outline introduction pathophysiology signs & symptoms complications diagnosis treatment...
TRANSCRIPT


Outline
• Introduction• Pathophysiology • Signs & Symptoms • Complications • Diagnosis• Treatment • Prevention • Ataxia Telangiectasia like Disorder

Introduction• Ataxia telangiectasia: (Multi-system Disease)
is a rare, progressive neurodegenerative, genetic disease characterized by cerebellar ataxia and ocular telangiectasia.
• Ataxia: is incoordination and lack of balance.
• Telangiectasia: are red eyes due to widening of small blood vessels in the conjunctiva.

IntroductionEpidemiology: • A-T is an extremely rare disease. It occurs in
both genders, among all races and on all continents around the world.
• 1 per 40,000 - 100,000 people worldwide suffer from ataxia-telangiectasia.
• Till now 18 cases of AT in 11 Saudi families were discovered.

Introduction Classification:So far they appear to be three forms of AT:• Pure AT where patients present with all/most of the diagnostic
symptoms. • Attenuated AT where patients do not possess all of the
diagnostic symptoms. • Carrier AT where individuals with a single ATM mutation show
an increased risk of cancer.
Sometimes they are classified into ‘types’ from I to IV:• Type I is the classic syndrome with all manifestations. • Type II lacks some of the typical findings but shows
radiosensitivity. • Type III has the classic clinical findings but is not radiosensitive. • Type IV shows only some clinical features and is not
radiosensitive.

IntroductionRelated Disorders: There are several other disorders with similar
symptoms that physicians may consider when diagnosing AT. These include:
• Ataxia oculomotor apraxia type 1 • Ataxia oculomotor apraxia type 2 • Cerebral palsy• Gaucher disease• Hartnup disease • Niemann-Pick disease • Refsum disease• Nijmegen breakage syndrome(NBS)• Ataxia telangiectasia like disorder(ATLD)

Pathophysiology• A-T is inherited as an autosomal recessive
diseases. Mutations in the ATM gene causes ataxia-telangiectasia. The ATM gene is located on the long (q) arm of chromosome 11 at position 23.3.

Pathophysiology• The ATM gene provides instructions for
making a protein that is located primarily in the nucleus of cells.
• Where it helps in:1. Controlling the rate at which cells grow
and divide.2. Normal development and activity of
several body systems, including the nervous system and the immune system.
3. Coordination in DNA repair by activating enzymes that fix the broken strands.
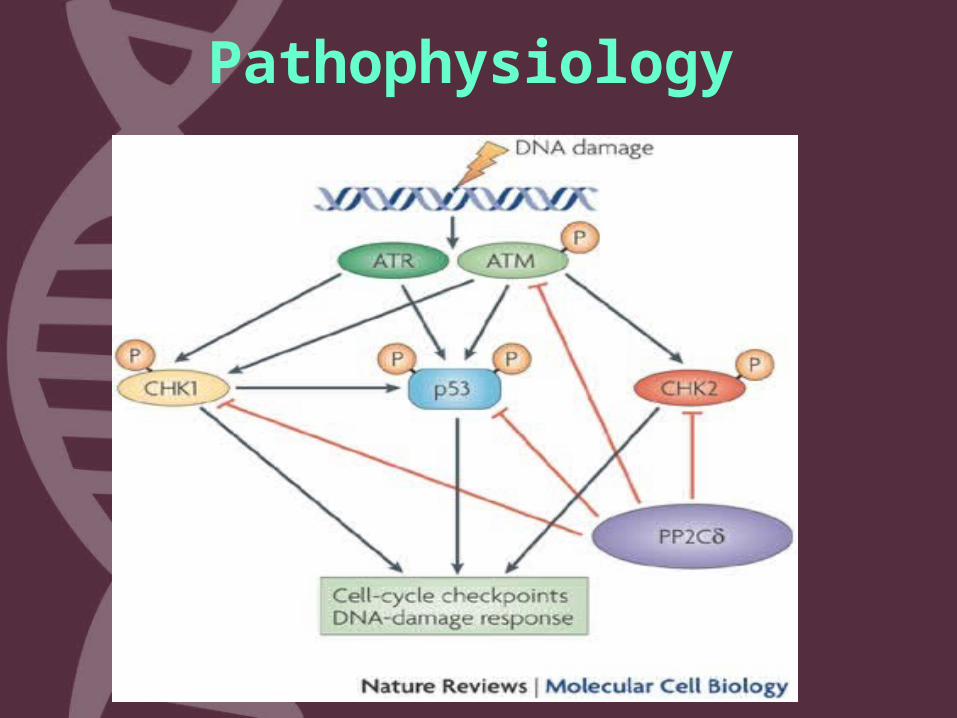
Pathophysiology

Pathophysiology• The mutated ATM gene makes a protein
that does not function properly. As a result:• The cells are hypersensitive to radiation .• Do not respond correctly to DNA
damage. • Death of cell inappropriately, particularly
in the cerebellum (the part of the brain involved in coordinating movements).


