3 遷移金属化合物 - fujimori groupwyvern.phys.s.u-tokyo.ac.jp/f/research/arch/lf.pdf3...
TRANSCRIPT

3 遷移金属化合物
3.1 クラスター・モデルと配置間相互作用
3.1.1 配位子場理論の適用限界
金属-絶縁体転移、高温超伝導、巨大磁気抵抗、スピン揺らぎなど、遷移金属化合物の多彩な物性の主役は遷移元素の d電子である。空間的に広がった s原子軌道、p原子軌道が幅の広い価電子帯、伝導帯を形成するのに対して、d軌道(特に第一遷移元素系列の 3d軌道)は固体中でも比較的よく原子の周りに局在している。そのため d軌道は、原子間の軌道の小さな重なりにより狭いバンド(dバンド)を形成するか、孤立イオンとほぼ同様に遷移金属イオンに局在する。これらの d軌道あるいは dバンドが部分的に電子で占められている場合に、d電子間の相互作用の効果が強くなり、その結果上記のような様々な物性が出現するのである。狭い d
バンド内を運動する電子は互いに避けあいながら結晶中を運動しており、その物性を完全に理解するには ∼1023個の電子のとてつもなく複雑な多体問題を解かなければならない。しかし、d電子が各遷移金属イオンに局在して絶縁体となっている場合には、各イオンに局在した数個の d電子を扱う多体問題となる。これとてまだ複雑ではあるが、かなり正確に取り扱う方法がある。それが 1954年に田辺・菅野により提唱された配位子場理論(強い結晶場の理論)である。以来、配位子場理論は遷移金属化合物絶縁体の光スペクトルや磁性の解析に適用され、輝かしい成功を収めて来た[1]。特に、光吸収スペクトルの基礎吸収端より低エネルギー側に(近赤外から近紫外領域にかけて)見られる鋭いピーク構造を、結晶場中に置かれた遷移金属イオン内の d → d遷移として、配位子場理論は見事に説明し、解析手法として確立されてきた。その配位子場理論が、1970年代以降いくつかの困難に直面した。その
一つが、半導体中の磁性不純物の光スペクトルである。カルコパイライト型と呼ばれる構造を持つ I-II-VI2族半導体(II-VI族半導体の II族元素が1:1の組成比で I族元素と III族元素に置き換わったもの)CuAlS2に鉄イオン(Al3+を置換し、d5電子配置を持つFe3+イオンとなっていると考えられている)をドープした物質の光スペクトルに、配位子場理論では説明できないくらい低いエネルギーの強い吸収、放出が観測されたのである。神原らはこれを説明するために、d5電子配置だけでなく、配位子 S原子の 3p軌道から鉄原子に電子が移動した d6L 電子配置(Lは配位子軌道に空いたホール)まで考慮してハミルトニアン行列を対角化した [2]。これが、配位子場の軌道もあらわに取り入れた “クラスター・モデル”を配置間相互作用(configuration interaction: CI)理論で取り扱い光スペクトルを解析した初めての例である。遷移金属イオンに局在した d5電子配置
1

ばかりでなく、電荷が 1個移動した状態 d6Lも考えた物理的背景は、配位子 S 原子の電気陰性度が低く(配位子 p軌道のエネルギーが高く)、配位子から遷移金属イオンに電子が移動しやすいことであった。それまで配位子場理論により光スペクトルが解析されてきた物質は、ほとんどが酸化物やハロゲン化物などのイオン結晶であり、配位子の電気陰性度は十分高く(配位子 p軌道のエネルギーが十分低く)、電荷移動をあらわに考慮する必要がなかったのである。配位子の p軌道の影響は、p-d混成を通じてd軌道が 10Dq(普通 1 eV程度)だけ結晶場分裂すること、p-d混成のために d電子間のクーロン積分・交換積分である “ラカー・パラメータ”が減少する(自由イオンに比べて 70 %程度に)ことを考えれば十分であったのである。
図 1: NiOの光電子スペクトル [3](a)、及びその配位子場理論(b)、CI理論(c)による解析。[5]
配位子場理論が直面したもう一つの困難が、NiO、Niハライドなどの光電子スペクトル [図 1(a)] の解析である。配位子場理論によれば、Ni2+
イオンに局在した d8電子状態から光電子が放出されると、終状態は d7電子配置になる(d8 + hν → d7 + e)。したがって、光電子スペクトルの形状は、d7電子配置の多重項構造を反映する。図 1(b)に示すように、d7多重項は酸素の pバンドの低結合エネルギー側に現れ、フェルミ準位のすぐ下に位置する。6個の酸素で正八面体状に囲まれたNi2+イオンの基底状態は、電子配置 t32g↑t
32g↓e
22g↑ を持ち、全スピンは S = 1である(3A2g 状
態)。ここから電子を1個光電子として放出すると、4T1g, 2T1g, 2Egの3つ終状態からなる多重項構造が出現することが配位子場理論から導かれる。しかし、配位子場理論で解析すると、ラカー・パラメータは自由イオンの半分程度の値になってしまうなどの問題点が出てきた。問題点が一層
2

明らかになったのが、1982∼3年に発表された、放射光を用いた NiO [3]や、Niハライドの共鳴光電子分光実験である。Ni 3d電子の放出強度を増大して観測しようと励起光エネルギーをNi 3p内殻吸収領域に合わせたところ、驚いたことに、フェルミ準位直下の d7多重項構造と思われていた(“主構造”)はむしろ弱くなり、酸素 pバンドよりさらに深く位置する “サテライト”の強度が共鳴的に増大したのである。ここで共鳴光電子分光とは、3p内殻吸収 3p63d8 + hν → 3p53d9とそれに続いて起こる内殻ホールのオージェ型崩壊 3p53d9 → 5p63d7 + eの量子力学的干渉効果により、3d電子の光電子放出断面積が共鳴的に増大する現象である。サテライトの方が共鳴増大を示したということは、主構造ではなくサテライト構造の方が d8 → d7 + e遷移によっていることを示している。この奇妙な共鳴光電子スペクトルの振る舞いをDavisは、1個の d軌道
と1個の p軌道からなる簡単化されたクラスター・モデルを用いて、CI理論的に説明した [4]。簡単のため、基底状態に1個のホールがあるとし、基底状態の全系の波動関数を
Ψg = α0|d〉+ β0|L〉 (1)
のように、2つの電子配置の線型結合で表わす。ここで、dは 3d軌道のホールである。これに対応して、光電子放出の終状態を
Ψf = αf |d2〉+ βf |dL〉+ γf |L2〉 (2)
と、やはり複数の電子配置の線型結合で表わす。|d〉, |d2〉はイオン的な電子配置で、配位子場理論で考える dn, dn−1電子配置にあたる。|L〉, |dL〉は配位子場理論では取り入れられていない電荷移動状態 dn+1L, dnLにあたる。つまり、d軌道と p軌道の混成は、電荷移動状態が混成するということで表現されている。d軌道のエネルギーを εd, p軌道のエネルギーをεpで表わすと、始状態の各電子配置のエネルギーは、それぞれ
E(d) = −εd, E(L) = −εp (3)
となる。ここで d軌道が p軌道より上にある εd > εpとすると、E(d) <
E(L)であるから基底状態の主要な電子配置は dとなる。終状態の各電子配置のエネルギーは、
E(d2) = −2εd + U,E(dL) = −εd − εp, E(L2) = −2εp (4)
で与えられる。ここで、U は d電子間の原子内クーロン・エネルギーであり、p電子間のクーロン・エネルギーは小さいとして省略する。終状態でのp軌道から d軌道への電荷移動 (d2 → dL)に要するエネルギー εd−εp−U
が正か負かは、∆ ≡ εd − εpと U の大小関係によって決まる。∆ > U で
3

