universidade federal do rio de janeiro...
TRANSCRIPT

UNIVERSIDADE FEDERAL DO RIO DE JANEIRO
INSTITUTO DE QUÍMICA
DEPARTAMENTO DE QUÍMICA INORGÂNICA
PREPARAÇÃO DE ANODOS PARA PILHAS DE CÉLULAS A COMBUSTÍVEL
USANDO CATALISADORES BIMETÁLICOS DE PLATINA
Monografia do projeto final de curso
Sidnei Valente de Paiva
Orientadoras: Profa. Dra. Ana Maria Rocco
Profa. Dra. Maria Iaponeide Fernandes Macêdo
Rio de Janeiro, 30 de outubro de 2006

ii
Esse Trabalho de Conclusão de Curso foi realizado com o apoio financeiro da
ANP - Agência Nacional do Petróleo, Gás Natural e Biocombustíveis.

iii
RESUMO
TÍTULO: Preparação de anodos para pilhas de células a combustível
usando catalisadores bimetálicos de platina
ALUNO: Sidnei Valente de Paiva
ORIENTADORAS: Profa. Dra. Ana Maria Rocco
Dra. Maria Iaponeide Fernandes Macêdo
Neste trabalho foi realizado um estudo exploratório da metodologia de
obtenção e caracterização de catalisadores bimetálicos de Pt-Co suportados
em carbono. Utilizou-se a técnica de Redução à Temperatura Programada
(TPR) para reduzir os íons metálicos adsorvidos no suporte e propiciar a
formação da liga, uma vez que trabalhos na literatura reportam que a formação
da mesma é essencial para a eficiência do processo de eletrocatálise e para
evitar o envenenamento do catalisador por CO em células a combustível.
Os catalisadores bimetálicos foram obtidos e caracterizados morfológica,
estrutural e eletrocataliticamente. Para tanto, foram preparados
eletrocatalisadores com diferentes concentrações de platina e cobalto variando-
se a temperatura e as condições de impregnação dos íons. Nos catalisadores
bimetálicos, a formação da liga foi comprovada através de pequenos
deslocamentos do pico de difração referente ao plano (111) no difratograma da
amostra com Pt. A ordem de adição dos precursores metálicos, assim como a
temperatura de dispersão do Co influencia na formação da liga. Os testes
eletrocatalíticos por voltametria cíclica em solução de H2SO4 com os
catalisadores preparados por adição simultânea mostraram maior atividade
para as amostras com 10% de cada metal. As áreas eletroativas para estes
sistemas foram menores que para o sistema Pt/C Electrochem e Pt/C
preparados no laboratório pelo método de redução com etileno glicol, em
função das partículas maiores.
Concluiu-se que um controle mais fino das condições de preparo são
necessárias para diminuir o tamanho das partículas, assim como a redução
prévia da Pt, via redução por etileno glicol, seguida da dispersão controlada do
segundo metal e redução por TPR para formação da liga.

iv
AGRADECIMENTOS
A orientadora Profa Ana Maria Rocco pela orientação científica ao longo
de tanto tempo trabalhando juntos.
A orientadora Profa. Maria Iaponeide Fernandes Macêdo pela orientação
ao longo de todas as etapas da monografia.
Aos meus pais e familiares pela educação, amor e incentivo aos estudos.
Aos amigos que conviveram ao meu lado ao longo de todo o trabalho no
laboratório, pela confiança e por terem tornado o dia-a-dia mais prazeroso.
A minha noiva, Rita de Cássia, por toda a ajuda, carinho e incentivo que
foram absolutamente indispensáveis para a conclusão da monografia.
A Márcia da Metalurgia/COPPE-RJ pelas imagens de microscopia
eletrônica de varredura.
Agradeço a ANP pela bolsa concedida para elaboração do projeto de final
de curso.
Aos NUCAT pelas análises de DRX.
Aos funcionários e professores do Instituto de Química da UFRJ, pelo
apoio e aprendizagem adquiridos durante o curso de química.
Ao Coração de Jesus por sempre pulsar de forma especial para mim,
pelas graças de cada dia e especialmente pela conclusão de mais esta etapa na
minha vida.
E, desde já, a banca examinadora por aceitar o convite, pelas críticas e
quaisquer contribuições que possam prestar.

v
ÍNDICE GERAL
1. Introdução ................................................................................................................... 01
1.1. Introdução geral ...................................................................................................... 01
1.2. Células a combustível. ............................................................................................ 04
1.3. Células a combustível do tipo PEM. ....................................................................... 06
1.4. Eletrodos de difusão gasosa. ................................................................................. 10
1.5. Empilhamento da célula. ........................................................................................ 11
1.6. Eficiência da célula a combustível. ......................................................................... 12
1.7. Célula a combustível a menbrana polimérica ........................................................ 14
1.8. Membrana polimérica condutora protônica ............................................................ 15
1.9. Eletrocatálise da reação de redução de oxigênio .................................................. 15
1.10. Eletrocatálise da reação de oxidação de hidrogênio............................................. 18
1.11. Envenenamento do catalisador por CO ................................................................ 19
1.12. Métodos de preparação de eletrocatalisadores .................................................... 19
1.12.1. Método do ácido fórmico .................................................................................... 20
1.12.2. Método de Bönnemann ...................................................................................... 21
1.12.3. Método da deposição espontânea ..................................................................... 22
1.12.4. Método de decomposição de precursores moleculares..................................... 24
1.12.5. Método de redução por álcool ............................................................................ 25
2. Objetivo ....................................................................................................................... 27
3. Materiais e Métodos ................................................................................................... 27
3.1. Precursores .............................................................................................................. 27
3.2. Procedimento experimental. .................................................................................... 27
3.2.1. Preparação da suspensão do carbono grafite e carbono Vulcan XC-72R. ......... 27
3.2.2. Preparação dos sistemas de Pt(NH3)4Cl2 nos suportes Cg e Cv. ......................... 28
3.2.2.1. Preparo da solução aquosa de Pt(NH3)4Cl2 ...................................................... 28
3.2.2.2. Adição da solução de Pt(NH3)4Cl2 à suspensão de Cv ..................................... 28
3.2.2.3. Adição da solução de Pt(NH3)4Cl2 à suspensão de Cg ..................................... 29
3.2.2.4. Redução a Temperatura Programada (TPR) .................................................... 29
3.2.3. Preparação do sistema Co(NO3)2.6H2O em Cv. ................................................... 29
3.2.3.1. Preparação do sistema Co(NO3)2.6H2O em Cv. ................................................ 29

vi
3.2.3.2. Adição da solução aquosa de Co(NO3)2.6H2O ao Cv ....................................... 29
3.2.3.3. Redução química do Co+2 ................................................................................. 30
3.2.3.4. Redução completa do Cobalto .......................................................................... 30
3.2.4. Sistema com adição simultânea dos sais de platina e cobalto ............................ 31
3.2.5. Adição e redução do segundo metal .................................................................... 32
3.2.6. Formação da liga Pt-Co ........................................................................................ 33
3.2.7. Método de redução empregando-se ácido fórmico .............................................. 34
3.3. Caracterização espectroscópica, estrutural e morfológica ..................................... 38
3.3.1. Espectoscopia Vibracional no Infravermelho por Transformada de Fourier ........ 38
3.3.2. Difração de Raios-X .............................................................................................. 38
3.3.3. Microscopia Eletrônica de Varredura ................................................................... 38
3.3.4. Redução a Temperatura Programada .................................................................. 38
3.3.5. Área Superficial..................................................................................................... 39
4. Resultados e Discussão. ............................................................................................ 39
4.1. Catalisadores Suportados em carbono grafite, Cg .................................................. 39
4.1.1. Caracterização por espectroscopia vibracional no infravermelho ....................... 39
4.1.2. Caracterização por difração de raios-X ................................................................ 40
4.1.3. Análise Termogravimétrica ................................................................................... 44
4.2. Catalisadores Suportados em carbono Vulcan XC-72R, Cv ................................... 45
4.2.1. Caracterização do Cv por espectroscopia vibracional no infravermelho ............. 45
4.2.2. Caracterização do Cv por difração de raios-X ...................................................... 45
4.2.3. Método da impregnação: Sistema Pt/Cv .............................................................. 46
4.2.3.1. Redução a Temperatura Programada ............................................................... 46
4.2.3.2. Difratogramas de Raios-X ................................................................................. 50
4.2.3.3. Microscopia Eletrônica de Varredura ................................................................ 55
4.2.4. Método da impregnação: Sistema Co/Cv ............................................................. 56
4.2.4.1. Redução a Temperatura Programada ............................................................... 56
4.2.4.2. Difratogramas de Raio-X ................................................................................... 61
4.2.4.3. Microscopia Eletrônica de Varredura ................................................................ 62
4.2.5. Método da impregnação: Sistemas PtCo/Cv ........................................................ 63
4.2.5.1. Redução a Temperatura Programada ............................................................... 63
4.2.5.2. Difratogramas de Raio-X ................................................................................... 65
4.2.5.3. Teste de eletrocatálise por voltametria cíclica................................................... 71

vii
4.3. Método do ácido fórmico: sistema Pt/Cv .................................................................. 76
4.3.1. Difratogramas de Raio-X ...................................................................................... 77
5. Conclusões ................................................................................................................. 80
6. Referências ................................................................................................................. 82

viii
ÍNDICE DE FIGURAS
Figura 1 - Desenho esquemático de uma célula a combustível do tipo PEM. .............. 7
Figura 2 - Configurações de veículos com motores de combustão interna e
veículos utilizando células a combustível. ...................................................................... 9
Figura 3 - Representação esquemática de um MEA mostrando em detalhes a
interface trifásica gás-líquido-sólido. ............................................................................ 11
Figura 4: Montagem de uma célula a combustível. ..................................................... 12
Figura 5- Dependência do rendimento teórico com a temperatura da conversão
eletroquímica do hidrogênio, do metano e no ciclo de Carnot. .................................... 13
Figura 6: Fluxograma do procedimento experimental ................................................ 36
Figura 7- Fluxograma do procedimento experimental do preparo das amostras
com adição inicial de Platina ........................................................................................ 37
Figura 8 - Espectro vibracional no infravermelho dos eletrocatalisadores
Pt(10%)Co(10%)/Cg, Pt(10%)/Cg e seus precursores. ................................................. 40
Figura 9 - Difratogramas de raios-X das amostras (a) Pt/Cg e (b) CoPt/Cg. ............... 44
Figura 10 - Curvas de TGA das amostras Pt+2/Cg, Pt/Cg, Co+2Pt/Cg e CoPt/Cg. ........ 45
Figura 11 - Espectro de FTIR do carbono Vulcan XC-72R. ........................................ 46
Figura 12 - Difratograma de raios X do Cv .................................................................. 47
Figura 13 - Curvas de TPR da amostra Pt+2(10%)/Cv_25ºC. ...................................... 48
Figura 14 - TPR realizado na amostra Pt+2(10%)/Cv_50ºC ......................................... 48
Figura 15 - TPR realizado na amostra Pt+2(15%)/Cv_50ºC. ........................................ 50
Figura 16 - TPR realizado na amostra Pt+2(20%)/Cv_50ºC. ........................................ 50
Figura 17- DRX da amostra Pt(10%)/Cv_50ºC ........................................................... 51
Figura 18- Difratogramas das amostras preparadas na temperatura de adição ao
carbono Vulcan de 50ºC em comparação com a ampstra comercial de
catalisador de platina em carbono ................................................................................ 53
Figura 19- Difratograma de raios-X com 2θ variando de 38 a 42o mostrando o
pico referente ao plano (111) de todas as amostras de Pt/Cv (platina
impregnadas em carbono vulcan) estudadas. ............................................................. 53

ix
Figura 20 - Detalhe da região entre 32 e 42 graus dos difratogramas de raios-x
das amostras de platina 10% em carbono vulcan preparadas em diferentes
temperaturas. ................................................................................................................ 55
Figura 21 - Imagem de Microscopia Eletrônica de Varredura da amostra
Pt(10%)/Cv_50ºC com um ampliação de 2000 vezes. ................................................ 56
Figura 22- Microscopia Eletrônica de Varredura da amostra Pt(10%)/Cv_50ºC
com um zoom de 20000 vezes. .................................................................................... 56
Figura 23- EDS da superfície da amostra Pt(10%)/Cv_50ºC. ..................................... 57
Figura 24- TPR realizado na amostra Co+2(10%)/Cv_25ºC......................................... 58
Figura 25-TPR realizado na amostra Co+2 (15%)/Cv_25ºC......................................... 58
Figura 26- TPR realizado na amostra Co+2(20%)/Cv_25ºC ........................................ 59
Figura 27- TPR realizado na amostra Co+2(10%)/Cv_50ºC. ........................................ 61
Figura 28- TPR realizado na amostra Co+2(15%)/Cv_50ºC......................................... 61
Figura 29- TPR realizado na amostra Co+2(20%)/Cv_ 50ºC........................................ 62
Figura 30 - Microscopia Eletrônica de Varredura da amostra Co(20%)/Cv_50ºC
com um zoom de 5000 vezes. ...................................................................................... 63
Figura 31- MEV da amostra Co(20%)/Cv_50ºC com um zoom de 20000 vezes. ....... 63
Figura 32- TPR realizado na amostra Pt+2(10%)Co+2(10%)/Cv_50ºC. ........................ 65
Figura 33- TPR realizado na amostra Pt+2(10%)_s_Co+2(10%)/Cv_50ºC. .................. 65
Figura 34- Difratogramas das amostras de Pt e Co com diferentes ordens de
adição dos precursores dos metais sobre carbono Vulcan. ........................................ 66
Figura 35- Gráfico da variação do parâmetro de rede em relação a concentração
dos precursores metálicos nas amostras de Pt e da liga preparada com adição
inicial de Pt, ambos preparados em carbono Vulcan e nas temperaturas de 25 e
50ºC. ............................................................................................................................. 68
Figura 36- Difratogramas de raios-X para as amostras de eletrocatalisadores
dispersos a 50oC, variando-se as concentrações dos metais de 10, 15 e 20%
(m/m) de Pt e Co sobre o carbono Vulcan. .................................................................. 69
Figura 37- Decomposição em funções primitivas Lorentzianas do pico referente
ao plano (111) no difratograma de raios-X das amostras bimetálicas Pt-Co em
três diferentes composições. ........................................................................................ 71
Figura 38 - Voltamograma Cíclico a 10 mV/s de -200 a 1000 mV versus ECS
para o eletrodo de Pt policristalino. Foi utilizado um eletrodo rotatório de Pt como

x
eletrodo de trabalho, como contra-eletrodo uma lâmina de Pt (1 cm2) e
calomelano saturado como o eletrodo de referência. .................................................. 72
Figura 39 - Voltametrias cíclicas a 10 mV/s de -200 a 1000 mV versus ECS para
filmes das amostras de PtCo/Cv com concentrações iguais dos dois metais
variando de 10, 15 e 20 % (m/m) sobre Cv. ................................................................. 74
Figura 40 - Voltamogramas cíclicos a 10mV/s de -200 a 1000 mV versus ECS
para 20% Pt/ carbono Vulcan XC-72R (Electrochem) (a), e 20% Pt/ carbono
Vulcan XC-72R obtido neste trabalho. Eletrodo de trabalho e contra-eletrodo de
Pt. .................................................................................................................................. 76
Figura 41 - Difratogramas das amostras de Platina (10%) em carbono vulcan
preparadas pelas técnicas da impregnação e método do ácido fórmico, com suas
respectivas variações metodológicas. .......................................................................... 78
Figura 42 - Comparação, em detalhe, do pico referente ao plano (111) da platina
em amostras preparadas pela técnica da impregnação e do ácido fórmico
respectivamente. ........................................................................................................... 79

xi
ÍNDICE DE TABELAS
Tabela 1 - Código das amostras precursoras dos catalisadores Pt/Cv. ...................... 29
Tabela 2 - Código das amostras precursoras das espécies Co/Cv............................. 31
Tabela 3 - Código das amostras das espécies Co/Cv. ................................................ 31
Tabela 4- Código das amostras precursoras dos catalisadores bimetálicos
Pt-s-Co/Cv. .................................................................................................................... 32
Tabela 5- Código das amostras precursoras dos catalisadores bimetálicos Pt-
Co/Cv ............................................................................................................................. 33
Tabela 6 - Código das amostras bimetálicas que receberam tratamento para
formação da liga. .......................................................................................................... 34
Tabela 7- Códigos das amostras preparadas pelo método do ácido fórmico. ............ 35
Tabela 8 - Tamanho médio de partículas e parâmetros de rede das amostras do
tipo Pt/Cv preparadas nas temperaturas de 25 e 50 ºC. .............................................. 54
Tabela 9 - Tamanho médio de partícula e o parâmetro de rede das amostras
preparadas pelo método da impregnação em diferentes temperaturas (25, 50 ºC)
com 10% (m/m) de platina em relação ao suporte. ...................................................... 55
Tabela 10- Tamanho de partícula e parâmetros de rede das amostras de Pt e Co
preparadas com ordem de adição de reagentes alterados. ......................................... 67
Tabela 11- Tamanhos de partícula e os parâmetros de rede para as amostras de
eletrocatalisadores com concentrações diferentes de Pt e Co. ................................... 72
Tabela 12- Área ativa eletrocatalíca em cm2 e tamanhos de partícula para as
amostras de eletrocatalisadores com mesma concentração de Pt e Co. .................... 77
Tabela 13- Tamanho médio de partícula e o parâmetro de rede das amostras
preparadas pelo método da redução com ácido fórmico. ............................................ 80

1
1 - Introdução
1.1 - Introdução Geral
De uma forma ou de outra, todas as atividades humanas sobre a Terra
provocam alterações ao meio ambiente. Dentre estas, várias alterações se
originam da geração, manuseio e do uso da energia. Em 1998, segundo as
Nações Unidas1, o consumo mundial de energia primária proveniente de fontes
não renováveis (petróleo, carvão, gás natural e nuclear) correspondeu a
aproximadamente 86% do total, cabendo apenas 14% às fontes renováveis. De
acordo com a Agência Internacional de Energia2 o consumo mundial de energia
primária passou de 283,629x1015 Btu no ano de 1980 para o valor de
446,442x1015 Btu para o ano de 2004, crescendo significantemente ano a ano.
Entre 2003 e 2004, o consumo global apresentou o maior aumento desde
1980. Além disso, mostra que os Estados Unidos continuam sendo os maiores
consumidores de energia primária, tendo um consumo anual sempre maior do
que toda a Europa.
A dependência de fontes não renováveis de energia desperta
preocupação permanente com o esgotamento das mesmas, além da
preocupação com a emissão de grandes quantidades de dióxido de carbono
(CO2) na atmosfera, as quais no ano de 1996 foram da ordem de 23 bilhões de
toneladas3, aproximadamente o dobro da quantidade emitida em 1965. A taxa
média de crescimento desta emissão verificada na década de 90 foi de 0,5%
ao ano. Como conseqüência, o teor de CO2 na atmosfera aumenta
progressivamente levando muitos especialistas a acreditarem que o aumento
da temperatura média da biosfera terrestre seja devido ao efeito estufa
provocado por este acréscimo de CO2 e de outros gases na atmosfera, já
denominados genericamente como gases de efeito estufa (GHG, Greenhouse
Gases). A preocupação com o crescimento do teor de GHG na atmosfera
começa a fazer parte de discussões internacionais (Rio-92, Kioto-97 e Bonn-
2001), a ponto de inúmeros países, notadamente aqueles que mais contribuem
com as emissões destes gases, já se comprometerem com algum tipo de
controle destas emissões. Até o momento estes compromissos permanecem
mais ao nível da retórica e não provocaram ações efetivas dos governos.

