mouse model of noonan syndrome reveals cell type– and gene dosage–dependent effects of ptpn11...
TRANSCRIPT

A R T I C L E S
Noonan syndrome, an autosomal dominant disorder with an inci-dence of 1:1,000–2,500 live births1–4, is characterized by multiple,variably penetrant defects, most frequently proportionate shortstature, abnormal face and congenital heart disease3,4. Facial abnor-malities include wide-set eyes, broad forehead and depressed nasalroot with a wide nasal base, downward-slanting palpebral fissures,and a ‘triangular face’. Noonan syndrome is the most commonnonchromosomal cause of congenital heart disease4,5. Pulmonicstenosis, resulting from dysplastic valve leaflets, is the most frequentmanifestation, but atrial, atrioventricular or ventricular septaldefects, mitral valve abnormalities and hypertrophic cardiomyopathyare also found5–8. Cryptorchidism, lymphatic abnormalities, skeletaldefects, mild mental retardation, hearing difficulties, and bleedingproblems occur variably. Individuals with Noonan syndrome oftenhave unexplained hepatosplenomegaly9. A small number developmyeloproliferative disease (MPD), which usually resolves but candevelop into leukemia10,11.
Germline missense mutations of human PTPN11 occur in ∼ 50%of individuals with Noonan syndrome12–15. The protein product,Shp2, is a ubiquitously expressed, non-transmembrane protein-tyrosine phosphatase (PTP) containing two N-terminal Src homol-ogy 2 (SH2) domains, a central PTP domain, and a C terminus withtyrosyl phosphorylation sites and a proline-rich domain. Shp2 is
essential for normal activation of the Ras-Erk cascade in most, ifnot all, receptor tyrosine kinase (RTK) and cytokine receptor sig-naling pathways16. The Shp2 SH2 domains bind some RTKs andnumerous scaffolding adapters (including IRS-, Gab-Dos- andFRS-family proteins)16. The PTP domain is required for all actionsof Shp2 (ref. 16), whereas the C terminus has a modulatory role17.In the basal state, the more N-terminal SH2 domain (N-SH2) inter-acts with the PTP domain to suppress catalytic activity18–20.Binding to phosphotyrosyl peptide ligands on RTKs or scaffoldingadapters relieves this inhibition, resulting in potent activation.Engineered mutations of key contacts between the N-SH2 and PTPdomains result in biochemically and biologically ‘activatedmutants’ of Shp220.
Most mutations causing Noonan syndrome affect residues at theN-SH2–PTP domain interface12–14, suggesting that Noonan syn-drome is due to activated, gain-of-function mutants of Shp2 (ref. 12).Recently, acquired somatic mutations in PTPN11 have been reportedin sporadic myeloid leukemia (especially juvenile myelomonocyticleukemia; JMML) and B cell acute lymphoblastic leukemia (B-ALL)21–23. Leukemia-associated mutations often involve the sameresidues as those associated with Noonan syndrome but result in lessconservative substitutions, suggesting they may cause even greaterShp2 activation.
1Cancer Biology Program, Division of Hematology-Oncology, Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, NRB1038, 330Brookline Ave., Boston, Massachusetts 02215, USA. 2Division of Cardiology, Department of Pediatrics, The Children’s Hospital of Philadelphia, Philadelphia,Pennsylvania 19104, USA. 3Rodent Histopathology Core, Harvard Medical School, Boston, MA 02115, USA. 4Department of Pathology, Emory University, Atlanta,Georgia 30322, USA. 5Department of Pathology and 6Division of Hematology-Oncology, Brigham and Women’s Hospital, Harvard Medical School, Boston,Massachusetts 02215, USA. 7Department of Medicine, Cardiology Division, University of Pennsylvania Health System, Philadelphia, Pennsylvania 19104, USA.Correspondence should be addressed to T.A. ([email protected]).
Published online 25 July 2004; doi:10.1038/nm1084
Mouse model of Noonan syndrome reveals cell type– andgene dosage–dependent effects of Ptpn11 mutationToshiyuki Araki1, M Golam Mohi1, Fraz A Ismat2, Roderick T Bronson3, Ifor R Williams4, Jeffery L Kutok5,Wentian Yang1, Lily I Pao1, D Gary Gilliland6, Jonathan A Epstein7 & Benjamin G Neel1
Noonan syndrome is a common human autosomal dominant birth defect, characterized by short stature, facial abnormalities,heart defects and possibly increased risk of leukemia. Mutations of Ptpn11 (also known as Shp2), which encodes the protein-tyrosine phosphatase Shp2, occur in ∼ 50% of individuals with Noonan syndrome, but their molecular, cellular anddevelopmental effects, and the relationship between Noonan syndrome and leukemia, are unclear. We generated mice expressingthe Noonan syndrome–associated mutant D61G. When homozygous, the D61G mutant is embryonic lethal, whereasheterozygotes have decreased viability. Surviving Ptpn11D61G/+ embryos (∼ 50%) have short stature, craniofacial abnormalitiessimilar to those in Noonan syndrome, and myeloproliferative disease. Severely affected Ptpn11D61G/+ embryos (∼ 50%) havemultiple cardiac defects similar to those in mice lacking the Ras-GAP protein neurofibromin. Their endocardial cushions haveincreased Erk activation, but Erk hyperactivation is cell and pathway specific. Our results clarify the relationship betweenNoonan syndrome and leukemia and show that a single Ptpn11 gain-of-function mutation evokes all major features of Noonansyndrome by acting on multiple developmental lineages in a gene dosage–dependent and pathway-selective manner.
NATURE MEDICINE VOLUME 10 | NUMBER 8 | AUGUST 2004 849
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
A R T I C L E S
The molecular, cellular and developmental effects of disease-asso-ciated Shp2 mutations remain unknown. It is not clear whetherNoonan syndrome–associated mutants are pure hypermorphs (gain-of-function mutants) or whether all features of Noonan syndromecan result from a single Ptpn11 mutation. Initial surveys of peoplewith Noonan syndrome have not identified strong genotype–phenotype correlations13–15,24, but the likelihood that modifier lociexist in the human population limits the strength of these negativedata. The relationship between Noonan syndrome and leukemia-associated Ptpn11 mutations also is unclear. To address these issues,we generated and characterized ‘knock-in’ mice expressing theNoonan syndrome–associated mutation D61G.
