle cells. IDO expression has been shown to
be induced by several cytokines such as interlukin 1 (IL-1), tumor necrosis factor alpha
(TNFα), IFNγ and cytotoxic T-lymphocyte associated protein 4 (CTLA-4)132,139,140

61
Role in immune regulation
Early studies suggested the main role of IDO was to halt microbial growth by
limiting their supply of tryptophan. Although some microorganisms have evolved to
synthesize tryptophan de novo, it is energetically unfavorable128 and therefore many
microbes take up tryptophan from the host microenvironment. This requirement for
external sources also makes them susceptible to its depletion, particularly those that are
closely reliant on the host cell for replication and survival. More recent literature,
however, suggests that IDO also participates in modulation of T cell responses,
particularly toward a tolerogenic lineage 130,135,141-144 by initiation of apoptosis, induction
of T cell anergy and limitations on the activity of effector T cells (Teff) by regulatory T
cells (Tregs) 135,143-147. IDO was first established in 1998 by Munn and Mellor to be
present at the maternal/fetal interface of pregnant mice. By inhibiting IDO activity
through the administration of 1-methyl tryptophan (MT), an IDO inhibitor, the authors
demonstrated IDO prevents T-cell mediated destruction of the allogeneic fetus148. Since
then, efforts to elucidate the mechanism of IDO-mediated T cell suppression have
focused on antigen presenting cells, mainly DCs and macrophages.
The immunomodulatory action of IDO involves various biochemical and
metabolic processes affecting an array of immune cells, but the focus remains primarily
on T cells. Two proposed mechanisms for IDO-mediated tolerance have emerged: (i)
depletion of tryptophan suppresses T cell proliferation by activating the general control
nonderepressible 2 (GCN2) stress-response kinase which functions to control
transcriptional and translational processes that couple cell growth to amino acid
availability 149-151 and (ii) downstream metabolites (collectively referred to as
kynurenines) directly interact with immune cells through the aryl hydrocarbon receptor

62
(AhR) 137,138 144 or inhibit IL-2 signaling, crucial T cell survival 152. Dendritic cells
expressing IDO can dictate T cell differentiation into a regulatory phenotype, thus
indirectly suppressing effector T cells. Thus, much of the literature describing IDO as an
tolerance-inducing immunomodulator has focused on overexpression of the gene in
transplant cells/tissues, shown to prolong graft survival in a number of models 145,153-156,
or inhibition of its enzymatic activity for the treatment of cancer 157-160.
The effects of IDO-expressing cells have been well characterized and
documented 86,130,143,148,161, however, little is known about any direct effect on DCs or
the effect of extracellular enzyme activity. In this study, we demonstrate that murine
DCs treated with exogenous human recombinant IDO maintain an immature phenotype
and provide robust suppression of antigen-specific T cell proliferation in vitro. The
mechanism of this suppression involves tryptophan depletion as well as kynurenine
accumulation. These results establish that IDO maintains its immunomodulatory
capacity in the extracellular environment and that extracellular IDO acts directly upon
DCs.
Materials and Methods
IDO Characteristics and Activity Assay
Recombinant human IDO (IDO) expressed in Escherichia coli was purchased
from R&D Systems (Minneapolis, MN) with a predicted molecular mass of 46 kDa,
purity >95% by SDS-PAGE, endotoxin levels <1 EU/µg of protein by the LAL method
and a specific activity of >500pmoles/min/µg as measured by its ability to oxidize L-
tryptophan to N-formyl-kynurenine. The specific activity was measured before
experiments to ensure maximal effect at the beginning of the assay following an
adapted procedure from Valladares et al. 162. The reaction substrate contained 200 µM

63
tryptophan, 20 mM ascorbic acid, 10 µM methylene blue, 225 U catalase and 50 mM
MES buffer (pH 6.5). Recombinant human IDO at 16 ng/mL was loaded onto a flat
bottom 96-well plate and the reaction started by mixture in 1:1 ratio with reaction
substrate. Absorbance was measured in kinetic mode for 5 minutes at 321 nm.
Dendritic Cell Culture and Extracellular Enzyme Treatment
Dendritic cells were generated by isolating the bone marrow from femurs and
tibias of 8-12-week-old C57BL6/j female mice euthanized by CO2 asphyxiation followed
by cervical dislocation in accordance with approved protocols by the University of
Florida. The marrow cells were obtained by flushing the shaft of the bones with a
25G5/8 needle using RPMI 1640 (Corning, Corning, NY) containing 10% fetal bovine
serum (Lonza, Walkersville, MD) and 1% penicillin-streptomycin (Hyclone, Logan, UT)
and mixed homogenously. The cell suspension was strained using a 70 µm cell strainer
(Becton, Dickinson, NJ, USA) and collected after centrifugation at 1600 rpm for 5
minutes at 4°C. The red blood cells were then removed by lysing with ACK Lysing buffer
(Lonza, Walkersville, MD) followed by centrifugation at 1600 rpm for 5 minutes at 4°C to
recover leukocyte progenitors. Remaining cells were re-suspended in DC media:
DMEM/F-12 with L-glutamine (Cellgro, Herndon, VA) containing 10% fetal bovine
serum, 1% sodium pyruvate (Lonza, Walkersville, MD), 1% non-essential amino acids
(Lonza, Walkersville, MD), 1% penicillin-streptomycin, 20 ng/ml of GM-CSF (R&D
Systems, MN, USA) and were plated on tissue culture flasks for 2 d in order to remove
adherent cells. After 2 d, floating cells were transferred to low attachment tissue culture
plates with fresh DC media for the expansion of DC precursors. Half media change was
performed on day 4 of isolation. On day 6 floating cells were carefully removed and
plated on to tissue culture plates at an appropriate cell density for the adhesion and

64
proliferation of DCs. On day 8 media was removed, cells washed with PBS and fresh
media provided. On day 10 dendritic cells were either treated with 15 µg/mL of human
IDO (R&D Systems, Minneapolis, MN) cultured in tryptophan free media supplemented
with 500 µM L-kynurenine (Sigma-Aldrich, St. Louis, MO) for 24 h at 37°C.
Dendritic Cell Phenotype and Maturation Resistance
Following IDO incubation, DCs were challenged with 1 µg/mL of
lipopolysaccharide (LPS) (Sigma-Aldrich, St. Louis, MO). DCs were then imaged to
determine physical characteristics using a Zeiss Axiovert 200M microscope and phase
contrast. DC viability was determined through microscopy using LIVE/DEAD imaging kit
(ThermoFisher Scientific, Waltham, VA) and confirmed via flow cytometry using
LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (ThermoFisher Scientific, Waltham,
VA) both techniques were used according to manufacturer’s instructions. DC maturation
was evaluated by the surface expression of stimulatory and co-stimulatory markers
(MHC II, CD80 and CD86) as well as cytokine release (IL-10 and IL-12p70) by flow
cytometry and enzyme-linked immunosorbent assay (ELISA), respectively. Following
enzyme incubation and LPS challenge, supernatant was collected and stored at -20°C
for subsequent analysis using BD OptEIA ELISA kits (BD Biosciences, Franklin Lakes,
NJ) following manufacturer’s instructions. Adherent cells were incubated with 5 mM
Na2EDTA in PBS solution at 37°C for 30 minutes and lifted using a cell scrapper. Cells
were then washed with 1% fetal bovine serum in PBS and incubated with LIVE/DEAD
Fixable Near-IR Dead Cell Stain kit for 10 mins at room temperature followed by
washing and incubation with antibodies (BD Bioscience, San Jose, CA) against
CD16/32 (Fcγ III/II receptor) (clone 2.4G2) for 30 mins on ice to block Fcγ receptors on

65
DCs. Cells were washed and stained with antibodies against CD11c (clone HL3) CD80
(clone 16-10A1), CD86 (clone GL1) and MHC II (clone M5/114.15.2).
T Cell Isolation and Proliferation Assay
T cells were isolated from splenocytes of OT-I and OT-II female mice (Jackson
Laboratories, Bar Harbor, ME) 8-12 weeks of age. The OT-I mouse model carries a
transgene insert for rearranged TCR α and β genes on CD8+ T cells, that assemble
allow recognition of ovalbumin peptide residue 257-264 in the context of H2kb. The OT-
II mouse model carries a transgene insert for rearranged TCR α and β genes on CD4+ T
cells, that are specific for ovalbumin 323-339 in the context of I-Ab. Animals were
euthanized by CO2 asphyxiation followed by cervical dislocation as a secondary method
in accordance with guidelines approved by the University of Florida. Spleens were
excised and homogenized with RPMI 1640 medium (MP Biomedicals, OH, USA)
containing 10% fetal bovine serum (Lonza, Walkersville, MD) and 1% penicillin-
streptomycin (Hyclone, Logan, UT). The cell suspension was then filtered through a 70
µm cell strainer and centrifuged at 1600 rpm for 5 minutes at 4°C. Red blood cells were
lysed with ACK Lysing buffer (Lonza, Walkersville, MD) followed by centrifugation to
recover lymphocytes. CD4+ and CD8+ T cells were purified using a negative selection
isolation kit (MACS Miltenyi Biotec, Bergisch Gladbach, Germany) following the
manufacturer’s instructions. Resulting T cells were then labeled with 10 µM
carboxyfluorescein succinimidyl ester (CFSE) to track proliferation. For antigen specific
proliferation, DCs were treated with extracellular IDO either in the presence or absence
of 1 mM 1-methyl-tryptophan (MT) for 24 h then pulsed with 1 µg/mL ovalbumin peptide,
323-339 for CD4+ T cell assays and 257-264 for CD8+ assays, for 3 h or cultured in
tryptophan free media supplemented with kynurenine. DCs and T cells were co-cultured

66
for 4 d. After day 4, cells were centrifuged and washed with 1% fetal bovine serum in
PBS and incubated with LIVE/DEAD Fixable Near-IR Dead Cell Stain kit for 10 mins at
room temperature. Dye was then removed and cells incubated with antibodies against
CD16/32 (Fcγ III/II receptor) (clone 2.4G2) for 30 mins on ice. Cells were washed and
stained with antibodies against either CD4 (clone RM4-5) or CD8 (clone 53-6.7) for 30
mins on ice.
Statistical Analysis
Statistical analyses were performed using ANOVA followed by Tukey’s
significance test to make pair-wise comparisons. Differences were considered
significant when p<0.05 using GraphPad Prism.
Results
IDO-treated DCs Resist LPS-induced Maturation
Murine bone marrow derived DCs were treated with recombinant human IDO at
15 µg/mL for 24 h. At this concentration with a specific activity of 500 pmoles/min/µg, it
was calculated that IDO would catabolize all tryptophan present in the cell culture media
within 18 hours. After 24 h DCs were challenged with LPS, a known DC stimulant,
overnight (>16 h). Following LPS challenge, DCs were imaged to characterize their
morphology and representative phase contrast images are shown in Figure 2-2.
Untreated DCs displayed characteristics of an immature phenotype marked by microvilli
(Figure 2-2A, arrow) which become veils upon maturation in response to LPS (Figure 2-
2B, arrow). When cells were treated with IDO (Figure 2-2C), immature morphology was
maintained, suggesting DCs did not recognize the enzyme as a damage associated
molecular pattern or a pathogen associated molecular pattern. Similar morphologies
were observed when DCs were cultured in tryptophan free media (Figure 2-2D), DC

67
media supplemented with kynurenine (Figure 2-2E) or tryptophan free media
supplemented with kynurenine (Figure 2-2F). Finally, when DCs were treated with IDO
for 24 h and then challenged with LPS overnight, an immature phenotype was observed
(Figure 2-2G) demonstrating that IDO-treated DCs resist LPS-induced maturation.
The effects of IDO supplementation on DC viability were inspected using
microscopy and quantified through flow cytometry, shown in Figure 2-3 (results an
average of three experiments with three replicates, total n=9). DCs were cultured in DC
media or with Triton X-100, to serve as an untreated and dead cell control respectively,
with IDO, in tryptophan free media, in DC media supplemented with kynurenine, or in
tryptophan free media supplemented with kynurenine (Figure 2-3A). Green staining in
Figure 2A represent viable cells, whereas the red color signifies cell death. Viability was
further quantified through flow cytometry (Figure 2-3B) where all groups, apart from
Triton X-100, maintained 60% viability after 24 h, equivalent to untreated controls. This
indicates that IDO, tryptophan depletion, and kynurenine formation do not induce
dendritic cell death.
To quantitatively asses the suppressive capacity of DC treatment with IDO, the
expression level of stimulatory (MHC II) and co-stimulatory (CD80, CD86) molecules
was analyzed by flow cytometry, shown in Figure 2-4 (results an average of three
experiments with three replicates, total n=9). Untreated DCs expressed low frequency of
the maturation markers CD80 (Figure 2-4A, 9% + 0.6), CD86 (Figure 2-4B, 3% + 0.2)
and MHC II (Fiure. 2-4C, 7% + 0.8). In contrast, upon LPS treatment expression of all
three activation markers significantly increased to 47% + 0.5 (CD80), 18% + 0.2 (CD86)
and 33% + 0.8 (MHCII) of cells. When DCs were treated with IDO for 24 h, maturation

68
markers remained comparable to immature or untreated cells with 10% + 0.5 (CD80),
3% + 0.2 (CD86) and 7% + 0.7 (MHC II). However, when DCs were cultured with IDO
followed by LPS challenge, maturation markers were lower than LPS alone, with 24% +
0.5 (CD80), 5% + 0.2 (CD86) and 13% + 0.7 (MHC II), confirming cells treated with IDO
resist LPS-induced maturation. Next, the release of inflammatory and anti-inflammatory
cytokines, IL-12p70 and IL-10, was evaluated through ELISA (Figure 2-5, results an
average of three experiments with three replicates, total n=9). IL-12p70 secretion levels
were determined at 38 + 8 pg/mL (untreated) and 1920 + 7 pg/mL (LPS treated). When
IDO was introduced to the culture IL-12p70 secretion remained low (61 + 8 pg/mL).
However, when cells were treated with IDO and challenged with LPS, IL-12p70
secretion significantly diminished compared to the LPS alone group (65 pg/mL, p < 0.05
(Figure 2-5A)). Conversely, IL-10 levels remained consistent, with significant changes
from untreated cells observed only in the LPS treated group at 52 + 13 pg/mL (Figure 2-
5B).
IDO-treated DCs Suppress Antigen Specific Proliferation, and Suppression is Active Enzyme Dependent
To evaluate whether IDO-treated DCs can attenuate antigen specific T cell
proliferation, we co-cultured these DCs with CFSE labeled T cells isolated from
splenocytes of T cell receptor (TCR) transgenic mice, OT-I and OT-II. The CD4+ and
CD8+ T cells of these mice proliferate in response to OVA peptide 323-339 and 257-
264, respectively. The data shown in Figure 2-6 (results an average of three
experiments with three replicates, total n=9) represent the percent proliferation of viable
CD4+ or CD8+ T cells, with the gating scheme used to quantify proliferation (Figure 2-
6A-D). In co-cultures where only DCs and T cells were present, or when IDO alone was

69
introduced, minimal proliferation was observed (11% + 1 and 4% + 1 for CD4+ T cells
and 17% + 1 and 35% + 3 for CD8+ T cells, respectively). When DCs were pulsed with
the corresponding peptide and co-cultured with T cells, T cells were activated and
proliferation was 95% + 1 for CD4+ and 83% + 0.8 for CD8+ after 4 d. However, when
DCs were cultured with IDO for 24 h, washed, and pulsed with peptide, T cell
proliferation was reduced to basal levels, 16% + 1 for CD4+ T cells and 36% + 2 for
CD8+ T cells. T cells only were also cultured and included as a control with proliferation
at 8% + 1 for CD4+ T cells and 11% + 2 for CD8+ T cells (Figure 2-6E-F). The
experiment was repeated, with the introduction of 1-methyl tryptophan (MT), a potent
IDO inhibitor 163, as a DC treatment (Figure. 2-6G). In the presence of MT-treated DCs,
both CD4+ and CD8+ T cell proliferation were restored to over 70%, indicating active
enzyme is required for the attenuation of proliferation. Lastly, the proposed mechanism
of action for IDO-induced suppression, tryptophan starvation and kynurenine
accumulation, were evaluated (Figure 2-7, results an average of three experiments with
three replicates, total n=9). The same co-culture was used, with variations in relevant
components of the culture media. DCs and T cells were co-cultured and percent
proliferating cells assessed in tryptophan containing media (31% + 0.5), in tryptophan
depleted media (21% + 2.3), or in tryptophan depleted media supplemented with
kynurenine (30% + 2.3) respectively. T cells cultured with OVA-pulsed DCs exhibited
proliferating cell frequencies of 81% + 2 in tryptophan containing media, 31% + 2.3 in
tryptophan free media, and 39% + 3 in tryptophan containing media supplemented with
kynurenine. Notably, the largest suppression of T cell proliferation, at 15% + 3, was
observed when DCs were pulsed in OVA and cultured in tryptophan free media

70
supplemented with kynurenine, suggesting the most dramatic response would be
provided by the active enzyme. T cells alone were cultured in all media conditions
mentioned (data not shown), and did not differ significantly from control T cells in
tryptophan containing media (5% + 2 proliferation).
Discussion
Suppression of inflammatory processes would be beneficial for the treatment of
autoimmune and inflammatory diseases as well as whole organ transplantation. Current
treatments are limited to non-specific systemic immunosuppressant drugs which carry
harmful off-target effects and the potential for opportunistic infections 164. To address
these limitations, scientists have focused on reprogramming patients own immune
systems to take advantage of naturally occurring tolerance mechanisms such as the
upregulation of IDO. Since Munn and Mellor demonstrated that the presence of IDO at
the maternal-fetal interface played a pivotal role in maternal T cell tolerance to fetal
tissue in mammals, 148 IDO is considered a potent immunomodulator and promoter of
tolerance.
Subsequent studies by Munn and Mellor indicated that suppression of T cell
proliferation was driven by depletion of the essential amino acid. T cells stimulated in
the presence of tryptophan activated normally, whereas T cells stimulated without
tryptophan experienced growth arrest at the G1 phase 142. The tryptophan starvation
theory was later challenged by Terness et al. who infected human DCs with
recombinant adenovirus harboring the human IDO gene and co-cultured them with
allogeneic T cells 144. The authors concluded suppression of T cell proliferation was
driven by the accumulation of tryptophan metabolites, particularly kynurenine, 3-
hydroxykunureine, 3-hydroxyanthranilic and quinolinic acid which largely induced cell

71
death. However, no studies to date have addressed the immunomodulatory properties
of IDO in the extracellular space or indicated a potential mechanism of action. This
study demonstrates IDO administered in the extracellular milieu metabolically
reprograms immune cells and maintains its potent suppressive capacity: inhibits the
maturation of DCs, even in the presence of inflammatory stimulus, and subsequently
modulates their immunostimulatory functions in vitro. Here, we demonstrate
suppression of T cell proliferation is mediated by a combination of both hypotheses
mentioned above. Tryptophan starvation and formation of kynurenines combine to
abrogate T cell responses in vitro.
Dendritic cells are key regulators of the immune system and although they play a
crucial role in the initiation of inflammatory responses they are also able to induce anti-
inflammatory outcomes. The potential for DCs to activate tolerance-inducing
mechanisms has been shown previously to be closely related to their maturation state.
T cell tolerance can be induced by immature DCs, which are characterized by low
expression of stimulatory and co-stimulatory molecules, and secretion of suppressive
cytokines 84,165,166. Multiple approaches have been used to generate tolerogenic DCs in
vitro and in vivo primarily by pharmacological agents 14,116,167-169 as well as gene
silencing of pro-inflammatory molecules and cytokines 170. This is the first study to
deliver extracellular IDO to DCs and induce a suppressive phenotype. Dendritic cells
treated with IDO showed significant resistance to LPS-induced upregulation of MHC II
and CD80/86 molecules. Additionally, IL-12p70 production was significantly diminished
while IL-10 was maintained in IDO + LPS conditions. While we did not define the
mechanism by which IDO acts upon DCs, kynurenines can engage the DC AhR and

72
induce the production of intracellular IDO through a positive feedback loop 171. Previous
reports showed AhR signaling is required for expression of IDO in DCs 137,138, which in
turn mediates DC maturation status through generalized reduction in cellular energetics,
generated by tryptophan starvation and lack of NAD production 172. Multiple reports
have shown IDO-mediated T cell arrest also induces apoptosis 143,173,174. However, in
this study no significant cell death was observed in either population. Dendritic cells
were cultured without tryptophan or in the presence of kynurenine to simulate conditions
generated by the enzyme and cell viability was on par with control cells, consistent with
in vitro extracellular IDO cultures.
Following maturation studies, IDO-treated DCs were co-cultured with CD4+ or
CD8+ T cells isolated from OT-I and OT-II mice, respectively, to evaluate the potential of
IDO-treated DCs to dictate immune responses. Suppression of antigen-induced T cell
proliferation was observed only in the groups where DCs were exposed to extracellular
IDO. Additionally, suppression of proliferation was confirmed to be mediated by the
active enzyme through use of MT. 1-Methyl tryptophan inhibits conversion of tryptophan
to kynurenines, likely preventing activation of the AhR and halting further IDO synthesis.
To further confirm depletion is mediated by the active enzyme, co-cultures were
performed in simulated enzymatic conditions. We demonstrated both tryptophan
depletion and kynurenine production are required for maximum T cell suppression
indicating future studies implementing IDO as a biological therapeutic must deliver it in
the active form.
In this study, we provided evidence to establish IDO as a potent
immunomodulator in the extracellular space. In recent years, therapeutic application of

73
IDO has primarily focused on overexpression of the gene in transplant cells/tissues,
shown to prolong graft survival in a number of models 145,153-156, or inhibition of its
enzymatic activity for the treatment of cancer 157-160. However, genetic overexpression
of IDO through the use of viral and non-viral vectors faces several obstacles, such as
viral vector-induced inflammation and malignancies175,176, which may hinder its clinical
application 177. For example, Tan et al. demonstrated DCs transduced with commonly
used high efficiency viral-vectors upregulate expression of stimulatory co-stimulatory
molecules and in the case of adenovirus, induced the production of Th1 and pro-
inflammatory cytokines 178. Additionally, infected cells demonstrated altered function
and inability to properly stimulate mixed lymphocytes reactions. The data presented
here lays the foundation for the use of extracellular IDO as a biologically active
therapeutic recombinant protein, as the enzyme enacts T cell suppression in vitro
utilizing the mechanisms established in studies of its native intracellular functions. By
demonstrating IDO can function as a potent immunomodulator in the extracellular
space, new delivery strategies and tissue targeting can be designed and used to induce
anti-inflammatory responses in autoimmunity, chronic inflammation and transplant
models.