2
Os estudos realizados em escala global para redução da taxa de
crescimento dos teores de GHG na atmosfera têm apontado para uma série de
procedimentos de curto, médio e longo prazo. Dentre estes procedimentos
pode-se citar (i) a substituição de combustíveis fósseis, (ii) a introdução de
medidas que tornam mais eficiente o uso da energia, (iii) a criação progressiva
de medidas legislativas de contenção de emissões nas grandes cidades (como
a da Califórnia, USA), (iv) investimentos pesados no desenvolvimento das
fontes renováveis de energia (como a energia eólica e a solar) e (v) a produção
de combustíveis denominados "limpos" (como os derivados da biomassa e o
hidrogênio), com baixa emissão de poluentes e/ou nenhuma liberação de CO2.
No caso específico do setor de transportes, sua participação no
consumo mundial de energia situa-se em torno de 20%, correspondendo
também a algo desta ordem sua participação no total das emissões globais de
CO2.
No Brasil, o uso de derivados do petróleo tem participação da ordem de
80% do total de emissões de CO2 do país 4 . O setor de transporte é o
responsável por mais da metade destes, ou seja, de 40% das emissões totais
brasileiras.
O setor de transportes é um dos principais emissores de GHG, e a
concentração de veículos nas grandes cidades faz com que localmente o
impacto ambiental causado pela queima dos combustíveis fósseis seja
significativo, principalmente em decorrência da concentração de poluentes no
perímetro urbano dos municípios. Este efeito é especialmente marcante na
região central das cidades onde, em geral existe pouca circulação do ar pela
presença de grande número de prédios e edifícios. O uso de combustíveis para
transporte no meio urbano tem sido o principal fator de comprometimento da
qualidade do ar das grandes cidades, embora nas duas últimas décadas tenha
havido reduções significativas na emissão de poluentes como monóxido de
carbono (CO), dióxido de enxofre (SO2), óxidos de nitrogênio (NOx)
provenientes da queima de derivados de petróleo em motores de veículos. No
caso do CO2, ao contrário dos demais, as emissões permaneceram com
índices crescentes.
Por tudo isto é que grandes cidades como Los Angeles, Cidade do
México, Tókio, São Paulo e outras vêm adotando e estudando a implantação

3
de diversas medidas para controlar este problema. Estas variam desde
alterações técnicas redutoras das emissões veiculares, tais como o uso de
aditivos especiais à gasolina, uso de catalisadores, até medidas de restrição ao
tráfego de veículos em determinadas áreas em certas épocas do ano, como o
denominado "rodízio" de veículos já implementados na cidade de São Paulo
desde 1995, pedágio eletrônico em determinados locais da cidade, controle
rigoroso do nível de emissão dos motores da frota com pesadas multas para
veículos fora de especificação, imposto adicional proporcional ao nível de
emissão do veículo. Estas últimas três ainda encontram-se em estudo.
Com o previsível esgotamento do petróleo nas próximas décadas, torna-
se urgente à busca por fontes energéticas alternativas, capazes de assegurar
ao mesmo tempo o suprimento diante de uma demanda mundial crescente e a
devida proteção ao meio ambiente. Sabe-se que os países em
desenvolvimento, ao contrário dos países desenvolvidos, ainda não atingiram
seu ápice na demanda por energia, o que constitui um bom motivo para que
este desenvolvimento ocorra de forma sustentável. Conseguir alcançá-lo exige
uma mudança profunda nos hábitos de produção e consumo da sociedade,
questão que envolve o acesso a tecnologias alternativas.
Desenvolvimento sustentável, segundo a Brundtland Commission
(Comissão Mundial para o Meio Ambiente e Desenvolvimento Sustentável), é
aquele capaz de satisfazer as necessidades do presente sem comprometer a
capacidade das futuras gerações de satisfazerem as suas próprias
necessidades5.
No Brasil, cerca de 41% da oferta interna de energia (OIE) é originada de
fontes renováveis, sendo que a geração de energia hidráulica corresponde a
14% da OIE e 27% obtém-se através de biomassa. O restante (59%) é oriunda
de combustíveis fósseis6.
1.2 - Células a Combustível
Considerando-se esta tendência, quanto ao esgotamento das reservas de
petróleo conhecidas, até o final do século XXI, o estudo de Pilhas de Células a
Combustível (CC) e, em especial de novos materiais, de modo a produzir
energia com custos menores para aplicações veiculares e estacionárias, vêm

4
recebendo grande destaque das empresas petrolíferas e de energia nos últimos
anos.
Alguns tipos de células a combustíveis são: a alcalina (AFC), a de
membrana de troca de próton (PEMFC), a de ácido fosfórico (PAFC), a de
carbonato fundido (MCFC), a de óxidos sólidos (SOFC) e a de metanol
(DMFC) 7.
Células a combustível baseadas em eletrólitos sólidos inorgânicos
requerem alta temperatura de operação, acima de 500oC. Em geral, tais células
são utilizáveis para grande geração de potência, enquanto as células a
combustível baseadas em eletrólitos sólidos poliméricos são destinadas
especialmente a veículos elétricos ou geradores de energia residenciais8.
Células a combustível baseadas em eletrólitos sólidos inorgânicos
requerem alta temperatura de operação, acima de 500oC. Em geral, tais células
são utilizáveis para grande geração de potência, enquanto as células a
combustível baseadas em eletrólitos sólidos poliméricos são destinadas
especialmente a veículos elétricos ou geradores de energia residenciais 9.
O combustível pode ser o hidrogênio, o gás natural, o metanol ou o etanol
e o oxidante pode ser o gás oxigênio ou o ar. O melhor combustível para as
células é o hidrogênio, que tem sido apontado como a maior fonte de energia do
futuro, devido à sua disponibilidade, flexibilidade de produção (pode ser obtido a
partir de diferentes matérias-primas e processos de produção) e versatilidade de
utilização (geração de eletricidade, portador de energia em fontes móveis e
estacionárias), além de ser não-tóxico, não-poluente e possuir alta densidade de
energia por unidade de massa.
O hidrogênio pode ser produzido a partir de diferentes fontes, divididas
em três classes principais: combustíveis fósseis, fontes renováveis e eletrólise da
água10. A energia requerida para a eletrólise pode ser de origem nuclear ou a
proveniente de fontes renováveis, como energia hidroelétrica, solar ou eólica.
Atualmente a eletrólise da água responde por apenas 4% da capacidade
mundial de produção de H2, devido ao alto custo e tecnologia ainda incipiente11.
A utilização de fontes renováveis, como biomassa e resíduos orgânicos, também
é altamente promissora, mas ainda se encontra em estágios iniciais de
desenvolvimento. Quanto aos combustíveis fósseis, o gás natural responde por
48% da produção mundial de H2, o petróleo por 30% e o carvão por 18%11. É

5
importante lembrar que, quando se utilizam combustíveis fósseis para produção
de hidrogênio, o CO2 é um importante subproduto e quanto maior o
hidrocarboneto, maior é a produção relativa de CO2, que é o principal causador
do efeito estufa. Portanto, entre os combustíveis fósseis, o gás natural é o mais
adequado à produção de H2 devido ao seu maior conteúdo relativo de hidrogênio
e também porque as reservas mundiais comprovadas de gás natural já excedem
as de petróleo e vem crescendo mais rapidamente do que estas tendências que
devem ser mantidas ao longo do século XXI12.
No Brasil, o gás natural tem tido uma participação discreta na matriz
energética. Em 1998, sua produção foi de 32 milhões de m3/dia, representando
menos de 3% do consumo total de energia primária. Desse total, cerca de 40%
era reinjetado nos poços das reservas associadas para aumentar a extração de
petróleo e outra grande parte queimada na boca-do-poço por falta de mercado13.
Este cenário está mudando e o aquecimento do mercado de gás natural é
visível, embora no momento, após a nacionalização dos recursos naturais da
Bolívia, o investimento realizado na construção do gasoduto Brasil-Bolívia em
dezembro de 1998, esteja sendo questionado. Além do gasoduto, as reservas
brasileiras de gás natural cresceram na última década a uma taxa média de 10%
ao ano, passando de 26 bilhões de m3 em 1975 para 225 bilhões em 1998, com
destaque para as reservas da bacia de Campos-RJ e na área de Urucu (região
amazônica).
O Relatório da Comissão do Gás Natural, do Ministério das Minas e
Energia, publicado em 1992, previu que a participação do gás na matriz de oferta
de energia deveria chegar a 12% em 201014.
Atualmente o gás natural representa cerca de 3% da energia primária
produzida no país, mais de 10 vezes menor que o petróleo. Há necessidade de
tecnologias, equipamentos, produtos e processos relacionados ao uso de gás
natural no país.
Portanto, em uma primeira fase, o gás natural poderia ser o combustível
para as Células a Combustível. Entretanto, para operar CC economicamente
viáveis é preciso avançar rapidamente no estudo de novos materiais inorgânicos
e orgânicos com propriedades adequadas à aplicação que aumentem a
eficiência e o tempo de vida desses dispositivos. Para tanto, é necessário o
conhecimento e domínio, dos fenômenos que definem as propriedades

6
observadas em nível macroscópico, isoladamente e durante o funcionamento da
CC. Apesar dos últimos avanços na área na última década do século XX, ainda
resta muita pesquisa a ser feita na área de catalisadores anódicos, oxidação
seletiva de CO, eletrólitos sólidos, estrutura de eletrodos e da engenharia da
pilha a combustível.
Protótipos de veículos com CC já foram mostrados pela Ford (P2000),
Daimler Chrysler (NECAR IV), Toyota (FCEV) 15 e outras indústrias. Nestes
veículos espera-se uma eficiência de conversão de energia, do tanque à roda,
de até 50%, enquanto para um veículo com motor de combustão convencional,
movido a gasolina e nas mesmas condições de teste, alcança-se apenas 23%
de eficiência.
Comparando-se também a emissão dos dois tipos de veículos, deve-se
salientar que a emissão de CO proveniente de um motor convencional é maior
que 100g/100Km e a de NOx é cerca de 8g/100Km, enquanto que as emissões
de CO e NOx de um automóvel com CC são praticamente nulas.
1.3 - Células a combustível do tipo PEM.
Dentre as tecnologias de CC a do tipo PEM (polymer electrolyte
membrane) é uma das mais convenientes em função de sua eficiência na faixa
de temperatura de 50 a 125 ºC para as aplicações de menor potência. Estes
dispositivos são, em princípio, baterias de funcionamento contínuo, que
produzem corrente contínua pela oxidação do combustível gasoso, geralmente
hidrogênio16.
A Figura 1 mostra o princípio de funcionamento desta célula com
membrana condutora protônica alimentada com H2/O2.
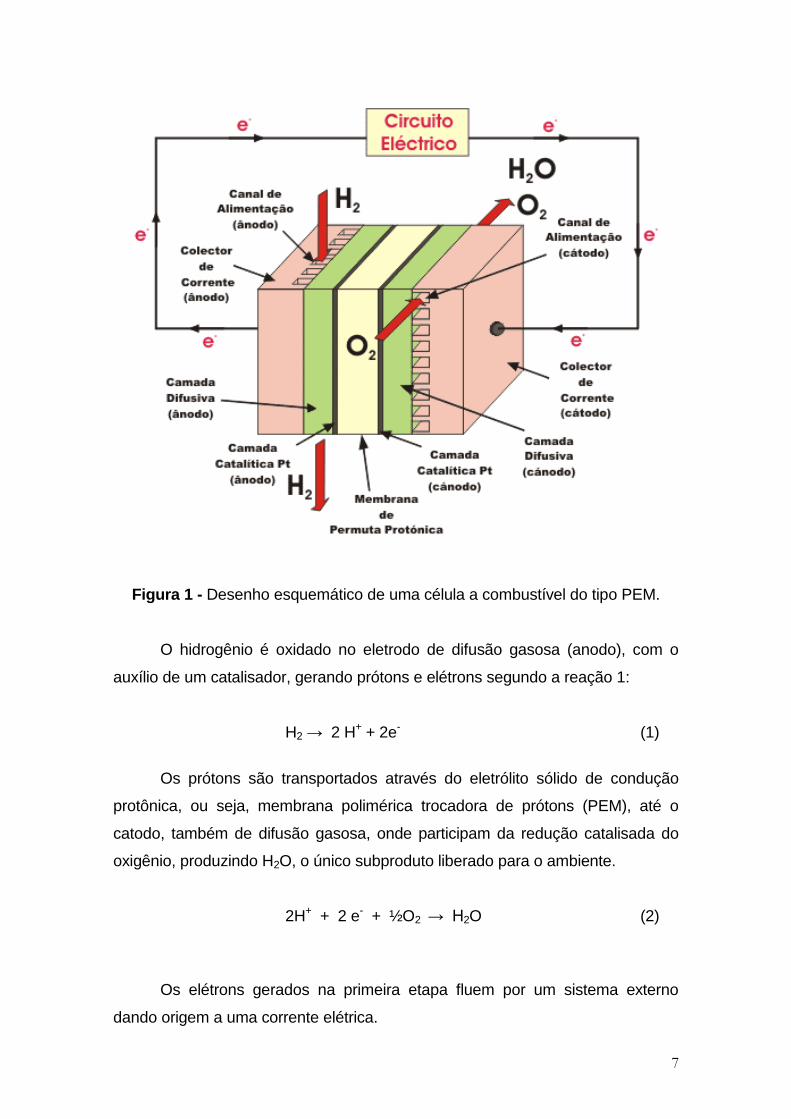
7
Figura 1 - Desenho esquemático de uma célula a combustível do tipo PEM.
O hidrogênio é oxidado no eletrodo de difusão gasosa (anodo), com o
auxílio de um catalisador, gerando prótons e elétrons segundo a reação 1:
H2 → 2 H+ + 2e- (1)
Os prótons são transportados através do eletrólito sólido de condução
protônica, ou seja, membrana polimérica trocadora de prótons (PEM), até o
catodo, também de difusão gasosa, onde participam da redução catalisada do
oxigênio, produzindo H2O, o único subproduto liberado para o ambiente.
2H+ + 2 e- + ½O2 → H2O (2)
Os elétrons gerados na primeira etapa fluem por um sistema externo
dando origem a uma corrente elétrica.

8
A reação global, que é acompanhada de liberação de calor, pode ser
descrita da seguinte forma:
H2 + ½O2 → H2O + energia (3)
A ruptura das moléculas diatômicas H2 e O2 requerem uma energia de
ativação da mesma ordem de grandeza de suas energias de formação, quando
as reações são homogêneas e ocorrem em fase gasosa. Em CC, ambas as
reações são heterogêneas e ocorrem na interface eletrodo/eletrólito, sendo
catalisadas na superfície do eletrodo. Em decorrência, utiliza-se nas células de
baixa temperatura de operação, platina como catalisador tanto na reação
anódica como na catódica16. A platina é dispersa, aleatoriamente, em partículas
nanométricas na superfície do suporte de catalisador que normalmente é
carbono Vulcan. O efeito catalítico no anodo resume-se na ruptura por adsorção
química da molécula de H2, enquanto no catodo somente no enfraquecimento da
ligação oxigênio/oxigênio, também por adsorção química da molécula de O2.
Entretanto, não se utiliza hidrogênio puro, mas sim uma mistura gasosa,
que contém além de hidrogênio, um pouco de vapor d’água, CO2 e CO. Este gás
é chamado de gás de reforma e provém da transformação catalítica heterogênea
de gás natural; hidrocarbonetos ou de também metanol, com vapor d’água, de
acordo com as reações totais (4, 5 e 6). Estas reações requerem considerável
energia térmica.
Reforma do gás natural (metano): CH4 + H2O → CO + 3H2 (4)
Reforma de hidrocarbonetos: CnH2m + 2nH2O → nCO2 + (2n + m) H2 (5)
Reforma de metanol: CH3OH + H2O → CO2 + 3H2 (6)
O desempenho das CC é limitado por perdas relacionadas à membrana,
aos eletrodos e ao sistema eletro-eletrônico.

9
Perdas significativas são originadas de: i) cinética reacional limitada, em
particular da reação de redução de oxigênio; ii) envenenamento do catalisador
anódico, particularmente quando se usa hidrogênio produzido por reforma; iii)
condutividade protônica limitada na camada catalisadora dos eletrodos; iv)
permeabilidade efetiva limitada do oxigênio ou hidrogênio na camada
catalisadora do eletrodo e v) limitação difusional do gás na camada difusora do
eletrodo.
Tendo em vista o objetivo deste trabalho, a seguir serão discutidos os
aspectos mais importantes dos fenômenos (i) e (ii) acima relacionados17.
Para a melhor compreensão da tecnologia envolvida em relação ao
emprego da CC no setor veicular é interessante traçar um comparativo entre a
configuração estrutural existente nos atuais veículos de combustão interna e as
possíveis configurações estruturais para um veículo utilizando CC, de acordo
com a Figura 2.
Figura 2 - Configurações de veículos com motores de combustão interna e
veículos utilizando células a combustível.

10
1.4 - Eletrodos de Difusão Gasosa
Eletrodos de difusão gasosa são camadas de alta porosidade, cuja
espessura depende do tipo de célula e pode variar de 20 μm até alguns
centésimos de milímetros. São separados um do outro por um eletrólito
(condutor iônico), de modo que os gases não se misturem. O eletrólito pode ser
um líquido, um polímero condutor de cátions saturado com um líquido.
Na célula do tipo PEM esses eletrodos são confeccionados pelo depósito
sobre uma folha de tecido de carbono, de uma camada fina de uma mistura
formada por partículas de carbono com nanopartículas de platina dispersas na
superfície. Essas partículas são recobertas por fibras microscópicas de PTFE ou,
no caso da PEM, também com o próprio material da membrana, formando um
agregado poroso. Uma parte dos poros entre os flocos de carbono é totalmente
preenchida pelo material do eletrólito, outra fração de poros é mantida
hidrofóbica, pois nestes poros as partículas estão recobertas com uma fina
camada de PTFE e estão disponíveis para a difusão dos gases no eletrodo.
A construção deste eletrodo tem como função à maximização da interface
trifásica gás-líquido-sólido, aumentando consideravelmente a velocidade dos
processos eletródicos. A figura 3 apresenta a representação esquemática de
uma unidade eletrodo/membrana/eletrodo (MEA).
Os eletrodos de difusão gasosa devem satisfazer no mínimo duas
exigências importantes: (i) devem possuir alta atividade catalítica, a fim de se
obter altas densidades de corrente e; (ii) os poros, durante a operação do
eletrodo, não podem apresentar forças capilares muito fortes, para não sugar
todo o eletrólito, e a pressão do gás não deve ser muito alta, para que o eletrólito
não seja totalmente expulso dos poros. Nestes dois extremos o eletrodo torna-se
ineficiente. A superfície interna dos poros do eletrodo é contatada por um filme
delgado do eletrólito, de modo que os poros relativamente grandes (diâmetros
entre 0,1 a 1 mm) fiquem livres para a circulação/difusão dos gases de trabalho.
Em células de baixa temperatura de operação (por exemplo: PEM), as
partículas do eletrocatalisador estão numa faixa de distribuição de tamanho
nanométrica. O eletrólito da célula PEM constitui-se de uma camada delgada
(100 a 150 μm de espessura) de um polímero condutor protônico (membrana
Nafion®), bastante resistente quimicamente18.

11
A fabricação destes eletrodos baseia-se, na maioria dos casos, na
fabricação de filmes precursores obtidos a partir de uma pasta, como nos
processos cerâmicos tradicionais (doctor-blade). Esta pasta contém, além do
catalisador, um formador de poros e um ligante orgânico apropriado, por
exemplo, um álcool polivinílico. O ligante dá sustentação intermediária ao filme,
sendo mais tarde evaporado por aquecimento.
A montagem da unidade MEA pode ser realizada por diferentes métodos,
prensagem a quente dos componentes preparados individualmente o usando as
seguintes técnicas: screen-printing, rolling, brushing, filtering e spraying19,20,21.
Figura 3 - Representação esquemática de um MEA mostrando em detalhes a
interface trifásica gás-líquido-sólido.
Após a montagem da unidade eletrodo/membrana, processa-se a retirada
do ligante orgânico polimérico da membrana, por aquecimento. Este processo
tem como efeito a fixação dos eletrodos na membrana formando uma célula.
1.5 Empilhamento da célula
Nestas células unitárias obtêm-se potenciais de trabalho para o sistema
hidrogênio/oxigênio entre 0,5 e 0,7 V. Potenciais de circuito aberto ficam entre

12
1,1 e 1,2 V. Estes valores são, sob o ponto de vista prático, muito baixos. A fim
de se obter potencial prático da ordem de 150 a 200 V torna-se necessário o
empilhamento em série de várias unidades de células (200 a 300), como
mostrado na Figura 4. As células são montadas eletricamente em série e
sobrepostas, normalmente dispostas verticalmente, formando um empilhamento
“stack”.
1: Placa bipolar; 2: moldura do anodo; 3: Anodo 4: Membrana; 5: Catodo; 6:moldura do catodo; 7: placa bipolar.
Figura 4: Montagem de uma célula a combustível.
Utiliza-se a placa bipolar para separar o anodo de uma célula do catodo
da célula seguinte. As placas bipolares devem ser estáveis quimicamente tanto
em atmosfera redutora quanto oxidante. Além disso, devem ser boas condutoras
de eletricidade e resistir ao ataque químico do eletrólito. Para células de baixa
temperatura de operação (80 a 200oC) utiliza-se carbono ou um compósito de
polímero e carbono.
1.6 - A eficiência das células a combustível
O princípio das células a combustível foi descoberto por Sir Grove já em
1835. No final do século passado, Wilhelm Ostwald e Walther Nernst
demonstraram a vantagem da combustão eletroquímica a frio em relação à
produção de eletricidade pela máquina de calor/mecânica, que funciona sob o
princípio de Carnot. A eficiência teórica “η” de qualquer processo de produção de
energia eletroquímica é obtida pelo quociente:
1 2 3 4 5 6 7

13
η: = ΔG/ΔH (7)
Na Figura 5 são mostrados valores de η para a reação de combustão
(reação 8) em fase gasosa em função da temperatura segundo o processo
eletroquímico e o ciclo de Carnot. A eficiência teórica eletroquímica diminui de 86
a 70% na faixa de temperaturas de 100 a 1000°C. A eficiência de Carnot, por
sua vez, eleva-se de 0 a 70% na mesma faixa e somente a temperaturas
superiores a 1000°C é maior que a eficiência teórica eletroquímica.
CH4 + 2O2 → 2 H2O + CO2 (8)
Portanto, células a combustível a hidrogênio apresentam uma eficiência
teórica significativamente maior que máquinas de Carnot, principalmente a
baixas temperaturas. Na Figura 5 também é mostrada, comparativamente, a
curva da conversão eletroquímica do metano. Neste caso, a eficiência teórica
encontra-se muito próxima de 100% em toda a faixa de temperaturas mostrada.
O metano torna-se, assim, o armazenador de energia primário de preferência
para células a combustível.
Figura 5- Dependência do rendimento teórico com a temperatura da conversão
eletroquímica do hidrogênio, do metano e no ciclo de Carnot.

