hypoxia-induced phosphorylation of chk2 in an ataxia ... filehypoxia-induced phosphorylation of chk2...
TRANSCRIPT

Hypoxia-Induced Phosphorylation of Chk2 in an Ataxia
Telangiectasia Mutated–Dependent Manner
Shannon L. Gibson,1,3Ranjit S. Bindra,
2,3and Peter M. Glazer
1,3
Departments of 1Genetics, 2Experimental Pathology, and 3Therapeutic Radiology, Yale University School of Medicine,New Haven, Connecticut
Abstract
Chk2 is a serine/threonine kinase that signals to cell cyclearrest, DNA repair, and apoptotic pathways following DNAdamage. It is activated by phosphorylation in response toionizing radiation, UV light, stalled replication forks, and othertypes of DNA damage. Hypoxia is a common feature of solidtumors and has been shown to affect the regulation of manygenes, including several DNA repair factors. We show here thatChk2 is phosphorylated on Thr68 and thereby activated in cellsin response to hypoxia, and that this phosphorylation isdependent on the damage response kinase ataxia telangiecta-sia mutated (ATM) but not on the related kinase ATM andRad3-related. Moreover, phosphorylation of Chk2 underhypoxia was attenuated in cells deficient in the repair factorsMLH1 or NBS1. Finally, Chk2 serves to protect cells fromapoptosis under hypoxic growth conditions. These resultsidentify hypoxia as a new stimulus for Chk2 activation in anATM-, MLH1-, and NBS1-dependent manner, and they suggest anovel pathway by which tumor hypoxia may influence cellsurvival and DNA repair. (Cancer Res 2005; 65(23): 10734-41)
Introduction
Chk2 is a tumor suppressor that is mutated in a variety of humantumor types (reviewed in ref. 1) and in a subset of Li-Fraumenipatients (2–4). Chk2 is a serine/threonine protein kinase that playsa key role in cell cycle arrest following DNA damage. After ionizingradiation or UV damage, the ataxia telangiectasia mutated (ATM)and the ATM and Rad3-related (ATR) protein kinases are activated,respectively. ATM and ATR both can phosphorylate Chk2 onThr68 (5, 6), which in turn leads to the phosphorylation of anumber of downstream proteins by Chk2, including p53 (on Ser20;refs. 7–9). On phosphorylation by Chk2, p53 is stabilized (10, 11),causing cell cycle arrest and regulating DNA repair.Other factors that participate in the activation of Chk2 under
various conditions include NBS1 (12, 13), 53BP1 (12, 14, 15), BRCA1(16), and MLH1 (17). In terms of ionizing radiation–inducedsignaling, NBS1 and 53BP1 have been placed in parallel pathwaysupstream of Chk2 (12). Both proteins affect the phosphorylation ofChk2 on Thr68. In addition, work by Brown et al. (17) suggests thatChk2 phosphorylation after ionizing radiation is MLH1 dependent.Whereas MLH1 is not suspected to be a kinase, the authors suggesta scaffolding role whereby it facilitates the interaction betweenATMandChk2. In addition tomodulation by these upstream factors,
Chk2 has been shown to autophosphorylate following exposure ofcells to ionizing radiation (14, 18–20).Solid tumors characteristically exhibit heterogeneity in vascu-
larization, perfusion, and oxygenation, with regions of moderate tosevere hypoxia. The pattern of hypoxia in tumors is dynamic withboth temporal and spatial variations. Clinical studies indicate thatextensive hypoxia in human tumors correlates with poor prognosis(21–24), reflecting not only the radioresistance of hypoxic cancercells but also a more aggressive phenotype.We and others have proposed that hypoxia may contribute to
tumor progression by promoting genetic instability. Increasedlevels of mutations have been detected in hypoxic cells (25–27) andhypoxia has been found to cause decreased expression of the DNArepair genes MLH1 , MSH2 , and Rad51 (28–30).Because MLH1 has been suggested to play a role in Chk2
activation, we initially hypothesized that the decrease in MLH1levels under hypoxia might cause an attenuation of Chk2 activationin response to ionizing radiation. Interestingly, however, we foundthat Chk2 phosphorylation is directly induced by hypoxia itself andthat this induction is substantially dependent on ATM. In addition,MLH1 and NBS1 were also found to play roles in Chk2 activation byhypoxia. Furthermore, a key downstream substrate of Chk2, p53,was found to be altered under hypoxic conditions in a Chk2-dependent manner, indicating that hypoxia-induced phosphoryla-tion of Chk2 leads to functional activation and downstreamsignaling. Finally, Chk2 seems to protect cells from hypoxia-inducedapoptosis and, thus, plays a role in cell survival under hypoxic stress.These results identify a new mechanism by which hypoxia caninfluence cell signaling and DNA damage response pathways.
Materials and Methods
Cell culture. HeLa, MCF-7, RKO, and HCT15 cells were maintained in
DMEM that was supplemented with 10% fetal bovine serum (FBS). HCT116/
2-3 and HCT116/3-6 cells (31) were grown in McCoy’s medium with 10%FBS and 0.8 mg/mL G418. HCT15 stable cell lines overexpressing wild-type
(WT) Chk2 were maintained in 0.8 mg/mL G418. LXIN (NBS1 mutant) and
NBS1 (LXIN corrected with NBS1 cDNA) cells were cultured in DMEM with
10% FBS and 0.4 mg/mL G418. NBS1 and LXIN cells were obtained fromDr. Patrick Concannon (Molecular Genetics Program, Benaroya Research
Institute at Virginia Mason, Seattle, WA) (ref. 32).