74
Figure 2-1. Tryptophan metabolism through the kynurenine pathway

75
Figure 2-2. Dendritic cells treated with IDO maintain an immature morphology even after
LPS challenge. Murine bone marrow derived DCs were cultured A) untreated, B) in the presence of LPS, C) with human recombinant IDO, D) in tryptophan free media, E) in tryptophan-containing media supplemented with kynurenine, F) in tryptophan-free media supplemented with kynurenine, or G) with human recombinant IDO for 24 h then challenged with LPS overnight. Arrows in panels A and B denote microvilli and veils characteristic of immature and mature DCs, respectively. Scale bar = 50 µm.

76
Figure 2-3. Treatment with soluble IDO does not induce dendritic cell death. Dendritic
cells were cultured in DC media or Triton X to serve as an untreated and dead cell control, respectively, in the presence of IDO, in tryptophan free media, in tryptophan-containing media supplemented with kynurenine or in tryptophan free media supplemented with kynurenine for 24 h. Following treatment, cells were A) stained with cell-permeant (green) and cell-impermeant dye (red) to denote live and dead cells, respectively, and imaged using AxioVision 200M. Scale bars = 50 µm or B) stained with live/dead fixable NIR dye and viability assessed through flowcytometry. n =3, mean + SEM with pair-wise significant difference from all other groups, ANOVA with Tukey’s post-hoc, denoted by * where p < 0.05.

77
Figure 2-4. Dendritic cells treated with soluble IDO resist LPS maturation. Dendritic cells
were incubated with soluble IDO for 24 h and challenged with LPS overnight. Untreated and LPS groups were included for comparison. Cells were immunostained for maturation markers A) CD80, B) CD86 and C) MHC II. Viable cells expressing CD11c and the marker of interest were assed via flow cytometry and shown as percent positive. n =3, mean + SEM with pair-wise significant difference from all other groups, ANOVA with Tukey’s post-hoc, denoted by * where p < 0.05.
Figure 2-5. Treatment of dendritic cells with exogenous IDO inhibits their IL-12 secretion
and maintains IL-10 production. Dendritic cells were incubated with IDO for 24 h and challenged with LPS overnight. Untreated and LPS groups were included for comparison. A) IL-12p70 secretion and B) IL-10 secretion were evaluated via ELISA of the supernatant for each condition. n =3, mean + SEM with pair-wise significant difference from all other groups, ANOVA with Tukey’s post-hoc, denoted by * where p < 0.05.

78
Figure 2-6. IDO-treated DCs suppress antigen specific T cell proliferation, and
suppression is active enzyme dependent. Dendritic cells were incubated with IDO in the presence or absence of 1-methyl tryptophan (MT) for 24 h, pulsed with the corresponding ovalbumin peptide (OVA) for 3 h. Dendritic cells were then co-cultured with CD4+ or CD8+ CFSE labeled T cells for 4 d. T cell proliferation was quantified through CFSE dilution via flowcytometry. Representative histograms for live T cells stimulated with A) non-treated DCs, B) DCs pulsed with OVA, C) IDO-treated DCs pulsed with OVA, D) IDO-MT-treated DCs pulsed with OVA. T cell proliferation was further quantified for E) CD4+ and F) CD8+ population where + and – represent presence or absence of specific component. G) CD4+ T cell proliferation assay with the introduction of MT during IDO treatment of DCs. n =3, mean + SEM with pair-wise significant difference from all other groups, ANOVA with Tukey’s post-hoc, denoted by * where p < 0.05.

79
Figure 2-7. Tryptophan depletion and kynurenine accumulation are needed to suppress
antigen specific proliferation. Dendritic cells were cultured in tryptophan (tryp) free media supplemented with kynurenine (kyn) or relevant controls for 24 h then pulsed with ovalbumin peptide (OVA) for an additional 3 h. Cells were washed and co-cultured with CD4+ CFSE labeled T cells isolated from OT-II mice for 4 d. + and – denote the presence or absence of a particular component during assay. Proliferation of live T cells was quantified through CFSE dilution via flowcytometry. n =3, mean + SEM with pair-wise significant difference from all other groups, ANOVA with Tukey’s post-hoc, denoted by * where p < 0.05.

80
CHAPTER 3 FUSION OF GALECTIN 3 WITH A MODEL ENZYME AT THE N-TERMINUS
PROLONGS RETENTION TIME IN VIVO
Background
Galectin 3
Galectins are evolutionarily conserved carbohydrate-binding proteins that share a
common structural fold and at least one carbohydrate recognition domain (CRD) of
approximately 130 amino acids179. The classification of all 15 galectins identified up to
date is based on structural similarities and includes: prototype galectins (galectin 1,
galectin 2, galectin 5, galectin 7, galectin 10, galectin 11, galectin 13, galectin 14 and
galectin 15), which have one CRD and exist as monomers or dimers; tandem repeat-
type galectins (galectin 4, galectin 6, galectin 8, galectin 9, galectin 12), which contain
two CRDs separated by a linker; and the chimera type galectin 3 (Gal3), which contains
a CRD connected to a non-lectin amino-terminal region179 (Figure 3-1). Within the
immune system, galectins are expressed by virtually all cells, either at a basal level or in
an inducible manner, and are significantly upregulated by activated B cells, T cells,
inflammatory macrophages, natural killer cells and CD4+CD25+ Tregs180. Particularly,
galectin 3 can be found within the nucleus, in the cytosol, on the cell surface and in the
extracellular compartment. It is well documented that Gal3 acts as an adhesion
molecule by cross-linking adjacent cells181. However, intracellularly, Gal3 is engaged in
several processes including pre-mRNA splicing182, regulation of cell growth183, cell cycle
progression and apoptosis184. Additionally, work by Demetriou et. al. has shown that
multivalent Gal3-N-glycan complexes can limit TCR clustering by restricting lateral TCR
movement within the plane of the membrane hindering T cell signaling184.

81
Galectin 3 as a retention strategy. Galectin 3 is member of the carbohydrate-
binding soluble lectin family and has the ability to oligomerize into a pentamer through
its flexible collagen-like domain on the N-terminus upon binding through the CRD to a
carbohydrate ligand181. Specifically, Gal3 has a strong affinity for N-Acetyl-D-
lactosamine (LacNAc) present in proteins of the extracellular matrix and certain surface
receptors on various immune cells184-187. Its pentameric architecture allows for cross-
linkage of cell surfaces and extracellular matrix proteins, triggering signals that dictate
the outcome of events such as cell adhesion, differentiation, and apoptosis188,189.
Galectin 3 has also been linked to tumor metastasis by triggering angiogenesis through
cross-linking of LacNAc presenting vascular endothelial growth factor (VEGF) receptor
2 and prolonging its retention at the cell surface190-194. Therefore, Gal3 has been
considered a target for cancer therapy with fusions of Gal3 proposed as drug delivery
tools in the context of tumor angiogenesis195-197. In this study, we show fusion of Gal3
with a model enzyme or IDO through the N-terminus alters its physiological
characteristics while conserving its binding affinity through the CRD.
NanoLuciferase
Bioluminescence methodologies are extraordinarily useful, particularly as
reporter molecules, due to their high sensitivity, broad dynamic range, and operational
simplicity198. These characteristics have incrementally been improved through
adaptations of native enzymes and substrates, originating from luminous organisms of
diverse evolutionary lineage199. Promega engineered both the enzyme: Nanoluciferase
(NanoLuc), from deep sea shrimp (Oplophorus gracilirostris), and the substrate:
furimazine, an imidazopyrazinone analog, to create a superior reporter system with
more efficient light emission200. The new enzyme produces signal (half-life > 2 h) with a

82
specific activity 150-fold greater than that of either firefly (Photinus pylaris) or Renilla
luciferases. Additionally, NanoLuc exhibits greater physical stability, retaining activity up
to incubations at 55°C or in culture medium for more than 15 h at 37°C. In this study
NanoLuc was used as a model enzyme to evaluate the ability of Gal3 to serve as a
tissue-targeting strategy.
Materials and Methods
Cloning of Galectin 3, Nanoluciferase and NanoLuc-Gal3
Genes encoding recombinant human Gal3 (Origene) and NanoLuc-Gal3 (IDT)
were inserted into pET-21d+ vectors between the NcoI and XhoI sites (Figure 3-2) by
Margaret Fettis, a PhD candidate in the laboratory of Dr. Greg Hudalla. The modified
plasmids were transformed into Top10 E.coli (Thermo Fisher) and selected on 100
μg/mL ampicillin-doped LB/agar plates overnight at 37°C. Positive clones were
sampled and used to inoculate 5 mL 100 μg/mL ampicillin-containing LB broth. Cultures
were grown overnight at 37°C, 220 rpm on an orbital shaker. Plasmids were isolated
from cultures via a plasmid mini-prep kit (Qiagen), according to manufacturer’s
instructions, and sequenced at the Interdisciplinary Center for Biotechnology Research
at the University of Florida. Using PCR, Nanoluciferase was amplified from the
NanoLuc-Gal3 gene and mutated to have NcoI and XhoI flank the gene using the
primers, 5’-CGC CTC GAG CGC CAG AAT GCG TT-3’ and 5’-GCT TAG CCA TGG
CGG TCT TCA CAC TCG AAG-3’. The PCR product was digested using NcoI and XhoI
and re-inserted into pET21d+. Plasmids were screened and sequenced as described
above.

83
Protein Expression and Purification
Origami B (DE3) E.coli (Novagen) were transformed with pET-21d-Gal3, pET-
21d-NanoLuc, or pET-21d-NanoLuc-Gal3 vectors and selected on 100 μg/mL ampicillin-
and 50 μg/mL kanamycin A-doped LB/agar plates overnight at 37°C. Positive clones
were picked and used to inoculate 10 mL 100 μg/mL ampicillin- and 50 μg/mL
kanamycin A-containing LB broth. Cultures were grown overnight at 37°C, 220 rpm on
an orbital shaker. Cultures were expanded into 1 L 100 μg/mL ampicillin- and 50 μg/mL
kanamycin A-containing 2xTY broth and grown at 37°C, 225 rpm on an orbital shaker
until an optical density of 0.6-0.8 (λ=600 nm) was reached. Protein expression was
induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and incubated for 18
h at 18°C, 225 rpm in an orbital shaker. Bacteria were pelleted by centrifugation, and
washed with PBS. Bacteria were lysed with B-PER (Thermo Fisher), a protease inhibitor
tablet (Thermo Fisher), 300 units DNAse I from bovine pancreas (Sigma), and 100 μg
lysozyme (Sigma) for 20 min. Lysed bacteria was cleared by centrifugation, and
supernatant containing recombinant proteins were loaded onto columns containing
HisPur cobalt resin (Thermo Fisher) equilibrated with PBS. Columns were washed with
20-30 column volumes and proteins were eluted with increasing concentration of
imidazole. Imidazole was removed by centrifugation, using Amicon filter tubes (MWCO
10kDa) (Millipore). For IDO-Gal3, a second purification step was required using size
exclusion chromatography in an AKTA pure chromatography system (GE Life
Sciences). Protein molecular weight and purity were analyzed with SDS-PAGE.
Endotoxin contaminants were removed by Detoxi-gel endotoxin removing columns
(Thermo Fisher) following manufacturer’s instructions. Endotoxin content was analyzed

84
using Pierce™ LAL Chromogenic Endotoxin Quantitation Kit (Thermo Fisher), and was
below 0.1 EU/mL in all stocks.
Mice and Cell Lines
C57BL/6 (B6) and B6.Cg-Tg(TcraTcrb)425Cbn/J (OT-II) mice were purchased
from Jackson Laboratory. Age-matched (6-12 weeks) female animals were used
throughout all experiments. All studies were performed in accordance with protocols
approved by the Institutional Animal Care and Use Committee (IACUC) at the University
of Florida. Experimental groups and sizes were approved by authorities for animal
welfare after being defined to balance statistical power, feasibility and ethical aspects.
All animals were cared for/monitored/inspected daily either by a member of the research
team or with an agreement by Animal Care Services personnel. Jurkat T cells were a
generous gift from the laboratory of Dr. Mark Atkinson purchased from the American
Type Culture Collection number TIB-152 clone E6-1.
Galectin 3 and NanoLucGal-3 Binding Affinity
Affinity of Gal3 and NanoLucGal3 for lactose was determined using affinity
chromatography in an AKTA Pure chromatography system (GE Life Sciences) equipped
with an α-lactose agarose column (Sigma-Aldrich) by Antonietta Restuccia, a PhD
candidate in the laboratory of Dr. Greg Hudalla. Proteins were eluted with a linear
gradient of β-lactose (Sigma-Aldrich) in phosphate buffer.
NanoLuc-Gal3 Binding Affinity to ECM Proteins
Non-tissue culture treated polystyrene plates were coated with either natural
mouse laminin (Invitrogen), collagen I from rat tail (Life Technologies), vitronectin from
human plasma (Corning Cellgro), IgG from mouse serum (Sigma-Aldrich) or α2-
macroglobulin from human plasma (Sigma-Aldrich) at various concentrations. Plates

85
were washed 3 times with PBS (ThermoFisher Scientific) and blocked with 1% heat
denatured bovine serum albumin (BSA) (ThermoFisher Scientific) for 30 min at room
temperature. Plates were washed as described earlier and incubated with 2nM
NanoLuc-Gal3 in the presence or absence of excess β-lactose (100mM) for 45 mins at
room temperature. Plates were washed and furimazine (Promega) added according to
manufacturer’s instructions. Bioluminescence was read using a Biotek Flx Fluorescent
Microplate Reader and compared to a standard curve to determine concentration of
bound protein.
NanoLuc-Gal3 Binding Affinity to Jurkat T Cells
Jurkat T cells were seeded at a density of 100,000 per well and incubated with
2nM NanoLuc-Gal3 in the presence or absence of excess β-lactose (100mM) for 1 hour,
washed 3 times with PBS and incubated with furimazine according to the
manufacturer’s instructions. Bioluminescence was read using a Biotek Flx Fluorescent
Microplate Reader. Results were compared to standard curve to determine
concentration of bound protein.
Jurkat T Cell Agglutination and Viability
Jurkat T cells were seeded at a density of 20,000 per well and incubated with 5
µM NanoLuc-Gal3 or 5 µM Gal3. After 1 hour, cells were imaged using a Zeiss Axiovert
200M with a 20X objective lens (Carl Zeiss Microscopy) to determine agglutination.
After 24 hours, viability was assessed using the LIVE/DEAD Fixable NIR Dead Cell
Stain Kit for 633 or 635 nm excitation (Life Technologies) according to the
manufacturer’s instructions and analyzed using flow cytometry.

86
Quantitative Precipitation Analysis of Galectin 3 and NanoLuc-Gal3
Precipitation analysis of galectin-3 and NanoLucGal-3 were completed following
protocols established elsewhere201 by Antonietta Restuccia. Briefly, 30 uM of galectin-3
or NanoLuc-Gal3 were incubated with asialofetuin (ASF, Sigma-Aldrich) at different
molar ratios in a 20 mM sodium phosphate buffer, pH 7.2, containing 10 mM β-
mercaptoethanol and 0.15 M sodium chloride. Proteins were incubated at room
temperature for 30 min, and light scattering was measured at 420 nm using
SpectraMax® M3 Multi-Mode Microplate Reader (Molecular Devices).
In Vivo Bioluminescence and Imaging
Bioluminescence images were acquired using an IVIS Spectrum In Vivo System
(IVIS). Living Image Software version 4.3.1 (Perkin Elmer, Waltham, MA) was used to
acquire the data immediately after furimazine administration. Exposure time for the
bioluminescence imaging was 1 second. Regions of interest were quantified as the
average radiance (photons/second/cm2/sr).
NanoLuc-Gal3 Tissue Distribution from Hock Injection
164 picomoles of NanoLuc-Gal3 in 40 µL of PBS were injected subcutaneously
into the hock of B6 mice. At the prescribed time points, animals were euthanized in
accordance with approved protocols. Organs and tissues of interest were harvested,
weighed, processed, incubated with furimazine and bioluminescence quantified by a
luminometer. Specific amount of protein in tissue was determined by comparison with a
standard curve of NanoLuc-Gal3 activity.
Alternative Injection Sites
164 ρmoles of NanoLuc-Gal3 or NanoLuc in 10 µL (subgingival), 100 µL
(intraperitoneal), 40 µL (intramuscular), 40 µL (subcutaneous – scruff of the neck) or 40

87
µL (subcutaneous – abdominal region) of PBS were administered. Furimazine (1:50
dilution in PBS at the same sites and volumes listed above) was administered daily until
no signal was observed for two consecutive days.
Statistical Analysis
Statistical analyses were performed using ANOVA followed by Tukey’s
significance test to make pair-wise comparisons. Differences were considered
significant when p<0.05 using GraphPad Prism.
Results
NanoLuc-Galectin 3 Retains Binding Affinity for Sugar Moiety when Compared to WT-Gal3
Equal concentrations of NanoLuc-Gal3 (red line) and wildtype galectin 3 (black
line) were injected into an α-lactose-agarose resin column and eluted with an increasing
β lactose gradient (Figure 3-3A). Wildtype and fusion proteins elute at the same β
lactose concentration (20 mM) indicating equivalent binding affinity for β lactose.
Galectin 3 Retains Binding Affinity to Sugar Moiety on ECM and Serum Proteins When Fused to an Enzyme on the N-terminus
Non-tissue culture treated polystyrene plates were coated with various
concentrations of proteins of the extracellular matrix known to bind Gal3 with various
degrees of affinity202-204. Namely, laminin, collagen type I, vitronectin, IgG and α2
macroglobulin were incubated with NanoLuc-Gal3 in the presence or absence of excess
β-lactose (Figure 3-3B-F). Plates were then washed and incubated with furimazine to
determine NanoLuc activity and correlated to specific protein mass through a standard
curve. Binding to all proteins is mediated by the carbohydrate recognition domain (CRD)
as determined by the incorporation of β-lactose which competes with immobilized
protein for the CRD. Binding follows a trend associated with the concentration of

88
immobilized protein, less protein present represents less binding of NanoLuc-Gal3,
consistent with expected LacNAc content. Additionally, binding to laminin is much
higher than collagen type I, vitronectin and both serum proteins, again consistent with
expected binding sites.
Galectin 3 Is Not Able to Self-assemble Into Pentamer and Precipitate Asialofetuin When Fused to NanoLuc on the N-terminus
Asialofetuin (ASF) is a glycoprotein containing complex carbohydrate chains
with terminal galactose and N-acetyl lactosamine (LacNAc) residues. As found
previously201, galectin-3 binds to terminal LacNAc, crosslinking and precipitating ASF as
seen by the increased light scatter. NanoLucGal-3, on the other hand, fails to crosslink
and precipitate ASF at any of the ratios of lectin to glycoprotein studied (Figure 3-4A).
Galectin 3 Binds to Jurkat T Cells but Does Not Induce Agglutination or Apoptosis
It has been well documented that WT Galectin 3 induces T cell agglutination and
apoptosis by binding CD7 and CD29 and inhibiting TCR clustering186,205. In this study,
Jurkat T cells were incubated with NanoLuc-Gal3 in the presence or absence of excess
β-lactose. First, binding was determined by washing cells and incubating them with
furimazine and a specific mass was determined by comparison to a standard curve (Fig.
2-3B). Jurkat T cells incubated with NanoLuc-Gal3 showed binding of 0.56 + 0.2 ng of
protein whereas Jurkat T cells incubated with NanoLuc-Gal3 and excess β-lactose
showed significantly decreased binding (p <0.05, 0.07 + 0.04 ng). Jurkat T cells were
then cultured untreated, with Gal3 or NanoLuc-Gal3 imaged and stained with Live/Dead
dye to assess agglutination and viability, respectively (Fig. 2-3C,D). Untreated cells
shown no agglutination and 53% + 4 viability, cells treated with Gal3 induced
agglutination determined by cells clusters and showed only 0.6% + 0.2 viability.