14
Células a combustível a membrana polimérica são células de baixa
temperatura de operação, que utilizam uma membrana polimérica como
eletrólito, também chamadas PEMFC (Próton Exchange Membran Fuel Cell),
são as mais promissoras como alternativa para motores a combustão, por ser
robustas e de fácil acionamento e desligamento, além das vantagens inerentes
como alta eficiência com baixa emissão de poluentes. Devido à baixa
temperatura de operação, e, mesmo utilizando-se ar como alimentação do
cátodo, tem-se emissão zero para NOx. As células de baixa temperatura também
se aplicam a unidades estacionárias.
Atualmente, o fator determinante para a sua entrada no mercado é, ainda,
o seu custo 22 . A célula do tipo PEM é a mais apropriada para a tração
automotiva e não se prevê, em curto prazo, sua substituição por nenhum outro
tipo de célula nesta aplicação.
A estratégia de desenvolvimento para as células estacionárias sofreu uma
grande mudança nos anos 90. Unidades das classes de 100 kW e de até alguns
MW eram preferenciais. Atualmente este desenvolvimento também está voltado
para unidades de alguns kW. A motivação vem da situação favorável do
mercado para a aplicação das células no fornecimento de energia elétrica em
residências. As células do tipo PEM são bastante promissoras para esta
aplicação devido ao seu avançado desenvolvimento para a indústria automotiva.
Pequenas instalações de CC para aplicações residenciais abrirão um
novo mercado no ramo de serviços de energia e de fornecimento de gás, se o
custo da instalação for suficientemente competitivo. Mesmo a preços um pouco
acima dos convencionais, esta tecnologia já pode assegurar um mercado
específico e promissor, onde suas características principais (como baixo impacto
ambiental e melhor aproveitamento do combustível) passam a ter um papel
relevante na escolha final.
Ainda assim, hoje o mercado principal da tecnologia de células a
membrana é o dos veículos elétricos não poluentes. O fato de que uma
importante montadora automobilística tem equipado não só ônibus, mas também
carros de passeio com células tipo PEMFC e, muito, além disso, tem uma meta
já anunciada de que em 8 anos 2% de sua produção serão veículos movidos a

15
PEMFC, não nos permite dúvidas quanto o futuro - e o mercado - deste tipo de
tecnologia7.
Para tornar viável a utilização comercial de células, alguns problemas são
motivo de intensos estudos. A geração de hidrogênio através da reforma
catalítica de um combustível líquido gera como subproduto o CO (100 a 2000
ppm). Esta espécie é particularmente danosa ao desempenho da célula a
combustível, pois adsorve irreversivelmente nos sítios de Pt (catalisador
normalmente utilizado) gerando um bloqueio físico que impede a oxidação do
combustível.
1.8 - Membrana polimérica condutora protônica
A maior parte das células a combustível relatadas na literatura utilizam
uma membrana polimérica de condução protônica comercial fluorada como a
DOW (DOW chemicals), a Flemion (Asashi Glass) e o NAFION (DuPont)23.
Entretanto, as membranas são de custo elevado dificultando os avanços na sua
comercialização e limitam as condições de temperatura de operação da célula
na faixa de 50 a 125 oC24 . Estas membranas são conhecidas genericamente por
Eletrólitos Sólidos Poliméricos (ESP).
1.9 - Eletrocátalise da reação de redução de oxigênio
A reação de redução de oxigênio (RRO) é considerada como uma das
reações eletrocatalíticas mais importantes devido a sua função em conversores
eletroquímicos de energia, vários processos industriais e em corrosão.
Conseqüentemente, há muitos anos é foco de interesse das pesquisas
eletroquímicas. A redução de oxigênio continua a ser um desafio para
eletroquímicos devido à sua complexidade cinética e à necessidade de melhores
eletrocatalisadores.
A redução de oxigênio é uma reação multieletrônica que usualmente inclui
várias etapas elementares no mecanismo reacional. A RRO em soluções
aquosas ácidas ocorre segundo dois mecanismos globais clássicos já
conhecidos25,26.

16
I) Mecanismo direto ou mecanismo 4-elétrons:
O2 + 4H+ + 4e- → 2H2O (9)
II) Mecanismo Peróxido ou mecanismo 2-elétrons
O2 + 2H+ + 2e- → H2O2 (10)
Sendo que o peróxido de hidrogênio pode ser reduzido em uma etapa posterior:
H2O2 + 2H+ + 2e- → 2H2O (11)
Ou sofrer decomposição química segundo:
2H2O2 → 2H2O + O2 (12)
O potencial Eo nestas equações corresponde ao valor padrão da reação
em função do eletrodo reversível de hidrogênio (ERH) a 25oC. A distinção entre
estes dois mecanismos é dificultada pelo fato de que a redução direta também
pode envolver a formação de peróxido, desde que este permaneça adsorvido na
superfície do eletrodo sem sofrer dessorção ou decomposição, e a sua redução
ocorra subseqüentemente. Sendo este processo também de 4 elétrons, porém,
chamado de mecanismo em série. Em potenciais mais catódicos juntamente
com o mecanismo via 4 elétrons ocorre o mecanismo via 2 elétrons, sendo
chamado de mecanismo paralelo.
Nas últimas décadas, muitos esforços têm sido dedicados ao estudo do
mecanismo da RRO em soluções aquosas ácidas e alcalinas sobre diferentes
materiais de eletrodo27. Em eletrólitos ácidos, platina e ligas de platina ainda são
consideradas as melhores opções para a redução de oxigênio, tanto em termos
dos mais baixos sobrepotenciais desejáveis para promover a reação, quanto da
estabilidade requerida. Assim, os eletrocatalisadores baseados em platina são
necessários para prover estabilidade no ambiente corrosivo da PEMFC e, neste
sentido, as ligas de platina favorecem a atividade catalítica desejada ao mesmo

17
tempo em que permitem a diminuição do teor de Pt, com conseqüente ganho
econômico.
Sobre platina e metais da família da platina ocorre o mecanismo paralelo,
mas a predominância é do mecanismo direto via 4 elétrons. Para estes metais
há duas propostas para a primeira etapa reacional. A primeira proposta é de
Damjanovic e colaboradores 28 em que a transferência de prótons ocorre
simultaneamente com a transferência de carga. A segunda visão é de que o
mecanismo mais comum, a redução via 4 elétrons sobre Pt, envolve adsorção
química seguida de dissociação da molécula de O2 sobre a superfície de platina
que, provavelmente, ocorre simultaneamente com a transferência de carga29.
Uma variedade de sistemas de ligas de Platina, binárias e ternárias, tem
sido discutidas e investigadas para a utilização em CC de ácido fosfórico (PAFC)
e de eletrólitos poliméricos (PEMFC) 30. A utilização de ligas de platina como
catalisadores catódicos tem atraído uma ampla atenção como um candidato
para alcançar uma alta performance, para o aumento na densidade do pó, e para
a redução do custo do componente31.
Ligas como Pt-Co, Pt-Cr, Pt-Ti, Pt-V e Pt-Fe suportadas em carbono
(Pt-M/C) têm sido muito estudadas para a redução de oxigênio em meio ácido.
Alguns estudos sobre estas ligas têm mostrado um aumento da atividade
eletrocatalítica para a RRO em relação à Pt pura29,32. Ligas de Pt-Co e Pt-Co-Cr
suportadas em carbono podem aliviar apreciavelmente o sobrepotencial da
redução de oxigênio em relação à platina pura33,34.
Ligas binárias de Pt-Fe exibem melhor resistência à sinterização das
partículas a elevadas temperaturas que catalisadores que contêm platina pura.
Este efeito é chamado de “efeito âncora” do ferro para a platina sobre substrato
de carbono. A mobilidade dos átomos de platina sobre substrato de carbono é
dificultada quando o ferro está presente.
Aricó e colaboradores35 realizaram estudos de difração de raios-X para Pt,
Pt-Co e Pt-Co-Cr e os resultados mostraram que a Pt suportada em carbono tem
estrutura cúbica de face centrada, porém, Pt-Co e Pt-Co-Cr suportadas em
carbono têm estrutura de face centrada tetragonal, o que está de acordo com a
proposta de Glass36. Outros estudos em ligas de Pt-Co suportadas em carbono
mostraram que uma estrutura cristalina desordenada é mais resistente à

18
corrosão que uma ordenada e mantém alta a atividade catalítica deste material
por um longo tempo37.
1.10 - Eletrocatálise da reação de oxidação de hidrogênio
Um outro problema muito importante relacionado à eficiência das células
a combustível está relacionado com a pureza do hidrogênio. Se o gás é
produzido por eletrólise da água, a pureza é elevada e o catalisador de platina
dispersa é plenamente satisfatório para promover com alta eficiência a reação de
oxidação de hidrogênio (ROH) que tem lugar no ânodo das células. Entretanto,
se o hidrogênio é produzido através da reforma de outros combustíveis, (gás
natural, biogás, metanol, etanol, etc.) o produto pode vir acompanhado de
impurezas tais como CO e compostos de enxofre (em níveis de 0,1%) que se
adsorvem fortemente sobre a platina, diminuindo de forma drástica a atividade
do eletrocatalisador em temperaturas abaixo de 150oC.
Através de estudos utilizando eletrodos de metais maciços, já se sabe que
a presença de um segundo elemento além da platina, como Ru, Sn, Os, Mo, etc,
formando ligas ou co-depósitos, leva a um incremento significativo na tolerância
ao CO se comparado à platina pura 38 , 39 , 40 . A busca do entendimento do
mecanismo de envenenamento tem se concentrado em uma variedade de
abordagens experimentais41, incluindo o uso de técnicas eletroquímicas, UV-
visível in situ, espectroscopia de infravermelho por reflectância, absorção de
raios-X, etc 42,38.
Diversos estudos realizados a 25oC em Pt ou ligas de Pt dispersa em
carbono indicam que o envenenamento ocorre devido à forte adsorção de CO na
superfície do catalisador, que bloqueia a adsorção da espécie reagente
(hidrogênio). Assim sendo, ocorre um forte efeito causado pela presença de CO
na curvas de polarização de uma célula PEMFC. No caso das ligas de Pt, um
mecanismo bifuncional tem sido proposto para explicar a melhor tolerância em
relação à presença do contaminante. De acordo com este modelo, a espécie
reagente adsorve-se preferencialmente nos átomos de Pt, enquanto que o outro
metal (Ru, Mo, etc) produz espécies oxigenadas ou óxidos hidratados que atuam
diretamente na oxidação do envenenador da reação. Nestas condições, a
corrente origina-se apenas da oxidação do reagente que ocorre nas zonas da

19
superfície da Pt livres de CO que se formam ao redor dos átomos do outro metal,
sendo que o restante da área permanece coberto pela compacta monocamada
de CO adsorvido.
Uma avaliação crítica dos resultados até agora obtidos permite concluir
que o desenvolvimento de novos materiais e estruturas eletródicas mais ativas
para a ROH na presença de CO é ainda um campo muito aberto para pesquisas.
Isto decorre do fato de que nem todos os fenômenos associados aos processos
envolvidos estão plenamente caracterizados, sendo ainda requerida muita
pesquisa de forma a superar as dificuldades envolvidas.
1.11 - Envenenamento do catalisador por CO
No caso da utilização de H2, a sua geração através da reforma catalítica
de um combustível líquido gera como subproduto o CO (100 a 2000 ppm). Esta
espécie é particularmente danosa ao desempenho da CC, pois adsorve
irreversivelmente nos sítios de Pt (catalisador normalmente utilizado) gerando
um bloqueio físico que impede a oxidação do hidrogênio. Sendo assim o
desempenho da PCC depende fortemente da concentração de CO e do tipo do
catalisador anódico.
1.12 - Métodos de preparação de catalisadores
O método convencional de preparação de catalisadores metálicos
suportados é a impregnação dos sais metálicos no suporte e posterior redução,
freqüentemente em fluxo de hidrogênio a alta temperatura (Redução a
Temperatura Programada – TPR).
Existem, porém, diversos métodos que mostram diferentes abordagens
na preparação de eletrocatalisadores, que oferecem maior ou menor grau de
controle da distribuição, composição e do tamanho das nanopartículas
produzidas. Alguns métodos permitem a preparação de sistemas de
nanopartículas metálicas sendo, portanto, vantajosos para o estudo isolado
dessas partículas e da interação com o suporte. Já outros métodos envolvem a
preparação direta do eletrocatalisador suportado. É fundamental destacar que as
características do eletrocatalisador obtido dependem não somente do método

20
utilizado, mas também das condições de preparação. Assim, o objetivo deve ser
sempre a produção de nanopartículas de tamanho adequado, monodispersas e
que apresentem uma distribuição homogênea no suporte. Estas características
têm demonstrado serem as mais adequadas para se obter o máximo
desempenho de um eletrocatalisador.
1.12.1 - Método do ácido fórmico
O método do ácido fórmico consiste na preparação de eletrocatalisadores
via redução química, visando alta atividade catalítica e quantidade reduzida de
metal nobre. Foi demonstrado que os eletrocatalisadores Pt/C preparados pelo
método do ácido fórmico apresentaram alta atividade catalítica, tanto para a
reação de oxidação de hidrogênio no ânodo, quanto para a reação de redução
de oxigênio no cátodo. Posteriormente, este método foi adaptado para
preparação de ligas de platina com certos metais de transição43.
A metodologia de preparação de eletrocatalisadores pelo método do ácido
fórmico consiste em adicionar o pó de carbono de alta área superficial (Vulcan
XC-72, 240 m2g-1) a uma solução de ácido fórmico e aquecer a mistura até 80ºC.
Uma solução contendo os sais de platina e do co-catalisador é adicionada em
etapas. Para o controle do andamento da redução, utiliza-se o iodeto de potássio
como reagente externo, com a finalidade de indicar a presença de platina em
solução (vermelho intenso). Após a redução total da platina não se observa mais
o tom avermelhado e novas adições da solução contendo os íons metálicos
podem ser realizadas. Após a redução total da platina, o catalisador é filtrado,
seco e triturado43.
Oliveira Neto e colaboradores43 prepararam eletrocatalisadores Pt:Ru pelo
método do ácido fórmico a partir de composições atômicas Pt:Ru de 90:10,
80:20, 70:30 e 60:40. Os eletrocatalisadores obtidos foram caracterizados pelas
técnicas de análise de espectroscopia de dispersão de energia de raios-X (EDX),
difração de raios-X (XRD) e microscopia eletrônica de transmissão (TEM). A
caracterização eletroquímica foi realizada por voltametria cíclica.
Os resultados de difração de raios-X mostraram que as partículas
metálicas encontram-se na forma de ligas e apresentam a estrutura cúbica de
face centrada da platina, que predomina nas ligas. As análises por EDX

21
mostraram que as composições atômicas das ligas Pt:Ru obtidas foram
semelhantes às composições de partida. As micrografias obtidas por TEM
apresentaram uma faixa estreita de distribuição de tamanho de partículas
variando de 1,6 a 6,4 nm, sendo que cerca de 50% das partículas encontram-se
na faixa de 3 nm.
1.12.2 - Método de Bönnemann
O método de Bönnemann, também conhecido como método coloidal,
pode ser utilizado para a obtenção de catalisadores mistos ou sistemas de
catalisadores, ternários e quaternários, sobre carvão ativo e carvão ativo
grafitizado. Esta metodologia pode ser aplicada com sucesso para a obtenção de
eletrocatalisadores à base de platina e ligas contendo metais (e/ou óxidos de
metais), em particular, Sn, V, W e Mo e demais elementos de transição, como
Cu, Fe, Co e Ni44,45,46.
Este método, numa versão modificada44 consiste na preparação de um
sistema coloidal em atmosfera inerte, utilizando-se solventes e sais
desidratados, o que encarece o método.Os sais anidros dos metais são
dissolvidos nas proporções desejadas em tetraidrofurano (THF) anidro ([H2O] <
0,005%) junto com uma quantidade apropriada de brometo de tetraoctilamônio
(N(oct)4Br). O agente redutor é preparado com a mistura de soluções de
N(oct)4Br em THF e uma solução de trietilidroborato de potássio (KHB(et)3) em
THF. Formase então uma solução de trietilidroborato de tetraoctilamônio
(N(oct)4HB(et)3), um forte redutor, como indicado pela Equação 13:
(oct)4Br + KHB(et)3 → N(oct)4HB(et)3 + KBr (13)
Para a redução dos íons metálicos utiliza-se uma vez e meia a quantidade
estequiométrica da solução de N(oct)4HB(et)3, que é adicionada à solução dos
sais dos metais a 40°C sob agitação. A redução pode ser observada pelo
escurecimento e geração de hidrogênio como mostrado na Equação 14:
MeXn + N(oct)4HB(et)3 → Me*[N(oct)4]+ + nB(et)3 + n/2 H2 + nX- (14)

22
A dispersão coloidal é, então, agitada à temperatura ambiente e o colóide
é formado pela adsorção do íon [N(oct)4]+ na superfície do metal/liga dos metais,
o que garante a dimensão nanométrica das partículas e sua grande solubilidade
em solventes orgânicos. Todas as etapas até a formação do colóide são
realizadas em atmosfera controlada.
Após a agitação, a dispersão coloidal é vagarosamente adicionada à
suspensão de pó de carbono (suporte) em THF44. Resíduos de íons [N(oct)4]+
ainda permanecem ligados às nanopartículas após a preparação do catalisador
suportado, prejudicando o desempenho eletroquímico. Segundo Schmidt e
colaboradores47. Pode-se eliminar os íons [N(oct)4]+ por oxidação, submetendo o
catalisador à passagem de um fluxo de 10% oxigênio/90% nitrogênio, seguido de
um fluxo de hidrogênio a 300°C.
Franco e colaboradores 48 prepararam eletrocatalisadores ternários
Pt:Ru:Mo com razões atômicas 1:1:1 e 1:1:0,5 pelo método de Bönnemann. As
análises por microscopia eletrônica de transmissão (Figura 3) mostraram que,
para ambos os eletrocatalisadores, as nanopartículas apresentavam-se bem
distribuídas e possuíam tamanhos de cerca de 2 nm.
1.12.3 - Método da deposição espontânea
Adzic e colaboradores 49 , 50 , relataram um método de preparação de
eletrocatalisador bimetálico de Platina, o qual reduz consideravelmente a
quantidade de platina empregada. Segundo os autores, somente um quarto de
uma monocamada de platina sobre nanopartículas de um segundo metal de
transição resulta em um eletrocatalisador com maior atividade e tolerância ao
envenenamento da Pt pelo monóxido de carbono, na oxidação de misturas
H2/CO (100 ppm de CO), em comparação com eletrocatalisadores comerciais
contendo ligas as mesmas ligas. O método de preparação deste
eletrocatalisador envolve a deposição espontânea de platina sobre
nanopartículas de rutênio suportadas em carbono, sem a aplicação de um
potencial externo. Inicialmente, as nanopartículas de rutênio suportadas no
carbono Vulcan XC-72 são tratadas em atmosfera de hidrogênio a 300ºC
por 2 h. Posteriormente, são esfriadas à temperatura ambiente e imersas em
uma solução contendo íons [PtCl6]2-.

23
O procedimento completo é realizado em atmosfera de hidrogênio e/ou
argônio e a quantidade de platina disponível para a deposição espontânea é
controlada pela concentração e volume da solução de imersão. Dessa forma
ocorre a formação de depósitos de platina desde frações de monocamadas até
multicamadas, sem a aplicação de um campo externo.
Em relação ao mecanismo da deposição espontânea de um metal nobre
sobre outro metal ainda não existe consenso. A deposição pode ser atribuída a
uma reação química com o hidrogênio adsorvido ou a um mecanismo
envolvendo a formação de espécies M-OH na superfície (Equações 15 e 16).
Ru0 + x(H2O) → RuOxHy + (2x-y)H+ + (2x-y)e- (15)
[PtCl6]2- + 4e- → Pt0 + 6Cl- (16)
A atividade catalítica dos eletrocatalisadores Pt:Ru, obtidos pela
deposição espontânea, foi determinada a partir de experimentos utilizando o
eletrodo rotativo de camada fina porosa. Na oxidação de H2 em H2SO4
0,5 mol L-1 a 25ºC, a atividade foi cerca de três vezes maior, considerando-se a
densidade de corrente por massa de platina, quando comparada ao
eletrocatalisador comercial E-TEK (liga Pt:Ru) 49.
Como conseqüência das propriedades eletrônicas modificadas da
monocamada de platina sobre o rutênio, espera-se que a ligação do CO49 com a
platina e o rutênio seja mais fraca quando comparada à dos metais em fases
puras. O mecanismo bifuncional é freqüentemente citado na literatura para
materiais Pt:Ru, devido à formação de espécies RuOH a baixos potenciais,
facilitando a oxidação do CO.
Wieckowski e colaboradores49 prepararam eletrocatalisadores através da
deposição espontânea de rutênio sobre nanopartículas de platina e testaram a
atividade na oxidação de metanol. A deposição espontânea envolveu a imersão
da platina em uma solução de cloreto de rutênio. Os eletrocatalisadores obtidos
foram duas vezes mais ativos, em termos de densidade de corrente por área
superficial de platina, que os catalisadores comerciais. Estes resultados ilustram
o efeito benéfico da adição de co-catalisadores à platina.

24
1.12.4 - Preparação de eletrocatalisadores a partir da decomposição de
precursores moleculares
Uma outra alternativa para o método convencional de preparação de
catalisadores bimetálicos suportados (impregnação dos sais metálicos no
suporte e posterior redução) é o uso de clusters ou complexos bimetálicos como
precursores. Dessa maneira, espera-se que a composição das nanopartículas
que resultam da ativação dessas moléculas sejam semelhantes à de seus
precursores, pois suas agregações e crescimento devem ser diferentes daqueles
feitos a partir da mistura de sais metálicos.
Nuzzo e Shapley51,52 descreveram a preparação de nanopartículas Pt:Ru
suportadas em carbono, preparadas a partir do cluster PtRu5C(CO)16. Os
resultados mostraram que a ativação do cluster precursor em atmosfera de
hidrogênio a 400ºC levou à formação de partículas bimetálicas, com distribuição
de partículas excepcionalmente estreita de cerca de 1,5 nm. A distribuição da
composição apresentouse centrada na relação atômica Pt:Ru de 1:5, indicando
uma coalescência uniforme do cluster precursor durante a ativação.
Posteriormente, foi também utilizado como precursor o cluster Pt2Ru4(CO)18 e,
também neste caso, as nanopartículas obtidas apresentavam uma composição
atômica Pt:Ru de 2:4 e diâmetros médios entre 1,0 e 1,5 nm.
Lukehart e colaboradores53,54 utilizaram como precursor o complexo (η-
C2H4)(Cl)Pt(μCl)2Ru(Cl)(η3:η3-C10H16). Este precursor foi incorporado em
diversos suportes de carbono e tratado em condições oxidantes e redutoras
apropriadas. As análises das partículas formadas revelaram que apresentavam
uma estequiometria Pt:Ru bem próxima de 1:1. Os materiais foram testados
como ânodo em células a combustível usando metanol diretamente como
combustível e, em alguns casos, mostraram-se mais ativos que os
eletrocatalisadores comerciais E-TEK.
Dickinson e colaboradores 55 descreveram a preparação de
eletrocatalisadores Pt:Ru a partir de uma mistura de clusters carbonilados. Neste
caso, os clusters carbonilados [Pt3(CO)3(μ2-CO)3]x-2 e Ru3(CO)12, na razão
atômica Pt:Ru de 1:1, e o suporte de carbono foram colocados em o-xileno e
refluxados a 143ºC por 24 h. O catalisador obtido apresentou nanopartículas
Pt:Ru pequenas (2,5 nm), homogeneamente distribuídas no suporte e com uma

25
distribuição de tamanho bem estreita (0,5 nm). No entanto, a deposição de
rutênio foi mais efetiva que a de platina nas condições utilizadas obtendo-se
valores de 62-67 átomo% para o rutênio e 33-38 átomo% para a platina nas
diferentes medidas feitas por EDX na amostra. A performance normalizada deste
eletrocatalisador na oxidação direta de metanol, quando comparada ao
eletrocatalisador comercial da E-TEK (Pt:Ru 50:50 átomo%), mostrou que a
baixos valores de corrente o catalisador da E-TEK apresenta melhor
performance, provavelmente, devido à razão atômica de 33:67 átomo% não ser
a mais adequada para oxidação de metanol. No entanto, para valores acima de
145 A g-1 de metal a tendência é inversa, provavelmente devido à maior área
superficial eletroquimicamente ativa (27,7 m2 g-1) que a do catalisador comercial
(22,2 m2 g-1).
1.12.5 - Método da redução por álcool
O método da redução por álcool foi desenvolvido por Toshima e
Yonezawa56, para preparar dispersões coloidais de nanopartículas apresentando
tamanho e distribuição bem uniformes. Neste método, o refluxo de uma solução
alcoólica contendo o íon metálico na presença de um agente estabilizante,
normalmente um polímero, fornece dispersões coloidais homogêneas das
nanopartículas metálicas correspondentes. O álcool funciona como solvente e
agente redutor, sendo oxidado a aldeídos e cetonas (Equação 17).
H2PtCl6 + 2 CH3OH ↔ Pt0 + 2 HCHO + 6 HCl (17)
Este método apresenta as seguintes vantagens: i) o procedimento é muito
simples e reprodutível, ii) as nanopartículas obtidas são pequenas e apresentam-
se bem distribuídas, iii) o tamanho das nanopartículas pode ser controlado
alterando as condições de preparação, como escolha do álcool, temperatura de
redução, quantidade e variedade do agente estabilizante, concentração do íon
metálico e uso de aditivos, iv) as dipersões coloidais das nanopartículas
apresentam alta atividade catalítica e v) as dispersões obtidas são bastante
estáveis.