あれば、光電子スペクトルにおける主構造は d(dn−1)終状態により、共鳴光電子分光で主構造の一方の強度が増大する。この場合、主構造のスペクトル形状は配位子場理論でそのまま解析できる。一方、∆ < U であれば、NiOのように主構造は L(dnL)終状態によるものとなり、共鳴光電子分光で強度が増大するのは d(dn−1)終状態によるサテライトとなる。この場合、配位子場理論はそのままでは適用できなくなる。藤森-南-菅野は、Davisの考え方を現実的なNiO6クラスター・モデルに拡張し、Ni 3d軌道の縮重と t2g、eg軌道への結晶場分裂、酸素 p軌道の対称性、d7、d8
電子配置の多重項構造などを取り入れて定量的な光電子スペクトルの解析を行った [5]。この解析では、電荷移動エネルギーを∆ =4.0 eV、原子内クーロン・エネルギーを U =7.5 eVに選ぶと、図 1(c)に示すように、主構造、サテライトともにスペクトル形状が再現された。∆ < U であるから、主構造は主に d8L電子配置からなる。配位子場理論で d7終状態多重項として解析できていたのはある程度偶然だったとも言える。しかしここで終状態の対称性は配位子場理論ですでに正しく与えられていたことは留意する必要がある。従来の解析では d8L電子配置部分空間の有効ハミルトニアンとして、d7電子配置の配意子場理論を用いていたと考えてもよい。
3.1.2 配置間相互作用クラスター・モデル
上で述べたように、配位子場理論の限界は、配位子の p軌道をあらわに取り扱わずに、結晶場パラメータ 10Dqを通じて d軌道に押し付けたことから来ていた。そこで、遷移金属イオンMp+ とそれを取り囲むm個の配位子Xq−からなるクラスターMX
(mq−p)−m の電子状態を考えること
にする。NiOの場合、Mp+ =Ni2+、Xq− =O2−なので、MX(mq−p)−m =
NiO10−6 を考えた。クラスターの全電荷を各イオンの形式電荷の和とする
ことによって、バンドギャップまで電子が詰まった絶縁体の電子状態を表現することができる。クラスターの全電荷がゼロでないのは、無限大の結晶から都合のよい有限のクラスターを切り出したためであり、クラスターはあくまで中性の状態を表わしていることに注意されたい。従って、このクラスターを “中性クラスター”あるいは “N -電子状態” と呼ぶことにする。光電子放出により電子が1個減ったクラスター(MX
(mq−p−1)−m )を
“正にイオン化した”クラスターあるいは “N − 1-電子状態”と呼び、逆光電子放出により電子が1個増えたクラスター(MX
(mq−p+1)−m )を “負に
イオン化したクラスター”あるいは “N + 1-電子状態”と呼ぶことにする。図 2に、中性および正・負にイオン化したクラスターの全エネルギー準位図を示す。図では中性クラスターはイオン的な dn電子配置が最もエネルギーが低く、配位子から遷移金属イオンに電子が1個移った dn+1L電
4

図 2: 中性(N -電子状態)および正・負にイオン化した(N − 1, N + 1-電子状態)クラスターのエネルギー準位図。Eh + Eeはバンドギャップの大きさに等しい。各電子配置は、多重項分裂、混成によるシフトを示している。
子配置がそれより電荷移動エネルギー∆(≡ E(dn → dn+1L))と呼ばれるエネルギーだけ高いエネルギーに位置している。それぞれの電子配置は、d電子間の交換相互作用と異方的クーロン相互作用によりエネルギー準位が分裂し、それぞれ固有の多重項構造を示す。多重項構造を無視すると、裸の d電子、p電子のエネルギーをそれぞれ εd、εp、2個の d電子間のクーロン・交換相互作用の平均値を U とすれば、dn電子配置、dn+1L
電子配置のエネルギーはそれぞれ
E(dn) = E0 + nεd +n(n− 1)
2U
E(dn+1L) = E0 + (n + 1)εd − εp +n(n + 1)
2U (5)
で与えられる。従って、電荷移動エネルギーは∆ = εd − εp + nU で表わされる。(5)式の E0は定数で、ここでも p電子間のクーロンエネルギーは無視できるとしている。p-d軌道混成により、dn電子配置と dn+1L電子配置は混成し、図 2に示したようにシフトする。p-d混成の結果、基底状態は純粋な dn電子配置ではなくなり、式 (1)のように dn+1Lの混成した状態になる。正にイオン化したクラスター(N − 1-電子状態)は電子が1個少ないか
ら、dn−1、dnL等の電子配置から構成される。
E(dn−1) = E0 + (n− 1)εd +(n− 1)(n− 2)
2U (6)
5

などから、電荷移動 dn−1 → dnLに要するエネルギーが∆− U であることがわかる。従って、∆ > U ならば dn−1の方が dnL よりもエネルギーが低く、光電子スペクトルの主構造を与える遷移は dn → dn−1 + eである。そして、光電子スペクトルの主構造は配位子場理論で解析できる。一方、∆ < U ならば dnLの方が dn−1よりもエネルギーが低くなり、主構造は dn → dnL + eになる。上記のNiOがこれにあたる。この場合、光電子スペクトルの解析は配位子場理論では不十分で、クラスター・モデルをCI理論で取り扱う必要がある。図 2は後者(∆ < U)の場合について描かれている。負にイオン化したクラスター(N + 1-電子状態)は、電子が1個多く、電荷移動 dn+1 → dn+2L に要するエネルギーは∆ + U と正の大きい量になる。従って、電子配置間の混成は弱くなり、逆光電子スペクトルでは、最も低エネルギーに現われる dn + e → dn+1の構造だけ考えればよいであろう。不純物準位や欠陥準位の電気伝導への寄与が無視できるときは、電気伝導度の活性化エネルギーから求まるバンドギャップの大きさは、十分に離れた位置に電子とホールを 1個ずつつくるのに要する最小のエネルギーで与えられる。従って、独立した2個のクラスターにそれぞれ電子とホールを作るのに要する最小エネルギーの和(図 2の Ehと Eeの和)がバンドギャップの大きさとなる。中性クラスターと正、負にイオン化したクラスター(すなわちN -電子、N − 1-電子、N + 1-電子状態)のそれぞれの基底状態エネルギーをEN,0、EN−1,0、EN+1,0とすると、バンドギャップの大きさは
Egap = (EN−1,0−EN,0)+(EN−1,0−EN,0) = EN−1,0 +EN+1,0−2EN−1,0
(7)で与えられる。従って、バンドギャップの大きさは、多重項分裂と p-d混成を無視すると、式 (5)、(6)より、∆ > U の場合Egap = U、∆ < U の場合Egap = ∆となることが簡単に示される。前者(∆ > U)の場合、バンドギャップ間の電子励起は、dn +dn → dn−1 +dn+1 のように遷移金属イオン間の電子移動で起こる。このような電子構造は、Mottが提唱しHubbardがモデル化したもの (ハバード・モデル)と同じで、モット・ハバード型と呼ばれる。後者(∆ < U)の電子構造をもつ場合、バンドギャップ間の電子励起は、dn + dn → dnL + dn+1のように非金属イオンからから遷移金属イオンへの電荷移動により起こり、電荷移動型と呼ばれる。図 2のエネルギー準位図にある各電子配置は、p-d混成によりシフトし、さらに多重項分裂による広がりを示しているが、dn電子配置の基底状態の、重心 E(dn)からの多重項分裂によるエネルギーの下がり ∆En(> 0)を多重項補正と呼ぶことにする [6]。多重項補正は、フント則に従ってス
6

図 3: 多重項補正。(a)Ueff , ∆eff の定義、(b)多重項補正によるモット・ハバード型、電荷移動型のバンドギャップへの補正(それぞれ Ueff − U ,∆eff −∆)[6]
ピン最大限に揃ったときに電子系が得する交換エネルギーが大部分である。多重項補正により、モット・ハバード型の場合はバンドギャップが2∆En −∆En−1 −∆En+1(≡ Ueff − U)だけ変化し、電荷移動型の場合は∆En −∆En+1(≡ ∆eff −∆) だけ補正を受ける(図 3(a))。これらの補正は正(安定化)、負(不安定化)両方の値を取りうる。ここで Ueff , ∆eff
は多重項補正を受けたクーロンエネルギーと電荷移動エネルギーである。n = 0, 1と n = 9, 10は多重項分裂がないから、∆En = 0である。n = 5でスピンが最大になり、∆Enは最大値をとる。従って、モット・ハバード型に対するバンドギャップの多重項補正は n = 5で鋭い正のピークをとり、他は小さな負の値をとる(図 3(b))。電荷移動型に対するバンドギャップの多重項補正は n ≥ 5で正、n ≤ 4で正の値をとる(同じく図 3(b))。図 2に示したN − 1-電子状態は、N -電子状態から電子が1個抜けた状
態で、N +1-電子状態はN -電子状態に電子が1個付加された状態である。従って、N -電子状態の基底状態をエネルギーの原点(フェルミ準位)として、N + 1-電子状態のエネルギーを上向きに、N − 1-電子状態のエネルギー準位を下向きにプロットすると、図 4のように通常のバンド構造の図に似たものが得られる。図ではさらに、実際の結晶での pバンド、dバンドの有限のエネルギー幅(それぞれWp,W)を考慮して、クラスター・モデルで得られる離散準位に幅をつけて描いている。モット・ハバード型では、上下に ∼ U だけ分裂した上部ハバード・バンドと下部ハバード・
7

