aanpak van metabole ziekten met acute neonatale … · 1 aanpak van metabole ziekten met acute...
TRANSCRIPT

1
AANPAK VAN METABOLE ZIEKTEN MET ACUTE
NEONATALE PRESENTATIE
Claudine De Praeter, Linda De Meirleir, update 5 december 2006
Inleiding
Erfelijke metabole ziekten (IMD) zijn een belangrijke oorzaak van neonatale
morbiditeit en mortaliteit, doch de diagnose wordt vaak slechts laattijdig vermoed.
Gebrek aan diagnostiek en laattijdig starten van een adequate therapie kan leiden tot
ernstige hersenschade of overlijden. Indien het genetisch risico niet geïdentificeerd
werd kunnen nog andere kinderen met dezelfde problemen geboren worden.
Presentatie van een IMD in de neonatale periode
Verschillende presentatievormen zijn mogelijk:
1. Diagnose op basis van neonatale screening (vb. phenylketonurie).
2. Acute ernstige aspecifieke ziekte, vaak geassocieerd met acidose en
encephalopathie. En epileptische encephalopathie of status epilepticus.
3. Cardiomyopathie.
4. Acuut leverlijden.
5. Associatie met dysmorfe kenmerken of multipele congenitale afwijkingen.
Sleutel tot de diagnose
De belangrijkste vereiste om een metabole ziekte te vinden is een hoge graad
van alertheid voor het vermoeden van een mogelijke IMD bij een ernstig zieke
neonaat. Belangrijke informatie wordt bekomen via de anamnese, klinisch onderzoek
en eenvoudige “bedside” testen. De diagnosestelling begint met een zorgvuldig
uitpluizen van de antenatale gegevens.
Volgende vragen dienen steeds gesteld te worden:
1. Was er reeds een vroegere neonatale sterfte of mors in utero?
2. Is er consanguiniteit? (overdracht meestal autosomaal recessief)
3. Was het neonataal verloop normaal?
4. Is de voeding veranderd sinds de geboorte? Nemen de symptomen toe sinds
starten voeding?

2
5. Zijn er tekenen van infectie?
6. Werd de neonaat onderworpen aan vasten of een chirurgische interventie?
7. Verbeterde de neonaat bij onderbreken van de melkvoeding?
8. Was er een deterioratie na herstarten van de voeding?
Identificatie van risico neonaten
De klinische diagnose kan geschematiseerd worden in 3 stappen.
1. Eerste aanblik: schijnbaar niet specifieke symptomen.
Respiratoire insufficiëntie, hypotonie, zwakke zuigreflex, braken, dehydratie,
lethargie of convulsies. Deze symptomen kunnen veroorzaakt worden door infectie of
andere neonatale ziekten. Braken met opgezet abdomen kan een obstructie mimeren.
2. Klinische context.
De onverwachte en “geheimzinnige” deterioratie van een neonaat na een
initiële normale periode is het belangrijkste signaal voor de aanwezigheid van een
IMD van het intoxicatietype.
3. Herevaluatie van het kind.
Neurologische deterioratie. Na een initiële symptoomvrije periode treden
voedingsmoeilijkheden op met achteruitgang van het zuigreflex. Het kind wordt
comateus ondanks ondersteunende maatregelen. Kunnen optreden: respiratoire
insufficiëntie, de hik, apnoe, bradycardie, hypothermie. In het comateus stadium is er
verandering van de musculaire tonus en treden onwillekeurige bewegingen op. Ook
trappelen, boksbewegingen en trage lidmaatbewegingen zowel spontaan als na
stimulatie worden gezien. Convulsies treden meestal later op en zijn niet constant. Het
EEG toont soms een “burst suppression” patroon. Soms wordt een abnormale geur
van de urine of van het lichaam vastgesteld (bv. zweetvoetgeur bij
isovaleriaanacidemie of glutaaracidurie type II, verbrande kandijsuiker bij MSUD,
…).
In geval van “energie deficiëntie” (bv. respiratoire keten deficienties) is er
meestal geen symptoomvrije periode. Men vindt ernstige veralgemeende hypotonie,
hypertrofische cardiomyopathie, en snel progressieve neurologische deterioratie.

3
Soms zijn er dysmorfische kenmerken of malformaties. Hyperlactacidemie, met of
zonder metabole acidose, is een frequent voorkomend teken.
Uitzonderlijk kunnen lysosomiale stapelingsziekten zich uiten in de neonatale
periode.
Bij peroxysomale ziekten zijn symptomen aanwezig vanaf de geboorte, vooral
dysmorfieën en ernstige neurologische dysfunctie (bv. Zellweger syndroom).
Een andere groep vormen de neonaten met hypoglycaemie, leverstoornissen
en hepatomegalie. Denk hier aan glycogenose type I of III, gluconeogenesedefecten,
galactosemie, fructose intolerantie, tyrosinaemie type I, α1–antitrypsinedeficiëntie of
neonatale hemochromatose.
In de Tabellen 1 tot 4 worden algoritmen voor screening, aanpak en
differentiaal diagnostiek weergegeven.
