yenİdoĞan dÖnemİnde kalitsal ... -...
TRANSCRIPT

YENİDOĞAN DÖNEMİNDE
KALITSAL METABOLİK HASTALIKLARA
YAKLAŞIM
Dr. Neslihan ÖNENLİ-MUNGAN
Çukurova Üniversitesi Tıp Fakültesi
Pediatri Anabilim Dalı

Tanım
* Kalıtsal Metabolik Hastalıklar;
Protein, karbohidrat veya yağ asitlerinin sentezi
ya da katabolizması ile ilgili olaylar sonucu gelişen
patolojik tablolar
* Birbiriyle ilintili veya farklı bir ya da daha
çok organ veya sistemi tutabilirler

Önemi
◘ Tedavi ile kalıcı hasarları önleyebilme olanağı
(PMR/katarakt)
◘ Özgün (MPS tip 1, 2, 6, Gaucher, Fabry
Pompe) tedavi olanağı
◘ Genetik danışma ve prenatal tanı olanağı

Tanıda gecikme
◘ Düşünülmüyor, ilk olarak akla iyi bilinen veya daha
sık görülen sepsis/intrakraniyal olaylar geliyor. Bu
nedenle özgün tanısal testler yapılmıyor ve tanı
konulamıyor
◘ Semptoma yaklaşılıp, semptomun bir kompleksin
parçası olabileceği gözden kaçıyor
◘ Çekiniliyor, tanı alsa da tedavi yok diye düşünülüyor
◘ Tarama programları yeterli hastalığı kapsamıyor
◘ Uygun ve zamanında örnek alınmıyor

◘ Kritik hastalıkta
◘ Nöbette
◘ Kusma ataklarında
◘ Parankimal karaciğer hastalığında
◘ Kardiyomiyopatide
◘ Metabolik asidozda
◘ Hiperamonemide
◘ Hipoglisemide
◘ Anne-baba akrabalığı,ailede benzer sorun
olduğunda, ailede zihinsel gerilik, nedeni
bilinmeyen çocuk ölümünde olduğunda
Ne zaman bir KMH düşünmeli

◘ Anormal saç yapısı
◘ Retinada kiraz kırmızısı leke, katarakt
◘ Hepatomegali/splenomegali
◘ Sarılık (direkt hiperbiluribinemi)
◘ Sensöronöral tip işitme kaybı
◘ Mikrosefali, hipotoni veya hipertoni
KMH düşündüren muayene bulguları

Fizyopatolojik olarak KMH
3 grupta incelenir
◘ İntoksikasyon tipi/intermediyer metabolizma
bozukluğu
◘ Kompleks bir molekülün sentez veya
katabolizmasında defekt
◘ Enerji üretim veya kullanımında defekt

İntermedier metabolizma
bozukluğu
◘ Eksik bir enzim ardında biriken
metabolitler fizyopatolojiden sorumlu
◘ Embryo/fötus gelişimine engel değil
◘ Genelde miyad, sorunsuz, normal
doğum, sepsis için risk taşımayan olgu
◘ Semptomsuz bir dönem sonrası
beslenmeyi takiben 2-3. günde başlayan
ve hızla ilerleyen klinik

◘ Aminoasit hastalıkları
(FKÜ, tirozinemi, MSUD)
◘ Organik asidemi (MMA, PA)
◘ Üre döngüsü enzim eksiklikleri
◘ Şeker intoleransları (Galaktozemi)
Hastalıkları

◘ Asemptomatik dönem – beslenme
akut bulgular/kusma, bilinç
değişiklikleri, nöbet, KC yetmezliği
Klinik
Atağı tetikleyen faktörler
Artmış katabolizma ve stres
◘ Enfeksiyon
◘ Gıda alımı

◘ Metabolik asidoz, ketoz, NH3↑, KŞ↓, LA↑
◘ Plazma ve idrar amino asit analizi
◘ İdrar organik asit analizi
◘ Tandem mass
Laboratuar
Tanısal Testler

