the utility of amplified fragment length polymorphisms in
TRANSCRIPT

Syst. Biol. 56(3):477-484, 2007Copyright © Society of Systematic BiologistsISSN: 1063-5157 print / 1076-836X onlineDOI: 10.1080/10635150701427077
The Utility of Amplified Fragment Length Polymorphisms in Phylogenetics:A Comparison of Homology within and between Genomes
DAVID M. ALTHOFF,1 MATTHEW A. GITZENDANNER,2 AND KARI A. SEGRAVES1
1 Department of Biology, Syracuse University, 130 College Place, Syracuse, NY 13244, USA; E-mail: [email protected] (DMA.)2 Department of Botany, University of Florida, P.O. Box 118526, Gainesville, FL 32611, USA
Abstract.—The amplified fragment length polymorphism (AFLP) technique is being increasingly used in phylogenetic stud-ies, especially in groups of rapidly radiating taxa. One of the key issues in the phylogenetic suitability of this technique iswhether the DNA fragments generated via the AFLP method are homologous within and among the taxa being studied.We used a bioinformatics approach to assess homology based on both chromosomal location and sequence similarity ofAFLP fragments. The AFLP technique was electronically simulated on genomes from eight organisms that represented arange of genome sizes. The results demonstrated that within a genome, the number of fragments is positively associatedwith genome size, and the degree of homology decreases with increasing numbers of fragments generated. The averagehomology of fragments was 89% for small genomes (<400 Mb) but decreased to 59% for large genomes (>2 Gb). Fragmenthomology for large genomes can be increased by excluding smaller fragments, although there is no clear upper limit forthe size of fragments to exclude. A second approach is to increase the number of selective nucleotides in the final selectiveamplification step. For strains of the same organism, homology based on chromosome location and sequence similarity offragments was 100%. Fragment homology for more distantly related taxa, however, decreased with greater time since diver-gence. We conclude that AFLP data are best suited for examining phylogeographic patterns within species and among veryrecently diverged species. [AFLP; bioinformatics; DNA sequence; genome; homology; phylogenetics; species radiations.]
Since its introduction by Vos et al. (1995), the ampli-fied fragment length polymorphism (AFLP) techniquehas been used extensively in studies ranging from pop-ulation genetics to phylogeography to phylogenetics(Mueller and Wolfenbarger, 1999; Bensch and Akesson,2005). Genomic DNA is first digested using two restric-tion endonucleases, typically one with a 6-bp recognitionsequence (usually EcoRI) and one with a 4-bp recognitionsequence (usually Msel). Adapters of known sequenceare then ligated to each end of the fragments and twosuccessive rounds of selective PCR amplification are per-formed. The first round of PCR (preselective or -f-1 am-plification) uses primers that match the adapters on theEcoRI end and Msel end of the fragments plus one extranucleotide. The second round (selective or +3 amplifi-cation) has an additional two nucleotides added to the+1 primers sequences. These rounds of selective ampli-fication reduce the resulting pool of DNA fragments toa size more manageable for analysis. Although the DNAfragments are anonymous, the method is remarkably re-liable and consistent (Vos et al., 1995). This technique isreadily adapted to new taxa because no taxon-specificinformation is needed and AFLPs are suitable for use inboth prokaryotic and eukaryotic organisms. Moreover,the technique surveys the entire genome, is relatively in-expensive, and generates many potential phylogeneticmarkers.
Although the AFLP technique offers many advantagesfor generating phylogenetic markers, the phylogeneticutility of the markers is limited by the assumptionof size homology—identifying the homology of thefragments is not simple or commonly done (Bussell etal., 2005). Homology, from a phylogenetic perspective,entails information about both the location of a fragmentin the genome and the degree of sequence similarity
of fragments of the same size. Because fragments areanonymous and sorted by size via electrophoresis, thereis the potential for homoplasy of fragments at severallevels. Within an individual, multiple fragments fromdifferent regions of the genome may comigrate. Thesefragments may or may not contain the same sequence.Comigrating fragments that have similar sequencesmay be orthologous copies of genes, paralogous copies,pseudogenes, or repeated sequences of unknownfunction. Furthermore, when making comparisonsamong taxa, fragments that are the same size may notnecessarily come from the same locus in the respectivegenome or have the same sequence. This uncertainty inhomology is a particularly important issue in phyloge-netic studies—especially because use of the techniqueis rapidly expanding to examine species radiations thathave been difficult to resolve based on other markerssuch as organellar and nuclear DNA sequences (e.g.,Albertson et al., 1999; Giannasi et al., 2001; DesprSset al., 2003; Brouat et al., 2004; Sullivan et al., 2004;Mendelson and Shaw, 2005). Although studies havedemonstrated that AFLPs can resolve phytogenies whenother markers have failed, and that AFLP phylogeniescorroborate morphology, many phylogeneticists, withgood rationale, remain concerned about the homologyof markers and their utility as phylogenetic characters.