RESULTSGeneration of D61G miceShp2 D61G occurs in several kindreds with Noonan syndrome, butnot in sporadic or Noonan syndrome–associated leukemia, and is
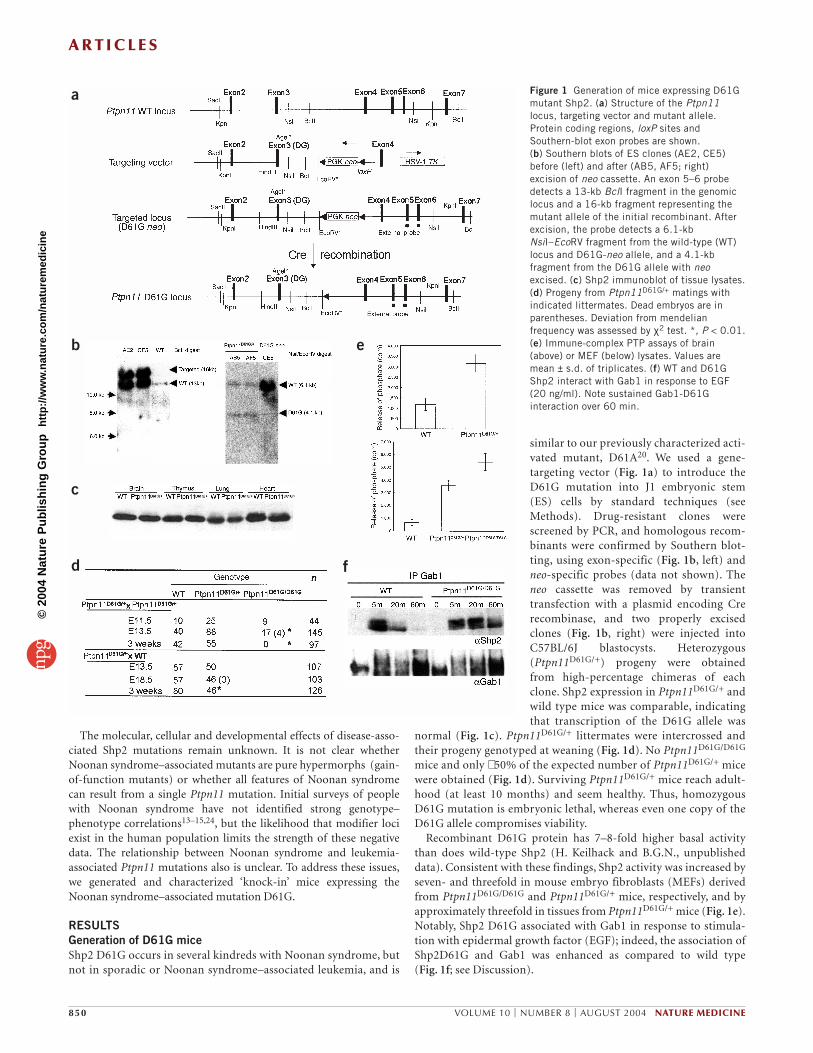
similar to our previously characterized acti-vated mutant, D61A20. We used a gene-targeting vector (Fig. 1a) to introduce theD61G mutation into J1 embryonic stem(ES) cells by standard techniques (seeMethods). Drug-resistant clones werescreened by PCR, and homologous recom-binants were confirmed by Southern blot-ting, using exon-specific (Fig. 1b, left) andneo-specific probes (data not shown). Theneo cassette was removed by transienttransfection with a plasmid encoding Crerecombinase, and two properly excisedclones (Fig. 1b, right) were injected intoC57BL/6J blastocysts. Heterozygous(Ptpn11D61G/+) progeny were obtainedfrom high-percentage chimeras of eachclone. Shp2 expression in Ptpn11D61G/+ andwild type mice was comparable, indicatingthat transcription of the D61G allele was
normal (Fig. 1c). Ptpn11D61G/+ littermates were intercrossed andtheir progeny genotyped at weaning (Fig. 1d). No Ptpn11D61G/D61G
mice and only ∼ 50% of the expected number of Ptpn11D61G/+ micewere obtained (Fig. 1d). Surviving Ptpn11D61G/+ mice reach adult-hood (at least 10 months) and seem healthy. Thus, homozygousD61G mutation is embryonic lethal, whereas even one copy of theD61G allele compromises viability.
Recombinant D61G protein has 7–8-fold higher basal activitythan does wild-type Shp2 (H. Keilhack and B.G.N., unpublisheddata). Consistent with these findings, Shp2 activity was increased byseven- and threefold in mouse embryo fibroblasts (MEFs) derivedfrom Ptpn11D61G/D61G and Ptpn11D61G/+ mice, respectively, and byapproximately threefold in tissues from Ptpn11D61G/+ mice (Fig. 1e).Notably, Shp2 D61G associated with Gab1 in response to stimula-tion with epidermal growth factor (EGF); indeed, the association ofShp2D61G and Gab1 was enhanced as compared to wild type (Fig. 1f; see Discussion).
850 VOLUME 10 | NUMBER 8 | AUGUST 2004 NATURE MEDICINE
a
b
c
d f
e
Figure 1 Generation of mice expressing D61Gmutant Shp2. (a) Structure of the Ptpn11locus, targeting vector and mutant allele.Protein coding regions, loxP sites andSouthern-blot exon probes are shown. (b) Southern blots of ES clones (AE2, CE5)before (left) and after (AB5, AF5; right)excision of neo cassette. An exon 5–6 probedetects a 13-kb BclI fragment in the genomiclocus and a 16-kb fragment representing themutant allele of the initial recombinant. Afterexcision, the probe detects a 6.1-kbNsiI–EcoRV fragment from the wild-type (WT)locus and D61G-neo allele, and a 4.1-kbfragment from the D61G allele with neoexcised. (c) Shp2 immunoblot of tissue lysates.(d) Progeny from Ptpn11D61G/+ matings withindicated littermates. Dead embryos are inparentheses. Deviation from mendelianfrequency was assessed by χ2 test. *, P < 0.01.(e) Immune-complex PTP assays of brain(above) or MEF (below) lysates. Values aremean ± s.d. of triplicates. (f) WT and D61GShp2 interact with Gab1 in response to EGF(20 ng/ml). Note sustained Gab1-D61Ginteraction over 60 min.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

A R T I C L E S
At embryonic day (E) 11.5, all genotypes were obtained atmendelian ratios. Fewer than expected Ptpn11D61G/D61G embryoswere recovered at E13.5, whereas Ptpn11D61G/+ embryos wereobtained at the expected frequency. At E18.5, some Ptpn11D61G/+
embryos were dead, and the overall number of heterozygotes was lessthan expected, but this difference was not statistically significant (Fig. 1d). Thus, Ptpn11D61G/D61G embryos die in mid-gestation,whereas the decreased viability of Ptpn11D61G/+ mice manifests late ingestation or perinatally.