Sekel
5. gün
Bilinç değişikliği, kusma
Komada 17. günde sevk
Asidoz yok
Hiperamonemi hafif
Ketozis, özel koku
Amino asit analizi MSUD
Diyaliz ve destek tedavi

Tirozinemili kardeş
5. günde beslenme
sonrası Aminoasit analizi
Tirozin artışı
Alfa-feto protein artışı
Organik asit analizi Süksinil aseton
Özel diyet, NTBC
12 aylık; 1,5 aydır NTBC temin edilemiyor
Ağrılı flask paralizi, polinöropati, Nörojenik krizi
Tirozinemi Tip I

Organik asidemi nedeniyle ex kardeş öyküsü
Anne-baba akraba, tek yumurta ikizi
Beslenme sonrası 7. günde supportif tedaviye yanıtsız ağır
metabolik asidoz, hiperamonemi
Periton diyalizi

Sekelsiz taburcu
Organik Asidemi
Geç alınan idrar örneği
nedeniyle özgün
tiplendirme yapılamadı

İlk atak 16. günde
Bilinç değişikliği
sık nefes alma
viral enfeksiyon
sonrası 4. atakta
komada başvuru
Ağır ketoasidoz, hiperamonemi
Periton diyalizi
Akut dönemde
Organik asit analiziyle tanı
Propionik Asidemi

2. günde kusma, beslenememe
Ensefalopati tablosu
Metabolik asidoz yok
Amonyak: 2000-3000
Önce periton diyalizi
Ardından hemodiyafiltrasyon
Amino asit analizi/Sitrülinemi

Kompleks bir molekülün sentez veya
katabolizmasında bozukluk
◘ Bulguları kalıcı ve ilerleyici
◘ Araya giren enfeksiyon,stres ve beslenme
değişikliğinden etkilenmez
◘ Asidoz, hiperamonemi gibi bir metabolik bozukluk
yapmaz
◘ YD’da nonimmün hidrops, sonra dismorfi ve yaygın
sistemik tutulum
◘ Peroksizomal hastalıklar, lizozomal depo hastalıkları
glikolizasyon ve kolesterol sentez defektleri

MPS I
MPS VI

Enerji üretim veya
kullanımında defekt
◘ Genellikle, KC, kalp, kas, beyin ayrı ayrı
veya beraber tutulur
◘ Sitoplazmik/mitokondrial olarak ayrılır
◘ Sitoplazmik olanlar glukoneogenez ve
glikojenoliz defektleri
◘ Mitokondrial olanlar daha ağır ve genelde
tedavisi olmayanlar (konj LA, solunum zincir
defektleri, YAOD)

◘ Klinik bulgular; hipotoni, miyopati
kardiyomiyopati, ani bebek ölümü
malformasyonlar
◘ Laboratuar bulguları; hipoglisemi
laktik asit yüksekliği
◘ Tanı için fonksiyonel testler, biyopsi
enzim analizi ve/veya moleküler
çalışma gerekli
◘ Tedavileri güç

Hipotoni
Hipertrofik KMP
Aile öyküsü
Enzim ve mutasyon
analizi
Pompe (GDH II)

17. günde ERT
Hipotoni çok az
Kardiyomiyopati yok

İlk ayda hipotoni ve
solunum problemi
nedeniyle başvuru
Ağır laktik asidoz
Kas biyopsisi ile kalıtsal kas
hastalığı dışlandı
Piruvat karboksilaz eksikliği

Temel Olarak 3 Sistem Tutulur
◘ Nörolojik
◘ Kardiyak
◘ Hepatik

Nörolojik bulgular
◘ Bilinç bozukluğu (letarji–koma)
◘ Hipotoni
◘ Nöbetler
◘ Hareket bozuklukları

Hepatik bulgular
◘ HM
◘ Hipoglisemi
◘ KC yetmezliği (sarılık, kanama, fonksiyon
bozukluğu, assit, ödem)
◘ Kolestatik sarılık