89
Conversely, cells treated with NanoLuc-Gal3 showed no appreciable clusters and 61%
+ 3 viability, comparable with untreated cultures.
NanoLuc-Gal3 Prevents Galectin 3-induced Cell Death
Jurkat T cells were incubated with various ratios of NanoLuc-Gal3 and Gal3
(Figure 3-5) to assess whether NanoLuc-Gal3 acts as an inhibitor of wild type Gal3.
Representative phase contrast images demonstrating Gal3-mediated agglutination of T
cells is shown in Fig. 3-5A. Untreated cells or cell treated with NanoLuc-Gal3 do not
exhibit appreciable clusters of cells, whereas cells treated with Gal3 severely
agglutinate after 4 h of culture. Remarkably, agglutination is alleviated upon incubation
with a 1:1 or a 2:1 ratio of NanoLuc-Gal3 to Gal3. Upon treatment with 3:1 ratio,
agglutination resembles that observed with untreated cells, trend is maintained at 4:1
and 5:1 ratios. Furthermore, viability following agglutination was evaluated (Fig. 3-5B).
Cultures of untreated cells showed 66% + 3 and 33% + 3 live and dead cells,
respectively. Upon treated with NanoLuc-Gal3 levels remained comparable to untreated
group with 63% + 4 live and 36% + 4 dead cells. Following incubation with Gal3, cell
cultures decreased to 30% + 0.5 live and increased to 68% + 1 dead. Treatment with
NanoLuc-Gal3 and Gal3 at a 1:1 ratio, increased viability to 54% + 2 and decreased cell
death to 44% + 3. An upward trend was observed in cell viability as the concentration of
NanoLuc-Gal3 was increased, with 63% + 6 live cells and 35% + 6 dead cells for the 2:1
ratio, 64% + 5 live cells and 29% + 3 dead cells for the 3:1 ratio, 69% + 1 live cells and
27% + 0.5 dead cells for the 4:1 ratio and 72% + 2 live cells and 25% + 2 dead cells for
the 5:1 ratio.

90
NanoLuc-Gal3 Binds Primary Splenocytes in a CRD Dependent Manner, Binding Does Not Act as a Damage Associated Molecular Pattern
NanoLuc-Gal3 was labeled with Fluorescein (FITC) and incubated with
splenocytes isolated from B6 mice in the presence or absence of excess β-lactose,
untreated cells were used as a negative control (Figure 3-6A). Approximately 30% of
CD3+CD4+ cells were bound to NanoLuc-Gal3 which significantly reduced to 10% when
treated with β-lactose; 25% of CD3+CD8+ decreased to 10%, 20% CD11c+ decreased to
8%, 40% B220 decreased to 10% and 25% of F4/80 decreased to 10%.
Galectin 3 Targeting in NanoLuc-Gal3 Provides Prolonged Retention Time When Administered Subcutaneously, Intraperitoneally and Intramuscularly
NanoLuc-Gal3 or NanoLuc was administered to B6 mice through various pre-
clinical relevant routes and imaged using an IVIS system (Figure 3-7). When
administered subcutaneously at the scruff of the neck, NanoLuc-Gal3 remained at the
injection site for 1 d, when introduced intraperitoneally 2 d, subcutaneously at the
abdominal region for 5 d, intramuscularly for 1 d, and subcutaneously at the hock for 7
d. Conversely, untargeted NanoLuc cleared from all injection sites within 24 h after
administration (Fig. 3-7A-E). Furthermore, the average radiance emitted was quantified
and as shown in the images, NanoLuc-Gal3 (black line) had a prolong local activity
when compared to untargeted NanoLuc (blue line) (Fig. 3-7F-J), demonstrating Gal3
serves as a retention strategy in vivo.
Galectin 3 Targeting in NanoLuc-Gal3 Provides Prolonged Retention Time When Administered Into the Hock with Minimal Drainage to Adjacent Tissues
NanoLuc-Gal3 was administered subcutaneously at the hock, mice euthanized
at various time points and tissues of interest harvested, processed, incubated with
furimazine and bioluminescence quantified by luminometer. The highest quantity of

91
protein was detected in the hock for 7 days with minimal drainage to the spleen,
bladder, kidneys and blood.
Discussion
Proteins have the most dynamic and diverse roles of any macromolecule in the
body, catalyzing biochemical reactions, forming receptors and channels on cell
membranes, transporting molecules within cells or organs, or aiding in cell to cell
communications 206. Because of their versatility, proteins are often used for replacement
therapies, to augment an existing pathway or to provide a novel function. For example,
enzyme replacement therapy, is a common practice for the treatment Goucher’s
disease, a genetic lysosomal storage disease characterized by deficiency of the
enzyme glucocerabrosidase 207. Deficit of the enzyme leads to accumulation of its
substrate, glucocerebroside, within the lysosomes of macrophages resulting in anemia,
thrombocytopenia, bone disease, hepatomegaly, and splenomegaly 208,209. In
Goucher’s disease the target cells are macrophages, which can efficiently uptake the
recombinant enzyme, imiglucerase, through the mannose-6-phosphate receptor
providing a unique targeting strategy 209. However, most enzymes do not make optimal
therapeutics as they are rapidly cleared from circulation by renal filtration and have half-
lives of minutes to a few hours leading to poor bioavailability and decreased activity 52,
which in many cases can render them marginal or unsuitable for clinical applications
210,211.
Fusion protein technologies aim at addressing rapid clearance limitation often
observed with enzyme therapeutic candidates. They exploit different properties of
protein domains to generate new molecular entities with desirable or enhanced
properties. The most clinically and commercially successful fusion therapeutics to date

92
contain the Fc fragment of immunoglobulins 212. Such Fc fusion can increase proteins
and peptides serum half-life to days and even weeks by binding to the salvage receptor,
FcRn 53,54. However, these technologies and platforms do not provide a tissue-retention
strategy and can often elicit an immune response213. In this study, we developed and
characterized a new fusion chimera, NanoLuc-Gal3, which demonstrated maintenance
of function for both the targeting and enzymatic moieties in vitro and prolonged retention
time of model enzyme in vivo.
Following expression and purification of the fusion, NanoLuc-Gal3, the activity of
both molecules was tested. Analysis of binding affinity for α-lactose agarose column
revealed fusion construct behaves almost identical to wild type Gal3 consistent with
published literature demonstrating proteins fused to Gal3 through the N-terminus have
no effect on Gal3 binding to its sugar ligand193,194. Additionally, NanoLuc-Gal3 binding to
immobilized proteins was also observed to be consistent with published works. Gal3
fusion binds components of the extracellular matrix with carbohydrate specificity, as
determined by the competitive assay with β-lactose, and binding to laminin is much
higher than collagen type I, vitronectin and serum proteins consistent with expected
LacNAc content 181,202,204,214.
Following ligand binding, wild type Gal3 undergoes conformational changes,
which enable its oligomerization into higher order structures 179. Therefore, we sought to
evaluate NanoLuc-Gal3 oligomerization capacity compared to Gal3 in collaboration with
Dr. Greg Hudalla and his student Antonietta Restuccia. Asialofetuin (ASF) is a
glycoprotein containing complex carbohydrate chains with terminal galactose and
LacNAc residues. As previously established 201, Gal3 binds to terminal LacNAc,

93
crosslinking and precipitating ASF as seen by the increased light scatter. NanoLucGal-
3, on the other hand, fails to crosslink and precipitate ASF at any of the ratios of lectin to
glycoprotein studied. Galectin 3 crosslinking of ASF molecules is driven by the
oligomerization of the lectin via the N-terminus. In the case of NanoLucGal-3, fusion is
achieved through the N-terminus likely making it unavailable for self-assembly into
higher ordered structures.
Much of the physiological processes involving Gal3 are due to its ability to
oligomerize into pentamers. With respect to immune cells, Gal3 acts in a dual manner,
either protecting T cells from apoptosis or promoting cell death, depending on whether it
functions intracellularly or is added exogenously to T cell cultures 179,186,215. Extracellular
Gal3, induces cell death in human T cells by binding to CD7 and CD29 186 or to CD45
and CD71 205 and mobilizing intracellular Ca2+ to promote the exposure of
phosphatidylserine on the cell surface, thus preparing the cell for phagocytic recognition
and subsequent removal 216. Additionally, Gal3-ligand lattices can limit T cell receptor
(TCR) clustering, thereby increasing the threshold for TCR signaling. In this study,
NanoLuc-Gal3 was exogenously administered to Jurkat T and determined to bind
specifically through the CRD to the surface of the cell. Whereas wild type Gal3
promotes Jurkat T cell agglutination and apoptosis driven by the N-terminal self-
association domain, fusion of NanoLuc at the N-terminus domain abrogates this activity,
presumably by blocking oligomerization. Additionally, we demonstrated NanoLuc-Gal3
can bind primary splenocytes in a CRD-dependent manner and binding to DCs
increases expression levels of stimulatory and co-stimulatory molecules. The data
presented here demonstrated NanoLuc-Gal3 does not act as a damage associated or

94
molecular associated pattern and does not initiate an inflammatory response, an
important characteristic for any therapeutic aiming to suppress inflammation.
Previous studies have demonstrated Gal3 with a truncated N-terminus can act as
an inhibitor of wild type Gal3 due to higher affinity for carbohydrate ligand 194. Although
no higher binding affinity was observed by fusion with NanoLuc, the ability of NanoLuc-
Gal3 to inhibit wild type Gal3 was evaluated. NanoLuc-Gal3 and Gal3 were
administered at various ratios to Jurkat T cells and agglutination and apoptosis
measured. Interestingly, the fusion construct competes with wild type Gal3 and even at
a 1:1 ration apoptosis is significantly reduced. This effect is attributed again to the
inability of the fusion protein to oligomerize into higher structured lattices often
associated with T cell apoptosis. Although not explored here, the data presented could
serve a new line of research for the development of Gal3 inhibitors sought after in
cancer metastasis applications 217.
With the work presented here, demonstrating fusion of Gal3 with a model
enzyme does not alter the binding affinity or activate immunological responses, we
sought to establish Gal3 as a tissue retention platform. In this study, we demonstrated
the Gal3 targeting domain in the NanoLuc-Gal3 fusion provides a prolonged retention
time for all sites tested whereas non-targeted NanoLuc is undetectable at all injection
sites after 24 h. Although an improvement is observed for all sites tested, a significantly
shorter retention time is observed for the intramuscular (IM) and intraperitoneal (IP)
groups. We hypothesize in the IM group this is due to injections of furimazine not
penetrating the tissue far enough limiting the necessary physical proximity between
enzyme and substrate, or the signal emitted is too deep into the tissue and below the

95
detection limit of the equipment used. For the IP group, although not tested here,
retention time is likely decreased due to the highly hydrodynamic environment and fast
turnover of proteins in the peritoneal fluid 218. Lastly, we established drainage of
NanoLuc-Gal3 administered at the hock to be minimal to major organs and clearance
through the renal system consistent with approved enzyme and fusion therapeutics.

96
Figure 3-1 Classification of galectins
Figure 3-2. Fusion protein construct. A) Schematic representation of wild-type galectin 3
(WT Gal3) and NanoLuciferase-Galectin 3 fusion protein. WT Gal3 forms a pentamer through self-assembly of the N-terminus domain. Upon fusion of NanoLuciferase at the N-terminus, self-assembly is disrupted while binding affinity for N-acetyl-D-lactosamine (LacNAc), present on proteins of the extracellular matrix and surface receptors on immune cells, is maintained. B) Gene construct for WT Gal3, NanoLuciferase, and NanoLuc-Gal3.

97
Figure 3-3. Galectin 3 retains binding affinity to sugar moiety when fused to an enzyme
on the N-terminus. A) Equal concentrations of NanoLuc-Gal3 (red line) and wildtype galectin 3 (black line) were injected into an α-lactose-agarose resin column and eluted with an increasing β lactose gradient. Wildtype and fusion proteins elute almost identically, indicating equivalent binding affinity. B) Polystyrene plates were coated at the specified concentrations with laminin, C) collagen type I, D) vitronectin, E) IgG, and F) α2 macroglobulin. NanoLuc-Gal3 was added either in the presence or absence of excess β lactose (to compete for Gal3 binding) and incubated 45 min. Plates were then washed, furimazine (NanoLuciferase substrate) added and bioluminescence quantified. Gal3 fusion binds extracellular matrix proteins with carbohydrate specificity, as β lactose blocks CRD activity; binding to laminin is much higher than collagen type I, vitronectin, and serum proteins consistent with expected LacNAc content.

98
Figure 3-4. Biology associated with Galectin 3 is disrupted when fused to an enzyme on
the N-terminus. A) Incubation of Galectin 3 and NanoLuc-Gal3 (30 uM) with increasing concentrations of asialofetuin (ASF), a glycoprotein containing carbohydrate chains with terminal LacNAc residues. Galectin 3 crosslinks and precipitates ASF molecules even at low ASF concentrations as determined by light scattering at 420 nm. NanoLuc-Gal3 fails to precipitate ASF, likely due to lack of the self-association domain that joins multiple CRDs and lead to the formation of large aggregates. B) Jurkat T cells were cultured with NanoLuc-Gal3 1 h in the presence or absence of excess β lactose. Cells were washed and incubated with furimazine to detect bioluminescence. NanoLuc-Gal3 binds to Jurkat through the CRD as demonstrated by inhibition with excess β lactose. T cells were cultured with NanoLuc-Gal3 1 h, C) imaged via phase contrast (scale bar = 50 µm) to evaluate agglutination, or D) live/dead stained and analyzed by flow cytometry to determine viability. Whereas wildtype galectin 3 promotes Jurkat T cell agglutination and apoptosis driven by the N-terminal self-association domain, fusion of NanoLuc to galectin 3 at the N-terminus abrogates this activity, presumably by blocking access to the self-association domain. N=3, mean + SEM with pair-wise significant difference (by ANOVA and Tukey’s post hoc) denoted by * where p < 0.05.

99
Figure 3-5. NanoLuc-Gal3 fusion prevents wild type Galectin 3-mediated agglutination
and apoptosis of Jurkat T cells. Jurkat T cells were cultured with NanoLuc-Gal3 (x µM), Galectin 3 (x µM) or different ratios of the two proteins for 4 h, A) imaged via phase contrast (scale bar = 50 µm) to evaluate agglutination, or B) live/dead stained and analyzed by flow cytometry to determine viability. Whereas wildtype galectin 3 promotes Jurkat T cell agglutination and apoptosis, NanoLuc-Gal3 competes with WT-Galectin 3 and prevents this activity in a concentration dependent manner. N=3, mean + SEM with pair-wise significant difference (by ANOVA and Tukey’s post hoc) denoted by * where p < 0.05.

100
Figure 3-6. NanoLuc-Gal3 fusion binds primary splenocytes in a CRD dependent
manner, binding to DCs does not act as a damage associated molecular pattern. A. Splenocytes were incubated with FITC-labeled NanoLuc-Gal3 fusion 45 min in the presence or absence of excess β lactose. Cells were immunostained and binding was assessed by flow cytometry of T cells (CD3, CD4 & CD8), dendritic cells (CD11c), B cells (B220) and macrophages (F4/80). Gal3 fusion binds leukocyte proteins with carbohydrate specificity. B. Murine bone marrow derived DCs were incubated with NanoLuc-Gal3 fusion in the presence or absence of excess β lactose. Binding to dendritic cells is CRD-mediated. C. Murine bone marrow derived DCs were treated with NanoLuc-Gal3 for 24 h. Cells were immunostained for activation markers (CD80, CD86 MHC II). Viable CD11c+ cells and the marker of interest were assed via flow cytometry, shown as % positive. NanoLuc-Gal3 does not activate DCs, suggesting foreign protein does not act as a damage associated molecular pattern. N=3, mean + SEM with pair-wise significant difference (by ANOVA and Tukey’s post hoc) denoted by * where p < 0.05.

101
Figure 3-7. Galectin 3 targeting in NanoLuc-Gal3 provides prolonged retention when
administered subcutaneously, intraperitoneally, and intramuscularly. NanoLuc or NanoLuc-Gal3 was administered to 8-week-old C57Bl/6 female mice. At specified times, furimazine was injected and animals imaged using in vivo imaging system (IVIS). Representative images of NanoLuc (left panel) and NanoLuc-Gal3 (right panel) administered A. subcutaneously into the scruff of the neck, B. intraperitoneally, C. subcutaneously into the abdominal region, D. intramuscularly or E. subcutaneously into the hock. F-J. Average radiance (p/s/cm2/sr) for each injection site was quantified. NanoLuc-Gal3 is retained subcutaneously for up to 7 d, intraperitoneally up to 3 d and intramuscularly up to 1 d whereas non-targeted NanoLuc is undetectable at 1 d for all administration routes. n=5 mice per administration route per protein, mean + SEM, with pair-wise significance (by ANOVA and Tukey’s post hoc) between both proteins denoted by ^, compared to 0 d for NanoLuc denoted by + and compared to 0 d for NanoLuc-Gal3 denoted by *, where p<0.05.

102
Figure 3-8. Galectin 3 targeting provides retention of a model enzyme at the injection
site with minimal drainage to adjacent tissues. NanoLuc-Gal3 was injected subcutaneous at the hock of 8-week-old C57BL/6 female mice and tissue distribution assessed over a 7 d period. To measure localized active NanoLuciferase, organs and tissues were excised, processed, incubated with furimazine and bioluminescence quantified by luminometer. n=5 mice per time point. NanoLuciferase remains active and at the injection site for up to 7 d, with minimal drainage to adjacent tissue and clearance through the renal system.
Sple
en
Liver
Lungs
Kid
ney
Inte
stin
e
Bla
dder
Inj.
hock
inj.
tibia
inj.
fem
ur
Con. h
ock
Con. t
ibia
Con. f
emur
Pla
sma
RBC
Urin
e
Feces
0.0
0.2
0.4
0.6
0.8
1 day 2 days 4 days 5 days 6 days 7 days
ug
pro
tein
/g t
issu
e

103
CHAPTER 4 FUSION OF INDOLEAMINE 2,3 DIOXYGENASE WITH GALECTIN 3 RETAINS
ENZYME AT INJECTION SITE AND HALTS INFLAMMATION IN VIVO
Background
Subcutaneous LPS-induced Inflammation
Lipopolysaccharide (LPS), found on the outer layer of gram-negative bacteria, is
often used to induce inflammation in vitro and in vivo in an array of pre-clinical
models219-222. Most studies have focused on systemic, either intravenous (IV), or
intraperitoneal (IP), administration for the induction of inflammation223. The dose
required to induce detectable cytokine levels in serum is 2 ng/kg in humans and 500
ng/kg in mice with peak concentrations at 2 h post injection224. In this study, we
administered a dose of 2 ng/g of body weight of a mouse subcutaneously to induce a
localized inflammatory response with no adverse systemic distribution.
Periodontal Disease
Periodontal diseases (PD) are a class of chronic inflammatory conditions of the
soft and hard tissues found in the oral cavity. Periodontal disease is highly prevalent
and begins with gingivitis, localized inflammation of the gingiva. If left untreated,
gingivitis can result in progressive loss of gingival tissue, the periodontal ligament and
eventually the adjacent supporting alveolar bone225. Chronic inflammation is initiated by
complex subgingival biofilms generally containing a portion of the gram negative
anaerobic commensal oral microbiota (dental plaque) as well as opportunistic
pathogens, including Porphyromonas gingivalis226. While microbes are key agents of
periodontal disease, they do not directly cause disease, but rather induce harmful
inflammatory responses in a susceptible host227-230. For example, in response to
pathogens, polymorphonuclear cells release reactive oxygen species and other factors

104
that can damage the gingival tissue. The secreted agents also enhance the production
of numerous proinflammatory cytokines that contribute to the disease, including IL-1β,
IL-6, and TNFα231. Periodontal disease may also worsen other inflammatory conditions
such as type 1 diabetes and atherosclerosis232,233.
Immunopathogenesis
Under conditions of health, gingival tissues contain low number of T lymphocytes
that assist in the maintenance of homeostasis between the host tissue and the bacterial
plaque. Disease occurs when the balance between the microbial biofilm and the host
are lost. This imbalance is still not fully understood but there are significant changes
observed to both the dental plaque and the host immune profile that results in a
heighted inflammatory state231.
The first line of defense to pathogens is provided by epithelial cells that function
as a physical barrier against microbes and elicit innate and adaptive immune
responses. Langerhans cells within the epithelium take up antigenic materials, transport
it to lymphoid organs and present it to lymphocytes. Tissue infiltration by neutrophils,
granulocytes and lymphocytes follows: neutrophils try to engulf and eliminate bacteria,
but in the case of periodontal disease, are overwhelmed by the persistence of the
microbial biofilm. Early lesions follow an acute neutrophil response and a slight increase
in lymphocyte infiltration. In chronic disease, an increase in DCs and macrophages as
well as lymphocytes is observed with higher presence of CD8+ T cells than CD4+ T
cells. The relevant importance and timing of Th cell subset involvement is unclear. Th1
cells might be important during the early stages of chronic periodontitis, whereas Th2
cell involvement appears to occur at later stages234. Lastly, this severe chronic

105
inflammatory response leads to alveolar bone resorption by osteoclasts, degradation of
ligament fibers and the formation of granulated tissue235.
Current treatments
Current treatment modalities for PD focus on mechanical approaches to reduce
the number of pathogens through non-surgical and surgical therapies. Non-surgical
approaches are used as a first line of treatment and consist of removal of supragingival
and subgingival dental plaque and calculus with scaling and root planing231. Non-
surgical therapies can often be combined with adjunctive therapies such as the local
and systemic delivery of antibiotics, or the systemic modulation of the host’s immune
responses. However, non-surgical approaches can be ineffective on their own,
particularly when moderate (4-6 mm) to deep (> 6mm) pockets have formed. In these
cases, several surgical approaches are available. Pocket reduction surgery includes
resection of soft and hard tissue using various techniques236,237, regenerative guided
surgery uses membranes to direct the growth of new periodontium preventing the
epithelium and connective tissue from growing in areas desired for bone and
ligament238, grafting239 and more recently laser-assisted attachment as a more
conservative approach240.
A more favorable approach to the treatment of PD should reduce the bacterial
challenge and favorably modulate the host response locally and in a non-invasive
fashion. Because of the multifactorial nature of the host immunological responses in
periodontal disease, a multimodal approach controlling the host response would be
advantageous. A key aspect of PD pathogenesis commonly overlooked is its localized
pathology, whereby the nature of the inflammation and tissue breakdown is not
localized to the oral cavity, but to the specific teeth and some cases to specific sites of

