induction of alloantigen-specific human t regulatory cells ... · 2 address correspondence and...
TRANSCRIPT

of February 3, 2019.This information is current as
PeptideRegulatory Cells by Vasoactive Intestinal Induction of Alloantigen-Specific Human T
David Pozo, Per Anderson and Elena Gonzalez-Rey
http://www.jimmunol.org/content/183/7/4346doi: 10.4049/jimmunol.0900400September 2009;
2009; 183:4346-4359; Prepublished online 4J Immunol
Referenceshttp://www.jimmunol.org/content/183/7/4346.full#ref-list-1
, 25 of which you can access for free at: cites 62 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2009 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 3, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

Induction of Alloantigen-Specific Human T Regulatory Cellsby Vasoactive Intestinal Peptide1
David Pozo,* Per Anderson,† and Elena Gonzalez-Rey2†‡
T regulatory cells (Tregs) are instrumental in the maintenance of immunological tolerance. Although Treg-based immunotherapyproved successful in preclinical autoimmunity and transplantation, factors involved in the generation of human Ag-specific Tregsare poorly known. In this study, we show that treatment of human CD4�CD25� T cells with the cytokine-like vasoactive intestinalpeptide (VIP) during in vitro stimulation induces an anergic FoxP3�CD4�CD25high T cell subset displaying potent regulatoryactivities against allospecific effector T cells, irrespective of the presence of naturally occurring Tregs. VIP-tolerant T cells arecharacterized by incapability to progress to S phase of cell cycle during stimulation with HLA-disparate APCs by negativelyaffecting the synthesis of cyclins D3 and E, the activation of cyclin-dependent kinases (cdk)2 and cdk4, and the down-regulationof the cdk inhibitor p27kip1. VIP interaction with the type 1 VIP receptor and subsequent activation of cAMP/protein kinase Apathway play a major role in all these effects. Moreover, VIP-tolerant T cells protect against acute graft-vs-host disease in a mousemodel of allogeneic bone marrow transplantation. The infusion of VIP-tolerant T cells together with the graft significantly reducesthe clinical signs and mortality rate typical of the graft-vs-host disease. These effects are mediated by impairing allogeneichaplotype-specific responses of donor CD4� cells in the transplanted animals. Our results suggest that including alloantigen-specific VIP-generated Tregs may be a valuable tool in therapeutic interventions to promote immunotolerance toward allogeneicgrafts and to reduce the need of general immunosuppressive drugs. The Journal of Immunology, 2009, 183: 4346–4359.
S afe induction of Ag-specific long-term tolerance is essen-tial to maintain immune homeostasis, to control autoreac-tive T cells in autoimmune diseases, and to achieve trans-
plantation tolerance (1, 2). Tolerance is mainly maintained throughthe intrathymic deletion of self-reactive T cells and through theinduction of T cell anergy and differentiation of T regulatory cells(Tregs).3
Two major populations of Tregs, with complementary and over-lapping functions in the control of immune response in vivo, exist,as follows: natural (or constitutive) and inducible (or adaptive)Tregs. Numerous studies have demonstrated the therapeutic use ofAg-specific Tregs in various experimental models of autoimmunediseases and allogeneic transplantation, providing long-term toler-ance by active and specific regulation of self-Ag and alloantigen-specific T cells (3–5). These findings have opened up excitingopportunities for new therapies in several human diseases that areassociated with Treg dysfunction. However, the translation of im-
portant biological findings about Tregs to the clinic has beenmainly limited by the inability to define their antigenic specificitiesand by the scarcity of circulating Tregs. The solution to this prob-lem might lie in expanding the Treg population in vitro, and mak-ing the Tregs Ag specific. However, although Tregs replicate ef-ficiently in vivo, they are anergic and refractory to stimulation invitro (3, 5). Thus, to efficiently expand Treg populations in vitrowhile maintaining their immunoregulatory properties in vivo, newprotocols must be developed that reproduce the conditions thatenable replication in vivo, including TCR occupancy, crucial co-stimulatory signals, and the presence of selective growth factors.However, although the ontogeny and mechanisms involved in thesuppressive action of Tregs on self- and alloreactive lymphocytesare widely described in the literature (2, 3, 5), the endogenousfactors and mechanisms controlling their peripheral generation orexpansion are mostly unknown.
Allogeneic bone marrow transplantation (BMT) is the treatmentof choice in many hematopoietic malignancies. Following high-dose chemotherapy or irradiation, the host is reconstituted withbone marrow cells, and the donor T cells are responsible for thegraft-vs-tumor effects that eliminate the remaining malignant cellsin the host. However, the same donor T cells, which recognizeMHC disparities in the recipient, expand and initiate a multiorgansystem distraction known as graft-vs-host disease (GvHD). In fact,acute GvHD is a major cause of morbidity and mortality in patientsundergoing allogeneic BMT (6). Most therapeutic approaches de-signed to reduce acute GvHD have focused on the development ofnonspecific immunosuppressive drugs and the ex vivo removal ofdonor T cells from the transplant (7). However, removal of theseT cells before grafting was shown to lead to transplant failure andleukemia relapse, and although successful in controlling T cellalloreactivity during transplantation, prolonged administration ofmany of the immunosuppressive drugs results in adverse side ef-fects derived from sustained immunosuppression (8). An alterna-tive approach to improve the allogeneic transplantation outcome is
*Andalusian Center for Molecular Biology and Regenerative Medicine, Seville,Spain; †Institute of Parasitology and Biomedicine, Consejo Superior de Investigacio-nes Cientificas, Granada, Spain; and ‡Department of Medical Biochemistry and Mo-lecular Biology, University of Seville, Seville, Spain
Received for publication February 5, 2009. Accepted for publication July 22, 2009.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by Spanish Ministry of Education and Science (to E.G.-R.), Spanish Ministry of Health (to D.P.), Junta de Andalucia, and Spanish Collab-orative Network on Multiple Sclerosis (ISCIII-RETICS-REEM).2 Address correspondence and reprint requests to Dr. Elena Gonzalez-Rey, Institutode Parasitologia y Biomedicina, Consejo Superior de Investigaciones Cientificas, PTCiencias Salud, Granada, 18100 Spain. E-mail address: [email protected] Abbreviations used in this paper: Treg, T regulatory cell; BMT, bone marrow trans-plantation; cdk, cyclin-dependent kinase; GvHD, graft-vs-host disease; int, interme-diate; PCCF, pigeon cytochrome c fragment; PKA, protein kinase A; Rb, retinoblas-toma gene product; pRb, phosphorylated Rb; TCD-BM, T cell-depleted bone marrowcell; VIP, vasoactive intestinal peptide; VPAC1, type 1 VIP receptor.
Copyright © 2009 by The American Association of Immunologists, Inc. 0022-1767/09/$2.00
The Journal of Immunology
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0900400
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

the induction of graft tolerance by selectively inactivating allo-reactive donor T cells in the absence of chronic immunosuppres-sion. Recently, the use of alloantigen-specific Tregs as a therapeu-tic strategy to limit the pathologic effects of donor-alloreactive Tcells has been proposed (1, 3, 9). Given the potential clinical im-portance of the induction of tolerance after the transplant, it iscrucial to identify immunosuppressive agents that do not interferewith the development of Tregs, and ideally improve the function orgeneration of the Treg compartment.
Vasoactive intestinal peptide (VIP), a neuropeptide releasedfrom the VIP-ergic innervation of immunocompetent tissues, suchas thymus, spleen, and lymph nodes as well as from Th2 cells inresponse to Ag stimulation and under inflammatory conditions, isa potent cytokine-like agent that affects both innate and adaptiveimmunity (10–12). Indeed, VIP has been used for the treatment ofvarious experimental models of inflammatory and autoimmunediseases (12, 13). The therapeutic effect of VIP was initially at-tributed to the down-regulation of a wide panel of inflammatorymediators and to the inhibition of autoreactive Th1 cells. However,recent evidence has demonstrated the involvement of Tregs in thebeneficial effect of VIP in immune disorders (14). VIP has beenshown to induce the emergence of Ag-specific Tregs in vivo withsuppressive activity on effector T cells (15–17). However, themechanisms involved in the generation or expansion of this Tregpopulation are not fully understood. Moreover, the tolerance/an-ergy-promoting potential of VIP in human T cells is still unknown.In this study, we investigate whether VIP is able to induce humanTregs with suppressive action on alloantigen-specific effector Tcells and the molecular mechanisms involved in such an effect. Wealso describe the therapeutic applicability of the VIP-tolerated Tcells in a murine model of histoincompatible BMT.
Materials and MethodsAbs and reagents
VIP, Sp-8-Br-cAMP, H-89, and histone H1 were purchased from Calbio-chem. The type 1 VIP receptor (VPAC1) antagonist (Ac-His1,D-Phe2,Lys15,Arg16,Leu27)VIP(3–7)-GRF(8–27) was previously described(18). FITC-, PerCP-, and PE-conjugated Abs against CTLA4, TGF-�1,IFN-�, IL-10, IL-17, IL-2, IL-4, V�3 (clone KJ25), CD154, CD25, andCD4, and Abs against pRb, CD3, and IL-10 were obtained from BDPharmingen. Human rIL-2 and rIL-15 were obtained from Roche Biomed-ical and PeproTech. Protein A/G agarose, and Abs against cyclin E, cyclinA, cyclin-dependent kinase (cdk)2, cyclin D2, cyclin D3, cdk4, cdk6, andp27kip1 were purchased from Santa Cruz Biotechnology. FITC- and Alexa-conjugated anti-mouse FoxP3 Ab (clone FKJ-16s) and anti-human FoxP3Abs (clones PCH101 and 236A/E7) were obtained from eBioscience. Pi-geon cytochrome c fragment (PCCF) was synthesized and purified in ourfacilities.
Cell isolation
PBMCs were isolated from buffy coat preparations derived from the wholeblood of healthy volunteers by density sedimentation on Ficoll-Hypaquegradients (20 min, 600 � g). Cells recovered from the gradient interfacewere washed twice in RPMI 1640 medium and immediately used for cul-ture or further purification. To isolate T cells (purity �96%), PBMCs wereincubated with anti-CD8, -CD14, -CD19, -CD20, and -CD56 mAbs(Coulter Immunotech) for 1 h at 4°C, followed by 1-h incubation at 4°Cwith anti-mouse IgG-coated magnetic beads, and bead-bound cells re-moved with a magnetic device. To minimize stimulation of cells, all thepurification steps were conducted in the absence of serum. CD4� T cells(purity of 94–98%) were isolated by negative selection from the PBMCsusing the CD4 isolation kit (Miltenyi Biotec). CD4�CD25� andCD4�CD25� T cell fractions were isolated (purity �96%) using theCD4�CD25� T Regulatory Cell Isolation kit from Miltenyi Biotec. Insome experiments, the different T cell populations (CD4�, CD4�CD25�,CD4�CD25�) were isolated by sorting using a FACSCalibur flow cytom-eter (BD Biosciences) after labeling with PE anti-CD25 and PerCP anti-CD4 Abs, as described below. All samples of CD4�CD25� T cells isolatedwith either beads or sorting used in the study were negative for FoxP3
expression, as assessed by flow cytometry (FoxP3� cells were �1%) andRT-PCR analysis. APCs were obtained from PBMCs after cell adhesion onplastic dishes for 2 h, followed by T cell depletion with anti-CD3-coatedmagnetic beads.
Murine CD4�CD25� and CD4�CD25� cells were isolated fromC57BL/6 mice (The Jackson Laboratory), as described previously (15).Spleens were gently minced in complete DMEM containing 10% FBS(BioWhittaker), and CD4� T cells were purified using a mouse CD4� Tcell column system (R&D Systems). T cell-depleted spleen cells (irradi-ated, 3000 rad) of BALB/c or C57BL/6 mice were used as APCs.
Cell cultures
All cultures were conducted in complete medium consisting of RPMI 1640medium supplemented with heat-inactivated human pooled serum (8%),L-glutamine (20 mM), sodium pyruvate (1%), nonessential amino acids(1%), and penicillin/streptomycin (1%) (all from Invitrogen) in a 5% CO2
humidified atmosphere at 37°C.