26
Wang e Hsing57 prepararam eletrocatalisadores Pt/C e Pt:Ru/C através do
método da redução por álcool. Para isto, uma solução metanol-água contendo
os sais de platina e de rutênio foi refluxada na presença de um agente
estabilizante (surfactante SB12) e do suporte de carbono. As análises por DRX,
TEM e XPS mostraram que as nanopartículas (tamanho médio de 2,5 nm)
apresentavam-se bem dispersas e suportadas no carbono. A caracterização
eletroquímica mostrou que, em comparação ao eletrocatalisador comercial da
ETEK, o eletrocatalisador Pt/C apresentou atividade catalítica similar para a
reação de redução de oxigênio, enquanto que o eletrocatalisador PtRu/C
apresentou maior tolerância ao envenenamento pelo monóxido de carbono.
Lee e colaboradores58 preparam o eletrocatalisador Pt/C pelo método da
redução por álcool. Uma solução aquosa de H2PtCl6.6H2O foi adicionada ao
álcool e uma pequena quantidade de solução aquosa de KOH foi adicionada
gota a gota. Uma razão molar KOH/Pt = 8 foi usada para induzir a formação de
nanopartículas pequenas e bem uniformes. O suporte de carbono foi disperso
nesta solução usando ultra-som e a mistura resultante foi aquecida em forno de
microondas por 60 s. A suspensão resultante foi filtrada, lavada com acetona e
seca. As medidas por EDX indicaram a incorporação de 9,5; 13,6 e 18,6% em
massa para as amostras preparadas a partir de 10, 15 e 20% em massa de
platina. As análises por TEM mostraram que as nanopartículas de platina obtidas
apresentavam-se bem uniformes e distribuídas na superfície do carbono. O
material contendo 18,6% em massa de platina apresentou nanopartículas na
faixa de 3,5 a 4,0 nm e poucas partículas maiores que 5,0 nm. O
eletrocatalisador comercial Pt/C E-TEK (20% em massa de Pt), usado para
comparação, apresentou tamanho médio de nanopartículas de 5,1 nm e uma
ampla distribuição entre 2 e 10 nm. As medidas eletroquímicas mostraram que o
eletrocatalisador Pt/C preparado desta forma apresentou maior atividade na
eletro-oxidação de metanol à temperatura ambiente que o catalisador comercial
da E-TEK.

27
2. Objetivos
2.1. Objetivo Geral
O Objetivo deste trabalho é realizar um estudo exploratório da
metodologia e caracterização de catalisadores de Pt-Co suportados em carbono.
2.2. Objetivos específicos
- Pretende-se utilizar a técnica de Redução a Temperatura Programada
para reduzir os íons metálicos adsorvidos no suporte e propiciar a formação
da liga.
- Deve-se obter e caracterizar morfológica, estrutural e eletroliticamente os
sistemas de catalizadores.
- Devem ser preparados eletrocatalisadores com diferentes concentrações
de platina e cobalto variando-se as condições de temperatura de
impregnação dos íons metálicos.

28
3. Materiais e Métodos
3.1. Precursores
Foram utilizados dois suportes para a preparação dos catalisadores:
carbono grafite (Cg) proveniente da Nacional de Grafite LTDA e carbono Vulcan
XC-72R (Cv) proveniente da Bandeirantes SA. Estes possuem área superficial
de 17 e 376 m2/g, respectivamente.
O sal de platina utilizado foi o cloreto de tetramin platina II, Pt(NH3)4Cl2, e
o ácido cloro platínico (H2PtCl6) Aldrich com 98% de pureza.
O sal de cobalto utilizado foi o nitrato de cobalto P.A., Co(NO3)2.14H2O,
Tedia.
3.2- Procedimento experimental
3.2.1- Preparação da suspensão do carbono grafite e carbono Vulcan XC-
72R.
Foram adicionados lentamente 20 mL de água a 1g de Cg ou Cv sob
agitação constante em aquecimento brando até que todo o pó de Cg ou Cv
estivessem úmidos. Adicionou-se mais 20 mL para suspender a mistura,
mantendo-se a temperatura a 50ºC e sonicação por 30 minutos.
3.2.2 - Preparação dos sistemas de Pt(NH3)4Cl2 nos suportes Cg e Cv
3.2.2.1 - Preparo da solução aquosa de Pt(NH3)4Cl2
Foram preparadas soluções aquosas de 10, 15 e 20% de platina por
sonicação do sal Pt(NH3)4Cl2 durante dez minutos. Em seguida cada uma
dessas soluções foram adicionadas à suspensão de Cv. Somente a solução de
10% de Pt foi adicionada à suspensão de Cg.

29
3.2.2.2 - Adição da solução de Pt(NH3)4Cl2 à suspensão de Cv
Com o auxílio de um funil de separação, adicionou-se as soluções
aquosas 10, 15 e 20% de Pt(NH3)4Cl2 (m/m) ao becher contendo a suspensão
de Cv nas temperaturas de 25 e 50ºC mantendo sob agitação nessa temperatura
por 2h. Utilizou-se ainda a temperatura de adição de 100ºC apenas para a
solução 10% (m/m) de platina. Esta adição foi realizada em balão acoplado a um
condensador de bolas para evitar perda de material durante o tempo de refluxo
de 2h. Em seguida a suspensão foi transferida para um becher. Esta amostra
recebeu o código Pt+2(10%)/Cv_100ºC.
Todos os sistemas permaneceram sob agitação e foram secos a
temperatura de 50ºC até massa constante.
Tabela 1 - Código das amostras precursoras dos catalisadores Pt/Cv.
Código da amostra*
Pt+2(10%)/Cv_25ºC
Pt+2(15%)/Cv_25ºC
Pt+2(20%)/Cv_25ºC
Pt+2(10%)/Cv_50ºC
Pt+2(15%)/Cv_50ºC
Pt+2(20%)/Cv_50ºC
Pt+2(10%)/Cv_100ºC
*Nos códigos mostra-se: O íon adicionado (composição da espécie iônica
metálica)/suporte de catalisador_temperatura de adição em oC.
3.2.2.3 - Adição da solução de Pt(NH3)4Cl2 à suspensão de Cg
Utilizou-se um procedimento semelhante ao descrito no tópico 3.2.2.2
para adicionar a solução aquosa de Pt(NH3)4Cl2 com 10% de platina à
suspensão de Cv. A adição foi realizada na temperatura de 25ºC. Esta amostra
recebeu o código de Pt(10%)/Cg_25ºC.

30
3.2.2.4 - Redução a Temperatura Programada (TPR)
Todos as amostras, previamente secas, foram transferidas para cadinhos
de porcelana e calcinadas em mufla por 1 h. Em seguida as amostras foram
reduzidas por TPR.
Para a redução o catalisador foi aquecido a taxa de aquecimento de
10 ºC/min, de 25 a 550ºC, permanecendo nesta temperatura por mais 1 h.
Durante esse procedimento, a amostra é mantida sob fluxo de 30 mL/min de
uma mistura 1,74% de H2/Ar. O consumo de H2 foi monitorado contra
temperatura, utilizando um detector de condutividade térmica.
3.2.3 - Preparação do sistema Co(NO3)2.6H2O em Cv
3.2.3.1 - Preparo da solução aquosa de Co(NO3)2.6H2O
Foram preparadas soluções aquosas 10, 15 e 20% de cobalto (m/m)
utilizando o sal Co(NO3)2.6H2O, por sonicação durante 10 minutos.
3.2.3.2 - Adição da solução aquosa de Co(NO3)2.6H2O ao Cv
Com o auxílio de um funil de separação, a adição da solução aquosa de
Co(NO3)2.6H2O com 10,15 e 20% (m/m) de cobalto foram adicionadas
lentamente ao becher contendo a suspensão de Cv na temperatura de 50ºC. A
adição da solução aquosa de Co(NO3)2.6H2O com 10% de cobalto também foi
adicionada a suspensão de Cv na temperatura de 25ºC. Esta amostra recebeu o
código Co+2(10%)C_25ºC.
Após a adição das soluções de cobalto, a temperatura dos sistemas foi
mantida a 50ºC sob agitação.

31
Tabela 2 - Código das amostras precursoras das espécies Co/Cv.
Código da amostra*
Co+2(10%)/Cv_50ºC
Co+2(15%)/Cv_50ºC
Co+2(20%)/Cv_50ºC
Co+2(10%)Cv_25ºC
*Nos códigos mostra-se: O íon adicionado (composição da espécie iônica
metálica)/suporte de catalisador_temperatura de adição em oC.
3.2.3.3 - Redução química do Co+2
Em cada um dos sistemas citados no tópico 3.2.3.2 foi adicionado 20 mL
da solução aquosa de sulfato de hidrazina 5% m/v. Em seguida, adicionou-se
NH4OH 10% v/v até pH 9 na solução.
3.2.3.4 - Redução completa do Cobalto
Todas as misturas resultantes do tópico 3.2.3.3 permaneceram sob
agitação e temperatura de 50 ºC até peso constante. As amostras
Co+2(10%)Cv_25ºC, Co+2(10%)/Cv_50ºC, Co+2(15%)/Cv_50ºC,
Co+2(20%)/Cv_50ºC foram tratadas por TPR na taxa de aquecimento de
10 º C/min, variando-se a temperatura de 25 ºC a 900ºC, e permanecendo nesta
temperatura por 1 h.
Tabela 3 - Código das amostras das espécies Co/Cv.
Código da amostra*
Co(10%)/Cv_50ºC
Co(15%)/Cv_50ºC
Co(20%)/Cv_50ºC
Co(10%)Cv_25ºC
*Nos códigos mostra-se: espécie metálica adicionada e reduzida (composição da
espécie iônica metálica)/suporte de catalisador_temperatura de adição em oC.

32
3.2.4 - Sistema com adição simultânea dos sais de platina e cobalto
As soluções de Pt(NH3)4Cl2 e Co(NO3)2.6H2O, ambas com 10, 15 e 20%
dos respectivos cátions, foram preparadas da mesma forma descrita nos tópicos
3.2.2.1 e 3.2.3.1 As soluções de platina e cobalto contendo as mesmas
concentrações foram misturadas sob sonicação por 10 minutos.
Cada uma das misturas foi adicionada a suspensões de carbono Cv de
forma semelhante à descrita anteriormente.
A adição da mistura das soluções de platina 10% e cobalto 10% à
suspensão de carbono Cv foi feita nas temperaturas de 25 e 50 ºC. A adição das
outras misturas das soluções de platina e cobalto, ambas com 15 e 20%, à
suspensão de carbono Cv foi realizada na temperatura de 50 ºC. Para a redução
dos íons metálicos de todas as amostras citadas acima seguiu-se o mesmo
procedimento adotado para a redução química do íon Co+2 no tópico 3.2.3.3 e
em seguida realizou-se um TPR nestas amostras. Para o TPR destas amostras
utilizou-se as mesmas condições descritas no tópico 3.2.3.4.
Tabela 4 – Código das amostras precursoras dos catalisadores bimetálicos
Pt-s-Co/Cv.
Código da amostra*
Pt+2(10%)_s_Co+2(10%)/Cv_25ºC
Pt+2(10%)_s_Co+2 (10%)/Cv_50ºC
Pt+2(15%)_s_Co+2(15%)/Cv_50ºC
Pt+2(20%)_s_Co+2(20%)/Cv_50ºC
*Nos códigos mostra-se: íon de platina (composição da espécie iônica
metálica)_s_íon de cobalto (composição da espécie iônica metálica)/suporte de
catalisador_temperatura de adição em oC. A letra “s” simboliza que a adição dos íons
ocorreu de forma simultânea.

33
3.2.5 - Adição e redução do segundo metal
Com exceção das amostras em que ocorreram adição simultânea, após a
redução completa da primeira espécie iônica metálica em cada amostra, iniciou-
se o procedimento de adição do segundo metal. Estes procedimentos foram
idênticos ao descrito anteriormente para os respectivos metais. Após adição do
segundo metal, realizou-se a redução do mesmo mediante os procedimentos já
descritos nos tópicos acima, para cada metal.
Exceto para uma dessas amostras, o segundo metal foi adicionado com a
mesma concentração em que o primeiro metal foi adicionado. A única exceção
foi realizada em uma das amostras Pt(10%)/Cv_25ºC que recebeu adição de
solução aquosa 20% de cobalto (m/m). Esta amostra recebeu o código de
Pt+2(10%)Co+2 (20%)/Cv_25ºC. As amostras com adição inicial de platina
receberam os códigos, de acordo com o mostrado na Tabela 5.
Tabela 5: Código das amostras precursoras dos catalisadores bimetálicos Pt-
Co/Cv
Código da amostra*
Pt+2(10%)Co+2(10%)/Cv_100ºC
Pt+2(10%)Co+2(10%)/Cv_25ºC
Pt+2(15%)Co+2(15%)/Cv_25ºC
Pt+2(20%)Co+2(20%)/Cv_25ºC
Pt+2(10%)Co+2(10%)/Cv_50ºC
Pt+2(20%)Co+2(20%)/Cv_50ºC
Pt+2 (10%)Co+2(20%)/Cv_25ºC
Pt+2Co+2(10%)/Cg_25ºC
*Nos códigos mostra-se: Primeiro íon adicionado (composição da espécie iônica
metálica) Segundo íon adicionado (composição da espécie iônica metálica)/suporte de
catalisador_temperatura de adição em oC.
Na amostra Co(10%)/Cv, ou seja, que recebeu adição de solução de 10%
de cobalto na temperatura de 50ºC e posterior redução, adicionou-se em
seguida a platina com a mesma concentração em que o primeiro metal foi
adicionado (10%). Utilizou-se o procedimento semelhante ao descrito nos

34
tópicos 3.2.2.2. e 3.2.3.4. Esta amostra recebeu o código
Co+2(10%)Pt+2(10%)/Cv_50ºC.
3.2.6 - Formação da liga Pt-Co
As amostras que já receberam a adição do segundo metal ou as amostras
que receberam adição simultânea de Pt+2 e Co+2 receberam tratamento no TPR
com o objetivo inicial de promover a redução do segundo metal ou,
respectivamente, promover a redução dos dois metais adicionados. Entretanto, o
objetivo principal do TPR nesta fase do processo foi promover a formação das
ligas Pt-Co em altas temperaturas e em ambiente inerte.
Após passar por este procedimento, cada uma das amostras recebeu um
novo código, onde o símbolo dos metais com o nox +2 foram alterados nos
códigos representando, assim, a redução do metal. Os códigos, seguindo a
mesma seqüência anterior passam a ser:
Tabela 6 - Código das amostras bimetálicas que receberam tratamento para
formação da liga.
Código da amostra*
Pt(10%)Co(10%)/Cv_100ºC
Pt(10%)Co(10%)/Cv_25ºC
Pt(15%)Co(15%)/Cv_25ºC
Pt(20%)Co(20%)/Cv_25ºC
Pt(10%)Co(10%)/Cg_25ºC
Pt(10%)Co(10%)/Cv_50ºC
Pt(20%)Co(20%)/Cv_50ºC
Pt(10%)Co(20%)/Cv_25ºC
Co(10%)Pt(10%)/Cv_50ºC
Pt(10%)_s_Co(10%)/C_25ºC
Pt(10%)_s_Co(10%)/C_50ºC
Pt(15%)_s_Co(15%)/C_50ºC
Pt(20%)_s_Co(20%)/C_50ºC

35
* Nos códigos mostra-se: Primeira espécie metálica adicionada e reduzida
(composição da espécie iônica metálica) Segundo espécie metálica adicionada e
reduzida (composição da espécie iônica metálica)/suporte de catalisador_temperatura
de adição das espécies iônicas metálicas em oC.
3.2.7- Método de redução empregando-se ácido fórmico
Foi adicionado 30 mL de ácido fórmico concentrado em um becher de 400
mL contendo 1g de carbono. O sistema foi submetido a sonicação por 5 minutos
e a seguir permaneceu em repouso, à temperatura ambiente, por 40 minutos.
Após o repouso, a suspensão de carbono foi agitada mecanicamente. A esta
suspensão foi adicionada uma solução aquosa 0,017 M (solubilização de
0,2108g em 30 mL de água) de ácido hexacloroplatínico (H2PtCl6), de coloração
amarela, preparada com auxílio do ultrasom. Durante a adição, e após a
descoloração da solução, a mistura permaneceu em agitação constante na
temperatura de 50ºC até a evaporação completa da água. Essa amostra foi
caracterizada por DRX para determinação de tamanho de partícula e parâmetro
de rede, para comparação. Esta amostra recebeu o código Pt(10%)/C –
Ac.fórmico.
Uma segunda amostra foi preparada procedendo-se de maneira
semelhante ao descrito acima. A diferença é que, nesta segunda, a área
superficial do carbono Vulcan foi ativada antes da adição do ácido fórmico. Para
promover a ativação, o carbono permaneceu por duas horas em aquecimento de
300ºC. Esta amostra recebeu o código Pt(10%)/C – Ac.fórmico_ativ.
Tabela 7- Códigos das amostras preparadas pelo método do ácido fórmico.
Código da amostra*
Pt(10%)Co(10%)/Cv_Ac.fórmico
Pt(10%)Co(10%)/Cv_Ac.fórmico_ativ.

36
A Figura 6 apresenta o fluxograma de forma resumida do procedimento
experimental.
Precursores do Carbono
Metodologias Metodologia
Ácido fórmico
Pt(NH3)4Cl2
Impregnação
Precursor da platina H2PtCl6
composição da espécie iônica metálica
10%
20% 10% 10% 100ºC 50ºC 25ºC
Co(NO3)2.14H2O
Carbono Vulcan XC72R
Impregnação
composição da espécie iônica metálica
Temperatura de Adição dos precursores
1º Pt 1º Co simultânea 15%
Ordem de adição dos precursores
Precursores dos metais
Grafite do Brasil
Redução completa das espécie
iônica metálica por TPR
Temperatura de secagem
50ºC
Figura 6: Fluxograma do procedimento experimental

37
A Figura 7 apresenta o fluxograma do procedimento experimental relativo
ao preparo das amostras que tiveram adição inicial de Pt.
Figura 7- Fluxograma do procedimento experimental do preparo das amostras
com adição inicial de Platina

38
3.3 - Caracterização espectroscópica, estrutural e morfológica
Os materiais foram caracterizados pelas seguintes técnicas:
3.3.1 - Espectoscopia Vibracional no Infravermelho por Transformada de
Fourier (FTIR)
As análises de FTIR foram realizadas usando um Espectrofotômetro
Nicolet Magna IR-760 na faixa de 4000 a 400 cm-1 com resolução de 1 cm-1 e
128 varreduras. As amostras foram preparadas utilizando-se pastilhas de KBr.
3.3.2 - Difração de Raios-X (DRX)
Análises de DRX foram realizadas em um Difratômetro de Raios-X
Miniflex da Rigaku (V= 15 kV, I= 30 mA) que utiliza radiação k de cobre
(=15418 Å). O intervalo angular, 2, de 5-90° foi varrido com passos de 1°,
utilizando tempo de contagem de 1s.
3.3.3 - Microscopia Eletrônica de Varredura (MEV)
Análises de MEV foram realizadas no aparelho Zeiss DSM 940. As
amostras foram dispersas em propanol e tratadas em ultrasom por 12 minutos.
Gotas desta dispersão foram colocadas sobre o porta amostra de alumínio e
seca a vácuo, seguida da metalização com ouro.
3.3.4 - Redução a Temperatura Programada (RTP)
Análises de Redução a Temperatura Programada (RTP) foram realizadas
em um equipamento em fluxo, que utiliza um microreator de quartzo em forma
de U. Antes do início da redução, o catalisador foi pré-tratado a 300oC por 2 h,
sob fluxo de argônio, de modo a remover toda a água para não interferir na
análise. Em seguida, o catalisador foi aquecido sob fluxo de 30 ml/min com uma
mistura 1,74% de H2/Ar e taxa de aquecimento de 10oC/min, de 25oC até 550oC,
permanecendo nesta temperatura por mais 1 h. O consumo de H2 foi monitorado
em função da temperatura, utilizando um detector de condutividade térmica.