106
these teeth. Thus, an adjunctive therapeutic approach would be most appropriate if it
can achieve sustained localized efficacy. In this study we develop, characterize and
utilize a chimera fusion protein containing an immunomodulatory moiety, indoleamine
2,3 dioxygenase (IDO), linked to a tissue-targeting molecule, Galectin 3 (Gal3), IDO-
Gal3, for the amelioration of inflammation in an LPS-induced model of inflammation and
a murine model of PD.
Materials and Methods
Cloning of IDO-Galectin 3
For the IDO-Gal3 gene, the IDO sequence was generously provided by the
laboratory of Dr. Carlos Gonzalez. Amplification and insertion of the BamHI restriction
site on the 3’, was done using the primers, 5’-CAG CTA CCA TGG CAC ACG CTA TGG
AAA-3’ and GAG AAC 5’-GGA TCC ACC TTC CTT CAA AAG-3’ by Antonietta
Restuccia. The NanoLuc-Gal3 gene was digested with NcoI and BamHI to remove
nanoluciferase gene and insert gal3 gene. Plasmids were screened and sequenced as
described in Chapter 3.
Protein Expression and Purification
Origami B (DE3) E.coli (Novagen) were transformed with pET-21d-galectin-3,
pET-21d-NanoLuc, pET-21d-NanoLuc-Gal3, or pET-21d-IDO-Gal3 vectors and selected
on 100 μg/mL ampicillin- and 50 μg/mL kanamycin A-doped LB/agar plates overnight at
37°C. Positive clones were picked and used to inoculate 10 mL 100 μg/mL ampicillin-
and 50 μg/mL kanamycin A-containing LB broth. Cultures were grown overnight at
37°C, 220 rpm on an orbital shaker. Plasmids were isolated from cultures via a plasmid
mini-prep kit (Qiagen), according to manufacturer’s instructions, and sequenced at the
Interdisciplinary Center for Biotechnology Research at the University of Florida. Cultures

107
of positive clones were then sub-cultured into 1 L 100 μg/mL ampicillin- and 50 μg/mL
kanamycin A-containing LB broth and grown at 37°C, 225 rpm on an orbital shaker until
an optical density of 0.6-0.8 (λ=600 nm) was reached. Protein expression was induced
with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and incubated for 18 h at
18°C, 225 rpm in an orbital shaker. Bacteria were pelleted by centrifugation, and
washed with PBS. Bacteria were lysed with B-PER (Thermo Fisher), a protease inhibitor
tablet (Thermo Fisher), 300 units DNAse I from bovine pancreas (Sigma), and 100 μg
lysozyme (Sigma) for 20 min. Lysed bacteria was cleared by centrifugation, and
supernatant containing recombinant proteins were loaded onto columns containing
HisPur cobalt resin (Thermo Fisher) equilibrated with PBS. Columns were washed with
20-30 column volumes and proteins were eluted with increasing concentration of
imidazole. Imidazole was removed by centrifugation, using Amicon filter tubes (MWCO
10kDa) (Millipore). A second purification step was required using size exclusion
chromatography in an AKTA pure chromatography system (GE Life Sciences). Protein
molecular weight and purity were analyzed with SDS-PAGE. Endotoxin contaminants
were removed by Detoxi-gel endotoxin removing columns (Thermo Fisher) following
manufacturer’s instructions. Endotoxin content was analyzed using Pierce™ LAL
Chromogenic Endotoxin Quantitation Kit (Thermo Fisher), and determined to be below
0.1 EU/mL in all stocks.
IDO Enzymatic Activity Assay
Recombinant human IDO (IDO) expressed in Escherichia coli was purchased
from R&D Systems (Minneapolis, MN) with a predicted molecular mass of 46 kDa,
purity >95% by SDS-PAGE, endotoxin levels <1 EU/µg of protein by the LAL method
and a specific activity of >500pmoles/min/µg as measured by its ability to oxidize L-

108
tryptophan to N-formyl-kynurenine. The specific activity of both proteins, IDO and IDO-
Gal3, was measured before experiments to ensure maximal effect at the beginning of
the assay following an adapted procedure from Valladares et al.162. The reaction
substrate contained 200 µM tryptophan, 20 mM ascorbic acid, 10 µM methylene blue,
225 U catalase and 50 mM MES buffer (pH 6.5). Equal moles of IDO and IDO-Gal3
were loaded onto a flat bottom 96-well plate and the reaction started by mixture in 1:1
ratio with reaction substrate. Absorbance was measured at 480 nm.
Galectin 3 and IDO-Gal3 Binding Affinity
Affinity of galectin-3 and NanoLucGal-3 for lactose was determined using affinity
chromatography in an AKTA Pure chromatography system (GE Life Sciences) equipped
with an alpha-lactose agarose column (Sigma-Aldrich) by Antonietta Restuccia. Proteins
were eluted with a linear gradient of beta-lactose (Sigma-Aldrich) in phosphate buffer.
Antibodies
The CD11c (PE-Cy7 hamster anti-mouse clone: HL3), CD80 (APC hamster anti-
mouse clone: 16-10A1), CD86 (FITC rat anti-mouse clone: GL1), MHC II (PE rat anti-
mouse clone: M5/114.15.2) and CD16/32 (Fc block purified rat anti-mouse clone:
2.4G2) antibodies used to define DCs and their maturation status were purchased from
BD Bioscience. The CD4 ( PE-Cy7 hamster anti-mouse clone: RM4-5) and CD8 (PE rat
anti-mouse clone: 53-6.7) antibodies were used to define T cells and also purchased
from BD Bioscience. The stained cells were analyzed using a Guava easyCyte 8HT
Benchtop Flow Cytometer (EMD Millipore), using the InCyte Software (version 3.1) for
analysis.

109
Mice and Cell Lines
C57BL/6 (B6) and B6.Cg-Tg(TcraTcrb)425Cbn/J (OT-II) mice were purchased
from Jackson Laboratory. Age-matched (6-12 weeks) female animals were used
throughout all experiments. All studies were performed in accordance with protocols
approved by the Institutional Animal Care and Use Committee (IACUC) at the University
of Florida. Experimental groups and sizes were approved by authorities for animal
welfare after being defined to balance statistical power, feasibility and ethical aspects.
All animals were cared for/monitored/inspected daily either by a member of the research
team or with an agreement by Animal Care Services personnel.
DC Maturation and Cytokine Release Profile
Dendritic cells were generated from murine, bone marrow isolated from the
femurs and tibias of B6 mice in the presence of granulocyte-macrophage colony
stimulating factor (GM-CSF) (R&D Systems) according to previously described
protocols72. Cells were cultured untreated, with 1 µg/mL LPS (Sigma-Aldrich) overnight,
0.32 µM IDO-Gal3 for 24 h, or with IDO-Gal3 for 24 h, washed, and challenged with
LPS overnight. Cells were stained for viability using LIVE/DEAD Fixable NIR Dead Cell
Stain Kit for 633 or 635 nm excitation, stimulatory (MHC II) and co-stimulatory
(CD80/86) markers and assessed via flowcytometry. Supernatants were collected and
analyzed for IL-10 and IL-12p70 cytokine expression using BD OptEIA ELISA kits (BD
Biosciences) following the manufacturer’s instructions.
Antigen Specific Co-cultures
Dendritic cells were generated as mentioned above and treated with IDO-Gal3 in
the presence or absence of 1 mM 1-methyl-DL-tryptophan (Sigma-Aldrich). T cells were
isolated from OT-II splenocytes and labeled with 1 µM Carboxyfluorescein succinimidyl

110
ester (CFSE) according to previously described methods (Chapter 2). Cells were co-
cultured at a 1:10 DC to T cell ratio and proliferation assessed by CFSE dilution after 4
days via flow cytometry.
LPS Administration at the Hock as a Model of Inflammation
B6 mice were administered 2.2 µg of IDO-Gal3 in 40 µL of PBS subcutaneously
at the region of the hock and challenged with 2 ng/g of lipopolysaccharide (LPS) in 40
µL 1 d, 3 d, or 5 d post IDO-Gal3 treatment. 2 h after LPS administration animals were
euthanized and the injection site collected. Soft tissue was macroscopically separated
from bone and submerged in in RNAlater RNA Stabilization Reagent (Qiagen) in
preparation for qPCR.
Quantitative PCR
Soft tissues were homogenized and RNA purified using RNeasy Protect Mini Kit
(Qiagen). cDNA was synthesized from RNA using the High-Capacity cDNA Reverse
Transcriptase Kit (ThermoFisher) for use in qPCR in accordance with manufacturers
instructions. qPCR analysis was ran with primers specific for pro-inflammatory
cytokines (I12a, Il12b, Il1b, Ifng and Il6). Results are presented as the ratio of gene
expression to Gapdh expression determined by the relative quantification method.
Treatment groups were normalized to PBS only group.
Mass Spectrometry
2.2µg of IDO-Gal3 in 40 µL of PBS or 40 µL of PBS alone was injected into the
hock site of B6 mice (n=3). 30 min after injection, the mice were sacrificed, the hock and
tibia tissue regions excised and flash frozen using liquid nitrogen. These samples were
then submitted to the Southeast Center for Integrated Metabolomics at the University of
for mass spectrometric analysis of kynurenine and tryptophan levels.

111
Hock Infiltration
B6 mice were injected into the hock with IDO-Gal3 and 1 or 5 d later challenged
with LPS (ipsilateral and contralateral) to assess modulation of inflammation locally and
systemically. 2 h after LPS challenge animals were euthanized and the injection sites
harvested. Tissues were fixed with 10% Neutral Formalin pH 7.4 overnight. After
fixation, tissues were washed in diH2O and decalcified by storing in 10%
Ethylenediaminetetraacetic acid (EDTA) at 4°C for 3 weeks. Samples were assessed
every 2-3 days for stiffness and EDTA solution replenished. Tissues were submitted in
70% ethanol to the University of Florida Molecular Pathology Core for processing,
paraffin embedding, sectioning, mounting and staining with Haemotoxylin and Eosin
(H&E). Tissues were imaged using a Zeiss Axiovert 200M with a 20X objective lens
through the multidimensional acquisition module. 11 images were taken per tissue and
scored by two blinded independent individuals based on cellular infiltration (0: absent,
1:mild, 2:moderate, 3:severe) and epidermis hypertrophy (0:0-20 µm in thickness, 1: 21-
40 µm, 2: 41-60 µm, 3: 61µm or above).
In vivo Bioluminescence and Imaging
Bioluminescence images were acquired using an IVIS Spectrum In Vivo System
(IVIS). Living Image Software version 4.3.1 (Perkin Elmer, Waltham, MA) was used to
acquire the data immediately after furimazine administration. Exposure time for the
bioluminescence imaging was 1 second. Regions of interest were quantified as the
average radiance (photons/second/cm2/sr).
Mucosal Induction of Inflammation
6-week-old B6 mice were lavaged with 0.12% chlorhexidine gluconate for 3 d
followed by an infection consisting of an oral lavage with 2.5x109 Porphorymonas

112
gingivalis strain 381 and 2.5x109 Aggregatibacter actinomycetemcomitans strain 29522
resuspended in 2% low viscosity carboxy-methyl-cellulose on 4 consecutive d, for 5
straight weeks. Mice received a dose of IDO-Gal3 each week either prophylactically or
therapeutically during the development of periodontal disease.
Subgingival Cytokine Production
The local immunological soluble mediator milieu was evaluated using
multiplexing technology by Fernanda Rocha, DDS, PhD in the laboratory of Dr.
Shannon Wallet. Briefly, the mandible and maxillae were subjected to ‘bead beating’ in
a cell-lysis buffer containing protease inhibitors. The resulting homogenates were
analyzed for various soluble mediators using multiplex technology.
Results
IDO Activity and Gal3 Affinity Are Not Altered Upon Fusion
The enzymatic activity of IDO and the binding affinity of Gal3 were determined
for both fused and unfused proteins (Figure 4-1). Unfused IDO was able to generate 0.1
µg/mL of kynurenine whereas fusion protein, IDO-Gal3, produced 0.3 µg/mL of
kynurenine demonstrating comparable activity regardless of fusion state. Wild type Gal3
eluded from α-lactose-agarose column at 15% β-lactose whereas IDO-Gal3 eluted at
65% indicative of stronger affinity.
IDO Retains Immunomodulatory Properties When Fused to Galectin 3
Murine bone marrow derived dendritic cells were incubated with IDO-Gal3 at 2
nM for 24 h and their maturation status determined (Figure 4-2). Expression of
stimulatory (MHC II) and co-stimulatory (CD80/86) molecules was determined through
flow cytometry (presented as mean + SEM of three experiments, total n=9) (Fig. 4-2A).
The percent of untreated DCs expressing CD80, CD86 and MHC II was 13% + 0.3, 6%

113
+ 0.1 and 23% + 0.4, respectively. In contrast, upon LPS treatment expression of three
activation markers significantly increased (p < 0.05) to 30% + 1, 33% + 3, and 27% +
0.3 of cells. When DCs were treated with IDO-Gal3 for 24 h, maturation markers
remained comparable to immature or untreated cells with 11% + 0.4, 5% + 0.2, and
14% + 2 of cells expressing CD80, CD86 and MHC II, respectively. However, when DCs
were cultured with IDO-Gal3 followed by LPS challenge maturation markers were lower
than LPS alone, with 12% + 0.5, 5% + 0.2 and 19% + 0.6 expressing CD80, CD86 and
MHC II, confirming cells treated with IDO-Gal3 resist LPS-induced maturation. Next, the
release profile of inflammatory and anti-inflammatory cytokines, IL-12p70 and IL-10
were analyzed via ELISA (Fig. 4-2B). IL-12p70 secretion levels were established at 17 +
9 pg/mL and 219 + 20 pg/mL for untreated and LPS-treated cells, respectively. When
IDO-Gal3 was introduced to the DC culture, IL-12p70 levels were determined at 12 + 5
pg/mL. Remarkably, when DCs were treated with IDO-Gal3 and challenged with LPS,
IL-12p70 levels were established at 3 + 1 pg/mL, comparable to untreated group.
Conversely, IL-10 production remained consistent, with no significant changes observed
between groups.
To evaluate whether IDO-Gal3 treated DCs can attenuate antigen specific CD4+
T cell proliferation, CFSE labeled T cells isolated from splenocytes of OT-II transgenic
mice were co-cultured with treated DCs. T cells from these mice proliferate in response
to OVA peptide 323-339 (discussed in detail in Chapter 2). The data shown in Figure 4-
3 represent the percent proliferation of viable CD4+ T cells stimulated with IDO-Gal3-
treated DCs. In co-cultures where only DCs and T cells were present, or when IDO-
Gal3 was introduced, minimal proliferation was observed at 33% + 0.9 and 35% + 1,

114
respectively. When DCs were pulsed with OVA323-339 and co-cultured with T cells, T
cells became activated and proliferation measured at 95% + 1 after 4 d. However, when
DCs were cultured with IDO-Gal3 for 24 h, washed, pulsed with OVA323-339, and co-
cultured with T cells, proliferation was reduced to basal levels measured at 33% + 2. T
cells only were also cultured for 4 d and included as a negative control with proliferation
at 5% + 0.7. (Fig. 4-3A) The experiment was repeated, with the introduction of the IDO
inhibitor MT as a DC treatment (Fig. 4-3B). In the presence of MT-treated DCs, CD4+ T
cell proliferation was restored to 58% + 2, indicating the active enzyme is required for
the attenuation of proliferation. A representative flow cytometry histogram for
proliferation of T cells stimulated with IDO-Gal3 treated DCs or IDO-Gal3 and MT
treated DCs is shown in Fig. 4-2C. Herein, we demonstrate fused IDO maintains its
metabolic reprogramming potential comparable to unfused enzyme as seen in Chapter
2.
IDO-Gal3 Reduces Inflammatory Cytokine Gene Expression at Various Time Points.
B6 mice were subcutaneously injected with IDO-Gal3 at the hock of the left hind
limb. 1 d, 3 d or 5 d after treatment mice were challenged with LPS as a model of local
inflammation and gene expression of pro-inflammatory cytokines (IL-12p35, IL-12p40,
IL-1β, IFN-γ, and IL-6) analyzed through qPCR. The data presented in Figure 4-4 is
representative of 5 animals per group normalized to cohort that received a PBS sham
injection. A schematic representation of the experimental schedule is provided in Fig. 4-
3A. Groups that showed no significant contribution to the modulation of inflammation at
early time points were dropped from further analysis. Treatment with LPS significantly
increased compared to PBS sham for IL-12p35 (4-fold), IL-12p40 (28-fold), IL-1β (2860-

115
fold), IFNγ (860-fold) and IL-6 (25400-fold). Treatment with untargeted IDO did not
increase IL-12p35 gene expression compared to sham PBS but increased IL-12p40
(14-fold), IL-1β (420-fold), INFγ (185-fold) and IL-6 (5556-fold). Challenge of untargeted
IDO with LPS injection did not increase IL-12p35 gene expression when compared to
sham but increased IL-12p40 (17-fold), IL-1β (1370-fold), IFNγ (370-fold) and IL-6
(15600-fold). Treatment with IDO-Gal3 only decreased IL-12p35 gene expression when
compared to sham (0.5-fold), and increased IL-12p40 (15-fold), IL-1β (1453-fold), IFNγ
(178-fold) and IL-6 (1400-fold). Remarkably, treatment with IDO-Gal3 challenged with
LPS 1 d later, decreased IL-12p35 expression compared to sham injection (0.2-fold),
and increased IL-12p40 (16-fold), IL-1β (1370-fold), IFNγ (156-fold) and IL-6 (6757-
fold). A different cohort of animals was again treated with IDO-Gal3 at D1, but the LPS
challenged was administered 3 d after (Fig. 4-3C). Similarly to 1 d data, large fold
changed were observed for all pro-inflammatory cytokines measured when mice were
treated with LPS only as a positive control. Following treatment with IDO-Gal3 at D1,
and LPS challenge at D3, all cytokines significantly decreased compared to LPS only
group. Lastly, a third cohort was administered IDO-Gal3 at D1 and challenged with LPS
at D5. Again, mice that received LPS only injections showed significant increase in
proinflammatory cytokine gene expression whereas mice that were prophylactically
treated with IDO-Gal3 and challenged with LPS showed a decreased in cytokine gene
expression when compared to LPS only group. Prophylactic treatment with IDO-Gal3
decreases LPS-mediated inflammatory cytokine gene expression at the hock.
IDO-Gal3 Modulates Local Tryptophan Catabolism
IDO-Gal3 or PBS was injected into the hock of B6 mice. Either 30 mins or 24 h
after injections animals were euthanized, the injection site and adjacent tissues excised

116
and flash frozen for mass spectrometry analysis of kynurenine and tryptophan levels
(Figure 4-5). 30 min after treatment (PBS) the kynurenine levels at the hock were 55 +
15 ng/g of protein while levels at the tibia were 48 + 4 ng/g of protein. For animals
injected with IDO-Gal3, kynurenine levels increased at the hock (1534 + 400 ng/g) and
the tibia (273 + 83 ng/g (Fig. 4-5A)). For the same cohort of mice, the tryptophan levels
in the PBS group were 3627 + 2000 ng/g of protein at the hock and 1714 + 400 ng/g
protein at the tibia. For the IDO-Gal3 injected groups, at the hock tryptophan levels were
4416 + 680 ng/g of protein and 2134 + 60 ng/g of protein at the region of the tibia (Fig.
4-5B). At 24 h post treatment, animals treated with PBS showed kynurenine levels at 14
+ 5 ng/g of protein at the hock, 13 + 5 ng/g of protein at the region of the tibia, and 23 +
9 ng/g of protein at the draining (popliteal) lymph node. When animals were injected
with IDO-Gal3, kynurenine levels were measured at 76 + 30 ng/g of protein at the hock,
73 + 30 ng/g of protein at the tibia and 57 + 25 ng/g of protein at the popliteal lymph
node (Figure 4-5C). Lastly, tryptophan levels were measured 24 h post treatment.
Tryptophan levels were established at 2153 + 929 ng/g of protein at the hock, 1608 +
900 ng/g of protein at the tibia, and 2609 + 600 ng/g of protein at the popliteal lymph
node. The IDO-Gal3 group showed Tryptophan levels for the IDO-Gal3 injected group
were 2244 + 700 ng/g of protein at the hock, 1884 + 600 ng/g of protein at the tibia and
1903 + 600 ng/g of protein at the popliteal lymph node (Fig. 4-5D).
IDO-Gal3 Modulates LPS-induced Inflammation Locally
To test the efficacy of the localization strategy in combating inflammation, B6
mice were injected with IDO-Gal3 or relevant controls as before and challenged with
LPS 1 d or 5 d after, in the ipsilateral (previously treated hind limb hock) and
contralateral (untreated opposite hind limb) site. 2 h post LPS challenge, animals were

117
euthanized and injection site excised (Figure 4-6). A schematic representation of the
injection schedule is shown in Fig. 4-6A. H&E stained tissues were scored based on
cellular infiltration and epidermal hypertrophy (enlargement of epidermis layer) following
LPS challenge either 1 d or 5 d post-treatment (Fig. 4-6B-E). Animals that were
administered PBS on both sides and received no LPS challenged showed low cellular
infiltration and average epidermal thickness (Fig 4-6panel). Animals that received PBS
injection followed by LPS challenge on both sides, demonstrated high cellular infiltration
and increased epidermal thickness (Fig 4-6panel). Animals that were treated with
untargeted IDO and received no LPS challenged showed characteristics comparable to
PBS group. When animals were treated with untargeted IDO and challenged with LPS,
H&E slides show cellular infiltration and epidermal hypertrophy similar to LPS group.
When IDO-Gal3 was administered with no LPS challenge, tissue resembled the PBS
only group (Fig 4-6panel). Remarkably, when IDO-Gal3 was administered and
challenged with LPS ipsilaterally, cellular infiltration and epidermis hypertrophy were
significantly reduced. Conversely, when LPS was administered contralaterally to IDO-
Gal3 treatment, tissue resembled the LPS only group with significant inflammation (Fig.
4-6F).
Gal3 Provides Retention of a Model Enzyme at Subgingival Tissue, Retention Time Is Not Altered by Infection State
NanoLuc or NanoLuc-Gal3 was administered to B6 mice subgingivally on 1 d
followed by daily administration of furimazine. Mice were sedated and imaged daily via
IVIS until no signal was detected for two consecutive days (Figure 4-7). NanoLuc alone
cleared from the injection site within 48 h. NanoLuc-Gal3 however, remained at the
injection site with detectable levels for 10 d. Quantification of signal obtained for both

