binding-induced stabilization and assembly of the phage p22 tail accessory factor gp4
TRANSCRIPT

doi:10.1016/j.jmb.2006.08.014 J. Mol. Biol. (2006) 363, 558–576
Binding-induced Stabilization and Assembly of thePhage P22 Tail Accessory Factor Gp4
Adam S. Olia1, Jawdat Al-Bassam2, Danella A. Winn-Stapley3
Lisa Joss4, Sherwood R. Casjens3 and Gino Cingolani1⁎
1Department of Biochemistryand Molecular Biology, SUNYUpstate Medical University,750, E. Adams Street, Syracuse,NY 13210, USA2Harvard Medical School,Harvard University, 250Longwood Avenue, Boston,MA 02115, USA3Department of Pathology,University of Utah School ofMedicine, Salt Lake City,UT 84112, USA4Department of Biochemistry,University of Utah MedicalSchool, Salt Lake City,UT 84112, USAPresent address: D. A. Winn-StapBiology, Massachusetts Institute of TCambridge, MA 02139, USA.Abbreviations used: gpx, protein p
isothermal titration calorimetry.E-mail address of the correspondi
0022-2836/$ - see front matter © 2006 E
To infect and replicate, bacteriophage P22 injects its 43 kbp genome acrossthe cell wall of Salmonella enterica serovar Typhimurium. The attachment ofphage P22 to the host cell as well as the injection of the viral DNA into thehost is mediated by the virion's tail complex. This 2.8 MDa molecularmachine is formed by five proteins, which include the portal protein gp1, theadhesion tailspike protein gp9, and three tail accessory factors: gp4, gp10,gp26. We have isolated the tail accessory factor gp4 and characterized itsstructure and binding interactions with portal protein. Interestingly, gp4exists in solution as a monomer, which displays an exceedingly lowstructural stability (Tm 34 °C). Unfolded gp4 is prone to aggregationwithin anarrow range of temperatures both in vitro and in Salmonella extracts. In thevirion the thermal unfolding of gp4 is prevented by the interaction with thedodecameric portal protein, which stabilizes the structure of gp4 andsuppresses unfolded gp4 from irreversibly aggregating in the Salmonellamilieu. The structural stabilization of gp4 is accompanied by theconcomitant oligomerization of the protein to form a ring of 12 subunitsbound to the lower end of the portal ring. The interaction of gp4 with portalprotein is complex and likely involves the distinct binding of two non-equivalent sets of six gp4 proteins. Binding of the first set of six gp4equivalents to dodecameric portal protein yields a gp(1)12:gp(4)6 assemblyintermediate, which is stably populated at 30 °C and can be resolved bynative gel electrophoresis. The final product of the assembly reaction is a bi-dodecameric gp(1)12:gp(4)12 complex, which appears hollow by electronmicroscopy, suggesting that gp4 does not physically plug the DNA entry/exit channel, but acts as a structural adaptor for the other tail accessoryfactors: gp10 and gp26.
© 2006 Elsevier Ltd. All rights reserved.
*Corresponding author
Keywords: bacteriophage P22; portal protein; gp4; gp10; gp26Introduction
P22 is double-stranded DNA (dsDNA) bacter-iophage that infects Salmonella enterica serovarTyphimurium. The mature phage virion is charac-terized by an icosahedral T=7 capsid approximately650 Å in diameter formed by 415 copies of coatprotein gp5.1–5 The icosahedral symmetry is inter-
ley, Department ofechnology,
roduct of gene x; ITC,
ng author:
lsevier Ltd. All rights reserve
rupted at a single vertex of the mature capsid by thetail apparatus, or “portal vertex structure”, thatprotrudes about 320 Å outside the coat protein shell6
and occupies the position that would have beenoccupied by five coat protein molecules in a perfectT=7 shell lattice (G. Lander, J. Johnson & S.R.C.,unpublished results). The tail apparatus is used byP22 to attach to the host cell wall and inject itsgenetic material into the host and contains only fiveproteins: a dodecameric ring of portal protein: gp1(12×83.5 kDa),7 six molecules of the trimerictailspike protein: gp9 (6×215.4 kDa),8 and severalcopies of each of the three tail accessory proteins(also called head completion proteins): gp4(18 .0 kDa) , gp10 (52 .3 kDa) , and gp26(24.7 kDa).9,10 Despite its relatively large size(about 2.8 MDa) the P22 tail complex is simpler
d.

559Assembly of Gp4 to P22 Portal Protein
than those observed in most other tailed-bacterio-phages such as T411,12 or T7.13
The assembly pathway of the P22 bacteriophagehas been extensively studied both in vivo and invitro.14 The mature P22 virus assembles via forma-tion of a T=7 meta-stable intermediate known as aprocapsid, which is formed by 415 molecules of thecoat protein gp5, 250−300 molecules of the scaffold-ing protein gp8 and one dodecameric ring of theportal protein gp1.15–17 Three minor non-structuralproteins: gp7, gp16, and gp20 are also present inthe procapsid and, although they are not requiredfor either gp1 incorporation or DNA packaging,they are essential for infectivity of the matureparticle.18 Concatameric P22 DNA is packaged intothe newly formed procapsid in a process thatrequires ATP hydrolysis in vitro.19,20 The 43 kbp P22chromosome is introduced into the virion throughthe portal protein ring with the aid of a terminasecomplex, formed by gp2 and gp3.21 Particlesmissing the portal protein do not package DNAor assemble a tail complex and form non-functionalprocapsid-like particles (PLP).18 Portal-less PLPsfail to mature into infectious particles and representdead-end products.19,20 Concomitantly with DNApackaging, the meta-stable procapsid coat proteinshell matures into a virion-like particle. Thisthermodynamically “down-hill” process22 is adramatic quaternary structural rearrangement inwhich the capsid shell undergoes an approximately10% expansion, and becomes more angular with athinner shell.16
After packaging, DNA is retained inside the viralcapsid in a quasi-crystalline state at concentrations inthe order of 500 mg/ml.23,24 To avoid chromosomeleakage, the portal vertex is stabilized by three P22-encoded tail accessory factors: gp4, gp10, and gp26,also known as portal closure or head completionproteins. gp4 is the first factor to bind to the growingtail structure after DNA is packaged, followed bygp10, and gp26.25 This is followed by the attach-ment of the six trimeric gp9 tailspikes,26,27 whichyields the mature infectious virion. Interestingly, allmutant phages gp4−, gp10− and gp26− encapsidateDNA efficiently and share a similar “DNA-leakage”phenotype, which results in a rapid loss of DNA.This indicates that gp4, gp10, and gp26 are notdirectly involved in DNA packaging, but are criticalto close the portal protein channel and retain thedsDNA inside the capsid.25,28
The framework of binding interactions within theP22 tail has been only partially elucidated usinggenetic analysis and electron microscopy. Forinstance, it is known that only tail accessory proteinsgp10 and gp4 are required for gp9 addition,18,25
which attaches efficiently to capsids that lackgp26.19 In contrast, gp10− capsids lack the tailspikeattachment site, suggesting that gp10, and notgp4, may physically connect the tailspike proteinto the virion.19 More recently it was reportedthat tail accessory factor gp4 possesses mureinhydrolase activity, which is essential to digest thepeptidoglycan layer present in the periplasm of
Salmonella.29 A 20 Å resolution cryo-EM reconstruc-tion of P22 tail extracted from mature phage wasalso recently reported.6 The structure reveals theoverall molecular architecture of the phage P22 tail.The positions of the gp1 portal protein and gp9tailspike were unambiguously localized in the cryo-EM reconstruction, by virtue of their size anddistinguishable 3D-topology. Likewise, based onthe biochemical data available for recombinantgp26,30 the elongated density emanating from theneck of the tail is thought to be gp26. In contrast, theother two tail accessory proteins gp4 and gp10, werenot seen as discrete structural units at the resolutionof the reconstruction, and were assigned to theelectron density between the portal protein ring andthe tailspikes. It was speculated that the oligomericstates of gp4 and gp10 were a dodecamer and ahexamer, respectively.6
Here we isolated the tail accessory factor gp4 andcharacterized its structural stability and bindinginteraction with the portal vertex structure. Our dataindicate that gp4 exists in solution as a monomer,which oligomerizes upon binding to the portal ring,generating a second molecular ring bound beneaththe portal ring.
Results
P22 tail accessory factor gp4 exists as anelongated monomer in solution
Genes 4, 10, and 26 map contiguously in the P22genome, where they are part of a single operon.31,32
The gene encoding gp4 was PCR amplified from P22DNA and cloned into a pET expression vector (seeMaterials and Methods), which yields recombinantgp4 fused to an N-terminal six-histidine tag (Mr20.2 kDa=18.02 kDa gp4 and 2.2 kDa of vector-encoded residues). The soluble protein was purifiedfrom the Escherichia coli extract by metal chelatingchromatography. It displays high solubility atneutral pH. To define the oligomeric state of gp4 insolution, pure gp4 was separated on a size exclusionchromatography (SEC) Superdex 200 column, whichresolves macromolecules in a size range of 10 to600 kDa. Under ionic strength conditions between 50and 250 mM NaCl at 4 °C gp4 is a monodispersespecies eluting as a globular protein having anapparent molecular mass of about 30 kDa (Figure1(a)), which could correspond to either a dimer or aslightly elongatedmonomer of gp4. To determine theexact oligomeric state of unassembled gp4 protein insolution, gel filtration-purified gp4 was subjected tosedimentation equilibrium analysis. This techniqueprovides a shape-unbiased measurement of the pro-tein mass.33 Figure 1(b) (bottom panel) shows theconcentration of gp4 protein as a function of positionin the rotor cell after equilibrium was attained. Theequilibrium data were fit at three different speedsusing non-linear least squares analysis programNONLIN.34 The final fit shown in Figure 1(b) (top

560 Assembly of Gp4 to P22 Portal Protein
panel) resulted from the simultaneous fitting of threedifferent concentration distributions. The gp4 pro-tein fits a monomer model very well with amolecular massMr=20.3(±1.3) kDa. Thus, at neutralpH and under physiological conditions gp4 proteinexists as a monomer in solution. The apparent gp4molecular mass of 30 kDa measured by gel filtrationlikely indicates that the protein adopts an elongatedshape in solution.
Gp4 is a helical protein that displays lowstructural stability
We next investigated the secondary structurecontent of gp4 in solution using circular dichroism(CD). Spectra recorded at 15 °C displayed a double
minimum at 208 and 222 nm and a maximum at196 nm, which is characteristic of α-helices (Figure 2(a)). The helical content estimated on the basis of thisspectrum is about 60%,which is consistentwith awellfolded, largely helical protein. This agrees well withthe helical content predicted by the secondarystructure prediction programPHD,35 which identifiesthree regions with high helical propensity within theN-terminal 125 amino acid residues of gp4 (see Figure8(a) below). In contrast, theC-terminal region of gp4 ispredicted to adopt a random-coil structure. This issupportedby the observation thatC-terminal residues125−166 of gp4 lack branched amino acids (valine,leucine and isoleucine), which are known to stabilizeβ-sheets, and contains five glycine and five prolineresidues, which are incompatible with helical folding.
Figure 1. Characterization of re-combinant gp4 protein. (a) Gelfiltration analysis of purified gp4.The Superdex 200 gel filtrationchromatography column was cali-brated using molecular mass mar-kers, whose elution volumes areindicated by the arrows. The appar-ent Mr for gp4 corresponds toabout 30 kDa. (b) Sedimentationequilibrium analysis of bacterioph-age P22 gp4 protein. The rightpanel shows the experimental datapoints for three different loadingconcentrations of N-terminally his-tidine-tagged gp4 protein, with thecorresponding calculated curve fit(continuous line). The gp4 proteinfits a monomer model with amolecular mass Mr=20.3(±1.3) kDa.

