klİnİk sİtogenetİk - gantep.edu.trgorucu/documents/ebe-238-7.pdf · 47, xxx (sÜper dİŞİ),...
TRANSCRIPT

KLİNİK SİTOGENETİK
OTOZOMAL KROMOZOMLARIN HASTALIKLARI

Down sendromu
Down sendromu ya da TRİZOMİ
21, en sık görülen kromozomal
hastalıklardan birisidir.
46, XX, +21 veya
46, XY, +21

Down sendromu
Fenotip, hastadan hastaya değişiklik
gösterir.
Doğum öncesi ve sonrasında tanımlanabilir.
Doğum sonrası en belirgin bulgu hipotonidir
(kas tonusunun düşük olması)
Belirgin yüz bulguları vardır.
Hastalarda boy kısadır.
Oksipital bölge düzdür.
Brakisefali
Boyun kısadır ve ensedeki deri
gevşektir
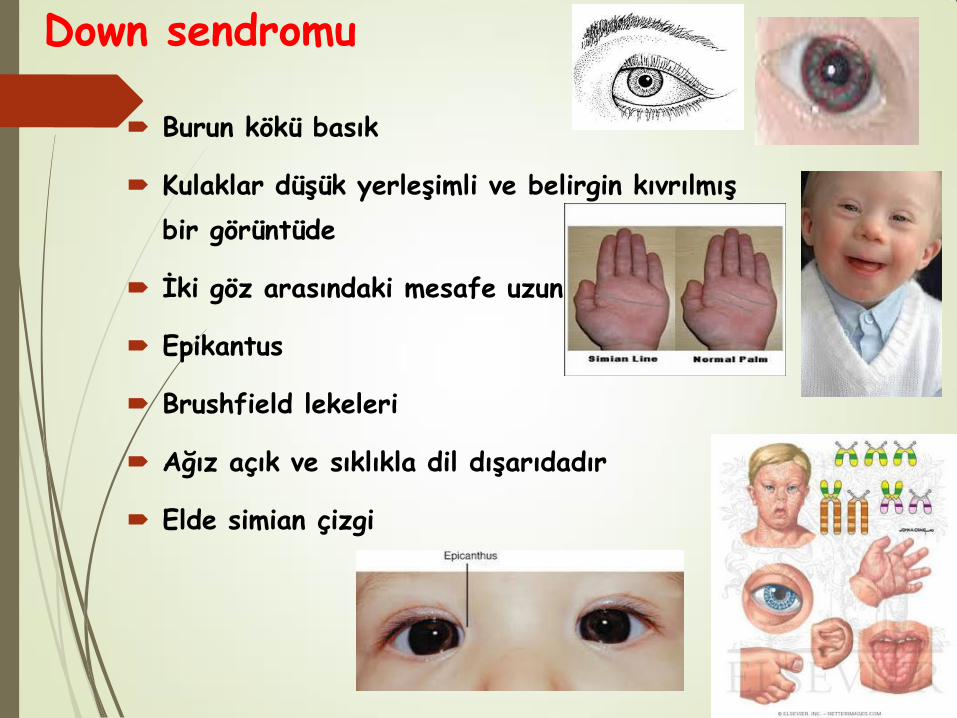
Down sendromu
Burun kökü basık
Kulaklar düşük yerleşimli ve belirgin kıvrılmış
bir görüntüde
İki göz arasındaki mesafe uzun
Epikantus
Brushfield lekeleri
Ağız açık ve sıklıkla dil dışarıdadır
Elde simian çizgi
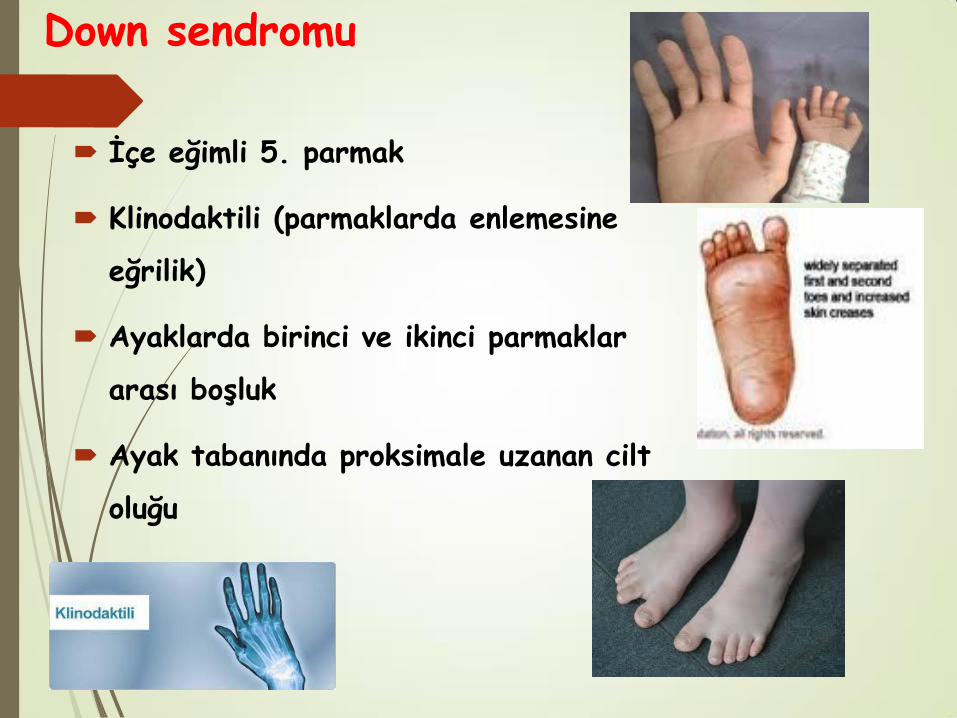
Down sendromu
İçe eğimli 5. parmak
Klinodaktili (parmaklarda enlemesine
eğrilik)
Ayaklarda birinci ve ikinci parmaklar
arası boşluk
Ayak tabanında proksimale uzanan cilt
oluğu