Allogeneic stimulation
Primary MLCs were performed in 96-well, round-bottom plates by stim-ulating responder PBMCs (105) with allogeneic HLA-mismatched 30 Gy�-irradiated stimulator PBMCs (105) in 200 �l of medium in the presenceor absence of VIP (10�7 M). Cells were pulsed with 0.5 �Ci (0.0185MBq)/well [3H]thymidine for the last 8 h of the culture and harvested ontomembranes, and proliferation was determined by measuring [3H]thymidineincorporation. Some primary MLCs were established with CD4�CD25� Tcells (5 � 104) and were stimulated with allogeneic HLA-mismatched�-irradiated PBMCs (5 � 104) without or with VIP. Secondary MLCs wereperformed to determine the memory response of allogeneic primed T cells.Cells were harvested after 6 days of primary stimulation, washed threetimes, and rested for 2 days. The viable cells (5 � 104) were recovered bydensity gradient centrifugation with Lymphoprep (Nycomed Pharma) andrestimulated in a secondary culture with HLA-mismatched �-irradiatedPBMCs (2 � 105) in the absence or presence of IL-2 (20 U/ml). [3H]Thy-midine incorporation was determined at different time points of secondaryMLC. When indicated, recovered viable cells (106/ml) from primary MLCwere stimulated in a secondary culture with anti-CD3/anti-CD28 mAb-coated magnetic beads (Invitrogen; 1 bead/cell). Cells were collected atdifferent time points after anti-CD3/CD28 restimulation and lysed for pro-tein and kinase activity determinations, as described below, and the pro-liferative response was determined as above.
T cell suppression assays
The suppressive capacity of VIP-treated cells was analyzed in a cocultureassay. After primary stimulation of responder cells (105 PBMCs orCD4�CD25� T cells, from donor A) in MLCs with HLA-mismatched 30Gy �-irradiated stimulator PBMCs (105, from donor B) in the presence orabsence of VIP for 6 days, the cells were harvested and allowed to recu-perate for 2 days. The recovered T cells were added at different ratios to anewly set-up primary MLC (consisting of 105 original responder PBMCsfrom donor A and 105 stimulator PBMCs from donor B), and proliferationwas measured by [3H]thymidine uptake. Some cultures were performed inthe presence of blocking anti-IL-10 (10 �g/ml), anti-TGF-�1 (10 �g/ml),and/or anti-CTLA4 (10 �g/ml) mAbs, or human rIL-2 (100 U/ml). Todetermine the cell-contact dependence of the suppressive response, weplaced responder PBMCs (5 � 104) with allogeneic stimulator PBMCs(5 � 104) in the bottom well of a Transwell system (Millipore; 0.4 �mpore), and the recovered T cells (2 � 104) with or without allogeneicstimulator PBMCs (5 � 104) in the upper Transwell insert. At day 4, theproliferative response of the responder PBMCs in the lower compartmentwas determined.
In similar experiments, responder PBMCs and T cells were labeled with2.5 �M CFSE (Molecular Probes) before setting up cocultures, and pro-liferating cells were determined by CFSE dilution by flow cytometry.
Assessment of apoptosis and cell viability
Quantitative determination of viability was performed using an annexinV-based apoptosis detection kit (R&D Systems) and subsequently analyzedby flow cytometry. Moreover, cell numbers were determined by countingcells excluding trypan blue after 72 h of culture.
Cell cycle analysis
Cells (106) were fixed in an ice-cold solution of 70% ethanol for at least 1 hand incubated with 0.1% RNase at 37°C for 45 min. Cells were then in-cubated with 50 �g/ml propidium iodide at 37°C for 30 min, before anal-ysis for DNA content by flow cytometry using CellQuest software.
4347The Journal of Immunology
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

Flow cytometry
Cells were incubated with various PerCP-, FITC-, and PE-labeled mAbsdiluted at optimal concentration for immunostaining, fixed in 1% parafor-maldehyde, and analyzed on a FACSCalibur flow cytometer. We usedisotype-matched Abs as controls, and IgG block (Sigma-Aldrich) to avoidthe nonspecific binding to Fc receptors. For analysis of intracellularCTLA4 and FoxP3, cells were stained first for surface CD4 and CD25 withPerCP anti-CD4 and PE anti-CD25, fixed with Cytofix/Cytoperm solution(BD Pharmingen), incubated with FITC anti-CTLA4 or FITC anti-FoxP3mAb diluted in 0.5% saponin, and analyzed by flow cytometry. BecauseTran et al. (19) recently reported that anti-FoxP3 mAb clone PCH101 couldresult in nonspecific staining of activated T cells, two different anti-FoxP3mAbs were used in this study (clones PCH101 and 236A/E7) to confirmour results. In addition, mRNA expression of FoxP3 was determined, asdescribed below.
For intracellular cytokine analysis, viable T cells were recovered afterstimulation by gradient centrifugation and stimulated at 106 cells/ml withPMA (10 ng/ml) plus ionomycin (50 ng/ml) for 6 h. Monensin (1.33 �M)was added for the last 4 h of culture. Cells were stained with PerCP anti-CD4 mAbs for 30 min at 4°C; washed; fixed/saponin permeabilized withCytofix/Cytoperm; stained with 0.5 �g/sample FITC-conjugated anti-IL-2,anti-IFN-�, anti-IL-4, anti-IL-17, anti-TGF-�1, anti-TNF-�, or anti-IL-10mAbs for 45 min at 4°C; and analyzed by flow cytometry.
Cytokine determination
Cytokine contents in the culture supernatants or sera were determined byspecific sandwich ELISAs using capture/biotinylated detection Abs fromBD Pharmingen.
Determination of gene expression
CTLA4 gene expression was determined by RT-PCR, as previously de-scribed (20). Primers used were designed to amplify the entire codingsequence of CTLA4 (5�-ATGGCTTGCCTTGGATTTCAGCGGCACAAGG-3� and 5�-TCAATTGATGGGAATAAAATAAGGCTGAAATTGC-3�). PCR conditions were as follows: 94°C for 5 min, 30 cycles at 94°C for30 s, 58°C for 30 s, and 72°C for 30 s, followed by a final extension at 72°Cfor 7 min. The amplified fragments were separated on 1.5% agarose gel andvisualized by ethidium bromide. RNA integrity and cDNA synthesis wereverified by amplifying �2-microglobulin cDNA. The intensity of the re-vealed bands was quantified by Image QuaNT software (Amersham Bio-sciences), normalized to those of �2-microglobulin, and expressed as ar-bitrary units. Human FoxP3 gene expression was quantitated by real-timePCR in triplicate according to the TaqMan Universal 2 master mix and runon the ABI/PRISM 7900 Sequence Detector System (Applied Biosystems)(20). The amount of FoxP3 mRNA expression was normalized with thehuman �-actin and calculated according to the comparative cycle thresholdmethod. FoxP3 primers and probe were as follows: forward primer, 5�-GCACCTTCCCAAATCCCAGT-3�; reverse primer, 5�-GGCCACTTGCAGACACCAT-3�; and fluorogenic probe (5-carboxyfluorescein/TAMRA),5�-CAGGAAGGACAGCACCCTTTCGGC-3�. Mouse FoxP3 expressionwas determined by RT-PCR, as described previously (21), by using the fol-lowing primers: FoxP3, 5�-CAGCTGCCTACAGTGCCCCTAG-3� and 5�-CATTTGCCAGCAGTGGGTAG-3�; hypoxanthine phosphoribosyltrans-ferase, 5�-GTTGGATACAGGCCAGACTTTGTTG-3� and 5�-GATTCAACTTGCTCTCATCTTAGGC-3�.
Western blot analysis
Whole-cell lysates were prepared by lysing 106 cells in lysis buffer (50 mMTris-HCl (pH 8.0), 100 mM NaCl, 5 mM EDTA, 50 mM glycerolphos-phate, 1% Triton X-100, 5% glycerol, 1 mM DTT, 5 mM benzamidine, 2�g/ml leupeptin, 5 �g/ml aprotinin, 1 mM PMSF, 5 mM NaF, 10 mMNa2HPO4, and 1 mM Na3VO4) for 30 min on ice and mixing gently. In-soluble fragments were pelleted by centrifugation for 30 min at 10,000 �g. Proteins from whole extracts (60–80 �g/lane) were separated by SDS-PAGE (12%; analysis of pRb: 8%) and blotted onto polyvinylidene diflu-oride membranes (Millipore) using a semidry system. Membranes wereblocked with TBS/Tween 20/3% nonfat dry milk for 1 h at room temper-ature and subsequently probed with the indicated primary Ab overnight at4°C. Immunodetection was performed by incubation with HRP-conjugatedAbs and visualized by ECL (Amersham Pharmacia). Equal protein loadingwas controlled by reprobing with anti-�-actin Abs. Cdk-associated kinaseactivities were determined, as described below.
Determination of cdk-associated kinase activity
To determine the Cdk2-, Cdk4-, cyclin E-, and cyclin D2-associated kinaseactivities, whole protein extracts (500 �g) were immunoprecipitated with
1 �g of the following Abs: anti-cdk2, anti-cyclin E, anti-cdk4, and anti-cyclin D2, for 2 h at 4°C under constant agitation. The immune complexeswere collected by incubation with 10 �g of protein A/G-Sepharose beadsfor 45 min at 4°C under constant agitation. The beads were extensivelywashed with the lysis buffer, and kinase activity was assayed by incubatingthe immunoprecipitates 30 min at 30°C with 50 mM HEPES (pH 7.5), 10mM MgCl2, 2.5 mM EGTA, 1 mM DTT, 10 mM �-glycerophosphate, 0.1mM Na3VO4, 1 mM NaF, 50 �M cold ATP, 10 �Ci of [�-32P]ATP (6000Ci/mmol; Amersham Biosciences), and 5 �g of histone H1 for cdk2 assayor 2 �g of retinoblastoma gene product (Rb)-GST for cdk4. The phosphor-ylated proteins were separated by SDS-PAGE, transferred to Immobilon Pmembranes (Millipore), and subsequently used for Western blot analysis.
GvHD model
All experiments were performed according to the Institutional Guidelinesfor the Care and Use of Laboratory Animals in Research and approved byConsejo Superior de Investigaciones Cientificas. CD4�CD25� T cells iso-lated from C57BL/6 mice (H-2b) were cultured with anti-CD3 (0.5 �g/ml)in the presence of syngeneic APCs in complete medium in the absence(Tcontrol) or presence of 10�7 M VIP (TVIP) for 4 days, then extensivelywashed and rested in medium containing IL-2 (10 U/ml) for 3 additionaldays. Viable Tcontrol or TVIP (106) recovered by gradient centrifugationwere injected i.v. into BALB/c mice (H-2d) that were lethally irradiated (8Gy total body irradiation with a 200 Kv x-ray source) and reconstituted i.v.with allogeneic T cell-depleted bone marrow cells (TCD-BM; 5 � 106)from C57BL/6 mice. The survival and appearance of the BALB/c hostswere monitored daily, and body weight was measured weekly. In otherexperiments, allogeneic transplantation was performed by a single i.v. in-jection of TCD-BM supplemented with 1.5 � 106 spleen mononuclearcells (1.5 � 107/mouse) isolated from C57BL/6 into recipient BALB/cmice lethally irradiated (10 Gy total body irradiation from a 200 Kv x-raysource). Two hours after transplantation, recipients received a single i.v.injection of CD4� T cells (106) from C57BL/6 mice that were stimulatedin a primary MLC with allogeneic spleen cells from BALB/c mice in theabsence or presence of 10�7 M VIP for 6 days, harvested, and rested for anadditional 2 days. Recipients were monitored from the day of transplanta-tion until they succumbed naturally to GvHD. Serum and spleen cells wereharvested 5 days following transplantation, and splenic donor H2-KbCD4�
and H2-KbCD8� T cells were isolated by immunomagnetic selection, asdescribed (22). Donor H2-KbCD4� cells (5 � 105) were stimulated withallogeneic splenic APCs (H-2d, 5 � 104), and the proliferative responsewas determined by [3H]thymidine incorporation and intracellular cytokineexpression by flow cytometry, as described above. Cytokine contents inserum were determined by ELISA, as above. The percentages of CD4�,CD25�, and CTLA4� T cells in the spleen of recipient mice were deter-mined by flow cytometry, as described above.
Transgenic mice
TCR-Cyt-5CC7-I/Rag1 transgenic (I-Ek) mice (PCCF TCR transgenic)were obtained from Taconic Farms. PCCF TCR transgenic mice were in-jected i.p. on days 0 and �2 with Ag (PCCF, 50 �g) and with or withoutVIP (5 nmol). At different times after initial Ag stimulation, spleen andlymph nodes (inguinal, mesenteric, and popliteal) were isolated and ana-lyzed for V�3, CD25, and FoxP3 expression by flow cytometry. In someexperiments, CD25 cells were depleted (in vivo depletion �98%) fromPCCF TCR transgenic mice by treating mice i.v. with anti-CD25 Ab (clonePC61, 1 mg) 3 days before Ag stimulation.
Data analysis
All values are expressed as mean � SD. Differences in survival of treat-ment groups were analyzed using the log-rank test. Differences in prolif-eration and cytokine production by cultures, serum cytokine levels, andpercentage of cells were analyzed using two-tailed Student’s t test. Valuesof p � 0.05 were considered significant.