A number of studies have begun to assess fragmenthomology via empirical and theoretical approaches (re-viewed in Koopman and Gort, 2004; Bussell et al.,2005). Empirical approaches have consisted of compar-ing fragment profiles from known crosses (Waugh et al.,1997) to extracting fragments from gels and sequenc-ing them to assessing sequence similarity across indi-viduals (Parsons and Shaw, 2001). O'Hanlon and Peakall(2000) outlined a simple method for detecting fragment
477
Dow
nloaded from https://academ
ic.oup.com/sysbio/article/56/3/477/1657880 by guest on 15 February 2022

478 SYSTEMATIC BIOLOGY VOL. 56
sequence similarity by comparing the fragment profilefrom the final round of selective amplification (usually 3selective nucleotides) to the profile from another roundwith additional selective nucleotides (such as 4 selec-tive nucleotides). The results from these approaches havesuggested that AFLPs are suitable for phylogenetic stud-ies because sequence similarity has ranged from 95% to100% in very closely related taxa. Empirical approaches,however, are labor intensive and only a small numberof fragments has been examined. Furthermore, stud-ies have shown that co-migration of fragments doesoccur (see Koopman and Gort, 2004; Mechanda et al.,2004).
Theoretical approaches have surveyed larger setsof markers by modeling the distribution of fragmentlengths and generating probabilities of comigration andhomoplasy. For example, Vekemans et al. (2002) used an-alytical theory, based on the work by Innan et al. (1999),and numerical simulations to determine the distributionof fragments generated by AFLPs and the relationshipbetween fragment size and homology. Based on MonteCarlo simulations and empirical results from two plantspecies, they found that homoplasy may be quite highand was likely to be even higher for shorter fragmentsizes. Koopman and Gort (2004) compared theoreticalpredictions of fragment length distributions to empiri-cal distributions generated from in silico AFLP for theArabidopsis thaliana genome. Based on the degree of ho-moplasy detected from this comparison, they developedsignificance tests and weighting values as a means tocorrect for size homoplasy when calculating similaritiesbased on band sharing.
Studies that have examined the homology of frag-ments have either used sequence similarity as a proxyfor homology or have examined the number of bandsthat may be comigrating for a given size. In most cases,only one or a few organisms have been examined. Con-sequently, we still do not have a good understandingof the homology of fragments within single individu-als, and the issue of homology of AFLP fragments acrossspecies remains unresolved. Furthermore, there has notbeen a comprehensive survey across taxa that differ ingenome size to assess the influence of genome size onthe degree of fragment homology.
In this study, we take a bioinformatics approach to as-sess the homology of fragments generated by the AFLPtechnique. Using a computer program that mimics theAFLP procedure, we examine fragment homology inwhole genomes, including organellar, from eight speciesacross the tree of life. We address the following questions:(1) What is the degree of positional and sequence homol-ogy of fragments generated from a single individual? (2)How does genome size influence the number of frag-ments produced and, subsequently, how does genomesize influence fragment homology? (3) What is the degreeof fragment homology between closely related strainsof the same species and between species that have di-verged within the last 7 million years? (4) Does the de-gree of homology change as a function of selective primersequence?
METHODS
Species Surveyed
We limited the species chosen for analysis accordingto several criteria: completeness of genome sequencing,assignment of all sequences to chromosomal location,taxonomic breadth, and genome size. Although there arecurrently many genomes being sequenced, most of theseare in the initial stages of assembly and remain incom-plete, especially in terms of assigning all sequence datato chromosomal location. We assessed within-organismfragment homology by surveying species along a con-tinuum from smaller to larger genomes. The genomesused were Bacillus anthracis strain A2012 from the NCBIEntrez Genome database, GenBank accession numberAAAC01000001 (ca. 5.23 Mb); Saccharomyces cerevisiaeRMll-la from the NCBI Entrez Genome database viathe Broad Institute (ca. 12.07 Mb); Caenorhabditis elegansfrom the NCBI Entrez Genome database (ca. 97 Mb); Ara-bidopsis thaliana from the NCBI Entrez Genome database(119.2 Mb); Drosophila melanogaster from the NCBI EntrezGenome database (ca. 180 Mb); Oryza sativa variety japon-ica from the International Rice Genome Sequence ProjectBuild 4.0 (ca. 389 Mb); Mus musculus Build 36 from theMouse Genome Sequencing Consortium via the Univer-sity of California genome browser (ca. 2.5 Gb); Homo sapi-ens Genome Build 36.1 from the International HumanGenome Project sequencing centers via the Universityof California genome browser (ca. 2.9 Gb). The mouseand human genome are not 100% complete in terms ofassigning all sequence data to chromosomal locations,but we included these genomes because they were muchlarger than the other genomes, and we were interested inwhether the pattern of homology changed for very largegenomes.