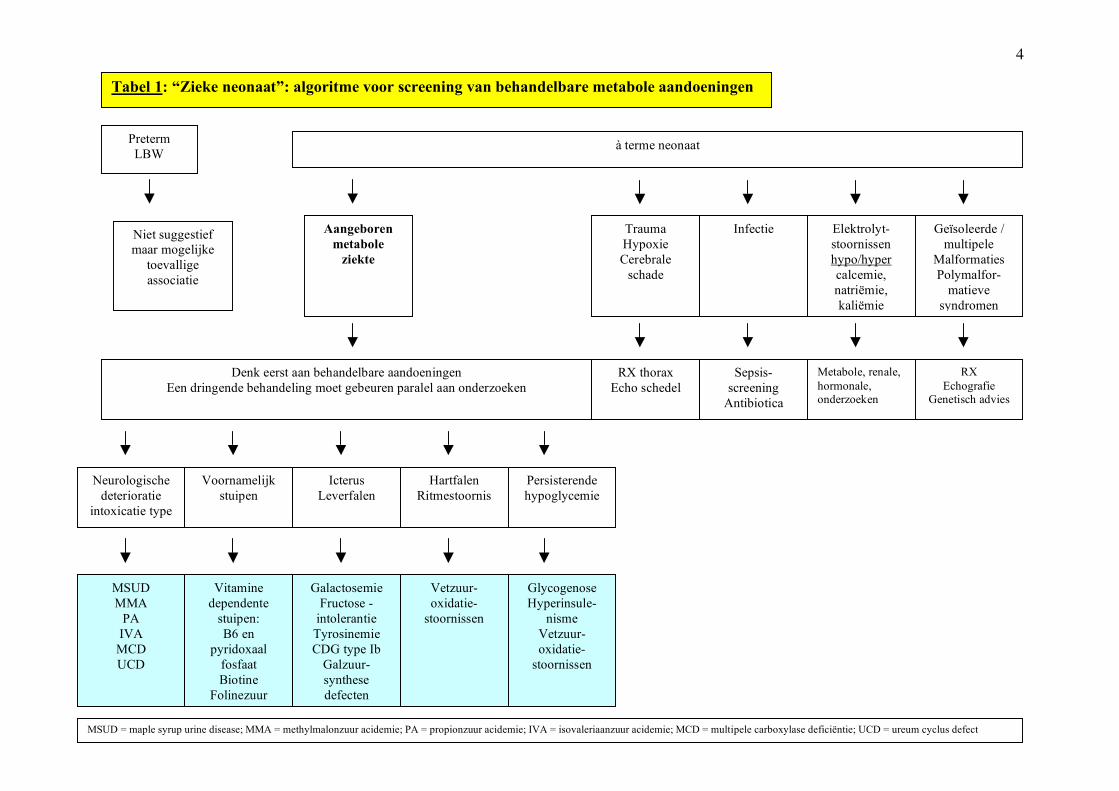
4
Preterm LBW
à terme neonaat
Aangeboren metabole
ziekte
Trauma Hypoxie Cerebrale
schade
Infectie Elektrolyt-stoornissen hypo/hyper calcemie, natriëmie, kaliëmie
Geïsoleerde / multipele
Malformaties Polymalfor-
matieve syndromen
Niet suggestief maar mogelijke
toevallige associatie
Denk eerst aan behandelbare aandoeningen Een dringende behandeling moet gebeuren paralel aan onderzoeken
Tabel 1: “Zieke neonaat”: algoritme voor screening van behandelbare metabole aandoeningen
Neurologische deterioratie
intoxicatie type
Voornamelijk stuipen
Icterus Leverfalen
Hartfalen Ritmestoornis
Persisterende hypoglycemie
MSUD MMA
PA IVA MCD UCD
Vitamine dependente
stuipen: B6 en
pyridoxaal fosfaat Biotine
Folinezuur Magnesium
Galactosemie Fructose -
intolerantie Tyrosinemie CDG type Ib
Galzuur-synthese defecten
Vetzuur-oxidatie-
stoornissen
Glycogenose Hyperinsule-
nisme Vetzuur-oxidatie-
stoornissen
RX thorax Echo schedel
Sepsis-screening
Antibiotica
Metabole, renale, hormonale, onderzoeken
RX Echografie
Genetisch advies
MSUD = maple syrup urine disease; MMA = methylmalonzuur acidemie; PA = propionzuur acidemie; IVA = isovaleriaanzuur acidemie; MCD = multipele carboxylase deficiëntie; UCD = ureum cyclus defect

5
Acute encephalopathie
Zuur-base evenwicht, lactaat, ammoniëmie, glycemie
Metabole acidose ± hyperammoniëmie
± lactaat acidose
Lactaat acidose Hyperammoniëmie Geen acidose
Hypoglycemie Geen acidose Geen hyperammoniëmie
Geen hypoglycemie
Metabole onderzoeken: screening organische zuren urine, aminozuren in plasma, acylcarnitines profiel, enz.
Organische zuren Vetzuuroxidatie-stoornissen
NKH SO deficiëntie PRS
Mitochondriale-, lactaatstoornissen
Ureumcyclus defecten THAN LPI PCD
Vetzuuroxidatie-stoornissen GSD, HFI, Galactosemie, F 1,6 diphosphaat deficiëntie MSUD
Aminozuur stoornissen
Convulsies
F 1,6 diphosphaat = fructose 1,6 diphosphaat, GSD = glycogeen stapelingsziekte, HFI = hereditaire fructose intolerantie, LPI = lysinurische eiwit intolerantie, MSUD = maple syrup urine disease, NKHG = niet ketotische hyperglycinemie, PCD = pyruvaat carboxylase deficiëntie, PRS = pyridoxine dependente stuipen, SO =sulfietoxidase, THAN = transiënte hyperammoniëmie van de neonaat.