ResultsVIP inhibits proliferation and induces cell cycle arrest inallogeneic activated human T cells
Anergy, the in vitro counterpart of tolerance in vivo, is defined asthe inability of T cells to expand after stimulation with fully com-petent APCs delivering TCR and costimulatory molecules (23). Toinvestigate the capacity of VIP to induce anergic human T cells,freshly isolated PBMCs were stimulated with �-irradiated HLA-mismatched stimulator PBMCs in the absence or presence of VIP,
4348 VIP-PROMOTED IMMUNE TOLERANCE TOWARD HUMAN ALLOANTIGENS
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
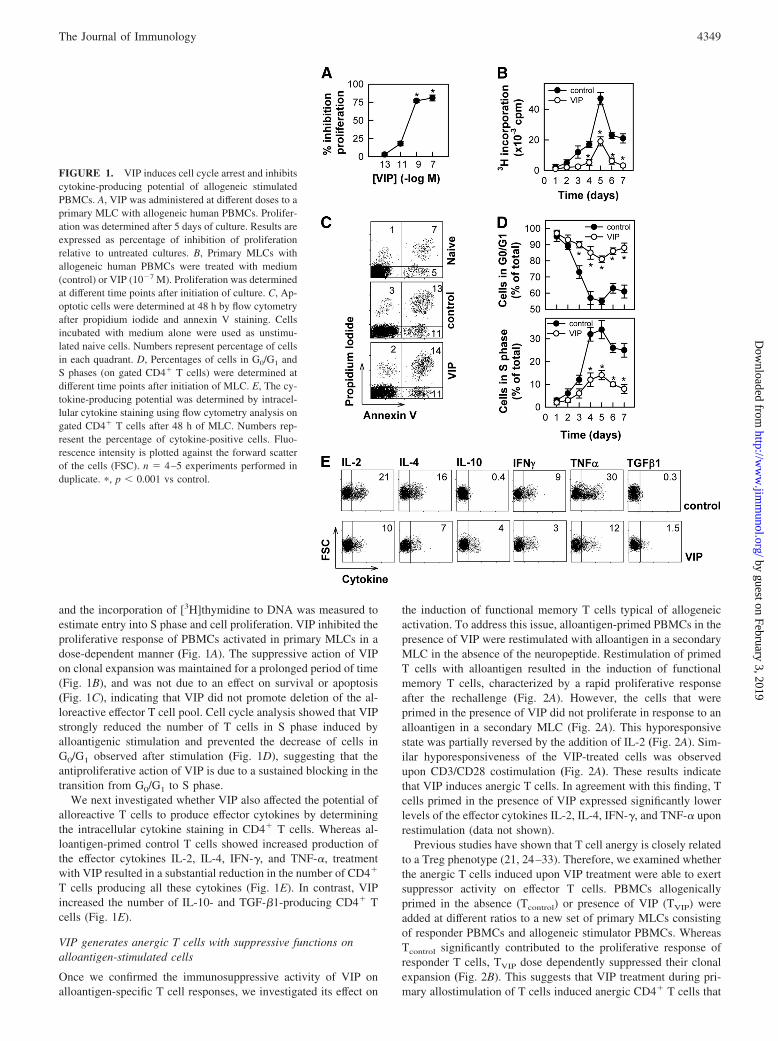
and the incorporation of [3H]thymidine to DNA was measured toestimate entry into S phase and cell proliferation. VIP inhibited theproliferative response of PBMCs activated in primary MLCs in adose-dependent manner (Fig. 1A). The suppressive action of VIPon clonal expansion was maintained for a prolonged period of time(Fig. 1B), and was not due to an effect on survival or apoptosis(Fig. 1C), indicating that VIP did not promote deletion of the al-loreactive effector T cell pool. Cell cycle analysis showed that VIPstrongly reduced the number of T cells in S phase induced byalloantigenic stimulation and prevented the decrease of cells inG0/G1 observed after stimulation (Fig. 1D), suggesting that theantiproliferative action of VIP is due to a sustained blocking in thetransition from G0/G1 to S phase.
We next investigated whether VIP also affected the potential ofalloreactive T cells to produce effector cytokines by determiningthe intracellular cytokine staining in CD4� T cells. Whereas al-loantigen-primed control T cells showed increased production ofthe effector cytokines IL-2, IL-4, IFN-�, and TNF-�, treatmentwith VIP resulted in a substantial reduction in the number of CD4�
T cells producing all these cytokines (Fig. 1E). In contrast, VIPincreased the number of IL-10- and TGF-�1-producing CD4� Tcells (Fig. 1E).
VIP generates anergic T cells with suppressive functions onalloantigen-stimulated cells
Once we confirmed the immunosuppressive activity of VIP onalloantigen-specific T cell responses, we investigated its effect on
the induction of functional memory T cells typical of allogeneicactivation. To address this issue, alloantigen-primed PBMCs in thepresence of VIP were restimulated with alloantigen in a secondaryMLC in the absence of the neuropeptide. Restimulation of primedT cells with alloantigen resulted in the induction of functionalmemory T cells, characterized by a rapid proliferative responseafter the rechallenge (Fig. 2A). However, the cells that wereprimed in the presence of VIP did not proliferate in response to analloantigen in a secondary MLC (Fig. 2A). This hyporesponsivestate was partially reversed by the addition of IL-2 (Fig. 2A). Sim-ilar hyporesponsiveness of the VIP-treated cells was observedupon CD3/CD28 costimulation (Fig. 2A). These results indicatethat VIP induces anergic T cells. In agreement with this finding, Tcells primed in the presence of VIP expressed significantly lowerlevels of the effector cytokines IL-2, IL-4, IFN-�, and TNF-� uponrestimulation (data not shown).
Previous studies have shown that T cell anergy is closely relatedto a Treg phenotype (21, 24–33). Therefore, we examined whetherthe anergic T cells induced upon VIP treatment were able to exertsuppressor activity on effector T cells. PBMCs allogenicallyprimed in the absence (Tcontrol) or presence of VIP (TVIP) wereadded at different ratios to a new set of primary MLCs consistingof responder PBMCs and allogeneic stimulator PBMCs. WhereasTcontrol significantly contributed to the proliferative response ofresponder T cells, TVIP dose dependently suppressed their clonalexpansion (Fig. 2B). This suggests that VIP treatment during pri-mary allostimulation of T cells induced anergic CD4� T cells that
FIGURE 1. VIP induces cell cycle arrest and inhibitscytokine-producing potential of allogeneic stimulatedPBMCs. A, VIP was administered at different doses to aprimary MLC with allogeneic human PBMCs. Prolifer-ation was determined after 5 days of culture. Results areexpressed as percentage of inhibition of proliferationrelative to untreated cultures. B, Primary MLCs withallogeneic human PBMCs were treated with medium(control) or VIP (10�7 M). Proliferation was determinedat different time points after initiation of culture. C, Ap-optotic cells were determined at 48 h by flow cytometryafter propidium iodide and annexin V staining. Cellsincubated with medium alone were used as unstimu-lated naive cells. Numbers represent percentage of cellsin each quadrant. D, Percentages of cells in G0/G1 andS phases (on gated CD4� T cells) were determined atdifferent time points after initiation of MLC. E, The cy-tokine-producing potential was determined by intracel-lular cytokine staining using flow cytometry analysis ongated CD4� T cells after 48 h of MLC. Numbers rep-resent the percentage of cytokine-positive cells. Fluo-rescence intensity is plotted against the forward scatterof the cells (FSC). n � 4–5 experiments performed induplicate. �, p � 0.001 vs control.
4349The Journal of Immunology
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
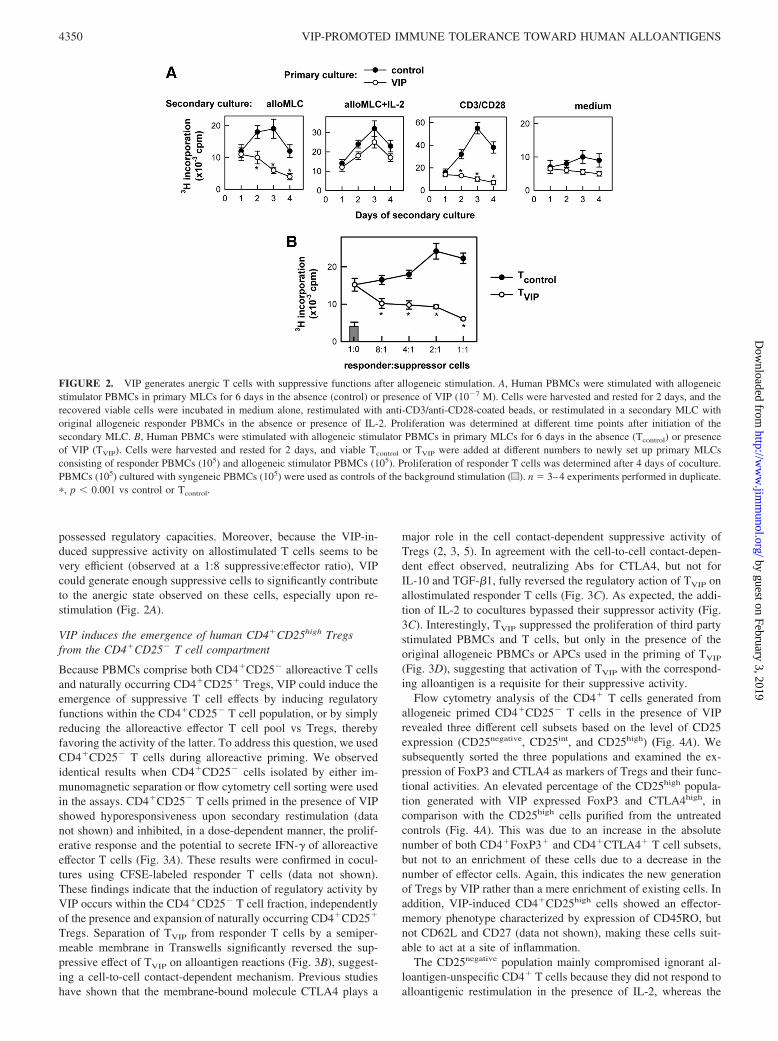
possessed regulatory capacities. Moreover, because the VIP-in-duced suppressive activity on allostimulated T cells seems to bevery efficient (observed at a 1:8 suppressive:effector ratio), VIPcould generate enough suppressive cells to significantly contributeto the anergic state observed on these cells, especially upon re-stimulation (Fig. 2A).
VIP induces the emergence of human CD4�CD25high Tregsfrom the CD4�CD25� T cell compartment
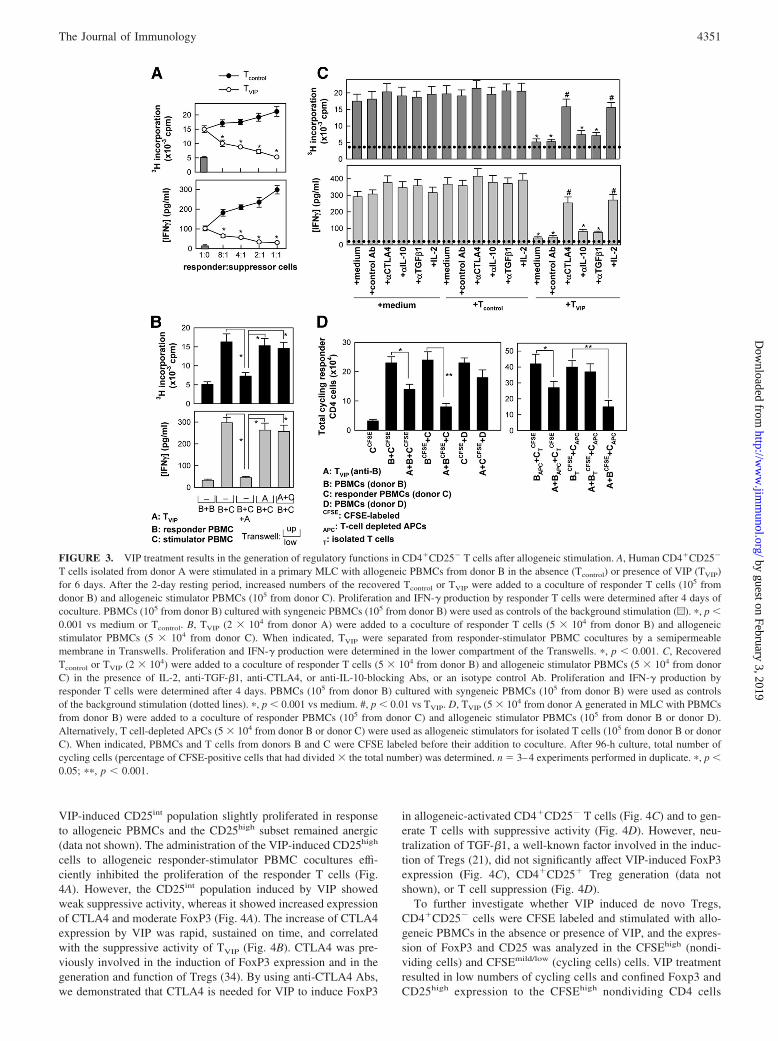
Because PBMCs comprise both CD4�CD25� alloreactive T cellsand naturally occurring CD4�CD25� Tregs, VIP could induce theemergence of suppressive T cell effects by inducing regulatoryfunctions within the CD4�CD25� T cell population, or by simplyreducing the alloreactive effector T cell pool vs Tregs, therebyfavoring the activity of the latter. To address this question, we usedCD4�CD25� T cells during alloreactive priming. We observedidentical results when CD4�CD25� cells isolated by either im-munomagnetic separation or flow cytometry cell sorting were usedin the assays. CD4�CD25� T cells primed in the presence of VIPshowed hyporesponsiveness upon secondary restimulation (datanot shown) and inhibited, in a dose-dependent manner, the prolif-erative response and the potential to secrete IFN-� of alloreactiveeffector T cells (Fig. 3A). These results were confirmed in cocul-tures using CFSE-labeled responder T cells (data not shown).These findings indicate that the induction of regulatory activity byVIP occurs within the CD4�CD25� T cell fraction, independentlyof the presence and expansion of naturally occurring CD4�CD25�
Tregs. Separation of TVIP from responder T cells by a semiper-meable membrane in Transwells significantly reversed the sup-pressive effect of TVIP on alloantigen reactions (Fig. 3B), suggest-ing a cell-to-cell contact-dependent mechanism. Previous studieshave shown that the membrane-bound molecule CTLA4 plays a
major role in the cell contact-dependent suppressive activity ofTregs (2, 3, 5). In agreement with the cell-to-cell contact-depen-dent effect observed, neutralizing Abs for CTLA4, but not forIL-10 and TGF-�1, fully reversed the regulatory action of TVIP onallostimulated responder T cells (Fig. 3C). As expected, the addi-tion of IL-2 to cocultures bypassed their suppressor activity (Fig.3C). Interestingly, TVIP suppressed the proliferation of third partystimulated PBMCs and T cells, but only in the presence of theoriginal allogeneic PBMCs or APCs used in the priming of TVIP
(Fig. 3D), suggesting that activation of TVIP with the correspond-ing alloantigen is a requisite for their suppressive activity.