We also assessed the homology of fragments betweenorganisms very closely related and more distantly re-lated. We compared the homology of fragments be-tween two strains of yeast, S. cerevisiae RMll-la andS. cerevisiae YJM789 (NCBI Entrez Genome database).This comparison would be analogous to a phylogeo-graphic study among populations of the same species.We also compared fragment homology between threefruit fly species D. melanogaster, D. simulans (D. simu-lans White 501, Genome sequencing center at Washing-ton University School of Medicine), and D. yakuba (NCBIEntrez Genome through Genome sequencing center atWashington University School of Medicine Build 2.1).These species have diverged 2 to 7 million of years ago(Russo et al., 1995). We caution that the results may notcompletely reflect the pattern of homology because theD. simulans and D. yakuba genomes still contain unas-signed sequence data. These three species, however, werethe best possible candidates for comparisons amongspecies.
Program Overview
We used a program written in Perl to first surveygenomes for sequence fragments containing specific
Dow
nloaded from https://academ
ic.oup.com/sysbio/article/56/3/477/1657880 by guest on 15 February 2022

2007 ALTHOFF ET AL.—HOMOLOGY OF AFLP FRAGMENTS 479
nucleotides on either end, and then to BLAST thosesequences against one another to assess sequence sim-ilarity. Specifically, the program conducts two nestedsearches for strings of nucleotides specified by the re-searcher. The first search identifies all sequence frag-ments that contain nucleotide sequences for an EcoRIrestriction site (GAATTC) on the 5' end and Msel re-striction site (TTAA) on the 3' end, and vice versa. Thissearch is analogous to the restriction digest in the AFLPtechnique. In the second search, the subset of fragmentsis reexamined for an additional three nucleotides adja-cent to the 3' end of each enzyme recognition sequence.This search is analogous to the +3 selective amplificationstep in which the polymerase chain reaction techniqueis used to selectively amplify fragments that have anadditional three nucleotides flanking the restriction siterecognition sequences. We searched for the same threeselective nucleotides (AAT) adjacent to the EcoRI recog-nition sequence and varied the selective nucleotides ad-jacent to the Msel recognition sequence. We used foursets of three selective nucleotides for the Msel recogni-tion sequence—CAA, CAC, CAG, and CAT. By usingdifferent Msel-associated selective nucleotides, differentgroups of fragments are detected. For the comparisonamong the Drosophila species we used an additional setof Msel-associated selective nucleotides (TTA, TTC, TTG,TTT) to increase the sample size and power of our testof the number of homologous bands shared among thespecies.
The program sorts the sequence fragments based onlength (bp). We only used fragments between 50 and 500bp, and binned fragments by their length with a 1-bpresolution. This is the standard size range and resolu-tion of AFLP fragments used in population genetic, phy-logeographic, and phylogenetic studies. The programcreates a set of folders based on the bin sizes foundamong the fragments and creates text files for each frag-ment detected. The text files contains a unique identifierand the DNA sequence. For example, there may be aset of folders titled 50, 150, and 250, and within thesefolders are text files with the DNA sequences of the frag-ments found. In some cases, there may be multiple frag-ments within a bin. The program then performs a BLASTsearch using BLAST 2.2.12 from NCBI to compare the se-quence similarity at the 100% and 95% similarity levelsfor the fragments within each bin. In this way, we as-sessed the sequence similarity of fragments of the samelength.
Analyses
For the within-organism analyses in which species hadthe genome packaged into chromosomes, we searchedfor fragments by chromosome. The resulting fragmentprofiles for each chromosome were then combined togive the organism-wide profile. The organism-wide pro-file represents the fragment profile that would be vi-sualized via electrophoresis in the laboratory. In casesin which the same fragment size was found multipletimes within or among more than one chromosome,
we assessed the sequence similarity of the fragments.The between-organism analyses were also conducted ona chromosome-by-chromosome basis. For the compar-ison between yeast genomes, only a very small num-ber of fragments were generated using three selectivenucleotides. Therefore, we used only two selective nu-cleotides after each restriction enzyme rather than threein order to increase the number of fragments for theanalyses.
Genomes were downloaded from the sources men-tioned above and were converted to standard GenBankformat. For the three Drosophila species, the genome datafrom NCBI are packaged into chromosome arms. Wecompared each arm separately across species and thenchecked the fragment pattern from the entire (both arms)chromosome to determine if fragments were homopla-sious or homologous. We also ran an additional set offour +3 selective nucleotide primer pairs to increase thesample size and power of our test of the number of ho-mologous bands shared among the Drosophila species.We used least squares regression to examine the rela-tionship between genome size and the overall numberof fragments produced and between the number of frag-ments produced and the number of homoplasious frag-ments both within and between chromosomes. We alsoconducted Wilcoxon tests to examine whether there weredifferences in the number of fragments produced by eachof the selective nucleotide combinations. Statistical anal-yses were conducted using JMP 5.0.1.2 (SAS Institute,Cary, NC).