Tabel 2
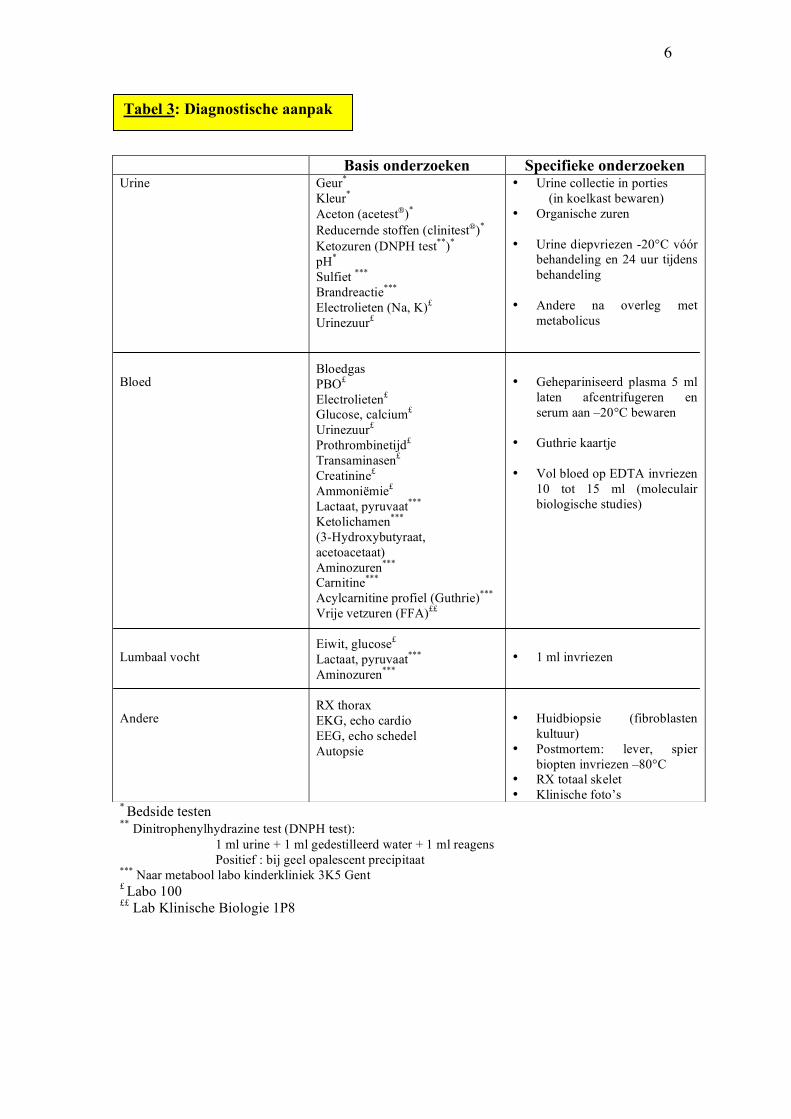
6
* Bedside testen
** Dinitrophenylhydrazine test (DNPH test): 1 ml urine + 1 ml gedestilleerd water + 1 ml reagens
Positief : bij geel opalescent precipitaat *** Naar metabool labo kinderkliniek 3K5 Gent £ Labo 100
££ Lab Klinische Biologie 1P8
Basis onderzoeken Specifieke onderzoeken Urine Bloed Lumbaal vocht Andere
Geur*
Kleur*
Aceton (acetest)*
Reducernde stoffen (clinitest)*
Ketozuren (DNPH test**)*
pH*
Sulfiet ***
Brandreactie***
Electrolieten (Na, K)£ Urinezuur£
Bloedgas PBO£
Electrolieten£
Glucose, calcium£
Urinezuur£
Prothrombinetijd£
Transaminasen£
Creatinine£
Ammoniëmie£
Lactaat, pyruvaat***
Ketolichamen***
(3-Hydroxybutyraat, acetoacetaat) Aminozuren***
Carnitine***
Acylcarnitine profiel (Guthrie)***
Vrije vetzuren (FFA)££
Eiwit, glucose£
Lactaat, pyruvaat***
Aminozuren***
RX thorax EKG, echo cardio EEG, echo schedel Autopsie
• Urine collectie in porties (in koelkast bewaren) • Organische zuren • Urine diepvriezen -20°C vóór
behandeling en 24 uur tijdens behandeling
• Andere na overleg met
metabolicus • Gehepariniseerd plasma 5 ml
laten afcentrifugeren en serum aan –20°C bewaren
• Guthrie kaartje • Vol bloed op EDTA invriezen
10 tot 15 ml (moleculair biologische studies)
• 1 ml invriezen • Huidbiopsie (fibroblasten