図 4: N − 1, N + 1-電子状態から構築された “1電子”準位図。モット・ハバード型 (a)および電荷移動型 (b)。
バンドがバンドギャップが形成していること、電荷移動型絶縁体では、上部ハバード・バンドと pバンドの間に ∼ ∆程度のギャップが形成されていることがわかる。電荷移動型の場合、下部ハバード・バンドは深い位置にあり、光電子スペクトルのサテライトを与えている。
3.2 電子物性を支配する因子
3.2.1 電子構造パラメータとバンドギャップ
図 4からもわかるように、クラスター・モデルで考慮されていなかった有限の酸素 pバンド幅、遷移金属 dバンド幅は、バンドギャップを減少させる方向に働く。dバンドの幅をW、pバンドの幅をWpとすると、モット・ハバード型絶縁体(∆ > U)のバンドギャップは ∼ U −W となり、電荷移動型絶縁体(∆ < U)のバンドギャップは∼ ∆− 1
2(W + Wp)となる。従って、モット・ハバード型絶縁体はU ∼ W で、電荷移動型絶縁体は∆ ∼ 1
2(W + Wp)でバンドギャップが閉じ、金属に転移する。これをもとに、∆-U 平面で描いた “相図”(図 5)を Zaanen-Sawatzky-Allen (ZSA)相図と呼ぶ [7]。“相図”の縦軸 U、横軸∆は、p軌道-d軌道間の混成相互作用を表わす p-d移動積分 Tpdで規格化してある。定義により直線∆ = U
の上側が電荷移動型、下側がモット・ハバード型となる。ただし、電荷移動型とモット・ハバード型の境界は相境界のようにはっきりしたものでは
8
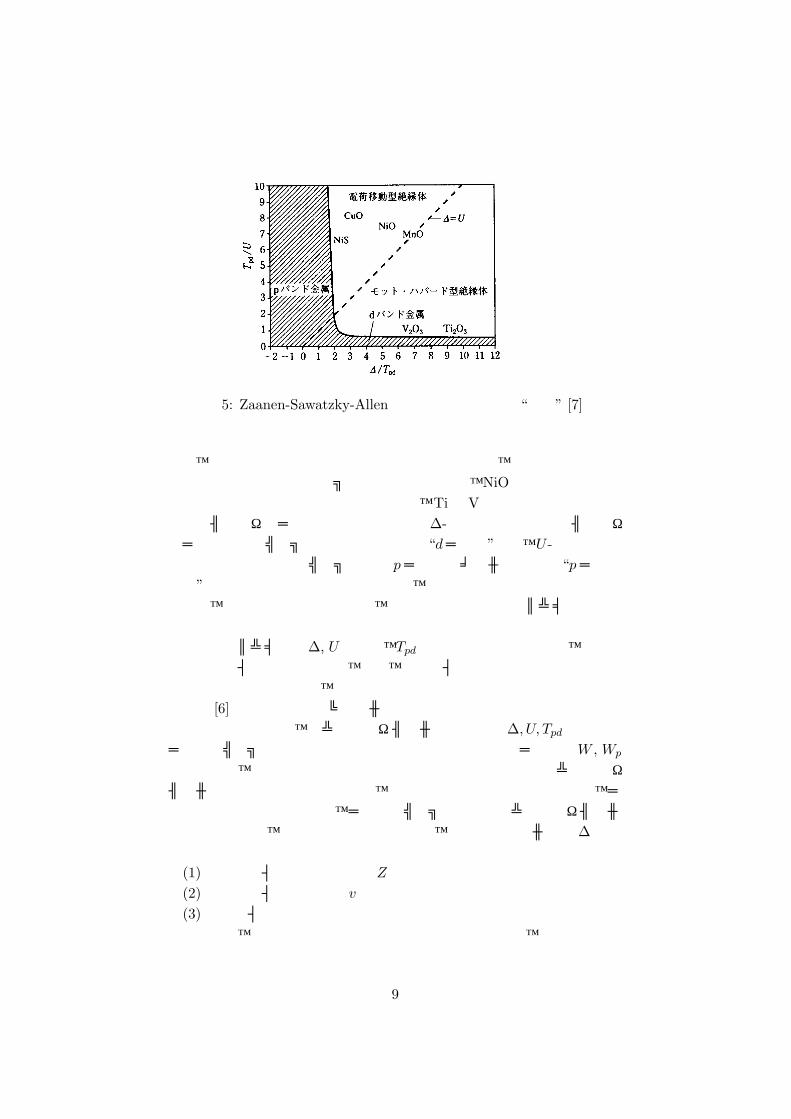
図 5: Zaanen-Sawatzky-Allenによって提唱された “相図” [7]
なく、徐々に性格が変わっていくものである。図には、酸化物を中心にいくつかの遷移金属化合物がプロットされているが、NiOや銅酸化物など重い遷移元素の酸化物は電荷移動型に属し、TiやVなど軽い遷移元素の酸化物はモット・ハバード型に属している。∆-軸付近の金属状態はモット・ハバード型のギャップが閉じた(通常の)“dバンド”金属、U -軸付近の金属状態は電荷移動型ギャップが閉じ pバンドにホールの入った “pバンド金属”と呼ぶことができる。この相図は、その後の研究によりやや変更されたが、それについて述べる前に、実際の物質の電子構造パラメータについて述べる。電子構造パラメータ∆, Uおよび、Tpdは物質によって異なるが、それら
は遷移金属イオンの原子番号、価数、非金属イオンの電気陰性度によって規則的に変化することが、光電子分光法を用いた研究により明らかになっている [6]。原子の実測スペクトルあるいは原子の計算から求まっている多重項補正を加えて、クラスター・モデルの範囲内で∆, U, Tpdを用いてバンドギャップを始めとする種々の物性量を予測できる。バンド幅W , Wp
の見積りは、周期的な結晶格子を考えなければならないのでクラスター・モデルの範囲では不可能であるが、類似した結晶構造の物質同士では、バンド幅は近いであろうから、バンドギャップの変化はクラスター・モデルに基づいて解釈、予測が可能である。まず、電荷移動エネルギー∆の物質依存性は次の規則に従う:
(1) 遷移金属イオンの原子番号 Z とともに減少する。(2) 遷移金属イオンの価数 vとともに大きく減少する。(3) 非金属イオンの電気陰性度と共に増加する。これらは、いずれも化学的な直感に合致したものであり、詳しく説明す
9

る必要はないであろう。次に、クーロン・エネルギー U の物質依存性は、(1) 遷移金属イオンの原子番号 Z とともにゆるやかに増加する。(2) 遷移金属イオンの価数 vとともにわずかに増加する。(3) 非金属イオンの分極率とともに減少する。(1), (2)の原因は、原子核やイオンの正の電荷の増加により、d軌道の
大きさが縮小することによる。上記の規則を半定量的にまとめると、酸化物に対して、
∆ ' 26− 0.6Z − 2.5v, U ' −2.5 + 0.3Z + 0.5v (8)
(単位 eV)となる。Tpdは 1∼2 eVの値をとるが、遷移金属イオンの原子番号 Zとともに減少する。これも、d軌道の広がりが縮小し、p軌道との重なりが減少することで説明される。例えば、NiOの Tpdは LaTiO3の約半分である。酸化物からカルコゲナイドに移ると、p軌道のエネルギーの上昇により∆が減少し、非金属イオンの分極率の増加により U が減少する。従って、硫化物の∆は酸化物より約 2.5 eV小さく、セレン化物、テルル化物と行くにしたがってさらに約 0.5 eV, 1.0 eV減少する。
図 6: 遷移金属酸化物のバンドギャップ。(a) クラスター・モデルによる計算値 [8]。(b)光学測定によるLaMO3のバンドギャップの実験値 [9]。“CT”は電荷移動型ギャップ、“Mott”はモット・ハバード型ギャップ。
上記の規則を用い、多重項補正も入れて、いろいろな遷移金属化合物のバンドギャップをクラスター・モデルで計算した結果を図 6(a)に示す [8]。計算値は n = 5で極大値、n = 4で極小値をとっているが、これは式 (8)のようなパラメータのスムーズな変化では説明できず、3.1.2で述べた多
10