118
proteins is shown as the average radiance (p/s/cm2/sr) over 10 d (Fig. 4-7A).
Representative images from animals injected with NanoLuc (left panel) and NanoLuc-
Gal3 (right panel) are shown in Fig. 4-7B. Furthermore, the retention time of NanoLuc-
Gal3 was evaluated in tissues with varying levels of infection (Fig. 4-7C). NanoLuc-Gal3
was administered to uninfected (black line), acutely infected (blue), or chronically
infected (red line) animals. No significant differences were observed.
IDO-Gal3 Suppresses Inflammatory Cytokine and Chemokine Production
IDO-Gal3 was administered to B6 mice, prophylactically or therapeutically to
polymicrobial infection (Figure 4-8), and the soluble mediators in the soft tissue
analyzed (Figure 4-9). Infected animals showed consistently high expression of IL-1β
(72 + 2 ρg/mL), IL-12p70 (26 + 1 ρg/mL), IL-33 (2505 + 271 ρg/mL), KC (101 + 9
ρg/mL), and IL-6 (24 + 1 ρg/mL) cytokines (Fig. 4-9A). Levels of IL-10 for infected group
were established at 8 + 0.7 ρg/mL. For animals treated prophylactically, IL-1β (66 + 1
ρg/mL), IL-12p70 (21 + 0.6 ρg/mL), IL-33 (1159 + 65 ρg/mL), KC (75 + 3 ρg/mL), and
IL-6 (21 + 0.4 ρg/mL) secretion levels significantly decreased, While IL-10 levels were
detected at 16 + 0.7 ρg/mL. When animals were treated therapeutically, significant
reduction was observed in the production of IL-1β (60 + 1 ρg/mL), IL-12p70 (13 + 0.8
ρg/mL), IL-33 (908 + 40 ρg/mL), KC (59 + 2 ρg/mL), and IL-6 (14 + 0.8 ρg/mL) and an
increase in IL-10 (23 + 0.8 ρg/mL) production. Uninfected animals were also treated
with IDO-Gal3 to determine any inflammation associated with administration of foreign
protein, IL-1β (56 + 0.5 ρg/mL), IL-12p70 (7 + 1 ρg/mL), IL-33 (752 + 29 ρg/mL), KC (51
+ 2 ρg/mL), IL-6 (8 + 0.7 ρg/mL), IL-10 (31 + 1 ρg/mL), levels were measured and
determined to not have major contributions to inflammatory response. Lastly, uninfected
animals were evaluated and levels of of IL-1β (51 + 1 ρg/mL), IL-12p70 (5 + 0.6 ρg/mL),

119
IL-33 (462 + 98 ρg/mL), KC (35 + 3 ρg/mL), IL-6 (5 + 0.6 ρg/mL) and IL-10 (25 + 0.6
ρg/mL) cytokines established. A similar trend was observed when chemokines (IP10,
MCP1 and MIP2) were evaluated (Fig. 4-9B) where statistically significant differences in
MCP1 and MIP2 were observed in animals treated therapeutically vs prophylactically.
Herein, we demonstrated treatment with IDO-Gal3 ameliorates local inflammation either
in an LPS-induced subcutaneous model of inflammation or in a polymicrobial murine
model of periodontal disease by decreasing cytokine gene expression and production
subsequently preventing cellular infiltration of the affected tissue.
Discussion
Inflammation is the protective reaction orchestrated by the immune system in
response to infection and/or injury241. Local and systemic inflammatory responses aim
to eliminate the inciting stimulus, promote tissue repair and healing, and in the case of
infection, establish immune memory such that the host mounts a faster response upon
repeated insult242. However, complications arise when the inflammatory response is
chronic and the balance between protective and destructive immunity favors
destruction. The inflammatory response is a complex but highly coordinated sequence
of events, involving molecular, cellular and physiological alterations. It begins with the
production of soluble mediators (e.g. complement, cytokines and chemokines) by
resident cells, including macrophages and DCs, in the injured or infected tissue. At the
same time, cell adhesion molecules are upregulated on circulating leukocytes and
endothelial cells, promoting the exudation of proteins and influx of granulocytes from the
blood243.This well-characterized phase of the inflammatory response is routinely
targeted using drugs such as non-steroidal anti-inflammatory drugs (NSAIDs)244-249 and
pro-inflammatory cytokine-blocking antibodies29,53,250-252. These drugs currently lead the

120
industry for the treatment of inflammatory diseases253. However, strategies that achieve
sustained, localized efficacy are still needed. In the present work, a fusion chimera,
IDO-Gal3, was developed, characterized and employed to suppress inflammation by
reprogramming local metabolism. This study demonstrates Gal3 fusion confers oral
submucosal retention for 10 days and IDO-Gal3 provides robust suppression of
inflammation, both in vitro and in vivo.
IDO-Gal3 was first characterized to guarantee no significant changes occurred
during fusion of both proteins. Previous published work by others as well as our analysis
of NanoLuc-Gal3 have demonstrated that fusion of a model enzyme at the N-terminus
has no effect on the carbohydrate affinity profile of Gal3194. Indeed, IDO-Gal3 bound an
α-lactose agarose column more tightly than wild type Gal3. Further studies will
investigate any conformational changes that may be responsible for this favorable
increased affinity. Chapter 2 established IDO as an immunomodulator in the
extracellular space by suppressing DC maturation and subsequent T cell responses.
The IDO-Gal3 fusion maintained the immunomodulatory capacity of unfused IDO. DCs
treated with exogenous IDO-Gal3 did not upregulate expression levels of stimulatory
and co-stimulatory molecules, exhibiting diminished IL-12p70 secretion while
maintaining IL-10 production. This study further solidifies the notion that IDO enacts an
anti-inflammatory response in the extracellular environment.
Following in vitro characterization, mice were treated with IDO-Gal3 at the hock
and challenged with LPS either 1 d, 3 d or 5 d post-treatment. The hock is a suitable
alternative to the more common footpad injection, and minimizes animal distress254.
Moreover, LPS-induced inflammation serves as a proof of concept platform and

121
establishes the fusion protein as a mediator of multiple inflammatory conditions.
Analysis of the injection site revealed IDO-Gal3 modulated innate and adaptive
immunity as determined by the low-expression of all pro-inflammatory cytokines tested.
IL-6 responds to immediate tissue damage while IL-12, expressed by macrophages and
DCs, works synergistically with IL-1β, produced by neutrophils, to regulate T cell
production of IFNγ and differentiation into Th1 T cells, key players in autoimmune
diseases such as type 1 diabetes, rheumatoid arthritis and psoriasis. Treatment with
IDO-Gal3 before an insult lessens potential for auto inflammatory damage and
establishes Gal3 as a retention strategy at the site of inflammation. Additionally, given
the complexity of the inflammatory milieu, technologies that simply target one soluble
mediator are often inefficient. Demonstrated here, IDO-Gal3 can alter genetic
expression of multiple soluble mediators involved in immunological responses with a
single dose.
As gene expression does not always translate to protein production and
subsequent cellular changes255, we performed experiments to verify that administration
of IDO-Gal3 was ideal for application in disease models of microbial inflammation. Upon
histological analysis of relevant tissues, it was determined IDO-Gal3 halts LPS-induced
inflammation by inhibiting cellular infiltration of the affected tissue. Anti-inflammatory
effects can only be observed in the tissue treated with IDO-Gal3 (ipsilateral to injection
site), indicating treatment is local and not systemic. Localized suppression is crucial for
decreasing susceptibility of the host to opportunistic infections, abnormal course of
disease and increased risk of lymphomas and cancers256 often observed with systemic
immunosuppression.

122
Periodontal disease (PD) is an ideal candidate for application of our novel fusion
protein. The initial response to polymicrobial insult, characteristic of PD, is an acute
inflammatory response with increased vascular dilation and blood flow. Then, an
increase in neutrophil and macrophage migration toward the lesion, due to chemokines
such as IP10, MCP1 and MIP2, originating from the microbial cells as well as host-
derived inflammatory mediators is observed. These cells then release pro-inflammatory
cytokines, IL-1β, IL-12 and IL-6, and KC (IL-8 murine homologue), at the site of injury
and recruit members of adaptive immunity. The character and intensity of the
inflammatory response determines whether the initial lesion resolves or becomes
chronic, leading to PD. Overproduction of IL-33, for example, has been shown to
significantly exacerbate periodontitis in mice leading to alveolar bone loss257.
In this study, Gal3 was established as a retention strategy in the subgingival
space. Studies using NanoLuc-Gal3 demonstrated a retention time of 10 d was
observed with no differences between healthy and infected tissues. Furthermore, the
state of disease did not alter retention time mediated by Gal3. These findings are of
extreme importance for the treatment of PD as the state of inflammation varies with the
patient. A larger clinical impact can be appreciated when treatment can be used across
an entire population.
Surprisingly, when compared to infected groups, or prophylactically treated
animals, subjects that received IDO-Gal3 therapeutically showed significantly less
production of pro-inflammatory cytokines and chemokines and an increase in the anti-
inflammatory cytokine, IL-10. Work with NanoLuc-Gal3 revealed a decrease in enzyme
activity over time. Under the assumption that IDO-Gal3 behaves similarly, animals

123
treated prophylactically received a decreased effective dose resulting in higher
production of pro-inflammatory cytokines. Further studies will need to be conducted to
determine the specific mechanism by which IDO-Gal3 is able to halt inflammation in a
polymicrobial model of periodontal disease.

124
Figure 4-1. Enzymatic activity of IDO is not altered upon fusion with Galectin 3, Galectin
3 retains binding affinity to sugar moiety upon fusion with IDO at N-terminus. A) Equal molar quantities of IDO-Gal3 and IDO were incubated with tryptophan and its conversion to kynurenine measured by addition of Ehrlich reagent. IDO-Gal3 shows comparable production of kynurenine to unfused IDO. B) Equal concentration of IDO-Gal3 (red line) and wild type Gal3 (black line) were injected into an α-lactose-agarose resin column and eluded with an increasing gradient of β-lactose. Fusion protein elutes at a higher concentration of β-lactose indicative of stronger binding affinity.

125
Figure 4-2. Indoleamine 2,3-dioxygenase maintains an immature dendritic cell
phenotype when fused to Galectin 3. Murine bone marrow derived dendritic cells were treated with IDO-Gal3 for 24 h, washed and and challenged with LPS overnight. A) Cells were co-stained for viability, CD11c and maturation markers: CD80, CD86 and MHC II and analyzed through flow cytometry. IDO-Gal3-treated dendritic cells resist LPS-induced maturation. B) Supernatants were collected and analyzed for the secretion of IL-12p70 and IL-10 through ELISA. Dendritic cells treated with IDO-Gal3 do not release IL-12p70 even when stimulated with LPS and maintain a steady IL-10 secretion. Values shown as percent positive. n =3, mean + SEM with pair-wise significant difference from all other groups, ANOVA with Tukey’s post-hoc, denoted by * where p < 0.05.

126
Figure 4-3. IDO-Gal3-treated DCs attenuate antigen specific T cell proliferation,
suppression is active enzyme dependant. Murine bone marrow derived DCs were cultured with IDO-Gal3 for 24 h, washed and pulsed with OVA peptide. OVA specific T cells were isolated from transgenic OT-II mice, labeled with CFSE and co-cultured with DCs for 4 d. A) Proliferation of viable CD4+ was assessed via CFSE dilution. IDO-Gal3-treated DCs attenuate antigen-specific T cell proliferation to levels comparable to no-antigen control. B) DCs were cultured with IDO-Gal3 in the presence or absence of a potent IDO inhibitor, 1-methyl tryptophan (MT), and co-cultured with T cells as described above. When IDO activity is inhibited T cell proliferation is re-stored to levels compared to antigen control. C) Representative flow plots of T cell proliferation with IDO-Gal3-treated DCs (left panel) and T cell proliferation with IDO-Gal3 and methyl-tryptophan-treated DCs (right panel). Values shown as mean + SEM, pair-wise significant difference (by ANOVA and Tukey’s significant test) is denoted by * where p < 0.05 for n=3.

127
Figure 4-4. IDO-Gal3 blocks subcutaneous LPS-induced inflammatory cytokine gene
expression. IDO-Gal3 or controls were injected in the hock of 8-week-old C57BL/6 female mice. A) Schematic representation of injection schedule. B) On day 1, C) day 3 and D) day 5, subcutaneous hock injection of LPS was administered as a local inflammatory challenge. Two hours post LPS challenge, soft tissue at the injection site was harvested, and inflammatory gene expression analyzed through real-time qPCR. Pair-wise significant difference (by ANOVA and Tukey’s post hoc) is denoted by * where p < 0.05; n=5 mice per group. IDO-Gal3 modulates inflammation subcutaneously at the hock by halting gene expression of inflammatory cytokines for at least 5 d. Values shown are normalized to PBS group. n =5, mean + SEM with pair-wise significant difference from all other groups, ANOVA with Tukey’s post-hoc, denoted by * where p < 0.05.

128
Figure 4-5. IDO-Gal3 modulates tryptophan metabolism at the injection site. IDO-Gal3
or PBS was administered subcutaneously at the hock, the injection site and adjacent tissues excised, flash frozen and submitted to the Biomedical Mass Spectrometry Core for analysis of kynurenine and tryptophan levels. A) 30 min post IDO-Gal3 treatment high amounts of kynurenine accumulate at the hock with minimal detection at the region of the tibia. B) Tryptophan levels at the hock and tibia do not change between groups, C) 24 h post treatment kynurenine levels are elevated compared to PBS group indicative of active IDO-Gal3 and metabolic reprogramming. D) Tryptophan levels between groups and tissues does not change. IDO-Gal3 promotes tryptophan metabolism and induces kynurenine production, but kynurenine likely diffuses away from site of injection making difference undetectable at 24 h. Values shown as percent positive. n =2, mean + SEM with significant difference from IDO-Gal3, t-test, denoted by * where p < 0.05, and significant difference from hock region, ANOVA, denote by + where p < 0.05.

129
Figure 4-6. IDO-Gal3 blocks subcutaneous LPS-induced inflammatory response,
suppression is not systemic. IDO-Gal3, or relevant controls, were injected into the hock of 8-week-old C57BL/6 female mice. On day 1 and day 5, a subcutaneous hock injection of LPS was administered as a local inflammatory challenge ipsilateral or contralateral to treatment. A) Schematic representation of injection schedule and experimental design. 2 h post LPS administration, the injection site was excised, demineralized, paraffin embedded and stained with Hematoxylin and Eosin (H&E). Sections were scored (0-3) based on B) cellular infiltration on day 1 or C) epidermis hypertrophy on day 1 AND D) cellular infiltration on day 5 or E) epidermis hypertrophy on day 5. F) Representative H&E images of injection sites IDO-Gal3 halts inflammation subcutaneously at the hock by preventing cellular infiltration locally (scale bar = 50 µm). n =5, mean + SEM with pair-wise significance (ANOVA with Tukey’s post hoc) denoted by * when compared to contralateral side, by + when compared to PBS + LPS ipsilateral side, and ^ when compared to PBS + LPS contralateral side.

130
Figure 4-7. Galectin 3 provides retention of a model enzyme in the subgingival space;
retention profile is not altered by disease state. NanoLuc or NanoLuc-Gal3 was administered to 8-week-old C57BL/6 female mice in the subgingival space. At specified times, furimazine was injected and animals imaged using in vivo imaging system (IVIS) A) Maximum relative luminescence was quantified over time. B) Representative images of NanoLuc (top row) and NanoLuc-Gal3 (bottom row) over time. C) NanoLuc-Gal3 administration into the subgingival space over time for no-infection (black line) early infection (blue line) and late infection (red line) cohorts. Galectin 3 provides retention of a model enzyme in the subgingival space for at least 10 days and retention profile is not altered by disease state. Values shown as percent positive. n =5, mean + SEM with pair-wise significant difference from all other groups, ANOVA with Tukey’s post-hoc.
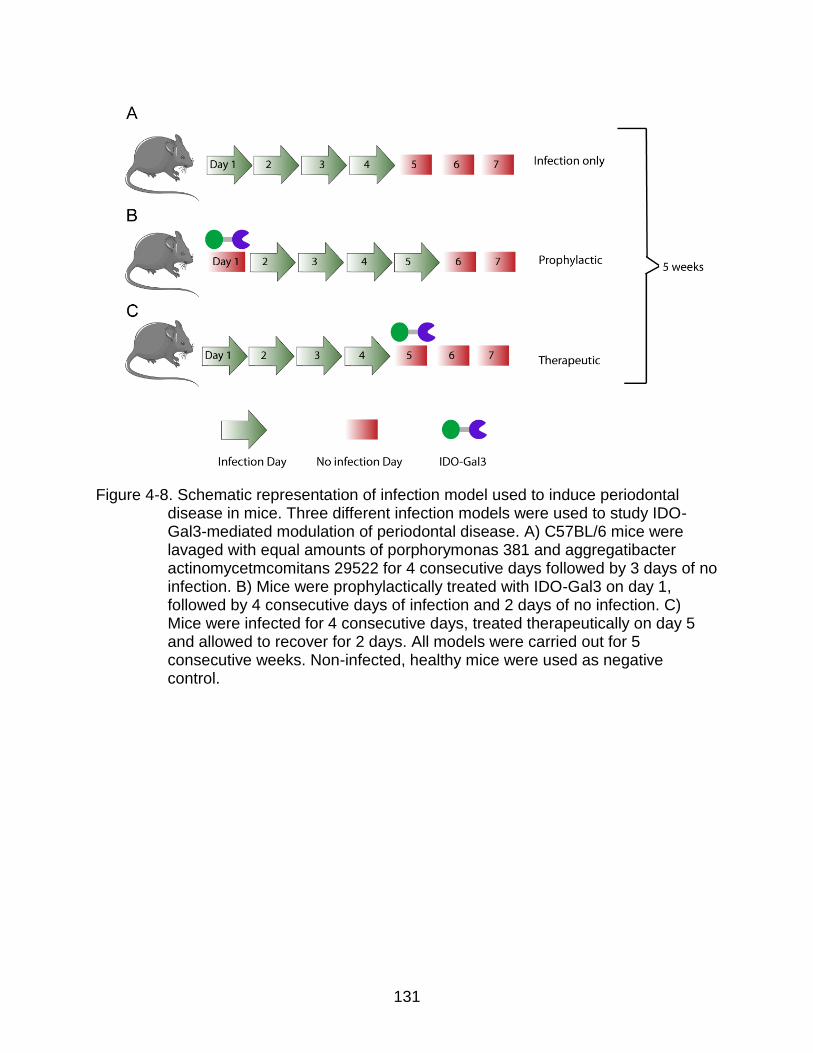
131
Figure 4-8. Schematic representation of infection model used to induce periodontal
disease in mice. Three different infection models were used to study IDO-Gal3-mediated modulation of periodontal disease. A) C57BL/6 mice were lavaged with equal amounts of porphorymonas 381 and aggregatibacter actinomycetmcomitans 29522 for 4 consecutive days followed by 3 days of no infection. B) Mice were prophylactically treated with IDO-Gal3 on day 1, followed by 4 consecutive days of infection and 2 days of no infection. C) Mice were infected for 4 consecutive days, treated therapeutically on day 5 and allowed to recover for 2 days. All models were carried out for 5 consecutive weeks. Non-infected, healthy mice were used as negative control.

132
Figure 4-9. Inflammatory cytokine and chemokine production in the subgingival space is
significantly reduced by administration of IDO-Gal3 both prophylactically and therapeutically. C57BL/6 mice were either infected with porphorymonas 381 and aggregatibacter actinomycetmcomitans 29522 (Infect) and treated with IDO-Gal3 prophylactically (IDO-Gal3-P) or therapeutically (IDO-Gal3-T); treated without presence of infection (IDO-Gal3-U) or left uninfected and untreated (unifect). After the 5 week study, animals were euthanized, mandibles and maxillae extracted and subjected to bead beating. The resulting homogenates were analyzed for A) cytokines and B) chemokines. * p value < 0.05 as indicated, ^ p value <0.05 IDO-Gal3-P vs IDO-Gal3-T, + p value < 0.05 IDO-Gal3-U vs uninfected as determined by oneway ANOVA with Bonferoni’s correction, n=7. Administration of IDO-Gal3 therapeutically significantly halts pro-inflammatory cytokine production in a polymicrobial model of periodontal disease.