kultuur) • Postmortem: lever, spier
biopten invriezen –80°C • RX totaal skelet • Klinische foto’s
Tabel 3: Diagnostische aanpak
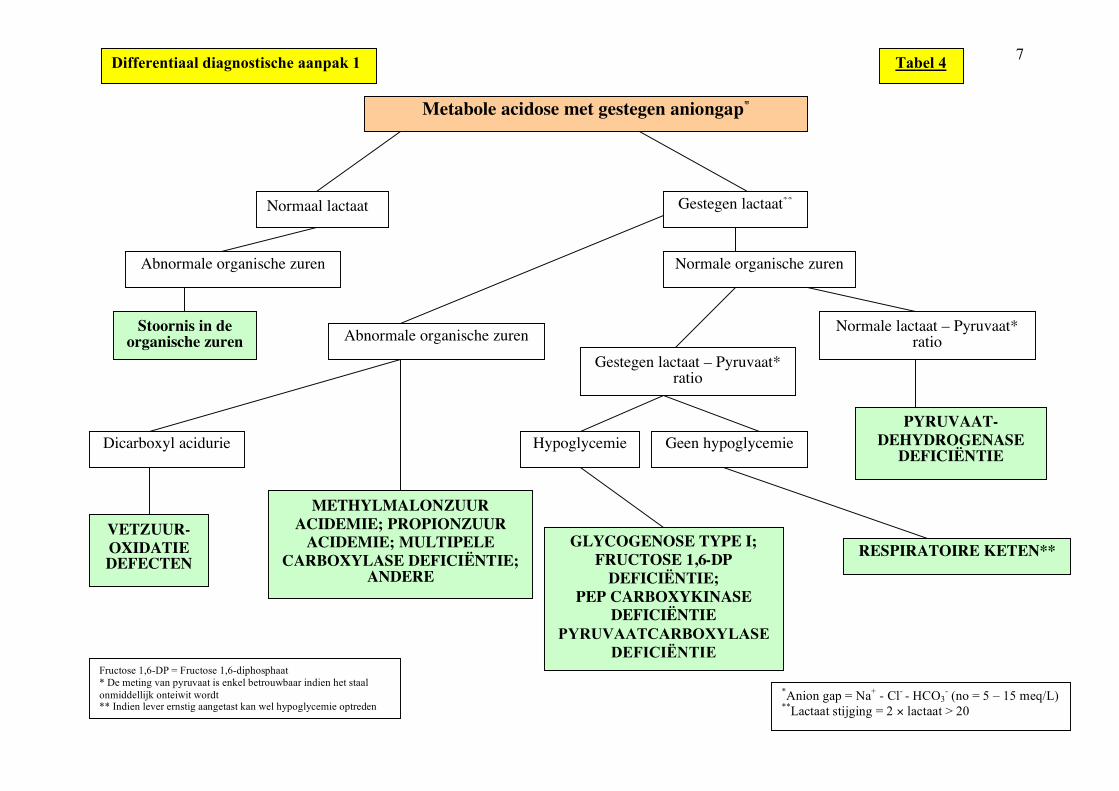
7
Metabole acidose met gestegen aniongap*
Normaal lactaat Gestegen lactaat**
Abnormale organische zuren Normale organische zuren
Stoornis in de organische zuren Abnormale organische zuren
Gestegen lactaat – Pyruvaat* ratio
Normale lactaat – Pyruvaat* ratio
Geen hypoglycemie Hypoglycemie PYRUVAAT-
DEHYDROGENASE DEFICIËNTIE
RESPIRATOIRE KETEN** GLYCOGENOSE TYPE I; FRUCTOSE 1,6-DP
DEFICIËNTIE; PEP CARBOXYKINASE
DEFICIËNTIE PYRUVAATCARBOXYLASE
DEFICIËNTIE
METHYLMALONZUUR ACIDEMIE; PROPIONZUUR
ACIDEMIE; MULTIPELE CARBOXYLASE DEFICIËNTIE;
ANDERE
Dicarboxyl acidurie
VETZUUR-OXIDATIE DEFECTEN
Differentiaal diagnostische aanpak 1
Fructose 1,6-DP = Fructose 1,6-diphosphaat * De meting van pyruvaat is enkel betrouwbaar indien het staal onmiddellijk onteiwit wordt ** Indien lever ernstig aangetast kan wel hypoglycemie optreden
Tabel 4
*Anion gap = Na+ - Cl- - HCO3- (no = 5 – 15 meq/L)
**Lactaat stijging = 2 × lactaat > 20
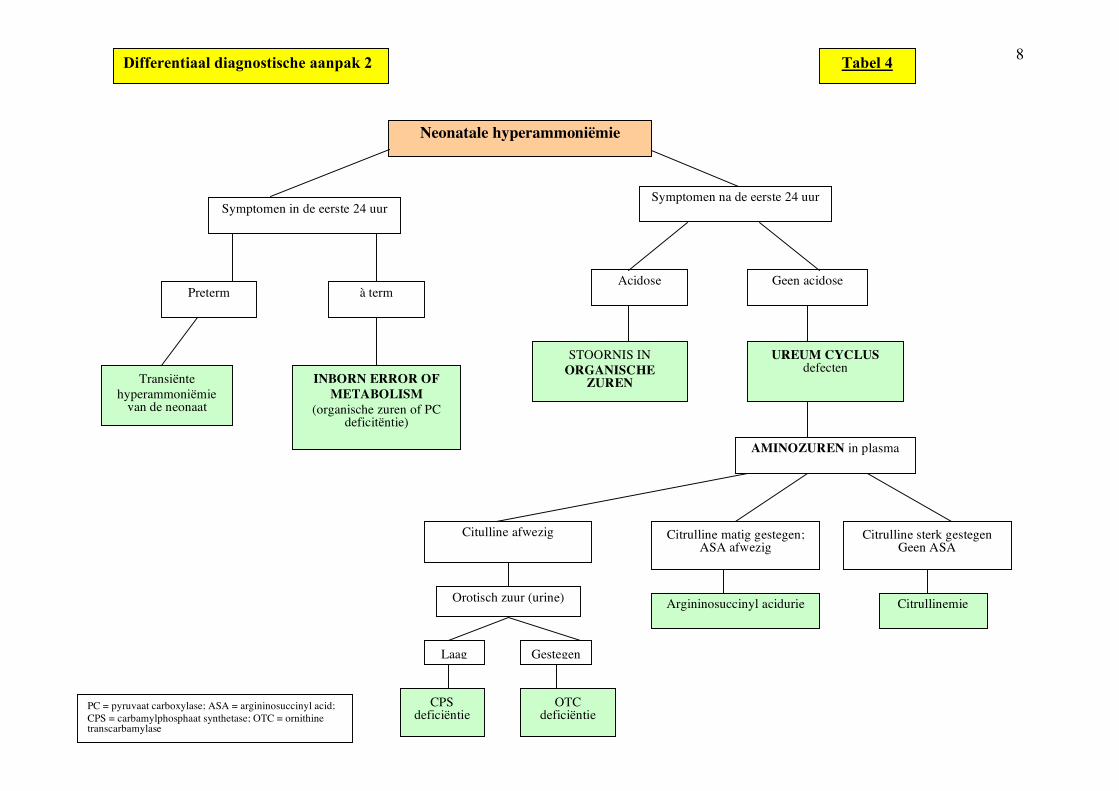
8
Tabel 4
Laag Gestegen
OTC deficiëntie
CPS deficiëntie
Orotisch zuur (urine) Argininosuccinyl acidurie Citrullinemie
Differentiaal diagnostische aanpak 2
Citrulline sterk gestegen Geen ASA