重項補正を受けたパラメータ∆eff , Ueff で初めて説明される(図 3参照)。現実の物質は p-d混成のため、どの物質もモット・ハバード型の性格と電荷移動型の性格を兼ね合わせているために、バンドギャップに対する多重項補正の効果は両者の特徴を反映している。光学測定により実験的に求めたペロブスカイト型酸化物 LaMO3のバンドギャップ(図 6(b))の遷移元素(M)依存性は [9]、このようにしてクラスター・モデル計算でかなりよく説明される。図 5のZSA相図は、その後の研究でいくつかの変更が加えられた。その
一つは、Ti, Vなど軽い遷移金属の化合物の多くが必ずしも典型的なモット・ハバード型(U < ∆)でなく、境界領域(U ∼ ∆)に属することがわかってきたことである。図 5の縦軸 U、横軸∆を Tpdでなく、“実質的な p-d混成の強さを与える量” Teff ≡
√10− nTpd(nは d電子数)で規格
化する方が p− d混成の強度を表すのに合理的である。Teff にかかる係数√10− nは、dnから dn+1Lへの電荷移動のチャンネル、すなわち、空の
d軌道の数が 10− n個あることによる。すると、周期律表の左側のTi, Vの化合物は Teff が大きくなり、ZSA相図の原点付近に集中する。大きなTeff のために、バンドギャップの大きさは電荷移動型の∆− 1
2(W +Wp)やモット・ハバード型の U −W ではなく、混成の大きな影響を受けて、ほぼ 2Teff に比例するようになる。∆と U の正確な値や微妙な大小関係は、バンドギャップの性格や大きさにとって重要ではなくなる。従って、初めに典型的なモット・ハバード型と考えられていた Ti, Vの化合物は、“中間型”あるいは “強混成型”と呼んだほうが適切であることがわかってきたのである。
図 7: 変更された Zaanen-Sawatzky-Allen相図 [10]。ここでは横軸∆、縦軸 U は Tpdで規格化していない。
11

ZSA相図のもう一つ重要な変更点は、図 7に示したように電荷移動型絶縁体の領域が上に述べた∆ ∼ 1
2(W +Wp)までではなく、∆ ∼ 0さらには∆ < 0まで広がっていることである [10]。∆が非常に小さくなってもバンドギャップが残るのは、エネルギーの近接した dnと dn+1Lが非常に強く混成し、中性クラスターの基底状態のエネルギーの下がりが大きくなる(従って図 2で Ee + Ehが大きくなる)ことによる。中性クラスターのエネルギー準位図(図 2中央)を見ると、連続準位である dn+1L電子配置が∆ < 0では基底状態になるので、系は金属になってしまうように思える。ところが、p-d混成が十分強いと、dn+1L連続準位の中から束縛状態が分離して、離散的な基底状態が出現するのである。このようにして系は絶縁体になる。∆ < 0の絶縁体の例として、Cuの原子価が異常に高い(Cu3+: d8)NaCuO2があり、その基底状態は主に d9Lからなっている。また、三角格子スピン系として知られる LiNiO2(Ni3+: d7。NiOのNi (111)面が 1枚おきに Liに置き換わった結晶構造を持つ)は、ほとんどゼロに近い ∆を持ち、同様なメカニズムで有限のバンドギャップを維持している。ここで、“∆ < 0型”絶縁体を考える際に、遷移金属イオンは一つのままで、p軌道をクラスターの分子軌道ではなく結晶全体に広がった pバンドとして考えた。このようなモデルはアンダーソン不純物モデルと呼ばれ、クラスター・モデルを現実に一歩近づけたものと考えられる。以下 3.2.4に述べる半導体中の遷移金属イオン不純物の電子構造にも、負の電荷移動エネルギーの概念とアンダーソン不純物モデルが用いられる。
3.2.2 磁性
これまでは、フント則に従って d電子のスピンが揃い、全スピンの大きさ S ができる限りの最大値をとる状態、いわゆる高スピン状態が各電子配置の基底状態であるとしていた。しかし配位子場理論によれば、結晶場10Dqが大きい時には、d電子はスピンを揃えるよりも、低いエネルギーの結晶場準位に入りたがるようになり、全スピンの大きさが小い低スピン状態が実現する。クラスター・モデルでも、以下に示すように、p-d混成が強くなると低スピン状態が実現しやすくなる。正八面体クラスターの場合、低スピン状態が可能なのは、配位子場理論と同様の d4, d5, d6, d7電子配置である。CuO4などの平面クラスターでは d3、d8電子配置でも低スピン状態の実現が可能である。図 2の中性クラスターの dn電子配置は、混成がなければ必ず高スピン状態が基底状態となるのに対して混成があると、より多くの eg 軌道が空の低スピン状態の方が p-d混成の影響を強く受け大きく下方にシフトし [Tpd(eg)/Tpd(t2g) ∼ 2なので、空の eg 軌道が
12

多い方が dn+1Lと強く混成する]、高スピン状態を追い越す可能性がある。そうなると低スピンの基底状態が実現する。低スピンの酸化物としては、LaCoO3 (Co3+: d6, S = 0)、NaCuO2 (S = 0)、LiNiO3 (S = 1/2) などがある。4d遷移金属の化合物では、d軌道が広がっているために原子内交換エネルギーが小さく p-d混成 Tpdも大きいため、低スピン状態が実現されやすい。例えば、SrRuO3(Ru4+: d4)は強磁性金属であるが、キューリー点より上で常磁性帯磁率は、Ru4+イオンが S = 1であることを示している。スピン S を持つ遷移金属イオンの磁気モーメントの大きさは、結晶場により軌道各運動量が消失していれば、2SµB(µB:電子のボーア磁子)である。p-d混成が存在すると、d電子のスピン密度の一部は配位子の p
軌道に移動する。しかし、反強磁性体では、隣の遷移金属イオンから移動してくる逆向きスピン密度が p軌道上で打ち消し合い、遷移金属イオン上にのみスピン密度が残る。残ったスピンの大きさは、クラスターの基底状態の波動関数をΨg = α|dn〉+ β|dn+1L〉とすると、(2|α|2 + |β|2)µBS で与えられる(n ≥ 5で高スピンの場合)。中性子回折の実験によれば、反強磁性体NiOのNiイオンの磁気モーメントは 1.8µBであるが、この値は光電子スペクトルの解析から求まった αで説明される。一方、ネール点やキューリー点より高温の常磁性帯磁率を与える有効磁子は 2
√S(S + 1)µB
で与えられ、p-d混成による減少は見られない。遷移金属イオンに残ったスピン密度と周りの配位子に移ったスピン密度が平行なままに、熱的にいろいろな方向に揺らいでいるためである。
図 8: Ni-O-Niクラスターにおける超交換相互作用の模式図。矢印はホールのスピンを表わす。ホールの入る軌道とスピンのうち、Ni 3dx2−y2は省略。 (a): スピンが平行な場合、 (b): 反平行な場合。
13

図 9: 組成MOを持つ酸化物の反強磁性ネール温度の計算値と実験値 [11]。
次に、多くの遷移金属化合物に見られる反強磁性の起源である超交換相互作用について述べる [11]。再び NiOを例にとり、直線状の3つのイオンからなるNi-O-Ni クラスター(Ni2O2+クラスター)、を考える。Niイオンのスピンが平行な場合と反平行な場合のエネルギーを比較し、スピン間の相互作用を論じる。Ni2+イオンは eg↓軌道(dx2−y2、d3z2−r2 , z軸をNi-O結合方向にとる)に 2個のホールを持つが、図 8に示すように、そのうちの d3z2−r2 軌道 (2個の Niイオンに対応して d1, d2と表わす)と酸素の pz 軌道(Lと表わす)だけが混成し、それらの間だけにホールが移動できる。対称性から、dx2−y2軌道は中央の酸素原子に混成できる p軌道を持たないので、dx2−y2 ホールは動かない。2個のNiのスピンが平行な場合(図 8(a))、基底状態の波動関数の主成分 |d1↑d2↑〉 に、Niのホールが酸素に移動した |d1↑L↑〉と |L↑d2↑〉 (いずれも相対エネルギー = ∆eff)が少し混成する摂動によりエネルギーが
∆E(↑, ↑) = −2t2
∆ eff(< 0) (9)
だけ変化する。ここで、tは d3z2−r2-pz間の移動積分で、NiO6クラスターの移動積分Tpd(eg)との間に t ≡ Tpd(eg)/
√3の関係がある。2個のNiのス
ピンが反平行な場合(図 8(b))、主成分 |d1↑d2↓〉 に混成するのは、|d1↑L↓〉と |L↑d2↓〉の他に、|d1↑d1↓〉、|d2↑d2↓〉(相対エネルギー= Ueff)と |L↑L↓〉(相対エネルギー= 2∆eff)もあり、エネルギーの変化は
∆E(↑, ↓) = − 2t2
∆eff(1 +
t2
∆2eff
+t2
Ueff∆eff)(< 0) (10)
14