Flow cytometry analysis of the CD4� T cells generated fromallogeneic primed CD4�CD25� T cells in the presence of VIPrevealed three different cell subsets based on the level of CD25expression (CD25negative, CD25int, and CD25high) (Fig. 4A). Wesubsequently sorted the three populations and examined the ex-pression of FoxP3 and CTLA4 as markers of Tregs and their func-tional activities. An elevated percentage of the CD25high popula-tion generated with VIP expressed FoxP3 and CTLA4high, incomparison with the CD25high cells purified from the untreatedcontrols (Fig. 4A). This was due to an increase in the absolutenumber of both CD4�FoxP3� and CD4�CTLA4� T cell subsets,but not to an enrichment of these cells due to a decrease in thenumber of effector cells. Again, this indicates the new generationof Tregs by VIP rather than a mere enrichment of existing cells. Inaddition, VIP-induced CD4�CD25high cells showed an effector-memory phenotype characterized by expression of CD45RO, butnot CD62L and CD27 (data not shown), making these cells suit-able to act at a site of inflammation.
The CD25negative population mainly compromised ignorant al-loantigen-unspecific CD4� T cells because they did not respond toalloantigenic restimulation in the presence of IL-2, whereas the
FIGURE 2. VIP generates anergic T cells with suppressive functions after allogeneic stimulation. A, Human PBMCs were stimulated with allogeneicstimulator PBMCs in primary MLCs for 6 days in the absence (control) or presence of VIP (10�7 M). Cells were harvested and rested for 2 days, and therecovered viable cells were incubated in medium alone, restimulated with anti-CD3/anti-CD28-coated beads, or restimulated in a secondary MLC withoriginal allogeneic responder PBMCs in the absence or presence of IL-2. Proliferation was determined at different time points after initiation of thesecondary MLC. B, Human PBMCs were stimulated with allogeneic stimulator PBMCs in primary MLCs for 6 days in the absence (Tcontrol) or presenceof VIP (TVIP). Cells were harvested and rested for 2 days, and viable Tcontrol or TVIP were added at different numbers to newly set up primary MLCsconsisting of responder PBMCs (105) and allogeneic stimulator PBMCs (105). Proliferation of responder T cells was determined after 4 days of coculture.PBMCs (105) cultured with syngeneic PBMCs (105) were used as controls of the background stimulation (u). n � 3–4 experiments performed in duplicate.�, p � 0.001 vs control or Tcontrol.
4350 VIP-PROMOTED IMMUNE TOLERANCE TOWARD HUMAN ALLOANTIGENS
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
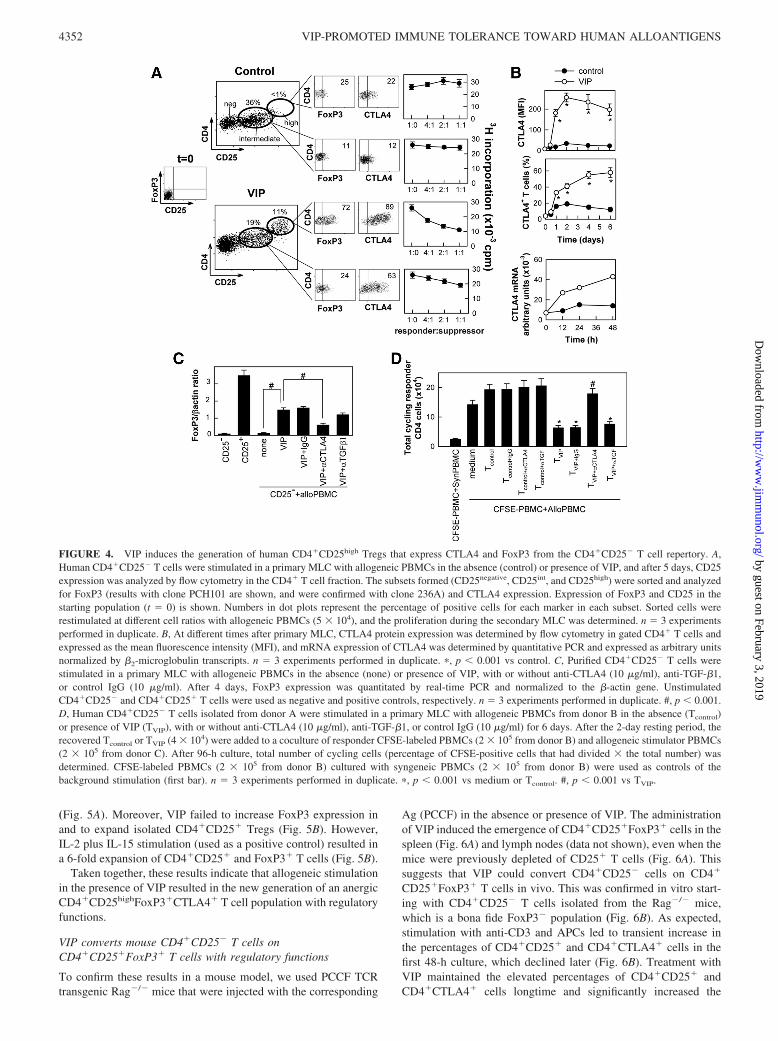
VIP-induced CD25int population slightly proliferated in responseto allogeneic PBMCs and the CD25high subset remained anergic(data not shown). The administration of the VIP-induced CD25high
cells to allogeneic responder-stimulator PBMC cocultures effi-ciently inhibited the proliferation of the responder T cells (Fig.4A). However, the CD25int population induced by VIP showedweak suppressive activity, whereas it showed increased expressionof CTLA4 and moderate FoxP3 (Fig. 4A). The increase of CTLA4expression by VIP was rapid, sustained on time, and correlatedwith the suppressive activity of TVIP (Fig. 4B). CTLA4 was pre-viously involved in the induction of FoxP3 expression and in thegeneration and function of Tregs (34). By using anti-CTLA4 Abs,we demonstrated that CTLA4 is needed for VIP to induce FoxP3
in allogeneic-activated CD4�CD25� T cells (Fig. 4C) and to gen-erate T cells with suppressive activity (Fig. 4D). However, neu-tralization of TGF-�1, a well-known factor involved in the induc-tion of Tregs (21), did not significantly affect VIP-induced FoxP3expression (Fig. 4C), CD4�CD25� Treg generation (data notshown), or T cell suppression (Fig. 4D).
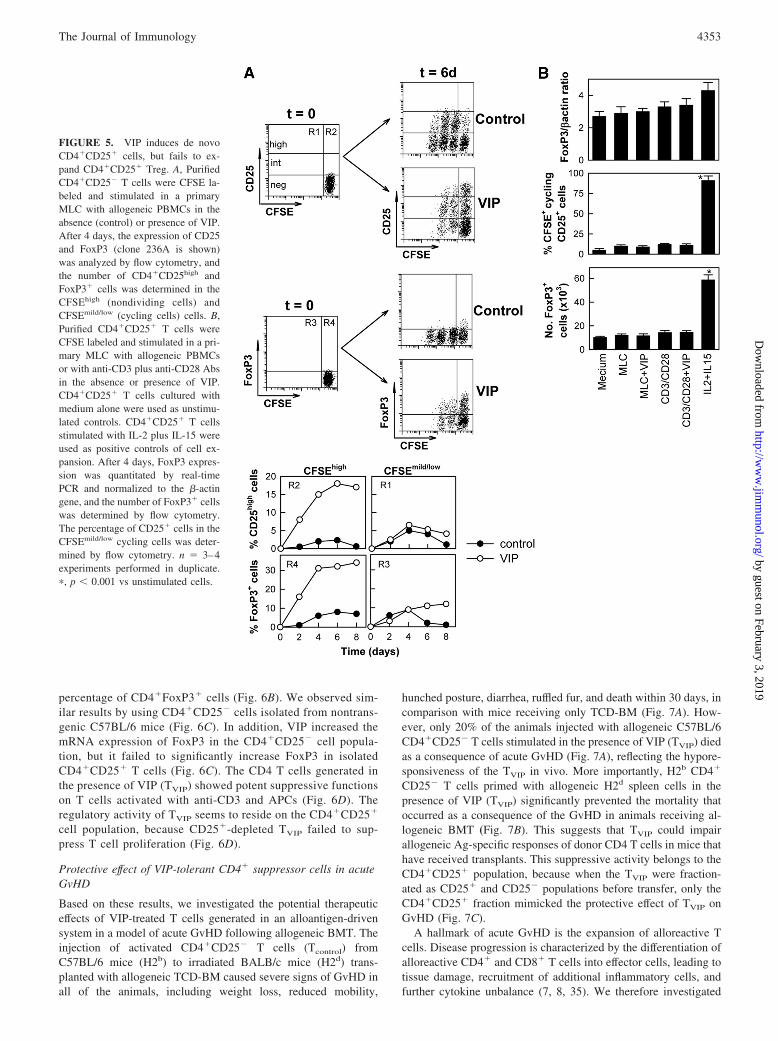
To further investigate whether VIP induced de novo Tregs,CD4�CD25� cells were CFSE labeled and stimulated with allo-geneic PBMCs in the absence or presence of VIP, and the expres-sion of FoxP3 and CD25 was analyzed in the CFSEhigh (nondi-viding cells) and CFSEmild/low (cycling cells) cells. VIP treatmentresulted in low numbers of cycling cells and confined Foxp3 andCD25high expression to the CFSEhigh nondividing CD4 cells
FIGURE 3. VIP treatment results in the generation of regulatory functions in CD4�CD25� T cells after allogeneic stimulation. A, Human CD4�CD25�
T cells isolated from donor A were stimulated in a primary MLC with allogeneic PBMCs from donor B in the absence (Tcontrol) or presence of VIP (TVIP)for 6 days. After the 2-day resting period, increased numbers of the recovered Tcontrol or TVIP were added to a coculture of responder T cells (105 fromdonor B) and allogeneic stimulator PBMCs (105 from donor C). Proliferation and IFN-� production by responder T cells were determined after 4 days ofcoculture. PBMCs (105 from donor B) cultured with syngeneic PBMCs (105 from donor B) were used as controls of the background stimulation (u). �, p �0.001 vs medium or Tcontrol. B, TVIP (2 � 104 from donor A) were added to a coculture of responder T cells (5 � 104 from donor B) and allogeneicstimulator PBMCs (5 � 104 from donor C). When indicated, TVIP were separated from responder-stimulator PBMC cocultures by a semipermeablemembrane in Transwells. Proliferation and IFN-� production were determined in the lower compartment of the Transwells. �, p � 0.001. C, RecoveredTcontrol or TVIP (2 � 104) were added to a coculture of responder T cells (5 � 104 from donor B) and allogeneic stimulator PBMCs (5 � 104 from donorC) in the presence of IL-2, anti-TGF-�1, anti-CTLA4, or anti-IL-10-blocking Abs, or an isotype control Ab. Proliferation and IFN-� production byresponder T cells were determined after 4 days. PBMCs (105 from donor B) cultured with syngeneic PBMCs (105 from donor B) were used as controlsof the background stimulation (dotted lines). �, p � 0.001 vs medium. #, p � 0.01 vs TVIP. D, TVIP (5 � 104 from donor A generated in MLC with PBMCsfrom donor B) were added to a coculture of responder PBMCs (105 from donor C) and allogeneic stimulator PBMCs (105 from donor B or donor D).Alternatively, T cell-depleted APCs (5 � 104 from donor B or donor C) were used as allogeneic stimulators for isolated T cells (105 from donor B or donorC). When indicated, PBMCs and T cells from donors B and C were CFSE labeled before their addition to coculture. After 96-h culture, total number ofcycling cells (percentage of CFSE-positive cells that had divided � the total number) was determined. n � 3–4 experiments performed in duplicate. �, p �0.05; ��, p � 0.001.