Citrulline matig gestegen; ASA afwezig
Citulline afwezig
AMINOZUREN in plasma
UREUM CYCLUS defecten
STOORNIS IN ORGANISCHE
ZUREN
Acidose Geen acidose
Symptomen na de eerste 24 uur Symptomen in de eerste 24 uur
à term Preterm
Neonatale hyperammoniëmie
INBORN ERROR OF METABOLISM
(organische zuren of PC deficitëntie)
Transiënte hyperammoniëmie
van de neonaat
PC = pyruvaat carboxylase; ASA = argininosuccinyl acid; CPS = carbamylphosphaat synthetase; OTC = ornithine transcarbamylase
Tabel 4
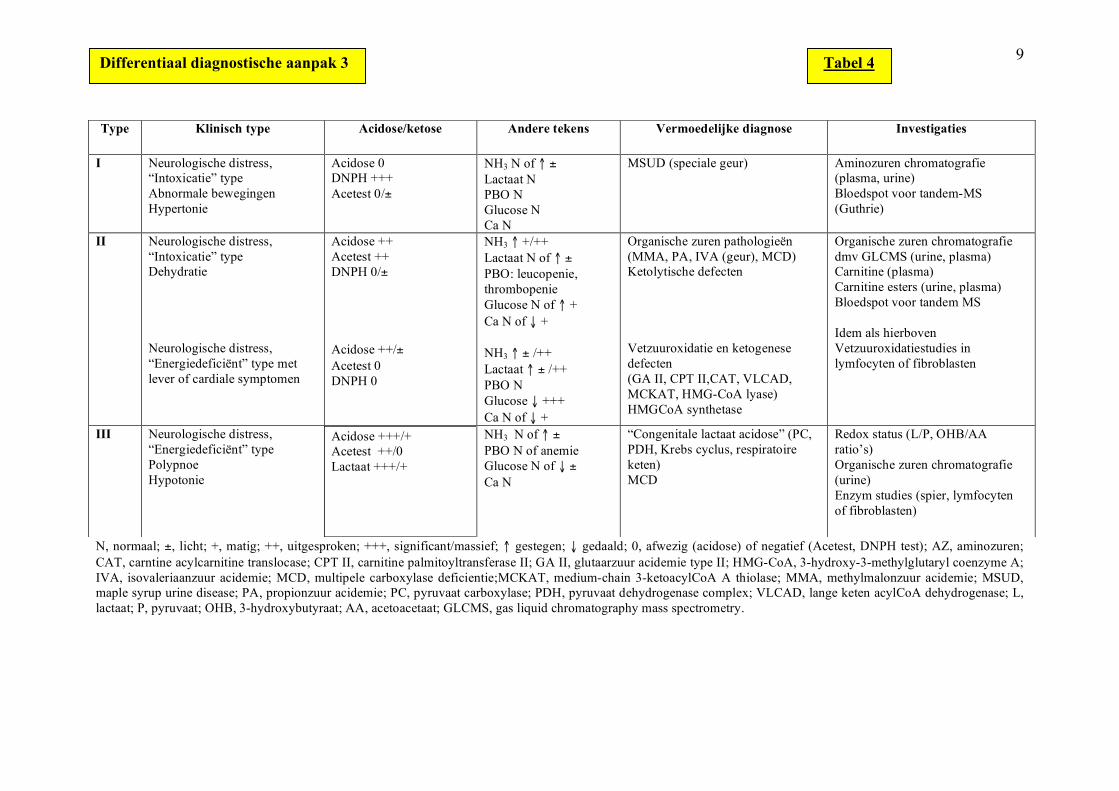
9
N, normaal; ±, licht; +, matig; ++, uitgesproken; +++, significant/massief; ↑ gestegen; ↓ gedaald; 0, afwezig (acidose) of negatief (Acetest, DNPH test); AZ, aminozuren; CAT, carntine acylcarnitine translocase; CPT II, carnitine palmitoyltransferase II; GA II, glutaarzuur acidemie type II; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; IVA, isovaleriaanzuur acidemie; MCD, multipele carboxylase deficientie;MCKAT, medium-chain 3-ketoacylCoA A thiolase; MMA, methylmalonzuur acidemie; MSUD, maple syrup urine disease; PA, propionzuur acidemie; PC, pyruvaat carboxylase; PDH, pyruvaat dehydrogenase complex; VLCAD, lange keten acylCoA dehydrogenase; L, lactaat; P, pyruvaat; OHB, 3-hydroxybutyraat; AA, acetoacetaat; GLCMS, gas liquid chromatography mass spectrometry.