となる。従って、スピンが反平行な場合の方がエネルギーが下がる。従って、ハイセンベルグ・モデルのハミルトニアンH = JS1 · S2に現れる交換相互作用定数 J は
J =t4
S2∆eff(
1∆2
eff
+1
Ueff∆eff) (11)
となり、この J を用いてネール温度が TN = 2JZS(S + 1)/3kB(kB: ボルツマン定数、Z: 金属イオンのまわりの最近接金属イオン数、NiOの場合Z = 6)で与えられる。NaCl型酸化物MO系列のネール温度の実験値と計算値を図 9に示す [11]。
図 10: RMnO3 のスピン・軌道整列。(a) LaMnO3 で実際に観測されるヤーン・テラー歪みのもとでのスピン・軌道整列。Mnイオンはほぼ単純立方格子を作っている。(b) ヤーン・テラー歪みのない場合。
NiOのNi2+イオンは eg↑軌道が2つとも占有されている。これに対して、Cu2+イオン(d9)、低スピン状態(S = 1
2)のNi3+イオン(t32g↑t32g↓e
1g↑)、
高スピン状態(S = 2)の Mn3+ イオン(t32g↑e1g↑)は、eg 軌道 dx2−y2、
d3z2−r2 のうちのどちらに電子が入るかという “軌道の自由度”がある。このような軌道の自由度を持つイオンの間の磁気的相互作用は、どの軌道に電子が入るかに依存しており、軌道整列とスピン整列の絡んだ複雑な磁性を示す。例として、負の巨大磁気抵抗を示す物質として、最近話題となっている R1−xSrxMnO3、R1−xCaxMnO3 (R3+ は希土類等の 3価のイオン)の母物質 RMnO3(Mn3+: d4)から切り出した直線状Mn-O-Mnクラスター(Mn2O4+クラスター)を考える。簡単のためにモット・ハバード型の電子構造を考えるが、定性的な結果は電荷移動型の場合も同様である。2つのMnの d3z2−r2軌道間の移動積分を t、dx2−y2軌道間の移動積分を t′ (|t′| << |t|) とすると、2個のMnイオンの eg電子がともに d3z2−r2
15

軌道を占める場合、NiOの場合と同様の機構でMnイオン間に反強磁性的相互作用が生じる。eg 電子がともに dx2−y2 軌道を占める場合は、移動積分 t′が非常に小さく反強磁性相互作用も弱いが、それぞれのMnイオンのt32g↑ 電子配置間の弱い反強磁性相互作用も加算される。eg 電子がMn(1)で d3z2−r2 軌道、Mn(2)で dx2−y2 軌道を占める場合、Mn(1)の d3z2−r2 電子がMn(2)の d3z2−r2 軌道に飛び移る摂動がある。Mn(1)とMn(2)のスピンが平行か、反平行かによって、中間状態として |d2,3z2−r2↑d2,x2−y2↑〉と |d2,3z2−r2↓d2,x2−y2↑〉が考えられるが、|d2,3z2−r2↑d2,x2−y2↑〉の方がフント則によりエネルギーが低い。従って、Mn(1)とMn(2)のスピンの間には強磁性的な相互作用が働くことになる。以上をまとめると、軌道が同じ隣接Mnイオン間には反強磁性的な相互作用が、軌道が異なる隣接Mnイオン間には強磁性的な相互作用が働く。図 10(a)に、この規則に従ったスピンと軌道の整列の様子を示す [12]。このような強磁性は BiMnO3 で実現されている可能性がある。軌道整列が起こるときは、特定の eg 軌道に電子またはホールが入るの
で、配位子イオンの位置がシフトして金属イオンの周りの対称性を下げるヤーン・テラー効果が起こることが多い [13]。LaMnO3で実際に観測されるヤーン・テラー歪は、MnO6八面体が交互に x方向, y方向に伸び、それぞれ d3x2−r2 軌道と d3y2−r2 軌道が電子で占められる。この構造では ab
面内で強磁性的に、c方向で反強磁性的にスピンが揃う(図 10(b))。このような軌道整列は、ヤーン・テラー歪があって初めてエネルギー的に安定し実現するものである。
3.2.3 光スペクトル
結晶イオン中の遷移金属イオンの光吸収スペクトル、特にイオン内のd → d遷移の解析は、配位子場理論が最も威力を発揮する場面である。一方、クラスター・モデルあるいはアンダーソン不純物モデルの CIによる取扱いでも、光スペクトルの解析を行うことができる。CI理論は配位子場理論に比べて計算にかなり労力を要するが、次の利点を持つ。
(1)配位子場理論では、物質ごとに実験に合わせるようにパラメータ(結晶場分裂 10Dq, ラカー・パラメータB, C)を決めており、それらの物理的意味づけが必ずしも明確でないことがあるが、クラスター・モデルでは、その起源や大きさの明確な 電子構造パラメータ(∆, U , Tpd)から、光スペクトルを導ける。これは、配位子場理論の基礎付けや、パラメータ10Dq、B、C の正当化にもつながる。
(2) 電荷移動エネルギー∆が小さく(あるいは負で)p-d混成が非常に強いために、配位子場理論を適用しにくい場合(例えば半導体中の遷移金
16

属イオンの光スペクトル)も、クラスター・モデルで解析が可能である。(3) pバンドから d軌道への光吸収(電荷移動吸収)を d → d光吸収と同じ枠内で取り扱え、電荷移動吸収端のエネルギーを予測できる。
図 11: NiO中のNi2+イオンのN -電子状態エネルギー準位図。図 3 の中性クラスターのエネルギー準位に当たる。p軌道はバンドを作っているとして、d9Lが連続準位として描かれている。
まずは、配位子場理論が成功している NiOの d → d光吸収をクラスター・モデルで調べて見る。Ni2+イオンの基底状態は S = 1の t62ge
2g電子
配置(3A2g状態)を持つが、スピンを変えずに電子を1個 t2g軌道から eg
軌道に励起した t52ge3g電子配置(
3T2g状態)への遷移が最もエネルギーが低く、hν =1.2 eVの吸収ピークに対応する。この遷移では、基底状態と終状態でスピン状態が同じために、エネルギー差はクーロン・交換積分(ラカー・パラメータB, C)を含まず、結晶場分裂 10Dqに等しくなる。同じ遷移をクラスター・モデルで見ると、図 11に示すように、Ni2+(d8)自由イオンの S = 1基底状態 3F が d9L 電子配置と混成し、3A2g、3T1g、3T2g
に分裂する。この分裂は、Lが正八面体クラスターの対称性を持っていることによっており、パラメータ 10Dqで表わされる結晶場分裂と定性的に同じである。3A2gでは2つの eg軌道が空なので、d9Lとの混成でエネルギーが∼ 2Tpd(eg)2/∆だけ下がるのに対し、3T2gは eg軌道が1つ空、t2g軌道が 1つ空なので、混成でエネルギーが∼ [Tpd(eg)2 +Tpd(t2g)2]/∆だけ下がる。Tpd(eg) ∼ 2Tpd(t2g)であるから、3T2g は 3A2g より∼ 3
4 [Tpd(eg)2]/∆だけエネルギーが高い。これが 10Dqに等しいと考えられる。(より正確に言うと、これに p-d軌道間の非直交性による egと t2gのエネルギー差約0.3 eVを加えたものが 10Dqに等しい。)カルコゲナイドは酸化物に比べて、∆がおよそ 2∼3 eV小さい。従っ
17

図 12: ZnS中に中性不純物として Znを置換した遷移金属イオン(N -電子状態)エネルギー準位図。光吸収の実験結果と比較している [15]。
て、ZnS、ZnSe、CdTe等の II-VI族化合物半導体の Zn2+、Cd2+位置を置換した 2価の遷移金属イオン(中性不純物)の光スペクトルは、強いp-d混成のために配位子場理論による解析が難しくなる。これらの物質の中には、発光材料、磁気光学材料などもあり、光スペクトルの解明は実用的にも重要な意味を持つ。強い p-d混成による困難を克服する一つの方法が、e軌道と t2軌道を区別したクーロン・交換積分を用いる解析である1。渡辺-上村は 10個の独立なクーロン・交換積分及び結晶場分裂 10Dqを第一原理計算により求めて、ZnS中の遷移金属イオンの光スペクトルの解析を行った [14]。クラスター・モデルを用いると、自由イオンと同じ dn電子配置と、結晶の空間対称性を反映した dn+1Lとの混成の結果、e軌道とt2軌道を区別したクーロン・交換積分を用いたのと同様な効果が取り入れられる [15]。図 12に示すように、クラスター・モデルで計算した中性クラスター(N -電子状態)の励起エネルギーと光吸収スペクトルとの一致は良い。
3.2.4 不純物準位
化合物半導体中の遷移金属イオンは、中性不純物でもすでに電荷移動エネルギー∆が小さく、配位子場理論が困難に直面したが、正にイオン化し
1化合物半導体中の遷移金属イオン位置は、配位子に正四面体状に4配位されていて空間反転対称性がないので、e, t2 のように “反転対称”を表す添え字 g が除かれている。
18