561Assembly of Gp4 to P22 Portal Protein
To assess the structural stability of gp4 in solution,we took advantage of the high helical content andstudied the thermal unfolding of the protein bycircular dichroism. Denaturation curves wererecorded following the variation in ellipticity at222 nm as a function of the temperature between10 °C and 80 °C. Under these conditions, monomericgp4 unfolded irreversibly as a single entity with anapparent thermal melting temperature (Tm) of 34 °C(Figure 2(b)). The slope of the transition between
Figure 2. Secondary structure content and structuralstability of purified gp4. (a) Characterization of thesecondary structure content of gp4 by circular dichroism.The CD scan of gp4 was recorded in PBS at 10 °C from196−260 nm. (b) Thermal denaturation of gp4 at pH 7.4.Changes in the mean ellipticity at 222 nm as a function oftemperature were measured at 1 deg.C increments atconstant intervals of 120 s. The gp4 protein shows a Tm of34 °C. Although temperature melts were performed to amaximum temperature of 80 °C, the horizontal axis in (b) isonly shown between 15 and 50 °C. (c) Chemical denatura-tion of gp4 in guanidine hydrochloride. Samples wereallowed to equilibrate over a period of 12 h at 15 °C in PBSat pH 7.4, after which the intrinsic fluorescence emission ofgp4 trypthophan residues was recorded at 370 nm.
native and denatured states is sigmoidal, suggestingthat the denaturation of the helices in gp4 is highlycooperative.36 Cooling from 80 °C back to 10 °Cresulted in incomplete refolding of gp4, indicatingthat under these conditions unfolding is irreversible.The apparent temperature of melting observed forgp4 (34 °C) is lower than that of most globularproteins,36 and likely reflects the intrinsic instabilityof the protein at 37 °C. To rule out the possibility thatsuch low stability was caused by the oligo-histidinetag, we also cloned gp4 fused to an intein tag. Thislarge tag is used for affinity purification on chitinbeads and self-cleaves in the presence of dithiothrei-tol, yielding native gp4 fused to only an N-terminalglycine. This gp4 displayed a low Tm identical to thatof His-tagged gp4 (data not shown), showing thatthe oligo-histidine moiety does not influence theoverall structural stability of the protein.In a complementary set of experiments, the
stability of gp4 was probed by measuring guanidinehydrochloride-induced equilibrium unfolding atneutral pH (Figure 2(c)). To prevent thermal unfold-ing, all equilibrium measurements were carried outat 15 °C, temperature at which gp4 appears fullyfolded by CD. Under these conditions, gp4 unfoldedirreversibly with a mid-point molarity of 1.3 Mguanidine. At comparable values of pH and ionicstrength, lysozyme, a murein hydrolase similar togp4 both in size and function, melts with amid-pointof 3.01 M guanidine.37 The denaturation profile seenfor gp4 (Figure 2(c)) fits a two-state model, whichwas used to calculate a ΔGH2O associated to theunfolding reaction to be about 3.5 kcal/mol. Theconformational stability (ΔGH2O) defines the energyneeded to convert native gp4 inwater from its foldedconformation into a random coil polypeptide chain.This value is lower than that observed for most smallmonomeric globular proteins like lysozyme orribonuclease A, whose thermodynamic stability atneutral pH ranges between 6 and 14 kcal/mol.38 TheΔGH2O calculated from gp4 GuHCl-equilibriumunfolding curve at 15 °C confirms that monomericgp4 adopts a folded yet poorly stable structure insolution. In contrast to “natively unfolded” pro-teins,39,40 which display poor folding and structuralstability under all experimental conditions (tempera-tures, ionic strength, concentration, etc), gp4 isfolded at room temperature but unfolds sponta-neously above 34 °C. This remarkable temperature-sensitivity in vitro may suggest that in its nativeenvironment, gp4 quickly unfolds if not promptlyassembled in the tail apparatus.
Gp4 aggregates in solution in a temperature andtime-dependent manner
To further characterize the structural stability ofgp4, we investigated if unfolded gp4 is susceptibleto aggregation and if this process takes place in atemperature-dependent manner. To address thisquestion, we developed an aggregation assay inSalmonella extract. In this assay folded gp4 that wasexpressed at 22 °C and purified at 4 °C, was added

562 Assembly of Gp4 to P22 Portal Protein
to a Salmonella extract at a final concentration of0.5 mg/ml. The extract, which has ionic strength,protein composition and concentration comparableto that of the host environment, was incubated for60 min at various temperatures between 24−44 °Cand then centrifuged at 4 °C to separate solubleproteins from high molecular mass aggregates.Pellet and supernatant fractions containing aggre-gated and folded gp4, respectively, were blottedonto a PVDF-membrane and His-tagged gp4 wasdetected using monoclonal anti-Penta-His antibody.As visible in Figure 3(a), the percentage of aggre-gated gp4 detected in the pellet fractions increasedlinearly in a temperature range 33−37 °C. In anarrow range of four degrees, the percentage ofsoluble gp4 detected in the supernatant fractionvaried from >80% at 33 °C (Figure 3(a), lane 5) to<20% at 37 °C (Figure 3(a), lane 9). This demon-strates that unfolded gp4 is highly prone toirreversible aggregation and such a process takes
incubated at various temperatures as in (a) and soluble and inanalyzed on 12.5% SDS-PAGE. The molecular mass markers arextract. gp4was expressed andpurified fromsolubleE.coli extracat 37 °C. Aggregation of gp4 as function of time was monitoredfraction (P) and soluble gp4 in the supernatant (Sn), were anmembrane. His-tag gp4 was detected using monoclonal α-Pensupernatant fraction, consistentlywith the high solubility of recoincubation approximately 50% of the protein is found in the pdecreases as a function of time: after 10min∼90%of the protein∼1 h, which agrees well with the data presented in (a) (see inte
place in Salmonella extracts at temperature close tothe apparent Tm∼34 °C measured by CD. At 37 °C,temperature at which phage P22 replicates in thehost, most gp4 is predicted to be unfolded andaggregates irreversibly to form large molecular massaggregates.The aggregation assay for gp4 was also repeated
in vitro using recombinant purified gp4. In thiscomplementary assay the concentration of gp4 wasleft at 0.5 mg/ml in PBS containing 150 mM sodiumchloride. As shown in Figure 3(b) lane 11, only25−30% of gp4 was found to aggregate at 37 °C,while more pronounced aggregation was observedat 44 °C (Figure 3(b), lane 15). Taken together thesedata indicate at the protein concentration used in theassay, 0.5 mg/ml, gp4 is less prone to aggregation inits purified form than in Salmonella extracts. This isconsistent with the notion that protein aggregationis enhanced at high protein concentration in thecrowded environment of the cell.41
Figure 3. Temperature-depen-dent aggregation of gp4. (a) Aggre-gation of gp4 in a Salmonellaextract. Purified recombinant gp4was added to a Salmonella extract ata final concentration of 0.5 mg/ml.The extract was incubated at var-ious temperatures between 24 °Cand 44 °C for 1 h, after whichinsoluble aggregates were pelletedby centrifugation at 18,000g for30 min at 4 °C. Pellet (P) andsupernatant (Sn) containing insolu-ble and soluble gp4, respectively,were dissolved in SDS-Loading buf-fer and separated on 15%acrylamideSDS-PAGE. Proteins were blottedonto a PVDF-membrane and His-tagged gp4 was detected using amonoclonal α-Penta-His antibody.Folded gp4 is largely present in thesoluble fraction between 24−33 °C(lanes 1 and 3, respectively).Between 35 °C and 39 °C most gp4unfolds and aggregates, which isdetected almost entirely in the pelletfraction (compare lane 6 to lane 12).The negative control at 44 °C alsoshows gp4 entirely aggregated in thepellet fraction (lane 14). (b) In vitroaggregation of gp4 monitored bySDS-PAGE. Gp4 at 0.5 mg/ml was
soluble fractions, indicated as Sn and P, respectively, weree in lane 1. (c) Time-course of gp4 aggregation in Salmonellats at 22 °C and added to a Salmonella extract pre-equilibratedbetween 0−60 min. Insoluble aggregates of gp4 in the pelletalyzed on a 12.5% SDS-PAGE and blotted onto a PVDFta-His antibody (Qiagen). At t=0, all gp4 is found in thembinant gp4 expressed and purified at 22 °C. After 1min ofellet fraction (lane 4) and the ratio soluble/aggregated gp4is fully aggregated (lane 10). This fraction is almost 99% afternsity of aggregated gp4 in (a), lane 10).