39
3.3.5 - Área Superficial (SBET)
A determinação das propriedades texturais dos suportes foi feita através
da técnica de isotermas de adsorção/dessorção de N2 a –196oC, em
equipamento Micromeritics ASAP (Accelerated Surface Area and Porosimetry)
modelo 2000. A amostra, previamente seca em estufa a 100oC por 2h, foi
submetida à pré-tratamento no próprio equipamento, que consistia no
aquecimento sob vácuo de 5x10-3 Torr de cerca de 250 mg de material, a 300oC
por 3h, para retirada da água superficial adsorvida. Após o tratamento a amostra
era pesada novamente para determinação exata da massa da amostra, sob
atmosfera de N2, após permitir o equilíbrio entre o gás e o sólido. A superfície
específica foi calculada pelo ajuste da isoterma de BET.

40
4. Resultados e Discussão
4.1. Catalisadores Suportados em carbono grafite, Cg
4.1.1 Infravermelho
A Figura 8 mostra os espectros vibracionais no infravermelho das
amostras Pt/C e CoPt/C antes e após o TPR e o do grafite para comparação. As
amostras antes do TPR apresentam em seus códigos os respectivos metais na
forma catiônica. O único espectro a apresentar picos é o da amostra
Pt(10%)Co+2(10%)/Cg, ou seja, antes do TPR. Neste, pode-se observar uma
banda centrada em 3153, que pode ser atribuída à deformação axial da ligação
OH de água, visto que o precursor do cobalto é muito hidrofílico, e uma outra
banda intensa em 1377 cm-1 referente ao nitrato. Em decorrência da alta
temperatura utilizada para a formação da liga, essas bandas desapareceram
após o TPR.
4000 3500 3000 2500 2000 1500 1000 500
16
24
Carbono Grafite
Pt+2
(10%)/Cg
Pt(10%)/Cg
Pt(10%)Co+2
(10%)/Cg
Pt(10%)Co(10%)/Cg
Número de onda (cm-1)
% T
ransm
itância
Figura 8 - Espectro vibracional no infravermelho dos eletrocatalisadores
Pt(10%)Co(10%)/Cg, Pt(10%)/Cg e seus precursores.

41
4.1.2 Difração de Raios-X
A difração de raios X pela técnica de pó permite uma avaliação do retículo
cristalino. O difratograma obtido depende da estrutura do cristal, que por sua vez
é determinada por fatores como: tipo de retículo, classe do cristal, parâmetros de
cela unitária e a distribuição dos vários tipos de íons, moléculas que estão na
cela. O número de reflexões e posição destas, em termos da posição angular
(2θ) ou distância interplanar (d) e o perfil do difratograma dependem dos fatores
citados anteriormente e do comprimento de onda dos raios X aplicado na
amostra para a coleta dos dados. Já a intensidade do pico depende dos tipos de
átomos presentes e suas respectivas posições e do tempo de exposição da
amostra aos raios X.
Como esta técnica depende de tantos parâmetros, pode-se dizer que
cada sólido cristalino apresenta um padrão difratométrico característico, como
uma impressão digital, permitindo sua identificação através das posições
angulares e intensidades relativas dos picos difratados.
Os difratogramas dos catalisadores bimetálicos são característicos de
sólidos cristalinos, o que permite qualificar possíveis alterações que essas
estruturas possam apresentar.
Para estimar a média do tamanho das nanopartículas de Pt formadas, foi
escolhido o pico em 2θ ~ 39,7° (111) do difratograma, utilizando a equação de
Scherrer (equação 18) descrita abaixo.
cos
9,0
Bd Equação (18)
Onde:
d(111) = tamanho médio das partículas em Ângstrons.
k= constante que depende do formato da partícula (0,9).
= comprimento de onda de radiação usado (Cu =1,5418Å).
= largura a meia altura do pico difratado da amostra em radianos.
= ângulo de difração de Bragg do ponto máximo do pico analisado.

42
Para uma célula cúbica de parâmetro de rede, a, o plano que intercepta
os três eixos nos vértices da célula (x=y=z=a) terá os índices de Miller (111).
Pelas distâncias interatômicas entre os segundos vizinhos, pode-se calcular o
parâmetro de rede. Para calcular o parâmetro de rede foi usada a equação 19:
21
222 lkh
ad hkl
Equação (19)
onde:
d = distância interatômica em Ângstrons
a = parâmetro de rede
h, k, l = índices de Miller, que são definidos pela intersecção de um plano
de átomos com os eixos do sistema de coordenadas da célula unitária.
À distância interatômica é calculada segundo a equação de Bragg (Equação 20):
dsen2 Equação (20)
A Pt pura apresenta picos59 em 39,7 (111), 46,3 (200), 67,4 (220), 81,9
(311) e 86,0 o (222) sendo estas características da estrutura cúbica de face
centrada (cfc) e parâmetro de rede 0,3912 nm.
A partir do pico referente ao plano (111) será estimada a média do
tamanho das nanopartículas. Ao fazer esta estimativa de tamanho considera-se
que, durante a redução por TPR, os átomos metálicos migram e formam
agregados dispersos no suporte. A distribuição dos tamanhos é fortemente
dependente dos detalhes da preparação e não são fáceis de predizer já que o
fenômeno superficial que ocorre durante a preparação não é bem compreendido.
O DRX é sensível à massa, uma pequena fração de partículas maiores poderia
produzir picos de difração mais estreitos 60.
A Figura 9 mostra os difratogramas de raios-X das amostras Pt/C e
CoPt/C após o TPR. Observa-se na amostra CoPt/C que o pico referente ao
plano (111) da Pt sofre um pequeno deslocamento de 2θ = 39,7 para 39,9 o. Em
função da razão dos dois metais na liga, e da própria quantidade de liga, esse
deslocamento pode ser maior. No trabalho Huang e colaboradores, o máximo
desse pico varia de 39,9o, para a uma razão Pt:Co(5:1), para 40,49 o para a

43
razão Pt:Co(1:1). Neste trabalho, observou-se um deslocamento de 2θ = 0,2 o.
Mesmo esse pequeno deslocamento indica que houve formação de liga com
incorporação de Co na estrutura fcc da Pt.
A presença de alguma quantidade de uma fase Co3O4 segregada é
evidenciada pelo aparecimento de um padrão de difração a 2θ = 54,8o. Nota-se
ainda o pico a 2θ = 36,7o. Lima e colaboradores61 atribuíram um pico para uma
amostra Pt-Co/Cv a 2θ = 59o à formação dessa fase segregada.
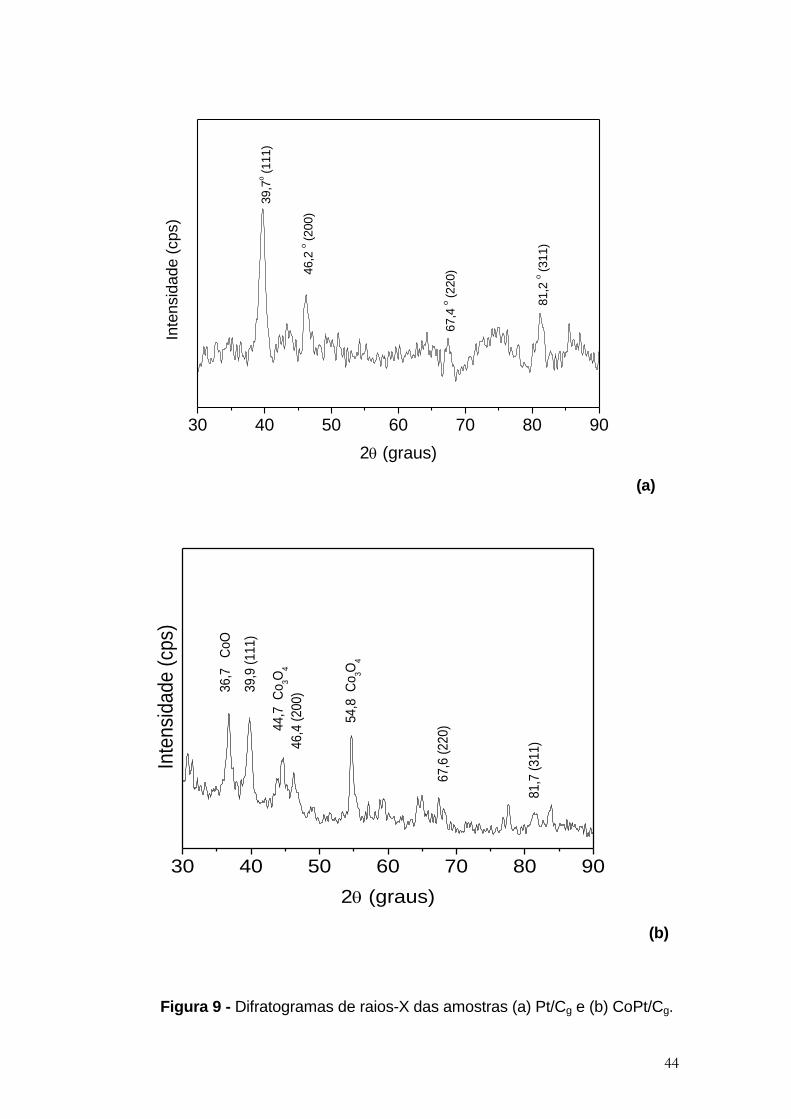
44
30 40 50 60 70 80 90
81,2
o (
311)
67,4
o (
220)46,2
o (
200)
39,7
o (
111)
Inte
nsid
ade (
cps)
2 (graus)
(a)
30 40 50 60 70 80 90
54,8
C
o3O
4
44,7
C
o3O
4
36,7
C
oO
81,7
(311)
67,6
(220)
46,4
(200)3
9,9
(111)
2 (graus)
Inte
nsi
da
de
(cp
s)
(b)
Figura 9 - Difratogramas de raios-X das amostras (a) Pt/Cg e (b) CoPt/Cg.

45
4.1.3 Termogravimetria
A Figura 10 mostra a curva TGA das amostras Pt/Cg e CoPt/Cg antes e
após TPR. Observam-se pelas curvas uma diferença no perfil da amostra
CoPt/C antes do TPR em relação ao perfil das demais amostras. Podemos
dividir a curva obtida da amostra CoPt/C em três etapas de decomposição: A
primeira, de 25 a 200oC, foi atribuída à perda de água superficial; a segunda, de
200 a 300oC, referente à perda de água interna no material; e a terceira, de 600
a 700 ºC, referente a perda de nitratos.
Na amostra CoPt/Cg após TPR observa-se um pico por volta de 736oC
sugerindo a formação de materiais voláteis provenientes de alguma reação
desenvolvida paralelamente.
0 200 400 600 800 1000
30
40
50
60
70
80
90
100
110
P t /C an tes do T P R
P t /C após T P R
C oP t /C an tes do T P R
C oP t /C após T P R
T em pera tu ra ( ºC )
Pe
rda
de
ma
ss
a (
%)
Figura 10 - Curvas de TGA das amostras Pt+2/Cg, Pt/Cg, Co+2Pt/Cg e CoPt/Cg.
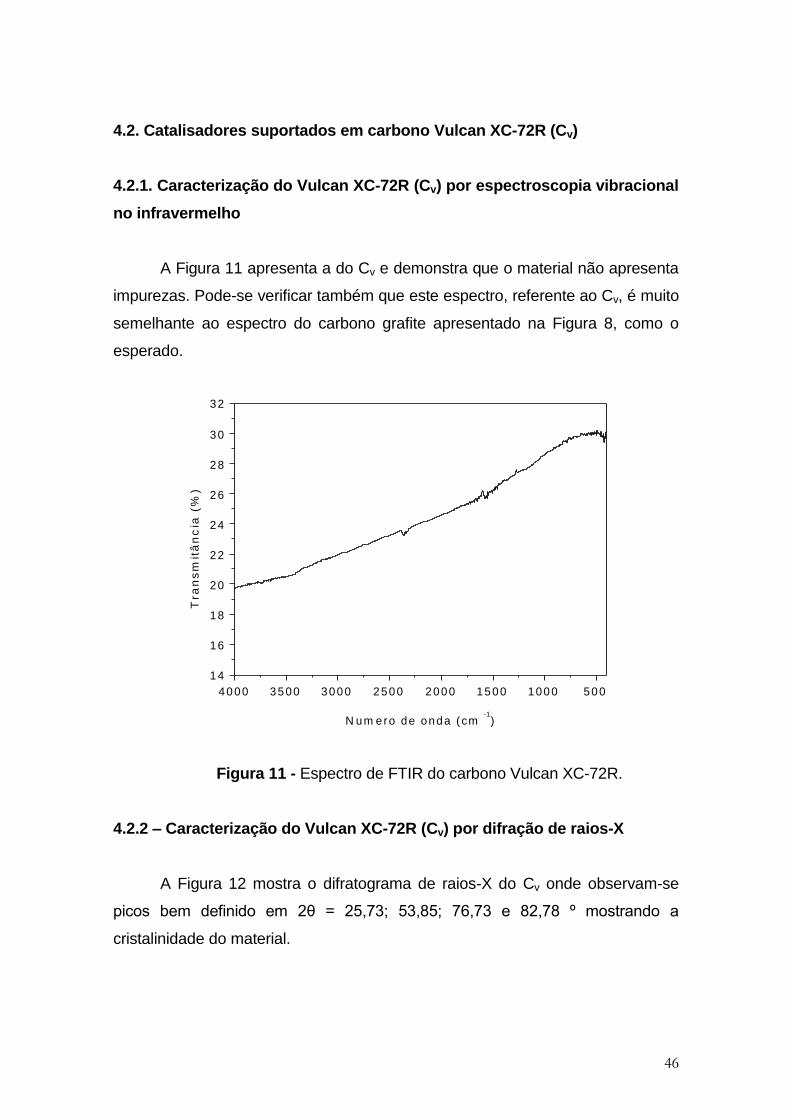
46
4.2. Catalisadores suportados em carbono Vulcan XC-72R (Cv)
4.2.1. Caracterização do Vulcan XC-72R (Cv) por espectroscopia vibracional
no infravermelho
A Figura 11 apresenta a do Cv e demonstra que o material não apresenta
impurezas. Pode-se verificar também que este espectro, referente ao Cv, é muito
semelhante ao espectro do carbono grafite apresentado na Figura 8, como o
esperado.
4000 3500 3000 2500 2000 1500 1000 500
14
16
18
20
22
24
26
28
30
32
Tra
ns
mit
ân
cia
(%
)
N um ero de onda (cm-1
)
Figura 11 - Espectro de FTIR do carbono Vulcan XC-72R.
4.2.2 – Caracterização do Vulcan XC-72R (Cv) por difração de raios-X
A Figura 12 mostra o difratograma de raios-X do Cv onde observam-se
picos bem definido em 2θ = 25,73; 53,85; 76,73 e 82,78 º mostrando a
cristalinidade do material.

47
1 0 2 0 3 0 4 0 5 0 6 0 7 0 8 0 9 0
Inte
ns
ida
de
(c
ps
)
2 (g raus )
Figura 12 - Difratograma de raios X do Cv
4.2.3. Método da impregnação: Sistema Pt/Cv
4.2.3.1 – Redução a Temperatura Programada
Para a redução dos íons de Pt+2 dispersos na matriz de Cv em diferentes
temperaturas foi utilizada a técnica de Redução a Temperatura Programada
(TPR).
As curvas de TPR apresentadas a seguir mostram os processos de
redução de Pt+2. As figuras 13 e 14 apresentam o TPR das amostras
Pt+2(10%)/Cv_25ºC e Pt+2(10%)/Cv_50ºC, respectivamente. Os perfis dos TPR
para as amostras com 10% (m/m) de Pt+2 disperso a 25 e 50 ºC não são
significantemente alterados e o consumo de H2 também permanece similar.
Existe um início de redução em torno de 100 ºC, provavelmente decorrente de
do processo inicial de nucleação e a redução com o conseqüente crescimento de
partícula torna-se mais intenso entre 200 e 350ºC. Não foi encontrado na
literatura indício de que o íon Pt2+ apresente dois estágios de redução
detectáveis por TPR.

48
100 200 300 400 500 600
-5
0
5
10
Consum
o d
e H
2
Temperatura (°C)
Figura 13 - Curvas de TPR da amostra Pt+2(10%)/Cv_25ºC.
200 400 600
-5
0
5
10
15
20
consum
o d
e H
2
temperatura (ºC)
Figura 14 - TPR realizado na amostra Pt+2(10%)/Cv_50ºC

49
As Figuras 15 e 16 apresentam as curvas de TPR das amostras
Pt+2(15%)/Cv_50ºC e Pt+2(20%)/Cv_50ºC.
Com o aumento da concentração de Pt+2 ocorre alteração no perfil das
curvas de TPR, como podemos verificar nas figuras 15 e 16, para as amostras
com 15 e 20% (m/m) de Pt+2, respectivamente. Existe um início de redução dos
íons com a formação dos primeiros núcleos e uma segunda e terceira etapas de
redução com máximos em 200 e 300ºC.
Não foram encontrados indícios de contaminação por outros metais,
entretanto, eventuais reações com moléculas adsorvidas não podem ser
descartadas, embora não tenha sido acoplado um massa ao TPR. Durante a
manipulação das amostras, em decorrência da dimensão nanométrica do
suporte, adsorção pode ter ocorrido em decorrência da exposição das amostras
na atmosfera do laboratório.
Os processos de redução e formação de nanopartículas de Pt são
afetados pela natureza do suporte que influencia fortemente no processo de
redução62,63,64. Nanopartículas de platina crescidas em zeólita Y utilizando-se a
mesma técnica de redução foram reduzidas a 100 ºC para 5% de Pt+2 (m/m) e
de 150 a 190oC para 1% de Pt+2 (m/m) 65,66. Aparentemente, concentração
menor de íons metálicos resultam em uma concentração maior de íons
fortemente ligados ao retículo, o que pode aumentar a temperatura de redução.

50
80 160 240 320 400 480 560
-5
0
5
10
consum
o d
e H
2
temperatura (ºC)
Figura 15 - TPR realizado na amostra Pt+2(15%)/Cv_50ºC.
100 200 300 400 500 600
-5
0
5
10
Consum
o d
e H
2
Temperatura (ºC)
Figura 16 - TPR realizado na amostra Pt+2(20%)/Cv_50ºC.

51
4.2.3.2 – Difratogramas de Raios-X
A Figura 17 apresenta o DRX da amostra Pt+2(10%)/C-50ºC onde são
mostrados os picos de difração da Pt a 39,95; 46,45; 67,70; 81,45 e 85,90. Estes
são atribuídos aos planos cristalográficos (111), (200), (220), (311) e (222),
respectivamente67.
O tamanho médio das partículas de platina pode ser calculado com base
no plano (111) de acordo com a fórmula de Scherrer68,69 como mostrado na
Equação 18. Onde λ é 0,154056 e B é o valor a meia altura do pico analisado,
em radianos.
Figura 17- DRX da amostra Pt(10%)/Cv_50ºC
30 35 40 45 50 55 60 65 70 75 80 85 90
(222)(311)(220)(111) (200)
inte
nsid
ade (
u.a
.)
2 (graus)

52
A Figura 18 mostra as curvas de DRX para as amostras Pt
(20%)/Cv_50°C, Pt (15%)/Cv_50 °C, Pt (10%)/Cv_ 50 °C, Pt (20%)/C – Comercial
para 2θ entre 35 e 55 graus, região dos picos referentes aos planos (111) e
(200) da platina. Observam-se esses picos em todas as amostras, entretantos,
estes são menos definidos e mais largos na amostra Pt (20%)/C – Comercial.
Normalmente, quanto menor a dimensão das partículas de determinado material,
menos definidos são os picos na curva de DRX. As partículas da amostra
Electrochem possuem tamanho médio de 2nm. Sendo assim, estima-se pelas
curvas de DRX que as partículas obtidas neste trabalho tenham maiores
dimensões.
35 40 45 50 55
(111) (200) Pt (20%)/C - 50 °C
Pt (15%)/C - 50 °C
Pt (10%)/C - 50 °C
Pt (20%)/C - Comercial
inte
nsid
ade (
u.a
.)
2 (graus)
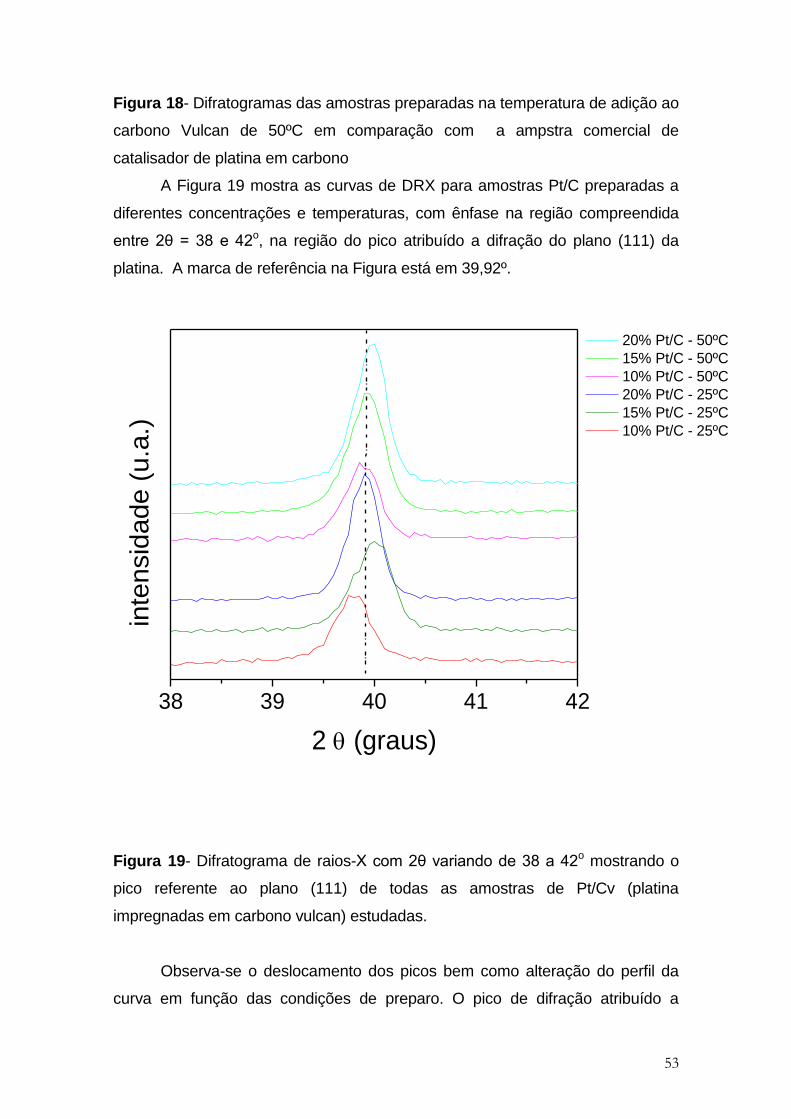
53
Figura 18- Difratogramas das amostras preparadas na temperatura de adição ao
carbono Vulcan de 50ºC em comparação com a ampstra comercial de
catalisador de platina em carbono
A Figura 19 mostra as curvas de DRX para amostras Pt/C preparadas a
diferentes concentrações e temperaturas, com ênfase na região compreendida
entre 2θ = 38 e 42o, na região do pico atribuído a difração do plano (111) da
platina. A marca de referência na Figura está em 39,92º.
Figura 19- Difratograma de raios-X com 2θ variando de 38 a 42o mostrando o
pico referente ao plano (111) de todas as amostras de Pt/Cv (platina
impregnadas em carbono vulcan) estudadas.
Observa-se o deslocamento dos picos bem como alteração do perfil da
curva em função das condições de preparo. O pico de difração atribuído a
38 39 40 41 42
20% Pt/C - 50ºC
15% Pt/C - 50ºC
10% Pt/C - 50ºC
20% Pt/C - 25ºC
15% Pt/C - 25ºC
10% Pt/C - 25ºC
inte
nsid
ade (
u.a
.)
2 (graus)