Analysis of Ptpn11D61G/D61G and Ptpn11D61G/+ embryosPtpn11D61G/D61G embryos (E13.5) were grossly hemorrhagic andedematous, whereas heterozygotes appeared normal (Fig. 2a).Histological analysis showed severe liver necrosis in Ptpn11D61G/D61G
embryos (Fig. 2b); decreased liver size also was evident in wholemounts (Fig. 2a). Although most heterozygotes had normal livers,some had mild liver damage (Fig. 2b). Ptpn11D61G/D61G embryos also
had severe cardiac defects, including atrial, atrioventricular or ventricular septal defects, double-outlet right ventricle (DORV) andmarkedly enlarged outflow tract and atrioventricular valve primor-dia (Fig. 2c,d). Their myocardium was markedly thinned, with histological evidence of hemorrhage, subcutaneous edema and pericardial and peritoneal effusions (Fig. 2a, c and data not shown).Thinned compact myocardium could reflect a primary myocardialdefect. Myocardial thinning is observed in many mouse models of structural heart disease, however, so it may be a nonspecific manifestation.
At E13.5, Ptpn11D61G/+ embryos fell into two groups, each repre-senting approximately half of embryos analyzed (n = 10). Severelyaffected embryos also had ventricular septal defects (VSD), DORVand enlarged valve primordia (Fig. 2c,d). Unlike homozygotes, how-ever, their myocardium was not thinned, nor did they show effu-sions or edema. The remaining Ptpn11D61G/+ mice had significantmitral valve enlargement (to an extent similar to that in the severely
NATURE MEDICINE VOLUME 10 | NUMBER 8 | AUGUST 2004 851
a c
d
b
Figure 2 Comparison of wild-type (WT) and D61G heterozygous andhomozygous embryos. (a) Gross appearance of E13.5 embryos. Arrowindicates hemorrhage. (b) H&E sections from WT (1), Ptpn11D61G/D61G (2)and Ptpn11D61G/+ (3 and 4) embryos. Arrows indicate hepatic necrosis. Bar,200 µm. (c) Representative transverse sections of Ptpn11D61G/+ andPtpn11D61G/D61G embryos. Note ventricular septal defect (VSD, blue arrows),atrial and atrioventricular septal defects (ASD, AVSD; blue arrowheads),double-outlet right ventricle (DORV), thinning of ventricular wall (red arrows)and hypertrophy of atrioventricular and outflow tract valves (black arrows andarrowheads). Bar, 100 µm. PA, pulmonary artery; Ao, aorta; PV, pulmonaryvalve; MV, mitral valve; RV, right ventricle; LV, left ventricle; P.E., pericardial effusion. (d) Cardiac phenotypes of Ptpn11D61G/+ mice. Frequency of indicatedcardiac defects and valve thickness in WT and Ptpn11D61G/+ embryos. Three E18.5 Ptpn11D61G/+ embryos were dead and degenerating, as indicated inparentheses. Statistical significance of differences in valve thickness between WT and Ptpn11D61G/+ was evaluated using two-tailed Student’s t-test.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

A R T I C L E S
affected group), but their hearts were otherwise normal. One ofthese mice had a small VSD (Fig. 2c), but defects of this magnitudeare seen occasionally in E13.5 wild-type mice (ref. 25, Fig. 2d anddata not shown). Notably, cardiac hypertrophy was not observed inPtpn11D61G/+ mice.
Cardiac valves develop from endothelial cells, which undergoepithelial-mesenchymal transformation toform endocardial cushions. Endocardialcushions proliferate, and then valve leafletsundergo morphogenesis, which involves substantial apoptosis. Cardiac neural crestcells also contribute to endocardial cushionformation. In the mouse, however, only outflow-tract endocardial cushions, whichgive rise to aortic and pulmonic valves,receive a neural crest contribution.
We assessed proliferation by BrdU stain-ing and apoptosis by TUNEL assay inendocardial cushions of Ptpn11D61G/D61G
and Ptpn11D61G/+ embryos. Wild-type endocardial cushions (atE13.5) showed low levels of proliferation (Fig. 3a) and substantialapoptosis (Fig. 3b); accordingly, their valve leaflets thinned con-siderably over succeeding days (Fig. 2d). In contrast,Ptpn11D61G/D61G endocardial cushions contained multiple BrdU+
cells (Fig. 3a). Proliferation was increased in some but not all
852 VOLUME 10 | NUMBER 8 | AUGUST 2004 NATURE MEDICINE
a b
Figure 3 Abnormal endocardial cushion homeostasis in Ptpn11D61G/D61G and Ptpn11D61G/+ embryos. (a) Proliferation, assessed by BrdU incorporation,in wild-type (WT; 1) Ptpn11D61G/D61G (2) and Ptpn11D61G/+ (3 and 4) embryos. Representative positive cells are indicated (arrows). Quantificationrevealed 14.3 ± 2.7, 27.5 ± 8.7, 10.6 ± 2.4 and 21 ± 4.1 positive cells per h.p.f, respectively, for groups 1–4. Values represent mean ± s.d. from 6embryos per group. (b) Apoptosis, detected by TUNEL assay in WT (1), Ptpn11D61G/D61G (2), and Ptpn11D61G/+ embryos (3 and 4). Representativepositive cells are indicated (arrowheads). Quantification revealed 30.6 ± 7.0, 12.6 ± 5.0, 30.6 ± 4.7 and 17 ± 4.3, positive cells per h.p.f., respectively.Values shown are mean ± s.d. of triplicate determinations. Note enhanced BrdU+ and decreased TUNEL+ cells in Ptpn11D61G/D61G and some, but not all,Ptpn11D61G/+ embryos.