Down sendromu Mental retardasyon (zeka geriliği)
IQ (Intelligence quotient-Zeka katsayısı) 30-60
arasındadır
Konjenital kalp defektleri; büyük bir kısmında
vardır. Düşüklerin daha fazlasında sebeptir.
Duodenal atrezi; duodenum, onikiparmak barsağı
Duodenum atrezisinde barsağın oluşumunda eksiklik
söz konusudur. Buna bağlı olarak barsakta pasaj
(geçiş) olmaz. Pasajın bazen az olduğu durumlar
duodenal atrezi adını alır. Duodenal atrezide
barsak darlığı vardır. Barsaktan geçiş vardır ancak
azalmıştır.

Down sendromu
Lösemi riskinde artış vardır.
Erken yaşlanma görülür
Down sendromlu gebeliklerde yaşam şansı en düşük olanlar
konjenital kalp hastalıklı olanlardır.
Klinik tanısı kolaydır. Tanının doğrulanması ve genetik
danışma için karyotipleme gereklidir.
Ayrıca kendilerine özel okullardan yaralanabilmeleri ve
kendilerine bakan bireylerin maddi yardım alabilmeleri için
yasal olarak genetik raporuna ihtiyaç vardır.
Tüm bu sorunlara rağmen; iletişim kurabilirler, kendi
kendilerine hayatlarını sürdürebilirler.

Down sendromu
21. Kromozomun mayotik kromozom ayrılmaması (non-
disjunction) sonucu trizomisinin görülmesidir.
Trizomili çocuk sahibi olma anne yaşıyla birlikte artar (özellikle
30 yaşından sonra)
Down sendromlu hastaların %4’ünde, kromozom 21q ile diğer
akrosentrik kromozomlardan birinin (14 veya 21) uzun kolu
arasında robertsonian translokasyon ile oluşan 46 kromozom
vardır.
46, XX, rob (14;21), +21
46, XY, rob (14;21), +21

Down sendromu
21q21q Translokasyon;
Oluşan gebelik ya down sendromludur ya da nadiren
yaşama şansı olan monozomi 21 olacaktır. 21q21q
kromozomu taşıyan kişinin normal çocuk sahibi olma şansı
olmayacaktır.
Mozaik Down sendromu;
Normal ve trizomi 21 karyotipli hücre gruplarına
sahiptir. Kişiler arasındaki farklılık mozaikliğin derecesine
bağlıdır.