563Assembly of Gp4 to P22 Portal Protein
The idea that gp4 is unstable under physiologicalconditions was further explored by determining therate at which gp4 aggregates at 37 °C in Salmonellaextract. In this assay, soluble His-gp4 expressed at22 °C and purified at 4 °C was added to a Salmonellaextract pre-equilibrated at 37 °C at a final concentra-tion of ∼0.5 mg/ml. The mixture was incubated at37 °C, and aliquots were taken after 1, 3, 5, 10, 30 and60 min. Aggregated gp4 was separated from solubleprotein by centrifugation at 4 °C. Pellet and super-natant fractions containing aggregated and solublegp4, respectively, were blotted onto a PVDF-mem-brane and detected usingmonoclonal anti-Penta-Hisantibody. As shown in Figure 3(c), gp4 displayed noaggregation at t=0 min, consistent with the highsolubility of recombinant gp4 expressed and purifiedat 22 °C. The time-course of aggregation alsoconfirmed that gp4 aggregation is not only tempera-ture-dependent, but also time-dependent. After1 min of incubation approximately 50% of theprotein was found in the pellet fraction (Figure 3(c),lane 4) and the ratio soluble/aggregated gp4 slowlydecreased as a function of time: after 10 min∼90% ofthe protein was fully aggregated (Figure 3(c), lane10). This fraction became almost 99% between 30 and60 min, which agrees well with the data presented inFigure 3(a) (see intensity of aggregated gp4 in lane10). This data confirmed that gp4 aggregation inSalmonella is a slow process that takes place at 37 °Cwithin approximately 30 min.
Gp4 binds an equimolar amount of portal protein
Tail accessory factor gp4 has previously beenshown to be the first tail accessory factor to be addedto newly DNA-filled capsid during P22 morpho-genesis.25 In in vitro complementation assays,Strauss and King25 observed that the formation ofinfectious P22 phages is highly dependent on theconcentration of gp4, which suggests multiplecopies of gp4 are incorporated into the newly filledcapsid, and Casjens and King42 measured 10(±5)molecules of gp4 in completed, infectious P22virions. To study the binding of gp4 to portalprotein and determine an accurate binding stoichio-metry between these two proteins, we used a nativebinding assay on agarose gel (Figure 4(a)). In thisassay, increasing molar equivalents of gp4 wereadded to dodecameric portal protein. The gp1:gp4mixture was then analyzed on a 2% agarose gel andprotein bands stained by Coomassie brilliant blue.To prevent thermal unfolding and aggregation ofgp4, all steps of the experiments were carried out at22 °C, a temperature at which gp4 exists as a well-folded monomeric protein. As shown in Figure 4(a),binding of gp4 to portal ring resulted in a significantshift in the electrophoretic mobility of gp1, whichis consistent with the formation of a larger Mrcomplex migrating slower on agarose gel. Thestoichiometry of the gp1:gp4 interaction was accu-rately monitored on agarose gels. Whereas sub-stoichiometric concentration of gp4 resulted in ashift of only part of the portal ring population
(Figure 4(a), lanes 2−6), shift of all portal rings wasobserved using an approximately 12-fold molarexcess of gp4 to portal rings (Figure 4(a), lane 7).This demonstrates that 12 copies of gp4 bind anequimolar amount of portal protein.To confirm the stoichiometry observed on native
agarose gel, we analyzed on 12.5% SDS-PAGEincreasing concentrations of purified dodecamericgp1 (Figure 4(b), lanes 1−5), monomeric gp4 (Figure4(b), lanes 6−10), and gel filtration purified gp1:gp4complex (Figure 4(b) lanes 11 and 12). After stainingwith Comassie brilliant blue R250 and extensivedestaining in 10% acetic acid, 45% methanol, theintensity of gp1 and gp4 bands was evaluated usingthe NIH image software and a calibration curve wasobtained by plotting the intensity of each proteinband as function of the protein concentration. Thecurve showed nearly perfect linearity up to 7 μg ofprotein loaded on the gel. The slope of these curveswas used to calculate the moles of gp1 and gp4present in the gel filtration purified complex in lanes11 and 12, respectively. Assuming that gp1 corre-sponds to a dodecamer, the quantification indicatesthat the gp4 band seen in the complex (Figure 4(b),lanes 11 and 12) contains 11.5(±0.3) equivalents,which agrees remarkably well with the 12:12stoichiometry seen on agarose (Figure 4(b), lane 11).The band-shift assay on agarose gel was also
repeated with partially assembled portal protein. Aspreviously reported,43 partially oligomerized portalprotein consists primarily of portal monomers andoligomers, which do not rapidly interconvert andcan be separated by size exclusion and ion exchangechromatography. On agarose gels partially oligo-merized portal protein migrates as two bandscorresponding to a faster migrating portal ring anda more diffuse band of monomeric gp1 (Figure 4(c),lane 1). Interestingly, upon addition of increasingequivalents of gp4 to partially oligomerized portalprotein no significant decrease in the gp1 monomerband was observed, suggesting that there is nobinding between portal monomer and gp4 (Figure4(c), lanes 2−8). In contrast, gp4 selectively asso-ciated with oligomeric portal ring, which wasshifted to form a slower-migrating gp1:gp4 com-plex. A nearly complete shift of portal protein wasobserved using a 12-fold molar excess of gp4 (Figure4(c), lanes 9). It should be noted that the concentra-tion of gp1 used in our assay is 25-fold lower thanthe critical concentration necessary for oligomeriza-tion of recombinant portal protein.43 Thus, theassembly of unreacted gp1 monomers to formportal ring, which may potentially become a newsubstrate for binding to gp4, is negligible under ourexperimental conditions. The quaternary structureof the oligomeric portal protein, and not monomericgp1, contains the binding determinants necessaryfor efficient binding and assembly of gp4. This couldbe explained either by gp4 binding at the interfacegenerated by two gp1 monomers or by a conforma-tional change in gp1 upon assembly into rings. Itremains unclear if the 12 molecules of gp4 contacteach other when bound to the portal ring.

Figure 4. Gp4 oligomerizeupon binding to 12-fold symmetricportal protein. (a) Native electro-phoresis mobility assay on 2% agar-ose. Increasing concentrations ofgp4 were added to purified dode-cameric P22 portal protein andsamples separated on agarose gel.Formation of a gp1:gp4 complex isobserved as a retarded bandmigrat-ing slower than gp1.Approximately12 equivalents of gp4, calculatedwith respect to dodecameric gp1,are necessary to observe a nearlycomplete shift of gp1 (lane 7). (b)Quantification of gp1:gp4 stoichio-metry. 1.5, 3.0, 4.5, 6.0 and 7.5 μg ofpurified dodecameric gp1 (lanes1−5) and monomeric gp4 (lanes6−10), as well as 7.5 and 9 μg of gelfiltration purified gp1:gp4 complex(lanes 11 and 12) were analyzed by12.5% acrylamide SDS-PAGE. Thegel was stained with Coomassiebrilliant blue R250 (Sigma) followedby extensive destaining in 10%acetic acid, 45% methanol. Theintensity of each band was evalu-ated using the NIH image softwareand used to calculate the moles ofgp1 and gp4 present in the complex.The quantification indicates that inlanes 11 and 12, the gp4 bandcontains 11.5(±0.3) equivalentswith respect of the gp1 band. (c)
Native electrophoresis mobility assay in 2% agarose with partially assembled gp1. Partially oligomerized portal proteinwas mixed with increasing molar ratios of gp4 and the samples were separated on an agarose gel. Gp4 selectively bindsoligomeric gp1, whose migration on agarose is significantly shifted (lanes 2−9). In both (a) and (c), free gp4 runs as asmeared band that is not visible in this gel (see also Figure 7, lane 3).
564 Assembly of Gp4 to P22 Portal Protein
We repeated the binding assay on agarose gelusing a truncated portal protein (named ΔC(1-602))that lacks C-terminal residues 603−725, whichassembles as a 12-fold symmetric ring like wild-type portal protein.6 This shorter version of gp1gave identical results as seen with full-length gp1 inFigure 4(a) (data not shown), indicating that the C-terminal portion of portal protein does not engage inbinding interactions with gp4. This is in agreementwith the observation that the C-terminal 50 aminoacid residues of portal protein are completelydispensible for its role in virus assembly andfunction44 and a model in which the C-terminaldomain of gp1 forms the “hat-like” domain on top ofP22 connector inside the capsid shell,6 while gp4binds the narrow bottom domain of the connectorthat protrudes from the shell.
Oligomeric portal protein suppressesaggregation of unfolded gp4 by stabilizingnative gp4
Since phage P22 assembles at 37 °C we wereinterested in understanding how gp4, which islargely unfolded and aggregated at this temperature
in vitro, can oligomerize upon binding to portalprotein. To address this point we needed todetermine the mechanisms by which portal proteininteracts with gp4. Two possible scenarios seemplausible. Portal protein may bind unfolded gp4 anddrive the protein to a folded conformation, whichthen assembles to form a dodecameric ring. Alter-natively, portal protein binds the native conforma-tion of gp4 that is in equilibrium with the unfoldedpolypeptide chain. Such an interaction leads to astructural stabilization of gp4 and suppression ofgp4 aggregation, which results in productive oligo-merization of 12 copies of gp4. To test these twohypotheses we repeated the aggregation assay inSalmonella cellular extract shown in Figure 3. Whengp4 pre-equilibrated to 37 °C was added to aSalmonella extract and incubated for 1 h at 37 °Cmost of the protein unfolded and aggregated (Figure5(a), lane 5). Consistent with the CD data, gp4 pre-heated at 37 °C is largely unfolded, and cellularchaperons present in the Salmonella extract fail tosuppress gp4 aggregation. To determine if portalprotein binds unfolded gp4, dodecameric portalprotein was pre-added to the Salmonella extract andgp4 pre-heated at 37 °C was added to the extract.

565Assembly of Gp4 to P22 Portal Protein
The mixture was then incubated at 37 °C for 1 h andcentrifuged to separate insoluble aggregates fromsoluble gp4. As shown in Figure 5(a), lane 7,addition of gp1 to the Salmonella extract afterincubation at 37 °C did not alter the pattern of gp4aggregation, which is detected primarily in thepellet fraction (Figure 5(a), lane 5). This indicatesthat portal protein does not bind unfolded gp4. Incontrast if 12-fold symmetric portal protein was pre-added to the purified gp4 at 22 °C, temperature atwhich most of gp4 is folded, and the complex wasadded to Salmonella extract at 37 °C for 1 h, most gp4was then detected in the soluble fraction (Figure5(a), lane 8). This indicates that gp1 binds the nativestate of gp4 and stabilizes its structure, thusreducing gp4 aggregation.In a complementary set of experiments carried out
entirely with purified proteins, oligomeric portalprotein was incubated at 37 °C with several samplesof gp4 that were pre-denatured at 37, 42, and 48 °C.The protein mixture was then equilibrated to 37 °Cand analyzed on an agarose gel at 37 °C. As apositive control for this reaction, gp1 was incubated
Figure 5. Dodecameric portalprotein stabilizes the native confor-mation of gp4. (a) His-gp4 wasadded to a Salmonella extract at37 °C and detected after SDS-PAGEusing monoclonal anti-Penta-Hisantibody (lane 1). No signal wasdetected from a Salmonella extractpreviously centrifugedat 18,000g for30 min to separate soluble (or Sn,supernatant) and insoluble fraction(or P, pellet), as indicated in lanes 2and 3, respectively. RecombinantHis-gp4 that was added to theSalmonella extract and incubated at37 °C readily aggregates and isdetected primarily in the pellet frac-tion (lanes 4 and 5). A small percen-tage of gp4 is detected in the solublefraction (lane 6).Addition of gp1 to aSalmonella extract pre-incubatedwith gp4 at 37 °C also results inaggregation of gp4, which is detec-ted primarily in the pellet fraction(lanes 6 and 7). In contrast, whenstoichiometric amounts of foldedgp4 and gp1 were added simulta-neously to the same extract atroom temperature and then incu-bated at 37 °C, most gp4 wasfound in the soluble fraction (lanes8 and 9), indicating that gp1suppresses gp4 aggregation by
stabilizing the native state of the protein. (b) Native electrophoresis mobility assay in 2% agarose at 37 °C. In lane1 dodecameric portal protein was pre-incubated at 37 °C for 1 h prior to analysis in gels. The ring is stable at thismperature and yields a sharp band. In lane 2, the gp1:gp4 complex was pre-formed at 33 °C using an excess of gp4,nd then equilibrated at 37 °C for 1 h. Excess gp4 unfolds at 37 °C and aggregates in the lane's well. In lanes 3, 4, and, gp4 was pre-equilibrated at 37 °C, 42 °C, and 48 °C, respectively for 1 h prior to mixing with dodecameric gp1. Innes 3, 4 and 5 a small amount of gp4 apparently refolds and binds gp1, which appears as a minor slower migratingand on agarose gel.
tea5lab
with folded gp4 at 22 °C and separated on agarosegel at 37 °C. The outcome of this experiment isshown in Figure 5(b). While the gp1 and gp4 formeda complex at 22 °C, which is perfectly stable at 37 °C(Figure 5(b), lanes 1 and 2), the majority of unfoldedgp4 failed to bind the portal ring. Only a smallamount of gp1:gp4 formed in the presence ofunfolded gp4, suggesting that the ability of mis-folded gp4 to refold in the presence of portal proteinwas limited to ∼5−10% of the gp4 population. Thisrefolding of gp4 was observed using gp4 heated at37, 42 °C and, to a lesser extent, 48 °C. Thisexperiment indicates that gp1 only marginally, if atall, improves the folding of unfolded gp4, but itexerts its primary function by stabilizing the nativeconformation of gp4.
P22 portal protein rings contain twonon-equivalent sets of binding sites for gp4
The thermodynamics of gp4 binding to portalprotein was analyzed by isothermal titration calori-metry (ITC). In a first set of experiments, a 245 μM
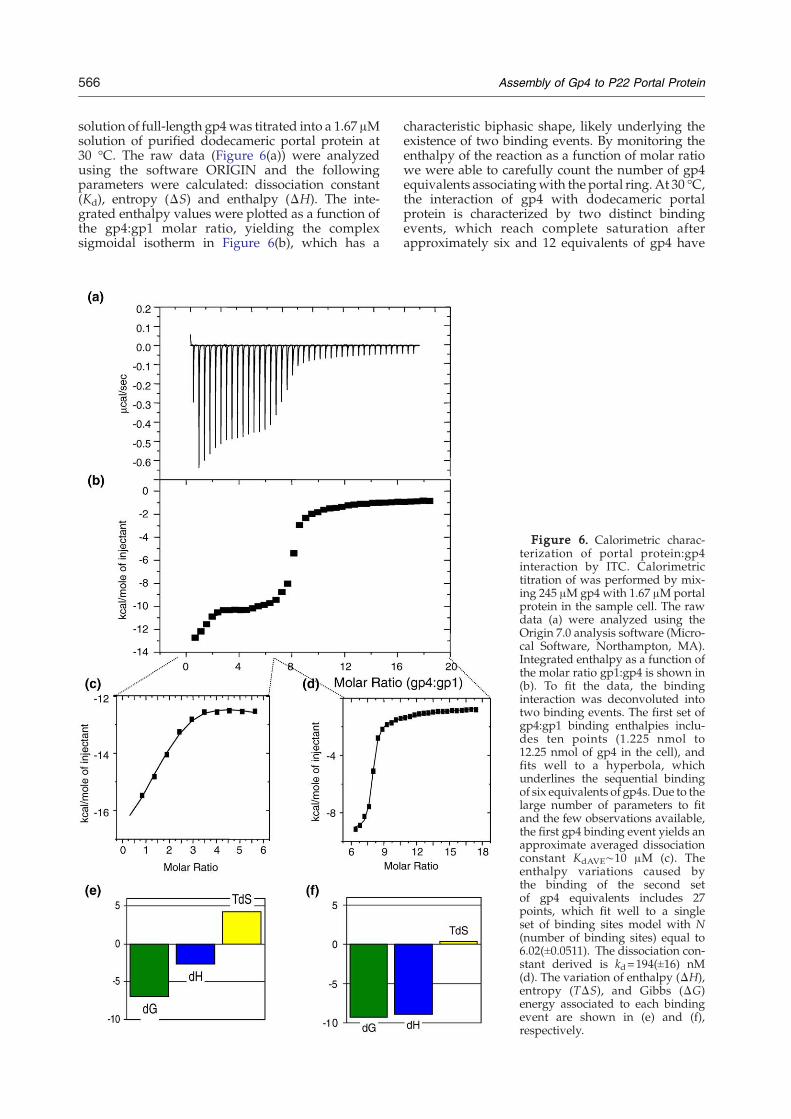
566 Assembly of Gp4 to P22 Portal Protein
solution of full-length gp4was titrated into a 1.67 μMsolution of purified dodecameric portal protein at30 °C. The raw data (Figure 6(a)) were analyzedusing the software ORIGIN and the followingparameters were calculated: dissociation constant(Kd), entropy (ΔS) and enthalpy (ΔH). The inte-grated enthalpy values were plotted as a function ofthe gp4:gp1 molar ratio, yielding the complexsigmoidal isotherm in Figure 6(b), which has a
characteristic biphasic shape, likely underlying theexistence of two binding events. By monitoring theenthalpy of the reaction as a function of molar ratiowe were able to carefully count the number of gp4equivalents associatingwith the portal ring. At 30 °C,the interaction of gp4 with dodecameric portalprotein is characterized by two distinct bindingevents, which reach complete saturation afterapproximately six and 12 equivalents of gp4 have
Figure 6. Calorimetric charac-terization of portal protein:gp4interaction by ITC. Calorimetrictitration of was performed by mix-ing 245 μM gp4 with 1.67 μMportalprotein in the sample cell. The rawdata (a) were analyzed using theOrigin 7.0 analysis software (Micro-cal Software, Northampton, MA).Integrated enthalpy as a function ofthe molar ratio gp1:gp4 is shown in(b). To fit the data, the bindinginteraction was deconvoluted intotwo binding events. The first set ofgp4:gp1 binding enthalpies inclu-des ten points (1.225 nmol to12.25 nmol of gp4 in the cell), andfits well to a hyperbola, whichunderlines the sequential bindingof six equivalents of gp4s. Due to thelarge number of parameters to fitand the few observations available,the first gp4 binding event yields anapproximate averaged dissociationconstant KdAVE∼10 μM (c). Theenthalpy variations caused bythe binding of the second setof gp4 equivalents includes 27points, which fit well to a singleset of binding sites model with N(number of binding sites) equal to6.02(±0.0511). The dissociation con-stant derived is kd=194(±16) nM(d). The variation of enthalpy (ΔH),entropy (TΔS), and Gibbs (ΔG)energy associated to each bindingevent are shown in (e) and (f),respectively.

567Assembly of Gp4 to P22 Portal Protein
bound the dodecameric portal ring (Figure 6(b)−(d)).Despite our efforts, all attempts to fit the biphasic-binding isotherm to a pre-defined binding modelwere unsuccessful, likely due to the complex natureof the gp4:gp1 interaction. To provide a quantitativedescription of the enthalpy released during associa-tion of gp4 to portal protein we decided to analyzethe enthalpy data in two batches and fit each batch ofdata to a knownmodel. Effectively, this is equivalentto deconvoluting a single binding isotherm into twobinding curves, each of which corresponds to adistinct gp4:gp1 binding event. The curve fitting ismade possible by the fact that each individualbinding event in the biphasic isotherm in Figure6(b) reaches complete saturation and the concentra-tion of both gp1 and gp4 in the cuvette are alwaysknown in the course of an ITC experiment. Theoutcome of this analysis is shown in Figure 6(c) and(d). Notably, the first binding event reaches a plateaubetween four and six equivalents of gp4 binding tothe portal ring. The binding isotherm for thisreaction is hyperbolic in nature (Figure 6(c)) andwas fit to a sequential binding of six equivalents ofgp4s to dodecameric portal protein. The six gp4shave comparable association constants, which cor-respond to an averaged dissociation constant ofKdAVE∼10 μM (Figure 6(c)). The few observationsavailable for this first binding event (only 11injections) and the large number of parameters tofit (six associations constants and six ΔHs) inevi-tably hamper the correct estimation of the dissocia-tion constant, whose value of 10 μM represents onlyan approximation. A control experiment where gp4was injected into reaction buffer at 30 °C showsminimal constant heat release (data not shown),confirming that the heat release observed upontitration of the first six gp4 equivalents into portalprotein is a genuine representation of the bindinginteraction taking place between the two proteinsand it is not owed to the heat of folding/unfolding ofgp4 injected into the cuvette at 30 °C. Consequently,the non-cooperative nature of this first binding event,together with the evidence that gp4 binds only theoligomeric form of gp1 (Figure 4(c)), supports theidea that six gp4 molecules occupy six binding siteson non-adjacent gp1:gp1 binding interfaces in thedodecameric ring (i.e. with a periodicity of 60°).The first low-affinity binding event is followed by
a second higher affinity interaction involving anadditional six copies of gp4. This second event(Figure 6(d)) starts after six copies of gp4 haveassociated with the portal ring and reaches satura-tion at a molar ratio gp4:gp1 close to 1:1 (or 12:12).The dissociation constant measured for the secondgp4 binding event is Kd=194(±16) nM, which is 50-fold higher affinity than the first type of bindingevent. The second binding event does not competewith the first event and instead takes place only afterthe first has reached complete saturation. Thissuggests a conformational change in portal proteinmay take place after the first set of gp4s have boundthe portal ring, thus exposing a new binding inter-face for the second set of gp4 equivalents. An
intriguing possibility is that the binding interfacesfor the second set of gp4 equivalents is formed by acombination of gp1 and gp4 binding determinants.However in the absence of a high resolutionstructure of portal protein:gp4 complex, this hypoth-esis remains speculative. Using the two thermody-namic equations ΔG=ΔH – TΔS and ΔG=– RT ln(Ka), we were able to compare the thermodynamicparameters associated to each of the two bindingevents observed by ITC. These events are thermo-dynamically distinct in that the first binding event islargely driven by entropy (Figure 6(e)), while thesecond binding event is largely dominated byenthalpic contributions (Figure 6(f)). The highentropic component of the first binding event isconsistent with hydrophobic interactions betweengp4 and portal protein. In contrast, the higherenthalpic gain of the second binding interactionsuggests that the second set of gp4s engages in morepolar contacts with portal protein and/or the first sixcopies of gp4.
Isolating the gp1:gp4 assembly intermediate onagarose gel
Binding assay on agarose gel at room temperature(22 °C) (Figure 4(a)) has shown that only two speciesare stably populated in the assembly reaction of gp4to portal protein, which correspond to free portalring gp(1)12, and fully decorated gp(1)12:gp(4)12complex. In contrast, the ITC titration at 30 °C(Figure 6) provided evidence for the existence of astably populated and saturable gp(1)12:gp(4)6 assem-bly intermediate, which plateaus after four to sixequivalents of gp4 have bound the portal ring. Toclarify this apparent discrepancy in our data, weinvestigated the role of temperature in the assemblyof gp4 to portal protein. In a first set of ITC titrationscarried out between 20−27 °C, we observed aremarkably reduced heat release during binding ofthe first six equivalents of gp4 to portal protein. Thiswas consistent with a significantly reduced firstbinding event (data not shown), likely owed to thepoor stability of the gp4:gp1 assembly intermediateat temperature below 27 °C. Likewise, it wasimpossible to obtain quantitative binding enthalpyvariations at temperatures >30 °C, because of gp4′stendency to severely aggregate in the syringe (at theconcentration of 0.25 mM or ∼5 mg/ml). Thissuggested that the gp(1)12:gp(4)6 assembly inter-mediate seen by ITC at 30 °C formed in atemperature-dependent manner and temperatureabove 27 °C was essential to stably populate thisintermediate. To test this hypothesis we repeated thenative gel by running the electrophoresis in a roomthermostatted at 30 °C. Remarkably, at this tempera-ture we found that the partially assembled gp(1)12:gp(4)6 intermediate migrates on gel as a slightlylowermobility band, clearly distinguishable from gp(1)12 and fully saturated gp(1)12:gp(4)12 complex(Figure 7(a), compare lanes 2 and 3 to lanes 1 and 4,respectively). Notably, the gp(1)12:gp(4)6 intermedi-ate was found to form quickly at 30 °C and remain

568 Assembly of Gp4 to P22 Portal Protein
stably populated on agarose gel. In Figure 7(a) lane 3,six molar equivalents of gp4 were added to gp(1)12,and themixture incubated at 30 °C for 30min prior toelectrophoretic analysis. Alternatively, in lane 2, sixmolar equivalents of gp4 were added to gp(1)12 at30 °C, and the mixture immediately separated onagarose gel. In both cases, the assembly intermediateformed and remained stably populated to beobserved on the gel. Addition of a higher fold excessof gp4 to portal resulted in both free gp(1)12 andgp(1)12:gp(4)12 complexes (Figure 7(a), lane 4). This isconsistent with the idea that at higher molarconcentration of gp4, the higher affinity binding ofthe second set of gp4 equivalents leads to fullsaturation of the portal ring, even in the presenceof substoichiometric concentrations of gp4.To monitor the formation (and disappearance) of
the gp(1)12:gp(4)6 intermediate with respect of themolar equivalents of gp4, we repeated the gp4
Figure 7. Isolating the gp1:gp4 assembly intermediate onstably populated gp(1)12:gp(4)6 assembly intermediate. In laneassembly intermediate in lanes 2 and 3migrates on gel as a slighsaturated decorated gp(1)12:gp(4)12 complex in lane 4 andintermediates in lanes 2 and 3were formed by adding six equivfor 30 s and 30min, respectively. In lane 4 approximately eight egp(1)12:gp(4)12 complex and free gp(1)12. (b) Titration of gp4 bboth gp(1)12:gp(4)6 assembly intermediate and fully decorattitration. The assembly intermediate appears at stoichiometrieswhere >six equivalents of gp4 are present.
titration on 2% agarose gel, but ran the agarose gelat 30 °C. As shown in Figure 7(b), the assemblyintermediate appeared at gp(1):gp(4) stoichiometriesequal to 12:(3-6) (lanes 3−6) and faded away in lane 7,where >six equivalents of gp4 are present. At higherconcentration of gp4, the gp(1)12:gp(4)12 complexstarts populating, which migrates clearly below theintermediate (see Figure 7(b), lanes 8 and 9). Takentogether these data indicate that: (1) the gp(1)12:gp(4)6 assembly intermediate is highly temperature-dependent and is stably populated at 30 °C; (2) inagreement with the ITC data, the gp(1)12:gp(4)6assembly intermediate is sufficiently stable to beisolated by native gel electrophoresis. Both nativeagarose gel and ITC data, however, do not conclu-sively allow the determination of whether we havefour or five or six gp4 equivalents bound to the portalring. Structural analysis by electron microscopy willbe necessary to elucidate this point.
agarose gel. (a) Native agarose gel run at 30 °C showing a1 is dodecameric portal protein gp(1)12. The gp(1)12:gp(4)6tly lower mobility band, clearly distinguishable from fullyfree dodecameric portal protein gp(1)12 in lane 4. Thealents of gp4 to gp(1)12 and incubating the complex at 30 °Cquivalents of gp4were added, yielding the fully decoratedinding to gp1 at 30 °C. By running the agarose gel at 30 °Ced gp(1)12:gp(4)12 portal protein are visible on the samegp(1):gp(4) equal to 12 (lanes 3−6) and fades away in lane 7,

569Assembly of Gp4 to P22 Portal Protein
The C-terminal domain of gp4 is required forhigh affinity binding to dodecameric portalprotein
To begin to map the binding site of gp4 for portalprotein we generated two C-terminal truncationmutants of gp4 that include residues 1−126 and 1−76, which were named ΔC126, and ΔC76, respec-tively. The mutants were designed based on thesecondary structure prediction of gp4 shown inFigure 8(a). The mutant ΔC126 lacks the predictednon-helical C-terminal region 127−166, while theshorter construct ΔC76 lacks both C-terminalmoiety 127-166 and the predicted helix spanningresidues 77 through 126. Attempts at generating adeletion construct in region 76-126 all yieldedinsoluble protein, suggesting that this region maybe solvent-exposed and so may be important for theoverall structural stability of gp4. Both ΔC126 andΔC76 mutants of gp4 were fused to an N-terminalsix-His tag (see Material and Methods) and purifiedfrom E. coli extracts by metal chelating chromato-graphy and gel filtration. In a native binding assayon agarose gels, we found that the larger constructΔC126 retains the ability to bind oligomeric portalrings (Figure 8(b), lane 4), while no significantinteraction was observed between the portal proteinand the shorter construct Δ76 (Figure 8(b), lane 6).To accurately determine the binding affinity ofΔC126 for portal protein, we repeated the ITCtitration described in the previous section. Thisbinding interaction displayed a dramatic reductionin the heat release and a loss of the two bindingevents seen for full-length gp4, yielding a simpler
single binding event (data not shown). Although wewere not able to fit these data to a model and obtainaccurate thermodynamic parameters, we estimate alow micromolar binding affinity between ΔC126-gp4 and oligomeric portal protein. Taken togetherthese data strongly suggest that the C-terminalregion of gp4 is critical for the high affinityinteraction with the portal vertex.
Electron microscopic analysis of tail accessoryfactor gp4 binding to portal rings
Negative stain electron microscopy was used tovisualize tail accessory factor gp4 bound to portalprotein. To isolate a homogeneous population ofportal protein decorated with gp4, we formed thegp1:gp4 complex using a fourfold excess of gp4 andseparated gp1:gp4 complex from unassembled gp4by a Sephacryl S-300 column chromatography at4 °C. The resulting particles were stained with 1%uranyl formate and visualized by transmissionelectron microscopy. Micrographs of the gp1:gp4complex were compared to gp4-free portal rings, asshown in Figure 9(a) and (b), respectively. Themajority of gp1:gp4 complexes seen on the micro-graphs displayed a preferential orientation on thegrid with the central hole perpendicular to the grid.In rare instances single complexes and head-to-head dimers of the gp1:gp4 complex were seen inside view (see higher magnifications in Figure 9(b)).Such side views are particularly informative, in thatthe dodecameric portal protein without bound gp4forms a mushroom-shaped structure with a channelthrough the center, and is composed of thin ring of
Figure 8. Mapping the bindingsite on gp4 for portal protein. (a)Topology of gp4 and two C-terminally truncated gp4s derivedfrom the secondary structure pre-diction program PHD.51 The grayboxes mark the three predicted α-helices. (b) Native electrophoresismobility assay on agarose showingthe interaction of gp4 deletionmutants with dodecameric portalprotein. The formation of a shiftedgp1 band indicates that both full-length gp4 and the deletionmutant Δ126 bind to oligomericgp1, but the short gp4 fragmentΔ76 fails to interact with P22portal rings. The short fragmentΔ76-gp4 is not visible on agarosegel consistent with its low mole-cular mass (∼10.4 kDa).

Figure 9. Negative stain electron microscopy of tail accessory factor gp4 bound to P22 portal protein rings. (a)Negative stain electron microscopy image of purified P22 portal protein rings. Many “donut-like” structures arevisible, which in most (rarer) side view cases adopt a head-to-head conformation. (b) Negative stain micrograph of gelfiltration purified portal protein in complex with gp4. In both (a) and (b) the scale bar corresponds to 50 nm. (c)Statistical analysis of negatively stained side views of gp1 ring and gp1:gp4 complexes indicates the particles haveaverage length of 118(±12) Å and 144(±12) Å, respectively (the uncertainties represent standard deviations). Binding ofgp4 to portal rings did not alter the size or morphology of top views. (d) Schematic model of portal protein bound to12 copies of gp4; dotted vertical lines denote the internal channel.
570 Assembly of Gp4 to P22 Portal Protein
density (crown domain) on top of a wider ring inthe center and a narrower bottom ring (Figure 9(a);see also Tang et al.6). In the presence of gp4, themushroom-like portal protein structure displays anadditional narrow layer of density bound to thebottom end of the portal ring (see bottom of Figure9(b)). Careful measurement of 97 negatively stainedgp1:gp4 particles (Figure 9(c)) revealed that thecomplex has an average length of 144(±12) Å. Incontrast, the average length of 77 portal ringsmeasured from a single grid is 118(±12) Å (Figure9(c)), indicating that 12 copies of gp4 form astructural ring at the bottom of the portal ringthat is approximately 26 Å in length. A schematic
model of the gp1:gp4 complex is presented inFigure 9(d).
Discussion
The assembly of the phage P22 tail is a complexevent that requires the sequential addition of foursoluble proteins (gp4, gp10, gp26, and gp9) to acapsid-bound dodecameric portal protein. The tailassembly is not simply the essential structural eventending the transformation of procapsids into maturecapsids, but also represents an essential step in thestabilization and retention of newly encapsidated

571Assembly of Gp4 to P22 Portal Protein
DNA inside the capsid.25,28 P22 particles lacking anyof the tail accessory factors are not completed, arevery unstable and are not infectious.While a numberof studies have been aimed at elucidating thestructure, assembly, and regulation of phage P22tailspike gp9 and portal protein gp1, the role andcomposition of tail accessory factors gp4, gp10 andgp26 have been only partially characterized. In anattempt to obtain a more detailed molecular descrip-tion of the events leading to portal vertex closure, wepreviously characterized tail accessory factor gp26.This factor was found to form a long trimeric coiledcoil protein.30 We hypothesize that gp26 forms theneedle (or fiber) emanating from the neck of the P22tail, which is also responsible for plugging the holethrough which DNA entered the capsid duringvirion assembly. These findings are strongly rein-forced by negative stain images of P22 virions45 andmore convincingly by the recent cryo-EM recon-struction of the P22 phage tail,6 which shows astriking needle protruding from the DNA channelthat we hypothesize is used by the phage to piercethe host cell membrane. In the work presented here,
we have expressed and isolated the tail accessoryprotein gp4 and characterized its structure andbinding interactions to the portal protein (Figure 10).
Gp4 is greatly stabilized upon binding to portalprotein
Gp4 exists in solution as an elongated monomer.We failed to detect gp4 aggregation or oligomeriza-tion even at elevated protein concentration(>15 mg/ml) and under low ionic strength condi-tions. In vitro, and in Salmonella extracts gp4 isextraordinarily sensitive to heat denaturation. Thepurified protein melts irreversibly with a Tm 34 °C,suggesting that at 37 °C, the temperature at whichSalmonella grows in the gut of warm-bloodedanimals, the equilibrium between folded andunfolded gp4 ((Unfoldedgp4)↔ (Foldedgp4)) may beshifted to the left and only a small population of theprotein adopts a folded conformation. In line withits poor stability, most gp4 aggregates in Salmonellaextracts to form large molecular mass aggregates.This pattern of aggregation is strongly dependent on
Figure 10. Model for gp1:gp4interaction. The (Unfoldedgp4)↔(Foldedgp4) equilibrium is largelyshifted to the left at 37 °C. Most ofthe gp4 population quickly aggre-gates in the Salmonella cellularenvironment. Dodecameric gp1binds the native state of gp4 andthus shifts the (Unfoldedgp4)↔(Foldedgp4) equilibrium to theright. A first set of six gp4 equiva-lents binds the portal oligomer in anon-cooperative manner, suggest-ing that the six gp4 equivalentsinteract with non-adjacent gp1:gp1binding interfaces. This transientintermediate (indicated in parenth-eses) undergoes a conformationalchangethatexposesahigheraffinity-binding surface for a second set ofsix gp4 equivalents. This secondbinding event is higher affinity(∼194) and largely dominated byenthalpic contribution. The gp1:gp4 complex is then bound bythe other tail accessory factorsgp10 and gp26, followed by tail-spike attachment.

572 Assembly of Gp4 to P22 Portal Protein
both temperature (over a narrow range between33−39 °C) and time. A time-course of aggregationreveals that at 37 °C gp4 aggregation takes place inSalmonella extract within 30 min, and a significantpopulation of soluble gp4 (approximately 10%)survives aggregation for ∼10 min.The temperature-dependent aggregation and the
lack of reversibility observed in the temperaturedenaturation experiment (Figure 2(b)) strongly sug-gest that the gp4 folding energy landscape is rough.Several folding intermediates are likely to exist andbe populated en route to the native state. Smallproteins (e.g. 100 amino –acid residues) usually havea relatively smooth folding landscape, and presentonly two stably populated species during thefolding reaction separated by a single transitionstate barrier, but gp4 likely adopts several partiallymisfolded conformations “funneling” toward themore stable native conformation via intramolecularcontacts. According to this model, the severetemperature-dependent aggregation of gp4 iscaused by the tendency of unfolded or partiallymisfolded gp4 intermediates to engage in intermo-lecular contacts.The unusually low structural stability of gp4 is in
striking contrast to the extreme thermostabilityobserved for the P22 tail complex extracted fromthe mature virus. The 2.8 MDa molecular machinecan be extracted frommature P22 phage using 1.5 Murea and prolonged heating at 70 °C.6 Gp4 is alsoless stable than several other individual componentsof P22 tail that display high degrees of thermo-stability even when isolated from the tail (Table 1).
A model for gp4 thermo-instability
The idea that gp4 is unstable under physiologicalconditions if not bound to gp1 raises the question ofhow gp4 can bind gp1, since presumably gp4 ispresent in the cell at 37 °C and waits to bind until thephage DNA is packaged. A plausible answer to thisquestion comes from the time-course of aggregationpresented in Figure 3(c). In Salmonella extract, at37 °C, temperature at which phage P22 infects thehost Salmonella, unfolded gp4 aggregates withinapproximately ∼30 min. The aggregation is slow,and after 10 min of incubation at 37 °C, as much as∼10% of gp4 is still present in the soluble monomeric(presumably active) conformation. Given the robust-ness of the DNA encapsidation, which in mostphages takes place in a time scale of minutes, 14DNA
Table 1. Structural stability of proteins forming thebacteriophage P22 tail
Tail protein Tm (°C) Ref
Gp1 (portal protein) 65 Moore and Prevelige20,43
Gp9 (tail-spike) 88 Goldenberg et al.52
Gp4 (tail accessory factor) 34 This article, Figure 2(b)Gp10 (tail accessory factor) n/oGp26 (tail accessory factor) 85 Andrews et al.30
Tm indicates the temperature of melting. The reference fortailspike is Goldenberg et al.52
packaging does not seem to be rate-limiting indetermining the fate of gp4 aggregation. Consider-ing that (i) gp4 is part of the late genes expressed afterP22 procapsids have been already assembled and (ii)the phage DNA packaging takes place in a time scaleof minutes, it is very reasonable to believe that thereis sufficient soluble gp4 in the cell immediately afterpackaging for assembling of gp4 into the phage tail.In this regard, the significantly lower thermody-
namic stability of monomeric gp4 compared to thatof other components of the P22 tail and its tendencyto slowly aggregate at 37 °C suggest that suchstructural instability may have a biological role. Gp4was recently reported to act as a murein hydrolase,which digests the pepdidoglycan layer present in theSalmonella cell wall between the inner and outermembranes.29 We speculate that the thermodynamicinstability of gp4 reflects the potential suicidal actionof this protein, which, if released in the host peri-plasm could digest the peptydoglycan layer, causingirreversible damage to the host cell wall. Althoughgp4 does not appear to be among the four P22-en-coded proteins injected into the host duringinfection,46 it is plausible that the protein disassem-bles from the tail apparatus once the virus haspenetrated the first lipid bilayer of Salmonella cellwalland helps to hydrolyze the thin layer of peptidogly-can present in the periplasmic compartment. If so, theintrinsic structural instability of the protein at 37 °Cwhen isolated from the phage tail may reflect amechanism of deactivation by unfolding.
The interaction between gp4 and portal proteinrings
Two common modes of portal−gp4 interactionoriginally seemed possible. First, gp1 could suppressgp4 aggregation by improving, or “facilitating” thefolding of gp4. This molecular function is usuallyexerted by molecular chaperones, which bind thenon-native state of proteins and assist them to reacha functional folded conformation.47 Second, portalprotein could suppress unfolding and aggregation ofgp4 by increasing the structural stability of gp4 bybinding its native state. In our in vitro and pseudo-invivo studies in Salmonella extracts we provide strongevidence that the latter model is correct. Portalprotein does not bind thermally unfolded gp4 andthus is not a molecular chaperone. In contrast, it canstabilize the structure of folded gp4 by engaging incontacts with the protein and providing a molecularsurface to stabilize the folded conformation.A striking feature of gp4:gp1 interaction resides in
the fact that gp4 binds exclusively dodecameric andnot monomeric portal protein. This may reflect theexistence of a “quality control mechanism” in theassembly of P22 tail. In fact, P22 portal protein formsdodecamers in vitro at concentration on the order of30−50 mg/ml, which is estimated to be greater than100 times that achieved during a wild-type phageinfection.43 The latter observation suggests that invivo portal ring oligomerization and phage procap-sid assembly are tightly coupled. While “free” 12-

573Assembly of Gp4 to P22 Portal Protein
fold symmetric portal protein does not sponta-neously form in Salmonella, gp1-rings assembleexclusively inside the virus procapsid. In this re-spect, the observation that gp4 assembles ondodecameric portal ring, and not on monomericportal protein, indicates that gp4 will assemble onlyin procapsids. If gp4 were able to interact with freeportal monomers, this may preclude the proteinfrom correctly assembling into the tail, with con-sequent loss of gp4 in abortive tail subcomplexes.Another striking feature of gp4:gp1 interactionresides in the fact that the structural stabilization ofgp4 is accompanied by oligomerization of gp4 toform a structural ring. This process is complex and,as shown using ITC, involves the binding of twonon-equivalent sets of six gp4 copies to a preformeddodecameric portal vertex. The first six copies of gp4bind with low affinity, perhaps at the portal proteinmonomer:monomer binding interface, followed bysixmore copies of gp4 to yield 12molecules of gp4, inan apparent ring attached to gp1. As shown in Figure6, the two sets of gp4s reach complete and indepen-dent saturations, suggesting that the two sets of gp4are non-equivalent and do not compete for the samesites on the portal protein ring. The binding of thefirst set of gp4 equivalent to portal protein is highlyinfluenced by temperature. At temperatures below30 °C, the first binding event is greatly reduced, andeven substoichiometric concentrations of gp4 yieldfully decorated gp(1)12:gp(4)12 complex. In contrast,at 30 °C the gp(1)12:gp(4)6 assembly intermediate issufficiently stable to be isolated on native gel.However, since the second binding event is ∼50-fold higher affinity than the first and yet it takes placeonly after the first event has reached complete satu-ration, it is plausible to postulate that a conforma-tional change in the gp(1)12:gp(4)6 intermediateexposes or creates a new higher affinity gp4 bindingsurface. In turn, the fact that the gp(1)12:gp(4)6intermediate has 50-fold higher affinity for mono-meric gp4 than free gp(1)12 suggests that the secondset of gp4s either interacts with both portal proteinand pre-bound gp4, or the first set of bound gp4scauses a conformational change in portal protein. Asshown in Figure 7, we have been able to isolate thegp(1)12:gp(4)6 assembly intermediate by formingand analyzing the complex at 30 °C. This indicatesthat the intermediate forms in a temperature-depen-dent manner and it is sufficiently populated andstable at 30 °C to survive migration on agarose gel.The fact that most likely sixmolecules of gp10 bind
to the finished portal−gp4 complex,6,25 fits nicelywith the notion of six molecules each of gp4s in twostates in the complex, since only one of the two typesof gp4 could bind gp10. It is fascinating to speculatethat the non-equivalence of gp4 binding sites ismediated through the folding of the C-terminaldomain of gp4. Our deletion analysis revealed thatremoval of this C-terminal region results in severelyreduced binding affinity for portal protein. Thisflexible, predicted random coil portion of gp4 maybecome exposed after the first set of six gp4s hasbound the portal oligomer, thus enhancing the
affinity of gp4 for itself. A higher-resolution three-dimensional structure of the gp1:gp4 complexwill berequired to unravel the detailed molecular basis ofgp4’s binding to the portal ring.
Materials and Methods
Molecular cloning of genes encoding gp4
The gene encoding the tail accessory factor gp4was PCRamplified from P22 DNAwith primers 5′-CACGATGCAT-ATGCAGATAAAGACTAAAGGCG and 5′-GTAGAGAG-GATCCGCCAGGGAATGAGCGGAG31,32 for thenucleotide sequence of phage P22. This DNAwas cleavedwith NdeI and BamHI and ligated into similarly cleavedplasmid expression vector pET-15b (Novagen, Madison,WI). The resulting plasmid, whose insert was completelysequenced to confirm its accuracy and structure, wasnamedpET15b-gp4. The cloned gene utilizes its native stopcodon so its expressed protein is identical to the native P22gene product, except it carries six histidine residues withinthe 20 vector-encoded amino acid residues that are fused toits N terminus. Gp4 C-terminally deleted mutants ΔC126andΔC76 were generated by introducing an “amber” stopcodon (TAG) at gp4position 77 and 127, respectively. Thesepoint mutations were introduced by PCR using theQuikChange site-directed mutagenesis kit (Stratagene).
Expression and purification of recombinant proteins
Recombinant gp4 protein was expressed from pET15b-gp4 in E. coli (strain BL21) cells (New England BioLabs,Ipswich, MA), in LB broth supplemented with 2.5 g/lglucose. After growth at 37 °C to an A600 nm of 0.6, gp4expression was induced with 0.5 mM IPTG and the culturewas incubated with shaking at 22 °C for 16 h. Cells werecollected and lysed by sonication in Lysis Buffer (200 mMNaCl, 20 mM Tris-Cl (pH 8.0), 3 mM β-mercaptoethanol(β-ME)) plus protease inhibitors (1 mM PMSF, 1 μg/mpepstatin, 1 μg/mL leupeptin). Recombinant gp4 fused toan N-terminal-6xHis tag was purified by metal chelatingchromatography using Qiagen Nickel-agarose beads. Theaffinity-purified protein was then concentrated to 1 mlusing a 5 kDa cutoff Millipore concentrator and furtherpurified by gel filtration on a Superdex S-200 column(Amersham Biosciences) equilibrated in Gel FiltrationBuffer (200 mM NaCl, 20 mM Tris-Cl (pH 8.0), 3 mM β-ME, 0.1 mM PMSF). Calibration of the S-200 column wascarried out using high molecular mass globular proteinstandards (BioRad, Richmond, CA) consisting of bovinegamma globulin (158 kDa), chicken ovalbumin (44 kDa),equine myoglobin (17 kDa), and vitamin B12 (1.3 kDa). Thegene encoding gp4 was also inserted into the pTYB4vector (New England BioLabs), which yields gp4 fused toan N-terminal self-splicing intein domain. Intein-fused-gp4 expressed in E. coli strain ER2566 (New EnglandBioLabs) was purified and the intein removed accordingto the manufacturer's instructions. Typically, one liter of E.coli yielded about 0.25 mg of pure protein, which wasconcentrated to approximately 15 mg/ml using a Milli-pore concentrator (Mr cut off 5 kDa).Recombinant full-length and C-terminally truncated
(AAs 1−602) P22 portal protein were expressed in E. colistrain BL21 cells and purified as described.6,48 To enrichthe sample for fully assembled dodecameric portal rings,0.5 M EDTAwas added to a final concentration of 55 mM,

574 Assembly of Gp4 to P22 Portal Protein
and samples were incubated at 37 °C for 2−3 h. Followingthis incubation, massive precipitation of portal proteinoccurred, which was removed from the sample bymultiple rounds of centrifugation. The resulting super-natant contained only fully oligomerized portal protein,which was then used for further studies.
Sedimentation equilibrium analysis
Sedimentation equilibrium analysis of P22 gp4 wasconducted in a Beckman Optima XL-A analytical ultra-centrifuge equipped with UV optics, at 4 °C, using a six-channel ANTi60 rotor with 12 mm thick, charcoal-eponcenterpieces. The three sample channels in each cellcontained three different loading concentrations of P22gp4 protein in 250 mM NaCl, 20 mM Tris-Cl buffer(pH 8.0), while reference channels contained the corres-ponding buffer only. Samples were centrifuged at30,000 rpm until sedimentation and chemical equilibriumwere attained. Cells were scanned radially in continuousmode, with data resulting from ten absorbance readingstaken at 0.001 cm intervals. Equilibrium was confirmed byno change in scans taken at 4 h intervals. Thepartial specificvolumeand theextinction coefficient forgp4was calculatedto be 0.712 ml/g (at 4 °C) from the amino acid sequenceusingdescribedmethods.49 Curve fitting and calculation ofmolecular mass were done with non-linear least squaretechniques using the computer software NONLIN.34
Circular dichroism, thermal and guanidinehydrochloride (GuHCl) melting curve
Circular dichroism spectra were collected on an AVIVcircular dichroism spectrometer model 62DS at 15 °C. Theconcentration of purified portal:gp4 complex was 0.2 mg/ml in phosphate-buffered saline (PBS) buffer: 0.02 Msodium phosphate buffer with 0.15 M sodium chloride,pH adjusted to 7.4. The molar ellipticity was monitoredusing a 1 mm path length cell and 1.0 nm wavelengthincrements from 195 to 250 nm. The resulting spectra werecorrected according to the base line by using the abovebuffer. For thermal unfolding curves the ellipticity at222 nm was monitored as a function of temperaturebetween 10−80 °C. The temperature was increased in 1 deg.C increments followed by 2 min equilibration. GuHClmelting curves were performed by adding increasingconcentrations of GuHCl to gp4 dissolved in PBS to afinal protein concentration of 0.2 mg/ml. Samples wereequilibrated overnight at 15 °C. Fluorescence scans weretaken from 300 nm to 450 nm in 1 nm steps with anexcitation of 295 nm on a SPEX Fluorolog-3-21 fluorometerat a constant temperature of 15 °C. Fluorescence emission at370 nm was then plotted as a function of GuHClconcentration and the data were fit to a model using thecomputer program Kalidegraph.
Aggregation assay in Salmonella extracts and nativegel electrophoresis
Extracts of Salmonella typhimurium DB7000 strain wereprepared as described,25 with the only exception that cellswere resuspended in PBS buffer. Concentrated, purifiedHis-tagged gp4 was added to 100 μl of extract at a finalconcentration of 0.5mg/ml. Samples of gp4were heated atvarious temperatures between 24−44 °C and subsequentlycentrifuged at 18,000g for 30 min at 4 °C. After centrifuga-tion the pellet fraction containing insoluble aggregates was
separated from the soluble fraction and analyzed on 12.5%(w/v) SDS-PAGE. The gel was then blotted onto a poly-vinylidene-fluoride (PVDF) membrane and His-tag gp4detected using monoclonal α-Penta-His antibody (Qia-gen). The in vitro aggregation assay was carried out usingpurified gp4 dissolved in PBS buffers. Native agarose gelelectrophoresis was carried out as described by Duda etal.50 The time-course of gp4 aggregation in Figure 3(c) wascarried out at 37 °C. Gp4 aliquots were taken after 1, 3, 5,10, 30 and 60 min and further analyzed as descried above.
Isothermal titration calorimetry (ITC)
All samples used in ITC experiments were dialyzed ex-tensively against 200 mMNaCl, 20 mM Tris-HCl (pH 8.0),2 mM β-ME buffer prior to each experiment. ITC ex-periments were carried out at 30 °C using a 300 μL syringewith a rotation speed of 295 rpm in the VP-ITC (Microcal).Gp4 was injected in 5 μl increments at a concentration of245 μM into 1.8 ml of portal protein at an oligomer con-centration of 1.67 μM with a spacing of 360 s betweenconsecutive injections. Titration data were analyzed usingthe Origin 7.0 data analysis software (Microcal Software,Northampton, MA). Data processing involved manualfitting of the peak baseline and background subtractionfrom the integrated heat peaks. All attempts to fit the datato predefined bindingmodelswere unsuccessful due to thecomplex nature of the gp4 interaction with portal protein.To fit the data we took the integrated corrected enthalpyvalue measured at 30 °C, and split the full isotherm in twobatches. The first included 11 injections, which corre-sponds to a gp4:gp1 molar ratio of 6:12. This can becalculated as we know that there are 3.00 nmol of portal inthe cuvette (1.8ml at 1.67 μM) and thatwe inject 1.225 nmolof gp4 per injection (5 μl at 245 μM), with the exception ofthe first small injection (only 3 μl). The first set of gp4:gp1binding enthalpies includes ten points (1.225 nmol to12.25 nmol of gp4 in the cell), and fits well to a hyperbola,which underlines the sequential binding of six equivalentsof gp4s. Ten points are obviously not sufficient to obtainaccurate values on all six association constants (ka), whichfit with values: ka1=1.12E5±2.2E5; ka2=8.12E4±4.9E4;ka3 = 5.08E4 ± 0; ka4 = 1.11E5 ± 0; ka5 = 1.20E5 ± 2.2E5;ka6=1.27E5±4.4E4. For the parameters with error equalto zero, the error could not be fit, due to a lack of obser-vations. Since all the binding sites seem to be on ap-proximately the same order these six kas were averaged toyield a value of kaAVE∼1.003E5. The dissociation constantfor the first gp4 binding event becomes kdAVE=1/kaAVE=1/1.003E5=9.97 μM (whichwe have approximatedto 10 μM). The kaAVE was also used in the ΔG and TΔScalculations. The second binding event is considerablybetter defined than the first and contains 27 injections,which corresponds to a gp4:gp1 molar ratio rangingbetween 6:12 to 30:12. The second binding event fits wellto a single set of binding sites model withN=6.02±0.0511,Ka=5.13e6±6.46e5 (KD=194(±16) nM). For both bindingevents, binding free energies and entropy were calculatedfrom the thermodynamic relationshipsΔG=ΔH–TΔS andΔG=–RT ln(Ka).
Negative stain electron microscopy
For electron microscopic examination, a 5−10 μl drop ofpurified portal or portal:gp4 complex at 0.1 mg/ml wasplaced on a Parafilm sheet and allowed to stand for1.0 min. A carbon-coated electron microscope gridpreviously glow discharged in a Polaron E5100 vacuum

575Assembly of Gp4 to P22 Portal Protein
evaporator was placed on top of the drop, and the proteinwas allowed to adsorb for 30 s. The grid was then stainedfor 30−60 swith 1%uranyl formate, blotted, and allowed toair dry. Specimens were observed and photographed in aPhilips CM120 electron microscope operated at 120 keV.Electron microscope images were collected on a GatanCCD camera at a magnification of 52,000x.
Acknowledgements
We thank Nancy Walker at SUNY UpstateMedical University for excellent technical support.We also thank Dr John Brandts at Microcal, for helpin processing and interpreting the ITC data. Thiswork was supported in part by NSF grant MCB-990526 to S.R.C.
References
1. Casjens, S. (1979). Molecular organization of thebacteriophage P22 coat protein shell. J. Mol. Biol. 131,1–14.
2. Prasad, B. V., Prevelige, P. E., Marietta, E., Chen, R. O.,Thomas, D., King, J. & Chiu, W. (1993). Three-dimensional transformation of capsids associatedwith genome packaging in a bacterial virus. J. Mol.Biol. 231, 65–74.
3. Thuman-Commike, P. A., Greene, B., Jakana, J.,Prasad, B. V., King, J., Prevelige, P. E., Jr & Chiu, W.(1996). Three-dimensional structure of scaffolding-containing phage P22 procapsids by electron cryo-microscopy. J. Mol. Biol. 260, 85–98.
4. Thuman-Commike, P. A., Greene, B., Malinski, J. A.,Burbea, M., McGough, A., Chiu, W. & Prevelige, P. E.,Jr (1999). Mechanism of scaffolding-directed virusassembly suggested by comparison of scaffolding-containing and scaffolding-lacking P22 procapsids.Biophys. J. 76, 3267–3277.
5. Thuman-Commike, P. A., Greene, B., Jakana, J.,McGough, A., Prevelige, P. E. & Chiu, W. (2000).Identification of additional coat-scaffolding interac-tions in a bacteriophage P22 mutant defective inmaturation. J. Virol. 74, 3871–3873.
6. Tang, L., Marion, W. R., Cingolani, G., Prevelige, P. E.& Johnson, J. E. (2005). Three-dimensional structure ofthe bacteriophage P22 tail machine. EMBO J. 24,2087–2095.
7. Bazinet, C., Benbasat, J., King, J., Carazo, J. M. &Carrascosa, J. L. (1988). Purification and organizationof the gene 1 portal protein required for phage P22DNA packaging. Biochemistry, 27, 1849–1856.
8. Goldenberg, D. & King, J. (1982). Trimeric intermedi-ate in the in vivo folding and subunit assembly of thetail spike endorhamnosidase of bacteriophage P22.Proc. Natl Acad. Sci. USA, 79, 3403–3407.
9. Casjens, S. & King, J. (1982). P22 morphogenesis.I: Catalytic scaffolding protein in capsid assembly.J. Supramol. Struct. 2, 202–224.
10. Hartweig, E., Bazinet, C. & King, J. (1986). DNAinjection apparatus of phage P22. Biophys. J. 49,24–26.
11. Leiman, P. G., Chipman, P. R., Kostyuchenko, V. A.,Mesyanzhinov, V. V. & Rossmann, M. G. (2004). Three-dimensional rearrangement of proteins in the tail of
bacteriophage T4 on infection of its host. Cell, 118,419–429.
12. Fokine, A. C. P., Leiman, P. G., Mesyanzhinov, V. V.,Rao, V. B. & Rossmann, M. G. (2004). Moleculararchitecture of the prolate head of bacteriophage T4.Proc. Natl Acad. Sci. USA, 101, 6003–6008.
13. Molineux, I. J. (2001). No syringes please, ejection ofphage T7 DNA from the virion is enzyme driven.Mol.Microbiol. 40, 1–8.
14. Casjens, S. & Weigele, P. (2005). Headful DNApackaging by bacteriophage P22. Viral Genome Packa-ging Machines: Genetics, Structure and Mechanism(Catalano, C. ed), pp. 80–88, Landes Publishing,Georgetown, TX, 80–88.
15. King, J., Lenk, E. V. & Botstein, D. (1973). Mechanismof head assembly and DNA encapsulation in Salmo-nella phage P22. II. Morphogenetic pathway. J. Mol.Biol. 80, 697–731.
16. Earnshaw, W., Casjens, S. & Harrison, S. C. (1976).Assembly of the head of bacteriophage P22: x-raydiffraction from heads, proheads and related struc-tures. J. Mol. Biol. 104, 387–410.
17. Thuman-Commike, P. A., Greene, B., Malinski, J. A.,King, J. & Chiu, W. (1998). Role of the scaffoldingprotein in P22 procapsid size determination suggestedby T=4 and T=7 procapsid structures. Biophys. J. 74,559–568.
18. Botstein, D., Waddell, C. H. & King, J. (1973).Mechanism of head assembly and DNA encapsula-tion in Salmonella phage p22. I. Genes, proteins,structures and DNA maturation. J. Mol. Biol. 80,669–695.
19. Poteete, A. R., Jarvik, V. & Botstein, D. (1979).Encapsulation of phage P22 DNA in vitro. Virology,95, 550–564.
20. Moore, S. D. & Prevelige, P. E., Jr (2002). Bacterioph-age p22 portal vertex formation in vivo. J. Mol. Biol.315, 975–994.
21. Casjens, S. & Weigele, P. (2004). Headful DNApackaging by bacteriophage P22. Viral Genome Packa-ging (Catalano, C. ed), Landes Publishing.
22. Galisteo, M. L. & King, J. (1993). Conformationaltransformations in the protein lattice of phage P22procapsids. Biophys. J. 65, 227–235.
23. Smith, D. E., Tans, S. J., Smith, S. B., Grimes, S.,Anderson, D. L. & Bustamante, C. (2001). Thebacteriophage straight phi29 portal motor can pack-age DNA against a large internal force. Nature, 413,748–752.
24. Earnshaw, W. C. & Casjens, S. R. (1980). DNApackaging by the double-stranded DNA bacterio-phages. Cell, 21, 319–331.
25. Strauss, H. & King, J. (1984). Steps in the stabilizationof newly packaged DNA during phage P22 morpho-genesis. J. Mol. Biol. 172, 523–543.
26. Israel, V. (1967). The production of inactive phage P22particles following induction. Virology, 33, 317–322.
27. Goldenberg, D. P. & King, J. (1981). Temperature-sensitive mutants blocked in the folding or subunit ofthe bacteriophage P22 tail spike protein: II. Activemutant proteins matured at 30 degrees C. J. Mol. Biol.145, 633–651.
28. Lenk, E., Casjens, S., Weeks, J. & King, J. (1975).Intracellular visualization of precursor capsids inphage P22 mutant infected cells. Virology, 68, 182–199.
29. Moak, M. & Molineux, I. J. (2004). Peptidoglycanhydrolytic activities associated with bacteriophagevirions. Mol. Microbiol. 51, 1169–1183.
30. Andrews, D., Butler, J. S., Al-Bassam, J., Joss, L.,

576 Assembly of Gp4 to P22 Portal Protein
Winn-Stapley, D. A., Casjens, S. & Cingolani, G.(2005). Bacteriophage P22 tail accessory factor GP26is a long triple-stranded coiled-coil. J. Biol. Chem. 280,5929–5933.
31. Sampson, L. & Casjens, S. (1993). Nucleotide sequenceof Salmonella bacteriophage P22 head completiongenes 10 and 26. Nucl. Acids Res. 21, 3326.
32. Pedulla, M. L., Ford, M. E., Karthikeyan, T., Houtz,J. M., Hendrix, R. W., Hatfull, G. F. et al. (2003).Corrected sequence of the bacteriophage p22 gen-ome. J. Bacteriol. 185, 1475–1477.
33. Yphantis, D. A. (1964). Equilibrium ultracentrifuga-tion of dilute solutions. Biochemistry, 47, 297–317.
34. Johnson, M. L., Correia, J. J., Yphantis, D. A. &Halvorson, H. R. (1981). Analysis of data from theanalytical ultracentrifuge by nonlinear least-squarestechniques. Biophys. J. 36, 575–588.
35. Przybylski, D. & Rost, B. (2002). Alignments grow,secondary structure prediction improves. Proteins:Struct. Funct. Genet. 46, 197–205.
36. Pace, C. N., Scholtz, J. M. & Creighton, T. E. (1997).Protein Structure: a Practical Approach, pp. 299–321,Oxford University Press, New York.
37. Greene, R. F., Jr & Pace, C. N. (1974). Urea andguanidine hydrochloride denaturation of ribonu-clease, lysozyme, alpha-chymotrypsin, and beta-lactoglobulin. J. Biol. Chem. 249, 5388–5393.
38. Neet, K. E. & Timm, D. E. (1994). Conformationalstability of dimeric proteins: quantitative studiesby equilibrium denaturation. Protein Sci. 3,2167–2174.
39. Dyson, H. J. & Wright, P. E. (2005). Intrinsicallyunstructured proteins and their functions. Nature Rev.Mol. Cell. Biol. 6, 197–208.
40. Wright, P. E. & Dyson, H. J. (1999). Intrinsicallyunstructured proteins: re-assessing the protein struc-ture-function paradigm. J. Mol. Biol. 293, 321–331.
41. Jahn, T. R. & Radford, S. E. (2005). The Yin and Yang ofprotein folding. FEBS J. 272, 5962–5970.
42. Casjens, S. & King, J. (1974). P22 morphogenesis:I. Catalytic scaffolding protein in capsid assembly.J. Supramol. Struct. 2, 202–224.
43. Moore, S. D. & Prevelige, P. E., Jr (2001). Structuraltransformations accompanying the assembly of bac-teriophage P22 portal protein rings in vitro. J. Biol.Chem. 276, 6779–6788.
44. Eppler, K., Wyckoff, E., Goates, J., Parr, R. & Casjens,S. (1991). Nucleotide sequence of the bacteriophageP22 genes required for DNA packaging. Virology, 183,519–538.
45. Berget, P. B. & Poteete, A. R. (1980). Structure andfunctions of the bacteriophage P22 tail protein. J. Virol.34, 234–243.
46. Israel, V. (1977). E proteins of bacteriophage P22: I.Identification and ejection from wild-type and defec-tive particles. J. Virol. 23, 91–97.
47. Bukau, B. & Horwich, A. L. (1998). The Hsp70 andHsp60 chaperone machines. Cell, 92, 351–366.
48. Cingolani, G., Moore, S. D., Prevelige, P. E., Jr &Johnson, J. E. (2002). Preliminary crystallographicanalysis of the bacteriophage P22 portal protein.J. Struct. Biol. 139, 46–54.
49. Laue, T. M. (1995). Sedimentation equilibrium asthermodynamic tool. Methods Enzymol. 259, 427–452.
50. Duda, R. L., Hempel, J., Michel, H., Shabanowitz,J., Hunt, D. & Hendrix, R. W. (1995). Structuraltransitions during bacteriophage HK97 head assem-bly. J. Mol. Biol. 247, 618–635.
51. Rost, B., Sander, C. & Schneider, R. (1994). PHD–anautomatic mail server for protein secondary structureprediction. Comput. Appl. Biosci. 10, 53–60.
52. Goldenberg, D. P., Berget, P. B. & King, J. (1982).Maturation of the tail spike endorhamnosidase ofSalmonella phage P22. J. Biol. Chem. 257, 7864–7871.
Edited by J. Karn
(Received 11 April 2006; received in revised form 4 August 2006; accepted 4 August 2006)Available online 12 August 2006