a b
c
Figure 4 Short stature and facial dysmorphia inPtpn11D61G/+ mice. (a) Growth curves of wild-type (WT) and Ptpn11D61G/+ male mice,measured from 3–16 weeks. Differences weresignificant (P < 0.05 for length and P < 0.01 forweight, assessed by repeated-measure ANOVA;post-hoc Fisher’s PLSD comparisons) at alltimes. Below, representative appearance of WTand Ptpn11D61G/+ mice at 6 weeks. Bar, 5 cm.(b) Gross appearance of faces (above) and AlcianBlue– and Alizarin Red–stained skulls (below) ofWT and Ptpn11D61G/+ mice. Dotted lines indicatedifferences in contours of lower face of WT andmutant mice. (c) Morphometric characteristics ofWT and Ptpn11D61G/+ skulls. Measurements wereobtained at the points indicated in lower panel ofb. Statistical significance was assessed by two-tailed Student’s t-test.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

A R T I C L E S
Ptpn11D61G/+ embryos (Fig. 3a). Moreover,Ptpn11D61G/D61G and some Ptpn11D61G/+
embryos showed decreased apoptosis (Fig. 3b). Although even mildly affectedPtpn11D61G/+ embryos had mitral valveenlargement at E13.5, there was no statisti-cally significant difference in valve size byE18.5 (Fig. 2d). The variation in valvethickness amongst mutant embryos atE18.5 was quite large, though, with someshowing clear valve enlargement even atthis age (see Supplementary Fig. 1 online).Nevertheless, the hearts of surviving adultsappeared normal (data not shown). Thedefective development of both atrioven-tricular and outflow-tract valves suggeststhat the D61G mutation perturbs valvulo-genesis through effects on endocardialendothelial cells.
Short stature and facial dysmorphia in Ptpn11D61G/+ miceLike humans with Noonan syndrome, male (Fig. 4a) and female (datanot shown) Ptpn11D61G/+ mice showed reduced length and weightwith preservation of overall body proportions. Some studies havereported abnormalities of the growth hormone–insulin-like growthfactor I (GH–IGF-1) axis in Noonan syndrome26. However, serumIGF-1 was comparable in wild-type and Ptpn11D61G/+ mice (503 and509 ng/ml, respectively). Consistent with their decreased body size,the skulls of Ptpn11D61G/+ mice were significantly shorter than wildtype (Fig. 4b). Skull width was unaffected, resulting in a significantlygreater length/width ratio. As in individuals with Noonan syndrome,the inner canthal distance was greater in Ptpn11D61G/+ mice (Fig. 4b,c). Ptpn11D61G/+ mice also tended to have a ‘triangular’ facialappearance, manifest as a wider and blunter snout (Fig. 4b).
Ptpn11D61G/+ mice develop mild myeloproliferative diseasePeripheral blood counts were normal in mice aged 6 weeks, but by 5 months, Ptpn11D61G/+ mice developed subtle but statistically significant leukocytosis, with normal hematocrit and platelet counts(Fig. 5a and data not shown). Absolute numbers of both neutrophils(2.2 × 103 ± 1.0 × 103/µl versus 3.6 × 103 ± 1.3 × 103/µl; P < 0.05) andlymphocytes (6.5 × 103 ± 1.1 × 103 /µl versus 9.5 × 103 ± 2.2 × 103 /µl;
P < 0.01) were increased in Ptpn11D61G/+ mice. Older Ptpn11D61G/+
mice developed splenomegaly, which could be marked, owing toextramedullary hematopoiesis comprised of maturing myeloid anderythroid elements (Fig. 5b,c). Mild myeloid hyperplasia was presentin bone marrow and spleen (Fig 5c). The bone marrow also containedunusual macrophages with eosinophilic crystalline-appearing mate-rial filling their cytosol (Fig. 5c). The mice showed splenic erythroidhyperplasia, which may help explain the preserved hematocrit (datanot shown). Flow cytometric analysis confirmed the myeloid expan-sion in bone marrow and spleen (Fig. 5d). Consistent with a cell-autonomous signaling defect in hematopoietic progenitors, bonemarrow from Ptpn11D61G/+ mice yielded factor-independent myeloidcolonies, and Ptpn11D61G/+ progenitors showed increased sensitivityto IL-3 and GM-CSF (Fig. 5e). Thus, all Ptpn11D61G/+ mice (examinedat 5 months) develop a subtle myeloproliferative phenotype, but thisdisorder is reasonably well tolerated and has not led to excess mortal-ity during up to 10 months of followup.
Cell context–dependent regulation of Erk activation by D61GPtpn11D61G/+ embryos showed increased immunostaining with phos-pho-Erk (p-Erk) antibodies (Fig. 6a,b). Notably, enhanced stainingwas restricted to regions where subsequent abnormalities wereobserved, such as the developing face and limb buds (Fig. 6a).
NATURE MEDICINE VOLUME 10 | NUMBER 8 | AUGUST 2004 853
a
c
e
d
bFigure 5 Ptpn11D61G/+ mice developmyeloproliferative syndrome. (a) White bloodcounts of wild-type (WT) and Ptpn11D61G/+ mice.(b) Spleen weights of WT and mutant mice. Insetshows typical WT spleen and markedly enlargedspleen from a 5-month Ptpn11D61G/+ mouse. (c) H&E-stained sections of bone marrow (BM; 1,WT and 2, Ptpn11D61G/+) and spleen(3,WT and 4, Ptpn11D61G/+). Representativemyeloid cells are indicated (yellow arrows). Inset:high-power view of Ptpn11D61G/+ BM, showingunusual macrophage containing eosinophiliccrystalline material (yellow arrowhead). (d) Flowcytometric analysis of bone marrow (above) andspleen (below). The percentage of each cellpopulation is indicated. (e) Myeloid colonyassays from WT and Ptpn11D61G/+ BM in theabsence or presence of the indicatedconcentrations of IL-3 (left) or GM-CSF (right).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

A R T I C L E S
Likewise, as compared to wild-type, 3 of 8 E11.5 embryos analyzedshowed increased numbers of p-Erk-reactive endocardial cushioncells, which ranged from a fairly small number of positive cells (Fig. 6b) to markedly increased staining (Fig. 6b). In contrast, thekinetics and magnitude of Erk activation in response to EGF (Fig. 6c),IGF-1, fibroblast growth factor (FGF) or platelet-derived growth fac-tor (PDGF) (data not shown) were unaltered in Ptpn11D61G/+ orPtpn11D61G/D61G MEFs. Yet the mutant Shp2 protein showed sus-tained association with Gab1 (Fig. 1f), even though Gab1 tyrosylphosphorylation was grossly unaffected (data not shown). These dataindicate that D61G acts as an Shp2 hypermorph to enhance Erk acti-vation, but in a manner dependent on pathway and/or cell context.
DISCUSSIONWe have characterized mice expressing the Noonan syndrome–associated mutation D61G. Whereas homozygous D61G mutationcauses mid-gestational lethality, ∼ 50% of Ptpn11D61G/+ mice survive toadulthood, where they show the common noncardiac features ofNoonan syndrome. The remaining heterozygotes succumb, most likelyowing to multiple cardiac defects similar to those in humans with
Noonan syndrome. The D61G mutation enhances Erk activation, butonly in some cell types and tissues. Furthermore, Ptpn11D61G/+ micedevelop a nonfatal MPD. Our data show that a single Shp2 mutationassociated with Noonan syndrome, acting predominantly if not exclu-sively as a hypermorph, has gene dosage- and cell/tissue- and/or signaling pathway-specific effects on mouse development.