Down sendromu
Kısmi Trizomi 21;
21. kromozomun sadece uzun kolunun 3 kopya bulunmasıdır. Nadir
görülür.
GENETİK DANIŞMA
Down sendromu prenatal dönemde amniyotik sıvı veya koriyonik
villus örneklerinden sitogenetik analizlerle tanımlanabilir.
Bunun için ilk olarak anne yaşının ileri, anne ve babanın
kromozomal olarak taşıyıcı veya aile öyküsünün bulunması
gereklidir.
Trizomi 21 tekrarlama riski; translokasyon tipi olanlarda
yüksektir. Genç annelerde risk daha fazladır. Yaşlı annelerde
yaşa bağlı olarak değişir.
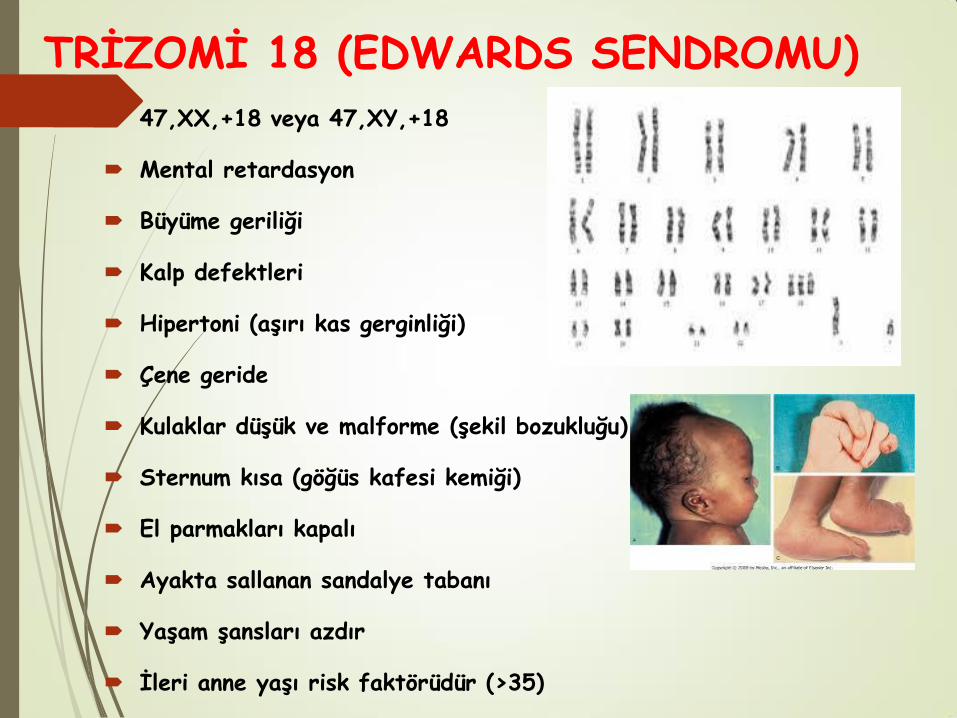
TRİZOMİ 18 (EDWARDS SENDROMU)
47,XX,+18 veya 47,XY,+18
Mental retardasyon
Büyüme geriliği
Kalp defektleri
Hipertoni (aşırı kas gerginliği)
Çene geride
Kulaklar düşük ve malforme (şekil bozukluğu)
Sternum kısa (göğüs kafesi kemiği)
El parmakları kapalı
Ayakta sallanan sandalye tabanı
Yaşam şansları azdır
İleri anne yaşı risk faktörüdür (>35)
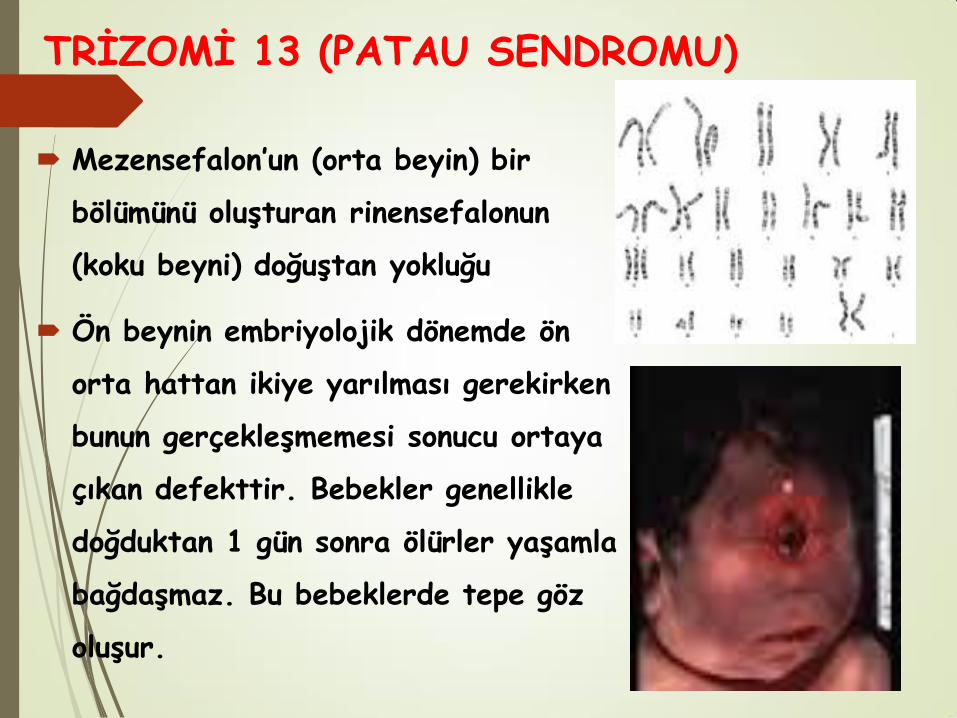
Mezensefalon’un (orta beyin) bir
bölümünü oluşturan rinensefalonun
(koku beyni) doğuştan yokluğu
Ön beynin embriyolojik dönemde ön
orta hattan ikiye yarılması gerekirken
bunun gerçekleşmemesi sonucu ortaya
çıkan defekttir. Bebekler genellikle
doğduktan 1 gün sonra ölürler yaşamla
bağdaşmaz. Bu bebeklerde tepe göz
oluşur.
TRİZOMİ 13 (PATAU SENDROMU)
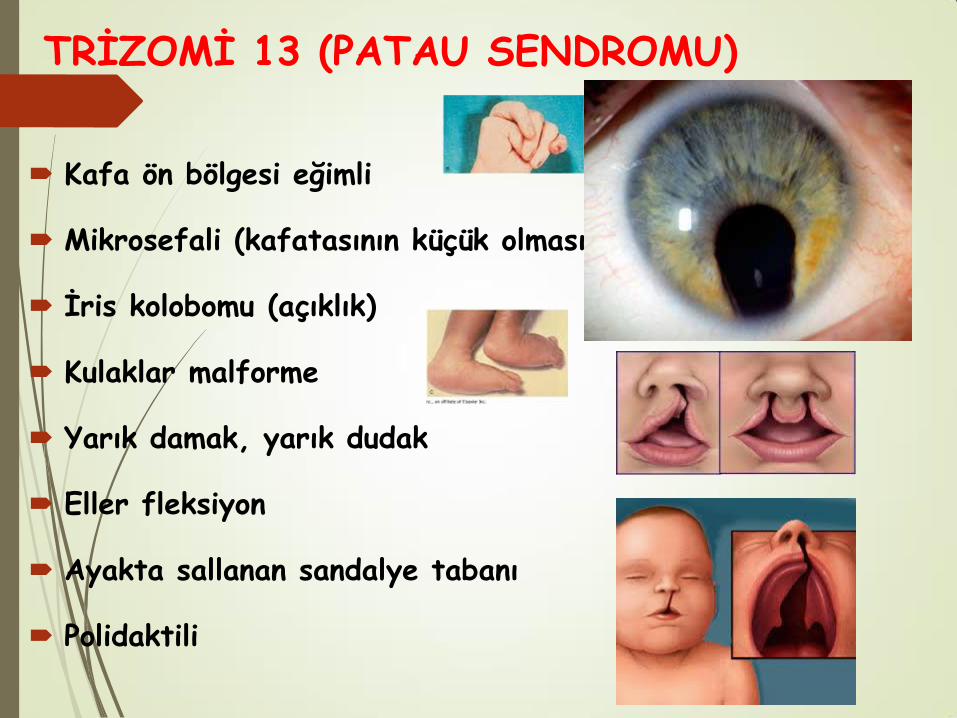
Kafa ön bölgesi eğimli
Mikrosefali (kafatasının küçük olması)
İris kolobomu (açıklık)
Kulaklar malforme
Yarık damak, yarık dudak
Eller fleksiyon
Ayakta sallanan sandalye tabanı
Polidaktili
TRİZOMİ 13 (PATAU SENDROMU)
Avuç içinde simian çizgi
Erkeklerde inmemiş testis (Kriptorşizidizm)
Kızlarda bikornuat uterus (kalp şeklinde
uterus)
Hipoplastik overler (gelişimi gecikmiş)
Polikistik böbrek hastalığı
Gebeliklerde tekrarlama riski düşüktür
Hastaların yarısı yaşamın ilk aylarında ölür
TRİZOMİ 13 (PATAU SENDROMU)