133
CHAPTER 5 CONCLUSIONS AND FUTURE DIRECTIONS
Enzyme therapeutics hold great promise for the treatment of inflammatory
conditions as they offer multiple advantages over small molecules. Enzymes serve a
highly specific and complex set of functions that cannot be mimicked by single chemical
compounds. Additionally, since the action of enzymes is so specific, there is less
potential for deleterious effects or interference with biological processes if administered
locally. The focused of this thesis was the development of a chimera protein with a
tissue-binding moiety fused to the enzyme indoleamine 2,3 dioxygenase (IDO) to modify
the immune microenvironment and ameliorate local inflammation.
The first set of studies conducted here established IDO as an
immunomodulator in the extracellular space. To date, therapeutic application of IDO has
primary focused on overexpression of the gene for transplant tolerance. However,
techniques used to induce IDO gene expression, such as treatment of APCs with IFNγ
or LPS and gene transfection have demonstrated harmful effects. Additionally, the cost
associated with gene therapy or ex vivo approaches prevent their worldwide application.
Herein, we demonstrate extracellular IDO prevents DC maturation in the presence of a
potent inflammatory stimuli, and subsequently suppress antigen specific T cell
proliferation. Although the effects of IDO on T cells is well known, future studies will
determine the specific mechanism by which proliferation is halted. DCs can induce
anergy or aid in the differentiation of naïve T cells into regulatory T cells (Tregs) causing
suppression of anti-inflammatory responses. Additionally, the role of the aryl
hydrocarbon receptor (AhR) and its activation through generated kynurenines should be
further explored. Here, we hypothesize the catabolism of tryptophan in the extracellular

134
space by IDO yield kynurenines that are transported into the cell by the Large
Amino Acid Transporter 1 (LAT-1) and bind the AhR inducing translocation into the
nucleus and binding to the dioxin-responsive enhancer (DRE) for the synthesis of
intracellular IDO. This potential mechanism is shown in Figure 5-1.
IDO was determined to be most efficacious when administered in the active form
which limits its delivery through commonly used biomaterials platforms such as particles
or hydrogels due to the harsh organic solvents used during the fabrication processes
and IDO’s susceptibility to denaturation. Additionally, it is well known bolus
administration of proteins often result in clearance through the renal system before any
therapeutic effect can be obtained. Therefore, a fusion protein, with Galectin 3 (Gal3)
was developed and characterized with Gal3 motif serving as a tissue-retention strategy.
As a complimentary reporter fusion, NanoLuciferase-Galectin 3 (NanoLuc-Gal3) was
also produced. Nanoluciferase reacts with its substrate, furimazine, to produce light
easily detectable during in vitro and in vivo assays. Characterization of the fusion
construct revealed Gal3 retains binding affinity to sugar moiety when fused through the
N-terminus but its biological activity is disrupted, likely due to its inability to oligomerize
into a higher-ordered structure. Furthermore, we established NanoLuc-Gal3 can bind
immune cells without triggering a DC-mediated inflammatory response. Retention was
significantly improved at multiple injection sites with renal filtration as a potential
clearance path for excess or unbound fusion administered. Future studies will
investigate clearance mechanism for sites other than the hock and processes in vivo
leading to protein degradation and inactivation, such as proteolytic degradation and
immunogenicity.

135
Upon establishment of Galectin 3 as a tissue-retention strategy it was fused with
IDO, IDO-Gal3, and their immunomodulatory and binding properties combined to target
inflammatory conditions. Using an LPS-induced subcutaneous inflammation model,
IDO-Gal3 was shown to inhibit cellular infiltration by decreased cytokine gene
expression at a localized tissue. Anti-inflammatory effects were observed for 5 days
post treatment at a single high dose. A proposed mechanism of action is depicted in
Figures 5-1. LPS binds to the toll-like receptor 4 (TLR4), resulting in conformational
changes and recruitment of the Myeloid Differentiation Primary Response Gene 88
(MyD88) and TIR Domain-Containing Adaptor Protein (TIRAP). The MyD88-TIRAP
complex regulates early nuclear factor kappa-light-chain-enhancer of activated B cell
(NF-κβ) activation and regulation of proinflammatory cytokines such as IL-12 and IL-1β
and anti-inflammatory cytokines such as IL-10. This is through the activation of the IL-1
Receptor-Associated Kinases (IRAKs) and the adaptor molecules TNF Receptor-
Associated Factor 6 (TRAF6). TRAF6 induces the activation of Transforming growth
factor-β-Activated Kinase 1 (TAK1) that leads to the activation of p38 Mitogen-Activated
Protein Kinase (p38 MAPK) and IκB Kinase (IΚΚ). IΚΚ signaling pathway leads to the
induction of the transcription factor NF-κβ whereas p38 leads to the activation of cAMP
response element binding protein (CREB). Figure 5-2 demonstrates the cytokine
network proposed in vivo and all the different cellular interactions IDO is believed to
alter. An initial injury is produced which results in the release of MCP1 and IP10 by
epithelial cells recruiting monocytes and NK cells. Monocytes then release MIP2 which
recruits polymorphonuclear leukocytes initiating the innate response. NK cells on the
other hand, release IFN-γ recruiting DCs and macrophages which play a crucial role in

136
the initiation of the adaptive immune system. IDO likely acts on NK cells in vivo given
the inflammatory signal is only provided for 2 h. Once IFN-γ production is diminished by
IDO, DCs do not release IL-12 halting the differentiation of naïve T cells into Th1 cells.
Furthermore, without the production of IL-1β, IL-6 or TNFα, Th17 or Th22 differentiation
is also not possible limiting the inflammation observed in the tissues evaluated. Future
work should focus on elucidating the specific mechanism and cytokine networks
involved in the suppression of inflammation.
Furthermore, work will be performed to determine minimal dose required to
induce desired conditions at early (1-5 d) and late time points (10-14 d). Additionally,
immunostaining of histological samples will be performed to correlate tissue retention
time and distribution of IDO-Gal3 with that of NanoLuc-Gal3. Lastly, antigenicity toward
the fusion protein should be evaluated as it might inhibit longevity and efficacy in vivo.
In terms of application to clinically relevant models, in this thesis IDO-Gal3 was
employed to halt inflammation in a polymicrobial model of periodontal disease.
NanoLuc-Gal3 confers an oral submucosal retention time of 10 d. Therapeutic
administration of IDO-Gal3 halts inflammation by decreasing the production of
proinflammatory cytokines and chemokines. Future studies will stain histological
samples with H&E to determine degree of cellular infiltration and correlate mechanism
of action to LPS-induced inflammation model. Furthermore, bone density analysis will
demonstrate whether treatment with IDO-Gal3 can prevent alveolar bone loss, a critical
component of new PD treatments.

137
Moving forward we hope to utilize the platformed engineered here for the
treatment of additional inflammatory conditions. We hope to introduce IDO-Gal3 into a
rat model of osteoarthritis for the modulation of inflammation in the intra articular space.
Efforts have already been dedicated to this space and an active collaboration with Dr.
Kyle Allen has shown Gal3 acts as a retention motif in an arthritic joint. Additionally, we
would like to test the efficacy of IDO-Gal3 in a murine model of psoriasis. NanoLuc-Gal3
administered subcutaneously remained at the injection site far longer than any of the
other spaces investigated (IP, IM). Psoriasis is a skin related autoimmune disorder with
cytokine release profiles shown here to be suppressed.
The work presented in this thesis shows IDO maintains its immunomodulatory
properties when administered to the extracellular space and can be retained at the site
of injection through fusion with Gal3 for 10 d. Subcutaneous and oral submucosal
administration of IDO-Gal3 halted inflammation by altering release profile of pro-
inflammatory cytokines preventing cellular infiltration.

138
Figure 5-1. Proposed mechanism for IDO-related suppression of inflammation by blockage of NF-κβ pathway initiated through binding of LPS to TLR4 on surface of immune cell.

139
Figure 5-2. Proposed in vivo cytokine network affected by the introduction of IDO. IDO likely primarily affects NK cells in vivo which halts initiation of adaptive immune system by cross-talk with DCs.

140
LIST OF REFERENCES
1 Khan, T. A. & Reddy, S. T. Immunological principles regulating
immunomodulation with biomaterials. Acta Biomaterialia 10, 1720-1727, doi:http://dx.doi.org/10.1016/j.actbio.2013.12.011 (2014).
2 Hlavaty, K. A., Luo, X., Shea, L. D. & Miller, S. D. Cellular and molecular
targeting for nanotherapeutics in transplantation tolerance. Clinical Immunology 160, 14-23, doi:http://dx.doi.org/10.1016/j.clim.2015.03.013 (2015).
3 Page, E. K., Dar, W. A. & Knechtle, S. J. Tolerogenic therapies in transplantation.
Front Immunol 3, 198, doi:10.3389/fimmu.2012.00198 (2012). 4 Bracho-Sanchez, E., Lewis, J. S. & Keselowsky, B. G. in Biomaterials in
Regenerative Medicine and the Immune System (ed Laura Santambrogio) Ch. 8, 139-156 (Springer International Publishing, 2015).
5 Lewis, J. S., Roy, K. & Keselowksy, B. G. Vol. 39 25-34 (Cambridge University
Press, MRS Bulletin, 2014). 6 Hippen, K. L. et al. Massive ex vivo expansion of human natural regulatory T
cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med 3, 83ra41, doi:10.1126/scitranslmed.3001809 (2011).
7 Clemente-Casares, X. et al. Expanding antigen-specific regulatory networks to
treat autoimmunity. Nature 530, 434-440, doi:10.1038/nature16962 (2016). 8 Klatzmann, D. & Abbas, A. K. The promise of low-dose interleukin-2 therapy for
autoimmune and inflammatory diseases. Nat Rev Immunol 15, 283-294, doi:10.1038/nri3823 http://www.nature.com/nri/journal/v15/n5/abs/nri3823.html#supplementary-information (2015).
9 Steenblock, E. R., Fadel, T., Labowsky, M., Pober, J. S. & Fahmy, T. M. An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response. J Biol Chem 286, 34883-34892, doi:10.1074/jbc.M111.276329 (2011).
10 Keselowsky, B. G., Xia, C. Q. & Clare-Salzler, M. Multifunctional dendritic cell-
targeting polymeric microparticles: engineering new vaccines for type 1 diabetes. Hum Vaccin 7, 37-44 (2011).

141
11 Butler, K. S. et al. Development of antibody-tagged nanoparticles for detection of transplant rejection using biomagnetic sensors. Cell Transplant 22, 1943-1954, doi:10.3727/096368912X657963 (2013).
12 Penno, E., Johnsson, C., Johansson, L. & Ahlström, H. Macrophage uptake of ultra-small iron oxide particles for magnetic resonance imaging in experimental acute cardiac transplant rejection. Acta Radiol 47, 264-271 (2006).
13 Alcantar, N. A., Aydil, E. S. & Israelachvili, J. N. Polyethylene glycol-coated
biocompatible surfaces. J Biomed Mater Res 51, 343-351 (2000). 14 Azzi, J. et al. Polylactide-cyclosporin A nanoparticles for targeted
immunosuppression. FASEB J 24, 3927-3938, doi:10.1096/fj.10-154690 (2010). 15 Azzi, J. et al. Targeted Delivery of Immunomodulators to Lymph Nodes. Cell
Rep, doi:10.1016/j.celrep.2016.04.007 (2016). 16 Shirali, A. C. et al. Nanoparticle delivery of mycophenolic acid upregulates PD-L1
on dendritic cells to prolong murine allograft survival. Am J Transplant 11, 2582-2592, doi:10.1111/j.1600-6143.2011.03725.x (2011).
17 Hlavaty, K. A. et al. Tolerance induction using nanoparticles bearing HY peptides
in bone marrow transplantation. Biomaterials 76, 1-10, doi:http://dx.doi.org/10.1016/j.biomaterials.2015.10.041 (2016).
18 Pan, Q. et al. Corticosteroid-loaded biodegradable nanoparticles for prevention of
corneal allograft rejection in rats. J Control Release 201, 32-40, doi:10.1016/j.jconrel.2015.01.009 (2015).
19 Lewis, J. S. et al. Combinatorial delivery of immunosuppressive factors to
dendritic cells using dual-sized microspheres. J Mater Chem B Mater Biol Med 2, 2562-2574, doi:10.1039/c3tb21460e (2014).
20 Lewis, J. S. et al. A combination dual-sized microparticle system modulates
dendritic cells and prevents type 1 diabetes in prediabetic NOD mice. Clin Immunol 160, 90-102, doi:10.1016/j.clim.2015.03.023 (2015).
21 Acharya, A. P., Clare-Salzler, M. J. & Keselowsky, B. G. A high-throughput
microparticle microarray platform for dendritic cell-targeting vaccines. Biomaterials 30, 4168-4177, doi:10.1016/j.biomaterials.2009.04.032 (2009).
22 Acharya, A. P. et al. A cell-based microarray to investigate combinatorial effects
of microparticle-encapsulated adjuvants on dendritic cell activation. Journal of Materials Chemistry B, doi:10.1039/C5TB01754H (2016).

142
23 Acharya, A. P., Lewis, J. S. & Keselowsky, B. G. Combinatorial co-encapsulation of hydrophobic molecules in poly(lactide-co-glycolide) microparticles. Biomaterials 34, 3422-3430, doi:10.1016/j.biomaterials.2013.01.032 (2013).
24 Larson, N. & Ghandehari, H. Polymeric conjugates for drug delivery. Chemistry of materials : a publication of the American Chemical Society 24, 840-853, doi:10.1021/cm2031569 (2012).
25 Dane, K. Y. et al. Nano-sized drug-loaded micelles deliver payload to lymph node
immune cells and prolong allograft survival. Journal of Controlled Release 156, 154-160, doi:http://dx.doi.org/10.1016/j.jconrel.2011.08.009 (2011).
26 Miki, K. et al. Combination therapy with dendritic cell vaccine and IL-2
encapsulating polymeric micelles enhances intra-tumoral accumulation of antigen-specific CTLs. Int Immunopharmacol 23, 499-504, doi:10.1016/j.intimp.2014.09.025 (2014).
27 Svenson, S. & Tomalia, D. A. Dendrimers in biomedical applications--reflections
on the field. Adv Drug Deliv Rev 57, 2106-2129, doi:10.1016/j.addr.2005.09.018 (2005).
28 Lundquist, J. J. & Toone, E. J. The cluster glycoside effect. Chem Rev 102, 555-
578 (2002). 29 Heimburg, J. et al. Inhibition of spontaneous breast cancer metastasis by anti-
Thomsen-Friedenreich antigen monoclonal antibody JAA-F11. Neoplasia 8, 939-948, doi:10.1593/neo.06493 (2006).
30 Baek, M.-G. & Roy, R. Synthesis and protein binding properties of T-antigen
containing GlycoPAMAM dendrimers. Bioorganic & Medicinal Chemistry 10, 11-17, doi:http://dx.doi.org/10.1016/S0968-0896(01)00248-6 (2002).
31 Cheng, Y., Zhao, L., Li, Y. & Xu, T. Design of biocompatible dendrimers for
cancer diagnosis and therapy: current status and future perspectives. Chem Soc Rev 40, 2673-2703, doi:10.1039/c0cs00097c (2011).
32 Ahmed, E. M. Hydrogel: Preparation, characterization, and applications: A
review. Journal of Advanced Research 6, 105-121, doi:http://dx.doi.org/10.1016/j.jare.2013.07.006 (2015).
33 Lim, F. & Sun, A. M. Microencapsulated islets as bioartificial endocrine pancreas.
Science 210, 908-910 (1980). 34 Neufeld, T. et al. The efficacy of an immunoisolating membrane system for islet
xenotransplantation in minipigs. PLoS One 8, e70150, doi:10.1371/journal.pone.0070150 (2013).

143
35 Zakrzewski, J. L., van den Brink, M. R. & Hubbell, J. A. Overcoming immunological barriers in regenerative medicine. Nat Biotechnol 32, 786-794, doi:10.1038/nbt.2960 (2014).
36 Schwendener, R. A. Liposomes as vaccine delivery systems: a review of the recent advances. Ther Adv Vaccines 2, 159-182, doi:10.1177/2051013614541440 (2014).
37 Brown, S. & Khan, D. R. The treatment of breast cancer using liposome
technology. J Drug Deliv 2012, 212965, doi:10.1155/2012/212965 (2012). 38 Xu, H., Li, Z. & Si, J. Nanocarriers in gene therapy: a review. J Biomed
Nanotechnol 10, 3483-3507 (2014). 39 de Kleer, I. et al. Autologous stem cell transplantation for autoimmunity induces
immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4+CD25+ immune regulatory network. Blood 107, 1696-1702, doi:10.1182/blood-2005-07-2800 (2006).
40 Mischler, R. & Metcalfe, I. C. Inflexal®V a trivalent virosome subunit influenza
vaccine: production. Vaccine 20, Supplement 5, B17-B23, doi:http://dx.doi.org/10.1016/S0264-410X(02)00512-1 (2002).
41 Bovier, P. A. Epaxal: a virosomal vaccine to prevent hepatitis A infection. Expert
Rev Vaccines 7, 1141-1150, doi:10.1586/14760584.7.8.1141 (2008). 42 Hirai, T. et al. A novel approach inducing transplant tolerance by activated
invariant natural killer T cells with costimulatory blockade. Am J Transplant 14, 554-567, doi:10.1111/ajt.12606 (2014).
43 Jiao, Q., Li, L., Mu, Q. & Zhang, Q. Immunomodulation of Nanoparticles in
Nanomedicine Applications. BioMed Research International 2014, 426028, doi:10.1155/2014/426028 (2014).
44 Zhao, L. et al. Nanoparticle vaccines. Vaccine 32, 327-337,
doi:http://dx.doi.org/10.1016/j.vaccine.2013.11.069 (2014). 45 Tao, W., Ziemer, K. S. & Gill, H. S. Gold nanoparticle-M2e conjugate
coformulated with CpG induces protective immunity against influenza A virus. Nanomedicine (Lond) 9, 237-251, doi:10.2217/nnm.13.58 (2014).
46 Xu, L. et al. Surface-engineered gold nanorods: promising DNA vaccine adjuvant
for HIV-1 treatment. Nano Lett 12, 2003-2012, doi:10.1021/nl300027p (2012). 47 Vega, R. A. et al. Modified gold nanoparticle vectors: a biocompatible intracellular
delivery system for pancreatic islet cell transplantation. Surgery 148, 858-865; discussion 865-856, doi:10.1016/j.surg.2010.07.036 (2010).

144
48 Villa, C. H. et al. Single-walled carbon nanotubes deliver peptide antigen into
dendritic cells and enhance IgG responses to tumor-associated antigens. ACS Nano 5, 5300-5311, doi:10.1021/nn200182x (2011).
49 Liu, Y., Zhao, Y., Sun, B. & Chen, C. Understanding the Toxicity of Carbon
Nanotubes. Accounts of Chemical Research 46, 702-713, doi:10.1021/ar300028m (2013).
50 Niut, Y. et al. Recent advances in the rational design of silica-based
nanoparticles for gene therapy. Ther Deliv 3, 1217-1237 (2012). 51 Xia, T. et al. Polyethyleneimine coating enhances the cellular uptake of
mesoporous silica nanoparticles and allows safe delivery of siRNA and DNA constructs. ACS Nano 3, 3273-3286, doi:10.1021/nn900918w (2009).
52 Carter, P. J. Introduction to current and future protein therapeutics: a protein
engineering perspective. Exp Cell Res 317, 1261-1269, doi:10.1016/j.yexcr.2011.02.013 (2011).
53 Huang, C. Receptor-Fc fusion therapeutics, traps, and MIMETIBODY technology.
Curr Opin Biotechnol 20, 692-699, doi:10.1016/j.copbio.2009.10.010 (2009). 54 Jazayeri, J. A. & Carroll, G. J. Fc-based cytokines : prospects for engineering
superior therapeutics. BioDrugs 22, 11-26 (2008). 55 Liu, F. et al. Fusion with extracellular domain of cytotoxic T-lymphocyte-
associated-antigen 4 leads to enhancement of immunogenicity of Hantaan virus DNA vaccines in C57BL/6 mice. Virology Journal 8, 448-448, doi:10.1186/1743-422X-8-448 (2011).
56 Santagostino, E. et al. Long-acting recombinant coagulation factor IX albumin
fusion protein (rIX-FP) in hemophilia B: results of a phase 3 trial. Blood 127, 1761 (2016).
57 Fahrer, J., Rausch, J. & Barth, H. A cell-permeable fusion protein based on
Clostridium botulinum C2 toxin for delivery of p53 tumorsuppressor into cancer cells. PLoS One 8, e72455, doi:10.1371/journal.pone.0072455 (2013).
58 Tan, Q. et al. Characterization and comparison of commercially available TNF
receptor 2-Fc fusion protein products. mAbs 4, 761-774, doi:10.4161/mabs.22276 (2012).
59 Mahler, S. Safety of biologics therapy: Monoclonal antibodies, cytokines, fusion
proteins, hormones, enzymes, coagulation proteins, vaccines, botulinum toxins. mAbs 9, 885-888, doi:10.1080/19420862.2017.1343709 (2017).