The cardiac defects in mice with D61G mutation(s) are gene-dosage dependent. D61G homozygotes have severe pan-valvularstenosis, large septal defects and DORV, which result in heart failureby E13.5. Approximately 50% of Ptpn11D61G/+ mice also showmarked, although less severe, cardiac defects; for example, heart fail-ure was not observed in Ptpn11D61G/+ mice. The remainingPtpn11D61G/+ mice have much milder abnormalities. Conceivably,modifier alleles segregate in this mixed background. Similar allelesmay exist in humans, given the substantial phenotypic differencesthat can occur among family members with Noonan syndrome1–5.Our mouse model may facilitate the identification of these loci.
Our results provide several new insights into the molecular patho-genesis of the cardiac defects seen in Noonan syndrome. Several linesof evidence indicate that the abnormal valve development is likely toreflect Shp2 gain of function. The spectrum of heart defects inPtpn11D61G/D61G and severely affected Ptpn11D61G/+ embryos ismarkedly similar to those in mice lacking the tumor-suppressor geneNf1 (refs. 25, 27–29). The Nf1 gene product, neurofibromin, is a Ras-GTPase-activating protein (Ras-GAP), and Nf1−/− endocardial cush-ions show increased activation of the Ras-Erk pathway25, increasedproliferation and decreased apoptosis29. Increased proliferation (Fig.3a), decreased apoptosis (Fig. 3b) and Erk hyperactivation (Fig. 6b)also are found in Ptpn11D61G/+ endocardial cushions. Shp2 hypomor-phism causes decreased cell proliferation and impaired Erk activationin multiple signaling pathways16. The opposite effects of D61G indi-cate that it acts as a hypermorph. The increased Erk activation in onlya subset of Ptpn11D61G/+ embryos, most likely those that will developsevere cardiac disease, suggests a causal role for Erk hyperactivation indefective cardiac valvulogenesis and indicates that the presumptivemodifier alleles may act upstream of Erk activation. Although neu-rofibromatosis-1 and Noonan syndrome are nonallelic disorders30,the strong similarity of a subset of their phenotypic characteristics inmouse and man, together with our biochemical analyses, suggest thatPtpn11 and Nf1 mutations affect similar signaling pathways.However, only some of the effects of Ptpn11 hypermorphic mutationresemble those of Nf1 loss of function. Shp2 and neurofibromin maybe limiting components for Ras-pathway regulation only in some celltypes, some but not all of which overlap. Alternatively, each may alsoregulate additional, distinct signaling pathways.
Cardiac valvulogenesis involves a complex interplay between endo-cardial and neural crest cells. In mice, neural crest cells contribute
854 VOLUME 10 | NUMBER 8 | AUGUST 2004 NATURE MEDICINE
a
b
c
Figure 6 Cell context–dependent enhancement of Erk activation by D61Gmutant of Shp2. (a) Whole mounts of E11.5 embryos stained with anti-pErkantibodies. Strong staining is visible in facial area and limb bud ofheterozygous embryo (arrows). (b) Section of outflow tracts of E11.5embryos stained with anti-pErk antibodies. Varyingly increased numbers ofpositive cells are seen in about half of Ptpn11D61G/+ embryos (middle andright). Analysis of serial sections revealed a mean of 7.4 Erk+ cells percushion per section in Ptpn11D61G/+ embryos as compared with 1.5 Erk+
cells per cushion per section in wild-type (WT) embryos (P < 0.002). (c) Normal Erk activation Ptpn11D61G/+ and Ptpn11D61G/D61G MEFs.Immunoblots of starved and EGF-stimulated MEF lysates probed with anti-pERK antibodies and then reprobed with anti-total Erk antibodies to controlfor loading. The kinetics and magnitude of Erk activation in MEFs areunaltered by hetero- or homozygous D61G mutation.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

A R T I C L E S
only to outflow tract valves25. Ptpn11D61G/+ embryos have atrioven-tricular and outflow-tract valvular stenosis, strongly suggesting thatD61G acts in endocardial cells. We cannot exclude additional actionsof D61G on cardiac neural crest, but we did not observe septationdefects such as are typically associated with neural crest defects31
(Fig. 2c and data not shown). Cardiac defects in Nf1−/− mice alsoreflect the absence of neurofibromin from endocardial cells29.Hypertrophic cardiomyopathy is reported in Noonan syndrome5, butit is uncommon in those with Ptpn11 mutations13,14,24,32. Thus, thenormal ventricular wall in Ptpn11D61G/+ mice does not differ signifi-cantly from the human Noonan syndrome phenotype.
By 5 months, Ptpn11D61G/+ mice develop mild MPD. Factor-inde-pendent colonies can be recovered, and hematopoietic progenitorsshow increased cytokine sensitivity. Thus, a Noonan syndrome–linkedShp2 mutation not previously associated with abnormalhematopoiesis enhances hematopoietic progenitor survival, prolifera-tion or both. Notably, this hematological disorder is similar to(although less severe than) the MPD that develops in Nf1+/− mice26 ormice with activated K-ras expressed selectively in hematopoietic pro-genitors33,34. Conceivably, Shp2 and Nf1 mutations may both affecthemangioblasts, the common progenitor of hematopoietic and someendothelial cells35,36. JMML patients without Ptpn11 mutations haveeither homozygous Nf1 or heterozygous oncogenic Ras mutations,indicating that D61G also acts as a hypermorph in the hematopoieticlineage. Notably, the hematopoietic effects of D61G provide the firstevidence for a pro-oncogenic PTP mutant.
Unlike Nf1- or Kras-associated MPD, the Ptpn11D61G/+ MPD is welltolerated. Notably, ∼ 25% of individuals with Noonan syndrome showpreviously unexplained hepatosplenomegaly9. In the limited number ofcases described, MPD has been demonstrated, which most often resolvesspontaneously. Our Ptpn11D61G/+ mice may provide a model for thisself-limited MPD. Rarely, individuals with Noonan syndrome developfatal MPD, typically JMML9. Additional genetic events, and/or specificShp2 mutations (for example, the somatic mutations associated withsporadic JMML or ALL), may be required to cause fatal MPD, and ourmice may provide a useful model for studying disease progression.
