the nitrogen assimilation control protein, nac, is a dna binding
TRANSCRIPT

JOURNAL OF BACTERIOLOGY, June 1995, p. 3546–3555 Vol. 177, No. 120021-9193/95/$04.0010Copyright q 1995, American Society for Microbiology
The Nitrogen Assimilation Control Protein, NAC, Is a DNABinding Transcription Activator in Klebsiella aerogenes
THOMAS J. GOSS AND ROBERT A. BENDER*
Department of Biology, The University of Michigan, Ann Arbor, Michigan 48109-1048
Received 22 September 1994/Accepted 11 April 1995
A 32-kDa polypeptide corresponding to NAC, the product of the Klebsiella aerogenes nac gene, was overex-pressed from a plasmid carrying a tac*-*nac operon fusion and purified to near homogeneity by takingadvantage of its unusual solubility properties. NAC was able to shift the electrophoretic migration of DNAfragments carrying the NAC-sensitive promoters hutUp, putPp1, and ureDp. The interaction between NAC andhutUp was localized to a 26-bp region centered approximately 64 bp upstream of the hutUp transcriptioninitiation site. Moreover, NAC protected this region from DNase I digestion. Mobility shift and DNase Iprotection studies utilizing the putP and ureD promoter regions identified NAC-binding regions of sizes andlocations similar to those found in hutUp. Comparison of the DNA sequences which were protected from DNaseI digestion by NAC suggests a minimal NAC-binding consensus sequence: 5*-ATA-N9-TAT-3*. In vitro tran-scription assays demonstrated that NAC was capable of activating the transcription of hutUp by s70-RNApolymerase holoenzyme when this promoter was presented as either a linear or supercoiled DNA molecule.Thus, NAC displays the in vitro DNA-binding and transcription activation properties which have beenpredicted for the product of the nac gene.
The enteric bacterium Klebsiella aerogenes can use a largenumber of nitrogen-containing compounds as its sole source ofnitrogen. In virtually every case, the expression of the operonsencoding these catabolic activities is regulated by the qualityand quantity of the nitrogen source present in the growthmedium (26). When ammonium, the preferred nitrogensource, is abundant, operons allowing the utilization of growthrate-limiting nitrogen sources are weakly expressed and oper-ons facilitating the assimilation of ammonium are derepressed.When cells are growth rate limited by the nitrogen source inthe medium, the converse is true: operons that allow the uti-lization of alternative nitrogen sources are derepressed andthose requiring elevated ammonium levels to effectively assim-ilate ammonium are repressed. In every case known, this reg-ulation in response to the supply of nitrogen requires theaction of a phosphorylated form of the transcription activatorNTRC (26). Phosphorylation of NTRC allows the activationand repression of operons in response to nitrogen limitation,and the lack of phosphorylation or lack of NTRC prevents theactivation and repression of such operons. The action of thisnitrogen regulatory (NTR) system and the path of signal trans-duction through this network are the subject of intense inves-tigation in a number of laboratories (1, 13, 36, 37).Several NTRC-dependent operons of K. aerogenes also re-
quire the product of the nac gene for nitrogen regulation (5,25, 42). The operons which facilitate the catabolism of histi-dine, proline, and urea are fully activated in response to nitro-gen limitation only if the nac gene is intact. Similarly, theoperons encoding glutamate dehydrogenase and glutamatesynthase are repressed in response to nitrogen limitation onlyif the nac gene is intact. The nac gene itself is activated inresponse to nitrogen limitation; however, its upper level ofactivity is limited only if the nac gene is intact (6). When theexpression of the nac gene was placed under the control of anisopropyl b-D-thiogalactopyranoside (IPTG)-inducible pro-
moter, the regulation of the NAC-dependent operons was en-tirely dependent on the presence or absence of IPTG (42). Insuch strains the NTR system was not necessary for expressionof this subset of nitrogen-regulated operons, and their physi-ological response to the nitrogen source present in the mediumwas eliminated. Thus, it appeared that NAC might be theactual transcription regulator for some of the nitrogen-regu-lated operons.The expression of the nac gene is tightly controlled by the
NTR system both in vivo and in vitro. Experiments with nac-lacZ fusions showed the clear dependence of nac expression onthe phosphorylated form of NTRC and on s54 (42). In vitrotranscription with purified components confirmed that RNApolymerase bearing the s54 subunit was essential (s70 was noteffective) and that the phosphorylated form of NTRC wasrequired for transcription of the nac gene (14). Thus, a modelemerged in which the NTR system was required for the ex-pression of the nac gene and the NAC protein was required forregulation of the NAC-dependent operons (4). Consequently,NTRC appeared to play an indirect role in the regulation ofsome operons. However, in vivo experiments could not clearlydistinguish between a model in which NAC was itself the reg-ulator of transcription and a model in which a product regu-lated by NAC was the actual transcription regulator.The promoters have been identified for two of the NAC-
dependent operons (hutUH and putP), and in both cases theseNAC-activated promoters (hutUp and putPp1) appear to bes70 dependent (9, 30, 33). This explains a need for an inter-mediate between the s54-dependent NTR system and the s70-dependent target operons. As such, NAC allows the recruit-ment of s70-dependent operons that had evolved for otherpurposes (e.g., degradation of histidine for use as a carbonsource) into a s54-dependent regulatory network.The DNA sequence of the nac gene predicted that NAC
would be a member of the LysR family of transcription regu-lators (41), all of which appear to act by binding near promot-ers to stimulate or repress transcription (40). Transcriptionfrom both hutUp and putPp1 can be activated by the cyclicAMP (cAMP)-catabolite gene activator protein (CAP) com-
* Corresponding author. Phone: (313) 936-2530. Fax: (313) 747-0884. Electronic mail address: [email protected].
3546

plex (in response to carbon-limited growth) and by a factorpresumed to be NAC (in response to nitrogen-limited growth).In vitro transcription analysis of the hutUp region, hut(P),revealed the presence of a second promoter, Pc, which is di-vergently oriented relative to hutUp. Although transcriptionfrom Pc is repressed by the cAMP-CAP complex in vitro (30,32, 33), its function in vivo, if any, is unknown. The hut(P)region has been extensively studied in this laboratory, makingit an attractive model system for use in the initial character-ization of the NAC protein from K. aerogenes.