Plasmids and RNA interference. The HA-Chk2 expression vector was
obtained from Dr. David F. Stern (Department of Pathology, Yale University,New Haven, CT) (ref. 18). RNA interference (RNAi) target sequences for ATM
were ligated into the pSUPER vector (OligoEngine, Seattle, WA). The
oligonucleotide sequences are as follows: ATM-1A, GATCCCCGCG-
CCTGATTCGAGATCCTAAGTTCTCTAGGATCTCGAATCAGG-CGCTTTTTGGAA; ATM-1B, AGCTTTTCCAAAAAGCGCCTGATT-
CGAGATCCTAGAGAACTTAGGATCTCGAATCAGGCGCGGG; ATM-2A,
GATCCCCTGGTGCTATTTACGGAGCTAAGTTCTCTAGCTCCGTAAATAG-CACCATTTTTGGAAA; and ATM-2B, AGCTTTTCCAAAAATGGTGCTATTT-
ACGGAGCTAGAGAACTTAGCTCCGTAAATAGCACCAGGG. Small interfer-
ing RNA (siRNA)–expressing stable cell lines were made by cotransfecting
Requests for reprints: Peter M. Glazer, Department of Therapeutic Radiology, YaleUniversity School of Medicine, P.O. Box 208040, New Haven, CT 06520-0804. Phone:203-737-2788; Fax: 203-785-6309; E-mail: [email protected].
I2005 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-05-1160
Cancer Res 2005; 65: (23). December 1, 2005 10734 www.aacrjournals.org
Research Article
Cancer Research. on September 23, 2017. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

pSUPER constructs with pcDNA3 (Invitrogen, Carlsbad, CA) at a ratio of 10:1using FuGENE 6 reagent as directed by the manufacturer (Roche, Indian-
apolis, IN). Stable lines were selected and maintained in 0.8 mg/mL G418.
RNAi directed against ATR (ATR-2 duplex, Dharmacon, Lafayette, CO), ATM
(siGENOME SMARTpool reagent, Dharmacon), or luciferase (Luciferase GL-2duplex, Dharmacon) was done transiently using Oligofectamine (Invitrogen).
Hypoxia and desferrioxamine treatments. Hypoxic cell culture
conditions were established as previously described (33) using a continuous
flow of a mixture of 95% N2 and 5% CO2 gas certified to contain <10 ppm O2
(Airgas Northeast, Cheshire, CT). Unless otherwise specified, experiments
were done with 0.01% O2. Hypoxic cell culture experiments in which in situ
lysis was done were carried out in an INVIVO2 400 chamber (Biotrace,
Cincinnati, OH) with glove box capabilities to allow manipulation underhypoxia. Desferrioxamine (Sigma, St. Louis, MO) treatments were carried
out by media supplementation to a final concentration of 250 Amol/L.
Ionizing radiation was done with a 137Cs irradiator at a dose rate of 165rad/min.
Phosphatase assay. Phosphatase treatments of protein samples were
carried out using E-phosphatase as per instructions of the manufacturer
(New England Biolabs, Beverly, MA). Briefly, 100 Ag of total cell lysatewere incubated with 1� reaction buffer, 2 mmol/L MnCl2, and 1 AL of
E-phosphatase. Reactions were carried out for 30 minutes at 30jC. Where
indicated, phosphatase inhibitors (50 mmol/L NaF and 10 mmol/L Na3VO4)
were added to the reactions before the introduction of enzyme.Western blot. Cells were lysed with radioimmunoprecipitation assay
buffer (150 mmol/L NaCl, 0.1% SDS) and 100 Ag of total protein per sample
were resolved by SDS-PAGE. Proteins were detected by standard
immunoblot protocol using the following primary antibodies: phospho-
Chk2(Thr68) and p53 (D0-1) from Santa Cruz Biotechnology (Santa Cruz,
CA); Chk2 (clone 7; Upstate Biotechnology, Lake Placid, NY); phospho-
p53(Ser20) (9287; Cell Signaling Technologies, Beverly, MA); tubulin (clone
B-5-1-2; Sigma); Hif-1a (clone 54) and NBS1 (clone 34; BD Transduction
Labs, Franklin Lakes, NJ); ATM (2c1) and ATR (clone 2B5; GeneTex,
San Antonio, TX); MLH1(clone G168-15; BD PharMingen, San Diego, CA);
and Glut-1 (GT12-A; Alpha Diagnostic International, San Antonio, TX) and
actin (Research Diagnostics, Inc., Flanders, NJ). Each experiment was
carried out at least thrice and representative Western blots are shown. Band
intensities were quantitated using the public domain NIH Image program
(version 1.63; developed at the U.S. NIH and available online at http://
rsb.info.nih.gov/nih-image/). Relative increases in Chk2 phosphorylation
were calculated as follows: (ratio of phospho-Chk2 in hypoxia versus
normoxia) / (ratio of total Chk2 in hypoxia versus normoxia).Northern blot. Total RNA was isolated using the TRIzol RNA isolation
reagent (Life Technologies, Carlsbad, CA). Northern blots were done as
previously described (28). The following primer pairs were used to
produce probes for Northern blot analysis by reverse transcription-PCR(RT-PCR) amplification of mRNA: Chk2, 5V-GCGGTCGTGATGTCTCGG-3V(sense) and 5V-TTCGTGTTCAAACCACGGA-3V (antisense); vascular endo-
thelial growth factor, 5V-CTTCACTGGATGTATTTGACTGCTGTGG-3V(sense) and 5V-GCTAGTGACTGTCACCGATCAGGGAG-3V (antisense). RT-
PCR products were confirmed by sequence analysis before use.
Cell cycle analysis. Cells cultured in normoxia or hypoxia were stained
with propidium iodide. DNA content was analyzed using a BectonDickinson FACS-Calibur flow cytometer. Histogram construction was done
using BD CellQuest Pro software (Becton Dickinson, San Jose, CA).
Experiments were conducted in triplicate.
Caspase activation assay. Caspase-3 activity was assayed using the
CaspACE Colorimetric Assay System according to the instructions of the
manufacturer (Promega Corp., Madison, WI). Absorbance readings for each
sample were determined at 405 nm using a SpectraMAX Gemini XS
microplate reader and SoftMax Pro software (Molecular Devices Corp.,
Sunnyvale, CA). The caspase specific activity for each sample was calculated
using the following formula: specific activity = pmol product per hour / Agprotein. P values were calculated based on unpaired two-tailed t tests using
the Microsoft Excel Plug-in Analyze-it (Analyze-it Software Ltd., Leeds,
United Kingdom). Caspase activation experiments were conducted in
replicates of six.