KEDİ AĞLAMASI (CRI DU CHAT - 5p
delesyonu sendromu)
5.Kromozomun kısa kolundaki büyük bir
delesyon sebep olur. Hasta bebekler
ağladıklarında kedi miyavlamasına benzer
ses çıkarırlar.
Derine yerleşik gözler
Düşük burun kemeri
Üst dişlerin sorunlu kapanması
Bel kemiğinde normal dışı yan kıvrım
OTOZOMAL DELESYON SENDROMLARI

Mikrosefali
Hipertelorizm (iki organ arası uzaklığın fazla olması)
Düşük doğum ağırlığı
Yutma ve emmede bozukluk, beslenememe
Hiperaktivite
Saldırganlık
Huysuzluk
Ense düşük
Yüksek ve dar damak yapısı
Parmaklar arasında kısmi perde
Etkilenen bireylerin üreme yeteneği vardır
Klinodaktili (parmaklarda enlemesine eğrilik)
Sindaktili (yapışık parmak)
KEDİ AĞLAMASI (CRI DU CHAT ) SENDROMU

Çocuklara erken dönemlerde konuşma ve davranış
terapileri uygulanmalıdır. Sendromu taşıyan
çocukların birçoğu eğitilebilir, kendilerine
bakabilecek kadar sosyal gelişim gösterebilir.
Bu sendroma sahip bireylerin ebeveynlerinin
genetik danışma alması ve çocuğa uygun eğitim
planının yapılması, aileye ve çocuğa daha kaliteli
bir yaşam sağlar.
KEDİ AĞLAMASI (CRI DU CHAT ) SENDROMU