RESULTS
The organellar genomes for all of the species did notyield any fragments. The number of fragments producedwas dependent on genome size (Table 1; Fig. la). Onaverage, larger genomes produced more fragments(Fig. la, y = 0.06* + 8.19; F U 6 = 545.18, P < 0.0001).There was no difference in the average number of frag-ments produced for each of the +3 selective nucleotidessets (Wilcoxon x2 = 1-85, df = 3, P < 0.60). The num-ber of homoplasious fragments both within and amongchromosomes was a function of the number of frag-ments produced (Fig. lb, c). Fragment homology de-creased (homoplasy increased) with increasing fragmentproduction both within chromosomes (y = 0.08* - 0.61;FI 26 = 71.82, P < 0.0001) and among chromosomes{y= 0.37* - 4.80; F1#26 = 456.64, P < 0.0001). The nu-cleotide sequence of the Msel +3 selective nucleotidesdid not influence fragment homology within a chromo-some (Wilcoxon x2 = 0.72, df = 3, P = 0.87) or withinan entire genome (Wilcoxon x2 = 0.80, df= 3, P = 0.85).Fragments of smaller sizes were more likely to be ho-moplasious than larger fragments (Fig. Id) across all ofthe genomes sampled. This pattern was driven by thelarge number of fragments generated from the mouseand human genomes (431 out of the 454 homoplasiousfragments from all genomes).
The comparison of homology between genomesyielded contrasting results (Table 2). The four EcoRI
Dow
nloaded from https://academ
ic.oup.com/sysbio/article/56/3/477/1657880 by guest on 15 February 2022
480 SYSTEMATIC BIOLOGY VOL. 56
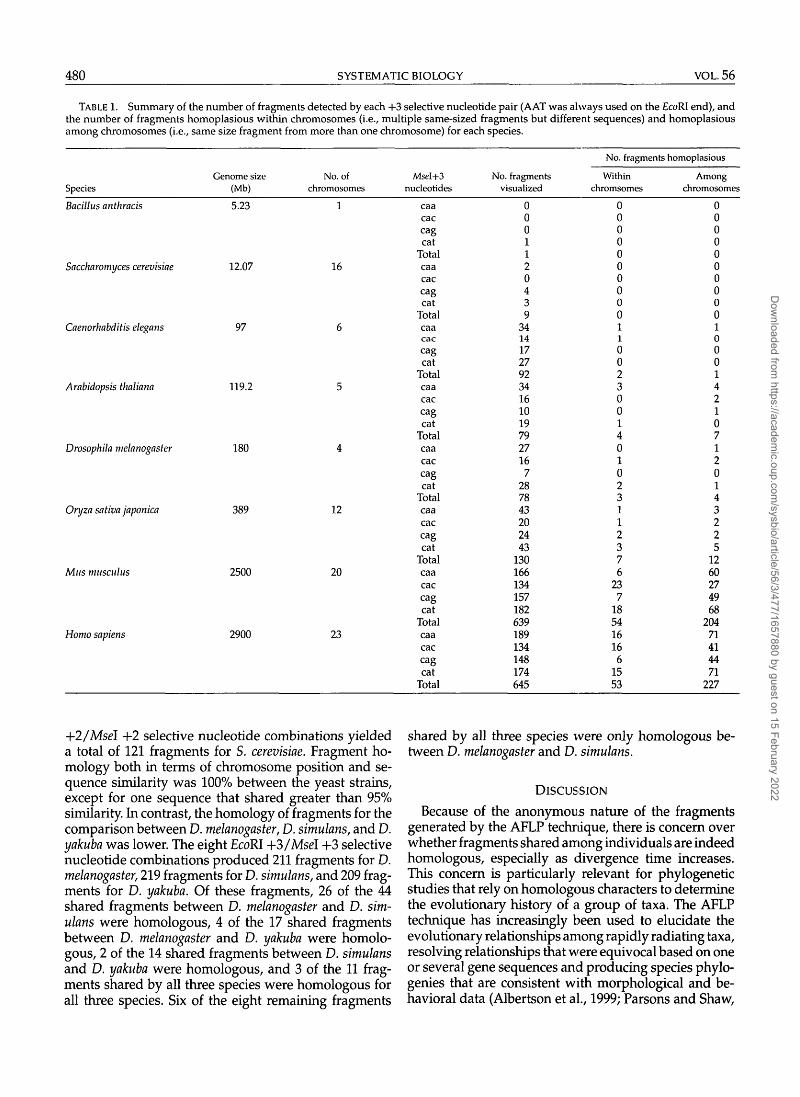
TABLE 1. Summary of the number of fragments detected by each +3 selective nucleotide pair (AAT was always used on the EcoRI end), andthe number of fragments homoplasious within chromosomes (i.e., multiple same-sized fragments but different sequences) and homoplasiousamong chromosomes (i.e., same size fragment from more than one chromosome) for each species.