Type Klinisch type
Acidose/ketose Andere tekens Vermoedelijke diagnose Investigaties
I Neurologische distress, “Intoxicatie” type Abnormale bewegingen Hypertonie
Acidose 0 DNPH +++ Acetest 0/±
NH3 N of ↑ ± Lactaat N PBO N Glucose N Ca N
MSUD (speciale geur) Aminozuren chromatografie (plasma, urine) Bloedspot voor tandem-MS (Guthrie)
II Neurologische distress, “Intoxicatie” type Dehydratie Neurologische distress, “Energiedeficiënt” type met lever of cardiale symptomen
Acidose ++ Acetest ++ DNPH 0/± Acidose ++/± Acetest 0 DNPH 0
NH3 ↑ +/++ Lactaat N of ↑ ± PBO: leucopenie, thrombopenie Glucose N of ↑ + Ca N of ↓ + NH3 ↑ ± /++ Lactaat ↑ ± /++ PBO N Glucose ↓ +++ Ca N of ↓ +
Organische zuren pathologieën (MMA, PA, IVA (geur), MCD) Ketolytische defecten Vetzuuroxidatie en ketogenese defecten (GA II, CPT II,CAT, VLCAD, MCKAT, HMG-CoA lyase) HMGCoA synthetase
Organische zuren chromatografie dmv GLCMS (urine, plasma) Carnitine (plasma) Carnitine esters (urine, plasma) Bloedspot voor tandem MS Idem als hierboven Vetzuuroxidatiestudies in lymfocyten of fibroblasten
III Neurologische distress, “Energiedeficiënt” type Polypnoe Hypotonie
Acidose +++/+ Acetest ++/0 Lactaat +++/+
NH3 N of ↑ ± PBO N of anemie Glucose N of ↓ ± Ca N
“Congenitale lactaat acidose” (PC, PDH, Krebs cyclus, respiratoire keten) MCD
Redox status (L/P, OHB/AA ratio’s) Organische zuren chromatografie (urine) Enzym studies (spier, lymfocyten of fibroblasten)
Differentiaal diagnostische aanpak 3 Tabel 4

10
IVa Neurologische distress, “Intoxicatie” type met matige lever symptomen Hypotonie Convulsies, coma
Acidose 0 (alkalose) Acetest 0 DNPH 0
NH3 ↑ +/+++ Lactaat N of ↑ + PBO N Glucose N Ca N
Ureum cyclus Triple H Vetzuur oxydatie defecten (GA II, CPT II, VLCAD, LCHAD, CAT)
AZ (plasma, urine) Orotisch zuur (urine) Lever, darm enzym studies (CPS, OTC)
IVb Neurologische distress Convulsies Myoclonieën Ernstige hypotonie
Acidose 0 Acetest 0 DNPH 0
NH3 N Lactaat N PBO N Glucose N
NKH, SO ± XO Pyridoxine- dependent Peroxisomale ziekten Trifunctioneel enzyme Respiratoire keten Neurotransmittor stoornissen CDG syndroom Cholesterol biosynthese
AZ (NKH, SO) VLCFA, fytaanzuur (plasma) Acylcarnitine profiel (plasma) Lactaat (plasma) Neurotransmittors (plasma, urine, CSF) Geglycolyleerd transferrine (plasma) Cholesterol (plasma)
N, normaal; ±, licht; +, matig; ++, uitgesproken; +++, significant/massief; ↑ gestegen; ↓ gedaald; 0, afwezig (acidose) of negatief (Acetest, DNPH test); AZ, aminozuren; CAT, carntine acylcarnitine translocase ; CPS, carbamylphosphaat synthetase, CPT II, carnitine palmitoyltransferase II; LCHAD, 3-hydroxy lange keten acylCoA dehydrogenase; NKH, niet ketotische hyperglycinemie; OTC, ornithine transcarbamylase; VLCAD, lange keten acylCoA dehydrogenase; VLCFA, zeer lange vetzuren XO, xanthine oxidase.