4351The Journal of Immunology
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
(Fig. 5A). Moreover, VIP failed to increase FoxP3 expression inand to expand isolated CD4�CD25� Tregs (Fig. 5B). However,IL-2 plus IL-15 stimulation (used as a positive control) resulted ina 6-fold expansion of CD4�CD25� and FoxP3� T cells (Fig. 5B).
Taken together, these results indicate that allogeneic stimulationin the presence of VIP resulted in the new generation of an anergicCD4�CD25highFoxP3�CTLA4� T cell population with regulatoryfunctions.
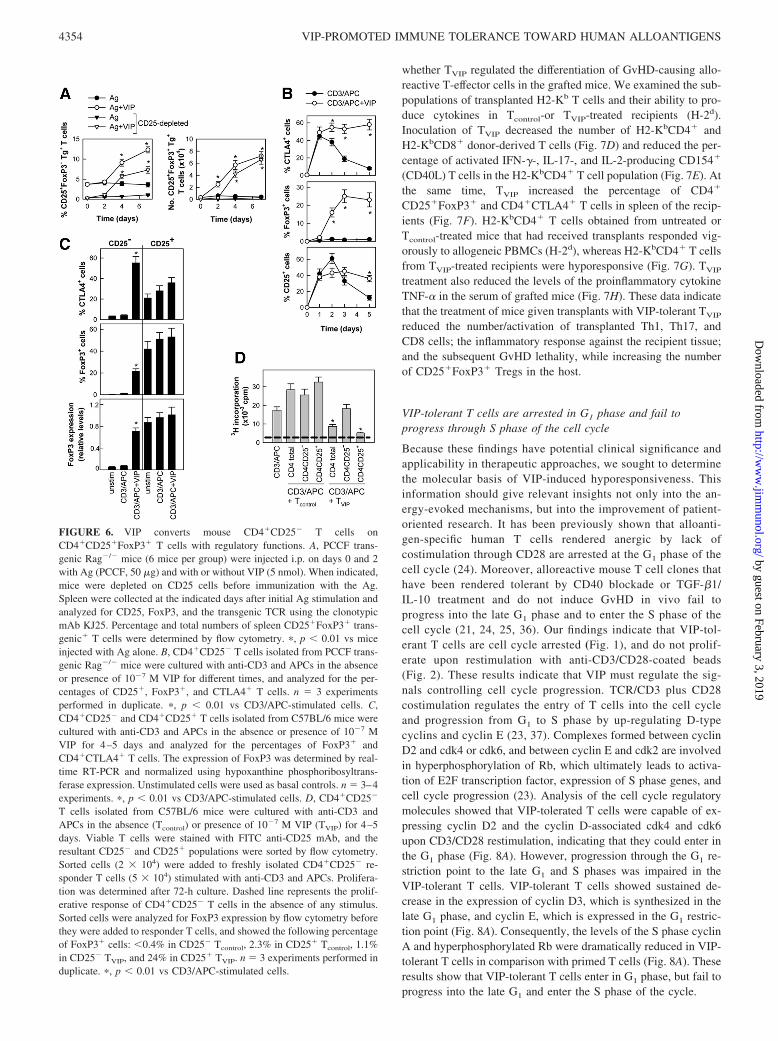
VIP converts mouse CD4�CD25� T cells onCD4�CD25�FoxP3� T cells with regulatory functions
To confirm these results in a mouse model, we used PCCF TCRtransgenic Rag�/� mice that were injected with the corresponding
Ag (PCCF) in the absence or presence of VIP. The administrationof VIP induced the emergence of CD4�CD25�FoxP3� cells in thespleen (Fig. 6A) and lymph nodes (data not shown), even when themice were previously depleted of CD25� T cells (Fig. 6A). Thissuggests that VIP could convert CD4�CD25� cells on CD4�
CD25�FoxP3� T cells in vivo. This was confirmed in vitro start-ing with CD4�CD25� T cells isolated from the Rag�/� mice,which is a bona fide FoxP3� population (Fig. 6B). As expected,stimulation with anti-CD3 and APCs led to transient increase inthe percentages of CD4�CD25� and CD4�CTLA4� cells in thefirst 48-h culture, which declined later (Fig. 6B). Treatment withVIP maintained the elevated percentages of CD4�CD25� andCD4�CTLA4� cells longtime and significantly increased the
FIGURE 4. VIP induces the generation of human CD4�CD25high Tregs that express CTLA4 and FoxP3 from the CD4�CD25� T cell repertory. A,Human CD4�CD25� T cells were stimulated in a primary MLC with allogeneic PBMCs in the absence (control) or presence of VIP, and after 5 days, CD25expression was analyzed by flow cytometry in the CD4� T cell fraction. The subsets formed (CD25negative, CD25int, and CD25high) were sorted and analyzedfor FoxP3 (results with clone PCH101 are shown, and were confirmed with clone 236A) and CTLA4 expression. Expression of FoxP3 and CD25 in thestarting population (t � 0) is shown. Numbers in dot plots represent the percentage of positive cells for each marker in each subset. Sorted cells wererestimulated at different cell ratios with allogeneic PBMCs (5 � 104), and the proliferation during the secondary MLC was determined. n � 3 experimentsperformed in duplicate. B, At different times after primary MLC, CTLA4 protein expression was determined by flow cytometry in gated CD4� T cells andexpressed as the mean fluorescence intensity (MFI), and mRNA expression of CTLA4 was determined by quantitative PCR and expressed as arbitrary unitsnormalized by �2-microglobulin transcripts. n � 3 experiments performed in duplicate. �, p � 0.001 vs control. C, Purified CD4�CD25� T cells werestimulated in a primary MLC with allogeneic PBMCs in the absence (none) or presence of VIP, with or without anti-CTLA4 (10 �g/ml), anti-TGF-�1,or control IgG (10 �g/ml). After 4 days, FoxP3 expression was quantitated by real-time PCR and normalized to the �-actin gene. UnstimulatedCD4�CD25� and CD4�CD25� T cells were used as negative and positive controls, respectively. n � 3 experiments performed in duplicate. #, p � 0.001.D, Human CD4�CD25� T cells isolated from donor A were stimulated in a primary MLC with allogeneic PBMCs from donor B in the absence (Tcontrol)or presence of VIP (TVIP), with or without anti-CTLA4 (10 �g/ml), anti-TGF-�1, or control IgG (10 �g/ml) for 6 days. After the 2-day resting period, therecovered Tcontrol or TVIP (4 � 104) were added to a coculture of responder CFSE-labeled PBMCs (2 � 105 from donor B) and allogeneic stimulator PBMCs(2 � 105 from donor C). After 96-h culture, total number of cycling cells (percentage of CFSE-positive cells that had divided � the total number) wasdetermined. CFSE-labeled PBMCs (2 � 105 from donor B) cultured with syngeneic PBMCs (2 � 105 from donor B) were used as controls of thebackground stimulation (first bar). n � 3 experiments performed in duplicate. �, p � 0.001 vs medium or Tcontrol. #, p � 0.001 vs TVIP.
4352 VIP-PROMOTED IMMUNE TOLERANCE TOWARD HUMAN ALLOANTIGENS
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
percentage of CD4�FoxP3� cells (Fig. 6B). We observed sim-ilar results by using CD4�CD25� cells isolated from nontrans-genic C57BL/6 mice (Fig. 6C). In addition, VIP increased themRNA expression of FoxP3 in the CD4�CD25� cell popula-tion, but it failed to significantly increase FoxP3 in isolatedCD4�CD25� T cells (Fig. 6C). The CD4 T cells generated inthe presence of VIP (TVIP) showed potent suppressive functionson T cells activated with anti-CD3 and APCs (Fig. 6D). Theregulatory activity of TVIP seems to reside on the CD4�CD25�
cell population, because CD25�-depleted TVIP failed to sup-press T cell proliferation (Fig. 6D).
Protective effect of VIP-tolerant CD4� suppressor cells in acuteGvHD
Based on these results, we investigated the potential therapeuticeffects of VIP-treated T cells generated in an alloantigen-drivensystem in a model of acute GvHD following allogeneic BMT. Theinjection of activated CD4�CD25� T cells (Tcontrol) fromC57BL/6 mice (H2b) to irradiated BALB/c mice (H2d) trans-planted with allogeneic TCD-BM caused severe signs of GvHD inall of the animals, including weight loss, reduced mobility,
hunched posture, diarrhea, ruffled fur, and death within 30 days, incomparison with mice receiving only TCD-BM (Fig. 7A). How-ever, only 20% of the animals injected with allogeneic C57BL/6CD4�CD25� T cells stimulated in the presence of VIP (TVIP) diedas a consequence of acute GvHD (Fig. 7A), reflecting the hypore-sponsiveness of the TVIP in vivo. More importantly, H2b CD4�
CD25� T cells primed with allogeneic H2d spleen cells in thepresence of VIP (TVIP) significantly prevented the mortality thatoccurred as a consequence of the GvHD in animals receiving al-logeneic BMT (Fig. 7B). This suggests that TVIP could impairallogeneic Ag-specific responses of donor CD4 T cells in mice thathave received transplants. This suppressive activity belongs to theCD4�CD25� population, because when the TVIP were fraction-ated as CD25� and CD25� populations before transfer, only theCD4�CD25� fraction mimicked the protective effect of TVIP onGvHD (Fig. 7C).
A hallmark of acute GvHD is the expansion of alloreactive Tcells. Disease progression is characterized by the differentiation ofalloreactive CD4� and CD8� T cells into effector cells, leading totissue damage, recruitment of additional inflammatory cells, andfurther cytokine unbalance (7, 8, 35). We therefore investigated
FIGURE 5. VIP induces de novoCD4�CD25� cells, but fails to ex-pand CD4�CD25� Treg. A, PurifiedCD4�CD25� T cells were CFSE la-beled and stimulated in a primaryMLC with allogeneic PBMCs in theabsence (control) or presence of VIP.After 4 days, the expression of CD25and FoxP3 (clone 236A is shown)was analyzed by flow cytometry, andthe number of CD4�CD25high andFoxP3� cells was determined in theCFSEhigh (nondividing cells) andCFSEmild/low (cycling cells) cells. B,Purified CD4�CD25� T cells wereCFSE labeled and stimulated in a pri-mary MLC with allogeneic PBMCsor with anti-CD3 plus anti-CD28 Absin the absence or presence of VIP.CD4�CD25� T cells cultured withmedium alone were used as unstimu-lated controls. CD4�CD25� T cellsstimulated with IL-2 plus IL-15 wereused as positive controls of cell ex-pansion. After 4 days, FoxP3 expres-sion was quantitated by real-timePCR and normalized to the �-actingene, and the number of FoxP3� cellswas determined by flow cytometry.The percentage of CD25� cells in theCFSEmild/low cycling cells was deter-mined by flow cytometry. n � 3–4experiments performed in duplicate.�, p � 0.001 vs unstimulated cells.
4353The Journal of Immunology
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
whether TVIP regulated the differentiation of GvHD-causing allo-reactive T-effector cells in the grafted mice. We examined the sub-populations of transplanted H2-Kb T cells and their ability to pro-duce cytokines in Tcontrol-or TVIP-treated recipients (H-2d).Inoculation of TVIP decreased the number of H2-KbCD4� andH2-KbCD8� donor-derived T cells (Fig. 7D) and reduced the per-centage of activated IFN-�-, IL-17-, and IL-2-producing CD154�
(CD40L) T cells in the H2-KbCD4� T cell population (Fig. 7E). Atthe same time, TVIP increased the percentage of CD4�
CD25�FoxP3� and CD4�CTLA4� T cells in spleen of the recip-ients (Fig. 7F). H2-KbCD4� T cells obtained from untreated orTcontrol-treated mice that had received transplants responded vig-orously to allogeneic PBMCs (H-2d), whereas H2-KbCD4� T cellsfrom TVIP-treated recipients were hyporesponsive (Fig. 7G). TVIP
treatment also reduced the levels of the proinflammatory cytokineTNF-� in the serum of grafted mice (Fig. 7H). These data indicatethat the treatment of mice given transplants with VIP-tolerant TVIP
reduced the number/activation of transplanted Th1, Th17, andCD8 cells; the inflammatory response against the recipient tissue;and the subsequent GvHD lethality, while increasing the numberof CD25�FoxP3� Tregs in the host.
VIP-tolerant T cells are arrested in G1 phase and fail toprogress through S phase of the cell cycle
Because these findings have potential clinical significance andapplicability in therapeutic approaches, we sought to determinethe molecular basis of VIP-induced hyporesponsiveness. Thisinformation should give relevant insights not only into the an-ergy-evoked mechanisms, but into the improvement of patient-oriented research. It has been previously shown that alloanti-gen-specific human T cells rendered anergic by lack ofcostimulation through CD28 are arrested at the G1 phase of thecell cycle (24). Moreover, alloreactive mouse T cell clones thathave been rendered tolerant by CD40 blockade or TGF-�1/IL-10 treatment and do not induce GvHD in vivo fail toprogress into the late G1 phase and to enter the S phase of thecell cycle (21, 24, 25, 36). Our findings indicate that VIP-tol-erant T cells are cell cycle arrested (Fig. 1), and do not prolif-erate upon restimulation with anti-CD3/CD28-coated beads(Fig. 2). These results indicate that VIP must regulate the sig-nals controlling cell cycle progression. TCR/CD3 plus CD28costimulation regulates the entry of T cells into the cell cycleand progression from G1 to S phase by up-regulating D-typecyclins and cyclin E (23, 37). Complexes formed between cyclinD2 and cdk4 or cdk6, and between cyclin E and cdk2 are involvedin hyperphosphorylation of Rb, which ultimately leads to activa-tion of E2F transcription factor, expression of S phase genes, andcell cycle progression (23). Analysis of the cell cycle regulatorymolecules showed that VIP-tolerated T cells were capable of ex-pressing cyclin D2 and the cyclin D-associated cdk4 and cdk6upon CD3/CD28 restimulation, indicating that they could enter inthe G1 phase (Fig. 8A). However, progression through the G1 re-striction point to the late G1 and S phases was impaired in theVIP-tolerant T cells. VIP-tolerant T cells showed sustained de-crease in the expression of cyclin D3, which is synthesized in thelate G1 phase, and cyclin E, which is expressed in the G1 restric-tion point (Fig. 8A). Consequently, the levels of the S phase cyclinA and hyperphosphorylated Rb were dramatically reduced in VIP-tolerant T cells in comparison with primed T cells (Fig. 8A). Theseresults show that VIP-tolerant T cells enter in G1 phase, but fail toprogress into the late G1 and enter the S phase of the cycle.
FIGURE 6. VIP converts mouse CD4�CD25� T cells onCD4�CD25�FoxP3� T cells with regulatory functions. A, PCCF trans-genic Rag�/� mice (6 mice per group) were injected i.p. on days 0 and 2with Ag (PCCF, 50 �g) and with or without VIP (5 nmol). When indicated,mice were depleted on CD25 cells before immunization with the Ag.Spleen were collected at the indicated days after initial Ag stimulation andanalyzed for CD25, FoxP3, and the transgenic TCR using the clonotypicmAb KJ25. Percentage and total numbers of spleen CD25�FoxP3� trans-genic� T cells were determined by flow cytometry. �, p � 0.01 vs miceinjected with Ag alone. B, CD4�CD25� T cells isolated from PCCF trans-genic Rag�/� mice were cultured with anti-CD3 and APCs in the absenceor presence of 10�7 M VIP for different times, and analyzed for the per-centages of CD25�, FoxP3�, and CTLA4� T cells. n � 3 experimentsperformed in duplicate. �, p � 0.01 vs CD3/APC-stimulated cells. C,CD4�CD25� and CD4�CD25� T cells isolated from C57BL/6 mice werecultured with anti-CD3 and APCs in the absence or presence of 10�7 MVIP for 4–5 days and analyzed for the percentages of FoxP3� andCD4�CTLA4� T cells. The expression of FoxP3 was determined by real-time RT-PCR and normalized using hypoxanthine phosphoribosyltrans-ferase expression. Unstimulated cells were used as basal controls. n � 3–4experiments. �, p � 0.01 vs CD3/APC-stimulated cells. D, CD4�CD25�
T cells isolated from C57BL/6 mice were cultured with anti-CD3 andAPCs in the absence (Tcontrol) or presence of 10�7 M VIP (TVIP) for 4–5days. Viable T cells were stained with FITC anti-CD25 mAb, and theresultant CD25� and CD25� populations were sorted by flow cytometry.Sorted cells (2 � 104) were added to freshly isolated CD4�CD25� re-sponder T cells (5 � 104) stimulated with anti-CD3 and APCs. Prolifera-tion was determined after 72-h culture. Dashed line represents the prolif-erative response of CD4�CD25� T cells in the absence of any stimulus.Sorted cells were analyzed for FoxP3 expression by flow cytometry beforethey were added to responder T cells, and showed the following percentageof FoxP3� cells: �0.4% in CD25� Tcontrol, 2.3% in CD25� Tcontrol, 1.1%in CD25� TVIP, and 24% in CD25� TVIP. n � 3 experiments performed induplicate. �, p � 0.01 vs CD3/APC-stimulated cells.
4354 VIP-PROMOTED IMMUNE TOLERANCE TOWARD HUMAN ALLOANTIGENS
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

VIP treatment prevents down-regulation of p27kip1 and resultsin defective activation of cdks
We next investigated whether the effect observed for VIP in thecell cycle progression was accompanied by impaired activation ofcdk2 and cdk4. VIP-tolerant T cells showed decreased levels ofboth cyclin D2-associated cdk4 and cyclin E-associated cdk2 ki-nase activities, in comparison with activated primed T cells (Fig.8B). These results confirm and extend the observation that VIPaffects the function of the molecular players involved in the G1
phase, thereby preventing the progression to the S phase.Besides cyclins and cdks, several cdk inhibitors also play a
prominent role in the regulation of the G1 phase. Because defectivecdk4 and cdk2 kinase activities were observed in VIP-treated Tcells, we investigated the possible regulation of cdk inhibitors byVIP. In T cells, the major inhibitory proteins of cdk4 and cdk2
activities are the members of the cip/kip (p21cip1, p27kip1, andp57kip2) family (37–42). As previously described (23), activationof control primed T cells down-regulated p27kip1 (Fig. 8C). Incontrast, VIP treatment not only prevented the degradation ofp27kip1, but increased its levels over the background expressionfound in unstimulated cells (Fig. 8C).
VIP induces tolerant T cells by elevating cAMP levels
The immunological actions of VIP are exerted through a family ofreceptors, consisting of VPAC1 and type 2 VPAC (10, 13, 43),coupled to adenylate cyclase and the elevation of intracellularcAMP levels and subsequent protein kinase A (PKA) activation.To determine whether the cAMP/PKA pathway mediates the sup-pressive actions for VIP on human T cells described in this work,
FIGURE 7. Protective effect of VIP-tolerant CD4 cells in a mouse model of acute GvHD. A, CD4�CD25� T cells isolated from C57BL/6 mice werecultured with anti-CD3 and APCs in the absence (Tcontrol) or presence of 10�7 M VIP (TVIP) for 4 days, extensively washed, and rested 3 days in mediumcontaining IL-2. Viable Tcontrol or TVIP (106) were injected with C57BL/6 TCD-BM (5 � 106) in irradiated BALB/c mice (10 per group). Mice transplantedwith TCD-BM alone were used as controls (None). Survival was monitored daily. B, CD4�CD25� T cells isolated from C57BL/6 mice were stimulatedin a primary MLC with allogeneic spleen cells from BALB/c mice in the absence (Tcontrol) or presence of 10�7 M VIP (TVIP) for 6 days, harvested, andrested for 2 days. Recovered Tcontrol or TVIP (106) were injected into irradiated BALB/c mice (5 per group) that received allogeneic transplants (BMT) ofTCD-BM supplemented with C57BL/6 splenocytes. Survival was monitored daily after BMT. C, Recovered TVIP (106) were sorted on CD25� and CD25�
populations before their injection into irradiated BALB/c mice (5 per group) that received allogeneic transplants (BMT) of TCD-BM supplemented withC57BL/6 splenocytes. Survival was monitored daily after BMT. D–F, Five days after BMT, the total number of donor H2-KbCD4� and H2-KbCD8� Tcells in lymph nodes per recipient mouse (D) and the percentages of CD154� and cytokine-producing T cells in the H2-KbCD4� T cell population (E) weredetermined. Moreover, the percentages of CD25�Foxp3� and CTLA4� CD4 T cells in lymph nodes were determined (F). Data in D–F represent the meanof pooled lymph nodes from two to three mice in each group. G, Donor H2-KbCD4� T cells isolated from spleen of recipient mice 5 days after BMT werestimulated with allogeneic BALB/c APCs (T cell/APC ratio 10:1), and proliferation was determined (n � 2). H, The TNF-� concentration in sera obtained5 days after BMT was determined by ELISA (n � 2). �, p � 0.001 vs Tcontrol in A and B; p � 0.001 vs BMT in C.
4355The Journal of Immunology
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

we assayed the effects of a VPAC1 antagonist, H89 (a PKA in-hibitor), and 8-Br-cAMP (a cell-permeable cAMP analog). Theeffects of VIP on the increase of FoxP3 and CTLA4 expression(Fig. 9A), the induction of T cell anergy (Fig. 9B), the generationof T cell-suppressive activity (Fig. 9C), and the modulation ofcyclin levels, cdk4 kinase activity, and expression of p27kip1 (Fig.9D) were reversed by the PKA inhibitor and the VPAC1 antago-nist. In agreement with this, 8-Br-cAMP mimicked the effects ofVIP in these events (Fig. 9). These findings indicate that the reg-ulatory effect of VIP on T cells is mainly mediated by its bindingto VPAC1 and the subsequent increase of intracellular cAMP andPKA activation.
DiscussionCellular therapy with in vitro induced/expanded Tregs is consid-ered a feasible approach to modulate effector T cells responsiblefor causing pathology in autoimmune diseases, allergies, allograftrejection or GvHD, and inflammatory diseases (44, 45). The abilityto translate preclinical studies with Tregs into the clinic requires anincreasing effort to identify immune factors that regulate the tol-erance/anergy state mediated by this cell population (45, 46). Inthis study, we have investigated the potential of the anti-inflammatory neuropeptide VIP to promote immune tolerance to-ward alloantigens. We focused on the effects of VIP on CD4� Tcells given the fact that these cells play an established role inallograft rejection as well as in immune regulation. Our data show
that VIP treatment of human CD4�CD25� T cells during in vitrostimulation induces an anergic CD4�CD25high T cell subset withregulatory activity. The phenotype of the Tregs induced by VIPwas further characterized by sustained expression of FoxP3 andCTLA4, both markers being associated with regulatory activity ofT cells (2, 3, 5, 47), and other markers characteristic of an effector-memory phenotype. Although the mechanisms involved in thegeneration of this Treg population are not fully understood, ourdata indicate that VIP directly programs the CD4�CD25� T cellrepertory toward a regulatory phenotype in the absence of natu-rally occurring CD4�CD25� Tregs. Some in vivo evidence sup-ports this hypothesis. VIP administration prevented disease pro-gression in CD25-depleted mice with experimental autoimmuneencephalomyelitis and arthritis by inducing the new emergence of
FIGURE 8. VIP-tolerant T cells are arrested at the early G1 phase of thecell cycle. Human PBMCs were stimulated in a primary MLC with allo-geneic PBMCs in the absence (control) or presence of VIP (10�7 M) for 6days. Viable T cells were recovered and restimulated with anti-CD3/anti-CD28-coated beads for the indicated times. A, After 48 h of restimulation,cell lysates were subjected to Western blot analysis using Abs againstcyclins, cdks, phosphorylated pRb, or actin. As a control, unstimulatedhuman T cells were used. B, After 36 h of restimulation, cell lysates wereimmunoprecipitated (IP) with Abs against cyclin D2 or cdk4 (upper pan-els) or against cyclin E or cdk2 (lower panels). The cyclin D2-cdk4 andcyclin E-ckd2 interactions were assayed by Western blot analysis of pre-cipitates with anti-cdk4 or anti-cdk2, respectively. Activities for cdk4/cy-clin D2- and cdk2/cyclin E-associated kinases were determined in in vitroreactions using pRb-GST and histone H1 as substrates, respectively. C,After 48 h of restimulation, cell lysates were subjected to Western blotanalysis using Abs against p27kip1 or actin. One representative experimentof three is shown.
FIGURE 9. VIP mediates the suppressive effect by elevating cAMP lev-els. Human PBMCs were stimulated with allogeneic stimulator PBMCs inprimary MLCs for 6 days in the absence (control) or presence of VIP (10�7
M), 8-Br-cAMP (0.1 mM), or VIP (10�7 M) with or without H89 (50ng/ml) or a VPAC1 antagonist (10�6 M). A, Cells were analyzed for FoxP3and CTLA4 expression by real-time RT-PCR and flow cytometry, respec-tively. B, Cells were harvested, rested for 2 days, and restimulated in asecondary MLC with original allogeneic responder PBMCs. Proliferationwas determined 3 days after initiation of the secondary culture. C, Recov-ered cells (5 � 104) from the primary MLC were added to newly set-upprimary MLCs consisting of responder PBMCs (105) and allogeneic stim-ulator PBMCs (105). Proliferation of responder T cells was determinedafter 4 days of coculture. D, Recovered cells from the primary MLC wererestimulated with anti-CD3/anti-CD28-coated beads, and after 36- to 48-hculture, cell lysates were subjected to Western blot analysis using Absagainst cyclins, p27kip1, or actin. Cyclin D2/cdk4-associated kinase activitywas determined using pRb-GST as substrate. As a control, unstimulatedhuman T cells were used. n � 3–4 experiments performed in duplicate.�, p � 0.001 vs control. #, p � 0.01 vs VIP treatment.
4356 VIP-PROMOTED IMMUNE TOLERANCE TOWARD HUMAN ALLOANTIGENS
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

peripheral CD4�CD25� Tregs (16, 17). Similarly, in this study weshow that VIP injection in TCR transgenic mice increased thenumbers of Ag-specific CD25�FoxP3� T cells in lymphoid or-gans, even in the initial absence of CD25� T cells. Moreover, invitro data show that VIP failed to expand isolated CD4�CD25� Tcells and to increase FoxP3 expression in these cells. Whether inour system the effect of VIP is uniform on all T cells, or only ona subset that differentiates into FoxP3�CD4�CD25� Tregs andsubsequently mediates anergy induction to the remaining T cells,needs to be further analyzed. In this sense, it has been describedthat certain CD25� regulatory cells that reside within the totalCD4�CD25� T cell repertory up-regulate CD25 and FoxP3 uponTCR triggering (48, 49). VIP could simply facilitate the expansionof this Treg-committed CD4�CD25� T cell population. Our re-sults show that VIP promotes the expression of CD25high andFoxP3 in the nondividing CD4�CD25� cells, supporting the hy-pothesis of de novo conversion of Tregs from CD4�CD25� cells.
Although VIP-induced Tregs produce IL-10 and some TGF-�1upon restimulation, none of these cytokines seem to play a majorrole in their suppressive action. In this sense, the VIP-tolerated Tcells clearly differ from the Tr1 cells generated in the presence ofIL-10 (50, 51), which suppress T cell activation through the sol-uble factors IL-10 and TGF-�1. However, a cell-to-cell contact-dependent mechanism mediated by the suppressive moleculeCTLA4 seems to be critically involved in the effect of VIP-tolerantT cells. CTLA4 has been widely identified as a membrane-boundmolecule with potent immunosuppressive effects that acts directlyon T cells and indirectly on APCs interfering with the costimula-tory signaling (2, 3, 5). Most importantly, a rapid and sustainedinduction of CTLA4 by VIP seems to be involved in the generationof Tregs by the neuropeptide. Induction of CTLA4 seems to be aprerequisite for VIP to generate CD4�FoxP3� T cells with regu-latory functions. Zheng et al. (34) also demonstrated that TGF-�requires CTLA4 early after activation to induce FoxP3 and gen-erate adaptive mouse CD4�CD25� Tregs. Considerable contro-versy exists regarding the regulation of FoxP3 expression in hu-man T cells, and some studies have suggested that TCRstimulation alone is sufficient to induce FoxP3 expression, at leasttransiently, and that the TGF-� produced by activated T cells andthe TGF-� present in the serum are critically involved in suchinduction (19, 49, 52, 53). FoxP3 expression could easily be in-duced in most naive T cells by the addition of exogenous TGF-�.However, in contrast to mouse CD4�CD25� naive T cells that areconverted by TGF-� to CD4�CD25� Tregs with suppressive ac-tivities, the human FoxP3� T cells induced with TGF-� in a singleround of stimulation were neither anergic nor suppressive (19, 21,49, 52, 53). Our data show that the VIP-tolerant CD4�CD25�
FoxP3� T cells are anergic and suppressive in both mouse andhuman systems. Whether FoxP3 induction is critical in the gener-ation of VIP-tolerant T cells and in their suppressive function re-mains unknown, but we know that both generation and functionare mostly TGF-� independent and CTLA4 dependent. In contrast,a secondary mechanism that could contribute to the suppressiveactivity of the VIP-tolerant T cells is the consumption of IL-2produced by the naive responders in the MLRs through the highlevels of CD25 expressed by these cells.
Cell cycle arrest and anergy seem to be critically related to thegeneration of Tregs (21, 24–33). The VIP-tolerant T cells share anumber of biochemical characteristics with anergic T cells gener-ated following other approaches, including the blockade of CD28costimulation or CD40L/CD40 interactions, the treatment withIL-10 and TGF-�1, or the treatment with synthetic immunosup-pressive agents typically used in transplantation (21, 24–26, 36).These common biochemical analyses may provide a powerful tool
in quantifying the degree of tolerance induction in individual pa-tients who receive T cells tolerized by different strategies. Ourresults and the aforementioned works indicate that one of the mostcritical events that occur during induction of T cell anergy follow-ing allostimulation is the alteration in the control of the expressionand activation of regulatory molecules of the cell cycle. The as-sociation of cyclins with specific cdks leads to activation of ho-loenzymes that regulate the progression through the differentphases of the cell cycle. Our data indicate that VIP-tolerant T cellsare capable of entering the G1 phase, but do not progress throughthe G1 restriction point to the late G1 and S phases. In agreementwith this, both cyclin D-cdk4 and cyclinE-cdk2 activities are im-paired in VIP-tolerant T cells, due to a reduced expression of cy-clins D3 and E and up-regulation of the cdk inhibitor p27kip1.Previous studies have shown that the cell cycle inhibitor p27kip1
acts during the late G1 phase by binding and inhibiting cdk2-cyclinE/A complexes (41). Upon stimulation, T cells can only progressthrough the cell cycle when p27kip1 is dissociated from the cdk2-cyclin E/A complexes. This is in general achieved by ubiquitina-tion and degradation of p27kip1, which is preceded by phosphor-ylation of p27kip1 (41, 42). Indeed, anergic and tolerant T cells arecharacterized by impaired degradation of p27kip1 and other toler-ance strategies pointed out to p27kip1 as a critical target of anergy(24–26). The increase of p27kip1 by VIP could be due to impairedp27kip1 degradation and/or to increased p27kip1 synthesis. Recentresults from our laboratory indicate that VIP prevents CD3/CD28-induced p27kip1 phosphorylation while increasing total p27kip1 lev-els (our unpublished observations). Various pathways are involvedin the phosphorylation of p27kip1, including our own formation ofcdk2/cyclin E enzymatic complex and the activation of the Ras-MAPK and PI3K-Akt pathways (41, 42, 54, 55). Our study dem-onstrates that VIP decreases the expression of cyclin E, and an-other recent study demonstrated the inhibition of the Ras-ERK1/2and PI3K-Akt pathways by VIP in human activated T cells (ourunpublished observations). These mechanisms are most likely re-sponsible for the inability of the VIP-tolerant T cells to down-regulate p27kip1.
Our results show that VIP’s tolerizing effect on T cells is me-diated by its binding to the VPAC1 receptor and subsequent in-crease in the intracellular levels of cAMP and activation of PKA.Indeed, some of the effects described for VIP in this study hadpreviously been mimicked by other cAMP-inducing agents, e.g., Tcell cycle arrest, up-regulation of p27kip1, and increase of CTLA4and FoxP3 expression (20, 25, 56–60). Moreover, Gavin et al.(61) described that the suppressive action of Tregs correlated withelevated intracellular cAMP levels, as a consequence of a dimin-ished expression of phosphodiesterase 3 (an enzyme that hydro-lyzes cAMP) in Tregs. The findings we report in this study haveimportant therapeutic implications. Due to its peptidic nature, VIPis very unstable and possesses a very short t1/2 time in vivo. There-fore, its potential use as an immunosuppressive drug in trans-plantation may be limited. The identification of VPAC1 as apotential target for the screening of more stable nonpeptidicagonists by drug discovery programs should overcome this lim-itation and extend the therapeutic use of VIP-based drugs inclinical transplantation.
It is worth noting that the strategies described in this work gen-erate Tregs with direct alloantigen specificity. These cells may beof particular benefit for patients receiving an HLA-mismatchedstem cell graft, in which alloantigen reactivity is important inGvHD. Although no effects have to date been reported, some clin-ical trials on Treg immunotherapy have been recently initiated onbone marrow-transplanted patients by infusing either CliniMACS-isolated CD4�CD25high Tregs or ex vivo manipulated CD4� T
4357The Journal of Immunology
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

cell lines containing induced regulatory Tr1 cells (44, 62). Ourstudy indicates that the infusion of VIP-tolerant T cells togetherwith the graft significantly reduces the clinical signs and mortalityrate typical of the acute GvHD response in mice reconstituted withallogeneic hematopoietic stem cells. These effects are mediatedby impairing allogeneic haplotype-specific responses of donorCD4� cells in the transplanted animals. Therefore, the inclusionof VIP-generated alloantigen-specific Tregs ex vivo in futuretherapeutic regimens may be a valuable aid in the applicabilityof BMT to minimize the dependence on nonspecific immuno-suppressive drugs currently used, to facilitate the successfultransplantation from mismatched donors, and to reduce the del-eterious consequences of acute GvHD.
DisclosuresThe authors have no financial conflict of interest.
References1. Brent, L., I. R. Cohen, P. C. Doherty, M. Feldmann, P. Matzinger, L. Ghost,
S. T. Holgate, P. Lachmann, N. A. Mitchison, G. Nossal, et al. 2007. Crystal-ballgazing: the future of immunological research viewed from the cutting edge. Clin.Exp. Immunol. 147: 1–10.
2. Mills, K. H. 2004. Regulatory T cells: friend or foe in immunity to infection? Nat.Rev. Immunol. 4: 841–855.
3. Bluestone, J. A. 2005. Regulatory T-cell therapy: is it ready for the clinic? Nat.Rev. Immunol. 5: 343–349.
4. Quintana, F. J., and I. R. Cohen. 2008. Regulatory T cells and immune compu-tation. Eur. J. Immunol. 38: 903–907.
5. Shevach, E. M. 2006. From vanilla to 28 flavors: multiple varieties of T regula-tory cells. Immunity 25: 195–201.
6. Wagner, J. E., A. D. Donnenberg, S. J. Noga, C. A. Cremo, I. K. Gao, H. J. Yin,G. B. Vogelsang, S. Rowley, R. Saral, and G. W. Santos. 1988. Lymphocytedepletion of donor bone marrow by counterflow centrifugal elutriation: results ofa phase I clinical trial. Blood 72: 1168–1176.
7. Drobyski, W. R., and D. Majewski. 1996. Treatment of donor mice with an ��T-cell receptor monoclonal antibody induces prolonged T-cell nonresponsivenessand effectively prevents lethal graft-versus-host disease in murine recipients ofmajor histocompatibility complex (MHC)-matched and MHC-mismatched donormarrow grafts. Blood 87: 5355–5369.
8. Cooke, K. R., W. Krenger, G. Hill, T. R. Martin, L. Kobzik, J. Brewer,R. Simmons, J. M. Crawford, M. R. van den Brink, and J. L. Ferrara. 1998. Hostreactive donor T cells are associated with lung injury after experimental alloge-neic bone marrow transplantation. Blood 92: 2571–2580.
9. Rutella, S., and R. M. Lemoli. 2004. Regulatory T cells and tolerogenic dendriticcells: from basic biology to clinical applications. Immunol. Lett. 94: 11–26.
10. Delgado, M., D. Pozo, and D. Ganea. 2004. The significance of vasoactive in-testinal peptide in immunomodulation. Pharmacol. Rev. 56: 249–290.
11. Pozo, D., and M. Delgado, M. 2004. The many faces of VIP in neuroimmunol-ogy: a cytokine rather a neuropeptide? FASEB J. 18: 1325–1334.
12. Gonzalez-Rey, E., A. Chorny, and M. Delgado. 2007. Regulation of immunetolerance by anti-inflammatory neuropeptides. Nat. Rev. Immunol. 7: 52–63.
13. Pozo, D., E. Gonzalez-Rey, A. Chorny, P. Anderson, N. Varela, and M. Delgado.2007. Tuning immune tolerance with vasoactive intestinal peptide: a new thera-peutic approach for immune disorders. Peptides 28: 1833–1846.
14. Gonzalez-Rey, E., and M. Delgado. 2007. Vasoactive intestinal peptide and reg-ulatory T-cell induction: a new mechanism and therapeutic potential for immunehomeostasis. Trends Mol. Med. 13: 241–251.