Unlike the cardiac defects, the facial abnormalities in Ptpn11D61G/+
mice, and by inference in Noonan syndrome, probably reflect D61Gaction in neural crest. All facial bones arise from cranial neuralcrest37,38; increased p-Erk staining in the developing face ofPtpn11D61G/+ mice is consistent with a neural crest signaling defect.The hematopoietic, craniofacial and growth defects in Ptpn11D61G/+
mice, unlike the cardiac defects, are completely penetrant. Cell- andpathway-specific differences in sensitivity to the action of D61G (orcell- and pathway-specific differences in the action of modifier loci)probably account for this difference in penetrance.
Ptpn11D61G/D61G mice develop severe liver necrosis, which almostcertainly contributes to embryonic lethality. Some Ptpn11D61G/+ micealso develop mild liver damage. Hepatic necrosis was observed only inPtpn11D61G/+ mice with severe cardiac defects, and right heart failurecan cause secondary congestive liver disease. But heart failure did notseem to be more severe in those heterozygotes with liver necrosis, sothe hepatic and cardiac abnormalities probably reflect independenteffects of Ptpn11 mutation. Other mouse models showing mid-gesta-tional hepatic death include those with defects in signaling from Met(the receptor for hepatocyte growth factor) or in NF-κB pathwaycomponents39,40. Shp2 is implicated as a positive signal transducer inthe Met and NF-κB pathways39,41,42. Conceivably, Shp2 mutationsmay have both gain-of-function and loss-of-function effects in differ-ent contexts. Alternatively, doubling the level of Shp2 phosphataseactivity, as occurs in Ptpn11D61G/D61G cells (Fig. 1e), may have toxic
effects through another mechanism. This may explain why the typesof Shp2 mutations found in sporadic leukemias (such as JMML orALL) are not usually seen in Noonan syndrome. Many of thesesomatic mutations result in higher basal Shp2 activity than doNoonan syndrome–associated mutations (ref. 21, and H. Keilhackand B.G.N., unpublished data), suggesting that this level of increasedPTP activity could be incompatible with embryonic viability. Indeed,hemizygous (Ptpn11D61G/–) mice are viable (see Supplementary Table1 online), which indicates that the lethality of Ptpn11D61G/D61G mice,and the greater penetrance and severity of their Noonansyndrome–like phenotypes, reflect increased dosage of D61G ratherthan a protective effect of wild-type Shp2.
Although several cells and tissue types are affected by germlineD61G mutation, most are not. Ptpn11D61G/+ or Ptpn11D61G/D61G
MEFs show normal Erk activation in response to multiple growth fac-tors, even though Shp2 is required for RTK-evoked Erk activation inthese cells16. A recent study also failed to observe increased Erk activa-tion in BaF3 cells expressing leukemogenic Shp2 mutants22. The rea-son for this differential responsiveness remains to be determined.Some Noonan syndrome–associated Shp2 mutants enhance Erk acti-vation in transient transfection assays, but only upon coexpression ofGab1 (refs. 21,43). Genetic evidence indicates that Shp2 action down-stream of many RTKs and cytokine receptors is mediated by a Gab1-Shp2 complex16. Although Erk activation is not enhanced inMEFs expressing D61G, the mutant Shp2 allele shows sustained asso-ciation with Gab1 (Fig. 1f), consistent with enhanced signaling fromGab1-Shp2. Presumably these cells can compensate for this increasedupstream signal, preventing excessive Erk activation; notably, a simi-lar absence of increased Erk activation is found in MEFs from miceexpressing activated Ras33,44. Perhaps the level of expression of Gab1and/or related scaffolding adapters determines whether or not a spe-cific cell responds to Noonan syndrome–associated mutants of Shp2.Consistent with this notion, Gab1 is expressed at particularly highlevels in the developing heart45.
In summary, Ptpn11D61G/+ mice provide an animal model fordelineating the pathogenesis of human Noonan syndrome. Futurestudies will be aimed at defining other signaling pathways deregulatedin these mice and the mechanisms by which these cause Noonan syn-drome–associated phenotypes.
METHODSGeneration of D61G mice. The D61G mutation and a unique AgeI site wereintroduced by site-directed mutagenesis. To construct the D61G targeting vec-tor, the long arm, which includes exons 2 and 3 (SacII–SpeI genomic fragment)and the short arm, containing exon 4 (BclI–ClaI fragment) were ligated intoPGK Neo-HSV-1 TK46. The targeting vector was linearized with SacII andused to electroporate J1 ES cells (129Sv background). DNA was isolated fromG418 and gancyclovir doubly resistant clones and screened by PCR. The 5′primer was positioned within the PGK promoter and the 3′ primer was out-side the targeting vector. Homologous recombination was confirmed bySouthern blotting using neo and exon probes. Correctly targeted ES cells weretransfected with Cre-expressing plasmid (pCMV Cre) to excise the neo cassetteand rescreened by PCR and Southern analysis. Additional details are availablefrom T.A. upon request. Two correctly targeted ES clones were microinjectedinto C57BL/6 (B6) blastocysts, and resulting chimeras were mated with B6mice. Both clones gave rise to germline transmission and produced similarphenotypes. Heterozygous offspring were intercrossed to produce homozy-gous mice, and mice with B6 × 129Sv hybrid background were used for allexperiments. For genotyping, genomic DNA was prepared from tails and thensubjected to PCR and digestion with AgeI, which marks the D61G allele.Concentrations of IGF-1 in serum (obtained from tail blood) were measuredby radioimmunoassay using a kit (Alpco Diagnostics). All animal studies wereapproved by the animal welfare committee at Harvard Medical School.
NATURE MEDICINE VOLUME 10 | NUMBER 8 | AUGUST 2004 855
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

A R T I C L E S
MEFs were prepared from E13.5 embryos and cultured in DMEM contain-ing 15% FBS, as described47. MEFs were starved in DMEM without FBS for atleast 16 h before stimulations. Experiments in Figure 1d used fibroblasts fromE13.5 embryos that were immortalized by the 3T3 protocol.
Protein analysis and PTP assays. Lysates were prepared from MEFs and tis-sues from 6-week mice as described48. For immune complex PTP assays,lysates were immunoprecipitated with anti-Shp2 antibodies (Santa Cruz),and immune complexes were washed twice with PTP reaction buffer (25 mM HEPES, pH 7.4, 150 mM NaCl, 100 µg/ml BSA, 5 mM EDTA and 10 mM DTT) without substrate. 32P-labeled RCM-lysozyme, prepared asdescribed20, was added (specific activity: 4 × 104 cpm/pmol; final concen-tration 56 nM), and phosphate released after 5 min at 37 °C was quantifiedby charcoal binding assay. For immunoprecipitations, lysates were incu-bated with anti-Gab1 antibodies (Upstate Biotechnology) and this was fol-lowed by immunoblotting of recovered immune complexes, as describedpreviously17.