Results
Hypoxia induces Chk2 phosphorylation. To examine theeffects of hypoxia on Chk2 phosphorylation, we cultured HeLaand MCF-7 cells under normoxic or hypoxic (0.01% O2)conditions for 48 hours. As a positive control, cells wereirradiated with 10 Gy of ionizing radiation. In both cell lines,hypoxia induced Chk2 phosphorylation on Thr68 (Fig. 1A). Chk2contains a number of SQ/TQ motifs, which are generally thoughtof as ATM/ATR consensus phosphorylation sites (34, 35). Ofthese sites, the most commonly studied is Thr68, which isknown to be phosphorylated and lead to Chk2 activation afterionizing radiation damage. Interestingly, although Chk2 phos-phorylation increased on exposure to hypoxia, total Chk2 proteinlevels decreased. By correcting for the decrease in total Chk2protein levels, we were able to quantitate the relative hypoxia-induced increases in Chk2 phosphorylation in HeLa and MCF-7cells as 3- and 5-fold, respectively. These relative increases inChk2 phosphorylation were comparable to changes in Chk2activation following exposure to ionizing radiation. We alsoobserved similar results on treatment of cells for 48 hours withthe iron chelator desferrioxamine, a chemical mimetic of hypoxia(Fig. 1B).Antibody specificity and Chk2 phosphorylation were confirmed
by phosphatase treatment (Fig. 1C). Samples from hypoxic orirradiated MCF-7 cells exhibited Chk2 phosphorylation wheneither added directly to the gel or when preincubated withreaction buffer alone (Fig. 1C, lanes 5, 6, 9 , and 10). However,incubation with E-phosphatase abolished this signal (Fig. 1C, lanes7 and 11). The signal was maintained by the presence of phos-phatase inhibitors to prevent Chk2 dephosphorylation (Fig. 1C,lanes 8 and 12).To confirm that Chk2 phosphorylation is induced by hypoxia
per se rather than during the brief reoxygenation that can occuron removal of cell culture dishes from a hypoxic chamber beforelysis, MCF-7 and RKO cells were incubated in a glove boxchamber at 0.1% O2. After 48 hours, the cells were lysed in situunder hypoxia and Chk2 phosphorylation was assessed in thelysates by Western blot (Fig. 1D). As shown, Chk2 is indeedphosphorylated on Thr68 under strictly hypoxic conditions; thephosphorylation cannot be attributed simply to a reoxygenationeffect. Similar to previous experiments, the relative increases inChk2 phosphorylation were determined to be 5-fold in MCF-7cells and 7-fold in RKO cells. Because of the limitations of theglove box chamber, this experiment was conducted under 0.1% O2,representing less severe hypoxia than the conditions for Figs. 1Aand C . However, the results are consistent, indicating thereproducibility of the Chk2 phosphorylation response over a rangeof hypoxic conditions.To determine the kinetics of the Chk2 phosphorylation and of
the decrease in total Chk2 protein, time-course experiments werecarried out with MCF-7 cells cultured in hypoxia and harvestedin situ within a glove box chamber. Increased phosphorylation onThr68 could be detected after 12 hours of exposure to hypoxia, withmaximum signal achieved at 72 hours (Fig. 2A). No further increasein Chk2 phosphorylation was detected at the 96-hour time point. Itis of note that cells harvested at all time points retained goodviability despite prolonged growth in low oxygen conditions. Thedecrease in total Chk2 levels was not observed until the 48-hourtime point, indicating that the protein is initially activated, but thatthere is a subsequent decrease in total Chk2 protein levels afterexposure to hypoxia.
Hypoxia-Induced Phosphorylation of Chk2
www.aacrjournals.org 10735 Cancer Res 2005; 65: (23). December 1, 2005
Cancer Research. on September 23, 2017. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

To explore the mechanism of Chk2 down-regulation, Northernblot analyses were done using MCF-7 and HeLa cells. The resultsindicate that Chk2 mRNA levels are decreased on exposure to either48 hours of hypoxia or 24 hours of desferrioxamine treatment(Fig. 2B).
Upstream activators involved in Chk2 phosphorylation. Asmentioned above, Chk2 is a substrate of the ATM kinase (5, 6). Toinvestigate the potential role of ATM in hypoxia-induced Chk2phosphorylation, MCF-7 cells were stably transfected with a vectordesigned to express siRNA directed against ATM (pSUPER-ATMi)or an empty vector control. The resulting cell lines, ATMi-32 andpSpr-7, respectively, were exposed to hypoxic (0.1% O2, 48 hours) ornormoxic growth conditions. At the time of harvest for analysis, thehypoxic cells were lysed in situ within the glove box chamber. Asshown in Fig. 3A , ATMi cells exhibited substantially reducedhypoxia-induced Chk2 phosphorylation when compared with theparental MCF-7 or pSpr-7 control cells. Due to the undetectablelevels of Chk2 phosphorylation in normoxic samples, we wereunable to quantitate the relative increases in Chk2 activation.However, by comparing the ratio of phospho-Chk2 to total Chk2under each condition, we were able to calculate a 3-fold decrease inChk2 phosphorylation in ATM-deficient cells in relation to controlMCF-7 and pSpr cells.Because the substrate targets for ATM can also be targets for the
related kinase, ATR, we sought to determine whether ATR playsany role in Chk2 phosphorylation under hypoxia. RKO and MCF-7cells were transfected with a synthetic siRNA duplex of a sequencepreviously shown to down-regulate ATR expression (36) or with aduplex targeted to the firefly luciferase gene as a control fornonspecific effects of RNAi. Sixteen hours after transfection, cellswere either maintained in normoxic conditions or placed inhypoxia (0.1% O2) for another 48 hours. Again, hypoxic cells werelysed in situ in the glove box chamber. Western blot analysis
Figure 1. Phosphorylation of Chk2 is induced by hypoxia and the hypoxiamimetic iron chelator desferrioxamine. A, Western blot analysis ofphospho-Chk2 (Thr68), total Chk2, Hif-1a (positive control for hypoxia),and tubulin (loading control) levels in HeLa and MCF-7 cells under normoxia(N), hypoxia (0.01% for 48 hours; H ), or after ionizing radiation exposure(10 Gy; IR ) as indicated. B, Western blot analysis, as in (A), of the proteinlevels in HeLa and MCF-7 cells with or without desferrioxamine treatment(250 Amol/L for 48 hours; DFX). C, effect of phosphatase treatment of celllysates on detection of phospho-Chk2 in samples from hypoxic or irradiated cells.MCF-7 cells were exposed to normoxia, hypoxia (48 hours), or ionizingradiation as above, and the respective lysates were treated as indicated withE-phosphatase or E-phosphatase plus phosphatase inhibitors. TCL, total celllysate directly loaded onto the gel. Lanes 2, 6, and 10, samples were treated withreaction buffer only. Phosphatase experiments were carried out four times.Representative Western blot. D, Western blot analysis of samples from MCF-7and RKO cells lysed in situ under hypoxia (0.1% O2, 48 hours) within a glovebox chamber. Figure 2. Chk2 phosphorylation precedes changes in protein levels and the
down-regulation seems to be transcriptional. A, MCF-7 cells were cultured inhypoxia (0.1% O2) for the indicated length of time. Cells were harvestedin situ within a glove box chamber. Chk2 phosphorylation and protein levels wereassayed by Western blot. B, MCF-7 and HeLa cells were grown in hypoxia(0.01% O2) and total RNA was harvested after 48 hours. MCF-7 cells werealso treated with 250 Amol/L desferrioxamine for 24 hours. Samples wereanalyzed by Northern blot. Experiments were conducted in triplicate.Representative Northern blots.