◘ Direkt hiperbilirubinemi;
Galaktozemi (bilateral katarakt)
Tirozinemi (kanda alfa feto-protein
yüksekliği, idrar organik asit analizinde
süksinil aseton artışı)

Kardiyak bulgular
◘ Kalp yetmezliği
◘ Dilate hipertrofik
◘ Ritm sorunları/perikardiyal efüzyon

◘ Sol ventrikül duvar kalınlaşması anlamlı
◘ Çoğunluğunda diğer kas tutulumları var
◘ Sistemik tutulum ve kardiyomiyopati
beraberse aksi ispatlanana dek tanı KMH
◘ Kardiyak tutuluma hepatomegali eşlik
ediyorsa glikojen depo hastalığı veya yağ asit
oksidasyon defekti düşünülür

Yeni doğanda başvuru tabloları
◘ Metabolik asidoz ve ensefalopati
◘ Metabolik asidoz olmadan ensefalopati
◘ Hepatik sendrom
◘ Kardiyak sendrom
◘ Nonimmün hidrops fetalis

◘ YD’da bulgu veren 100’den fazla KMH var
◘ Genellikle neden ne olursa olsun hasta bebekte
görülen ilk bulgu letarji, emmenin azalması veya
kaybolması
◘ Sepsis için risk faktörü taşımayan zamanında
doğan bir bebekte bu bulgular mutlaka KMH’ı
düşündürmeli
◘ Ancak unutulmamalı ki kalıtsal metabolik
hastalığı olan bebekler kolay sepsise girer
◘ Sepsis tanısı konulması KMH’ı ekarte ettirmez

Hasta yenidoğana yaklaşım
PM/DDA
Hasta Bebek
■ KMH için
tipik değil
■ BÇ ↓
maternal FKÜ
Tanıyla beraber acil tedavi
Miad Hasta Bebek
■ Hipoksi
■ İskemi
■ Kanama
■ Öykü
■ PA AC
■ Kraniyal
USG
■ Enfeksiyon
■ Kültür
■ Akut faz
reaktanları
Elektrolit sorunları
Hipo/Hiper Ca
Hipo/Hiper Na
Hipo/Hiper K
İzole multipl
malformasyonlar
Sendromlar
Biyokimyasal
testler
Hormonal/renal
tetkikler
Radyoloji
EKO
Kromozom

Nörolojik
bozulma
intoksikasyon
KMH
Ön
planda
nöbet
Glikojenozis D
YAOD Galaktozemi
Tirozinemi
CDG
Safra asit
sentez defektleri
LCHAD
Porfiri
B 6 ↓
PNPO
MCD
Folinik A ↓
3 P GD
GLUT- I
Sarılık
KC yetmezliği
Kalp yetmezliği
Ritm sorunları
Persistan
hipoglisemi
MSUD
MMA
PA
İVA
MCD
ÜDED
GA II
YAOD
Laktik
asidoz
Solunum
zincir D
YAOD
Mitokondri
hast

◘ Çok ağır hipotoni ve dirençli nöbet nadiren
KMH lehine, bu tür nöbet molibden kofaktör
eksikliği, NKHG, piridoksin, folinik asit ve biotin
eksikliğinde sık. Metabolik asidoz, hipoglisemi
ve hiperamonemi olmaz
◘ Ağır hipotoni konjenital laktik asidemi ve
solunum zincir defektlerinde olabilir
◘ Genellikle YD döneminde nörolojik bulgu
veren KMH’lar intoksikasyon veya enerji
eksikliği tipi olanlar
Nöbetler ve Hipotoni

◘ İntoksikasyon tipinde semptomsuz dönem
sonrasında solunum, nabız, tonus, bilinç
değişiklikleri, hipotermi, tremor ve jerkler
◘ Enerji eksikliği tipinde klinik hemen
başlar, daha az tipik ve daha ağır, hipotoni
dismorfi, malformasyonlar. Bilinç değişikliği
nadir. Metabolik asidoz olmadan LA
yüksekliği tipik

Tanıda 3 Laboratuar Bulgusu
◘ Hipoglisemi
◘ Metabolik ve/veya
Laktik asidoz
◘ Hiperamonemi

Metabolik Asidoz
◘ YD ve prematürde glomerül filtrasyon hızı
düşük, tübülüslere geçen fosfat yükü az
idrarla hidrojen iyon atılımı yetersiz
olduğundan kolay gelişir

Metabolik asidozlu hastaya yaklaşımda
ilk basamak anyon açığının hesaplanması
KMH’da anyon açığı 16’dan fazla
Anyon açığı: [Na+] [K+] – [Cl-] [HCO-3]

METABOLİK ASİDOZ
Normal anyon açığı
(hiperkloremik) Artmış anyon açığı
Organik asit
birikimi
Böbrekten azalmış
H+iyon atılımı • RTA
• DKA • Dehidrasyon
• İshal
• Açlık
• Laktik asidoz
• Salisilat
zehirlenmesi
• Etilen glikol
zehirlenmesi
• Aminoasidüri
• KMH
• Böbrek
yetmezliği
• YD ve PM

◘ Organik anyonların birikimi ile
karakterize metabolik asidozda ilk
planda KMH düşünülmeli
◘ Bu durumda çoğunlukla biriken
organik anyona spesifik anormal bir
idrar ve/veya ter kokusu olur

◘ Metabolik asidoza spesifik bir klinik bulgu
yok
◘ Örnekleme mutlaka asidoz sırasında
yapılmalı
◘ Asidozlu her hastada LA/PA, keton
amonyak ve KŞ’i bakılmalı

◘ Plazma NH3 düzeyi 70 µg/dl altında olmalı
◘ Kalıtsal metabolik hastalıklar dışında
hiperamonemi yapan nedenlerde amonyak
nadiren 180 µg/dl’i aşar
◘ Kan alırken zorlanma, hemoliz, laboratuara
geç ulaştırma, buz içinde transport olmaması
sigara, el teması gibi birçok faktör ve bazı
ilaçlar kan NH3 düzeyinde artışa yol açar
Hiperamonemi

◘ Kusma hiperamonemi yapan KMH’ların ilk
bulgularından
◘ Protein intoleransını işaret eder
◘ Pilor stenozunu düşündürecek kadar ağır olur
◘ Nedeni açıklanamayan her kusma ve
bilinç değişikliğinde amonyak bakılmalı
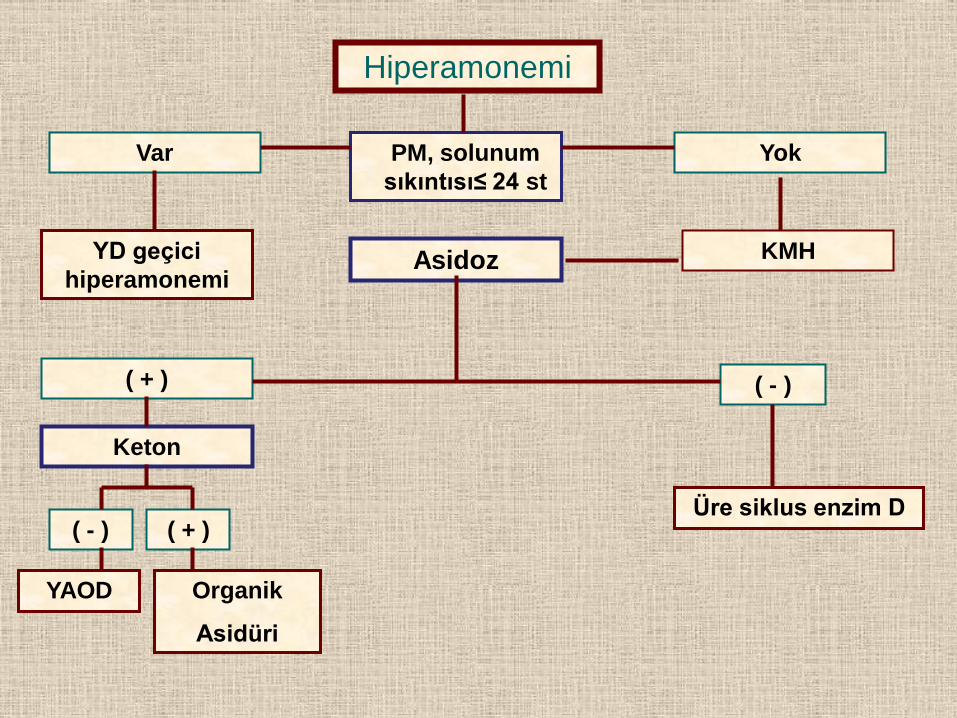
Hiperamonemi
PM, solunum
sıkıntısı≤ 24 st
Yok Var
YD geçici
hiperamonemi Asidoz KMH
( + ) ( - )
Keton
( - ) ( + )
YAOD Organik
Asidüri
Üre siklus enzim D
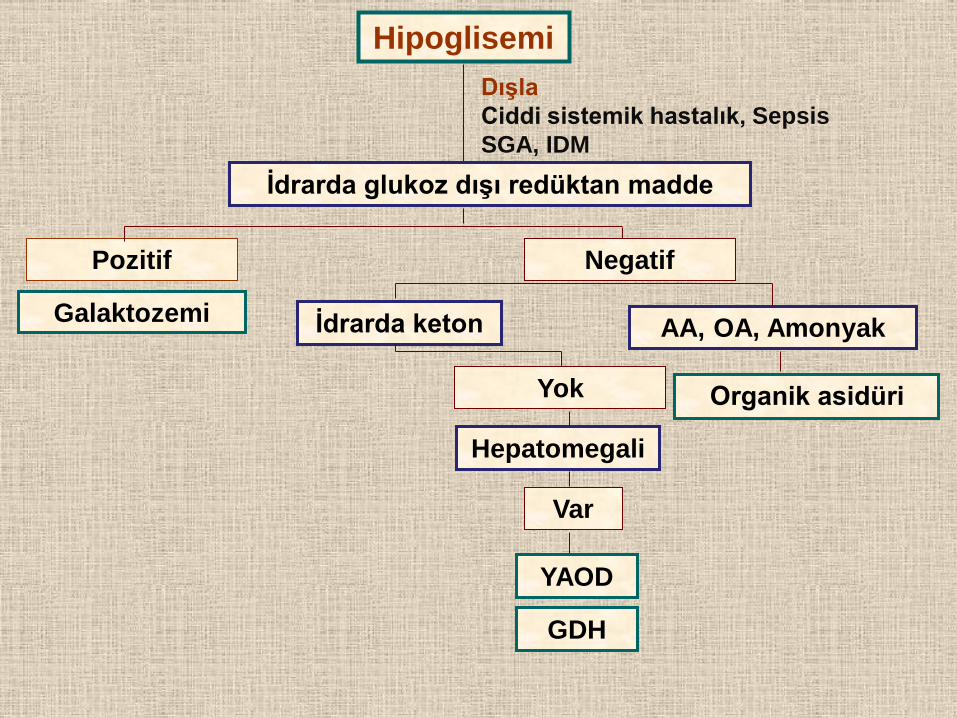
Hipoglisemi
Dışla
Ciddi sistemik hastalık, Sepsis
SGA, IDM
İdrarda glukoz dışı redüktan madde
Pozitif Negatif
AA, OA, Amonyak İdrarda keton
Organik asidüri Yok
Hepatomegali
Var
YAOD
Galaktozemi
GDH

Örnekleme
Hemen Yapılanlar
İDRAR Koku
Görünüş
Keton
Redüktan madde
pH
Sülfi test
Elektrolit
KAN CBC
Elektrolit, anyon
açığı, KŞ, Ca
KG
Ürik asit
Koagülasyon
ALT, AST
NH3
LA/PA
Keton

Örnek Saklama
İDRAR Taze 10-20 cc dondur / -20oC
(Org A, AA, orotik asit, porfirinler)
KAN Plazma/5 ml/heparinize / -20oC
Guthrie kağıdına en az 2 nokta
EDTA’lı tüp / tam kan / 10 cc / dondur / genetik
Serum / Tandem/ HPLC / A.A, açil karnitin

DİĞER
◘ LP
◘ PA AC/ Telegrafi
◘ EKO, EKG
◘ Kraniyal USG / EEG
◘ Cilt biyopsi (fibroblast kültürü)
◘ BOS (1 ml)
◘ Postmortem (K.C. kas)

Acil tedavide temel prensipler
◘ Geç kalınırsa mortalite ve morbidite yüksek
◘ Supportif (ilk basamak yeterli solunum ve dolaşım desteği,
olayı başlatan/arttıran enfeksiyonu araştırma dehidrasyon,
elektrolit dengesizliğini düzeltme)
◘ Beslenme (parenteral, enteral-protein ve yağ kesilir-max
48 saat, yüksek kalori)
◘ Spesifik tedavi (vit, alternatif yol sağlama)
◘ Toksin uzaklaştırılma (ekschange, periton diyalizi-özellikle
2,5 kg altında, devamlı hemofiltrasyon, hemodiyaliz)

◘ Özellikle ketoasidozda intrasellüler dehidrasyon
gözden kaçabilir, yetersiz sıvı tedavisi kadar hızlı
rehidrasyon, hipotonik sıvı kullanımı ve alkalizasyon
da beyin ödemini arttırır
◘ Hidrasyon 48 saate yayılmalı, günlük sıvı 3 L/m2’i
geçmemeli
◘ Hipoglisemi önlenmeli. KŞ’i 150 mg/dl üzerinde
tutulmaya çalışılmalı, gerektiğinde insülin infüzyonu
verilebilir. Tek istisna laktik asitin çok yüksek olduğu
mitokondriyal hastalıklar

◘ Fazla bikarbonat tedavisi hipopotasemi
beyin kanaması, hipernatremi yapabilir
◘ Bollus tarzında bikarbonat ancak pH
7’nin altında veya kardiyovasküler yan
etki varsa verilir
◘ Hedef, plazma bikarbonatını 15 mEq/L’e
çıkarmak

◘ Hiperamonemi tedavisinde protein alımı kesilir
yüksek kalori sağlanır
◘ Sodyum benzoat (250-500 mg/kg/gün oral/iv)
sodyum fenilbütirat, L-arjinin
◘ Hastayı krize sokan durumun (enfeksiyon gibi)
tedavisi
◘ Kafa içi basınç izlemi
◘ Valproik asitten sakınma

MESAJ
◘ Kritik hastalığı olan yenidoğanda mutlaka
sepsisle beraber KMH düşünülmeli
◘ Tüm hastalıklarda görülen emmeme, letarji
hipotoni ve solunum problemi gibi özgün
olmayan semptomlar KMH’nın da ilk bulguları
◘ Yenidoğanda bulgu veren KMH’lar çoğunlukla
intoksikasyon tipi
◘ KMH’ların çoğu otozomal resesif olarak kalıtılır
ancak sporadik olgular veya ailenin ilk bebeği
olabileceği de unutulmamalı

◘ Sorunlar PM’e bağlanmamalı, çünkü hastalar genelde miyad bebekler ◘ KMH tanısı koyabilmek için zamanında, yani akut atak sırasında ve uygun şekilde örnek almak çok önemli, aksi takdirde özgün tanı için ikinci bir atağı beklemek gerekir
◘ Zamanında müdahale ile sekelsiz iyileşmeyi
sağlamak tedavideki en önemli prensip
◘ Aile bilgilendirilirken; akut ve kronik sorunlar
hastalığın kalıtım şekli ve prenatal tanı mutlaka
anlatılmalı