Species
Bacillus anthracis
Saccharomyces cerevisiae
Caenorhabditis elegans
Arabidopsis thaliana
Drosophila melanogaster
Oryza sativa japonica
Mus musculus
Homo sapiens
Genome size(Mb)
5.23
12.07
97
119.2
180
389
2500
2900
No. ofchromosomes
1
16
6
5
4
12
20
23
MseI+3nucleo tides
caacaccagcat
Totalcaacaccagcat
Totalcaacaccagcat
Totalcaacaccagcat
Totalcaacaccagcat
Totalcaacaccagcat
Totalcaacaccagcat
Totalcaacaccagcat
Total
No. fragmentsvisualized
0001120439
3414172792341610197927167
287843202443
130166134157182639189134148174645
No. fragments homoplasious
Withinchromsomes
0000000000
I-H
1002300
I-H
401023112376
237
185416166
1553
Amongchromosomes
00000000001000142107120143225
1260274968
20471414471
227
+2/Msel +2 selective nucleotide combinations yieldeda total of 121 fragments for S. cerevisiae. Fragment ho-mology both in terms of chromosome position and se-quence similarity was 100% between the yeast strains,except for one sequence that shared greater than 95%similarity. In contrast, the homology of fragments for thecomparison between D. melanogaster, D. sitnulans, and D.yakuba was lower. The eight EcoRI +3/Msel +3 selectivenucleotide combinations produced 211 fragments for D.melanogaster, 219 fragments for D. simulans, and 209 frag-ments for D. yakuba. Of these fragments, 26 of the 44shared fragments between D. melanogaster and D. sim-ulans were homologous, 4 of the 17 shared fragmentsbetween D. melanogaster and D. yakuba were homolo-gous, 2 of the 14 shared fragments between D. simulansand D. yakuba were homologous, and 3 of the 11 frag-ments shared by all three species were homologous forall three species. Six of the eight remaining fragments
shared by all three species were only homologous be-tween D. melanogaster and D. simulans.
DISCUSSION
Because of the anonymous nature of the fragmentsgenerated by the AFLP technique, there is concern overwhether fragments shared among individuals are indeedhomologous, especially as divergence time increases.This concern is particularly relevant for phylogeneticstudies that rely on homologous characters to determinethe evolutionary history of a group of taxa. The AFLPtechnique has increasingly been used to elucidate theevolutionary relationships among rapidly radiating taxa,resolving relationships that were equivocal based on oneor several gene sequences and producing species phylo-genies that are consistent with morphological and be-havioral data (Albertson et al, 1999; Parsons and Shaw,
Dow
nloaded from https://academ
ic.oup.com/sysbio/article/56/3/477/1657880 by guest on 15 February 2022

2007 ALTHOFF ET AL.—HOMOLOGY OF AFLP FRAGMENTS 481
a)
"S 175
15 150-
•g 125-CO
c 100-(U
a)•*= 50 •dZ 25 •
0 •
1
c)
80-
« 70-© CO
F a) 6 0 -
5 8 50-w go | 4 0 "S o 30-9- °>g § 20-o E•c ca io -62 0-
•
•f *• 1
10 100 1000
genome size (Mb)
•
m
•
• • • • • • •%•*% •
b)2!5 •
I 20-03 w
E CD
o) £5 §15"(0 O
2 E.2 o
opia
sin
chr o
O •> 3
d2 0 -
*
* *
• • • •
•I 10 100
No. fragments visualized
d)
100-
CO
cE 7 5
5*"^ 50-oz
25 -
1 10 100No. fragments visualized
50 100 150 200 250 300 350 400 450fragment size (bp)
FIGURE 1. Relationship between genome size and homoplasy of AFLP fragments. Only genomes that produced bands are displayed, (a) Thenumber of fragments produced as a function of genome size, (b) The number of homoplasious fragments (i.e., comigrating) within a chromosomeas a function of the number of fragments detected, (c) The number of homoplasious fragments (i.e., comigrating) from across the entire genomeas a function of number of fragments detected, (d) Frequency distribution of the fragment sizes that were homoplasious from the eight genomessampled (431 of 454 fragments were from the mouse and human genomes).
2001; Ogden and Thorpe, 2002; Despres et al., 2003; Sul-livan et al., 2004; Althoff et al., 2006). Many researchers,including the authors, have operated under the assump-tion that AFLP fragments are homologous or that onlya very small percentage of the fragments are homopla-sious. Moreover, another assumption is that the resultingphylogenetic signal from homologous fragments wouldovercome any weak signal from homoplasious mark-ers. Koopman (2005) surveyed the phylogenetic signalpresent in AFLP datasets by comparing AFLP phylo-genetic analyses to analyses of ITS sequence data in awide range of taxa (plants, fungi, and bacteria). He foundthat ample phylogenetic signal is present within AFLPdatasets and the phylogenetic results were largely con-sistent with those derived from sequence data.