Signs and Symptoms
• Abnormal gait (ataxia) and posture.• Slurred speech (dysarthria) and drooling.• Difficulty with eye movements (oculomotor
apraxia).• Abnormal swallowing (dysphagia).• Intellect.• Immune system.• Predisposition to develop cancer.• Growth and endocrine system.• Skin.

Signs and Symptoms

Complications
• Cancer such as lymphoma, leukemia. • Diabetes.• Progressive movement disorder that leads to
wheelchair use .• Scoliosis.• Severe, recurrent lung infections.• Death usually occurs by later childhood .

DiagnosisClinically: • The clinical diagnosis becomes most apparent
after age 10 years, when ataxia, apraxia, telangiectasia, and dysarthria are fully expressed. By this age, cerebellar atrophy is also apparent on magnetic resonance imaging (MRI) studies.
• In contrast, in very young infants the diagnosis can be elusive and easily confused with mild cerebral palsy, acute infectious or episodic ataxia, ataxia with oculomotor apraxia malignancy, or even other rare genetic or mitochondria disorders. Cerebellar atrophy is usually not apparent on MRI in young patients.

Diagnosis
DiagnosisAncillary Tests• Elevated serum alpha-fetoprotein (AFP)
levels which are found in 95% of patients.
• Serum immunoglobulin levels can also be abnormal and reveal marked deficiencies of IgE (in 80% of patients), IgG (in 80%), and IgA (in 60%).
• B and T cell screening shows that the: T-cell levels are usually lowB-cell levels are normal or slightly elevated.

DiagnosisDNA and Cell Based Testing• DNA sequencing can confirm the presence
of mutations can also be used to identify carriers.
• Karyotyping usually reveals characteristic translocations between chromosomes 7 & 14.

DiagnosisDNA and CelNBased Testing• Measurement of cellular damage (cell death
or chromosomal breakage) after exposure of cells to x-rays in the laboratory
• Radiosensitivity is also characteristic of A-T cells done by colony survival assay (CSA)

Diagnosis
• Those tests are not sufficiently sensitive or specific for A-T , have long turnaround times, or require large blood samples. This prompted them to develop a new flow cytometry method for the diagnosis of A-T based on the measurement of histone H2AX phosphorylation.

Treatment• There is currently no cure for A-T. While there is no
treatment that will slow its progress, various forms of physical, speech, and occupational therapy can help patients adapt to the symptoms.
• With A-T, injections of gamma globulin, may strengthen the weakened immune system. The physician may also prescribe high doses of vitamins.
• For some patients, arm weights can help control the arm movements. Other symptoms, such as heart arrhythmias and diabetes, are treated with appropriate drugs.

Prevention
• Couples with a family history of this condition who are considering pregnancy may consider genetic counseling.
• Parents of a child with this disorder may have a slight increased risk of cancer. They should have genetic counseling and more intensive cancer screenings.

Ataxia Telangiectasia –Like Disorder
• Ataxia telangiectasia-like disorder (ATLD) is a very rare variant of ataxia telangiectasia (AT). Mre11 mutations are the underlying cause of ATLD.

Introduction
ATLD is very rare, at present, only 16 confirmed cases, four in the UK, two in Italy and 10 in Saudi Arabia. The patients were either homozygous or compound heterozygous for 5 different mutations in the Mre11 gene.