54
amostra Pt(15%)/Cv_25ºC apresentou maior deslocamento em relação aos
demais. Esta mesma amostra apresentou o menor parâmetro de rede e o menor
tamanho de partícula entre as amostras em questão. Esses parâmetros são
mostrados na Tabela 8.
Tabela 8: Tamanho médio de partículas e parâmetros de rede das amostras do tipo Pt/Cv preparadas nas temperaturas de 25 e 50 ºC.
Amostra Tamanho de partícula (nm)
Parâmetro de rede (nm)
Pt(20%)/Cv – 50ºC 22,328 3,907
Pt(15%)/Cv – 50ºC 19,298 3,911
Pt(10%)/Cv – 50ºC 20,465 3,915
Pt(20%)/Cv – 25ºC 23,666 3,913
Pt(15%)/Cv – 25ºC 17,962 3,905
Pt(10%)/Cv – 25ºC 18,800 3,924
A Figura 20 mostra as curvas de DRX com 2 entre 32 e 42o, na região do
pico referente ao plano (111) para as amostras preparadas pelo método da
impregnação em diferentes temperaturas (25, 50 e 100ºC) com 10% (m/m) de
platina em relação ao suporte.
Observa-se que de forma geral, menores concentrações de Pt2+ e menores
temperaturas na etapa de impregnação resultam em partículas de menores
dimensões.
A Tabela 9 mostra os tamanhos médios das partículas e os parâmetros
de rede calculados com base no pico atribuído ao plano (111) para estas
amostras, exceto para a amostra preparada a 100ºC.
Na Figura 20, o difratograma da amostra obtida com refluxo a 100°C
mostra um o pico a 2θ = 34,74°. Esse pico foi atribuído à formação de PtO2 por
Zhi Qun Tian e colaboradores70, em trabalho similar a este. Os autores atribuem
à formação do óxido a inexistência do pico a 2 = 39,92° referente ao plano
(111). Portanto, conclui-se que a temperatura de refluxo de 100 oC favoreceu a
formação do óxido que não foi reduzido por TPR na temperatura empregadas. A
formação de óxido não foi observada para nenhuma outra amostra em nenhuma
outra concentração.

55
Figura 20 - Detalhe da região entre 32 e 42 graus dos difratogramas de raios-x
das amostras de platina 10% em carbono vulcan preparadas em diferentes
temperaturas.
Tabela 9 - Tamanho médio de partícula e o parâmetro de rede das amostras
preparadas pelo método da impregnação em diferentes temperaturas (25, 50 ºC)
com 10% (m/m) de platina em relação ao suporte.
Amostra Tamanho de partícula (nm)
Parâmetro de rede
Pt(20%)/C - comercial 2,279 3,913
Pt(10%)/Cv_25ºC 18,800 3,924
Pt(10%)/Cv_50ºC 20,465 3,915
35 40
(111)
PtO2
Pt(10%)/C - 25ºC
Pt(10%)/C - 50ºC
Pt(10%)/C - 100ºC
inte
nsid
ade (
u.a
.)
2 (graus)

56
4.2.3.3 – Microscopia Eletrônica de Varredura
As Figuras 21 e 22 mostram imagens de microscopia eletrônica de
varredura da amostra Pt(10%)/Cv_ 50ºC em duas ampliações diferentes. As
partículas com tamanho médio de 20 nm não podem ser observadas na
resolução do MEV. Entretanto, a eventual formação de agregados maiores do
metal pode ser observada na Figura 21 e com melhor resolução na Figura 22.
Figura 21 - Imagem de Microscopia Eletrônica de Varredura da amostra
Pt(10%)/Cv_50ºC com um ampliação de 2000 vezes.
Figura 22- Microscopia Eletrônica de Varredura da amostra Pt(10%)/Cv_50ºC
com um zoom de 20000 vezes.
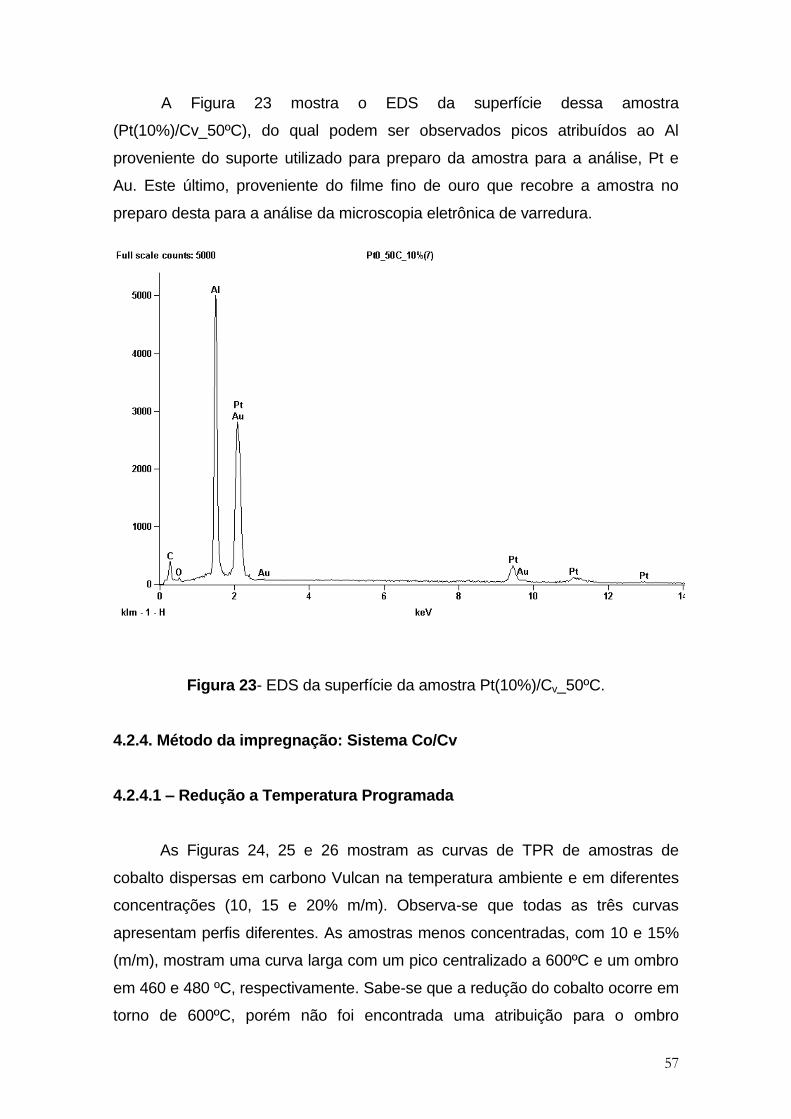
57
A Figura 23 mostra o EDS da superfície dessa amostra
(Pt(10%)/Cv_50ºC), do qual podem ser observados picos atribuídos ao Al
proveniente do suporte utilizado para preparo da amostra para a análise, Pt e
Au. Este último, proveniente do filme fino de ouro que recobre a amostra no
preparo desta para a análise da microscopia eletrônica de varredura.
Figura 23- EDS da superfície da amostra Pt(10%)/Cv_50ºC.
4.2.4. Método da impregnação: Sistema Co/Cv
4.2.4.1 – Redução a Temperatura Programada
As Figuras 24, 25 e 26 mostram as curvas de TPR de amostras de
cobalto dispersas em carbono Vulcan na temperatura ambiente e em diferentes
concentrações (10, 15 e 20% m/m). Observa-se que todas as três curvas
apresentam perfis diferentes. As amostras menos concentradas, com 10 e 15%
(m/m), mostram uma curva larga com um pico centralizado a 600ºC e um ombro
em 460 e 480 ºC, respectivamente. Sabe-se que a redução do cobalto ocorre em
torno de 600ºC, porém não foi encontrada uma atribuição para o ombro

58
observado. Provavelmente, este corresponde a uma reação decorrente de
substâncias adsorvidas nas partículas de suporte ou catalisador durante a
manipulação das amostras, ou seja, a impurezas decorrentes de falhas na
manipulação.
Figura 24- TPR realizado na amostra Co+2(10%)/Cv_25ºC
Figura 25-TPR realizado na amostra Co+2 (15%)/Cv_25ºC
100 200 300 400 500 600 700 800 900
0
20
40
60
80
100
Consum
o d
e H
2
Temperatura
100 200 300 400 500 600 700 800 900
0
20
40
60
80
100
Consum
o d
e H
2
Temperatura (ºC)

59
Observa-se da Figura 26 que o perfil da curva de TPR da amostra com
20% de Co (m/m) é semelhante ao perfil da amostra com 10%, porém a curva
apresenta apenas a banda com máximo em 600ºC na amostra mais
concentrada, como seria esperado também para as concentrações mais baixas.
100 200 300 400 500 600 700 800 900
0
20
40
60
80
100
C
onsum
o d
e H
2
Temperatura (ºC)
Figura 26- TPR realizado na amostra Co+2(20%)/Cv_25ºC .
As Figuras 27, 28 e 29 mostram as curvas de TPR das amostras de Co2+
10, 15 e 20% (m/m), respectivamente, dispersos em carbono Vulcan
impregnadas a 50ºC. Pode-se observar que o perfil da curva de TPR da amostra
menos concentrada em Co, de código Co+2(10%)/Cv_50ºC, mostra um perfil
diferente das outras duas curvas, porém mais aproximado do esperado para
esse tipo de amostra. As curvas de TPR das amostras Co+2(15%)/Cv_50ºC e
Co+2(20%)/Cv_50ºC apresentam um maior consumo de H2 no máximo das
respectivas curvas, o que é coerente com a maior concentração do íon metálico
a ser reduzida. Da mesma forma, como ocorre com as curvas das amostras
preparadas à temperatura ambiente, observa-se que a amostra preparada com
15% de Co apresenta um ombro, em torno de 470ºC, de maior intensidade em
relação as demais curvas das amostras também preparadas a 50ºC, atribuído
também a reações ocorridas paralelamente.As figuras 27,28 e 29 mostram as

60
curvas de TPR das amostras de Cobalto 10, 15 e 20% (m/m), respectivamente,
dispersos em carbono Vulcan na temperatura de impregnação igual a 50ºC.
Pode-se observar que o perfil da curva de TPR da amostra menos
concentrada em Co, de código Co+2 (10%)/Cv_50ºC, mostra um perfil diferente
das outras duas curvas.
As curvas de TPR das amostras Co+2(15%)/Cv_50ºC e
Co+2(20%)/Cv_50ºC apresentam um maior consumo de H2 no máximo das
respectivas curvas. Da mesma forma como ocorre com as curvas das amostras
preparadas na temperatura de ambiente, podemos observar que a amostra
preparada com 15% de Co apresenta um ombro, em torno de 470 ºC, de maior
intensidade em relação às demais curvas das amostras também preparadas a
50 ºC, atribuído também a reações ocorridas paralelamente.
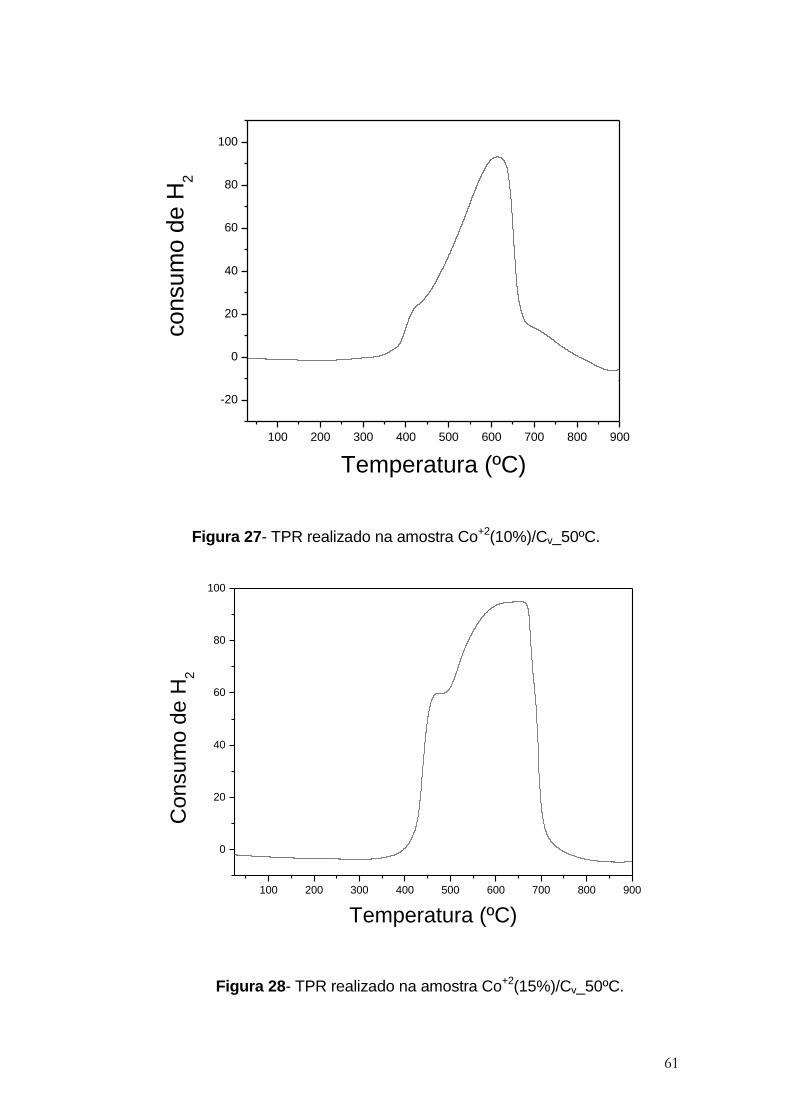
61
100 200 300 400 500 600 700 800 900
-20
0
20
40
60
80
100
consum
o d
e H
2
Temperatura (ºC)
Figura 27- TPR realizado na amostra Co+2(10%)/Cv_50ºC.
100 200 300 400 500 600 700 800 900
0
20
40
60
80
100
Consum
o d
e H
2
Temperatura (ºC)
Figura 28- TPR realizado na amostra Co+2(15%)/Cv_50ºC.

62
100 200 300 400 500 600 700 800 900
-10
0
10
20
30
40
50
60
70
80
90
100
Consum
o d
e H
2
Temperatura (ºC)
Figura 29- TPR realizado na amostra Co+2(20%)/Cv_ 50ºC.
4.2.4.2 - Difratogramas de Raios-X
Embora se tenha comprovado a redução do íon cobalto pelo estudo por
TPR, com o estudo realizado por difração de raios-X, não foi possível atribuir os
picos do Co. Segundo Xin e colaboradores, o Co puro sobre partículas de Vulcan
apresenta três picos de baixa intensidade no difratograma, a 2θ igual a 44,8;
52,0 e 76o atribuídos aos planos (111), (200) e (220) do Co71.
Nos difratogramas obtidos neste trabalho não foi possível separar o sinal
desses picos do ruído, provavelmente, por problemas no equipamento que
impediram a acumulação de sinal suficiente para a realização precisa da análise.
Essas amostras estão sendo refeitas e os resultados de sua caracterização
serão apresentados em trabalho futuro.
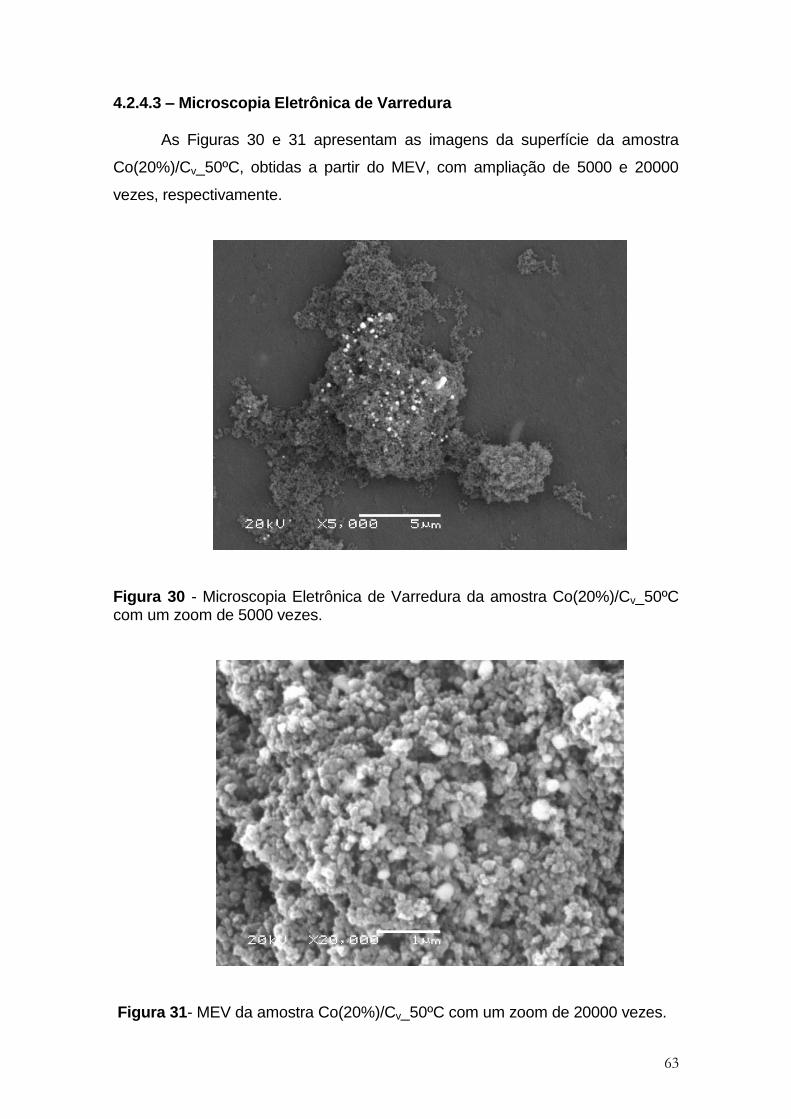
63
4.2.4.3 – Microscopia Eletrônica de Varredura As Figuras 30 e 31 apresentam as imagens da superfície da amostra
Co(20%)/Cv_50ºC, obtidas a partir do MEV, com ampliação de 5000 e 20000
vezes, respectivamente.
Figura 30 - Microscopia Eletrônica de Varredura da amostra Co(20%)/Cv_50ºC com um zoom de 5000 vezes.
Figura 31- MEV da amostra Co(20%)/Cv_50ºC com um zoom de 20000 vezes.

64
Observam-se regiões de coloração branca que são atribuídas a grandes
agregados de Co, já que a resolução do MEV não permite detectar a presença
de nanopartículas. Pode-se verificar que o Co não foi bem disperso como a
platina. Sugere-se para trabalhos futuros submeter a amostra à tratamento por
sonificação com aquecimento na impregnação do Cv com a solução do sal de
Co, ou utilizar a técnica de codeposição de íons de cobalto e platina para
aumentar a homogeneidade da dispersão.
4.2.4. Método da impregnação: Sistemas PtCo/Cv
4.2.4.1 – Redução a Temperatura Programada
As Figuras 32 e 33 mostram as curvas de TPR das amostras
Pt+2(10%)Co+2(10%)/Cv_50ºC e Pt+2(10%)_s_Co+2(10%)/Cv_50ºC, onde a
primeira amostra foi preparada com adição inicial de platina e a segunda com
adição simultânea de platina e cobalto. Observa-se que para a primeira amostra,
Figura 32, a curva mostra um primeiro processo de redução com baixo consumo
de H2 e máximo em torno de 250ºC, seguido de um segundo processo com
consumo grande de H2 entre 500 e 800ºC. O primeiro processo pode ser
atribuído à redução da platina e o segundo à redução do cobalto que para as
amostras anteriores apresentava consumo de H2 máximo por volta de 600ºC.
Como o segundo processo se estende a aproximadamente 800oC, isso pode ser
indicativo da formação da liga Co/Pt, que requer maior energia. Na Figura 33
porém, não foi observado o mesmo comportamento de consumo de H2.
Observou-se o sinal em torno de 550ºC e dois ombros em torno de 400ºC e 530
oC. A ordem de adição dos íons metálicos precursores parece influenciar no
perfil das curvas de TPR e, portanto, na extensão da formação da liga Pt/Co.
Caso haja formação de liga, esta normalmente é evidenciada nos difratogramas
de raios-x da amostra que mostra um ombro ou pequenos deslocamentos do
pico (111) da platina. A força da interação do íon com o substrato influencia na
temperatura de redução.