CİNSİYET KROMOZOMU HASTALIKLARI

47,XXY
Hastalar uzun boylu, ince ve uzun bacaklı
Kollar uzun
Bireyler püberteye kadar normaldir
Hipogonadizm (testislerin testosteron veya sperm ya da her ikisini birden üretmedeki yetersizliği) ile ilk belirtiler başlar
Testisler küçüktür
İkincil seks karakterleri (sakal, bıyık,..) az gelişir
İnfertildir
KLINEFELTER SENDROMU

Paternal mayoz I’deki hata sonucu oluşabilir
Maternal mayoz I’deki hata sonucu oluşabilir
(anne yaşı ileridir)
Mayoz II’deki hata sonucu oluşabilir
Zigot oluşumundan sonraki mitotik hatalar
sebebiyle görülebilir
KLINEFELTER SENDROMU

Erkek bireyin iki Y kromozomu taşımasına neden
olan, eşey kromozomlarında meydana gelen
anöploidi durumudur
Erkek bireyde gamet oluşumu sırasında mayozun
ikinci evresinde Y kromozomlarının ayrılmamasıyla
YY kromozomu taşıyan spermler oluşabilir. Eğer
bu spermler normal bir yumurta hücresini
döllerse, embriyo 44+XYY kromozom setini taşır.
Birey 47, XYY karyotipli olarak doğar.
47, XYY (SÜPER ERKEK) SENDROMU

Boy ortalamaları normalin 7 cm’nin üzerindedir
Gelişme geriliği ve davranış sorunları vardır
Zeka düzeyleri biraz düşüktür
Bir dönem bireylerin cinayete eğilimli oldukları
düşünülmüştür. Fakat bu eğilimin kromozom yapısından
değil düşük zeka düzeyinden kaynaklandığı söylenmektedir.
Harvard üniversitesi bir çalışma başlatmış; yeni
doğanlarda kromozom analizi. Fakat halkın tepkisini
çekmiş ve son verilmiştir
47, XYY SENDROMU
47, XXX (SÜPER DİŞİ), TRIPLE X SENDROMU
Maternal mayoz I’deki hatalar
sonucu oluşur
Ortalamadan biraz uzun
Genellikle fertil
IQ düşük
Öğrenme güçlüğü
Mikrosefali
Epikantal katlanma

TURNER SENDROMU
45, X
Eşey organları ve eşey hücreleri gelişmez
İnfertil bireylerdir
Konjenital böbrek rahatsızlıkları,
Kalp anomalileri,
Kistik higroma (boyun bölgesinde
saptanabilen birden fazla odacıklı kistik
yapılar)
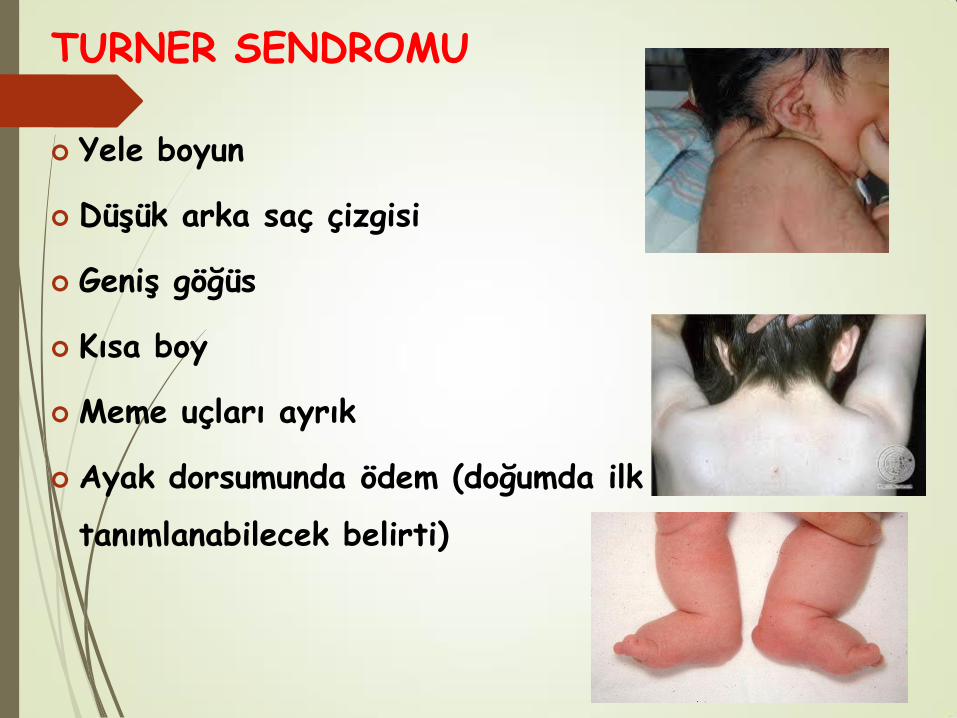
TURNER SENDROMU
Yele boyun
Düşük arka saç çizgisi
Geniş göğüs
Kısa boy
Meme uçları ayrık
Ayak dorsumunda ödem (doğumda ilk
tanımlanabilecek belirti)

ENZİM BOZUKLUKLARI

AMİNOASİDOPATİLER
HİPERFENİLALANİNEMİLER
Kan fenilalanin düzeyinin artmasına neden olan
anormalliklerdir.
Fenilalanin hidroksilazı sentezleyen gendeki
mutasyonlar sonucu oluşur.

FENİLKETANURİ
Konjenital bir metabolizma hastalığıdır
Otozomal resesif geçişlidir
Fenilalanini parçalayamadıkları için fenilketanurili
çocukların vücut sıvılarında bu aminoasit birikir
Beyine zarar verir
Yenidoğan taraması ile tespit edilebilir

PÜRİN METABOLİZMASI BOZUKLUKLARI
LESCH-NYHAN SENDROMU
X’e bağlı bir enzim olan hipoksantin guanin
fosforiboziltransferazı kodlayan HPRT lokusundaki
mutasyonlar sonucu oluşur.
Enzim eksikliğinde hipoksantin ve guanin
katabolizmasının artmasından dolayı ürik asit düzeyi
yükselmektedir.

PÜRİN METABOLİZMASI
BOZUKLUKLARI
LESCH-NYHAN SENDROMU
Hastalık, 2-3 yaşlarında
nörolojik belirtilerle ortaya
çıkmaktadır.
Çocuklar saldırgan davranışlıdır,
el ve ayak parmaklarıyla
dudaklarını ısırarak kendilerine
zarar verirler.

PÜRİN METABOLİZMASI BOZUKLUKLARI
LİZOZOMAL DEPO HASTALIKLARI
Lizozomal enzimleri kodlayan genlerdeki bir
mutasyon nedeniyle lizozomal enzim eksikliklerine
bağlı ortaya çıkan bir grup hastalıktır
Lizozomal Depo Hastalıklarında yıkılamayan
moleküller lizozomlar içerisinde depolanmaya
başlar. Bu durum bir süre sonra hücrelerin
şişmesine, hücre fonksiyonlarının bozulmasına ve
hücre ölümüne neden olur.

PÜRİN METABOLİZMASI BOZUKLUKLARI
LİZOZOMAL DEPO HASTALIKLARI
Mukopolisakkaridozlar (MPS)
Gaucher hastalığı
Lizozomal Depo Hastalıklarının çoğu otozomal
resesif kalıtılır, birkaç tanesi ise X-e bağlıdır

LİZOZOMAL DEPO HASTALIKLARI
Anormal yüz görünümü
Korneal bulutlanma ya da benzer
oküler anomaliler
Anjiyokeratom (genişlemiş kan
damarlarından oluşan vasküler
lezyonlar)
Umblikal ve inguinal herniler (karın
duvarı ve kasık fıtığı)

LİZOZOMAL DEPO HASTALIKLARI
Kısa boy
Gelişme geriliği
Eklem ve iskelet deformiteleri
Organomegali (özellikle karaciğer ve dalak)
Kas güçsüzlüğü ve kontrol bozukluğu (Ataksi,
nöbetler vs)
Nörolojik zayıflık, kazanılmış yetilerde kayıp

TAY-SACHS HASTALIĞI
Otozomal resesif bir hastalıktır
GM2 gangliosidosis veya hegzozaminidaz a
eksikliği
Çok aşırı miktarda yağ dokusu dokuda ve
beyindeki sinir hücrelerinde birikir. Bu birikim
sinir hücrelerini parçalayarak akli ve fiziki
problemlere neden olur.

TAY-SACHS HASTALIĞI
Tay-Sachs Hastalığı olan bebekler doğumdan
sonraki ilk ayda normaldirler ve daha sonra
sinir hücreleri fazla yağ deposu nedeniyle
şişmeye başlar, akli ve fiziki problemlere
neden olur. Çocuk kör ve sağır olur ve yutma
zorluğu başlar. Kaslarda atrofi (erime) ve felç
başlar. En iyi tedavi ile bile Tay – Sachs
hastalığı olan çocuklar en fazla 4 yaşına kadar
yaşayabilirler.

ADENOZİN DEAMİNAZ (ADA) EKSİKLİĞİ
Otozomal resesif kalıtılır.
T ve B lenfositlerde ileri derecede fonksiyon
bozukluğu vardır.
ADA eksikliği olan çocuklar, steril bir ortamda
yaşamadıkları sürece genellikle 2 yaşından önce
bir enfeksiyon nedeniyle kaybedilirler.

PÜRİN NÜKLEOZİD FOSFORİLAZ EKSİKLİĞİ
Otozomal resesif kalıtılır.
Pürin nükleozid fosforilaz eksikliğinden dolayı
pürin nükleotidlerinin katabolizması azalmıştır
T lenfositlerde fonksiyon bozukluğu vardır. B
lenfositler normaldir.

ÜRE DÖNGÜSÜ BOZUKLUKLARI
Üre döngüsü amonyağın üreye dönüştürüldüğü enzim
mekanizmasıdır. Üre döngüsündeki bozukluklar üre
yapımında görevli 6 enzimin herhangi birindeki genetik
defekt sonucunda ortaya çıkar.
Belirtileri;
- Artmış plazma amonyak düzeyi
- Kan glukozu, keton, pH ve CO2 düzeyinde
anormallikler
- Artmış plazma sitrülini
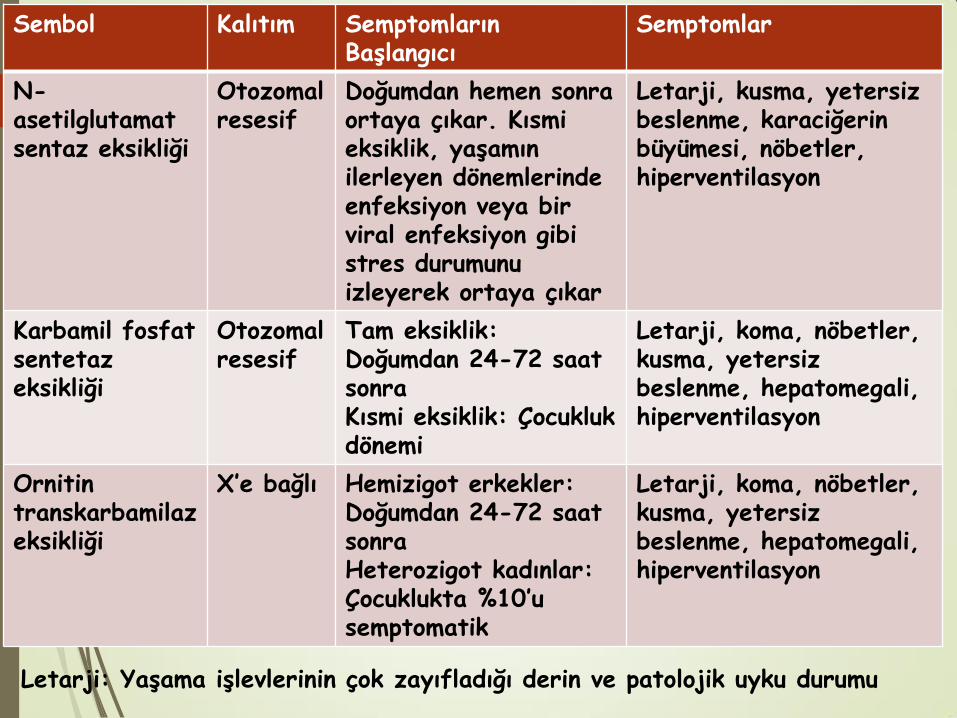
Sembol Kalıtım Semptomların Başlangıcı
Semptomlar
N-asetilglutamat sentaz eksikliği
Otozomal resesif
Doğumdan hemen sonra ortaya çıkar. Kısmi eksiklik, yaşamın ilerleyen dönemlerinde enfeksiyon veya bir viral enfeksiyon gibi stres durumunu izleyerek ortaya çıkar
Letarji, kusma, yetersiz beslenme, karaciğerin büyümesi, nöbetler, hiperventilasyon
Karbamil fosfat sentetaz eksikliği
Otozomal resesif
Tam eksiklik: Doğumdan 24-72 saat sonra Kısmi eksiklik: Çocukluk dönemi
Letarji, koma, nöbetler, kusma, yetersiz beslenme, hepatomegali, hiperventilasyon
Ornitin transkarbamilaz eksikliği
X’e bağlı Hemizigot erkekler: Doğumdan 24-72 saat sonra Heterozigot kadınlar: Çocuklukta %10’u semptomatik
Letarji, koma, nöbetler, kusma, yetersiz beslenme, hepatomegali, hiperventilasyon
Letarji: Yaşama işlevlerinin çok zayıfladığı derin ve patolojik uyku durumu

Sembol Kalıtım Semptomların Başlangıcı
Semptomlar
Argininosüksinat sentetaz eksikliği (sitrülinemi)
Otozomal resesif
Tam eksiklik: Doğumdan 24-72 saat sonra Kısmi eksiklik: Çocukluk dönemi
Letarji, koma, nöbetler, kusma, yetersiz beslenme, hepatomegali, hiperventilasyon
Argininosüksinat liyaz eksikliği (argininosüksinik asidüri)
Otozomal resesif
Tam eksiklik: Doğumdan 24-72 saat sonra Kısmi eksiklik: Çocukluk dönemi
Letarji, koma, nöbetler, kusma, yetersiz beslenme, hepatomegali, hiperventilasyon
Arginaz eksikliği
Otozomal resesif
Diğer üre döngüsü bozukluklarından daha yavaş başlangıç
Gelişimsel gecikme, protein intoleransı, spastisite, nöbetler, irritabilite, iştahsızlık, kusma

MİTOKONDRİYAL HASTALIKLAR

YAĞ ASİDİ OKSİDASYON BOZUKLUKLARI
Yağ asitleri, mitokondriyal yağ asidi oksidasyon
enzimlerinden birinin eksikliği nedeniyle biriken,
potansiyel olarak toksik türevlerdir.
Yağ asidi oksidasyonu, enerji üretiminin giderek
artan oranda yağ metabolizmasına bağlı olduğu,
uzamış açlık ve/veya artmış enerji ihtiyacı
(ateş, stres) dönemlerinde ortaya çıkar.

NASIL TANI KONULUR?
Artmış plazma açil-karnitinleri ve idrar organik
asitleri ile tanı konulur. Tanı, özgül genler için
mutasyon analizi veya cilt biyopsisi ile elde
edilen fibroblast kültüründen enzim incelemesi
ile doğrulanır.

ORGANİK ASİDEMİLER
Çeşitli enzim eksiklikleri nedeniyle vücut sıvılarında
organik asitlerin birikerek toksik etki yapmalarıyla
yaşamın ilk günlerinde yaşamı tehdit eden, ciddi
asidoz atakları başta olmak üzere çok farklı klinik
bulgularla kendini gösteren otozomal resesif geçişli
kalıtsal metabolizma bozukluklarıdır.

KİSTİK FİBROZİS
Akciğer, pankreas, bağırsak, ter bezlerinde
görülen hastalık CF transmembran regülatörü
kodlayan gende mutasyonların neden olduğu
otozomal resesif bir hastalıktır.
Ter testi ve genetik olarak tanı konulabilir.
Ter testi ile hastanın ön kolundaki ter bezleri
uyarılarak toplanan terde klor ölçümü yapılır.
Genetik olarak tanı CFTR genindeki F ve T
mutasyonları ile konulur.

Pankreatik yetmezlik
Malnütrisyon
Metabolizmanın artışı ile sonuçlanan
akciğerde kronik enfeksiyonlar
Genetik bozukluğa bağlı artmış
metabolizma
Kronik akciğer hastalığı
CF ilişkili diyabet
KİSTİK FİBROZİS

Solunum yollarında bulunan salgılar ile
akciğerin temiz ve sağlıklı kalması sağlanır.
Solunum yollarındaki tozlar, yabancı cisimler,
mikroplar bu akıcı salgı ile dışarıya atılırlar.
Kistik fibroziste bu salgıların kıvamı artar ve
koyulaşarak akıcı özelliğini kaybeder.
Koyulaşan salgılar akciğerlerdeki bronşlarda
tıkanmalara neden olur ve akciğerlerin içine
hava giriş-çıkışı bozulur. Bu ortamda akciğer
enfeksiyonu riski oluşur.
KİSTİK FİBROZİS

Solunum sisteminde;
Uzun süren ve devam eden öksürük
Hırıltılı solunum
Tekrarlayan akciğer enfeksiyonu
Bronşlarda genişleme ve koyu kıvamlı balgam
Egzersiz sırasında ortaya çıkan nefes darlığı
Oksijensizliğe bağlı olarak parmaklarda çomaklaşma
KİSTİK FİBROZİS

YAPISAL PROTEİN BOZUKLUKLARI
Duchenne Muscular Dystophy (DMD)
Distrofin genindeki mutasyonlar sonucu oluşur
X’e bağlı resesif geçişlidir
Distrofin, X kromozomunun Xp21 bölgesinde kodlanan
bir proteindir. Bu gendeki bozukluk sonucu distrofin hiç
üretilemez. Hastalık çekinik olarak kalıtıldığından
yalnızca erkek çocuklarda görülür. Kızlar genellikle
taşıyıcıdır, hastalık belirtisi göstermezler. Çünkü
kadınlarda iki adet X kromozomu vardır. Birinde
gerçekleşen mutasyon diğer kromozomla telafi edilebilir

Hasta çocuğun giderek normal yolda yürümesi
zorlaşır, sıklıkla düşer. Omuz çevresindeki kaslar
etkilendiği zaman kollarda zayıflık ortaya çıkar.
Hastalar 10 yaşına geldiklerinde yürüyememe başlar
ve tekerlekli sandalyeye bağımlılık olur. En son
olarak solunum ve gövde kasları etkilenir. Geçmişte
20 yaşlarında genelde hasta akciğer enfeksiyonuyla
kaybedilirken bugün iyi bir bakımla yaşam süresi
uzamıştır. Hastalık kalp kasını da tutabilir ama bu
genellikle ciddi bir belirti ortaya çıkarmaz
Duchenne Muscular Dystophy (DMD)