145
60 Rogers, B., Dong, D. & Li, Z. Recombinant human serum albumin fusion proteins and novel applications in drug delivery and therapy. Curr Pharm Des 21, 1899-1907 (2015).
61 Blair, H. A. & Keating, G. M. Albiglutide: a review of its use in patients with type 2
diabetes mellitus. Drugs 75, 651-663, doi:10.1007/s40265-015-0370-5 (2015). 62 Lyseng-Williamson, K. A. Coagulation Factor IX (Recombinant), Albumin Fusion
Protein (Albutrepenonacog Alfa; Idelvion(R)): A Review of Its Use in Haemophilia B. Drugs 77, 97-106, doi:10.1007/s40265-016-0679-8 (2017).
63 Foged, C., Brodin, B., Frokjaer, S. & Sundblad, A. Particle size and surface
charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm 298, 315-322, doi:10.1016/j.ijpharm.2005.03.035 (2005).
64 Sahay, G., Alakhova, D. Y. & Kabanov, A. V. Endocytosis of Nanomedicines.
Journal of controlled release : official journal of the Controlled Release Society 145, 182-195, doi:10.1016/j.jconrel.2010.01.036 (2010).
65 Champion, J. A., Katare, Y. K. & Mitragotri, S. Particle shape: a new design
parameter for micro- and nanoscale drug delivery carriers. J Control Release 121, 3-9, doi:10.1016/j.jconrel.2007.03.022 (2007).
66 Champion, J. A. & Mitragotri, S. Role of target geometry in phagocytosis. Proc
Natl Acad Sci U S A 103, 4930-4934, doi:10.1073/pnas.0600997103 (2006). 67 Schaeublin, N. M. et al. Surface charge of gold nanoparticles mediates
mechanism of toxicity. Nanoscale 3, 410-420, doi:10.1039/c0nr00478b (2011). 68 Naha, P. C., Davoren, M., Lyng, F. M. & Byrne, H. J. Reactive oxygen species
(ROS) induced cytokine production and cytotoxicity of PAMAM dendrimers in J774A.1 cells. Toxicol Appl Pharmacol 246, 91-99, doi:10.1016/j.taap.2010.04.014 (2010).
69 Kwon, Y. J., Standley, S. M., Goh, S. L. & Fréchet, J. M. J. Enhanced antigen
presentation and immunostimulation of dendritic cells using acid-degradable cationic nanoparticles. Journal of Controlled Release 105, 199-212, doi:http://dx.doi.org/10.1016/j.jconrel.2005.02.027 (2005).
70 Acharya, A. P., Dolgova, N. V., Clare-Salzler, M. J. & Keselowsky, B. G.
Adhesive substrate-modulation of adaptive immune responses. Biomaterials 29, 4736-4750, doi:10.1016/j.biomaterials.2008.08.040 (2008).

146
71 Acharya, A. P., Dolgova, N. V., Xia, C. Q., Clare-Salzler, M. J. & Keselowsky, B. G. Adhesive substrates modulate the activation and stimulatory capacity of non-obese diabetic mouse-derived dendritic cells. Acta Biomater 7, 180-192, doi:10.1016/j.actbio.2010.08.026 (2011).
72 Lewis, J. S., Zaveri, T. D., Crooks, C. P., 2nd & Keselowsky, B. G. Microparticle
surface modifications targeting dendritic cells for non-activating applications. Biomaterials 33, 7221-7232, doi:10.1016/j.biomaterials.2012.06.049 (2012).
73 Acharya, A. P. et al. The modulation of dendritic cell integrin binding and
activation by RGD-peptide density gradient substrates. Biomaterials 31, 7444-7454, doi:10.1016/j.biomaterials.2010.06.025 (2010).
74 Zaveri, T. D., Lewis, J. S., Dolgova, N. V., Clare-Salzler, M. J. & Keselowsky, B.
G. Integrin-directed modulation of macrophage responses to biomaterials. Biomaterials 35, 3504-3515, doi:10.1016/j.biomaterials.2014.01.007 (2014).
75 Sionkowska, A. Current research on the blends of natural and synthetic polymers
as new biomaterials: Review. Progress in Polymer Science 36, 1254-1276, doi:http://dx.doi.org/10.1016/j.progpolymsci.2011.05.003 (2011).
76 Winter, M. et al. Activation of the inflammasome by amorphous silica and TiO2
nanoparticles in murine dendritic cells. Nanotoxicology 5, 326-340, doi:10.3109/17435390.2010.506957 (2011).
77 Lutz, M. B. & Schuler, G. Immature, semi-mature and fully mature dendritic cells:
which signals induce tolerance or immunity? Trends Immunol 23, 445-449 (2002).
78 Hwu, P. et al. Indoleamine 2,3-dioxygenase production by human dendritic cells
results in the inhibition of T cell proliferation. J Immunol 164, 3596-3599 (2000). 79 Banchereau, J. et al. Immunobiology of dendritic cells. Annu Rev Immunol 18,
767-811, doi:10.1146/annurev.immunol.18.1.767 (2000). 80 Akira, S., Uematsu, S. & Takeuchi, O. Pathogen Recognition and Innate
Immunity. Cell 124, 783-801, doi:http://dx.doi.org/10.1016/j.cell.2006.02.015 (2006).
81 Iwasaki, A. & Medzhitov, R. Toll-like receptor control of the adaptive immune
responses. Nat Immunol 5, 987-995, doi:10.1038/ni1112 (2004). 82 Broere, F., Apasov, S. G., Sitkovsky, M. V. & Eden, W. v. Principles of
Immunopharmacology: 3rd revised and extended edition. (Springer, 2011).

147
83 Korn, T., Bettelli, E., Oukka, M. & Kuchroo, V. K. IL-17 and Th17 Cells. Annu Rev Immunol 27, 485-517, doi:10.1146/annurev.immunol.021908.132710 (2009).
84 Steinman, R. M., Hawiger, D. & Nussenzweig, M. C. Tolerogenic dendritic cells.
Annu Rev Immunol 21, 685-711, doi:10.1146/annurev.immunol.21.120601.141040 (2003).
85 Hoeffel, G. et al. Adult Langerhans cells derive predominantly from embryonic
fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 209, 1167-1181, doi:10.1084/jem.20120340 (2012).
86 Huang, L., Baban, B., Johnson, B. A., 3rd & Mellor, A. L. Dendritic cells,
indoleamine 2,3 dioxygenase and acquired immune privilege. Int Rev Immunol 29, 133-155, doi:10.3109/08830180903349669 (2010).
87 Jones, K. S. Biomaterials as vaccine adjuvants. Biotechnol Prog 24, 807-814,
doi:10.1002/btpr.10 (2008). 88 Sharp, F. A. et al. Uptake of particulate vaccine adjuvants by dendritic cells
activates the NALP3 inflammasome. Proc Natl Acad Sci U S A 106, 870-875, doi:10.1073/pnas.0804897106 (2009).
89 Ali, O. A., Huebsch, N., Cao, L., Dranoff, G. & Mooney, D. J. Infection-mimicking
materials to program dendritic cells in situ. Nat Mater 8, 151-158, doi:10.1038/nmat2357 (2009).
90 Sneh-Edri, H., Likhtenshtein, D. & Stepensky, D. Intracellular targeting of PLGA
nanoparticles encapsulating antigenic peptide to the endoplasmic reticulum of dendritic cells and its effect on antigen cross-presentation in vitro. Mol Pharm 8, 1266-1275, doi:10.1021/mp200198c (2011).
91 El-Sayed, M. E., Hoffman, A. S. & Stayton, P. S. Smart polymeric carriers for
enhanced intracellular delivery of therapeutic macromolecules. Expert Opin Biol Ther 5, 23-32, doi:10.1517/14712598.5.1.23 (2005).
92 Flanary, S., Hoffman, A. S. & Stayton, P. S. Antigen Delivery with
Poly(Propylacrylic Acid) Conjugation Enhances MHC-1 Presentation and T-Cell Activation. Bioconjug Chem 20, 241-248, doi:10.1021/bc800317a (2009).
93 Schlosser, E. et al. TLR ligands and antigen need to be coencapsulated into the
same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses. Vaccine 26, 1626-1637, doi:10.1016/j.vaccine.2008.01.030 (2008).

148
94 Yoshida, M., Mata, J. & Babensee, J. E. Effect of poly(lactic-co-glycolic acid) contact on maturation of murine bone marrow-derived dendritic cells. J Biomed Mater Res A 80, 7-12, doi:10.1002/jbm.a.30832 (2007).
95 Beaudette, T. T. et al. In vivo studies on the effect of co-encapsulation of CpG
DNA and antigen in acid-degradable microparticle vaccines. Mol Pharm 6, 1160-1169, doi:10.1021/mp900038e (2009).
96 Elamanchili, P., Diwan, M., Cao, M. & Samuel, J. Characterization of poly(D,L-
lactic-co-glycolic acid) based nanoparticulate system for enhanced delivery of antigens to dendritic cells. Vaccine 22, 2406-2412, doi:10.1016/j.vaccine.2003.12.032 (2004).
97 Ali, O. A., Tayalia, P., Shvartsman, D., Lewin, S. & Mooney, D. J. Inflammatory
cytokines presented from polymer matrices differentially generate and activate DCs. Adv Funct Mater 23, 4621-4628, doi:10.1002/adfm.201203859 (2013).
98 Singh, A., Suri, S. & Roy, K. In-situ crosslinking hydrogels for combinatorial
delivery of chemokines and siRNA-DNA carrying microparticles to dendritic cells. Biomaterials 30, 5187-5200, doi:10.1016/j.biomaterials.2009.06.001 (2009).
99 Pradhan, P. et al. The effect of combined IL10 siRNA and CpG ODN as
pathogen-mimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials 35, 5491-5504, doi:10.1016/j.biomaterials.2014.03.039 (2014).
100 Hudalla, G. A. et al. A self-adjuvanting supramolecular vaccine carrying a folded
protein antigen. Adv Healthc Mater 2, 1114-1119, doi:10.1002/adhm.201200435 (2013).
101 Goodman, J. et al. Vol. Volume 31, issue 12 1269-1280 (Particle & Particle
Systems Characterization, 2014). 102 Haughney, S. L., Ross, K. A., Boggiatto, P. M., Wannemuehler, M. J. &
Narasimhan, B. Effect of nanovaccine chemistry on humoral immune response kinetics and maturation. Nanoscale 6, 13770-13778, doi:10.1039/c4nr03724c (2014).
103 Vela-Ramirez, J. E. et al. Safety and biocompatibility of carbohydrate-
functionalized polyanhydride nanoparticles. Aaps j 17, 256-267, doi:10.1208/s12248-014-9699-z (2015).
104 Fischer, S. et al. The preservation of phenotype and functionality of dendritic
cells upon phagocytosis of polyelectrolyte-coated PLGA microparticles. Biomaterials 28, 994-1004, doi:10.1016/j.biomaterials.2006.10.034 (2007).

149
105 Rogers, T. H. & Babensee, J. E. The role of integrins in the recognition and response of dendritic cells to biomaterials. Biomaterials 32, 1270-1279, doi:10.1016/j.biomaterials.2010.10.014 (2011).
106 Volchenkov, R., Karlsen, M., Jonsson, R. & Appel, S. Type 1 regulatory T cells
and regulatory B cells induced by tolerogenic dendritic cells. Scand J Immunol 77, 246-254, doi:10.1111/sji.12039 (2013).
107 Schwartz, R. H. T cell anergy. Annu Rev Immunol 21, 305-334,
doi:10.1146/annurev.immunol.21.120601.141110 (2003). 108 Sakaguchi, S. et al. Immunologic tolerance maintained by CD25+ CD4+
regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev 182, 18-32 (2001).
109 Woltman, A. M. et al. The effect of calcineurin inhibitors and corticosteroids on
the differentiation of human dendritic cells. Eur J Immunol 30, 1807-1812, doi:10.1002/1521-4141(200007)30:7<1807::aid-immu1807>3.0.co;2-n (2000).
110 Hackstein, H. et al. Aspirin inhibits in vitro maturation and in vivo
immunostimulatory function of murine myeloid dendritic cells. J Immunol 166, 7053-7062 (2001).
111 Mehling, A. et al. Mycophenolate mofetil impairs the maturation and function of
murine dendritic cells. J Immunol 165, 2374-2381 (2000). 112 Penna, G. & Adorini, L. 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation,
maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol 164, 2405-2411 (2000).
113 Jhunjhunwala, S., Raimondi, G., Thomson, A. W. & Little, S. R. Delivery of
rapamycin to dendritic cells using degradable microparticles. J Control Release 133, 191-197, doi:10.1016/j.jconrel.2008.10.011 (2009).
114 Yamaguchi, Y., Tsumura, H., Miwa, M. & Inaba, K. Contrasting effects of TGF-
beta 1 and TNF-alpha on the development of dendritic cells from progenitors in mouse bone marrow. Stem Cells 15, 144-153, doi:10.1002/stem.150144 (1997).
115 Steinbrink, K., Wolfl, M., Jonuleit, H., Knop, J. & Enk, A. H. Induction of tolerance
by IL-10-treated dendritic cells. J Immunol 159, 4772-4780 (1997). 116 Hackstein, H. et al. Rapamycin inhibits IL-4--induced dendritic cell maturation in
vitro and dendritic cell mobilization and function in vivo. Blood 101, 4457-4463, doi:10.1182/blood-2002-11-3370 (2003).

150
117 Bryant, J. et al. Nanoparticle delivery of donor antigens for transplant tolerance in allogeneic islet transplantation. Biomaterials 35, 8887-8894, doi:10.1016/j.biomaterials.2014.06.044 (2014).
118 Powell, J. D., Pollizzi, K. N., Heikamp, E. B. & Horton, M. R. Regulation of
immune responses by mTOR. Annu Rev Immunol 30, 39-68, doi:10.1146/annurev-immunol-020711-075024 (2012).
119 Maldonado, R. A. et al. Polymeric synthetic nanoparticles for the induction of
antigen-specific immunological tolerance. Proc Natl Acad Sci U S A, doi:10.1073/pnas.1408686111 (2014).
120 Babensee, J. E. & Paranjpe, A. Differential levels of dendritic cell maturation on
different biomaterials used in combination products. J Biomed Mater Res A 74, 503-510 (2005).
121 Leclerc, C. et al. Immobilized cytokines as biomaterials for manufacturing
immune cell based vaccines. J Biomed Mater Res A 86, 1033-1040, doi:10.1002/jbm.a.31751 (2008).
122 Yuba, E. et al. Gene delivery to dendritic cells mediated by complexes of
lipoplexes and pH-sensitive fusogenic polymer-modified liposomes. J Control Release 130, 77-83, doi:10.1016/j.jconrel.2008.05.007 (2008).
123 Palama, I. E., Cortese, B., D'Amone, S. & Gigli, G. Vol. 3 144 (Biomaterials
Science, 2015). 124 Machen, J. et al. Antisense oligonucleotides down-regulating costimulation
confer diabetes-preventive properties to nonobese diabetic mouse dendritic cells. J Immunol 173, 4331-4341 (2004).
125 Wang, J., Lu, Z., Wientjes, M. G. & Au, J. L. S. in Aaps j Vol. 12 492-503
(2010). 126 Giannoukakis, N., Phillips, B., Finegold, D., Harnaha, J. & Trucco, M. Phase I
(safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 34, 2026-2032, doi:10.2337/dc11-0472 (2011).
127 Phillips, B. et al. A microsphere-based vaccine prevents and reverses new-onset
autoimmune diabetes. Diabetes 57, 1544-1555, doi:10.2337/db07-0507 (2008). 128 Yeung, A. W., Terentis, A. C., King, N. J. & Thomas, S. R. Role of indoleamine
2,3-dioxygenase in health and disease. Clin Sci (Lond) 129, 601-672, doi:10.1042/cs20140392 (2015).

151
129 Takikawa, O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated l-tryptophan metabolism. Biochemical and Biophysical Research Communications 338, 12-19, doi:https://doi.org/10.1016/j.bbrc.2005.09.032 (2005).
130 Fallarino, F., Grohmann, U. & Puccetti, P. Indoleamine 2,3-dioxygenase: from
catalyst to signaling function. Eur J Immunol 42, 1932-1937, doi:10.1002/eji.201242572 (2012).
131 Yanagida, O. et al. Human L-type amino acid transporter 1 (LAT1):
characterization of function and expression in tumor cell lines. Biochim Biophys Acta 1514, 291-302 (2001).
132 Babcock, T. A. & Carlin, J. M. Transcriptional activation of indoleamine
dioxygenase by interleukin 1 and tumor necrosis factor alpha in interferon-treated epithelial cells. Cytokine 12, 588-594, doi:10.1006/cyto.1999.0661 (2000).
133 Carlin, J. M., Borden, E. C., Sondel, P. M. & Byrne, G. I. Interferon-induced
indoleamine 2,3-dioxygenase activity in human mononuclear phagocytes. J Leukoc Biol 45, 29-34 (1989).
134 Guillemin, G. J., Smythe, G., Takikawa, O. & Brew, B. J. Expression of
indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 49, 15-23, doi:10.1002/glia.20090 (2005).
135 Pallotta, M. T. et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-
term tolerance by dendritic cells. Nat Immunol 12, 870-878, doi:10.1038/ni.2077 (2011).
136 Efron, P. A. et al. Differential maturation of murine bone-marrow derived dendritic
cells with lipopolysaccharide and tumor necrosis factor-alpha. J Endotoxin Res 11, 145-160, doi:10.1179/096805105x46583 (2005).
137 Mezrich, J. D. et al. An interaction between kynurenine and the aryl hydrocarbon
receptor can generate regulatory T cells. J Immunol 185, 3190-3198, doi:10.4049/jimmunol.0903670 (2010).
138 Nguyen, N. T. et al. Aryl hydrocarbon receptor negatively regulates dendritic cell
immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A 107, 19961-19966, doi:10.1073/pnas.1014465107 (2010).
139 Grohmann, U. et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat
Immunol 3, 1097-1101, doi:10.1038/ni846 (2002).

152
140 Braun, D., Longman, R. S. & Albert, M. L. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood 106, 2375-2381, doi:10.1182/blood-2005-03-0979 (2005).
141 Lim, J. Y., Lee, S. E., Park, G., Choi, E. Y. & Min, C. K. Inhibition of indoleamine
2,3-dioxygenase (IDO) by stereoisomers of 1-methyl tryptophan in an experimental graft-versus-tumor model. Exp Hematol, doi:10.1016/j.exphem.2014.06.006 (2014).
142 Munn, D. H. et al. Inhibition of T cell proliferation by macrophage tryptophan
catabolism. J Exp Med 189, 1363-1372 (1999). 143 Sun, J. et al. Upregulated expression of indoleamine 2, 3-dioxygenase in CHO
cells induces apoptosis of competent T cells and increases proportion of Treg cells. J Exp Clin Cancer Res 30, 82, doi:10.1186/1756-9966-30-82 (2011).
144 Terness, P. et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-
dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med 196, 447-457 (2002).
145 do Prado, K. M. et al. Indoleamine 2,3-dioxygenase (IDO) activity in placental
compartments of renal-transplanted pregnant women. Am J Reprod Immunol 72, 45-56, doi:10.1111/aji.12233 (2014).
146 Johnson, T. S. & Munn, D. H. Host indoleamine 2,3-dioxygenase: contribution to
systemic acquired tumor tolerance. Immunol Invest 41, 765-797, doi:10.3109/08820139.2012.689405 (2012).
147 Stubbendorff, M. et al. Immunological properties of extraembryonic human
mesenchymal stromal cells derived from gestational tissue. Stem Cells Dev 22, 2619-2629, doi:10.1089/scd.2013.0043 (2013).
148 Munn, D. H. et al. Prevention of allogeneic fetal rejection by tryptophan
catabolism. Science 281, 1191-1193 (1998). 149 Zhang, P. et al. The GCN2 eIF2α Kinase Is Required for Adaptation to Amino
Acid Deprivation in Mice. Molecular and Cellular Biology 22, 6681-6688, doi:10.1128/MCB.22.19.6681-6688.2002 (2002).
150 Fallarino, F. et al. The combined effects of tryptophan starvation and tryptophan
catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol 176, 6752-6761 (2006).
151 Mellor, A. L. & Munn, D. H. Tryptophan catabolism and T-cell tolerance:
immunosuppression by starvation? Immunol Today 20, 469-473 (1999).

153
152 Dagenais-Lussier, X. et al. Kynurenine Reduces Memory CD4 T-Cell Survival by Interfering with Interleukin-2 Signaling Early during HIV-1 Infection. J Virol 90, 7967-7979, doi:10.1128/jvi.00994-16 (2016).
153 Brandacher, G. et al. Non-invasive monitoring of kidney allograft rejection
through IDO metabolism evaluation. Kidney Int 71, 60-67, doi:10.1038/sj.ki.5002023 (2007).
154 Cook, C. H. et al. Spontaneous renal allograft acceptance associated with
"regulatory" dendritic cells and IDO. J Immunol 180, 3103-3112 (2008). 155 Miki, T. et al. Blockade of tryptophan catabolism prevents spontaneous
tolerogenicity of liver allografts. Transplant Proc 33, 129-130 (2001). 156 Guillonneau, C. et al. CD40Ig treatment results in allograft acceptance mediated
by CD8CD45RC T cells, IFN-gamma, and indoleamine 2,3-dioxygenase. J Clin Invest 117, 1096-1106, doi:10.1172/jci28801 (2007).
157 Friberg, M. et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion
of T cell-mediated rejection. Int J Cancer 101, 151-155, doi:10.1002/ijc.10645 (2002).
158 Johnson, B. A., 3rd, Baban, B. & Mellor, A. L. Targeting the immunoregulatory
indoleamine 2,3 dioxygenase pathway in immunotherapy. Immunotherapy 1, 645-661, doi:10.2217/imt.09.21 (2009).
159 Muller, A. J. & Prendergast, G. C. Marrying immunotherapy with chemotherapy:
why say IDO? Cancer Res 65, 8065-8068, doi:10.1158/0008-5472.can-05-2213 (2005).
160 Munn, D. H. & Mellor, A. L. Indoleamine 2,3-dioxygenase and tumor-induced
tolerance. Journal of Clinical Investigation 117, 1147-1154, doi:10.1172/JCI31178 (2007).
161 Funeshima, N. et al. Inhibition of allogeneic T-cell responses by dendritic cells
expressing transduced indoleamine 2,3-dioxygenase. J Gene Med 7, 565-575, doi:10.1002/jgm.698 (2005).
162 Valladares, R. et al. Lactobacillus johnsonii inhibits indoleamine 2,3-dioxygenase
and alters tryptophan metabolite levels in BioBreeding rats. Faseb j 27, 1711-1720, doi:10.1096/fj.12-223339 (2013).
163 Cady, S. G. & Sono, M. 1-Methyl-DL-tryptophan, beta-(3-benzofuranyl)-DL-
alanine (the oxygen analog of tryptophan), and beta-[3-benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch Biochem Biophys 291, 326-333 (1991).

154
164 Wiseman, A. C. Immunosuppressive Medications. Clinical Journal of the American Society of Nephrology : CJASN 11, 332-343, doi:10.2215/CJN.08570814 (2016).
165 Barratt-Boyes, S. M. & Thomson, A. W. Dendritic cells: tools and targets for
transplant tolerance. Am J Transplant 5, 2807-2813, doi:10.1111/j.1600-6143.2005.01116.x (2005).
166 Li, H. & Shi, B. Tolerogenic dendritic cells and their applications in
transplantation. Cell Mol Immunol 12, 24-30, doi:10.1038/cmi.2014.52 (2015). 167 Sauma, D. et al. Cyclosporine preconditions dendritic cells during differentiation
and reduces IL-2 and IL-12 production following activation: a potential tolerogenic effect. Transplant Proc 35, 2515-2517 (2003).
168 Fischer, R., Turnquist, H. R., Taner, T. & Thomson, A. W. Use of rapamycin in
the induction of tolerogenic dendritic cells. Handb Exp Pharmacol, 215-232, doi:10.1007/978-3-540-71029-5_10 (2009).
169 Barragan, M., Good, M. & Kolls, J. K. Regulation of Dendritic Cell Function by
Vitamin D. Nutrients 7, 8127-8151, doi:10.3390/nu7095383 (2015). 170 Li, M. et al. Immune modulation and tolerance induction by RelB-silenced
dendritic cells through RNA interference. J Immunol 178, 5480-5487 (2007). 171 Li, Q., Harden, J. L., Anderson, C. D. & Egilmez, N. K. Tolerogenic Phenotype of
IFN-gamma-Induced IDO+ Dendritic Cells Is Maintained via an Autocrine IDO-Kynurenine/AhR-IDO Loop. J Immunol 197, 962-970, doi:10.4049/jimmunol.1502615 (2016).
172 Mbongue, J. C. et al. Induction of Indoleamine 2, 3-Dioxygenase in Human
Dendritic Cells by a Cholera Toxin B Subunit—Proinsulin Vaccine. PLoS ONE 10, e0118562, doi:10.1371/journal.pone.0118562 (2015).
173 Fallarino, F. et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ 9,
1069-1077, doi:10.1038/sj.cdd.4401073 (2002). 174 Hayashi, T. et al. Inhibition of experimental asthma by indoleamine 2,3-
dioxygenase. J Clin Invest 114, 270-279, doi:10.1172/jci21275 (2004). 175 Hacein-Bey-Abina, S. et al. in N Engl J Med Vol. 348 255-256 (2003). 176 Couzin, J. & Kaiser, J. in Science Vol. 307 1028 (2005).

155
177 Rezakhanlou, A. M. et al. Highly Efficient Stable Expression of Indoleamine 2,3 Dioxygenase Gene in Primary Fibroblasts. Biological Procedures Online 12, 107-112, doi:10.1007/s12575-010-9028-6 (2010).
178 Tan, P. H. et al. Modulation of human dendritic-cell function following
transduction with viral vectors: implications for gene therapy. Blood 105, 3824-3832, doi:10.1182/blood-2004-10-3880 (2005).
179 Rabinovich, G. A. & Toscano, M. A. Turning 'sweet' on immunity: galectin-glycan
interactions in immune tolerance and inflammation. Nat Rev Immunol 9, 338-352, doi:10.1038/nri2536 (2009).
180 van Kooyk, Y. & Rabinovich, G. A. Protein-glycan interactions in the control of
innate and adaptive immune responses. Nat Immunol 9, 593-601, doi:10.1038/ni.f.203 (2008).
181 Dumic, J., Dabelic, S. & Flogel, M. Galectin-3: an open-ended story. Biochim
Biophys Acta 1760, 616-635, doi:10.1016/j.bbagen.2005.12.020 (2006). 182 Dagher, S. F., Wang, J. L. & Patterson, R. J. Identification of galectin-3 as a
factor in pre-mRNA splicing. Proc Natl Acad Sci U S A 92, 1213-1217 (1995). 183 Wang, J. L., Gray, R. M., Haudek, K. C. & Patterson, R. J. Nucleocytoplasmic
lectins. Biochim Biophys Acta 1673, 75-93, doi:10.1016/j.bbagen.2004.03.013 (2004).
184 Demetriou, M., Granovsky, M., Quaggin, S. & Dennis, J. W. Negative regulation
of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 409, 733-739, doi:10.1038/35055582 (2001).
185 Rapoport, E. M. & Bovin, N. V. Specificity of Human Galectins on Cell Surfaces.
Biochemistry (Mosc) 80, 846-856, doi:10.1134/s0006297915070056 (2015). 186 Fukumori, T. et al. CD29 and CD7 mediate galectin-3-induced type II T-cell
apoptosis. Cancer Res 63, 8302-8311 (2003). 187 MacKinnon, A. C. et al. Regulation of alternative macrophage activation by
galectin-3. J Immunol 180, 2650-2658 (2008). 188 Hernandez, J. D. & Baum, L. G. Ah, sweet mystery of death! Galectins and
control of cell fate. Glycobiology 12, 127r-136r (2002). 189 Swarte, V. V., Mebius, R. E., Joziasse, D. H., Van den Eijnden, D. H. & Kraal, G.
Lymphocyte triggering via L-selectin leads to enhanced galectin-3-mediated binding to dendritic cells. Eur J Immunol 28, 2864-2871, doi:10.1002/(sici)1521-4141(199809)28:09<2864::aid-immu2864>3.0.co;2-u (1998).

156
190 Markowska, A. I., Liu, F. T. & Panjwani, N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J Exp Med 207, 1981-1993, doi:10.1084/jem.20090121 (2010).
191 Croci, D. O. et al. Glycosylation-dependent lectin-receptor interactions preserve
angiogenesis in anti-VEGF refractory tumors. Cell 156, 744-758, doi:10.1016/j.cell.2014.01.043 (2014).
192 Funasaka, T., Raz, A. & Nangia-Makker, P. Galectin-3 in angiogenesis and
metastasis. Glycobiology 24, 886-891, doi:10.1093/glycob/cwu086 (2014). 193 Kupper, C. E. et al. Fluorescent SNAP-tag galectin fusion proteins as novel tools
in glycobiology. Curr Pharm Des 19, 5457-5467 (2013). 194 Bocker, S. & Elling, L. Binding characteristics of galectin-3 fusion proteins.
Glycobiology 27, 457-468, doi:10.1093/glycob/cwx007 (2017). 195 John, C. M., Leffler, H., Kahl-Knutsson, B., Svensson, I. & Jarvis, G. A.
Truncated galectin-3 inhibits tumor growth and metastasis in orthotopic nude mouse model of human breast cancer. Clin Cancer Res 9, 2374-2383 (2003).
196 Johnson, K. D. et al. Galectin-3 as a Potential Therapeutic Target in Tumors
Arising from Malignant Endothelia. Neoplasia (New York, N.Y.) 9, 662-670 (2007).
197 Pieters, R. J. Inhibition and detection of galectins. Chembiochem 7, 721-728,
doi:10.1002/cbic.200600011 (2006). 198 He, S.-X., Song, G., Shi, J.-P., Guo, Y.-Q. & Guo, Z.-Y. Nanoluciferase as a
novel quantitative protein fusion tag: Application for overexpression and bioluminescent receptor-binding assays of human leukemia inhibitory factor. Biochimie 106, 140-148, doi:https://doi.org/10.1016/j.biochi.2014.08.012 (2014).
199 Thorne, N., Inglese, J. & Auld, D. S. Illuminating insights into firefly luciferase and
other bioluminescent reporters used in chemical biology. Chemistry & biology 17, 646-657, doi:10.1016/j.chembiol.2010.05.012 (2010).
200 Hall, M. P. et al. Engineered Luciferase Reporter from a Deep Sea Shrimp
Utilizing a Novel Imidazopyrazinone Substrate. ACS Chemical Biology 7, 1848-1857, doi:10.1021/cb3002478 (2012).
201 Ahmad, N. et al. Galectin-3 precipitates as a pentamer with synthetic multivalent
carbohydrates and forms heterogeneous cross-linked complexes. J Biol Chem 279, 10841-10847, doi:10.1074/jbc.M312834200 (2004).

157
202 Barboni, E. A., Bawumia, S. & Hughes, R. C. Kinetic measurements of binding of galectin 3 to a laminin substratum. Glycoconj J 16, 365-373 (1999).
203 Friedrichs, J., Manninen, A., Muller, D. J. & Helenius, J. Galectin-3 Regulates
Integrin α2β1-mediated Adhesion to Collagen-I and -IV. Journal of Biological Chemistry 283, 32264-32272, doi:10.1074/jbc.M803634200 (2008).
204 Cederfur, C. et al. Different affinity of galectins for human serum glycoproteins:
galectin-3 binds many protease inhibitors and acute phase proteins. Glycobiology 18, 384-394, doi:10.1093/glycob/cwn015 (2008).
205 Stillman, B. N. et al. Galectin-3 and galectin-1 bind distinct cell surface
glycoprotein receptors to induce T cell death. J Immunol 176, 778-789 (2006). 206 Leader, B., Baca, Q. J. & Golan, D. E. Protein therapeutics: a summary and
pharmacological classification. Nat Rev Drug Discov 7, 21-39 (2008). 207 Lachmann, R. H. Enzyme replacement therapy for lysosomal storage diseases.
Curr Opin Pediatr 23, 588-593, doi:10.1097/MOP.0b013e32834c20d9 (2011). 208 Barton, N. W. et al. Replacement Therapy for Inherited Enzyme Deficiency —
Macrophage-Targeted Glucocerebrosidase for Gaucher's Disease. New England Journal of Medicine 324, 1464-1470, doi:10.1056/NEJM199105233242104 (1991).
209 Rosenbloom, B. E. & Weinreb, N. J. Gaucher disease: a comprehensive review.
Crit Rev Oncog 18, 163-175 (2013). 210 Kontermann, R. E. Strategies to Extend Plasma Half-Lives of Recombinant
Antibodies. BioDrugs 23, 93-109, doi:10.2165/00063030-200923020-00003 (2009).
211 Kontermann, R. E. Strategies for extended serum half-life of protein therapeutics.
Current Opinion in Biotechnology 22, 868-876, doi:https://doi.org/10.1016/j.copbio.2011.06.012 (2011).
212 Walsh, G. Biopharmaceutical benchmarks 2014. Nat Biotech 32, 992-1000,
doi:10.1038/nbt.3040 http://www.nature.com/nbt/journal/v32/n10/abs/nbt.3040.html#supplementary-information (2014).
213 Golding, B., Jiang, H., Sauna, Z. & Levin, D. Immune responses to chimeric
human-FIX-mouse-Fc fusion protein in a hemophilia B mouse model (THER2P.963). The Journal of Immunology 194, 67.14 (2015).

158
214 Nangia-Makker, P., Balan, V. & Raz, A. Galectin-3-Binding and Metastasis. Methods in molecular biology (Clifton, N.J.) 878, 251-266, doi:10.1007/978-1-61779-854-2_17 (2012).
215 Yang, R. Y., Hsu, D. K. & Liu, F. T. Expression of galectin-3 modulates T-cell
growth and apoptosis. Proc Natl Acad Sci U S A 93, 6737-6742 (1996). 216 Stowell, S. R. et al. Differential roles of galectin-1 and galectin-3 in regulating
leukocyte viability and cytokine secretion. J Immunol 180, 3091-3102 (2008). 217 Ahmed, H. & AlSadek, D. M. M. Galectin-3 as a Potential Target to Prevent
Cancer Metastasis. Clinical Medicine Insights. Oncology 9, 113-121, doi:10.4137/CMO.S29462 (2015).
218 Edelstam, G. A., Laurent, U. B., Lundkvist, O. E., Fraser, J. R. & Laurent, T. C.
Concentration and turnover of intraperitoneal hyaluronan during inflammation. Inflammation 16, 459-469 (1992).
219 Catorce, M. N. & Gevorkian, G. LPS-induced Murine Neuroinflammation Model:
Main Features and Suitability for Pre-clinical Assessment of Nutraceuticals. Current Neuropharmacology 14, 155-164, doi:10.2174/1570159X14666151204122017 (2016).
220 Juskewitch, J. E. et al. LPS-induced murine systemic inflammation is driven by
parenchymal cell activation and exclusively predicted by early MCP-1 plasma levels. Am J Pathol 180, 32-40, doi:10.1016/j.ajpath.2011.10.001 (2012).
221 Ghorani, V., Boskabady, M. H., Khazdair, M. R. & Kianmeher, M. Experimental
animal models for COPD: a methodological review. Tob Induc Dis 15, 25, doi:10.1186/s12971-017-0130-2 (2017).
222 Kell, D. B. & Pretorius, E. On the translocation of bacteria and their
lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr Biol (Camb) 7, 1339-1377, doi:10.1039/c5ib00158g (2015).
223 Copeland, S. et al. Acute Inflammatory Response to Endotoxin in Mice and
Humans. Clinical and Diagnostic Laboratory Immunology 12, 60-67 (2005). 224 Ianaro, A., Tersigni, M. & D'Acquisto, F. New insight in LPS antagonist. Mini Rev
Med Chem 9, 306-317 (2009). 225 Oz, H. S. & Puleo, D. A. Animal models for periodontal disease. J Biomed
Biotechnol 2011, 754857, doi:10.1155/2011/754857 (2011).

159
226 Holt, S. C. & Ebersole, J. L. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the "red complex", a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000 38, 72-122, doi:10.1111/j.1600-0757.2005.00113.x (2005).
227 Blinkhorn, A. et al. Is there a role for triclosan/copolymer toothpaste in the
management of periodontal disease? Br Dent J 207, 117-125, doi:10.1038/sj.bdj.2009.669 (2009).
228 Page, R. C. & Kornman, K. S. The pathogenesis of human periodontitis: an
introduction. Periodontol 2000 14, 9-11 (1997). 229 Madianos, P. N., Bobetsis, Y. A. & Kinane, D. F. Generation of inflammatory
stimuli: how bacteria set up inflammatory responses in the gingiva. J Clin Periodontol 32 Suppl 6, 57-71, doi:10.1111/j.1600-051X.2005.00821.x (2005).
230 Gaffar, A., Scherl, D., Afflitto, J. & Coleman, E. J. The effect of triclosan on
mediators of gingival inflammation. J Clin Periodontol 22, 480-484 (1995). 231 Kinane, D. F., Stathopoulou, P. G. & Papapanou, P. N. Periodontal diseases. Nat
Rev Dis Primers 3, 17038, doi:10.1038/nrdp.2017.38 (2017). 232 Gotsman, I. et al. Periodontal destruction is associated with coronary artery
disease and periodontal infection with acute coronary syndrome. J Periodontol 78, 849-858, doi:10.1902/jop.2007.060301 (2007).
233 Khader, Y. S., Dauod, A. S., El-Qaderi, S. S., Alkafajei, A. & Batayha, W. Q.
Periodontal status of diabetics compared with nondiabetics: a meta-analysis. J Diabetes Complications 20, 59-68, doi:10.1016/j.jdiacomp.2005.05.006 (2006).
234 Gemmell, E. & Seymour, G. J. Immunoregulatory control of Th1/Th2 cytokine
profiles in periodontal disease. Periodontol 2000 35, 21-41, doi:10.1111/j.0906-6713.2004.003557.x (2004).
235 Gemmell, E., Marshall, R. I. & Seymour, G. J. Cytokines and prostaglandins in
immune homeostasis and tissue destruction in periodontal disease. Periodontol 2000 14, 112-143 (1997).
236 Everett, F. G., Waerhaug, J. & Widman, A. Leonard Widman: surgical treatment
of pyorrhea alveolaris. J Periodontol 42, 571-579, doi:10.1902/jop.1971.42.9.571 (1971).
237 Ramfjord, S. P. & Nissle, R. R. The modified widman flap. J Periodontol 45, 601-
607, doi:10.1902/jop.1974.45.8.2.601 (1974).

160
238 Nyman, S., Lindhe, J., Karring, T. & Rylander, H. New attachment following surgical treatment of human periodontal disease. J Clin Periodontol 9, 290-296 (1982).
239 Larsson, L. et al. Regenerative Medicine for Periodontal and Peri-implant
Diseases. J Dent Res 95, 255-266, doi:10.1177/0022034515618887 (2016). 240 Yukna, R. A., Carr, R. L. & Evans, G. H. Histologic evaluation of an Nd:YAG
laser-assisted new attachment procedure in humans. Int J Periodontics Restorative Dent 27, 577-587 (2007).
241 Weiss, U. Inflammation. Nature 454, 427-427 (2008). 242 Fullerton, J. N. & Gilroy, D. W. Resolution of inflammation: a new therapeutic
frontier. Nat Rev Drug Discov 15, 551-567, doi:10.1038/nrd.2016.39 (2016). 243 Gabay, C. & Kushner, I. Acute-Phase Proteins and Other Systemic Responses to
Inflammation. New England Journal of Medicine 340, 448-454, doi:10.1056/NEJM199902113400607 (1999).
244 Wang, L. et al. Efficacy evaluation of low-dose aspirin in IVF/ICSI patients
evidence from 13 RCTs: A systematic review and meta-analysis. Medicine (Baltimore) 96, e7720, doi:10.1097/md.0000000000007720 (2017).
245 Taneja, A., Della Pasqua, O. & Danhof, M. Challenges in translational drug
research in neuropathic and inflammatory pain: the prerequisites for a new paradigm. Eur J Clin Pharmacol, doi:10.1007/s00228-017-2301-8 (2017).
246 Munoz Olmo, L., Juan Armas, J. & Gomariz Garcia, J. J. [Risk of fatal/non-fatal
events in patients with previous coronary heart disease/acute myocardial infarction and treatment with non-steroidal anti-inflammatory drugs]. Semergen, doi:10.1016/j.semerg.2017.07.004 (2017).
247 Nimmo, S. M., Foo, I. T. H. & Paterson, H. M. Enhanced recovery after surgery:
Pain management. J Surg Oncol, doi:10.1002/jso.24814 (2017). 248 Patel, N. U., Vera, N. C., Shealy, E. R., Wetzel, M. & Feldman, S. R. A Review of
the Use of Secukinumab for Psoriatic Arthritis. Rheumatol Ther, doi:10.1007/s40744-017-0076-0 (2017).
249 Hayta, E. & Elden, H. Acute spinal cord injury: A review of pathophysiology and
potential of non-steroidal anti-inflammatory drugs for pharmacological intervention. J Chem Neuroanat, doi:10.1016/j.jchemneu.2017.08.001 (2017).
250 Herold, K. C. et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes
mellitus. N Engl J Med 346, 1692-1698, doi:10.1056/NEJMoa012864 (2002).

161
251 Keymeulen, B. et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 352, 2598-2608, doi:10.1056/NEJMoa043980 (2005).
252 Kotsovilis, S. & Andreakos, E. Therapeutic human monoclonal antibodies in
inflammatory diseases. Methods Mol Biol 1060, 37-59, doi:10.1007/978-1-62703-586-6_3 (2014).
253 Laveti, D. et al. Anti-inflammatory treatments for chronic diseases: a review.
Inflamm Allergy Drug Targets 12, 349-361 (2013). 254 Kamala, T. Hock immunization: a humane alternative to mouse footpad
injections. J Immunol Methods 328, 204-214, doi:10.1016/j.jim.2007.08.004 (2007).
255 Vogel, C. & Marcotte, E. M. Insights into the regulation of protein abundance
from proteomic and transcriptomic analyses. Nat Rev Genet 13, 227-232, doi:10.1038/nrg3185 (2012).
256 Kowal, A., Warminska, J. & Krasowska, D. Immunosuppressive drugs in
dermatology--benefits and threats. Ann Univ Mariae Curie Sklodowska Med 58, 14-21 (2003).
257 Malcolm, J. et al. IL-33 Exacerbates Periodontal Disease through Induction of
RANKL. Journal of Dental Research 94, 968-975, doi:10.1177/0022034515577815 (2015).

162
BIOGRAPHICAL SKETCH
Evelyn Bracho Sanchez was born in Caracas, Venezuela in 1989. She moved to
Titusville, FL in 2002 and attended middle and high school until 2007 when she returned
to Venezuela. Evelyn obtained her high school degree from Escuela Comunitaria and
soon thereafter returned to Gainesville, FL. In 2008, Evelyn graduated from Eastside
High School and in 2010 from Santa Fe College. Evelyn transferred to the University of
Florida where she obtained her Bachelor of Science in Materials Science and
Engineering. During her undergraduate tenure, Evelyn conducted research under the
mentorship of Dr. Brian Sorg, who introduced her to biomaterials for immune
applications. The work with Dr. Sorg, lead to an interest in researching potential cures
for complex immunological diseases which directed her interest to the laboratory of Dr.
Benjamin Keselowsky. In August 2012, Evelyn joined the doctoral program in
Biomedical Engineering at the University of Florida and received her PhD in December
2017 under the mentorship of Dr. Keselowsky.
During her doctoral studies, Evelyn worked on numerous projects, largely
focused on biomaterials for immunomodulation. She began with the production and
modification of synthetic microparticles which serve as a vaccine delivery system for
type 1 diabetes. Soon after, through collaboration with Dr. Greg Hudalla, her focus
shifted to engineering of a cell and tissue targeting fusion enzyme to direct localized
cellular metabolism for the treatment of inflammation.
Following her studies, Evelyn hopes to pursue a career as a Medical Science
Liaison in biotechnology where she can apply her skills developed at the University of
Florida to disseminate knowledge and expand strategies to combat immunological
diseases.