MATERIALS AND METHODS
Strains and plasmids. Escherichia coli K-12 strain EB2567 harbors plasmidpEC205.1 carrying the tac9-9nac operon fusion in which the expression of the nacgene is under the control of the IPTG-inducible tac promoter (42). PlasmidspOS1 and pOS2, carrying the K. aerogenes hut(P) region, and their RD and LDdeletion derivatives have been described previously (35). Plasmids pOSLD113and pOSLD113* carry deletions of the first 112 nucleotides of the 250-bp insertcarried by pOS1. In addition, pOSLD113* carries an internal deletion of 10 bpfrom nucleotides 136 to 145 (35). Plasmid pAM1202, used as a supercoiledtemplate for in vitro transcription studies, consists of 1.24-kb BglII-PvuII frag-ment from pCB102 (7) cloned into the BamHI-HincII site of pTE103 (12) suchthat transcripts originating from hutUp terminate at the factor-independent T7terminator located approximately 350 nucleotides downstream from the hutUptranscription initiation site. For the analysis of the interaction between NAC andthe K. aerogenes ureD promoter region, religation of NaeI-HincII-digested plas-mid pKAU22 (22) generated the deletion derivative pKAU22D1, which retainsthe ureD promoter region from nucleotides 1 to 258. A similar analysis wascarried out with the K. aerogenes putAP region utilizing plasmid pKC9 (9).NAC induction and purification. NAC synthesis from the plasmid-borne tac9-
9nac operon fusion carried by strain EB2567 was induced in mid-log-phasecultures (ca. 2 3 108 CFU/ml) growing in LB medium supplemented with 50 mgof kanamycin per ml at 378C by the addition of IPTG to 10 mM. The cells wereharvested at late-log or early stationary phase (ca. 8 3 108 CFU/ml) by centrif-ugation. In preparing NAC, three grades of this protein, which differ in the purityof NAC, have been studied: crude extracts, partially purified NAC, and purifiedNAC, the later referred to simply as NAC. To obtain crude extracts, harvestedcells were washed in buffer 4 (250 mM NaCl, 100 mM NaH2PO4 [pH 7.0] withNaOH, 2.5 mM MgCl2, 1 mM 2-mercaptoethanol), concentrated 50-fold inbuffer 4, and disrupted by passage through a French pressure cell two times at12,000 lb/in2 at 48C. Particulate cell debris was removed from the pressate bycentrifugation (30 min at 48C, 30,000 3 g), and the supernatant fluid, whichcontained the NAC protein, was mixed with an equal volume of glycerol andstored at 2208C. To obtain partially purified NAC, harvested cells were washed,concentrated 50-fold in buffer 2 (50 mM imidazole [pH 7.0] with HCl, 2.5 mMMgCl2, 1 mM 2-mercaptoethanol), and disrupted as described above. The ma-terial sedimented from the disrupted cell suspension by centrifugation (30 min at48C, 30,000 3 g) was resuspended in buffer 2 and dialyzed against 500 volumesof buffer 4 for at least 21⁄2 h at 48C. The contents of the dialysis bag wererecovered, clarified by centrifugation (30 min at 48C, 30,000 3 g), and stored at48C. For partially purified NAC used in the molecular sieve analysis, the clarifiedfluid was mixed with one-ninth volume of glycerol and stored at 48C. For purifiedNAC, the clarified fluid was dialyzed against 500 volumes of buffer 3 (250 mMKCl, 50 mM imidazole [pH 7.0] with HCl, 2.5 mM MgCl2, 1 mM 2-mercapto-ethanol) overnight at 48C. The contents of the dialysis bag were recovered andmost of the fine sediment material was recovered by brief centrifugation at 7503 g at 48C. The supernatant fluid was carefully removed, and the pellet wasgently resuspended in 1/200th the original culture volume of ice-cold buffer 4.After 2 h at 08C, the resuspended material was clarified by centrifugation (30 minat 48C, 30,000 3 g), and the supernatant fluid was either mixed with an equalvolume of glycerol and stored at 2208C or further concentrated by overnightdialysis at 48C against 500 volumes of buffer 5 (50% glycerol, 125 mM NaCl, 50mM NaH2PO4 [pH 7.0] with NaOH, 1.25 mM MgCl2, 0.5 mM 2-mercaptoetha-nol) and stored at 2208C. The purified NAC preparations obtained by thisprocedure contained 20 to 84 pmol of NAC dimer or 1.25 to 5.25 mg of proteinper ml as determined by the method of Lowry et al. (24) after correction for theglycerol content in buffer 6.When the influence of nitrogen availability on the DNA-binding activity of
NAC was investigated, NAC was partially purified from strain EB2567 grown inminimal medium under conditions of nitrogen excess or nitrogen limitation byusing the same partial purification procedure described above except that the LBmedium was replaced by W4 minimal salts medium (25) supplemented withglucose (0.4%), thiamine (0.001%), kanamycin (50 mg/ml), and either ammo-nium chloride plus glutamine (both at 0.2%) for nitrogen excess or glutaminealone (0.04%) for nitrogen limitation.Denaturing polyacrylamide gel electrophoresis. Analysis of polypeptides syn-
thesized by EB2567 was carried out using denaturing slab gels according to themethod of Laemmli (21) with separating gel acrylamide concentrations between
10 and 15%. Bovine serum albumin (BSA) (66 kDa), egg albumin (45 kDa),pepsin (34.7 kDa), carbonic anhydrase (29 kDa), trypsinogen (24 kDa), b-lacto-globulin (18.4 kDa), and lysozyme (14.3 kDa) were used as molecular massstandards. The intensities of the protein bands were quantitated by densitometerscanning.Characterization of NAC protein. Both Sephadex G-100 medium and Bio-Gel
A-1.5m molecular sieve matrices were used to estimate the native molecularweight of NAC. Protein samples contained in buffer 4 supplemented with glyc-erol to 10% were layered onto columns, and protein elution profiles were de-termined by densitometer scanning of Coomassie-stained sodium dodecyl sulfate(SDS)-polyacrylamide displays of column fractions. Columns were calibratedwith bromophenol blue (670 Da), cytochrome c (12.4 kDa), carbonic anhydrase(29 kDa), BSA (66 kDa), yeast alcohol dehydrogenase (150-kDa tetramer com-posed of 44-kDa subunits), and blue dextran (2,000 kDa) molecular mass stan-dards. The elution profiles of the standards and NAC polypeptide obtained withboth molecular sieve matrices were similar and were not significantly alteredwhen standards and NAC were simultaneously or independently analyzed.The isoelectric point (pI) of the NAC polypeptide was determined by the
method of Giulian (16) on a vertical polyacrylamide slab gel system employing anacrylamide concentration of 5.5% and a pH range from 3.5 to 10.0. Soybeantrypsin inhibitor (pI of 4.6), bovine erythrocyte carbonic anhydrase (pI of 5.9),horse heart myoglobin (pIs of 6.8 and 7.2), rabbit muscle L-lactate dehydroge-nase (pIs of 8.3, 8.4 and 8.6), and bovine pancreas trypsinogen (pI of 9.3) wereused as standards.DNA manipulations. In general, the methods outlined by Maniatis et al. (27)
were used in manipulating the DNA.Mobility shift assay. The binding of NAC to DNA fragments carrying either
hutUp, ureDp, or putPp1 was determined by the mobility shift assay. Basically, 1ml of NAC in buffer 5, dilutions of NAC in buffer 6 (buffer 5 supplemented with1.0 mg of BSA per ml), or buffer 6 alone was mixed with 0.1 pmol of restrictionenzyme-digested plasmid DNA contained in 9 ml of 0.1-strength Tris-EDTA(TE) buffer (1 mM Tris HCl [pH 7.4], 0.1 mM EDTA). The mixture wasincubated at room temperature for 5 to 15 min, after which 1.5 ml of gel loadingsolution (40 mM Tris HCl [pH 8.4], 4 mM EDTA, 0.2% bromophenol blue, 0.2%xylene cyanol, 25% glycerol) was added, and the mixture was loaded onto a 4%polyacrylamide-TE (pH 8.4) slab gel and subjected to electrophoresis at 15 V/cmfor 2 h at 48C. The gels were stained with ethidium bromide (0.05 mg/ml), andthe electrophoretic mobilities of the DNA fragments were determined by UVfluorescence.In vitro transcription. Restriction fragments used as templates for in vitro
transcription runoff assays were purified by agarose gel electrophoresis andelectroelution. The templates carrying the hut(P) region were a 250-bp SalIfragment purified from pOS1, a 140-bp EcoRI-SalI fragment purified frompOSLD113, and a 130-bp EcoRI-SalI fragment purified from pOSLD113*. The invitro transcription procedure has been described previously (32, 33); however, inthis study, NAC was used in place of CAP and, where indicated, potassiumglutamate was used in place of potassium chloride as the predominant salt in thetranscription buffer. Basically, the templates (ca. 0.4 pmol) were incubated witheither 25% glycerol, dilutions of NAC in buffer 6, or buffer 6 alone for 5 to 20 minat room temperature prior to the addition of 0.4 U (ca. 1 pmol) of E. coli RNApolymerase holoenzyme carrying the s70 subunit. s70-RNA polymerase prepa-rations supplied by New England Nuclear, Boehringer Mannheim Biochemicals,and Epicentre Technologies yielded generally similar results, although somedifferences were seen when transcription was carried out in the presence of 25%glycerol and when NAC activated transcription from the linear LD113* template.Open complex formation was allowed to occur for 30 min at 308C, heparin andribonucleotide triphosphates plus [a-32P]UTP were added, and the mixture wasincubated at 378C for 10 min. Transcription reactions were terminated by theaddition of 2 volumes of a solution containing 100 mM sodium acetate (pH 5.5),0.4% SDS, and 1 mg of yeast tRNA per ml. The terminated reaction mixtureswere extracted once with an equal volume of phenol-chloroform and once withchloroform. The aqueous phase (50 ml) recovered from the extraction procedurewas mixed with 25 ml of 7.5 M ammonium acetate–5 ml of 100 mM MgCl2–150ml of ethanol, and the polynucleotides were precipitated after overnight incuba-tion at 2208C. The precipitated material was resuspended in 80% form-amide–13 Tris-borate-EDTA (TBE) (89 mM Tris, 89 mM boric acid [pH 8.3]with HCl, 2 mM EDTA), boiled, separated by electrophoresis on an 8% acryl-amide–7 M urea–TBE gel, and subjected to autoradiography. Supercoiled plas-mid templates were purified by the alkaline lysis method, subjected to equilib-rium density centrifugation on CsCl-ethidium bromide gradients (27), andanalyzed by the same procedure as that used for the linear templates.DNase I footprint analysis. Linearized plasmid DNA was 39 end labeled with
a DNA polymerase Klenow fragment, precipitated, and redigested to generatetwo end-labeled fragments, one less than 25 bp and the other a longer DNAfragment carrying the promoter region of interest. A 7-ml solution containingapproximately 0.16 pmol of end-labeled DNA dissolved in 1.293 in vitro tran-scription buffer (38.6 mM Tris [pH 7.9], 129 mMKCl, 12.9 mMMgCl2, 0.129 mMEDTA, 0.129 mM dithiothreitol, 34.7 mg of BSA per ml) was mixed with 1 ml ofeither NAC, dilutions of NAC in buffer 6, or buffer 6 alone, all of which werediluted at a ratio of 1/3 in 13 transcription buffer (32, 33) prior to addition to theDNA samples. Where indicated, potassium glutamate was substituted for potas-sium chloride as the predominant salt in the transcription buffer. The DNA-
VOL. 177, 1995 KLEBSIELLA AEROGENES NAC PROTEIN 3547

protein mixtures were incubated for 15 min at room temperature, 1 ml of E. coliRNA polymerase holoenzyme carrying the s70 subunit (0.44 U, ca. 1 pmol) orRNA polymerase dilution buffer (10 mM Tris [pH 8.0], 100 mM KCl, 1 mMEDTA, 50% glycerol) was added, and the mixture was incubated for 30 min at308C. Limited digestion of the protein-bound DNA was initiated by the additionof 1 ml of DNase I solution (used at 2.33 1023 U/ml after dilution in 10 mM Tris[pH 8.0]–10 mM MgCl2–5 mM CaCl2–0.1 mM dithiothreitol). Digestion wascarried out for 1 min at room temperature and terminated by the addition offormamide to 50% and boiling for 5 min. DNase I digestion patterns wereanalyzed on 6% acrylamide–7 M urea–TBE gels. To facilitate the alignment ofDNase I digestion patterns with DNA sequences, A1G sequence ladders weregenerated for each end-labeled DNA substrate by limited acid depurinationfollowed by piperidine cleavage (3).