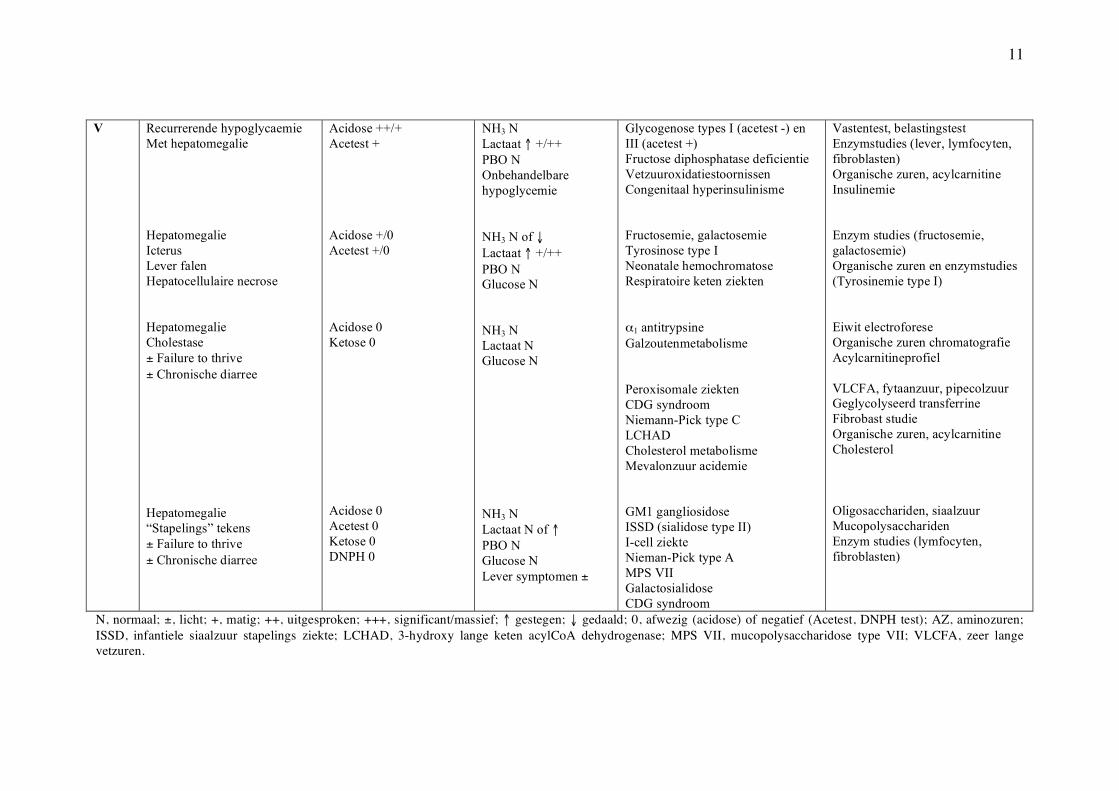
11
V Recurrerende hypoglycaemie Met hepatomegalie Hepatomegalie Icterus Lever falen Hepatocellulaire necrose Hepatomegalie Cholestase ± Failure to thrive ± Chronische diarree Hepatomegalie “Stapelings” tekens ± Failure to thrive ± Chronische diarree
Acidose ++/+ Acetest + Acidose +/0 Acetest +/0 Acidose 0 Ketose 0 Acidose 0 Acetest 0 Ketose 0 DNPH 0
NH3 N Lactaat ↑ +/++ PBO N Onbehandelbare hypoglycemie NH3 N of ↓ Lactaat ↑ +/++ PBO N Glucose N NH3 N Lactaat N Glucose N NH3 N Lactaat N of ↑ PBO N Glucose N Lever symptomen ±
Glycogenose types I (acetest -) en III (acetest +) Fructose diphosphatase deficientie Vetzuuroxidatiestoornissen Congenitaal hyperinsulinisme Fructosemie, galactosemie Tyrosinose type I Neonatale hemochromatose Respiratoire keten ziekten α1 antitrypsine Galzoutenmetabolisme Peroxisomale ziekten CDG syndroom Niemann-Pick type C LCHAD Cholesterol metabolisme Mevalonzuur acidemie GM1 gangliosidose ISSD (sialidose type II) I-cell ziekte Nieman-Pick type A MPS VII Galactosialidose CDG syndroom
Vastentest, belastingstest Enzymstudies (lever, lymfocyten, fibroblasten) Organische zuren, acylcarnitine Insulinemie Enzym studies (fructosemie, galactosemie) Organische zuren en enzymstudies (Tyrosinemie type I) Eiwit electroforese Organische zuren chromatografie Acylcarnitineprofiel VLCFA, fytaanzuur, pipecolzuur Geglycolyseerd transferrine Fibrobast studie Organische zuren, acylcarnitine Cholesterol Oligosacchariden, siaalzuur Mucopolysacchariden Enzym studies (lymfocyten, fibroblasten)
N, normaal; ±, licht; +, matig; ++, uitgesproken; +++, significant/massief; ↑ gestegen; ↓ gedaald; 0, afwezig (acidose) of negatief (Acetest, DNPH test); AZ, aminozuren; ISSD, infantiele siaalzuur stapelings ziekte; LCHAD, 3-hydroxy lange keten acylCoA dehydrogenase; MPS VII, mucopolysaccharidose type VII; VLCFA, zeer lange vetzuren.

12
Initiële aanpak in afwachting van de resultaten
Na diagnosestelling van een erfelijke metabole ziekte van het intoxicatie type,
wordt de therapie gericht op 1) onderdrukken van de productie van toxische
metabolieten door katabolisme van endogene eiwitten en eiwitten uit voedingen 2)
eliminatie van toxische producten stimuleren via extrarenale procedures en specieke
alternatieve wegen, indien deze bestaan. DUS stop voeding en geef toch genoeg
calorieën om anabolisme te bestrijden.
Ondersteunende therapie
De meeste van deze zieke neonaten zullen ventilatoire en circulatoire
ondersteuning nodig hebben. Corrigeer stoornissen in electrolieten, calcium en
phosphor.
Hydratie
Bij metabole decompensatie zal, als gevolg van voedingsmoeilijkheden en
verhoogd renaal vochtverlies, een hypovolemie optreden met prerenale
nierinsufficiëntie. Rehydratie en op peil houden van de vochtbalans is vaak
noodzakelijk. Een efficiënte diurese is belangrijk in situaties met renale eliminatie
van toxische producten en hun bijproducten zoals methylmalonaat, acylcarnitines.