Cancer Research
Cancer Res 2005; 65: (23). December 1, 2005 10736 www.aacrjournals.org
Cancer Research. on September 23, 2017. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

(Fig. 3B) revealed that ATR down-regulation by RNAi in either RKOor MCF-7 cells does not reduce Chk2 phosphorylation in responseto hypoxia.To further test the role of ATR in hypoxia-induced Chk2
activation, we examined the effect of ATR knockdown in anATM-deficient cell background. ATMi cells (constitutivelyexpressing siRNA directed against ATM) were transfected withsynthetic siRNA duplexes directed against either ATM or ATR.siRNA targeted to luciferase was used as a control. As shown inFig. 3C , additional reduction of ATM expression resulted in evengreater suppression of Chk2 phosphorylation under hypoxicconditions. However, knockdown of ATR expression had no effecton Chk2 activation in ATM-deficient cells. Thus, residual Chk2phosphorylation in ATMi cells seems to be the result of theincomplete knockdown of ATM and not the result of compen-satory ATR function. In fact, these results further indicate thatATR has no apparent role in Chk2 phosphorylation in responseto hypoxia.Because MLH1 has been shown to play a role in Chk2 activation
following ionizing radiation exposure (17), we examined MLH1-deficient and complemented cells (31) with regard to Chk2 phos-
phorylation in response to hypoxia. HCT116/3-6 (MLH1+) andHCT116/2-3 (MLH1�) were grown in normoxia or hypoxia for 48hours. The MLH1-deficient cells exhibited reduced Chk2 phosphor-ylation under hypoxia compared with the MLH1-complementedcells, which showed a 6-fold higher ratio of phospho-Chk2 to totalChk2 (Fig. 3D). A similar dependency on MLH1 was also seen afterdesferrioxamine treatment (data not shown).NBS1, a factor in DNA double-strand break repair, also signals
to Chk2 after ionizing radiation exposure (12, 13). We examinedNBS1-deficient and cDNA-complemented human cells (32) to testthe NBS1 dependence of Chk2 phosphorylation after exposure to48 hours of hypoxia. Cells deficient in NBS1 exhibited 3-fold lowerChk2 phosphorylation than their complemented counterparts(Fig. 3E). Under the electrophoresis conditions used, a highermobility species of Chk2 is visible in hypoxic samples usingan antibody to total Chk2 (Fig. 3E, lanes 2 and 4). Such a shiftin mobility is indicative of Chk2 hyperphosphorylation. Notably,although hypoxia induces Chk2 down-regulation in both NBS1-proficient and -deficient cells, a greater proportion (f50%) ofthe remaining Chk2 exists as the higher mobility species incells expressing NBS1. This observation reinforces the data
Figure 3. ATM, MLH1, and NBS1 mediateChk2 phosphorylation in response tohypoxia. A, MCF-7 cells were stablytransfected with an empty pSUPER vector(pSpr ) or with a vector designed to expresssiRNA directed against ATM mRNA(ATMi). Cells were exposed to 48 hoursof normoxia or hypoxia (0.1% O2) andhypoxic cells were lysed in situ . Westernblot analysis was done to evaluate levels ofphospho-Chk2 (Thr68), total Chk2, Glut-1(as a positive control for hypoxia), andtubulin (as a loading control; top ). ATMlevels were confirmed by Western blot foreach cell line (bottom ). Dashed lines,separate lanes from the same gel. B, RKOcells (left) and MCF-7 cells (right ) weretransfected with synthetic RNAi duplexesdirected against ATR (ATR) or luciferase(Luc ) or mock transfected (None ). Hypoxiccells were grown in 0.1% O2 for 48 hoursand lysed in situ . Western blot analysis wascarried out to visualize levels of theindicated proteins. Equal loading wasconfirmed by Western blot analysis oftubulin in RKO cells and of actin in MCF-7cells. Dashed lines, nonadjacent lanesfrom the same gel. C, MCF-7 cells stablyexpressing siRNA directed against ATMwere transiently transfected with syntheticRNA duplexes targeting luciferase, ATM,or ATR. Parental MCF-7 cells transfectedwith siRNA against luciferase were usedas controls for WT protein levels. Hypoxiccells were grown in 0.1% O2 for 48 hoursand lysed in situ . Cell lysates wereanalyzed by Western blot for the indicatedfactors. Dashed lines, separate lanesfrom the same gel. D, derivatives of theMLH1-deficient colon cancer cell lineHCT116, HCT116/3-6 (MLH1+) orHCT116/2-3 (MLH1�), were grown innormoxia or hypoxia (48 hours) and celllysate samples were analyzed byWestern blot for the indicated proteins.E, a matched pair of NBS1-deficient orcomplemented cell lines (NBS1� andNBS1+) were exposed to normoxia orhypoxia for 48 hours and cell lysateswere analyzed by Western blot as above.