Our results from using bioinformatics tools to performin silico AFLP procedures on entire genomes demon-strated that fragment homology within individuals canbe quite high, but is dependent on genome size. For thegenomes under 400 Mb, on average 11% (range 0% to18.65%) of the fragments comigrating were homopla-
sious. This result suggests that 89% of the fragments gen-erated by the AFLP technique are homologous. For themouse and human genomes that are over 2 Gb in size,however, fragment homology was considerably lower.On average, 41.5% (range 33% to 49%) of the fragmentswere homoplasious. This pattern is driven by the numberof fragments produced per genome. The proportion ofhomologous fragments was negatively correlated withthe number of AFLP fragments, and larger genomes pro-duced more fragments. There are two potential ways toreduce homoplasy in these larger genomes. One is to ex-clude smaller sized fragments from analysis. Vekemanset al. (2002) demonstrated that smaller sized fragmentsare more likely to be homoplasious. Our results con-firm this observation, although there was no clear cutoff point for fragment size and a decrease in homo-plasy (Fig. Id). For example, eliminating fragments lessthan 125 bp from the genomes analyzed would helpreduce the levels of homoplasy by approximately onethird on average, but there are still markers greaterthan 125 bp that are homoplasious. A second way to
Dow
nloaded from https://academ
ic.oup.com/sysbio/article/56/3/477/1657880 by guest on 15 February 2022

482 SYSTEMATIC BIOLOGY VOL. 56
TABLE 2. (a) Summary of the homology of fragments detected by each MseI+2 selective nucleotide set (AA was always used on the EcoRIend), for two strains of Saccharomyces cerevisiae. Each shared fragment between S. cerevisiae strains originated from the same chromosome, (b)Fragment homology for each MseI+3 selective nucleotide set (AAT was always used on the EcoRI end) for Drosophila melanogaster, D. simulans,and D. yakuba. In contrast with S. cerevisiae, many shared fragments originated from different chromosomes and were not homologous.
Msel+2nucleotides
cacccgctTotal
Msd+3nucleotides
caacaccagcatttattc
"Sttt
Total
(a) Saccharomyces cerevisiae strain RMll-la compared to strain YMJ789
No. fragments visualized
RMll-lc
37202539
121
i YMJ789
37202539
121
No. fragmentsshared
37202539
121
Fragments with sequence similarity of
100%
37202538
120
(b) Homology of fragments detected in Drosophila melanogaster, D. simulans, and D. yakuba
No. fragments visualized
melanogaster
27177
2823174349
211
simulans
27147
2131164261
219
yakuba
2516152220183756
209
melanogaster andsimulans
7(6)2(2)03(2)7(4)2(2)
11(6)12(4)
44 (26)
>95%
———11
No. shared fragments between (no. homologous)
melanogasterand yakuba
2(1)003(2)01(0)5(1)6(0)
17(4)
simulans andyakuba
1(1)0004(1)02(0)7(0)
14(2)
All species
1 (1 mel. & sim., 0 for all)00
2 (2 for all)0
1 (1 for all)4 (2 mel. & sim., 0 for all)3 (3 mel. & sim., 0 for all)11 (6 mel. & sim., 3 for all)
reduce homoplasy is to use more stringent primers (suchas four selective nucleotides). Indeed, a simulation us-ing a four selective nucleotide primer pair (aatt andcatt) on the mouse genome yielded 33 fragments, onlyone of which was homoplasious. There is a tradeoffin that the number of phylogenetic markers decreaseswith more selective conditions, but for large genomesthis may be the only means of increasing markerhomology.
The comparisons of fragment homology betweenclosely related taxa suggested that homology and thephylogenetic usefulness of AFLPs will vary, dependingon time since divergence. We used time since diver-gence as a proxy for whole-genome divergence, underthe assumption that increasing time since divergenceequates with an increase in whole genome divergence.Comparisons among baker's and brewer's yeast strainsdemonstrated complete homology of the AFLP frag-ments generated. If we assume that the evolutionarydistance among the yeast strains approximates evolu-tionary distance among populations within a species,AFLP markers should be quite useful in determiningphylogeographic patterns. We must highlight the caveatthat the high homology of fragments for our comparisonof yeast may be biased by the fact that yeast has a smallgenome. The comparison between D. melanogaster, D.simulans, and D. yakuba, however, indicates that there isan upper limit to fragment homology among divergedtaxa. Drosophila melanogaster and D. yakuba are estimatedto have diverged approximately 6 million years ago,and D. melanogaster and D. simulans about 2 millionyears ago (Russo et al., 1995). The D. melanogaster-D.simulans genomes shared more bands (44) than eithershared with D. yakuba (17 for melanogaster-yakuba and 14
for simulans-yakuba), and the proportion of homologousbands was also higher. Although more than 200 bandswere generated for each species, only a small numberof these were shared among all of them. This findinghas two implications. First, many of the fragments willbe unique to a taxon rather than shared among taxa.Second, only a very small proportion of the fragmentswill determine the relationships among taxa and aportion of these will be homoplasious. The results fromthis study fit well with the caution given by othersthat AFLPs should only be used to assess relationshipsof very closely related taxa (O'Hanlon and Peakall,2000; Bussell et al., 2005; Koopman, 2005). We alsosuggest that AFLPs should be used only in instanceswhere DNA sequence data are insufficiently variableto resolve the phylogeny. For larger genomes (greaterthan 400Mb), fragment homology can be increased bydiscarding smaller fragments and using more stringentselective primers in the final selective amplificationstep.