65
100 200 300 400 500 600 700 800 900
-20
0
20
40
60
80
100
consum
o d
e H
2
Temperatura (ºC)
Figura 32- TPR realizado na amostra Pt+2(10%)Co+2(10%)/Cv_50ºC.
100 200 300 400 500 600 700 800 900
-20
0
20
40
60
80
100
consum
o d
e H
2
temperatura (ºC)
Figura 33- TPR realizado na amostra Pt+2(10%)_s_Co+2(10%)/Cv_50ºC.

66
4.2.5.2 – Difratogramas de Raios-X
Na Figura 34, são mostrados os difratogramas das amostras preparadas
para 10% (m/m) de platina e cobalto em relação ao carbono Vulcan e nas
mesmas temperaturas de adição (50ºC). A diferença no preparo das mesmas foi
a adição dos íons metálicos, que se encontra descrita na figura.
30 35 40 45 50 55 60 65 70 75 80 85
(311)(220)(111) (200)
(10%)PtCo/C - 50ºC - adição inicial de Pt
(10%)PtCo/C - 50ºC - adição inicial de Co
(10%)PtCo/C - 50ºC - adição simultânea
inte
nsid
ade (
u.a
.)
2 (graus)
Figura 34- Difratogramas das amostras de Pt e Co com diferentes ordens de
adição dos precursores dos metais sobre carbono Vulcan.
A tabela 10 apresenta o tamanho de partícula de todas as amostras
citadas na figura 34, bem como os seus respectivos parâmetros de rede.

67
Tabela 10: Tamanho de partícula e parâmetros de rede das amostras de Pt e
Co preparadas com ordem de adição de reagentes alterados.
Amostra Tamanho de partícula (nm)
Parâmetro de rede (nm)
Pt/Co(10%)/Cv_50 °C simultaneo 15.293 3.915
Pt/Co(10%)/Cv_50 °C (1º Co) 18.197 3.909
Pt/Co(10%)/Cv_50 °C (1º Pt) 21.570 3.911
Como já evidente da análise dos difratogramas de raios-X, a amostra com
adição simultânea de precursores apresentou os menores tamanhos de
partícula, de aproximadamente 15 nm. Aparentemente, a adição simultânea
favorece a formação de partículas menores.
A Figura 35 apresenta a variação do parâmetro de rede em relação a
concentração de metais para cada amostra que contém platina, preparadas nas
temperaturas de 25 e 50ºC. Observa-se que nas amostras preparadas a 50ºC os
parâmetros de rede obtidos apresentaram valores relativamente estáveis, em
relação ao perfil da curva que representa a variação do parâmetro de rede de
Pt/C (25oC). Observa-se também que ocorre uma queda no valor do parâmetro
de rede da platina ao adicionarmos o cobalto no sistema. Nas duas temperaturas
de preparo, ocorre um valor menor para as amostras PtCo/C em relação a Pt/C,
sendo detectada uma diferença maior para as amostras preparadas a
temperatura ambiente. O perfil da curva muda significativamente para as
amostras preparadas a 25ºC. Nota-se que as amostras nesta temperatura
apresentam variações aleatórias, enquanto que, as amostras preparadas na
temperatura de 50ºC apresentam um parâmetro de rede inversamente
proporcional à concentração de platina.
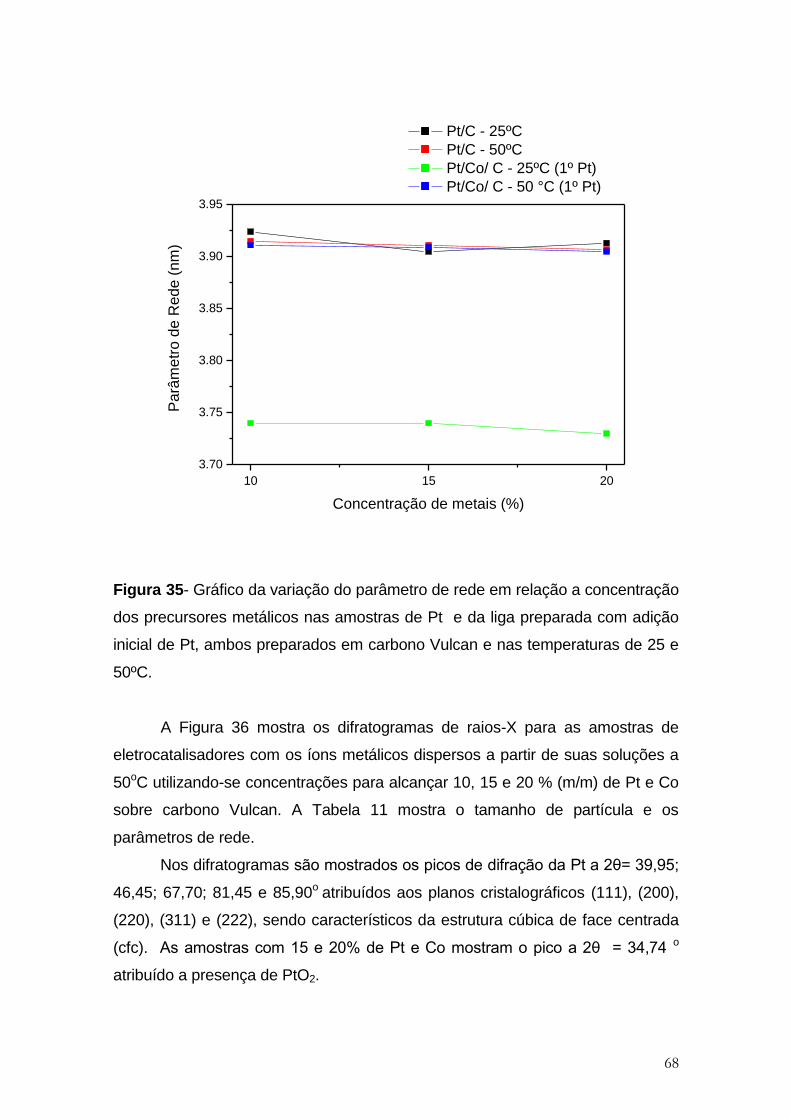
68
10 15 20
3.70
3.75
3.80
3.85
3.90
3.95
P
arâ
me
tro
de
Re
de
(n
m)
Concentração de metais (%)
Pt/C - 25ºC
Pt/C - 50ºC
Pt/Co/ C - 25ºC (1º Pt)
Pt/Co/ C - 50 °C (1º Pt)
Figura 35- Gráfico da variação do parâmetro de rede em relação a concentração
dos precursores metálicos nas amostras de Pt e da liga preparada com adição
inicial de Pt, ambos preparados em carbono Vulcan e nas temperaturas de 25 e
50ºC.
A Figura 36 mostra os difratogramas de raios-X para as amostras de
eletrocatalisadores com os íons metálicos dispersos a partir de suas soluções a
50oC utilizando-se concentrações para alcançar 10, 15 e 20 % (m/m) de Pt e Co
sobre carbono Vulcan. A Tabela 11 mostra o tamanho de partícula e os
parâmetros de rede.
Nos difratogramas são mostrados os picos de difração da Pt a 2θ= 39,95;
46,45; 67,70; 81,45 e 85,90o atribuídos aos planos cristalográficos (111), (200),
(220), (311) e (222), sendo característicos da estrutura cúbica de face centrada
(cfc). As amostras com 15 e 20% de Pt e Co mostram o pico a 2θ = 34,74 o
atribuído a presença de PtO2.

69
Nos difratogramas da Figura 36 não pôde ser observado visualmente
ombro no pico referente ao plano (111). Procedeu-se a decomposição do pico
em duas funções primitivas Lorentzianas. Estas são mostradas na Figura 37 (a),
(b) e (c) e trazem o ponto máximo da contribuição em graus e a porcentagem em
área de cada contribuição. Nota-se que os três picos podem ser decompostos
em duas contribuições sendo a diferença entre os máximos das duas
contribuições de 0,14o (10 %), 0,10o (15%) e 0,12 o(20%). Essa diferença é
suficiente para se inferir a formação de liga, entretanto, é importante considerar
que a resolução do difratograma não foi a adequada para esse tipo de
tratamento, em função do equipamento utilizado. É de consenso nos trabalhos
da literatura que a redução por aquecimento em atmosfera de H2 é essencial
para, e via de regra, resulta na formação de liga. Os trabalhos em que se
procede a redução via química para obter partículas com dimensões variando de
2 a 7 nm, mencionam a não formação da liga e a necessidade de aquecimento a
altas temperaturas para obtê-la, com um custo adicional do aumento do tamanho
de partícula médio.
40 60 80
(222)(311)(220)(111) (200)
PtCo(10%)/Cv - 50ºC
PtCo(15%)/Cv - 50ºC
PtCo(20%)/Cv - 50ºC
inte
nsid
ade (
u.a
.)
2 (graus)
Figura 36- Difratogramas de raios-X para as amostras de eletrocatalisadores
dispersos a 50oC, variando-se as concentrações dos metais de 10, 15 e 20%
(m/m) de Pt e Co sobre o carbono Vulcan.

70
Observou-se da Tabela 11 que existe uma tendência ao aumento do
tamanho de partícula com o aumento da concentração dos metais, o que é
esperado já que um número significativamente maior de íons pode migrar para a
superfície das partículas com posterior redução.
A dimensão maior das partículas a 15% deve-se, provavelmente, a falha
humana no controle do tempo de dispersão dos íons metálicos ou aumento do
aquecimento acidental. Estes experimentos já foram refeitos em trabalho de
continuidade deste projeto e os resultados mostram a tendência esperada. Para
as três amostras estudadas as dimensões de partículas calculadas são maiores
do que as esperadas. Erros grandes são esperados nesses cálculos de tamanho
de partícula por DRX, de até 100% de dimensão a mais para o cálculo quando
comparado com a dimensão média de partícula obtida por microscopia eletrônica
de transmissão. Mesmo considerando a possibilidade de tais erros a dimensão
de 8 a 15 nm já resultaria em material de propriedades catalíticas ineficientes
para a aplicação. Para menores tamanhos de partículas conclui-se ser mais
conveniente inicialmente a redução química de platina, obtendo-se partículas
variando de 2 a 4 nm com posterior dispersão e redução do segundo metal.
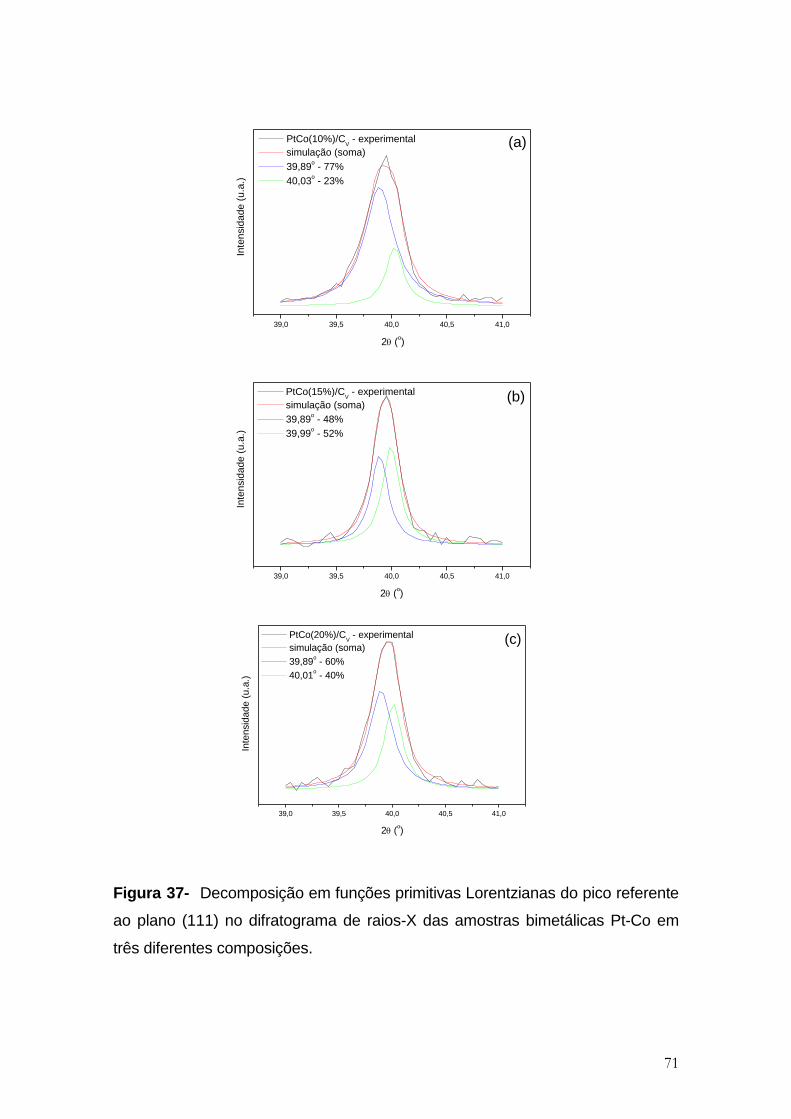
71
39,0 39,5 40,0 40,5 41,0
(a)
PtCo(10%)/CV - experimental
simulação (soma)
39,89o - 77%
40,03o - 23%
Inte
nsid
ade (
u.a
.)
2 (o)
39,0 39,5 40,0 40,5 41,0
(b)
PtCo(15%)/CV - experimental
simulação (soma)
39,89o - 48%
39,99o - 52%
Inte
nsid
ade
(u
.a.)
2 (o)
39,0 39,5 40,0 40,5 41,0
(c)
PtCo(20%)/CV - experimental
simulação (soma)
39,89o - 60%
40,01o - 40%
Inte
nsid
ade (
u.a
.)
2 (o)
Figura 37- Decomposição em funções primitivas Lorentzianas do pico referente
ao plano (111) no difratograma de raios-X das amostras bimetálicas Pt-Co em
três diferentes composições.

72
Tabela 11- Tamanhos de partícula e os parâmetros de rede para as amostras de
eletrocatalisadores com concentrações diferentes de Pt e Co.
Amostra Tamanho de partícula
(nm)
Parâmetro de rede
(nm)
Pt(10%)_s_Co(10%)/C_50ºC 15,3 3,9154
Pt(15%)_s_Co(15%)/C_50ºC 29,2 3,9092
Pt(20%)_s_Co(20%)/C_50ºC 18,0 3,9051
4.2.5.3 – Teste de eletrocatálise por voltametria cíclica
Para facilitar a discussão dos resultados de voltametria cíclica (VC) dos
testes de eletrocatálise em solução aquosa de ácido sulfúrico 1 mol/L, é
mostrada inicialmente na Figura 38 a voltametria cíclica realizada em um
eletrodo de platina policristalino, nas mesmas condições de análise das
amostras.
-200 0 200 400 600 800 1000
-3
-2
-1
0
1
2
Co
rre
nte
(m
A)
Potencial (mV)
Hads
H+
Pt Pt óxido
H+ H
ads
Pt óxido Pt
Região de dupla camada
Figura 38 - Voltamograma Cíclico a 10 mV/s de -200 a 1000 mV versus ECS
para o eletrodo de Pt policristalino. Foi utilizado um eletrodo rotatório de Pt como
eletrodo de trabalho, como contra-eletrodo uma lâmina de Pt (1 cm2) e
calomelano saturado como o eletrodo de referência.

73
O perfil do voltamograma obtido mostra a formação de óxido de Pt na
região anódica de 700 a 1000 mV, ocorrido devido a traços de oxigênio
presentes na solução que foi deaerada com Ar. A redução do óxido formado
ocorre em aproximadamente 500 mV, sentido de varredura catódica. A adsorção
e desorção de H2 ocorre na região entre -200 e 0 mV. Portanto, mostram-se os
potenciais em que devem ocorrer os picos de adsorção e desorção de H2 nas
amostras estudadas neste trabalho.
Na Figura 39 mostram-se os VC das amostras de eletrocatalisadores
bimetálicos com 10,15 e 20 % de Pt e de Co (m/m) sobre Vulcan.
O perfil dos voltamogramas obtidos mostram também a formação de
óxido de Pt na região anódica de 700 a 1000 mV, ocorrido devido a traços de
oxigênio presentes na solução que foi deaerada com Ar. A redução do óxido
formado ocorre na faixa de corrente entre 300 e 600 mV, sentido de varredura
catódica. A adsorção e desorção de H2 ocorre na região entre -200 e 0 mV.
Com o aumento da concentração dos metais nas amostras observa-se
um aumento da corrente de pico anódico e de pico catódico progressivo,
resultado coerente com o aumento de massa. Entretanto, esses valores são
baixos comparados aos valores esperados, indicando que uma baixa área ativa
é alcançada em decorrência das partículas terem dimensões variando em torno
de 20 nm. Para todas as amostras pode ser observada uma corrente capacitiva
significativa.
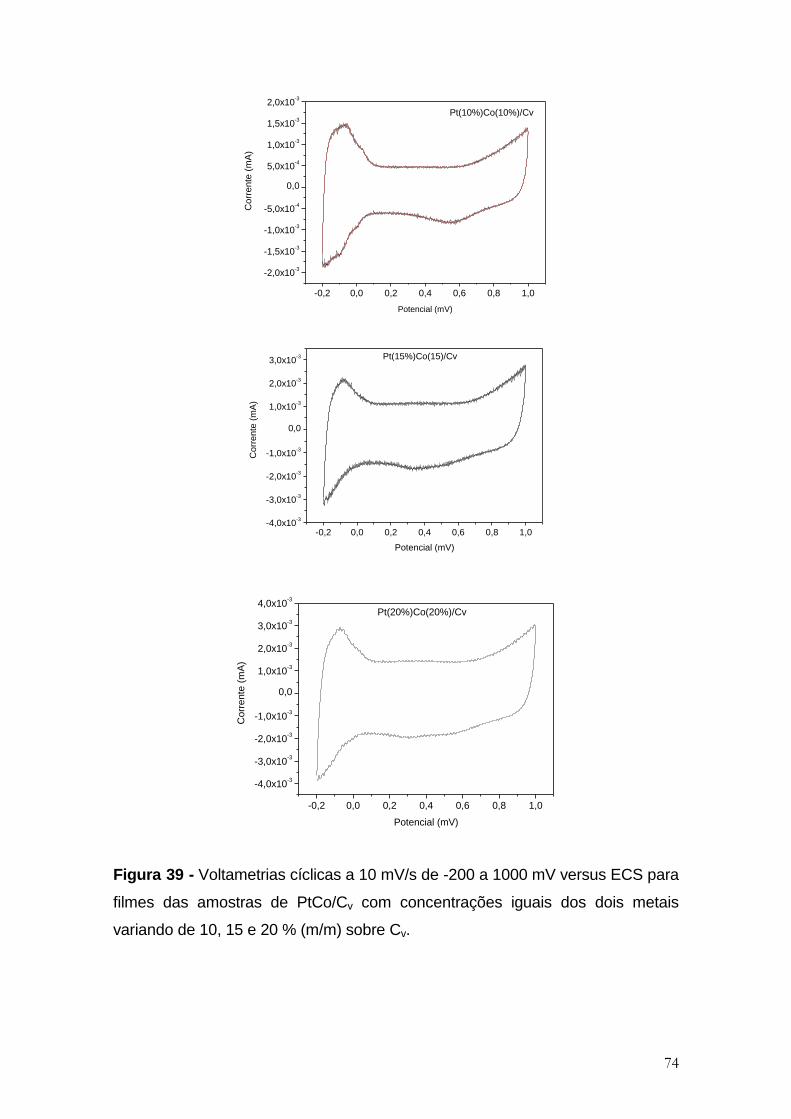
74
-0,2 0,0 0,2 0,4 0,6 0,8 1,0
-2,0x10-3
-1,5x10-3
-1,0x10-3
-5,0x10-4
0,0
5,0x10-4
1,0x10-3
1,5x10-3
2,0x10-3
Corr
ente
(m
A)
Potencial (mV)
Pt(10%)Co(10%)/Cv
-0,2 0,0 0,2 0,4 0,6 0,8 1,0-4,0x10
-3
-3,0x10-3
-2,0x10-3
-1,0x10-3
0,0
1,0x10-3
2,0x10-3
3,0x10-3
Corr
ente
(m
A)
Potencial (mV)
Pt(15%)Co(15)/Cv
-0,2 0,0 0,2 0,4 0,6 0,8 1,0
-4,0x10-3
-3,0x10-3
-2,0x10-3
-1,0x10-3
0,0
1,0x10-3
2,0x10-3
3,0x10-3
4,0x10-3
Corr
ente
(m
A)
Potencial (mV)
Pt(20%)Co(20%)/Cv
Figura 39 - Voltametrias cíclicas a 10 mV/s de -200 a 1000 mV versus ECS para
filmes das amostras de PtCo/Cv com concentrações iguais dos dois metais
variando de 10, 15 e 20 % (m/m) sobre Cv.

75
A Figura 40 mostra os voltamogramas cíclicos obtidos para eletrodos de
Pt recobertos com amostra comercial (Electrochem) e a de carbono Vulcan/Pt
obtida pelo grupo com redução por TPR para comparação com os resultados
obtidos para as amostras de bimetálicos. A resposta eletroquímica para a
amostra não comercial é inferior a amostra Electrochem, como observado nas
correntes de pico. Isto sugere um maior tamanho médio de partículas, bem como
menor dispersão, a qual fornece uma menor área de contato entre a Pt e a
solução, desfavorecendo a formação do óxido de Pt. Como se pode observar o
pico de redução do óxido é diminuído com relação à amostra Electrochem
sugerindo uma menor superfície de contato na amostra preparada no laboratório.
A região de -200 a 0 mV correspondente a adsorção e desorção de H2
apresenta pico com valores de corrente de aproximadamente metade do valor
observado para a amostra Electrochem. Esses resultados são coerentes com os
dados previamente observados para o tamanho médio das partículas de Pt
dispersas em carbono Vulcan, de aproximadamente 9 nm (de acordo com os
resultados de DRX apresentados previamente) comparativamente ao valor
médio de tamanho de partícula de Pt na amostra Electrochem de 2 nm.
A área ativa eletrocatalíca em cm2 para a amostra de carbono Vulcan foi
calculada a partir da Equação 4, considerando um valor de correspondência.