Hypoxia-Induced Phosphorylation of Chk2
www.aacrjournals.org 10737 Cancer Res 2005; 65: (23). December 1, 2005
Cancer Research. on September 23, 2017. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

obtained using the phospho-Chk2–specific antibody and furtherindicates that Chk2 phosphorylation is attenuated in NBS1-deficient cells.Overall, the data in Fig. 3 indicate that ATM, MLH1, and NBS1 all
participate in Chk2 phosphorylation in response to hypoxia. Incontrast, hypoxia-induced Chk2 phosphorylation is independent ofATR. Likewise, two other mediators of Chk2 activation, 53BP1 andBRCA1, did not affect Chk2 phosphorylation after exposure tohypoxia (data not shown).p53 phosphorylation in response to hypoxia is dependent on
Chk2 and ataxia telangiectasia mutated. To show that hypoxia-induced Chk2 phosphorylation leads to functional activation, weexamined the phosphorylation of p53 as a known Chk2 target.MCF-7 cells were grown in normoxia or hypoxia for 48 hours andcell lysates were assayed by Western blot for phospho-p53 (Ser20)and total p53. As shown in Fig. 4A , p53 is phosphorylated on Ser20after exposure to hypoxia and this phosphorylation correlates withan increase in total p53. This phosphorylation event is knownto be mediated by Chk2 in response to ionizing radiation (7–9) andits occurrence in hypoxia indicates that Chk2 is functionally active.To determine whether Chk2 is, in fact, responsible for p53
phosphorylation, a comparison was made between HCT15 cells(Chk2-deficient) and HCT15 subclones complemented with a vectorexpressing WT Chk2 cDNA. The parental HCT15 cells exhibitedminimal hypoxia-induced changes in p53 phosphorylation (Fig. 4B).However, in HCT15 cells complemented with a WT Chk2 cDNAconstruct, phosphorylation of p53 on Ser20 was substantiallyincreased on exposure to 48 hours of hypoxia (Fig. 4B).Because we have shown that ATM contributes to Chk2
phosphorylation, we sought to determine whether ATM also affectsp53 phosphorylation on Ser20. MCF-7 and ATMi cell lines werecultured in normoxia or hypoxia for 48 hours. Whereas MCF-7 cellsexhibited increased p53 phosphorylation after exposure to eitherhypoxia or ionizing radiation, ATMi cells displayed attenuated p53phosphorylation in response to both stimuli (Fig. 4C), consistentwith a role for ATM in p53 activation under hypoxia. These dataindicate that both ATM and Chk2 can be placed in a pathwayleading to p53 phosphorylation in response to hypoxia.Chk2 protects cells from hypoxia-induced apoptosis. Chk2 is
known to affect a number of cellular processes, including cell cyclearrest and apoptosis. To address the possibility that Chk2activation in hypoxia might influence cell cycle progression,
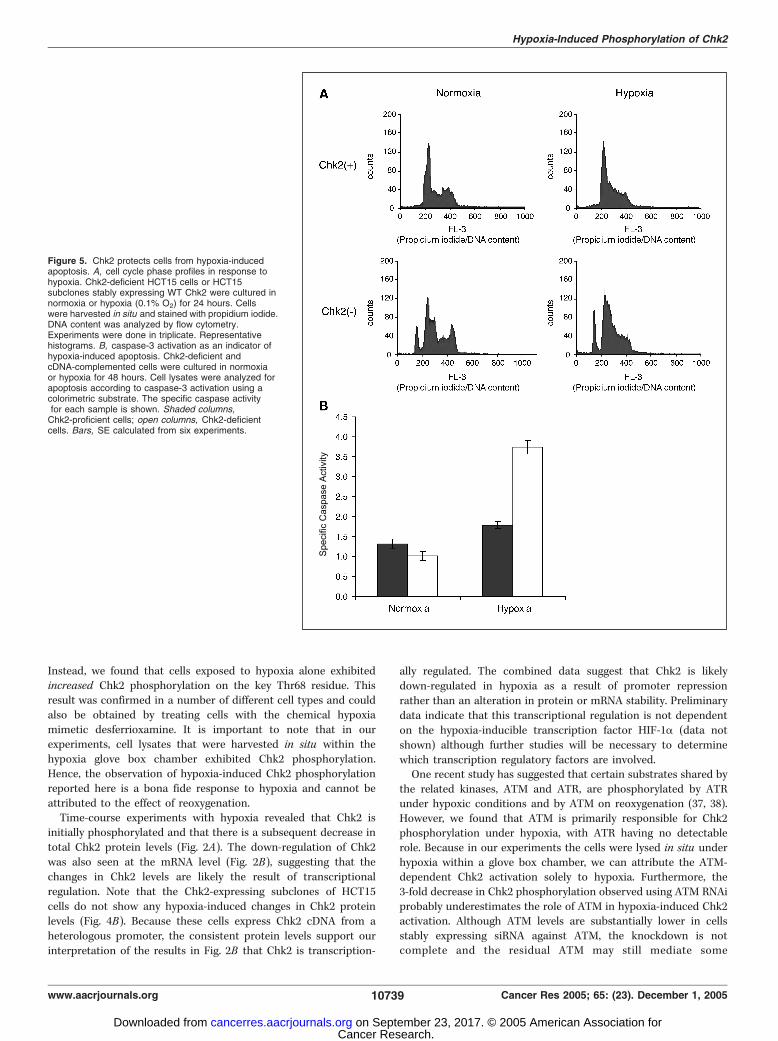
Chk2-deficient and Chk2-proficient cells were cultured in normoxiaor hypoxia (0.1%) for 24 hours and stained with propidium iodideto assay cellular DNA content. Hypoxic cells were treated andharvested within the hypoxia glove box chamber. We did notobserve major differences in cell cycle phase redistribution inresponse to hypoxia in a comparison of Chk2-deficient and Chk2-proficient cells (Fig. 5A) except for a 30% increase in the sub-G1
population in Chk2-deficient cells that were exposed to hypoxia.Essentially no sub-G1 DNA content cells were seen in the Chk2-proficient cells.Because sub-G1 DNA content is often associated with apoptosis,
we examined caspase-3 activation as a measure of cellularapoptosis. Chk2-deficient HCT15 cells and HCT15 subclonescomplemented with WT Chk2 cDNA were cultured undernormoxic or hypoxic conditions for 48 hours and caspase-3activation was assayed. As shown in Fig. 5B , Chk2-deficient cellsexposed to hypoxia exhibit a 3.7-fold increase in caspase activation(P < 0.0001), and thus apoptosis, when compared with theirnormoxic counterparts. Cells expressing WT Chk2, however, didnot show a significant hypoxia-induced increase in caspase activity,indicating that Chk2 serves to protect cells from apoptosis underhypoxic conditions.