The Future of AFLPs in Phylogenetics
The appeal of AFLPs for phylogenetics is that the tech-nique generates large numbers of markers and surveysthe entire genome rather than just a single gene or asmall number of genes. The issue of fragment homology,however, is a major concern in using AFLPs in phyloge-netic studies. The current study, combined with previ-ous work, suggests that this issue is one that will not beresolved easily without considerable effort to comparehomology for organisms with small and large genomesand at all hierarchical levels—from within individuals
Dow
nloaded from https://academ
ic.oup.com/sysbio/article/56/3/477/1657880 by guest on 15 February 2022

2007 ALTHOFF ET AL.—HOMOLOGY OF AFLP FRAGMENTS 483
to between species within genera. The completion ofongoing genome sequencing projects of closely relatedspecies, especially Drosophila, will be extremely valu-able in testing the homologous nature of AFLP mark-ers as a function of time since divergence among taxa.The current results demonstrate that not all AFLP mark-ers of a given length are homologous, and we sug-gest that researchers should approach the interpretationof phylogenetic trees with caution, especially withoutthe added benefit of congruent relationships from otherdata such as morphology, behavior, or other molecularmarkers.
Another issue confronting the use of AFLPs in phylo-genetics is the limited avenues of phylogenetic analysis.Currently, AFLPs are analyzed in predominately twoways—by using parsimony on the presence and absenceof fragments or converting the AFLP fragment patternsinto a distance measure and using minimum evolutionor neighbor joining search algorithms to determinethe best phylogenetic tree. Both approaches assumethat fragments of the same size are homologous acrossall the taxa analyzed. Our results clearly demonstratethat this assumption is going to be violated for sometaxonomic comparisons. An additional, and muchmore tenuous assumption that we have not tested hereis that the absence of a fragment is homologous as well.Alternatively, converting the fragment patterns into adistance matrix assumes that genetic distance is a goodmeasure of evolutionary divergence and evolutionarydivergence translates into evolutionary history. The useof both parsimony and distance approaches have beena source of contention in phylogenetics and have ledto the use of alternative analytical approaches. We aresomewhat constrained in using likelihood and Bayesianphylogenetic approaches because there are currentlyvery few models of evolution describing the patternof marker gain and loss, and these models are basedon restriction fragment patterns rather than the AFLPtechnique per se. For example, the F81 model for binarydata is available for use in maximum likelihood analysesimplemented in TreePuzzle (Schmidt et al., 2002), andthe RESTRICTION model that allows different ratesof fragment gain and loss, and rate differences acrossmarkers is available in Bayesian methods implementedin MrBayes v3.1 (Ronquist and Huelsenbeck, 2003).Models based on restriction site patterns represent a sim-plified picture of AFLP fragment evolution (Felsenstein,2004) and to date there has been no empirical evaluationof their performance for AFLPs. Moreover, as the resultsdemonstrated, genome size may be a critical factorin determining the homology of fragments, and theirgain and loss. In addition, other issues such as markernonindependence and clustering of markers based onG+C content need to be evaluated and incorporated intoevolutionary models of AFLP markers. The continuingdevelopment of such models will advance the utilityof AFLP markers in phylogenetic studies, althoughthe current work indicates that homology assessmentwill continue to be an essential component of anystudy.
ACKNOWLEDGMENTS
We thank Jack Sullivan for spurring us to think critically about thehomology of AFLP fragments, Jason Machacek for generating a pre-liminary version of the software used in the manuscript, and CelesteBrown for facilitating the interaction between biologists and computerprogrammers. We also thank the IBEST program at the University ofIdaho for computer time and support. Frank (Andy) Anderson and twoanonymous reviewers provided very valuable criticism that greatlyimproved the manuscript. This work was funded by NSF grant DEB0321293 to David Althoff and Olle Pellmyr and NSF grant DBI0306164to Kari Segraves.
REFERENCES
Albertson, R. C, J. A. Markert, P. D. Danley, and T. D. Kocher. 1999. Phy-logeny of a rapid evolving clade: The cichlid fishes of Lake Malawi,East Africa. Proc. Natl. Acad. Sci. 96:5107-5110.