76
-200 0 200 400 600 800 1000
-1,0
-0,8
-0,6
-0,4
-0,2
0,0
0,2
0,4
0,6 (a) 20% Pt/C preparado no laboratório
(b)20% Pt/C vulcan XC-72R (Electrochem)
Corr
ente
(m
A)
Potencial (mV)
Figura 40 - Voltamogramas cíclicos a 10mV/s de -200 a 1000 mV versus ECS
para 20% Pt/ carbono Vulcan XC-72R (Electrochem) (a), e 20% Pt/ carbono
Vulcan XC-72R obtido neste trabalho. Eletrodo de trabalho e contra-eletrodo de
Pt.
Para a amostra Electrochem a área superficial estimada foi de 36,72 m2 e
6,61 m2 para a amostra não comercial. Este valor é coerente com o tamanho
médio das nanopartículas de Pt, conhecidas para os dois sistemas de carbono.
SEL (cm2) = QH/0.21 mC Equação (4)
Onde:
SEL = área real estimada eletroquimicamente.
QH = Carga estimada através da integração da área sob o pico de desorção de
H2 sobre o eletrodo de Pt.
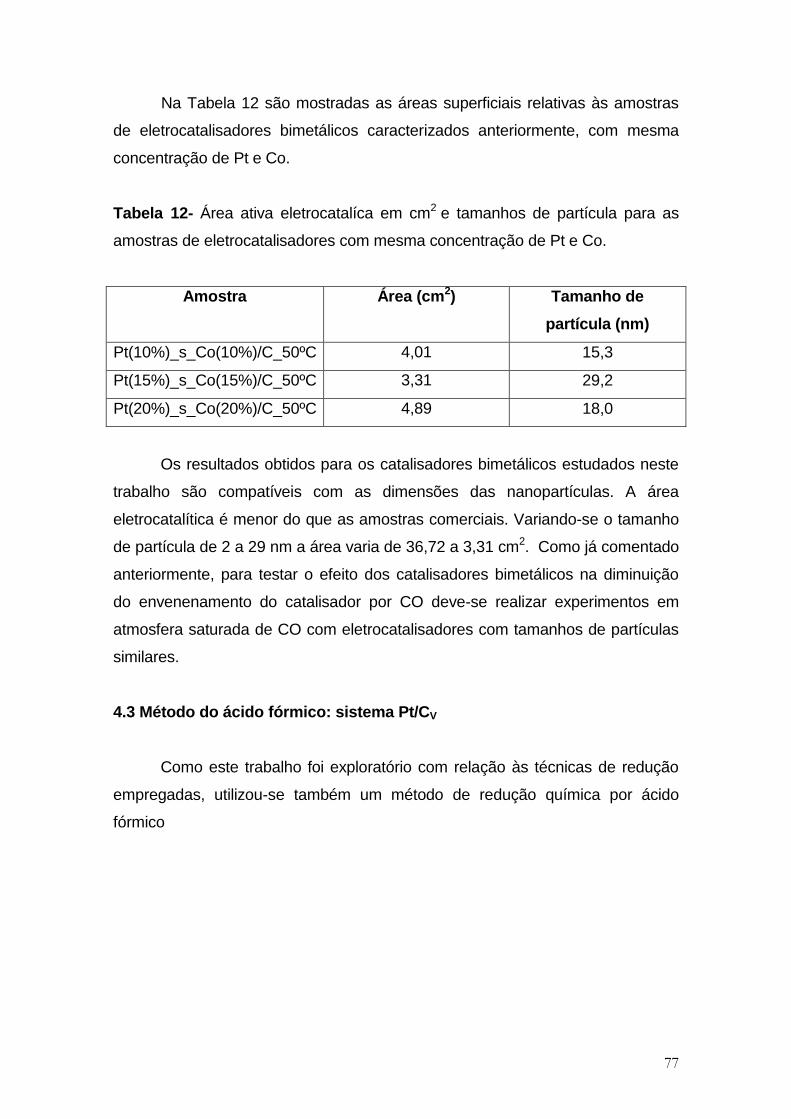
77
Na Tabela 12 são mostradas as áreas superficiais relativas às amostras
de eletrocatalisadores bimetálicos caracterizados anteriormente, com mesma
concentração de Pt e Co.
Tabela 12- Área ativa eletrocatalíca em cm2 e tamanhos de partícula para as
amostras de eletrocatalisadores com mesma concentração de Pt e Co.
Amostra Área (cm2) Tamanho de
partícula (nm)
Pt(10%)_s_Co(10%)/C_50ºC 4,01 15,3
Pt(15%)_s_Co(15%)/C_50ºC 3,31 29,2
Pt(20%)_s_Co(20%)/C_50ºC 4,89 18,0
Os resultados obtidos para os catalisadores bimetálicos estudados neste
trabalho são compatíveis com as dimensões das nanopartículas. A área
eletrocatalítica é menor do que as amostras comerciais. Variando-se o tamanho
de partícula de 2 a 29 nm a área varia de 36,72 a 3,31 cm2. Como já comentado
anteriormente, para testar o efeito dos catalisadores bimetálicos na diminuição
do envenenamento do catalisador por CO deve-se realizar experimentos em
atmosfera saturada de CO com eletrocatalisadores com tamanhos de partículas
similares.
4.3 Método do ácido fórmico: sistema Pt/CV
Como este trabalho foi exploratório com relação às técnicas de redução
empregadas, utilizou-se também um método de redução química por ácido
fórmico

78
4.3.1 Caracterização por Difração de raios-X
Na Figura 41 são mostrados comparativamente os difratogramas de raios-X
para as amostras reduzidas por TPR, preparadas por dispersão do sal a 25 e 50
oC e amostras reduzidas quimicamente, todas contendo a razão Pt(10%)/Cv.
40 50 60 70 80 90
Pt(10%)/C - 25ºC
Pt(10%)/C - 50ºC
Pt(10%)/C - ác. fórmico
Pt(10%)/C - ác. fórmico ativ.
inte
nsid
ade (
u.a
.)
2 (graus)
Figura 41 - Difratogramas das amostras de Platina (10%) em carbono vulcan
preparadas pelas técnicas da impregnação e método do ácido fórmico, com suas
respectivas variações metodológicas.

79
A Figura 42 apresenta uma ampliação da região do pico relativo ao plano
de difração (111). Ao comparar os picos podemos perceber diferenças nos perfis
dos mesmos, de acordo com as respectivas técnicas. Os picos das amostras
preparadas pelo método da impregnação seguida de redução por TPR são mais
estreitos, provavelmente, representativos de um sistema com partículas maiores
do que as observadas para as amostras preparadas pelo método do ácido
fórmico. Na Tabela 13 são mostrados os tamanhos médios de partículas para as
amostras reduzidas quimicamente.
38,5 39,0 39,5 40,0 40,5 41,0
Pt(10%)/C - 25ºC
Pt(10%)/C - 50ºC
Pt(10%)/C - ác. fórmico
Pt(10%)/C - ác. fórmico ativ.
inte
nsid
ade (
u.a
.)
2 (graus)
Figura 42 - Comparação, em detalhe, do pico referente ao plano (111) da platina
em amostras preparadas pela técnica da impregnação e do ácido fórmico
respectivamente.

80
A Tabela 13 apresenta o tamanho médio de partícula e o parâmetro de
rede das amostras preparadas pelo método da redução com ácido fórmico.
Mostra-se comparativamente os dados para a amostra comercial Electrochem.
Tabela 13 - Tamanho médio de partícula e o parâmetro de rede das amostras
preparadas pelo método da redução com ácido fórmico.
Amostra Tamanho de partícula (nm)
Parâmetro de rede (nm)
Pt(20%)/C – comercial 2,279 3,913
Pt(10%)/C – Ac.fórmico 11.188 3.920
Pt(10%)/C – Ac.fórmico ativ. 15.166 3.9140
Embora as amostras reduzidas com ácido fórmico tenham apresentado
menores tamanhos de partícula, ainda são bem maiores que as partículas
comerciais. Provavelmente alguma falha no controle do tempo ou temperatura
de redução ocorreu, já que em trabalho recente o mesmo método forneceu
dimensão de partículas de aproximadamente 7 nm (DRX). Sabe-se, entretanto,
que a técnica de DRX aplicada à análise de tamanho de partículas com menos
que 10 nm pode resultar em até 100 % de erro, mostrando valores maiores,
portanto, pode-se ter alcançado dimensões de aproximadamente 5 nm na
amostra preparada neste trabalho, com ácido fórmico.
Em outro trabalho recente realizado no grupo utilizando o método químico
de redução com etileno glicol obteve-se partículas com diâmetro de
aproximadamente 5 nm por microscopia eletrônica de transmissão. Entretanto, a
utilização de métodos químicos de redução, embora resultem em dimensões
menores de partículas, geralmente produzem partículas com misturas de fases e
não formação de liga. Esta normalmente é formada após tratamento térmico a
altas temperaturas, o que resulta em um aumento da dimensão das partículas.

81
5 - Conclusões
Neste trabalho foram obtidos eletrocatalisadores bimetálicos CoPt/C
variando-se as condições experimentais de impregnação e redução. Foi
observado pelo TPR que a redução da platina se dá em uma só etapa, ou seja,
Pt2+ Pt por volta de 300oC, e a redução do cobalto, Co2+
Co, por volta de
597oC. Entretanto, pequenas variações nas temperaturas de redução e do
consumo de hidrogênio foram observadas em função das concentrações dos
íons metálicos precursores e da ordem de adição dos reagentes, para os
eletrocatalisadores bimetálicos.
A análise por difração de raios-X identificou a existência de platina na
forma cfc, entretanto, nas amostras bimetálicas não foi possível identificar por
DRX os picos de difração do Co. Estes são pouco intensos e foram confundidos
com ruído.
Nos catalisadores bimetálicos, a formação da liga foi comprovada apenas
através de pequenos deslocamentos de 2 de 39,90 a 39,97o no pico referente
ao plano (111) da Pt, na amostra Pt(10%)Co(10%)/Cv_50ºC - adição inicial de Pt.
Isso indica que a ordem de adição dos precursores metálicos, assim como a
temperatura de dispersão do Co influencia na formação da liga. Entretanto, é
importante salientar que o baixo número de acumulação de sinal no difratômetro
de raios-X utilizado, pode comprometer a visualização de um maior
deslocamento ou observação de um ombro nesse pico a maiores valores de 2,
já que a temperatura de redução no TPR alcançou valores de até 900o C,
temperatura suficiente para formação de liga identificável por DRX.
Foi observado que o suporte utilizado também influencia nas propriedades
das partículas obtidas. Isso é coerente com trabalhos da literatura, já que íons
adsorvem com energias diferentes em diferentes suportes, o que pode alterar as
condições de temperatura e velocidade de redução.
Os tamanhos de partícula calculados por DRX variaram de acordo com a
alteração das condições de preparo. Diferença considerável foi observada para
os eletrocatalisadores preparados por adição simultânea dos íons metálicos
(10%Pt10%Co), os quais apresentaram menores dimensões. Isto pode ser
atribuído ao menor tempo de exposição do sistema a energia de ativação alta no
aquecimento para a redução por TPR. Observou-se também para essas

82
amostras com adição simultânea dos íons metálicos que o tamanho de partícula
aumenta com a concentração dos íons, de acordo com o esperado.
Os testes eletrocatalíticos por voltametria cíclica em solução de H2SO4
com os catalisadores preparados por adição simultânea, mostraram atividade
maior para as amostras com 10% de cada metal. A área eletroativa e portanto,
as correntes de pico foram maiores para as menores concentrações, e
correspondem às menores partículas. Portanto, a adição do segundo metal
permite atingir valores de conversão de H2 maiores com massas reduzidas de
Pt. As áreas eletroativas para esses sistemas foram menores que para o
sistema Pt/C Electrochem e Pt/C preparados no laboratório pelo método de
redução com etileno glicol, em função das partículas maiores. Um controle mais
fino das condições de preparo são necessárias para diminuir o tamanho das
partículas, assim como a redução prévia da Pt, via redução por etileno glicol,
seguida da dispersão controlada do segundo metal e redução por TPR para
formação da liga.
Utilizando a técnica de dispersão para os metais estudados recomenda-
se a adição simultânea dos metais nas concentrações de 10% e na temperatura
de adição de 50 ºC. Como continuidade deste trabalho recomenda-se variar as
condições do TPR.

83
8. Referências
1. World Energy Assessment: Energy and the Challenge of Sustainability. UNDP, UN, 2000 2 http://www.eia.doe.gov/pub/international/iealf/tablee1.xls (acessada em 16/10/2006). 3 . Relatório sobre o Desenvolvimento Mundial 2000/2001. Banco Mundial, 2001, págs. 302/303. 4 . Inventário Brasileiro de emissões antrópicas de gases de efeito estufa http://www.mct.gov.br/upd_blob/8812.pdf (acessada 16/10/2006). 5. C. Song., Catalysis Today 77 (2002) 17. 6 . MME (Ministério de Minas e Energia), Plano de Longo Prazo Projeção da
Matriz - 2022, Sumário Executivo, dezembro de 2002, p. 47-76. Disponível em: www.mme.gov.br Acessado em: 01 agosto 2004.
7. Wendt H.; Götz M.; Linardi M.; Química Nova, 2000, Vol. 23 , nº 4, 538-546. 8. Kobayashi T.; Rikukawa M.; Sanui K.; Ogata N.;, Solid State Ionics, 1998, 106, 219. 9. Acres G. J. K.; Frost J.C.; Hards G.A.; Potter R.J.; Ralph T.R.; Thompsett D.; Burtein G.T.; Hutchings G.J.; Catalysis Today, 1997, 38, 393. 10. Conte M.; Iacobazzi A.; Rochetti M.; Vellone R.; Journal Power Sources
2001, 100, 171. 11. Armor J.N.; Applied Catalysis A General 1999, 176. 159. 12. Lunsford J.H.; Catalysis Today 2000, 63, 165. 13. Turdera E.M.V.; Guerra S.M.; Almeida R.A.; Revista Brasileira de Energia,
1997, Vol. 6, No 2, pp.57. 14. Bajay S.V.; Rodrigues M.G.; Revista Brasileira de Energia, 1996,Vol. 5, No 1, pp.24-47, 15. Hoogers G.; Thompsett D.; Chemistry & Industry, 1999, 18, 796. 16. Appleby A. J.; Foulkes F. R.; Fuel Cell Handbook; Ed Van Nostrand Reinhold; New York, EUA, 1989. 17. Ticianelli E. A.; Camara G. A.; Santos L.G.R.A.; Quimica Nova, 2005, Vol. 28, No. 4, 664-669.

84
18 . Appleby A. J.; Fuel Cells: Trends in Research and Application; Ed. Hemisphere/ Springer; Washington, EUA, 1987. 19. Giorgi L.; Antolini E.;. Pozio A.; Passalacqua E.; Electrochimica Acta., 1998, 43, 3675. 20. Antolini E., Giorgi L., Pozio A., Passalacqua E.; Journal of Power Source 1999, 77, 136. 21. Pozio A.; Giorgi L.; Antolini E.; Passalacqua E.; Electrochimica Acta, 1998, 46, 555.
22. Fuel Cell Seminars: Proc. Fuel Cell Seminar Orlando, Florida, USA, 1996; Courtesy Associates Inc.; Washington DC, EUA, 1996.
23. Poltarzewski Z.; Wiekzorek W.; Przyluski J.; Antonucci V.; Solid State Ionics, 1999, 119, 301. 24. Kordesch K.V.; Simader G.R.; Chemistry Reviews., 1995, 95, 191. 25. Yeager E. B.; Electrochimica Acta, 1984, 29, 1527. 26. Yeager E. B.; Journal of Molecular Catalysis., 1986, 38, 5. 27. Xiong, L.; Kannan A. M.; Manthiram, A.; Electrochemistry Communications.
2002, 4, 898 28. Damjanovic, A.; Brusic V.; Bockris, J. O. M.; The Journal of Physical Chemistry. 1967, 71, 2471. 29. Adzic, R. R.; Lipkowski J.; Ross, P. N.; Electrocatalysis.; Ed.Wiley-VCH: New
York, 1998, p. 209. 30 . Vad, T.; Hajbolouri, F.; Haubold, H.-G; Scherer, G.G.; Wokaun A.; The Journal of Physical Chemistry B., 2002, 106, 4181-4191. 31. Yu P., et al., Journal of Power Sources, 2005, 144, 11–20. 32. Min, M. K.; Cho, J. H.; Cho, K. W..; Kim, H.; Electrochimica Acta., 2000, 45,
4211. 33 . Watanabe, M.; Tsurumi, K.; Mizukami, T.; Nakamura, T.; Stonehart P.;
Journal of The Electrochemical Society., 1994, 141, 2659. 34. Freund A., Lang J., Lehman T., Starz K. A.; Catalysis Today, 1996, 27, 279. 35. Aricò A. S.; Shukla A. K.; Kim H.; Park S.; Min M.; Antonucci V.; Applied
Surface Science, 2001, 172, 33.

85
36. Glass J. T.; Cahen G. L.; Stoner G. E.; Journal of The Electrochemical Society., 1987, 134, 58.
37 . Toda, T.; Igarashi, H.; Uchida, H.; Watanabe, M.; Journal of The
Electrochemical Society., 1999, 146, 3750. 38. Lin W. F.; Iwasita T.; Vielstich W.; The Journal of Physical Chemistry B., 1999, 103, 3250. 39. Liu R., et al.; The Journal of Physical Chemistry. B, 104, (2000), 3518. 40. Min M. K.; Cho J. H.; Cho K. W.; Kim H.; Electrochimica Acta 2000, 45, 4211. 41 . Santiago E. I.; Giz M. J.; Ticianelli E. A.; The Journal of Solid State
Electrochemistry. 2003, 7, 607. 42. Wang K.; Gasteiger H. A.; Markovic N. M.; Ross P. N.; Electrochimica Acta,
1996, 41, 2587 43. Oliveira Neto, A.; Tese de Doutorado, Universidade de São Paulo, Brasil,
2001. 44. Bönnemann H.; Brijoux W.; Brinkmann R.; Dinjus E.; Jouben T.; Korall B.;
Angewandte Chemie, 1991, 30, 1312. 45. Götz M.; Wendt H.; Electrochimica Acta, 1998, 43, 3637. 46. Radmilovic, V.; Gasteiger, H. A.; Ross Jr, P. N.; Journal of Catalysis. 1995, 154, 98. 47 Schmidt, T; J., Gasteiger, H. A.; Behm, R. J.; Eletrochemistry
Communications 1999, 1, 1. 48. Franco, E. G., Neto, A.O.; Linardi, M.; Arico, E.; Journal of the Brazilian
Chemical Society., 2002, 13, 516. 49 . Brankovic S. R.; McBreen J.; Adzic R. R.; Journal of Electroanalytical
Chemistry 2001, 503, 99. 50. Brankovic S. R.; Wang J. X.; Adzic R. R.; Electrochemical and Solid-State
Letters 2001, 4, A217 51. Nashner, M. S.; Frenkel, A. I.; Adler, D. L.; Shapley, J. R.; Nuzzo, R.G.;
Journal of the American Chemical Society 1997, 119, 7760. 52. Hills C. W.; Nashner M. S.; Frenkel A. I.; Shapley J. R.;. Nuzzo R. G;
Langmuir, 1999,15, 690.

86
53. Boxall, D. L.; Deluga, G. A.; Kenik, E.A.; King W. D.;.Lukehart, C. M; Journal of Materials Chemistry., 2001, 13, 891.
54. Steigerwalt, E. S.; Deluga, G. A.; Cliffel, D. E.; Lukehart, C. M.; The Journal of
Physical Chemistry B, 2001, 105, 8097 55. Dickinson A. J.; Carrette, L. P. L.; Collins, J. A. ; Friedrich, K. A.; Stimming,
U.; Electrochimica Acta, 2002, 47,3733. 56. Toshima, N.; Yonezawa, T.; New Journal of Chemistry 1998,22, 1179 57. Wang, X.; Hsing, I. M.; Electrochimica Acta, 2002, 47, 2981. 58. Chen, W. X.; Lee, J. Y. ; Lui, Z . ; Chemical Communications. 2002, 2588. 59 . Joint Committee on Powder Diffraction Standards – (JCPDS) 1-1190. 60.Huang, H. ; Yang, Y. ; Tang, T. ; Akins D. ; Electrochemistry Communications, 2006, 8, 1220 61 . Lima, F.H.B. ; Lizcano-Valbuena, W.H. ; Teixeira-Neto, E. ; Nart, F.C. ; Gonzalez, E.R. ; Ticianelli, E.A. ;Electrochimica Acta, 2006, xxx (on line). 62. Gonzalez, E. R.; Spinacé, E. V.; Neto, A. O.;. Franco, E. G; Linardi, M.; Química Nova, 2004, Vol. 27, No. 4, 648-654,. 63. Gates, B. C.; Catalytic Chemistry, John Wiley & Sons, Inc.: New York, 1992,
cap. 6, p. 378. 64. Salgado, J. R. C.; Gonzalez, E. R.; Eclética Química, 2003, Vol 28, nº 2. 65. Oliveira, R. C. S.; Macêdo, M. I. F.; Mota, C. J. A.; Herbst, M. H.; Rocco A. M.; Resumos da 29ª Reunião anual da Sociedade Brasileira de Química – SBQ, Águas de Lindóia, 2006. 66. Oliveira, R. C. S.; Macêdo, M. I. F.; Mota, C. J. A.; Herbst, M. H.; Rocco A. M.; Resumos da 28ª Reunião anual da Sociedade Brasileira de Química – SBQ, Poços de Caldas, 2005. 67. Tian, Z.Q.; Xie, F.Y.; Shen, P.K.; Journal of Materials Science 2004, 39, 507-1509. 68. Xu, H.; Lin, Z.; Qiu, Y.; Tang, Q. ; Chinese Journal of Catalysis, 2003, 24, 143 69. Radamilovic, V.; Gasteiger, H.A.; Jr. Ross, P. N.; Journal of Catalysis.; 1995, 154, 98 70. Tian, Z.Q.; Xie, F.Y.; Shen, P.K.; Journal of Materials Science 2004, 39,1507-1509.

87
71. Zhang, X; Chan, K-Y; Journals of Materials Chemistry, 2002, 12, 1203. .