た不純物では、電荷移動 dn−1 → dnLに要するエネルギー∆− U は負になり、配位子場理論では取り扱いが難しい状況になる(図 2のN −1-電子状態エネルギー準位図参照)。すなわち、不純物は 3.2.1で述べたNaCuO2
中の Cu3+イオンのような、負の電荷移動エネルギー∆を持つ遷移金属不純物イオンと類似の電子状態にあり、連続準位 dnLから p-d混成により分離した束縛状態にある。この基底状態に束縛エネルギー以上のエネルギーを与えると、系は dnL連続準位に励起される。したがって、ホールの束縛準位が、母体の価電子体頂上より束縛エネルギーだけ上のバンドギャップ内にあるように見える。
図 13: ZnS中の中性遷移金属不純物に対するホール、電子の束縛準位。実験値とも比較している [15]。
束縛エネルギーの見積りには、価電子帯頂上のエネルギーや波動関数を正確に知る必要があるので、クラスター・モデルではなくアンダーソン不純物モデルを用いる必要がある。中性イオンの光吸収スペクトルを計算した時(図 12)と同じパラメータを用いて計算した ZnS中のホールの束縛準位、電子の束縛準位を図 13に示す。ホールの束縛準位から伝導帯の底までのエネルギーが、中性不純物を正にイオン化するのに要するエネルギー(ドナーのイオン化エネルギー)であり、価電子帯の頂上から電子の束縛準位までのエネルギーが、中性不純物を負にイオン化するのに要するエネルギー(アクセプタのイオン化エネルギー)である。これらのドナー準位、アクセプタ準位は、遷移元素の原子番号の関数として単調ではなく、一見不規則な変化をしているように見える。これは3.1.2で述べた多重項補正のためである。例えば、多重項効果により安定化している d5イオンを正にイオン化するには大きなエネルギーを要する
19

ので、Mn2+(d5)イオンのドナー準位が深くなっている。d4イオンに電子を付加し安定した d5イオンをつくるとエネルギーを得するのでCr2+(d4)イオンのアクセプタ準位が深くなっている。
3.3 金属-絶縁体転移
3.3.1 バンド幅制御
バンドギャップの大きさが物質によってどう系統的に変化するかを 3.2.1で見た。そこで小さなバンドギャップを持つとされた物質は、金属と絶縁体の境界近くにあると考えられる。実際、クラスター・モデル計算でギャップがほとんどゼロと予測されたAFeO3 (A: 2価のアルカリ土類イオン)(図 6)は、SrFeO3が金属で、CaFeO3が温度によって金属-絶縁体転移を起こす物質である。金属と絶縁体の境界付近にある物質は、圧力、化学組成などの条件を微妙に変えただけで、金属-絶縁体転移を起こす可能性が高い。本節では、化学組成の変化とは、Caと Srを置き換えるような、遷移金属イオンの価数 (dバンドのフィリング)を変えないで格子定数や結合角のみを変えるものを指す。このような原子置換は、圧力と似た効果を与えるので “化学的圧力” とも呼ばれる。圧力、化学組成の変化は原子軌道間の重なりを変化させ、バンド幅W を変化させるので、このようにして引き起こされる金属-絶縁体転移をバンド幅制御型金属-絶縁体転移と呼び、このような物質系をバンド幅制御系と呼ぶ。例にあげた AFeO3はペロブスカイト型構造 (図 14)をもつが、A サイトに Ca2+のようなイオン半径の小さいイオンが入ると、立方晶のペロブスカイト型構造が図 14に示したように正方晶に歪み、Fe-O-Fe結合角が 180度から減少して酸素 p
軌道を介した Fe 3d軌道間の重なりが減少する。バンド幅制御によって金属-絶縁体転移を起こす例としては、V原子を Crや Tiに置換したV2O3,NiS2−xSex, NiS1−xSexなどが古くから知られている。また、AFeO3と同じペロブスカイト型結晶構造を持ち、同じメカニズムでバンド幅W が制御される系として、モット・ハバード型Ca1−xSrxVO3、Y1−xLaxTiO3が挙げられる。この2つの系は、組成(x)を変えてもそれぞれ金属側、絶縁体側のみしかカバーせず、金属-絶縁体転移は見られないが、バンド幅W とクーロン相互作用 U の比 U/W による物性の系統的な変化を研究するのに絶好の物質系である。まず、NiSおよびその置換体 NiS1−xSexに於ける金属-非金属転移について述べる [16]。(ここで “非金属相”と呼んだのは、絶縁体か半金属かが明らかでないからである。)NiOは約 4 eVのバンドギャップを持つ絶縁体であったが、NiOの酸素 2pに比べて NiSの硫黄 3p軌道はエネルギーが約 2.5 eV程高いので、ギャップの大きさを与える d8 → d9L 電荷移動
20

図 14: ペロブスカイト型遷移金属酸化物AMO3の結晶構造 (a)とその正方晶歪み (b)
エネルギー∆が NiSでは小さくなる。さらに、六方晶 NiAs型結晶構造を持つNiSでは、短いNi-Ni原子間距離のために dバンド幅W が(そしておそらく pバンド幅Wpも)広がっているため、バンドギャップがゼロ付近まで減少しているものと考えられる。0.14 eV程度のバンドギャップ(擬ギャップ?)を持つ NiSの反強磁性非金属相の伝導帯の底は、クラスター・モデル計算によれば d9状態であり、t32g↑t
32g↓e
2g↑基底状態に eg↓電子
を付け加えた状態(2Eg 状態)である。価電子帯頂上は d8L状態であるが、Niの eg↑軌道と同じ対称性を持つ S 3p分子軌道から電子を取り去った状態(これも 2Eg状態)である。従って、NiSが非金属から金属に1次相転移するのは、eg↓ 伝導帯の底と、eg↑ 価電子帯の頂上が重なった時である。この非金属→金属転移は、(1) 温度を 260 K以上に上げる、(2) Sを Seに置換する、(3) 高圧をかける、のいずれかによって引き起こすことができる。(1)では、高温でエントロピーの高い金属相を安定化させている。(2)では、S→Seの置換で∆を減少させ、同時に p-d混成の増大でdバンドの幅も広げギャップを閉じている。 (3) では、原子軌道間の重なりを増大させてバンド幅を広げている。図 15 は、温度と Se量を変えた場合のNiS1−xSex系の相図を示す。(3)のみが厳密な意味での “バンド幅制御”であるが、図 15の相図は横軸を圧力にとった相図とぴったりと重なる。非金属から金属へ転移する圧力は、絶対零度で 20 kbarである。金属-絶縁体転移近傍の物質の電子状態は、金属相でも、クラスター・モデルや配位子場理論のような局在電子モデルで表現される電子状態がある程度意味を持っている。例えば、NiSの非金属相の光電子スペクトルは、NiOと同様 d7終状態からなるサテライトと d8L終状態からなる主構
21

図 15: NiS1−xSexの相図 [16]。
造を示し、NiS6クラスター・モデルでよく説明できるが、金属相になり反強磁性が消失しても、スペクトル形状はフェルミ準位の極く近傍を除いてほとんど変化しない [17]。これは、多重項構造の原因となっている原子内 d-dクーロン・交換相互作用、サテライト構造の原因となっている p-d混成が、金属-非金属転移に際してほとんど変化しないためと考えられる。また、バンドギャップが閉じたばかりの金属の基底状態は非常に電子相関が強く、電荷の揺らぎが抑えられていて、普通の金属には程遠い電子状態にあると考えられる。すなわち、NiS6クラスターに注目すると、金属相の基底状態は、非金属体相の基底状態 α|d8〉+ β|d9L〉+ ...(N -電子 3A2g
状態)に d9(N + 1-電子 1Eg状態), d8L(N − 1-電子 2Eg状態)がわずかに混った電子状態であると考えられる。電荷移動型なので U が大きく、電荷の揺らぎが有効に抑えられている。ギャップが閉じても、依然としてdバンドが上下に U だけハバード分裂しているので(図 4参照)、金属になっても絶縁体と比べて大きな変化がスペクトルに起こらないのである。
NiS2とその混晶系NiS2−xSexの相図は、図 16に示すように反強磁性金属相、常磁性絶縁体相も示し、NiS1−xSexの相図に比べてかなり複雑である [18]。ここでもNiS1−xSexと同様、横軸を圧力としても、全く同じ相図が描ける。NiS2の結晶構造はパイライト型と呼ばれ、Ni2+イオンと S2−
2
分子イオンがNaCl型格子を作っている。NiS2 は、バンドギャップ 0.3 eVを持つ反強磁性絶縁体である。Sを Seで置換していくと x ' 0.5で反強磁性金属に1次転移し、x ' 1.0で常磁性金属に2次転移する。NiS1−xSexが反強磁性非金属から常磁性金属に一気に1次転移するのと対照的である。反強磁性絶縁体-反強磁性金属転移では、金属側でキャリアー数が転移に向かって減少し絶縁体になる [19]。反強磁性秩序は転移で変わらずに、Ni
22