Histology and immunohistochemistry. For routine histology, embryos andtissues were fixed with Bouin’s fixative and embedded in paraffin, and sec-tions were stained with hematoxylin and eosin (H&E). Valve thickness wasmeasured at its biggest diameter in serial sections. Immunostaining wasdone as described29 on embryos fixed in 4% paraformaldehyde or 70%ethanol. For proliferation assays, 5′-bromo-2′-deoxyuridine (BrdU, Sigma)was injected into pregnant mice (50 µg/g body weight) for 2 h and thenkilled. Apoptosis was assessed using the In Situ Cell Death Detection kit(Roche) according to the manufacturer’s instructions. Cells were counter-stained with basic fuchsin (100 µg/ml). The numbers of BrdU+ or TUNEL+
cells per high-power field (h.p.f. = 0.02 µm2) were quantified as an index ofproliferation or apoptosis, respectively. Whole-mount p-Erk staining wasdone as described49.
Flow cytometric analyses and colony assays. Single-cell suspensions wereprepared from bone marrow and spleen. Erythrocytes were lysed withNH4Cl, Fc receptors blocked by incubating with 2.4G2 hybridoma (anti-CD16/CD32) supernatant, and remaining cells stained with allophycocyanin-conjugated anti–Gr-1 and phycoerythrin-conjugatedanti–Mac-1 (PharMingen). After washing, 7-aminoactinomycin D solutionwas added to facilitate exclusion of dead cells. Data were acquired by usingFACSCalibur flow cytometer (Becton Dickinson). Bone marrow colonyassays were performed as described50.
Note: Supplementary information is available on the Nature Medicine website.
ACKNOWLEDGEMENTSWe thank C.J. Rosen and J. Burgess for measuring IGF-I levels in serum and W. Pufor helpful discussions. This work was supported by US National Institutes ofHealth (NIH) R01 CA49152 and DK66600 and a Translational Research Grantfrom the Leukemia and Lymphoma Society (to B.G.N.), NIH R01 HL62974 andHL61475 (to J.A.E.), N.I.H. P01 DK50654 (to B.G.N., D.G.G. and J.L.K.) and NIH R01 DK64730 (to I.R.W.). Flow cytometric studies were partially supportedby Digestive Disease Research and Development Center grant NIH DK 64399.D.G.G. is an Investigator of the Howard Hughes Medical Institute. T. A. andM.G.M. were supported by fellowships from The Leukemia and LymphomaSociety. L.P. was supported by NIH training grant T32CA81156 and F.A.I. by NIH training grant T32HL007915.
COMPETING INTERESTS STATEMENTThe authors declare competing financial interests; see the Nature Medicine websitefor details.
Received 3 May; accepted 28 June 2004Published online at http://www.nature.com/naturemedicine/
1. Noonan, J.A. Hypertelorism with Turner phenotype. A new syndrome with asso-ciated congenital heart disease. Am. J. Dis. Child. 116, 373–380 (1968).
2. Nora, J.J., Nora, A.H., Sinha, A.K., Spangler, R.D. & Lubs, H.A. The Ullrich-Noonan syndrome (Turner phenotype). Am. J. Dis. Child. 127, 48–55 (1974).
3. Allanson, J.E. Noonan syndrome. J. Med. Genet. 24, 9–13 (1987).4. Noonan, J.A. Noonan syndrome. An update and review for the primary pediatri-
cian. Clin. Pediatr. (Phila.) 33, 548–555 (1994).
5. Marino, B., Digilio, M.C., Toscano, A., Giannotti, A. & Dallapiccola, B.Congenital heart diseases in children with Noonan syndrome: an expanded car-diac spectrum with high prevalence of atrioventricular canal. J. Pediatr. 135,703–706 (1999).
6. Marino, B. et al. Noonan syndrome: structural abnormalities of the mitral valvecausing subaortic obstruction. Eur. J. Pediatr. 154, 949–952 (1995).
7. Digilio, M.C. et al. Noonan syndrome and aortic coarctation. Am. J. Med. Genet.80, 160–162 (1998).
8. Bertola, D.R. et al. Cardiac findings in 31 patients with Noonan’s syndrome.Arq. Bras. Cardiol. 75, 409–412 (2000).
9. Bader-Meunier, B. et al. Occurrence of myeloproliferative disorder in patientswith Noonan syndrome. J. Pediatr. 130, 885–889 (1997).
10. Johannes, J.M., Garcia, E.R., De Vaan, G.A. & Weening, R.S. Noonan’s syn-drome in association with acute leukemia. Pediatr. Hematol. Oncol. 12,571–575 (1995).
11. Side, L.E. & Shannon, K.M. Myeloid disorders in infants with Noonan syndromeand a resident’s “rule” recalled. J. Pediatr. 130, 857–859 (1997).
12. Tartaglia, M. et al. Mutations in PTPN11, encoding the protein tyrosine phos-phatase SHP-2, cause Noonan syndrome. Nat. Genet. 29, 465–468 (2001).
13. Kosaki, K. et al. PTPN11 (protein-tyrosine phosphatase, nonreceptor-type 11)mutations in seven Japanese patients with Noonan syndrome. J. Clin.Endocrinol. Metab. 87, 3529–3533 (2002).
14. Maheshwari, M. et al. PTPN11 mutations in Noonan syndrome type I: detectionof recurrent mutations in exons 3 and 13. Hum. Mutat. 20, 298–304 (2002).
15. Tartaglia, M. et al. PTPN11 mutations in Noonan syndrome: molecular spec-trum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J.Hum. Genet. 70, 1555–1563 (2002).
16. Neel, B.G., Gu, H. & Pao, L. The ‘Shp’ing news: SH2 domain-containing tyro-sine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293(2003).
17. Araki, T., Nawa, H. & Neel, B.G. Tyrosyl phosphorylation of Shp2 is required fornormal ERK activation in response to some, but not all, growth factors. J. Biol.Chem. 278, 41677–41684 (2003).
18. Barford, D. & Neel, B.G. Revealing mechanisms for SH2 domain mediated reg-ulation of the protein tyrosine phosphatase SHP-2. Structure 6, 249–254(1998).