Introduction• Clinically
A-T ATLD
Progressive cerebellar ataxia
Progressive cerebellar ataxia
Translocation between 7 & 14
Translocation between 7 & 14
Hyper sensitivity to radiation
Hyper sensitivity to radiation
Immunodeficiency mostly thorough lowering of IgA, IgG and IgE levels.
Normal levels of total IgG, IgA and IgM
Telangiectasia No Telangiectasia
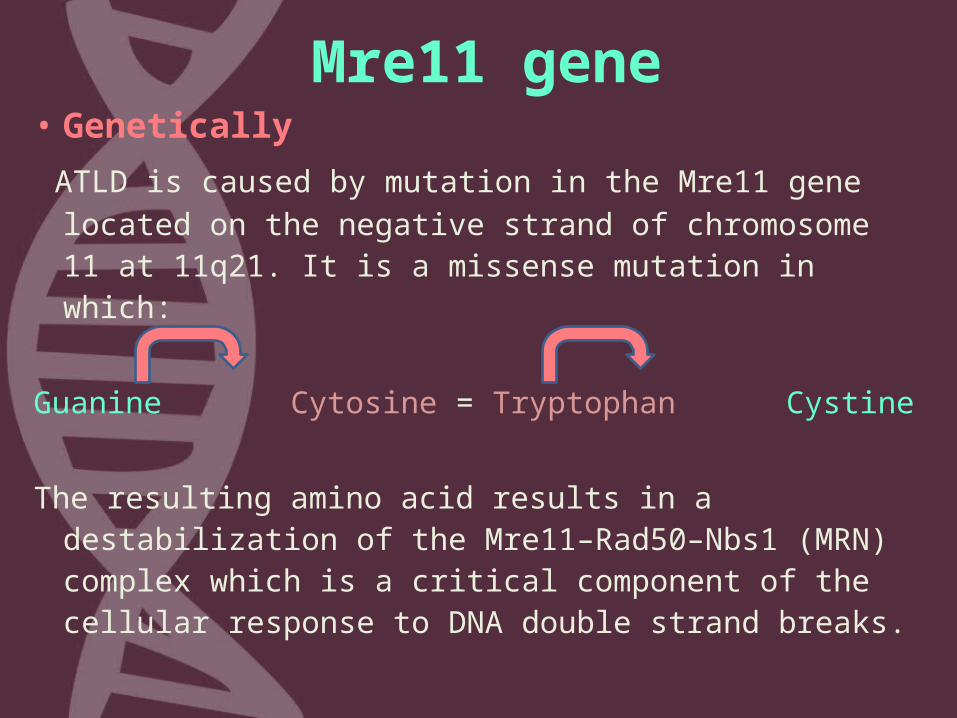
Mre11 gene• Genetically ATLD is caused by mutation in the Mre11 gene
located on the negative strand of chromosome 11 at 11q21. It is a missense mutation in which:
Guanine Cytosine = Tryptophan Cystine The resulting amino acid results in a
destabilization of the Mre11–Rad50–Nbs1 (MRN) complex which is a critical component of the cellular response to DNA double strand breaks.

Mre11 gene

Mre11 Protein• Mre11 protein is involved in different responses
to cellular damage induced by ionizing radiation and radiomimetic chemicals, including complexing with chromatin and with other damage response proteins, and the induction of different cell cycle checkpoints.

Assessment of Carriers' Frequency of a Novel MRE11 Mutation
Responsible for the Rare Ataxia Telangiectasia-Like Disorder
• A cohort of 284 individuals was included in this study. There were 67 females and 217 males.

Methodology
1. Genomic DNA was extracted .
2. Relevant segments of DNA were amplified by thermocycler.
3. The amplified fragment was directly sequenced.

Results
• A• G• A• A• C• T• C• T• T• G• G• T• T• T• A• A• C• T• T• A
• Wild-type:
• Heterozygous:
• A• G• A• A• C• T• C• T• T• G• C• T• T• T• A• A• C• T• T• A• Condon 210 W/C
• 630 G/C
• 630 G/G
• Codon 210 W/W
Out of the 284 individuals, only two were heterozygous for the missense mutation, while the remaining 282 were all wild-type .

Conclusion• The presence of two heterozygous individuals with
mutation in the Mre11 gene indicates the existence of this rare mutation in our local population. The diagnosis of Mre11 ATLD should be in the mind of physicians whenever they encounter an A-T like disorder and genetic study should be carried out to confirm the diagnosis.
• The fact that the 10 previously described Saudi ATLD patients are from the central region of Saudi Arabia could suggest higher frequency of this mutation in geographically isolated families. Testing for the presence of Mre11 mutation could ultimately be incorporated to pre-marriage or pre-implementation screening to reduce the risk of genetic diseases.