Discussion
The work reported here shows that Chk2 phosphorylation onThr68 is induced by hypoxia in a manner dependent on ATM,MLH1, and NBS1. p53, a well-documented target of Chk2, is alsophosphorylated under hypoxia, and this was found to occur in aChk2-dependent manner, consistent with functional Chk2 phos-phorylation and activation under these conditions. In addition, thepresence of Chk2 was found to suppress hypoxia-induced apoptosis.Taken together, these results delineate a novel signaling pathway inresponse to hypoxic stress that affects cell survival. Because hypoxiais a common feature of solid tumors, this pathway may play a keyrole in cancer cell growth regulation and DNA damage response.This line of investigation was prompted by work implicating
MLH1 in the ATM-Chk2 signaling cascade that is initiated byionizing radiation (17). We had previously shown that MLH1 levelsare down-regulated by exposure of human cancer cells to hypoxia,and so we hypothesized that any MLH1-dependent response toionizing radiation might consequently be compromised by hypoxia.
Figure 4. p53 is phosphorylated inresponse to hypoxia in a Chk2-dependentmanner. A, MCF-7 cells were exposed tonormoxia or hypoxia (0.01% O2 for 48hours). Cell lysates were analyzed byWestern blot for the indicated factors.B, Chk2-deficient HCT15 cells weretransfected with a vector designed toexpress WT Chk2. Stable cell lines wereestablished for vector transfection and thecells were exposed to normoxia or hypoxiafor 48 hours, followed by Western blotanalysis of the indicated factors. C, MCF-7cells were stably transfected with apSUPER vector designed to expresssiRNA against ATM. Cells were cultured innormoxia or hypoxia for 48 hours andcell lysates were analyzed by Westernblot for the indicated factors.
Cancer Research
Cancer Res 2005; 65: (23). December 1, 2005 10738 www.aacrjournals.org
Cancer Research. on September 23, 2017. © 2005 American Association forcancerres.aacrjournals.org Downloaded from
Instead, we found that cells exposed to hypoxia alone exhibitedincreased Chk2 phosphorylation on the key Thr68 residue. Thisresult was confirmed in a number of different cell types and couldalso be obtained by treating cells with the chemical hypoxiamimetic desferrioxamine. It is important to note that in ourexperiments, cell lysates that were harvested in situ within thehypoxia glove box chamber exhibited Chk2 phosphorylation.Hence, the observation of hypoxia-induced Chk2 phosphorylationreported here is a bona fide response to hypoxia and cannot beattributed to the effect of reoxygenation.Time-course experiments with hypoxia revealed that Chk2 is
initially phosphorylated and that there is a subsequent decrease intotal Chk2 protein levels (Fig. 2A). The down-regulation of Chk2was also seen at the mRNA level (Fig. 2B), suggesting that thechanges in Chk2 levels are likely the result of transcriptionalregulation. Note that the Chk2-expressing subclones of HCT15cells do not show any hypoxia-induced changes in Chk2 proteinlevels (Fig. 4B). Because these cells express Chk2 cDNA from aheterologous promoter, the consistent protein levels support ourinterpretation of the results in Fig. 2B that Chk2 is transcription-
ally regulated. The combined data suggest that Chk2 is likelydown-regulated in hypoxia as a result of promoter repressionrather than an alteration in protein or mRNA stability. Preliminarydata indicate that this transcriptional regulation is not dependenton the hypoxia-inducible transcription factor HIF-1a (data notshown) although further studies will be necessary to determinewhich transcription regulatory factors are involved.One recent study has suggested that certain substrates shared by
the related kinases, ATM and ATR, are phosphorylated by ATRunder hypoxic conditions and by ATM on reoxygenation (37, 38).However, we found that ATM is primarily responsible for Chk2phosphorylation under hypoxia, with ATR having no detectablerole. Because in our experiments the cells were lysed in situ underhypoxia within a glove box chamber, we can attribute the ATM-dependent Chk2 activation solely to hypoxia. Furthermore, the3-fold decrease in Chk2 phosphorylation observed using ATM RNAiprobably underestimates the role of ATM in hypoxia-induced Chk2activation. Although ATM levels are substantially lower in cellsstably expressing siRNA against ATM, the knockdown is notcomplete and the residual ATM may still mediate some
Figure 5. Chk2 protects cells from hypoxia-inducedapoptosis. A, cell cycle phase profiles in response tohypoxia. Chk2-deficient HCT15 cells or HCT15subclones stably expressing WT Chk2 were cultured innormoxia or hypoxia (0.1% O2) for 24 hours. Cellswere harvested in situ and stained with propidium iodide.DNA content was analyzed by flow cytometry.Experiments were done in triplicate. Representativehistograms. B, caspase-3 activation as an indicator ofhypoxia-induced apoptosis. Chk2-deficient andcDNA-complemented cells were cultured in normoxiaor hypoxia for 48 hours. Cell lysates were analyzed forapoptosis according to caspase-3 activation using acolorimetric substrate. The specific caspase activityfor each sample is shown. Shaded columns,Chk2-proficient cells; open columns, Chk2-deficientcells. Bars, SE calculated from six experiments.
Hypoxia-Induced Phosphorylation of Chk2
www.aacrjournals.org 10739 Cancer Res 2005; 65: (23). December 1, 2005
Cancer Research. on September 23, 2017. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

phosphorylation of Chk2. In this regard, MCF-7 cells constitutivelyexpressing siRNA against ATM, which were transfected withsynthetic RNAi duplexes against ATM, showed further reductionin ATM levels and a corresponding additional suppression of Chk2phosphorylation under hypoxia. In contrast, knockdown of ATR inATM RNAi–expressing cells showed no further suppression of Chk2phosphorylation under hypoxia, consistent with the lack of effect ofthe individual ATR knockdown above.We also tested the roles of MLH1 and NBS1, both of which have
been implicated by other studies in ionizing radiation–inducedsignaling to Chk2 (12, 13, 17). We found that both can influence Chk2phosphorylation in response to hypoxia with substantial decreasesin hypoxia-induced Chk2 phosphorylation in cells deficient in eitherfactor.Following activation by exposure to ionizing radiation or other
DNA damaging agents, Chk2 phosphorylates the downstreameffector p53 (7–9). On phosphorylation by Chk2, p53 is stabilized(10, 11), causing cell cycle arrest and regulating DNA repair.Although hypoxia-induced p53 stabilization has been previouslyreported (39), we have shown here in a comparison of Chk2-deficient and Chk2-proficient cells that p53 phosphorylation andstabilization under hypoxia are both Chk2 dependent. These dataindicate that Chk2 phosphorylation in hypoxia is associated withfunctional activation and downstream consequences. Furthermore,we have shown that p53 phosphorylation is also dependent onATM, allowing us to place Chk2 and ATM in a hypoxia-inducedpathway leading to p53 phosphorylation.Chk2 shares many substrates with the functionally related kinase
Chk1. Although Chk1 has been implicated in posthypoxia cellsurvival (40), we have been unable to detect Chk1 phosphorylationin our cells under either hypoxia or desferrioxamine exposure (datanot shown).
Chk2 is known to affect several cellular processes, including cellcycle arrest, apoptosis, and DNA repair. We have shown here thatcells deficient in Chk2 are especially sensitive to low oxygen levelsand are more susceptible to hypoxia-induced apoptosis (Fig. 5).We hypothesize that Chk2 either functions to stabilize stalledreplication forks or contributes to the repair of DNA lesions,thereby limiting the accumulation of DNA damage and precludingthe induction of apoptosis.Based on the work reported here, we propose a model in
which hypoxia can lead to ATM and/or NBS1 activation, eitherthrough altered DNA metabolism or changes in chromatinstructure. ATM, which may be activated either directly or as aresult of communication with NBS1, then phosphorylates Chk2.Because MLH1 has not been shown to possess kinase activity, itis likely that MLH1 acts as a scaffold to properly orient ATM andChk2. Such a scaffolding role to position ATM together withChk2 was previously proposed for MLH1 as a response to ion-izing radiation (17).Chk2 is constitutively phosphorylated on Thr68 in many human
tumors, especially those in which p53 is mutated (41). However, thecause of this constitutive phosphorylation is unknown. Our resultsshowing that Chk2 is phosphorylated under hypoxic conditionsraise the possibility that prolonged tumor hypoxia and/or cycles ofhypoxia and reoxygenation may provide a mechanism that leads topersistent Chk2 activation.
Acknowledgments
Received 4/5/2005; revised 9/4/2005; accepted 9/20/2005.Grant support: NIH grant ES05775 (P.M. Glazer).The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Z. Yun, D. Campisi Hegan, C. Brdlik, and L. Cabral for their help.
References
1. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpointcontrol and cancer. Cancer Cell 2003;3:421–9.
2. Bell DW, Varley JM, Szydlo TE, et al. Heterozygousgerm line hCHK2 mutations in Li-Fraumeni syndrome.Science 1999;286:2528–31.
3. Haruki N, Saito H, Tatematsu Y, et al. Histologicaltype-selective, tumor-predominant expression of a novelCHK1 isoform and infrequent in vivo somatic CHK2mutation in small cell lung cancer. Cancer Res 2000;60:4689–92.
4. Lee SB, Kim SH, Bell DW, et al. Destabilization ofCHK2 by a missense mutation associated with Li-Fraumeni Syndrome. Cancer Res 2001;61:8062–7.
5. Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K,Elledge SJ. Ataxia telangiectasia-mutated phosphory-lates Chk2 in vivo and in vitro . Proc Natl Acad Sci U S A2000;97:10389–94.
6. Chaturvedi P, Eng WK, Zhu Y, et al. MammalianChk2 is a downstream effector of the ATM-dependentDNA damage checkpoint pathway. Oncogene 1999;18:4047–54.
7. Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD.Phosphorylation of Ser-20 mediates stabilization ofhuman p53 in response to DNA damage. Proc NatlAcad Sci U S A 1999;96:13777–82.
8. Hirao A, Kong YY, Matsuoka S, et al. DNA damage-induced activation of p53 by the checkpoint kinaseChk2. Science 2000;287:1824–7.
9. Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The humanhomologs of checkpoint kinases Chk1 and Cds1 (Chk2)phosphorylate p53 at multiple DNA damage-induciblesites. Genes Dev 2000;14:289–300.
10. Chehab NH, Malikzay A, Appel M, Halazonetis TD.Chk2/hCds1 functions as a DNA damage check-point in G(1) by stabilizing p53. Genes Dev 2000;14:278–88.
11. Dumaz N, Milne DM, Jardine LJ, Meek DW. Criticalroles for the serine 20, but not the serine 15,phosphorylation site and for the polyproline domainin regulating p53 turnover. Biochem J 2001;359:459–64.
12. Mochan TA, Venere M, DiTullio RA, Jr., HalazonetisTD. 53BP1 and NFBD1/MDC1-Nbs1 function in parallelinteracting pathways activating ataxia-telangiectasiamutated (ATM) in response to DNA damage. CancerRes 2003;63:8586–91.
13. Buscemi G, Savio C, Zannini L, et al. Chk2 activationdependence on Nbs1 after DNA damage. Mol Cell Biol2001;21:5214–22.
14. Wang B, Matsuoka S, Carpenter PB, Elledge SJ.53BP1, a mediator of the DNA damage checkpoint.Science 2002;298:1435–8.
15. Ward IM, Minn K, van Deursen J, Chen J. p53 Bindingprotein 53BP1 is required for DNA damage responsesand tumor suppression in mice. Mol Cell Biol2003;23:2556–63.
16. Foray N, Marot D, Gabriel A, et al. A subset of ATM-and ATR-dependent phosphorylation events requiresthe BRCA1 protein. EMBO J 2003;22:2860–71.
17. Brown KD, Rathi A, Kamath R, et al. The mismatchrepair system is required for S-phase checkpointactivation. Nat Genet 2003;33:80–4.
18. Xu X, Tsvetkov LM, Stern DF. Chk2 activation andphosphorylation-dependent oligomerization. Mol CellBiol 2002;22:4419–32.
19. Schwarz JK, Lovly CM, Piwnica-Worms H. Regulationof the Chk2 protein kinase by oligomerization-mediated
cis - and trans -phosphorylation. Mol Cancer Res 2003;1:598–609.
20. Wu X, Chen J. Autophosphorylation of checkpointkinase 2 at serine 516 is required for radiation-inducedapoptosis. J Biol Chem 2003;278:36163–8.
21. Brizel DM, Scully SP, Harrelson JM, et al. Radiationtherapy and hyperthermia improve the oxygenation ofhuman soft tissue sarcomas. Cancer Res 1996;56:5347–50.
22. Nordsmark M, Overgaard M, Overgaard J. Pretreat-ment oxygenation predicts radiation response inadvanced squamous cell carcinoma of the head andneck. Radiother Oncol 1996;41:31–9.
23. Loncaster JA, Harris AL, Davidson SE, et al. Carbonicanhydrase (CA IX) expression, a potential new intrinsicmarker of hypoxia: correlations with tumor oxygenmeasurements and prognosis in locally advancedcarcinoma of the cervix. Cancer Res 2001;61:6394–9.
24. Williams KJ, Cowen RL, Brown LM, Chinje EC, Jaffar M,Stratford IJ. Hypoxia in tumors: molecular targets for anti-cancer therapeutics. Adv Enzyme Regul 2004;44:93–108.
25. Yuan J, Narayanan L, Rockwell S, Glazer PM.Diminished DNA repair and elevated mutagenesis inmammalian cells exposed to hypoxia and low pH.Cancer Res 2000;60:4372–6.
26. Li CY, Little JB, Hu K, et al. Persistent geneticinstability in cancer cells induced by non-DNA-damagingstress exposures. Cancer Res 2001;61:428–32.
27. Paquette B, Little JB. In vivo enhancement ofgenomic instability in minisatellite sequences of mouseC3H/10T1/2 cells transformed in vitro by X-rays. CancerRes 1994;54:3173–8.
28. Mihaylova VT, Bindra RS, Yuan J, et al. Decreasedexpression of the DNA mismatch repair gene Mlh1
Cancer Research
Cancer Res 2005; 65: (23). December 1, 2005 10740 www.aacrjournals.org
Cancer Research. on September 23, 2017. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

under hypoxic stress in mammalian cells. Mol Cell Biol2003;23:3265–73.
29. Bindra RS, Schaffer PJ, Meng A, et al. Down-regulation of Rad51 and decreased homologous recom-bination in hypoxic cancer cells. Mol Cell Biol 2004;24:8504–18.
30. Koshiji M, To KK, Hammer S, et al. HIF-1a inducesgenetic instability by transcriptionally down-regulatingMutSa expression. Mol Cell 2005;17:793–803.
31. Koi M, Umar A, Chauhan DP, et al. Humanchromosome 3 corrects mismatch repair deficiencyand microsatellite instability and reduces N -methyl-NV-nitro-N -nitrosoguanidine tolerance in colon tumor cellswith homozygous hMLH1 mutation. Cancer Res 1994;54:4308–12.
32. Cerosaletti KM, Concannon P. Nibrin forkhead-associated domain and breast cancer C-terminal
domain are both required for nuclear focus forma-tion and phosphorylation. J Biol Chem 2003;278:21944–51.
33. Reynolds TY, Rockwell S, Glazer PM. Geneticinstability induced by the tumor microenvironment.Cancer Res 1996;56:5754–7.
34. O’Neill T, Dwyer AJ, Ziv Y, et al. Utilization of orientedpeptide libraries to identify substrate motifs selected byATM. J Biol Chem 2000;275:22719–27.
35. Kim ST, Lim DS, Canman CE, Kastan MB. Substratespecificities and identification of putative substrates ofATM kinase family members. J Biol Chem 1999;274:37538–43.
36. Casper AM, Nghiem P, Arlt MF, Glover TW. ATRregulates fragile site stability. Cell 2002;111:779–89.
37. Hammond EM, Dorie MJ, Giaccia AJ. ATR/ATMtargets are phosphorylated by ATR in response to
hypoxia and ATM in response to reoxygenation. J BiolChem 2003;278:12207–13.
38. Hammond EM, Giaccia AJ. The role of ATM and ATRin the cellular response to hypoxia and re-oxygenation.DNA Repair (Amst) 2004;3:1117–22.
39. Graeber TG, Peterson JF, Tsai M, Monica K, FornaceAJ, Jr., Giaccia AJ. Hypoxia induces accumulation of p53protein, but activation of a G1-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol CellBiol 1994;14:6264–77.
40. Hammond EM, Dorie MJ, Giaccia AJ. Inhibition ofATR leads to increased sensitivity to hypoxia/reoxyge-nation. Cancer Res 2004;64:6556–62.
41. DiTullio RA, Jr., Mochan TA, Venere M, et al. 53BP1functions in an ATM-dependent checkpoint pathwaythat is constitutively activated in human cancer. NatCell Biol 2002;4:998–1002.
Hypoxia-Induced Phosphorylation of Chk2
www.aacrjournals.org 10741 Cancer Res 2005; 65: (23). December 1, 2005
Cancer Research. on September 23, 2017. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

2005;65:10734-10741. Cancer Res Shannon L. Gibson, Ranjit S. Bindra and Peter M. Glazer
Dependent Manner−Telangiectasia Mutated Hypoxia-Induced Phosphorylation of Chk2 in an Ataxia
Updated version
http://cancerres.aacrjournals.org/content/65/23/10734
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/65/23/10734.full#ref-list-1
This article cites 40 articles, 31 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/65/23/10734.full#related-urls
This article has been cited by 20 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Cancer Research. on September 23, 2017. © 2005 American Association forcancerres.aacrjournals.org Downloaded from