19. Hof, P., Pluskey, S., Dhe-Paganon, S., Eck, M.J. & Shoelson, S.E. Crystal struc-ture of the tyrosine phosphatase SHP-2. Cell 92, 441–450 (1998).
20. O’Reilly, A.M., Pluskey, S., Shoelson, S.E. & Neel, B.G. Activated mutants ofSHP-2 preferentially induce elongation of Xenopus animal caps. Mol. Cell. Biol.20, 299–311 (2000).
21. Tartaglia, M. et al. Somatic mutations in PTPN11 in juvenile myelomonocyticleukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet.34, 148–150 (2003).
22. Loh, M.L. et al. Somatic mutations in PTPN11 implicate the protein tyrosinephosphatase SHP-2 in leukemogenesis. Blood 103, 2325–2331 (2003).
23. Tartaglia, M. et al. Genetic evidence for lineage- and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in child-hood acute leukemia. Blood 104, 307–313 (2004).
24. Musante, L. et al. Spectrum of mutations in PTPN11 and genotype-phenotypecorrelation in 96 patients with Noonan syndrome and five patients with cardio-facio-cutaneous syndrome. Eur. J. Hum. Genet. 11, 201–206 (2003).
25. Gitler, A.D. et al. Nf1 has an essential role in endothelial cells. Nat. Genet. 33,75–79 (2003).
26. Ahmed, M.L. et al. Noonan’s syndrome: abnormalities of the growthhormone/IGF-I axis and the response to treatment with human biosyntheticgrowth hormone. Acta Paediatr. Scand. 80, 446–450 (1991).
27. Jacks, T. et al. Tumour predisposition in mice heterozygous for a targeted muta-tion in Nf1. Nat. Genet. 7, 353–361 (1994).
28. Brannan, C.I. et al. Targeted disruption of the neurofibromatosis type-1 geneleads to developmental abnormalities in heart and various neural crest-derivedtissues. Genes Dev. 8, 1019–1029 (1994).
29. Lakkis, M.M. & Epstein, J.A. Neurofibromin modulation of ras activity isrequired for normal endocardial-mesenchymal transformation in the developingheart. Development 125, 4359–4367 (1998).
30. Bahuau, M. et al. Exclusion of allelism of Noonan syndrome and neurofibro-matosis-type 1 in a large family with Noonan syndrome-neurofibromatosis asso-ciation. Am. J. Med. Genet. 66, 347–355 (1996).
31. Epstein, J.A. Developing models of DiGeorge syndrome. Trends Genet. 17,S13–S17 (2001).
32. Sarkozy, A. et al. Correlation between PTPN11 gene mutations and congenitalheart defects in Noonan and LEOPARD syndromes. J. Med. Genet. 40,704–708 (2003).
33. Braun, B.S. et al. Somatic activation of oncogenic Kras in hematopoietic cellsinitiates a rapidly fatal myeloproliferative disorder. Proc. Natl Acad. Sci. USA101, 597–602 (2004).
34. Chan, I.T. et al. Conditional expression of oncogenic K-ras from its endogenouspromoter induces a myeloproliferative disease. J. Clin. Invest. 113, 528–538(2004).
35. Dieterlen-Lievre, F. Hematopoiesis: progenitors and their genetic program. Curr.Biol. 8, R727–R730 (1998).
36. Gitler, A.D. et al. Tie2-Cre-induced inactivation of a conditional mutant Nf1allele in mouse results in a myeloproliferative disorder that models juvenilemyelomonocytic leukemia. Pediatr. Res. 55, 581–584 (2004).
856 VOLUME 10 | NUMBER 8 | AUGUST 2004 NATURE MEDICINE
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

A R T I C L E S
37. Couly, G.F., Coltey, P.M. & Le Douarin, N.M. The triple origin of skull in highervertebrates: a study in quail-chick chimeras. Development 117, 409–429(1993).
38. Kontges, G. & Lumsden, A. Rhombencephalic neural crest segmentation is pre-served throughout craniofacial ontogeny. Development 122, 3229–3242(1996).
39. Birchmeier, C., Birchmeier, W., Gherardi, E. & Vande Woude, G.F. Met, metasta-sis, motility and more. Nat. Rev. Mol. Cell Biol. 4, 915–925 (2003).
40. Ghosh, S. & Karin, M. Missing pieces in the NF-κB puzzle. Cell 109, S81–S96(2002).
41. Kapoor, G.S., Zhan, Y., Johnson, G.R. & O’Rourke, D.M. Distinct domains in theSHP-2 phosphatase differentially regulate epidermal growth factor receptor/NF-κB activation through Gab1 in glioblastoma cells. Mol. Cell. Biol. 24, 823–836(2004).
42. You, M., Flick, L.M., Yu, D. & Feng, G.S. Modulation of the nuclear factor kappaB pathway by Shp-2 tyrosine phosphatase in mediating the induction of inter-leukin (IL)-6 by IL-1 or tumor necrosis factor. J. Exp. Med. 193, 101–110(2001).
43. Fragale, A., Tartaglia, M., Wu, J. & Gelb, B.D. Noonan syndrome–associatedSHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and
sustained ERK2/MAPK1 activation. Hum. Mutat. 23, 267–277 (2004).44. Tuveson, D.A. et al. Endogenous oncogenic K-rasG12D stimulates proliferation
and widespread neoplastic and developmental defects. Cancer Cell 5, 375–387(2004).
45. Itoh, M. et al. Role of Gab1 in heart, placenta, and skin development andgrowth factor- and cytokine-induced extracellular signal-regulated kinase mito-gen-activated protein kinase activation. Mol. Cell. Biol. 20, 3695–3704(2000).
46. Su, I.H. et al. Ezh2 controls B cell development through histone H3 methylationand Igh rearrangement. Nat. Immunol. 4, 124–131 (2003).
47. Zhang, S.Q. et al. Shp2 regulates SRC family kinase activity and Ras/Erk acti-vation by controlling Csk recruitment. Mol. Cell 13, 341–355 (2004).
48. Klaman, L.D. et al. Increased energy expenditure, decreased adiposity, and tis-sue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficientmice. Mol. Cell. Biol. 20, 5479–5489 (2000).
49. Corson, L.B., Yamanaka, Y., Lai, K.M. & Rossant, J. Spatial and temporal pat-terns of ERK signaling during mouse embryogenesis. Development 130,4527–4537 (2003).
50. Sattler, M. et al. Critical role for Gab2 in transformation by BCR/ABL. CancerCell 1, 479–492 (2002).
NATURE MEDICINE VOLUME 10 | NUMBER 8 | AUGUST 2004 857
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine