universita’ degli studi di catania -...
TRANSCRIPT

UNIVERSITA’ DEGLI STUDI DI CATANIA
FACOLTA’ DI INGEGNERIA
DIPARTIMENTO DI METODOLOGIE FISICHE E CHIMICHE PER
L’INGEGNERIA
============================================
DOTTORATO DI RICERCA IN MATERIALI POLIMERICI PER
USI SPECIALI
XXIII CICLO
Dott. VINCENZO CARLO ASARISI
________________________
SINTESI E CARATTERIZZAZIONE DI
POLIETERISOLFONI SOLFONATI
PER APPLICAZIONI SPECIALI
_______________
RELAZIONE FINALE SULLE ATTIVITA’ SVOLTE
________________
RELATORE:
Ch.mo Prof. Gianluca Cicala
COORDINATORE :
Ch.mo Prof FRANCESCO BOTTINO
============================================
TRIENNIO 2007-2010

- 2 -
INDICE
1. INTRODUZIONE PAG. 6
1.1 Cenni storici generali sulle Fuel Cells PAG. 6
1.2 Membrane innovative a scambio protonico
ad alte prestazioni basate su nanocompositi
poli (eteresulfone) PAG. 14
1.3 Caratteristiche e proprietà strutturali PAG. 32
1.4 La reazione di solfonazione dei poliarilati PAG. 29
1.5 Metodiche di sintesi di poli(arilen eteri
solfoni)solfonati PAG. 35
2. SCOPO DEL LAVORO DI RICERCA PAG. 41
3. PARTE SPERIMENTALE PAG. 43

- 3 -
3.1 Materiali PAG. 43
3.2 Sintesi dei prepolimeri 4K e 7K PAG. 44
3.3 Monomeri impiegati PAG. 44
3.4 Tecniche per la caratterizzazione dei Polimeri PAG. 46
3.5 Solfonazione del monomero DCDPS PAG. 48
3.6 Sintesi dei polimeri S-PES a diversi gradi di
solfonazione PAG. 54
3.7 Reazione di POST-POLIMERIZZAZIONE PAG. 58
3.8 Sviluppi degli studi nel prossimo futuro PAG. 59
3.9 Studio della cinetica reazione di sintesi del
prepolimero PES solforato “perfettamente
alternato” PAG. 62
3.10 Caratterizzazione dei Polimeri PAG. 64

- 4 -
3.10.1 Spettrometria di massa MALDI-TOF PAG. 64
3.10.2 Spettroscopia NMR PAG. 73
3.10.3 Viscosimetria-SEC PAG. 74
3.10.4 Viscosimetria PAG. 76
3.10.5 Spettroscopia infrarossa (FT-IR) PAG. 77
3.10.6 DSC (Calorimetria a Scansione
Differenziale) PAG. 93
3.10.7 Termogravimetria PAG. 95
4. RISULTATI E DISCUSSIONE PAG. 98
4.1 Spettrometria di massa MALDI-TOF MS PAG. 103
4.2 Caratterizzazione NMR PAG.118
4.3 SEC-viscometria PAG.132

- 5 -
4.4 Spettroscopia FT-IR PAG. 138
4.5 Caratterizzazione termica PAG. 140
4.6 Analisi dinamo meccaniche (DMA) PAG. 157
5. CONCLUSIONI PAG. 161
6. ELENCO TABELLE, FIGURE E SCHEMI PAG. 169
7. RINGRAZIAMENTI PAG. 176
8. BIBLIOGRAFIA PAG. 177

- 6 -
1. INTRODUZIONE
1.1 Cenni storici generali sulle Fuel Cells
Sebbene le prime elementari celle a combustibile siano state
inventate e realizzate a metà del 19°secolo, queste non hanno
trovato applicazioni utili fino al tempo dell'esplorazione
spaziale nel 1960. Da allora, lo sviluppo della tecnologia della
pila a combustibile è passata attraverso diversi cicli di intensa
attività, ciascuno alternato da un periodo di ridotto interesse.
Tuttavia, nel corso degli ultimi due decenni, è aumentato
notevolmente lo sforzo a livello mondiale per sviluppare
materiali adatti e sistemi innovativi per l’utilizzo di celle a
combustibile.
Tutto ha origine dalla crescente esigenza di fonti di energia
pulita e rinnovabile; tale esigenza di primaria importanza nella
società moderna include in se altre necessità. Tra queste la

- 7 -
richiesta di sistemi a risparmio energetico per il trasporto, la
volontà da parte dei governi degli più industrializzati di ridurre
le emissioni di CO
2
e di altri agenti le cui emissioni in atmosfera
son potenzialmente nocive per l’equilibrio ambientale.
Nel settore elettronico vi è inoltre la domanda di fonti di
energia ad alta densità di potenza per applicazioni portatili
elettroniche.
A causa del livello elevato di interesse verso la tecnologia
legata alle celle a combustibile, negli ultimi dieci anni ci sono
stati numerosi articoli di sintesi e simposi focalizzati a porre
ordine sullo stato dell'arte legato allo sviluppo di tale tecnologia.
Il centro nevralgico di una cella a combustibile è costituito dal
sistema di membrane polielettrolitiche (PEM) il quale deve avere
caratteristiche ideali a fornire l’energia primaria poi convertita
successivamente in energia utile al trasporto o all’alimentazione
portatile in base alle applicazioni auspicate.
Il nucleo di questa tecnologia è una membrana polimerica che
conduce protoni ma separa il combustibile dall’ossidante. Il
materiale utilizzato storicamente e più frequentemente nei

- 8 -
sistemi a combustibile delle PEM è Nafion, un polimero a base
di perfluorocarburi che possiede residui di acido solfonico nella
sua struttura molecolare.
Il Nafion è un materiale commerciale è stato negli ultimi anni
ampiamente studiato in quanto considerato da più parti il
materiale ad oggi più idoneo all’utilizzo come substrato delle
membrane PEM nelle celle a combustibile.
Mauritz e Moore hanno preparato una raccolta delle attuali
conoscenze acquisite in tale settore allo scopo di ottimizzare e
comprendere in modo più organico lo stato dell’arte.
In seguito alla crescente esigenza di fonti di energia pulita e
rinnovabile altri sistemi polimerici che avrebbero prestazioni
ancora migliori di Nafion e/o avrebbero costi più bassi sono stati
oggetto di numerosi studi per un possibile futuro utilizzo come
membrane a scambio protonico.
Questi tipi di materiali possiedono complesse proprietà di
trasporto che non coinvolgono solo il movimento di protoni, ma
anche il movimento di acqua.

- 9 -
Una trattazione teorica dei meccanismi di trasporto e dei
processi in questi conduttori protonici è stata condotta da Kreuer,
Paddison, Spohr, e Schuster e da Weber e Newman.
Nelle membrane polielettrolitiche PEM delle celle a
combustibile, l'attività e l'efficienza del catalizzatore sono ancora
problemi significativi. Russell e Rose hanno riassunto il lavoro
fondamentale che coinvolge la spettroscopia di assorbimento di
raggi X sui catalizzatori nei sistemi di celle a combustibile a
bassa temperatura. Questi tipi di studi sono estremamente utili
per sviluppare una comprensione dettagliata dei meccanismi di
reazione sulla superficie del catalizzatore e potrebbe portare allo
sviluppo di nuovi sistemi catalitici volti a migliorare l’efficienza.
Importanti nello sviluppo della tecnologia delle celle a
combustibile sono i modelli matematici di alcuni aspetti
ingegneristici dei sistemi utilizzati nelle celle a combustibile.
Wang
20,22,25
scrive alcuni lavori relativi a questo argomento.
Infine è importante considerare che affinchè i sistemi di celle a
combustibile PEM siano adattabili per le applicazioni portatili,
una fonte di combustibile ad alta densità di energia deve essere

- 10 -
considerata. Tothis, Holladay, Wang, e Jones hanno preparato un
lavoro riepilogativo sugli ultimi sviluppi dei lavori legati
all’utilizzo della tecnologia a microreattore per convertire i
combustibili liquidi in idrogeno per l'alimentazione diretta nella
PEM della cella a combustibile.
Un altro sistema basato su celle a combustibile che è oggetto
di intenso lavoro di ricerca è il tipo ad ossido solido.
Adler presenta i fattori che regolano la limitazione della
velocità di reazione di riduzione di ossigeno all'interno del catodi
di tali Fuel Cells.
McIntosh e Gorte invece trattano l'anodo della cella
combustibile ad ossido solido, esaminando l’ossidazione
catalitica diretta degli idrocarburi impiegati.
Infine Calabrese, Barton, Gallaway, e Atanossov nei loro
studio proiettano la loro attenzione a sviluppi prossimi futuri.
Nel loro articolo, presentano lavori sperimentali basati sullo
studio di alcuni innovativi sistemi a base di celle a combustibile
biologiche a meccanismo enzimatico che si stanno sviluppando e
che potrebbero trovare applicazioni in dispositivi impiantabili.

- 11 -
Una sezione trasversale schematica di una singola cella di una
PEMFC con i singoli componenti mostrati è schematizzata nella
Figura 1.
Una singola cella è costituita da un catodo, un anodo, e da una
membrana separatrice polielettrolitica. Ogni elettrodo ha uno
strato electrocatalitico, ed uno strato adibito alla diffusione del
gas (GDL). Gli strati di catalizzatore possono essere collegati a
una membrana o, a volte al materiale GDL (il cosiddetto
elettrodo di diffusione dei gas, GDE).
FigurFigura 1- Sezione trasversale schematica di una singola cella
di u di una PEMFC

- 12 -
Ogni singola cella produce una tensione di circa 0,6-0,7V. Per
produrre una tensione adeguata, le singole celle sono "impilate"
per formare una batteria di celle a combustibile.
Le singole celle sono collegate elettricamente in serie
mediante piatti bipolari, e piatti con speciali terminazioni
vengono poste ad entrambe le estremità in modo da fornire la
forza di compressione necessaria per mantenere l'integrità
strutturale.
Le piastre bipolari forniscono i percorsi per la conduzione
degli elettroni tra le celle, distribuiscono il gas reagente su tutta
la superficie attiva della membrana elettrolitica (MEA)
(attraverso i canali di flusso integrato nelle piastre), eliminano il
calore in eccesso (attraverso canali di raffreddamento), e fornire
integrità strutturale nonché le barriere ai gas di anodo e catodo.
Direttamente adiacente ai piatti bipolari sono gli strati di
diffusione del gas (GDLs), essi di solito sono costituiti da due
strati; un substrato macroporoso e uno strato microporoso
(MPL). I GDLs sono gas permeabili e aiutano a distribuire gli

- 13 -
altri gas al livello catalizzatore, conducono la corrente elettrica, e
forniscono anche una rete di passaggi di acqua allo stato liquido.
La porzione di substrato macroporoso è costituito da un
matrice in fibra di carbonio con un grande volume di vuoto, di
solito il 75-85%, e un MPL prevalentemente idrofobico formato
da carbonio nero miscelato con un polimero fluorurato.
Il GDL al catodo ha normalmente una sua MPL correlata
mentre il GDL all'anodo può averla o anche non averla.
I processi elettrochimici che avvengono in una cella a
combustibile si verificano nella maggior parte dei casi nel più
interno dei tre strati della cella, comunemente conosciuto come
la MEA o in alternativa come una membrana rivestita dal
catalizzatore (CCM).
Nei primi anni di sviluppo delle PEMFC (la seconda metà
degli anni’60), 10 ricercatori definirono una MEA come una
membrana costituita da due elettrodi a diffusione di gas (GDEs)
più una membrana polimerica a conduzione protonica, oppure
ionomero. Questa forma di MEA racchiudeva i substrati GDL

- 14 -
con uno strato elettrocatalitico depositato su ogni superficie
GDL al posto di quelli che ora sono i MPLs.
I moderni strati elettrocatalitici di PEMFC sono solitamente
composti da strutture a conduzione protonica costituite da
materiale ionomero e catalizzatori a base di metalli nobili
(platino) supportati su carbonio.
1.2 Membrane innovative a scambio protonico ad
alte prestazioni basate su nanocompositi di
poli(etere sulfone)
Negli ultimi anni, a causa della esaurimento delle risorse non
rinnovabili e le riserve di combustibili fossili, la domanda di
fonti energetiche alternative ha acquisito una crescente
attenzione.
E ' prevedibile che il nostro pianeta nel prossimo futuro potrà
attraversare una crisi energetica dovuta al progressivo
esaurimento delle fonti non rinnovabili.

- 15 -
Ciò avverrà a meno che non si procederà effettuando un
approvvigionamento energetico sostenibile sfruttando soprattutto
le fonti rinnovabili a nostra disposizione.
D'altra parte l'uso continuato di idrocarburi è tra le principali
cause di inquinamento del nostro pianeta e quindi del
cambiamento climatico e del surriscaldamento globale.
Quindi per tali ragioni è sempre crescente in tutti i settori ad
elevato impatto energetico e ambientale la richiesta di
approvvigionamento di energia mediante fonti rinnovabili,
sostenibili e soprattutto pulite e da ciò nasce il crescente
interesse scientifico e tecnologico verso il raggiungimento di tale
obiettivo.
Tra le fonti energetiche alternative, la tecnologia delle celle a
combustibile è particolarmente studiata ed il suo interesse deriva
dalla sue caratteristiche eco-friendly e all’alta efficienza della
resa energetica.
Le celle a combustibile infatti sono in grado di convertire
l'energia chimica di un combustibile in elettricità attraverso un

- 16 -
processo di ossidoriduzione con ridotta emissione di inquinanti.
Nel corso degli ultimi anni, la compatibilità delle celle a
membrana elettrolitica polimerica (PEMFCs) come fonte di
energia in automobili e dispositivi portatili elettrici è stata
studiata e si è evoluta.
Tra i diversi tipi di celle a combustibile le PEMFCs e quelle a
metanolo (DMFC) richiedono l'uso di membrane polimeriche a
conduzione protonica; ciò ha portato ad un crescente interesse
per lo sviluppo di numerose classi di materiali polimerici per usi
ingegneristici da utilizzare come elettroliti polimerici.
Il ruolo della membrana a scambio protonico è quello di
permettere il trasferimento di protoni dall'anodo al catodo e
anche di essere un materiale non elettron-conduttore.
Le DMFC hanno molti vantaggi rispetto gli altri tipi di celle a
combustibile, comprese maggiore densità di energia, maggiore
semplicità di sistema, basse emissioni di agenti inquinanti
nonchè la possibilità di lavorare a temperatura ambiente e di
avere uno start-up rapido.

- 17 -
Tali proprietà favorevoli hanno reso la DMFC appropriata per
il trasporto veicolare e varie applicazioni che richiedono energia
silenziosa, pulita e portatile.
Le esigenze principali delle membrane polimeriche a
conduzione protonica nelle applicazioni DMFC sono alta
conducibilità protonica, bassa permeabilità al metanolo, buona
stabilità meccanica / termica e anche il basso costo.
Attualmente, i materiali più comuni utilizzati come membrana
per entrambe le applicazioni (PEMFC e DMFC) sono degli
ionomeri perfluorurati come il Nafion. Tali materiali polimerici
possiedono alta stabilità chimica, resistenza allo sforzo a lungo
termine e una buona conducibilità protonica. Tuttavia, a causa
del costo relativamente elevato di fabbricazione dell’alta
permeabilità al metanolo, della bassa conducibilità e delle scarse
proprietà meccaniche a temperature oltre gli 80°C, l'utilizzo di
membrane in Nafion per il sistema DMFC è limitato e lo
sviluppo di materiali polimerici alternativi ad elevata
conducibilità protonica è auspicabile.

- 18 -
Un approccio per ottenere materiali ionomeri aventi alte
prestazioni e basso costo è la sintesi e la modifica dei polimeri a
base idrocarburica. A questo proposito, diversi polimeri sono
stati proposti e valutati finora.
Tra i polimeri non fluorurati, i polieteri-solfoni (PES) sono
stati considerati tra i materiali polimerici più adatti alla
preparazione di tali membrane a scambio protonico grazie alle
loro caratteristiche.
Tale classe di materiali termoplastici ha riscosso negli ultimi
anni un notevole interesse scientifico, tecnologico ed economico
per via delle sue proprietà uniche che trovano giustificazione
nella natura chimica delle macromolecole.
Questo materiale infatti fa parte di una famiglia di composti
macromolecolari aventi simile composizione chimica; le catene
polimeriche infatti hanno una unità ripetitiva costituita in gran
parte da strutture aromatiche nei differenti concatenamenti
poliammidico, poliestere, polietere, poliimmidico, etc.

- 19 -
Queste classi di polimeri possiedono ottime proprietà
meccaniche, buona resistenza agli agenti atmosferici e se
opportunamente additivati o chimicamente modificati buone
proprietà termiche e proprietà ignifughe con resistenza alla
degradazione ad elevate temperature, ed elevati valori di
conducibilità elettrica.
La possibilità di ottenere polimeri con proprietà controllate
consente l’utilizzo per applicazioni in svariati settori industriali
quali, ad esempio nella costruzione di parti strutturali di
apparecchiature, o anche nella produzione di membrane per celle
a combustibile o dissalazione delle acque, o come agenti
tenacizzanti di resine termoindurenti.
Tra questi tipi di polimeri, trovano un ampio spettro di
applicazioni i poliarilati la cui struttura può essere scritta
genericamente come:
–Ar–X–Ar–Y–Ar–Z–

- 20 -
in cui Ar rappresenta la parte aromatica, che può essere un
semplice anello benzenico o un gruppo più complesso tipo
naftene, difenile, etc., e X, Y e Z i gruppi di giunzione, che
possono essere tutti uguali o differenti (ad esempio, gruppi
carbonato, solfone, ammide, immide, etere, tioetere od estere), in
posizione –orto, -meta e/o –para.
L’ampia di scelta tra i gruppi funzionali summenzionati
porta ad una grande varietà di strutture polimeriche, ciascuna
contraddistinta da ben precise caratteristiche in termini di
solubilità, stabilità ai solventi, temperatura di transizione vetrosa
(Tg), morfologia, reologia, processabilità, etc..
Tra le famiglie che fanno parte di questa classe di
materiali, presentano particolare interesse ed applicazione
tecnologica i poliarilensolfoni (Union Carbide, 1965; PSU, PES)
ed i poliarileterechetoni (ICI, 1978; PEK, PEEK), che per
l’elevata tenacità, leggerezza, inflessibilità, stabilità
dimensionale alle alte temperature, e resistenza alla
degradazione termo- e foto-ossidativa, sono studiati per

- 21 -
applicazioni nei settori aerospaziali, automobilistico e delle celle
a combustibile.
Per ogni materiale commerciale sono riportate in letteratura
le proprietà meccaniche, chimiche e fisiche in funzione della
composizione, tenendo conto che, come nel caso dei più comuni
poli(bisfenolo-A carbonato)(PC), polietilentereftalato (PET),
polibutilentereftalato (PBT), polimeri cristallo-liquidi
termotropici, poliammidi e poliimmidi, anche per i
polieteresolfoni ed poli(etere-etere-chetone) il successo di
mercato è legato all’ottimizzazione tra le caratteristiche
desiderate ed i costi di produzione.
1.3 Caratteristiche e proprietà strutturali
I polimeri aromatici, rispetto ai corrispondenti polimeri
alifatici, mostrano vantaggi significativi in particolari
applicazioni in campo ingegneristico e in applicazioni per
materiali con elevate prestazioni, possiedono infatti stabilità
termica e resistenza alla fiamma superiori ai corrispondenti
polimeri alifatici.

- 22 -
La struttura di questa classe di polimeri può essere amorfa o
parzialmente cristallina con elevate temperature di transizione
vetrosa (T
g
) e di fusione (T
m
) e con un ampio range di
temperatura di utilizzo.
Le proprietà fondamentali che distinguono questa famiglia di
polimeri dalle altre possono essere spiegate prendendo in
considerazione la struttura molecolare, ed in particolare la
maggiore stabilità termica dei residui aromatici o l’assenza di
gruppi sostituenti oppure terminali attivi che possono indurre
reazioni di degradazione della catena. La rigidità strutturale
dovuta alla elevata percentuale di componente aromatica
comporta una limitata possibilità di rotazione delle catene lungo
gli assi dei legami carbonio-carbonio e ciò limita la possibilità di
libero movimento nello spazio da parte delle catene; a questo
effetto va aggiunta la presenza di legami ad idrogeno che induce
una maggior stabilità conformazionale e rigidità delle molecole
1
.
Un elemento che determina notevoli cambiamenti di proprietà
tra i vari materiali è la natura del gruppo che unisce gli anelli
aromatici. Ad esempio, nella Tabella 1 sono comparate le

- 23 -
temperature di transizione vetrosa (T
g
), fusione (T
m
) e
degradazione (PDT) dei composti polimerici in funzione del
gruppo di giunzione.
STRUTTURA Tg (°C) Tm (°C) PDT (°C)
( - Ф - )
n
--- 530 500
( - Ф - O - )
n
80 298 400
( - Ф - S -)
n
90 295 350
( - Ф - SO
2
-)
n
350 520 500
Tabella 1
2
: proprietà termiche di alcuni polimeri aromatici
Dalla Tabella 1 risulta evidente come il para-fenilene
( [--]
n
) sia caratterizzato da un’eccezionale stabilità termica
(fino a circa 500°C non subisce alcuna degradazione) ma per
contro, l’alta temperatura di fusione (circa 530°C) rende il
materiale polimerico non processabile per via termica in quanto
decompone prima di fondere.

- 24 -
L’inserimento in catena dei ponti difenil-etere modifica
drasticamente le proprietà del prodotto; infatti il materiale
diventa amorfo o parzialmente cristallino con una temperatura di
transizione vetrosa intorno agli 80°C, una temperatura di
degradazione termica di circa 400°C e tra le caratteristiche più
interessanti, una buona processabilità a temperature prossime ai
300°C e quindi ben al di sotto della temperatura di
decomposizione.
La sostituzione dell’ossigeno con lo zolfo porta a polisolfuri
([--S-]
n
) che presentano una T
g
confrontabile ma anche, a
causa della maggiore cristallinità, una struttura più rigida e una
minore temperatura di decomposizione (il solfuro infatti tende ad
essere ossidato a solfone).
Il passaggio alla struttura polisolfonica ([--SO
2
-]
n
)
determina un forte aumento della T
g
del materiale che è di circa
350 °C. Aumenta la resistenza all’attacco dei solventi ma il
materiale diventa meno lavorabile in quanto la temperatura di
fusione risulta più alta di quella di decomposizione.

- 25 -
Da questi esempi si deduce che si possono modificare le
proprietà del materiale variando opportunamente il numero e la
posizione relativa dei gruppi etere-chetone ed etere-solfone
all’interno della catena polimerica. Ad esempio, è stato
verificato che se diminuisce la percentuale di gruppi chetonici in
catena aumenta la T
g
, la rigidità e la polarità delle
macromolecole. Allo stesso tempo diminuisce la cristallizzabilità
del polimero con un indebolimento della resistenza agli agenti
chimici e ai solventi.
Nel progettare un nuovo materiale occorre quindi, sulla base
della conoscenza delle relazioni che intercorrono tra struttura
molecolare, microstruttura e proprietà fisiche e meccaniche,
trovare un adeguato compromesso tra i parametri caratteristici
del polimero (T
g
, T
m
, PDT, cristallizzabilità, resistenza ai
solventi e processabilità) valutando anche i costi di produzione.
Un tentativo in tal senso è stato fatto da Carlier
3
che
esaminando il comportamento termico di policondensati arilici
(eteri-solfori-chetoni) ha progettato e sintetizzato un polimero

- 26 -
con la cristallinità, e dunque la resistenza ai solventi, di un
PEEK e la temperatura di transizione vetrosa di un PES.
Esaminando un ampia gamma di polieterichetoni e
polieterisolfoni è stata messa a punto una legge teorica che
correla la natura dei gruppi in catena con le caratteristiche
termiche del polimero. Questa teoria è paragonabile al metodo di
Van Krevelen
4
, ma rispetto ad esso risulta più specifico e quindi
coerente per i poliaromatici.
La temperatura di transizione vetrosa di un materiale dipende
sia dalla rigidità della catena polimerica e sia dalla sua polarità.
Nei PES i gruppi solfonici possiedono una forte
elettronegatività che induce una consistente delocalizzazione
delle nuvole elettroniche dei residui aromatici; da ciò si origina
un parziale carattere π dei legami carbonio–zolfo che limita la
possibilità di rotazione lungo l’asse del legame. Questo si
ripercuote in un aumento della rigidità molecolare che giustifica
i più alti valori della T
g
rispetto ai corrispondenti PEK.

- 27 -
Sono stati osservati inoltre differenti comportamenti in
funzione della posizione del legame etereo; più precisamente i
legami in ‘para’ del gruppo solfonico portano a polimeri con una
T
g
maggiore rispetto a quelli con legami in meta. Tutto ciò si
spiega sulla base delle possibili forme di risonanza che sono due
nel caso del legame in para e una sola in quella dei legami in
meta
5
che porta, in questo ultimo caso, ad una maggiore
flessibilità della catena e quindi ad una T
g
minore.
Riguardo alla diversa elettronegatività dei gruppi solfonici e
chetonici il Carlier definisce la PRCL (Percentage Rigid Chain
Lenght) attribuendo valore 100% di rigidità (cioè massimo
impedimento alla rateazione) ai due legami S-C ai lati dell’SO
2
,
una parziale restrizione rotazionale del 33% dei legami connessi
al gruppo chetonico, e 0% di rigidità (cioè massima libera
rotazione) ai due legami C-O del gruppo etere. Riportando in
grafico (Figura 1), per i vari polimeri tipo PEK e PES, i valori di
T
g
e T
m
(dati di letteratura) rispetto ai valori di PRCL si ottiene
una andamento lineare.
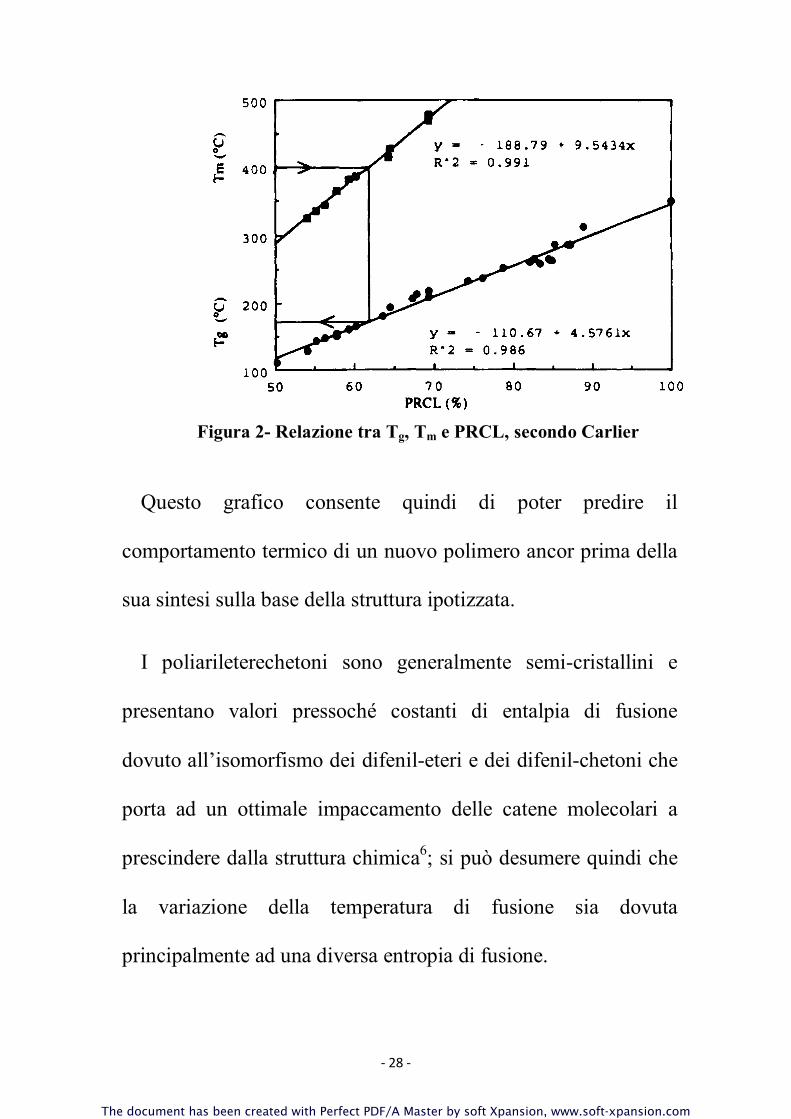
- 28 -
Questo grafico consente quindi di poter predire il
comportamento termico di un nuovo polimero ancor prima della
sua sintesi sulla base della struttura ipotizzata.
I poliarileterechetoni sono generalmente semi-cristallini e
presentano valori pressoché costanti di entalpia di fusione
dovuto all’isomorfismo dei difenil-eteri e dei difenil-chetoni che
porta ad un ottimale impaccamento delle catene molecolari a
prescindere dalla struttura chimica
6
; si può desumere quindi che
la variazione della temperatura di fusione sia dovuta
principalmente ad una diversa entropia di fusione.
Figura 2- Relazione tra T
g
, T
m
e PRCL, secondo Carlier

- 29 -
Il PRCL regola quindi sia la T
m
che la T
g
e come mostrato
nella Figura 2 la pendenza della retta relativa ai valori di T
m
è
significativamente più alta di quella dei corrispondenti valori di
T
g
. In Figura 3 è mostrato come varia la temperatura di
transizione vetrosa in funzione di quella di fusione. Poiché la
massima temperatura di lavorazione dei poliaromatici, senza
rischiare la degradazione termica del materiale, è di circa 400°C,
è possibile prevedere da questo grafico una massima T
g
del
polimero di 170–200 °C.
Figura 3- Relazione tra Tg e Tm

- 30 -
Considerato che i PES spesso presentano strutture
simmetriche, risulta comprensibile la loro scarsa tendenza a
cristallizzare. A tale proposito, Blackadder e Ghavamikia
7-9
hanno provato che il PES può cristallizzare in presenza di
opportuni solventi come diclorometano e cloroformio. Essi,
utilizzando la diffrazione dei raggi X, hanno potuto identificare
morfologie simili a cristalli che crescono durante il trattamento
ma che scompaiono all’allontanamento dei solventi.
In condizioni normali quindi i PES, diversamente dai poli(eteri
chetoni) (PEK), devono essere considerati fondamentalmente
amorfi (comportamento evidenziato anche da misure termiche ed
ottiche).
Le differenze morfologiche fra PES e PEK vengono attribuite
a fattori sterici di maggior ingombro dei gruppi solfonici e ai
differenti angoli di legame in corrispondenza del gruppo di
giunzione che nel caso dell’etere difenilico è di circa 124°
mentre nel corrispondente difenilsolfone è di 105°
10
. Si ritiene
che entrambi i fattori, provocando un minore impiccamento

- 31 -
delle catene all’interno della massa polimerica, siano la causa un
abbassamento dell’entalpia del sistema che porta ad un più basso
valore della temperatura di fusione dei PES. Per contro nei PES
si produce anche un contemporaneo irrigidimento delle catene
che è responsabile dell’innalzamento delle T
g
; in definitiva si
assiste ad una diminuzione dell’intervallo T
m
–T
g
e quindi una
minore cristallizzabilità del polimero.
Queste ipotesi sono avvalorate dall’aver mostrato come sia
possibile cristallizzare dei PEES (polietere-etere solfone) dopo
aver diminuito, tramite aggiunta di solventi, la T
g
del polimero
11
.
Si è già accennato alla ristretta gamma di possibili reazioni
che portano alla sintesi di queste classi polimeriche; il dover
progettare policondensazioni porta ad affrontare varie
problematiche, non sempre facilmente risolvibili. La scelta dei
monomeri risulta di cruciale importanza, infatti le reazioni
nucleofile sono sensibili ad ogni possibile impurezza presente
nell’ambiente di reazione, è altresì d’obbligo lavorare con
miscele perfettamente stechiometriche, per non inficiare la

- 32 -
reattività dei monomeri.
L’elevato costo dei monomeri di questo tipo è uno dei
principali svantaggi di questo approccio alla sintesi di
poliaromatici. Sono stati effettuati vari tentativi al fine di poter
utilizzare monomeri di partenza aventi prezzo minore; Ullman
12
nel 1904 riportò un metodo per formare ponti etere dalla
condensazione di un’alide aromatica con il sale di un fenolo,
usando sali metallici, in particolare di rame, per catalizzare la
razione ma ottenendo macromolecole a bassa massa molare.
13-17
1.4 La reazione di solfonazione dei poliarilati
Gli anelli aromatici dei poliarilati possono essere
modificati con vari agenti funzionalizzanti e tra le reazioni di
funzionalizzazione, la più studiata, è la già citata introduzione di
gruppi solfonici (-SO
3
H) in catena (solfonazione) che può essere
effettuata in vari modi per reazioni dei polimeri con Acido

- 33 -
clorosolfonico, acido solforico fumante, trimetil silil-
clorosolfonato, SO
3
ecc..
Probabilmente la prima reazione di solfonazione di poli(arilen
eteri-solfoni) (PES) è stata riportata in letteratura da Quentin
18-19
.
La reazione avviene a temperatura ambiente e l’agente
solfonante impiegato è l’acido clorosolfonico (ClSO
3
H) che
mediante sostituzione elettrofila introduce gruppi –SO
3
H in
catena a livello dell’anello aromatico e viene utilizzato per la
produzione di un polimero commerciale (Udel
®
)
20
. La reazione è
stata condotta per diversi intervalli di tempo ottenendo così
differenti gradi di solfonazione.
Sono stati effettuati molti studi con l’obiettivo di migliorare le
rese del prodotto solforato. L’acido clorosolfonico causa però
reazioni secondarie indesiderate (ramificazioni, scissioni di
catena, cross-linking ecc.) che condizionano sensibilmente le
rese finali. Uno studio volto a minimizzare la reazione di
scissione della catena polimerica è stato compiuto utilizzando
acido trimetilsilil clorosolfonato
21
in cui il gruppo silil- riduce le
reazioni secondarie.

- 34 -
Altri lavori scientifici descrivono invece procedure di
funzionalizzazione svolte utilizzando sia l’ossido SO
3
20
che
complessi SO
3
-trietil fosfato (TPE) come agente solfonante della
catena poliarilica
22
; l’SO
3
infatti ha il vantaggio di avere una
struttura molecolare in cui i tre atomi di ossigeno sono legati
mediante altrettanti doppi legami all’atomo di zolfo centrale, e
ciò rende la molecola (SO
3
) un agente solfonante ancor più forte
dell’acido solforico concentrato tanto da poter dare reazioni di
sostituzione sull’anello aromatico a temperature comprese tra 0-
10°C; in questo modo le reazioni secondarie di ‘cross-linking’
sono state ridotte notevolmente.
L’attività di ricerca in questo campo è ovviamente tesa a
modellare la struttura dei poliarilati in funzione delle possibili
applicazioni e mettere a punto adeguati ed innovativi processi
produttivi per migliorarne le proprietà.

- 35 -
1.5 Metodiche di sintesi di poli(arilen eteri solfoni)
solfonati
Sono stati riportati in letteratura molti tentativi di
funzionalizzare con gruppi solfonici delle strutture poliariliche
aventi ponti solfonici ed eterei in catena; uno dei primi tentativi
fu quello riportato da Ueda
23
; in questo lavoro è riportato che la
sintesi del polimero viene effettuata utilizzando come reagenti di
partenza il monomero di-alogenato diclorodifenilsolfone
(DCDPS) , lo stesso monomero precedentemente funzionalizzato
con gruppi solfonici (SDCDPS), e il bisfenolo A.
Il lavoro di Ueda è stato poi ripreso e sviluppato da Robeson e
Matzner che proposero di utilizzare i polimeri solfonati da loro
ottenuti (con lo stesso metodo descritto precedentemente) come
additivi di resine poliestere per migliorarne le proprietà ritardanti
di fiamma.
McGrath
24-26
nei suoi lavori scientifici
mise a punto un altro
metodo di solfonazione del DCDPS ottenendo rese finali più alte

- 36 -
rispetto ai precedenti tentativi. Inoltre per variare la percentuale
di componente aromatica in catena utilizzò vari tipi di bisfenoli;
tra i principali utilizzi che egli propose nel suo lavoro per questa
famiglia di poliarilati c’è l’applicazione come membrane a
scambio protonico per celle a combustibile.
Tra le altre principali applicazioni, i PES solfonati trovano
ampio spazio in applicazioni come: adesivi per strati metallici
27
,
membrane per separare gas e solidi da soluzione
28
e matrici per
materiali compositi e come agenti rinforzanti per resine
termostabili
29
.
Il problema che si pone nei summenzionati processi di
solfonazione dei poli(arilen eteri-solfoni) è determinato dalle
difficoltà di controllo della cinetica e della % di solfonazione
degli anelli aromatici. Questo comporta spesso, la preparazione
di materiali con proprietà non riproducibili poi nelle stesse
condizioni di solfonazione.

- 37 -
I polieteri solfoni sono una classe di poliaromatici di notevole
importanza per speciali applicazioni che includono agenti per
l’indurimento delle resine epossidiche, adesivi per legami tra
metallo-metallo, membrane per la separazione di sostanze
gassose da substrati solidi e membrane per celle a combustibile.
L’ultima applicazione è di crescente interesse per via dello
sviluppo di celle a combustibile con proprietà sempre più
sofisticate.
Grandi sforzi sono stati dedicati allo sviluppo di polimeri ad
alte prestazioni nella conducibilità protonica che possano
superare i limiti delle membrane polimeriche per fluorurate.
I poli(arilen eteri-solfoni) come già detto in precedenza
solfonati costituiscono una classe promettente di materiali che
possono costituire una valida alternativa alle membrane a base
per fluorurata grazie alle loro elevate proprietà meccaniche e alla
stabilità chimica e termica e all’ossidazione.
Sono stati compiuti parecchi studi sul problema
dell’introduzione di gruppi solfonici nell’ossatura principale dei

- 38 -
polieteri solfoni.
Tra le differenti vie di sintesi le più diffuse sono:
- Post-solfonazione di polieterisolfoni commerciali mediante
reazione di sostituzione elettrofila aromatica o sostituzioni
nucleofile;
- Sintesi di polimeri solfonati a partire da monomeri o
oligomeri solfonati fatti reagire con monomeri standard.
Il primo approccio è il più economico ma può condurre a
rotture di catena e abbassamento della massa molare media
durante la reazione di sostituzione nucleofila.
Il secondo approccio basato sulla sintesi a partire da monomeri
presenta il vantaggio di poter operare il controllo della struttura
finale della macromolecola e delle principali proprietà del
polimero (ad esempio la distribuzione della massa
molare media, la struttura chimica, i gruppi terminali, il grado
di solfonazione della catena macromolecolare).

- 39 -
Un altro vantaggio dell’approccio sintetico a partire da
monomeri ed oligomeri è la possibilità di ottenere strutture multi
blocco controllate che portano alla formazione di domini ben
separati di strutture idrofobiche (in corrispondenza dei blocchi
non solfonati) e di strutture idrofiliche in corrispondenza dei
blocchi solfonati.
La creazione di tali strutture morfologiche è considerata un
vantaggio chiave per controllare le proprietà di rigonfiamento
mediante assorbimento di acqua da parte della membrane.
L’esatta conoscenza della struttura macromolecolare è di
fondamentale importanza nello studio di nuove famiglie di
polimeri per valutare le loro proprietà chimiche, fisiche,
meccaniche ecc. e dallo studio di tali proprietà, trovare le
potenziali applicazioni.
Tra le tecniche utilizzate nella caratterizzazione
macromolecolare le tecniche spettroscopiche come l’NMR e il
MALDI-TOF MS (Matrix Assisted Laser Desorption/Ionization

- 40 -
- Time of Flight Mass Spectrometry) costituiscono due tra le
tecniche più potenti in utilizzo oggi per via della grande quantità
e precisione delle informazioni che riescono a fornire.
Recentemente, per ottenere una caratterizzazione fine e
dettagliata dei copolieterisolfoni le due tecniche sono state
utilizzate in maniera combinata e complementare
30
.
Grazie all’uso combinato dell’NMR e dell’FT-IR
31-35
MALDI-
TOF MS
36-40
ed sono state messe in luce caratteristiche
strutturali ed informazioni sulla composizione chimica
macromolecolare di copolimeri complessi e sulla natura di
alcune reazioni secondarie che avvengono durante la sintesi
mediante lo studio della cinetica di reazione.

- 41 -
2. SCOPO DEL LAVORO DI RICERCA
Lo scopo del lavoro sperimentale della presente tesi di Dottorato è
stato focalizzato allo studio della reazione di sintesi di un’innovativa
classe di tecnopolimeri a base aromatica aventi una struttura
parzialmente polare; i poli(arilen eteri-solfoni) (PES).
Tale studio sperimentale avrà l’obiettivo, a partire dai dati di
letteratura e dall’esperienza maturata dal gruppo di ricerca nel
settore, di studiare la cinetica della reazione di policondensazione, e
partendo dai risultati di tale studio riuscire a mettere a punto un
metodo innovativo di sintesi per questa classe di macromolecole, in
alternativa al metodo già utilizzato. Ciò sarebbe particolarmente utile
per riuscire ad ottenere materiali idonei ad essere utilizzati in
determinati campi applicativi, dato che con le tecniche tradizionali di
sintesi i polimeri non garantiscono le proprietà chimiche, termiche e
meccaniche richieste.
Il requisito di filmabilità del materiale è strettamente legato
all’ottenimento di catene polimeriche ad elevata massa molare
media.

- 42 -
Ciò infatti garantirebbe una rigidità dimensionale del sistema.
Contemporaneamente, ottenere un elevato grado di solfonazione
aumenterebbe il potere polielettrolitico del materiale.
I poli(arilen eteri-solfoni) aventi diversi gradi di solfonazione
ottenuti mediante reazione di policondensazione nucleofila aromatica
saranno caratterizzati sotto molteplici aspetti, al fine di studiarne le
loro proprietà chimiche, termiche e meccaniche e successivamente i
loro possibili campi applicativi.
La letteratura relativa ai poli(arilen eteri-solfoni) solfonati,
oggetto del nostro studio indica tale classe di polimeri, come
possibile alternativa ai materiali tutt’ora in uso nella preparazione di
membrane per ‘fuel-cells’. Ciò risulterebbe possibile a causa
dell’elevata polarità delle catene polimeriche di questa classe di
macromolecole. Si potrebbero inoltre ipotizzare possibili
applicazioni come membrane per la separazione di gas, grazie anche
all’utilizzo di nano particelle inorganiche (silice funzionalizzata,
zeoliti ecc.) tale classe di materiali potrebbe inoltre trovare
applicazione come potenziali composti da utilizzare nel campo delle
resine (epossidiche ecc.) ad esempio come agenti indurenti.

- 43 -
3. PARTE SPERIMENTALE
3.1 Materiali
Tutti i reagenti ed i solventi utilizzati per le sintesi sono stati
acquistati dalla Sigma-Aldrich: 4,4’-di-clorodifenilsulfone
(DCDPS), 4,4’-di-idrossidifenilsolfone (DHDPS),
tetrametilensulfone (solfolano), 4-4’-bis-(4-
clorofenilsolfone)difenile (LCDC), m-amminofenolo (mAP),
carbonato di potassio e metanolo. Il DCDPS è stato
ricristallizzato due volte da toluene; il solfolano è stato distillato
a pressione ridotta e quindi conservato in essiccatore fino
all'utilizzo, in modo da minimizzare la presenza di tracce
d'acqua., tutti gli altri prodotti sono stati utilizzati senza altri
pretrattamenti, esclusa la permanenza in stufa da vuoto per una
notte.

- 44 -
3.2 Sintesi dei prepolimeri 4K e 7K
Il metodo standard utilizzato per sintetizzare le catene
poliaromatiche dei poli(arilen eteri-solfoni) sintetizzati e studiati
nel presente lavoro di tesi è la reazione nucleofila di
policondensazione tra unità bis-fenoliche attivate in ambiente
basico e unità di-alogenure aromatiche
30
:
3.3 Monomeri impiegati
Per realizzare la sintesi dei poli(arilen eteri-solfoni) si
preferisce utilizzare il dicloro-difenil solfone (DCDPS),
nonostante il corrispondente di-fluoruro (DFDPS) sia più
reattivo in quanto l’atomo di fluoro sostiuito nell’anello
aromatico è un miglior gruppo uscente rispetto al cloro.
I reagenti cloro-derivati sono tuttavia abbastanza reattivi
poiché l’effetto elettron-attrattore del gruppo solfonico,
polarizzando i legami C-Cl ne abbassa l’energia di legame e

- 45 -
facilita quindi l’uscita del cloro e la reazione di
policondensazione.
Viene utilizzato NMP (N-metil pirrolidone) al 99% di purezza
come solvente per le reazioni di polimerizzazione, in quanto i
reagenti utilizzati presentano solubilità buona solubilità in questo
solvente; inoltre questo solvente presenta un alto punto di
ebollizione (sopra la temperatura a cui avviene la reazione).
E’ qui riportato l’elenco dei reagenti utilizzati nelle reazioni di
polimerizzazione e funzionalizzazione:
4,4
I
-dicloro-difenilsolfone (DCDPS) micronizzato
SO
2
Cl Cl
DCDPS
4,4
I
-diidrossi-difenilsolfone (DIDPS) micronizzato
OH SO
2
OH
DIDPS

- 46 -
3.4 Tecniche per la caratterizzazione dei Polimeri
Tutti i campioni sintetizzati sono stati sottoposti ad
un’accurata caratterizzazione molecolare, volta a determinare il
peso molecolare, volta a determinare il peso molecolare,
confermare la struttura chimica, la natura dei gruppi terminali, il
grado di solfonazione effettivo, e termica allo scopo di correlare
tali proprietà alle caratteristiche strutturali ed alla composizione.
Due Poli(arilen eteri-solfoni) con una massa molare media di
4000 g/mol e 7000 g/mol rispettivamente, sono stati preparati
mediante reazione di policondensazione nucleofila aromatica tra
il DCDPS con un eccesso di DHDPS come sottolineato nello
Schema 1. La reazione di sintesi dei suddetti polieterisolfoni è
stata condotta in un pallone a tre colli da 250 ml corredato di
piastra riscaldante ed agitatrice, per garantire il mescolamento
continuo della miscela di reazione. L'ambiente di reazione è
inoltre mantenuto inerte per tutta la durata della reazione
mediante un flusso di azoto in ingresso nel pallone mediante

- 47 -
opportuno raccordo in vetro. Il pallone è corredato inoltre di un
refrigerante a bolle posto in corripondenza del collo centrale, per
garantire la condensazione dei vapori.
Per ottenere il pre-polimero PES-OH avente una massa molare
media teorica (Mn) di 4K,in un pallone a tre colli da 250ml sono
stati aggiunti 5.11g di DCDPS (1.78*10-2 moli) e 4.89g di
DHDPS (1.95*10-2 moli, il 9.5% in moli di eccesso). Sono
quindi stati aggiunti 50ml di solvente N-metil pirroliodone
(NMP). Quando la miscela di reazione ha raggiunto i 90-100°C è
stato infine aggiunto il carbonato di potassio (1.83*10-2 moli di
K
2
CO
3
, il 2.8% di eccesso in moli rispetto al DCDPS).
La temperatura di reazione è stata portata a 190°C la
formazione e la miscela fatta reagire a questa temperatura per 30
minuti (tempo in cui si dovrebbe avere la formazione dei gruppi
fenati terminali), quindi per un'ora a 205°C e per 4 ore a 225°C,
il tutto in ambiente inerte garantito da un flusso di azoto
continuo per tutta la durata della reazione. La miscela così fatta
reagire è stata dapprima raffreddata fino a temperatura ambiente

- 48 -
e quindi è stata aggiunta ad una miscela di acqua distillata ed
etanolo fresco (95% v/v) in rapporto 1/1, di un volume pari a
circa 5 volte il volume della miscela di reazione ottenendo un
precipitato bianco costituito dai micro-granuli di polimero
ottenuti. Il precipitato bianco viene così filtrato e lavato 4 volte
con acqua ed etanolo in miscela 1:1 ed infine il prodotto filtrato
viene posto in stufa da vuoto a 120°C per 24ore.
Il prepolimero PES-OH avente massa molare media teorica di
7K è stato sintetizzato usando un rapporto molare
DHDPS/DCDPS di 1.05, seguendo la stessa procedura di
reazione utilizzata per l'analogo prepolimero avente Mn=4K.
3.5 Solfonazione del monomero DCDPS
La seguente procedura porta alla funzionalizzazione degli
anelli aromatici del monomero DCDPS con gruppi solfonici in
posizione ‘orto’ al gruppo cloro-terminale
41
.

- 49 -
Qui di seguito nello Schema 2 è riportato lo schema della
reazione di solfonazione del monomero DCDPS:
Qui di seguito i reagenti utilizzati nella reazione di
solfonazione:
Diclorodifenilsolfone (DCDPS)
Acido solforico fumante al 20% in SO
3
Idrossido di sodio
Cloruro di sodio
Schema 2- La reazione di solfonazione del DCDPS

- 50 -
WATER OUT
WATER IN
NITROGEN IN NITROGEN OUT
VACUUM
Figura 4- Disegno schematico dell’apparato di reazione
utilizzato
per le reazioni di sintesi.

- 51 -
Si effettua uno step preliminare di trattamento del monomero
di partenza mediante essiccazione in stufa da vuoto a 80 °C per
circa 12 ore. Viene pesata la quantità opportuna di DCDPS che
si vuole funzionalizzare e si preleva la quantità corrispondente di
acido solforico fumante al 20% in SO
3
; si utilizza un rapporto ½
tra (gr)DCDPS/(ml)H
2
SO
4
.
Il DCDPS da funzionalizzare viene introdotto in un pallone in
vetro da 100ml a tre colli; attraverso uno dei due colli laterali
viene introdotto goccia a goccia mediante un imbuto da carico
l’acido solforico fumante; dato che la reazione avviene ad alta
temperatura è necessario disporre di una piastra riscaldante
dotata di sistema di agitazione e di un bagno d’olio siliconico in
cui viene immerso il pallone in cui avviene la reazione. La
miscela di reazione viene riscaldata a 110°C per 6 ore, quindi il
prodotto ottenuto è raffreddato fino a temperatura ambiente e
successivamente viene precipitato in 400 ml di acqua distillata a
0°C. Alla soluzione così ottenuta vengono aggiunti 180gr di
NaCl e successivamente il sale sodiato del diclorodifenilsolfone

- 52 -
solfonato precipita come polvere bianca. Il precipitato così
ottenuto viene filtrato e ridisciolto in 400ml di una soluzione
acquosa 2N di NaOH a 10°C, fino a pH 6/7 ed infine è
aggiunto altro NaCl in eccesso per essere sicuri di aver
precipitato completamente la forma sodiata del monomero
solfonato.
Il precipitato viene quindi filtrato e ricristallizzato in una
miscela metanolo/acqua in rapporto 9/1 ottenendo rese di
prodotto intorno al 90%. In figura 5 è rappresentato lo spettro
1
H-NMR del monomero SDCDPS così funzionalizzato.
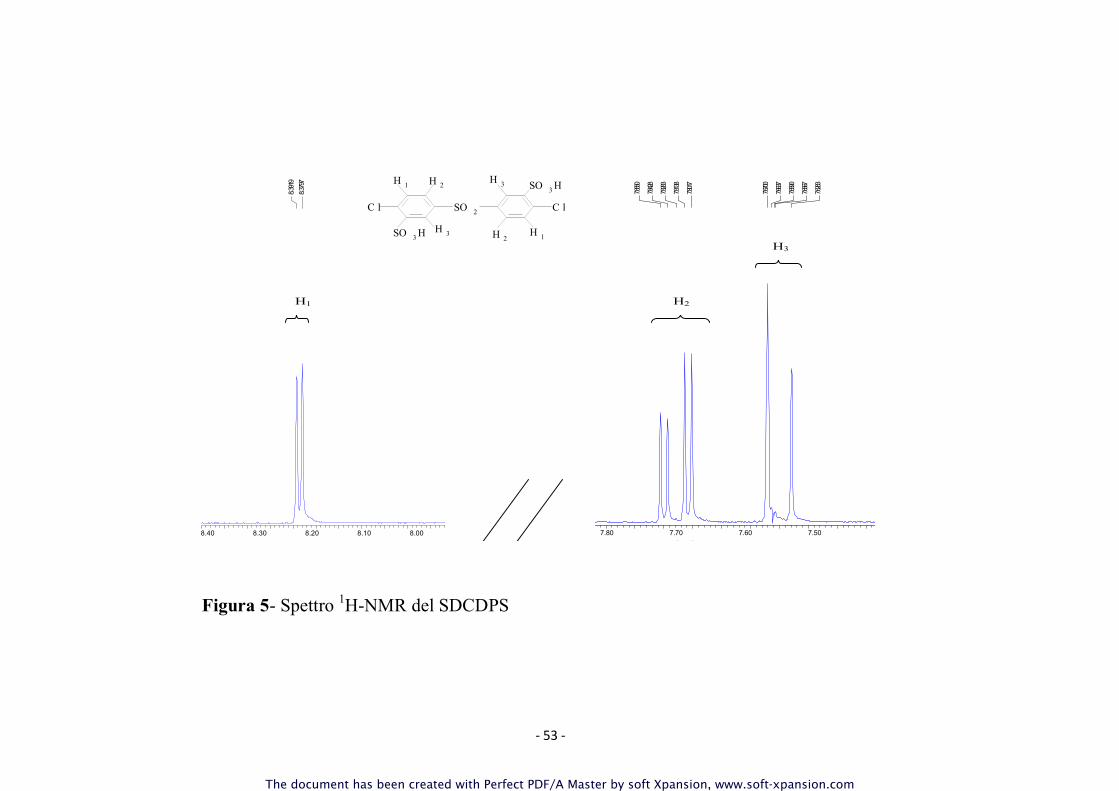
- 53 -
8.3919
8.3797
7.8550
7.8428
7.8283
7.8138
7.8017
7.6700
7.6637
7.6590
7.6567
7.6283
(ppm)
7.107.207.307.407.507.607.707.807.908.008.108.208.308.408.50
SDCDPS
H
1
H
2
H
3
8.3919
8.3797
7.8550
7.8428
7.8283
7.8138
7.8017
7.6700
7.6637
7.6590
7.6567
7.6283
(ppm)
7.107.207.307.407.507.607.707.807.908.008.108.208.308.408.50
SDCDPS
SO
2
SO
3
H
C lC l
SO
3
H
H
1
H
3
H
3
H
1
H
2
H
2
Figura 3: Spettro
1
H-NMR del diclorodifenil solfone di solfonato
ppm
Figura 5- Spettro
1
H-NMR del SDCDPS

- 54 -
3.6 Sintesi dei polimeri S-PES a diversi gradi di
solfonazione
Sono stati ottenuti poli(arilen eteri-solfoni) di-solfonati (S-
PES) mediante reazione di policondensazione nucleofila
aromatica tra il pre-polimero PES oligomero a massa 4k o 7K
utilizzato come agente telechelico ed il monomero di-solfonato
(SDCDPS) ottenuto precedentemente utilizzando vari rapporti
molari PES-OH/SDCDPS in base alla percentuale di
solfonazione teorica del PES solfonato (S-PES) che si vuole
ottenere. La miscela di reazione è stata fatta reagire per 12 ore a
200°C sotto agitazione, in un pallone a tre colli sormontato da
refrigerante a bolle, ambiente inerte garantito da un flusso di
azoto per tutta la durata della reazione.
La quantità di SDCDPS fatta reagire è stata calcolata in
proporzione alla quantità di gruppi terminali idrossi presenti nel
PES-OH pre-polimero.

- 55 -
In particolare vengono utilizzati 4 rapporti molari PES-
OH/SDCDPS: 1.20, 1.05, 1.00 e 0.90) in relazione al grado di
solfonazione teorico (DS) calcolato per ciascuna reazione e alle
distribuzioni di massa molare (MMD).
La reazione di policondensazione nucleofila aromatica è stata
realizzata utilizzando un pallone a tre colli da 250ml sormontato
da un refrigerante a bolle ed equipaggiato con una piastra
riscaldante provvista di sistema di agitazione magnetica.
L'ambiente di reazione viene mantenuto inerte mediante flusso
di azoto per tutta la durata della reazione. Il solvente utilizzato è
tetrametilensolfone e per garantire l'assenza di acqua
nell'ambiente di reazione viene utilizzato toluene come agente
azeotropico. La miscela di reazione viene riscaldata fino a 140°C
per circa 3 ore, in modo da garantire la totale disidratazione
dell'ambiente. La presenza di acqua all'interno dell'ambiente di
reazione favorisce reazioni secondarie di transeterificazione; in
tali reazioni l'acqua rompe le catene polimeriche in crescita al
livello dei ponti eterei abbassando così la massa molare media

- 56 -
delle catene polimeriche.
La temperatura viene quindi aumentata a 200°C e la reazione
viene portata avanti per 24 ore. La miscela reagita è stata quindi
precipitata in una soluzione acquaosa 1M di HCl. Il precipitato
così ottenuto sotto forma di polvere bianca è stato lavato 5 volte
con una miscela acqua distillata/etanolo 1:1. Il prodotto viene
quindi essiccato in stufa da vuoto a 120°C per 24 ore. I campioni
di copolimero sono riportati come S-PES-mr (12h) nella tabella
x, dove mr indica il rapporto molare PES-OH/SDCDPS. Qui di
seguito è illustrato lo schema della reazione di sintesi degli S-
PES.

- 57 -
SO
2
SO
3
Na
O
SO
3
Na
SO
2
O
x
SO
2
O
y
SO
2
OHSO
2
O
n
+
+ K
2
CO
3
HO SO
2
ClCl
SO
3
Na
SO
3
Na
1
SO
2
SO
3
H
O
SO
3
H
SO
2
O
x
SO
2
O
y
HCl
Scheme 3 – Typical synthesis of sulfonated poly(arylen ether-sulfone)s.
Schema 3- Reazione di sintesi del PES solfonato

- 58 -
3.7 Reazione di POST-POLIMERIZZAZIONE
La necessità di prolungare il tempo di reazione a posteriori, in
un secondo step consecutivo alle prime 12 ore è giustificata
dadai risultati ottenuti dalla caratterizzazione chimica (NMR,
MALDI, Viscosimetria-SEC, FT-IR) effettuata sui campioni di
S-PES ottenuti dopo 12h di reazione.
Gli spettri NMR ottenuti hanno infatti evidenziato come la
reazione dopo 12 ore fosse ancora parzialmente incompleta; ciò
si evince dai dati ottenuti sulla massa molare media che risulta
più bassa delle previsioni teoriche e dalla presenza di un residuo
rilevante di catene aventi gruppi –Cl terminali; i calcoli
stechiometrici sui campioni di S-PES preparati dopo 12 ore di
reazione infatti, sono stati effettuati ponendo in leggero eccesso
il pre-polimero PES-OH in modo tale da garantire una pressoché
totale terminazione delle catene di polimero S-PES finale con
gruppi fenolici e la presenza residua di gruppi terminali –Cl è
indice di possibili altre reazioni di policondensazione nucleofila

- 59 -
aromatica tra i terminali di catena con allungamento delle stesse
mediante meccanismo di “chain coupling”.
La reazione è stata condotta ad una temperatura di 200°C in
ambiente inerte per flusso di azoto in solvente NMP per ulteriori
24 e 48h, in presenza di K
2
CO
3
(0.01g). Il polimero ottenuto
viene lavato 5 volte in miscela acqua distillata/etanolo 1:1. Il
prodotto è stato essiccato in stufa da vuoto a 150°C per 24 ore. I
campioni ottenuti sono nominati in Tabella 2 come S-PES-
mr(12+xh) (con x=24 o 48).
3.8 Sviluppi degli studi nel prossimo futuro
Le reazioni di sintesi dei PES solfonati realizzate fin qui hanno
dimostrato che il metodo utilizzato, che prevede la sintesi di un
pre-polimero a bassa massa (3K-7K) fatto reagire
con il monomero solfonato SDCDPS, non ci ha permesso di
ottenere catene polimeriche aventi un grado di solfonazione
maggiore del 10-12% (il grado di solfonazione è espresso in % di

- 60 -
moli di gruppi solfonici –SO
3
H sostituiti ai protoni aromatici
nella catena principale, rispetto ai protoni aromatici totali).
La massa molare media inoltre non supera i 10K a causa di
reazioni secondarie di trans-eterificazione inter e intramolecolari
che limitano la lunghezza delle catene polimeriche verso valori
più alti. Questi risultati costituiscono un limite se si considerano
le potenziali applicazioni dei materiali sintetizzati (fuel cells,
film membrana per gas ecc.). Un grado di solfonazione più
elevato infatti, aumentando il numero di gruppi altamente polari
presenti in catena accrescerebbe l’effetto polielettrolitico del
materiale con conseguente incremento della conducibilità
protonica. La concentrazione di gruppi solfonici non può però
superare una soglia limite (÷35-40%), in quanto il materiale
diventerebbe troppo polare e ciò lo renderebbe solubile in tutti i
solventi polari compresa l’acqua.
Un aumento della massa molare media invece avrebbe effetti
positivi sulle proprietà meccaniche, in quanto renderebbe il
materiale più rigido e resistente.

- 61 -
Inoltre, è stato visto nella letteratura e confermato da prove
sperimentali svolte presso i nostri laboratori che il sotto un certo
valore indicativo di massa molare media corrispondente a circa
20K il materiale non possiede le auspicate proprietà di rigidità
dimensionale sufficienti ad essere ridotto in film abbastanza
sottili per i test di conducibilità protonica..
E’ quindi condizione necessaria per poter utilizzare tale classe
di materiali polimerici per gli scopi sopraelencati, ottenere catene
polimeriche di una certa lunghezza.
Un’alternativa innovativa alla metodica di sintesi descritta che
prende spunto dagli ultimi lavori pubblicati su tale problematica,
prevede l’utilizzo di gruppi un agente telechelico tri-funzionale,
il 3,5 difluoro-clorobenzene.
Il vantaggio innovativo che dovrebbe fornire l’utilizzo di
questa molecola nella reazione di policondensazione consiste
nella possibilità di utilizzare condizioni di reazione più blande
(temperatura più bass, circa 130-140°C) e tempi di reazione più
brevi. Ciò è reso possibile dalla maggiore reattività del gruppo
terminale –F rispetto al terminale –Cl tradizionalmente utilizzato

- 62 -
fin qui. Quest’ultimo approccio è ancora in una fase di studio per
la messa a punto delle condizioni ideali allo svolgimento della
reazione.
3.9 Studio della cinetica reazione di sintesi del prepolimero
PES solfonato “perfettamente alternato “
Riuscire ad ottenere delle masse molari medie più elevate di
quelle ottenute con la metodica fin qui utilizzata garantirebbe
una maggiore rigidità strutturale e proprietà meccaniche tali da
permettere la filmabilità del materiale sintetizzato.
Al fine di superare i limiti fin qui raggiunti è stato messo a
punto un innovativo metodo di sintesi di S-PES.
Tale metodo prevede la sintesi iniziale di un pre-polimero
idrossi-terminato a bassa massa (1-2K) ottenuto a partire dal
monomero solfonato SDCDPS con il DHDPS non solfonato.
Il pre-polimero così ottenuto viene indicato con l’espressione
“perfettamente alternato” in quanto in catena si alternano i

- 63 -
residui monomerici non-solfonati ai residui di-solfonati
provenienti dal monomero SDCDPS.
Il pre-polimero ottenuto è quindi fatto ulteriormente reagire
con il monomero DCDPS la fine di far crescere la massa molare
media.
Chimicamente il monomero solfonato SDCDPS ha una
reattività inferiore rispetto all’analogo monomero non solfonato,
per motivi di ingombro sterico del gruppo solfone laterale ma
anche perché tale gruppo probabilmente “disattiva” parzialmente
il terminale –Cl rendendolo un peggior gruppo uscente nella
reazione di policondensazione nucleofila aromatica. Questa
considerazione teorica è avvalorata dall’evidenza sperimentale.
E’ stato intrapreso lo studio della cinetica di questa innovativa
metodica sintesi. Sono stati effettuati dei prelievi a tempi
determinati (3H, 6H, 9H, 24H, 70H) della miscela di reazione
mirati a monitorare il grado di avanzamento della reazione.
Ciascuna aliquota prelevata è quindi dovrà essere caratterizzata
chimicamente (NMR, MALDI ed FT-IR Viscosità-SEC) e
termicamente (DSC e TGA).

- 64 -
3.10 Caratterizzazione dei Polimeri
Tutti i campioni sintetizzati sono stati sottoposti ad un’accurata
caratterizzazione molecolare - volta a determinare il peso
molecolare, confermare la struttura chimica, la natura dei gruppi
terminali, il grado di solfonazione effettivo - e termica - allo
scopo di correlare tali proprietà alle caratteristiche strutturali ed
alla composizione.
Per la caratterizzazione dei polimeri ottenuti sono state
utilizzate le tecniche di seguito descritte.
3.10.1 Spettrometria di massa MALDI-TOF
Gli spettri di massa MALDI-TOF sono stati registrati mediante
uno spettrometro di massa Voyager–DE STR (Perseptive
Biosystem), sia in modo lineare che con il Reflectron.

- 65 -
Questo strumento è dotato di un laser ad N
2
con λ=337 nm ed
un’ampiezza d’impulso di 3 ns. Si è preferito lavorare nella
modalità a ioni positivi. La differenza di potenziale
d’accelerazione è di 25 KV; il voltaggio della griglia e il tempo
di ritardo sono stati ottimizzati per ogni campione per realizzare
la migliore risoluzione delle masse; l’intensità del raggio laser è
stato mantenuto leggermente al di sopra del limite minimo
necessario per il processo desorbimento/ionizzazione.
I campioni destinati al MALDI sono stati preparati nel
seguente metodo, che prevede la deposizione in piastrina
seguendo uno schema “multilayer” ossia deponendo successivi
strati di soluzione di matrice, e successivamente di soluzione del
campione. Le due soluzioni sono preparate separatamente e
deposte sulla piastrina non appena la soluzione precedentemente
aggiunta è cristallizzata in seguito all’evaporazione del solvente.
Così facendo è stato possibile utilizzare anche solventi più
affini ai campioni come il DMSO per solubilizzare in toto il
campione.

- 66 -
Ciò che si evince dagli spettri è una totale solubilizzazione del
campione polimerico esaminato, che si riscontra in una maggiore
e più ragionevole gamma di segnali registrati dallo strumento, in
un più ampio intervallo di masse (800÷15000 Da).
- Cenni sulla spettroscopia MALDI
La spettrometria di massa MALDI è una tecnica che ha
acquistato sempre più importanza nella caratterizzazione dei
materiali polimerici.
Fra i principali vantaggi di questa tecnica ricordiamo:
gli alti valori di massa molecolare analizzabili
facilità di preparazione dei campioni
brevi tempi d’analisi
ottimo grado di accuratezza nella determinazione delle
masse molecolari

- 67 -
L’acronimo MALDI (Matrix Assisted Laser Desorption
Ionization ) è spesso seguito da TOF, che indica l’impiego di un
analizzatore a tempo di volo (TOF: time of flight).
In un tipico esperimento, il campione viene mescolato con un
largo eccesso di una matrice (rapporto circa 1 a 2000, o maggiore
a seconda della massa molare media del materiale polimerico)
scelta sulla base dell’affinità chimico–fisica con l’analita e della
capacità di assorbire la luce del laser adoperato (elevato
coefficiente di estinzione molare alla lunghezza d’onda del
laser); è richiesta in pratica un’ottima miscibilità tra il campione
(in presenza di un adeguato solvente) e la matrice in modo da
ottenere, evaporato il solvente una omogenea struttura solida
cristallina della miscela.
Il desorbimento dipende dal laser e dalla giusta scelta della
matrice. Questa deve assorbire l’energia della luce laser pulsata e
promuoverne il trasferimento all’analita. In questo modo
l’energia arriva alle molecole di campione in forma indiretta
riducendo al minimo i fenomeni di frammentazione.

- 68 -
Dopo ogni impulso laser la parte di miscela colpita desorbe,
formando una nuvola gassosa, costituita da ioni molecolari della
matrice e dell’analita stesso, che, a causa del vuoto all’interno
dello strumento (10
-7
Torr), si espande liberamente.
Normalmente il processo di ionizzazione è coadiuvato dalla
cattura di ioni alcalini (Li
+
Na
+
K
+
) o H
+
che portano alla
rivelazione di addotti rispettivamente protonati, sodiati etc..
Cationi specifici (es. Ag
+
) vengono aggiunti per l'analisi di
polimeri come il polistirene.
Gli ioni in libera espansione vengono accelerati da uno stesso
potenziale (stessa energia, E
k
) e inviati nell’analizzatore TOF in
cui si produce la separazione delle specie in funzione del tempo
impiegato a percorrere una distanza fissa (tubo di volo) e quindi
separati in base al rapporto massa carica (m/z). La relazione che
esprime matematicamente questo concetto è:
t
volo
= K (m/z)
1/2

- 69 -
dove:
t
volo
= tempo di volo dello ione con carica
K = costante caratteristica dello strumento
m = massa dello ione molecolare
z = carica dello ione
In particolare, il TOF è un tubo mantenuto sotto alto vuoto
collocato orizzontalmente o verticalmente attaccato ad
un’estremità alla camera di ionizzazione e dal lato opposto al
rivelatore.
Gli ioni formati nella sorgente vengono “sparati” nel tubo di
volo da un elevato potenziale di accelerazione applicato tra il
porta campione ed una griglia soprastante; l’effetto del
potenziale è quello di conferire la stessa energia cinetica (E
k
) a
tutti gli ioni in modo che essi vengono separati in base al loro
tempo di percorrenza del tubo che è direttamente dipendente
dalla massa.

- 70 -
In particolare, gli ioni a massa inferiore avranno un minor
tempo di volo e saranno i primi a raggiungere il rivelatore.
Il potere risolutivo del TOF di pende da alcuni parametri:
lunghezza del tubo
entità del vuoto
potenziale di accelerazione
dispersione delle velocità iniziali degli ioni prodotti in
sorgente
L’ultimo punto è molto importante ai fini di una buona riuscita
dell’analisi; ciò che conta è che non vi siano differenze
nell’energia degli ioni formati nella sorgente. Questo obiettivo è
raggiunto nell’analisi MALDI mediante la produzione di
pacchetti di ioni (positivi o negativi) in seguito al
bombardamento del campione con brevi impulsi di laser.
E’ comunque presente una certa dispersione energetica durante
le fasi di desorbimento–ionizzazione, ciò provoca una minore
omogeneità e quindi una bassa risoluzione con allargamento dei
picchi nel spettro.

- 71 -
Per compensare ed ottimizzare la risoluzione dello strumento
sono state messe a punto delle tecniche di focalizzazione; le
principali sono la Delay Extraction (DE) ed il Reflector che
hanno la caratteristica di poter essere usate sia singolarmente che
in coppia.
Entrambe provvedono a compensare le differenze posizionali o
di energia cinetica esistenti fra ioni ad uguale rapporto m/z.
Nella DE si agisce provocando un ritardo nell’accensione del
potenziale di accelerazione; ciò provoca una omogeneizzazione
delle energie degli ioni prima che essi vengano accelerati dal
campo elettrico a causa delle ripetute e reciproche collisioni che
livellano eventuali iniziali differenze con compattazione degli
ioni con la stessa massa che, avendo un tempo di volo uguale,
vengono rivelate simultaneamente.
Il Reflector, invece, fa sì che gli ioni, prima di uscire
dall’analizzatore, siano convogliati in un opportuno sistema di
griglie (specchio elettrostatico) a cui è applicato un potenziale
dello stesso segno degli ioni.

- 72 -
In questo modo specie con uguale m/z ma diversa E
k
avranno
diversa capacità di penetrare il Reflector, prima di essere riflessi;
in particolare, ioni a maggiore energia percorreranno una
maggiore distanza in modo che una volta fuori dal sistema tutti
gli ioni a uguale m/z saranno compattati e raggiungeranno
contemporaneamente l’analizzatore.
Il rivelatore che viene normalmente utilizzato è un
fotomoltiplicatore di elettroni a diodi separati; esso ha il
vantaggio oltre a fungere da rivelatore, di amplificare la corrente
ionica in arrivo (Vedi Figura 6).
Figura 6- Qui rappresentato lo schema di un fotomoltiplicatore di
elettroni a diodi separati

- 73 -
3.10.2 Spettroscopia NMR
Gli spettri NMR riportati in questo lavoro di tesi sono stati
registrati utilizzando lo strumento Bruker 200 A-CF. Come
solvente adoperato per la registrazione degli spettri si usa
dimetil-solfossido deuterato (DMSOd
6
) commerciale della
Aldrich Chemical Co.
I chemical shift dei segnali protonici, espressi in ppm, sono
stati attribuiti utilizzando come riferimento il quintupletto ben
distinguibile del (DMSOd
6
), a 2.5 ppm.
Gli spettri dei polimeri funzionalizzati sono stati registrati in
DMSOd
6
alla temperatura di 60°C (333°K) e i dati sono stati
elaborati con il software 1D Win-NMR applicando le funzioni di
Lorentz-Gauss per migliorare la risoluzione dei picchi.
I valori dei chimica shift sono stati accuratamente assegnati ai
protoni specifici utilizzando i dati riportati in letteratura
30,42
.

- 74 -
3.10.3 Viscosimetia-SEC
La caratterizzazione molecolare (MMD ovvero distribuzione
di massa molare, la viscosità intrinseca e le proprietà
conformazionali) dei prepolimeri PES-OH e dei copolimeri S-
PES è stata condotta mediante un sistema cromatografico multi
detector ad esclusione dimensionale (SEC) utilizzando N,N-
dimetilacetammide come fase mobile.
L’apparato strumentale utilizzato consiste di un sistema
cromatografico integrato GPCV2000 Waters (Milford, MA)
collegato on-line a due detectors: (1) Viscosimetro differenziale
(DV); (2) rifrattometro differenziale (DRI modello 2414)
utilizzato come rivelatore di concentrazione.
Tale sistema cromatografico multidetector SEC-DV utilizzato
è stato descritto nei precedenti lavori
43
.
Il set di colonne cromatografiche impiegato è composto da due
colonne miste Styragel (HR5E e HR4E con dimensioni delle
particelle pari a 5 lm) della Waters.

- 75 -
Le condizioni sperimentali utilizzate sono; DMAc þ 0.01 M,
LiBr come fase mobile, 80 °C di temperatura, 0.8 mL/min di
portate del flusso.
E’ ben noto che il rivelatore DV on-line da indicazioni sulla
viscosità relativa η
r
delle frazioni polimeriche che vengono
analizzate mediante eluizione.
Dalla combinazione dei valori di η
r
e di concentrazione
determinata mediante il rivelatore DRI on-line, si ottiene la
funzione sperimentale [η]= ƒ(V) dove [η] indica la viscosità
intrinseca e V il volume di eluizione.
Mediante il suddetto apparato SEC-DV è possibile ottenere
delle importanti informazioni sulle proprietà conformazionali
mediante l’equazione Mark-Houwink-Sakurada (MHS) [η]=
ƒ(M) dove con M si indica la massa molare.

- 76 -
3.10.4 Viscosimetria
Per l’analisi viscosimetrica è stato utilizzato un viscosimetro
Ubbelohde a livello sospeso immerso in bagno d’acqua
termostatato a 25 °C.
E’ stata calcolata la viscosità ridotta di ogni campione
polimerico in dimetil-formammide (DMF); sono state preparate
soluzioni alla stessa concentrazione 0.5M (0.125gr/25ml) di
ciascun polimero in DMF. Sono stati quindi registrati con
l’ausilio di un cronometro (con precisione al decimo di secondo)
i tempi di eluizione delle soluzione e del solvente.
Dalle medie sui tempi di eluizione del solvente e della
soluzione contenente il polimero si risale alla viscosità ridotta
attraverso i seguenti passaggi:
η (relativa) = T
1
/T
0
(T
1
=tempo di eluizione soluzione)
(T
0
=tempo di eluizione solvente)
η (specifica) = η
REL
– 1 (C = concentrazione)
η (ridotta) = η
SPEC
/ C

- 77 -
3.10.5 Spettroscopia infrarossa (FT-IR)
Questa tecnica spettroscopica è stata utilizzata per
caratterizzare i gruppi funzionali presenti in catena; ciascun
gruppo presente nel polimero infatti è riconoscibile mediante
segnali di assorbimento che cadono a valori di frequenza ben
precisi.
E’ inoltre possibile misurare il grado di solfonazione delle
catene polimeriche, comparando un picco di assorbimento
caratteristico dei gruppi solfonici con uno standard caratteristico
della catena.
Per effettuare le misure vengono preparate pasticche
semitrasparenti di campioni in KBr a concentrazione 0.5% in
analita. Le pasticche sono state preparate mediante una
pasticcatrice che consente di ottenere le pasticche nell’opportuno
diametro e spessore effettuando una pressione di circa 10 ton in
un’opportuna pressa.

- 78 -
Gli spettri FT-IR sono stati registrati, mediante uno
spettrometro Perkin Elmer Spectrum One, dotato di rivelatore al
litio tantalato (LiTaO
3
). Gli spettri FTIR sono stati acquisiti nella
regione del medio infrarosso (4000-400 cm
-1
) ed elaborati
attraverso il Software Spectrum One.
- Cenni generali sulla spettroscopia IR
I dispositivi strumentali oggi a disposizione per ottenere spettri
nel medio e lontano IR (lo studio e le applicazioni del vicino
infrarosso sono relativamente recenti) sono sostanzialmente di
due tipi:
- SPETTROFOTOMETRI A DISPERSIONE
- SPETTROFOTOMETRI A INTERFERENZA

- 79 -
I primi sono senz’altro quelli maggiormente diffusi nei
moderni laboratori, soprattutto per motivi di costo; i secondi
offrono invece prestazioni senz’altro superiori ma i costi sono
decisamente più elevati.
L’intervallo di lunghezze d’onda coperto dagli strumenti è
generalmente compreso tra 4000 e 625 cm
-1
, corrispondente al
medio IR, ma sono anche molto diffusi quelli con intervallo
spettrale esteso verso λ più elevate, fino a 400 e 200 cm-1, che
include anche il lontano IR.
Gli spettrofotometri a dispersione
Figura 7- Spettrofotometro IR a dispersione

- 80 -
Questi strumenti lavorano per lo più nella configurazione a
doppio raggio: un dispositivo (chopper) ripartisce la radiazione
continua emessa dalla sorgente in due fasci di uguale intensità.
Uno dei fasci viene fatto passare attraverso il campione, l'altro
serve come riferimento ed attraversa di solito l'aria e, nel caso di
soluzioni, una cella contenente il solvente puro. Dopo
l'azzeramento ottico, i due fasci sono nuovamente riuniti.
Il monocromatore (prisma o reticolo) scompone la radiazione
risultante nelle sue componenti spettrali. Queste possono quindi
essere analizzate, secondo le varie lunghezze d'onda, dal
rilevatore, nel quale è registrata istante per istante la radiazione
monocromatica (scansione).
Dopo amplificazione i segnali sono espressi, mediante un
registratore, sotto forma di spettro e in genere la registrazione di
uno spettro richiede mediamente circa 10 minuti.
In questo strumento il monocromatore di trova a valle del
comparto celle in quanto le radiazioni IR hanno energia minore

- 81 -
rispetto a quelle UV, ed è quindi necessario sfruttarle al massimo
per ottenere uno spettro.
Da notare che se usassimo una radiazione UV non scomposta
la sua elevata energia causerebbe la decomposizione del
campione.
Tipi di sorgenti
- Globar: filamento di carburo di silicio (richiede eccessiva
potenza).
- Filamento di Nernst: costituito da una miscela di
ossidi fusi (troppo fragile).
- Filamento di Nichel–Cromo: il più usato perché poco
costoso, resistente e assorbe poca potenza
- Filamento di Wolframio: per il vicino IR

- 82 -
Sistema fotometrico
Composto da:
- Sistema di specchi + chopper: servono a portare nello
stesso cammino i due raggi (riferimento e campione)
separatamente.
- Cuneo ottico (o pettine) che, collocato sul raggio di
riferimento, assorbe la stessa quantità di energia
che il campione assorbe dal raggio che lo attraversa.
Monocromatore
Formato essenzialmente da:
- fenditura d’ingresso per la radiazione policromatica.
- dispositivo di dispersione (reticolo di riflessione), che separa
le componenti della radiazione policromatica.

- 83 -
- dispositivo di focalizzazione (filtro) che preleva dall’insieme
delle radiazioni disperse un sottile intervallo di lunghezze
d’onda.
- fenditura d’uscita.
I movimenti del reticolo e del filtro consentono di far passare
dalla fenditura di uscita, in successione, la sequenza ordinata di
tutte le bande che nel loro insieme compongono la radiazione
policromatica. Tale movimento realizza la cosiddetta scansione
delle lunghezze d’onda.
Rivelatore
Si tratta del dispositivo in grado di convertire la radiazione
termica (IR) in un segnale elettrico, che viene poi inviato al
sistema di elaborazione e di registrazione.
Come abbiamo detto, il chopper indirizza sul rivelatore il
raggio campione ed il raggio di riferimento in maniera alternata

- 84 -
(ciò permette di effettuare un confronto continuo fra l’intensità
dei due raggi).
Fintantoché tali intensità si mantengono uguali (cioè il
campione non assorbe) il rivelatore e il registratore rimangono in
equilibrio.
Quando invece l’intensità del raggio campione diminuisce (a
seguito del fenomeno di assorbimento) il segnale elettrico
generato dal rivelatore, e opportunamente elaborato, muove un
motore che insinua un cuneo ottico sul cammino del raggio di
riferimento.
L’ampiezza dell’attenuazione operata dal cuneo per
equalizzare le intensità dei due raggi è direttamente
proporzionale alla variazione di intensità del raggio campione.
L’intensità residua del raggio dopo la variazione viene
espressa come percentuale rispetto all’intensità iniziale, e
rappresenta la trasmittanza percentuale %T del campione.

- 85 -
L’intera operazione viene effettuata in continuo per tutto
l’intervallo spettrale.
Spettrofotometria in Trasformata di Fourier (FT-IR)
Questa tecnica strumentale è basata sulla spettroscopia
infrarossa classica. Si tratta di una tecnica recente creata grazie
alla computerizzazione del laboratorio strumentale.
Il suo principio di base è rappresentato dalla possibilità di
cogliere contemporaneamente tutte le frequenze dello spettro IR
nel rilevatore, il che rende superflua la scansione della lunghezza
d'onda.
Questo è possibile trasformando, per mezzo di un
interferometro, la radiazione IR policromatica emessa dalla
sorgente (istante per istante con la medesima intensità) in un
interferogramma, dove l’assorbimento non è più funzione della
frequenza, ma del tempo (cioè si passa da dominio delle

- 86 -
frequenze a dominio dei tempi).
Contrariamente agli spettrofotometri tradizionali, quindi, in
questa apparecchiatura non si ha un monocromatore a
dispersione, ma viene utilizzato l’interferometro di Michelson, il
quale produce nel corso di una speciale scansione
l’interferogramma della sostanza in esame.
Dopo il passaggio della radiazione così "trattata" attraverso il
campione, l'interferogramma viene trasformato dal calcolatore
collegato allo strumento in un tradizionale spettro infrarosso
mediante un'operazione matematica, la cosiddetta trasformata di
Fourier.
In questa maniera si passa perciò dall’interferogramma, un
grafico dello spazio o del tempo, a uno spettro comune, che
rappresenta però la variazione dell’intensità del segnale in
funzione del numero d’onda (o della lunghezza d’onda) della
radiazione.
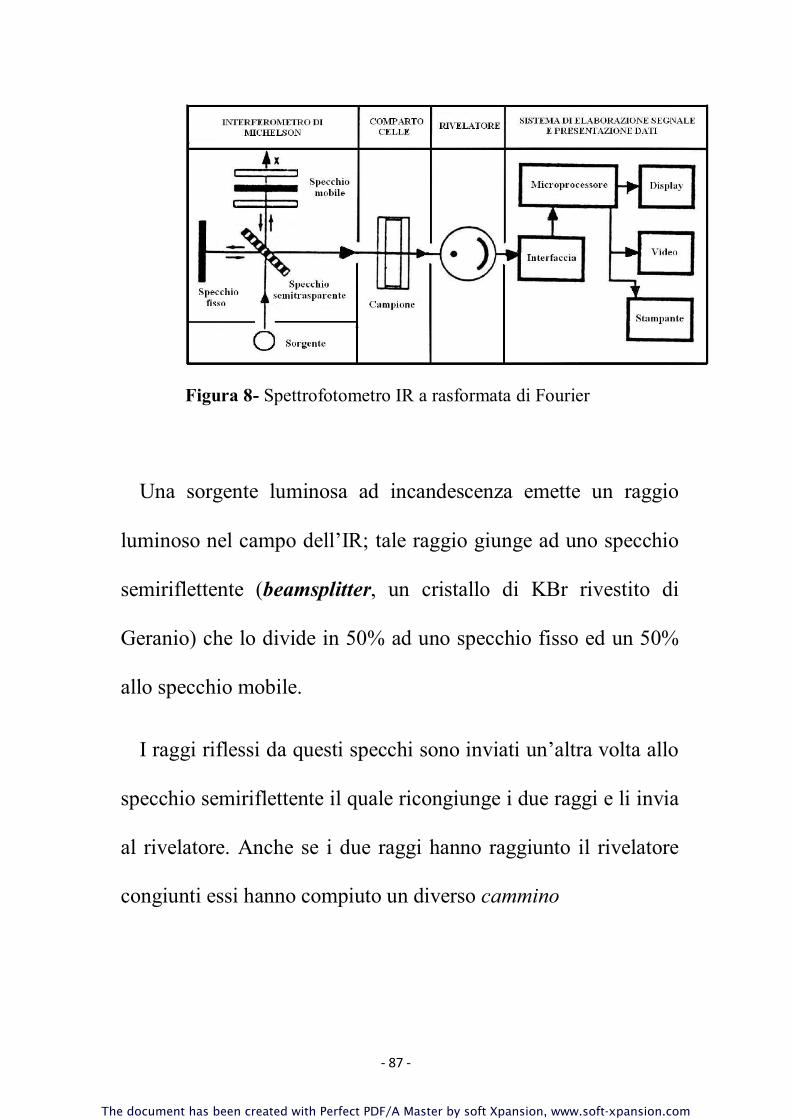
- 87 -
Una sorgente luminosa ad incandescenza emette un raggio
luminoso nel campo dell’IR; tale raggio giunge ad uno specchio
semiriflettente (beamsplitter, un cristallo di KBr rivestito di
Geranio) che lo divide in 50% ad uno specchio fisso ed un 50%
allo specchio mobile.
I raggi riflessi da questi specchi sono inviati un’altra volta allo
specchio semiriflettente il quale ricongiunge i due raggi e li invia
al rivelatore. Anche se i due raggi hanno raggiunto il rivelatore
congiunti essi hanno compiuto un diverso cammino
Figura 8- Spettrofotometro IR a rasformata di Fourier

- 88 -
ottico: a seconda della differenza del cammino ottico dei due
raggi si creano delle interferenze costruttive o distruttive che
creano un segnale al rilevatore proporzionale alla differenza di
cammino ottico dei due raggi e quindi dalla posizione dello
specchio mobile in quell’istante.
In base al movimento del suddetto specchio tutte le radiazioni
monocromatiche contenute nella luce emessa dalla lampada
danno luogo ad un segnale complessivo di interferenza
(interferogramma) che contiene in sé le informazioni riguardanti
la frequenza e l’intensità della radiazione emessa.
La trasformata di Fourier, effettuata dal calcolatore dello
strumento una volta che il raggio è arrivato al detector, mostra
sullo schermo un tradizionale spettro infrarosso, trasformando il
segnale di intensità luminosa in funzione del tempo (spostamento
dello specchio) in segnale di intensità in funzione del numero
d’onda.

- 89 -
Un requisito fondamentale del FT-IR è quello di ottenere un
buon interferogramma, dal quale dipende la precisione dello
spettro ottenuto: in altre parole lo specchio mobile deve avere
una velocità costante e la sua posizione deve essere nota in
maniera esatta in ogni istante. Lo specchio deve inoltre
mantenere una planarità costante durante tutto il suo spostamento
Rispetto alla tecnica convenzionale la spettroscopia FT-IR
offre tre vantaggi:
1) Un notevole risparmio di tempo: siccome la radiazione di
tutte le lunghezze d'onda viene registrata contemporaneamente
dal rilevatore, il tempo di misura si riduce a pochi secondi
rispetto ai 10 minuti circa degli strumenti tradizionali
2) Un miglior rapporto segnale-rumore: rispetto alla tecnica
a scansione, dove è registrata sempre una sola lunghezza d'onda
(mentre tutto il resto va perso in intensità),
la potenza complessiva della sorgente di radiazione rimane
costantemente disponibile.

- 90 -
Al rivelatore arriva dunque una maggiore potenza rispetto agli
strumenti a dispersione
3) Elevata precisione dei numeri d'onda: è possibile
sovrapporre al segnale come standard interno la radiazione
monocromatica di una sorgente laser, in cui la frequenza è nota
con estrema precisione.
4) Nessun effetto di riscaldamento del campione: la sorgente
è infatti sufficientemente lontana dal campione
5) Possibilità di interfacciare un gascromatografo
6) Assenza di luce diffusa
La tecnica FT rende inutile pure la suddivisione delle
radiazioni in un raggio di misura ed in uno di riferimento,
operazione che è soggetta a disturbi; gli spettrometri FT-IR sono
monoraggio.
Campione e riferimento sono supportati su una slitta, che li
porta entrambi nel cammino ottico uno dopo l'altro (se si fa
riferimento all'aria, si lascia semplicemente vuoto il relativo

- 91 -
contenitore).
Gli spettri sono raccolti e memorizzati separatamente ed infine
lo spettro di riferimento (background) viene sottratto
numericamente dello spettro del composto.
La tecnica FT-IR consente di ottenere importanti risposte
strutturali con brevi tempi di analisi e semplicità delle procedure
strumentali. Tali vantaggi hanno reso questa tecnica un punto di
forza nella caratterizzazione dei composti organici.
Analisi in Riflettanza Totale Attenuata (ATR)
Figura 9- Rappresentazione schematica del sistema ATR

- 92 -
La sostanza viene deposta in polvere al di sopra di un prisma
costituito da un materiale ad elevato indice di rifrazione per tutte
le radiazioni infrarosse (AgCl o KRS-5, una miscela di bromuri e
ioduri di tallio).
Ad ogni riflessione il raggio penetra per qualche frazione di
micron nella sostanza, venendone in piccola parte assorbito (o
attenuato). Dopo alcune riflessioni (9-10, ma anche 25 con i
sistemi ottici più complessi) la diminuzione dell’intensità del
raggio è sufficiente per essere rilevata dallo spettrofotometro,
dando uno spettro IR in Riflettanza Totale Attenuata (ATR).
La tecnica ATR però determina uno spostamento delle
lunghezze d’onda di assorbimento e una certa distorsione delle
bande; di conseguenza per l’interpretazione qualitativa degli
spettri bisogna fare riferimento a cataloghi registrati in ATR.
Inoltre gli spettri delle sostanze sono confrontabili solo se
registrati con lo stesso angolo di incidenza.

- 93 -
3.10.6 DSC (Calorimetria a Scansione Differenziale)
La temperatura di transizione vetrosa (T
g
) dei polimeri è stata
determinata con l’ausilio di un TA Instrument Q100 Differential
Scanning Calorimetry (DSC).
Le tarature dell’entalpia e della temperatura del nostro
apparato sono state effettuate attraverso due programmi
incorporati, secondo la procedura suggerita dal costruttore
44
.
La variazione di Entalpia è stata calibrata mediante un
programma incorporato, basato sull’entalpia di fusione dell’indio
(3,267 kJ mol
-1
), in ottimo accordo con quella di (3,273±0.022)
kJ mol
-1
da una raccolta di materiali di riferimento della IUPAC
utilizzate per le misure di entalpia
45
. La calibrazione della
temperatura è stata fatta attraverso un altro programma
incorporato in base al punto di fusione dei tre standard
selezionati (indio, piombo e zinco). I punti di fusione di questi
tre metalli (156,6 °C, 327,5°C e 419,5°C rispettivamente),

- 94 -
utilizzati dalla Mettler nel programma del processore, sono in
ottimo accordo con quelli raccomandati dalla IUPAC
45
. Il
programma mette a confronto la risposta strumentale con i valori
di temperatura di riferimento e fornisce un opportuno set di
parametri di calibrazione nel range di temperatura di interesse
(150°C ÷ 420°C), che coprono completamente il range dei valori
di Tg. dei materiali polimerici qui studiati.
Delle scansioni effettuate con campioni di Indio nuovo, hanno
mostrato un accordo con il valore di entalpia standard entro un
errore dello 0,25%.
La precisione con cui lo strumento determina la temperatura di
analisi, è stata misurata mediante varie scansioni con standard di
Indio e Stagno ad una velocità di scansione di 10°Cmin
-1
; il
valore ottenuto è entro lo 0,08% così come descritto in
letteratura
45
.
Le tarature dello strumento del calorimetro sono state ripetute
a distanza di due settimane.

- 95 -
I valori di Tg sono stati determinati in atmosfera inerte per
azoto. Prima d’ogni esperimento viene registrata la linea di base
mediante un crogiolo d’alluminio vuoto.
I valori di T
g
sono stati presi nel punto medio della transizione
vetrosa in ogni termogramma. Per l’analisi sono stati pesati circa
6 x 10
-3
g di ogni campione e posti in crogiuoli di alluminio
sigillati; l’analita è stato mantenuto in atmosfera d’aria e la
scansione è stata impostata con un innalzamento di temperatura
di 10 ° C al minuto.
3.10.7 Termogravimetria
La stabilità termica dei polimeri è stata valutata mediante una
termobilancia TA Instruments Q-500, munita di crogiolo in
platino.
La temperatura della fornace è stata calibrata seguendo la
procedura riportata nel manuale d'uso del materiale
44
, sulla base

- 96 -
del cambiamento delle proprietà magnetiche di tre campioni di
metallo (Isatherm, Nichel e Trafoperm) e i loro
punti di Curie (142,5°C , 357,0°C e 749,0°C rispettivamente),
considerati tra gli standard più precisi dalla letteratura attuale
46
.
La calibrazione della temperatura è stata ripetuta ogni due
settimane.
Le prove di termodegradazione sui campioni polimerici sono
state effettuate in condizioni di riscaldamento dinamico, sia in
ambiente inerte sotto corrente d'azoto (0,02 L min
-1
) che in
atmosfera ossidativa per la presenza di aria.
Le prove termogravimetriche sono state condotte in un range
di temperatura che va da 35°C a 800°C utilizzando diverse
velocità di riscaldamento (Φ= 2, 5, 7.5, 10, 12.5, 15, 17.5 e 20°C
min
-1
). In ciascuna prova termogravimetrica sono stati utilizzati
4-6 mg di polimero effettuando la scansione sotto un flusso di
azoto di 60 ml/min. I termogrammi ottenuti sono stati registrati
nel database del programma di acquisizione. I dati sperimentali a
diverse sono quindi stati elaborati dal processore TC 10A alla

- 97 -
fine di ogni esperimento.
I dati ottenuti forniscono le informazioni necessarie per
determinare la percentuale di polimero degradato (1-D)% in
funzione della temperatura, dove D=(Wo-W)/Wo, dove con Wo
e W vengono indicati rispettivamente il peso iniziale del
campione e quello durante la scansione.
La temperatura di massima velocità di decomposizione termica
del polimero (PDT) corrisponde alla temperatura alla quale i
polimeri studiati presentano la massima velocità di perdita in
peso.
Pertanto le curve relative alla velocità di perdita in peso in
funzione della temperatura (DTG) ed i loro massimi (PDT)
forniscono una indicazione sulla stabilità termica dei campioni
utilizzati.

- 98 -
4. RISULTATI E DISCUSSIONE
Nel presente lavoro di tesi sperimentale è stato affrontato lo
studio della reazione di sintesi di una particolare famiglia di co-
polimeri aventi in catena principale una base costituita da anelli
aromatici e ponti eterei e solfonici alternati.
In modo particolare è stata focalizzata l’attenzione sulla
reazione di solfonazione delle catene poliaromatiche mediante
introduzione di gruppi -SO
3
H come sostituenti ai protoni
aromatici.
I co-poli(eteri solfoni) oggetto del nostro studio sono stati
ottenuti mediante reazione di policondensazione nucleofila
aromatica del sale bi-sodico del dicloro difenil-solfone solfonato
(SDCDPS) ottenuto in uno step preventivo (vedi parte
sperimentale) con un omopolimero terminato idrossi ad entrambe
le estremità di catena (PES-OH) utilizzato come agente

- 99 -
telechelico (vedi schema 3 a pag.53 della parte sperimentale).
Sono state preparate due serie distinte di poli(arilen eteri
solfoni) solfonati (S-PES). Una a partire da un pre-polimero PES
telechelico di massa molare media ÷ 4000g/mol ed una in cui
tale prepolimero ha una massa di ÷7000g/mol.
Tale differenziazione è motivata dall’esigenza di studiare la
cinetica della reazione di policondensazione nucleofila aromatica
su due importanti aspetti. Il primo riguarda lo studio degli effetti
che derivano dall’utilizzo di un pre-polimero a diversa lunghezza
sulla reazione stessa; il secondo interessa invece lo studio delle
proprietà fisiche, chimiche termiche e meccaniche delle catere
polimeriche ottenute al variare del numero di gruppi solfonici
laterali presenti in catena, ovvero al variare del grado si
solfonazione (DS).
Le reazioni di policondensazione nucleofila aromatica sono
state condotte alla temperatura 200°C per 12 ore sotto costante
agitazione ed in ambiente inerte.

- 100 -
Sono stati utilizzati quattro ben definiti rapporti molari tra i
due reagenti principali PES-OH/SDCDPS.
I rapporti molari sono stati calcolati in base alla massa molare
media numerica (Mn) del prepolimero telechelico PES-OH (vedi
Tabella 3 a pag 104 dei risultati) e del grado di solfonazione che
in ciascuna reazione si programma di ottenere.
I copolimeri sintetizzati saranno indicati da ora in avanti con la
sigla S-PES-MR (xh), dove “MR” è l’acronimo inglese di
“Molar Ratio” ovvero il rapporto molare trai due blocchi
prepolimerici di partenza (PESOH)/SDCDPS e “xh” il tempo di
reazione.
La struttura chimica dei poli(arilen eteri solfoni) preparati
durante il presente lavoro, è stata studiata mediante le seguenti
tecniche di indagine strutturale:
1
H-NMR, FT-IR con sistema
ATR e MALDI-TOF MS.
Lo studio delle proprietà termiche e termo-meccaniche di tali
materiali è stato svolto mediante l’utilizzo di tecniche delle

- 101 -
tecniche di analisi qui di seguito elencate: termogravimetria
(TGA), calorimetria a scansione differenziale (DSC) ed analisi
dinamo-meccanica DMA.
Il confronto incrociato dei risultati ottenuti dall’utilizzo di
ciascuna delle tecniche analitiche utilizzate per la
caratterizzazione dei polimeri oggetto del presente studio ha
come obiettivo quello di rafforzare i risultati raggiunti.
L’indagine chimica condotta mediante la tecnica
spettroscopica
1
H-NMR ha permesso di ottenere fondamentali
informazioni sia sulla composizione delle catene polimeriche,
come ad esempio il grado di solfonazione delle catene stesse
ottenute durante la sintesi (DS) che informazioni di carattere
strutturale come la stima del valore di massa molare media
numerica (Mn) dei copolimeri solfonati ottenuti.
Un altro potente strumento di indagine chimica che ha fornito
importanti risultati in merito alla caratterizzazione strutturale e
composizionale dei materiali polimerici sintetizzati è la tecnica

- 102 -
viscosimetrica mediante Cromatografia ad Esclusione
Dimensionale (viscosimetria SEC-DV). Questa tecnica
d’indagine, a partire da misure di viscosità di soluzioni
polimeriche opportunamente ottenute ci ha consentito di
ottenere ulteriori informazioni circa la massa molare media
viscosimetrica (Mv) delle catene polimeriche dei campioni
caratterizzati.
Al fine di individuare quante unità PES disolfonate e quali tipi
di gruppi terminali sono presenti per ogni catena, nonchè per
permettere una più profonda comprensione del meccanismo e
della cinetica della reazione principale di policondensazione
nucleofila aromatica che coinvolge le catene polimeriche PES e
di eventuali reazioni secondarie, è stato condotto uno studio
approfondito basato sulla spettrometria di massa MALDI-TOF
MS come tecnica di indagine composizionale e strutturale.
In primo luogo i due agenti telechelici pre-polimerici PES-OH
con una massa molare media (Mn) di 4000 g/mol e 7000 g/mol,

- 103 -
(qui da ora in avanti menzionato univocamente come PES-OH
(4K) e PES-OH (7K) rispettivamente) sono stati sintetizzati
utilizzando un rapporto molare DHDPS/DCDPS di 1,09/1 e
1.05/1, rispettivamente, come suggerito dalla letteratura corrente
ed indicato in modo semplificato nello schema 1 (pag.40 parte
sperimentale).
4.1 Spettrometria di massa MALDI-TOF MS
I risultati dell’indagine MALDI-TOF MS indicano la
diminuzione del numero di catene di copolimeri terminati con
gruppi cloro rispetto a quelli aventi gruppi fenolici ad entrambe
le estremità di catena. Tali dati sperimentali confermano
l’avanzamento della reazione di policondensazione dopo le
prime 12 ore di reazione. Spettri di massa MALDI TOF ottenuti
mostrano cluster omogenei di picchi che vanno da m/z 2000 a
m/z 26000, indicando l’aumento della distribuzione di massa

- 104 -
molare media (MMD) come auspicato in precedenza. Negli
spettri di massa MALDI-TOF i picchi più intensi corrispondono
alle famiglie terminate con i gruppi OH ad entrambe le estremità
(specie A
n,
con =2 nella tabella 2), mentre picchi intensi
corrispondenti a valori di =3 e =4 sono stati osservati per un
range di massa superiore a m/z 4000.
Le intensità delle serie di picchi osservati nelle famiglie di co-
polimeri post-reagiti è molto inferiore rispetto a quelle osservate
negli spettri di massa dei campioni di copolimero ottenute dopo
12 ore di reazione (figure 3 e 4). Risulta evidente quindi che
l’intensità delle serie di picchi B
n,
e C
n,
aumenta in modo
inversamente proporzionale al tempo di reazione.
Confrontando gli spettri di massa MALDI-TOF dei copolimeri
ottenuti per reazione di post-condensazione con quelli
appartenenti ai copolimeri ottenuti dopo 12h di reazione, nel
range di massa m/z 2.000-5.000 abbiamo anche osservato

- 105 -
incremento dell'intensità relativa dei cicli co-oligomerici (specie
n,
in Tabella 2 pag.105 dei risultati).
Questi dati suggeriscono che i cicli possono essere ottenuti da
reazioni di post-condensazione che coinvolgono i gruppi
terminali OH e Cl appartenenti alla stessa catena
macromolecolare (specie B della Tabella 2) o di due catene
differenti (specie B
n,
e C
n,
in Tabella 2).
Lo spettro di massa MALDI-TOF del pre-polimero PES-OH
(4K) mostrato in Figura 10 presenta una singola famiglia di
picchi di massa, nel range di massa che va da 1.000-8.000 Da.
Tale famiglia corrisponde agli ioni sodiati delle catene PES, e
aventi gruppi terminali idrossili ad entrambe le estremità.

- 106 -
Figura 10- Spettro di massa MALDI-TOF del pre-polimero PES-OH (4K)

- 107 -
Lo spettro di massa del pre-polimero PES-OH (7K) mostra una
sola intensa serie di picchi, così come di PES-OH (4K), avente
massa corrispondente a quella degli ioni sodiati (Na
+
) delle
catene di PES terminate di-idrossi con un numero di unità
ripetitive (n) che vanno da 9 (m/z = 2539) fino ad un valore
massimo di 46 (m/z = 11.944), e che mostra il picco più intenso
ad un valore del rapporto m/z =7.469 corrispondente ad
oligomeri con n = 28.
I valori calcolati sono riportati nella Tabella 2 rappresentata
nella pagina seguente.

- 108 -
Tabella 2- Assegnamenti strutturali dei picchi presenti negli spettri MALDI-TOF degli S-PES.

- 109 -
A partire dai campioni di pre-polimero PES-OH ottenuti in un
primo step di sintesi abbiamo sintetizzato quattro copolimeri
solfonati PES che si differenziano fondamentalmente per il grado
di solfonazione. Essi sono: S-PES-0.9 (12h), S-PES-1.0 (12h), S-
PES-1.05 (12h) ed S-PES-1.2 (12h) elencati in base al grado
solfonazione ottenuto, in ordine decrescente. Anche per i
copolimeri PES solfonati ottenuti per via sintetica, così come
visto in precedenza per i pre-polimeri PES-OH sono state
effettuate tutte le caratterizzazioni opportune a determinare la
struttura e la composizione macromolecolare dei campioni
studiati. Mediante la spettroscopia di massa MALDI-TOF MS e
l’indagine spettroscopica
1
H-NMR sono state ottenute importanti
informazioni circa la composizione chimica ed anche la natura
dei gruppi terminali. E’stato inoltre possibile determinare con
buona precisione il numero medio di unità solfonate presenti
lungo le catene macromolecolari.
Lo spettro di massa MALDI-TOF del campione S-PES-0.9
(12h) è mostrato in Figura 11.

- 110 -
Figura 11- Spettro di massa MALDI-TOF di S-PES-0.9(12h) (a) e sezione ampliata dello stesso spettro
di massa (b) e dello spettro del S-PES-1.2(12h) (c).

- 111 -
Esso mostra un cluster complesso di picchi che parte da un
valore di m/z 1300 fino a m/z 9600. La differenza di massa tra
due picchi omologhi appartenenti a due cluster contigui è di 232
uma (esattamente corrispondente alla massa dell’unità ripetitiva
del PES non-solfonato).
Lo spettro di massa del S-PES-1.2 (12h) è molto simile.
Informazioni dettagliate sulla composizione chimica delle catene
e dei loro gruppi terminali si possono ottenere estendendo le
regioni di massa da m/z 2300 fino a m/z 3000, come riportato nel
riquadro in Figura 11 (Figure 11-b e 11-c).
Entrambi gli spettri mostrano che ogni cluster presente nello
spettro di massa è costituito da quattro famiglie di picchi che
corrispondono ad altrettante famiglie di catene polimeriche
aventi differenti gruppi terminali (specie A
n,
, X
n,
,
n,
, B
n,
); i
loro assegnamenti sono riassunti nella Tabella 3, dove con n
sono indicate le unità PES non solfonate e con le unità di-
solfonate.

- 112 -
Per il campione SPES-0.9 (12h), i picchi di massa più intensi
corrispondono alle unità con 2 co-oligomeri di-solfonati PES
(DS-PES) lungo le catene e terminate con gruppi idrossili su
entrambe le estremità di catena (specie A
n,=2
), mentre i picchi
corrispondenti alle famiglie di co-oligomeri con due unità di DS-
PES e terminate con i gruppi -OH in una delle estremità e con
gruppi cloro all’altra estremità (specie B
n,
) dominano gli
spettri del copolimero S-PES-1.2 (12h).
Si deduce dalle caratterizzazioni effettuate e dalle conclusioni
ottenute sulla presenza e distribuzione dei gruppi terminali di
catena che nelle catene di co-oligomero che appartengono alle
specie B
n,
una unità monometrica di-solfonata con buona
probabilità si trova collocata come terminale di catena.
Lo studio effettuato mediante analisi spettroscopica MALDI-
TOF ha permesso di osservare anche i co-oligomeri ciclici
(specie
n,
).

- 113 -
Tale specie presenta due unità d-S-PES nel range di massa m/z
1300 ÷ 4000 degli spettri di massa di entrambi i copolimeri.
In accordo con i risultati ottenuti dallo studio degli spettri
ampliati nelle Figure 11b e 11c si può affermare con buona
sicurezza che la formazione di cicli avvenga con maggiore
frequenza nell’ambiente di reazione quando il rapporto molare
PES-OH/SDCDPS superi il valore limite di 1.0.
Negli spettri di entrambi i campioni si osserva anche la
presenza di co-oligomeri aventi tre unità di d-S-PES (X
n,
) in
catena; in questa specie, una unità di d-S-PES appare come sale
mono sodiato.
La presenza di queste famiglie di picchi è sintomo
dell’incompleta trasformazione della forma sodiata dei
copolimeri S-PES sintetizzati sotto forma dell’acido
corrispondente. Tale trasformazione è stata ottenuta mediante
trattamento con una soluzione di HCl 1M (vedi parte
sperimentale).

- 114 -
Gli spettri di massa dei due copolimeri, nel range di massa
superiore a m/z 3400 hanno picchi corrispondenti ai co-oligomeri
con tre unità di d-S-PES appartenenti alle famiglie A
n,
e B
n,
.
Le stesse famiglie di co-oligomeri (A
n,
, B
n,
, Φ
n,
e X
n,
) sono
osservate anche negli spettri di massa MALDI-TOF di
copolimeri di-solfonati preparati a partire da PES-OH (7K) e
indicati come S-PES 1.00 (12h) e S-PES 1.05 (12h).
Lo spettro di massa MALDI-TOF di S-PES 1.05 (12h), come
pure quello del S-PES1.0 (12h), mostrato nella figura nella
pagina seguente (Figura 12) presentano l’omologo cluster di
picchi da m/z 1500 a m/z 10000 e i picchi più intensi sono
intorno a 5000Da.

- 115 -
Figura 12- Spettro MALDI-TOF del S-PES-1.0(12h) (a). Il riquadro a destra riporta la sezione
ampliata degli spettri S-PES-1.0(12h) (b) ed S-PES-1.05(12h) (c).

- 116 -
Confrontando questo spettro con quello del S-PES0.9 (12h) in
Figura 11, risulta evidente che i copolimeri ottenuti dal pre-
polimero telechelico di partenza PES-OH (7K) hanno una massa
molare media (Mw e Mn) superiore a quella dei corrispondenti
d-S-PES sintetizzati da PES-OH (4K). Ciò peraltro è confermato
dai dati viscosimetrici e SEC-DV riportati in seguito (vedi
Tabella 5 a pag.131 dei risultati).
L’indagine delle sezioni ingrandite degli spettri di massa di S-
PES1.0 e S-PES1.05 nell’intervallo di massa (m/z) tra 4.150 e
4.850, riportato nelle Figure 12b e 12c rispettivamente, evidenzia
che questi copolimeri, parallelamente alla specie co-oligomerica
discussa precedentemente, presentano anche una porzione di
catene terminate con gruppi cloro ad entrambe le estremità,
(specie C
n,
in Figura 12 e nella Tabella 2).

- 117 -
a
Calcolato mediante spettroscopia
1
H-NMR.
b
Calcolato mediante analisi DMA.
c
Determinato mediante analisi TGA a 800°C.
d
Grado di solfonazione.
e
Ottenuto per reazione di post-condensazione dei corrispondenti S-PES-12h
Tabella 3- Riepilogo dei risultati della caratterizzazione effettuata sui campioni di PES-OH ed S-PES-X

- 118 -
4.2 Caratterizzazione NMR
Mediante spettroscopia
1
H-NMR sono state ottenute
informazioni fondamentali per uno studio chiaro ed esaustivo
della composizione e della struttura macromolecolare dei
campioni di PES sintetizzati.
Grazie alla caratterizzazione spettroscopica effettuata è stato
possibile rispondere ad importanti domande circa la struttura
molecolare, la composizione chimica, i gruppi terminali, e anche
il grado di solfonazione dei copolimeri S-PES sintetizzati.
Le assegnazioni complete degli spettri
1
H-NMR sono state
effettuate dal confronto con i dati di letteratura [27,41] e con gli
spettri di PES-OH (4K) (vedi Figura 13a a pag.118 dei risultati) e
del monomero SDCDPS.
I chemical shifts dei gruppi caratteristici della struttura
molecolare sono riassunti nella Tabella 4.

- 119 -
Gli spettri
1
H-NMR registrati per i campioni di pre-polimero
PES-OH confermano i risultati riportati qui di seguito.
La Figura 13 riporta lo spettro del pre-polimero PES-OH (4K)
confrontato con gli spettri NMR protonici dei PES solfonati (S-
PES) sintetizzati.
Lo spettro ottenuto mostra la presenza di due doppietti centrati
a 6,82 ppm e 7,67 ppm (indicati come a'' e b'' rispettivamente)
dovuti alla risonanza dei protoni aromatici in orto e in meta
rispetto al gruppo terminale idrossile.
Sia i protoni aromatici in orto ai gruppi solfonici che quelli in
orto all'ossigeno dei gruppi difenil-etere (rispettivamente protoni
b ed a in Figura 13a e nella tabella 4) dell’ossatura principale
delle catene di PES-OH, presentano nello spettro di risonanza
due doppietti a valori di chemical shift pari rispettivamente a
7,98 ppm e 7,28 ppm.

- 120 -
La massa molare media numerica (Mn) dei due polimeri PES-
OH è stata calcolata mediante lo studio dello spettro
1
H-NMR
attraverso il calcolo matematico delle aree degli integrali definiti
dei picchi di risonanza riferiti ai protoni aromatici H
b''
che
appartengono ai terminali di catena e le risonanze dovute ai
protoni aromatici presenti nella catena macromolecolare
principale H
a
, utilizzando la seguente equazione:
Xn = 1 + (I
b''
/ I
a
)
Gli spettri
1
H-NMR dei copolimeri PES di-solfonati ottenuti
dopo 12 ore e le reazioni di PES-OH (4K) sono sovrapposti in
Figura 13.
La Figura 13 riporta anche l'assegnazione di tutti i picchi in
base alle assegnazioni dei chemical shifts.

- 121 -
Negli spettri NMR protonici dei campioni di S-PES a diverso
grado di solfonazione riportati in Figura 13 risulta evidente come
i picchi dei segnali relativi ai protoni adiacenti al gruppo –SO
3
H
pendente (ad esempio H
3
ed H
3’
) aumentino di intensità con
l’aumentare del grado di solfonazione delle catene, e siano
completamente assenti nel prepolimero PES-OH. Ciò è
un’ulteriore prova che la reazione di solfonazione è avvenuta
correttamente come previsto e che tale reazione può essere
controllata e riproducibile con buona precisione, ponendosi nelle
le condizioni di reazione sperimentali idonee (rapporto tra i
reagenti, grado di purezza dei solventi e dei reagenti, temperatura
e ambiente di reazione controllato).
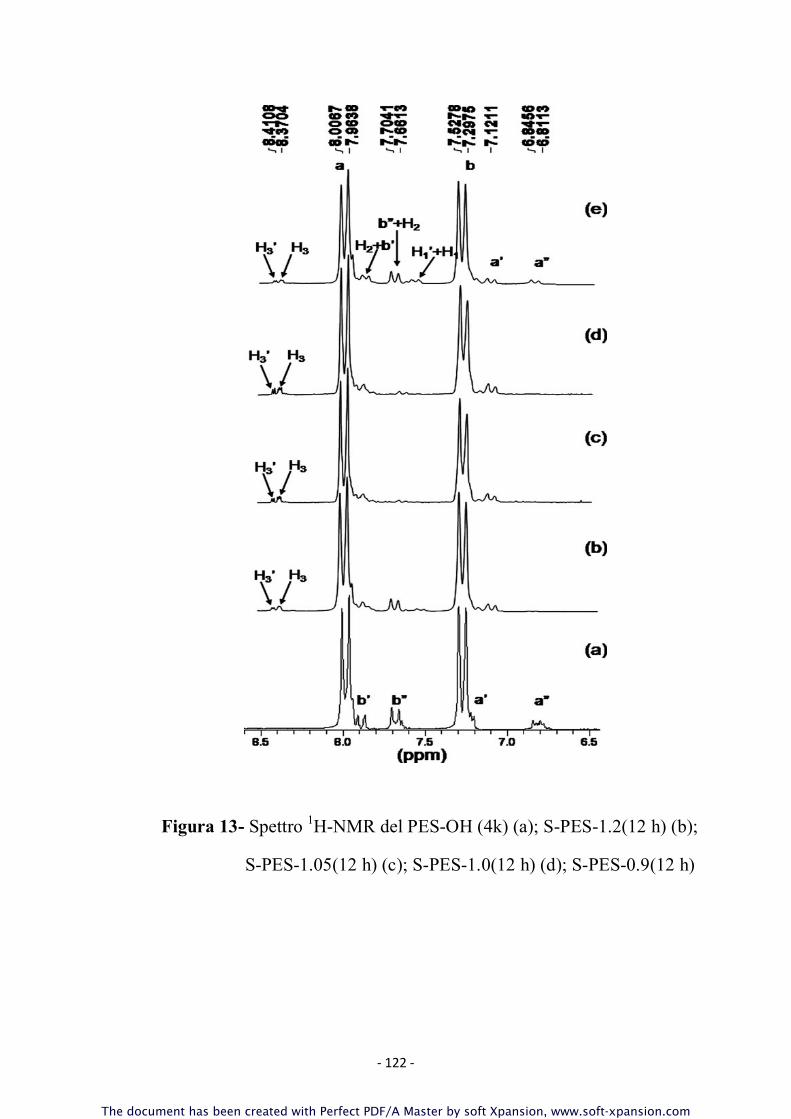
- 122 -
Figura 13- Spettro
1
H-NMR del PES-OH (4k) (a); S-PES-1.2(12 h) (b);
S-PES-1.05(12 h) (c); S-PES-1.0(12 h) (d); S-PES-0.9(12 h)
(e).

- 123 -
Ciò permette di distinguere le unità PES di-solfonate dai
blocchi PES non solfonati. Questi ultimi danno infatti due
doppietti centrati a 7,98 ppm (protoni Ha) e a 7,27 ppm (protoni
Hb), mentre le unità di-solfonate PES danno segnali nelle
regioni: 8,35-8,41 ppm (protoni e H
3
H
3'
), 7,84-7,91 ppm
(protoni H
2
e H
2'
), 7-50-7.62 ppm (protoni H
1
e H
1'
) e di 7,12-
7,20 ppm (protoni H
1
).
Una buona risoluzione dei picchi permette di distinguere anche
i segnali appartenenti alle unità di S-PES collocate all’interno
della catena principale da quelle posizionate come terminali di
catena. Ad esempio i picchi H
3
appartengono alle unità di-
solfonate nella catena principale che uniscono due blocchi PES
non solfonati, mentre i picchi H
3'
appartengono alle stessa unità
nei terminali di catena.
Dallo studio degli spettri di segnali protonici in Figura 13b-d
possiamo anche caratterizzare i gruppi terminali fenolici (picchi
H
a"
e H
b''
).

- 124 -
E’ interessante notare come le intensità dei picchi dovuti ai
gruppi terminali diminuiscono con l'aumento della massa molare
media (Mn) di copolimeri S-PES ottenuti dopo 12 ore di reazione
(vedi parte sperimentale).

- 125 -
- Chemical shifts misurati con un’accuratezza di ±0.01 ppm
- Abbeviazioni utilizzate: d, doppietto; dd, doppio doppietto.
Tabella 4- Assegnamenti dei segnali dovuti ai protoni aromatici appartenenti alle sequenze ed ai gruppi
terminali presenti nei copolimeri S-PES.

- 126 -
Dalle misure delle aree degli integrali (I) dei protoni
appartenenti ai blocchi PES non solfonati (protoni Ha) e di quelli
dovuti alle unità S-PES (H
3
protoni e H
3'
) sono state ottenute
indicazioni sulle frazioni molari delle unità PES solfonate (C
S -
PES
) e sulle unità di PES non solfonate (C
PES
):
C
PES
= (I
HA
/ 4) / (I
HA
/ 4 + + I
H3
I
H3'
)
C
S-PES
= 1 - C
PES
E’ stato inoltre calcolato sulla base dei dati sperimentali anche
il grado di solfonazione (DS) dei copolimeri mediante la
seguente equazione:
DS = 2 ∙ C
S-PSE
Importanti informazioni sono state ottenute anche sul grado di
polimerizzazione (Xn) ed è stata inoltre calcolata la massa
molare media (Mn) dei copolimeri S-PES.

- 127 -
Xn = (I
HA
/ 4 + I
H3
/ 2) / (C
PES
∙ I
HA"
/ 2 + C
S-PES
∙ I
H3'
)
Mn = ∙ Xn (232,2 + 376,3 C
PES
∙ C
S-PES
)
I valori 232,2 e 376,3 corrispondono rispettivamente alla
massa delle unità ripetitive non solfonate e solfonate in catena.
Il grado di solfonazione DS, che definisce il numero medio di
gruppi solfonici (-SO
3
H) nelle catene di copolimero S-PES,
corrisponde al doppio della frazione molare C
S-PES
, dal momento
che ogni unità ripetitiva solfonata presenta due gruppi SO
3
H
(DS = 2 ∙ C
S-PES
).
Dallo studio degli spettri
1
H-NMR possiamo anche calcolare la
percentuale in moli (mol%) di unità solfonate (S-PES) presenti
come unità terminali di catena (C
S-PES-end
) in riferimento alla
quantità totale di guppi solfonici lungo le catene
macromolecolari, mediante l’equazione seguente:
C
S-PES-end
= I
H3'
/ (I
H3
+ I
H3'
) ∙ 100

- 128 -
Poiché i risultati derivati dallo studio condotto mediante
spettrometria di massa MALDI-TOF MS e
1
H-NMR sui
campioni di S-PES ottenuti dopo 12 ore di reazione hanno
rivelato che le catene co-polimeriche possiedono una percentuale
di gruppi terminali fenolici e cloro elevata, è stata continuata la
reazione di sostituzione nucleofila al fine di favorire in tal modo
ulteriori reazioni di poli-condensazione tra le catene ottenute fino
a quel tempo di reazione mediante meccanismo di chain-
coupling tra le catene intermedie formatesi.
Estendendo il tempo di reazione oltre le 12h le catene co-
polimeriche terminate con i suddetti gruppi (-OH e -Cl) hanno
maggiore probabilità di urtare con i corispondenti gruppi
terminali partner di reazione e formare un’unica grande catena
macromolecolare a partire dalle due catene di partenza; ciò
avviene mediante il suddetto meccanismo di chain-coupling.
L’evidenza sperimentale dell’avvenuta reazione dovrebbe
essere un conseguente spostamento verso valori più alti della

- 129 -
distribuzione di massa molare (MMD) dei copolimeri reagiti e di
conseguenza una diminuzione negli spettri dei campioni
analizzati, delle intensità dei segnali relativi ai gruppi terminali
di catena.
Seguendo questo ragionamento abbiamo effettuato a posteriori
una post-reazione di polimerizzazione per due aliquote di
campioni fin qui ottenuti nelle prime sintesi S-PES0.9(12h) ed S-
PES1.2(12h).
Le durate delle nuove reazioni sono state di 24 e 48 ore, e la
temperatura di reazione è stata mantenuta a 200°C (vedi parte
sperimentale).
L’avanzamento della reazione di post-condensazione è stato
monitorato così come per i campioni precedentemente
sintetizzati mediante indagine spettroscopica
1
H-NMR e
spettrometria di massa MALDI-TOF MS. La cinetica della
reazione di post-condensazione è stata inoltre seguita mediante lo
studio degli spettri
1
H-NMR dei copolimeri S-PES sintetizzati.

- 130 -
Come esempio, sono sovrapposti in Figura 14 spettri di S-
PES0.9 (12h), S-PES0.9 (12+24h) e S-PES0.9 (12+48h). Con gli
altri picchi normalizzati, si può osservare che come aumentando
il tempo di post-reazione, l'intensità dei segnali dovuta ai protoni
aromatici appartenenti ai gruppi terminali (protoni: H
3'
a 8,38
ppm, H
b''
a 7,68 ppm, H
a''
a 7,69 ppm, vedi la tabella 4 per i loro
assegnamenti), a poco a poco diventano più piccoli, a causa
dell'aumento della massa molare media, come confermano i
valori di Mn calcolati mediante le integrazioni dei picchi
protonici negli spettri
1
H-NMR (vedi Tabella 1). Questi risultati
sono stati confermati anche da studi comparati di viscosimetria
SEC ed off-line (vedere i dati nella Tabella 5 e la discussione in
seguito).
Il grado di solfonazione dei copolimeri S-PES ottenuti
mediante reazione di post-condensazione sono stati calcolati dai
loro spettri
1
H-NMR. Per ogni copolimero il valore DS non
cambia rispetto a quella del corrispondente S-PES (12h) da cui
proviene.

- 131 -
In Figura 14 è riportato il confronto tra gli spettri
1
H-NMR del
campione di S-PES-0.9 a 12h e dello stesso campione cui è stata
effettuata la post-condensazione a due tempi di reazione (36h e
60h).
Figura 14- Spettro
1
H NMR del S-PES-0.9-12h (a), 36h (b), and 60 h (c).

- 132 -
4.3 SEC-viscometria
Il riepilogo dei risultati ottenuti dalla caratterizzazione
molecolare mediante analisi viscometrica SEC del pre-polimero
PES-OH (4K) e dei quattro campioni S-PES è mostrato nella
Tabella 5.
La tabella riporta la massa molare del picco cromatografico
(Mp), le masse molari medie più significative per lo studio
condotto (Mn, Mw, Mz), gli indici polidispersità (Mw / Mn, Mz /
Mw), la viscosità intrinseca [] in solvente DMAC + LiBr
0.05M a 80 °C di temperatura ed infine i coefficienti di MHS
(intercetta k e pendenza a).
Wang et al.
47
hanno evidenziato che i dati di viscosità
intrinseca per i copolimeri solfonati PES non erano paragonabili
a quelli dei polimeri non solfonati, dal momento che le catene
polimeriche elettrolitiche interagiscono attraverso i gruppi
solfonati.

- 133 -
Per sopprimere gli effetti dovuti al comportamento delle catene
come polielettrolita, in accordo con alcuni autori al tradizionale
solvente NMP è stato agiunto LiBr 0,05 M; la piccola quantità di
sale ha il ruolo di mitigare efficacemente l'effetto polielettrolitico
dei gruppi solfonici permettendo una migliore caratterizzazione
dei materiali contenenti gruppi fortemente polari per la presenza
di ioni.
I dati riportati nella Tabella 5 confermano chiaramente le
precedenti conclusioni circa la reazione di polimerizzazione dei
S-PES. Aumentando il tempo di polimerizzazione a partire dal
pre-polimero PES-OH e passando al S-PES 12h fino ad arrivare
al S-PES 60h la massa molare media aumenta in modo evidente.
In particolare per i campioni di S-PES0.9 la massa molare
media ponderale (Mw) aumenta da 8,1 kg/mol (PES-OH) a 21,4
kg/mol (S-PES0.9 (60h).

- 134 -
Molto interessante è anche il risultato di polidispersità dei
campioni ottenuto. I risultati ottenuti sono compresi tra 1,3 e 1,4
il che corrisponde ad un range di masse molto ristretto.
Questa tendenza è evidente anche per i campioni di S-PES1.2.
La Figura 15 mostra il confronto tra i differenziali di MMD di
quattro campioni: PES-OH (4K), S-PES0.9 (12h), S-PES0.9
(36h) e S-PES0.9 (60h). Le MMD dei campioni mostrano
chiaramente la tendenza crescente in massa molare.
Figura 15- Confronto tra il differenziale della massa molare (MWD)
di PES-OH(4k) (a), S-PES-0.9(12h) (b), S-PES-O.9(36h) (c)
e S-PES-0.9(60h) (d).

- 135 -
L'aumento della massa molare è evidente tra il pre-polimero
PES-OH ed il PES0.9 (12h) ed ancora fino al PES0.9 (36h); non
vi è invece differenza significativa in massa molare tra PES0.9
(36h) e il PES0.9 (60h).
La Figura 16 mostra il confronto dell’andamento di MHS,
[]= k * M
a
, di quattro campioni: PES-OH (4K), S-PES 0.9
(12h), S-PES 0.9 (36h) e S-PES 0.9 ( 60h). La pendenza del
grafico di MHS per i campioni PES-OH e PES 0.9 (12h) è molto
simile (a = 0,65).
Figura 16- Confronto delle curve di MHS in campioni di PES-OH(4K)
(a), S-PES-0.9(12h), S-PES-0.9(36h) (c) e S-PES-0.9(60h) (d).

- 136 -
L’andamento di MHS per S-PES 0.9 (36h) ed S-PSE 0,9 (60h)
campioni è completamente diverso (a = 0,85). In altre parole, i
campioni S-PES 0.9 (36h) e S-PES 0.9 (60h) hanno una
maggiore lunghezza delle catene polimeriche e più gruppi
funzionali solfonici nelle catene polimeriche (e di conseguenza
catene più rigide).

- 137 -
Tabella 5- Dati riepilogativi della caratterizzazione mediante analisi SEC-DV del prepolimero PES-OH e dei
campioni di S-PES sintetizzati.

- 138 -
4.4 Spettroscopia FT-IR
Gli spettri FT-IR con sistema ATR sono stati utilizzati per
confermare la presenza di gruppi pendenti SO
3
H presenti nelle
catene di copolimero S-PES. Gli spettri IR sono stati ottenuti
ponendo in ascisse il numero d’onda in unità di misura cm
-1
,
contro la trasmittanza (%) posta in ordinata.
La Figura 17 mostra gli spettri del PES-OH telechelico (4K)
genitore e dei copolimeri solfonati S-PES0.9 (12h) e S-PES1.2
(12h) sintetizzati partire da questo. Gli spettri FT-IR di campioni
solfonati S-PES mostrano bande di assorbimento caratteristico a
1030 cm
-1
grazie alle vibrazioni di stretching simmetrico dei
gruppi -SO
3
H (indicata con il simbolo in Figura 17).
Le vibrazioni di stretching asimmetrico dei gruppi-SO
3
H sono
presenti a circa 1180 cm
-1
,
48
ma non sono facilmente osservabili
a causa della sovrapposizione con altri fenomeni di trasmittanza
che si sovrappongono parzialmente alla prima.

- 139 -
Tutti i copolimeri S-PES hanno dato spettri FT-IR simili a
quelli delle figure 6b-c.
Figura 17- Spettro FT-IR del pre-polimero PES- OH(4K) (a),
di S-PES-1.2(12h) e S-PES-0.9(12h).

- 140 -
4.5 Caratterizzazione termica
E’ stato effettuato lo studio del comportamento termico dei
poli(arilen eteri-solfoni) solfonati sintetizzati mediante scansione
termica in un ambiente statico, sia in corrente d'azoto che in aria.
Il comportamento termico dei materiali studiati nei due
ambienti è risultato paragonabile.
Ogni campione prima di essere analizzato è stato essiccato a
180°C per 12 ore.
La curva TGA rappresentata nella Figura18 mostra che i
copolimeri S-PES analizzati nel range di temperatura studiato
(35°C - 700 °C) subiscono tre fenomeni degradativi disti tinti
l’uno dall’altro ciascuno e caratterizzato da un preciso range di
temperatura. Le curve di termodegradazione sono state ottenute
con una velocità di scansione di 10°C /min
-1
sia in ambiente
inerte che in atmosfera ossidativa.

- 141 -
In Figura 19 e 20 sono mostrati due dei termogrammi ottenuti
nel presente studio.
Figura 19- Curve di termodegradazione in ambiente inerte (N2) del
PES-OH (a),
SPES-1.2 (b), S_PES-1.05 (c), S-PES-1.0 (d) e S-PES-0.9 (e)
a
e
d
c
b
Figura 18- Curve TGA del S-PES-1.2(12h) (a), S-PES- 1.05(12h)
(b), S-PES-1.00(12h) (c) ed S-PES-0.9(12h) (d).

- 142 -
Dal momento che la stabilità termica dei polimeri è collegata
sia con la temperatura di decomposizione iniziale (T
i
) che con la
velocità di degradazione, i dati sperimentali ottenuti mediante le
analisi termo gravimetriche (TG) sono stati utilizzati per
determinare le energie di attivazione apparente associate al primo
step degradativo (÷400 °C). Allo stesso modo sono state le
temperature di decomposizione iniziale dei nostri polimeri.
Figura 20- Curve di termodegradazione in ambiente ossidativo (aria) del
PES-OH (a), S-PES-1.2 (b), S-PES-1.05 (c), S-PES-1.0 (d) ed S-PES-0.9(e)
a
b
c
d
e

- 143 -
Qui di seguito nella Figura 21 sono rappresentati graficamente
i risultati dello studio della temperatura di inizio degradazione in
funzione del grado di solfonazione dei materiali studiati.
La seconda e la terza fase di degradazione non sono state
considerate perché la perdita di in peso dopo il primo step di
degradazione termica è diminuito fino ad arrivare a circa l’11-
13%. Tale dato ci indica che il primo step di degradazione è
quello in cui avvengono i principali fenomeni di
decomposizione.
Figura 21- Temperatura di inizio decomposizione (Ti) in funzione del grado
di solfonazione sotto flusso di azoto ( ) e in atmosfera statica di aria ( )

- 144 -
I valori di energia di attivazione (E
a
) del processo di termo
degradazione delle catene sono stati ottenuti mediante il metodo
Kissinger classico
49
, che si basa sulla seguente equazione:
dove F è la velocità di riscaldamento e T
m
è la temperatura alla
massima velocità di perdita in peso. Come valori di temperatura
T
m
sono stati quelli corrispondenti al primo picco
termodegradativo (DTG).
Diverse scansioni sono state effettuate e sono stati determinati
i valori medi (T
m
) ad ogni velocità di scansione (Tabelle 6 e 7).
I dati nelle Tabelle 6 e 7 sono stati quindi ricalcolati in base
all’Equazione Kissinger.

- 145 -
Tabella 6- Temperature alla a cui corrisponde la massima velocità di perdita
in peso (Tm) per il primo stadio di degradazione del PES-OH e degli S-PES
in atmosfera inerte per azoto e a varie velocità di riscaldamento.
Tabella 7- Temperature alla a cui corrisponde la massima velocità di
perdita in peso (Tm) pe il primo stadio di degradazione del PES-OH e
degli S-PES in atmosfera di aria e a varie velocità di riscaldamento.

- 146 -
I valori di energia di attivazione che riguardano il processo di
termodegradazione studiato sono stati ottenuti sia in ambiente
inerte per (N
2
) che in quello ossidante (aria).
I valori di tali energie corrispondono alle pendenze delle
equazioni lineari mostrate nelle Figure 22 e 23.
Figura 22- Energia di attivazione apparente del processo di degradazione (Ea)
in funzione del grado di solfonazione sotto flusso di azoto ( ) e in aria ( ).
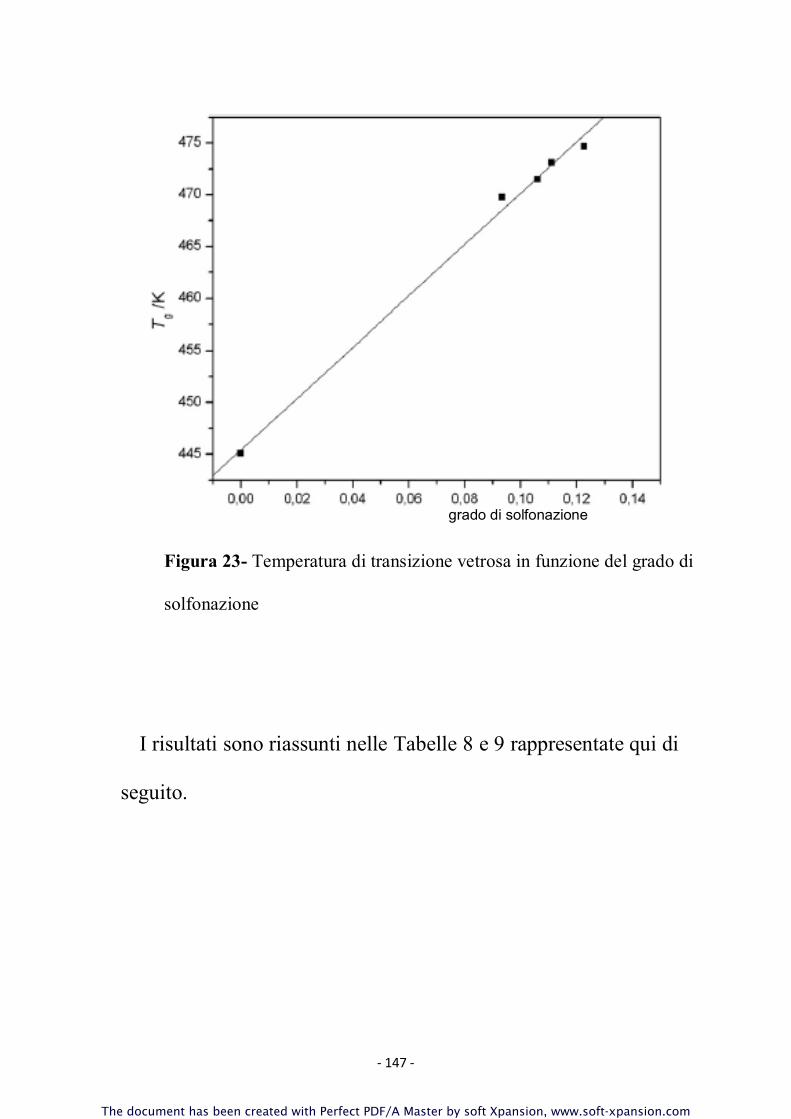
- 147 -
I risultati sono riassunti nelle Tabelle 8 e 9 rappresentate qui di
seguito.
Figura 23- Temperatura di transizione vetrosa in funzione del grado di
solfonazione
grado di solfonazione

- 148 -
Tabella 8- Coefficiente di regressione ed energia di attivazione apparente
del processo di degradazione (Ea) in azoto, calcolata mediante l’equazione
di Kissinger per i campioni di PES sintetizzati
Tabella 9- Coefficiente di regressione ed energia di attivazione apparente
del processo di degradazione (Ea) in aria, calcolata mediante l’equazione di
Kissinger per i campioni di PES sintetizzati

- 149 -
Le temperature di inizio decomposizione (onset), ottenute a
diverse velocità di riscaldamento sono state calcolate a partire
dalle curve di termodegradazione (TG).
Tale temperatura è stata determinata graficamente come
l’ascissa del punto di intersezione tra la retta tangente nel tratto
iniziale (prima che sia avvenuto il processo di degradazione) alla
curva di perdita in peso e una retta tangente alla stessa curva nel
punto di massima velocità di perdita in peso e quindi avente
pendenza massima.
I valori temperatura di inizio degradazione (T
i
) per ciascun
campione studiato a varie velocità di riscaldamento sono risultati
diversi tra loro, ma la tendenza osservata tra vari i campioni
studiati è stata la stessa indipendentemente dalla velocità di
scansione usata.
Per motivi di semplicità sono stati esaminati e sono riportati,
solo i valori a 10°C/min nella Tabella 10.
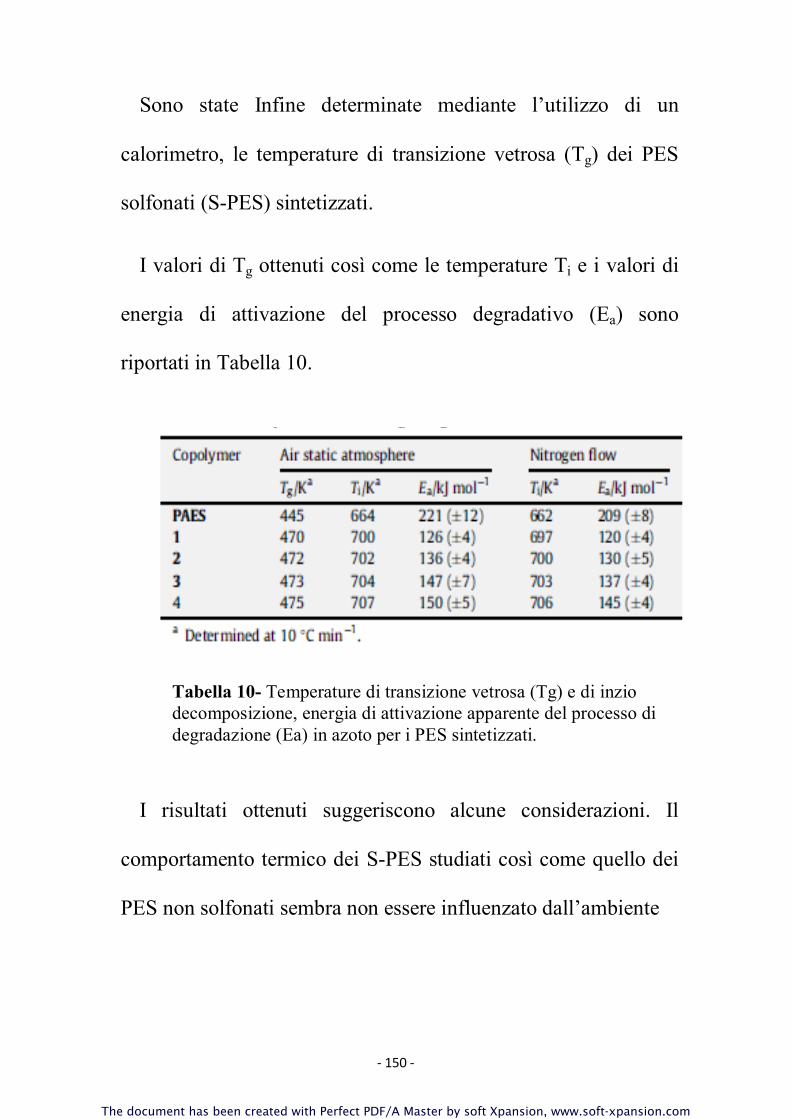
- 150 -
Sono state Infine determinate mediante l’utilizzo di un
calorimetro, le temperature di transizione vetrosa (T
g
) dei PES
solfonati (S-PES) sintetizzati.
I valori di T
g
ottenuti così come le temperature T
i
e i valori di
energia di attivazione del processo degradativo (E
a
) sono
riportati in Tabella 10.
I risultati ottenuti suggeriscono alcune considerazioni. Il
comportamento termico dei S-PES studiati così come quello dei
PES non solfonati sembra non essere influenzato dall’ambiente
Tabella 10- Temperature di transizione vetrosa (Tg) e di inzio
decomposizione, energia di attivazione apparente del processo di
degradazione (Ea) in azoto per i PES sintetizzati.

- 151 -
in cui viene condotto l’esperimento termodegradativo (inerte o
ossidativo).
Inoltre, non solo la forma delle curve ottenute nei
termogrammi TG nelle due condizioni sperimentali utilizzate
sono molto simili, ma anche le temperature di decomposizione
iniziale sotto flusso di azoto sono molto prossime a quelle in
atmosfera ossidativa per aria (Tabella 10), suggerendo così che la
presenza di ossigeno non influenza il meccanismo iniziale di
decomposizione.
I polimeri PES funzionalizzati che sono stati oggetto del nostro
studio, subiscono processi degradativi nell'intervallo di
temperatura che va da ~400°C a ~700°C ed hanno una
percentuale di residuo carbonioso alla fine del processo di
degradazione (~800°C) che varia dal ~5 al ~10% a seconda dei
campioni esaminati.

- 152 -
Come si può osservare dalla Figura 18 (pag.134) in cui sono
riportate le curve di degradazione termica TG dei polimeri S-
PES studiati, i tracciati presentano tre picchi di massima velocità
di perdita in peso (PDT) evidenti nel grafico come altrettanti
gradini nelle curve di degradazione e corrispondenti ai tre
meccanismi di degradazione.
Il primo step di decomposizione che potrebbe essere attribuito
alla decomposizione dei gruppi del-SO
3
H pendenti si trova nella
zona di temperatura che va da 420-450°C e corrisponde alla
rottura delle catene polimeriche in corrispondenza di ponti eterei
(-Φ-O-/-Φ-).
Il secondo processo degradativo avviene in un range di
temperatura che va dai 520 ai 580°C e provoca l’eliminazione di
gruppi –SO
2
- della catena e la formazione di unità di-feniliche;
infine il terzo meccanismo avviene intorno ai 630-680°C ed
avviene con l’eliminazione di molecole di idrogeno e formazione
di strutture dibenzofuraniche in accordo con la letteratura
50
.

- 153 -
È evidente inoltre che i polimeri PES ad elevato grado di
solfonazione (a percentuali che vanno dal 7% al 13%) danno
una percentuale di residuo a 800 °C leggermente superiore
(~10%) ai PES funzionalizzati con diversi gruppi terminali per i
quali il residuo a 800°C risulta simile per tutti (3-5%).

- 154 -
Il seguente schema descrive il primo dei tre meccanismi di degradazione che avvengono nelle
catene polimeriche PES studiate nel range di temperatura che va da 420°C a 450°C:
Schema 4
O
O
.
.
estrazione
H
OH
H
+
+
Ricombinazione
(a)
O H
O
OO
O
O
H
H
O
O
O
H
H
+
estrazione
H
O
H
O
HO
(b)

- 155 -
Nel seguente schema è riportato il secondo meccanismo di degradazione che avviene nel range di
temperatura che va da 520°C e 580°C:
Schema 5
SO
2
.
.
estrazione
H
OH
H
+
Ricombinazione
SO
2
O O
SO
2
Estrazione
.
SO
2
.
+
SO
2
- SO
2

- 156 -
Il seguente schema rappresenta infine il terzo ed ultimo meccanismo di degradazione (630°C-
680°C):
Schema 6
O HO
O
O
(b)
OH
O
- H
2
(a)
OO O
OH
OH
OH
- 2 H
2
O
O
OH

- 157 -
4.6 Analisi dinamo meccaniche (DMA)
Le temperature di transizione vetrosa (Tg) di tutti i copolimeri
solfonati sono state determinate mediante analisi dinamo
meccanica DMA attraverso cicli di riscaldamento e
raffreddamento già discussi nella parte sperimentale.
I risultati sono stati elencati nella Tabella 3. Come confronto
nella Tabella 3 sono stati riportati anche i dati di Tg di entrambi i
prepolimeri telechelici PES-OH utilizzati durante le sintesi dei S-
PES. Si può osservare che per i copolimeri S-PES ottenuti allo
stesso tempo di reazione (12h), si ha l'aumento dei valori di Tg
sia all’aumentare del grado di solfonazione (DS) che del grado di
polimerizzazione (Xn) del PES-OH genitore, da cui sono stati
sintetizzati . I valori di Tg dei copolimeri preparati per reazione
di post-condensazione a partire dal S-PES0.9 (12h) (Figura 24)
ed S-PES1.2 (12h) (Figura 25) aumentano se confrontati con i
corrispondenti valori di Tg dei copolimeri iniziale, suggerendo
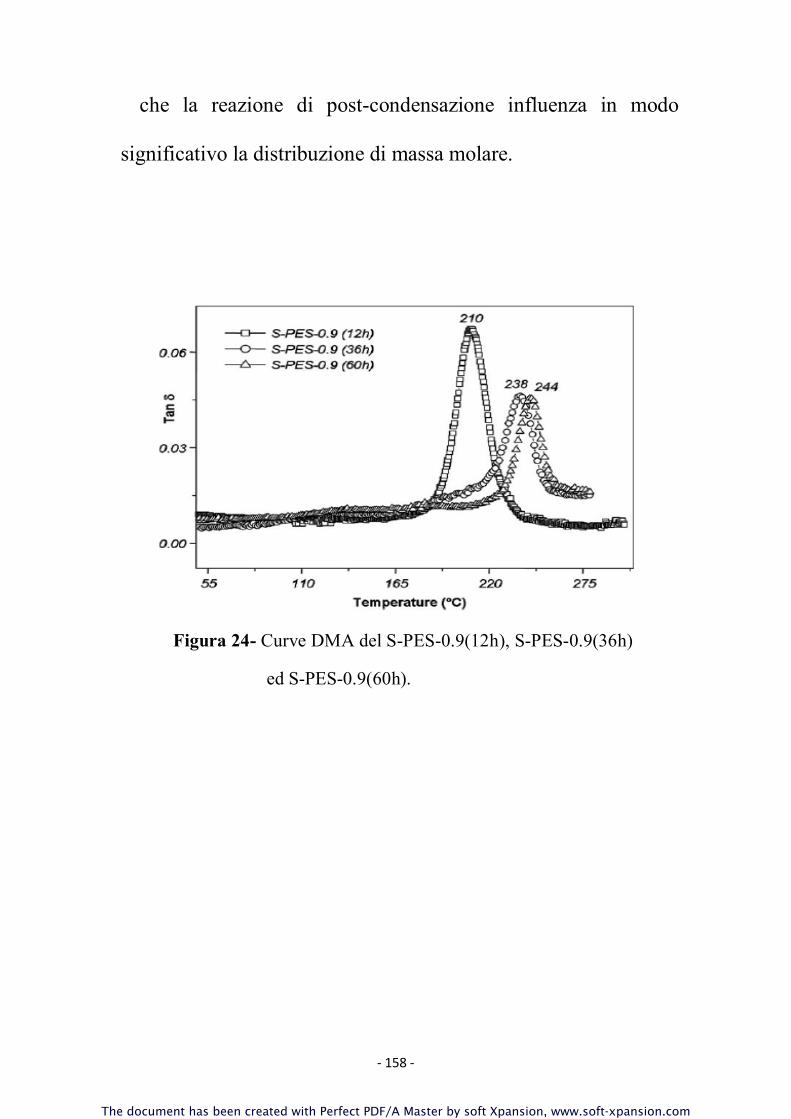
- 158 -
che la reazione di post-condensazione influenza in modo
significativo la distribuzione di massa molare.
Figura 24- Curve DMA del S-PES-0.9(12h), S-PES-0.9(36h)
ed S-PES-0.9(60h).

- 159 -
Per entrambe le serie di copolimeri S-PES si osserva che esiste
una chiara tendenza per la temperatura di transizione vetrosa ad
aumentare con il tempo di post-condensazione.
L'aumento di Tg è il risultato dell'aumento della massa molare,
che è indicato nella Tabella 3 e dai dati SEC-viscometria nella
Tabella 5.
Figura 25- Curve DMA del S-PES-1.2(12h), S-PES-1.2(36h) ed
S-PES-1.2(60h).

- 160 -
I dati nella Tabella 3 indicano che il valore di soglia della
temperatura di transizione vetrosa per questa classe di copolimeri
aventi un DS di 10-11 mol% è tra 244-246 °C.

- 161 -
5. CONCLUSIONI
La composizione chimica dei S-PES sintetizzati è stata determinata
a partire alle indicazioni tratte dalla letteratura corrente e dai passati
studi condotti dal gruppo di ricerca di cui faccio parte, mediante le
più comuni tecniche di indagine utilizzate in chimica organica
macromolecolare, ovvero NMR ed FT-IR. Tali tecniche spesso non
riescono a rispondere a tutti i quesiti riguardo alla composizione e
delle macromolecole studiate e alla natura delle terminazioni di
catena che possono influenzare le proprietà polielettrolitiche del
materiale studiato. In particolare, mediante le tecniche citate non è
particolarmente semplice distinguere i copolimeri a blocchi dai
corrispondenti omopolimeri presenti nel materiale studiato. Inoltre,
per campioni con massa molare media (Mn) elevata, con le tecniche
NMR i gruppi terminali non possono essere determinati in maniera
quantitativa.

- 162 -
MALDI-TOF MS è una tecnica potente che ci ha consentito di
rilevare e fornire indicazioni circa le caratteristiche chimiche delle
macromolecole oggetto del nostro studio, anche di quelle frazioni
meno abbondanti nella miscela analizzata. Tale tecnica ha permesso
l'identificazione delle unità ripetitive co-polimeriche, delle
terminazioni di catena, della presenza oligomeri ciclici e anche di
specie presenti in quantità minoritaria ma pur sempre significative
nello studio della cinetica di reazione.
In relazione alla classe di polimeri studiata, i poli(arilen eteri
solfoni) solfonati i lavori fin qui pubblicati dalla comunità scientifica
non avevano fino ad ora indagato la metodica di sintesi da noi
riportata e pochissimi studi così approfonditi e basato su molteplici
tecniche d’indagine chimica, termica e meccanica (MALDI-TOF MS,
1
H-NMR, SEC-DV, FT-IR, DSC, DMA, TGA) erano stati fin qui
svolti in tal modo.
Pertanto, in questo lavoro abbiamo concentrato i nostri studi per
dimostrare l'enorme potenzialità di combinare le tecniche suddette
per una caratterizzazione basata su molteplici aspetti, completa e
inequivocabile dei copolimeri PES solfonati.

- 163 -
Particolare importanza in tale studio sperimentale è stata prestata
nella valutazione della cinetica di reazione e quindi dell’avanzamento
del grado di polimerizzazione e del grado di solfonazione delle
catene polimeriche al variare del tempo di reazione, del rapporto
utilizzato tra i monomeri e tra i pre-polimeri e di tutte le condizioni al
contorno (temperatura di reazione, purezza dei reagenti e dei solventi
impiegati, composizione chimica dell’ambiente di reazione ed
eventuale presenza di umidità).
Nel presente lavoro lo studio della reazione di policondensazione
nucleofila aromatica è stato affrontato anche mediante l'uso della
tecnica cromatografica SEC e dal relativo calcolo della viscosità.
Tale approccio sperimentale permette di ottenere ulteriori prove che
avvalorino ancora di più i risultati ottenuti mediante l’utilizzo delle
tecniche di caratterizzazione chimica precedentemente discusse.
I materiali polimerici sintetizzati sono inoltre stati studiati con
opportune tecniche di indagine analitica (Termogravimetria ed analisi
dinamo-meccanica) per definire le loro proprietà termiche.

- 164 -
Tali proprieà dei S-PES sintetizzati sono state comparate con
campioni di PES non solfonati ed aventi gli stessi gruppi terminali e
massa molare media paragonabile.
I poli(arilen eteri solfoni) solfonati mostrano una più
elevata temperatura di transizione vetrosa rispetto ai corrispondenti
PES non solfonati e una temperatura di inizio decomposizione più
alta pur avendo masse molari paragonabili. Ai fini pratici tale
proprietà consente un campo di applicazione più ampio, conseguente
all’aumento della resistenza termica allo stato solido. Inoltre,
l'aumento del grado di solfonazione dà luogo ad un ulteriore
incremento dei valori di Tg e Ti. E’ inoltre interessante notare il forte
incremento della velocità di degradazione dei copolimeri PES
solfonati se confrontato con gli omologhi PES non solfonati, come
conseguenza del calo drammatico della loro energia di attivazione
apparente di degrado. Tuttavia la loro stabilità termica molto più
bassa dal punto di vista cinetico appare in questo caso irrilevante
nella valutazione della stabilità termica globale, se si considerano i
valori di Ti molto più elevati per gli S-PES rispetto ai PES tal quali
51
.

- 165 -
Le prove sperimentali mostrano una dipendenza lineare di Ti, Ea e
dei valori di Tg rispetto al grado di solfonazione degli S-PES.
L’influenza lineare del gruppo solfone (-SO
3
H) verso tali proprietà
dei materiali studiati è tanto più evidente per valori di massa molare
media dei compresi tra 7 e 10K.
Tale correlazione verrà approfondita in lavori futuri, una volta
sintetizzati poli(arilen eteri solfoni) aventi una massa molare media
più elevata dei copolimeri PES studiati.
I termogrammi ottenuti evidenziano che i poli(arilen-eteri
solfoni) solfonati (S-PES) studiati possiedono una temperatura di
transizione vetrosa (Tg) che raggiunge i 244-246°C nei campioni
con più elevata massa molare media e grado di solfonazione
(DS) prossimo a 10-11mol%.
Poiché la composizione chimica, i gruppi terminali, il grado di
solfonazione e la distribuzione di massa molecolare sono fattori
che incidono in modo primario su alcune determinanti proprietà
del materiale ai fini applicativi (ad esempio

- 166 -
l’assorbimento d'acqua la conducibilità protonica planare, la
resistenza meccanica) e di conseguenza sulle applicazioni (vedi
membrane a scambio protonico a base di copolimeri S-PES), nel
lavoro successivo verranno approfondite queste correlazioni
portando avanti nuove varianti alle ormai consolidate metodiche
di sintesi fin qui utilizzate con l’obiettivo di ottenere catene
polimeriche con massa molare media (Mn) e grado di
solfonazione (DS) più elevati.
Il controllo delle condizioni della reazione di
policondensazione consente effettuare previsioni sui possibili
gruppi terminali di catena, sulla composizione chimica e sul
grado di solfonazione.
Tali previsioni, basate fondamentalmente sullo stato dell’arte
attuale e su conoscenze acquisite a partire da studi propedeutici
al presente hanno condotto alla preparazione di una serie di
campioni polimerici costituiti da macromolecole aventi proprietà
chimiche, termiche e meccaniche secondo certi aspetti superiori,
se confrontate con quelle dei PES solfonati preparati con

- 167 -
metodiche di sintesi tradizionali e già studiate in passato.
Le caratteristiche chimiche, le proprietà termiche e meccaniche
dei poli(arilen eteri solfoni) studiati nel presente lavoro di tesi
danno importanti spunti di riflessione su alcuni dei possibili
campi applicativi in cui potrebbero trovare utilizzo tali materiali
in scala industriale: tra i principali vi potrebbe essere quello
dell’utilizzo di tali materiali per la produzione di membrane
polielettrolitiche a scambio protonico, vista la natura altamente
polare delle catene macromolecolari vista la presenza dei ponti
solfonici ed eterei lungo la catena macromolecolare principale e
dei gruppi solfonici in prevalenza acidi in catena laterale.
Tali membrane trovano spazio in diversi settori industriali:
quello automobilistico e dei trasporti in generale è uno dei
principali per via dello sviluppo sempre crescente dei motori
alimentati ad idrogeno e a metanolo in cui si ha l’utilizzo di
membrane polielettrolitiche per le celle a combustibile.

- 168 -
I risultati del presente lavoro scientifico sono stati: presentati
in congressi Nazionali; pubblicati su riviste scientifiche
internazionali ad alto valore nel campo dei materiali polimerici.
52

- 169 -
6. ELENCO FIGURE, TABELLE E SCHEMI
ELENCO FIGURE
Figura 1. Sezione trasversale schematica di una singola cella
di una PEMFC.
Figura 2. Relazione tra Tg,Tm e PRCL, secondo Carlier.
Figura 3. Relazione tra Tg e Tm.
Figura 4. Disegno schematico dell’apparato di reazione
utilizzato per le reazioni di sintesi.
Figura 5. Spettro
1
H-NMR del SDCDPS.
Figura 6. Schema di un fotomoltiplicatore di elettroni a diodi
separati.
Figura 7. Spettrofotometro IR a dispersione.

- 170 -
Figura 8. Spettrofotometro IR in Trasformata di Fourier.
Figura 9. Rappresentazione schematica del sistema ATR.
Figura 10. Spettro di massa MALDI-TOF del pre-polimero
PES-OH (4K)
Figura 11. Spettro di massa MALDI-TOF di S-PES-0.9(12h)
(a) e sezione ampliata dello stesso spettro di massa
(b)e dello spettro del S-PES-1.2(12h) (c).
Figura 12. Spettro MALDI-TOF del S-PES-1.0(12h) (a). Il
riquadro a destra riporta la sezione ampliata degli
spettri S-PES-1.0(12h) (b) ed S-PES-1.05(12h) (c).
Figura 13. Spettro
1
H-NMR del PES-OH (4k), S-PES-1.2(12
h) (b), S-PES-1.05(12 h)(c), S-PES-1.0(12 h) (d);
S-PES-0.9(12 h) (e).
Figura 14. Spettro
1
H NMR del S-PES-0.9-12h (a), 36h (b),
and 60 h (c).

- 171 -
Figura 15. Confronto tra il differenziale della massa molare
(MWD) di PES-OH(4k) (a), S-PES-0.9(12h) (b),
S-PES-O.9(36h) (c) e S-PES-0.9(60h) (d).
Figura 16. Confronto delle curve di MHS in campioni di PES-
OH(4K) (a), S-PES-0.9(12h), S-PES-0.9(36h) (c) e
S-PES-0.9(60h) (d).
Figura 17. Spettro FT-IR del pre-polimero PES- OH(4K) (a),
di S-PES-1.2(12h) e S-PES-0.9(12h).
Figura 18. Curve TGA del S-PES-1.2(12h) (a), S-PES-
1.05(12h) (b), S-PES-1.00(12h) (c) ed S-PES-
0.9(12h) (d).
Figura 19. Curve di termodegradazione in ambiente inerte
(N2) del PES-OH (a), SPES-1.2 (b), S_PES-1.05
(c), S-PES-1.0 (d) ed S-PES-0.9 (e).
Figura 20. Curve di termodegradazione in ambiente ossidativo
(aria) del PES-OH (a), SPES-1.2 (b), S_PES-1.05
(c), S-PES-1.0 (d) e S-PES-0.9 (e).

- 172 -
Figura 21. Temperatura di inizio decomposizione (Ti) in
funzione del grado di solfonazione sotto flusso di
azoto e in atmosfera statica di aria.
Figura 22. Energia di attivazione apparente del processo di
degradazione (Ea) in funzione del grado di
solfonazione sotto flusso di azoto e in aria .
Figura 23. Temperatura di transizione vetrosa in funzione del
grado di solfonazione
Figura 24. Curve DMA del S-PES-0.9(12h), S-PES-0.9(36h)
ed S-PES-0.9(60h).
Figura 25. Curve DMA del S-PES-1.2(12h), S-PES-1.2(36h)
ed S-PES-1.2(60h).

- 173 -
ELENCO TABELLE
Tabella 1. Proprietà termiche di alcuni polimeri aromatici
Tabella 2. Assegnamenti strutturali dei picchi presenti negli
spettri MALDI-TOF degli S-PES
Tabella 3. Riepilogo dei risultati della caratterizzazione
effettuata sui campioni di PES-OH ed S-PES-X
Tabella 4. Assegnamenti dei segnali dovuti ai protoni
aromatici appartenenti alle sequenze ed ai gruppi
terminali presenti nei copolimeri S-PES.
Tabella 5. Dati riepilogativi della caratterizzazione mediante
analisi SEC-DV del prepolimero PES-OH e dei
campioni di S-PES sintetizzati.
Tabella 6. Temperature alla a cui corrisponde la massima
velocità di perdita in peso (Tm) per il primo stadio
di degradazione del PES-OH e degli S-PES in
atmosfera inerte per azoto e a varie velocità di
riscaldamento.

- 174 -
Tabella 7. Temperature alla a cui corrisponde la massima
velocità di perdita in peso (Tm) pe il primo stadio
di degradazione del PES-OH e degli S-PES in
atmosfera di aria e a varie velocità di
riscaldamento.
Tabella 8. Coefficiente di regressione ed energia di attivazione
apparente del processo di degradazione (Ea) in
azoto, calcolata mediante l’equazione di Kissinger
per i campioni di PES sintetizzati.
Tabella 9. Coefficiente di regressione ed energia di attivazione
apparente del processo di degradazione (Ea) in aria,
calcolata mediante l’equazione di Kissinger per i
campioni di PES sintetizzati.
Tabella 10. Temperature di transizione vetrosa (Tg) e di inzio
decomposizione, energia di attivazione apparente
del processo di degradazione (Ea) in azoto per i
PES sintetizzati.

- 175 -
ELENCO SCHEMI
Schema 1. Sintesi del PES-OH telechelico.
Schema 2. La reazione di solfonazione del DCDPS.
Schema 3. Reazione di sintesi del PES solfonato.
Schema 4. Primo meccanismo di degradazione delle catene
polimeriche PES nel range di temperatura che va da
420°C a 450°C.
Schema 5. Secondo meccanismo di degradazione delle catene
polimeriche PES nel range di temperatura che va da
525°C a 580°C.
Schema 6. Terzo meccanismo di degradazione delle catene
polimeriche PES nel range di temperatura che va da
630°C-680°C.

- 176 -
7. RINGRAZIAMENTI
Al termine di questo percorso di studi post laurea sono tante le persone
che meritano i miei ringraziamenti e la mia riconoscenza, per il traguardo
raggiunto.
Per il sostegno umano e morale e per avermi dato sempre la forza e i
mezzi di andare avanti e raggiungere questo importante traguardo
ringrazio la mia famiglia, mio padre Ernesto, mia madre Antonia e mio
fratello Marco.
Ringrazio il mio relatore Prof. Gianluca Cicala per aver messo a mio
servizio le sue conoscenze mostrando grande disponibilità ed umanità
durante tutto il mio percorso didattico.
Un ringraziamento particolare al Dott. Filippo Samperi, ricercatore del
CNR di Catania per esser stato per me in tutti questi anni un punto di
riferimento come uomo ed una fonte di conoscenza ed esperienza che mi
ha aiutato a crescere professionalmente.
Infine vorrei rivolgere un ringraziamento generale a tutte quelle persone
(colleghi, collaboratori, amici, parenti) che direttamente o indirettamente
mi sono state vicine e mi hanno aiutato a raggiungere questo importante
obiettivo.

- 177 -
8. BIBLIOGRAFIA
1) Yee, A. F.; Du, J.; Thouless, D. In Polymer Blends: Performance. Paul, D.
R.; Bucknall, C. B., Eds.; Wiley: Toronto, 2000; Vol. 2, Chapter 26, pp
225–269.
2) Scamporrino, E.; Mineo, P.; Scamporrino, A.; Dattilo, S.;Vitalini, D.;
Alicata, R. J Polym Sci Part A: Polym Chem 2009, 47, 5682–5689.
3) Sugama, T.; Carciello, N. R. Int J Adhes 1993, 13, 257–266.
4) Pinnau, I.; Koros, W. J. J Appl Polym Sci 1991, 43, 1491–1502.
5) Hickner, M. A.; Ghassemi, H.; Kim, Y. S.; Einsla, B. R.; McGrath, J. E.
Chem Rev 2004, 104, 4587–4611.
6) Higashihara, T.; Matsumoto, K.; Ueda, M. Polymer 2009, 50, 5341–5357.
7) Iojoiu, C.; Sanchez, J-Y. High Perform Polym 2009, 21, 673–692.
8) Rikukawa, M.; Sanui, K. Prog Polym Sci 2000, 25, 1463–1502.
9) Yang, Y.; Holdcroft, S. Fuel Cells 2005, 5, 171–186.
10) Genova-Dimitrova, P.; Baradie, B.; Foscallo, D.; Poisignon, C.; Sanchez,
J. Y. J Membr Sci 2001, 185, 59–71.
11) Iojoiu, C.; Genova-Dimitrova, P.; Marechal, M.; Sanchez, J. Y.
Elettrochim Acta 2006, 51, 4789–4801.
12) Blaco, J. F.; Nguyen, Q. T.; Schatzel, P. J Appl Polym Sci 2002, 84,
2461–2473.

- 178 -
13) Keres, J. A.; Cui, W.; Reichle, S. J Polym Sci Part A: Polym Chem 1996,
43, 2421–2438. Vallejo, E.; Pourcelly, G.; Gavach, C.; Mercier, R.;
Pineri, M.J Membr Sci 1999, 160, 127–137.
14) Lee, H. S.; Badama, A. S.; Roy, A; McGrath, J. E. J Polym Sci Part A:
Polym Chem 2007, 45, 4879–4890.
15) Yu, X.; Roy, A.; Dunn, S.; Yang, J.; McGrath, J. E. Macromol Symp
2006, 245, 439–449.
16) Ghassemi, H.; McGrath, J. E.; Zawodzinski, T. A. Polymer 2006, 47,
4132–4139.
17) Lee, H. S.; Roy, A.; Lane, O.; Dunn, S.; McGrath, J. E. Polymer 2008,
49, 715–723.
18) Kim, Y. S.; Dong, L.; Hickner, M. A.; Pivovar, B. S.; McGrath,
J.E.Polymer 2003, 44, 5729–5736.
19) Wang, Z.; Ni, H.; Zhao, C.; Li, X.; Zhang, G.; Shao, K.; Na, H. J Membr
Sci 2006, 285, 239–248.
20) Weisse, H.; Keul, H.; Hocker, H. U.S. Patent 6,451,921 B2, 2002.
21) Wang, F.; Hickner, M.; Ji, Q.; Harrison,W.;Mecham, J.; Zawodzinski, T.
A.; McGrath, J. E. Macromol Symp 2001, 175, 387–395.
22) Ueda, M.; Toyota, H.; Ochi, T.; Sugiyama, J.; Yonetake, K.; Masuko,
T.; Teramoto, T., J. Polym. Sci., Polym. Chem. Ed.,1993, 31,85.

- 179 -
23) Li, Y.; Wang, F.; Yang, J.; Liu, D.; Roy, A.; Case, S.; Lesko, J.;
McGrath, J. E. Polymer 2006, 47, 4210–4217.
24) Lee, M.; Park, J. K.; Lee, H. S.; Lane, O.; Moore, R. B.; McGrath, J. E.;
Baird, D. G. Polymer 2009, 50, 6129–6138.
25) Wang, F.; Hickner, M.; Kim, Y. S.; Zawodzinski, T. A.; McGrath, J. E. J
Membr Sci 2002, 197, 231–242.
27) Sugama T, Carciello NR. Int J Adhesion & Adhesives 1993;13:257
28) Pinnau I, Koros JW. Journal of Appl. Polymer Science 1991;43:1491
29) McGrail PT. Polym Int 1996;41:103
30) Puglisi, C.; Samperi, F.; Cicala, G.; Recca, A.; Restuccia, C. L. Polymer
2006, 47, 1861–1874.
31) Guan, R.; Zou, H.; Lu, D.; Gong, C.; Liu, Y. Eur Polym J 2005, 41,
1554–1560.
32) Muthu Lakshmi, R. T. S.; Vyas, M. K.; Brar A. S.; Varma, I. K. Eur
Polym Mater 2006, 42, 1423–1432.
33) Pang, J.; Zhang, H.; Li, X.; Jiang, Z. Macromolecules 2007, 40, 9435–
9442.
34) Matsumoto, K.; Higashihara, T.; Ueda, M. Macromolecules 2009, 42,
1161–1166.
35) Yoshimura, K.; Iwasaki, K. Macromolecules 2009, 42, 9302–9306.

- 180 -
36) Montaudo, G.; Montaudo, M. S.; Samperi, F. In Mass Spectrometry of
Polymers; Montaudo, G.; Lattimer, R. P., Eds.; CRC Press: Boca Raton,
2002; Chapters 2, 10.
37) Puglisi, C.; Samperi, F.; Di Giorgi, S.; Montaudo, G. Polym Degrad Stab
2002, 78, 369–378.
38) Montaudo, G.; Montaudo, M. S.; Samperi, F. Rev Prog Polym Sci 2006,
31, 277–357.
39) Montaudo, G.; Montaudo, M. S.; Puglisi, C.; Samperi, F.Rapid Commun
Mass Spectrom 1995, 9, 453–460.
40) Pasch, H.; Schrepp, W. In MALDI-TOF Mass Spectrometry of Synthetic
Polymers; Springer-Verlag: Berlin, 2003; 298
41) Sankir, M.; Bhanu, V. A.; Harrison, W. L.; Ghassemi, H.;Wiles, K. B.;
Glass, T. E.; Brink, A. E.; Brink, M. H.; McGrath,J. R. J Appl Polym
Sci 2006, 100, 4595–4602.
42) Maes, C.; Devaux, J.; Legras, R.; Parsons, W.; McGrail, P. T.J Polym Sci
Part A: Polym Chem 1994, 32, 3171–3182.
43) Mendichi, R.; Giacometti Schieroni, A. In Current Trends in Polymer
Science; Pandalai, S.G., Ed.; TWR Network: Trivandrum India, 2001;
Vol. 6, pp 17–32.
44) User’s manual TA 3000 system. Greifensee: Mettler Instrument, AG;
1984.

- 181 -
45) Della Gatta, G.; Richardson, MJ.; Sarge, SM.; Stølen, S.; Standards,
calibration, and guidelines in microcalorimetry. Part 2. Calibration
standards for differential scanning calorimetry (IUPAC Technical
Report). Pure Appl. Chem 2006, 78 (7), 1455-76.
46) Csaba, N.; Handbook of thermal analysis and calorimetry. In: Gallagher
Patrick K, Brown Michael E, editors. Principles and practice, Vol. 1.
Elsevier; 1998.
47) Wang, F.; Glass, T.; Li, X.; Hickner, M.; Kim, Y. S.; McGrath J. E.
Polym Prepr 2002, 43, 492–493.
48) Kim, I.C.; Yoon, H.G.; Lee, K.; J Appl Polym Sci 2002, 84, 1300–1307.
49) Kissinger, HE.; Reaction kinetics in differential thermal analysis. Anal.
Chem. 1957, 29(11), 1702-6.
50) Samperi, F.; Puglisi, C.; Ferreri, T.; Messina, R.; Cicala, G.; Recca, A.;
Restuccia, C. L.; Scamporrino, A. Polym Degrad Stab 2007, 95, 1304–
1315.
51) Abate, L.; Blanco, I.; Cicala, G.; Mamo, A.; Recca, G.; Scamporrino, A.;
The influence of chain rigidity on the thermal properties of some novel
random copolyethersulfones. Polym Degrad Stab 2010, 95(5), 798-802.
52) Samperi, F.; Battiato, S.; Puglis, C.; Asarisi, V.; Recca, A.; Cicala, G.;
Mendichi, R. ; Synthesis and Characterization of Sulfonated
Copolyethersulfones. J. Polym Sci. Part A: Polym Chem. 2010, 48, 3010-
3023.