Zuur-base evenwicht
Bij ernstige acidose (pH < 7.15) is intraveneuse correctie met
natriumbicarbonaat (0,5 - 2 mEq/uur of mmol/uur) noodzakelijk. Volg hierbij strikt
de natriëmie om een hypernatriëmie te voorkomen.
Infectie
Bij neonaten in metabole crisis komt sepsis frequent voor. Dit resulteert in een
persisterend katabolisme met therapiefalen. Men zal naar een geassocieerde infectie
zoeken, deze behandelen of voorkomen.
Centraal veneuse lijn
Een centraal veneuse lijn moet voorzien worden om de hoge energie
behoeften te kunnen nakomen.

13
Specifieke therapeutische interventies
1. Voeding
We moeten zorgen voor anabolisme. Indien mogelijk wordt het liefst voor
enterale voeding gekozen. Gezien de voedingsintolerantie en de nood aan invasieve
technieken zullen we vaak totale parenterale nutritie moeten gebruiken. Gebruik
hypertoon glucose (15-20%) en lipiden (2-3 g/kg/dag = C 10 – 15 ml/kg/dag) behalve
als een vetzuuroxidatiestoornis wordt vermoed. Start zo snel mogelijk opnieuw met
eiwit (initieel 0,25-0,5 gr/kg/dag) om aminozuur deficiënties of langdurige negatieve
stikstofbalans te voorkomen. Schakel zo snel mogelijk over naar enterale voeding.
2. Wisseltransfusie / peritoneaal dialyse / continue hemofiltratie / hemodialyse
Wisseltransfusie is absoluut zinloos en gecontraïndiceerd bij de meeste
metabole ziekten zoals organische aciduriën, hyperammoniëmie, ureumcyclus
stoornis of MSUD. Het is absoluut ineffectief, en verslechtert meestal nog de
toestand. Hemodialyse of hemofiltratie, en vooral counter current flow, zijn meest
aangewezen (peritoneaal dialyse is 5-20 maal minder effectief).
Adjuverende therapie
1. Zorg voor anabolisme
Insuline inhibeert de endogene eiwitafbraak en zorgt voor anabolisme. Start
insuline aan lage dosis 0,02 tot 0,05 E/kg/uur.
2. Vitamine therapie
Sommige metabole ziekten reageren op vitamine therapie (vb. Vit B12 bij
methylmalonzuuracidemie, biotine bij propionzuuracidemie, carnitine bij organische
abituriënt, pyridoxine of pyridoxaal fosfaat). Tijdens een metabole crisis is het blind
gebruik van een multivitamine cocktail weinig succesvol. Het is beter de
laboresultaten af te wachten en bij diagnosestelling het specifieke vitamine te
gebruiken.

14
Bij vermoeden van een metabole ziekte blijft de belangrijkste boodschap:
1. Verzorging in een gespecialiseerd neonataal centrum.
2. Vraag advies aan een specialist in metabole ziekten.
Procedure bij overlijden van een neonaat met vermoedelijke metabole ziekte
1. Huidbiopt en weefsels voor enzymatisch onderzoek.
Huidbiopt voor fibroblasten kweek.
Werk in steriele omstandigheden. Voor een huidbiopsie ontsmetten met
alcohol 90°; de huid vastgrijpen met een niet getand krom pincet waarbij 2-3 mm huid
boven de bovenrand uitkomt over een lengte van 4-5 mm; snij met een wegwerp
scalpel de uitstekende huid af langs het pincet; plaats het biopt onmiddellijk en steriel
in het geschikte kultuurmedium∗; in nood in steriel fysiologisch water voor 12 tot
maximaal 24 uur en bewaren op kamertemperatuur.
Weefsels (o.a. lever, skeletspier) onmiddellijk diepvriezen in vloeibare stikstof en
bewaren bij –80°C (diepvries 3K5). In wachtsituatie vloeibare stikstof te verkrijgen
op APD of spoedopname. Stalen in diepvries aan – 20°C en tijdens werkuren zo snel
mogelijk naar op diepvries 3K5 laten brengen.
∗Optimem 1 (Gibco BRL) cataloognr. 31985-047
Foetal Calf Serum (JRH Biosciences) cataloognr. S-00001aP
Ultroser (Gibco BRL) cataloognr. 091-25950
Penicillin-Streptomycin (Gibco BRL) (10000IU/ml) cataloognr. 15140-106
L-Glutamine (Gibco BRL) cataloognr. 25030-032
Kanamycin (Gibco BRL) cataloognr. 15160-021
2-Mercaptoethanol (Gibco BRL) cataloognr. 31350-010
Fungizone (Gibco BRL) cataloognr. 15280-019
Procedure
1. Op aseptische wijze 25 ml FCS, 10 ml Ultroser, 5 ml L-Glutamine, 5 ml Penicillin-Streptomycin, 5 ml
Kanamycin, 0,5 ml 2-Mercaptoethanol en 0,5 ml Fungizone toevoegen aan het Optimem.
2. Mengen zodat alles oplost.

15
2. Huidbiopt en weefsels voor chromosomen en/of DNA onderzoek.
3. Lichaamsvochten voor chemisch onderzoek.
Liefst af te nemen vóór overlijden.
Bloed: 10-20ml op heparine, onmiddellijk afcentrifugeren en plasma
invriezen per 5 ml.
Urine: 20 ml diepvriezen in porties van 5 ml.
Lumbaal vocht: 1-2 ml invriezen.
Bloed en galvochten voor carnitine en acylcarnitines profiel.
4. Foto’s, RX, echografie
5. Volledige autopsie.