図 16: NiS2−xSexの相図 [18]。
サイトの磁気モーメントが Se置換により徐々に減少しているだけである。絶縁体相の伝導帯と価電子帯がわずかに重なった他は、バンド構造に変化がないのであろう。反強磁性金属-常磁性金属転移付近では、転移に向かってスピン揺らぎが激しくなり、伝導電子の質量が重くなる。NiS2−xSexの光電子スペクトルは NiS1−xSexとよく似ている。Niの価数、Sイオンの配位数が同じなので、クラスター・モデル計算の立場からは、類似のスペクトルが得られるのは自然である。金属-絶縁体転移を起こすモット・ハバード型の物質として数 10年来研究されてきたのが V2O3である。その相図は NiS2−xSex と似て、反強磁性絶縁体、常磁性絶縁体金属、常磁性絶縁体、常磁性絶縁体金属とバラエティーに富んでいる。Vを Tiに置換すると圧力をかけるのと同じ相図となり、Vを Crに置換すると負の圧力をかけたことになる。モット・ハバード型絶縁体のギャップが閉じるのはU ∼ W であるから、電荷移動型と異なり、金属側での U は小さい。従って金属相では、多重項構造やサテライト構造の明瞭でない、金属らしい光電子スペクトルが得られる可能性が高い。d1電子配置を持つ遷移金属酸化物について、U/W
比を少しずつ変えることによって実現した金属-絶縁体転移の近傍の光電子スペクトルを図 17に示す。U/W が臨界値(∼1)を下回り金属に転移すると、フェルミ準位上に有限の状態密度が現われ、U/W の減少とともにその強度が増す。一方、絶縁相で存在した、ハバード・バンドが金属相でも生き残り、その強度は U/W の減少とともに連続的に減少する。
23

図 17: d1電子配置を持つ遷移金属酸化物のフェルミ準位近傍の光電子スペクトル [20]。実線はバンド計算。
3.3.2 フィリング制御
モット・ハバード型あるいは電荷移動型絶縁体を金属にするには、圧力や化学的圧力によりギャップをつぶすバンド幅制御絶縁体→金属転移の他に、遷移金属イオンの平均価数を変化させ、伝導帯あるいは価電子帯に電子またはホールをキャリアーとして導入するフィリング制御型絶縁体→金属転移がある。dバンドを占める電子数 n(遷移金属イオンあたり)とU/W (電荷移動型については∆/W)を軸とする2次元平面上の金属-絶縁体相図を図 18に示す。n =整数の軸上では、臨界値 U/W ∼1で金属-絶縁体転移が起きるが、電子あるいはホールが余分なキャリアーとして導入されると nが整数軸を外れ、ついには金属となる。結晶が完全な周期性を保ち、キャリアーが不純物ポテンシャルに束縛されたり、格子歪みやスピン分極と強く相互作用するなどして束縛されなければ、整数軸からの無限小のずれで系は金属となるはずである。しかし、実際の物質では、キャリアーを導入するためには母体と価数の異なるイオンを導入しなければならない。例えば、ペロブスカイト型 RMO3の希土類イオン R3+を Sr2+
またはCa2+イオンに置換することによって、電荷中性の条件を満たすように、ホールがM イオンの dバンドに導入される。Sr2+、Ca2+イオンは母体の平均的な電荷分布から見て負の電荷を持つように見え、ホール濃度
24

の低い極限では、ホールは不純物に束縛され水素原子のようになる。不純物状態の軌道半径 aB は、水素原子のボーア半径との類推で、m∗をキャリアーの有効質量、ε0 を母体の誘電率として、aB = h2ε0/m∗e2 で与えられる。不純物濃度 x(キャリアー濃度に等しい)が大きくなり、不純物軌道間の重なりが大きくなると、系は金属に転移する。この絶縁体→金属転移の臨界濃度は、aBx1/3 = 0.25で与えられる。U/W ∼ 1 付近にある絶縁体ならば、バンドギャップは小さいから、ε0は大きく、m∗ は小さいので、aB は大きくなり xは小さくなる。すなわち、少量のキャリアードーピングにより系は金属に転移する。U/W À 1ならば、逆に aB が小さく、xが大きくなる。すなわち、金属になるには高いホール濃度を必要とする。フィリング制御系での絶縁相の安定性には、さらに軌道整列や電荷整列の安定性が重要な寄与する場合もある。電荷整列がおこるには、電荷整列の周期と格子の周期との整合性が重要であり、x = 0.5、x = 0.33など特定の組成でそれぞれ2倍周期、3倍周期などの絶縁相が安定化される。
図 18: n-U/W(または∆/W)平面上での金属-絶縁体相図。
図 18の概念的な n-U/W(n-∆/W)相図を、化学組成 AMO3 (A:希土類またはアルカリ土類イオン)を持つペロブスカイト型酸化物全体について具体的に描いたのが図 19である。ここで、縦軸はU/W (∆/W)そのものでなく Aイオンの価数及びサイズであり、これによって間接的にU/W(∆/W)が制御されている。Aイオンの価数が下がれば遷移金属イオンM の価数が上がり、電荷移動型では∆が小さくなり(3.2.1参照)、モット・ハバード型では d軌道と p軌道の混成が増すので実質的な U が
25

図 19: 化学組成AMO3を持つペロブスカイト型遷移金属酸化物の相図。
小さくなる。また、Aイオンの大きさが小さくなると、正方晶の歪み(図14)が増し、W が減少する。n = 5の周辺で絶縁体の領域が広くなっているのは、3.1.2で述べた多重項補正の寄与でバンド・ギャップが大きくなっているからである。図 19に示す物質のうち、代表的なフィリング制御型金属-絶縁体転移を示す物質として La1−xSrxTiO3が挙げられる [21]。LaTiO3(d1) は反強磁性絶縁体であるが、La3+をわずかに(x ∼ 0.03)Sr2+に置換すると常磁性金属になる。また、SrTiO3(d0)は dバンドが空の普通の絶縁体(半導体)であるが、Sr2+をわずかに La3+に置換すると金属(超伝導体)になる。従って、0 < x < 1の大部分の領域が金属である。金属相でのホール係数の測定よれば、キャリアーは常に電子で、キャリアー濃度は n(≡ 1−x)に等しい。x ∼ 0.03における反強磁性絶縁体-常磁性金属転移(いわゆるモット転移)に向かって電子比熱(比熱の γT 項)から求めた伝導電子の質量m∗が発散的に増大する。従って、電子数 nが 1に近づくと、質量が発散し絶縁体になるという描像が得られる。一方、高温超伝導体La2−xSrxCuO4で見られるフィリング制御型金属-絶縁体転移は、La1−xSrxTiO3とは全く異なった振る舞いを示す。La2CuO4
はやはり反強磁性絶縁体で、x ∼ 0.05で絶縁体から常磁性金属(超伝導体)に転移する。ところがホール係数からは、金属相でのキャリアーはホールで、キャリアー濃度は xに等しいことがわかっている。電子比熱γT はモット転移(x ∼ 0.05)に向かって減少する [22]。これはホール数が減少し絶縁体に転移するという描像で説明される。類似の物質に見える La1−xSrxTiO3と La2−xSrxCuO4 が、なぜこのような劇的な差を示すのか、その原因はまだよくわかっていない。
26

再び図 18に戻って、現在話題になっている負の巨大磁気抵抗を示すMn酸化物La1−xSrxMnO3の金属-絶縁体転移に注目する。LaMnO3が軌道の整列した反強磁性体であることは 3.2.2で述べたが、これに La3+ →Sr2+
置換でホールを導入すると x ∼ 0.1で強磁性金属へ転移し、x = 0.3付近で、キューリー温度は Tc=350Kに達する。x < 0.2では、キューリー温度より上で電気抵抗が半導体的な振る舞いをする(図 20)。この常磁性絶縁体-強磁性金属転移点の近傍の電気抵抗は、磁場をかけると急に減少し、いわゆる負の巨大磁気抵抗を示す。
図 20: La1−xSrxMnO3の電気抵抗 [23]。
3.3.3 その他の金属-絶縁体転移
フィリング制御系では、遷移金属イオンあたりの電子数が整数個に近づくと絶縁体になりやすい傾向が見られる(図 18 参照)。ところが、La2−xSrxNiO4、R1−xAxMnO3、La1−xSrxFeO3などでは、xが 1/2, 1/3などの分数に近づくいた時も、電気抵抗が上昇し絶縁体(半導体)的な温度依存性を示す現象が見られる。このとき、電子線や中性子線の回折実験から、2倍、3倍などの超周期構造が見られ、La2−xSrxNiO4 では Ni2+
と Ni3+が、R1−xAxMnO3ではMn3+とMn4+が周期的に配列する電荷整列が起こっていることがわかっている。前節で述べた、常磁性絶縁体高温相から強磁性金属低温相に転移する
La1−xSrxMnO3では、電荷整列は見られないが、LaをNdに置き換えたNd0.5Sr0.5MnO3では、強磁性金属相からさらに温度を下げると、Tco =160
27