Althoff, D. M., K. A. Segraves, J. Leebens-Mack, and O. Pellmyr. 2006.Patterns of speciation in the yucca moths: Parallel radiations withinthe Tegeticula yuccasella species complex. Syst. Biol. 55:398-410.
Bensch, S., and M. Akesson. 2005. Ten years of AFLP in ecology andevolution: Why so few animals? Mol. Ecol. 14:2899-2914.
Brouat, C, D. McKey, and E. J. P. Douzery. 2004. Differentiation ina geographical mosaic of plants coevolving with ants: Phylogenyof the Leonardoxa africana complex (Fabaceae:Caesalpinioideae) us-ing amplified fragment length polymorphism markers. Mol. Ecol.13:1157-1171.
Bussell, J. D., M. Waycott, and J. A. Chappill. 2005. Arbitrarily ampli-fied DNA markers as characters for phylogenetic inference. Perspect.Plant Ecol. Evol. Syst. 7:3-26.
Despres, L., L. Gielly, W. Redoutet, and P. Taberlet. 2003. Using AFLP toresolve phylogenetic relationships in a morphologically diversifiedplant species complex when nuclear and chloroplast sequences failto reveal variability. Mol. Phylogenet. Evol. 27:185-196.
Giannasi, N., R. Thorpe, and A. Malhotra. 2001. The use of amplifiedfragment length polymorphism in determining species trees at finetaxonomic levels: Analysis of a medically important snake, Trimeresu-rus albolabris. Mol. Ecol. 10:419-426.
Innan, H., R. Terauchi, G. Kahl, and F. Tajima. 1999. A method forestimating nucleotide diversity from AFLP data. Genetics 151:1157-1164.
Koopman, W. J. M. 2005. Phylogenetic signal in AFLP data sets. Syst.Biol. 54:197-217.
Koopman, W. J. M., and G. Gort. 2004. Significance tests and weightedvalues for AFLP similarities, based on Arabidopsis in silico AFLPfragment length distributions. Genetics 167:1915-1928.
Mechanda, S. M., B. R. Baum, D. A. Johnson, and J. T. Arnason.2004. Sequence assessment of comigrating AFLP (TM) bands inEchinacea—implications for comparative biological studies. Genome47:15-25.
Mendelson, T. C, and K. L. Shaw. 2005. Rapid speciation in an arthro-pod. Nature 433:375.
Mueller, U. G., and L. L. Wolfenbarger. 1999. AFLP genotyping andfingerprinting. Trends Ecol. Evol. 14:389-394.
Ogden, R., and R. S. Thorpe. 2002. The usefulness of amplified frag-ment length polymorphism markers for taxon discrimination acrossgraduated fine evolutionary levels in Caribbean Anolis lizards. Mol.Ecol. 14:437^45.
O'Hanlon, P. C, and R. Peakall. 2000. A simple method for the detectionof size homoplasy among amplified fragment length polymorphismfragments. Mol. Ecol. 9:815-816.
Parsons, Y. M., and K. L. Shaw. 2001. Species boundaries and geneticdiversity among Hawaiian crickets of the genus Laupala identifiedusing amplified fragment length polymorphism. Mol. Ecol. 10:1765-1772.
Russo, C. A. M., N. Takezaki, and M. Nei. 1995. Molecular phylogenyand divergence times of Drosophilid species. Mol. Biol. Evol. 12:391—404.
Sullivan, J. P., S. Lavoue, M. E. Arnegard, and C. D. Hopkins.2004. AFLPs resolve phylogeny and reveal mitochondrial introgres-sion within a species flock of African electric fish (Mormyroidea:Teleostei). Evolution 58:825-841.
Dow
nloaded from https://academ
ic.oup.com/sysbio/article/56/3/477/1657880 by guest on 15 February 2022

484 SYSTEMATIC BIOLOGY VOL. 56
Vekemans, X., T. Beauwens, M. Lemaire, and I. Roldan-Ruiz. 2002. Waugh, R., N. Bonar, E. Baird, B. Thomas, A. Graner, P. Hayes,Data from amplified fragment length polymorphism (AFLP) mark- and W. Powell. 1997. Homology of AFLP products in threeers show indication of size homoplasy and of a relationship between mapping populations of bark'y. Mol. Gen. Genet. 255:311-degree of homoplasy and fragment size. Mol. Ecol. 11:139-151. 321.
Vos, P., R. Hogers, M. Bleeker, M. Reijans, T. van de Lee, M. Homes, A.Frijters, J. Pot, J. Peleman, M. Kuiper, and M. Zabeau. 1995. AFLP: A First submitted 27 September 2006; reviews returned 24 November 2006;new technique for DNA fingerprinting. Nucleic Acids Res. 23:4407- final acceptance 8 March 20074414. Associate Editor: Frank Anderson
Dow
nloaded from https://academ
ic.oup.com/sysbio/article/56/3/477/1657880 by guest on 15 February 2022