RESULTS
Purification of NAC. NAC was overexpressed and purifiedfrom E. coli EB2567, which carries the tac9-9nac operon fusionon plasmid pEC205.1. Growth of EB2567 in the presence ofIPTG resulted in the accumulation of large amounts of a 32-kDa polypeptide (Fig. 1, lanes 1 and 2) previously shown to beNAC (42). NAC represented about 20% of the total protein ofsuch induced cells. When cells were lysed in a high-salt buffer(buffer 4), the bulk of the NAC was soluble and was recoveredin the supernatant fraction following centrifugation to removecellular debris (Fig. 1, lanes 2 and 4). However, when the cellswere lysed in a low-salt buffer (buffer 2), much of the NAC wasinsoluble and was recovered in the pellet following centrifuga-tion (Fig. 1, lane 6). This insoluble material was enriched forNAC over the starting material (cf. Fig. 1, lanes 2 and 6).Resuspension of the insoluble material in high-salt buffer re-sulted in a rather specific extraction of NAC from the precip-itate (Fig. 1, lane 8). The resolubilized material shown in lane8 of Fig. 1 is referred to hereafter as partially purified NAC.The yield of NAC in such partially purified preparations wasestimated to be about 70%.For a more highly purified NAC preparation, the partially
purified NAC was dialyzed against a medium salt buffer (buffer3), resulting in the formation of a fine precipitate. The materialextracted from this precipitate in high-salt buffer was a highlypurified preparation of NAC hereafter referred to as NAC(Fig. 1, lane 10). Although this final step resulted in a nearly
homogeneous preparation of NAC, the recovery was poor andthe final yield of NAC was only about 5 to 10% relative to thestarting material. Despite the poor recovery, a 100-ml culturegenerally yielded about 1 mg of NAC.A sample of NAC was subjected to N-terminal amino acid
sequence analysis at the University of Michigan Protein andCarbohydrate Structure Facility. The reported sequence was[M/W]NLRRL[K/W]YFV (the identity of the bracketed ami-no acids could not be unambiguously resolved), in completeagreement with the sequence MNLRRLKYFV predicted fromthe nucleotide sequence of the nac gene (41). Molecular sieveanalysis of partially purified NAC showed that NAC eluted ata volume similar to that of BSA (data not shown), suggestingthat NAC exists largely as a homodimer under these high-saltconditions. Isoelectric focusing of NAC and partially purifiedNAC showed a major band with a pI of approximately 9 (datanot shown), in good agreement with the abundance of arginineresidues (20 per 305-residue monomer) and the value inferredfrom the DNA sequence of the nac gene.Interaction of NAC with hut(P). The DNA-binding activity
of NAC was tested by using the mobility shift assay and re-striction enzyme digests of plasmids bearing portions of thehut(P) region. NAC preferentially retarded the mobility of theDNA fragment from pOSLD116, which carries the hutUp re-gion, and the amount of this fragment that was shifted in-creased with increasing amounts of NAC (Fig. 2). When highlevels of NAC were used, the mobilities of certain vector-derived fragments were also retarded, whereas those of severalother fragments were unaffected (Fig. 2, lane 4), suggestingthat even the retardation of vector-derived fragments was se-quence specific. The NAC-dependent mobility shift was dis-rupted when heparin (0.1 mg/ml) was present in the incubationmixture or when NAC was boiled prior to incubation (data notshown).Partially purified NAC from cells grown under nitrogen lim-
itation or nitrogen excess was equally active in shifting DNAfragments bearing hut(P) (data not shown). Moreover, inclu-sion of the potential nitrogen effectors glutamine, glutamate,2-oxoglutarate, or ammonium in the incubation mixture at 10mM had no effect.To identify the sequences necessary for NAC binding, we
analyzed the NAC-mediated mobility shift of DNA fragments,
FIG. 1. Identification and purification of NAC polypeptide. Odd-numberedlanes, material obtained from uninduced cultures of strain EB2567; even-num-bered lanes, material obtained from IPTG-induced cultures of EB2567. Lanes: 1and 2, 103 whole cells; 3 and 4, crude extracts prepared in buffer 4 (high-saltbuffer); 5 and 6, homogenized insoluble material recovered from crude extractsprepared in buffer 2 (low-salt buffer); 7 and 8, material from lanes 5 and 6 whichwas resolubilized by extraction with buffer 4; 9 and 10, material from lanes 7 and8 which was precipitated by dialysis against buffer 3 (medium salt buffer) andresolubilized in buffer 4; M, pepsin (34.7 kDa) and carbonic anhydrase (29 kDa)molecular mass standards. Samples were separated on a 15% polyacrylamide-SDS gel and visualized with Coomassie stain.
FIG. 2. Mobility shift assay of the interaction of NAC with hutUp. pOSLD116was digested with EcoRI, HindIII, and HpaII. The resulting DNA fragments (0.1pmol) were preincubated with buffer 6 (lane 1) or NAC (2.22, 6.67, and 20 pmol,lanes 2, 3 and 4, respectively), separated on a 4% polyacrylamide gel, stainedwith ethidium bromide, and visualized by UV fluorescence. The position of theunbound 158-bp hut insert is indicated on the left.
3548 GOSS AND BENDER J. BACTERIOL.
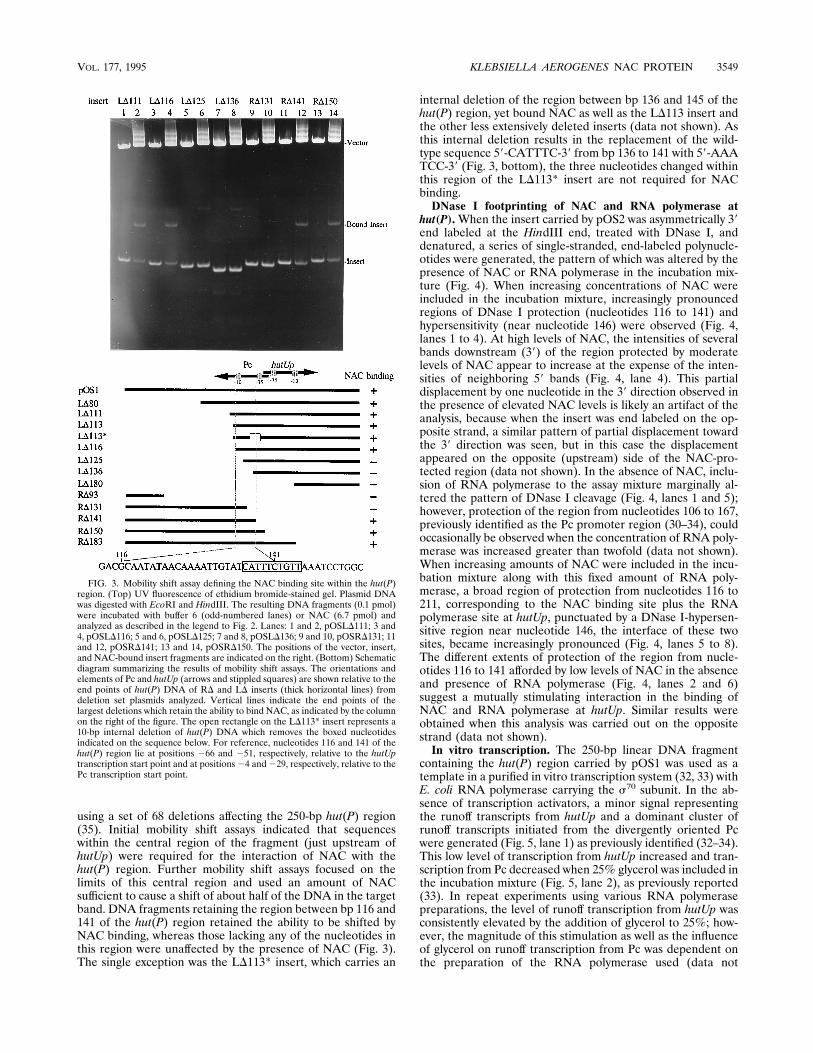
using a set of 68 deletions affecting the 250-bp hut(P) region(35). Initial mobility shift assays indicated that sequenceswithin the central region of the fragment (just upstream ofhutUp) were required for the interaction of NAC with thehut(P) region. Further mobility shift assays focused on thelimits of this central region and used an amount of NACsufficient to cause a shift of about half of the DNA in the targetband. DNA fragments retaining the region between bp 116 and141 of the hut(P) region retained the ability to be shifted byNAC binding, whereas those lacking any of the nucleotides inthis region were unaffected by the presence of NAC (Fig. 3).The single exception was the LD113* insert, which carries an
internal deletion of the region between bp 136 and 145 of thehut(P) region, yet bound NAC as well as the LD113 insert andthe other less extensively deleted inserts (data not shown). Asthis internal deletion results in the replacement of the wild-type sequence 59-CATTTC-39 from bp 136 to 141 with 59-AAATCC-39 (Fig. 3, bottom), the three nucleotides changed withinthis region of the LD113* insert are not required for NACbinding.DNase I footprinting of NAC and RNA polymerase at
hut(P).When the insert carried by pOS2 was asymmetrically 39end labeled at the HindIII end, treated with DNase I, anddenatured, a series of single-stranded, end-labeled polynucle-otides were generated, the pattern of which was altered by thepresence of NAC or RNA polymerase in the incubation mix-ture (Fig. 4). When increasing concentrations of NAC wereincluded in the incubation mixture, increasingly pronouncedregions of DNase I protection (nucleotides 116 to 141) andhypersensitivity (near nucleotide 146) were observed (Fig. 4,lanes 1 to 4). At high levels of NAC, the intensities of severalbands downstream (39) of the region protected by moderatelevels of NAC appear to increase at the expense of the inten-sities of neighboring 59 bands (Fig. 4, lane 4). This partialdisplacement by one nucleotide in the 39 direction observed inthe presence of elevated NAC levels is likely an artifact of theanalysis, because when the insert was end labeled on the op-posite strand, a similar pattern of partial displacement towardthe 39 direction was seen, but in this case the displacementappeared on the opposite (upstream) side of the NAC-pro-tected region (data not shown). In the absence of NAC, inclu-sion of RNA polymerase to the assay mixture marginally al-tered the pattern of DNase I cleavage (Fig. 4, lanes 1 and 5);however, protection of the region from nucleotides 106 to 167,previously identified as the Pc promoter region (30–34), couldoccasionally be observed when the concentration of RNA poly-merase was increased greater than twofold (data not shown).When increasing amounts of NAC were included in the incu-bation mixture along with this fixed amount of RNA poly-merase, a broad region of protection from nucleotides 116 to211, corresponding to the NAC binding site plus the RNApolymerase site at hutUp, punctuated by a DNase I-hypersen-sitive region near nucleotide 146, the interface of these twosites, became increasingly pronounced (Fig. 4, lanes 5 to 8).The different extents of protection of the region from nucle-otides 116 to 141 afforded by low levels of NAC in the absenceand presence of RNA polymerase (Fig. 4, lanes 2 and 6)suggest a mutually stimulating interaction in the binding ofNAC and RNA polymerase at hutUp. Similar results wereobtained when this analysis was carried out on the oppositestrand (data not shown).In vitro transcription. The 250-bp linear DNA fragment
containing the hut(P) region carried by pOS1 was used as atemplate in a purified in vitro transcription system (32, 33) withE. coli RNA polymerase carrying the s70 subunit. In the ab-sence of transcription activators, a minor signal representingthe runoff transcripts from hutUp and a dominant cluster ofrunoff transcripts initiated from the divergently oriented Pcwere generated (Fig. 5, lane 1) as previously identified (32–34).This low level of transcription from hutUp increased and tran-scription from Pc decreased when 25% glycerol was included inthe incubation mixture (Fig. 5, lane 2), as previously reported(33). In repeat experiments using various RNA polymerasepreparations, the level of runoff transcription from hutUp wasconsistently elevated by the addition of glycerol to 25%; how-ever, the magnitude of this stimulation as well as the influenceof glycerol on runoff transcription from Pc was dependent onthe preparation of the RNA polymerase used (data not
FIG. 3. Mobility shift assay defining the NAC binding site within the hut(P)region. (Top) UV fluorescence of ethidium bromide-stained gel. Plasmid DNAwas digested with EcoRI and HindIII. The resulting DNA fragments (0.1 pmol)were incubated with buffer 6 (odd-numbered lanes) or NAC (6.7 pmol) andanalyzed as described in the legend to Fig. 2. Lanes: 1 and 2, pOSLD111; 3 and4, pOSLD116; 5 and 6, pOSLD125; 7 and 8, pOSLD136; 9 and 10, pOSRD131; 11and 12, pOSRD141; 13 and 14, pOSRD150. The positions of the vector, insert,and NAC-bound insert fragments are indicated on the right. (Bottom) Schematicdiagram summarizing the results of mobility shift assays. The orientations andelements of Pc and hutUp (arrows and stippled squares) are shown relative to theend points of hut(P) DNA of RD and LD inserts (thick horizontal lines) fromdeletion set plasmids analyzed. Vertical lines indicate the end points of thelargest deletions which retain the ability to bind NAC, as indicated by the columnon the right of the figure. The open rectangle on the LD113* insert represents a10-bp internal deletion of hut(P) DNA which removes the boxed nucleotidesindicated on the sequence below. For reference, nucleotides 116 and 141 of thehut(P) region lie at positions 266 and 251, respectively, relative to the hutUptranscription start point and at positions24 and229, respectively, relative to thePc transcription start point.
VOL. 177, 1995 KLEBSIELLA AEROGENES NAC PROTEIN 3549

shown). When the incubation mixture was supplemented withNAC, a shift in the pattern of runoff transcripts similar to thatresulting from the addition of 25% glycerol was generated,with the level of transcription from hutUp increasing and thatfrom Pc decreasing with increasing amounts of NAC (Fig. 5,lanes 3 and 4). Although the magnitude of the NAC-depen-dent shifts in runoff transcription patterns varied with the prep-aration of RNA polymerase used, the direction of the shifts
remained unaffected (data not shown). The amount of glycerolpresent in the NAC preparation was well below that needed toinfluence transcription (cf. Fig. 5, lanes 1 and 5).The LD113 insert carries the entire hutUp RNA polymerase
binding site and the maximum region required for NAC bind-ing but lacks the Pc transcription initiation site and down-stream sequences (34, 35). As expected, only the runoff tran-scripts originating from hutUp, synthesized at a low basal level,could be detected when the LD113 insert was used as a tem-plate in this assay in the absence of activators (Fig. 5, lane 6).As seen with the undeleted pOS1 template, NAC and glycerolwere able to stimulate transcription from hutUp present on theLD113 template (Fig. 5, lanes 7 to 9), although the magnitudeof this stimulation varied with the preparation of RNA poly-merase. Like LD113, the LD113* template carries an intacthutUp RNA polymerase binding site which directed a low levelof runoff transcription in the absence of transcription activa-tors and which can be stimulated by high concentrations ofglycerol (Fig. 5, lane 10 and 11). However, in contrast to theresults with the LD113 template, NAC was consistently less
FIG. 4. DNase I footprinting of NAC and RNA polymerase on the hut(P)region. pOS2 was digested with HindIII, 39 end labeled, and redigested withHaeIII. The resulting DNA (0.16 pmol) was dissolved in 13 in vitro potassiumglutamate transcription buffer and preincubated with control buffer 6 (lanes 1, 5,and 9) or NAC (lanes 2 and 6, 3 pmol; lanes 3 and 7, 9 pmol; lanes 4 and 8, 27pmol) and either RNA polymerase dilution buffer (lanes 1 to 4 and 9) or 0.44 Uof RNA polymerase provided by Boehringer Mannheim Biochemicals (lanes 5 to8) as described in Materials and Methods. Protein-DNA complexes were par-tially digested with DNase I (0.0023 U) prior to being boiled in the presence offormamide, separation on a 6% polyacrylamide–TBE–urea gel, and autoradiog-raphy. The nucleotide sequence and features of the hut(P) region, as previouslydetermined (30, 33), were aligned to those of the footprinted regions by using theA1G sequence ladder (lane 10). Lines to the left of the figure indicates theapproximate extents of the regions protected by NAC and RNA polymerase. Thearrow indicates the region of DNase I hypersensitivity induced by the binding ofNAC and RNA polymerase.
FIG. 5. Runoff transcripts synthesized in vitro from the hut(P) region. Tran-scripts from the 250-bp pOS1 template (lanes 1 to 5), the 140-bp LD113 template(lanes 6 to 9), and the 130-bp LD113* template (lanes 10 to 13) were synthesizedby using RNA polymerase provided by New England Nuclear, as described inMaterials and Methods, after preincubation with the following amendments: noaddition (lane 1), 25% glycerol (lanes 2, 7, and 11), 20 pmol of NAC (lanes 3, 8,and 12), 2.22 pmol of NAC (lanes 4, 9, and 13) or buffer 6 (lanes 5, 6, and 10).The positions of the runoff transcripts from hutUp and Pc displayed on theautoradiogram are indicated.
3550 GOSS AND BENDER J. BACTERIOL.
effective at stimulating the level of runoff transcription fromhutUp present on the LD113* template. Depending on thepreparation of RNA polymerase used, the influence of NACon transcription from the LD113* template varied from nostimulation (Fig. 5, lanes 10, 12, and 13) to significant stimu-lation but less than that seen with the LD113 template (datanot shown). In addition, the basal levels of transcription from
the LD113 and LD113* templates varied with the preparationof RNA polymerase used (data not shown).The RD183 deletion removes nucleotides lying downstream
from the 29 position of hutUp, relative to its start site, abol-ishing the ability of hutUp to function as a promoter in vitro(34). When this template was used in the in vitro transcriptionassay, Pc was repressed by NAC (data not shown). A similardirect repression of transcription from the Pc promoter byNAC in the absence of a functional hutUp was observed invitro by using the 177-bp SphI fragment from pOS1, whichlacks the region of hutUp downstream of the 235 hexamer(data not shown).In vitro transcription analysis was also carried out with a
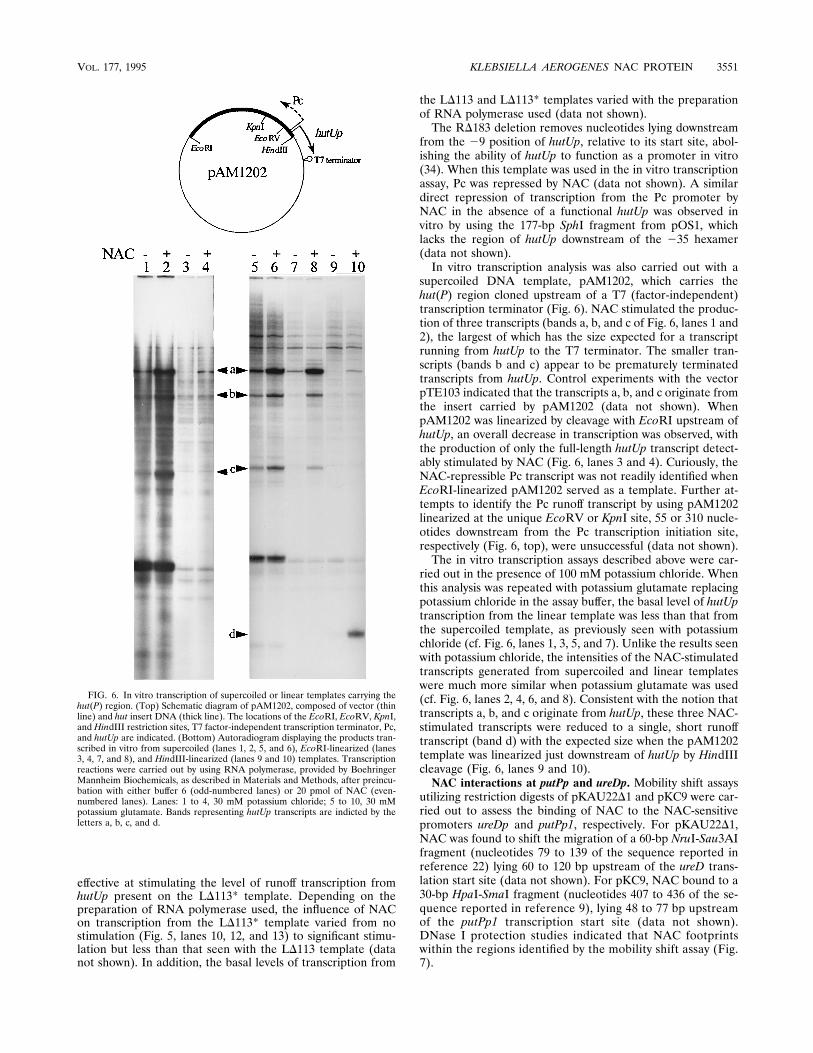
supercoiled DNA template, pAM1202, which carries thehut(P) region cloned upstream of a T7 (factor-independent)transcription terminator (Fig. 6). NAC stimulated the produc-tion of three transcripts (bands a, b, and c of Fig. 6, lanes 1 and2), the largest of which has the size expected for a transcriptrunning from hutUp to the T7 terminator. The smaller tran-scripts (bands b and c) appear to be prematurely terminatedtranscripts from hutUp. Control experiments with the vectorpTE103 indicated that the transcripts a, b, and c originate fromthe insert carried by pAM1202 (data not shown). WhenpAM1202 was linearized by cleavage with EcoRI upstream ofhutUp, an overall decrease in transcription was observed, withthe production of only the full-length hutUp transcript detect-ably stimulated by NAC (Fig. 6, lanes 3 and 4). Curiously, theNAC-repressible Pc transcript was not readily identified whenEcoRI-linearized pAM1202 served as a template. Further at-tempts to identify the Pc runoff transcript by using pAM1202linearized at the unique EcoRV or KpnI site, 55 or 310 nucle-otides downstream from the Pc transcription initiation site,respectively (Fig. 6, top), were unsuccessful (data not shown).The in vitro transcription assays described above were car-
ried out in the presence of 100 mM potassium chloride. Whenthis analysis was repeated with potassium glutamate replacingpotassium chloride in the assay buffer, the basal level of hutUptranscription from the linear template was less than that fromthe supercoiled template, as previously seen with potassiumchloride (cf. Fig. 6, lanes 1, 3, 5, and 7). Unlike the results seenwith potassium chloride, the intensities of the NAC-stimulatedtranscripts generated from supercoiled and linear templateswere much more similar when potassium glutamate was used(cf. Fig. 6, lanes 2, 4, 6, and 8). Consistent with the notion thattranscripts a, b, and c originate from hutUp, these three NAC-stimulated transcripts were reduced to a single, short runofftranscript (band d) with the expected size when the pAM1202template was linearized just downstream of hutUp by HindIIIcleavage (Fig. 6, lanes 9 and 10).NAC interactions at putPp and ureDp. Mobility shift assays
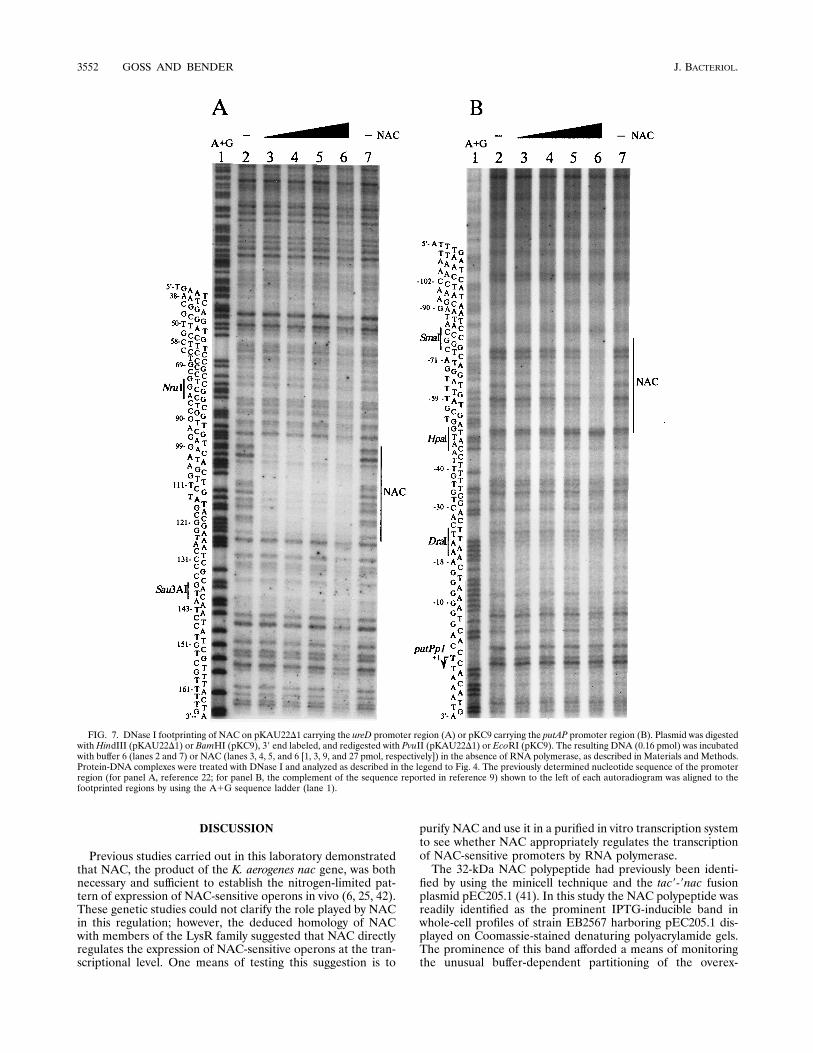
utilizing restriction digests of pKAU22D1 and pKC9 were car-ried out to assess the binding of NAC to the NAC-sensitivepromoters ureDp and putPp1, respectively. For pKAU22D1,NAC was found to shift the migration of a 60-bp NruI-Sau3AIfragment (nucleotides 79 to 139 of the sequence reported inreference 22) lying 60 to 120 bp upstream of the ureD trans-lation start site (data not shown). For pKC9, NAC bound to a30-bp HpaI-SmaI fragment (nucleotides 407 to 436 of the se-quence reported in reference 9), lying 48 to 77 bp upstreamof the putPp1 transcription start site (data not shown).DNase I protection studies indicated that NAC footprintswithin the regions identified by the mobility shift assay (Fig.7).
FIG. 6. In vitro transcription of supercoiled or linear templates carrying thehut(P) region. (Top) Schematic diagram of pAM1202, composed of vector (thinline) and hut insert DNA (thick line). The locations of the EcoRI, EcoRV, KpnI,and HindIII restriction sites, T7 factor-independent transcription terminator, Pc,and hutUp are indicated. (Bottom) Autoradiogram displaying the products tran-scribed in vitro from supercoiled (lanes 1, 2, 5, and 6), EcoRI-linearized (lanes3, 4, 7, and 8), and HindIII-linearized (lanes 9 and 10) templates. Transcriptionreactions were carried out by using RNA polymerase, provided by BoehringerMannheim Biochemicals, as described in Materials and Methods, after preincu-bation with either buffer 6 (odd-numbered lanes) or 20 pmol of NAC (even-numbered lanes). Lanes: 1 to 4, 30 mM potassium chloride; 5 to 10, 30 mMpotassium glutamate. Bands representing hutUp transcripts are indicted by theletters a, b, c, and d.
VOL. 177, 1995 KLEBSIELLA AEROGENES NAC PROTEIN 3551
DISCUSSION
Previous studies carried out in this laboratory demonstratedthat NAC, the product of the K. aerogenes nac gene, was bothnecessary and sufficient to establish the nitrogen-limited pat-tern of expression of NAC-sensitive operons in vivo (6, 25, 42).These genetic studies could not clarify the role played by NACin this regulation; however, the deduced homology of NACwith members of the LysR family suggested that NAC directlyregulates the expression of NAC-sensitive operons at the tran-scriptional level. One means of testing this suggestion is to
purify NAC and use it in a purified in vitro transcription systemto see whether NAC appropriately regulates the transcriptionof NAC-sensitive promoters by RNA polymerase.The 32-kDa NAC polypeptide had previously been identi-
fied by using the minicell technique and the tac9-9nac fusionplasmid pEC205.1 (41). In this study the NAC polypeptide wasreadily identified as the prominent IPTG-inducible band inwhole-cell profiles of strain EB2567 harboring pEC205.1 dis-played on Coomassie-stained denaturing polyacrylamide gels.The prominence of this band afforded a means of monitoringthe unusual buffer-dependent partitioning of the overex-
FIG. 7. DNase I footprinting of NAC on pKAU22D1 carrying the ureD promoter region (A) or pKC9 carrying the putAP promoter region (B). Plasmid was digestedwith HindIII (pKAU22D1) or BamHI (pKC9), 39 end labeled, and redigested with PvuII (pKAU22D1) or EcoRI (pKC9). The resulting DNA (0.16 pmol) was incubatedwith buffer 6 (lanes 2 and 7) or NAC (lanes 3, 4, 5, and 6 [1, 3, 9, and 27 pmol, respectively]) in the absence of RNA polymerase, as described in Materials and Methods.Protein-DNA complexes were treated with DNase I and analyzed as described in the legend to Fig. 4. The previously determined nucleotide sequence of the promoterregion (for panel A, reference 22; for panel B, the complement of the sequence reported in reference 9) shown to the left of each autoradiogram was aligned to thefootprinted regions by using the A1G sequence ladder (lane 1).
3552 GOSS AND BENDER J. BACTERIOL.

pressed NAC protein between soluble and particulate fractionsof disrupted cell suspensions. Analysis of the overexpressedNAC suggested that it exists as a polydisperse aggregate inlow-ionic-strength solutions and as a soluble homodimer insolutions with elevated salt concentrations. Presently, it is un-clear whether NAC’s tendency to aggregate in low-ionic-strength solutions is an artifact of NAC overexpression or anintrinsic property of the NAC polypeptide. Reducing the rateof NAC synthesis by lowering both the culturing temperatureto 278C and the concentration of IPTG to 1 mM apparently ledto the formation of smaller NAC aggregates in a low-salt so-lution, the bulk of which could be sedimented only when cen-trifugation was extended to 90 min. Despite our lack of under-standing as to its cause, the unusual solubility properties of theoverexpressed NAC have facilitated the purification of NAC,allowing an assessment of NAC’s role in the transcription ofNAC-sensitive promoters to be made.The in vitro transcription studies reported here demonstrate
that NAC regulates the transcription from the hut(P) region ina manner similar to that previously reported for CAP (30,32–34). Moreover, the activation of hutUp by NAC occursregardless of the linear or supercoiled conformation of thehut(P) template. These observations verify the notion drawnfrom in vivo studies that NAC is both necessary and sufficientto activate the transcription of the hutUH operon by s70-RNApolymerase in response to nitrogen limitation.In the course of analyzing the ability of NAC to activate
hutUp carried on a supercoiled template, it appeared that boththe basal and NAC-activated levels of transcription fromhutUp were influenced by the template’s conformation. Theinfluence of template conformation on the NAC-activatedlevel of hutUp transcription was largely eliminated when po-tassium glutamate was substituted for potassium chloride inthe in vitro transcription assay buffer. Curiously, the basal levelof hutUp transcription was marginally, if at all, influenced bythis substitution. Mobility shift assays indicated that the bind-ing of NAC to linear DNA fragments was relatively insensitiveto the substitution of potassium glutamate for potassium chlo-ride and that NAC bound equally well to hut(P) carried onsupercoiled and linear plasmids (18). Moreover, the footprintsof NAC or NAC plus RNA polymerase at hut(P) were alsounaffected by this substitution (18). Studies by other groupshave suggested that the substitution of potassium glutamatefor potassium chloride alters the effective concentrations ofmagnesium and potassium ions (23, 45). The mechanism bywhich the substitution of potassium glutamate for potassiumchloride influences the interactions of NAC, RNA polymerase,and hut(P) under these in vitro conditions is presently un-known. As the influence of glutamate on transcription activa-tion of hutUp by NAC is apparent only with linear templates,this curiosity is unlikely to be physiologically significant.Previous in vitro studies on the regulation of transcription
from the hut(P) region have consistently identified transcriptsoriginating from Pc (32, 34). In this analysis, both basal andrepressed levels of transcription from Pc were observed withthe 250-bp hut(P) template derived from pOS1; however, no Pctranscripts could be detected with pAM1202 templates. More-
over, RNA polymerase was found to inconsistently protect Pcfrom DNase I digestion, whereas RNA polymerase consis-tently footprinted at hutUp in the presence of NAC. Theseobservations suggest that RNA polymerase does not readilybind at Pc, especially in the presence of competing vectorsequences. Taken together with the sluggish kinetics of opencomplex formation (33), Pc appears to be an intrinsically weakpromoter in vitro.The LD113 and LD113* constructs allowed a preliminary
assessment of the spacing requirements for NAC’s transcrip-tional activation of hutUp to be made. When the NAC bindingsite was moved 10 bp closer to the transcription start site,maintaining the relative phasing of bound NAC and RNApolymerase with respect to the axis of the DNA helix, NACbecame less effective in stimulating transcription from hutUpcarried on the linear template in vitro. Although this result wasconsistently observed upon repeat analysis, the magnitude ofthe difference varied with the preparation of RNA polymerase.In contrast, preliminary analysis of plasmid-borne operon fu-sions in vivo suggests that the 10-bp deletion carried by LD113*only partially interferes with the activation of transcriptionfrom hutUp by NAC (18). Thus, it is possible that the spacingrequirements for NAC’s activation of hutUp observed in vitromay differ from those required in vivo.Previous studies concerning the interaction of CAP with the
hut(P) region identified two CAP binding sites centered 281.5and 241.5 with respect to the hutUp transcription start site(34). The presence of a NAC binding site between and partiallyoverlapping these CAP sites raises the question of whetherthese two activators might act synergistically to stimulate tran-scription from hutUp under certain conditions. Simultaneousoccupancy of NAC and CAP sites at hut(P) is unlikely to beachieved under normal growth conditions; however, whenNAC expression was artificially induced during growth underconditions of carbon/energy limitation, the level of expressionfrom hutUp was less than that expected for synergistic activa-tion by these two activators (19).The conditions which allow NAC to substantially activate
transcription from putPp1 and ureDp carried on linear tem-plates in vitro have not yet been identified. Moreover, DNaseI protection studies indicated that the stable binding of RNApolymerase to these promoters was not detectably stimulatedby NAC. Like hutUp, putPp1 appears to be a s70-dependentpromoter, which is repressed in the absence of substrate andcan be independently activated in response to carbon or nitro-gen limitation in vivo (9). Moreover, the transcription startsites for both hutUp and putPp1 are unaffected by the level oftranscription or the nature of the activator (9, 30, 34), and bothpromoters have associated NAC binding sites which are cen-tered 64 bp upstream of the transcription start sites. Despitethese similarities, differences in the number and locations ofCAP sites associated with these promoters (9, 34) and theinteraction of these promoters with RNA polymerase in thepresence of NAC in vitro suggest that the activation of putPp1differs from that of hutUp in vivo. In contrast to hutUp andputPp1, the K. aerogenes ure operon does not appear to beregulated by either carbon or substrate (urea) availability.
FIG. 8. Alignment of regions footprinted by NAC at the hutUp, ureDp, and putPp1 regions. The sequences of the nontemplate strands for the respective promotersare indicated. The approximate extents of the regions footprinted by NAC are underlined. Identical nucleotides within these regions are given in bold type, and thosenucleotides symmetrically positioned are boxed.
VOL. 177, 1995 KLEBSIELLA AEROGENES NAC PROTEIN 3553

Moreover, the nature and location of ureDp have not yet beenclearly ascertained. Studies on the K. pneumoniae ure operon(10) and fusions of the K. aerogenes ureD control region to thelacZ reporter gene (18) suggest that the region used in thisstudy contains all the promoter elements essential for nitrogenregulation. We are presently attempting to define the condi-tions which allow NAC to fully activate the ureDp and putPp1promoters in vitro.Comparison of the extents of protection from DNase I di-
gestion afforded by moderate amounts of NAC at the hut(P),putAP, and ure control regions suggests that there may besignificant differences in the affinities of NAC for these bindingsites. The in vivo studies reported by Schwacha and Bender(42) as well as preliminary qualitative assessments obtainedfrom competition mobility shift experiments (18) support thenotion that the affinity of NAC for the ure control region isgreater than that for the hut(P) and putAP control regions. Wehope to verify this notion by quantitatively assessing the affin-ities of NAC for various DNA binding sites.Many LysR family transcriptional regulators bind to their
cognate promoters either in a nonextended form, bound to apromoter-distal recognition site, or in an extended form, in-teracting with both recognition and promoter-proximal activa-tion sites (reviewed in reference 40). The extent and positionof the footprints of NAC on the hut(P), putAP, and ureDcontrol regions resemble those of other LysR family regulatorsbound to their recognition sites in the nonextended form. Incontrast, the extent and complexity of the footprint of NAC atnacp (14) are more representative of the extended LysR bind-ing interaction. Although the footprint of NAC on hut(P) issignificantly different from that on nacp, it is possible that theweak DNase I-hypersensitive site near nucleotide 146 of hut(P)induced by NAC binding (Fig. 4) is due to a minority of hut(P)fragments bound by NAC in the extended form, the footprintat the activation site being masked by the cleavage patterncorresponding to the majority of promoter fragments bound byNAC in the nonextended form. The notion that the binding ofNAC to hut(P) might occur at sites in addition to the recog-nition site is suggested by the occurrence of multiply shiftedbands in mobility shift assays using excess NAC and DNAtargets carrying both recognition and activation regions ofhutUp (18). Curiously, such multiply shifted bands are not asevident in similar mobility shift experiments with the putAPand ureD control regions (18). The apparent mutual stimula-tion of binding of NAC and RNA polymerase to hut(P) issimilar to that recently reported for the binding of OxyR atoxySp (20).DNA binding studies verified that NAC binds to the hut(P),
putAP, and ureD control regions, as expected of a factor whichdirectly regulates the transcription of these promoters. In eachcase the binding of NAC to these control regions protected a25- to 30-bp region from DNase I digestion. One of manypossible alignments of these footprinted regions reveals thecommon sequence 59-ATA-N6-TNGTAT-39 (Fig. 8), whichcontains the motif 59-T-N11-A-39 proposed to be the core rec-ognition sequence common to LysR family member bindingsites (17). The sequence 59-ATA-N9-TAT-39 is an attractivecandidate for the minimal NAC-binding consensus sequence,as it displays the hyphenated dyad symmetry commonly asso-ciated with sites bound by homodimeric DNA binding proteins(15). This symmetry consideration limits the minimal NAC-binding consensus sequence to only six common nucleotides;however, it is possible that recognition elements other thannucleotide sequence per se may be important in conferringspecificity on the interaction of NAC with DNA in vivo. Curi-ously, the region of nacp footprinted by NAC (14) is distin-
guished from those identified at hutUp, ureDp, and putPp1, notonly in that it is larger and more complex but also in that itlacks a perfect match to the minimal NAC-binding consensussequence.A computer-assisted search of reported enteric DNA se-
quences identified a large number of occurrences of the min-imal NAC-binding consensus sequence. Of these, three ap-peared to be of some significance: one within the promoterregion of the E. coli codBA operon coding for cytosine utiliza-tion (11) (locus ECCODAB, accession number X63656), asecond within the E. coli gdhA promoter region (44) (locusECGDHAK, accession number K02499), and a third withinthe E. coli asnAC control region coding for asparagine synthe-sis (29) (locus ECASNA, accession number V00263). The E.coli cytosine deaminase operon is activated in response tonitrogen limitation (2), whereas both glutamate dehydroge-nase and the ammonium-dependent asparagine synthetase ofK. aerogenes are repressed in response to nitrogen limitation(8, 38). Conflicting reports can be found concerning the influ-ence of nitrogen limitation on the expression of glutamatedehydrogenase in E. coli (39, 43). Preliminary studies on thecod control regions cloned from both E. coli and K. aerogenesindicate the presence of associated NAC binding sites (28).Previous in vivo studies have demonstrated that NAC is both
necessary (4, 25) and sufficient (42) for the repression of gdhApand gltBp and the autorepression of nacp in K. aerogenes. Fenget al. (14) have demonstrated that NAC plays a direct role inthe autorepression of nacp in vitro. We are presently investi-gating the repression of the K. aerogenes gdhA and gltBD oper-ons by NAC.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant GM 47156for the National Institutes of Health to R.A.B.We are grateful to A. Macaluso for constructing pAM1202 and to
W. B. Muse and B. K. Janes for allowing us to discuss their unpub-lished results.
REFERENCES
1. Alvarez-Morales, A., R. Dixon, and M. Merrick. 1984. Positive and negativecontrol of the glnA ntrBC regulon in Klebsiella pneumoniae. EMBO J. 3:501–507.
2. Andersen, L., M. Kilstrup, and J. Neuhard. 1989. Pyrimidine, purine andnitrogen control of cytosine deaminase synthesis in Escherichia coli K12.Involvement of the glnLG and purR genes in the regulation of codA expres-sion. Arch. Microbiol. 152:115–118.
3. Bencini, D. A., G. A. O’Donovan, and J. R. Wild. 1984. Rapid chemicaldegradation sequencing. BioTechniques 2:4–5.
4. Bender, R. A. 1991. The role of the NAC protein in the nitrogen regulationof Klebsiella aerogenes. Mol. Microbiol. 5:2575–2580.