15. Delgado, M., A. Chorny, E. Gonzalez-Rey, and D. Ganea. 2005. Vasoactiveintestinal peptide generates CD4�CD25� regulatory T cells in vivo. J. LeukocyteBiol. 78: 1327–1338.
16. Fernandez-Martin, A., E. Gonzalez-Rey, A. Chorny, D. Ganea, and M. Delgado.2006. Vasoactive intestinal peptide induces regulatory T cells during experimen-tal autoimmune encephalomyelitis. Eur. J. Immunol. 36: 318–326.
17. Gonzalez-Rey, E., A. Fernandez-Martin, A. Chorny, and M. Delgado. 2006. Va-soactive intestinal peptide induces CD4�,CD25� T regulatory cells with thera-peutic effect in collagen-induced arthritis. Arthritis Rheum. 54: 864–876.
18. Delgado, M., and D. Ganea. 2001. Vasoactive intestinal peptide and pituitaryadenylate cyclase-activating polypeptide inhibit nuclear factor-�B-dependentgene activation at multiple levels in the human monocytic cell line THP-1.J. Biol. Chem. 276: 369–380.
19. Tran, D. Q., H. Ramsey, and E. M. Shevach. 2007. Induction of FOXP3 expres-sion in naive human CD4�FOXP3 T cells by T-cell receptor stimulation is trans-forming growth factor-� dependent but does not confer a regulatory phenotype.Blood 110: 2983–2990.
20. Vendetti, S., A. Riccomi, A. Sacchi, L. Gatta, C. Pioli, and M. T. De Magistris.2002. Cyclic adenosine 5�-monophosphate and calcium induce CD152 (CTLA-4)up-regulation in resting CD4� T lymphocytes. J. Immunol. 169: 6231–6235.
21. Chen, W., W. Jin, N. Hardegen, K. J. Lei, L. Li, N. Marinos, G. McGrady, andS. M. Wahl. 2003. Conversion of peripheral CD4�CD25� naive T cells toCD4�CD25� regulatory T cells by TGF-� induction of transcription factorFoxp3. J. Exp. Med. 198: 1875–1886.
22. Chorny, A., E. Gonzalez-Rey, A. Fernandez-Martin, D. Ganea, and M. Delgado.2006. Vasoactive intestinal peptide induces regulatory dendritic cells that preventacute graft-versus-host disease while maintaining the graft-versus-tumor re-sponse. Blood 107: 3787–3794.
23. Schwartz, R. H. 1997. T cell clonal anergy. Curr. Opin. Immunol. 9: 351–357.24. Boussiotis, V. A., Z. M. Chen, J. C. Zeller, W. J. Murphy, A. Berezovskaya,
S. Narula, M. G. Roncarolo, and B. R. Blazar. 2001. Altered T-cell receptor �CD28-mediated signaling and blocked cell cycle progression in interleukin 10and transforming growth factor-�-treated alloreactive T cells that do not inducegraft-versus-host disease. Blood 97: 565–571.
25. Boussiotis, V. A., G. J. Freeman, P. A. Taylor, A. Berezovskaya, I. Grass,B. R. Blazar, and L. M. Nadler. 2000. p27kip1 functions as an anergy factorinhibiting interleukin 2 transcription and clonal expansion of alloreactive humanand mouse helper T lymphocytes. Nat. Med. 6: 290–297.
26. Kreijveld, E., H. J. Koenen, L. B. Hilbrands, H. J. van Hooff, and I. Joosten. 2007.The immunosuppressive drug FK778 induces regulatory activity in stimulatedhuman CD4� CD25� T cells. Blood 109: 244–252.
27. Li, L., W. R. Godfrey, S. B. Porter, Y. Ge, C. H. June, B. R. Blazar, andV. A. Boussiotis. 2005. CD4�CD25� regulatory T-cell lines from human cordblood have functional and molecular properties of T-cell anergy. Blood 106:3068–3073.
28. Lee, S. M., B. Gao, and D. Fang. 2008. FoxP3 maintains Treg unresponsivenessby selectively inhibiting the promoter DNA-binding activity of AP-1. Blood 111:3599–3606.
29. Kubach, J., P. Lutter, T. Bopp, S. Stoll, C. Becker, E. Huter, C. Richter,P. Weingarten, T. Warger, J. Knop, et al. 2007. Human CD4�CD25� regulatoryT cells: proteome analysis identifies galectin-10 as a novel marker essential fortheir anergy and suppressive function. Blood 110: 1550–1558.
30. Ng, W. F., P. J. Duggan, F. Ponchel, G. Matarese, G. Lombardi, A. D. Edwards,J. D. Isaacs, and R. I. Lechler. 2001. Human CD4�CD25� cells: a naturallyoccurring population of regulatory T cells. Blood 98: 2736–2744.
31. Tsang, J. Y., N. O. Camara, E. Eren, H. Schneider, C. Rudd, G. Lombardi, andR. I. Lechler. 2006. Altered proximal T cell receptor (TCR) signaling in humanCD4�CD25� regulatory T cells. J. Leukocyte Biol. 80: 145–151.
32. Jiang, S., N. Camara, G. Lombardi, and R. I. Lechler. 2003. Induction of al-lopeptide-specific human CD4�CD25� regulatory T cells ex vivo. Blood 102:2180–2186.
33. Jago, C. B., J. Yates, N. O. Camara, R. I. Lechler, and G. Lombardi. 2004.Differential expression of CTLA-4 among T cell subsets. Clin. Exp. Immunol.136: 463–471.
34. Zheng, S. G., J. H. Wang, W. Stohl, K. S. Kim, J. D. Gray, and D. A. Horwitz.2006. TGF-� requires CTLA-4 early after T cell activation to induce FoxP3 andgenerate adaptive CD4�CD25� regulatory T cells. J. Immunol. 176: 3321–3329.
35. Herrera, C., A. Torres, J. M. Garcia-Castellano, J. Roman, C. Martin, J. Serrano,M. Falcon, M. A. Alvarez, P. Gomez, and F. Martinez. 1999. Prevention ofgraft-versus-host disease in high risk patients by depletion of CD4� and reduc-tion of CD8� lymphocytes in the marrow graft. Bone Marrow Transplant. 23:443–450.
36. Blazar, B. R., P. A. Taylor, R. J. Noelle, and D. A. Vallera. 1998. CD4� T cellstolerized ex vivo to host alloantigen by anti-CD40 ligand (CD40L:CD154) an-tibody lose their graft-versus-host disease lethality capacity but retain nominalantigen responses. J. Clin. Invest. 102: 473–482.
37. Appleman, L. J., A. Berezovskaya, I. Grass, and V. A. Boussiotis. 2000. CD28costimulation mediates T cell expansion via IL-2-independent and IL-2-depen-dent regulation of cell cycle progression. J. Immunol. 164: 144–151.
38. Nakayama, K. I., S. Hatakeyama, and K. Nakayama. 2001. Regulation of the cellcycle at the G1-S transition by proteolysis of cyclin E and p27Kip1. Biochem.Biophys. Res. Commun. 282: 853–860.
39. Assoian, R. K. 2004. Stopping and going with p27kip1. Dev. Cell 6: 458–459.40. Coqueret, O. 2003. New roles for p21 and p27 cell-cycle inhibitors: a function for
each cell compartment? Trends Cell Biol. 13: 65–70.41. Miller, J. P., N. Yeh, A. Vidal, and A. Koff. 2007. Interweaving the cell cycle
machinery with cell differentiation. Cell Cycle 6: 2932–2938.42. Vidal, A., and A. Koff. 2000. Cell-cycle inhibitors: three families united by a
common cause. Gene 247: 1–15.43. Gonzalez-Rey, E., and M. Delgado. 2007. Anti-inflammatory neuropeptide re-
ceptors: new therapeutic targets for immune disorders? Trends Pharmacol. Sci.28: 482–491.
44. Roncarolo, M. G., and M. Battaglia. 2007. Regulatory T-cell immunotherapy fortolerance to self antigens and alloantigens in humans. Nat. Rev. Immunol. 7:585–598.
45. Sakaguchi, S. 2008. Regulatory T cells in the past and for the future. Eur. J. Im-munol. 38: 901–937.
46. Sakaguchi, S., and F. Powrie. 2007. Emerging challenges in regulatory T cellfunction and biology. Science 317: 627–629.
47. Joffre, O., T. Santolaria, D. Calise, T. Al Saati, D. Hudrisier, P. Romagnoli, andJ. P. van Meerwijk. 2008. Prevention of acute and chronic allograft rejection withCD4�CD25�Foxp3� regulatory T lymphocytes. Nat. Med. 14: 88–92.
48. Stephens, L. A., and D. Mason. 2000. CD25 is a marker for CD4� thymocytesthat prevent autoimmune diabetes in rats, but peripheral T cells with this functionare found in both CD25� and CD25� subpopulations. J. Immunol. 165:3105–3110.
49. Walker, M. R., D. J. Kasprowicz, V. H. Gersuk, A. Benard, M. Van Landeghen,J. H. Buckner, and S. F. Ziegler. 2003. Induction of FoxP3 and acquisition of Tregulatory activity by stimulated human CD4�CD25� T cells. J. Clin. Invest.112: 1437–1443.
4358 VIP-PROMOTED IMMUNE TOLERANCE TOWARD HUMAN ALLOANTIGENS
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

50. Groux, H., M. Bigler, J. E. de Vries, and M. G. Roncarolo. 1996. Interleukin-10induces a long-term antigen-specific anergic state in human CD4� T cells. J. Exp.Med. 184: 19–29.
51. Groux, H., A. O’Garra, M. Bigler, M. Rouleau, S. Antonenko, J. E. de Vries, andM. G. Roncarolo. 1997. A CD4� T-cell subset inhibits antigen-specific T-cellresponses and prevents colitis. Nature 389: 737–742.
52. Shevach, E. M., D. Q. Tran, T. S. Davidson, and J. Anderson. 2008. The criticalcontribution of TGF-� to the induction of Foxp3 expression and regulatory T cellfunction. Eur. J. Immunol. 38: 915–917.
53. Horwitz, D. A., S. G. Zheng, J. Wang, and J. D. Gray. 2008. Critical role of IL-2and TGF-� in generation, function and stabilization of Foxp3�CD4� Treg. Eur.J. Immunol. 38: 912–915.
54. Kawada, M., S. Yamagoe, Y. Murakami, K. Suzuki, S. Mizuno, and Y. Uehara.1997. Induction of p27Kip1 degradation and anchorage independence by Rasthrough the MAP kinase signaling pathway. Oncogene 15: 629–637.
55. Liang, J., and J. M. Slingerland. 2003. Multiple roles of the PI3K/PKB (Akt)pathway in cell cycle progression. Cell Cycle 2: 339–345.
56. Grader-Beck, T., A. A. van Puijenbroek, L. M. Nadler, and V. A. Boussiotis.2003. cAMP inhibits both Ras and Rap1 activation in primary human T lym-phocytes, but only Ras inhibition correlates with blockade of cell cycle progres-sion. Blood 101: 998–1006.
57. Heijink, I. H., H. F. Kauffman, D. S. Postma, J. G. de Monchy, and E. Vellenga.2003. Sensitivity of IL-5 production to the cAMP-dependent pathway in humanT cells is reduced by exogenous IL-2 in a phosphoinositide 3-kinase-dependentway. Eur. J. Immunol. 33: 2206–2215.
58. Ramstad, C., V. Sundvold, H. K. Johansen, and T. Lea. 2000. cAMP-dependentprotein kinase (PKA) inhibits T cell activation by phosphorylating Ser-43 of raf-1in the MAPK/ERK pathway. Cell. Signal. 12: 557–563.
59. Van Oirschot, B. A., M. Stahl, S. M. Lens, and R. H. Medema. 2001. Proteinkinase A regulates expression of p27kip1 and cyclin D3 to suppress proliferationof leukemic T cell lines. J. Biol. Chem. 276: 33854–33860.
60. Baratelli, F., Y. Lin, L. Zhu, S.-C. Yang, N. Heuze-Vourc�h, G. Zeng,K. Reckamp, M. Dohadwala, S. Sharma, and S. M. Dubinett. 2005. ProstaglandinE2 induces FOXP3 gene expression and T regulatory cell function in humanCD4� T cells. J. Immunol. 175: 1483–1490.
61. Gavin, M. A., S. R. Clarke, E. Negrou, A. Gallegos, and A. Rudensky. 2002.Homeostasis and anergy of CD4�CD25� suppressor T cells in vivo. Nat. Im-munol. 3: 33–41.
62. Levings, M. K., R. Sangregorio, and M. G. Roncarolo. 2001. HumanCD25�CD4� T regulatory cells suppress naive and memory T cell proliferationand can be expanded in vitro without loss of function. J. Exp. Med. 193:1295–1302.
4359The Journal of Immunology
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from