図 21: Sm0.5Sr0.5MnO3における電荷、スピン、軌道の配列構造 [24]。
Kで電荷整列した反強磁性絶縁体に転移する [24]。この電荷整列相では、スピンと軌道が図 21のような、CE型反強磁性構造と呼ばれる状態に整列する。スピンと軌道の配列の規則は 3.2.2で見たが、ここではMn4+(t32g↑電子配置のため異方性がない)とMn3+(t32g↑e
1g↑電子配置のため eg軌道の
異方性を持つ)の間のスピンと軌道の結合の法則も必要である。Mn3+ から eg軌道が伸びた方向にあるMn4+には、eg電子が飛び移る摂動と飛び移り先のイオン内でのフント結合により、強磁性的な相互作用が働く。eg軌道が伸びた方向と直角方向にあるMn4+とは、この相互作用は弱く、t32gスピン間の超交換相互作用による反強磁性的な相互作用が勝つ。また、eg軌道が特定の方向に伸びることによって、Mn3+イオンの周りの酸素位置がシフトし、ヤーン・テラー歪を起こす。この歪みは 3.2.2で述べたLaMnO3
に於けるヤーン・テラー歪と同じものと考えられる。La0.5Sr0.5MnO3が電荷整列を起こさず、Aサイトの平均イオン半径の小さいNd0.5Sr0.5MnO3
は電荷整列を起こす。さらにイオン半径の小さい Pr0.5Ca0.5MnO3では転移温度Tcoがさらに上昇し、常磁性絶縁体高温相から(強磁性金属を経ないで)一気に電荷整列した反強磁性絶縁体相に転移する。La0.5Sr0.5MnO3 →Sm0.5Sr0.5MnO3 →Pr0.5Ca0.5MnO3の順番で電荷整列がおこりやすくなる原因として、Aサイトのイオン半径の減少により正方晶の歪みが増大しMn-O-Mn結合角が減少して、egバンド幅が減少するために強磁性金属相の安定性が減ることが考えられている。
La1−xSrxFeO3のx =2/3での電荷配列は、MnやNi酸化物から推測されるようなFe3+ : Fe4+ = 1 : 2の電荷整列でなく、Fe3+ : Fe5+ = 2 : 1の電荷整列であることが、メスバウアー効果、中性子回折の実験よりわかっている [25, 26]。つまり、本来存在すべきFe4+イオンが、2Fe4+ → Fe3++Fe5+
28

のように電荷不均化しているのである。これは、CaFeO3(Fe4+: d4)が低温で電荷不均化を起こし半導体になるのと同じ原因と考えられる。酸化物中の Fe4+イオンがこのような不均化に対して不安定なのは、図 6に示したように、バンドギャップがゼロに近いことが原因である。バンドギャップの大きさEgapは式 (7)で与えられるから、Egap < 0の場合はギャップが閉じて金属になる以外に、電荷不均化を起こすこともあり得るのである。ここで、“Fe4+”の形式的電子配置は d4であるが、実際の電子配置は d5L
が支配的であることに注意されたい [(3.3.1)]。さらに “Fe5+”も、d4ではなく d5L2に近い。つまり、Fe4+の電荷不均化は、d電子の分布はほとんど d5のままで、主に pバンドで起こっている現象であると言える。そのためか、形式価数の異なる Feイオンがほとんど同じイオン半径を持っているように見え、電荷不均化は格子変形を伴わない。以上のように、電荷整列による金属→絶縁体転移では、電子状態と格子変形との結合が重要で転移は格子変形に助けられているが、やはり転移の引き金は電子系のスピンと軌道の整列である。一方、電子-格子結合自体が引き金となる金属→絶縁体転移もある。磁気整列の見られないVO2
(V4+: d1)の金属-絶縁体転移(転移温度 Tt = 350 K)がその1例である[27]。VO2はルチル型という結晶構造を持ち、VO6クラスターがO-O辺を共有して一方向に繋がっている。t2g軌道のうちの隣接V原子方向に伸びた dxy 軌道に電子が入り、高温相では金属、低温相では2個の V原子が2量体を作った絶縁体となる。金属-絶縁体の問題は高温超伝導、巨大磁気抵抗、電子-格子相互作用など、実に多くの問題と関連した奥の深い問題である。ここで挙げた例はそのごく一部に過ぎないが、その一端を垣間見て頂けたならば幸いである。
参考文献
[1] 上村洸、菅野暁、田辺行人、配位子場理論とその応用(裳華房、1969年)
[2] T. Kambara, K. Suzuki and K. Gondaira, J. Phys. Soc. Jpn. 39,764 (1975)
[3] M. R. Thuler, R. L. Benbow, and Z. Hurych, Phys. Rev. B 27, 2082(1983).
[4] L. C. Davis, Phys. Rev. B 25, 1912 (1982).
[5] A. Fujimori, F. Minami, and S. Sugano, Phys. Rev. B 29, 5225(1984); A. Fujimori and F. Minami, Phys. Rev. B 30, 957 (1984).
29

[6] 津田惟雄、那須奎一郎、藤森淳、白鳥紀一:電気伝導性酸化物(裳華房、1993年)
[7] J. Zaanen, G. A. Sawatzky and J. W. Allen, Phys. Rev. Lett. 55,418 (1985).
[8] T. Saitoh, A. E. Bocquet, T. Mizokawa, and A. Fujimori, Phys.Rev. B 52, 7934 (1995).
[9] T. Arima, Y. Tokura, and J. B. Torrance, Phys. Rev. B 48, 17006(1993).
[10] T. Mizokawa, A. Fujimori, H. Namatame, K. Akeyama, and N.Kosugi, Phys. Rev. B 49, 7193 (1994).
[11] J. Zaanen and G. A. Sawatzky, Can. J. Phys. 62, 1262 (1987).
[12] T. Mizokawa and A. Fujimori, Phys. Rev. B 56, R686 (1997).
[13] K. I. Kugel and D. I. Khomskii, Sov. Phys.-JETP 37, 725 (1973).
[14] 渡辺聡:固体物理 24, 737 (1989).
[15] T. Mizokawa and A. Fujimori, Phys. Rev. B 48, 14150 (1993).
[16] M. Matoba, S. Anzai and A. Fujimori, J. Phys. Soc. Jpn. 60, 4230(1991).
[17] M. Nakamura, A. Sekiyama, H. Namatame, H. Kino, A. Fujimori,A. Misu, H. Ikoma, M. Matoba, and S. Anzai, Phys. Rev. Lett. 73,2891 (1994).
[18] G. Czyzek, J. Fink, H. Schmidt, G. Krill, M.F. Lapierre, P. Paissod,F. Gautier and C. Robert, J. Magn. Magn. Mater. 3, 58 (1976).
[19] S. Miyasaka, H. Takagi, Y. Sekine, H. Takahashi, N. Mori, and R.J. Cava, submitted to Phys. Rev. B (1997)
[20] A. Fujimori, I. Hase, H. Namatame, Y. Fujishima, Y. Tokura, H.Eisaki, S. Uchida, K. Takegahara, and F.M.F. de Groot, Phys. Rev.Lett. 69, 1796 (1992).
[21] Y. Tokura, Y. Taguchi, Y. Okada, Y. Fujishima, T. Arima, K. Ku-magai and Y. Iye, Phys. Rev. Lett. 70, 2126 (1993).
30

[22] N. Momono, M. Ido, T. Nakano, M. Oda, Y. Okajima and K. Ya-maya, Physica C 233, 395 (1994).
[23] A. Urushibara, Y. Moritomo, T. Arima, A. Asamitsu, G. Kido andY. Tokura, Phys. Rev. B 51, 11103 (1995).
[24] 富岡泰秀、吉沢英樹、三浦登、固体物理 31, 124 (1997).
[25] M. Takano, J. Kawachi, N. Nakanishi and Y. Tokura, J. Solid StateChem. 39, 75 (1981).
[26] P.D. Battle, T.C. Gibb and P. Lightfoot, J. Solid State Chem. 84,271 (1990).
[27] A. Zylbersztejn and N.F. Mott, Phys. Rev. B 11, 4383 (1975).
31