5. Bender, R. A., P. M. Snyder, R. Bueno, M. Quinto, and B. Magasanik. 1983.Nitrogen regulation system of Klebsiella aerogenes: the nac gene. J. Bacteriol.156:444–446.
6. Best, E. A., and R. A. Bender. 1990. Cloning of the Klebsiella aerogenes nacgene, which encodes a factor required for nitrogen regulation of the histidineutilization (hut) operons in Salmonella typhimurium. J. Bacteriol. 172:7043–7048.
7. Boylan, S. A., L. J. Eades, K. A. Janssen, M. I. Lomax, and R. A. Bender.1984. A restriction enzyme cleavage map of the histidine utilization (hut)genes of Klebsiella aerogenes and deletions lacking regions of hut DNA. Mol.Gen. Genet. 193:92–98.
8. Brenchley, J. E., M. J. Prival, and B. Magasanik. 1973. Regulation of thesynthesis of enzymes responsible for glutamate formation in Klebsiella aero-genes. J. Biol. Chem. 248:6122–6128.
9. Chen, L.-M., and S. Maloy. 1991. Regulation of proline utilization in entericbacteria: cloning and characterization of the Klebsiella put control region. J.Bacteriol. 173:783–790.
10. Collins, C. M., D. M. Gutman, and H. Laman. 1993. Identification of anitrogen-regulated promoter controlling expression of Klebsiella pneumoniaeurease genes. Mol. Microbiol. 8:187–198.
11. Danielsen, S., M. Kilstrup, K. Barilla, B. Jochimsen, and J. Neuhard. 1992.Characterization of the Escherichia coli codAB operon encoding cytosine
3554 GOSS AND BENDER J. BACTERIOL.

permease and cytosine deaminase. Mol. Microbiol. 6:1335–1344.12. Elliott, T., and E. P. Geiduschek. 1984. Defining a bacteriophage T4 late
promoter: absence of a ‘‘235’’ region. Cell 36:211–219.13. Feng, J., M. R. Atkinson, W. McCleary, J. B. Stock, B. L. Wanner, and A. J.
Ninfa. 1992. Role of phosphorylated metabolic intermediates in the regula-tion of glutamine synthetase synthesis in Escherichia coli. J. Bacteriol. 174:6061–6070.
14. Feng, J.-L., T. J. Goss, R. A. Bender, and A. J. Ninfa. Characterization of thenac promoter of Klebsiella aerogenes. Submitted for publication.
15. Gicquel-Sanzey, B., and P. Cossart. 1982. Homologies between differentprocaryotic DNA-binding regulatory proteins and between their sites ofaction. EMBO J. 1:591–595.
16. Giulian, G. 1990. Isoelectric focusing of mouse ascites fluid and myosin inthe SE 600 vertical slab gel unit, p. 128–130. In Hoefer electrophoresiscatalog and exercises, 1990–1991. Hoefer Scientific Instruments, San Fran-cisco.
17. Goethals, K., M. Van Montagu, and M. Holsters. 1992. Conserved motifs ina divergent nod box of Azorhizobium caulinodans ORS571 reveal a commonstructure in promoters regulated by LysR-type proteins. Proc. Natl. Acad.Sci. USA 89:1646–1650.
18. Goss, T. J., and R. A. Bender. Unpublished data.19. Janes, B. K., and R. A. Bender. Unpublished data.20. Kullik, I., M. B. Toledano, L. A. Tartaglia, and G. Storz. 1995. Mutational
analysis of the redox-sensitive transcriptional regulator OxyR: regions im-portant for oxidation and transcriptional activation. J. Bacteriol. 177:1275–1284.
21. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4. Nature (London) 227:680–685.
22. Lee, M. H., S. B. Mulrooney, M. J. Renner, Y. Markowicz, and R. P. Haus-inger. 1992. Klebsiella aerogenes urease gene cluster: sequence of ureD anddemonstration that four accessory genes (ureD, ureE, ureF, and ureG) areinvolved in nickel metallocenter biosynthesis. J. Bacteriol. 174:4324–4330.
23. Leirmo, S., C. Harrison, D. S. Cayley, R. R. Burgess, and M. T. Record, Jr.1987. Replacement of potassium chloride by potassium glutamate dramati-cally enhances protein-DNA interactions in vitro. Biochemistry 26:2095–2101.
24. Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Proteinmeasurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275.
25. Macaluso, A., E. A. Best, and R. A. Bender. 1990. Role of the nac geneproduct in the nitrogen regulation of some operons of Klebsiella aerogenes. J.Bacteriol. 172:7249–7255.
26. Magasanik, B. 1982. Genetic control of nitrogen assimilation in bacteria.Annu. Rev. Genet. 16:135–168.
27. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: alaboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor,N.Y.
28. Muse, W. B., M. Kilstrup, S. Danielsen, J. Neuhard, and R. A. Bender.Unpublished data.
29. Nakamura, M., M. Yamada, Y. Hirota, K. Sugimoto, A. Oka, and M. Taka-nami. 1981. Nucleotide sequence of the asnA gene coding for asparagine
synthetase of E. coli K-12. Nucleic Acids Res. 9:4669–4676.30. Nieuwkoop, A. J., S. A. Baldauf, M. E. S. Hudspeth, and R. A. Bender. 1988.
Bidirectional promoter in the hut(P) region of the histidine utilization (hut)operons from Klebsiella aerogenes. J. Bacteriol. 170:2240–2246.
31. Nieuwkoop, A. J., and R. A. Bender. 1988. RNA polymerase as a repressor oftranscription in the hut(P) region of mutant Klebsiella aerogenes histidineutilization operons. J. Bacteriol. 170:4986–4990.
32. Nieuwkoop, A. J., S. A. Boylan, and R. A. Bender. 1984. Regulation of hutUHoperon expression by the catabolite gene activator protein-cyclic AMP com-plex in Klebsiella aerogenes. J. Bacteriol. 159:934–939.
33. Osuna, R., S. A. Boylan, and R. A. Bender. 1991. In vitro transcription of thehistidine utilization (hutUH) operon from Klebsiella aerogenes. J. Bacteriol.173:116–123.
34. Osuna, R., B. K. Janes, and R. A. Bender. 1994. Roles of catabolite activatorprotein sites centered at 281.5 and 241.5 in the activation of the Klebsiellaaerogenes histidine utilization operon hutUH. J. Bacteriol. 176:5513–5524.
35. Osuna, R., A. Schwacha, and R. A. Bender. 1994. Identification of the hutUHoperator (hutUo) from Klebsiella aerogenes by DNA deletion analysis. J.Bacteriol. 176:5525–5529.
36. Porter, S. C., A. K. North, A. B. Wedel, and S. Kustu. 1993. Oligomerizationof NTRC at the glnA enhancer is required for transcriptional activation.Genes Dev. 7:2258–2273.
37. Ray, L., F. Claverie-Martin, P. Weglenski, and B. Magasanik. 1990. Role ofthe promoter in activation of transcription by nitrogen regulator I phosphatein Escherichia coli. J. Bacteriol. 172:818–823.
38. Reitzer, L. J., and B. Magasanik. 1982. Asparagine synthetases of Klebsiellaaerogenes: properties and regulation of synthesis. J. Bacteriol. 151:1299–1313.
39. Riba, L., B. Becerril, L. Servin-Gonzales, F. Valle, and F. Bolivar. 1988.Identification of a functional promoter for the Escherichia coli gdhA geneand its regulation. Gene 71:233–246.
40. Schell, M. A. 1993. Molecular biology of the LysR family of transcriptionalregulators. Annu. Rev. Microbiol. 47:597–626.
41. Schwacha, A., and R. A. Bender. 1993. The nac (nitrogen assimilation con-trol) gene from Klebsiella aerogenes. J. Bacteriol. 175:2107–2115.
42. Schwacha, A., and R. A. Bender. 1993. The product of the Klebsiella aero-genes nac (nitrogen assimilation control) gene is sufficient for activation ofthe hut operons and repression of the gdh operon. J. Bacteriol. 175:2116–2124.
43. Streicher, S. L., A. B. Deleo, and B. Magasanik. 1976. Regulation of enzymeformation in Klebsiella aerogenes by episomal glutamine synthetase of Esch-erichia coli. J. Bacteriol. 127:184–192.
44. Valle, F., E. Sanvicente, R. Seeburg, A. Covarrubias, R. L. Rodriguez, and F.Bolivar. 1983. Nucleotide sequence of the promoter and amino-terminalcoding region of the glutamate dehydrogenase structural gene of Escherichiacoli. Gene 23:199–209.
45. Zou, L., and J. P. Richardson. 1991. Enhancement of transcription termi-nation factor rho activity with potassium glutamate. J. Biol. Chem. 266:10201–10209.
VOL. 177, 1995 KLEBSIELLA AEROGENES NAC PROTEIN 3555